zero valent molybdenum and tungsten ethylene isocyanide complexes: synthesis and structural...
TRANSCRIPT

Po~yhafrm Vol. 7, No. 19/u), pp. 1831-1840, 1988 Printed in Great Britain
0277~5387/88 S3.00+.00 0 1988 Pergamon Ptms plc
ZERO VALENT MOLYBDENUM AND TUNGSTEN ETHYLENE ISOCYANIDE COMPLEXES: SYNTHESIS AND
STRUCTURAL PROPERTIES*
ERNEST0 CARMONA,? AGUSTIN GALINDO and JOSE M. MARIN
Departamento de Quimica Inorganica-Instituto de Ciencia de Materiales, Facultad de Quimica, Universidad de Sevilla-CSIC, Apartado 553,41071 Sevilla, Spain
and
ENRIQUE GUTIERREZ, ANGELES MONGE and CARIDAD RUIZ
Instituto de Quimica Inorganica Elhtiyar y Departamento de Quirnica Inorganica, Universidad Complutense de Madrid-CSIC, 28040 Madrid, Spain
Abstract--Compounds of composition trans,mer-[M(C,H,),(CNR)(PMe3)3] (1) and truns,trans,trans-[M(C2H4)2(CNR)2(PMe3)2] (2) for various combinations of M = MO, W and R = CHMe*, CMe, and C6H 1 ,, have been obtained by the stepwise reaction of truns- w(C2H&(PMeS)4] with the corresponding isocyanide. Compounds 2 can also be obtained by treatment of 1 with CNR, a reaction that allows the synthesis of the mixed isocyanide truns,trans-[Mo(CZH4),(CNCMe,)(CNC,HL ,)(PMe,),] (2g). A similar reaction, starting with traqmer-[M(C,H,),(CO)(PMe&], produces the mixed isocyanide-carbonyl deriva- tives trans,trans-[M(C,H,),(CNR)(CO)(PMe3),1(3). X-ray structural studies on complexes 2b and 3b (M = MO, R = CMe,) have been carried out. Both compounds are monoclinic, P *,,“, but while the structure of 3b has been refined to a conventional R value of 0.034 by using 2976 observed reflections (unit cell constants: a = 10.972(3), b = 18.923(4), c = 10.544(3) A; #I = 98.89(2)“) ex ensive disorder problems have prevented anisotropic t refinement of 2b, and a final isotropic R value of 0.097 has been obtained. For compound 3b, the Mo-C bond distances are 1.959(5) (Ma-CO), 2.137(4) (Mo-CNR) and 2.283(9) A (av. Mo-C,H,).
The field of isocyanide complexes of transition metals has considerably developed over the past few years.’ A number of studies aimed at the synthesis of new species,’ their structural characterization3 and the ascertaining of their bonding capability, in particular the comparison of their u-donor and rr- acceptor properties with those of carbonyl and car- bene ligands,4 has been reported recently. Earlier studies on molybdenum and tungsten isocyan- ide chemistry allowed the preparation of mixed phosphine-isocyanide complexes, e.g. tram-[M (CNR)2(dppe)2]S” (dppe = Ph2CH2CH2PPh2) and
*Dedicated to Professor Sir Geoffrey Wilkinson on the occasion of his retirement.
t Author to whom correspondence should be addressed.
[M(CNR),(PMe2Ph),_,,lSb (n = 2, 3, 4), and of phosphinecarbonyl-isocyanide compounds [MO (CO)2(CNR)2(PR’~)2].6 Using the ethylene com- plexes trans-[M(C2H4),(PMe,)J7 as starting ma- terials, we have now enlarged the range of com- plexes of this type. Herein we report the synthesis of the mixed ethylene-isocyanid+phosphine com- pounds trurqmer-[M(C2H4),(CNR)(PMe,),] (1) and truns,truns,truns-[M(C2H4)2(CNR)2(PMe3)2] (2), as well as that of the mixed ethylene-carbonyl- isocyanide-phosphine species, truns,truns-[M(C, H4)2(CO)(CNR)(PMe,)2] (3). The reactions studied are shown in Scheme 1 and spectroscopic data for the new compounds in Tables 1 and 2. X-ray struc- tural analyses on two of these complexes, 2b and 3b (M = MO, R = CMe,), are also reported.
1831

1832 E. CARMONA et al.
i( RNC-M-CNR
-
z 2CNR
\ 4,
CNR
t
-
\ co
2 atm
co -
/ 2-3atm
co 2-3 atm
4P 4
OC-M-CNR
=: CzH,
P : PMe3 M: Mo,W
R : 'Pr,'Bu,Cy,CH1Ph
Scheme 1.
RESULTS AND DISCUSSION
Compounds of composition trans,mer-[M(C,H&
(CNWPMedd (1)
The ethylene complexes trans-[M(C2H,& (PMe,),] (M = MO, W) have been shown to undergo a facile substitution of one of the co- ordinated PMe3 groups by carbon monoxide, with formation’ of the carbonyl derivatives trans,mer-p(C2H.,),(CO)(PMe,),]. A similar reac- tion has now been found to take place with iso- cyanides. Low temperature addition of an alkyl isocyanide, CNR, to petroleum ether solutions of truns-[M(C2HJ2(PMe&] produces compounds of composition [M(C,H,),(CNR)(PMe&] (l), (M = MO ; R = CHMe,, la, CMe,, lb. M = W ; R = CMe,, lc), as shown in eq. (1).
-+ [M(C2H4)2(CNR)(PMe3)31+PMe3. (1)
Other combinations of M = MO, W and R = CHMe2, CMe3, CbHll have been attempted, and although the reactions seem to proceed similarly, only in the above cases have crystalline products been isolated. Compounds 1 are air-sen- sitive solids, very soluble in common organic sol- vents from which they can only be crystallized in
very concentrated solutions. Their IR spectra dis- play a strong, broad absorption in the range 1990- 1960 cm- ‘, which can be attributed to vcN of the coordinated isocyanide ligand. This value is lower by ca 15&180 cm- ’ than that of the free ligands, and this suggests an appreciable n-bonding inter- action with the metal centre. The shift to low fre- quency is not however so pronounced as in the compounds trans-[M(CNR)z(dppe)z]5a and truns- [M(CNR),(PMe,Ph),lsb (Av N 215385), but it is much higher than that found in the related carbon dioxide complexes, truqmer-[Mo(CO&(CNR) (PMe,),] which display values’ of vcN very simi- lar to those of the free isocyanide ligands (e.g. 2100 cm-’ for R = CMe3, Av N 35 cm-‘). This is in accord with a larger contribution of reson- ance structure II in compounds 1 than in the
/R M-C--N-R- M-C=%
I II
CO2 derivatives, which can, in turn, be inter- preted in terms of a higher electron density at the metal centre in the “Mo(CzH&(PMe&” core than in the “Mo(C0J,(PMe3)3” core, in agreement with the strong x-acceptor capability attributed to the side-on coordinated carbon dioxide ligand. 9
Zero valent MO and W ethylene isocyanide complexes
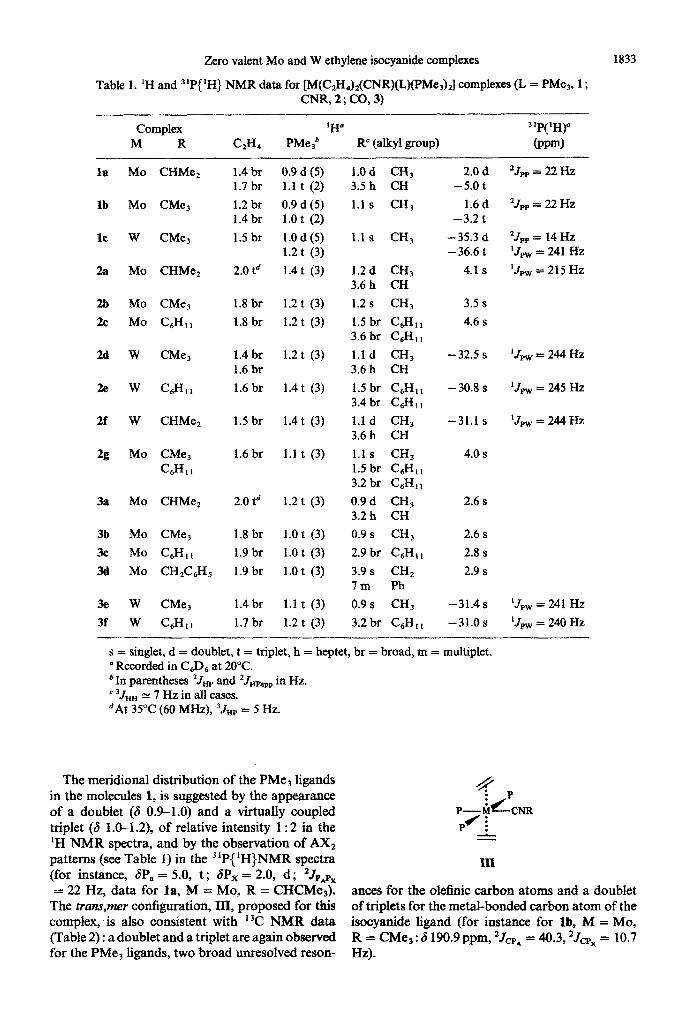
Table 1. ‘H and 31F(1H) NMR data for [M(C,H,)&NR)(L)(PMe,)J complexes (L = PMe3, 1; CNR, 2 ; CO, 3)
1833
Complex 1 d H 3’P( ‘H)” M R CzHd PMe3 R’ (apry gtoup) @pm)
0.9 d (5) 1.1 t (2)
0.9 d (5) 1.0t (2)
l.Od(5) 1.2 t (3)
1.4 t (3)
1.2 t (3)
1.2 t (3)
1.2 t (3)
1.4t (3)
1.4 t (3)
I.1 t (3)
1.2 t (3)
1.0t (3)
1.0 t (3)
1.0t (3)
1.1 t (3)
1.2 t (3)
1.0d CH3 3.5 h CH
1.1 s CH,
1.1 s CH,
1.2d CH3 3.6 h CH
1.2 s CH3
1.5 br C&I,, 3.4 br C&I1 1
1.1 d CH, 3.6 h CH
1.5 br CsHll 3.4 br C,H, 1
1.1 d CH, 3.6 h CH
1.1 s CH3 1.5 br CsHll 3.2 br CsHl,
0.9d CH, 3.2 h CH
0.9 s CH,,
2.9 br &HI,
3.9 s CH2 7m Ph
0.9 s CH3
3.2 br C&H,,
2.0 d -5.0 t
1.6d -3.2 t
-35.3 d - 36.6 t
4.1 s
‘Jpp = 22 Hz
2Jpp = 22 Hz
‘Jpp = 14 Hz
‘JPW = 241 Hz
‘Jpw = 215 Hz
3.5 s
4.6 s
-32.5 s
- 30.8 s
-31.1 s
r&v = 244Hz
‘Jpw = 245 Hz
‘Jpw = 244 Hz
4.0 s
2.6 s
2.6 s
2.8 s
2.9 s
-31.4 s ‘JPW =241Hz
-31.0 s ‘Jpw = 240 Hz
la MO CHMe* 1.4 br 1.7 br
1.2 br 1.4 br
1.5 br
2.0 td
1.8 br
1.8 br
1.4 br 1.6 br
1.6 br
1.5 br
1.6 br
2.0 P
1.8 br
1.9 br
1.9 br
1.4 br
1.7 br
lb MO CMe,
1C W CMe,
238 MO CHMe,
2b MO CMe3
2c MO GHII
2d
W GH,, 2e
W CHMe,
CMe3
&HI I
3a MO CHMez
3h MO
362 MO
3d MO
3e
3f
W CMe3
W GHLi
s = singlet, d = doublet, t = triplet, h = beptet, br = broad, m = multiplet. “Recorded in C6D6 at 20°C. ‘In parentheses 2Jwp and *Jmapp in Hz. ’ 3JHH N 7 Hz in all cases. ‘At 35°C (60 MHz), 3JHp = 5 Hz.
The meridional distribution of the PMe3 ligands in the molecules 1, is suggested by the appearance of a doublet (6 09-1.0) and a virtually coupled triplet (6 l&-1.2), of relative intensity 1:2 in the ‘H NMR spectra, and by the observation of AX2 patterns (see Table 1) in the “P~‘H~NMR spectra (for instance, 6P, = 5.0, t ; SP, = 2.0, d; *Jpnpx = 22 Hz, data for la, M = MO, R = CHCMe,).
The mns,mer contiguration, III, proposed for this complex, is also consistent with ’ 3C NMR data (Table 2) : a doublet and a triplet are again observed for the PMe, ligands, two broad unresolved reson-
antes for the olefinic carbon atoms and a doublet of triplets for the metal-bonded carbon atom of the isocyanide ligand (for instance for lb, M = MO, R = CMe, : 6 190.9 ppm, ‘JcpA = 40.3, 2Jcpx = 10.7
Hz).
1834 E. CARMONA et al.
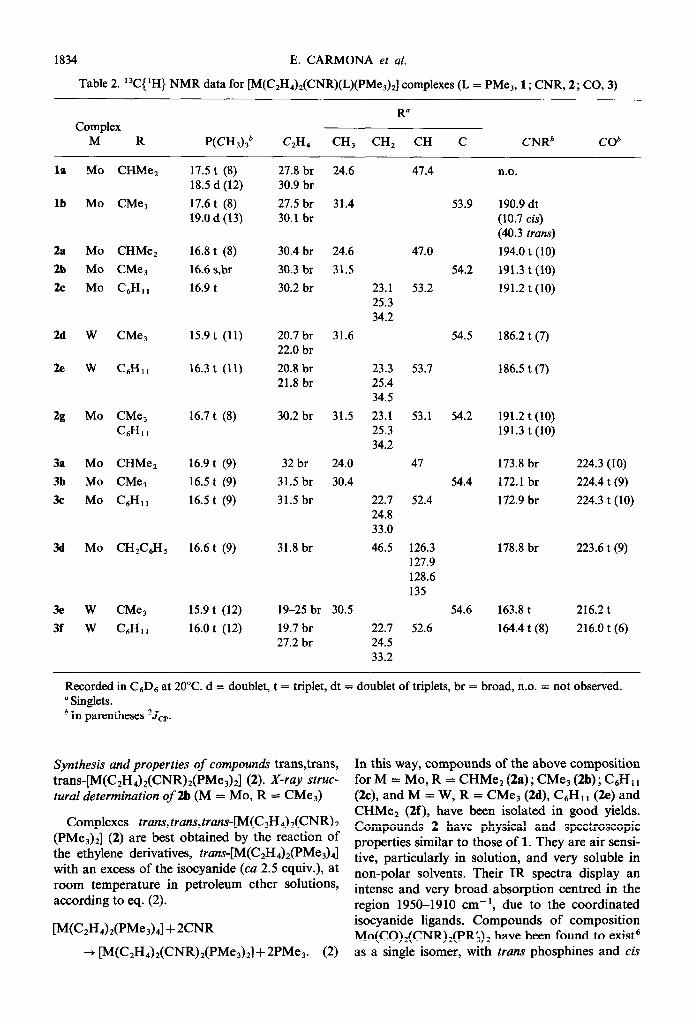
Table 2. 13C{ ‘H) NMR data for [M(C,HJ,(CNR)(L)(PMeJJ complexes (L = PMe,, 1; CNR, 2; CO, 3)
R” Complex
M R P(CH3)36 CzH., CH, CH, CH C CNRb cob
la
lb
MO CHMe,
MO CMe,
2a MO CHMe,
2b MO CMe,
2c Mo GH,,
2d
2e
W CMe,
W C,HII
2g
3a
3b
3c
MO CMe,
CsH,,
MO CHMe,
MO CMe,
Mo &HI,
3d
3e
3f
MO CH&H,
W CMe, 15.9 t (12)
W C,H,, 16.0 t (12)
17.5 t (8) 18.5 d (12)
17.6 t (8) 19.0 d (13)
16.8 t (8)
16.6 s,br
16.9 t
15.9 t (11)
16.3 t (11)
16.7 t (8)
16.9 t (9)
16.5 t (9)
16.5 t (9)
16.6 t (9)
27.8 br 30.9 br
27.5 br 30.1 br
30.4 br
30.3 br
30.2 br
20.7 br 22.0 br
20.8 br 21.8 br
30.2 br
32 br
31.5 br
31.5 br
31.8 br
19-25 br
19.7 br 27.2 br
24.6 47.4 n.0.
31.4 53.9
24.6 47.0
31.5 54.2
23.1 53.2 25.3 34.2
31.6 54.5
23.3 53.7 25.4 34.5
31.5 23.1 53.1 54.2 25.3 34.2
24.0 47
30.4 54.4
22.7 52.4 24.8 33.0
46.5 126.3 127.9 128.6 135
30.5 54.6
22.7 52.6 24.5 33.2
190.9 dt (10.7 cis) (40.3 trum)
194.0 t (10)
191.3 t (10)
191.2 t (10)
186.2 t (7)
186.5 t (7)
191.2 t (10) 191.3 t (10)
173.8 br
172.1 br
172.9 br
178.8 br
224.3 (10)
224.4 t (9)
224.3 t (10)
223.6 t (9)
163.8 t 216.2 t
164.4 t (8) 216.0 t (6)
Recorded in CsDs at 20°C. d = doublet, t = triplet, dt = doublet of triplets, br = broad, n.o. = not observed. a Singlets. b In parentheses *Jcp.
Synthesis and properties of compounds trans,trans, trans-[M(C,H,),(CNR),(PMe&j (2). X-ray struc- tural determination of 2b (M = MO, R = CMe,)
Complexes trans,trans,trans-[M(CzH&(CNR), (PMe,)J (2) are best obtained by the reaction of the ethylene derivatives, trans-[M(C,H&(PMe&] with an excess of the isocyanide (ca 2.5 equiv.), at room temperature in petroleum ether solutions, according to eq. (2).
l?W&>~PMe3>41 +XNR
--, [M(C2H4)*(CNR)2(PMe3)21+2PMe3. (2)
In this way, compounds of the above composition for M = MO, R = CHMe* (2a) ; CMe, (2b) ; C6H1, (2c), and M = W, R = CMe3 (2d), C6H1, (2e) and CHMe, (2f), have been isolated in good yields. Compounds 2 have physical and spectroscopic properties similar to those of 1. They are air sensi- tive, particularly in solution, and very soluble in non-polar solvents. Their IR spectra display an intense and very broad absorption centred in the region 1950-19 10 cm- ‘, due to the coordinated isocyanide ligands. Compounds of composition Mo(CO),(CNR),(PR& have been found to exist6 as a single isomer, with trans phosphines and cis

Zero valent MO and W ethylene isocyanide complexes 1835
carbonyl and alkyl isocyanide ligands. Chemical or electrochemical oxidation of these species is fol- lowed by a rapid isomerization to the all trans cations, but re-reduction produces the original iso- mers. For the analogous aryl isocyanide derivatives, a mixture of isomers is generally observed.6 Inter- estingly, compounds 2 seem to exist as single iso- mers with trans phosphine, ethylene and isocyanide ligands, and this is clearly inferred from spec- troscopic data (Tables 1 and 2) and from an X- ray study carried out on complex 2b (M = MO, R = CMe3). The ‘H NMR spectra of complexes 2 show, in addition to signals due to the coordinated CNR groups, a virtually coupled triplet for the PMe, ligands and a broad, unresolved signal for the ethylene protons, indicating restricted rotation around the M-olefin bond axis. For compound 2a rotation is rapid at room temperature and a triplet is observed at 6 1.8 ppm (3Ji.ip = 5 Hz). While 31P NMR data do not provide useful information on the geometrical distribution of the PMe3 ligands in these complexes, additional evidence for the trans arrangement of the two RNC and the two PMe, groups comes from i3C NMR studies. In addition to the resonances associated with the phosphine and ethylene groups, which are listed in Table 2, a triplet is observed at 6 191-194 ppm due to the metal-bonded carbon atom of the CNR ligands. This and the values of ca 7-10 Hz found for the carbon to phosphorus coupling constant in these complexes, which are typical of cis C-P coupling, ’ O are clearly indicative of an all trans distribution of the ethylene, isocyanide and phosphine groups, as shown in IV. To further confirm this proposal, an
< :dP
RNC-M-CNR p('
I -
IV
X-ray study of the molybdenum complex, 2b has been undertaken. The structure has been resolved by Patterson and Fourier techniques (see Experi- mental), but due to extensive disorder problems it could not be refined anisotropically. Nevertheless, the positions of all but the ethylene atoms could be fixed without ambiguity, although the values obtained for the bond distances and angles should be regarded and used with caution when attempting to establish precise comparisons. The molecule is centrosymmetric, with the molybdenum atom at the (O,O,O) inversion centre, coordinated to two mutu- ally trans phosphines, at a Mo-P distance of 2.450(6) A (av.), to two trans isocyanide ligands,
with a Ma-C separation of 2.04(3) A (av.), and to two trans ethylene groups whose location cannot be precisely determined, as already indicated. By comparison with the structures found for other related ethylene complexes, e.g. trans#4(C2H&
U’Med41,7 trans-mer-[Mo(C,H&(CO)(PMe3)31’ and trans,trans-[Mo(C2H&(CO)(CNCMe3) (PMe,),] (uide infia), a trans, mutually perpen- dicular distribution can be proposed for the ethylene ligands, as shown schematically in IV.
Compounds 2 can also be obtained by the reac- tions of precursors 1 with the corresponding iso- cyanide [eq. (3)]
[M(C,H,),(CNR)(PMe3),I + CNR
+ [M(C,H,)2(CNR),(PMe3),1+PMe3. (3)
If the second isocyanide, CNR’, is different from the one already coordinated, mixed complexes [Mo(C,H&(CNR)(CNR’)o,l can be ob- tained. As an example of species of this type, the compound [Mo(CJI&(CNCMe3)(CNC6H, ,) (PMe,),] (2g) has been obtained. For 2g a singlet at 4.0 ppm is observed in the 3’P{ ‘H} NMR spec- trum, and two triplets at 191.2 and 191.3 ppm (Mo-CNR) in the 13C{‘H) NMR spectrum (‘Jcr = 10 Hz), in excellent agreement with a structure of type IV.
Compounds trans,trans-[M(C,H,),(CO)(CNR) (PMe,),] (3). Preparation and properties and the molecular structure of 3b (M = MO, R = CMe3)
Previous work from this laboratory7b has shown that the complexes trans,mer-[M(C2HJZ(CO) (PMe,),] react with carbon monoxide under pressure (2-3 atm), with formation of the bis(car- bonyl) derivatives trans,trans,trans-[M(C,H,),(C0)2 (PMe,)J. A similar reaction with the isoelectronic isocyanide ligand provides the mixed carbonyl- isocyanide derivatives, traqtrans-w(C,H,),(CO) (CNR)(PMe,),] (M = MO, R = CHMe,, 3a;
CMe3, 3b; C6Hl ,, 3~; CH2C6Hs, 3d. M = W, R = CMe,, 3e ; C6H, i, 3f), as depicted in eq. (4).
[M(C2H32(Co)(PMe3)31 + CNR
-+ [M(C2Hd2(Co>(CNR)(PMe,),l+PMe3. (4)
Compounds 3 are pale-yellow or white-yellowish crystalline materials, somewhat more stable in air than 1 and 2, and also very soluble in common organic solvents. Preparation of these derivatives can also be achieved from compounds 1 and CO under pressure [eq. (5)], but yields are generally lower than those obtained following the route speci-

1836 E. CARMONA et al.
fied in eq. (4).
-+ ~(C,H,),(Co)(CNR)(PMe,),l+PMe3. (5)
The formation of all these compounds and their interconversion reactions are summarized in Scheme 1.
Examination of the IR spectra of complexes 3 shows that they all display a strong absorption in the region 1870-I 830 cn- i, due to vco of the carbon monoxide ligand, and a second intense band at 21 IO-2070 cm- ’ associated with the isocyanide ligand which occupies a position tram with respect to the carbonyl. The former appears almost at the same frequency as that in the complexes, trans,mer-[M(C,H,),(CO)(PMe3)3] (1870, M = MO ; 1855 cm- ‘, M = W), and this seems to indicate that the major role in the withdrawing of rr-electron density from the metal centre in these compounds is being played by the carbonyl ligand, with little participation of the isocyanide group, which would, at best, behave as a very weak x- acid ligand. This is in agreement with the previous observations by other workers,4a who have found that the bonding of the CNR ligand in carbonyl isocyanide carbene complexes of tungsten is domi- nated by the 0 component, with the CNR ligand acting as a stronger Lewis base than CO and as a much weaker n-acid. Also in accord with this, is the high value found for vCN in these complexes, with very low shifts with respect to the free ligands
Fig. 1.
(Av N 80 cm- I), as compared with shifts of ca 150- 200 cm- ’ found for compounds 1 and 2. It is clear from these arguments, that the M-CNR bond in 3 is predominantly c in character, with little or no contribution of a-back donation, i.e. with a larger contribution of resonance structure I, and this is also substantiated by the long Mo-CNR bond distance found for complex 3b and by the almost linear C-N-C angle of the coordinated iso- cyanide ligand (see below) in this complex.
The all tram structure proposed for compounds 3 is supported by NMR data (Tables 1 and 2). Thus, they all exhibit a singlet in the 31P{ ‘H} NMR spectrum and a triplet in the ‘H NMR spectrum for the coordinated phosphine ligands. The 13C res- onance of the carbonyl ligand appears also as a triplet (2Jrc = 6-10 Hz) while the metal bonded carbon atom of the isocyanide group provides a broad, unresolved signal in the molybdenum derivatives and a triplet (‘Jcp = 8 Hz) in the tung- sten compounds. This is clearly in agreement with structure V.
g :dP
OCTCNR - - V
Figure 1 shows an ORTEP illustration of com- plex 3b, relevant bond distances and angles are col-
Molecular structure and atom labelling scheme for truns,trans-wo(C,H4),(CO)(CNCMe3)
PMeAl W-4.

Zero valent MO and W ethylene isocyanide complexes 1837
lected in Table 3. The complex is essentially octa- [Mo(C2H&(PMe&] and truns,mer-[Mo(C2H& hedral, with the ethylene ligands mutually (CO)(PMe,),]. The strong interaction between the perpendicular, each eclipsing the truns P-MO-P metal centre and the carbonyl group, previously or C-Me-C vectors of the plane perpendicular alluded to, is also shown by the short Ma-C to the Mo-C2H4 bond axis. This orientation, first (carbonyl) distance of 1.959(5) A, which is shorter predicted by Osbom in the complex fruns- by ca 0.18 A than the Ma-C (isocyanide)
PWC&h(&wGl” and then supported by bond length. The latter distance of 2.137(4) & theoretical calculations,‘* is similar to that found is appreciably longer than that found in in other related ethylene complexes.’ The MO-C [Mo(~$-C~H~P(M~)C,H,)(CNCM~,)(PM~P~~)~]~~ and the C-C bond distances for the MO-&H, (1.984(13) A) d an in trans,mer-[Mo(CO&(CNR)
units average 2.283(9) and 1.412(5) A, respectively, (PMe,),]* (2.07(2) A, R = CHMe2; 2.03(l) & R
and compare well with the values found’ in trans- = CHzC6HS). Finally, the C6NX7 bond angle
Table 3. Selected bond distances (A) and angles (“) for truns,trum-[Mo(C,H& (CWCNCMeXMGl (W
Bond distances
Mo-P( 1) Mo-P(2)
Ma-C(l) Ma--C(2) Ma-C(3) Ma-C(4) Ma--C(5) Ma-c(6)
2.486( 1) 0-w) 1.167(6) 2.470(2) N<(6) 1.153(6) 1.959(5) N--c(7) l&1(6) 2.262(S) c(2w(3) 1.416(7) 2.264(5) C(4)--c(5) 1.407(7) 2.294(5) 2.310(5) 2.137(4)
Angles
P(l)-Mo-P(2)
P(l)-Ma-C(l) P(l)--Ma--c(2) P(l)-Mo-C(3) P(l)-Ma-C(4) P(l)-Ma--c(5) P(l)-Mo-c(6) P(2)--Ma--C(l) P(2)-Mo-C(2) P(2)-Mo-c(3) P(2)-Mo-c(4) P(2k-Mo--c(5) P(2)-Mo-C(6) C(l)_Mo-C(2) C(l)_Mo--C(3) C(l)_Mo--C(4) C(l)-Mo-CXs) C(l)-Mo-c(6) c(2)-Mo-c(3) ~(2)-Mo--~(4) c(2>-Mo-c(5) C(2I-Mo--c(6)
171.1(l) 92.9(2) 76.4( 1)
112.9(l) 85.7(l) 86.4(l) 88.3(l) 91.2(2)
111.5(l) 75.1(l) 85.6(l) 87.3(l) 89.1(l) 88.8(2) 87.5(2)
108.3(2) 72.7(2)
169.5(2) 36.5(2)
155.9(2) 154.2(2) 81.4(2)
C(3)-Mo-C(4) ‘J3)_Mo-(J5) C(3)-Ma--c(6) C(4)-Mo-C(5) C(4)-Me--C(6) C(5)-Mo-c(6)
Mo-P(l)-C(ll) 116/t(2) Mo-P(l)-C(12) 117.3(2) Mo-P(ljC(13) 121.2(2)
c(ll>-P(l)--c(l2) 101.6(3)
C(l l)-P(l)-c(l3) 98.6(2)
c(l2>-P(lw(l3) 98.0(3)
Mo-P(2)-C( 14) Mo-P(2)-C( 15) Mo-P(2)-C( 16)
C(l4>-P(2w(l5) C(l4)-P(2w(16) C(l5)_P(2~(16)
(X6)-N-c(7) Mo-C(l+O Mo+2w(3) Mo--C(3w(2) Mo-C(4-C(5) Mo--C(5w(4) Ma-C(6)-N
155.4(2) 153.4(2) 82.5(2) 35.6(2) 82.2(2)
117.7(2)
116.7(2) 117.8(2) 120.1(2) 100.8(3) 98.7(3) 99.0(3)
176.1(5) 178.9(5) 71.8(3) 71.7(3) 72.8(3) 71.6(3)
174.2(4)

1838 E. CARMONA et al.
of 176.1(S)“, almost linear, is in accord with the considerations outlined above concerning the contribution of resonance form II to the total structure of these complexes.
EXPERIMENTAL
Microanalyses were carried out by Pascher Microanalytical Laboratory, Germany. IR spectra were recorded on a Perkin-Ehner model 684 instru- ment. ‘H, 3’P and ’ 3C NMR spectra were run on a Varian XL-200 instrument. 3’P NMR shifts were measured with respect to external 85% H3P04_ ‘II and 13C NMR spectra were referenced using the ‘H and 13C resonance, respectively, of the sol- vent as an internal standard but are reported with respect to SiMe4.
All preparations and other operations were car- ried out under oxygen-free nitrogen, following con- ventional Schlenk techniques. Solvents were dried and degassed before use. The petroleum ether used had b.p. 40-6O”C. The compounds, trcdns-
~(C~H~)*~Me3)~ and ~~~,~~-~(C*H~)*(CO) (PMe,),]? and the ligand PMe313 were prepared by pub~shed methods. Isocyanides were from commercial sources or prepared by standard methods. ’ 4
Synthesis of trans,mer-[M(C,H,J,(CNR)(PMe,),] complexes, (1)
Into a solution of trans-[M(CzH4),(PMe3)J (1 mmol), in petroleum ether (50 cm3), at - 2O”C, the corresponding isocyanide (1 mmol) is added. The reaction mixture is stirred and allowed to reach room t~~rat~e in about 20 min. The stirring is continued for another 4-5 h and the solvent then removed in uacuo. The yellow residue is extracted with petroleum ether (15-20 cm3), centrifuged and concentrated. Very concentrated solutions are required to crystallize these complexes. Upon cooling at -20°C yellow crystals of the desired compounds are obtained. Yields 70-75%. Complex la (M = MO, R = CHMeJ : IR (Nujol) : vCN 1960 s, br cm-’ . Complex lb (M = MO, R = CMe,) : IR (Nujol): vcN 1990 s, br cm-‘. Found: C, 46.4; H, 9.4. Calc. for C, 8H44P3NM~ : C, 46.6 ; H, 9.5. Complex lc (M = W, R = CMe,) : IR ~ujol) : VCN 1990 s, br cm-‘.
Synthesis of trans,trans,trans-[M(CzH4),(CNR), (PMe,)J complexes, (2)
A solution of truns-[M(C,H,),(PMe,),] (ca 1 mmol) in 20-25 cm3 of petroleum ether is treated with an excess (I : 2.5) of the isocyanide, CNR
(R = CHMe2, CMe,, C6H, *), at room temperature with stirring. After 15-20 h (or ca 30 h for M = W) the resulting solution is evaporated to dryness. Extraction with 20 cm3 of petroleum ether and cen- trifugation gives a yellow solution, which upon con- centration and cooling at -20°C provides yellow crystals of the corresponding complexes. Yields are between 70% (complexes 2c and 2e) and 85% (com- plex 2a). Complex 2a (M = MO, R = CHMe*) : IR (Nujol) : vcN 1930 s (very broad) cm- ‘. Found : C, 48.4;H,8.9.Calc.forC,~H~~P~N~Mo:~,48.9;H, 9.1. Complex 2b (M = MO, R = CMe3) : IR (Nujol) : VCN 1930 s (very broad) cm-‘. Found : C, 51.4; H, 9.6. Calc. for C20H44P2N2M~: C, 51.1; H, 9.4. Complex 2c (M = MO, R = C6H1i): IR (Nujol) : vCN 1950 s (very broad) cm- ‘. Found : C, 55.2; H, 9.3. Calc. for C2,H4,P,N,Mo: C, 55.2; H, 9.2. Complex 2d (M = W, R = CMe3) : IR (Nujol) : vCN 1930 s (very broad) cm-‘. Found: C, 43.1; H, 8.1. Calc. for C20H44P2N2W: C, 43.0; H, 7.9. Complex 2e (M = W, R = C6H1 ,) : IR (Nujol) : vCN 1940 s (very broad) cm- ’ . Found : C, 47.5 ; H, 7.9. Calc. for C24H48PZN2W : C, 47.2 ; H, 7.9. Complex 2f (M = W, R = CHMe*): IR (Nujol): vcN 1910 s (very broad) cm- ‘.
Preparation of trans,trans-[Mo(C,H4)2(CNCMe3)
@NC&I I JPMe3hl CW
Complex lb (0.46 g, 1 mmol), dissolved in pet- roleum ether (25 cm3), is reacted with a two-fold excess of CNC6H 11 (2 mmol) at room temperature for 20 h. The resulting solution is evaporated to dryness and the residue extracted with 20 cm3 of petroleum ether. Cent~ugation, partial evapor- ation of the solvent and cooling at -20°C affords yellow crystals of the compounds. Yield 68%. IR (Nujol) : vcN 1950 s (very broad) cm- ‘. Found : C, 52.6 ; H, 9.1. Calc. for C22H46P2NZM~ : C, 53.2 ; H, 9.3.
Synthesis of trans,trans-[M(C,H4)2(CO)(CNR) (PMe 3) ;I complexes, (3)
An excess of the isocyanide (2 mmol) is added to a solution of the complexes, rrans-[M(CzH4)z (CO)(PMe3)3] (M = MO, W) (cu I mmol) in pet- roleum ether (25 cm3). The mixture is stirred at room temperature for 16-20 h (for the tungsten compounds 30-35 h are required). The volatile materials are removed by pumping in vucuo and the residue extracted with petroleum ether (20 cm3). The solution is centrifuged, concentrated and cooled at - 20°C. White or whiteyellowish crystals are obtained in 80-90% yield (complex 3d in 65% yield). Complexes 3 can also be obtained by reac-

Zero valent MO and W ethylene isocyanide complexes 1839
tion of compounds 1 with CO under pressure (3- 4 atm), but the yields are generally lower and the reaction times longer. Complex 3a (M = MO, R = CHMeJ : IR (Nujol) : vCN 2070 s, vco 1840 s cn-‘. Found: C, 45.9 ; H, 8.3. Calc. for C15H33PZNOM~: C, 44.9; H, 8.2. Complex 3b (M = MO, R = CMe,): IR (Nujol): vcN 2100 s, vco 1830 cm-‘. Found : C, 46.4 ; H, 8.6. Calc. for C,,H,,P,NOMo: C, 46.3; H, 8.4. Complex 3e (M = MO, R = C6H1 1) : IR (Nujol) : vCN 2120 s, vco 1870 s cm- I. Found : C, 48.9 ; H, 8.6. Calc. for CL8H37P2NOM~: C, 49.0; H, 8.4. Complex 3d (M = MO, R = CH2CbH5) : IR (Nujol) : vCN 2110 s, vco 1840 s cm-‘. Found: C, 51.1; H, 7.4. Calc. for Ci9H,,P,NOMo : C, 50.8 ; H, 7.4. Complex 3e (M = W, R = CMe,) : IR (Nujol) : vCN 2100 s, vco 1870 s cm-‘. Found: C, 38.3; H, 7.2. Calc. for C16H35P2NOW: C, 38.2; H, 7.0. Complex 3f (M = W, R = CsHl ,) : IR (Nujol) : vCN 2110 s, vco 1850 s cm- I. Found : C, 40.8 ; H, 7.1. Calc. for C18H37P2NOW: C, 40.8; H, 7.0.
Crystallographic studies
Yellow prismatic single crystals of tranqtrans, trans-[Mo(C,H&(CNCMe&(PMe& (2b) and trans,tr~-~o(C,H,),(CO)(~CMe3) (3V were introduced into a Lyndeman capillaries, sealed in an N2 atmosphere and mounted on a Nonius CAD-4 diffractometer.
Crystal data. Compound 2b : C2,,H+,PZN2Mo, A4 = 470.47, monoclinic, space group P2,/n, a = 11.182(4), b = 13.210(9), c = 10.312(3) A, /I = 116&l(3)“, U = 1364.1 A’,2 = 2, D, = 1.14gcm3, &MO-&) = 5.9 cn- ‘, &MO-&) = 0.71069 A.
Crystal data. Compound 3b : C, 6H35P,NOMo, M = 415.35, monoclinic, space group P2, n, a = 10.972(3), b = 18.923(4), c = 10.544(3) 8, B= 98.89(2)“, U = 2189.2 A3, 2 = 4, D, = 1.28 g’cm3, ~(Mo-K,) = 7.38 cm’, ~(Mo-KJ = 0.71069 A.
Data collection. Unit cell dimensions were deter- mined by least-squares refinement of the observed setting angles of 25 accurately centred reflections. Intensity data collection employed the w-28 scan technique. Measured data: compound 2b, 2392 reflections and compound 3b, 3798 reflections (28 < SO”). The data were corrected for Lorentz and polarization effects. Scattering factors for neutral atoms and anomalous dispersion corrections for MO and P were taken from International Tables
for X-ray Crystallography. l5
*Atomic coordinates have also been deposited with the Cambridge Crystallographic Data Centre.
Structure solution and rejinements. Compound 2b : The structure was solved by conventional Pat- terson and Fourier techniques, but due to extensive disorder problems it could not be refined aniso- tropically. Because of this, after checking the space group again, the refinement has been carried out in an isotropic mode. An empirical absorption cor- rection16 was applied using unit weights. The final residual R value was 0.097. Nevertheless, the posi- tion of all, but the ethylene atoms, could be fixed with clarity, although the values obtained for their bond distances and angles should be regarded with some caution in order to establish precise com- parisons. Compound 3b : The structure was solved by conventional Patterson and Fourier techniques. An empirical absorption correction, l6 applied at the end of the isotropic refinement using unit weights, led to a conventional R value of 0.060. After mixed full matrix least-squares refinement with isotropic thermal parameters for C(8), C(9) and C( lo), minimizing Zo((F,( - IF,l)* gave an R value of 0.047. A difference synthesis calculated with reflec- tions having a sin f?/n < 0.5 A-’ showed all H atoms as the highest peaks of the map. Final refinement with fixed isotropic temperature factors for H atoms and unit weights led to R = 0.034. Most of the calculations were performed with XRAY80. I7
For both complexes bond distances and angles, final atomic coordinates, tables of thermal par- ameters and listing of observed and calculated structure factors have been deposited with the Editor as supplementary material.*
Acknowledgement-We are grateful to the Comision AX- sora de Investigation Cientifica y T&mica for support of this work (EC. and E.G.).
1.
2.
3.
4.
5.
REFERENCES
P. M. Treichel, Adv. Organomet. Chem. 1973,11,21; E. Singleton and H. Oosthuizen, Ado. Organomet. Chem. 1983,22,209; Y. Yamamoto, Coord. Chem. Rev. 1980,32, 193. L. B. Kool, M. D. Rausch, M. Herberhold, H. G. Alt, V. Thewalt and B. Honold, Organometallics 1986, 4, 2465 ; L. B. Anderson, T. J. Barder, F. A. Cotton, K. R. Dumbar, L. R. Falvello and R. A. Walton, Znorg. Chem. 1986,25,3629. (a) W. D. Jones, G. P. Foster and J. M. Putinas, Znorg. Chem. 1987,26,2120; (b) R. L. Luck, R. H. Morris and J. F. Sawyer, Organometallics 1984, 3, 247. (a) M. P. Guy, J. T. Guy and D. W. Bennett, Organo- metalfics 1986,5, 1696; (b) D. B. Beach, R. Barton- cello, G. Granozzi and W. L. Jolly, Organo- metallics 1985,4,3 11. (a) J. Chatt, C. M. Elson, A. J. L. Pombeiro, R. L.

1840 E. CARMONA et al.
Richards and G. H. D. Royston, J. Gem. Sot., Dalton Trans. 1978, 165 ; (b) A. J. L. Pombeiro, J. Chatt and R. L. Richards, J. Organometal. Chem. 1980,190,297.
6. J. C. Deaton and R. A. Walton, J. Organometal. Chem. 1981, 219, 187; K. A. Conner and R. A. Walton, Znorg. Chem. 1986,25,4422.
7. (a) E. Carmona, J. M. Marin, M. L. Poveda, J. L. Atwood and R. D. Rogers, J. Am. Chem. Sot. 1983, 105,3014; (b) E. Carmona, A. Galindo, M. L. Poveda and R. D. Rogers, Znorg. Chem. 1985,24,4033.
8. R. Alvarez, E. Carmona, J. M. Marin, M. L. Poveda, E. GutiCrrez-Puebla and A. Monge, J. Am. Chem. Sot. 1986,108,2286.
9. C. Mealli, R. Hoffmann and A. Stockis, Znorg. Chem. 1984,23, 56; S. Sakaki, K. Kitaura and K. Moro- kuma, Znorg. Chem. 1982,21, 760.
10. B. E. Mann and B. F. Taylor, “C NMR Data for Organometallic Compounds (Edited by P. M. Maitlis,
F. G. A. Stone and R. West), p. 23. Academic Press, New York (1981).
11. J. W. Byrne, H. U. Blaser and J. A. Osbom, J. Am. Chem. Sot. 1975,97,3871.
12. T. A. Albright, R. Hoffmann, J. C. Thibeault and D. L. Thorn, J. Am. Chem. Sot. 1979,101,3801; C. Bachman, J. Demynck and A. Veillard, J. Am. Chem. Sac. 1978,100,2366.
13. W. Wolfsberger and H. Schmidbaur, Synth. React. Znorg. Met. Org. Chem. 1974,4, 149.
14. G. W. Gokel, R. P. Widera and W. P. Weber, Org. Synth. 1976, 55, 96.
15. International Tables for X-ray Crystallography, Vol. IV, pp. 72-98. Kynoch Press, Birmingham (1974).
16. N. Walker and D. Stuart, Acta Cryst. 1983, A39, 158.
17. J. M. Stewart, The XRAY80 System. Computer Science Center, University of Maryland, College Park (1985).