wellcome trust workshop working with pathogen genomes module 2 gene prediction
TRANSCRIPT

Wellcome Trust Workshop
Working with Pathogen Genomes
Module 2 Gene Prediction
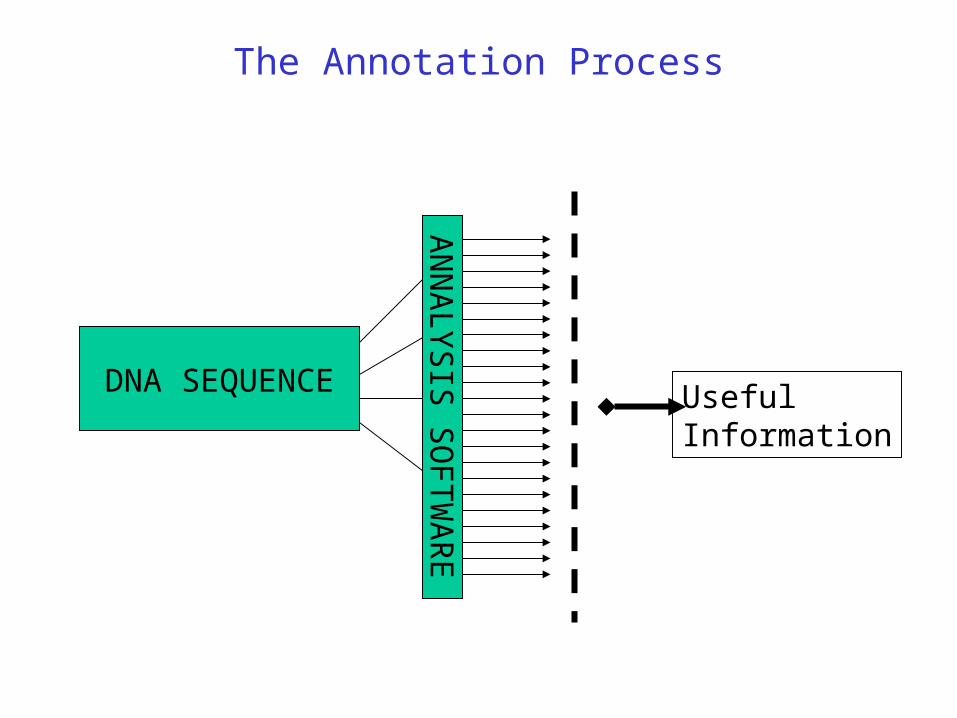
The Annotation Process
DNA SEQUENCE
AN
NA
LY
SIS
SO
FT
WA
RE
UsefulInformation
Annotator
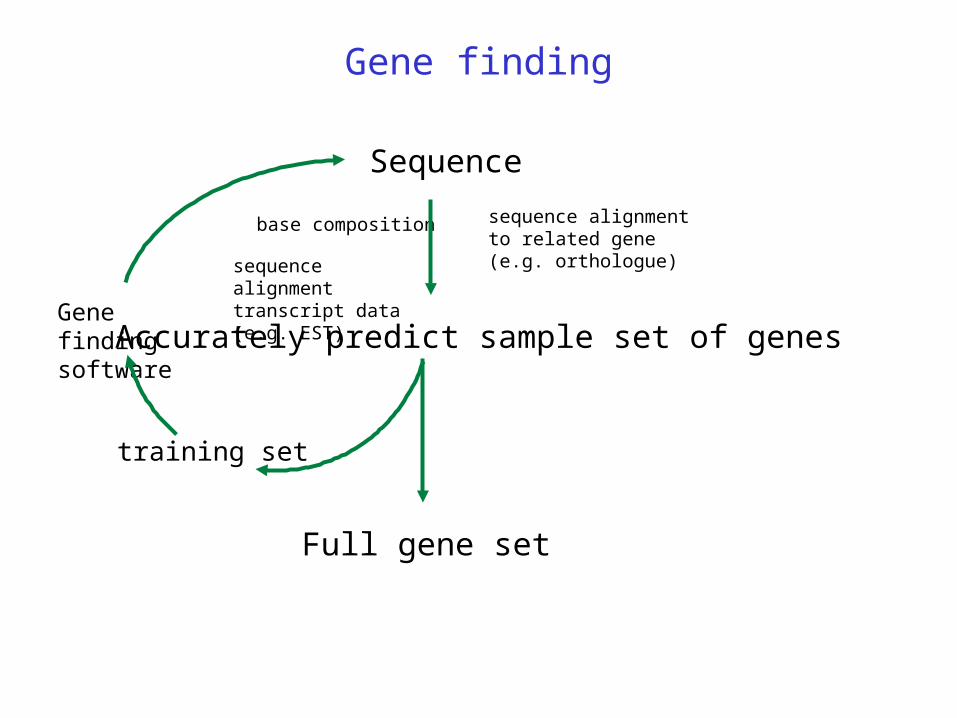
Gene finding
Accurately predict sample set of genes
Sequence
base composition sequence alignment to related gene (e.g. orthologue) sequence alignment
transcript data (e.g. EST)
training set
Gene finding software
Full gene set
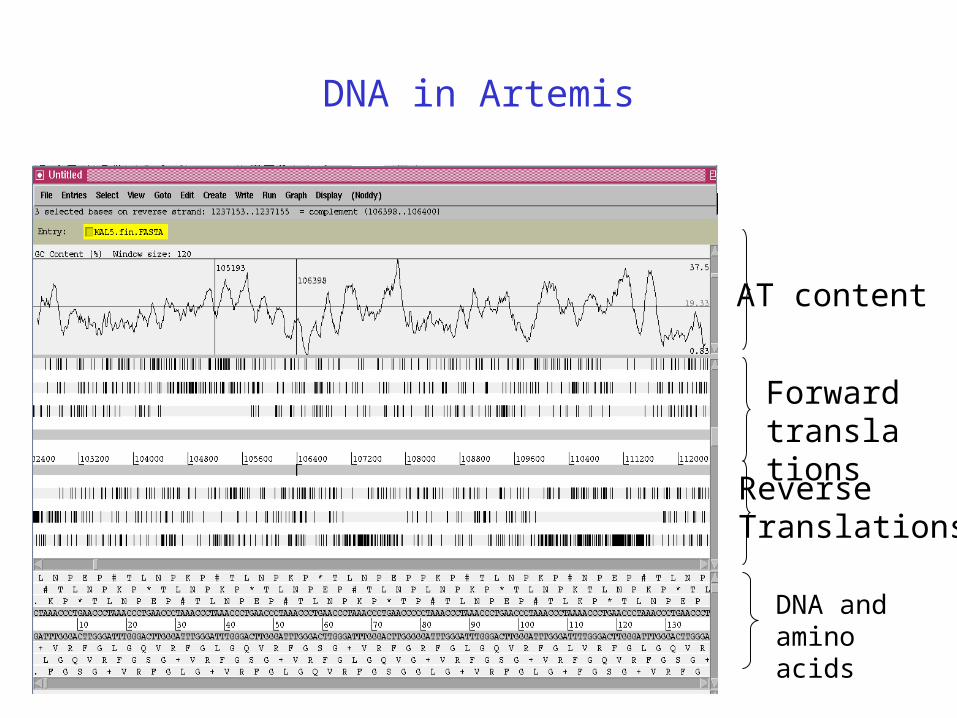
AT content
Forward translations
Reverse Translations
DNA and aminoacids
DNA in Artemis

Gene prediction programs:ORFs and CDSs
ORFs are not equivalent to CDSs
Not all open reading frames are coding sequences

GC content
• Coding regions have higher GC content in AT-rich genomes
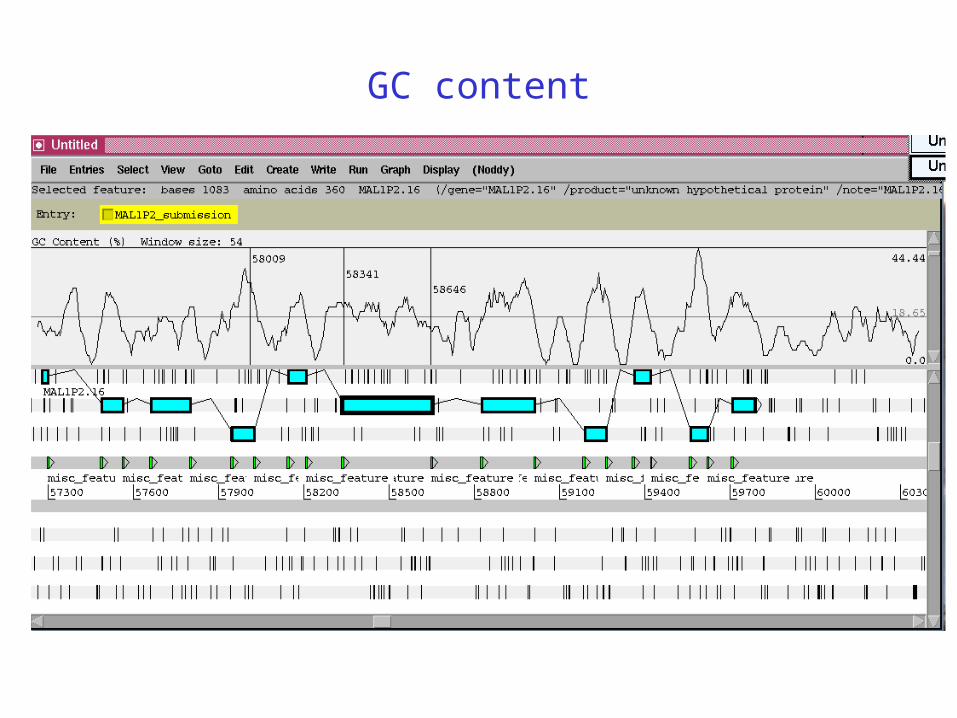
GC content

CODON USAGE
• Codon bias is different for each organism.• DNA content in coding regions is restricted
– but it is not restricted in non coding regions.
• The codon usage for any particular gene can influence expression.

Codon usage
• All organisms have a preferred set of codons.
Malaria TrypanosomaGUU 0.41 GUU 0.28
GUC 0.06 GUC 0.19
GUA 0.42 GUA 0.14 GUG 0.11 GUG 0.39
Codon Usage
• http://www.kazusa.or.jp/codon/
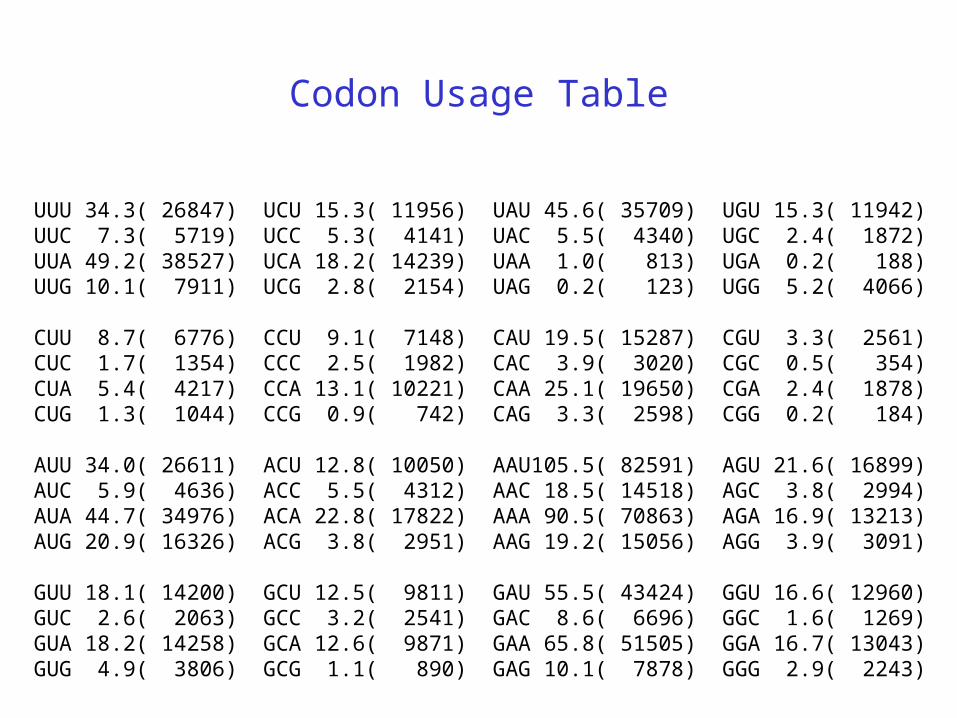
Codon Usage Table
UUU 34.3( 26847) UCU 15.3( 11956) UAU 45.6( 35709) UGU 15.3( 11942)UUC 7.3( 5719) UCC 5.3( 4141) UAC 5.5( 4340) UGC 2.4( 1872)UUA 49.2( 38527) UCA 18.2( 14239) UAA 1.0( 813) UGA 0.2( 188)UUG 10.1( 7911) UCG 2.8( 2154) UAG 0.2( 123) UGG 5.2( 4066)
CUU 8.7( 6776) CCU 9.1( 7148) CAU 19.5( 15287) CGU 3.3( 2561)CUC 1.7( 1354) CCC 2.5( 1982) CAC 3.9( 3020) CGC 0.5( 354)CUA 5.4( 4217) CCA 13.1( 10221) CAA 25.1( 19650) CGA 2.4( 1878)CUG 1.3( 1044) CCG 0.9( 742) CAG 3.3( 2598) CGG 0.2( 184)
AUU 34.0( 26611) ACU 12.8( 10050) AAU105.5( 82591) AGU 21.6( 16899)AUC 5.9( 4636) ACC 5.5( 4312) AAC 18.5( 14518) AGC 3.8( 2994)AUA 44.7( 34976) ACA 22.8( 17822) AAA 90.5( 70863) AGA 16.9( 13213)AUG 20.9( 16326) ACG 3.8( 2951) AAG 19.2( 15056) AGG 3.9( 3091)
GUU 18.1( 14200) GCU 12.5( 9811) GAU 55.5( 43424) GGU 16.6( 12960)GUC 2.6( 2063) GCC 3.2( 2541) GAC 8.6( 6696) GGC 1.6( 1269)GUA 18.2( 14258) GCA 12.6( 9871) GAA 65.8( 51505) GGA 16.7( 13043)GUG 4.9( 3806) GCG 1.1( 890) GAG 10.1( 7878) GGG 2.9( 2243)

Codon Usage in Artemis
Forward frames
Reverseframes

Gene prediction: Amino acid usage: Correlation scores
Within each window, plots correlation between amino acid usage in window and global amino-acid usage in EMBL
“Magic number” = 52.7
Arbitrary units

Gene prediction: Correlation scores
M. tuberculosis NADH dehydrogenase operon

Gene prediction: Positional base preference (FramePlot)
Plots the GC content in each position of each reading frame of the DNA sequence.
In G+C-rich organisms the GC content of the 3rd base is often higher; in A+T rich organisms it is lower.
Good prediction of coding in malaria and trypanosomes and G+C-rich prokaryotes.
G+C content of chromosome
Frame-specific G+C content
1
2
3


Genefinding programs
• Genefinding software packages use Hidden Markov Models.• Predict coding, intergenic and intron sequences• Need to be trained on a specific organism.• Never perfect!

What is an HMM
• A statistical model that represents a gene.• Similar to a “weight matrix” but one that can recognise gaps and
treat them in a systematic way.• Has a different “states” that represent introns, exons, intergenic
regions, etc• Considers the “state” of preceding sequence

A typical HMM
http://linkage.rockefeller.edu/wli/gene/krogh98.pdf

Gene prediction programs: Problems
• ORFs are not equivalent to CDSs• Gene prediction programs find new genes that share properties
with a given set of genes. • They can be confounded by:
– Sequence constraints (ribosomal proteins etc.)
– Sequence biases
– Sequence quality
– Different sets of genes
– Horizontal gene transfer
– Non-coding DNA

Gene prediction programs: ProblemsSequence composition variation
Y. pestis ribosomal proteins
glimmer
orpheus
final

Gene prediction programs: Problems
Non-protein coding regions: S. typhi ribosomal RNA genes
glimmer
genefinder
final
orpheus
glimmer
genefinder
final
orpheus
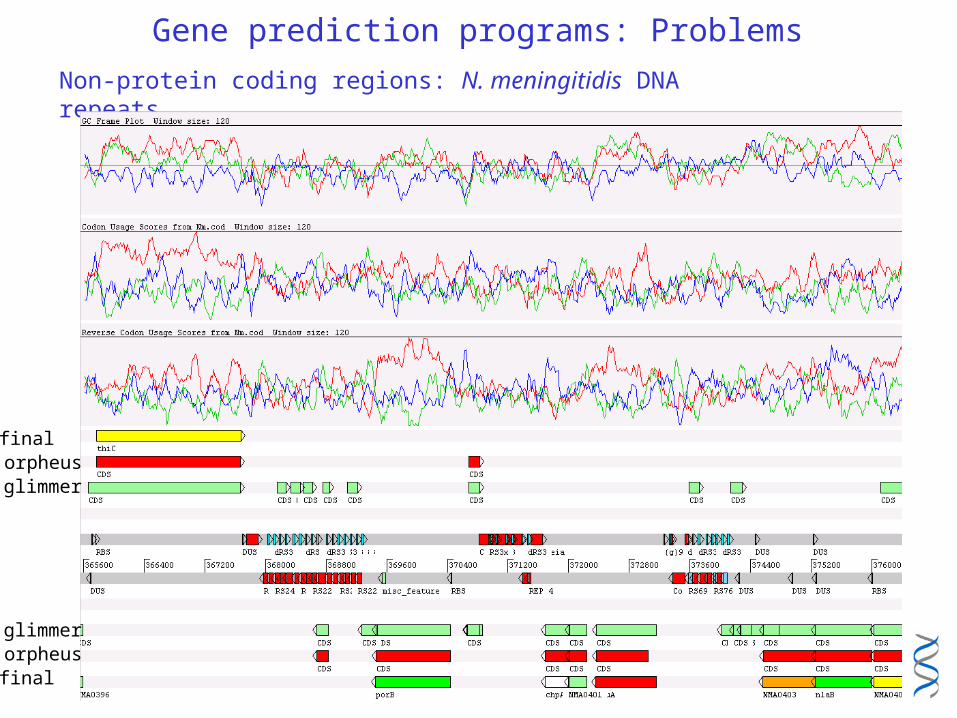
Gene prediction programs: Problems
Non-protein coding regions: N. meningitidis DNA repeats
glimmerorpheusfinal
glimmerorpheusfinal
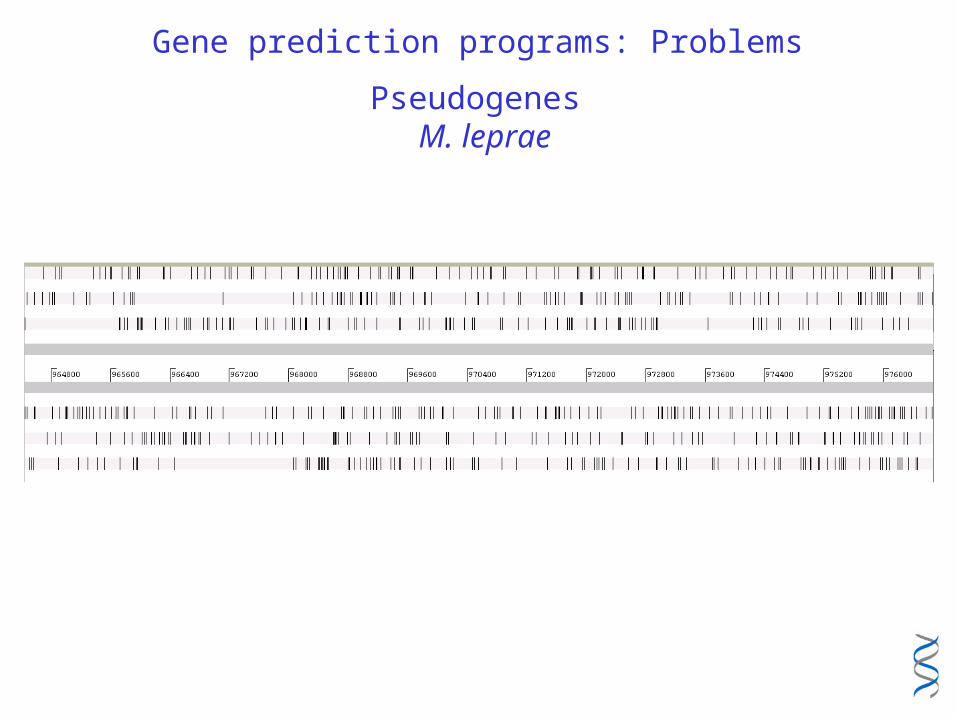
Gene prediction programs: Problems
Pseudogenes M. leprae

Gene prediction programs: Problems
Pseudogenes: M. leprae
Glimmer

Gene prediction programs: Problems
Pseudogenes: M. lepraePseudogenes: M. leprae
ORPHEUS
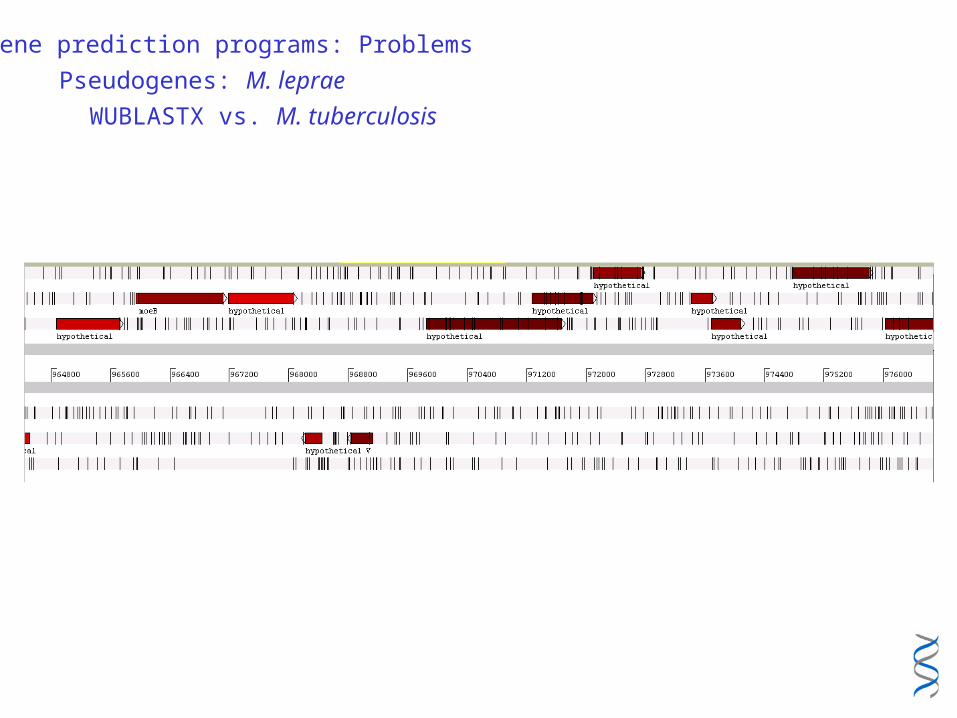
Gene prediction programs: Problems
Pseudogenes: M. leprae
WUBLASTX vs. M. tuberculosis

Gene prediction programs: Problems
Pseudogenes: M. leprae
Final annotation
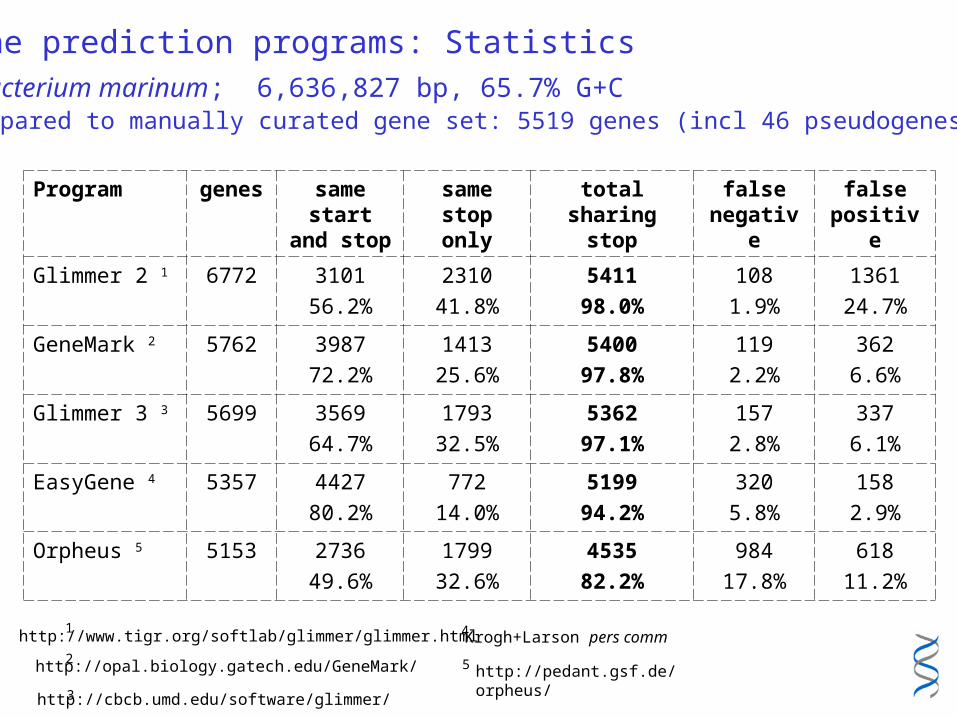
Gene prediction programs: Statistics
Krogh+Larson pers comm
5 http://pedant.gsf.de/orpheus/
3
http://www.tigr.org/softlab/glimmer/glimmer.html1
Program genes same start and stop
same stop only
total sharing stop
false negative
false positive
Glimmer 2 1 6772 3101
56.2%
2310
41.8%
5411
98.0%
108
1.9%
1361
24.7%
GeneMark 2 5762 3987
72.2%
1413
25.6%
5400
97.8%
119
2.2%
362
6.6%
Glimmer 3 3 5699 3569
64.7%
1793
32.5%
5362
97.1%
157
2.8%
337
6.1%
EasyGene 4 5357 4427
80.2%
772
14.0%
5199
94.2%
320
5.8%
158
2.9%
Orpheus 5 5153 2736
49.6%
1799
32.6%
4535
82.2%
984
17.8%
618
11.2%
Mycobacterium marinum; 6,636,827 bp, 65.7% G+C compared to manually curated gene set: 5519 genes (incl 46 pseudogenes)
http://cbcb.umd.edu/software/glimmer/
4
2 http://opal.biology.gatech.edu/GeneMark/

Gene prediction programs: Problems
splicing Plasmodium falciparum
Original annotation
Updated annotation

Homology Data
• Coding regions are more conserved than non coding regions due to selective pressure.
• Comparing all possible translations against all known proteins will give clues to known genes.
• Blastx

BLASTX

Blastx on frame lines

EST sequencing
AAAAAAAAAACAP
AAAAAAAAAACAP
TTTTTTTTT
TTTTTTTTT
intron exon5’UTR Mstop 3’UTR
EST
EST
cDNA
mRNA

ESTs
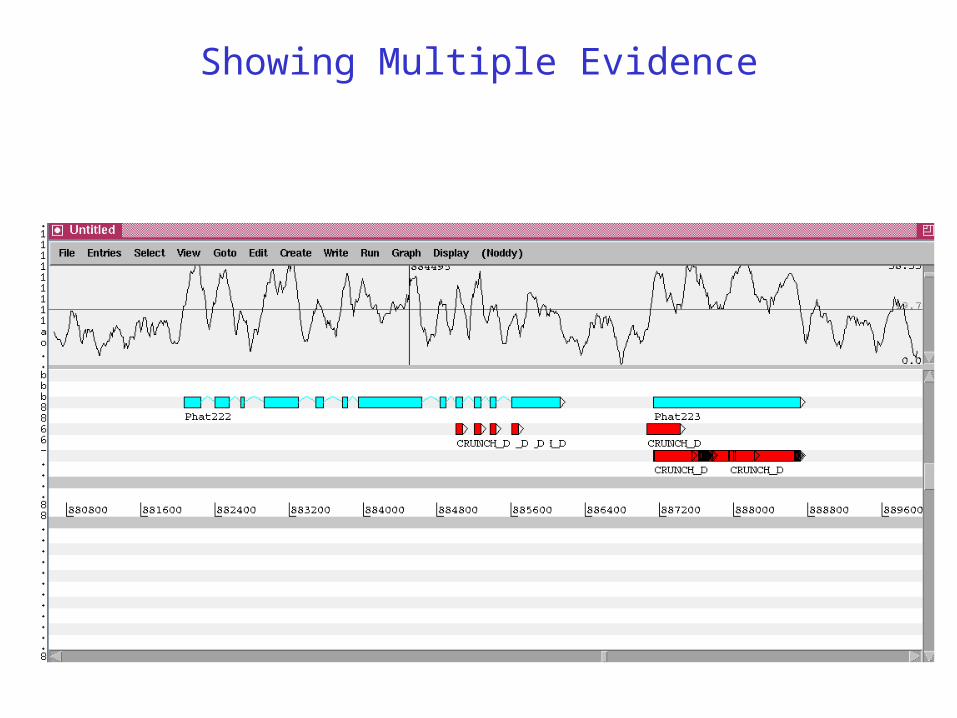
Showing Multiple Evidence

Schistosoma mansoni expression
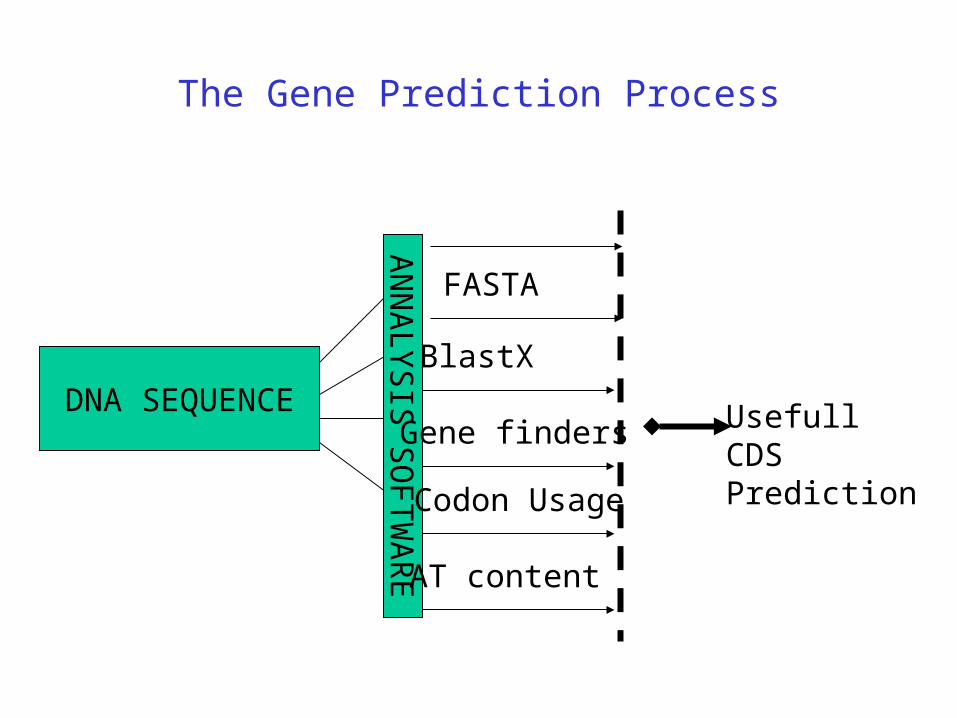
The Gene Prediction Process
DNA SEQUENCE
AN
NA
LY
SIS
SO
FT
WA
RE
UsefullCDSPrediction
Annotator
AT content
Gene finders
Codon Usage
BlastX
FASTA
ESTs

QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
highlighted manually reviewed gene structure
pale brown hit to H. contortus EST cluster in Nembase found using PASAbrown-green hit to H.contortus individual ESTs in NCBI database found using PASApink/red blocks hits to Uniprotbright green twinscan prediction (homology based)pale pink snap prediction (ab initio)yellow hmmgene prediction (ab initio)pale blue genscan prediction (ab initio)red genefinder (ab initio)dark blue fgenesh prediction (ab initio)jade green augustus hints prediction (homology based)orange augustus prediction (ab initio)purple genewise prediction (homology based)
Gene prediction in eukaryotes: HMMs
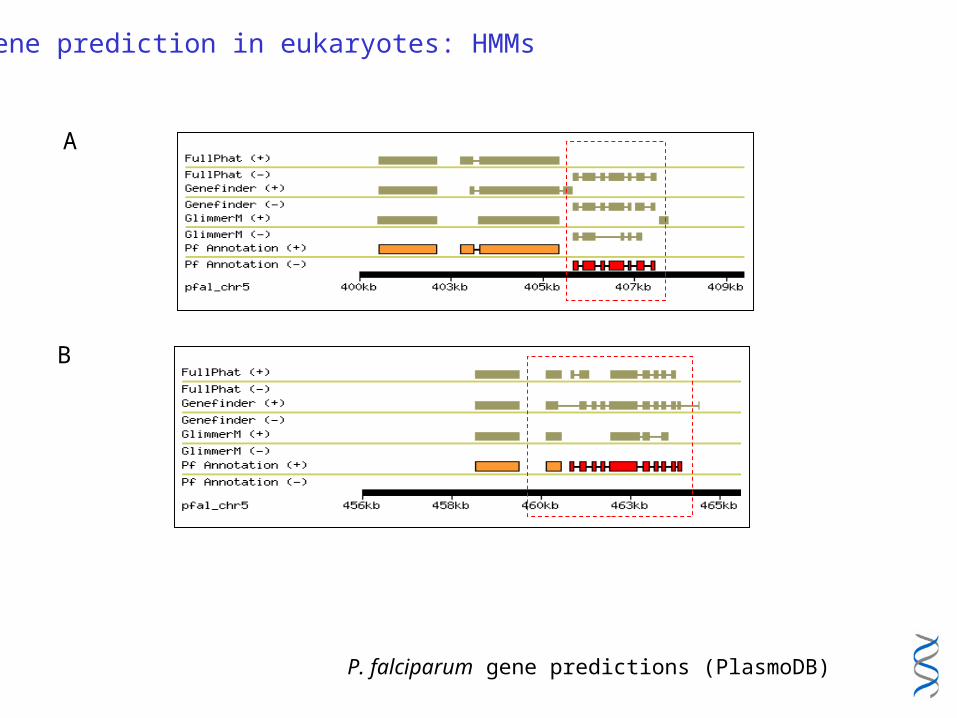
A
B
Gene prediction in eukaryotes: HMMs
P. falciparum gene predictions (PlasmoDB)

Gene prediction in eukaryotes: HMMs
Dictyostelium discoideum gene predictions
BartfinderhmmgenegeneidPhatEST(contig)combined prediction

QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
Manual refinement
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
P. falciparum
P. knowlesi

Ongoing manual annotation e.g. PF14_0021, PF14_0022
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
P. falciparum
P. vivax
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
Revised annotation(back to Two genes!)

Using FASTA Results
• FASTA is a global alignment tool
BLAST
FASTA
• Reduces sensitivity increases specificity