researchrepository.ucd.ie · web viewduring normoxia, when sufficient oxygen is available, o 2,...
TRANSCRIPT

The hypoxia mimetic protocatechuic acid ethyl ester inhibits synaptic signaling and plasticity in the rat hippocampus
Sinead M. Lanigan and John J. O’Connor*UCD School of Biomolecular and Biomedical Science, UCD Conway Institute of Biomolecular and Biomedical Research, Belfield, Dublin 4, Ireland.
*Corresponding Author: John J. O’Connor, UCD School of Biomolecular and Biomedical Science, UCD Conway Institute of Biomolecular and Biomedical Research, Belfield, Dublin 4, Ireland. Email: [email protected]; Tel.: + 353 1 716 6765
AbbreviationsEDHB, protocatechuic acid ethyl ester (also known as ethyl 3,4,dihydroxybenzoate); 2-OG, 2-oxoglutarate; PHD, proyl-4-hydroxylase domains; NMDAR, N-methyl-D-aspartate receptor; AP-V, (2R)-amino-5-phosphonovaleric acid; GABAR, γ-Aminobutyric acid recptor; mDG, medial dentate gyrus; CA1, Cornu Ammonis 1; HIF, hypoxia-inducible factor; HRE, hypoxia-responsive element; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; DMOG, dimethyloxaloylglycine; DFO, desferrioxamine; aCSF, artificial cerebrospinal fluid; fEPSPs, field excitatory postsynaptic potentials; PPR, paired pulse ratio; LTP, long-term potentiation; HFS, high frequency stimulation; DIV, days in vitro; PI, propidium iodide; DMSO, Dimethyl sulfoxide;
AbstractDuring hypoxia a number of physiological changes occur within neurons including the stabilisation of hypoxia-inducible factors (HIFs). The activity of these proteins is regulated by O2, Fe2+, 2-OG and ascorbate-dependant hydroxylases which contain prolyl-4-hydroxylase domains (PHDs). PHD inhibitors have been widely used and have been shown to have a preconditioning and protective effect against a later and more severe hypoxic insult. In this study we have investigated the neuroprotective effects of the PHD inhibitor, protocatechuic acid ethyl ester (ethyl 3,4,dihydroxybenzoate: EDHB) as well as its effects on synaptic transmission and plasticity in the rat hippocampus using electrophysiological techniques. We report for the first time, an acute concentration-dependent and reversible inhibitory effect of EDHB (10 to 100M) on synaptic transmission in the dentate gyrus but not CA1 region which does not affect cell viability. This effect was attenuated through the application of the NMDA or GABAA
receptor antagonists, AP-5 and picrotoxin in the dentate gyrus. There were no changes in the ratio of paired responses after EDHB application suggesting a post-synaptic mechanism of action. EDHB (100M), was found to inhibit synaptic plasticity in both the dentate gyrus and CA1 regions. Application of exogenous Fe2+ (100M) or digoxin (100nM) did not reverse EDHB’s inhibitory effect on synaptic transmission or plasticity in both regions, suggesting that its effects may be HIF-independent. These results highlight a novel modulatory role for the PHD inhibitor EDHB in hippocampal synaptic transmission and plasticity. A novel post-synaptic mechanism of action may be involved possibly involving NMDA and GABAA receptor activation.
Keywords: Protocatechuic acid ethyl ester/Ethyl 3,4,dihydroxybenzoate, Prolyl Hydroxylase, EPSP, Hypoxia, LTP, Hippocampus.
IntroductionHypoxic stress is one of the main components of an ischemic insult and occurs when oxygen demand exceeds supply. However an ischemic insult also results in over-excitation of the neuronal tissue, mitochondrial dysfunction and oxygen radical release, hypercapnia, tissue acidosis, damage of the blood-brain-barrier and recruitment of inflammatory mediators. The hippocampus is particuarly sensitive to hypoxia and thus an ischemic episode, the result of which may trigger a number of events including, depolarization of neurons and glia and subsequent increases in the concentration of glutamate in the extracellular space and over-activation of N-methyl-D-aspartic acid (NMDA) receptors (Dirnagl et al., 1999; Kreisman et al., 2000). During hypoxia a number of physiological changes may also occur including synaptic arrest whereby synaptic transmission is reduced, as well as a change in hypoxia-inducible factor (HIF) stabilisation. HIFs are key transcriptional regulators and play a major role in oxygen homeostasis in the body. They are composed of a heterodimer consisting of a constitutively expressed β subunit and one of two α subunits, HIF-1α or HIF-2α, which are mainly regulated by oxygen at the protein level. The activity of these HIFs are regulated by oxygen-dependant hydroxylases which contain prolyl-4-hydroxyase domains (PHDs; Corcoran et al., 2013). These PHDs are the enzymatic gatekeepers of the adaptive response to hypoxia and thus qualify as oxygen sensors (Kaelin and Ratcliffe, 2008).
During normoxia, when sufficient oxygen is available, O2, iron (Fe2+), 2-oxoglutarate (2-OG) and ascorbate bind to these PHDs and can constitutively hydroxylate HIF-1α on two conserved proline residues, Proline 402 and 564 (Epstein et al., 2001) so that it can be recognised for ubiquitination by E3 ubiquitin ligase von Hippel Lindau protein and degraded (Siddiq et al., 2007). However during hypoxia, when there is a lack of oxygen, PHDs no longer bind to HIF-1α and its degradation ceases, allowing the stable protein to build up and accumulate in the cytosol. HIF-1α can then translocate to the nucleus where it dimerises with HIF-1β, forming a heterodimeric complex which acts as a transcriptional factor, activating gene transcription at hypoxia-responsive elements (HREs). HREs include genes involved in the promotion of cell survival, angiogenesis and anaerobic metabolism. HIF-1-regulated genes include vascular endothelial growth factor (VEGF), erythropoietin (EPO; Lee & Percy, 2011), and endothelial nitic oxide synthase (eNOS; Coulet et al., 2003). Recently it has been suggested the PHDs might have other HIF-independent mechanisms of action, including the regulation of AMPAR recycling (Park et al., 2012). Inhibitors of the HIF-PHDs, which permit the activation of hypoxic adaptation under conditions of normoxia, have therefore become an attractive therapeutic target for ischemia and stroke.
Protocatechuic acid ethyl ester (also known as ethyl 3,4,dihydroxybenzoate (EDHB)) is a lipophilic compound that acts as a pharmacological hypoxic
1

mimetic and PHD inhibitor. Similar to other PHD inhibitors such as dimethyloxaloylglycine (DMOG), EDHB is an analogue of 2-OG, however it also acts as an Fe2+ chelator similar to the actions of desferrioxamine (DFO; Wang et al., 2002). DMOG and DFO have been widely used as preconditioning agents providing a protective effect against a later ischemic insult (Hanson et al., 2009, Ogle et al., 2012, Zhang et al., 2015). Although preconditioning may be a well-established neuroprotective modality it is dependent on the level of insult depths caused to the neurons (Tauskela et al., 2016). Evidence has emerged suggesting that preconditioning with EDHB significantly improves cellular viability in cultured astrocytes after H2O2 injury treatment (Chu et al., 2010), as well as having a protective effect against hypoxia-induced oxidative damage in L6 myoblast cells (Nimker et al., 2015). EDHB has also shown protective activity by promoting endogenous enzyme antioxidants, preventing free radical formation, and hydrogen peroxide (H2O2) induced oxidative damage on cultured PC12 cells (Shi et al., 2006). EDHB may inhibit cell death in a model of neurotrophin deprivation that involves depriving sympathetic neurons of nerve growth factor (NGF; Lomb et al., 2009), as well as attenuating acute hypobaric hypoxia mediated vascular leakage in the rat brain (Singh et al., 2016). Because of the lack of electrophysiological data on the actions of EDHB (as well as other PHD inhibitors) in the CNS and to gain insight into the roles that PHDs and PHD inhibitors may have in the brain, we have used electrophysiological techniques to examine any acute effect of EDHB on synaptic signaling in the rat hippocampus.
Experimental ProceduresPreparation of Hippocampal SlicesAll experimental procedures carried out were approved by the Animal Research and Ethics Committee of the Biomedical Facility at University College Dublin. Wistar rats aged between p21 and p28 (60-120 g) were used and were anaesthetised using 5% isoflurane before decapitation using a guillotine. The brain was removed immediately and placed in ice-cold cutting artificial cerebrospinal fluid (aCSF) comprising 120mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 10 mM glucose, 2 mM MgSO4, and 2 mM CaCl2. The solution was kept bubbling with 95% O2/5% CO2 on ice. Using a blade the cerebellum and forebrain were removed from the cerebrum. 350µm transverse hippocampal slices were then prepared using a Leica VT1000S Vibroslice in ice-cold cutting solution perfused with 95% O2/5% CO2. The slices were transferred to a holding chamber containing oxygenated cutting solution and left to recover for 1 hr at room temperature. Hippocampal slices were placed in a submerged recording chamber connected to a 50ml reservoir of recording aCSF comprising 120 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 10 mM glucose, 1.2 mM MgSO4, and 2 mM CaCl2, with bubbling of 95% O2/5% CO2 at a maintained temperature of 31–33°C with a flow rate of 4-5 ml/min. The slice was allowed to recover in the recording chamber for a minimum of 20 min prior to any recordings.
Electrophysiology In these experiments we stimulated either the medial perforant pathway of the dentate gyrus (mDG) or the Schaffer-collateral pathway of the CA1 region of the hippocampus to evoke field excitatory
postsynaptic potentials (fEPSPs) using aCSF-filled monopolar glass electrodes. fEPSPs were elicited every 30 s and recorded from the dendritic field of the mDG granule or the CA1 pyramidal neurons. Stimulation of responses was carried out using a S48 Stimulator (Grass Instrument; Massachusetts, USA) via a Grass SIU5 stimulus isolation unit. fEPSPs were recorded using an Axopatch 1D and associated CV-4 head stage, which amplified evoked responses 1000-fold. Signals were low passed filtered at 5KHz and sampled at 20 KHz using WinWCP software (J. Demspter, Strathclyde, UK). On occasions where a DAM 50 differential amplifier (World Precision Instruments, USA) was used, fEPSPs were filtered at 0.1 Hz to 10 KHz and also sampled at 20 KHz using WinWCP software. The slope of the fEPSP was measured 1.75 ms to 3.5 ms post stimulus artifact. Stimulus strength was adjusted to give 40% maximal response, determined by an input/output curve.
Hypoxic Exposure and PO2 MeasurementSlices were exposed to hypoxia by switching the gas that passed over the slice chamber from 95% O2/5% CO2 to 95% N2/5% CO2 (Batti & O’Connor, 2010). Re-oxygenation was obtained by reversing this procedure, switching the gas from 95% N2/5% CO2 back to 95% O2/5% CO2. In three experiments during delivery of the hypoxia and re-oxygenation, oxygen levels on the surface and within the hippocampal slice were analysed using a pO2 monitor (OXILITETM) performing fluorescence-quenching oxymetry. The oxygen probe was positioned inside the hippocampal slice at a depth of approximately 100 µm form the surface of the slice. Changes in pO2 were monitored every 5 s. The detectable range of the probe was 0-200mmHg (see Figure 3C and D).
Stimulation ProtocolsThe slices were stimulated at 40% maximal fEPSP slope for a minimum of 20 min in order to acquire a baseline. That is every 30 s a pair of stimuli were applied separated by 50ms. Paired pulse stimulation of the Schaffer collaterals in the CA1 region gives rise to an increase in the size of the second fEPSP (paired pulse facilitation) whilst paired stimulation of the medial perforant pathway in the dentate gyrus gives rise to a decrease in the size of the second fEPSP (paired pulse depression; see O’Leary et al, 1997 for full details). The paired-pulse ratio (PPR) was quantified as the ratio of fEPSP2/fEPSP1 slope. Long-term potentiation (LTP) was elicited by high frequency stimulation (HFS), 3 trains at 100Hz (1s duration) separated by 20s intervals. For LTP to be observed in the mDG, picrotoxin was added to the perfusing solution and the stimulation voltage was increased from 40% to 70% during the induction protocol. For LTP in the CA1 the stimulation voltage stimulation was set between 40 and 70% during the induction protocol.
Excitability of SlicesIn all electrophysiological experiments, the brain slices were not spontaneously active and only evoked fEPSPs when electrically stimulated. In our experiments slices do not spontaneously fire or normally show spontaneous activity. On occasion in the dentate gyrus, in the presence of picrotoxin, after the induction of LTP a small number of population spikes may appear within the fEPSP. These do not
2

affect the measurement of the fEPSP slope, which is measured between 1.75 and 3.5 ms post stimulus. In these experiments the nerve volley was also measured post stimulus artifact to peak amplitude of the nerve volley (usually occurring within 1ms).
Statistical Analysis in Electrophysiological StudiesUsing WinWCP software all fEPSP slopes are given as a percentage of the initial mean baseline slope. The mean baseline slope is an average of the first 20 min of fEPSPs slopes recorded prior to any drug administration, hypoxia or LTP induction. Data was analysed using Prism software (GraphPad). All data was also analysed for normality and for each experiment at least 95% of the area under the curve was within 1.96 SD of the mean. To further confirm this we carried out a two sample Kolmogorov–Smirnov (K-S) test for both control and EDHB baseline and control and EDHB LTP experiments. K-S found both data sets consistent with a normal distribution (P=0.3 and P=0.9 respectively). Bar chart values were analysed using one-way ANOVA with a post-Bonferroni test. For time course data 5 min readings (10 recordings) were taken from critical time points during the experiments, that is the baseline, the end of acute hypoxic exposure, recovery and also from 55 to 60 min post LTP induction. These values were analysed using one-way ANOVA with a post-Bonferroni test in order to compare the different sets of data. All data was expressed as means±SEM. P<0.05 was considered to be statistically significant. The n values correspond to the number of experiments carried out on different animals.
Organotypic Hippocampal Cell CultureOrganotypic hippocampal cultures were prepared from post-natal day 7-9 Wistar rats according to Stoppini et al. (1991). The rats were rapidly decapitated and their brain removed and placed in ice-cold Earle’s balanced salt solution (EBSS). The brain was then dissected along the midline to separate the hemispheres and the hippocampi were rolled out. The hippocampi were transferred to the Teflon stage of a McIlwain tissue chopper and cut into 400µm slices. The slices were washed into a 6-well plate containing EBSS and separated under a light microscope. In a laminar flow hood the slices were then arranged onto organotypic inserts (Millipore). The inserts were then placed into sterile 6-well culture plates and maintained with an air/media interface in a humidified incubator at 35ºC and 5% CO2 for at least 8 days in vitro (DIV) before experimentation. The organotypic medium consisted of 50% minimum essential medium (MEM, Gibco), 25% Earle’s balanced salt solution (EBSS, Gibco), 25% heat-inactivated horse serum (Sigma), 1mM L-glutamine, 28mM D-glucose, 25mM HEPES and 100U/ml penicillin/ streptomycin.
Cell Viability AnalysisExperiments presented in this study were at 9 days in vitro (DIV). The cultures were transferred to treatment media containing 100μM EDHB for 2h or 24h before imaging. The control cultures were exposed to media changes at the same time points as those, which underwent EDHB exposure. In order to assess cell death within the hippocampus following EDHB exposure, 2 μM PI was added to the medium. Cultures were washed in PBS and imaged using a custom built fluorescence microscope using point grey grasshopper 3 sensor (4x lens). PI was excited at 488nm and the emission wavelength was
detected with the 562-588nm band pass filter. The degree of fluorescence corresponds to the degree of cell death in the tissue, both neuronal and non-neuronal. In order to normalize against total cell area the cultures were placed in 20% methanol for 30 min at 4ºC in order to permeabilize the cells and were then re-exposed to 2µM PI for 10min to stain total cell area and re-imaged.
Glutamate Application and Pre-conditioning Effects of EDHB Treatment on Cell ViabilityAt DIV 9-10 the cultures were exposed to fresh media containing 100 µM EDHB for 24h. The inserts were then allowed to recover for 24h in fresh media before being exposed to 2 mM glutamic acid for 24h (DIV 11-12). Other authors have used similar concentrations of glutamate (3.5 mM; Ziobro et al., 2011). They were then transferred to fresh media containing 2 µM PI for a recovery of 46-48h (DIV 13-14). The controls were subjected to media changes at the same time. The amount of PI uptake into the dead or dying cells was then analyzed for each treatment (as above) in order to determine the neuroprotective effects of EDHB pre-conditioning against glutamate-induced excito-toxicity.
Statistical Analysis in Culture ImagesThe images were analyzed using the EBImage plugin for the statistical analysis program R (Batti et al., 2010). A threshold image was generated to remove any non-specific background fluorescence. The proportion of pixels that were above the threshold level was then calculated. This was then repeated for the images captured post-permeabilization, in order to obtain the proportion of pixels that occupied cellular space. The proportion of pixel area occupied by dead cells was expressed as a percentage of the total pixel area of all cells for each of the corresponding images. Controls were normalized to 100% and all other treatments were compared relative to this. Bar graphs of cell death/PI+ cells were normalized to control cultures. Bar chart values were analysed using one way ANOVA with a post-Bonferroni test. All data was expressed as means±SEM. All data in Figure 9 represents the data from 3 individual cultures. In Figure 10 data was collected from between 8 to 12 cultures.
Drugs Protocatechuic acid ethyl ester (EDHB), Picrotoxin, Fe (II) sulfate heptahydrate and Digoxin were all obtained from Sigma, UK. (2R)-amino-5-phosphonovaleric acid (AP-5) was obtained from Tocris Bioscience, UK. All agents except Fe (II) and AP-5 were dissolved in dimethyl sulfoxide (DMSO) with a final concentration <0.02% when diluted in aCSF. AP-5 was dissolved in sterile filtered PBS and added to the aCSF reservoir. Fe (II) was dissolved in aCSF and added to the aCSF reservoir. DMSO controls were carried out at the same dilution (<0.02% V/V). Control data was gathered on different days or weeks throughout the time course of these experiments.
ResultsEffect of EDHB on synaptic signaling in the hippocampusApplication of EDHB at 10M for 30min had no effect of fEPSP slope in the medial dentate gyrus, 30M caused a small (but not significant) decrease and 100M had a significant inhibitory effect on
3

synaptic transmission (mDG; 98.0±3.9%; n=5, 91.0±2.6%; n=4, and 70.5±4.5%, n=9, P<0.001, respectively; Figure 1A, B). EDHB was found to have no effects on cell excitability, determined by graphing nerve volley amplitude versus EPSP amplitude before and following 30min 100µM EDHB perfusion (Figure 1C). The effect of EDHB in the mDG was reversible (89.1±4.1% 1 hr after washout, n=8; Figure 2A) but did not modulate paired pulse depression when 2 stimuli were applied at 50ms intervals (before 0.85±0.05; during 0.83±0.05; 1 hr after EDHB 0.83±0.06, all n=5; Figure 2B). Application of a hypoxic event to slices of mDG gave rise to a depression of fEPSP slope to 31.1±2.3%, (n=5, P<0.001) and a recovery after 30 min to 75.3±3.6% (n=5). In contrast to the effects of EDHB, hypoxia gave rise to a significant change from PPD to PPF as would be expected for a pre-synaptic change in transmitter release (before 0.89±0.04; during 1.46±0.09 and after hypoxia 0.92±0.06, all n=4, P<0.001; Figure 2C, D).
Figure. 1. Application of EDHB in hippocampal slices reduces baseline synaptic transmission in the mDG. A. In the mDG, EDHB had no significant effect on fEPSP slope at 10µM (blue circles; 98.0±3.9%; n=5). 30µM EDHB caused a small decrease in fEPSP (green square; 91.0±2.6%; at 30min; n=4), but 100µM EDHB significantly depressed fEPSP slope (red triangles; 70.5±4.5%; at 30 min; n=9). Insets are representative traces showing the effect of EDHB on fEPSP slope at the times indicated. B. Vertical scatter plot summarising the effects of EDHB at 10, 30 and 100M, 30 min after perfusion. C. EDHB was found to have no effects on excitability, determined by input-output curves before (open circles) and following 30min 100µM EDHB perfusion (red circles). The fibre volley amplitude (FV) was plotted against the fEPSP amplitude. As the input voltage increased, both the fibre volley and the fEPSP increased in linear fashion. There is no change in FV-EPSP relation in the presence of 100M EDHB.
Figure 2. Comparison of the effects of EDHB and hypoxia on fEPSP slope in the mDG. A. The effects of 30 min EDHB (100M) treatment on fEPSP slope are reversible (65.0±4.0% at 30 min and 89.1±4.1 at 1 hr washout; n=8). B. There was no significant effect of EDHB on the paired pulse ratio (PPD) in the mDG. PPD 5min before: 0.85±0.05, n=5, 5min at end of EDHB perfusion: 0.83±0.05, n=5; after 1h washout: 0.83±0.06, n=5. C. 30 min hypoxia (95%N2./5%CO2) reversibly decreased baseline fEPSP in the mDG (31.1±2.3% at 30 min and 75.3±3.6 at 30 min recovery; n=5). In A and C insets to the right show representative fEPSPs before, during and after washout of EDHB or hypoxia D. During hypoxia the ratio of paired pulses increases indicating a presynaptic mechanism of action. PPD 5min before: 0.89±0.04, n=4, 5min at end of hypoxia: 1.46±0.09, n=4; after 30 min recovery: 0.92±0.06, n=4. All data is expressed as mean±SEM, n=4-8 for all experiments.
We next investigated the effect of EDHB in the CA1 region of the hippocampus. Application of EDHB at 100M had no significant effect on fEPSP slope over 30 min (98.3±3.2%, n=7; Figure 3A).
However as has previously been demonstrated 30 min hypoxia gave rise to a near complete inhibition of fEPSP slope (4.6±1.2%, P<0.001) and recovery to 103.5± 9.1% at 30 min (n=5; Figure 3B). These results highlight the regional differences with application of EDHB and hypoxia in the mDG and CA1 region.
Figure 3. Hypoxia but not EDHB reduces baseline fEPSP in the CA1 region. A. Application of EDHB had no effect on fEPSP slope in the CA1 region (98.3±3.2%, n=7). B. 30 min hypoxia (95%N2./5%CO2) reversibly decreased baseline fEPSP in the CA1 (4.6±1.2% at 30 min and 103.5±9.1% at 30 min recovery; n=5). In A and B insets at the top show representative fEPSPs before, during and after washout of either EDHB or hypoxia. All data is expressed as mean±SEM, n=5-7 for all experiments. C. Measurement of the PO2
100µm below the hippocampal slice indicated a significant decrease in oxygen tension within a 5 min (300 s) exposure to 95%N2./5%CO2 (blue line). Oxygen tension exceeded 200 mmHg 3 min after re-oxygenation. D. Expanded view of the initial decline of PO2 for recordings on the surface (filled squares) and 100 µm below the surface of the slice (open circles). Vertical axis indicates both O2 percentage and PO2 in mmHg. The average of n=3 is illustrated in C &D).
A role for NMDA and GABAA receptors in the inhibitory effect of EDHBWe further examined if the inhibitory effects of EDHB in the mDG were due to its Fe2+ chelating and HIF-stabilizing properties. Perfusion of 100µM EDHB in the presence of 100µM Fe2+ gave rise to an inhibition of fEPSP after 30 min that was not different from EDHB application alone (76.8±4.1%, n=5 versus 70.5±4.5% after 30 min, n=9, respectively). Similar results were obtained after perfusion of the HIF-1 inhibitor, digoxin (100nM; 74.3±5.1%, n=5; Figure 4A, B), suggesting the effects may be mediated through non HIF-stabilizing mechanisms of action. Previously we have reported that the acute inhibitory effects of another PHD inhibitor, DMOG, on fEPSP slope were partially NMDAR-mediated (Batti et al., 2010). Here we report that AP-5 (25M), the NMDA receptor antagonist, partially but significantly reversed the effects of EDHB on fEPSP slope (86.3±2.8%, n=6 compared to 70.5±4.5%, n=9, P<0.05) after 30 min.
Figure 4. The inhibitory effect of EDHB may involve NMDA and GABAA receptorsA. Time course showing that 20 min prior application of exogenous Fe2+ (100µM; 76.8±4.1%, n=5) or digoxin (100nM; 74.3±5.1%; n=5) does not significantly alter the inhibitory effect of EDHB on fEPSP slope (100µM; 70.5±4.5%; n=9). Insets are representative traces showing the effect of treatment on fEPSP. B. Summary of the data in A showing effects of agents at 30 min. C. 20min prior application of AP-5 (25 µM; 86.3±2.8%; n=6) or picrotoxin (100µM; 83.7±2.7%; n=8) significantly attenuated the inhibitory effect of EDHB on fEPSP slope (70.5±4.5%; n=9). Combined
4

application of picrotoxin and AP-5 gave rise to a further attenuation of the inhibitory effects of EDHBs (95.5±0.8%; n=5). Insets are representative traces showing the effect of AP-5 and picrotoxin and EDHB on fEPSP. D. Summary of the data in C showing effects of agents at 30 min. All data is expressed as mean±SEM. *P<0.05, ***P<0.001.
We also investigated a role for the GABAA receptor due to the greater inhibitory tone in the mDG compared to the CA1. Prior application of picrotoxin (100M) partially but significantly reversed the effect of EDHB at 30 min (83.7±2.7%, P<0.05, n=8; Figure 4C, D). Finally combined application of picrotoxin and AP-5 had a more significant effect in reversing EDHBs depressive actions when compared to picrotoxin and AP-5 applied alone (95.5±0.8%; n=5; P<0.001).
Effects of EDHB on LTP in the mDG and CA1 hippocampusFinally we investigated the effects of EDHB on LTP in both the mDG and CA1 regions of the hippocampus. Robust control LTP was induced in both the mDG and CA1 (application of picrotoxin was required in the mDG to achieve reliable LTP; 152.1±10.4% and 143.2±8.2%, respectively, n=5 for both after 60 min). Slices were treated with EDHB for 30 min and the final 20 min of the fEPSP slopes were normalized to 100% before induction of LTP. Application of 30µM and 100M but not 10M EDHB significantly impaired LTP in the mDG (118.2±4.8%; n=4, P < 0.05 and 111.7±5.6%, n=5, P<0.01 respectively, compared to controls at 60 min; Figure 5A-B).
Figure 5. EDHB impairs synaptic plasticity in both the mDG and CA1 regions. A. Time course showing changes in fEPSP slope for control (black; 152.1±10.4%; n=5), 10µM EDHB (blue; 133.6±5.2%; n=6), 30µM (118.2±4.8%; n=4) and 100µM EDHB (red; 111.7± 5.6%; n=5) on LTP in the mDG region. B. Summary of the data in A showing LTP after 1 hr. C. Time course showing changes in fEPSP slope in the CA1 region for control (grey; 143.2±8.2%; n=5) and 100µM EDHB (pink; 112.0±5.1%, n=4) on LTP. D. Summary of the data in A showing LTP after 1 hr. Baseline fEPSP slope was recorded for 20 min followed by HFS. LTP was monitored for 1 hr post induction. Arrows in A & C indicate high frequency stimulation to induce LTP. Insets in A and C show sample fEPSP traces, 60 min following HFS superimposed over a representative fEPSP 5 min prior to HFS. All data is expressed as mean±SEM. *P<0.05, **P<0.01 compared to controls.
100M EDHB was also found to significantly impair LTP in the CA1 region (112.0±5.1%, n=5, P<0.01 compared to controls at 60 min; Figure 5C-D). We further analyzed whether the effects of EDHB were primarily mediated through PHD inhibition. In the mDG application of Fe2+ alone (100µM) had no effects on LTP (141.1±6.5%, n=5) and failed to reverse the inhibitory effects of EDHB on LTP (112.2±5.3%, n=5; Figure 6A, B). Application of digoxin at a concentration that had no effect on LTP on its own (100nM; 137.7±6.7%, n=5) also failed to reverse the effect of EDHB on LTP (119.1±7.9%, n=5; Figure 6C, D). Similarly, in the CA1 application of Fe2+ alone (100µM) had no effects on LTP (136.7±9.4%, n=3) and failed to
reverse the inhibitory effects of EDHB on LTP (115.3±2.1%, n=4; Figure 7A, B). Application of digoxin at a concentration that had no effect on LTP on its own (100nM; 139.2±5.4%, n=4) also failed to reverse the effect of EDHB on LTP (112.2±3.3%, n=4; Figure 7C, D). The inhibitory effects of 100µM EDHB on LTP in the CA1 and mDG were reversible with full LTP restored after 40min or 60min washout (140.0±8.7%, n=4 and 147.9±7.6%, n=4, respectively, P<0.05; Figure 8A-D).
Figure 6. EDHB impairment of synaptic plasticity in the mDG appears to be HIF-independent. A. Time course showing changes in fEPSP slope of control (black; 152.1±10.4%, n=5), 100µM EDHB (red; 111.7±5.6%, n=5), exogenous Fe2+ alone (yellow; 141.1±6.5%, n=5) and Fe2+ & EDHB (purple; 112.2±5.3%, n=5) on LTP in the mDG region. B. Summary of the data in A showing LTP after 1 hr. C. Time course showing changes in fEPSP slope of control (black; 152.1±10.4%, n=5), 100µM EDHB (red; 111.7±5.6%, n=5), Digoxin alone (green; 137.7±6.7%, n=4) and Digoxin & EDHB (brown; 119.1±7.9%, n=5) on LTP in the mDG region. D. Summary of the data in C showing LTP after 1 hr. Baseline fEPSP slope was recorded for 20 min followed by HFS. LTP was monitored for 1 hr post induction. Arrows in A & C indicate high frequency stimulation to induce LTP. Insets show sample fEPSP traces, 60 min following HFS superimposed over a representative fEPSP 5 min prior to HFS. All data is expressed as mean ± SEM. *P<0.05.
Figure 7. EDHB impairment of synaptic plasticity in the CA1 also appears to be HIF-independent. A. Time course showing changes in fEPSP slope in control (grey; 143.2±8.2%, n=5), 100µM EDHB (red; 110.0±4.4%, n=5), exogenous Fe2+ alone (yellow; 136.7±9.4%, n=3) and Fe2+ & EDHB (purple; 115.3±2.1%, n=4) on LTP in the CA1 region. B. Summary of the data in A showing LTP after 1 hr. C. Time course showing changes in fEPSP slope of control (grey; 143.2±8.2%, n=5), 100µM EDHB (red; 110.0±4.4%, n=5), Digoxin alone (green; 139.2±5.4%, n=4) and Digoxin & EDHB (brown; 112.2±3.3%, n=4) on LTP in the mDG region. D. Summary of the data in C showing LTP after 1 hr. Baseline fEPSP slope was recorded for 20 min followed by HFS. LTP was monitored for 1 hr post induction. Arrows in A & C indicate high frequency stimulation to induce LTP. Insets show sample fEPSP traces, 60 min following HFS superimposed over a representative fEPSP 5 min prior to HFS. All data is expressed as mean ± SEM. *P<0.05, **P<0.01.
Pre-conditioning effects of EDHB on cell viability in the hippocampusIn order to establish if EDHB pretreatment results in a neuroprotective effect, the PI assay was used to investigate its pre-conditioning effects on glutamate-mediated excitotoxicity. Application of EDHB (100M) alone for 2h or 24h had no effect on the viability of cells in the hippocampus compared to control (109.6±20.1%, 124.5±14.6% versus 98.1±16.4%, n=3, respectively; Figure 9A, B).
5
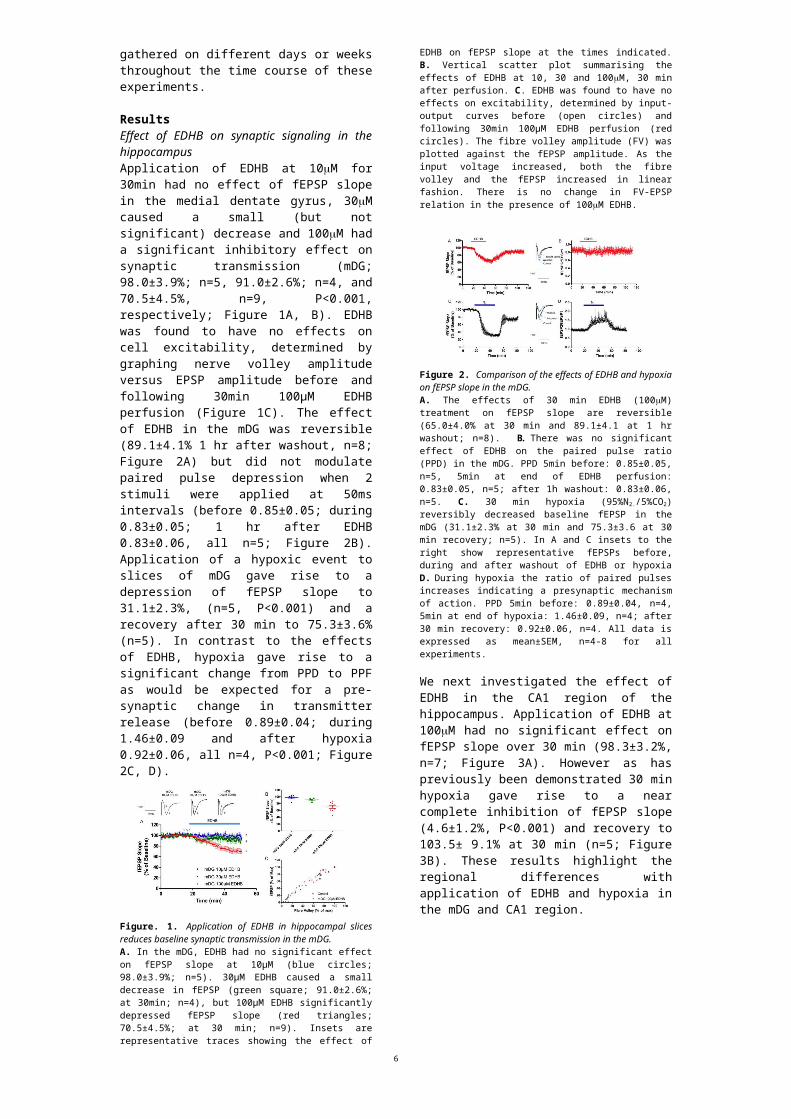
Figure 8. The inhibition of synaptic plasticity by EDHB in the mDG and CA1 regions is reversible. A. Time course showing changes in fEPSP slope for control (Black; 152.1±10.4%; n=5), 100µM EDHB (Red; 111.7± 5.6%; n=5) and washout (Green; 147.9±7.6%; n=4) on LTP in the mDG region. B. Summary of the data in A showing LTP after 1 hr. C. Time course showing changes in fEPSP slope in the CA1 region for control (Grey; 143.2±8.2%; n=5), 100µM EDHB (Red; 112.0±5.1%, n=4) and washout (Green; 140.0±8.7%; n=4) on LTP in the CA1 region. D. Summary of the data in A showing LTP after 1 hr. Baseline fEPSP slope was recorded for 20 min followed by HFS. LTP was monitored for 1 hr post induction. Arrows in A & C indicate high frequency stimulation to induce LTP. Insets in A and C show sample fEPSP traces, 60 min following HFS superimposed over a representative fEPSP 5 min prior to HFS. All data is expressed as mean±SEM. *P < 0.05 compared to controls; #P < 0.05 compared to 100µM EDHB.
Treatment of hippocampal cultures with 24h 2mM glutamate induced significant cell death compared to control cultures (390.2±53.6% n=12 versus 100.0±14.9% n=9, respectively; p<0.001). Pretreatment of the cultures with 100M EDHB for 24h prior to glutamate exposure protected the hippocampus against glutamate-induced cell death, with EDHB pre-conditioning significantly attenuating the amount of cell death caused by glutamate treatment alone (204.0±40.9% n=8 and 390.2±53.6% n=12, respectively; p<0.05; Figure 10A, B).
Figure. 9. The effects of EDHB on cell viability in organotypic hippocampal cultures. A. 9-10 DIV cultures were exposed to 100µM EDHB for 2h or 24h before imaging. For each slice 4X fluorescent images were taken before and after fixation and additional incubation with PI (2µM). Data resulting from the PI staining before fixation was counted as “cell death” values (Live – top panel), while those from post-fixation and further PI incubation we counted as “total cell” values (Total – bottom panel). B. Bar graph showing cell death expressed as a percentage of control. 2h and 24h treatment with 100µM EDHB had no significant effects on cell death/viability (109.6±20.1%, 124.5±14.6% versus 98.1±16.4%, n=3, respectively). Scale bar=500µm.
Discussion Despite the extensive research carried out on the neuroprotective effects of inhibiting PHDs in-vitro (Lomb et al., 2007, 2009) and in-vivo (Baranova et al., 2007, Singh et al., 2016), there has been very little insight into the modulatory role that PHD inhibition may have on synaptic signaling in the brain. In this study, we observed a pre-conditioning and neuroprotective effect with the PHD inhibitor, EDHB, against glutamate-induced excitotoxicity. We also observed that acute treatment with EDHB, significantly decreased synaptic transmission in the mDG but not the CA1, suggesting regional differences of action, similar to what is seen with the effects of hypoxia. Importantly this effect of EDHB did not seem to involve an increase in
neuronal excitability. Unlike hypoxia, which involves the activation of adenosine A1
autoreceptors, EDHB’s effects appear to be mediated post-synaptically.
Figure. 10. The effects of EDHB pre-conditioning and glutamate-induced excitotoxicity. A. 9-10 DIV cultures were exposed to fresh media containing 100µM EDHB or vehicle for 24h. Inserts were then allowed to recover for 24h in fresh media before being exposed to 2mM glutamic acid for 24h (DIV 11-12). They were then transferred to fresh media containing 2µM PI for a recovery of 46-48h (DIV 13-14). The controls were subjected to media changes at the same time. B. Bar graph showing cell death expressed as a percentage of control. 24h treatment with 2mM glutamic acid caused a significant increase in hippocampal cell death compared to controls (390.2±53.6%, n=12 versus 100.0±14.9%, n=9, respectively; ***P<0.001). Prior exposure of cultures to EDHB significantly attenuated glutamates detrimental effects on cell viability (204.0±40.9%, n=8 and 390.2±53.6%, n=12, respectively; #
P<0.05). Scale bar=500µm.
In addition, we have demonstrated that EDHB has a concentration-dependent inhibitory effect on synaptic plasticity in both the mDG and CA1, which is reversible upon washout. Furthermore, we provide evidence that EDHB’s modulatory effects in the hippocampus may be HIF-independent. We have previously reported that the application of another PHD inhibitor, DMOG, which acts as a competitive 2-OG antagonist, also causes a reversible inhibition of synaptic signaling and reduces the isolated NMDA fEPSP in the rat hippocampus (Batti et al., 2010). We previously reported a role for the specific PHD2 isoform in modulating synaptic plasticity (Corcoran et al., 2013). Others have reported that the PHD inhibitor, DFO, an iron chelator, also impairs synaptic signaling and plasticity in an NMDAR-dependent manner (Muñoz et al., 2011). Together these results suggest a role for PHD inhibitors in modulating hippocampal synaptic signaling and highlight a novel and unexplored function of EDHB in the CNS.
We found that acute application of EDHB (100µM) in the mDG caused a reversible decrease in the fEPSP similar in time-course and magnitude to that observed during hypoxia. This decrease was not due to cell death as EDHB had no effect on cell viability and was reversible upon washout. During hypoxia when there is a decrease in oxygen supply, the neurons quickly adapt to the lack of energy supply (ATP) and increased extracellular adenosine by activating adenosine A1 autoreceptors (Fowler et al., 1989; Dale et al., 2000; Frenguelli et al., 2003). This results in a profound depression of synaptic transmission, primarily by pre-synaptic inhibition of glutamate release. It is now thought that the inhibition in transmission may be an adaptive neuroprotective response against metabolic insults (Dux et al., 1992, Rudolphi et al., 1992), protecting against excessive glutamate released from apoptotic and necrotic cells, although recent work suggests that synaptic failure may also contribute to cell death (Le Feber et al., 2016). Our hypoxia experiments show decreased fEPSP and changes in
6

paired-pulse ratio (PPR) during hypoxia indicating a pre-synaptic action. However the inhibitory effect of EDHB did not have any effect on PPR. Also the effects of EDHB were dissimilar in the mDG and CA1 regions. Although EDHB causes a reversible inhibition in synaptic transmission in the mDG, this depressive effect was not seen in the CA1 region of the hippocampus. Using young animals in our experiments during hypoxia, in the CA1 the fEPSP decreases to 100% but to only 31% in the mDG. The reasons for these region-specific differences are still largely unknown with regards to hypoxia and EDHB. We have used young animals, which may have different responses to hypoxia than older animals. It may also be due to physiological differences between the pyramidal and granule cells. Previous experiments have found that the pyramidal cells of the CA1 region are highly vulnerable to damage from a hypoxia-insult, whereas the neurons of the DG are more resistant (Kreisman et al., 2000; Yao et al., 1998). Also granule cell neurons have been found to maintain calcium homeostasis and to express brain-derived neurotrophic factor (BDNF) mRNA at higher levels than CA1 neurons (Yao et al., 1998). BDNF is important for the survival of neurons and the activity of synapses. Another regional difference between the CA1 and mDG is the greater inhibitory tone and GABAergic modulation in the DG, which was shown in a morphological study by Hörtnagl et al. (1991). They showed a higher activation of GABA and glutamic acid decarboxylase (GAD) in the rat hippocampal DG compared to the CA3 and CA1.
We found that addition of picrotoxin, a GABAA
receptor antagonist, in the mDG prior to EDHB exposure, significantly attenuated its inhibitory effects on baseline transmission, suggesting that EDHB may have a GABAergic effect that has not been previously reported and may explain the regional differences seen in this study. During excitatory synaptic transmission, simultaneous stimulation of post-synaptic GABAA receptors as a result of inter-neuronal signaling may result in depression of excitatory synaptic transmission by reducing the generation of post-synaptic potentials (Vida et al., 2006). However GABA may also act in an autocrine or paracrine manner to reduce GABAergic neurotransmission resulting in a disinhibition and overall depression of inhibitory signaling (Tóth et al., 1997). Our laboratory has previously reported that the application of DMOG also causes a reversible inhibition of synaptic signaling and reduces the isolated NMDA fEPSP in the rat hippocampus (Batti et al., 2010). We have observed a similar EDHB-induced depression of basal synaptic transmission in the mDG, which required functional NMDA receptors, as the response was attenuated by prior application of the NMDA receptor antagonist, AP-5. A similar NMDA-dependent response was observed in hippocampal slices treated with DFO (Muñoz et al., 2011). Interestingly when picrotoxin and AP-5 were added together there was a more significant attenuation of the effect of EDHB on synaptic signaling (additive effect). Taken together these data suggest that EDHB may have a novel post-synaptic effect, which may involve the activation of GABAA
and/or NMDA receptors.
Recently there has been some evidence for a novel HIF-independent function for PHDs in the CNS. In C.elegans neurons it has been shown that hypoxia
regulates the trafficking of the glutamate receptor GLR-1. Hypoxia or mutations in egl-9, a PHD oxygen sensor, results in the internalization of GLR-1 and the reduction of glutamate-activated currents. Surprisingly, HIF-1 is not required for this effect (Park et al., 2012). Siddiq et al., (2009) has also shown that selective inhibition of PHD1 isoform using DMOG and DFO mediated neuroprotection against oxidative death in-vitro via HIF- and CREB-independent pathways. Unlike the PHD inhibitors DMOG and DFO, EDHB has a dual action as a substrate analogue of 2-oxoglutarate and it also functions as an iron chelator (Wang et al., 2002). It has been reported that DFO, which has a higher affinity for ferric iron, causes a similar decrease in basal transmission in the CA1 that was reversible upon application of exogenous iron (Muñoz et al., 2011). However this was not seen in our study when exogenous iron was added prior to EDHB in the mDG, suggesting that although having similar chelation properties, DFO and EDHBs functional effects may be different. This finding also suggests that EDHBs effects on synaptic transmission may be independent of its ability to stabilize HIF. Further confirming this hypothesis, it was found that prior application of the HIF-1α inhibitor, digoxin, had no significant effect on EDHBs inhibition of fEPSP. Digoxin inhibits HIF translation from mRNA and since HIF protein is rapidly turned over in normoxic conditions, the net effect is to reduce protein expression of HIF-1α. It was first identified as a HIF inhibitor in cancer therapeutic drug screens (Zhang et al., 2008). Since then it has been shown to be a pre-conditioning agent with neuroprotective effects against ischemic injury (Peng et al., 2016).
We also found that EDHB significantly inhibited LTP in both mDG and CA1 regions. Interestingly, this LTP inhibition was reversible in both the CA1 and the mDG once EDHB was washed out, suggesting EDHB has an acute and direct effect on synaptic plasticity in the mDG. Picrotoxin was present for all mDG LTP experiments in order to produce robust and reliable LTP. However it was not present in CA1 LTP experiments. If the reduction in LTP by EDHB in the CA1 region is a similar mechanism of action as in the dentate gyrus the presence of picrotoxin in CA1 slices should not change the effect of EDHB on LTP. Application of exogenous Fe2+ was not able to reverse the effect of EDHB on LTP in the mDG or CA1 unlike that previously observed with DFO (Munoz et al., 2011) suggesting that EDHBs effects on synaptic plasticity may be mediated through a different mechanism. Fe2+ is known to be important for neuron development and memory function (Carlson et al., 2009). However EDHB’s affinity for Fe (III) has been shown to be substantially lower than that of DFO, although it is capable of promoting effective iron deficiency in cells, and the cellular responses to EDHB closely resembled the responses elicited by DFO (Wang et al 2002). Previously Shaminder et al., (2009) have shown that the HIF inhibitor digoxin shows cerebral protection against ischemia/reperfusion injury in mice. However we found that prior application of digoxin, did not reverse the effects of EDHB on LTP in the mDG or CA1. The effects of EDHB on LTP are more likely to be mediated through a HIF-independent mechanism of action, perhaps by affecting intracellular calcium levels or kinases like CamKII and PKC which phosphorylate AMPA and NMDA
7

receptors (Grosshans and Browning, 2001) and play an intrinsic role in synaptic plasticity.
The effect of mild hypoxia on synaptic transmission has been largely described as an adaptive neuroprotective mechanism, established to prevent the over-activation of NMDA receptors and thus excitotoxicity (Sebastiao et al., 2001). Since a number of previous studies have shown pre-conditioning and neuroprotective effects for a mild transient ischemic attack (TIA; Moncaye et al., 2000; Grabb and Choi, 1999; Liu et al., 2000), and because of the similarities reported here between the effects of PHD inhibition on the fEPSPs and hypoxic depression, we wanted to investigate the potential therapeutic implications of PHD inhibition on a glutamate-induced ischemic insult. Using an in-vitro model of glutamate-induced excitotoxicity, based on the observation that overstimulation of glutamate receptors promotes neuronal cell death (Pellegrini-Giampietro et al., 1999; Rothman and Olney, 1986), we assessed the action of pre-conditioning with EDHB in organotypic hippocampal cultures. Using a viability assay to assess the integrity of the hippocampal cells following glutamate excitotoxicity, we report a significant increase in cell death in glutamate alone treated cultures, whilst EDHB (100µM) pre-treatment significantly attenuated the detrimental effects of glutamate on cell viability. It will be of interest in future experiments to determine if this prevention of excitotoxicity shares common signaling pathways with that shown for the effects of EDHB on synaptic signalling. Finally DeFazio et al., (2009) has indicated that neuroprotection conferred by preconditioning may depend on functional modifications of GABA synapses.
In conclusion we have demonstrated a neuroprotective effect and a novel modulatory role for the PHD inhibitor, EDHB, in hippocampal synaptic function. We found that EDHB inhibits basal synaptic transmission in the medial dentate gyrus, an effect that was not seen in the CA1, suggesting like hypoxia, EDHB has regional differences. We also report for the first time a post-synaptic mechanism of action for EDHB, which requires GABAA and NMDA receptor activation. EDHB was also found to block long-term potentiation in both mDG and CA1 regions. Further research will be required to elucidate the exact mechanism of action of EDHB on synaptic signaling in the CNS.
AcknowledgementsThis research did not receive any specific grant from funding agencies in the public, commercial, ornot-for-profit sectors. We would like to thank University College Dublin for financial support.
FundingThis work was supported by University College Dublin
ReferencesBaranova, O., Miranda, L.F., Pichiule, P., Dragatsis, I., Johnson, R.S. and Chavez, J.C., 2007. Neuron-specific inactivation of the hypoxia inducible factor 1α increases brain injury in a mouse model of transient focal cerebral ischemia. Journal of Neuroscience, 27(23), pp.6320-6332.
Batti L, O'Connor JJ. 2010. Tumor necrosis factor-alpha impairs the recovery of synaptic transmission from hypoxia in rat hippocampal slices. J Neuroimmunol. 218(1-2), pp21-27.
Batti, L., Taylor, C.T. and O'Connor, J.J., 2010. Hydroxylase inhibition reduces synaptic transmission and protects against a glutamate-induced ischemia in the CA1 region of the rat hippocampus. Neuroscience, 167(4), pp.1014-1024.
Carlson, E.S., Tkac, I., Magid, R., O'Connor, M.B., Andrews, N.C., Schallert, T., Gunshin, H., Georgieff, M.K. and Petryk, A., 2009. Iron is essential for neuron development and memory function in mouse hippocampus. The Journal of nutrition, 139(4), pp.672-679.
Chu, P.W., Beart, P.M. and Jones, N.M., 2010. Preconditioning protects against oxidative injury involving hypoxia-inducible factor-1 and vascular endothelial growth factor in cultured astrocytes. European journal of pharmacology, 633(1), pp.24-32.
Corcoran, A., Kunze, R., Harney, S.C., Breier, G., Marti, H.H. and O'connor, J.J., 2013. A role for prolyl hydroxylase domain proteins in hippocampal synaptic plasticity. Hippocampus, 23(10), pp.861-872.
Coulet, F., Nadaud, S., Agrapart, M. and Soubrier, F., 2003. Identification of hypoxia-response element in the human endothelial nitric-oxide synthase gene promoter. Journal of Biological Chemistry, 278(47), pp.46230-46240.
Dale, N., Pearson, T. and Frenguelli, B.G., 2000. Direct measurement of adenosine release during hypoxia in the CA1 region of the rat hippocampal slice. The Journal of physiology, 526(1), pp.143-155.
DeFazio RA1, Raval AP, Lin HW, Dave KR, Della-Morte D, Perez-Pinzon MA. 2009. GABA synapses mediate neuroprotection after ischemic and epsilonPKC preconditioning in rat hippocampal slice cultures. J Cereb Blood Flow Metab. 29(2), pp375-84.
Dirnagl U, Iadecola C, Moskowitz MA. 1999. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22(9), pp391-397.
Dux, E., Schubert, P. and Kreutzberg, G.W., 1992. Ultrastructural localization of calcium in ischemic hippocampal slices: the influence of adenosine and theophylline. Journal of Cerebral Blood Flow & Metabolism, 12(3), pp.520-524.
Epstein, A.C., Gleadle, J.M., McNeill, L.A., Hewitson, K.S., O'Rourke, J., Mole, D.R., Mukherji, M., Metzen, E., Wilson, M.I., Dhanda, A. and Tian, Y.M., 2001. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell, 107(1), pp.43-54.
Fowler, J.C., 1989. Adenosine antagonists delay hypoxia-induced depression of neuronal activity in hippocampal brain slice. Brain research, 490(2), pp.378-384.
Frenguelli, B.G., Llaudet, E. and Dale, N., 2003. High‐resolution real‐time recording with microelectrode biosensors reveals novel aspects of adenosine release during hypoxia in rat hippocampal slices. Journal of neurochemistry, 86(6), pp.1506-1515.
Grabb, M.C. and Choi, D.W., 1999. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. Journal of Neuroscience, 19(5), pp.1657-1662.
Grosshans, D.R. and Browning, M.D., 2001. Protein kinase C activation induces tyrosine phosphorylation of the NR2A and NR2B subunits of the NMDA receptor. Journal of neurochemistry, 76(3), pp.737-744.
Hanson, L.R., Roeytenberg, A., Martinez, P.M., Coppes, V.G., Sweet, D.C., Rao, R.J., Marti, D.L., Hoekman, J.D., Matthews, R.B., Frey, W.H. and Panter, S.S., 2009. Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. Journal of Pharmacology and Experimental Therapeutics, 330(3), pp.679-686.
Hörtnagl, H., Berger, M.L., Sperk, G. and Pifl, C.H., 1991. Regional heterogeneity in the distribution of neurotransmitter markers in the rat hippocampus. Neuroscience, 45(2), pp.261-272.
Kaelin, W.G. and Ratcliffe, P.J., 2008. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular cell, 30(4), pp.393-402.
Kreisman, N.R., Soliman, S. and Gozal, D., 2000. Regional differences in hypoxic depolarization and swelling in hippocampal slices. Journal of neurophysiology, 83(2), pp.1031-1038.
le Feber J, Tzafi Pavlidou S, Erkamp N, van Putten MJ, Hofmeijer J. 2016. Progression of Neuronal Damage in an In Vitro Model of the Ischemic Penumbra. PLoS One. 11(2): e0147231.
Lee, F.S. and Percy, M.J., 2011. The HIF pathway and erythrocytosis. Annual Review of Pathology: Mechanisms of Disease, 6, pp.165-192.
Liu, J., Ginis, I., Spatz, M. and Hallenbeck, J.M., 2000. Hypoxic preconditioning protects cultured neurons against hypoxic stress
8

via TNF-α and ceramide. American Journal of Physiology-Cell Physiology, 278(1), pp.C144-C153.
Lomb, D.J., Straub, J.A. and Freeman, R.S., 2007. Prolyl hydroxylase inhibitors delay neuronal cell death caused by trophic factor deprivation. Journal of neurochemistry, 103(5), pp.1897-1906.
Lomb, D.J., Desouza, L.A., Franklin, J.L. and Freeman, R.S., 2009. Prolyl hydroxylase inhibitors depend on extracellular glucose and hypoxia-inducible factor (HIF)-2α to inhibit cell death caused by nerve growth factor (NGF) deprivation: evidence that HIF-2α has a role in NGF-promoted survival of sympathetic neurons. Molecular pharmacology, 75(5), pp.1198-1209.
Moncayo, J., De Freitas, G.R., Bogousslavsky, J., Altieri, M. and Van Melle, G., 2000. Do transient ischemic attacks have a neuroprotective effect? Neurology, 54(11), pp.2089-2094.
Muñoz, P., Humeres, A., Elgueta, C., Kirkwood, A., Hidalgo, C. and Núñez, M.T., 2011. Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. Journal of Biological Chemistry, 286(15), pp.13382-13392.
Nimker, C., Kaur, G., Revo, A., Chaudhary, P. and Bansal, A., 2015. Ethyl 3, 4-dihydroxy benzoate, a unique preconditioning agent for alleviating hypoxia-mediated oxidative damage in L6 myoblasts cells. The Journal of Physiological Sciences, 65(1), pp.77-87.
Ogle, M.E., Gu, X., Espinera, A.R. and Wei, L., 2012. Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1α. Neurobiology of disease, 45(2), pp.733-742.
O'Leary, D.M., Cassidy, E.M. and O'Connor, J.J. 1997. Group II and III metabotropic glutamate receptors modulate paired pulse depression in the rat dentate gyrus in vitro. Eur J Pharmacol. 340(1), pp. 35-44.
Park, E.C., Ghose, P., Shao, Z., Ye, Q., Kang, L., Xu, X.S., Powell‐Coffman, J.A. and Rongo, C., 2012. Hypoxia regulates glutamate receptor trafficking through an HIF‐independent mechanism. The EMBO journal, 31(6), pp.1379-1393.
Peng, K., Tan, D., He, M., Guo, D., Huang, J., Wang, X., Liu, C. and Zheng, X., 2016. Studies on cerebral protection of digoxin against hypoxic–ischemic brain damage in neonatal rats. NeuroReport, 27(12), pp.906-915.
Rudolphi, K.A., Schubert, P., Parkinson, F.E. and Fredholm, B.B., 1992. Neuroprotective role of adenosine in cerebral ischaemia. Trends in pharmacological sciences, 13, pp.439-445.
Sebastiao, A.M., de Mendonca, A., Moreira, T. and Ribeiro, J.A., 2001. Activation of synaptic NMDA receptors by action potential-dependent release of transmitter during hypoxia impairs recovery of synaptic transmission on reoxygenation. Journal of Neuroscience, 21(21), pp.8564-8571.
Shaminder, K.A.U.R., Rehni, A.K., Singh, N. and Jaggi, A.S., 2009. Studies on cerebral protection of digoxin against ischemia/reperfusion injury in mice. Yakugaku Zasshi, 129(4), pp.435-443.
Shi, G.F., An, L.J., Jiang, B., Guan, S. and Bao, Y.M., 2006. Alpinia protocatechuic acid protects against oxidative damage in vitro and reduces oxidative stress in vivo. Neuroscience letters, 403(3), pp.206-210.
Siddiq, A., Aminova, L.R. and Ratan, R.R., 2007. Hypoxia inducible factor prolyl 4-hydroxylase enzymes: center stage in the battle against hypoxia, metabolic compromise and oxidative stress. Neurochemical research, 32(4-5), pp.931-946.
Siddiq, A., Aminova, L.R., Troy, C.M., Suh, K., Messer, Z., Semenza, G.L. and Ratan, R.R., 2009. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF-and CREB-independent pathways. Journal of Neuroscience, 29(27), pp.8828-8838.
Singh, D.P., Nimker, C., Paliwal, P. and Bansal, A., 2016. Ethyl 3, 4-dihydroxybenzoate (EDHB): a prolyl hydroxylase inhibitor attenuates acute hypobaric hypoxia mediated vascular leakage in brain. The Journal of Physiological Sciences, 66(4), pp.315-326.
Stoppini, L., Buchs, P.A. and Muller, D., 1991. A simple method for organotypic cultures of nervous tissue. Journal of neuroscience methods, 37(2), pp.173-182.
Tóth, K., Freund, T.F. and Miles, R., 1997. Disinhibition of rat hippocampal pyramidal cells by GABAergic afferents from the septum. The Journal of physiology, 500(2), pp.463-474.
Vida, I., Bartos, M. and Jonas, P., 2006. Shunting inhibition improves robustness of gamma oscillations in hippocampal interneuron networks by homogenizing firing rates. Neuron, 49(1), pp.107-117.
Wang, J., Buss, J.L., Chen, G., Ponka, P. and Pantopoulos, K., 2002. The prolyl 4‐hydroxylase inhibitor ethyl‐3, 4‐dihydroxybenzoate generates effective iron deficiency in cultured cells. FEBS letters, 529(2-3), pp.309-312.
Yao, H., Huang, Y.H., Liu, Z.W., Wan, Q., Ding, A.S., Zhao, B., Fan, M. and Wang, F.Z., 1998. The different responses to anoxia in cultured CA1 and DG neurons from newborn rats. Sheng li xue bao:[Acta physiologica Sinica], 50(1), pp.61-66.
Zhang, H., Qian, D.Z., Tan, Y.S., Lee, K., Gao, P., Ren, Y.R., Rey, S., Hammers, H., Chang, D., Pili, R. and Dang, C.V., 2008. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proceedings of the National Academy of Sciences, 105(50), pp.19579-19586.
Zhang, X.Y., Cao, J.B., Zhang, L.M., Li, Y.F. and Mi, W.D., 2015. Deferoxamine attenuates lipopolysaccharide-induced neuroinflammation and memory impairment in mice. Journal of neuroinflammation, 12(1), p.20.
Ziobro JM, Deshpande LS, Delorenzo RJ. 2011. An organotypic hippocampal slice culture model of excitotoxic injury induced spontaneous recurrent epileptiform discharges. Brain Res. 1371, pp110-20.
9