vitamin b6 reduces hippocampal apoptosis in experimental ... · research article open access...
TRANSCRIPT

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393http://www.biomedcentral.com/1471-2334/13/393
source: http://boris.unibe.ch/52654/ | downloaded: 13.3.2017
RESEARCH ARTICLE Open Access
Vitamin B6 reduces hippocampal apoptosis inexperimental pneumococcal meningitisDenise C Zysset-Burri1,2,3, Caroline L Bellac4, Stephen L Leib1,4* and Matthias Wittwer1
Abstract
Background: Bacterial meningitis caused by Streptococcus pneumoniae leads to death in up to 30% of patients andleaves up to half of the survivors with neurological sequelae. The inflammatory host reaction initiates the inductionof the kynurenine pathway and contributes to hippocampal apoptosis, a form of brain damage that is associatedwith learning and memory deficits in experimental paradigms. Vitamin B6 is an enzymatic cofactor in thekynurenine pathway and may thus limit the accumulation of neurotoxic metabolites and preserve the cellularenergy status.The aim of this study in a pneumococcal meningitis model was to investigate the effect of vitamin B6 onhippocampal apoptosis by histomorphology, by transcriptomics and by measurement of cellular nicotine amideadenine dinucleotide content.
Methods and results: Eleven day old Wistar rats were infected with 1x106 cfu/ml of S. pneumoniae andrandomized for treatment with vitamin B6 or saline as controls. Vitamin B6 led to a significant (p > 0.02) reductionof hippocampal apoptosis. According to functional annotation based clustering, vitamin B6 led to down-regulationof genes involved in processes of inflammatory response, while genes encoding for processes related to circadianrhythm, neuronal signaling and apoptotic cell death were mostly up-regulated.
Conclusions: Our results provide evidence that attenuation of apoptosis by vitamin B6 is multi-factorial includingdown-modulation of inflammation, up-regulation of the neuroprotective brain-derived neurotrophic factor andprevention of the exhaustion of cellular energy stores. The neuroprotective effect identifies vitamin B6 as a potentialtarget for the development of strategies to attenuate brain injury in bacterial meningitis.
Keywords: Bacterial meningitis, Streptococcus pneumoniae, Kynurenine pathway, Vitamin B6
BackgroundBacterial meningitis (BM) caused by S. pneumoniae is alife-threatening disease associated with high mortalityand morbidity rates. In spite of effective antimicrobialtherapy and intensive care, about 50% of survivors sufferfrom long-term sequelae, including hearing loss, neuro-functional problems, seizure disorders, sensory-motordeficits, and persisting learning and memory difficulties[1-3].Two pathophysiologically different forms of brain inju-
ry, namely hippocampal apoptosis and cortical necrosis,
* Correspondence: [email protected] Division, Spiez Laboratory, Federal Office for Civil Protection,Austrasse, CH-3700, Spiez, Switzerland4Neuroinfection Laboratory, Institute for Infectious Diseases, University ofBern, Friedbühlstrasse 51, CH-3010, Bern, SwitzerlandFull list of author information is available at the end of the article
© 2013 Zysset-Burri et al.; licensee BioMed CeCreative Commons Attribution License (http:/distribution, and reproduction in any medium
have been demonstrated in patients [4] and in corre-sponding experimental animal models of BM. Damageto the hippocampal formation has been associated withlearning and memory impairments [3,5].Inflammatory conditions in the brain induce trypto-
phan (TRP) degradation through the kynurenine (KYN)pathway, resulting in several neuroactive metaboliteswhich can be both, neurotoxic or neuroprotective(Figure 1). The KYN pathway may be involved in themechanisms leading to brain damage associated with in-flammatory brain diseases, such as multiple sclerosis orcerebral malaria [6,7]. The pathophysiology of pneumo-coccal meningitis is initiated by activation of the im-mune system of the host, leading to the induction ofmetabolic pathways in the brain [6]. Increased TRP deg-radation caused by the activation of the KYN pathwaymay also be involved in the processes that result in
ntral Ltd. This is an Open Access article distributed under the terms of the/creativecommons.org/licenses/by/2.0), which permits unrestricted use,, provided the original work is properly cited.

Tryptophan (TRP)
Kynurenine (KYN)
Indoleamine 2,3-dioxygenase
3-Hydroxykynurenine (3-HKYN) Kynurenic acid (KYNA)Anthranilic acid (AA)
Kynurenine 3-hydroxylaseKynurenine aminotransferase
Kynureninase
3-Hydroxyanthranilic acid (3-HAA)
KynureninaseAnthranilate 3-hydroxylase
Quinolinic acid
3-Hydroxyanthranilate 3,4-dioxygenase
Nicotinamide adenine dinucleotide (NAD+)
Figure 1 Schematic of the kynurenine pathway in the rat brain. Tryptophan is metabolized over multiple steps into quinolinic acid, finallyresulting in de novo synthesis of NAD+. Several neuroactive intermediates are included in this pathway: neuroprotective kynurenic acid,neurotoxic 3-hydroxykynurenine and neurotoxic 3-hydroxyanthranilic acid. Neurotoxic intermediates are written in red, neuroprotective ones ingreen [6,7].
Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 2 of 15http://www.biomedcentral.com/1471-2334/13/393
neuronal damage observed in pneumococcal meningitis[2,6,8]. The neurotoxic effect of the intermediates3-hydroxykynurenine and 3-hydroxyanthanilic acid in-volves the generation of superoxide and hydrogen pe-roxide that contribute to oxidative processes implicatedin the pathophysiology of meningitis. In contrast, neu-roprotective kynurenic acid (KYNA), an antagonist ofthe excitotoxic N-methyl-D-aspartate (NMDA) receptor,protects from excitotoxic brain damage in experimentalBM [6]. Furthermore, the catabolism of TRP over theKYN pathway is the exclusive de novo synthesis pathwayfor nicotine amide adenine dinucleotide (NAD+) ineukaryotic cells [6]. NAD+ fuels the poly(adenosine5′-diphosphate (ADP)-ribose) polymerase whose over-activation during neuro-inflammatory diseases may de-plete intracellular NAD+ levels and thus, resulting innecrotic cell death [9]. Therefore, the KYN pathway in-duced in pneumococcal meningitis may influence thefate of neuronal tissue over NAD+ supply [6,9].Pyridoxal 5′-phosphate, the active form of vitamin B6,
optimizes the substrate flux in the KYN pathway by act-ing as cofactor for two key enzymes, KYN aminotrans-ferase and kynureninase [10]. Administration of vitaminB6 may attenuate neuronal cell death in BM by pre-venting both, the accumulation of neurotoxic intermedi-ates of the KYN pathway and cellular energy depletionby enhancing the de novo synthesis of NAD+.In the present study, we evaluated the mode of action
of vitamin B6 by microarrays. We interpreted thetranscriptomic data using biological system based ana-lysis rather than a “gene by gene” approach. The GeneOntology (GO) [11] and the Kyoto Encyclopedia ofGenes and Genomes (KEGG) pathway [12] database
provide a basis for grouping genes according to theirmolecular functions, biologic processes and cellular com-ponents, and their involvement in concordant cellularpathways, respectively.Histopathological analysis showed that vitamin B6 sig-
nificantly reduced hippocampal apoptosis in pneumo-coccal meningitis. Furthermore, based on fluorescencemeasurements of hippocampal NAD+ levels, an effectof vitamin B6 in preserving cellular energy stores wasfound.
MethodsEthics statementAll animal studies were approved by the Animal Careand Experimentation Committee of the Canton of Bern,Switzerland (Nr. 26/07), and followed the Swiss nationalguidelines for the performance of animal experiments.
Model of experimental pneumococcal meningitisWe used an established model of experimental pneumo-coccal meningitis in infant rats [13]. On postnatal day11, Wistar rats (n = 28) were infected by intracisternalinjection of 10μl of saline solution containing 1 × 106
cfu/ml of S. pneumoniae (serotype 3). At time of in-fection, animals (n = 14) received 360μl of vitaminB6 subcutaneously (s.c.; 600mg/kg; Streuli, Uznach,Switzerland). Placebo-treated animals (n = 14) wereinjected s.c. with 360 μl of 0.85% NaCl. Eighteenhours after infection, all animals were treated s.c. with100 mg/kg of the antibiotic ceftriaxone (Roche Pharma,Reinach, Switzerland) and a second dose of vitamin B6or 0.85% NaCl was administered. At the same timepoint, infection was documented by quantitative culture

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 3 of 15http://www.biomedcentral.com/1471-2334/13/393
of 5 μl of cerebrospinal fluid (CSF) and all rats wereweighed and clinically assessed using the following scoresystem: 1 for comatose animals, 2 for rats that do notturn upright after positioning on the back, 3 for animalsthat turn within 30 s, 4 for animals that turn within lessthan 5 s and 5 for rats with normal activity [13]. Twentyfour hours after infection, the rats were sacrificed by anoverdose of intraperitoneal (i.p.) pentobarbital (100 mg/kg,Esconarkon, Streuli & Co. AG, Uznach, Switzerland).For NAD+ measurements, Wistar rats (n = 15) were
infected by intracisternal injection of 10 μl of 1×106 cfu/mlof S. pneumoniae. At time of infection, animals were ran-domized for treatment with vitamin B6 (600 mg/kg s.c. 0and 18 h p.i., n = 6) or an equal volume (360 μl) of 0.85%NaCl (s.c., n = 9). Three saline-treated rats were sacrificedat the same time point (0h p.i.). The antibiotic therapywas started 18 h post-infection (100 mg/kg ceftriaxone,s.c.). At the same time point, 3 vitamin B6- and 3 saline-treated rats were sacrificed (18 h p.i.), and a second appli-cation of vitamin B6 and 0.85% NaCl, respectively, wereadministered to the remaining 6 animals. These animalswere sacrificed 24 h after infection (24 h p.i.).
Tissue processingImmediately after sacrifice, the animals were perfusedvia the left cardiac ventricle with 30 ml of RNAse-freeice-cold phosphate buffered saline (PBS). The brainswere dissected followed by removal of the meninges andsegmentation of the brains into the 2 hemispheres. Theright hemisphere was fixed in 4% paraformaldehyde(Grogg, Stettlen-Deisswil, Switzerland) in PBS for 3 daysat 4°C and then cryo-protected in 18% sucrose at 4°Cuntil further processing for histopathological assessmentof brain injury. From the left hemisphere the hippocam-pus was dissected in ice-cold PBS, stored in RNAlater®(Ambion Europe Ltd., Huntingdon, UK) for 1 day at 4°Cand subsequently at −80°C until isolation of RNA. ForNAD+ measurements, the hippocampus of the lefthemisphere was frozen on dry ice and stored at −80°C.
HistopathologyTo assess the brain damage caused by BM the brainswere analyzed histomorphologically. The cryo-protectedbrains were frozen in 2-methylbutane (−50°C), and fromeach animal four 45 μm cryo-sections of the dentategyrus were cut using a Cryostat (Leica CM1850 cryostat)and transferred onto a gelatin/chrom alum-coated glassslide. The slides were put in Xylol, hydrated, Nisslstained with cresyl violet, dehydrated and mounted withEntellan® (Merck, Darmstadt, Germany). The amount ofapoptotic cells in the dentate gyrus of the hippocampusand of the extent of damage to the cerebral cortex wereevaluated using bright-field microscopy. Neurons of thedentate granule cell layer with morphological changes
characteristic for apoptosis (condensed, fragmented nu-clei and/or apoptotic bodies) were counted in 3 visualfields (400× magnification) in each of the 2 blades of thedentate gyrus. An average score per animal was calcu-lated from all sections evaluated, applying the followingscoring system: 0–5 cells = 0, 6–20 cells = 1 and >20cells = 2 [14]. The cortical damage was assessed as theamount of damage of the total volume of the cortex aspreviously reported [15].
RNA isolation, quality control and chip hybridizationFrom tissue samples of the hippocampus total RNA wasisolated using the magnetic beads based EZ1 RNA Uni-versal Tissue Kit (Qiagen, Basel, Switzerland) and EZ1BioRobot (Qiagen). Tissue stabilized in RNAlater® wasmixed with 750 μl QIAzol® Lysis reagent. Samples wereimmediately homogenized by a rotor-stator homogenizer(TissueRuptor®, Qiagen). After incubation for 5 min atroom temperature, 150 μl chloroform (Grogg) was addedto the homogenized tissue samples. A centrifuging stepfor 15 min at 4°C and 12’000 rpm resulted in the separ-ation of the sample into 3 phases. 300 μl of the upperphase containing RNA was used as starting material forRNA isolation using the EZ1 BioRobot, following themanufacturer’s protocol.Quantification of RNA was performed on the Agilent
2100 Bioanalyzer platform (RNA 6000 Nano, AgilentTechnologies, Waldbronn, Germany) and validated onthe NanoDrop® (NanoDrop, Wilmington, USA) device.From 28 histopathologically evaluated rat brains with
evidence for apoptosis, RNA extracts from 5 vitamin B6-and 5 saline-treated animals were selected randomly forarray hybridization. Chip hybridization was performed incooperation with the Lausanne DNA Array Facility(University of Lausanne, Switzerland). Double-strandedcDNAs were synthesized from 100ng of total RNA usingT7 promoter-(N) 6 primers (Affymetrix, Santa Clara,CA) and the Whole Target Transcript cDNA synthesiskit (Affymetrix). Quantification and quality control ofcDNA was performed by NanoDrop® and Agilent 2100Bioanalyzer platform, respectively. Three microgram offragmented, biotinylated cDNA was hybridized in aHybe Oven (GeneChip® 640) overnight onto com-mercially available GeneChip® Rat Gene 1.0 ST Array(Affymetrix) containing over 27’000 rat genes. The hy-bridized samples were stained with streptavidin phyco-erythrin and the signal was amplified by a biotinylatedanti-streptavidin antibody. Washing, staining and ampli-fication were performed in an Affymetrix GeneChip®Fluidics Station 450. The components required for thesesteps were provided by the GeneChip® Hybridization,Wash, and Stain kit (Affymetrix). Microarrays werescanned in an Affymetrix GeneArray® scanner 3000.Resulting image files served as basis for the calculation

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 4 of 15http://www.biomedcentral.com/1471-2334/13/393
of signal intensities with the Affymetrix GeneChip®Operating Software (GCOS).
DataminingAll data is MIAME compliant and has been deposited inthe ArrayExpress database of the European Bioinformat-ics Institute (http://www.ebi.ac.uk/arrayexpress, acces-sion number E-MEXP-3555).Chip data analysis was carried out on the R platform
for statistical programming using packages from theBioconductor project [16]. Because of the asymmetricdistribution of microarray data, expression values werelog2 transformed. Background correction, quantile nor-malization and probe set summary (robust regression,only perfect matches) were performed with non linearmethods based on the robust multi average (RMA) func-tion of the Bioconductor affy package [17]. Chip qualitycontrol was explorative evaluated using box plots of theraw log scale intensities and MA-plots visualizing signalintensity dependent effects on the log-ratios (affypackage).To reduce the number of hypothesis to be tested in
the adjacent significance tests, genes were filtered basedon the following criteria: all genes that were expressedunder the estimated background intensity of 26 fluores-cent units on at least 4 of the 10 arrays and genes withan interquantile range of less than 0.001 were excluded.Differentially expressed genes were identified by usingthe linear models for microarray data (limma) package[18] which implements a moderated t-statistic for signifi-cance testing. The type 1 error rate was adjusted to 1%using the Benjamini-Hochberg false discovery rate algo-rithm [19]. Genes which had cross-hybridized on thechip or which had no annotation in any existing databank were excluded from further analysis.The transcriptomic data were evaluated by the func-
tional annotation clustering tool of DAVID (Databasefor Annotation, Visualization and Integrated Discovery)bioinformatics (http://david.abcc.ncifcrf.gov/) for GOstatistics [11] and by biological system based analysisusing the KEGG pathway database (http://www.genome.jp/kegg) for pathway analysis [12].
Table 1 TaqMan® gene expression assays (Applied Biosystem
Name
Brain-derived neurotrophic factor (BDNF)
Neuronal PAS domain protein 4 (Npas4)
Lysozyme 2 (Lyz2)
Platelet-activating factor acetylhydrolase 2 (Pafah2)
Nuclear receptor subfamily 4, group A, member 1 (Nr4a1)
Ribosomal protein L24 (Rpl24)
The genes selected for chip validation by real time PCR and their corresponding refRpl24 was used as housekeeping gene.
Chip validation by real time PCRcDNA was synthesized from 1.5 μg of total RNA usingthe High-Capacity cDNA Reverse Transcription Kit(Applied Biosystems, Foster City, CA), according to themanufacturer’s protocol. The cDNA samples were di-luted 1:5 with RNAse-free water and aliquots werestored at −20°C. Real time PCR was performed using theQuantiFast Probe PCR kit from Qiagen (composed ofHot Star Taq Plus DNA polymerase and dNTP mixin PCR buffer) and TaqMan® Gene Expression Assays(Applied Biosystems).All reactions were carried out as duplicates. Template
cDNA was amplified with the Rotor-Gene Q platform(Corbet RESEARCH) operating with the Run on Soft-ware version “Rotor-Gene 1.7.87”.The primers used for this PCR and their reference se-
quences (RefSeq) as well as their ordering numbers ofApplied Biosystems are listened in Table 1. The delta Ctvalues were calculated based on normalization to thehousekeeping gene ribosomal protein L24 (Rpl24).
Hippocampal NAD+ levelsFor assessment of cellular energy status NAD+ levelswere measured in hippocampal tissue from rats with BMtreated with vitamin B6 or saline at 0, 18 and 24 hoursafter infection (n=3 for each experimental group andtime).Frozen dissected hippocampi were homogenized 1:10
(wt/vol) in ice-cold assay buffer (50 mM Tris and 2 mMMgCl2, pH 8.0) and 50 μl of the homogenates weretransferred into a 96-well fluorescence plate. NAD+was quantified according to the method of Putt andHegenrother [20]. The plate was read on a SpectraMaxPlus (Molecular Devices, Sunnyvale, CA) with an excita-tion of 360 nm and an emission of 445 nm, and valueswere plotted against a NAD+ calibration curve (Sigma,St. Louis, MO).
ResultsClinical parameters of meningitisBy 18 h after infection, all rats infected with S. pneu-moniae had meningitis, as evidenced by positive bac-terial titers in the CSF (log10 6.8 - log10 8.0 cfu/ml)
s)
RefSeq Assay number
NM_012513.3 Rn01484924_m1
NM_153626.1 Rn00596522_m1
NM_012771.2 Rn00562794_m1
NM_177932.2 Rn00710058_m1
NM_024388.1 Rn01533237_m1
NM_001007637.1 Rn01455518_g1
erence sequences (RefSeq) and ordering numbers of Applied Biosystems.
Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 5 of 15http://www.biomedcentral.com/1471-2334/13/393
and reduced weight gain. Between 18 h and 24 h post-infection, the animals treated with vitamin B6 lost 0.50 ±0.04 g of body weight, whereas the weight of placebo-treated animals was reduced by 0.74 ± 0.02 g (p < 0.07,unpaired t test).
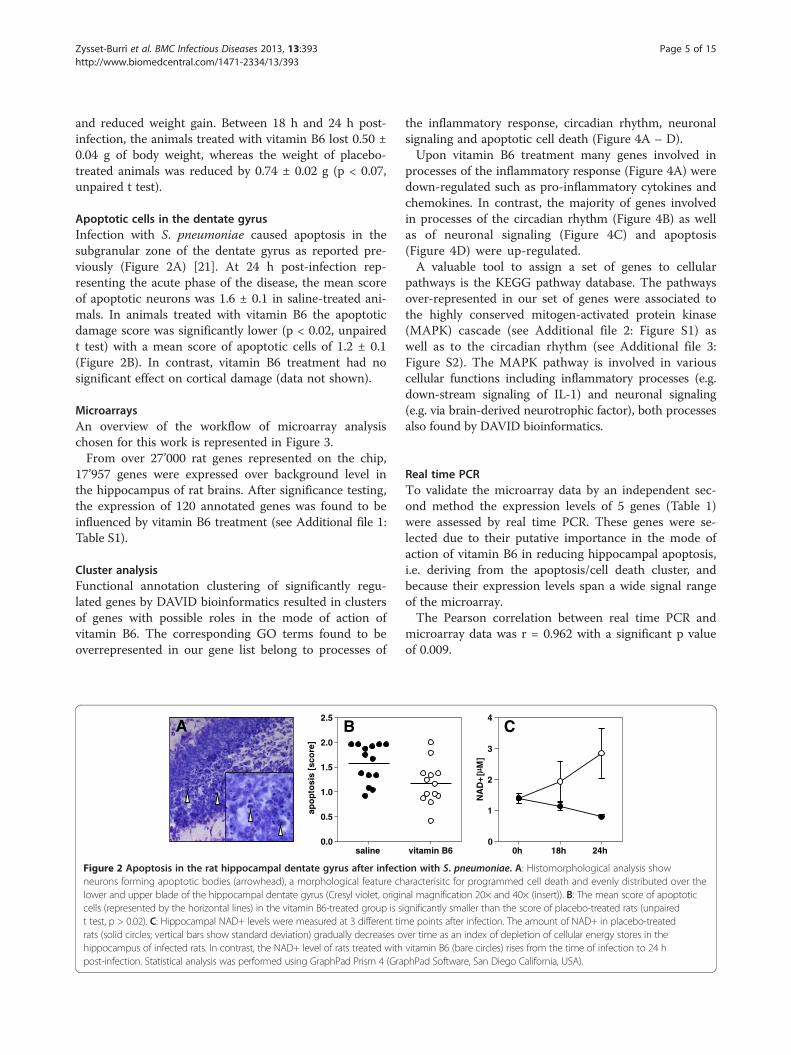
Apoptotic cells in the dentate gyrusInfection with S. pneumoniae caused apoptosis in thesubgranular zone of the dentate gyrus as reported pre-viously (Figure 2A) [21]. At 24 h post-infection rep-resenting the acute phase of the disease, the mean scoreof apoptotic neurons was 1.6 ± 0.1 in saline-treated ani-mals. In animals treated with vitamin B6 the apoptoticdamage score was significantly lower (p < 0.02, unpairedt test) with a mean score of apoptotic cells of 1.2 ± 0.1(Figure 2B). In contrast, vitamin B6 treatment had nosignificant effect on cortical damage (data not shown).
MicroarraysAn overview of the workflow of microarray analysischosen for this work is represented in Figure 3.From over 27’000 rat genes represented on the chip,
17’957 genes were expressed over background level inthe hippocampus of rat brains. After significance testing,the expression of 120 annotated genes was found to beinfluenced by vitamin B6 treatment (see Additional file 1:Table S1).
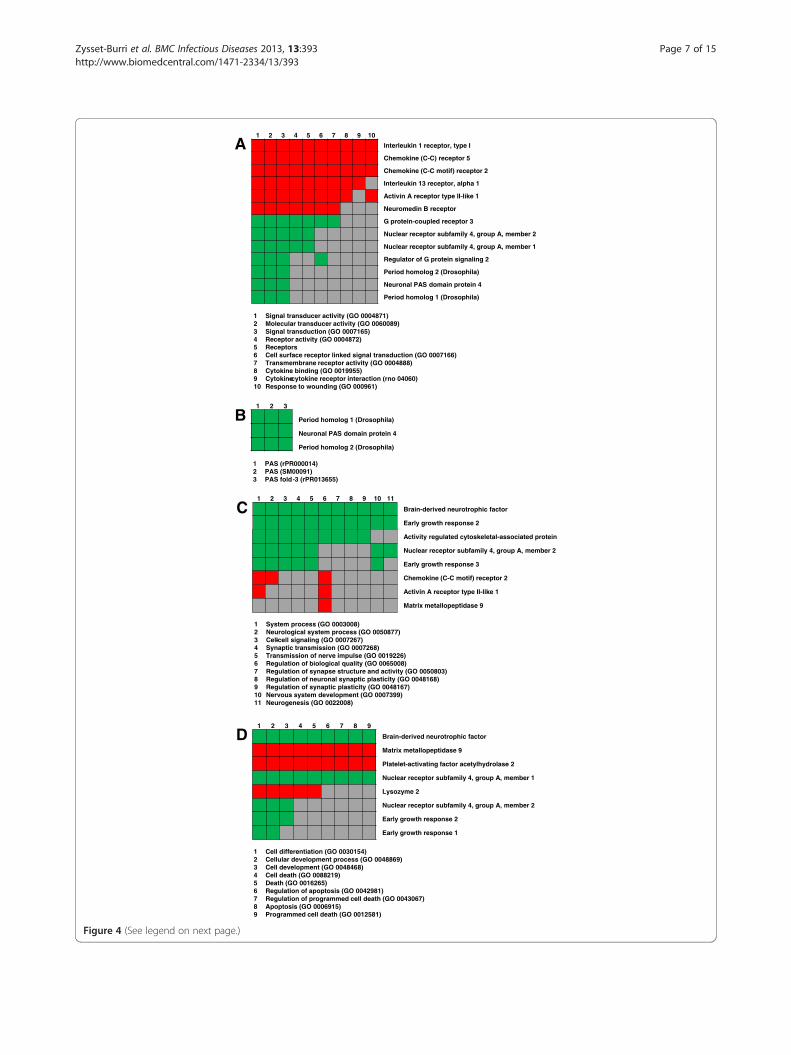
Cluster analysisFunctional annotation clustering of significantly regu-lated genes by DAVID bioinformatics resulted in clustersof genes with possible roles in the mode of action ofvitamin B6. The corresponding GO terms found to beoverrepresented in our gene list belong to processes of
saline0.0
0.5
1.0
1.5
2.0
2.5
apo
pto
sis
[sco
re]
A B
Figure 2 Apoptosis in the rat hippocampal dentate gyrus after infectneurons forming apoptotic bodies (arrowhead), a morphological feature chlower and upper blade of the hippocampal dentate gyrus (Cresyl violet, origincells (represented by the horizontal lines) in the vitamin B6-treated group is st test, p > 0.02). C: Hippocampal NAD+ levels were measured at 3 different timrats (solid circles; vertical bars show standard deviation) gradually decreases ohippocampus of infected rats. In contrast, the NAD+ level of rats treated withpost-infection. Statistical analysis was performed using GraphPad Prism 4 (Gra
the inflammatory response, circadian rhythm, neuronalsignaling and apoptotic cell death (Figure 4A – D).Upon vitamin B6 treatment many genes involved in
processes of the inflammatory response (Figure 4A) weredown-regulated such as pro-inflammatory cytokines andchemokines. In contrast, the majority of genes involvedin processes of the circadian rhythm (Figure 4B) as wellas of neuronal signaling (Figure 4C) and apoptosis(Figure 4D) were up-regulated.A valuable tool to assign a set of genes to cellular
pathways is the KEGG pathway database. The pathwaysover-represented in our set of genes were associated tothe highly conserved mitogen-activated protein kinase(MAPK) cascade (see Additional file 2: Figure S1) aswell as to the circadian rhythm (see Additional file 3:Figure S2). The MAPK pathway is involved in variouscellular functions including inflammatory processes (e.g.down-stream signaling of IL-1) and neuronal signaling(e.g. via brain-derived neurotrophic factor), both processesalso found by DAVID bioinformatics.
Real time PCRTo validate the microarray data by an independent sec-ond method the expression levels of 5 genes (Table 1)were assessed by real time PCR. These genes were se-lected due to their putative importance in the mode ofaction of vitamin B6 in reducing hippocampal apoptosis,i.e. deriving from the apoptosis/cell death cluster, andbecause their expression levels span a wide signal rangeof the microarray.The Pearson correlation between real time PCR and
microarray data was r = 0.962 with a significant p valueof 0.009.
vitamin B6 0h 18h 24h0
1
2
3
4
NA
D+
[M
]
C
ion with S. pneumoniae. A: Histomorphological analysis showaracterisitc for programmed cell death and evenly distributed over theal magnification 20× and 40× (insert)). B: The mean score of apoptoticignificantly smaller than the score of placebo-treated rats (unpairede points after infection. The amount of NAD+ in placebo-treated
ver time as an index of depletion of cellular energy stores in thevitamin B6 (bare circles) rises from the time of infection to 24 hphPad Software, San Diego California, USA).

GeneChip® Rat Gene 1.0 ST Array (Affymetrix) containing over 27`000 genes
(rat genome)
Subtraction of all genes expressed below background and with a small signal variance
Number of expressed genes in the rat brain: 17`957
Significance testing / type I error correction using limma package
Number of differentially expressed genes between vitamin B6- and placebo-treated
rats: 120 Datamining
Analysis tools and chip validation
Cluster analysis
KEGG: pathway analysis DAVID bioinformatics
Apoptosis/cell death cluster
Circadian rhythm cluster
Chip validation based on RNA level
Apoptosis parameters
PCR
Inflammatory response cluster
Neuronal signaling cluster
Correlation between array and apoptosis score
Figure 3 Data analysis flowchart. Overview of the workflow of data analysis chosen for the present work. Analysis was carried out onthe R platform for statistical programming, cluster analysis was performed according to DAVID bioinformatics and pathway analysis wasbased on KEGG.
Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 6 of 15http://www.biomedcentral.com/1471-2334/13/393
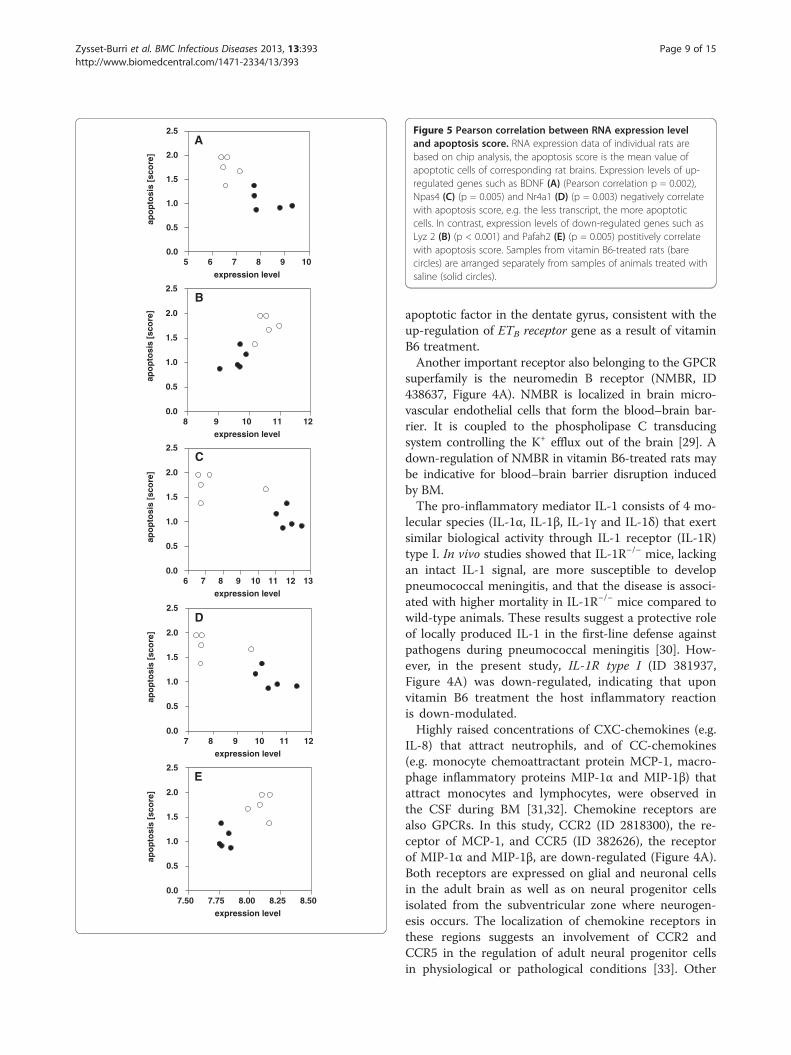
Correlation between RNA expression level and apoptosisscorePearson correlation between RNA expression levels ofselected genes and apoptosis score was highly significantwith p values between < 0.001 and 0.005 (Figure 5). Thetranscripts chosen for the correlation analysis were thesame as assessed by real time PCR.Due to these highly significant correlations between
expression level and apoptosis score, the selected genesare candidates for apoptosis markers, although the timepoint is late (24 h post-infection) in regard to the notionthat pro- and anti-apoptotic factors change at 12-16 hafter infection [22]. However, further experiments areneeded to determine the application of these genes aspotential apoptosis markers.
NAD+ levels in hippocampal tissueHippocampal NAD+ levels of rats with BM droppedafter infection, indicating a decrease of cellular energy inthe course of the disease. In contrast to the placebogroup, the amount of NAD+ in the hippocampus of ratstreated with vitamin B6 increased during the same timeperiod (Figure 2C). Thus, an effect of vitamin B6 to pre-serve cellular energy stores, likely by optimizing the sub-strate flux in the kynurenine pathway, was found duringthe acute phase of BM when hippocampal apoptosisdevelops.
DiscussionApoptosis of cells in the hippocampal dentate gyrus is acharacteristic form of brain damage in BM [3,21]. In
1 2 3 4 5 6 7 8 9 10
Interleukin 1 receptor, type I
Chemokine (C-C) receptor 5
Chemokine (C-C motif) receptor 2
Interleukin 13 receptor, alpha 1
Activin A receptor type II-like 1
Neuromedin B receptor
G protein-coupled receptor 3
Nuclear receptor subfamily 4, group A, member 2
Nuclear receptor subfamily 4, group A, member 1
Regulator of G protein signaling 2
Period homolog 2 (Drosophila)
Neuronal PAS domain protein 4
Period homolog 1 (Drosophila)
1 Signal transducer activity (GO 0004871)2 Molecular transducer activity (GO 0060089)3 Signal transduction (GO 0007165)4 Receptor activity (GO 0004872)5 Receptors6 Cell surface receptor linked signal transduction (GO 0007166)7 Transmembrane receptor activity (GO 0004888)8 Cytokine binding (GO 0019955)9 Cytokine-cytokine receptor interaction (rno 04060)10 Response to wounding (GO 000961)
A
B
1 System process (GO 0003008)2 Neurological system process (GO 0050877)3 Cell-cell signaling (GO 0007267)4 Synaptic transmission (GO 0007268)5 Transmission of nerve impulse (GO 0019226)6 Regulation of biological quality (GO 0065008)7 Regulation of synapse structure and activity (GO 0050803)8 Regulation of neuronal synaptic plasticity (GO 0048168)9 Regulation of synaptic plasticity (GO 0048167)10 Nervous system development (GO 0007399)11 Neurogenesis (GO 0022008)
1 2 3
Period homolog 1 (Drosophila)
Neuronal PAS domain protein 4
Period homolog 2 (Drosophila)
D
1 PAS (rPR000014)2 PAS (SM00091)3 PAS fold -3 (rPR013655)
1 2 3 4 5 6 7 8 9
Brain-derived neurotrophic factor
Matrix metallopeptidase 9
Platelet-activating factor acetylhydrolase 2
Nuclear receptor subfamily 4, group A, member 1
Lysozyme 2
Nuclear receptor subfamily 4, group A, member 2
Early growth response 2
Early growth response 1
1 Cell differentiation (GO 0030154)2 Cellular development process (GO 0048869)3 Cell development (GO 0048468)4 Cell death (GO 0088219)5 Death (GO 0016265)6 Regulation of apoptosis (GO 0042981)7 Regulation of programmed cell death (GO 0043067)8 Apoptosis (GO 0006915)9 Programmed cell death (GO 0012581)
C1 2 3 4 5 6 7 8 9 10 11
Brain-derived neurotrophic factor
Early growth response 2
Activity regulated cytoskeletal-associated protein
Nuclear receptor subfamily 4, group A, member 2
Early growth response 3
Chemokine (C-C motif) receptor 2
Activin A receptor type II-like 1
Matrix metallopeptidase 9
Figure 4 (See legend on next page.)
Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 7 of 15http://www.biomedcentral.com/1471-2334/13/393

(See figure on previous page.)Figure 4 Functional annotation cluster. The 120 significantly expressed genes were clustered into functionally related groups such asinflammatory response (A), circadian rhythm (B), neuronal signaling (C) and apoptosis / cell death (D) based on DAVID bioinformatics. The genesbelonging to the individual clusters are listened on the right side. The suitable functions with the corresponding GO terms are mentioned belowthe clusters. Green marked squares state that the gene is up-regulated in that GO term, red marked squares stand for down-regulated genes, andgrey filled squares mean that the gene is not involved in that process.
Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 8 of 15http://www.biomedcentral.com/1471-2334/13/393
experimental models an association of injury to the den-tate gyrus with learning and memory deficits has beenshown [5]. The present study demonstrates that treat-ment with vitamin B6 reduces the number of apoptoticcells in the hippocampal dentate gyrus. We investigatedthe mechanisms underlying this neuroprotective effectby studying the influence of vitamin B6 on the transcrip-tome and on cellular energy stores.In the model used, hippocampal apoptosis starts to
occur during the acute phase of BM (18-24 h) with apeak in the sub-acute phase at 36 h, and reaches controllevels in the late phase of the disease (46-72 h). Recentstudies in our lab showed that in the acute phase of BM,genes that are significantly expressed in the hippocam-pus and cortex are mainly involved in processes of theimmune and inflammatory response [23]. The innate im-mune system has a predominant role in immune defensein the otherwise immune privileged brain tissue. How-ever, neurological complications secondary to BM [3]suggest that the host defense mechanisms are inefficientin eliminating the pathogen and furthermore, that thehost inflammatory reactions contribute considerably tothe brain damage observed in the disease. As shown pre-viously, the major events on transcriptional level con-cerning the regulation of the host immune responseoccur within the first 3 days after infection [23].A promising candidate for pre-treatment of BM is vita-
min B6 administered during the acute phase of the dis-ease. A prerequisite for the application of a therapeutictarget is the understanding of the processes leading to thedesired effects, in the case of vitamin B6 to a reduction ofhippocampal apoptosis. Thus, in the present study, we fo-cused on the mechanisms that take place as a conse-quence of vitamin B6 treatment in the acute phase of BM.The time point for the second application of vitaminB6 (18 h post-infection) and the endpoint at 24 h post-infection were chosen during the acute phase of BMaccording to recent findings which define a time window fortherapeutic interventions during this phase of the disease.In order to handle the huge data mass resulting from a
microarray study, we clustered the significantly regulatedtranscripts according to their involvement in given bio-logical processes (Figure 4).
Inflammatory responseThe first step in immune activation is the recognition ofbacterial products by pattern recognition receptors, such
as Toll-like receptors (TLRs), expressed on resident cellsof the central nervous system (CNS) [3]. Experimentalstudies showed that primary mouse microglial cells, theresident macrophages of the CNS, are activated uponstimulation with agonists of several TLRs. Activatedmicroglia release inflammatory mediators such as nitricoxide (NO) or activin A. Activin A is a member of thetransforming growth factor-β (TGF-β) family whichplays a central role in different aspects of cell growthand differentiation [24]. Previous studies showed thatactivin A is increased in the CSF of patients sufferingfrom BM. Furthermore, activin A may decrease the secre-tion of NO and pro-inflammatory mediators by activatedmicroglia, and enhances microglial proliferation, sugges-ting that activin A may counteract the activation of micro-glial cells in BM [25]. The biological functions of membersof the TGF-β family are mediated by 2 types of transmem-brane serine/threonine kinase receptors (type I and II) thatare expressed on activated microglial cells. Therefore,microglia is not only the source but also the target cells ofactivin A in the CNS [25]. The down-regulation of theactivin A receptor type II-like 1 (ID 382555, Figure 4A)upon vitamin B6 treatment may indicate a reduced activa-tion of microglia and thus, a decreased release of tissue-destructive mediators.G protein coupled receptors (GPCRs) change their
conformation upon stimulation by an activating signal.This activated state is controlled by regulators of G pro-tein signaling (RGS) [26,27]. RGS2 (ID 377853) as wellas GPCR3 (ID 435019) are up-regulated in vitamin B6-treated rats compared to saline-treated animals, sug-gesting an involvement of G protein signaling in cellularprocesses leading to a reduction of hippocampal apop-tosis (Figure 4A). Recent studies documented thatamong the RGS family, RGS2 plays a prominent role inregulating synaptic transmission and plasticity in hippo-campal neurons [26]. It is therefore likely that increasedRGS2 levels modulate synaptic output, probably leadingto an elevated survival of neurons upon vitamin B6treatment.The GPCR3 endothelinB (ETB) has been found to be
important for the physiological reduction of neuronalapoptosis in the dentate gyrus during postnatal deve-lopment. Furthermore, ETB receptors are involved inpathological apoptosis in the dentate gyrus, especially ina rabbit model of pneumococcal meningitis [28]. Thesedata suggest a role for the ETB receptor as an anti-
7.50 7.75 8.00 8.25 8.500.0
0.5
1.0
1.5
2.0
2.5
expression level
apo
pto
sis
[sco
re]
E
7 8 9 10 11 120.0
0.5
1.0
1.5
2.0
2.5
expression level
apo
pto
sis
[sco
re]
D
6 7 8 9 10 11 12 130.0
0.5
1.0
1.5
2.0
2.5
expression level
apo
pto
sis
[sco
re]
C
8 9 10 11 120.0
0.5
1.0
1.5
2.0
2.5
expression level
apo
pto
sis
[sco
re]
B
A
5 6 7 8 9 100.0
0.5
1.0
1.5
2.0
2.5
expression level
apo
pto
sis
[sco
re]
Figure 5 Pearson correlation between RNA expression leveland apoptosis score. RNA expression data of individual rats arebased on chip analysis, the apoptosis score is the mean value ofapoptotic cells of corresponding rat brains. Expression levels of up-regulated genes such as BDNF (A) (Pearson correlation p = 0.002),Npas4 (C) (p = 0.005) and Nr4a1 (D) (p = 0.003) negatively correlatewith apoptosis score, e.g. the less transcript, the more apoptoticcells. In contrast, expression levels of down-regulated genes such asLyz 2 (B) (p < 0.001) and Pafah2 (E) (p = 0.005) postitively correlatewith apoptosis score. Samples from vitamin B6-treated rats (barecircles) are arranged separately from samples of animals treated withsaline (solid circles).
Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 9 of 15http://www.biomedcentral.com/1471-2334/13/393
apoptotic factor in the dentate gyrus, consistent with theup-regulation of ETB receptor gene as a result of vitaminB6 treatment.Another important receptor also belonging to the GPCR
superfamily is the neuromedin B receptor (NMBR, ID438637, Figure 4A). NMBR is localized in brain micro-vascular endothelial cells that form the blood–brain bar-rier. It is coupled to the phospholipase C transducingsystem controlling the K+ efflux out of the brain [29]. Adown-regulation of NMBR in vitamin B6-treated rats maybe indicative for blood–brain barrier disruption inducedby BM.The pro-inflammatory mediator IL-1 consists of 4 mo-
lecular species (IL-1α, IL-1β, IL-1γ and IL-1δ) that exertsimilar biological activity through IL-1 receptor (IL-1R)type I. In vivo studies showed that IL-1R−/− mice, lackingan intact IL-1 signal, are more susceptible to developpneumococcal meningitis, and that the disease is associ-ated with higher mortality in IL-1R−/− mice compared towild-type animals. These results suggest a protective roleof locally produced IL-1 in the first-line defense againstpathogens during pneumococcal meningitis [30]. How-ever, in the present study, IL-1R type I (ID 381937,Figure 4A) was down-regulated, indicating that uponvitamin B6 treatment the host inflammatory reactionis down-modulated.Highly raised concentrations of CXC-chemokines (e.g.
IL-8) that attract neutrophils, and of CC-chemokines(e.g. monocyte chemoattractant protein MCP-1, macro-phage inflammatory proteins MIP-1α and MIP-1β) thatattract monocytes and lymphocytes, were observed inthe CSF during BM [31,32]. Chemokine receptors arealso GPCRs. In this study, CCR2 (ID 2818300), the re-ceptor of MCP-1, and CCR5 (ID 382626), the receptorof MIP-1α and MIP-1β, are down-regulated (Figure 4A).Both receptors are expressed on glial and neuronal cellsin the adult brain as well as on neural progenitor cellsisolated from the subventricular zone where neurogen-esis occurs. The localization of chemokine receptors inthese regions suggests an involvement of CCR2 andCCR5 in the regulation of adult neural progenitor cellsin physiological or pathological conditions [33]. Other

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 10 of 15http://www.biomedcentral.com/1471-2334/13/393
studies showed that CCR2 is one of the most prominentchemokine receptor associated with neuro-inflammatorydiseases such as multiple sclerosis and experimental auto-immune encephalomyelitis [34]. However, the down-regulation of CCR2 and CCR5 following vitamin B6treatment may result in a reduced production of neuro-inflammatory mediators by glial or neuronal cells. Further-more, recruitment of monocytes and lymphocytes to theCSF may also be reduced. Finally, it could also influencethe neurogenetic processes observed in the hippocampaldentate gyrus (see below).Following inflammation, microglial cells become acti-
vated and produce inflammatory mediators causingbrain damage in a variety of neurodegenerative dis-orders. Since inflammation may exacerbate braindamage, the control and reduction of brain inflamma-tion is pathophysiologically important. IL-13 is ananti-inflammatory cytokine which minimizes the pro-duction of inflammatory mediators from activatedmicroglia (e.g. TNFα and MIP-1α) [35]. Moreover, ex-perimental studies showed that exogenous IL-13 se-lectively induces apoptotic death of activated microglia[36]. Another study demonstrated that neurons andmicroglia cooperatively down-regulate brain inflam-mation by inducing endogenous IL-13 expression inmicroglia, resulting in microglial death and elevationof neuronal survival [35]. Suggesting a reduced inflam-matory reaction as assessed by a down-regulation of pro-inflammatory cytokines (IL-1R type I) and chemokines(CCR2 and CCR5) in vitamin B6-treated rats, the require-ment for anti-inflammatory cytokines such as IL-13 isreduced. This suggestion is consistent with the down-modulation of the IL-13 receptor alpha 1 (ID 432716,Figure 4A) gene upon vitamin B6 treatment.In summary, vitamin B6 down-modulates the inflam-
matory response as evidenced by reduced RNA levelsencoding for pro-inflammatory cytokines and chemo-kines, and by transcriptional indication for diminishedactivation of microglia. Because the brain damage ob-served in BM, including hippocampal apoptosis, ismainly due to the host inflammatory reaction [37], adown-modulated immune reaction may decisively con-tribute to diminished hippocampal apoptosis observedin vitamin B6-treated rats. Evidence for strong anti-inflammatory effects of vitamin B6 in patients with sys-temic inflammatory symptoms has also been providedby others [38-40].
Circadian rhythmThe circadian rhythm is generated by a set of interactinggenes and proteins (see Additional file 3: Figure S2). Forexample in mammals, the protein products of the clockand Bmal1 genes act together to induce the expressionof other clock genes including period (PER) [41]. The
up-regulation of period homolog transcripts (PER1, ID395923, and PER2, ID 392428) in vitamin B6- comparedto placebo-treated rats suggests an involvement of thecircadian rhythm in the regulation of apoptotic pro-cesses (Figure 4B).Recent studies demonstrated a circadian periodicity of
the TRP metabolism via the KYN pathway [42]. How-ever, TRP metabolism in the brain mainly occurs via 2different pathways, the methoxyindole and the KYNpathway. In experimental models as well as in humans,melatonin, the main metabolite of the methoxyindolepathway, acts as neuroprotective agent. It inhibits theNMDA receptor and thus, protects the neurons fromexcitotoxic damage. The same effect is mediated byKYNA, a neuroprotective metabolite of the KYN path-way. The inhibition of the NMDA receptor activity par-tially depends on the reduction of the NO synthaseactivity, therefore decreasing the amount of NO pro-duced as a result of NMDA activation. Melatonin alsofollows a circadian rhythmic pattern, mainly determinedby the pineal gland that increases the production ofmelatonin upon physiological stimuli such as darkness.Activation of either the methoxyindole or the KYN path-way reaches an equilibrium in normal conditions by anincrease in the TRP degradation via the KYN pathwayduring the day and via the methoxyindole pathway dur-ing the night [43]. This equilibrium is lost under condi-tions of stress including febrile and epileptic seizuresand probably also in other pathological situations [44].BM displaying a stress situation could influence theequilibrium between the methoxyindole and the KYNpathway. Because vitamin B6 acts as a cofactor for 2 keyenzymes of the KYN pathway and also positively affectsthe pineal production of melatonin [43], administrationof vitamin B6 could restore this equilibrium. Therefore,melatonin as a immunomodulatory agent could play animportant role in neuroinflammation and subsequentbrain injury [45].The elevation of cellular NAD+ levels through the
vitamin B6 induced activation of the KYN pathway ob-served in this study, may also have an influence on fac-tors involved in the circadian rhythm described above.NAD+ has been shown to act as a central circadianregulator. Concerning the role of NAD+ in cellular en-ergy stores, a molecular coupling between the circadianrhythm and energy metabolism has been proposed[46-48]. Moreover, a link between disruption of circadianrhythm and hippocampal learning and memory has beenreported in rats using the water maze task. Chronicstress, sleep deprivation and decreases in melatonin se-cretion are some of the many side effects of circadiandisruption. By its anti-oxidant and neuroprotective rolein the brain, melatonin deprivation may contributeto brain damage in individuals suffering from chronic

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 11 of 15http://www.biomedcentral.com/1471-2334/13/393
circadian disruption. In transgenic mouse models ofAlzheimer’s disease, melatonin treatment may reducethe deposition of β-amyloid and protects against oxida-tive stress. A possible speculation is that with decreasinglevels of melatonin, individuals suffering from chroniccircadian disruption become more vulnerable to braindamage associated with learning and memory impair-ment [49]. Another study showed that the clock genecould have an important role on spatial learning in mice,as assessed by water maze [50]. Furthermore, experi-mental mouse models suggest that cell cycle and apop-totic processes may be regulated by circadian clockgenes in bone marrow [41].
Neuronal signalingNeurogenesis, the continuous production of new neu-rons from a population of dividing neural progenitorcells, occurs in the hippocampal dentate gyrus. It isinfluenced by pathological situations such as ischemia orinflammation. BM may affect the production of neuronalsurvival factors such as brain-derived neurotrophic factor(BDNF, ID 382982, Figure 4C) gene, thereby promotingthe survival of neuronal cells and thus, having an impacton neurogenetic processes [51].Recent studies demonstrated that the expression of
BNDF and its receptor TrkB is increased in mature neu-rons during the acute phase of pneumococcal meningitis[1]. BDNF protein co-localizes with cells expressingTrkB in the hippocampal CA3/4 region and the hilus ad-jacent to the subgranular zone of the dentate gyruswhere the proliferation of progenitor cells is increased.These findings indicate an involvement of endogenousBDNF and TrkB signaling in neurogenesis after BM [23].However, the persistence of neurological sequelae in upto 50% of survivors from BM [1,3] suggests that en-dogenous mechanisms responsible for neuroregenerationare inefficient.Since treatment with exogenous BDNF results in the
reduction of various forms of cell death in experimentalpneumococcal meningitis [52], one can speculate thatthe up-regulated expression level of BDNF in vitaminB6-treated animals plays an important role in dimini-shing hippocampal apoptosis.BDNF induces the expression of many genes in hippo-
campal cells in culture, including activity regulated cyto-skeletal-associated protein (ARC, ID 382171, Figure 4C)gene. ARC itself is involved in memory consolidationand long-term potentiation [53]. Because injury to thehippocampal dentate gyrus is associated with learningand memory deficits [5], the up-regulation of ARC RNAin our study provides further evidence for a role ofBDNF in the reduction of hippocampal apoptosis.Another gene involved in neuronal signaling processes
is early growth response 2 (EGR2, ID 438557, Figure 4C).
EGR2 is an important mediator of the growth-suppressivesignal of phosphatase and tensin homolog (PTEN) andplays a key role in the PTEN-induced apoptotic path-way. It alters the permeability of mitochondrial mem-branes, resulting in the release of cytochrome c whichin turn activates caspase-3, -8 and −9. As an alternativeroute, EGR2 may directly induce the expression ofpro-apoptotic factors of the Bcl-2 family [54]. In thepresent study, EGR2 is up-regulated by vitamin B6treatment. This result is not consistent with a reductionof apoptotic cell death by vitamin B6. This discrepancybetween an induction of apoptosis by EGR2 and an up-regulation of EGR2 under circumstances that have beenproven to diminish apoptosis may be due to differentexperimental conditions. In both studies, the molecularmechanisms of the apoptotic pathway were analyzed bymicroarrays, but we used an in vivo model system ofBM, whereas cancer-derived cells served as in vitro cul-ture system for the study performed by Unoki andNakamura [54]. Furthermore, posttranslational mecha-nisms such as phosphorylation, important for thebiological activity of PTEN, are not considered inmicroarray experiments.Members of the nuclear receptor subfamily 4 group A
(NR4A) are classified as early response genes expressedin a wide variety of metabolically demanding and energydependent tissues such as the brain. They are inducedby a broad range of signals, including stress, growth fac-tors, inflammatory cytokines, hormones, calcium, neuro-transmitters and physical stimuli. Consistent with thepleiotropic physiological stimuli inducing the NR4Amembers, these receptors have been implicated in cellcycle regulation, apoptosis, neurological disease, inflam-mation, carcinogenesis and atherogenesis [55]. Since BMis an inflammatory disease associated with brain damagedue to hippocampal apoptosis and often leads to neu-rological deficits, the NR4A subfamily may play an es-sential role in this disease. In the present study, bothmember 1 and 2 of the NR4A family (NR4A1, ID382110, and NR4A2, ID 438446) are up-regulated, sug-gesting an involvement in apoptotic processes (Figure 4C).Recent studies showed that the role of the Nr4A membersin cancer is largely defined by the implication of the sub-family in the regulation of apoptosis [55]. Furthermore,experimental studies with macrophages demonstrated aninvolvement of NR4A1 in modulating apoptosis in the in-flammatory response. Recent work also suggested that incertain cell lines NR4A1 translocates to the mitochondriato release cytochrome c [56].
Apoptosis/cell deathPlatelet activating factor (PAF) is an extremely potentactivator of inflammatory cells owing to the expressionof its receptor by numerous cells of the innate immune

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 12 of 15http://www.biomedcentral.com/1471-2334/13/393
system. Accordingly, hydrolysis of PAF by extracellularor intracellular PAF acetylhydrolases is predicted to in-hibit inflammatory signaling. Indeed, expression ofplasma PAF acetylhydrolase is increased by stimulationwith inflammatory agonists such as LPS, and decreasedby anti-inflammatory drugs [57]. Given the possible anti-inflammatory effect of vitamin B6 as suggested by reducedlevels of pro-inflammatory mediators (e.g. cytokines andchemokines) and diminished activation of inflammatorycells (e.g. microglia), vitamin B6 may down-regulatethe expression of PAF hydrolase. This hypothesis wastested by the vitamin B6 induced attenuation of PAFacetylhydrolase 2 (PAFAH2, ID 549642, Figure 4D)levels in our study.PAF induces apoptosis independent of its receptor, but
the mechanism underlying this ability is not fully under-stood [57]. However, PAFAH2 hydrolyzes not only PAFbut also short-chain phospholipids [58]. These subs-trates are pro-apoptotic, pointing to an essential role ofPAFAH2 as anti-apoptotic agent [57]. Recent studiesreported that a transfection of the plasma PAFAH2 genereduces glutamate-induced apoptosis in cultured rat cor-tical neurons. Moreover, studies using a mouse model offocal cerebral ischemia showed that PAFAH2 exertsstrong neuroprotective effects against ischemic injury inthe CNS by protecting neurons against oxidative stress[58]. In this context, it seems that down-regulatedPAFAH2 does not contribute to the processes leadingto the reduced hippocampal apoptosis in vitamin B6-treated rats.Beside the role of matrix metalloproteinases (MMPs)
in blood–brain barrier disruption and extravasation ofinflammatory cells into the CNS [3,59], recent studiessuggested an involvement of MMPs in glial and neuronalcell death. Furthermore, an excessive increase of MMP-9(ID 382260, Figure 4D) in BM has been identified as arisk factor for the development of neurological sequelae[59]. Therefore, the down-regulation of MMP-9 uponvitamin B6 treatment indicates a long-term effect ofvitamin B6 in terms of reduced learning and memoryimpairments.MMPs are also increased by antimicrobial peptides.
Antimicrobial peptides are effector molecules of the in-nate immune system with antibiotic function. Asidefrom their antibiotic functions, they may be involved inimmune responses and inflammatory disease. For ex-ample, they may amplify inflammation by activation ofcytokine and chemokine expression in immune cells[60]. Lysozyme (Lyz) is an antimicrobial protein belong-ing to the defensin family of host-defense proteins whichare distributed widely in biological fluids and tissues. Ex-perimental studies with transgenic mice showed that Lyzraises the levels of antioxidant reserves that are requiredto manage non-pathological amounts of reactive oxygen
species. These antioxidant properties are partly mediatedvia negative regulation of stress response genes (e.g.c-Jun) and also involve the blockade of cellular apoptosisin vitro [61]. However, Brandenburg et al. reported thatthere is no increase of Lyz in the CSF and serum sam-ples from patients with meningitis [60]. In the presentstudy, we found a down-regulation of Lyz 2 (ID 378051,Figure 4D) in vitamin B6-treated rats when compared tosaline-treated animals. This down-regulation could be afurther indication of a reduced inflammation and inthis context, would explain the reduced levels of pro-inflammatory cytokines and chemokines.Recent studies showed that adjuvant BDNF protects
the brain from caspase 3-dependent hippocampal apop-tosis in experimental BM [13]. In the present study,up-regulated endogenous BDNF is also involved inapoptotic processes as indicated by the apoptotic celldeath cluster (Figure 4D). This result provides furtherevidence for a crucial role of BDNF in reducing hippo-campal apoptosis upon vitamin B6 treatment.But how does vitamin B6 induce BDNF expression?
Several studies showed that BDNF expression in neur-onal cells is induced by activation of calcium channelsand recruitment of calcium-sensitive transcription fac-tors [62]. The excitatory amino acid glutamate which isincreased in interstitial brain fluid in BM [63] induces acalcium influx by binding to the NMDA receptor andthus, may stimulate the production of BDNF [64]. Onthe contrary, KYNA, the neuroprotective intermediate ofthe KYN pathway, is an antagonist of the NMDA recep-tor and therefore, inhibits calcium influx. Moreover,in vitro studies using rat cerebral cortex nerve terminalsshowed that vitamin B6 inhibits glutamate releasethrough the suppression of calcium influx [65].However, other studies reported that high levels of
IL-1β decrease BDNF mRNA expression in the rathippocampus [66]. Thus, the increased amount of BDNFtranscripts in vitamin B6-treated rats may result fromdecreased levels of IL-1β. This suggestion is also sup-ported by the down-regulation of the IL-1R type I geneas discussed previously.A related phenomenon could be observed in the
brains of rats administered an antibiotic plus dexa-methasone. Given the up-regulation of BDNF RNA andprotein in this study, Li et al. hypothesize that the adju-vant therapy with dexamethasone might have a benefi-cial effect on BM via up-regulation of neuroprotectiveBDNF. In addition, this study demonstrated a dose-dependent down-regulation of BDNF RNA and proteinin rats treated with antibiotics alone. A possible reasonfor this finding is the lysis of bacteria caused by the anti-biotic treatment, resulting in the release of bacterialcomponents that stimulate the expression of pro-inflammatory mediators such as IL-1β [66].

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 13 of 15http://www.biomedcentral.com/1471-2334/13/393
ConclusionsPre-treatment with vitamin B6 in BM exerts neuropro-tective effects in terms of reduced apoptosis in the hip-pocampal dentate gyrus of infant rats. Although theprocesses required for this effect need more investiga-tion, preservation of cellular energy stores, reduction ofthe inflammatory response and up-regulation of BDNFexpression may, at least partially, explain the neuro-protective properties of vitamin B6 in models of pneu-mococcal meningitis.
Additional files
Additional file 1: Table S1. Gene list. Green marked genes are up-regulated, red marked genes are down-regulated. The genes arearranged according to decreasing fold change.
Additional file 2: Figure S1. MAPK signaling pathway. Pathway analysisof significantly regulated genes according to KEGG database. The genesare stained according to their regulation level: Green marked genes areup-regulated, red marked genes are down-regulated and light greenstained genes are on control level.
Additional file 3: Figure S2. Circadian rhythm. Pathway analysis ofsignificantly regulated genes according to KEGG database. The genes arestained according to their regulation level: Green marked genes areup-regulated, red marked genes are down-regulated and light greenstained genes are on control level.
AbbreviationsARC: Activity regulated cytoskeletal-associated protein; BDNF: Brain-derivedneurotrophic factor; BM: Bacterial meningitis; CNS: central nervous system;CSF: Cerebrospinal fluid; DAVID: Database for annotation, visualization andintegrated discovery; EGR2: Early growth response 2; ETB: EndothelinB;GCOS: GeneChip® operating software; GO: Gene ontology; GPCRs: G proteincoupled receptors; IL-1R: IL-1 receptor; i.p.: Intraperitoneal; KEGG: KyotoEncyclopedia of genes and genomes; KYN: Kynurenine; KYNA: Kynurenic acid;Limma: Linear models for microarray data; LYZ: Lysozyme; MAPK: Mitogen-activated protein kinase; MCP: Monocyte chemoattractant protein;MIP: Macrophage inflammatory protein; MMP: Matrix metalloproteinase;NAD+: Nicotine amide adenine dinucleotide; NMBR: Neuromedin B receptor;NMDA: N-methyl-D-aspartate; NO: Nitric oxide; Npas4: Neuronal PAS domainprotein 4; NR4A: Nuclear receptor subfamily 4 group A; PAF: Plateletactivating factor; PAFAH2: PAF acetylhydrolase 2; PBS: Phosphate bufferedsaline; PER: Period; PTEN: Phosphatase and tensin homolog;RefSeq: Reference sequences; RGS: Regulators of G protein signaling;RMA: Robust multi average; Rpl24: Ribosomal protein L24; s.c.:Subcutaneously; TGF-β: Transforming growth factor-β; TLRs: Toll-likereceptors; TRP: Tryptophan.
Competing interestsAll authors declared that they have no competing interests.
Authors’ contributionsMW and SLL conceived and designed the study. DCZ and CLB performedthe experiments. DCZ and MW analyzed the data. DCZ, MW and SLL wrotethe paper. All authors read and approved the final manuscript.
AcknowledgmentsSpecial thanks go to Roney S. Coimbra for participation in the initiation ofthe project, Kevin Oberson and Angela Bühlmann for excellent technicalassistance, as well as to the Lausanne Genomic Technologies Facility(University of Lausanne, Switzerland) for chip hybridization with technicalsupport from Otto Hagenbüchle.The experiments of this work were done at the Institute for InfectiousDiseases of the University of Bern.
FundingThe study was supported by the UBS Optimus Foundation and by the SwissNational Science Foundation (31003A-138094). The funders had no role instudy design, data collection and analysis, decision to publish, or preparationof the manuscript.
Author details1Biology Division, Spiez Laboratory, Federal Office for Civil Protection,Austrasse, CH-3700, Spiez, Switzerland. 2Institute of Parasitology University ofBern, Länggassstrasse 122, CH-3012, Bern, Switzerland. 3Graduate School forCellular and Biomedical Sciences, University of Bern, Bern, Switzerland.4Neuroinfection Laboratory, Institute for Infectious Diseases, University ofBern, Friedbühlstrasse 51, CH-3010, Bern, Switzerland.
Received: 23 January 2013 Accepted: 21 August 2013Published: 27 August 2013
References1. Grimwood K, Anderson VA, Bond L, Catroppa C, Hore RL, Keir EH, Nolan T,
Roberton DM: Adverse outcomes of bacterial meningitis in school-agesurvivors. Pediatrics 1995, 95:646–656.
2. Bellac CL, Coimbra RS, Christen S, Leib SL: Inhibition of the kynurenine-NAD+ pathway leads to energy failure and exacerbates apoptosis inpneumococcal meningitis. J Neuropathol Exp Neurol 2010, 69:1096–1104.
3. Koedel U, Scheld WM, Pfister HW: Pathogenesis and pathophysiology ofpneumococcal meningitis. Lancet Infect Dis 2002, 2:721–736.
4. Nau R, Soto A, Bruck W: Apoptosis of neurons in the dentate gyrus inhumans suffering from bacterial meningitis. J Neuropathol Exp Neurol1999, 58:265–274.
5. Loeffler JM, Ringer R, Hablutzel M, Tauber MG, Leib SL: The free radicalscavenger alpha-phenyl-tert-butyl nitrone aggravates hippocampalapoptosis and learning deficits in experimental pneumococcalmeningitis. J Infect Dis 2001, 183:247–252.
6. Bellac CL, Coimbra RS, Christen S, Leib SL: Pneumococcal meningitiscauses accumulation of neurotoxic kynurenine metabolites in brainregions prone to injury. Neurobiol Dis 2006, 24:395–402.
7. Guillemin GJ, Cullen KM, Lim CK, Smythe GA, Garner B, Kapoor V,Takikawa O, Brew BJ: Characterization of the kynurenine pathway inhuman neurons. J Neurosci 2007, 27:12884–12892.
8. de Souza FR, Fontes FL, da Silva TA, Coutinho LG, Leib SL, Agnez-Lima LF:Association of kynurenine aminotransferase II gene C401Tpolymorphism with immune response in patients with meningitis.BMC Med Genet 2011, 12:51.
9. Ha HC, Snyder SH: Poly(ADP-ribose) polymerase-1 in the nervous system.Neurobiol Dis 2000, 7:225–239.
10. van de Kamp JL, Smolen A: Response of kynurenine pathway enzymes topregnancy and dietary level of vitamin B-6. Pharmacol Biochem Behav1995, 51:753–758.
11. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP,Dolinski K, Dwight SS, Eppig JT, et al: Gene ontology: tool for theunification of biology. The Gene Ontology Consortium. Nat Genet 2000,25:25–29.
12. Kanehisa M, Goto S: KEGG: kyoto encyclopedia of genes and genomes.Nucleic Acids Res 2000, 28:27–30.
13. Leib SL, Clements JM, Lindberg RL, Heimgartner C, Loeffler JM, Pfister LA,Tauber MG, Leppert D: Inhibition of matrix metalloproteinases andtumour necrosis factor alpha converting enzyme as adjuvant therapy inpneumococcal meningitis. Brain 2001, 124:1734–1742.
14. Gianinazzi C, Grandgirard D, Imboden H, Egger L, Meli DN, Bifrare YD,Joss PC, Tauber MG, Borner C, Leib SL: Caspase-3 mediates hippocampalapoptosis in pneumococcal meningitis. Acta Neuropathol 2003,105:499–507.
15. Leib SL, Heimgartner C, Bifrare YD, Loeffler JM, Taauber MG:Dexamethasone aggravates hippocampal apoptosis and learningdeficiency in pneumococcal meningitis in infant rats. Pediatr Res 2003,54:353–357.
16. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B,Gautier L, Ge Y, Gentry J, et al: Bioconductor: open software developmentfor computational biology and bioinformatics. Genome Biol 2004, 5:R80.
17. Gautier L, Cope L, Bolstad BM, Irizarry RA: affy–analysis of AffymetrixGeneChip data at the probe level. Bioinformatics 2004, 20:307–315.

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 14 of 15http://www.biomedcentral.com/1471-2334/13/393
18. Kooperberg C, Aragaki A, Strand AD, Olson JM: Significance testing forsmall microarray experiments. Stat Med 2005, 24:2281–2298.
19. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I: Controlling the falsediscovery rate in behavior genetics research. Behav Brain Res 2001,125:279–284.
20. Putt KS, Hergenrother PJ: An enzymatic assay for poly(ADP-ribose)polymerase-1 (PARP-1) via the chemical quantitation of NAD(+):application to the high-throughput screening of small molecules aspotential inhibitors. Anal Biochem 2004, 326:78–86.
21. Bifrare YD, Gianinazzi C, Imboden H, Leib SL, Tauber MG: Bacterialmeningitis causes two distinct forms of cellular damage in thehippocampal dentate gyrus in infant rats. Hippocampus 2003,13:481–488.
22. Coimbra RS, Voisin V, de Saizieu AB, Lindberg RL, Wittwer M, Leppert D,Leib SL: Gene expression in cortex and hippocampus during acutepneumococcal meningitis. BMC Biol 2006, 4:15.
23. Wittwer M, Grandgirard D, Rohrbach J, Leib SL: Tracking the transcriptionalhost response from the acute to the regenerative phase of experimentalpneumococcal meningitis. BMC Infect Dis 2010, 10:176.
24. Ebert S, Zeretzke M, Nau R, Michel U: Microglial cells and peritonealmacrophages release activin A upon stimulation with Toll-like receptoragonists. Neurosci Lett 2007, 413:241–244.
25. Wilms H, Schwark T, Brandenburg LO, Sievers J, Dengler R, Deuschl G,Lucius R: Regulation of activin A synthesis in microglial cells:pathophysiological implications for bacterial meningitis. J Neurosci Res2010, 88:16–23.
26. Han J, Mark MD, Li X, Xie M, Waka S, Rettig J, Herlitze S: RGS2 determinesshort-term synaptic plasticity in hippocampal neurons by regulatingGi/o-mediated inhibition of presynaptic Ca2+ channels. Neuron 2006,51:575–586.
27. Taymans JM, Wintmolders C, Te Riele P, Jurzak M, Groenewegen HJ, LeysenJE, Langlois X: Detailed localization of regulator of G protein signaling 2messenger ribonucleic acid and protein in the rat brain. Neuroscience2002, 114:39–53.
28. Ehrenreich H, Nau TR, Dembowski C, Hasselblatt M, Barth M, Hahn A,Schilling L, Siren AL, Bruck W: Endothelin b receptor deficiency isassociated with an increased rate of neuronal apoptosis in the dentategyrus. Neuroscience 2000, 95:993–1001.
29. Vigne P, Feolde E, Van Renterghem C, Breittmayer JP, Frelin C: Propertiesand functions of a neuromedin-B-preferring bombesin receptor in brainmicrovascular endothelial cells. Eur J Biochem 1995, 233:414–418.
30. Zwijnenburg PJ, van der Poll T, Florquin S, Roord JJ, Van Furth AM:IL-1 receptor type 1 gene-deficient mice demonstrate an impairedhost defense against pneumococcal meningitis. J Immunol 2003,170:4724–4730.
31. Spanaus KS, Nadal D, Pfister HW, Seebach J, Widmer U, Frei K, Gloor S,Fontana A: C-X-C and C-C chemokines are expressed in the cerebrospinalfluid in bacterial meningitis and mediate chemotactic activity onperipheral blood-derived polymorphonuclear and mononuclear cellsin vitro. J Immunol 1997, 158:1956–1964.
32. Tauber MG, Moser B: Cytokines and chemokines in meningealinflammation: biology and clinical implications. Clin Infect Dis 1999,28:1–11. quiz 12.
33. Ji JF, He BP, Dheen ST, Tay SS: Expression of chemokine receptorsCXCR4, CCR2, CCR5 and CX3CR1 in neural progenitor cells isolatedfrom the subventricular zone of the adult rat brain. Neurosci Lett2004, 355:236–240.
34. Banisadr G, Gosselin RD, Mechighel P, Rostene W, Kitabgi P,Melik Parsadaniantz S: Constitutive neuronal expression of CCR2chemokine receptor and its colocalization with neurotransmitters innormal rat brain: functional effect of MCP-1/CCL2 on calciummobilization in primary cultured neurons. J Comp Neurol 2005,492:178–192.
35. Shin WH, Lee DY, Park KW, Kim SU, Yang MS, Joe EH, Jin BK: Microgliaexpressing interleukin-13 undergo cell death and contribute to neuronalsurvival in vivo. Glia 2004, 46:142–152.
36. Yang MS, Park EJ, Sohn S, Kwon HJ, Shin WH, Pyo HK, Jin B, Choi KS, Jou I,Joe EH: Interleukin-13 and −4 induce death of activated microglia.Glia 2002, 38:273–280.
37. Pfister HW, Scheld WM: Brain injury in bacterial meningitis: therapeuticimplications. Curr Opin Neurol 1997, 10:254–259.
38. Huang SC, Wei JC, Wu DJ, Huang YC: Vitamin B(6) supplementationimproves pro-inflammatory responses in patients with rheumatoidarthritis. Eur J Clin Nutr 2010, 64:1007–1013.
39. Ulvik A, Midttun O, Ringdal Pedersen E, Nygard O, Ueland PM: Associationof plasma B-6 vitamers with systemic markers of inflammation beforeand after pyridoxine treatment in patients with stable angina pectoris.Am J Clin Nutr 2012, 95:1072–1078.
40. Lotto V, Choi SW, Friso S: Vitamin B6: a challenging link between nutritionand inflammation in CVD. Br J Nutr 2011, 106:183–195.
41. Granda TG, Liu XH, Smaaland R, Cermakian N, Filipski E, Sassone-Corsi P,Levi F: Circadian regulation of cell cycle and apoptosis proteins in mousebone marrow and tumor. FASEB J 2005, 19:304–306.
42. Rapoport MI, Beisel WR: Circadian periodicity of tryptophan metabolism.J Clin Invest 1968, 47:934–939.
43. Munoz-Hoyos A, Molina-Carballo A, Macias M, Rodriguez-Cabezas T,Martin-Medina E, Narbona-Lopez E, Valenzuela-Ruiz A, Acuna-Castroviejo D:Comparison between tryptophan methoxyindole and kynureninemetabolic pathways in normal and preterm neonates and in neonateswith acute fetal distress. Eur J Endocrinol 1998, 139:89–95.
44. Munoz-Hoyos A, Molina-Carballo A, Rodriguez-Cabezas T, Uberos-FernandezJ, Ruiz-Cosano C, Acuna-Castroviejo D: Relationships betweenmethoxyindole and kynurenine pathway metabolites in plasma andurine in children suffering from febrile and epileptic seizures. ClinEndocrinol (Oxf ) 1997, 47:667–677.
45. Spreer A, Gerber J, Baake D, Hanssen M, Huether G, Nau R:Antiinflammatory but no neuroprotective effects of melatonin underclinical treatment conditions in rabbit models of bacterial meningitis.J Neurosci Res 2006, 84:1575–1579.
46. Masri S, Zocchi L, Katada S, Mora E, Sassone-Corsi P: The circadian clocktranscriptional complex: metabolic feedback intersects with epigeneticcontrol. Ann N Y Acad Sci 2012, 1264:103–109.
47. Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P: Circadiancontrol of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009,324:654–657.
48. Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B,Hong HK, Chong JL, Buhr ED, Lee C, et al: Circadian clock feedback cyclethrough NAMPT-mediated NAD+ biosynthesis. Science 2009, 324:651–654.
49. Craig LA, McDonald RJ: Chronic disruption of circadian rhythms impairshippocampal memory in the rat. Brain Res Bull 2008, 76:141–151.
50. Sei H, Oishi K, Sano A, Seno H, Ohmori T, Morita Y, Ishida N: Clock mutantmice with Jcl/ICR background shows an impaired learning ability inwater maze, but not in passive avoidance, at the beginning of darkphase. Congenit Anom (Kyoto) 2006, 46:81–85.
51. Grandgirard D, Bifrare YD, Pleasure SJ, Kummer J, Leib SL, Tauber MG:Pneumococcal meningitis induces apoptosis in recently postmitoticimmature neurons in the dentate gyrus of neonatal rats. Dev Neurosci2007, 29:134–142.
52. Bifrare YD, Kummer J, Joss P, Tauber MG, Leib SL: Brain-derivedneurotrophic factor protects against multiple forms of brain injury inbacterial meningitis. J Infect Dis 2005, 191:40–45.
53. Larsen MH, Rosenbrock H, Sams-Dodd F, Mikkelsen JD: Expression of brainderived neurotrophic factor, activity-regulated cytoskeleton proteinmRNA, and enhancement of adult hippocampal neurogenesis in ratsafter sub-chronic and chronic treatment with the triple monoaminere-uptake inhibitor tesofensine. Eur J Pharmacol 2007, 555:115–121.
54. Unoki M, Nakamura Y: EGR2 induces apoptosis in various cancer celllines by direct transactivation of BNIP3L and BAK. Oncogene 2003,22:2172–2185.
55. Maxwell MA, Muscat GE: The NR4A subgroup: immediate early responsegenes with pleiotropic physiological roles. Nucl Recept Signal 2006, 4:e002.
56. Pei L, Castrillo A, Tontonoz P: Regulation of macrophage inflammatorygene expression by the orphan nuclear receptor Nur77. Mol Endocrinol2006, 20:786–794.
57. McIntyre TM, Prescott SM, Stafforini DM: The emerging roles of PAFacetylhydrolase. J Lipid Res 2009, 50(Suppl):S255–S259.
58. Umemura K, Kato I, Hirashima Y, Ishii Y, Inoue T, Aoki J, Kono N, Oya T,Hayashi N, Hamada H, et al: Neuroprotective role of transgenic PAF-acetylhydrolase II in mouse models of focal cerebral ischemia. Stroke2007, 38:1063–1068.
59. Leppert D, Leib SL, Grygar C, Miller KM, Schaad UB, Hollander GA: Matrixmetalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during

Zysset-Burri et al. BMC Infectious Diseases 2013, 13:393 Page 15 of 15http://www.biomedcentral.com/1471-2334/13/393
bacterial meningitis: association with blood–brain barrier damage andneurological sequelae. Clin Infect Dis 2000, 31:80–84.
60. Brandenburg LO, Varoga D, Nicolaeva N, Leib SL, Wilms H, Podschun R,Wruck CJ, Schroder JM, Pufe T, Lucius R: Role of glial cells in the functionalexpression of LL-37/rat cathelin-related antimicrobial peptide inmeningitis. J Neuropathol Exp Neurol 2008, 67:1041–1054.
61. Liu H, Zheng F, Cao Q, Ren B, Zhu L, Striker G, Vlassara H: Amelioration ofoxidant stress by the defensin lysozyme. Am J Physiol Endocrinol Metab2006, 290:E824–E832.
62. Viviani B, Bartesaghi S, Corsini E, Villa P, Ghezzi P, Garau A, Galli CL,Marinovich M: Erythropoietin protects primary hippocampal neuronsincreasing the expression of brain-derived neurotrophic factor.J Neurochem 2005, 93:412–421.
63. Leib SL, Kim YS, Ferriero DM, Tauber MG: Neuroprotective effect ofexcitatory amino acid antagonist kynurenic acid in experimentalbacterial meningitis. J Infect Dis 1996, 173:166–171.
64. Mattson MP: Glutamate and neurotrophic factors in neuronal plasticityand disease. Ann N Y Acad Sci 2008, 1144:97–112.
65. Yang TT, Wang SJ: Pyridoxine inhibits depolarization-evoked glutamaterelease in nerve terminals from rat cerebral cortex: a possibleneuroprotective mechanism? J Pharmacol Exp Ther 2009, 331:244–254.
66. Li L, Shui QX, Zhao ZY: Regulation of brain-derived neurotrophic factor(BDNF) expression following antibiotic treatment of experimentalbacterial meningitis. J Child Neurol 2003, 18:828–834.
doi:10.1186/1471-2334-13-393Cite this article as: Zysset-Burri et al.: Vitamin B6 reduces hippocampalapoptosis in experimental pneumococcal meningitis. BMC InfectiousDiseases 2013 13:393.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit