vasopressin-induced intracellular redistribution of protein kinase d in intestinal epithelial cells
TRANSCRIPT

JOURNAL OF CELLULAR PHYSIOLOGY 196:483–492 (2003)
Vasopressin-Induced Intracellular Redistributionof Protein Kinase D in Intestinal Epithelial Cells
OSVALDO REY, ELENA ZHUKOVA, JAMES SINNETT-SMITH, AND ENRIQUE ROZENGURT*
Unit of Signal Transduction and Gastrointestinal Cancer,Division of Digestive Diseases, Department of Medicine,
UCLA-CURE Digestive Diseases Research Center and Molecular Biology Institute,David Geffen School of Medicine,
University of California at Los Angeles, California
The spatio-temporal changes of signaling molecules in response to G protein-coupled receptors (GPCR) stimulation is a poorly understood process in intestinalepithelial cells. Here we investigate the dynamic mechanisms associated withGPCR signaling in living rat intestinal epithelial cells by characterizing the in-tracellular translocation of protein kinase D (PKD), a serine/threonine proteinkinase involved in mitogenic signaling in intestinal epithelial cells. Analysis of theintracellular steady-state distribution of green fluorescent protein (GFP)-taggedPKD indicated that in non-stimulated IEC-18 cells, GFP-PKD is predominantlycytoplasmic.However, cell stimulationwith theGPCR agonist vasopressin inducesa rapid translocation of GFP-PKD from the cytosol to the plasma membrane that isaccompanied by its activation via protein kinase C (PKC)-mediated process andposterior plasma membrane dissociation. Subsequently, active PKD is importedinto the nuclei where it transiently accumulates before being exported into thecytosol by a mechanism that requires a competent Crm1 nuclear export pathway.These findings provide evidence for a mechanism by which PKC coordinates inintestinal epithelial cells the translocation and activation of PKD in response tovasopressin-induced GPCR activation. J. Cell. Physiol. 196: 483–492, 2003.� 2003 Wiley-Liss, Inc.
The sequential proliferation and lineage specificdifferentiation, migration, and cell death of epithelialcells of the intestinal mucosa is a tightly regulatedprocess modulated by a broad spectrum of regulatorypeptides (Santos et al., 1997; Burgess, 1998; Kanai et al.,1998). IEC-18 cells, a non-transformed cell line derivedfrom rat ileal crypts (Quaroni et al., 1979), have provideda valuable in vitro model to examine the effect of someof those regulatory peptides on permeability, migra-tion, differentiation, and proliferation (Dignass andPodolsky, 1993; Santos et al., 1997; Goke et al., 1998;Ray et al., 1999). For example, we recently demon-strated that the neurohypophysial non-peptide argininevasopressin (AVP), a G protein-coupled receptor (GPCR)agonist also known as antidiuretic hormone, inducesproliferation and migration of intestinal epithelial cellsby a signal transduction pathway that requires proteinkinase C (PKC) activity (Chiu et al., 2002). However, themechanisms by which the AVP-generated signals in theplasma membrane are propagated to critical down-stream targets remain largely undefined.
Protein kinase D (PKD) (Johannes et al., 1994;Valverde et al., 1994) is a serine/threonine proteinkinases that can be activated in intact IEC-18 cells bytumor promoting phorbol esters, lysophosphatidic acidand AVP by a PKC-dependent signal transductionpathway (Chiu and Rozengurt, 2001; Chiu et al.,2002). The salient features of PKD structure include
the presence of a catalytic domain distantly related toCa2þ-regulated kinases, a pleckstrin homology (PH)domain that regulates PKD activity and a highlyhydrophobic stretch of amino acids in its N-terminalregion (Johannes et al., 1994; Valverde et al., 1994;Iglesias and Rozengurt, 1998; Waldron et al., 1999). TheN-terminal region of PKD contains in addition to the PHdomain, a cysteine-rich domain (CRD) that confers highaffinity binding to phorbol esters and diacylglycerolDAG (Rozengurt et al., 1997; Iglesias et al., 1998a;Iglesias and Rozengurt, 1999).
� 2003 WILEY-LISS, INC.
Enrique Rozengurt is the Ronald S. Hirshberg Professor ofTranslational Pancreatic Cancer Research.
Contract grant sponsor: National Institute of Health/NIDDK;Contract grant numbers: DK 55003, DK 56930, DK 17294;Contract grant sponsor: National Institute of Health/NCI;Contract grant number: K01CA097956-01.
*Correspondence to: Enrique Rozengurt, 900 Veteran Avenue,Warren Hall Room 11-124, Department of Medicine, School ofMedicine, University of California at Los Angeles, Los Angeles,CA 90095-1786. E-mail: [email protected]
Received 11 February 2003; Accepted 13 March 2003
DOI: 10.1002/jcp.10323

PKD has been implicated in the regulation of a varietyof cellular functions including DNA synthesis and cellproliferation (Zhukova et al., 2001), Golgi organizationand function (Jamora et al., 1999; Liljedahl et al., 2001),EGF receptor and c-Jun signaling (Bagowski et al.,1999; Hurd and Rozengurt, 2001; Hurd et al., 2002),NFkb-mediated gene expression (Johannes et al., 1998)and cell migration (Bowden et al., 1999). Our previousresults suggest that the regulation of these diversefunctions is associated to the dynamic intracellulardistribution ofPKD (Rey andRozengurt, 2001; Reyet al.,2001a,b). In particular, we recently demonstrated thatmitogenic GPCR agonists induce a rapid translocation ofPKD from the cytosol to the nucleus in Swiss 3T3fibroblasts (Rey et al., 2001a). Despite its potentialimportance, no information is available on ligand-induced PKD nuclear translocation in any other systemincluding intestinal epithelial cells.
In the present study, we examined the effect of theGPCR agonist AVP on the intracellular distribution ofPKD in living intestinal epithelial IEC-18 cells. Ourresults show that in non-stimulated cells, PKD ispredominantly cytoplasmic. This steady-state distribu-tion of PKD results from its continuous shuttlingbetween the nucleus and the cytoplasm by a mechanismthat requires a competent Crm1-nuclear export path-way. The mitogenic GPCR agonist AVP induces a rapidand reversible translocation of the green fluorescenttagged-PKD to the plasma membrane of IEC-18 cells,which is PKC-dependent. Our results also demonstratethat stimulation with this agonist induce a transitorynuclear accumulation of PKD. These findings demon-strate, for the first time, that the intracellular distribu-tion of PKD in intestinal epithelial cells is rapidlymodified by AVP stimulation and provide furtherevidence for a mechanism where the activation andintracellular distribution of PKD is coordinated by PKC.
MATERIALS AND METHODSCells
IEC-18 cells were purchased from American TypeCulture Collection (Manassas, VA). Stock cultures ofthese cells were maintained in Dulbecco’s modifiedEagle’s medium (DMEM) supplemented with 10% fetalbovine serum (FBS) in a humidified atmosphere con-taining 10% CO2 and 90% air at 378C. For experimentalpurposes, cells were plated onto 15-mm no. 1 round glasscoverslips (Warner Instrument Corporation, Hamden,CT) inside 33-mm dishes at 7�104 cells/dish and trans-fected 18–20 h later for imaging. For Western blot ex-periments, cells were plated in 86-mm dishes at 3�105 cells/dish and transfected 18–20 h later. Cells weretransfected with 1 mg DNA/33 mm dish or 3 mg DNA/86-mm dish using LipofectAMINE PLUS (Life Techno-logies, Inc., Gaithersburg, MD) according to the manu-facturer’s suggested conditions. Transfected cells wereincubated for 18–20 h before agonist stimulation.
For immunocytochemistry, 2�105 IEC18-PKD.GFPcells were plated in 33 mm dishes and grown in DMEMcontaining 10% fetal bovine serum for 7–9 days until thecells become confluent and quiescent (Seufferlein et al.,1996).
Phoenix packaging cells (kindly provided by Dr. G.Nolan, Stanford University, Stanford, CA) were cul-
tured in the same media in a humidified atmospherecontaining 5% CO2.
Production of retrovirus and cDNA constructs andretrovirus production
pMSCVneo retroviral vector was engineered toinclude a single cassette that expresses the murinePKD and green fluorescent protein (GFP) from the samepromoter. The MSCV-IRES-GFP plasmid was con-structed by substituting the neo gene in the vector withthe IRES-GFP fragment. PKD cDNA (spanning fromposition þ50 to þ2910, from the published GenBankTM
sequence was inserted into EcoRI site of the MSCV-IRES-GFP plasmid, upstream of IRES. This fragment ofPKD cDNA, which contains all coding sequences butlacks the polyadenylation signal, was amplified bypolymerase chain reaction. Polymerase chain reactionwas initiated from bipartite primers 5’-CTGGAAT-TCCTCCCGGAAAGTTTGGTGGTT (sense) and 5’-AT-CGAATTCGTGTTTTGACAGATTAGAGG (antisense)that introduced EcoRI restriction sites at 5’- and 3’-endsof the fragment. The nucleotide sequence of theamplified PKD coding region was confirmed by sequen-cing. For retrovirus production, logarithmically growingPhoenix ecotropic cells were transfected with MSCV-PKD-IRES-GFP using Fugene 6 Transfection Reagentas per the manufacturer’s protocol. Virus-containingsupernatants were collected 48 h after transfection andused immediately. Logarithmically growing IEC-18cells were incubated with the virus-containing super-natants in the presence of 5 g/ml Polybrene for 5 h. Cellswere collected 48–72 h later, and GFP-positive fractionswere FACS-sorted using a Becton Dickinson FACStarPLUS machine. GFP-positive cells were propagated,and multiple aliquots were frozen. A fresh batch of cellswas restarted every 2 months. Following sorting, GFP-positive IEC-18 cells were maintained as describedabove. The vectors encoding the chimeric fusion proteinbetween green (GFP) or red fluorescent protein (RFP)and PKD were previously described (Matthews et al.,2000; Rey et al., 2001b).
Cell imaging
Live cell imaging. In order to maintain a constanttemperature of 378C during the experimental pro-cedures, cells were grown on the 15-mm glass cover-slips and subsequently mounted in a perfusion chamber(RC-25 Warner Instrument Corporation) and perfusedwith medium preheated at 378C by a SH-27B solutionin-line heater (Warner Instrument Corporation). Themedium was supplemented with 10 mM HEPES, pH 7.2.The perfusion chamber was mounted on an epifluores-cence microscope (Zeiss Axioskop) and cells imagedwith a Zeiss water-immersion objective (Achroplan 40�/0.75 w, Carl Zeiss, Inc., Jena, Germany). Images werecaptured as uncompressed 24-bit TIFF files with a SPOTcooled (�128C) single CCD color digital camera (threepass method) driven by SPOT version 2.1 software(Diagnostic Instruments, Inc., Sterling Heights, MI).GFP or RFP fluorescence was observed with a HI Q filterset for FITC or Rhodamine/Tritc, respectively (ChromaTechnology, Brattleboro, VT). Fifty cells were analyzedper experiment and each experiment was performed in
484 REY ET AL.

triplicate. The selected cells displayed in the appro-priate figures were representative of 90% of the popula-tion of positive cells.
Indirect immunofluorescence. Indirect immuno-fluorescence of IEC18-PKD.GFP cells was performedin 10% buffered formalin phosphate fixed/0.3% TritonX-100-PBS permeabilized cultures. After extensive PBSwashing, fixed cells were incubated for 18 h at 258C inblocking buffer (PBS-1% gelatin-0.05% Tween 20) (BB)and then stained at 258C for 60 min with a rabbitpolyclonal antibody anti-PKD/PKCm diluted in BB.Subsequently, the cells were washed with PBS-0.05%Tween 20 at 258C and stained at 258C for 60 min withTexas Red-conjugated goat-anti rabbit diluted in BB andwashed again with PBS-0.05% Tween 20. After severalPBS washes, the samples were mounted with a gelvatol-glycerol solution containing 2.5% 1,4-diazobicyclo-[2.2.2]octane (Rey et al., 2001b). The samples wereimaged with an epifluorescence Zeiss Axioskop and aZeiss oil-immersion objective (Plan-Apochromat 40�/1.0, Carl Zeiss, Inc.). Images were captured as uncom-pressed 24-bit TIFF files with a SPOT cooled (�128C)single CCD color digital camera (three pass method)driven by SPOT version 2.1 software. Texas Red signalswere observed with HI Q filter sets for Rhodamine/Tritc(Chroma Technology). The selected cells displayed in theappropriate figures was representative of 90% of thepopulation of positive cells.
In vitro kinase assays
Immune complexes were washed twice with lysisbuffer, then twice with kinase buffer consisting of30 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 mMdithiothreitol. Autophosphorylation reactions were ini-tiated by combining 20 ml of immune complexes with 5 mlof a phosphorylation mixture containing 100 mM [g-32P]-ATP (specific activity, 400–600 cpm/pmol) in kinasebuffer. Following incubation at 308C for 10 min, thereactions were terminated by addition of 1 ml ice-coldkinase buffer and placed on ice. Immune complexes wererecovered by centrifugation, and the proteins extractedfor SDS–PAGE analysis by addition of 2� SDS–PAGEsample buffer (200 mM Tris-HCl, pH 6.8, 0.1 mM sodiumorthovanadate, 1 mM EDTA, 6% SDS, 2 mM EDTA, 4%2-mercaptoethanol, 10% glycerol). Dried SDS–PAGEgels were subjected to autoradiography to visualizeradiolabeled protein bands. The obtained results arerepresentative of three independent experiments.
Western blot analysis
Samples of cell lysates were directly solubilized byboiling in SDS–PAGE sample buffer. Following SDS–PAGE on 10% gels, proteins were transferred toImmobilon-P membranes (Millipore Corp., Bedford,MA) and blocked by overnight incubation with 5% non-fat dried milk in PBS, pH 7.2. Membranes wereincubated at room temperature for 3 h with rabbitantiserum specifically recognizing the C-terminus ofPKD (PA-1) diluted in phosphate-buffered saline con-taining 3% non-fat dried milk. To determine thephosphorylation/activation state of PKD, the mem-branes were incubated with an antibody, phospho-PKD Ser-744/748 that specifically recognizes the phos-phorylated state of Ser744 and Ser748 within the
activation loop of PKDs (Waldron et al., 2001). Immu-noreactive bands were visualized using horseradishperoxidase-conjugated anti-rabbit or anti-mouse IgGand subsequent enhanced chemiluminescence Westernblotting ECL reagent (Amersham Pharmacia, Piscat-away, NJ). The obtained results are representative ofthree independent experiments.
Materials
[g-32P]-ATP (370MBq/ml) and horseradish peroxi-dase-conjugated donkey anti-rabbit IgG were fromAmersham Pharmacia. The anti-PKD C-terminus anti-body PA-1 was produced as previously described (VanLint et al., 1995). The anti-PKD (clone C20) antibodywas obtained from Santa Cruz Biotechnology, Inc.(Santa Cruz, CA). The anti-phospho-PKD Ser-744/748antibody was obtained from Cell Signaling Technology(Beverly, MA). Texas Red conjugated goat-anti rabbitimmunoglobulins were obtained from Molecular Probes(Eugene, OR). Vasopressin was obtained from SigmaChemical Company (St. Louis, MO). pMSCVneo retro-viral vector was from Clontech (Palo Alto, CA). Restric-tion enzymes were purchased from New EnglandBiolabs (Beverly, MA). Fugene 6 Transfection Reagentwas obtained from Roche Molecular Biochemicals(Indianapolis, IN). Leptomycin B was a generous giftfrom Dr. Minoru Yoshida, Department of Biotechnology,University of Tokyo. All the other reagents were thehighest grade commercially available.
RESULTSIntracellular distribution of PKD in unstimulated
intestinal epithelial cells
In order to examine the dynamic distribution of PKDin live IEC-18 cells, we used a fusion protein between theGFP of Aequorea victoria and PKD (GFP-PKD) thatdisplays similar biological properties to that of nativePKD (Matthews et al., 1999). Specifically, the fusion of aGFP tag to PKD did not produce any detectable effect onits basal catalytic activity or stimulation (Matthewset al., 1999; Waldron et al., 2001). In addition, theinherent fluorescence of GFP allowed us to visualize thetranslocation of GFP-PKD in living cells.
IEC-18 cells were transiently transfected withpEGFP-C3 (empty vector) or pGFP-PKD and the in-tracellular distribution of the encoded proteins exam-ined in unstimulated live cells 18 h post-transfection(Fig. 1). We found that whereas GFP was distributedthroughout the cytoplasm and nucleus of IEC-18 cells,GFP-PKD was localized in the cytoplasm. In addition,we detected that in some IEC-18 cells the GFP-PKDfluorescence was more pronounced at the perinucleararea, consistent with a partial localization of this proteinto the Golgi compartment as we and others previouslydemonstrated for other cell systems (Prestle et al., 1996;Jamora et al., 1999; Liljedahl et al., 2001; Rey andRozengurt, 2001; Rey et al., 2001b). To rule out possibleinterference with the normal intracellular distributionof PKD in IEC-18 cells due to the presence of thefluorescent tag at its N-terminus, we analyzed thelocalization of a chimeric protein between the RFP fromDiscosoma sp. fused to the C-terminus of PKD. Nodifference was detected in the distribution of PKD-RFPcompared to GFP-PKD (Fig. 1). The morphology of
PKD DISTRIBUTION IN INTESTINAL CELLS 485
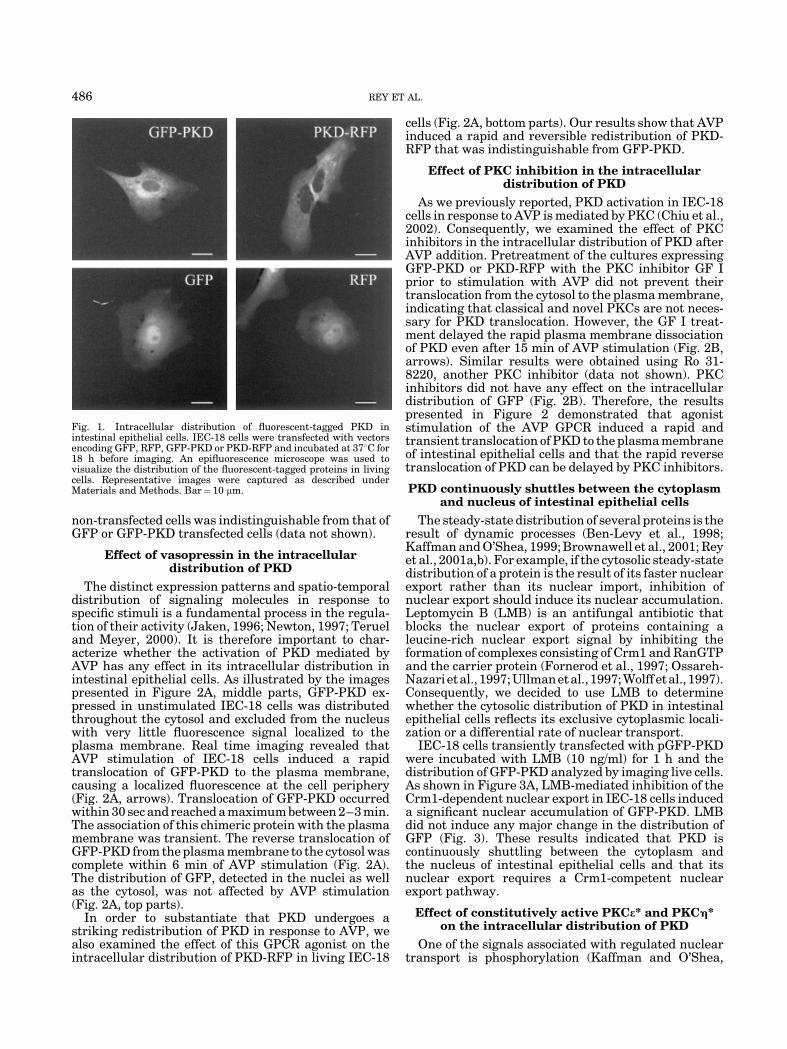
non-transfected cells was indistinguishable from that ofGFP or GFP-PKD transfected cells (data not shown).
Effect of vasopressin in the intracellulardistribution of PKD
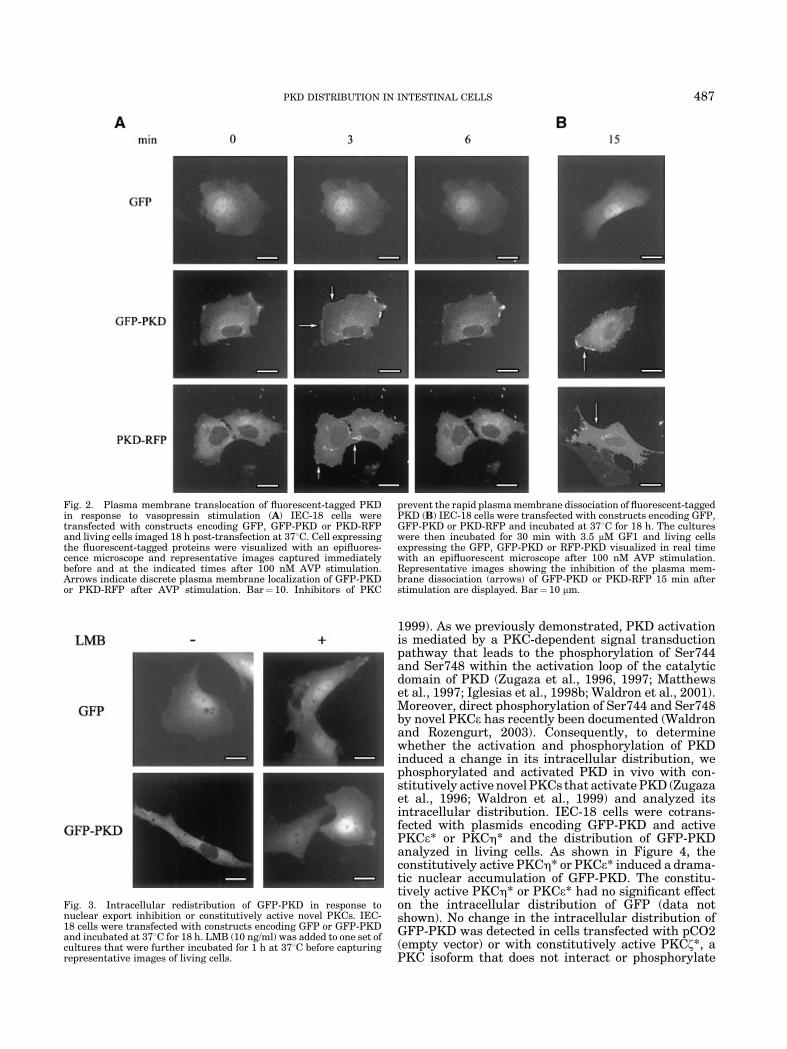
The distinct expression patterns and spatio-temporaldistribution of signaling molecules in response tospecific stimuli is a fundamental process in the regula-tion of their activity (Jaken, 1996; Newton, 1997; Terueland Meyer, 2000). It is therefore important to char-acterize whether the activation of PKD mediated byAVP has any effect in its intracellular distribution inintestinal epithelial cells. As illustrated by the imagespresented in Figure 2A, middle parts, GFP-PKD ex-pressed in unstimulated IEC-18 cells was distributedthroughout the cytosol and excluded from the nucleuswith very little fluorescence signal localized to theplasma membrane. Real time imaging revealed thatAVP stimulation of IEC-18 cells induced a rapidtranslocation of GFP-PKD to the plasma membrane,causing a localized fluorescence at the cell periphery(Fig. 2A, arrows). Translocation of GFP-PKD occurredwithin 30 sec and reached a maximum between 2–3 min.The association of this chimeric protein with the plasmamembrane was transient. The reverse translocation ofGFP-PKD from the plasma membrane to the cytosol wascomplete within 6 min of AVP stimulation (Fig. 2A).The distribution of GFP, detected in the nuclei as wellas the cytosol, was not affected by AVP stimulation(Fig. 2A, top parts).
In order to substantiate that PKD undergoes astriking redistribution of PKD in response to AVP, wealso examined the effect of this GPCR agonist on theintracellular distribution of PKD-RFP in living IEC-18
cells (Fig. 2A, bottom parts). Our results show that AVPinduced a rapid and reversible redistribution of PKD-RFP that was indistinguishable from GFP-PKD.
Effect of PKC inhibition in the intracellulardistribution of PKD
As we previously reported, PKD activation in IEC-18cells in response to AVP is mediated by PKC (Chiu et al.,2002). Consequently, we examined the effect of PKCinhibitors in the intracellular distribution of PKD afterAVP addition. Pretreatment of the cultures expressingGFP-PKD or PKD-RFP with the PKC inhibitor GF Iprior to stimulation with AVP did not prevent theirtranslocation from the cytosol to the plasma membrane,indicating that classical and novel PKCs are not neces-sary for PKD translocation. However, the GF I treat-ment delayed the rapid plasma membrane dissociationof PKD even after 15 min of AVP stimulation (Fig. 2B,arrows). Similar results were obtained using Ro 31-8220, another PKC inhibitor (data not shown). PKCinhibitors did not have any effect on the intracellulardistribution of GFP (Fig. 2B). Therefore, the resultspresented in Figure 2 demonstrated that agoniststimulation of the AVP GPCR induced a rapid andtransient translocation of PKD to the plasma membraneof intestinal epithelial cells and that the rapid reversetranslocation of PKD can be delayed by PKC inhibitors.
PKD continuously shuttles between the cytoplasmand nucleus of intestinal epithelial cells
The steady-state distribution of several proteins is theresult of dynamic processes (Ben-Levy et al., 1998;Kaffman and O’Shea, 1999; Brownawell et al., 2001; Reyet al., 2001a,b). For example, if the cytosolic steady-statedistribution of a protein is the result of its faster nuclearexport rather than its nuclear import, inhibition ofnuclear export should induce its nuclear accumulation.Leptomycin B (LMB) is an antifungal antibiotic thatblocks the nuclear export of proteins containing aleucine-rich nuclear export signal by inhibiting theformation of complexes consisting of Crm1 and RanGTPand the carrier protein (Fornerod et al., 1997; Ossareh-Nazarietal., 1997;Ullmanetal., 1997;Wolff etal., 1997).Consequently, we decided to use LMB to determinewhether the cytosolic distribution of PKD in intestinalepithelial cells reflects its exclusive cytoplasmic locali-zation or a differential rate of nuclear transport.
IEC-18 cells transiently transfected with pGFP-PKDwere incubated with LMB (10 ng/ml) for 1 h and thedistribution of GFP-PKD analyzed by imaging live cells.As shown in Figure 3A, LMB-mediated inhibition of theCrm1-dependent nuclear export in IEC-18 cells induceda significant nuclear accumulation of GFP-PKD. LMBdid not induce any major change in the distribution ofGFP (Fig. 3). These results indicated that PKD iscontinuously shuttling between the cytoplasm andthe nucleus of intestinal epithelial cells and that itsnuclear export requires a Crm1-competent nuclearexport pathway.
Effect of constitutively active PKCe* and PKCh*on the intracellular distribution of PKD
One of the signals associated with regulated nucleartransport is phosphorylation (Kaffman and O’Shea,
Fig. 1. Intracellular distribution of fluorescent-tagged PKD inintestinal epithelial cells. IEC-18 cells were transfected with vectorsencoding GFP, RFP, GFP-PKD or PKD-RFP and incubated at 378C for18 h before imaging. An epifluorescence microscope was used tovisualize the distribution of the fluorescent-tagged proteins in livingcells. Representative images were captured as described underMaterials and Methods. Bar¼ 10 mm.
486 REY ET AL.
1999). As we previously demonstrated, PKD activationis mediated by a PKC-dependent signal transductionpathway that leads to the phosphorylation of Ser744and Ser748 within the activation loop of the catalyticdomain of PKD (Zugaza et al., 1996, 1997; Matthewset al., 1997; Iglesias et al., 1998b; Waldron et al., 2001).Moreover, direct phosphorylation of Ser744 and Ser748by novel PKCe has recently been documented (Waldronand Rozengurt, 2003). Consequently, to determinewhether the activation and phosphorylation of PKDinduced a change in its intracellular distribution, wephosphorylated and activated PKD in vivo with con-stitutively active novel PKCs that activate PKD (Zugazaet al., 1996; Waldron et al., 1999) and analyzed itsintracellular distribution. IEC-18 cells were cotrans-fected with plasmids encoding GFP-PKD and activePKCe* or PKCh* and the distribution of GFP-PKDanalyzed in living cells. As shown in Figure 4, theconstitutively active PKCh* or PKCe* induced a drama-tic nuclear accumulation of GFP-PKD. The constitu-tively active PKCh* or PKCe* had no significant effecton the intracellular distribution of GFP (data notshown). No change in the intracellular distribution ofGFP-PKD was detected in cells transfected with pCO2(empty vector) or with constitutively active PKCz*, aPKC isoform that does not interact or phosphorylate
Fig. 2. Plasma membrane translocation of fluorescent-tagged PKDin response to vasopressin stimulation (A) IEC-18 cells weretransfected with constructs encoding GFP, GFP-PKD or PKD-RFPand living cells imaged 18 h post-transfection at 378C. Cell expressingthe fluorescent-tagged proteins were visualized with an epifluores-cence microscope and representative images captured immediatelybefore and at the indicated times after 100 nM AVP stimulation.Arrows indicate discrete plasma membrane localization of GFP-PKDor PKD-RFP after AVP stimulation. Bar¼ 10. Inhibitors of PKC
prevent the rapid plasma membrane dissociation of fluorescent-taggedPKD (B) IEC-18 cells were transfected with constructs encoding GFP,GFP-PKD or PKD-RFP and incubated at 378C for 18 h. The cultureswere then incubated for 30 min with 3.5 mM GF1 and living cellsexpressing the GFP, GFP-PKD or RFP-PKD visualized in real timewith an epifluorescent microscope after 100 nM AVP stimulation.Representative images showing the inhibition of the plasma mem-brane dissociation (arrows) of GFP-PKD or PKD-RFP 15 min afterstimulation are displayed. Bar¼ 10 mm.
Fig. 3. Intracellular redistribution of GFP-PKD in response tonuclear export inhibition or constitutively active novel PKCs. IEC-18 cells were transfected with constructs encoding GFP or GFP-PKDand incubated at 378C for 18 h. LMB (10 ng/ml) was added to one set ofcultures that were further incubated for 1 h at 378C before capturingrepresentative images of living cells.
PKD DISTRIBUTION IN INTESTINAL CELLS 487

PKD (Zugaza et al., 1996; Waldron et al., 1999). Theseresults suggest that the activation and phosphorylationof PKD is a potent stimulus for its nuclear translocationin intestinal epithelial cells.
Establishment and characterization of IEC-18cells stably expressing PKD
Our results suggested that the activation and phos-phorylation of PKD via constitutively active PKCh* orPKCe* modified the ratio between PKD nuclear importand export in intestinal epithelial cells. Consequently,our next objective was to determine whether a regulatednuclear transport of PKD could be achieved underphysiological conditions, such as, via PKD activationand phosphorylation by agonist stimulation of theendogenous AVP receptor. This was important becauseregulated transport mechanisms are crucial in modulat-ing signaling information by targeting proteins todifferent cellular compartments in response to specificcell stimuli (Ben-Levy et al., 1998; Kaffman and O’Shea,1999; Brownawell et al., 2001). Quiescent cells are anexcellent cell system model extensively used to eluci-date the signal transduction pathways activated by ago-nists that act through endogenously expressed GPCRs(Rozengurt, 1986, 1998). Because the cell cultureconditions (medium containing 10% fetal bovine serum)that we used to transiently express the fluorescent-tagged PKD prevented us from inducing G0/G1 cell cyclearrest in IEC-18 cells, we instead used a cell line termedIEC18-PKD.GFP. We established this cell line in ourlaboratory by infecting IEC-18 cells with a retrovirusencoding PKD linked via an internal ribosome entry siteto GFP. This bicistronic retroviral vector drives theexpression of PKD and GFP as two separate proteinsunder the control of the long terminal repeat of themurine stem cell virus (MSCV). Since the same pro-moter drives the transcription of both genes, cellsexpressing higher levels of GFP also express higherlevels of PKD. Consequently, GFP was used as a markerfor selection of PKD-positive cells (IEC18-PKD.GFP).After infection, cells expressing higher levels of GFPwere sorted by FACS, collected, and propagated forfurther studies. As a control, parallel cultures of IEC-18cells were infected with MSCV retrovirus encoding onlyGFP and then FACS-sorted, collected, and propagatedto generate IEC18-GFP cells.
The expression of PKD in the FACS-sorted cells wasanalyzed by SDS–PAGE and Western blotting with thePA-1 antibody against the C-terminal region of PKD. As
shown in Figure 5A, lysates of IEC18-PKD.GFP cellsexhibited a marked increase (approximately 5–6 fold) inthe expression of an immunoreactive band of 115 KDa,which corresponds to the molecular mass of PKD(Valverde et al., 1994), as compared with the endogen-ous PKD expressed IEC18-GFP cells. The detection ofthis band was completely blocked by the EEREMKAL-SERVSIL peptide that corresponds to the C-terminalregion of PKD. Thus, using high efficiency retroviralmediated transfection we generated IEC-18 cells over-expressing PKD.
We reported that IEC-18 cell stimulation with AVPinduces rapid PKC-dependent conversion of PKD froman inactive to an active state (Chiu et al., 2002).Consequently, we examined whether the activity ofPKD overexpressed in IEC-18 cells is regulated in asimilar manner. Intact IEC18-PKD.GFP cells weretreated with AVP, lysed and PKD was immunoprecipi-tated with PA-1 antiserum. The resulting immunocom-plexes were incubated with [g-32P]ATP, and theincorporation of 32P into PKD was analyzed by SDS–PAGE and autoradiography. As shown in Figure 5B,PKD isolated from unstimulated IEC18-PKD.GFP cellshad very low catalytic activity, indicating that over-expression did not lead to constitutive activation of thisenzyme. Stimulation of these cells with AVP induced astriking increase in PKD kinase activity that wasmaintained during cell lysis and immunoprecipitation.Endogenous PKD expressed in control IEC18-GFP cellswas also activated by AVP as previously reported (Chiuet al., 2002).
We had previously proposed that phosphorylation ofSer744 and Ser748 within the PKD activation loop playsa critical role in its activation (Iglesias et al., 1998b;Yuan et al., 2000, 2002; Waldron et al., 2001; Waldronand Rozengurt, 2003). Recently, a novel antibodyrecognizing predominantly the phosphorylated state ofSer 744 (pS744/pS748) in PKD was successfully used inour laboratory to monitor activation loop phosphoryla-tion in response to PDB, bombesin or NT (Waldronet al., 2001; Guha et al., 2002; Waldron and Rozengurt,2003). Therefore, we used the pS744/pS748 antibody toexamine whether AVP treatment of intact IEC18-PKD.GFP cells induces the phosphorylation of Ser744and Ser748 within the activation loop of PKD. IEC18-PKD.GFP cells transiently were stimulated for 10 minwith 100 nM AVP and lysed. The solubilized proteinswere resolved by SDS–PAGE and Western blotted usingthe pS744/pS748 antibody. As shown in Figure 5C, AVP
Fig. 4. Intracellular redistribution of GFP-PKD in response to constitutively active novel PKCs. IEC-18cells were transiently cotransfected with pGFP-PKD and either pCO2 containing the cDNAs encoding theconstitutively active PKCe*, PKCh*, or PKCz* as indicated. After 18 h, representative images of livingcells were captured as described under Materials and Methods. Bar¼10 mm.
488 REY ET AL.
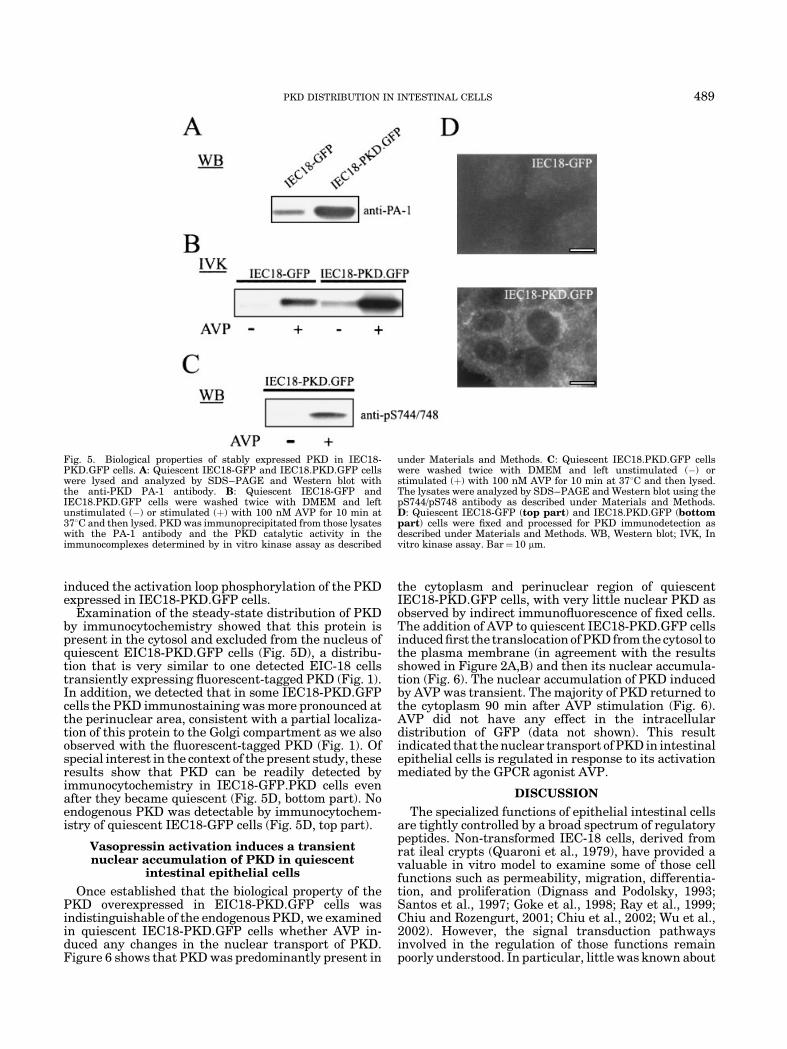
induced the activation loop phosphorylation of the PKDexpressed in IEC18-PKD.GFP cells.
Examination of the steady-state distribution of PKDby immunocytochemistry showed that this protein ispresent in the cytosol and excluded from the nucleus ofquiescent EIC18-PKD.GFP cells (Fig. 5D), a distribu-tion that is very similar to one detected EIC-18 cellstransiently expressing fluorescent-tagged PKD (Fig. 1).In addition, we detected that in some IEC18-PKD.GFPcells the PKD immunostaining was more pronounced atthe perinuclear area, consistent with a partial localiza-tion of this protein to the Golgi compartment as we alsoobserved with the fluorescent-tagged PKD (Fig. 1). Ofspecial interest in the context of the present study, theseresults show that PKD can be readily detected byimmunocytochemistry in IEC18-GFP.PKD cells evenafter they became quiescent (Fig. 5D, bottom part). Noendogenous PKD was detectable by immunocytochem-istry of quiescent IEC18-GFP cells (Fig. 5D, top part).
Vasopressin activation induces a transientnuclear accumulation of PKD in quiescent
intestinal epithelial cells
Once established that the biological property of thePKD overexpressed in EIC18-PKD.GFP cells wasindistinguishable of the endogenous PKD, we examinedin quiescent IEC18-PKD.GFP cells whether AVP in-duced any changes in the nuclear transport of PKD.Figure 6 shows that PKD was predominantly present in
the cytoplasm and perinuclear region of quiescentIEC18-PKD.GFP cells, with very little nuclear PKD asobserved by indirect immunofluorescence of fixed cells.The addition of AVP to quiescent IEC18-PKD.GFP cellsinduced first the translocation of PKD from the cytosol tothe plasma membrane (in agreement with the resultsshowed in Figure 2A,B) and then its nuclear accumula-tion (Fig. 6). The nuclear accumulation of PKD inducedby AVP was transient. The majority of PKD returned tothe cytoplasm 90 min after AVP stimulation (Fig. 6).AVP did not have any effect in the intracellulardistribution of GFP (data not shown). This resultindicated that the nuclear transport of PKD in intestinalepithelial cells is regulated in response to its activationmediated by the GPCR agonist AVP.
DISCUSSION
The specialized functions of epithelial intestinal cellsare tightly controlled by a broad spectrum of regulatorypeptides. Non-transformed IEC-18 cells, derived fromrat ileal crypts (Quaroni et al., 1979), have provided avaluable in vitro model to examine some of those cellfunctions such as permeability, migration, differentia-tion, and proliferation (Dignass and Podolsky, 1993;Santos et al., 1997; Goke et al., 1998; Ray et al., 1999;Chiu and Rozengurt, 2001; Chiu et al., 2002; Wu et al.,2002). However, the signal transduction pathwaysinvolved in the regulation of those functions remainpoorly understood. In particular, little was known about
Fig. 5. Biological properties of stably expressed PKD in IEC18-PKD.GFP cells. A: Quiescent IEC18-GFP and IEC18.PKD.GFP cellswere lysed and analyzed by SDS–PAGE and Western blot withthe anti-PKD PA-1 antibody. B: Quiescent IEC18-GFP andIEC18.PKD.GFP cells were washed twice with DMEM and leftunstimulated (�) or stimulated (þ) with 100 nM AVP for 10 min at378C and then lysed. PKD was immunoprecipitated from those lysateswith the PA-1 antibody and the PKD catalytic activity in theimmunocomplexes determined by in vitro kinase assay as described
under Materials and Methods. C: Quiescent IEC18.PKD.GFP cellswere washed twice with DMEM and left unstimulated (�) orstimulated (þ) with 100 nM AVP for 10 min at 378C and then lysed.The lysates were analyzed by SDS–PAGE and Western blot using thepS744/pS748 antibody as described under Materials and Methods.D: Quiescent IEC18-GFP (top part) and IEC18.PKD.GFP (bottompart) cells were fixed and processed for PKD immunodetection asdescribed under Materials and Methods. WB, Western blot; IVK, Invitro kinase assay. Bar¼ 10 mm.
PKD DISTRIBUTION IN INTESTINAL CELLS 489

the effect of GPCR agonists in the dynamic distributionof signaling molecules involved in intestinal epithelialcell regulation, a fundamental event in the regulation ofsignaling molecules activity (Jaken, 1996; Newton,1997; Teruel and Meyer, 2000). In the present study,we used visualization of fluorescent tagged-PKD in liveIEC-18 cells and immunocytochemistry of quiescentIEC-18 cells stably expressing non-fluorescent taggedPKD to demonstrate that AVP regulates the intracel-lular distribution of PKD in intestinal epithelial cells.
The steady-state distribution of PKD in non-stimu-lated IEC-18 cells was predominantly cytoplasmic.Stimulation of IEC-18 cells with the mitogenic GPCRagonist AVP, which activates PKD (Chiu et al., 2002),induced a rapid and reversible plasma membranetranslocation of PKD. PKC inhibitors did not preventthe translocation from the cytosol to the plasmamembrane of PKD in response to AVP. However, theysubstantially retarded its rapid reverse translocationfrom the plasma membrane to the cytosol, indicatingthat the plasma membrane dissociation of PKD inintestinal epithelial cells is coupled to their PKC-mediated activation. Based on these and our previousresults (Rey et al., 2001b) we conclude that PKCs notonly play a critical role in the activation of PKD but alsoin its rapid reverse translocation from the plasmamembrane.
In recent years, an increasing number of regulatoryprotein kinases including PKCl, Bruton’s tyrosinekinase, Abl and mitogen-activated protein kinase(MAPK/ERK), have been shown to shuttle between thenucleus and the cytoplasm (Chen et al., 1992; Lewiset al., 1996; Buchner, 2000; Mohamed et al., 2000; Cyert,2001; Gama-Carvalho and Carmo-Fonseca, 2001; Per-ander et al., 2001). Our results show that inhibition ofthe Crm1-dependent nuclear export pathway caused ashift in the steady-state distribution of PKD in IEC-18cells from predominantly cytoplasmic to nuclear andcytoplasmic, indicating that PKD shuttles between bothcompartments.
Several proteins that continuously shuttle betweenthe cytoplasm and the nucleus are subject to regulatednuclear transport in response to specific cellular signals,including phosphorylation (Ben-Levy et al., 1998; Kaff-man and O’Shea, 1999; Brownawell et al., 2001). Thesesignals can modify the relative rates of nuclear importand export of these proteins and consequently, theirsubcellular localization. We previously demonstratedthat PKD can be activated and phosphorylated by
constitutively active novel PKCs (Zugaza et al., 1996;Waldron et al., 1999; Waldron and Rozengurt, 2003).However, no information was available in any cellsystem on the intracellular distribution of PKD result-ing from its activation by constitutively active novelPKCs. Interestingly, our results showed that in IEC-18cells the activation of GFP-PKD mediated by consti-tutively active PKCe* or PKCh* was associated withits striking nuclear accumulation. This observationstrongly supports the conclusion that the nuclearlocalization of PKD in response to its activation is notan exclusive property associated to fibroblasts (Reyet al., 2001a).
In order to obtain further support for this conclusion,we examined the distribution of PKD constitutivelyexpressed in quiescent epithelial cells, since quiescentcells are extensively used to elucidate the signaltransduction pathways activated by agonists that actthrough endogenously expressed GPCRs (Rozengurt,1986, 1998). AVP stimulation of quiescent IEC18 cellsalso resulted in the nuclear accumulation of PKD.However, in contrast with the nuclear accumulationmediated by constitutively active novel PKCs, thenuclear accumulation mediated by AVP stimulationwas transient with PKD returning to the cytosol after90 min. Interestingly, the nuclear accumulation of PKDin response to the mitogenic signals evoked by AVP inEIC-18 cells is similar to the distinct spatial andtemporal localization of MAPK/ERK in response togrowth factors. In quiescent cells, the mitogenic stimu-lation elicited by growth factors also induces thetransient nuclear accumulation of MAPK/ERK (Chenet al., 1992; Gonzalez et al., 1993; Lenormand et al.,1993; Moriguchi et al., 1996; Fukuda et al., 1997;Khokhlatchev et al., 1998). The mechanism mediatingthe transient nuclear accumulation of active PKD asresult of AVP stimulation may reflect a low affinity forthe nuclear export machinery that is modified at a laterstage. Alternatively, the transient nuclear retention ofactive PKD can result from its transient interaction witha nuclear anchoring protein.
Given that in fibroblasts PKD activation mediated bymitogenic GPCR agonists induces its nuclear accumula-tion and potentiates cell proliferation (Rey et al., 2001a;Zhukova et al., 2001), it is tempting to speculate that thenuclear accumulation of active PKD in intestinalepithelial cells in response to AVP stimulation contri-butes to the transduction of the mitogenic signal inducedby this GPCR agonist.
Fig. 6. Vasopressin induce a transient redistribution of PKD from the cytoplasm to the nucleus ofquiescent intestinal epithelial cells. Quiescent IEC18-GFP.PKD cells incubated at 378C during 9 dayswere processed for immunocytochemistry before and at the indicated times after 100 nM AVPstimulation. PKD was detected in fixed cells as described under Materials and Methods. Bar¼ 10 mm.
490 REY ET AL.

ACKNOWLEDGMENTS
We are grateful to Dr. Minoru Yoshida, from theDepartment of Biotechnology, University of Tokyo, forthe generous gift of Leptomycin B, and to the Morphol-ogy/Imaging Core of CURE.
LITERATURE CITED
Bagowski CP, Stein-Gerlach M, Choidas A, Ullrich A. 1999. Cell-typespecific phosphorylation of threonines T654 and T669 by PKDdefines the signal capacity of the EGF receptor. EMBO J 18:5567–5576.
Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. 1998.Nuclear export of the stress-activated protein kinase p38 mediatedby its substrate MAPKAP kinase-2. Curr Biol 8:1049–1057.
Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC. 1999. Aninvasion-related complex of cortactin, paxillin and PKCmu associ-ates with invadopodia at sites of extracellular matrix degradation.Oncogene 18:4440–4449.
Brownawell AM, Kops GJ, Macara IG, Burgering BM. 2001. Inhibitionof nuclear import by protein kinase B (Akt) regulates the subcellulardistribution and activity of the forkhead transcription factor AFX.Mol Cell Biol 21:3534–3546.
Buchner K. 2000. The role of protein kinase C in the regulation of cellgrowth and in signalling to the cell nucleus. J Cancer Res Clin Oncol126:1–11.
Burgess AW. 1998. Growth control mechanisms in normal andtransformed intestinal cells. Philos Trans R Soc Lond B Biol Sci353:903–909.
Chen RH, Sarnecki C, Blenis J. 1992. Nuclear localization andregulation of erk- and rsk-encoded protein kinases. Mol Cell Biol12:915–927.
Chiu T, Rozengurt E. 2001. PKD in intestinal epithelial cells: Rapidactivation by phorbol esters, LPA, and angiotensin through PKC.Am J Physiol Cell Physiol 280:C929–C942.
Chiu T, Wu SS, Santiskulvong C, Tangkijvanich P, Yee HF, Jr.,Rozengurt E. 2002. Vasopressin-mediated mitogenic signaling inintestinal epithelial cells. Am J Physiol Cell Physiol 282:C434–C450.
Cyert MS. 2001. Regulation of nuclear localization during signaling.J Biol Chem 276:20805–20808.
Dignass AU, Podolsky DK. 1993. Cytokine modulation of intestinalepithelial cell restitution: Central role of transforming growth factorbeta. Gastroenterology 105:1323–1332.
Fornerod M, Ohno M, Yoshida M, Mattaj IW. 1997. CRM1 is an exportreceptor for leucine-rich nuclear export signals. Cell 90:1051–1060.
Fukuda M, Gotoh Y, Nishida E. 1997. Interaction of MAP kinase withMAP kinase kinase: Its possible role in the control of nucleocyto-plasmic transport of MAP kinase. EMBO J 16:1901–1908.
Gama-Carvalho M, Carmo-Fonseca M. 2001. The rules and roles ofnucleocytoplasmic shuttling proteins. FEBS Lett 498:157–163.
Goke M, Kanai M, Podolsky DK. 1998. Intestinal fibroblasts regulateintestinal epithelial cell proliferation via hepatocyte growth factor.Am J Physiol 274:G809–G818.
Gonzalez FA, Seth A, Raden DL, Bowman DS, Fay FS, Davis RJ. 1993.Serum-induced translocation of mitogen-activated protein kinase tothe cell surface ruffling membrane and the nucleus. J Cell Biol 122:1089–1101.
Guha S, Rey O, Rozengurt E. 2002. Neurotensin induces proteinkinase C-dependent protein kinase D activation and DNA synthesisin human pancreatic carcinoma cell line PANC-1. Cancer Res 62:1632–1640.
Hurd C, Rozengurt E. 2001. Protein kinase D is sufficient to suppressEGF-induced c-Jun Ser 63 phosphorylation. Biochem Biophys ResCommun 282:404–408.
Hurd C, Waldron RT, Rozengurt E. 2002. Protein kinase D complexeswith C-Jun N-terminal kinase via activation loop phosphorylationand phosphorylates the C-Jun N-terminus. Oncogene 21:2154–2160.
Iglesias T, Rozengurt E. 1998. Protein kinase D activation by muta-tions within its pleckstrin homology domain. J Biol Chem 273:410–416.
Iglesias T, Rozengurt E. 1999. Protein kinase D activation by deletionof its cysteine-rich motifs. FEBS Lett 454:53–56.
Iglesias T, Matthews S, Rozengurt E. 1998a. Dissimilar phorbol esterbinding properties of the individual cysteine-rich motifs of proteinkinase D. FEBS Lett 437:19–23.
Iglesias T, Waldron RT, Rozengurt E. 1998b. Identification of in vivophosphorylation sites required for protein kinase D activation.J Biol Chem 273:27662–27667.
Jaken S. 1996. Protein kinase C isozymes and substrates. Curr OpinCell Biol 8:168–173.
Jamora C, Yamanouye N, Van Lint J, Laudenslager J, VandenheedeJR, Faulkner DJ, Malhotra V. 1999. Gbetagamma-mediated re-gulation of Golgi organization is through the direct activation ofprotein kinase D. Cell 98:59–68.
Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. 1994.PKCu is a novel, atypical member of the protein kinase C family.J Biol Chem 269:6140–6148.
Johannes FJ, Horn J, Link G, Haas E, Siemienski K, Wajant H,Pfizenmaier K. 1998. Protein kinase Cmu downregulation of tumor-necrosis-factor-induced apoptosis correlates with enhanced expres-sion of nuclear-factor-kappaB-dependent protective genes. Eur JBiochem 257:47–54.
Kaffman A, O’Shea EK. 1999. Regulation of nuclear localization: A keyto a door. Ann Rev Cell Dev Biol 15:291–339.
Kanai M, Mullen C, Podolsky DK. 1998. Intestinal trefoil factorinduces inactivation of extracellular signal-regulated protein kinasein intestinal epithelial cells. Proc Natl Acad Sci USA 95:178–182.
Khokhlatchev AV, Canagarajah B, Wilsbacher J, Robinson M,Atkinson M, Goldsmith E, Cobb MH. 1998. Phosphorylation of theMAP kinase ERK2 promotes its homodimerization and nucleartranslocation. Cell 93:605–615.
Lenormand P, Sardet C, Pages G, L’Allemain G, Brunet A, PouyssegurJ. 1993. Growth factors induce nuclear translocation of MAPkinases (p42mapk and p44mapk) but not of their activator MAPkinase kinase (p45mapkk) in fibroblasts. J Cell Biol 122:1079–1088.
Lewis JM, Baskaran R, Taagepera S, Schwartz MA, Wang JY. 1996.Integrin regulation of c-Abl tyrosine kinase activity and cytoplas-mic-nuclear transport. Proc Natl Acad Sci USA 93:15174–15179.
Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V.2001. Protein kinase D regulates the fission of cell surface destin-ed transport carriers from the trans-Golgi network. Cell 104:409–420.
Matthews SA, Pettit GR, Rozengurt E. 1997. Bryostatin 1 inducesbiphasic activation of protein kinase D in intact cells. J Biol Chem272:20245–20250.
Matthews S, Iglesias T, Cantrell D, Rozengurt E. 1999. Dynamic re-distribution of protein kinase D (PKD) as revealed by a GFP-PKDfusion protein: Dissociation from PKD activation. FEBS Lett 457:515–521.
Matthews SA, Iglesias T, Rozengurt E, Cantrell D. 2000. Spatial andtemporal regulation of protein kinase D (PKD). EMBO J 19:2935–2945.
Mohamed AJ, Vargas L, Nore BF, Backesjo CM, Christensson B,Smith CI. 2000. Nucleocytoplasmic shuttling of Bruton’s tyrosinekinase. J Biol Chem 275:40614–40619.
Moriguchi T, Gotoh Y, Nishida E. 1996. Roles of the MAP kinasecascade in vertebrates. Adv Pharmacol 36:121–137.
Newton AC. 1997. Regulation of protein kinase C. Curr Opin Cell Biol9:161–167.
Ossareh-Nazari B, Bachelerie F, Dargemont C. 1997. Evidence for arole of CRM1 in signal-mediated nuclear protein export. Science278:141–144.
Perander M, Bjorkoy G, Johansen T. 2001. Nuclear import and exportsignals enable rapid nucleocytoplasmic shuttling of the atypicalprotein kinase C lambda. J Biol Chem 276:13015–13024.
Prestle J, Pfizenmaier K, Brenner J, Johannes FJ. 1996. Proteinkinase C mu is located at the Golgi compartment. J Cell Biol 134:1401–1410.
Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. 1979. Epithelioidcell cultures from rat small intestine. Characterization by morpho-logic and immunologic criteria. J Cell Biol 80:248–265.
Ray RM, Zimmerman BJ, McCormack SA, Patel TB, Johnson LR.1999. Polyamine depletion arrests cell cycle and induces inhibitorsp21(Waf1/Cip1), p27(Kip1), and p53 in IEC-6 cells. Am J Physiol276:C684–C691.
Rey O, Rozengurt E. 2001. Protein kinase D interacts with Golgi via itsCysteine-rich domain. Biochem Biophys Res Commun 287:21–26.
Rey O, Sinnett-Smith J, Zhukova E, Rozengurt E. 2001a. Regulatednucleocytoplasmic transport of protein kinase D in response to Gprotein-coupled receptor activation. J Biol Chem 276:49228–49235.
Rey O, Young SH, Cantrell D, Rozengurt E. 2001b. Rapid proteinkinase D translocation in response to G protein-coupled receptoractivation: Dependence on protein kinase C. J Biol Chem 276:32616–32626.
PKD DISTRIBUTION IN INTESTINAL CELLS 491

Rozengurt E. 1986. Early signals in the mitogenic response. Science234:161–166.
Rozengurt E. 1998. Signal transduction pathways in the mitogenicresponse to G protein-coupled neuropeptide receptor agonists. J CellPhysiol 177:507–517.
Rozengurt E, Sinnett-Smith J, Zugaza JL. 1997. Protein kinase D:A novel target for diacylglycerol and phorbol esters. Biochem SocTrans 25:565–571.
Santos MF, McCormack SA, Guo Z, Okolicany J, Zheng Y, JohnsonLR, Tigyi G. 1997. Rho proteins play a critical role in cell migrationduring the early phase of mucosal restitution. J Clin Invest 100:216–225.
Seufferlein T, Withers DJ, Mann D, Rozengurt E. 1996. Dissociation ofmitogen-activated protein kinase activation from p125 focal adhe-sion kinase tyrosine phosphorylation in Swiss 3T3 cells stimulatedby bombesin, lysophosphatidic acid, and platelet-derived growthfactor. Mol Biol Cell 7:1865–1875.
Teruel MN, Meyer T. 2000. Translocation and reversible localizationof signaling proteins: A dynamic future for signal transduction. Cell103:181–184.
Ullman KS, Powers MA, Forbes DJ. 1997. Nuclear export receptors:From importin to exportin. Cell 90:967–970.
Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. 1994.Molecular cloning and characterization of protein kinase D: A targetfor diacylglycerol and phorbol esters with a distinctive catalyticdomain. Proc Natl Acad Sci USA 91:8572–8576.
Van Lint JV, Sinnett-Smith J, Rozengurt E. 1995. Expression andcharacterization of PKD, a phorbol ester and diacylglycerol-stimulated serine protein kinase. J Biol Chem 270:1455–1461.
Waldron RT, Rozengurt E. 2003. Protein Kinase C PhosphorylatesProtein Kinase D Activation Loop Ser744 and Ser748 and ReleasesAutoinhibition by the Pleckstrin Homology Domain. J Biol Chem278:154–163.
Waldron RT, Iglesias T, Rozengurt E. 1999. The pleckstrin homologydomain of protein kinase D interacts preferentially with the etaisoform of protein kinase C. J Biol Chem 274:9224–9230.
Waldron R, Rey O, Iglesis T, Tugal T, Cantrell D, Rozengurt E. 2001.Activation loop Ser744 and Ser748 in protein kinase D are trans-phosphorylated in vivo. J Biol Chem 276:32606–32615.
Wolff B, Sanglier JJ, Wang Y. 1997. Leptomycin B is an inhibitorof nuclear export: Inhibition of nucleo-cytoplasmic translocation ofthe human immunodeficiency virus type 1 (HIV-1) Rev protein andRev-dependent mRNA. Chem Biol 4:139–147.
Wu SS, Chiu T, Rozengurt E. 2002. ANG II and LPA induce Pyk2tyrosine phosphorylation in intestinal epithelial cells: Role of Ca2þ,PKC, and Rho kinase. Am J Physiol Cell Physiol 282:C1432–C1444.
Yuan J, Slice L, Walsh JH, Rozengurt E. 2000. Activation of proteinkinase D by signaling through the alpha subunit of the hetero-trimeric G protein G(q). J Biol Chem 275:2157–2164.
Yuan J, Slice LW, Gu J, Rozengurt E. 2002. Cooperation of Gq, Gi andG12/13 in protein kinase D activation and phosphorylation inducedby lysophosphatidic acid. J Biol Chem 10:10.
Zhukova E, Sinnett-Smith J, Rozengurt E. 2001. Protein kinase Dpotentiates DNA synthesis and cell proliferation induced bybombesin, vasopressin or phorbol esters in Swiss 3Te cells. J BiolChem 276:40298–40305.
Zugaza JL, Sinnett-Smith J, Van Lint J, Rozengurt E. 1996. Proteinkinase D (PKD) activation in intact cells through a protein kinaseC-dependent signal transduction pathway. EMBO J 15:6220–6230.
Zugaza JL, Waldron RT, Sinnett-Smith J, Rozengurt E. 1997.Bombesin, vasopressin, endothelin, bradykinin, and platelet-derived growth factor rapidly activate protein kinase D through aprotein kinase C-dependent signal transduction pathway. J BiolChem 272: 23952–23960.
492 REY ET AL.