university of groningen total syntheses of (–)-borrelidin … 1 catalytic asymmetric total...
TRANSCRIPT

University of Groningen
Total syntheses of (–)-Borrelidin and (–)-Doliculide and the development of the catalyticasymmetric addition of Grignard reagents to ketonesMadduri Venkata, Ashoka Vardhan Reddy
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2012
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Madduri Venkata, A. V. R. (2012). Total syntheses of (–)-Borrelidin and (–)-Doliculide and the developmentof the catalytic asymmetric addition of Grignard reagents to ketones Groningen: University of Groningen
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 14-06-2018

Chapter 1
Catalytic Asymmetric Total Synthesis of Macrocyclic Polyketides and Methods for the Enantioselective Preparation of Tertiary Alcohols
This chapter gives an introduction on the asymmetric synthesis of important macrocyclic polyketides and an overview of the methods currently available for enantioselective synthesis of tertiary alcohols. Non-catalytic methods are briefly summarized, followed by an overview of catalytic asymmetric methods.
A part of this chapter has been submitted for publication: Madduri, A. V. R.; Harutyunyan, S. R.; Minnaard, A. J. Drug Discovery Today: Technologies 2012.

2
Chapter 1
1.1 Asymmetric Synthesis
Asymmetric synthesis of organic molecules is important in the fields of biology, pharmacy, and agriculture.1,2,3 As is often the case, only one of the enantiomers of a biologically active compound has the desired activity, whereas the other enantiomer is inactive or has even adverse effects. Examples of property differentiation within enantiomer pairs are numerous. Dopa,4 Thalidomide5 and Ethambutol6,7 emphasize the necessity to produce these drugs in an enantiomerically pure fashion (Figure 1).
Figure 1: Different enantiomers of a drug can have different effects
The field of asymmetric synthesis is not only relevant for biologically active molecules, but includes other functional molecules as well. Examples in the field of supramolecular chemistry and nanotechnology include the molecular motor of Feringa et al.8 and the molecular elevator of Stoddart et al.9 (Figure 2).

3
Introduction
Figure 2: Examples of chiral molecules that have mechanical properties a) a unidirectional molecular motor b) a molecular elevator
The production of enantiomerically pure compounds generally presents significant challenges and several methods to obtain enantiopure compounds are utilized:10
1) Application of natural enantiopure compounds, the so-called chiral pool: amino acids, hydroxy acids, carbohydrates, terpenes, alkaloids etcetera.
2) Separation of racemates: by resolution of diastereomers, (enzymatic) kinetic resolution and resolution by preferential crystallization.
3) Synthesis from prochiral compounds: asymmetric synthesis (enzymatic as well as non-enzymatic).
Of these possible approaches, asymmetric synthesis, and especially catalysis, is arguably one of the most elegant methods. Asymmetric catalysis is an efficient method to prepare enantiopure compounds in a large quantity. Therefore, remarkable efforts have been devoted to develop new catalytic asymmetric reactions and to improve the enantioselectivity and the rate of the existing ones.11-
14 To achieve maximum chiral amplification, chiral catalysts should efficiently differentiate between enantiotopic groups or faces of prochiral molecules. For that purpose, homogeneous metal complexes have become the most powerful and general tools available. Indeed, metal-mediated asymmetric catalysis has been intensively studied during the last four decades, since the first report of

4
Chapter 1
homogeneous copper catalyzed asymmetric cyclopropanation.15 Although initially only modest enantioselectivities were realized, enormous advances have been achieved since then. Rhodium-phosphine complex catalyzed asymmetric hydrogenation of dehydroamino acids16,17 and titanium-tartrate complex catalyzed asymmetric epoxidation of allylic alcohols were early milestones in metal catalyzed asymmetric catalysis,18 providing high enantioselectivity and accessing broad substrate scope. These reactions demonstrated the high practicality of asymmetric catalysis in the laboratory as well as in industry and the importance of asymmetric catalysis was recognized in the award of a Nobel Prize to Knowles, Noyori and Sharpless for their inspiring work on asymmetric catalytic hydrogenation and oxidation reactions.19
Natural products, often but not always chiral, play a crucial role in the development of novel drugs.20 Amongst the enormous amount of natural products known, macrocyclic polyketides clearly stand out. They often display remarkable biological activities and many of them (or their derivatives) have been successfully developed into drugs.21,22 The chemical diversity of macrocyclic polyketides is immense and provides an invaluable source of inspiration for innovative drug design. With their semi-rigid backbone, the appended substituents are placed in well-defined positions in space. 21,23 It is assumed that semi-rigid scaffolds possess some of the favorable binding properties of rigid molecules (a small loss of entropy upon binding), and yet are flexible enough to adapt their conformation during a binding event. An example of a synthetic macrocycle with improved binding (target selectivity) as compared to its linear analogue is the cyclic compound 1 (not a ketide), which is a 17-fold more potent inhibitor of the matrix metalloproteinase MMP-8 than its linear analogue 2 (Figure 3a).24 Well-known examples of macrocyclic polyketides which shows anticancer, antifungal, immuno-suppressive and antibiotic activity are Epothilone B,25,26 Amphotericin B,27,28 Rapamycin,29,30 and Erythromycin,31,32 respectively (Figure 3b).

5
Introduction
Figure 3: a) Compounds 1 and 2 are inhibitors of MMP-8, which is a member of the family of MMPs needed for maintenance of the extracellular matrix. The macrocyclic compound 1 is a 17-fold more potent inhibitor than the linear analogue 2 b) Structures of the naturally occurring macrocyclic drugs epothilone B, amphotericin B, rapamycin, and erythromycin.
Borrelidin33,34 and Doliculide35-38 are also macrocyclic polyketides with distinct bioactivity, though have not been developed into drugs. Borrelidin, discovered as a metabolite of Streptomyces rochei, is a biologically interesting and structurally unique 18-membered macrolide (Figure 4).39 Its anti-angiogenesis activity (the inhibition of blood vessel formation) is of significance to biological and pharmacological research.40 (–)-Doliculide, a 16-membered depsipeptide (Figure 4) was isolated by Yamada et al. in 1994 from the Japanese sea hare Dolabella auricularia.35 It exhibited exceedingly potent cytotoxicity against HeLa-S3 cells with an IC50 value of 0.005 μg/mL.36,41 Doliculide is structurally somewhat distinct from other cyclic depsipeptides in both the peptide and polyketide region. The dense arrangement of stereogenic centers in these macrocycles has attracted considerable attention from organic chemists as well.34,38

6
Chapter 1
Figure 4: Structures of natural products Borrelidin and Doliculide
A part of this thesis deals with an improved catalytic synthesis of the above mentioned macrocycles Borrelidin and Doliculide. Two reactions-types, used as key steps in these syntheses are the copper-catalyzed enantioselective 1,4-addition of methylmagnesium bromide, and the ruthenium-catalyzed asymmetric hydrogenation of -ketoesters and ketones. These strategies were the basis of a significant gain in synthetic efficiency, compared to the ones existing in literature.
1.2 Catalytic asymmetric 1,4-addition
When organometallic reagents are used in addition reactions to , -unsaturated ketones, the regioselectivity of the reaction i.e. 1,4-addition (conjugate addition) versus 1,2-addition (direct addition) must be taken into consideration (Scheme 1a). Given the substrate, the principal factor that determines the regioselectivity is the nature of the nucleophile. In general, whilst soft organometallics such as cuprates result in a high selectivity for 1,4-addition; hard nucleophiles such as Grignard reagents tend to afford 1,2-addition.
Scheme 1: a) 1,4-addition versus 1,2-addition. b) Catalytic asymmetric conjugate addition reaction on thioesters

7
Introduction
The asymmetric copper catalyzed conjugate addition of organometallic reagents such as diorganozinc, triorganoaluminum and organomagnesium compounds to unsaturated carbonyl compounds is entrenched synthetic methodology for the preparation of enantiopure compounds.42-44 Earlier work by our group has demonstrated that the Cu/Josiphos-catalyzed iterative asymmetric conjugate addition of MeMgBr leads to excellent stereochemical control in the synthesis of oligo-1,3-methyl and oligo-1,5-methyl arrays (Scheme 1b).45 This has been successfully exploited in the first total synthesis of -D-Mannosyl Phosphomycoketide46 and Phthiocerol Dimycocerosate A, both from Mycobacterium tuberculosis47 (Figure 5). Using this efficient and robust methodology, also the deoxypolypropionate units in Borrelidin and Doliculide have been prepared (see Chapter 2 and Chapter 3).
Figure 5: -D-Mannosyl Phosphomycoketide and Phthiocerol Dimycocerosate A from Mycobacterium tuberculosis
1.3 Catalytic asymmetric hydrogenation
Knowles and co-workers at Monsanto,16 developed a new synthesis of L-DOPA (Scheme 2a). With the use of the bidentate phosphine (R,R)-DiPAMP, enamide 3 was hydrogenated to the protected amino acid 4, that was converted to L-DOPA by acid catalyzed hydrolysis. This reaction was brought to commercial operation, making it the first commercialized catalytic asymmetric synthesis using a chiral transition metal complex.48 Following the pioneering work of Knowles, Noyori and Takaya in 1980 discovered BINAP (Scheme 2b), a new chiral diphosphine that

8
Chapter 1
turned out to be an extremely versatile ligand for various enantioselective catalytic processes.49 A classic example of its success is the hydrogenation of alkene 5 with the use of 0.5 mol% of a (S)-BINAP-Ru(OAc)2 complex, to give the anti-inflammatory agent (S)-naproxen 6 in 92% yield with 97% ee (Scheme 2b). Further research revealed that functionalized ketones were also reduced with the use of BINAP-ruthenium complexes that contain halides.
Scheme 2: a) The Monsanto synthesis of L-DOPA. b) Catalytic asymmetric synthesis of (S)-Naproxen using a (S)-BINAP-Ru complex
Some remarkable effects were found during this research, e.g. the addition of a small amount of a diamine completely changed the chemoselectivity from reduction of a C=C double bond to reduction of a carbonyl group.50 The reduction was achieved with turnover numbers of nearly 100.000. The development of these highly chemo- and enantioselective carbonyl reductions has been described in detail by Noyori in a recent review.51 These very successful “Noyori catalysts” have been further modified and applied to key chiral elements in Borrelidin and Doliculide, such as enantiomerically enriched -hydroxy esters and chiral secondary alcohols, see Chapter 2 and Chapter 3.52,53

9
Introduction
1.4 Chiral tertiary alcohols: Existing methods and new developments
The second part of this thesis is devoted to the development of a novel method to access chiral tertiary alcohols.
Chiral enantiopure alcohols represent a key class of organic molecules due to their widespread occurrence in natural products, pharmaceuticals and their application as chiral building blocks in synthetic chemistry (Figure 6a).54-56 Different strategies for the preparation of single enantiomers of chiral secondary alcohols comprise kinetic resolution, enantioselective desymmetrization, asymmetric hydrogenation (both with enzymes and transition-metal catalysts) and, recently, enantioselective conjugate addition of water.57-59 Generating chiral tertiary alcohols is however particularly challenging.60 Asymmetric hydrogenation is obviously not applicable for the formation of tertiary alcohols and the resolution of racemic tertiary alcohols with lipases or esterases is mostly not efficient. Therefore, methods for the synthesis of chiral tertiary alcohols rely mainly on the construction of carbon-carbon bonds.12,14,61
The catalytic asymmetric addition of organometallic reagents to aldehydes and in particular to ketones is one of the most straightforward methods for carbon-carbon bond formation (Figure 6b).62,63 Over the years, the catalytic asymmetric addition of organometallic reagents to aromatic aldehydes affording secondary alcohols has been well developed.64 Catalytic asymmetric addition of organometallic reagents to ketones is considerably more challenging. The difficulties are associated with a significantly diminished reactivity of ketones compared to aldehydes, and a decreased enantiodiscrimination due to the smaller steric and electronic differences between the two substituents on the carbonyl group. To overcome the lower reactivity of ketones, the use of highly reactive organometallic compounds such as Grignard and organolithium reagents has been explored.62,65,66 However, these reactive organometallics are intrinsically nucleophilic and can add to carbonyl compounds without the aid of a catalyst, resulting in a background reaction providing racemic alcohols. In addition, these reagents are strong bases as well and give rise to the formation of side products due to enolisation.

10
Chapter 1
NH
O
F3C
O
Cl
O
OH
OPh
HNS
O O
NCF3
O
O
N
O
N HO
Me OH
O OHOHP
HO
NaO OO
O
Efavirenz
Tripanavir
Camptothecin
Fostriecin
NHO
N
NOH
key precoursor of anantifungal drug
F
F
a)
R1 R2
O
R1 R2
RHO
*1,2-addition
b)chiral catalyst
+ R M
chiral tertiary alcoholsRM = R2Zn, RMgX, RLiR1, R2 = Ar, AlkAr, ArAlk, Alk
R1 R2
O
+ MgBrH
R3R
organometalicreagent
Mg
Br
R1
R2
O
H R3R
R3
R
H2O
R1 R2
OMgBr
R1 R2
OH
1,2-reductionproduct
cyclic six-memberedtransition state
c)
O
OMeMe
OH
Ar Ar
OH
Ar Ar
R1 R2
O
+ RMgX or RLi
1.2 eq
R1
9 to 84% yield25 to 98% ee
2 eq
R1, R2 & R = aryl & alkyl
100 oC, THF
Taddol
Seebach et al. 1992, 1994
R2
HO Rd) L1
Figure 6: a) Pharmaceutically active compounds bearing a chiral tertiary alcohol moiety. b) Asymmetric addition of an organometallic to a ketone. c) Plausible mechanism for the 1,2-redution (side-reaction) of carbonyl compounds during organometallic addition. d) Asymmetric addition of Grignard and organolithium reagents to ketones

11
Introduction
For alkyl Grignard and lithium compounds containing -hydrogens, also reduction of the carbonyl group is a serious complication (Figure 6c). Therefore the enantioselective addition of these reagents to both ketones and aldehydes required, until recently, at least one equivalent of a chiral ligand (Figure 6d).67 A variety of organotitanium reagents, prepared in situ by transmetalation of ClTi(O-iPr)3 using Grignard and organolithium reagents, has been used successfully as well, after the strict removal of magnesium and lithium salts.68,69 However, the development of a catalytic reaction has been hampered by the high reactivity of these organometallic reagents.
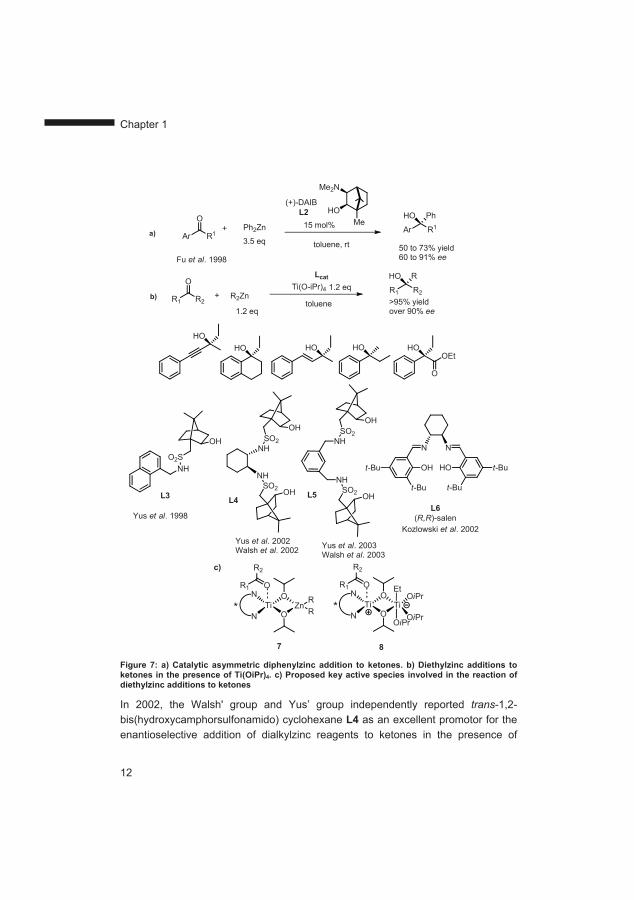
An important alternative to reduce the amount of chiral ligand is the use of, much less reactive, organozinc reagents.70 In order to add to ketones, organozinc reagents require Lewis acid activation of the carbonyl group, chiral Lewis base activation of the organozinc reagent, or a combination of both strategies. In 1998, Dosa and Fu reported the first example of a catalytic asymmetric addition of diphenylzinc to ketones, catalyzed by 3-exo-(dimethylamino)isoborneol (DAIB, L2, Figure 7a).71-73 Shortly thereafter, Yus and co-workers reported the first catalytic asymmetric addition of dialkylzinc reagents catalyzed by hydroxycamphorsulfonamide ligand L3 (Figure 7b) in the presence of titanium tetraisopropoxide.74
12
Chapter 1
Ar R1
O+ Ph2Zn
3.5 eq
Me2N
HOMe
(+)-DAIB
toluene, rt
Ar R1
HO Ph15 mol%
Fu et al. 199850 to 73% yield60 to 91% ee
Ti(O-iPr)4+ R2Zn
toluene
1.2 eq
1.2 eqR1 R2
OR1>95% yieldover 90% ee
R2
HO R
a)
b)
OH
NHO2S
NH
NH
SO2
OH
SO2 OH
NH
NH
SO2
OH
SO2 OH
Yus et al. 1998
Yus et al. 2003Walsh et al. 2003
Yus et al. 2002Walsh et al. 2002
Lcat
L3 L5L4
N
OH
t-Bu
t-Bu
N
HO
t-Bu
t-Bu
L6(R,R)-salen
L2
HOHO HO
O
OEtHOHO
Kozlowski et al. 2002
TiN
N
O
OZn
RR*
O
R2
R1
TiN
N
O
OTi*
O
R2
R1OiPr
OiPr
Et
OiPr
7 8
c)
Figure 7: a) Catalytic asymmetric diphenylzinc addition to ketones. b) Diethylzinc additions to ketones in the presence of Ti(OiPr)4. c) Proposed key active species involved in the reaction of diethylzinc additions to ketones
In 2002, the Walsh' group and Yus’ group independently reported trans-1,2-bis(hydroxycamphorsulfonamido) cyclohexane L4 as an excellent promotor for the enantioselective addition of dialkylzinc reagents to ketones in the presence of

13
Introduction
titanium tetraisopropoxide (Figure 7b).75,76 In addition, diarylzinc reagents as well as , -unsaturated ketones were successfully employed using this ligand. These results inspired the design of the improved ligand L5 (Figure 7b) for the asymmetric 1,2-addition of dialkylzinc reagents to ketones.77,78 The catalytically active species for dialkylzinc addition reactions using sulphonamide ligands has been postulated to be a dinuclear titanium complex or in some cases a titanium-zinc complex in which one titanium atom bears the chiral ligand and the ketone and the other titanium atom (or zinc atom) the alkyl moiety (Figure 7c, 7 and 8). The two metal centres are connected by two isopropoxy bridges (Figure 7c).
A common feature of these reactions is the necessity to employ equimolar amounts or an excess of titanium tetraisopropoxide in order to achieve good selectivities. It is believed that addition of organozinc reagents to non-reactive ketones proceeds via in situ formation of alkyltitanum species which are in principle less reactive than Grignard reagents however superior to dialkylzinc reagents (Figure 7c). This hypothesis is supported by a recent report from Li et al. on the direct addition of alkyltitanium reagents to aldehydes catalyzed by a chiral titanium catalyst derived from H8-BINOL.79 In one case it was possible to carry out the reaction using 60 mol% of titanium tetraisopropoxide.80 In another case it was reported that Ti(OiPr)4-free 1,2-addition to aromatic ketones is possible when a 3-fold excess of organozinc reagent is used.81
In the case of -ketoesters, which are more reactive than normal ketones, catalytic amounts of titanium tetraisopropoxide are sufficient. Kozlowski and co-workers designed a chiral salen ligand L6 (Figure 7b) capable of catalyzing the addition of organozinc reagents to ketoesters in the presence of 10 mol% of titanium tetraisopropoxide.82 It was proposed that this chiral salen ligand is able to chelate at the same time one titanium atom, following alkoxy interchange on titanium tetraisopropoxide, and one zinc atom, through coordination of the nitrogen atom by diethylzinc thus providing bifunctional alkylation.
These discoveries have permitted the development of an array of methods for the addition of organozinc reagents to aromatic ketones, enones and ketoesters.63,64,83 Nevertheless, diorganozinc reagents have an inherent drawback critical to practical applications.84,85 In their standard method of preparation, zinc oxidatively adds to alkyl iodides leading to alkylzinc iodides, which after distillation or sublimation, provide dialkylzinc reagents. Due to the thermal instability of their higher homologs, this method is applicable only to diorganozinc reagents with small alkyl groups. Therefore, only few organozinc reagents are commercially available. To circumvent this intrinsic problem, several methods for preparing diorganozinc reagents,

14
Chapter 1
including functionalized analogues, have been developed by the groups of Knochel84 and Charett.85
The successful 1,2-addition of organozinc reagents to ketones contrasts strongly with the few examples of the addition of organoaluminium reagents to ketones. The examples are limited mostly to arylation reactions (Figure 8a).86-88 In 2007 Gao and coworkers reported the first example of the asymmetric addition of arylaluminum reagents to ketones catalyzed by a titanium catalyst of (S)-BINOL L8 (Figure 8b).
Good yields and enantioselectivities up to 97% were achieved for a variety of alkyl aryl ketones. Also in this case, an excess of organoaluminium reagent as well as a superstoichiometric use of titanium tetraisopropoxide is required.
Figure 8: Asymmetric Ti(IV)-catalyzed addition on aryl methyl ketones using a) Arylaluminum reagents & b) Alkenylaluminum reagents
Readily available Grignard reagents have only been used in combination with stoichiometric amounts of a chiral ligand (Figure 6c). This is not surprising, as the uncatalyzed addition of the Grignard reagent is a formidable competitor. Indeed, catalytic non-asymmetric addition of Grignard reagents to ketones has become possible only recently using Zn(II) salts as catalyst.89,90 In 2008 Harada et al. reported that highly enantioselective catalytic addition of Grignard reagents to aldehydes is possible, albeit requiring the use of excess titanium tetraisopropoxide, most probably via formation of organotitanium reagents as nucleophiles.91,92

15
Introduction
1.5 Outline of this thesis
In this thesis, the application of copper-catalyzed asymmetric conjugate addition and ruthenium-catalyzed asymmetric hydrogenation reactions are described in the total synthesis of the macrocyclic polyketides Borrelidin and Doliculide. A second topic of the thesis is the development of a novel catalytic asymmetric synthesis of chiral tertiary alcohols. Next to these main topics, an asymmetric amplification in 1,2-addition reactions has been observed, and procedures for copper-catalyzed conjugate addition reactions to -substituted enones, and iridium catalyzed epimerization of alcohols and amines have been developed.
In Chapter 2, the asymmetric catalytic formal synthesis of (–)-Borrelidin is described. Iterative copper-catalyzed asymmetric conjugate addition and asymmetric hydrogenation are key strategic elements in this synthesis of Borrelidin.
In Chapter 3 the catalytic asymmetric synthesis of (–)-Doliculide is described, using the same key elements as in the synthesis of Borrelidin. Furthermore, the synthesis of two analogues of Doliculide is described, that should reveal its close relationship to other natural depsipeptides.
In Chapter 4, the first enantioselective copper-catalyzed 1,2-addition of Grignard reagents to , -unsaturated ketones is described. Careful selection of copper catalyst and reaction parameters allows the complete discrimination between 1,4 and 1,2 addition of Grignard reagents to enones, with very high stereoselectivity.
In Chapter 5 the first copper-catalyzed asymmetric 1,2-addition of Grignard reagents to aryl alkyl ketones is described. This method leads to benzylic tertiary alcohols in up to 97% yield and 99% ee.
In Chapter 6 an asymmetric amplification phenomenon in the copper catalyzed 1,2-addition of Grignard reagents to carbonyl compounds is described. This is explained by the solid-solution equilibrium of the copper ligand complexes.
In Chapter 7 the first copper-catalyzed asymmetric conjugate addition of Grignard reagents to -methyl cyclopentenone and -methyl cyclohexenone is described.
Finally in Chapter 8, a novel method for the selective epimerization of 1,2-amino-alcohols using half-sandwich cationic iridacycles is described. This is applied in the epimerization of hydroxy and amino groups in complex molecules.

16
Chapter 1
1.6 References
(1) Fischer, H. P.; Buser, H. P.; Chemla, P.; Huxley, P.; Lutz, W.; Mirza, S.; Ramos Tombo, G. M.; Van Lommen, G.; Sipido, V. Bull. Soc. Chim. Belg. 1994, 103, 565.
(2) Ramos Tombo, G. M.; Bellus, D. Angew. Chem. 1991, 103, 1219. (3) Noyori, R. Chem. Tech. 1992, 22, 366. (4) Cotzias, G. C.; Papavasiliou, P. S.; Gellene, R. New Engl. J. Med. 1969, 281, 272. (5) Ando, Y.; Fuse, E.; Figg, W. D. Clin. Cancer. Res. 2002, 8, 1964. (6) Wilkinson, R. G.; Shepherd, R. G.; Thomas, J. P.; Baughn, C. J. Am. Chem. Soc.
1961, 83, 2212. (7) Wilkinson, R. G.; Shepherd, R. G.; Thomas, J. P.; Baughn, C. Am. Rev. Respir.
Dis. 1961, 83, 891. (8) Koumura, N.; Geertsema, E. M.; Meetsma, A.; Feringa, B. L. J. Am. Chem. Soc.
2000, 122, 12005. (9) Badji , J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F. Science 2004, 303,
1845. (10) Crosby, J. Tetrahedron 1991, 47, 4789. (11) Catalytic asymmetric synthesis; 2nd edn ed.; Ojima, I., Ed.; Wiley: New York
2000. (12) Walsh, P. J.; Kozlowski, M. C., Eds.; University Science Books: California, 2009. (13) Noyori, R., Ed.; John Wiley and Sons: New York, 1994. (14) Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H., Eds.; Springer-Verlag: Berlin, 2004. (15) Nozaki, H.; Moriuti, S.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1966, 7, 5239. (16) Knowles, S.; Sabacky, M. J. Chem. Commun. 1968, 1445. (17) Dang, T. P.; Kagan, H. B. Chemm. Commun. 1971, 481. (18) Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 5974. (19) Nobel lectures can be read on http://nobelprize.org/nobel_prizes/chemistry/
laureates/2001. (20) von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Angew.
Chem. Int. Ed. 2006, 45, 5072. (21) Wessjohann, L. A.; Ruijter, E.; Garcia-Rivera, D.; Brandt, W. Mol. Divers. 2005, 9,
171. (22) Newman, D. J.; Cragg, G. M.; Snader, K. M. J. Nat. Prod. 2003, 66, 1022. (23) Zhang, M. Q.; Wilkinson, B. Curr. Opin. Biotechnol. 2007, 18, 478. (24) Cherney, R. J.; Wang, L.; Meyer, D. T.; Xue, C.-B.; Wasserman, Z. R.; Hardman,
K. D.; Welch, P. K.; Covington, M. B.; Copeland, R. A.; Arner, E. C.; DeGrado, W. F.; Decicco, C. P. J. Med. Chem. 1998, 41, 1749.
(25) Höfle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H. Angew. Chem. Int. Ed. 1996, 35, 1567.
(26) Altmann, K.-H.; Pfeiffer, B.; Arseniyadis, S.; Pratt, B. A.; Nicolaou, K. C. ChemMedChem 2007, 2, 396.
(27) Nicolaou, K. C.; Daines, R. A.; Chakraborty, T. K.; Ogawa, Y. J. Am. Chem. Soc. 1987, 109, 2821.
(28) Volmer, A. A.; Szpilman, A. M.; Carreira, E. M. Nat. Prod. Rep. 2010, 27, 1329. (29) Nicolaou, K. C.; Chakraborty, T. K.; Piscopio, A. D.; Minowa, N.; Bertinato, P. J.
Am. Chem. Soc. 1993, 115, 4419. (30) Graziani, E. I. Nat. Prod. Rep. 2009, 26, 602. (31) Woodward, R. B.; Logusch, E.; Nambiar, K. P.; Sakan, K.; Ward, D. E.; Au-Yeung,
B. W.; Balaram, P.; Browne, L. J.; Card, P. J.; Chen, C. H. J. Am. Chem. Soc. 1981, 103, 3210.
(32) Katz, L.; Ashley, G. W. Chem. Rev. 2005, 105, 499. (33) Madduri, A. V. R.; Minnaard, A. J. Chem. Eur. J. 2010, 16, 11726.

17
Introduction
(34) Nagamitsu, T.; Harigaya, Y.; Omura, S. Proc. Jpn. Acad. Ser. B-Phys. Biol. Sci. 2005, 81, 244.
(35) Ishiwata, H.; Nemoto, T.; Ojika, M.; Yamada, K. The Journal of Organic Chemistry 1994, 59, 4710.
(36) Ishiwata, H.; Sone, H.; Kigoshi, H.; Yamada, K. Tetrahedron 1994, 50, 12853. (37) Ghosh, A. K.; Liu, C. Org. Lett. 2001, 3, 635. (38) Hanessian, S.; Mascitti, V.; Giroux, S. PNAS. 2004, 101, 11996. (39) Berger, J.; Jampolsky, L. M.; Goldberg, M. W. Arch. Biochem. 1949, 22, 476. (40) Wakabayashi, T.; Kageyama, R.; Naruse, N.; Tsukahara, N.; Funahashi, Y.; Kitoh,
K.; Watanabe, Y. J. Antibiot. 1997, 50, 671. (41) Bai, R.; Covell, D. G.; Liu, C.; Ghosh, A. K.; Hamel, E. J. Biol. Chem. 2002, 277,
32165. (42) Alexakis, A.; Bäckvall, J.-E.; Krause, N.; Pamies, O.; Dieguez, M. Chem. Rev.
2008, 108, 2796. (43) Harutyunyan, S. R.; den Hartog, T.; Geurts, K.; Minnaard, A. J.; Feringa, B. L.
Chem. Rev. 2008, 108, 2824. (44) Jerphagnon, T.; Pizzuti, G. M.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev.
2009, 38, 1039. (45) Lopez, F.; Harutyunyan, S. R.; Minnaard, A. J.; Feringa, B. L. J. Am. Chem. Soc.
2004, 126, 12784. (46) van Summeren, R. P.; Moody, D. B.; Feringa, B. L.; Minnaard, A. J. J. Am. Chem.
Soc. 2006, 128, 4546. (47) Casas-Arce, E.; ter Horst, B.; Feringa, B. L.; Minnaard, A. J. Chem. Eur. J 2008,
14, 4157. (48) Merck Index, 12th ed., 1996, p. 933. (49) Miyashita, A.; Yasuda, A.; Takaya, H.; Toriumi, K.; Ito, T.; Souchi, T.; Noyori, R. J.
Am. Chem. Soc. 1980, 102, 7932. (50) Ohkuma, T.; Ooka, H.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1995, 117,
10417. (51) Noyori, R.; Ohkuma, T. Angew. Chem. Int. Ed. 2001, 40, 40. (52) Noyori, R.; Ikeda, T.; Ohkuma, T.; Widhalm, M.; Kitamura, M.; Takaya, H.;
Akutagawa, S.; Sayo, N.; Saito, T.; Taketomi, T.; Kumobayashi, H. J. Am. Chem. Soc. 1989, 111, 9134.
(53) Kramer, R.; Brückner, R. Angew. Chem. Int. Ed. 2007, 46, 6537. (54) Nicolaou, K. C.; Montagnon, T. Molecules that Changed the World; Wiley-VCH:
Weinheim, 2008. (55) Corey, E. J.; Guzman-Perez, A. Angew. Chem. Int. Ed. 1998, 37, 388. (56) Shibasaki, M.; Kanai, M. Chem. Rev. 2008, 108, 2853. (57) Kourist, R.; Domínguez de María, P.; Bornscheuer, U. T. ChemBioChem 2008, 9,
491. (58) Paravidino, M.; Holt, J.; Romano, D.; Singh, N.; Arends, I. W. C. E.; Minnaard, A.
J.; Orru, R. V. A.; Molinari, F.; Hanefeld, U. J. Mol. Cat. B: Enzymatic 2010, 63, 87.
(59) Boersma, A. J.; Coquière, D.; Geerdink, D.; Rosati, F.; Feringa, B. L.; Roelfes, G. Nat. Chem. 2010, 2, 991.
(60) Stymiest, J. L.; Bagutski, V.; French, R. M.; Aggarwal, V. K. Nature 2008, 456, 778.
(61) Organotransition metal chemistry: From bonding to catalysis; Hartwig, J. F., Ed.; University Science Books: Sausalito, California, 2010.
(62) Luderer, M. R.; Bailey, W. F.; Luderer, M. R.; Fair, J. D.; Dancer, R. J.; Sommer, M. B. Tetrahedron: Asymmetry 2009, 20, 981.
(63) Pu, L.; Yu, H.-B. Chem. Rev. 2001, 101, 757. (64) Binder, C. M.; Singaram, B. Org. Prep. Proc. Int. 2011, 43, 139.

18
Chapter 1
(65) Hatano, M.; Ishihara, K. Synthesis 2008, 11, 1647. (66) Mukaiyama, T.; Soai, K.; Sato, T.; Shimizu, H.; Suzuki, K. J. Am. Chem. Soc.
1979, 101, 1455. (67) Weber, B.; Seebach, D. Angew. Chem. 1992, 104, 96. (68) von dem Bussche-Huennefeld, J. L.; Seebach, D. Tetrahedron 1992, 48, 5719. (69) Weber, B.; Seebach, D. Tetrahedron 1994, 50, 7473. (70) Ramón, D. J.; Yus, M. Angew. Chem. Int. Ed. 2004, 43, 284. (71) Dosa, P. I.; Fu, G. J. Am. Chem. Soc. 1998, 120, 445. (72) Yus, M.; Ramón, D. J.; Prieto, O. Tetrahedron: Asymmetry 2002, 13, 2291. (73) Gilman, H.; Straley, J. M. Recl. Trav. Chim. Pays-Bas 1936, 55, 821. (74) Ramón, D. J.; Yus, M. Tetrahedron Lett. 1998, 39, 1239. (75) García, C.; LaRochelle, L. K.; Walsh, P. J. J. Am. Chem. Soc. 2002, 124, 10970. (76) Yus, M.; Ramón, D. J.; Prieto, O. Tetrahedron: Asymmetry 2003, 14, 1103. (77) García, C.; Walsh, P. J. Org. Lett 2003, 5, 3641. (78) Walsh, P. J. Acc. Chem. Res. 2003, 36, 739. (79) Li, Q.; Gau, H.-M. Chirality 2011, 23, 929. (80) Li, H.; García, C.; Walsh, P. J. PNAS 2004, 101, 5425. (81) Hatano, M.; Miyamoto, T.; Ishihara, K. Org. Lett. 2007, 9, 4535. (82) DiMauro, E. F.; Kozlowski, M. C. J. Am. Chem. Soc. 2002, 124, 12668. (83) Yus, M.; González-Gómez, J. C.; Foubelo, F. Chem. Rev. 2011, 111, 7774. (84) Côté, A.; Charette, A. B. J. Am. Chem. Soc. 2008, 130, 2771. (85) Kneisel, F. F.; Dochnahl, M.; Knochel, P. Angew. Chem. Int. Ed. 2004, 43, 1017. (86) Chen, C.-A.; Wu, K.-H.; Gau, H.-M. Angew. Chem. 2007, 119, 5469. (87) Biradar, D. B.; Zhou, S.; Gau, H.-M. Org. Lett. 2009, 11, 3386. (88) Biradar, D. B.; Gau, H.-M. Org. Lett. 2009, 11, 499. (89) Hatano, M.; Suzuki, S.; Ishihara, K. J. Am. Chem. Soc. 2006, 128, 9998. (90) Jansen, J. F. G. A.; Feringa, B. L. J. Org. Chem 1990, 55, 4168. (91) Muramatsu, Y.; Harada, T. Angew. Chem. Int. Ed. 2008, 47, 1088. (92) Muramatsu, Y.; Kanehira, S.; Tanigawa, M.; Miyawaki, Y.; Harada, T. Bull. Chem.
Soc. Jpn. 2010, 83, 19.