tungsten alkyl alkylidyne and bis(alkylidene) complexes
TRANSCRIPT

University of Tennessee, Knoxville University of Tennessee, Knoxville
TRACE: Tennessee Research and Creative TRACE: Tennessee Research and Creative
Exchange Exchange
Doctoral Dissertations Graduate School
8-2005
Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. Their Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. Their
Unusual Inter-Conversion and Reactions with Phosphines, Unusual Inter-Conversion and Reactions with Phosphines,
Dioxygen and Water Dioxygen and Water
Laurel Anne Morton University of Tennessee - Knoxville
Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss
Part of the Chemistry Commons
Recommended Citation Recommended Citation Morton, Laurel Anne, "Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. Their Unusual Inter-Conversion and Reactions with Phosphines, Dioxygen and Water. " PhD diss., University of Tennessee, 2005. https://trace.tennessee.edu/utk_graddiss/2258
This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected].

To the Graduate Council:
I am submitting herewith a dissertation written by Laurel Anne Morton entitled "Tungsten Alkyl
Alkylidyne and Bis(Alkylidene) Complexes. Their Unusual Inter-Conversion and Reactions with
Phosphines, Dioxygen and Water." I have examined the final electronic copy of this dissertation
for form and content and recommend that it be accepted in partial fulfillment of the
requirements for the degree of Doctor of Philosophy, with a major in Chemistry.
Ziling (Ben) Xue, Major Professor
We have read this dissertation and recommend its acceptance:
Craig E. Barnes, Charles D. Feigerle, X. Peter Zhang, John R. Collier
Accepted for the Council:
Carolyn R. Hodges
Vice Provost and Dean of the Graduate School
(Original signatures are on file with official student records.)

To the Graduate Council: I am submitting herewith a dissertation written by Laurel Anne Morton entitled "Tungsten Alkyl Alkylidyne and Bis(Alkylidene) Complexes. Their Unusual Inter-Conversion and Reactions with Phosphines, Dioxygen and Water." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Chemistry.
Ziling (Ben) Xue Major Professor
We have read this dissertation and recommend its acceptance: Craig E. Barnes__________________ Charles D. Feigerle_______________ X. Peter Zhang___________________ John R. Collier___________________
Accepted for the Council:
Anne Mayhew_______________ Vice Chancellor and Dean of the Graduate School
(Original signatures are on file with official student records.)

TUNGSTEN ALKYL ALKYLIDYNE AND BIS(ALKYLIDENE)
COMPLEXES. THEIR UNUSUAL INTER-CONVERSION AND
REACTIONS WITH PHOSPHINES, DIOXYGEN AND WATER
A Dissertation
Presented for the
Doctor of Philosophy Degree
The University of Tennessee, Knoxville
Laurel Anne Morton
August 2005

ii
DEDICATION
This dissertation is dedicated to my loving husband, Sam, for his constant
patience and understanding, and to my immediate family: Mom, Dad, Kim,
Shannon, and Sara, for their unending love and support. I would not have made
it through the past few years of graduate studies without their support.

iii
ACKNOWLEDGEMENTS
There are numerous people whom I would like to thank that have, in some
way or another, had an impact on my life during my doctoral studies. First and
foremost, I would like to thank my research advisor, Professor Zi-Ling (Ben) Xue,
for his immense help and patience with me over the past few years. He was an
excellent mentor and advisor and has really gone above and beyond what I
expected from a research advisor. I do not think I could have finished without his
constant support and encouragement.
I would like to acknowledge my committee members: Dr. Craig Barnes,
Dr. Peter Zhang, Dr. Charles Feigerle, and Dr. John Collier, for providing much
needed guidance and reviews of this work.
The following individuals extended technical support for various aspects of
my research at the University of Tennessee, for which I am grateful: Jason Clark
and Dr. Hongjun Pan for their help with the 400 MHz NMR instrument, Dr. Chuck
Campana at Bruker for his help in solving the crystal structure of the
bis(alkylidene) complex 3b, Dr. Al Tuinman for his wonderful mass spectrometry
analyses, and finally Dr. John F. C. Turner whose guidance and expertise
effected me in so many ways. I will be ever grateful for his technical support and
friendship.
I extend thanks as well to the support staff in the chemistry department:
Marilyn Ownby, Art Pratt, Rachelle Allen, Kelly Preston, Bill Gurley, Johnny

iv
Jones, John Nelson, Carol Moulton, and Tray Allen. You have all made my life
much more enjoyable over the past several years.
Of course I would like to thank all of my co-workers and colleagues, both
past and present, for their friendships, for putting up with me, for many helpful
discussions about my research, and for making the lab a productive and pleasant
environment in which to work. Special thanks goes to Dr. Xianghua (Bruce) Yu
for all his help with my crystal structures and many friendly discussions, to Lynn
Rodman and Nathan Carrington, whose lunch banter will be missed, to Dr. Jaime
Blanton for all her friendly female conversations which were greatly missed after
she left, to He (Steve) Qiu and Dr. Shujian (Jim) Chen for the nice laboratory
companionship. To former group members, Drs. Tianniu (Rick) Chen and Ruitao
(Ray) Wang, you are missed, and I hope we meet again soon. To the newest
Xue-group recruits, Jay Chocklett, Michael Bleakley, and Kristie Carter, thank
you for the companionship and good luck to you.
I would like to thank other noteworthy colleagues in the chemistry
department that for their friendships: Kathleen Giesfeldt and Marco de Jesus, for
the much needed afternoon breaks and friendly encounters, and Megan Bragg,
who I could always count on when I needed extra NMR time.
I would like to extend warm thanks and gratitude to my undergraduate
research advisors and professors at Tennessee Tech for their wonderful tutelage
and encouragement to attend graduate school: Dr. Dale Ensor, Dr. Dan
Swartling, Dr. Jeff Boles, Dr. Ed Lisic, Dr. Tom Furtsch, Dr. Barbara Jackson, Dr.
Scott Northrup, Dr. David Crouse, Dr. Bob Glinski, and Ms. Kathy Rust. Without

v
my foundation and experiences at TTU I would not have been inspired to be a
professor or be where I am today.
Lastly, I am inexpressibly grateful to my family for their loving support and
willingness to listen to my endless diatribes. Thanks to my parents, Anne and
Mike Ray, for encouraging me to follow my dreams and supporting me through
the years. Thanks to my wonderful sisters, Kim, Shannon, and Sara, who have
been my best friends growing up and especially now as we are all “grown up”
together. I will miss our Saturdays together. I would especially like to thank my
husband, Sam, who constantly kept me on track and encouraged me to
persevere, even from afar. I would also like to thank my mother and father-in-
law, Mary and Sammy Morton, for their phone calls of encouragement; you have
become like a second set of parents to me.
This project was supported by the National Science Foundation and the
Camille Dreyfus Teacher-Scholar program. Their support is greatly appreciated.

vi
ABSTRACT
This dissertation focuses on the unique chemistry of d0 alkylidyne and
bis(alkylidene) complexes with β-silicon atoms. The goals of this dissertation
research were to prepare and characterize these new d0 transition metal
complexes, to study their reactivities including thermodynamics and kinetics of
the inter-conversion between the alkylidyne and bis(alkylidene) complexes, and
to explore the mechanism of the reactions of O2 or water with d0 alkylidyne
complexes.
A summary of the research in this dissertation is provided in Chapter 1.
Chapter 2 reports the study of an unusual exchange between new alkyl
alkylidyne and bis(alkylidene) complexes promoted by phosphine coordination.
A novel d0 tungsten alkylidyne complex (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) was
prepared from (Me3SiCH2)3W≡CSiMe3 (4a) and PMe3, and found to undergo a
rare exchange with its bis(alkylidene) tautomer
(Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b). The thermodynamics of this equilibrium
were investigated by variable-temperature 31P NMR to give the thermodynamic
parameters ΔHo and ΔSo of the exchange. Kinetic studies show that the α-
hydrogen exchange between 3a and 3b follows first-order reversible kinetics.
Activation parameters ΔH≠ and ΔS≠ for the forward (3a → 3b) and reverse
reactions (3b → 3a) are reported. Reaction of 4a with PMe2Ph to give alkyl
alkylidyne 5a and bis(alkylidene) 5b tautomeric mixture has also been studied,

vii
and the exchange here is compared with that involving 3a and 3b.
Preparation and characterization of two new alkyl alkylidene alkylidyne
complexes, and kinetic studies of their formation are presented in Chapter 3.
The 3a º 3b equilibrium mixture under heating in the presence of excess PMe3
was found to undergo α-hydrogen abstraction and convert to
(Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe3)2 (8a-b). This reaction follows
second-order kinetics – first order with respect to 3 and PMe3. In the presence of
excess PMe3, pseudo first-order kinetics was observed to give the activation
parameters ΔH≠ and ΔS≠ for the reaction. Preparation of
(Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe2Ph)2 (9a-b) is also reported and
compared with the formation of 8a-b.
Chapter 4 describes the synthesis of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3
(11) and its reaction with O2 to yield an unusual oxo-siloxy complex
O=W(OSiMe3)(CH2SiMe3)3 (12). Unexpected –SiMe3 migration from an
alkylidyne to an oxo ligand occurs in the reaction. In the absence of O=PMe3,
the reaction of (Me3SiCH2)3W≡CSiMe3 (4a) and O2 did not yield 12. 12 was
isolated as (Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3 (13), an adduct
with (Me3SiCH2)3W≡CSiMe3 (4a). Studies with 18O-labelled 18O2 have been
conducted to determine whether the oxygen atoms in 12 come from 18O2. High-
resolution mass spectrometry (HRMS) was used to analyze the products.
Chapter 5 reports the study of the reaction of (Me3SiCH2)3W≡CSiMe3 (4a)
with H2O and the unexpected formation of CH4 and 12 in this reaction. The

viii
reaction of 4a with D2O showed α-hydrogen scrambling during the reaction and
formation of methane isotopomers, which were studied by HRMS. Reactions of
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with H2O were also studied, and found
to yield the oxo complex 12 as well.

ix
TABLE OF CONTENTS
Chapter Page
1. Introduction………………………………………………………………………1
1.1 Foreword ………………………………………………………………...1
1.1.1 Fischer and Schrock Carbene and Carbyne Ligands………..1
1.1.2 Dioxygen………………………………………………………….3
1.1.3 Reactions of d0 Transition Metal Complexes with O2……..…7
1.2 Current Dissertation ……………………………………………………8
1.2.1 Chapter 2 ………………………………………………………..8
1.2.2 Chapter 3 ………………………………………………………..9
1.2.3 Chapter 4 ………………………………………………………10
1.2.4 Chapter 5 ………………………………………………………10
1.2.5 Chapter 6 ………………………………………………………11
2. New Tungsten Alkyl Alkylidyne and Bis(alkylidene) Complexes and Kinetic
and Thermodynamic Studies of Their Unusual Inter-Conversions.…………...…12
2.1 Introduction …………………………………………………………….12
2.2 Results and Discussion ………………………………………………16
2.2.1 Synthesis and Characterization of the Tungsten Alkyl
Alkylidyne Complex (Me3SiCH2)3W(≡CSiMe3)(PMe3)
3a)……………………………………………………………….16

x
2.2.2 Synthesis and Characterization of the Tungsten
Bis(alkylidene) Complex (Me3SiCH2)2W(=CHSiMe3)2(PMe3)
(3b)…………………………………………….………………..18
2.2.3 Synthesis and Characterization of (Me3SiCH2)3W(≡CSiMe3)-
(PMe2Ph) (5a) and (Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph)
(5b)………………………………………….…………………..29
2.2.4 Kinetic and Thermodynamic Studies of the 3a º 3b
Exchange….…………………………………………………....31
2.2.5 Thermodynamic and Kinetic Studies of the 5a º 5b
Exchange. A Comparison of PMe3 and PMe2Ph………..…38
2.2.6 Theoretical Studies of the 3a º 3b Exchange….………..…39
2.3 Concluding Remarks ………………………………………………….44
2.4 Experimental Section …………………………………………………45
2.4.1 General Procedures ………………………………………..…45
2.4.2 NMR Experiments………………………………………….….48
2.4.3 Preparation of (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a)………48
2.4.4 Preparation of (Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b)...…49
2.4.5 Preparation of (Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a) ..…50
2.4.6 Preparation of (Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph)
(5b)………………………………………………………………51
2.4.7 Kinetic Study of the Conversion of
(Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) to 3b…………………52

xi
2.4.8 Thermodynamic Study of the Equilibrium between 3a
and 3b ………………………………………………………….52
2.4.9 Kinetic and Thermodynamic Studies of the Conversion of
(Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a) to 5b at 303 K .…..53
2.4.10 Reaction of (ButCH2)3W≡CBut (1) with PMe3 at Room
Temperature in Benzene-d6…………………………………..53
2.4.11 Determination of X-ray Crystal Structure for 3b……………54
2.4.12 Computational Details…………………………………………55
3. Synthesis of Unusual Tungsten Alkyl Alkylidene Alkylidyne Complexes
and Kinetic Studies of Their Formation……………………………………..…….…56
3.1 Introduction.…………………………………………………………....56
3.2 Results and Discussion.………………………………………...…….60
3.2.1 Synthesis and Characterization of
(Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe3)2 (8a-b)…….…60
3.2.2 Synthesis and Characterization of
(Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe2Ph)2 (9a-b)..…..67
3.2.3 Kinetic Study of the Conversion of the 3a º 3b Mixture
to 8a-b………………..…………………………………...….…72
3.2.4 Kinetic Studies of the 5 → 9 Conversion and a Comparison
of Its Rate with the Rate of the Formation of 8....................73
3.2.5 Attempted Reactions of (Me3SiCH2)3W≡CSiMe3 (4a) with

xii
PCy3 and PPh3…………………………………………..…..…79
3.3 Concluding Remarks…………………………………………..………79
3.4 Experimental Section ………………………………………………....81
3.4.1 General Procedures ………………………………………..…81
3.4.2 Preparation of (Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe3)2
(8a-b)……………………………………………………………81
3.4.3 Preparation of
(Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe2Ph)2 (9a-b)……82
3.4.4 Kinetic Studies of the Formation of 8a-b and
9a-b.…………………………………………….………………84
3.4.5 Attempted Synthesis of the Adducts between PCy3 or PPh3
and 4a……………………………………………………..……85
4. Synthesis of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) and Studies of Its
Reaction with Dioxygen………………………………………………………….……86
4.1 Introduction …………………………………………………………….86
4.2 Results and Discussion ………………………………………………91
4.2.1 Synthesis and Characterization of
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11)……………..….…91
4.2.2 Reaction of 11 with O2. Synthesis and Characterization of
the Tungsten Oxo Siloxy Complex
O=W(OSiMe3)(CH2SiMe3)3 (12)…..…………………..…..…98

xiii
4.2.3 HRMS Study of CO2 Produced from the Reaction of 11
with O2 and Determination of Its Yield by 13CO2
Analysis…………………………………………………..……105
4.2.4 Mass Spectrometric Studies of the Reaction of 11 with
18O2………………………….…………..…………………..…105
4.2.5 Reaction of O2 with the Equilibrium Mixture 3a º 3b….....106
4.3 Concluding Remarks.……………………………………………..…110
4.4 Experimental Section …………………………………………..……110
4.4.1 General Procedures …………………………………………110
4.4.2 Preparation of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3
(11)……………………………………………….….…...……111
4.4.3 Preparation of O=W(OSiMe3)(CH2SiMe3)3 (12) and
(Me3SiCH2)3W(≡CSiMe3)←O=W(OSiMe3)(CH2SiMe3)3
(13)…………………..……………………………………...…111
4.4.4. Reaction of (Me3SiCH2)3W≡CSiMe3 (4a) with O2…………113
4.4.5 Reaction of O2 with the Equilibrium Mixture
(Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) º
(Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b)……..…………….113
4.4.6 Determination of X-ray Crystal Structures of 11 and
13……………………………...............................................113
4.4.7 Quantitative Determination of CO2 Yielded from the Reaction
of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with O2…..…114

xiv
4.4.8 Determination of Isotopomers of CO2 from the Reaction
of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with 18O2...…115
5. Reactions of Tungsten Alkylidyne Complexes with Water………………116
5.1 Introduction ……………………………………………………..……116
5.2 Results and Discussion ………………………………….…………117
5.2.1 Reaction of (Me3SiCH2)3W(≡CSiMe3) (4a) with H2O.
Preparation of O=W(OSiMe3)(CH2SiMe3)3 (12) and
Study of the CH4 Formation……………………..……….…117
5.2.2 Reaction of (Me3SiCH2)3W(≡CSiMe3) (4a) with D2O…..…120
5.2.3 Reaction of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with
H2O ..…………………………………………………….….…125
5.3 Concluding Remarks.……………………………………………..…128
5.4 Experimental Section ……………………………………………..…128
5.4.1 General Procedures …………………………………………128
5.4.2 Preparation of
(Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3 (13)
via the Reaction of 4a with H2O………………………….…129
5.4.3 Quantitative Determination of CH4 Yielded from the
Reaction of (Me3SiCH2)3W≡CSiMe3 (4a) with H2O……....129
5.4.4 Reaction of (Me3SiCH2)3W≡CSiMe3 (4a) with D2O.………130

xv
5.4.5 2H NMR Studies of the Reaction of (Me3SiCH2)3W≡CSiMe3
(4a) with D2O……………………………………….…………131
5.4.6. The Analysis of Volatile Products by Mass Spectrometry
(MS) and High-Resolution Mass Spectrometry
(HRMS)……………………………………………….……….131
5.4.7. Reaction of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with
H2O……………………………………………………………132
5.4.8. Decomposition of 12 by H2O…………………………….…132
6. Future Studies ………………………………………………………………133
References……. …………………………………………………………………….135
Appendix……………………………………………………………………………...146
VITA ……………………………………………………………………………..……181

xvi
LIST OF TABLES
Table Page
2.1 Crystal data and structure refinement for 3b…………………………… 25
2.2. Selected bond distances (D) and angles (°) for 3b…………………….. 27
2.3. Equilibrium (Keq) and rate constants (k1 and k-1) of the 3a º 3b
exchange…………………………………………………………………… 33
3.1. NMR resonances of 8a and 8b (ppm)…………………………………… 64
3.2. NMR resonances of 9a and 9b (ppm)…………………………………... 68
3.3. Rate constant (k2) of the formation of 8a-b……………………………… 75
3.4. Activation parameters in reactions through cyclometalation
transition state……………………………………………………………… 77
4.1. Crystal data and structure refinement for
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11)……………………………… 95
4.2. Selected bond distances (D) and angles (°) for 11……………………... 97
4.3. Crystal data and structure refinement for 13……………………………. 100
4.4. Selected bond distances (D) and angles (°) for 13……………………... 102
4.5. Bond distance comparison………………………………………………... 104
5.1. List of HRMS peaks and their intensities in the 15-20 Dalton
region ……………………………………………………………………….. 122
A1. Atomic coordinates (× 104) and equivalent isotropic
displacement parameters (Å2 × 103) for 3b.…………………………….. 147

xvii
A2. Bond distances (Å) for 3b..................................................................... 148
A3. Bond angles (°) in 3b……………….……………………………………... 150
A4. Anisotropic displacement parameters (Å2 × 103) for 3b……………….. 154
A5. Hydrogen coordinates (× 104) and isotropic displacement parameters
(Å2 × 103) for 3b…………………………………………………………….. 155
A6. Atomic coordinates (× 104) and equivalent isotropic
displacement parameters (Å2 × 103) for 11……………………………… 157
A7. Bond distances (Å) for 11…………………………………………………. 159
A8. Bond angles (°) for 11………….………………………………………….. 160
A9. Anisotropic displacement parameters (Å2 × 103) for 11……………….. 161
A10. Hydrogen coordinates (× 104) and isotropic displacement
parameters (Å2 × 103) for 11………………………………………………. 163
A11. Atomic coordinates (× 104) and equivalent isotropic
displacement parameters (Å2 × 103) for 13……………………………… 166
A12. Bond distances (Å) for 13...................................................................... 169
A13. Bond angles (°) for 13........................................................................... 171
A14. Anisotropic displacement parameters (Å2 × 103) in 13…………………. 174
A15. Hydrogen coordinates (× 104) and isotropic displacement
parameters (Å2 × 103) in 13……………………………………………….. 177

xviii
LIST OF FIGURES
Figure Page
1.1. A qualitative molecular orbital diagram of dioxygen …………………… 4
2.1. Upper: 13C{1H} NMR (62.896 MHz) spectrum of a mixture of 3a
and 3b at 23 °C. Lower: 1H-gated-decoupled 13C NMR
(62.896 MHz) spectrum of a mixture of 3a and 3b at 23°C………….... 17
2.2. Upper: 1H NMR (400.11 MHz) spectrum of
(Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b) at –20 °C. Lower:
Expansion of the –0.1-1.4 ppm region of the spectrum………………... 19
2.3. 13C{1H} NMR (100.63 MHz) spectrum of 3b at –40 °C.
Upper: −2-60 ppm region. Lower: 246-261 ppm region……………….. 20
2.4. Upper: 1H NMR (400.11 MHz) spectrum of
(Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) at –50 °C. Lower: 13C{1H} NMR
(100.63 MHz) of 3a at −50 °C…………………………………………...... 22
2.5. Newman projections of 3a and 3b showing diastereotopic protons….. 23
2.6. An ORTEP view of bis(alkylidene) 3b showing 30% probability
thermal ellipsoids…………………………………..…………………..…..24
2.7. Newman projections of 5a and 5b showing the diastereotopic
protons………………………………………………………………………. 30
2.8. A plot of ln Keq vs 1/T of the equilibrium 3a º 3b………………………. 34
2.9. Kinetic plots of the reversible reactions 3a º 3b.………………….…… 36

xix
2.10. Eyring plot for the reversible reactions 3a º 3b………………………… 37
2.11. Kinetic plot of the formation of 5b from 5a at 303 K……………………. 40
2.12. Spatial plots of three LUMOs for alkylidyne and bis(alkylidene)
tautomers with orbital energies (au).…………………………………….. 43
3.1. 1H NMR spectrum of a mixture of 8a and 8b (toluene-d8)…………….. 61
3.2. 13C NMR spectrum of a mixture of 8a and 8b (toluene-d8)…………..... 62
3.3. Spatial drawings of complexes 8a, 8b, 9a, and 9b showing
stereochemistry around the W=C bond………………………………….. 66
3.4. 31P NMR spectrum of 9a and 9b.……………..………………….………. 69
3.5. 1H NMR spectrum of 9a-b. (Top) Alkylidene W=CHSiMe3 region for 9a
and 9b. (Bottom) Alkyl and phosphine region…………………………. 70
3.6. The Newman projection of 9a-b and the 1H NMR spectrum showing the
two methyl resonances in PMe2Ph with virtual 31P-H coupling……….. 71
3.7. Kinetic plots of the conversion 3a-b → 8a-b……………………………. 74
3.8. Eyring plot of the conversion 3a-b → 8a-b……………………………… 76
3.9. Kinetic plot for the formation of 9a-b at 348.2 K.……………………….. 78
4.1. An ORTEP view of complex 11…………………………………………. 94
4.2. An ORTEP view of complex 13…………………………………………. 99
4.3. Mass spectra (38-51 Dalton region) of the isotopomers of carbon
dioxide: …………………………………………………………………….. 107
4.4. Mass spectra of dioxygen isotopomers (16O=16O; 16O=18O;
18O=18O).…………...............………………………………………………. 108
5.1. MS data of volatile products in the reaction of 4a with H2O…………… 119

xx
5.2. The HRMS mass spectrum for the methane isotopomers in the 15-20
Dalton region.………………………………………………………………. 121
5.3. 2H NMR spectrum of the α-H region in 12…………………………....…. 124

xxi
LIST OF SCHEMES
Scheme Page
2.1. Proposed intermediates in alkylidene/alkylidyne scrambling
processes…………………………………………………………………… 13
2.2. Calculated relative energies for model complexes 3a’, 3b’, 4a’,
and 4b’………………………………………………………………………. 42
3.1. Some reported d0 complexes containing metal-carbon single, double,
and triple bonds…………………………………………………………….. 58
3.2. Formation of complexes 8a-b and 9a-b from equilibrium mixtures of
3a-b and 5a-b………………………………………………………………. 59
4.1. Formation of metal alkoxide complexes from oxygen insertion
reactions……………………………………………………………………. 87
4.2. Oxygen reactions with d0 complexes……………………………………. 89
4.3. Preparation of complexes 12 and 13…………………………………….. 90
4.4. Two pathways leading to 13………………………………………………. 109
5.1. The different routes to form 12 through reactions of H2O……………… 118
5.2. Reaction of D2O with 4a and the formation of methane isotopomers… 123
5.3. Formation of 12 from the reaction of 11 with H2O……………………… 127

xxii
NUMBERING SCHEME FOR COMPOUNDS IN THE TEXT
1-13C (ButCH2)3W≡13CBut
1 (ButCH2)3W≡CtBu
2a (ButCH2)2W(≡CBut)(SiButPh2)
2b (ButCH2)W(=CHBut)2(SiButPh2)
3a (Me3SiCH2)3W(≡CSiMe3)(PMe3)
3a’ (CH3)3W(≡CH)(PMe3)
3b (Me3SiCH2)2W(=CHSiMe3)2(PMe3)
3b’ (CH3)2W(=CH2)2(PMe3)
4a (Me3SiCH2)3W≡CSiMe3
4a’ (CH3)3W≡CH
4b (Me3SiCH2)2W(=CHSiMe3)2
4b’ (CH3)2W(=CH2)2
5a (Me3SiCH2)3W(≡CSiMe3)(PMe2Ph)
5b (Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph)
6 (ButCH2)W(≡CtBu)(=CHtBu)(PMe3)2
7 (ButCH2)W(≡CtBu)(=CHtBu)(dmpe)
8a syn-(Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe3)2
8b anti-(Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe3)2
9a syn-(Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe2Ph)2

xxiii
9b anti-(Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe2Ph)2
10 (ButCH2)2W(=O)([=CR(SiPhBut)]
11 (Me3SiCH2)3(Me3SiC≡)W←O=PMe3
12 O=W(OSiMe3)(CH2SiMe3)3
13 (Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3

1
CHAPTER 1
Introduction
1.1. Foreword
A current research focus of synthetic inorganic chemistry is the controlled
activation of dioxygen such as single oxygen atom transfer to substrates that
mimics biological systems where such reactions are catalyzed enzymatically.
Among the types of multiply bonded ligands, oxo complexes were the first
discovered and have been most extensively studied. Much work has been done
on the chemistry and biochemistry of dioxygen activation and reactivity with dn
transition metal complexes largely to understand oxygenase enzymes and to
develop model systems for their functions in biological systems.1 Oxidation of
the metals is often involved in these reactions. The chemistry of dioxygen with d0
early-transition-metal complexes including those with metal-carbon triple bonds
remains a largely unexplored area.
1.1.1. Fischer and Schrock Carbene and Carbyne Ligands
The history of the study of metal-carbon multiple bonds and how they are
formed is rich. Fischer and coworkers discovered the first carbene complexes in
1964 and the first carbyne complexes in 1973 (Eqs. 1 and 2).2 Fischer-type
carbene and carbyne ligands are often observed in low-oxidation-state metal
complexes. Typically, they are found in the chemistry of late transition metals

2
Eq. 1.1
Eq. 1.2
bearing π-acceptor ligands, and the carbene ligands often contain π-donor
substituents such as –OCH3. Eqs. 1.1 and 1.2 exemplify these characteristics.
In contrast to the Fischer carbenes, in 1973, an attempt to form
pentaneopentyltantalum resulted in the synthesis of the first carbene ligand in a
high-oxidation-state metal complex, now known as the Schrock-type alkylidene
(Eq. 1.3).3 In 1978, the first Schrock alkylidyne was reported (Eq. 1.4).3
Eq. 1.3
Eq. 1.4
Schrock-type alkylidene and alkylidyne ligands are typically found in high-
oxidation-state early transition metal complexes containing non-π-acceptor
ligands and non-π-donor R groups.4 With the discovery of two new classes of
compounds in such a short time-span, the field of organometallic chemistry
(CO)5M COCH3
RM
CO
CX
CO
CO
OC
R+ BX3
M = Cr, Mo, WX = Cl, Br, IR = Me, Et, Ph
W(CO)6
(1) LiR/Et2O
(2) [(CH3)3O]BF4W
CO
COCH3
ROC
CO
OCCO
(ButCH2)3TaCl2+ 2 ButCH2Li
(ButCH2)3Ta=CHtBu
WCl66 ButCH2Li
(ButCH2)3W CtBu (1)

3
underwent a rapid expansion. Fischer and Schrock complexes proved to be
useful in many different areas of catalysis, and in doing so widened fundamental
understanding of these new classes of compounds. Investigations into the
chemistry of compounds that contain metal-carbon multiple bonds have
continued steadily since.5,2b
1.1.2. Dioxygen
Dioxygen has a triplet ground state with two unpaired electrons. Both the
unpaired electrons have parallel spins and the half-filled antibonding molecular
orbitals of 3O2 can accommodate two additional electrons as shown in Figure
1.1.6 The direct reaction of 3O2 with singlet compounds to give singlet products is
a spin-forbidden process with a very low rate of reaction. One way to circumvent
this energy barrier is via a free radical pathway.7 The reaction of a singlet
compound with 3O2 forming two doublets (free radicals) is a spin-allowed
process. The process is highly endothermic and is observed typically with
reactive substrates that form resonance stabilized radicals.
Another method to overcome the high spin conservation barrier is by
combining 3O2 with a paramagnetic transition metal ion, as shown in Eq. 1.5.
Eq. 1.5
Mn + 3O2Mn+1
OO

4
1s 1s
2s 2s
2p 2p
σ
σ∗
σ∗
σ∗
σ
σ
π
π∗ π∗
π
Energy
O OO=O
Figure 1.1. A qualitative molecular orbital diagram of dioxygen.

5
The resulting metal dioxygen complex may react selectively with singlet
compounds under moderate conditions. This concept forms the basis for the
extensive studies of oxygen activation by metal complexes. Since in most
oxidation reactions there is no resonance stabilizer, this mechanism is the most
accepted. It has been found that nearly all transition metals will bind dioxygen,
resulting in a wide variety of complex formations.
The interaction of metal complexes with dioxygen has been the subject of
numerous studies for its importance in many oxidation reactions, especially those
involved in biological respiration processes.7e A large interdisciplinary effort has
been mounted to understand oxygenase enzymes of all kinds by isolating and
studying the pure enzymes, and by developing model systems.7e The first
reported dioxygen complex was Werner’s7e CoIII complex [{(NH3)5-Co}2O2]4+ in
1898. A renewed interest in dioxygen chemistry began in 1936 when Pauling
and Coryell discovered that “the oxygen molecule undergoes a profound change
in electronic structure on combination with hemoglobin”.7f It has since been
found that nature utilizes metal-oxo complexes in numerous important enzymes.
An unprecedented amount of research has focused on the cytochrome P-450
family of enzymes, which contain an oxo-iron porphyrin system and have been
found to be involved in a wide range of biological oxidation processes. A second
class of oxo-metal based enzymes are the molybdenum- or tungsten-containing
“oxo-transferases”, which like P-450, are involved in both oxidative and reductive
processes. Significant advances have been made in the development of model
systems that mimic the enzymatic transformations in the body.5b

6
The mechanism of reactions of organometallic complexes is of
fundamental importance to understanding the role of transition metals in catalytic
oxidation processes. Little is known, however, about how dioxygen reacts with
metal-carbon multiple bonds. The study of transition metal organometallics and
their reactions with dioxygen began when Frankland reported the autoxidation of
dimethyl zinc in 1853. Forty years later, Demuth and Meyer clearly defined the
reaction and its product, zinc peroxide.8 Studies of reactions between O2 and
early transition metal organometallics began in the early 1970’s. Zucchini and
coworkers investigated the reaction of O2 with Zr(CH2Ph)4 which was found to
react two equiv of O2 per mole.9a Brindley and Hodgson studied the reaction of
O2 with titanium, zirconium, dimolybdenum, and tungsten alkyl complexes.9b
Their measurement of the total absorption of oxygen per alkyl ligand suggested
metal alkoxides were the stable end products. Their findings and those that
followed supported oxygen insertion into the metal-alkyl and other metal-ligand
bonds.
In general, it is now known that oxygen insertion to the alkyl-metal bond
gives mainly alkoxides and some peroxides. When the central metal reacts with
dioxygen, an oxo complex is often obtained. It is now accepted that most of the
oxygenation reactions occur by a radical mechanism to overcome the spin-
forbidden energy barrier.1

7
1.1.3. Reactions of d0 Transition Metal Complexes with O2
Reactions of d0 transition metal complexes with O2 have been studied in
the synthesis of metal oxide materials used in the microelectronics industry.10−12
Oxygen insertion into Zr-Si and Zr-R bonds in the reactions of Cp2ZrCl(SiMe3),
Cp2ZrRCl, and Cp2ZrR2 with O2 has been reported by Tilley,10g Schwartz,10a
Gibson,10b Brindley10d and coworkers. The Cp-free complexes (RO)2TiMe2,
(ArO)2TaMe3, (ArN=)2MoMe2, and LY(μ-Me)2AlMe2 (L = porphyrin) have been
shown to undergo O insertion in their reactions with O2, as reported by
Wolczanski,11a,b Rothwell,11c Gibson,11d Schaverien11m and coworkers,
respectively. In recent years, reactions of O2 with d0 complexes such as
Ta(NR2)5 and (Et2N)3Ta=NBut have been used in the preparation of
microelectronic metal oxide thin films as new gate materials.12 The nature of
these CVD reactions is largely unknown, and mechanistic pathways to the
microelectronic MOn materials are not understood.
Reactions of d0 transition metal complexes with dioxygen have attracted
much attention and have been used to make metal oxides, known as high
dielectric constant (κ) gate materials, and used as microelectronic gate materials
in VLSI devices. High-κ MOn oxide thin films have been explored to replace
SiO2, a relatively low-κ solid (κ = 3.9), as the next generation of gate materials.
These materials have also been utilized as new capacitor materials for DRAM
(dynamic random access memory) devices.13

8
Our research has been mostly focused on the fundamental chemistry of
d0 transition metal alkylidyne complexes and their reactions with O2. Reactions
of H2O with transition metal complexes, an alternative route to MOn materials,
have also been investigated to reveal unique chemistry and provides a
comparison to the reactions involving O2.
1.2. Current Dissertation
The preparation and characterization of novel d0 tungsten alkyl alkylidyne
and bis(alkylidene) complexes, mechanistic pathways in the formation of these
compounds, and studies of their reactions towards O2 and H2O and related
chemistry of these complexes are the foci of this Ph.D. dissertation.
1.2.1. Chapter 2
Preparation, characterization, and X-ray crystal structure of novel alkyl
alkylidyne (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a), bis(alkylidene)
(Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b), and studies of the interconversion
between alkylidyne 3a and bis(alkylidene) 3b complexes are reported. An
adduct between PMe3 and alkyl alkylidyne (Me3SiCH2)3W≡CSiMe3 (4a),
(Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a), was found to undergo a rare reversible
transformation to its bis(alkylidene) tautomer (Me3SiCH2)2W(=CHSiMe3)2(PMe3)
(3b). The bis(alkylidene) tautomer 3b is favored in the 3a º 3b
[(Me3SiCH2)3W(≡CSiMe3)(PMe3) º (Me3SiCH2)2W(=CHSiMe3)2(PMe3)]

9
equilibrium with Keq ranging from 12.3(0.2) at 278(1) K to 9.37(0.12) at 303(1) K,
giving the thermodynamic parameters for the equilibrium: ΔHo = –1.8(0.5)
kcal/mol and ΔSo = –1.5(1.7) eu. The α-hydrogen exchange between 3a and 3b
follows first-order reversible kinetics. The activation parameters are ΔH≠ =
16.2(1.2) kcal/mol and ΔS≠ = –22(4) eu for the forward reaction (3a → 3b), and
ΔH≠ = 18.0(1.3) kcal/mol and ΔS≠ = –21(4) eu for the reverse reaction (3b → 3a).
The role of PMe3 in the exchange 3a º 3b has been studied by density functional
theory calculations through the collaboration with researchers at the Hong Kong
University of Science and Technology. A paper about the chemistry in this
chapter has been published in J. Am. Chem. Soc. 2004, 126, 10208-10209.14
1.2.2. Chapter 3
Heating the 3a º 3b equilibrium mixture in the presence of excess PMe3
caused an α-hydrogen abstraction and conversion to
(Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe3)2 (8a-b) (Eq. 1.6).
(Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) º (Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b)
→ (Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe3)2 (8a-b) Eq. 1.6
This reaction was found to follow second-order kinetics – first order with respect
to 3 and PMe3, respectively. In the presence of excess PMe3, pseudo first-order
kinetics was observed and activation parameters are ΔH≠ = 27.4(1.5) kcal/mol

10
and ΔS≠ = –1(4) eu. Preparation of
(Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe2Ph)2 (9a-b) is also reported and
compared with the formation of 8a-b.
1.2.3. Chapter 4
Synthesis of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11), an adduct between
(Me3SiCH2)3W≡CSiMe3 (4a) and O=PMe3, is reported in Chapter 4. 11 was
found to react with O2 to give CO2 and O=W(OSiMe3)(CH2SiMe3)3 (12) which
was isolated as an adduct with 4a,
(Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3 (13). In this reaction, the
–SiMe3 group in the ≡CSiMe3 ligand undergoes an unprecedented migration to
form the –OSiMe3 ligand in 12, and the ≡C– atom is oxidized to CO2. O=PMe3 is
believed to serve as a mediator for the 1,3-silyl migration, and the mechanism
may be radical in nature. Studies with 18O-labelled 18O2 have been conducted to
determine whether the oxygen atoms in 12 come from 18O2. High-resolution
mass spectrometry (HRMS) was used to analyze the products.
1.2.4. Chapter 5
Reaction of (Me3SiCH2)3W≡CSiMe3 (4a) with H2O was studied, and found
to yield the oxo complexes 12 as well and CH4. In this reaction, a silyl migration
in the ≡CSiMe3 ligand was also observed, and the ≡C− atom is converted to CH4.
These studies show that two different alkylidyne complexes, 4a and 11, react

11
with O2 and H2O, respectively, to yield the same unusual oxo complex 12. The
reaction of 4a with D2O showed α-hydrogen scrambling during the reaction and
formation of methane isotopomers, which were studied by HRMS. Reactions of
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with H2O were also studied, and found
to yield the oxo complex 12 as well.
1.2.5. Chapter 6
Based on the work reported in the current dissertation, some future
studies are suggested in this chapter.

12
CHAPTER 2
New Tungsten Alkyl Alkylidyne and Bis(alkylidene) Complexes
and Kinetic and Thermodynamic Studies of Their Unusual Inter-
Conversions
2.1. Introduction
The reactivity of α-hydrogen atoms in alkyl ligands free of β-hydrogen
atoms (e.g., Me3CCH2 and Me3SiCH2) has been studied due to their role in the
formation of high-oxidation-state alkylidene and alkylidyne complexes.3a,5b,5d,15−17
d0-Alkyl alkylidyne (RCH2)3W≡CR’ and alkylidene (ButCH2)3Ta=CDBut complexes
are known to undergo α-hydrogen migrations among the α-carbon atoms.3c,18 In
previous work, bis(alkylidene) complexes are believed to be intermediates in
alkyl-alkylidyne scrambling through α-hydrogen transfer in alkylidyne complexes
(ButCH2)3W≡13CCMe3 (1-13C) and (ButCH2)3W≡CSiMe3.18 Such α-hydrogen
exchanges in, for example, (ButCH2)3W≡13CCMe3 (1-13C) at 60 °C, lead to
scrambling of the 13C labeled atom between the alkyl and alkylidene ligands and
the formation of (ButCH2)2(But-13CH2)W≡CBut (Scheme 2.1a). A statistical
distribution of the 13C-labeled α-carbon atoms in (But*CH2)3W≡*CBut (*C: 25%
13C) is reached in 24 hours.18c In other words, the 13C label is present in the α-
carbon atoms in the 13CH2 : ≡13C groups in a 3 : 1 ratio. Another case of such
alkyl-alkylidene scrambling has been reported in d0 (ButCH2)3W≡CSiMe3,

13
WCSiMe3
ButCH2
ButCH2
CH2But
WButCH2
ButCH2
CHBut
CHSiMe3
WCH2SiMe3
ButCH2
ButCH2
CBut
OsCHButButCD2
ButCD2
CHBut
OsButCD2
ButCD2
C
CH2But
But
OsCH2ButButCD2
ButCD
CDBut
W13CButButCH2
ButCH2
CH2But
WButCH2
ButCH2
CHBut
13CHButW
13CH2ButButCH2
ButCH2
CBut
(a)
(b)
(c)
1-13C
Scheme 2.1. Proposed intermediates in alkylidene/alkylidyne scrambling
processes.18,19

14
leading to the formation of (ButCH2)2(Me3SiCH2)W≡CBut (Scheme 2.1b).
Deuterium labeling studies using (ButCHD)3W≡CSiMe3 and (ButCD2)3W≡CSiMe3
as well as kinetic studies of the α-hydrogen exchanges are consistent with
unimolecular and stepwise transfer of two H atoms in one alkyl ligand to the
alkylidyne ligand in (ButCH2)3W≡CSiMe3.18a A bis(alkylidene)
"(ButCH2)2W(=CHSiMe3)(=CHBut)" was proposed as an intermediate in the
transfer (Scheme 1b), although it was not observed.18a It is reasonable to
assume that there is a similar bis(alkylidene) intermediate in the alkyl-alkylidyne
scrambling in (ButCH2)3W≡13CBut (1-13C, Scheme 2.1a).18c In the d2 osmium
bis(alkylidene) complex Os(=CHBut)2(CD2But)2, H/D atoms were found to
scramble among the α-carbon atoms (Scheme 2.1c).19c This exchange is
believed to occur through the alkyl alkylidyne intermediate "(ButCH2)3Os≡CBut."
Although the exchange of α-hydrogen atoms is a fundamental dynamic
process in these archetypical d0 alkylidene and alkylidyne complexes, there is, to
our knowledge, only one direct observation of such an exchange between
bis(alkylidene) and alkylidyne tautomers.3b,5c,5d,15a,20,21 Alkylidyne complex
(Me3CCH2)2W(≡CCMe3)(SiButPh2) (2a), a silyl analog of 1, was found to be in an
equilibrium with its silyl bis(alkylidene) tautomer
(Me3CCH2)W(=CHCMe3)2(SiButPh2) (2b) (Eq. 2.1).22

15
(Eq. 2.1)
(ButCH2)W(=CHBut)2(SiButPh2) (2b) is one of the rare known d0
bis(neopentylidene) complexes; the only other known examples involve tantalum
and niobium d0 bis(neopentylidene) complexes.16e,19c,23 Theoretical studies
reveal that the silyl ligand plays a critical role in the relative stabilities of the d0
tungsten bis(alkylidene) tautomer (2b) through the d-p π interaction between the
silyl ligand and the electron density in the metal-alkylidyne/alkylidene bonds.22b
We recently observed that (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a), an
adduct between PMe3 and (Me3SiCH2)3W≡CSiMe3 (4a), undergoes an exchange
with its bis(alkylidene) tautomer (Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b, Eq.
2.2).14
(Eq. 2.2)
In the absence of the phosphine, the bis(alkylidene) tautomer
“(Me3SiCH2)2W(=CHSiMe3)2 (4b)” was not observed. Unlike the exchange
involving silyl alkylidyne and bis(alkylidene) complexes 2a and 2b, this is an
WCButButCH2
Ph2ButSi
CH2But
WButCH2
Ph2ButSi
CHBut
CHBut
Keq
2a 2b
W
R
RR
R
PMe3 PMe3
W
R
R RH
HR
k1
k-1
Keq
R = SiMe3, 3a 3b

16
unusual phosphine-induced exchange. Assuming (Me3SiCH2)3W≡CSiMe3 (4a)
undergoes an alkyl-alkylidyne scrambling involving “(Me3SiCH2)2W(=CHSiMe3)2
(4b)” as an intermediate similar to that in Scheme 2.1b, the current work
suggests that PMe3 coordination making (Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b)
significantly stabilizes 4a to make its phosphine adduct 3b observable at room
temperature. The synthesis of analogues (Me3SiCH2)3W(≡CSiMe3)(PMe2Ph)
(5a) and (Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph) (5b) was also studied and the
tautomeric mixture was found to undergo a similar equilibrium. Our preparation
and characterization of 3a, 3b, 5a, and 5b as well as thermodynamic and kinetic
studies of the 3a º 3b exchange in Eq. 2.2 are reported here.
2.2. Results and Discussion
2.2.1. Synthesis and Characterization of the Tungsten Alkyl Alkylidyne
Complex (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a)
Addition of PMe3 to a solution of (Me3SiCH2)3W≡CSiMe3 (4a) in toluene-d8
leads to an immediate color change from yellow to red, and the formation of the
PMe3 adduct (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a). NMR spectroscopic
characterization [1H, 13C, 31P, 29Si, 1H-gated-decoupled 13C (Figure 2.1) and
HMQC] of 3a at –50 °C suggests that the PMe3 ligand coordinates cis to the
alkylidyne ligand. Two alkyl resonances were observed in the 1H (in a 1 : 2 ratio),
13C and 29Si NMR spectra of 3a at –50 °C, as expected from the structure of 3a

17
Figure 2.1. Upper: 13C NMR (62.896 MHz) spectrum of a mixture of 3a and 3b
at 23 °C. Lower: 1H-gated-decoupled 13C NMR (62.896 MHz) spectrum of a
mixture of 3a and 3b at 23 °C.

18
in Eq. 2.2. The coupling constant 2JP-C-axial of 36.5 Hz for the axial –CH2R ligand
is, as expected, larger than 2JP-C-equatorial of 7.2 Hz for the equatorial –CH2R
ligand. The resonance of the alkylidyne C atom in the PMe3 adduct
(Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) at 358.81 ppm appeared as a doublet (2JP-C
= 14.5 Hz) in both 13C and 1H-gated-decoupled 13C NMR spectra, and is down-
field shifted from that of (Me3SiCH2)3W≡CSiMe3 (4a) at 343.27 ppm.
2.2.2. Synthesis and Characterization of the Tungsten Bis(alkylidene)
Complex (Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b)
Upon warming the solution of 3a in toluene-d8 to room temperature, 3a
was found to undergo alkyl-to-alkylidyne α-hydrogen migration to give
bis(alkylidene) (Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b), which was characterized
by 1H (Figure 2.2), 13C (Figure 2.3), 31P, 29Si, 1H-gated-decoupled 13C (Figure
2.1) and HMQC NMR spectroscopy. The two tautomers are found to be in
equilibrium (3a º 3b). The two alkylidene ligands are inequivalent; the alkylidene
C resonances in 3b are observed as doublet of doublets at 256.43 (1JC-H = 123.5
Hz) and 254.71 ppm (1JC-H = 102.6 Hz) in the 1H-gated-decoupled 13C NMR
spectrum at –50 °C (Figure 2.1). The coupling constants 2JP-C-axial and 2JP-C-
equatorial of 32.3 and 0 Hz for the axial and equatorial –CH2R ligands, respectively,
and 2JP-C of 11.8 and 12.6 Hz for the two alkylidene ligands suggest that the two
alkylidene and one alkyl ligands coordinate cis to the PMe3 ligand. The 1H NMR
resonances of the alkylidene H atoms in 3b were observed as

19
Figure 2.2. Upper: 1H NMR (400.11 MHz) spectrum of
(Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b) at –20 °C. Lower: Expansion of the –0.1-
1.4 ppm region of the spectrum.

20
Figure 2.3. 13C NMR (100.63 MHz) spectrum of 3b at –40 °C. Upper: −2-60
ppm region. Lower: 246-261 ppm region.

21
doublets at 7.985 (3JP-H = 5.6 Hz) and 7.1982 (3JP-H = 4.0 Hz) ppm, respectively.
The presence of two inequivalent alkylidene ligands suggest that 3b adopts an
anti, syn configuration (Eq. 2.2), and it is unlikely that the two ligands are
involved in a fast rotation about the W=C bonds. The prochiral W atom in 3a
gives rise to diastereotopic α-hydrogen atoms (CHaHb-SiMe3) for the equatorial
alkyl ligands observed as doublet of doublets at 0.751 ppm (both 2JHa−Hb and
3JP−H = 14.4 Hz) and 0.213 ppm (3JP-H = 28.2 Hz) in its 1H NMR spectrum at –50
°C (Figure 2.4). The presence of the prochiral W atom in 3b similarly leads to
diastereotopic α-hydrogen atoms (CHeHf-SiMe3, Figure 2.5) for the equatorial
alkyl ligand observed as doublet of doublets at 0.917 (3JP-H = 17.7 Hz, 2JHa-Hb =
11.3 Hz) and 0.876 (3JP-H = 32.2 Hz) ppm in the 1H NMR spectrum at –50 °C.
The chemical shift differences between the diastereotopic Ha and Hb atoms in 3a
(0.538 ppm) is larger than that (0.041 ppm) in 3b. The chemical shift difference
in alkylidyne 3a is, however, smaller than 4.56 ppm in pseudo-tetrahedral silyl
alkylidyne complex (Me3CCH2)2W(≡CSiMe3)[Si(SiMe3)3].20j
The equilibrium mixture of 3a and 3b is stable in solution for several days
and cooling the solution to –30 °C yielded crystals of 3b that were suitable for X-
ray diffraction studies. A representation of the molecular structure,
crystallographic refinement data, and selected bond distances and angles of 3b
are given in Figure 2.6, and Tables 2.1 and 2.2, respectively. Low-temperature
1H NMR spectra of the crystals showed that the crystals are those of 3b,
indicating that 3b preferentially crystallized from the mixture. Elemental analysis

22
ppm (t1)
0.000.501.00
Figure 2.4. Upper: 1H NMR (400.11 MHz) spectrum of 3a at –50 °C.
Lower: 13C NMR (100.63 MHz) of 3a at −50 °C.
ppm (t1) 050100150200250300350
358.0359.081.082.0
49.0049.50

23
HH
R(a)
CR
CH2R(e)(e)RCH2
HaHb
R(e) CH2R(a)(e)RCH2
PMe3RC
HcHd
R(a)
CH2R(e)
CR
HC
R
H
HeHf
R(e)C
H
R
PMe3(a)RCH2
CR
H
W
(a)R
R(e)R
(e)R
PMe3
W
(a)R
(e)R C H
R
C R
H
PMe3
down the axial alkyl ligand down the equatorial alkyl ligand
down the axial alkyl ligand down the equatorial alkyl ligand
R = SiMe3; (a) = axial; (e) = equatorial
3a
3b
Figure 2.5. Newman projections of 3a and 3b showing diastereotopic protons.

24
Figure 2.6. An ORTEP view of bis(alkylidene) 3b showing 30% probability
thermal ellipsoids. This is a disordered structure. See page 28 for details.

25
Table 2.1. Crystal data and structure refinement for 3b
Compound No. 3b
Empirical formula (formula weight) C21H53O1P1Si5W1 (676.90)
Temperature –100(2) °C
Wavelength 0.71073 Å
Crystal system Hexagonal
Space group P6(3)
Unit Cell Dimensions a = 12.1310(5) Å α = 90°
b = 12.1310(5) Å β = 90°
c = 12.1091(5) Å γ = 120°
Volume 1543.25(11) Å3
Z 2
Density (calculated) 1.457 g/cm3
Absorption coefficient 4.000 mm−1
F(000) 692
Crystal size 0.35 × 0.25 × 0.25 mm3
θ range for data collection 1.68 to 27.42°
Index ranges -15 ≤ h ≤ 15, -15 ≤ k ≤ 15, -15 ≤ l ≤ 15
Reflections collected 15949

26
Table 2.1. Continued
Compound No. 3b
Independent reflections 2352 [R(int) = 0.0371
Completeness to θ = 27.42° 100.0%
Absorption correction Semi-empirical from equivalents
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 2352 / 112 / 86
Goodness-of-fit on F2 1.207
Final R indices [I > 2σ(I)] R1 = 0.0624, wR2 = 0.1235
R indices (all data) R1 = 0.0655, wR2 = 0.1255
Absolute structure parameter 0.44(11)
Largest diff. peak and hole 1.881 and −2.401 e.Å−3
a R1 = Σ||Fo| – | Fc|| / Σ|Fo|; wR2 = [Σ w(Fo2 – Fc
2)2 / Σ w(Fo2)2]1/2;
w = 1/[σ2(Fo) + (aP)2 + bP]; P = [2Fc2 + Max(Fo
2, 0)]/3
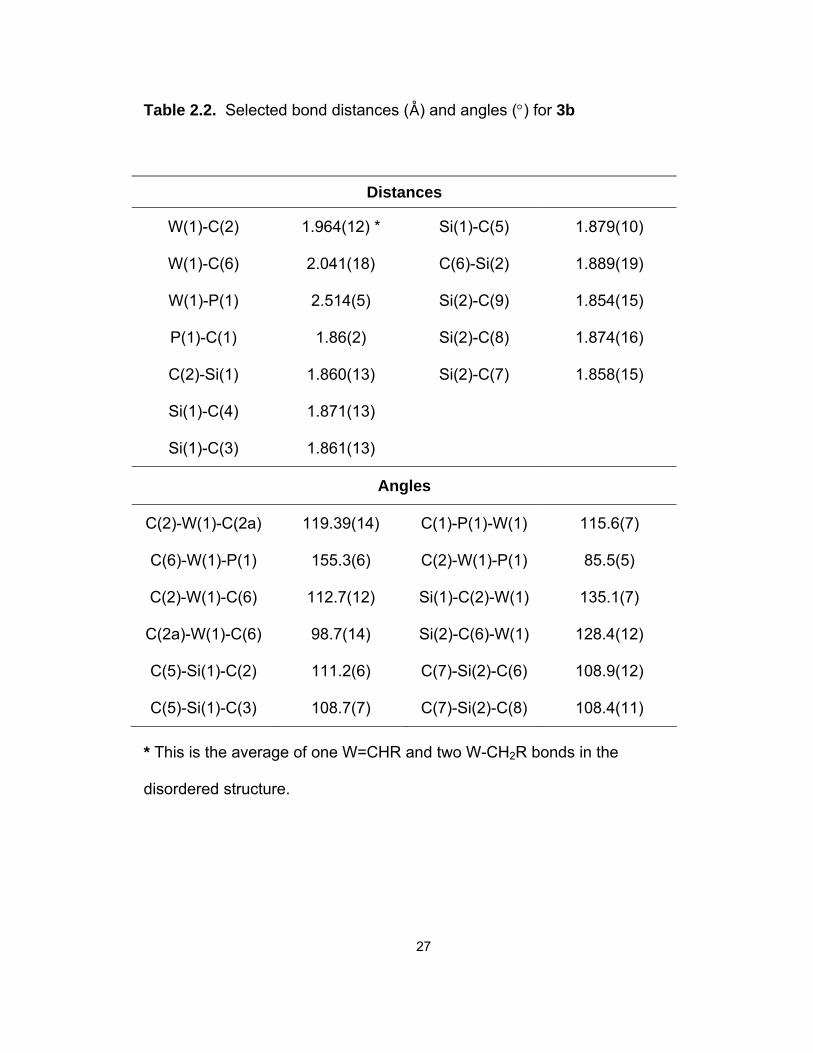
27
Table 2.2. Selected bond distances (D) and angles (°) for 3b
Distances
W(1)-C(2) 1.964(12) * Si(1)-C(5) 1.879(10)
W(1)-C(6) 2.041(18) C(6)-Si(2) 1.889(19)
W(1)-P(1) 2.514(5) Si(2)-C(9) 1.854(15)
P(1)-C(1) 1.86(2) Si(2)-C(8) 1.874(16)
C(2)-Si(1) 1.860(13) Si(2)-C(7) 1.858(15)
Si(1)-C(4) 1.871(13)
Si(1)-C(3) 1.861(13)
Angles
C(2)-W(1)-C(2a) 119.39(14) C(1)-P(1)-W(1) 115.6(7)
C(6)-W(1)-P(1) 155.3(6) C(2)-W(1)-P(1) 85.5(5)
C(2)-W(1)-C(6) 112.7(12) Si(1)-C(2)-W(1) 135.1(7)
C(2a)-W(1)-C(6) 98.7(14) Si(2)-C(6)-W(1) 128.4(12)
C(5)-Si(1)-C(2) 111.2(6) C(7)-Si(2)-C(6) 108.9(12)
C(5)-Si(1)-C(3) 108.7(7) C(7)-Si(2)-C(8) 108.4(11)
* This is the average of one W=CHR and two W-CH2R bonds in the
disordered structure.

28
of the crystals is consistent with the composition of 3b. The X-ray crystal
structure of 3b was found to be severely disordered with a crystallographically
imposed 3-fold axis through the Si(2), W(1) and P(1) atoms. The structure was
solved with the help from Dr. Charles F. Campana of Bruker AXS, Inc. The C(6)
atom was located in three equivalent positions: C(6), C(6a) and C(6b), each with
a partial occupancy of 1/3. Only C(6) is shown in the ORTEP view of 3b (Figure
2.6). 3b adopts a pseudo trigonal bipyramidal structure with C(6) and P(1) in the
axial positions and the C(6)-W(1)-P(1) angle of 155.3(6)°. The axial W(1)-C(6)
bond distance of 2.041(18) D is similar to 2.096(5) D in (ButCH2)3W≡CSiMe3,18a
but shorter than 2.112(9) D and 2.118(9) D in
(ButCH2)2W(=O)[=C(SiMe3)SiButPh2]22a,c and 2.258(8) D in the alkyl alkylidene
alkylidyne complex W(CH2But)(=CHBut)(≡CBut)[Me2P(CH2)2PMe2].24 The 3-fold
disorder leads to an average of the three equatorial W-C bond distance [W(1)-
C(2)] of 1.964(12) D. This average is smaller than the W(1)-C(6) bond distance
of 2.041(18) D, suggesting that the two alkylidene W=C bonds are in the
equatorial positions. This observation is consistent with the NMR data of 3b
discussed earlier in this section. The equatorial C(2)-Si(1) bond distance of
1.860(13) D is slightly shorter than the axial C(6)-Si(2) distance of 1.889(19) D.
The W(1)-P(1) bond distance of 2.514(5) D is similar to those [2.450(3) and
2.577(3) D] in W(CH2But)(=CHBut)(≡CBut)[Me2P(CH2)2PMe2].24

29
2.2.3. Synthesis and Characterization of (Me3SiCH2)3W(≡CSiMe3)(PMe2Ph)
(5a) and (Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph) (5b)
(Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a) and
(Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph) (5b) were prepared by a procedure similar
to that used to prepare complexes 3a and 3b. Upon addition of PMe2Ph to a
solution of (Me3SiCH2)3W(≡CSiMe3) (4a) in toluene-d8 at 23 °C, an immediate
color change from yellow to orange-red was observed. NMR spectroscopic data
at −50 °C, vide infra, were consistent with the formation of the PMe2Ph adduct
(Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a). NMR spectroscopic characterization
(1H, 13C, 31P, 29Si, and HMQC) of 5a suggests that the PMe2Ph ligand
coordinates cis to the alkylidyne ligand as in 3a. Upon warming the solution of
5a in toluene-d8 to 23 °C, 5a was found to undergo alkyl-to-alkylidyne α-
hydrogen migration to give bis(alkylidene) (Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph)
(5b), which was characterized by 1H, 13C, 31P, 29Si, and HMQC NMR
spectroscopy. As in 3a º 3b, the two tautomers reach an equilibrium (5a º 5b)
[Keq (303 K) = 4.65(0.11)].
5a and 5b also exhibit diastereotopic α-hydrogen atoms on the equatorial
alkyl ligands, as shown in the Newman projections in Figure 2.7. The 1H NMR
resonances of the alkylidene H atoms in 5b were observed as doublets at 8.217
(3JP-H = 5.3 Hz) and 7.621 (3JP-H = 3.6 Hz) ppm, respectively. Similarly, the 13C
NMR resonances of the alkylidene ligands in 5b appeared as doublets at 257.8
and 256.0 ppm, respectively. These observations suggest that, as in 3b, there
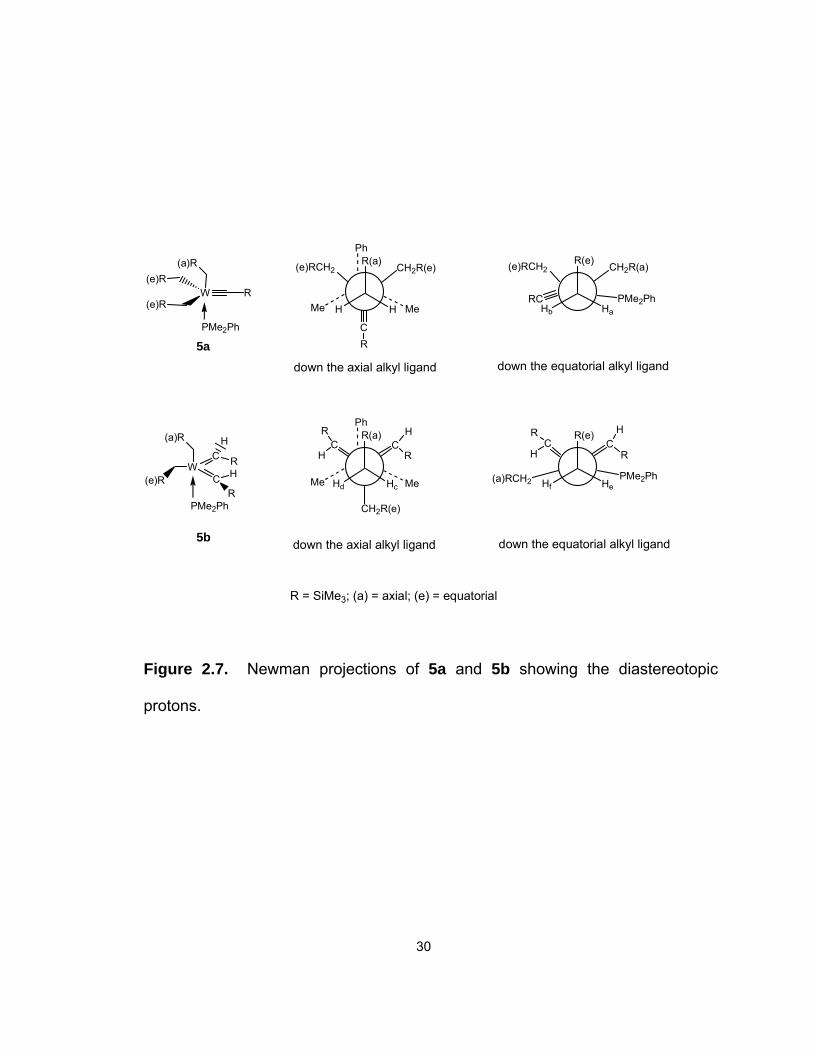
30
HH
R(a)
MeMe
Ph
CR
CH2R(e)(e)RCH2
HaHb
R(e) CH2R(a)(e)RCH2
PMe2PhRC
HcHd
R(a)
MeMe
Ph
CH2R(e)
CR
HC
R
H
HeHf
R(e)C
H
R
PMe2Ph(a)RCH2
CR
H
W
(a)R
R(e)R
(e)R
PMe2Ph
W
(a)R
(e)R C H
R
C R
H
PMe2Ph
down the axial alkyl ligand down the equatorial alkyl ligand
down the axial alkyl ligand down the equatorial alkyl ligand
R = SiMe3; (a) = axial; (e) = equatorial
5a
5b
Figure 2.7. Newman projections of 5a and 5b showing the diastereotopic
protons.

31
are two inequivalent alkylidene ligands in 5b. The coupling constants 2JP-C-axial
and 2JP-C-equatorial of 32.2 and 0 Hz for the axial and equatorial –CH2R ligands,
respectively, and 2JP-C of 12.1 and 11.1 Hz for the two alkylidene ligands suggest
that the two alkylidene and one alkyl ligands coordinate cis to the PMe3 ligand.
Two inequivalent alkylidene ligands suggest that 5b adopts an anti, syn
configuration similar to that of 3b. The prochiral W atom in 5a gives rise to
diastereotopic α-hydrogen atoms (CHaHb-SiMe3) for the equatorial alkyl ligands
as doublet of doublets at 0.750 ppm (2JHa-Hb = 3JP-H = 14.8 Hz) and 0.530 ppm in
the 1H NMR spectrum at –50 °C.
The equilibrium mixture of 5a and 5b in solution was found thermally
unstable. They are converted to (Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe2Ph)2
(9a-b), which is discussed in Chapter 3. Repeated attempts to purify 5a-b
through crystallization gave thermally unstable, oily liquids which failed to give
satisfactory C elemental analysis, although the H and P elemental analyses were
good.25 The characterization of 5a-b was thus based on their 1H, 13C, 31P, 29Si,
1H-decoupled 13C, and HMQC NMR spectra.
2.2.4. Kinetic and Thermodynamic Studies of the 3a º 3b Exchange
In the current work, (Me3SiCH2)3W(≡CSiMe3)(PR3) (3a and 5a), adducts
between phosphines PR3 and alkyl alkylidyne (Me3SiCH2)3W≡CSiMe3 (4a), were
found to undergo exchanges with their bis(alkylidene) tautomers
(Me3SiCH2)2W(=CHSiMe3)2(PR3) (3b and 5b). Given the unusual nature of the

32
exchange processes of 3a-b and 5a-b in the presence of phosphines, further
kinetic and thermodynamic studies were performed with the goal of elucidating
the mechanism. These studies of the 3a º 3b and 5a º 5b exchanges are
presented here and in the subsequent section (Section 2.2.5), respectively.
Variable-temperature NMR spectra of the tautomerization 3a º 3b were
studied, and the equilibrium constants, Keq = [3b] / [3a], measured between 278
and 303 K are listed in Table 2.3. A plot of ln Keq vs 1/T (Figure 2.8) gave ΔH° =
–1.8(0.5) kcal/mol and ΔS° = –1.5(1.7) eu.14 The equilibrium constants (Keq)
range from 12.3(0.2) at 278 K to 9.37(0.12) at 303 K, indicating that the
alkylidene isomer 3b is favored. Decreasing the temperature shifts the
equilibrium towards 3a. The process 3a º 3b is slightly exothermic with ΔH° =
–1.8(0.5) kcal/mol. It is interesting to note that the d0 bis(alkylidene) complex 3b
is thermodynamically close in energy to its alkylidyne isomer 3a [ΔG°298K =
–1.3(1.0) kcal/mol], although 3b is slightly more stable. If there is an alkyl-
alkylidyne scrambling process in (Me3SiCH2)3W≡CSiMe3 (4a) as in
(ButCH2)3W≡CSiMe3 º (ButCH2)2W(CH2SiMe3)(≡CBut), the proposed
bis(alkylidene) intermediate “(Me3SiCH2)2W(=CHSiMe3)2 (4b)” is much higher in
energy than 4a, and coordination with PMe3 to give 3b significantly lowers its
energy so that 3a º 3b equilibrium is observed.18,22
In the previous study of the exchange of silyl complexes
(ButCH2)2W(≡CBut)(SiButPh2) (2a) º (ButCH2)W(=CHBut)2(SiButPh2) (2b), only

33
Table 2.3. Equilibrium (Keq) and rate constants (k1 and k−1) of the 3a º 3b
exchange a
T (K) Keq b k1 x 105 (s−1)c k−1 x 106 (s−1)c
278 ± 1 12.3 ± 0.2 1.42 ± 0.02 1.160 ± 0.018
283 ± 1 11.52 ± 0.08 2.47 ± 0.13 2.14 ± 0.11
288 ± 1 10.941 ± 0.012 4.16 ± 0.04 3.80 ± 0.04
293 ± 1 10.43 ± 0.07 7.6 ± 0.3 7.3 ± 0.3
298 ± 1 9.80 ± 0.05 10.55 ± 0.10 10.71 ± 0.10
303 ± 1 9.37 ± 0.12 17.5 ± 0.5 18.6 ± 0.6
a Solvent: toluene-d8 b The largest random uncertainty is σKeq(ran)/Keq = 0.2/12.3 = 1.6%. The total
uncertainty σKeq/Keq of 5.2% was calculated from σKeq(ran)/Keq = 1.6% and the
estimated systematic uncertainty σKeq(sys)/Keq = 5% by σKeq/Keq = [(σKeq(ran)/Keq)2
+ (σKeq(sys)/Keq)2]1/2. c The largest random uncertainties are δk1(ran)/k1 = 0.13/2.47 = 5.3% and
δk−1(ran)/k−1 = 0.11/2.14 = 5.1%. The total uncertainties δk1/k1 = 0.0726 and
δk−1/k−1 = 0.0717 were calculated from δk(ran)/k and the estimated systematic
uncertainty δk(sys)/k = 5% by δk/k = [(δk(ran)/k)2 + (δk(sys)/k)2]1/2.

34
1000/T3.3 3.4 3.5 3.6
ln K
eq
2.2
2.3
2.4
2.5
slope = -0.7521intercept = 0.9063R² = 0.999
Figure 2.8. A plot of ln Keq vs 1/T of the equilibrium 3a º 3b.

35
the thermodynamic properties were reported. We have conducted kinetic studies
of the unusual alkyl alkylidyne and bis(alkylidene) exchange between 3a and 3b.
Variable-temperature 1H NMR experiments for the 3a º 3b exchange
between 278 and 303 K show that the α-hydrogen migrations between 3a and 3b
follow first-order reversible kinetics (Eqs. 2.3 and 2.4),26
(Eq. 2.3)
(Eq. 2.4)
where I3b-0, I3b-t and I3b-e are the integrations of 3b at time t = 0, t = t, and
equilibrium, respectively; k1 and k−1 are the rate constants for the forward and
reverse reactions, respectively. The kinetic plots for the exchange are shown in
Figure 2.9, and the rate constants from Eq. 2.3 are given in Table 2.3. Eyring
plots (Figure 2.10) lead to the activation parameters of the exchange: ΔH1≠ =
16.2(1.2) kcal/mol, ΔS1≠ = –22(4) eu for the forward reaction 3a → 3b, and ΔH2
≠
= 18.0(1.3) kcal/mol, ΔS2≠ = –21(4) eu for the reverse 3b → 3a reaction.
The 3a º 3b exchange is significantly slower than the
(ButCH2)2W(≡CBut)(SiButPh2) (2a) º (ButCH2)W(=CHBut)2(SiButPh2) (2b)
exchange; The latter was observed in the 2D-NOESY spectra (tmix = 3 s) at 23
°C.22a The 3a º 3b exchange is, however, much faster than the alkyl-alkylidene
scrambling in (ButCH2)3W≡CSiMe3 which was observed at >70 °C.18a The
( )( ) ( )
[ ][ ]3a3b
eq ==
+−=⎥⎦
⎤⎢⎣
⎡−−
−
−
−−
−−
1
1
11
0e
teln
kkK
tkkIIII
3b3b
3b3b

36
Time (min)0 200 400 600 800 1000
-3
-2
-1
0
ln [(
I 3b-e
- I 3b
-t) /
( I 3b -
e - I 3
b -0)
]
278 K
283 K
288 K
293 K298 K
303 K
Figure 2.9. Kinetic plots of the reversible reactions 3a º 3b.

37
1000/T3.2 3.3 3.4 3.5 3.6 3.7
ln (k
1/T) o
r ln
(k-1
/T)
-20
-19
-18
-17
-16
-15
-14
k1
k-1
Figure 2.10. Eyring plots for the reversible reactions 3a º 3b.

38
activation free energy ΔG1≠
298K of 23(2) kcal/mol for the forward reaction 3a → 3b
is 28.1(1.1) kcal/mol lower than that for the alkyl-alkylidene scrambling in
(ButCH2)3W≡CSiMe3.18a
It is interesting to note that (ButCH2)3W≡CtBu (1) reacts with neat PMe3 in
a sealed tube at 100 °C, giving (ButCH2)W(≡CtBu)(=CHtBu)(PMe3)2 (6) through
α-hydrogen abstraction and CMe4 elimination, as Schrock and Clark have
reported (and discussed in Chapter 3).15a When ca. 1 equiv of PMe3 was added
to a solution of 1 in benzene-d6 at room temperature, a similar reaction giving 6
and CMe4 occurred. No adduct between 1 and PMe3 was observed. The studies
here reveal the differences in the reactivities of (Me3SiCH2)3W≡CSiMe3 (4a) and
its analog (ButCH2)3W≡CBut (1) toward PMe3. The role of PMe3 in the 3a º 3b
exchange has been studied by density functional theory calculations and will be
discussed in Section 2.2.6.
2.2.5. Thermodynamic and Kinetic Studies of the 5a º 5b Exchange. A
Comparison of PMe3 and PMe2Ph
PMe2Ph is bulkier than PMe3, and the phenyl group often acts as an
electron withdrawing group. Both yielded alkylidyne adducts in their reactions
with 4a: (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) and
(Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a). The thermodynamic and kinetic studies
were conducted for the (Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a) º
(Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph) (5b) exchange at 303 K. The 5a º 5b

39
equilibrium [Keq′ = 4.65(0.11) at 303 K] is shifted more to the left (5a) than the 3a
º 3b equilibrium [Keq = 9.37(0.12) at 303 K]. Our theoretical studies presented in
the next section (Section 2.2.6) indicate that PMe3 ligands prefer to bind to the
bis(alkylidene) tautomers, and this coordination plays a critical role in the
stabilization of the bis(alkylidene) tautomers. Perhaps the bulkier PMe2Ph
ligands with an electron-withdrawing phenyl group donate less electron density to
the metal centers in 5a and 5b, shifting the equilibrium to alkyl alkylidyne 5a.
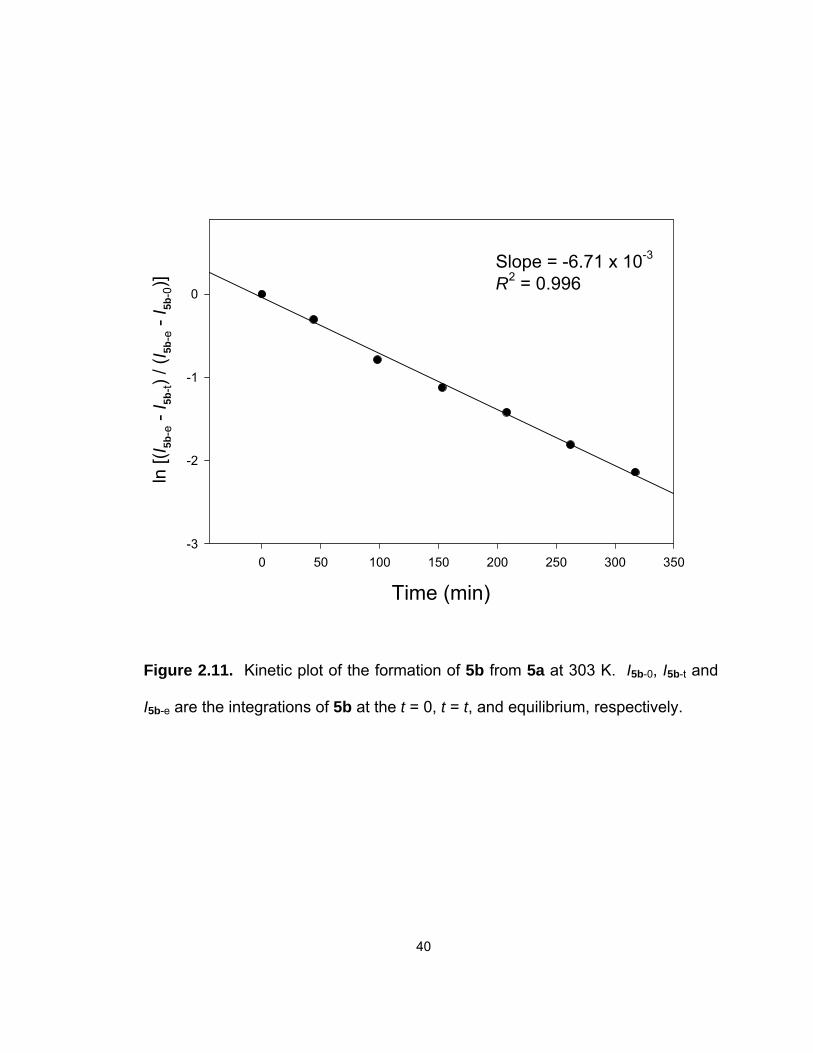
Kinetic studies using a kinetic equation similar to Eq. 2.3 give k1′ = (1.0 ±
0.1) x 10−4 s−1 and k−1′ = (2.2 ± 0.2) x 10−5 s−1 for the 5a → 5b and the reverse 5b
→ 5a conversions at 303 K, respectively (Figure 2.11). In comparison, the rates
for the 3a º 3b exchanges in the PMe3 complexes are k1 = (1.75 ± 0.05) x 10−4
s−1 and k−1 = (1.86 ± 0.06) x 10−5 s−1. The forward 5a → 5b conversion in the
PMe2Ph complexes is slower than the 3a → 3b conversion, whereas the reverse
5b → 5a conversion is slightly faster than the 3b → 3a conversion. The 3a º 3b
equilibrium is more shifted to 3b [Keq = 9.37(0.12)] than in the 5a º 5b equilibrium
[Keq′ = 4.65(0.11)], resulting in a higher kinetic barrier for the reverse 3b → 3a
conversion. It is not clear what leads to the higher kinetic barrier.
2.2.6. Theoretical Studies of the 3a º 3b Exchange
To understand the effect of the phosphine ligand on the relative stabilities
of the tautomers, DFT calculations were carried out by Professors Zhen Yang Lin
40
Time (min)0 50 100 150 200 250 300 350
ln [(
I 5b-e
- I 5b
-t) /
( I 5b-e
- I 5b
-0)]
-3
-2
-1
0
Slope = -6.71 x 10-3
R2 = 0.996
Figure 2.11. Kinetic plot of the formation of 5b from 5a at 303 K. I5b-0, I5b-t and
I5b-e are the integrations of 5b at the t = 0, t = t, and equilibrium, respectively.

41
and Yun-Dong Wu’s research groups at the Hong Kong University of Science
and Technology. The results of the DFT calculations on the corresponding
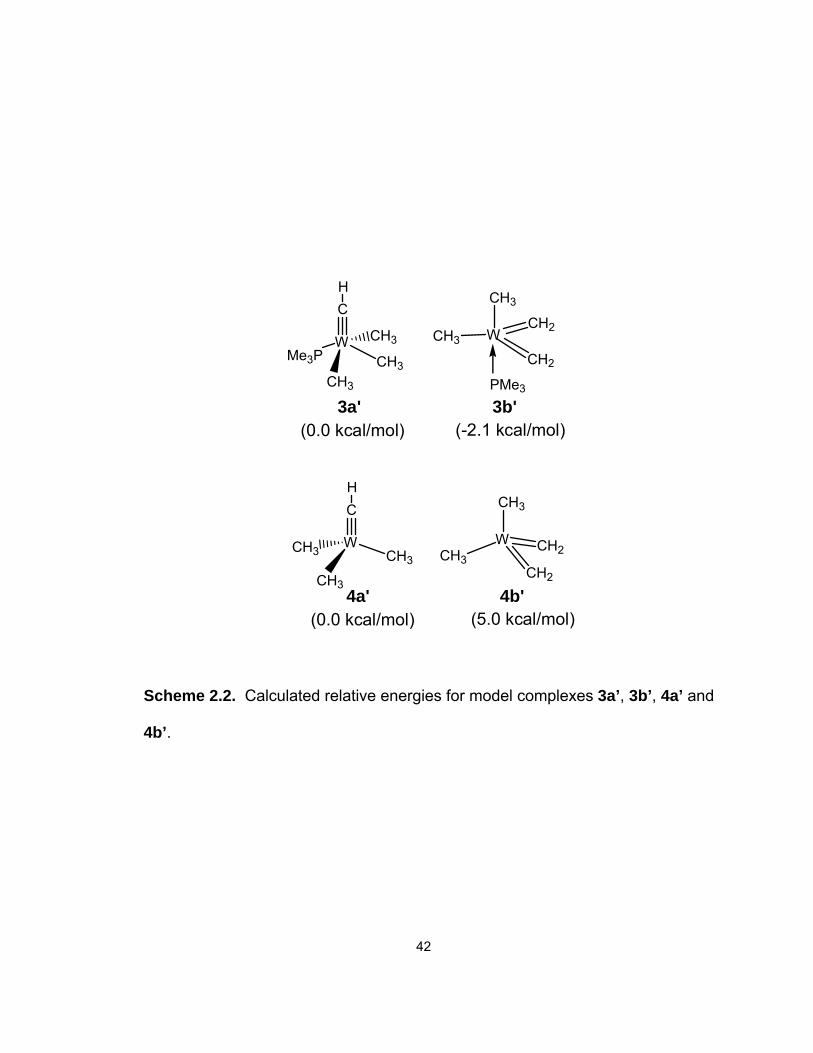
model complexes are given in Scheme 2.2. The results show that PMe3
promotes the conversion of the alkylidyne complex to the bis(alkilidene)
tautomer. 3b’ is calculated to be more stable than 3a’ by 2.1 kcal/mol, consistent
with the experimentally measured ΔH° (–1.8 kcal/mol) for the 3a → 3b
conversion.
The relative energies shown in Scheme 2.2 suggest that PMe3 binds with
the bis(alkylidene) tautomer relatively more strongly than with the alkylidyne
tautomer, reversing the relative stability in the two adduct isomers. To
understand how PMe3 coordinates to 4a’ and 4b’, their lowest unoccupied
orbitals (LUMOs), which are responsible for interaction with PMe3, were
examined in the frontier region. The structures of 3a’ and 3b’ together with the
relative binding ability of 4a’ and 4b’ are closely related to the orbital feature and
orbital energies of the LUMOs. Figure 2.12 shows the plots of the relevant
LUMOs for both 4a’ and 4b’. The maximum amplitudes of the LUMOs determine
the coordination sites for the PMe3 ligand. In both structures, the vacant sites
are in the axial positions (pointing to the bottom). In the case of 4a’, the
maximum amplitudes of the LUMO are in the equatorial positions and
approximately 90° to the alkylidyne ligand. In 4b’, the maximum amplitude of
one lobe in the LUMO is in the vacant site and is also approximately 90° to the
two alkylidene ligands. Thus, in both 4a’ and 4b’, PMe3 prefers to occupy the
42
W
C
CH3
CH3
CH3
H
W
CH3
CH3CH2
CH2
W
C
CH3CH3
CH3
H
Me3PW
CH3
CH3
CH2
CH2
PMe3
4a' 4b'(0.0 kcal/mol) (5.0 kcal/mol)
3a' 3b'(0.0 kcal/mol) (-2.1 kcal/mol)
Scheme 2.2. Calculated relative energies for model complexes 3a’, 3b’, 4a’ and
4b’.

43
H3C WCH2
CH2
CH3
CH3W
H3CH3C
H
4a' 4b'
-0.05779
LUMO
LUMO+1
LUMO+2
LUMO
LUMO+1
LUMO+2
-0.05773
-0.02259
-0.07230
-0.03562
-0.02774
Figure 2.12. Spatial plots of three LUMOs for alkylidyne and bis(alkylidene)
tautomers with orbital energies (au). The results were obtained from B3LYP/BSII
calculations.

44
coordination sites trans to W-CH3 instead of W=CH2 or W≡CH, reflecting the
stronger trans influence properties of the W=CH2 and W≡CH bonds. The LUMO
of 4b’ is much lower in energy than that of 4a’ (Figure 2.12), giving greater ligand
binding energy with PMe3. Therefore, 3b’ is more stable than 3a’. Clearly, the
PMe3 coordination affects the thermal stabilities of the tautomers significantly.
2.3. Concluding Remarks
In this chapter, an adduct between PMe3 and alkyl alkylidyne
(Me3SiCH2)3W≡CSiMe3 (4a), (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a), was found to
undergo a rare exchange with its bis(alkylidene) tautomer
(Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b). Thermodynamic studies show that the
bis(alkylidene) tautomer 3b is favored in the 3a º 3b equilibrium. The α-
hydrogen exchange between 3a and 3b follows first-order reversible kinetics.
Bis(alkylidene) complexes are believed to be intermediates in alkyl-alkylidyne
scrambling through α-hydrogen transfer in alkylidyne complexes. The role of
PMe3 in the 3a º 3b exchange has been studied by density functional theory
calculations. The current system provides a rare direct observation of an α-
hydrogen migration product 3b and thermodynamic, kinetic, and theoretical study
of its formation. Synthesis and characterization of complexes 5a and 5b
containing PMe2Ph ligands was also studied. Their exchange was found to be
shifted to 5a and slower than that of the 3a º 3b exchange.

45
2.4. Experimental Section
2.4.1. General Procedures
All manipulations were performed under a dry nitrogen atmosphere with
the use of either a dry box or standard Schlenk techniques. Solvents were
purified by distillation from potassium/benzophenone ketyl. Benzene-d6 and
toluene-d8 were dried over activated molecular sieves and stored under N2.
WCl6 was freshly sublimed under vacuum. (Me3SiCH2)3W≡CSiMe3 (4a)15g was
prepared from W(OMe)3Cl3 and six equiv of Me3SiCH2MgCl by a procedure
similar to that used in the preparation of (Me3CCH2)3W≡CCMe3 (1).15h 1H and
13C NMR spectra were recorded on a Bruker AC-250 or AMX-400 spectrometer
and referenced to solvent (residual protons in the 1H spectra). 31P, 29Si, and
HMQC (Heteronuclear Multiple Quantum Coherence) and 1H-gated-decoupled-
13C spectra were recorded on a Bruker AMX-400 spectrometer. Elemental
analysis was performed by Complete Analysis Laboratories Inc., Parsippany,
New Jersey.
For the thermodynamic studies, the equilibrium constants Keq were
obtained from at least two separate experiments at a given temperature, and
their averages are listed in Table 2.3. The maximum random uncertainty in the
equilibrium constants was combined with the estimated systematic uncertainty of
ca. 5%. The total uncertainties in the equilibrium constants were used in the
ln Keq vs. 1/T plot in Figure 2.8 and error propagation calculations.
The estimated uncertainty in the temperature measurements for an NMR probe
was 1 K. The enthalpy (ΔH°) and entropy (ΔS°) changes were calculated from an

46
unweighted nonlinear least-squares procedure contained in the SigmaPlot
Scientific Graph System. The uncertainties in ΔH° and ΔS° were computed from
the following error propagation formulas which were derived from –RT ln Keq =
ΔH° – TΔS°.27
( )
2
eq
eq2
minmax
2min
2max
2
22
(min)eq
(max)eq4
minmax
4max
2min
4min
2max
22
)(2
ln)(
)(
⎟⎟⎠
⎞⎜⎜⎝
⎛
−+
⎟⎠⎞
⎜⎝⎛
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛
−+
=°Δ
KK
TTTTR
TT
KK
TTTTTTRH
σ
σσ
(Eq. 2.4)
2
eq
eq2
minmax
2min
2max
2
22
(min)eq
(max)eq4
minmax
2max
2min
22
)()(
ln)(
2)(
⎟⎟⎠
⎞⎜⎜⎝
⎛
−+
+
⎟⎠⎞
⎜⎝⎛
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛
−=°Δ
KK
TTTTR
TT
KK
TTTTRS
σ
σσ
(Eq. 2.5)
Tmin and Tmax are the minimum and maximum temperatures in the current
studies; T is the mean temperature in the current studies. Keq(min) and Keq(max) are
the minimum and maximum equilibrium constants, respectively. σKeq/Keq is given
in Table 2.3. For the kinetic studies, the rate constants k1 and k−1 were obtained
from at least two separate experiments at a given temperature, and their
averages are listed. The estimated uncertainty (σT) in the temperature
measurements for an NMR probe was 1 K. The enthalpy (ΔH≠) and entropy
(ΔS≠) were calculated from an unweighted nonlinear least-squares procedure
contained in the SigmaPlot Scientific Graph System. The uncertainties in ΔH≠

47
and ΔS≠ were computed from the following error propagation formulas (Eqs. 2.7
and 2.8)27 which were derived from the Eyring Equation (Eq. 2.6) below.
⎥⎦
⎤⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛−
Δ+
Δ−=⎟
⎠⎞
⎜⎝⎛ ≠≠
bkh
RS
RTH
Tk lnln (Eq. 2.6)
⎪⎩
⎪⎨⎧
⎪⎭
⎪⎬⎫
⎟⎠⎞
⎜⎝⎛+
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠⎞
⎜⎝⎛ ++⎟
⎠⎞
⎜⎝⎛ +⎟
⎠⎞
⎜⎝⎛
×=≠
22
max
2
min
2
2
2min
2max
22
σ2ΔΔ1
ΔΔ1σ
Δ)Δσ(
kk
TLT
TLT
TT
TTTR
H
(Eq. 2.7)
⎪⎩
⎪⎨⎧
⎪⎭
⎪⎬⎫
+⎟⎠⎞
⎜⎝⎛+
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠⎞
⎜⎝⎛ ++⎟
⎠⎞
⎜⎝⎛ +⎟
⎠⎞
⎜⎝⎛
×=≠
)(σΔΔ1
ΔΔ1σ
Δ)Δσ(
2min
2max
22
max2
min
2
min2
max
2
2
22
TTkk
TLTT
TLTT
TT
TRS
(Eq. 2.8)
⎟⎟⎠
⎞⎜⎜⎝
⎛−⎟⎟
⎠
⎞⎜⎜⎝
⎛=Δ
min
min
max
max lnlnTk
Tk
L
minmax TTT −=Δ
kb and h are the Boltzman and Plank constants, respectively. The values of σk/k
are given in Table 2.3.

48
2.4.2. NMR Experiments
Complete and unambiguous assignments of all proton and carbons
resonances were achieved on the basis of chemical shift considerations, as well
as NMR experiments, namely HMQC and 1H-gated-decoupled-13C experiments.
HMQC (Heternuclear Multiple Quantum Coherence), the simplest form of an
inverse H,X correlation technique,28a,c was used to assign diastereotopic protons
in complexes 3 and 5. 1H-gated-decoupled-13C experiments were utilized to
determine the number of hydrogen atoms that are bonded to an α-carbon atom
and to calculate the 1JC-H coupling constants.28b,c
2.4.3. Preparation of (Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a)
3a was prepared by the vacuum transfer of PMe3 (25 mmHg, 0.104 mmol)
to a J. R. Youngs NMR tube containing 4a (48 mg, 0.0904 mmol) in toluene-d8.
The reaction mixture is kept frozen in liquid nitrogen until placed in the pre-cooled
NMR probe at −50 °C. 3a: 1H NMR (toluene-d8, 400.11 MHz, −50 °C, J in Hz) δ
0.904 (d, 9H, PMe3, 2JP-H = 7.2), 0.751 (dd, 2H, eq-CHaHb-SiMe3, 2JH-H = 3JP-H =
14.4), 0.503 (s, 9H, ≡CSiMe3), 0.487 (d, 2H, ax-CH2SiMe3, 3JP-H = 10.7), 0.329 (s,
9H, ax-CH2SiMe3), 0.316 (s, 18H, eq-CH2SiMe3), 0.213 (dd, 2H, eq-CHaHb-
SiMe3, 3JP-H = 28.2, 2JH-H = 14.1); 13C{1H} NMR (toluene-d8, 100.63 MHz, −50 °C,
J in Hz) δ 358.81 (d, ≡CSiMe3, 2JP-C = 14.5, 1JW-C = 159.1), 81.69 (d, ax-
CH2SiMe3, 2JP-C = 36.5, 1JW-C = 117.9), 49.45 (d, eq-CH2SiMe3, 1JH-C = 111.6, 2JP-
C = 7.2, 1JW-C = 59.9), 17.84 (d, PMe3, 1JH-C = 128.2, 1JP-C = 22.8), 4.23 (s,

49
≡CSiMe3, 1JH-C = 117.8, 3JW-C = 50.1), 3.55 (s, ax-CH2SiMe3, 1JH-C = 117.1, 3JW-C
= 33.2), 3.48 (s, eq-CH2SiMe3, 1JH-C = 117.1, 3JW-C = 25.0); 31P{1H} (toluene-d8,
161.97 MHz, −50 °C, J in Hz) δ −5.64 (s, 1JW-P = 28.9); 29Si{1H} (toluene-d8,
79.49 MHz, −50 °C, J in Hz) δ −0.269 (d, ax-CH2SiMe3, 3JP-Si = 2.0), −2.482 (d,
eq-CH2SiMe3, 3JP-Si = 4.9, 2JW-Si = 50.2), −21.129 (d, ≡CSiMe3, 3JP-Si = 2.3, 2JW-Si
= 52.3). The assignments of the 1H and 13C NMR were confirmed by low-
temperature HMQC experiments.
2.4.4. Preparation of (Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b)
A solution of (Me3SiCH2)3W≡CSiMe3 (4a, 95 mg, 0.179 mmol) in pentane
was frozen by liquid nitrogen in a 125-ml Schlenk flask. The flask headspace
was evacuated. PMe3 (100 mmHg, 0.358 mmol) was then vacuum transferred to
the flask submerged in liquid nitrogen. The PMe3 was then condensed in the
flask, and the flask was warmed to room temperature. The flask was then added
nitrogen, and stirred overnight at room temperature. Cooling the solution at –30
°C overnight gave crystals of 3b (87 mg, 0.143 mmol, 80% yield). 1H NMR
(toluene-d8, 400.11 MHz, −20 °C, J in Hz) δ 7.985 (d, 1H, =CHSiMe3, 3JP-H = 5.6),
7.192 (d, 1H, =CHSiMe3, 3JP-H = 4.0), 1.024 (d, 9H, PMe3, 2JP-H = 7.8), 0.917 (dd,
1H, eq-CHaHb-SiMe3, 3JP-H = 17.7, 2JH-H = 11.3), 0.876 (dd, 1H, eq-CHaHb-SiMe3,
3JP-H = 32.2, 2JH-H = 11.3; overlapped with eq-CHaHb-SiMe3 peak), 0.388 (s, 9H,
ax-CH2SiMe3, 2JSi-H= 6.0), 0.388 (d, 2H, ax-CH2SiMe3, 3JP-H = 11.4; overlapped
with ax-CH2SiMe3 peak), 0.342 (s, 9H, eq-CH2SiMe3), 0.280 (s, 9H, =CHSiMe3),

50
0.128 (s, 9H, =CHSiMe3); 13C{1H} (toluene-d8, 100.63 MHz, −40 °C, J in Hz) δ
256.43 (d, =CHSiMe3, 1JH-C = 123.5, 2JP-C = 11.8), 254.71 (d, =CHSiMe3, 1JH-C =
102.6, 2JP-C = 12.6), 50.71 (s, eq-CH2SiMe3, 1JH-C = 105.3, 1JW-C= 43.0), 38.01 (d,
ax-CH2SiMe3, 1JH-C = 111.0, 2JP-C = 32.3), 18.80 (d, PMe3, 1JH-C = 126.4, 1JP-C =
26.4), 4.82 (s, ax-CH2SiMe3, 1JH-C = 118.1), 3.08 (s, eq-CH2SiMe3, 1JH-C = 118.8),
1.76 (s, =CHSiMe3, 1JH-C = 117.3), 1.64 (s, =CHSiMe3, 1JH-C = 117.3); 31P{1H}
(toluene-d8, 161.97 MHz, −50 °C, J in Hz) δ 0.61 (s, 1JW-P = 115.5); 29Si{1H}
(toluene-d8, 79.49 MHz, −50 °C, J in Hz) δ −0.73 (d, ax-CH2SiMe3, 3JP-Si = 19.2),
−1.96 (d, eq-CH2SiMe3, 3JP-Si = 4.1), −8.63 (d, =CHSiMe3, 3JP-Si = 2.9), −9.89 (d,
=CHSiMe3, 3JP-Si = 4.8). 1H and 13C assignments were confirmed by low-
temperature HMQC experiments. Anal. Calcd. C, 37.61, H, 8.47. Found C,
37.43, H, 8.66.
2.4.5. Preparation of (Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a)
A solution of (Me3SiCH2)3W≡CSiMe3 (4a, 50 mg, 0.094 mmol) in toluene-
d8 was added ca. 10-fold excess PMe2Ph via cannula transfer to a J. R. Youngs
NMR tube. An immediate color change from yellow to orange-red was observed
signifying the formation of 5a. At −50 °C the solution of 5a was stable. 5a: 1H
NMR (toluene-d8, 399.97 MHz, −50 °C, J in Hz) δ 7.4-7.0 (m, 5H, PMe2Ph), 1.30
(d, 6H, PMe2Ph, 2JP-H = 6.0), 0.750 (dd, 2H, eq-CHaHb-SiMe3, 2JH-H = 3JP-H =
14.8), 0.506 (s, 9H, ≡CSiMe3), 0.599 (broad singlet, 2H, ax-CH2SiMe3), 0.363 (s,
9H, ax-CH2SiMe3), 0.243 (s, 18H, eq-CH2SiMe3), 0.530 (broad, 2H, eq-CHaHb-

51
SiMe3); 13C{1H} NMR (toluene-d8, 100.59 MHz, −60 °C, J in Hz) δ 358.91 (d,
≡CSiMe3, 2JP-C = 14.1), 83.86 (d, ax-CH2SiMe3, 2JP-C = 34.2), 50.38 (d, eq-
CH2SiMe3, 2JP-C = 7.0, 16.74 (d, PMe2Ph, 1JP-C = 22.1), 3.59 (s, ≡CSiMe3, 3JW-C =
51.3), 2.97(s, ax-CH2SiMe3), 2.82 (s, eq-CH2SiMe3); 31P{1H} (toluene-d8, 161.92
MHz, −60 °C, J in Hz) δ 6.57 (s); 29Si{1H} (toluene-d8, 79.46 MHz, −60 °C, J in
Hz) δ 0.677 (d, ax-CH2SiMe3, 3JP-Si = 1.0), −1.22 (d, eq-CH2SiMe3, 3JP-Si = 4.8),
−19.28 (d, ≡CSiMe3, 3JP-Si = 1.9, 2JW-Si = 51.0).
2.4.6. Preparation of (Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph) (5b)
Upon warming a solution of 5a to room temperature it was converted to
5b. 5b was stable for several days in solution at room temperature. Attempts to
obtain pure crystals of 5 yielded a dark brown oil, which was thermally unstable.25
5b: 1H NMR (toluene-d8, 399.97 MHz, −20 °C, J in Hz) δ 8.22 (d, 1H, =CHSiMe3,
3JP-H = 5.3), 7.62 (d, 1H, =CHSiMe3, 3JP-H = 3.6), 7.5-7.0 (m, PMe2Ph), 1.38 (d,
6H, PMe2Ph, 2JP-H = 7.6), 0.728 (dd, 1H, eq-CHaHb-SiMe3, 3JP-H = 18.6, 2JH-H =
11.2), 1.01 (dd, 1H, eq-CHaHb-SiMe3, 3JP-H = 33.0, 2JH-H = 11.2), 0.331 (s, 9H, ax-
CH2SiMe3), 0.372 (d, 2H, ax-CH2SiMe3, 3JP-H =9.2), 0.326 (s, 9H, eq-CH2SiMe3),
0.137 (s, 9H, =CHSiMe3), 0.056 (s, 9H, =CHSiMe3); 13C{1H} (toluene-d8, 100.59
MHz, −20 °C, J in Hz) δ 257.8 (d, =CHSiMe3, 2JP-C = 12.1), 256.0 (d, =CHSiMe3,
2JP-C = 11.1), 53.5 (s, eq-CH2SiMe3), 37.4 (d, ax-CH2SiMe3, 2JP-C = 32.2), 15.5 (d,
PMe2Ph, 1JP-C = 26.2), 4.25 (s, ax-CH2SiMe3), 2.90 (s, eq-CH2SiMe3), 1.86 (s,
=CHSiMe3), 1.67 (s, =CHSiMe3); 31P{1H} (toluene-d8, 161.92 MHz, −60 °C, J in

52
Hz) δ 13.9 (s, 1JW-P = 97.1); 29Si{1H} (toluene-d8, 79.46 MHz, −20 °C, J in Hz) δ
1.09 (s, ax-CH2SiMe3), −1.75 (d, eq-CH2SiMe3, 3JP-Si = 5.6), −7.85 (d, =CHSiMe3,
3JP-Si = 2.9), −9.16 (d, =CHSiMe3, 3JP-Si = 4.0).
2.4.7. Kinetic Study of the Conversion of (Me3SiCH2)3W(≡CSiMe3)(PMe3)
(3a) to 3b
At least two experiments for each temperature were conducted. A mixture
of (Me3SiCH2)3W≡CSiMe3 (4a), 4,4’-dimethylbiphenyl (an internal standard), and
toluene-d8 in J. R. Youngs NMR tubes in liquid nitrogen were added ca. 2-21
equiv of PMe3 through vacuum transfer. The samples were kept below –78 °C
before insertion into the NMR probe. The NMR probe was pre-cooled or pre-
heated to the set temperature. After the NMR tubes were inserted into the probe,
the 1H NMR spectra were taken after the temperature was stabilized, and the
integrations of 3b, relative to those of the internal standard, were used as I3b-0 at t
= 0. Once the equilibrium between 3a and 3b was reached, the integration of 3b
was used as I3b-e.
2.4.8. Thermodynamic Study of the Equilibrium between 3a and 3b
Three samples of 3a-b were prepared with at least 10 equiv of PMe3, and
kept at room temperature for over 24 hours to ensure equilibrium was
established. The samples were then placed in a circulation bath at 278.0(0.1),
283.0(0.1), 288.0(0.1), 293.0(0.1), 298.0(0.1), or 303.0(0.1) K for at least 6 hours.

53
31P NMR spectra were taken at −50 °C with a relaxation delay of 10 s. Keq = [3b]
/ [3a] = I3b / I3a were calculated from the integrations of the two tautomers.
2.4.9. Kinetic and Thermodynamic Studies of the Conversion of
(Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a) to 5b at 303 K
A mixture of (Me3SiCH2)3W≡CSiMe3 (4a, 35 mg, 0.066 mmol), 4,4’-
dimethylbiphenyl (an internal standard), and toluene-d8 in J. R. Youngs NMR
tubes were added ca. 10-fold excess PMe2Ph via cannula transfer. Kinetic
measurements were taken in duplicate on a Bruker 400 MHz NMR at 303 K.
Integration of the 1H PMe2− peak of 5b was used in the kinetic plot. For
thermodynamic measurements, two samples of 5a-b were prepared with at least
10 equiv of PMe2Ph, and kept at room temperature for over 24 hours to ensure
that equilibrium was established. The samples were placed in a circulation bath
at 303.0(0.1) K for at least 6 hours. 31P NMR spectra were taken at −50 °C with
a relaxation delay of 10 s. Keq = [5b] / [5a] = I5b / I5a were calculated from the
integrations of the two tautomers.
2.4.10. Reaction of (ButCH2)3W≡CBut (1) with PMe3 at Room Temperature in
Benzene-d6
This experiment was conducted by Ruitao Wang, a post-doctoral
researcher in our group, as part of the current project.

54
Schrock and Clark have reported that the reaction of 1 with neat PMe3 in a
sealed tube at 100 °C gave (ButCH2)W(≡CtBu)(=CHtBu)(PMe3)2 (6).15a The
current studies were conducted at room temperature in a solution to see if an
adduct formed between 1 and PMe3. 1 (100 mg, 0.214 mmol) in benzene-d6 in a
J. R. Youngs NMR tube in liquid nitrogen was added ca. 1 equiv of PMe3 through
vacuum transfer. A slow reaction at room temperature occurred to give 6. After
3 days at 23 °C, a mixture of unreacted 1 (and PMe3) and 6 in ca. 1 : 2 ratio was
observed. No adduct between 1 and PMe3 in the mixture was observed.
2.4.11. Determination of the X-ray Crystal Structure of 3b
The crystal structure was determined on a Bruker AXS Smart 1000 X-ray
diffractometer equipped with a CCD area detector, and fitted with an upgraded
Nicolet LT-2 low temperature device. Suitable crystals were coated with
Paratone oil and mounted under a stream of nitrogen at −100 °C. Intensity data
were measured with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å).
Background counts were measured at the beginning and the end of each scan
with the crystal and counter kept stationary. The structure was solved by direct
method, and then a 2-fold twinning law (TWIN 0 1 0 1 0 0 0 0 –1, and BASF
0.45126) was used to solve the twinning by merohdry. All non-hydrogen atoms
but four C’s in both axial ligands were refined with anisotropic displacement
coefficients. All hydrogen atoms were placed in calculated positions and
introduced into the refinement as fixed contributors with an isotropic U value of
0.008 Å2.

55
2.4.12. Computational Details
These calculations were performed by the research groups of Professors
Zhenyang Lin and Yun-Dong Wu at the Hong Kong University of Science and
Technology through collaboration with our research group.
All calculations were performed using the Gaussian 03 package29a with
density functional theory at the B3LYP level. Full geometry optimizations were
carried out with basis set I (BSI): LanL2DZ with f polarization functions for W29b
and 6-31G* for the rest of elements. Single point energies were calculated with
basis set II (BSII): SDDAll for W29c and 6-311++G** for other elements. Vibration
frequency calculations were performed for all the structures with the BSI. The
calculated relative energies shown in the text have been corrected with zero-
point-energy (ZPE).

56
CHAPTER 3
Synthesis of Unusual Tungsten Alkyl Alkylidene Alkylidyne
Complexes and Kinetic Studies of Their Formation
3.1. Introduction
Tungsten and molybdenum alkylidene and alkylidyne complexes have
been extensively studied in part for their potential applications in olefin
metathesis. (Me3CCH2)3W≡CCMe3 (1), one of the first alkyl alkylidyne
complexes, was originally prepared in ca. 25% yield from WCl6 and six equiv of
LiCH2CMe3.32a It is now prepared from W(OMe)3Cl3 and six equiv of
Me3CCH2MgCl in ca. 50% yield.32b This method to prepare trialkyl alkylidyne
complexes is probably limited to those containing bulky alkyls free of β-hydrogen
atoms, as these are unlikely to undergo β-hydrogen elimination. Other trialkyl
alkylidyne complexes include (Me3SiCH2)3M≡CSiMe3 [M = Mo, W (4a)] and
(Me3CCH2)3W≡CSiMe3.15g,18a The reactivity of (Me3SiCH2)3W≡CSiMe3 (4a) has
been extensively studied throughout this dissertation.
High-oxidation-state alkylidyne complexes such as (Me3SiCH2)3W≡CSiMe3
(4a) contain unoccupied low-lying metal d orbitals, and, as a result, they are
generally stabilized by the presence of σ-donor ligands (e.g., PMe3). The first
complex containing alkyl, alkylidene, and alkylidyne ligands was reported by
Schrock and Clark in 1978.15a Upon heating a solution of (Me3CCH2)3W≡CCMe3
(1) in liquid PMe3 at 100 °C, the transition metal complex undergoes α-hydrogen

57
abstraction to yield (Me3CCH2)(Me3CCH=)(Me3CC≡)W(PMe3)2 (6) (Scheme 3.1).
An analogous complex, (Me3CCH2)(Me3CCH=)(Me3CC≡)W(dmpe) (7), which
contained the chelating phosphine dmpe (Me2PCH2CH2PMe2), was also reported
by Schrock and Clark.15a The crystal structure of this complex was later reported
by Churchill and Youngs in 1979.24 Complex 7, unlike the bis-PMe3 complex 6,
exhibited cis coordination of the chelating phosphine and the alkylidyne ligand
occupied the axial site (Scheme 3.1). It is hypothesized that the phosphine
ligands in complex 6 are coordinated trans to one another, and the other ligands
occupy the equatorial sites to form a trigonal bipyramidal configuration. d0 Alkyl
alkylidene alkylidyne complexes Re(CH2CMe3)2(=CHCMe3)(≡CCMe3),
(Me3CCH2)(Me3CCH=)(Me3CC≡)Re(py)2(OTf), and their derivatives have also
been reported (Scheme 3.1b-c).19
We reported earlier unusual reactions of d0 tantalum bis(alklyidene)
complexes such as (Me3SiCH2)Ta(=CHSiMe3)2(PMe3)2 with silanes.23d,e The
reactivities of the d0 tungsten complexes containing Me3SiCH= and Me3SiC≡
ligands toward silanes were of interest to us. We have recently prepared
(Me3SiCH2)(Me3SiCH=)(Me3SiC≡)W(PMe3)2 (8a-b) and
(Me3SiCH2)(Me3SiCH=)(Me3SiC≡)W(PMe2Ph)2 (9a-b) through heating the 3a º
3b and 5a º 5b equilibrium mixtures in the presence of excess phosphine, PMe3
and PMe2Ph, respectively (Scheme 3.2).

58
CCMe3Me3CCH2
Me3CCH2 W
CH2CMe3
WCCMe3
CHCMe3
Me3CCH2
PMe3
PMe3
W
C
C
CH2CMe3
Me2P
PMe2
CMe3
CMe3
H
Re
C
C
Me3CCH2 py
pyOTf
CMe3
Me3C
H
6
7
Re(CCMe3)(CH2CMe3)3(OTf)2 py
(b)
(a)
syn isomer
Re
C
CMe3CCH2
CMe3
CMe3
HMe3CCH2
(c)
[Re(=CHCMe3)(CCMe3)(NH2CMe3)Cl2]2(THF)
[Re(=CHCMe3)(CCMe3)Cl2]x
PMe3, 100 oC
dmpe, 100 oC
Me3CCH2MgCl
Me3CCH2MgClTHF, -40 oC
THF, -40 oC
Et2O, 25 oC
Scheme 3.1. Some reported d0 complexes containing metal-carbon single,
double, and triple bonds.15a,19

59
W
Me3Si
SiMe3
Me3Si
Me3Si
PR3
excess PR3
W
Me3Si
Me3Si SiMe3
H
H
SiMe3
PR3
-SiMe4
WCSiMe3
CHSiMe3
Me3SiCH2
PR3
PR3
k1 k-1Keq
3a/5a
3b/5b
8a-b, 9a-b
R3 = Me3 (3 and 8) Me2Ph (5 and 9)
heat
Scheme 3.2. Formation of complexes 8a-b and 9a-b from the equilibrium
mixtures of 3a-b and 5a-b.

60
Both complexes 8 and 9 exist as mixtures of two isomers, 8a-b and 9a-b, as
observed by NMR spectroscopy. Our preparation and characterization of 8a-b
and 9a-b as well as kinetic studies of their formation are reported here.
3.2. Results and Discussion
3.2.1. Synthesis and Characterization of
(Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe3)2 (8a-b)
We reported in Chapter 2 the synthesis of complexes
(Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) and (Me3SiCH2)2W(=CHSiMe3)2(PMe3)
(3b),14 as well as (Me3SiCH2)3W(≡CSiMe3)(PMe2Ph) (5a) and
(Me3SiCH2)2W(=CHSiMe3)2(PMe2Ph) (5b), which undergo α-hydrogen migrations
to establish the exchange equilibria, 3a º 3b, and 5a º 5b. Upon heating these
equilibrium systems in the presence of excess PMe3 or PMe2Ph, respectively, the
mixtures were found to yield alkyl alkylidene alkylidyne complexes
(Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe3)2 (8a-b) and
(Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe2Ph)2 (9a-b) through α-hydrogen
abstraction and elimination of SiMe4.
The 1H, 13C, and 31P NMR spectral analysis of complex 8 revealed two
distinct isomers, 8a and 8b, in solution (Figures 3.1 and 3.2). The ratio of the two
isomers (8a : 8b) is 7 : 1 based on 1H NMR integration. NMR spectroscopic
characterization (1H, 13C, 31P, 29Si, 1H-gated-decoupled 13C and HMQC) of the

61
ppm (f1)-0.500.000.501.001.502.002.503.00
13.35013.40013.45013.500
10.4010.5010.6010.7010.80
Figure 3.1. 1H NMR spectrum of a mixture of 8a and 8b (toluene-d8).
8a =CH-
8b =CH-
PMe3 8b -SiMe3
8a -SiMe3
SiMe4
8a –CH2– 8b –CH2–
Free PMe3
Internal Standard

62
ppm (t1)
01020304050
339.0340.0341.0342.0343.0
273.50274.00274.50275.00275.50
Figure 3.2. 13C NMR spectrum of a mixture of 8a and 8b (toluene-d8).
8b ≡C- 8a ≡C-
8a =CH- 8b =CH-
8a –SiMe3 8b –SiMe3
SiMe4
8a –CH2– 8b –CH2–

63
more abundant 8a suggests that the PMe3 ligands coordinate trans to one
another. One resonance in the 31P NMR spectrum for 8a was observed at −2.21
ppm (1JP-W = 124.7 Hz). The two trans-PMe3 ligands exhibit virtual coupling and
appear as a pseudo-triplet in the 1H and 13C NMR spectra at 1.26 and 20.74
ppm, respectively.4b Three singlet resonances of –SiMe3 groups were observed
in the 1H, 13C and 29Si NMR spectra of 8a; their chemical shifts are listed in Table
3.1. The 1H resonances of the α-hydrogen atoms in –CH2SiMe3 appear as a
triplet at −0.036 ppm with a large 2JP-H = 22.4 Hz. In the Ta bis(alkylidene)-
bis(phosphine) complex, (Me3SiCH2)Ta(=CHSiMe3)2(PMe3)2, where the
phosphines are trans to one another, a large coupling constant (2JP-H = 19.8 Hz)
was observed as well.31 The resonance of the alkylidyne C atom in 8a appears
at 339.0 ppm as a triplet (2JP-C = 11.1 Hz; 1JW-C = 161.8 Hz) due to its coupling
with two equivalent P atoms, and is shifted up-field from that of
(Me3SiCH2)3W≡CSiMe3 (4a) at 343.27 ppm.20h The alkylidene C and alkyl α-C
atoms appear as triplets as well at 275.0 ppm (2JP-C = 11.1 Hz; 1JW-C = 101.5 Hz)
and 25.63 ppm (2JP-C = 6.2 Hz; 1JW-C = 36.3 Hz), respectively. Most of the 1H,
13C, and 31P resonances of complex 8b are only slightly shifted from those of 8a,
and their chemical shifts are given in Table 3.1. One exception in the 1H NMR
spectrum, the alkylidene proton, W=CHSiMe3, is significantly shifted in 8b to
13.46 ppm, from 10.54 ppm for 8a. Likewise, the W−CH2SiMe3 resonance in the
13C NMR spectrum at 34.0 ppm is shifted in 8b from 25.6 ppm in 8a.

64
Table 3.1. NMR resonances of 8a and 8b (ppm)
Ligand 8a 8b
1H resonances
−CHxSiMe3, x = 0, 1, 2 0.331, 0.231, 0.185 0.323, 0.217, 0.060
=CHSiMe3 10.54 13.46
−CH2SiMe3 −0.036 −0.373
PMe3 1.26 1.29
13C resonances
−CHxSiMe3, x = 0, 1, 2 5.2, 3.7, 2.4 6.4, 3.1, 2.0
=CHSiMe3 275.0 273.8
≡CSiMe3 339.0 343.5
−CH2SiMe3 25.6 34.0
PMe3 20.74 20.7
31P resonances
PMe3 −2.21 −2.40
29Si resonances
=CHSiMe3 −4.66 n/a* (not available)
≡CSiMe3 −23.08 n/a
−CH2SiMe3 −2.06 n/a

65
Attempts to isolate pure crystals of 8a-b for elemental analysis or X-ray
diffraction were unsuccessful. 8a-b are unstable without excess PMe3. Once
excess PMe3 and other volatiles were removed, 8a-b was found to decompose.
The 1H NMR spectrum of the solid, dissolved in toluene-d8, reveals unknown
decomposed products. Numerous attempts to isolate 8a-b failed in the presence
of PMe3. Phosphine ligands are believed to be labile in 8a-b and dissociate
readily from the complexes, leading to the decomposition of 8a-b.
The two isomers 8a and 8b are likely syn- and anti- rotamers (Figure
3.3).19b,32 In the former, the –SiMe3 group on the alkylidene ligand points toward
the alkylidyne ligand. Such rotameric mixtures were also observed in alkylidene
alkylidyne complexes Re(=CHCMe3)(≡CCMe3)(OR)2.32a In the dmpe complex
(Me3CCH2)(Me3CCH=)(Me3CC≡)W(dmpe) (7), the only isomer observed in the X-
ray crystal structure is the syn isomer (Scheme 3.1a). Only one isomer of
(Me3CCH2)(Me3CCH=)(Me3CC≡)Re(py)2(OTf) in an octahedral environment was
observed, and it was believed to be the syn isomer (Scheme 3.1c).19b 8a-b,
while in a trigonal bipyramidal environment, are sterically hindered. It is likely
that the syn 8a is the major rotamer (Figure 3.3).

66
WC
CMe3SiCH2
PMe3
SiMe3
H
SiMe3
PMe3
WC
CMe3SiCH2
PMe3
H
SiMe3
SiMe3
PMe3
WC
CMe3SiCH2
PMe2Ph
SiMe3
H
SiMe3
PMe2Ph
WC
CMe3SiCH2
PMe2Ph
H
SiMe3
SiMe3
PMe2Ph
8a - syn 8b - anti
9a - syn 9b - anti
ratio 8a : 8b = 7 : 1
ratio 9a : 9b = 27 : 1
Figure 3.3. Spatial drawings of complexes 8a, 8b, 9a, and 9b showing
stereochemistry around the W=C bond.

67
3.2.2. Synthesis and Characterization of
(Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe2Ph)2 (9a-b)
(Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe2Ph)2 (9a-b) was prepared by
heating the 5a º 5b mixture in the presence of excess PMe2Ph where it
undergoes an α-hydrogen abstraction reaction to form 9a-b. Complex 9 also
exists as two isomers in solution, 9a º 9b, in a ratio of 9a : 9b = 27 : 1 based on
their 1H NMR spectra. The 1H and 31P NMR resonances of 9a and 9b, and 13C
NMR resonances for 9a are presented in Table 3.2. The 1H and 31P NMR
spectra of complexes 9a and 9b are given in Figures 3.4 and 3.5. Attempts to
obtain 13C and 29Si NMR spectra of the minor isomer 9b failed due to its very low
concentration in solution. 13C NMR spectra of a mixture of 9a and 9b (prepared
from 5a-b), after 48 hours of data acquisition at 23 °C on a Bruker AMX-400
spectrometer, revealed only the resonances of the major isomer 9a. Repeated
attempts to grow crystals in the presence of excess PMe2Ph were unsuccessful,
as were the attempts to grow crystals of 8a-b. Without excess PMe2Ph, 9a-b
was found to be unstable and decompose, as in the case of 8a-b.
The methyl groups on the PMe2Ph ligand in complex 9a-b are
diastereotopic. Figure 3.6 shows the Newman projection of 9a down a W−P
bond. The methyl groups of 9a thus appear as two pseudo triplets in the 1H and
13C NMR spectra. The 1H NMR resonance of one of the methyl groups on 9b
overlaps with those of 9a.

68
Table 3.2. NMR resonances of 9a and 9b (ppm)
Ligand 9a 9b
1H resonances
−CHxSiMe3, x = 0, 1, 2 0.487, 0.307, −0.266 0.323, 0.217, −0.060
=CHSiMe3 10.89 13.73
−CH2SiMe3 −0.025 −0.134
PMeaMebPh 1.687, 1.659 1.614, 1.640
13C resonances
−CHxSiMe3, x = 0, 1, 2 3.76, 3.20, 2.45 n/a* (not available)
=CHSiMe3 277.02 n/a
≡CSiMe3 340.72 n/a
−CH2SiMe3 27.71 n/a
PMeaMebPh 22.44, 20.01 n/a
31P resonances
PMeaMebPh 12.58 10.85
29Si resonances
=CHSiMe3 −3.79 n/a* (not available)
≡CSiMe3 −22.10 n/a
−CH2SiMe3 −2.82 n/a

69
ppm (f1) 10.011.012.013.014.0
9a
9b
5a/5b* *
Figure 3.4. 31P NMR spectrum of 9a and 9b. A small amount of 5a-b is present
in solution as a broad peak overlapping with 9b. (* indicates 183W satellites)
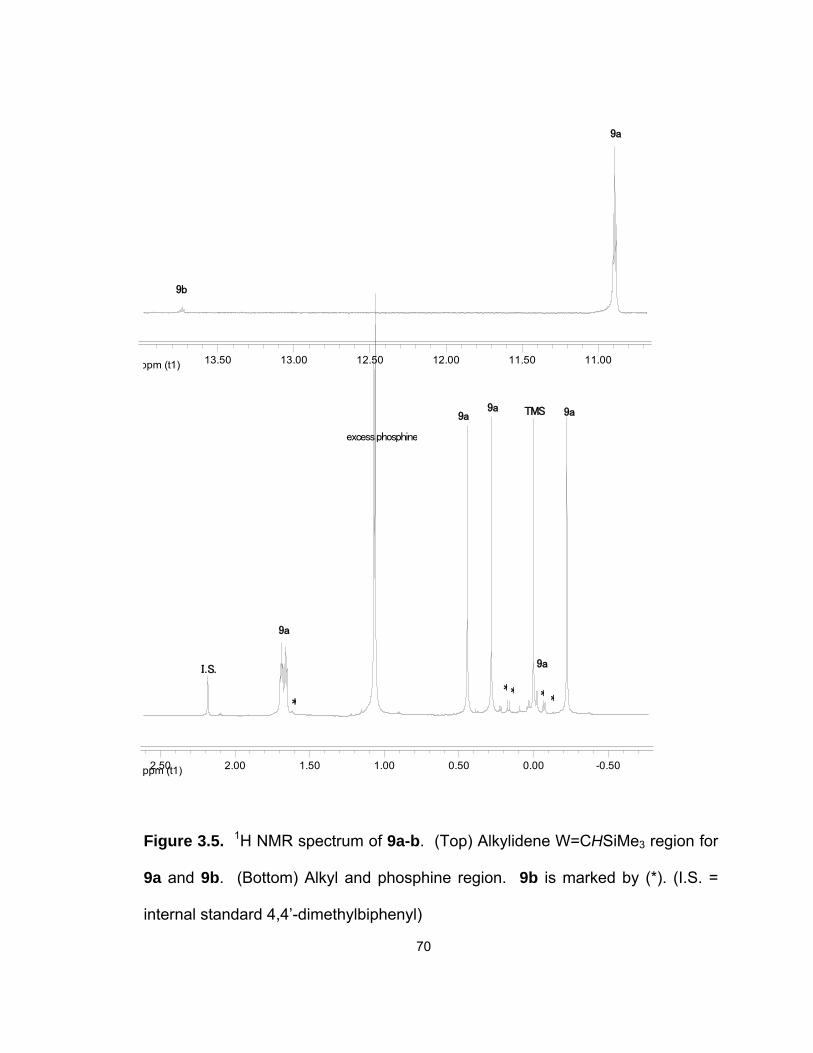
70
ppm (t1) 11.0011.5012.0012.5013.0013.50
9a
9b
ppm (t1) -0.500.000.501.001.502.002.50
TMS
I.S.
***
**
9a9a 9a
9a
excess phosphine
9a
Figure 3.5. 1H NMR spectrum of 9a-b. (Top) Alkylidene W=CHSiMe3 region for
9a and 9b. (Bottom) Alkyl and phosphine region. 9b is marked by (*). (I.S. =
internal standard 4,4’-dimethylbiphenyl)

71
ppm (t1) 1.6001.6501.700
9a
9b
Figure 3.6. The Newman projection of 9a-b and the 1H NMR spectrum showing
the two methyl resonances in PMe2Ph with virtual 31P-1H coupling.
MeaMeb
Ph
CH2R
RC CHR

72
3.2.3. Kinetic Study of the Conversion of the 3a º 3b Mixture to 8a-b
In the previous chapter, we reported the exchange between
(Me3SiCH2)3W(≡CSiMe3)(PR3) (3a and 5a) and (Me3SiCH2)2W(=CHSiMe3)2(PR3)
(3b and 5b, R = Me3 or Me2Ph). In the presence of excess phosphine, kinetic
studies were conducted to study the α-hydrogen abstraction reactions to yield
alkyl alkylidene alkylidyne complexes 8a-b and 9a-b, as discussed in the current
chapter.
The α-hydrogen abstraction reaction to give 8a-b follows second-order
kinetics. In the presence of excess PMe3, the α-hydrogen abstraction reaction of
3a-b to give 8a-b was found to follow pseudo first-order kinetics (Eqs. 3.1 and
3.2),26 as the 1H NMR spectra of the conversion revealed.
(Eq. 3.1)
(Eq. 3.2)
(Eq. 3.3)
where I30 and I8t are integrations of the PMe3 ligands in 3a-b (total) at t = 0, and
in 8a-b (total) at t = t in the 1H NMR spectra.
excess PR3+ WCSiMe3
CHSiMe3
Me3SiCH2
PMe3
PMe3SiMe4
3
8
[ ] [ ] [ ] [ ] [ ] [ ] [ ]
tkI
I I
kkk kdt
d
'ln
PMe',PMewhen;'PMe
23
83
3223232
0
t0−=⎟
⎠⎞
⎜⎝⎛ −
=>>== 3 3- 3- 3

73
The kinetic studies for the conversion of 3 → 8 were conducted between
333.2(0.1) and 358.2(0.1) K. The kinetic plots and rate constants (k2) are given
in Figure 3.7 and Table 3.3, respectively. The Eyring plot (Figure 3.8) gives the
activation parameters of the conversion: ΔH≠ = 29.2(1.5) kcal/mol and ΔS≠ =
4.2(1.3) eu.
It is not clear whether alkyl alkylidyne 3a, bis(alkylidene) 3b or both
undergo the α-hydrogen abstraction to give 8a-b. The process may involve a
cyclometalation transition state. The activation parameters of the conversion ΔH≠
= 29.2(1.5) kcal/mol and ΔS≠ = 4.2(1.3) eu are similar to other reported reactions
through cyclometalation transition states for complexes containing −CH2CMe3
and/or −CH2SiMe3 ligands (Table 3.4).33,18a,16c
3.2.4. Kinetic Studies of the 5 → 9 Conversion and a Comparison of Its
Rate with the Rate of the Formation of 8
PMe2Ph is bulkier than PMe3, and the phenyl group often acts as an
electron withdrawing group. In their reactions with 3 and 5, both yielded alkyl
alkylidene alkylidyne complexes: (Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe3)2 (8)
and (Me3SiCH2)(Me3SiCH=)W(≡CSiMe3)(PMe2Ph)2 (9). The kinetics of the
formation of 9a-b was also studied at 348.2 K (Figure 3.9) and analyzed with a
kinetic equation similar to Eq. 3.3. These kinetic studies yield the rate constant
k3 = 1.5 × 10−5 M−1s−1 at 348.2(0.1) K. In comparison, the rate constant (k2) for
the formation of the PMe3 analog 8a-b at 348.2(0.1) K is 3.0(0.2) × 10−5 M−1s−1.

74
Time (min)0 500 1000 1500 2000
ln [
( I 3- 0
- I 8-
t) / I
3-0 ]
-2
-1
0
358.2 K 353.2 K348.2 K 343.2 K
338.2 K
333.2 K
Figure 3.7. Kinetic plots for the conversion of 3a-b → 8a-b. I3-0 and I8-t are total
integrations of the PMe3 ligands in both 3a and 3b at t = 0, and in both 8a and 8b
at t = t in the 1H NMR spectra.

75
Table 3.3. Rate constant (k2) of the formation of 8a-b.a
T (K) k2 x 105 (M−1 s−1)b
333.2 ± 0.1 0.35 ± 0.03
338.2 ± 0.1 0.85 ± 0.06
343.2 ± 0.1 1.53 ± 0.07
348.2 ± 0.1 3.0 ± 0.2
353.2 ± 0.1 5.1 ± 0.5
358.2 ± 0.1 8.6 ± 0.6
a Solvent: toluene-d8. b The largest random uncertainty is δk2(ran)/k2 = 0.5/5.1 = 9.8%. The total
uncertainty δk2/k2 = 0.1100 was calculated from δk2(ran)/k2 and the estimated
systematic uncertainty δk2(sys)/k2 = 5% by δk2/k2 = [(δk2(ran)/k2)2 + (δk2(sys)/k2)2]1/2.

76
1000/T2.75 2.80 2.85 2.90 2.95 3.00 3.05
ln (k
/T)
-19
-18
-17
-16
-15
-14
Slope = -14.70Intercept = 25.89R2 = 0.994
Figure 3.8. Eyring plot of the conversion 3a-b → 8a-b.

77
Table 3.4. Activation parameters in reactions through cyclometalation transition
states
Reactions ΔH≠ (kcal/mol) ΔS≠ (eu)
CpTaCl2(CH2CMe3)2 → 21 ± 2 −4 ± 10
CpTaCl2(=CHCMe3) + CMe4 33
(Me3CCH2)3W≡CSiMe3 →
(Me3CCH2)2(Me3SiCH2)W≡CCMe3 18a 27.5 ± 0.6 −2.0 ± 1.7
(Me3CCH2)2(Me3SiCH2)W≡CCMe3 →
(Me3CCH2)3W≡CSiMe3 18a 25.4 ± 0.8 −9.5 ± 1.9
(Me3SiCH2)5Ta → (Me3SiCH2)3Ta=CHSiMe3 16c 21.6 ± 1.4 −5 ± 5
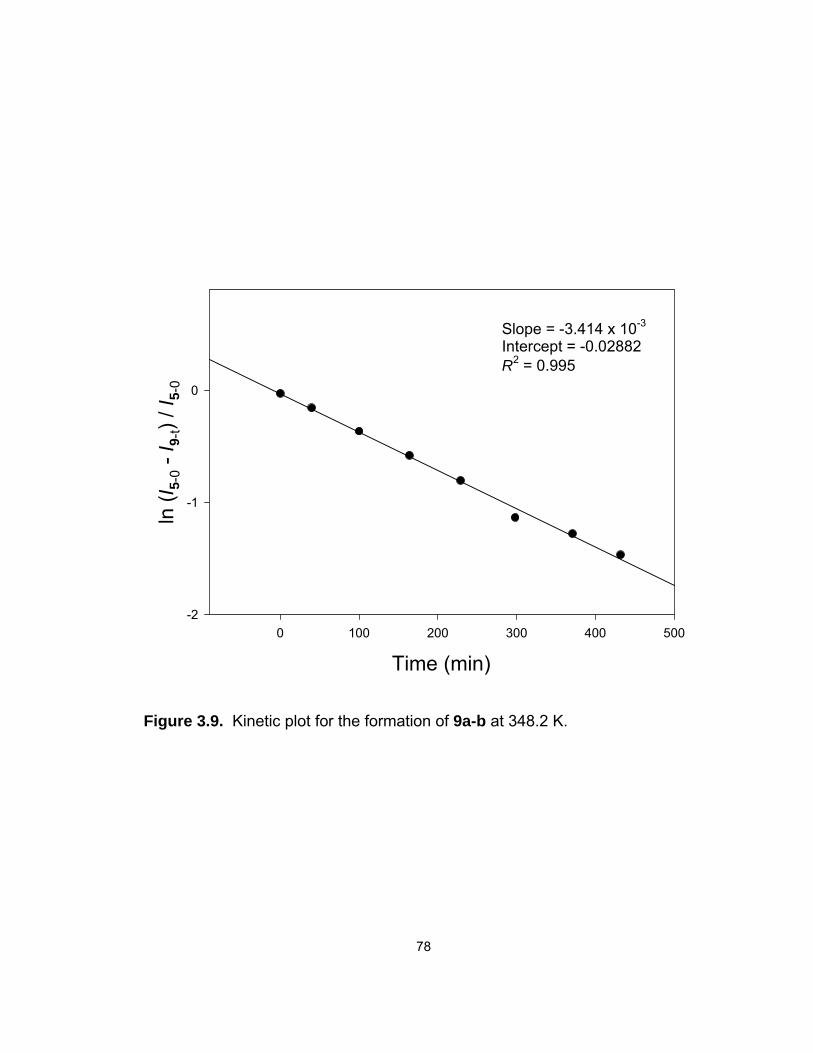
78
Time (min)0 100 200 300 400 500
ln (I
5 -0 -
I 9-t)
/ I 5 -
0
-2
-1
0
Slope = -3.414 x 10-3
Intercept = -0.02882R2 = 0.995
Figure 3.9. Kinetic plot for the formation of 9a-b at 348.2 K.

79
In other words, the rate of the formation of the bulkier 9a-b is about a half of the
rate in the formation of the PMe3 adduct 8a-b. This is similar to the slower rate of
the 5a º 5b inter-conversion reported in Chapter 2.
3.2.5. Attempted Reactions of (Me3SiCH2)3W≡CSiMe3 (4a) with PCy3 and
PPh3
Attempts were made to prepare compounds analogous to 8a-b and 9a-b
using phosphines other than PMe3 and PMe2Ph. The addition of excess PCy3 or
PPh3 to solutions of 4a in toluene, and heating the solutions for two days at 100
°C yielded no products. No complexation was observed between PPh3 or PCy3
and 4a. It is not clear why these two phosphines do not form alkyl alkylidene
alkylidyne complexes.
3.3. Concluding Remarks
In this chapter, equilibrium mixtures (Me3SiCH2)3W(≡CSiMe3)(PR3) (3a/5a)
º (Me3SiCH2)2W(=CHSiMe3)2(PR3) (3b/5b), were shown to convert to alkyl
alkylidene alkylidyne complexes (Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PR3)2 (8a-b
and 9a-b). Two isomers of complexes 8 (8a and 8b) and 9 (9a and 9b) were
observed in solution. Kinetic studies show that the α-hydrogen abstraction
reactions to form 8a-b and 9a-b follow second-order kinetics. The presence of
excess phosphine (PMe3 or PMe2Ph) in solution has been proven necessary for
the stability of both 8a-b and 9a-b.

80
It is interesting to note the difference in the reactivities of
(Me3SiCH2)3W≡CSiMe3 (4a) and its analog (Me3CCH2)3W≡CCMe3 (1) toward
PMe3. (Me3CCH2)3W≡CCMe3 (1) reacts with neat PMe3 in a sealed tube at 100
°C, giving alkyl alkylidene alkylidyne complex
(Me3CCH2)W(≡CCMe3)(=CHCMe3)(PMe3)2 (6) through α-hydrogen abstraction
and CMe4 elimination, as Schrock and Clark have reported.15a When a solution
of 1 in benzene-d6 was added ca. 1 equiv of PMe3 at room temperature, a similar
reaction giving 6 and CMe4 occurred (Chapter 2, Section 2.4.10). No adduct
between alkylidyne (Me3CCH2)3W≡CCMe3 (1) and PMe3 was observed. In
comparison, PR3 coordinates readily to (Me3SiCH2)3W≡CSiMe3 (4a), an analog
of 1, to give the phosphine alkylidyne adducts (Me3SiCH2)3W(≡CSiMe3)(PR3) (3a
and 5a). These phosphine alkylidyne adducts then undergo α-hydrogen
migration to give the bis(alkylidene) tautomers (Me3SiCH2)2W(=CHSiMe3)2(PR3)
(3b and 5b). Coordination of the second phosphine ligands to the tautomeric
mixtures leads to α-hydrogen abstraction to give the alkyl alkylidene alkylidyne
complexes 8a-b and 9a-b reported in the current chapter. In other words, in the
current case involving the –CH2SiMe3 and ≡CSiMe3 ligands, there are
intermediates [alkyl alkylidyne-PR3 º bis(alkylidene)-PR3 tautomeric mixtures]
before the formation of two alkyl alkylidene alkylidyne complexes. The current
work exemplifies the differences in −CH2CMe3 and −CH2SiMe3 ligand systems.

81
3.4. Experimental Section
3.4.1. General Procedures
All manipulations were performed under a dry nitrogen atmosphere with
the use of either a glovebox or standard Schlenk techniques. Solvents were
purified by distillation from potassium benzophenone ketyl. NMR solvents were
dried and stored over 5 Å molecular sieves. 1H and 13C{1H} NMR spectra were
recorded on a Bruker AC-250 or AMX-400 spectrometer and referenced to
solvent (residual protons in the 1H spectra). 31P, 29Si, and HMQC (Heteronuclear
Multiple Quantum Coherence) spectra were recorded on a Bruker AMX-400
spectrometer. 29Si chemical shifts were referenced to SiMe4. 3 and 5 were
prepared by procedures in Chapter 2.
3.4.2. Preparation of (Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe3)2 (8a-b)
8a-b was prepared by heating an equilibrium mixture of 3a º 3b with
excess PMe3. A sample was prepared by vacuum transferring at least a 10-fold
excess of PMe3 to a J.R. Youngs tube containing 4a (50.0 mg, 0.0942 mmol) and
toluene-d8 (0.5 mL). The mixture was kept at 23 °C overnight and then heated at
75 °C overnight. The formation of the 8a º 8b mixture is 100% based on 1H
NMR studies. When the volatiles (SiMe4, PMe3, and solvent) from the reaction
were removed, 8a and 8b were found to quickly decompose to unknown species.
Repeated attempts to grow crystals of 8a-b in the presence of excess PMe3
failed. 8a was thus characterized by 1H, 31P, 29Si, 13C, DEPT, HMQC, and 1H-

82
gated-decoupled-13C NMR. 8a: 1H NMR (toluene-d8, 399.97 MHz, 23 °C, J in
Hz) δ 10.54 (t, 1H, =CHSiMe3, 3JP-H = 4.2), 1.26 (m, 9H, PMe3), 0.331 (s, 9H, -
SiMe3), 0.231 (s, 9H, -SiMe3), 0.185 (s, 9H, -SiMe3), −0.036 (t, 2H, CH2-SiMe3,
3JP-H = 22.4); 13C{1H} NMR (toluene-d8, 100.59 MHz, 23 °C, J in Hz) δ 339.0 (t,
≡CSiMe3, 2JP-C = 11.1, 1JW-C = 161.8), 275.0 (t, =CHSiMe3, 2JP-C = 11.1, 1JW-C =
101.5), 25.63 (t, CH2SiMe3, 2JP-C = 6.2, 1JW-C = 36.3), 20.74 (m, PMe3), 5.21 (s,
-SiMe3), 3.66 (s, -SiMe3), 2.38 (s, -SiMe3); 31P{1H} NMR (toluene-d8, 161.92 MHz,
23 °C, J in Hz) δ −2.21 (s, 1JW-P = 124.7); 29Si{1H} NMR (toluene-d8, MHz, °C, J
in Hz) δ −2.06 (s, -CH2SiMe3), −4.66 (s, =CHSiMe3), −23.08 (s, ≡CSiMe3). 1H
and 13C assignments were confirmed by HMQC experiments. 8b: 1H NMR
(toluene-d8, 399.97 MHz, 23 °C, J in Hz) δ 13.46 (t, 1H, =CHSiMe3, 3JP-H = 5.6),
1.29 (m, 9H, PMe3), 0.323 (s, 9H, -SiMe3), 0.217 (s, 9H, -SiMe3), 0.060 (s, 9H,
-SiMe3), −0.373 (t, 2H, CH2-SiMe3, 3JP-H = 21.4); 13C{1H} NMR (toluene-d8,
100.59 MHz, 23 °C, J in Hz) δ 343.5 (t, ≡CSiMe3, 2JP-C = 10.5), 273.8 (t,
=CHSiMe3, 2JP-C = 11.1) 34.0 (t, CH2SiMe3, 2JP-C = 6.2), 20.7 (m, overlapping with
8a and toluene-d8 peaks, PMe3), 6.4 (s, -SiMe3), 3.1 (s, -SiMe3), 2.0 (s, -SiMe3);
31P{1H} NMR (toluene-d8, 161.92 MHz, 23 °C, J in Hz) δ −2.4 (s).
3.4.3. Preparation of (Me3SiCH2)W(≡CSiMe3)(=CHSiMe3)(PMe2Ph)2 (9a-b)
9a-b were prepared by a procedure similar to that used to prepare 8, by
heating an equilibrium mixture of 5a º 5b with excess PMe2Ph. PMe2Ph (ca. 10-
fold excess) was added via cannula transfer to a J.R. Youngs tube containing 4a

83
and toluene-d8. The solution was kept for 24 hours at room temperature to
establish the 5a º 5b equilibrium. The solution was then heated at 75 °C for 24
hours to give the 9a º 9b mixture in 100% yield based on 1H NMR. 9a was
characterized by 1H, 31P, 29Si, 13C, and HMQC NMR. Upon removal of excess
PMe2Ph, 9a-b were found to decompose to unknown species. The ratio of 9a :
9b in solution was 27 : 1. Repeated attempts to grow crystals of 9a-b in the
presence of PMe2Ph were unsuccessful. 9a: 1H NMR (toluene-d8, 399.97 MHz,
−20 °C, J in Hz) δ 10.89 (t, 1H, =CHSiMe3, 3JP-H = 3.7), 7.4-7.0 (m, 5H, PMe2Ph),
1.640 (m, 3H, PMeaMebPh), 1.622 (m, 3H, PMeaMebPh), 0.441 (s, 9H, -SiMe3),
0.279 (s, 9H, -SiMe3), −0.226 (s, 9H, -SiMe3), −0.025 (t, 2H, CH2-SiMe3, 3JP-H =
21.6); 13C{1H} NMR (toluene-d8, 100.59 MHz, −20 °C, J in Hz) δ 340.72 (t,
≡CSiMe3, 2JP-C = 9.9), 277.02 (t, =CHSiMe3, 2JP-C = 11.7), 130-124 (m, PMe2Ph),
27.71 (s, CH2SiMe3, 2JP-C = 5.6), 22.44 (m, PMeaMeb), 20.01 (m, PMeaMeb), 3.76
(s, -SiMe3), 3.20 (s, -SiMe3), 2.45 (s, -SiMe3); 31P{1H} NMR (toluene-d8, 161.92
MHz, 23 °C, J in Hz) δ 12.58 (s, 1JW-P = 126.4); 29Si{1H} NMR (toluene-d8, 79.46
MHz, −20 °C, J in Hz) δ −2.82 (s, -CH2SiMe3), −3.79 (s, =CHSiMe3), −22.10 (s,
≡CSiMe3). 1H and 13C assignments were confirmed by HMQC experiments. 9b:
1H NMR (toluene-d8, 399.97 MHz, −20 °C, J in Hz) δ 13.73 (t, 1H, =CHSiMe3, 3JP-
H = 4.9), 1.61 (m, 3H, PMeaMebPh), 1.64 (m, 3H, PMeaMebPh, overlapping with
9a), 0.175 (s, 9H, -SiMe3), 0.159 (s, 9H, -SiMe3), −0.066 (s, 9H, -SiMe3), −0.134
(t, 2H, CH2-SiMe3, overlapping with 9a); 31P{1H} NMR (toluene-d8, 161.92 MHz,
23 °C, J in Hz) δ 10.85 (s, 1JW-P = 125.4).

84
3.4.4. Kinetic Studies of the Formation of 8a-b and 9a-b
In the kinetic studies of the formation of 3a-b, a mixture of
(Me3SiCH2)3W≡CSiMe3 (29.8-37.8 mg, 0.0562-0.0712 mmol) 4,4’-
dimethylbiphenyl (an internal standard), and toluene-d8 in J. R. Young’s NMR
tubes were added at least a 10-fold excess PMe3 ([PMe3]: 1.42-2.31 M) via
vacuum transfer. The sample was kept at 23 °C overnight to establish the 3a º
3b equilibrium. The sample was then placed in a circulation bath between 60 °C
(333.2 K) and 85 °C (358.2 K). After a measured period of time, the experiment
was quenched by placing the sample in a dry ice/ethanol bath at −78 °C, and 1H
NMR was acquired at room temperature. Integration of the 1H –PMe3
resonances at 1.26-1.29 ppm for 8a-b versus an internal standard was used to
give the kinetic plots in Figure 3.7. Both isomers were integrated together since
the peaks overlap in the 1H NMR spectra. The average slope of two experiments
was used to calculate k2. The enthalpy (ΔH≠) and entropy (ΔS≠) were calculated
from an unweighted nonlinear least-squares procedure contained in the
SigmaPlot Scientific Graph System. The uncertainties in ΔH≠ and ΔS≠ were
computed from the error propagation formulas, Eqs. 2.7 and 2.8,27 which were
presented in Chapter 2.
Similar to 8a-b, the kinetics of the conversion of 5a-b to 9a-b was
monitored by 1H NMR. A mixture of (Me3SiCH2)3W≡CSiMe3 (33.2 mg), PMe2Ph
([PMe2Ph] = 3.79 M), and 4,4’-dimethylbiphenyl (an internal standard) in toluene-
d8 in J. R. Young’s NMR tube was heated in a circulation bath at 75.0 °C (348.2

85
K) for a measured amount of time, and the reaction was then quenched in dry
ice/ethanol bath at −78 °C. 1H NMR was acquired at room temperature, and the
integration of the 1H –PMe2Ph resonances at 1.61-1.64 ppm for 9a-b versus an
internal standard was used to give the kinetic plot in Figure 3.9.
3.4.5. Attempted Synthesis of the Adducts between PCy3 or PPh3 and 4a
Two separate experiments were conducted with 50 mg of
(Me3SiCH2)3W≡CSiMe3 (4a), 4,4’-dimethylbiphenyl (an internal standard), and
toluene-d8 in J. R. Youngs NMR tubes. PCy3 or PPh3, respectively, was added in
at least a 10-fold excess. The solution was heated for two days at 100 °C. No
reaction or adducts were observed in 1H NMR spectroscopy.

86
CHAPTER 4
Synthesis of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) and Studies
of Its Reaction with Dioxygen
4.1. Introduction
Reactions of dioxygen with transition metal complexes have been
extensively investigated in part because they are essential steps in some
biological and catalytic processes.1,34 Such studies have focused mainly on
dn (n ≠ 0) transition metal complexes, and oxidation of the metals by dioxygen are
often involved in these reactions. In comparison, there are relatively fewer
studies of reactions of d0 transition metal complexes with dioxygen.10,11
Schwartz,10a,b Gibson,10c and coworkers reported oxygen-atom insertion into the
Zr-R bond in the reaction of Cp2ZrRCl with dioxygen. Similar reactions between
Cp2ZrR2 and dioxygen were shown to give Cp2Zr(OR)2.10d Bercaw and
coworkers studied the conversion of Cp*2Hf(R)(O-OBut) to Cp*2Hf(OR)(OBut).10e,f
Tilley reported oxygen-atom insertion into the Zr-Si bond in the reaction of
Cp2ZrClSiMe3 with dioxygen to give Cp2MCl(OSiMe3).10g In Cp-free complexes,
Wolczanski, Rothwell, Gibson and their coworkers reported reactions of dioxygen
with (RO)2TiMe2,11a,b (ArO)2TaMe3,11c and (ArN=)2MoMe211d to yield
(RO)2Ti(OMe)2, (ArO)2Ta(OMe)3, and [(ArN=)2Mo(Me)(µ-OMe)]2, respectively
(Scheme 4.1). Autoxidation of M(CH2R)4 by dioxygen was found to be rapid.11e
Reactions of dioxygen with diamides (N~N)TiMeX (X = Me, Cl; N~N = silacyclo-

87
(RO)2TiMe
Me
O2
(ArO)2TaMe
MeMe
O2
(ArN=)2MoMe
Me
O2
(RO)2TiOMe
OMe
(ArO)2TaOMe
OMeOMe
(ArN=)2MoO
Mo(=NAr)2O
Me
MeMe
Me
(a)
(b)
(c)
Scheme 4.1. Formation of metal alkoxide complexes from oxygen insertion
reactions.11a,b,c,d

88
alkane or -alkene bridge) gave [(N~N)Ti(X)(µ-OMe)]2.11f In contrast, the reaction
of alkyl-free (N~N)TiCl2 with O2 led to diamide ligand oxidation. LY(µ-Me)2AlMe2
(L = porphyrin) was found to activate O2 to give LY(µ-OMe)2AlMe2.11g,h In recent
years, reactions of O2 with d0 complexes such as Ta(NR2)5 and (Et2N)3Ta=NBut
have been of intense interest in the preparation of microelectronic metal oxide
thin films.12 The mechanistic pathways in these chemical vapor deposition
reactions are, however, not clear.
We have been intrigued by our recent observations of (a) the formation of
an oxo alkylidene (ButCH2)2W(=O)[=CR(SiPh2But)] (10) in the reaction of
dioxygen with d0 silyl alkylidyne (ButCH2)2W(≡CBut)(SiPh2But) (2a);22a (b) O
insertion into Ta-Si and Ta-N bonds in the reaction of O2 with d0 (Me2N)4Ta-
Si(SiMe3)3 (Scheme 4.2);22b and (c) the formation of rare trinuclear oxo
aminoxide complexes M3(NMe2)6(μ-NMe2)3(μ3-O)(μ3-ONMe2) (M = Zr, Hf) from
the reactions of d0 amides M(NMe2)4 (M = Zr, Hf) with O2.22c,d In particular, the
formation of the oxo alkylidene 10 was the result of an unusual silyl to alkylidyne
migration promoted by the reaction with O2.22a We have prepared
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11), an adduct between d0 alkyl alkylidyne
(Me3SiCH2)3W≡CSiMe3 (4a) and trimethylphosphine oxide (O=PMe3). As part of
our studies of metal silyl complexes and similarities/differences between M-Si
(silyl) and M-C (alkyl) bonds, we have investigated the reaction of 11 with O2, and
were surprised to find that this reaction gave an oxo siloxy complex
O=W(OSiMe3)(CH2SiMe3)3 (12) which was isolated as an adduct (13) with
(Me3SiCH2)3W≡CSiMe3 (4a, Scheme 4.3). The –SiMe3 group in the ≡CSiMe3

89
WCtBu
SiPh2But
ButCH2
ButCH2
O2
O2Ta NMe2Me2N
Me2NNMe2
Si(SiMe3)3
Ta ONMe2Me2N
Me2NNMe2
OSi(SiMe3)3
WC
O
SiPh2But
But
ButCH2
ButCH2
(a)
(b)
2a 10
Scheme 4.2. Oxygen reactions with d0 complexes.23

90
SiMe3
CH2SiMe3Me3SiCH2 Me3SiCH2 W
C2 O2
OPMe3 CO2
CH2SiMe3Me3SiCH2 Me3SiCH2
WO
OSiMe3
SiMe3
CH2SiMe3Me3SiCH2 Me3SiCH2
WC
CH2SiMe3Me3SiCH2 Me3SiCH2
WO
OSiMe3
11 12
+ 4a
13
- O=PMe3
Scheme 4.3. Preparation of complexes 12 and 13.

91
ligand in 11 underwent an unusual migration to give the -OSiMe3 ligand in
12.5b,15c,20a The preparation of 11 and 12 and our studies of their formation are
reported in this chapter.
4.2. Results and Discussion
4.2.1. Synthesis and Characterization of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3
(11)
(Me3SiCH2)3W≡CSiMe3 (4a)15g was found to readily form the adduct
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11, Eq. 4.1) with O=PMe3. Mixing the two
compounds in Et2O, and cooling the solution to –30 °C yielded pale yellow
crystals of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) in 57.9% yield.
(Eq. 4.1)
11 was characterized by 1H, 13C, 29Si, and 31P NMR and X-ray diffraction.
The 1H NMR resonance of the α-H atoms (1.30 ppm, benzene-d6) in the
–CH2SiMe3 ligands in 11 is down-field shifted from that (0.710 ppm) in its parent
complex (Me3SiCH2)3W≡CSiMe3 (4a). The 13C resonances of the α-C atoms (the
alkylidyne ≡CSiMe3 and alkyl -CH2SiMe3 ligands) in 11 at 327.39 and 69.56 ppm,
respectively, in benzene-d6, are both up-field shifted from those (343.67 and
SiMe3
CH2SiMe3Me3SiCH2
Me3SiCH2 W
C
O
PMe3114a
+ O=PMe3
SiMe3
CH2SiMe3Me3SiCH2
Me3SiCH2 W
C

92
75.85 ppm) in (Me3SiCH2)3W≡CSiMe3 (4a). The NMR shift differences of both α-
H and α-C atoms are perhaps a reflection of increased electron densities in 14e
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11), as a result of O=PMe3 coordination to
12e (Me3SiCH2)3W≡CSiMe3 (4a).
The 1H NMR resonances of the γ-H atoms in the -CH2SiMe3 and ≡CSiMe3
ligands in 11 at 0.533 and 0.303 ppm, respectively, are shifted down-field from
those (0.427 and 0.162 ppm) in 4a. The 13C resonances of the γ-C atoms at 3.26
and 2.19 ppm for -CH2SiMe3 and ≡CSiMe3 ligands in 11, respectively, are both
down-field shifted from those (2.32 and 2.15 ppm) in (Me3SiCH2)3W≡CSiMe3
(4a), although the resonances of the ≡CSiMe3 ligands in both complexes are
close. It is not clear why these resonances are down-field shifted.
The 31P NMR resonance of 11 at 41.95 ppm for the O=PMe3 ligand in 11
is down-field shifted from that (31.68 ppm) of O=PMe3. The 1H and 13C NMR
resonances of γ-H and γ-C atoms of the O=PMe3 ligands in 11 at 0.842 and
17.28 ppm, respectively, are both slightly up-field shifted from those (0.87 and
18.03 ppm) in O=PMe3. The 29Si NMR resonances of the β-Si atoms at –3.42
and –23.17 ppm for the –CH2SiMe3 and ≡CSiMe3 ligands in 14e 11, respectively,
are both up-field shifted from those (–0.837 and –17.57 ppm) in phosphine oxide-
free, 12e (Me3SiCH2)3W≡CSiMe3 (4a).
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) yielded crystals that were
suitable for X-ray diffraction studies. A representation of the molecular structure,
crystallographic refinement data, and selected bond distances and angles of 11

93
are given in Figure 4.1, and Tables 4.1 and 4.2, respectively.
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) adopts the pseudo trigonal bipyramidal
structure (Figure 4.1). The O=PMe3 ligand and the alkylidyne ≡CSiMe3 ligand
are in the axial positions with the C(4)-W(1)-O(1) angle of 179.5(3)°. The P(1)-
O(1)-W(1)-C(4)-Si(4) linkage is nearly linear. The W(4)≡C(4) bond distance of
1.763(8) Å is slightly longer than that [1.739(8) Å] in (Me3CCD2)3W≡CSiMe3,18
which will be discussed later. As expected, the C(4)-W(1)-C(n) (n = 1, 2, 3)
angles of 96.4(3)-97.3(4)° in the trigonal bipyramidal 11 are significantly smaller
than that [106.59(13)°] in the tetra-coordinated (Me3CCD2)3W≡CSiMe3.18 The
W(1)-C(n) (n = 1, 2, 3) bond distances of 2.097(7)-2.115(1) Å are similar to those
[2.096(5) Å] in (Me3CCD2)3W≡CSiMe3.18
It is interesting to note that the current reaction of d0
(Me3SiCH2)3W≡CSiMe3 (4a) with O=PMe3 yields the adduct 11. In comparison,
Wolczanski and coworkers recently found that, in the reaction of a d2 metal
complex with O=PR3, the oxygen atom transfers from the P atom to the metal
complex and oxidizes the metal (Eq. 4.2).35
(Eq. 4.2)
M(OSiBut)3
OM(OSiBut)3 + O=PR'3
M = Ta, V; R' = Me, Ph
+ PR'3

94
Figure 4.1. An ORTEP view of complex 11.

95
Table 4.1. Crystal data and structure refinement for
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11).
Compound No. 11
Empirical formula (Formula weight) C19H51O1P1Si4W1 (622.78)
Temperature −100(2) °C
Wavelength 0.71073 Å
Crystal system Orthorhombic
Space group Pnma
Unit cell dimensions a = 17.903(4) Å α = 90°
b = 10.516(2) Å β = 90°
c = 17.191(4) Å γ = 90°
Volume 3236.6(12) Å3
Z 4
Density (calculated) 1.278 g/cm3
Absorption coefficient 3.773 mm−1
F(000) 1272
Crystal size 0.30 × 0.30 × 0.20 mm3
θ range for data collection 2.25 to 28.34°
Index ranges -23 ≤ h ≤ 23, -13 ≤ k ≤ 13, -22 ≤ l ≤ 22
Reflections collected 32436
Independent reflections 7734 [Rint = 0.0526]
Completeness to θ = 28.34° 97.9%
Max. and min. transmission 0.5191 and 0.3973

96
Table 4.1. Continued
Compound No. 11
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 7734 / 1 / 250
Goodness-of-fit on F2 0.973
Final R indices [I > 2σ(I)] R1 = 0.0373, wR2 = 0.0746
R indices (all data) R1 = 0.0669, wR2 = 0.0866
Absolute structure parameter 0.023(11)
Largest diff. peak and hole 1.845 and −0.761 eÅ−3
a R1 = Σ||Fo| – | Fc|| / Σ|Fo|; wR2 = [Σ w(Fo2 – Fc
2)2 / Σ w(Fo2)2]1/2;
w = 1/[σ2(Fo) + (aP)2 + bP]; P = [2Fc2 + Max(Fo
2, 0)]/3
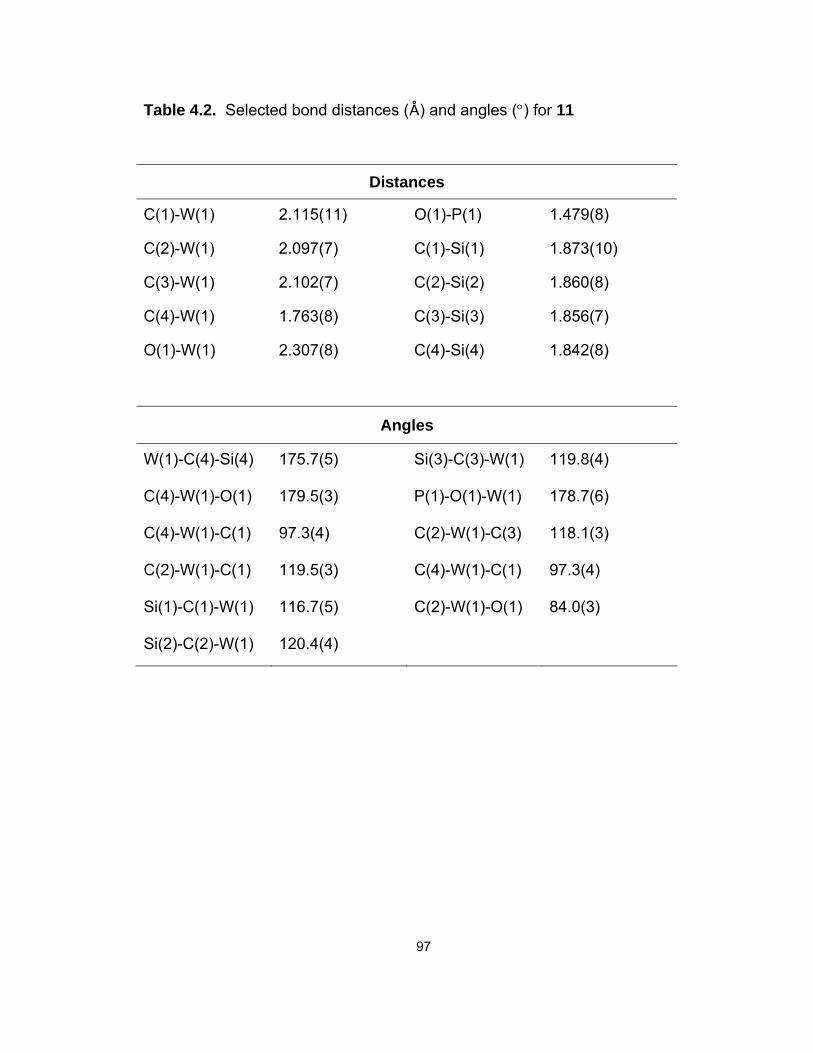
97
Table 4.2. Selected bond distances (D) and angles (°) for 11
Distances
C(1)-W(1) 2.115(11) O(1)-P(1) 1.479(8)
C(2)-W(1) 2.097(7) C(1)-Si(1) 1.873(10)
C(3)-W(1) 2.102(7) C(2)-Si(2) 1.860(8)
C(4)-W(1) 1.763(8) C(3)-Si(3) 1.856(7)
O(1)-W(1) 2.307(8) C(4)-Si(4) 1.842(8)
Angles
W(1)-C(4)-Si(4) 175.7(5) Si(3)-C(3)-W(1) 119.8(4)
C(4)-W(1)-O(1) 179.5(3) P(1)-O(1)-W(1) 178.7(6)
C(4)-W(1)-C(1) 97.3(4) C(2)-W(1)-C(3) 118.1(3)
C(2)-W(1)-C(1) 119.5(3) C(4)-W(1)-C(1) 97.3(4)
Si(1)-C(1)-W(1) 116.7(5) C(2)-W(1)-O(1) 84.0(3)
Si(2)-C(2)-W(1) 120.4(4)

98
4.2.2. Reaction of 11 with O2. Synthesis and Characterization of the
Tungsten Oxo Siloxy Complex O=W(OSiMe3)(CH2SiMe3)3 (12)
When (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) was exposed to O2, 11
was found to convert to O=W(OSiMe3)(CH2SiMe3)3 (12) containing a siloxy
(−OSiMe3) and an oxo ligand. A mass spectrometric (MS) analysis of the
gaseous products revealed the formation of CO2 in the reaction (Scheme 4.3),
which is discussed below. When (Me3SiCH2)3W≡CSiMe3 (4a) was exposed to O2
in the absence of O=PMe3, in comparison, decomposition of 4a was observed,
giving unknown species.
Addition of (Me3SiCH2)3W≡CSiMe3 (4a) to the reaction mixture and
crystallization at –30 °C gave crystals of
(Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3 (13) that were suitable for
X-ray diffraction studies. A representation of the molecular structure of 13, a
1 : 1 adduct between 12 and electron deficient 4a, is shown in Figure 4.2.
Crystallographic refinement data and selected bond distances and angles are
listed in Tables 4.3 and 4.4, respectively. 1H, 13C and 29Si NMR spectra of 13
showed the resonances of the –OSiMe3 ligand at 0.283, 1.99, and 10.74 ppm,
respectively. The 29Si NMR resonance of –O–SiMe3 at 10.74 ppm is down-field
shifted from those of –C≡W–CH2SiMe3 at –1.50 ppm, O=W−CH2SiMe3 at –2.97
ppm, and ≡CSiMe3 at –19.76 ppm in 13. The NMR resonances of
(Me3SiCH2)3(Me3SiC≡)W moiety in 13 were found to be slightly shifted from those
of (Me3SiCH2)3W≡CSiMe3 (4a).

99
Figure 4.2. An ORTEP view of complex 13.

100
Table 4.3. Crystal data and structure refinement for 13.
Compound No. 13
Empirical formula (Formula weight) C31H84O2Si8W2 (1081.40)
Temperature −100(2) °C
Wavelength 0.71073 Å
Crystal system Hexagonal
Space group R-3
Unit cell dimensions a = 16.1077(7) Å α = 90°
b = 16.1077(7) Å β = 90°
C = 68.397(4) Å γ = 120°
Volume 15368.5(13) Å3
Z 12
Density (calculated) 1.402 Mg/m3
Absorption coefficient 4.697 mm−1
F(000) 6552
Crystal size 0.29 × 0.25 × 0.15 mm3
θ range for data collection 1.58 to 22.50°
Index ranges -17 ≤ h ≤ 17, -17 ≤ k ≤ 17, -73 ≤ l ≤ 73
Reflections collected 30361
Independent reflections 4488 [R(int) = 0.0392]
Completeness to θ = 22.50° 99.9%

101
Table 4.3. Continued
Compound No. 13
Max. and min transmission 0.5393 and 0.3428
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 4488 / 0 / 275
Goodness-of-fit on F2 0.889
Final R indices [I > 2σ(I)] R1 = 0.0228, wR2 = 0.0559
R indices (all data) R1 = 0.0396, wR2 = 0.0706
Largest diff. peak and hole 1.533 and −0.458 eÅ−3
a R1 = Σ||Fo| – | Fc|| / Σ|Fo|; wR2 = [Σ w(Fo2 – Fc
2)2 / Σ w(Fo2)2]1/2;
w = 1/[σ2(Fo) + (aP)2 + bP]; P = [2Fc2 + Max(Fo
2, 0)]/3

102
Table 4.4. Selected bond distances (D) and angles (°) for 13
Distances
C(1)-W(1) 1.775(11) O(1)-W(1) 2.578(6)
C(3)-W(1) 2.100(5) O(2)-W(2) 1.924(6)
C(7)-W(2) 2.098(5) O(1)-W(2) 1.735(6)
O(2)-Si(4) 1.630(6) C(3)-Si(2) 1.872(6)
C(7)-Si(3) 1.895(5) C(1)-Si(1) 1.846(11)
C(2)-Si(1) 1.871(6) C(5)-Si(2) 1.852(6)
C(11)-Si(4) 1.851(6) C(10)-Si(3) 1.866(6)
Angles
W(1)-C(1)-Si(1) 180.0 Si(3)-C(7)-W(2) 119.0(3)
W(2)-O(1)-W(1) 180.0 Si(2)-C(3)-W(1) 123.7(3)
Si(4)-O(2)-W(2) 180.0 C(1)-W(1)-O(1) 180.000(1)
O(2)-W(2)-C(7) 87.67(14) O(1)-W(2)-O(2) 180.000(1)
C(1)-W(1)-C(3) 100.45(15)

103
In the reaction of O2 with d0 silyl alkylidyne (ButCH2)2W(≡CBut)(SiPh2But)
(2a), the silyl ligand migrates to the alkylidyne ligand to give an oxo alkylidene
(ButCH2)2W(=O)[=CR(SiPh2But)] (10).22a In contrast, in the reaction of O2 with d0
alkyl alkylidyne 11, the silyl moiety in the Me3SiC≡W ligand migrates to the newly
formed oxo ligand. These are two rare examples of the reactions of d0
alkylidynes with O2,1,10,11,34 and they demonstrate rich chemistry in the reactions
of d0 complexes with O2. Silicon is known to be oxophilic, and this perhaps
drives the unusual silyl migration in the reaction of 11 with O2. Theoretical
calculations, performed by Professor Yun-Dong Wu’s research group at the Hong
Kong University of Science and Technology, indicate that O=PMe3 mediates and
significantly lowers the activation of the migration.
The X-ray structure of 13 reveals a crystallographically imposed C3
symmetry through the C≡W←O=W-O-Si bonds, giving thus a linear W(2)-O(2)-
Si(4) linkage. 4a and 11 are bonded through a W=O→W dative bond [2.578(6)
D]. The W≡C bond distance of 1.775(11) D in 13 is longer than those in
(Me3CCD2)3W≡CSiMe3 [1.739(8) D]18 and 11 [1.763(8) D] (Table 4.5). In the
structures of both 11 and 13, the ≡CSiMe3 ligands are trans to oxygen atoms that
use their lone pair electrons to form dative bonds to the W atoms in (W←O). The
longer W≡C bond distances in 11 and 13 may reflect trans influence of the O
ligands.4 The C(1)-W(1)-C(3) bond angle of 100.45(15)E in 13 is smaller than
that [106.59(13)E] in (Me3CCD2)3W≡CSiMe3,18 as a result of the coordination of

104
Table 4.5. Bond distance comparison.18
Complex
C
ButCH2
ButCH2W
CH2But
SiMe3
CH2RRCH2
RCH2 W
C
R
O
PMe3
11
WRCH2
RCH2
O
O
SiMe3
CH2R
W CH2R
C
RCH2
RCH2
R
13
Bond Bond Distance (D)
W≡C 1.739(8) 1.763(8) 1.796(8)
W-C 2.096(5) 2.104(8) (avg) 2.100(5)
O→W 2.307(8) 2.572(5)

105
12 to (Me3SiCH2)3W≡CSiMe3 (4a). The W=O bond distance of 1.735(6) D in 13
is similar to those in other W oxo complexes.5b,36
4.2.3. HRMS Study of CO2 Produced from the Reaction of 11 with O2 and
Determination of Its Yield by 13CO2 Analysis
We devised a method to quantitatively determine the molar amount of CO2
produced during the reaction of 11 with O2. An HRMS analysis of the gaseous
products using 13CO2 as the calibration revealed the formation of CO2 (CO2 : 12 ≈
0.91 : 1). Given the yield of 12, it is not clear if CO2 is indeed a byproduct of the
formation of 12. It is, however, reasonable to assume that, in the oxidation of 11
to give 12, the alkylidyne C atom in (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) was
thus oxidized by 2 equiv of O2 from the former –437 to +4 oxidation state, and
removed as CO2.
4.2.4. Mass Spectrometric Studies of the Reaction of 11 with 18O2
Mass spectrometric studies were also carried out to determine, during the
reaction with 18O2, whether oxygen scrambling occurs between the O=PMe3
ligand and 18O2. MS analysis was performed to determine whether C18O16O,
C16O16O and/or C18O18O were present in the product mixture. The mass spectra
are shown in Figures 4.3 and 4.4. In the reaction of 11 with excess 18O2, an MS
analysis of the gaseous products revealed the formation of 16O=C=18O (25%)
and 18O=C=18O (75%), suggesting that the O atom in the O=PMe3 ligand in 11

106
was transferred to CO2 during the reaction. The observations of the 46 and 48
Dalton peaks in Figure 4.3 indicate that both 16O=12C=18O and 18O=12C=18O were
products from the reaction. Using the intensities of the 40 (40Ar) peaks in Figure
4.3 as reference, the ratio of the 46 (16O=12C=18O) and 48 (18O=12C=18O) Dalton
peaks in the sample (Figure 4.3, Bottom) is 1 : 3. Because the ratios of the 40
and 44 Dalton peak intensities in both sample (Bottom) and background (Top)
are similar, it is unlikely that 16O=12C=16O was yielded from the reaction. The
nature of the exchange(s) is not clear.
The mass spectra (30-38 Dalton region) of dioxygen isotopomers
(16O=16O; 16O=18O; 18O=18O) in the gaseous products and background prior to
the introduction of gases to the mass spectrometer are shown in Figure 4.4.
These spectra suggest that there was little 16O=16O or 16O=18O in the sample that
would have contributed to the formation of 16O=12C=18O.
4.2.5. Reaction of O2 with the Equilibrium Mixture of 3a º 3b
The first synthesis of 12 occurred serendipitously through unwanted
exposure to air during the reaction of 4a with PMe3. Complex 13 crystallized
from a solution containing 3a-b, 4a, PMe3, and solvent. Controlled experiments
later confirmed this reaction was reproducible in approximately 10% yield based
on 1H NMR. Scheme 4.4 shows both routes to form complex 13.

107
Figure 4.3. Mass spectra (38-51 Dalton region) of the isotopomers of carbon dioxide: (Top) The background prior to
the introduction of the gases into the spectrometer; (Bottom) The gaseous products from the reaction of 11 with 18O2.

108
Figure 4.4. Mass spectra of dioxygen isotopomers (16O=16O; 16O=18O; 18O=18O): (Top) The background spectrum;
(Bottom) The gaseous products from the reaction of 11 with 18O2. The 30-35 Dalton region was enlarged by 50 times.

109
WRCH2
RCH2
O
O
SiMe3
CH2R
W CH2R
C
RCH2
RCH2
R
13
W
CH2R
CRRCH2
RCH2
W
CH2R
CRRCH2
RCH2
PMe3
Keq
R
CH2RRCH2
RCH2W
C
O
PMe3
CHRRCH2
W
CH2R
RCH
PMe33b
4a
+ PMe3
+ O=PMe3
k1
k-1
3a
11
1. O2
2. 4a
1. O2
2. 4a
Scheme 4.4. Two pathways leading to 13.

110
4.3. Concluding Remarks
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11), an adduct between
(Me3SiCH2)3W≡CSiMe3 (4a) and O=PMe3, was found to react with O2 to give
O=W(OSiMe3)(CH2SiMe3)3 (3) and CO2. In the absence of O=PMe3, 12 was not
observed. 12 was isolated as an adduct with (Me3SiCH2)3W≡CSiMe3 (4a),
(Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3 (13), which was
characterized by X-ray crystallography.
4.4. Experimental Section
4.4.1. General Procedures
All manipulations were performed under a dry nitrogen atmosphere with
the use of either a glovebox or standard Schlenk techniques. Solvents were
purified by distillation from potassium benzophenone ketyl. NMR solvents were
dried and stored over 5 Å molecular sieves. NMR spectra were recorded on
Bruker AC-250 and AMX-400 Fourier transform spectrometers. 1H and 13C NMR
spectra were referenced to solvents (residual protons in the 1H spectra). 29Si
chemical shifts were referenced to SiMe4. Elemental analyses were carried out
by E+R Microanalytical Laboratory, Inc., Parsippany, New Jersey.
(Me3SiCH2)3W≡CSiMe3 (4a)15g was prepared from WCl3(OMe)3 and
ClMgCH2SiMe3 using a procedure similar to that for the preparation of
(Me3CCH2)3W≡CCMe3.15h O=PMe3 was purchased from Organometallics, Inc.
(East Hampstead, New Hampshire) and used as received. O2 (99.6% purity)

111
and 18O2 (>99% 18O, Isotec, Miamisburg, Ohio) were used as received. 13CO2
(99% 13C, <1% 18O) was purchased from Cambridge Isotope Laboratories, Inc.
(Andover, Massachusetts) and used as received.
4.4.2. Preparation of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11)
Solids of (Me3SiCH2)3W≡CSiMe3 (4a, 200 mg, 0.377 mmol) and O=PMe3
(34.7 mg, 0.377 mmol) were placed in a Schlenk flask, and were then dissolved
in Et2O (ca. 15 mL). The mixture was stirred until all O=PMe3 was dissolved in
ca. 10 min. Cooling the solution at −30 °C overnight gave crystals of 11 (136 mg,
0.218 mmol, 57.9% yield). 1H NMR (benzene-d6, 400.04 MHz, 23 °C, J in Hz) δ
1.30 (-CH2SiMe3), 0.842 (d, O=PMe3 2JP-H = 12.7), 0.533 (≡CSiMe3), 0.303
(-CH2SiMe3). 13C{1H} NMR (benzene-d6, 100.6 MHz, 23 °C) δ 327.39 (≡CSiMe3),
69.56 (CH2SiMe3), 17.38 (d, O=PMe3, 2JP-C = 70.4), 3.26 (-CH2SiMe3), 2.19
(≡CSiMe3). 31P{1H} NMR (benzene-d6, H3PO4 internal standard, 161.94 MHz, 23
°C) δ 41.95. 29Si{1H} NMR (benzene-d6) δ −23.17 (≡CSiMe3), −3.42 (CH2SiMe3).
Anal. Calcd. C 36.64, H 8.25. Found C 36.45, H 8.06.
4.4.3. Preparation of O=W(OSiMe3)(CH2SiMe3)3 (12) and
(Me3SiCH2)3W(≡CSiMe3)←O=W(OSiMe3)(CH2SiMe3)3 (13)
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11, 86.4 mg, 0.139 mmol) was
dissolved in toluene-d8 (ca. 0.5 mL) in a Youngs NMR tube. Three aliquots of
1000 mmHg (0.325 mmol) O2 were added over a period of 4 days, and product

112
formation was monitored by 1H NMR spectroscopy. The yield of 12 was ca. 30%
as estimated by NMR. When the reaction was completed, the solution was
transferred to a Schlenk flask, and all volatiles were removed in vacuo. The
remaining solid was dissolved in pentane and filtered into a solution of
(Me3SiCH2)3W≡CSiMe3 (4a) in pentane. Cooling the solution to –30 °C gave
crystals of (Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3 (13, 25.8 mg,
0.0238 mmol, 17.1%). 1H NMR (toluene-d8, 400.04 MHz, 23 °C) δ 1.640 (6H,
CH2−W=O; 2JW-H = 10.8 Hz), 0.697 (6H, CH2−W≡C; 2JW-H = 9.8 Hz), 0.403 (9H,
Me3SiC≡), 0.283 (9H, Me3Si-O), 0.241 (27H, Me3SiCH2-W=O), 0.160 (27H,
Me3SiCH2-W≡C). 13C{1H} NMR (toluene-d8, 100.6 MHz, 23 °C) δ 343.49
(Me3SiC≡), 75.77 (CH2W−), 62.90 (CH2W=O), 2.39 (Me3SiCH2W≡C), 1.58
(Me3SiCH2W=O), 2.07 (Me3SiC≡), 1.99 (O-SiMe3). 29Si{1H} NMR (toluene-d8,
79.49 MHz, 23 °C) δ 10.74 (Me3Si-O), −1.50 (Me3SiCH2-W≡C), −2.97 (Me3SiCH2-
W=O), −19.76 (Me3SiC≡). Anal. Calcd. for C31H84O2Si8W2: C 34.43, H 7.83, Si
20.78, P 0.00. Found: C 34.42, H 7.86, Si 21.03, P <0.02. The elemental
analysis of phosphorus was conducted to rule out that the –OSiMe3 ligand
observed in the X-ray crystal structure of 13 was O=PMe3.
In a separate study, (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11, 86.4 mg,
0.139 mmol) in toluene-d8 (ca. 0.5 mL) in a Youngs NMR tube was added O2.
Formation of O=W(OSiMe3)(CH2SiMe3)3 (12) was observed, and subsequent
analyses with mass spectrometry (MS) of the gaseous products were conducted.

113
4.4.4. Reaction of (Me3SiCH2)3W≡CSiMe3 (4a) with O2
(Me3SiCH2)3W≡CSiMe3 (4a, 50 mg, 0.094 mmol) in benzene–d6 (ca. 0.5
mL) in a Youngs NMR tube was frozen by liquid nitrogen and all volatiles were
removed from the NMR tube. O2 (1200 mmHg, ca. 0.1 mmol) was introduced
into the Youngs tube and allowed to react at 5 °C. The reaction was monitored
by 1H NMR. Only unidentifiable decomposition products and SiMe4 were
observed.
4.4.5. Reaction of O2 with the Equilibrium Mixture of
(Me3SiCH2)3W(≡CSiMe3)(PMe3) (3a) º (Me3SiCH2)2W(=CHSiMe3)2(PMe3) (3b)
O2 (1200 mmHg, 0.1 mmol) was added to an equilibrium mixture of 3a º
3b prepared from 33 mg of (Me3SiCH2)3W(≡CSiMe3) (4a) in excess PMe3 (0.062
mmol) in benzene-d6. The reaction was monitored by NMR, and the yield of
O=W(OSiMe3)(CH2SiMe3)3 (12) estimated by NMR was 10%.
4.4.6. Determination of X-ray Crystal Structures of 11 and 13
The X-ray crystal structures of 11 and 13 were determined on a Bruker
AXS Smart 1000 X-ray diffractometer equipped with a CCD area detector and a
graphite-monochromated Mo source (Kα radiation, 0.71073 Å) and fitted with an
upgraded Nicolet LT-2 low temperature device. Suitable crystals were coated
with paratone oil (Exxon) and mounted on a glass fiber under a stream of
nitrogen at –100(2) °C. The structures of 11 and 13 were solved by direct

114
method. All non-hydrogen atoms were anisotropically refined. Empirical
absorption correction was performed with SADABS. Global refinements for the
unit cells and data reductions were performed under the Saint program (Version
6.02). All calculations were performed using SHELXTL (Version 5.1) proprietary
software package.
4.4.7. Quantitative Determination of CO2 Yielded from the Reaction of
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with O2
In a Youngs NMR tube, (Me3SiCH2)3W≡CSiMe3 (4a, 66 mg, 0.12 mmol),
O=PMe3 (11.2 mg, 0.12 mmol), and 4,4’-dimethylbiphenyl (5.9 mg, 0.032 mmol,
internal standard) were dissolved in ca. 0.5 mL of toluene-d8. 1H NMR spectrum
of the solution revealed the nearly quantitative formation of
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11). The Youngs tube (headspace = 2.8
mL) was then attached to a gas manifold, and the solution was frozen in liquid
nitrogen, while most of the Youngs tube was immersed in liquid nitrogen. The N2
gas in the Youngs tube was removed in vacuo. Approximately 1.3 mmol of O2
(0.5 atm) was introduced into a gas manifold (65 mL). The valve on the Youngs
NMR tube was opened for approximately 5 sec to introduce O2 gas into the tube.
Since the boiling point of O2 (90.2 K) is high than that (77.4 K) of N2, O2 was
condensed into the Youngs tube. An HRMS analysis of the production mixture
after the reaction showed the presence of excess O2.
The solution was warmed to room temperature and shaken. 1H NMR
spectrum revealed the formation of O=W(OSiMe3)(CH2SiMe3)3 (12, 0.011 mmol,

115
9.4% yield based on 11). On a calibrated vacuum line with a volume of 170.7
mL, 13CO2 (0.0145 atm, 0.101 mmol) was introduced. The Youngs tube
containing the product, attached to the line previously, was opened and allowed
to mix with the 13CO2 gas in the manifold for approximately 15 min. The Youngs
tube was frozen by liquid nitrogen to condense the CO2 gas (13CO2 and 12CO2)
and then thawed. The tube was then frozen by liquid nitrogen, and was thawed.
This process was repeated for a total of 3 times to allow a good mixing of 13CO2
and 12CO2. The tube was frozen again and the NMR valve was closed. After the
tube was thawed, an HRMS analysis gave 13CO2 : 12CO2 = 100.00 : 10.14. Thus
the yield of CO2 from the reaction was 0.010 mmol, or ca. 91% of
O=W(OSiMe3)(CH2SiMe3)3 (12) from the reaction.
4.4.8. Determination of Isotopomers of CO2 from the Reaction of
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with 18O2
A Youngs NMR tube containing 11 (30 mg in toluene-d8) was frozen in
liquid nitrogen, and evacuated. Excess 18O2 (>99% 18O) was added, and the
NMR tube was warmed to room temperature. After the 1H NMR of the solution
indicated that the reaction was complete, the gaseous products were analyzed
by MS (Figures 4.3 and 4.4).

116
CHAPTER 5
Reactions of Tungsten Alkylidyne Complexes with Water
5.1. Introduction
Oxo ligands play a major role in the chemistry of metals, particularly the
high valent, hard early transition metals.38 With their large electronegativity, oxo
ligands are effective to bring out the high oxidation states. Many early-transition-
metal complexes are water-sensitive,4b,39 and their reactions with H2O usually
lead to the formation of oxo complexes.40-44 The nature of these reactions and
their applications in, for example, the formation of microelectronic metal oxide
materials45 have attracted much recent attention.
Reactivities of early-transition-metal organometallic complexes toward
water are of particular interest. Hillhouse and Bercaw reported that water reacts
in a clean, stepwise manner with d0 Cp*2MH2 (Cp* = C5Me5; M = Zr, Hf) to yield
Cp*2M(H)(OH), (Cp*MH)2O, Cp*2M(OH)2, and finally Cp*2M(OH)2.H2O, through
the conversion of M-H bonds to M-O bonds accompanied by H2 evolution.40
Rosenthal and coworkers studied the reaction of d2 Cp2Zr(THF)(η2-
Me3SiC≡CSiMe3) with H2O, and found it to yield the zirconoxane complex
[Cp2ZrC(SiMe3)=CH(SiMe3)]2O.41 Reactions of 1 equiv of H2O with alkylidene
complexes d0 Cp*2Ta(H)(=X) (X = CH2, C=CH2) yield XH2 (CH4, and CH2=CH2,
respectively) and Cp*2Ta(H)(=O).42 Schrock, Lippard, Osborn and coworkers
investigated the reactions of water with Group 6 neopentylidyne complexes, and

117
found that the reactions of H2O with d0 (ButCH2)3W≡CBut (1), (ButO)3W≡CBut,
and (ButCH2)2Mo(=CHBut)(=NBut) gave W2O3(CH2But)6,43a,44a
[NEt4][WO3(CH2But)],44a and [{Mo(=NBut)(CH2But)3}2(MoO4)],44b respectively, and
CMe4. Legzdins and coworkers reported that the reactions of H2O/O2 with d4
Cp*M(NO)(CH2SiMe3)2 (M = Mo, W) afford Cp*M(=O)2(CH2SiMe3).44c
We reported in Chapter 4 that the reaction of O2 with d0
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) surprisingly yields a siloxide
O=W(OSiMe3)(CH2SiMe3)3 (12). We have also found that the reactions of H2O
with (Me3SiCH2)3W≡CSiMe3 (4a), or 11 unexpectedly yielded CH4 and 12
(Scheme 5.1). As in the reaction of O2 with 11, one -SiMe3 group undergoes an
unusual migration in these reactions with H2O to give the -OSiMe3 ligand in
12.42,45 When 4a reacted with D2O, analysis of the methane isotopomers by
high-resolution mass spectrometry (HRMS) revealed the formation of CD4, CHD3
and CH2D2. Our studies of these reactions with water are presented.
5.2. Results and Discussion
5.2.1. Reaction of (Me3SiCH2)3W(≡CSiMe3) (4a) with H2O. Preparation of
O=W(OSiMe3)(CH2SiMe3)3 (12) and Study of the CH4 Formation
We were surprised to find that, when 3.4 equiv of H2O was added to
(Me3SiCH2)3W≡CSiMe3 (4a) in THF-d8, subsequent reaction afforded 12 in ca.
60% yield by NMR. HRMS analyses of the gaseous products using 13CH4 as the
calibration showed the formation of 1.7 equiv (excess) of CH4 (Figure 5.1).

118
R
CH2RRCH2 RCH2 W
C
2 H2O
CH4
O
PMe3
R
CH2RRCH2
RCH2 WC
CH4
2 H2O
CH2RRCH2
RCH2 W
O
OSiMe3
1112
4a
R = SiMe3
4a
CH2RRCH2
RCH2 W
O
OSiMe3
R
CH2RRCH2
RCH2 WC
13
Scheme 5.1. The different routes to form 12 through reactions of H2O.

119
Figure 5.1. MS data of volatile products in the reaction of 4a with H2O.

120
12 was also found to decompose to CH4 and unknown species in the presence of
excess H2O. It is thus likely that 12 reacted with the excess H2O to yield
additional CH4.
5.2.2. Reaction of (Me3SiCH2)3W(≡CSiMe3) (4a) with D2O
In order to probe the mechanistic pathways in the formation of 12 and
CH4, (Me3SiCH2)3W≡CSiMe3 (4a) in THF-d8 was added ca. 5 equiv of D2O
(99.9% D). An HRMS analysis of the methane isotopomers gave the following
ratios of CH4 : CH3D : CH2D2 : CHD3 : CD4 = 0 : 3 : 100 : 19 : 32(± 5)% (Figure
5.2, Table 5.1). If the observation of CH3D is ignored, as its HRMS signal is
below the estimated error of 5%, the observations of CH2D2 and CHD3 were
surprising. 2H NMR of the reaction of 4a with D2O revealed the formation of
O=W(OSiMe3)(CH2-nDnSiMe3)3 (12-dn). The mechanistic pathway in the
formation of 12 is not clear. In addition, 12 is likely decomposed by excess D2O
to give the methane isotopomers as well. However, the formation of CH2D2 and
CHD3, and the 2H NMR studies are consistent with suggested pathways in
Scheme 5.2 (Figure 5.3). The first step involves the addition of D–OD to the
W≡C– bond to give A. The migration of the second D atom in W−OD to the
W=CD− bond affords the tetraalkyl W oxo intermediate C. In addition to this
pathway, A apparently undergoes an unexpected α-hydrogen migration from a
W–CH2– to the W=CD– bond to yield B containing a W=CH– bond. Similar α-
hydrogen migrations have been reported.14,18,19c,22a,b Subsequent D migration

121
Figure 5.2. The HRMS mass spectrum for the methane isotopomers in the 15-
20 Dalton region. Isotopomers of methylene (CX2), methyl (CX3) and methane
(CX4) are given in blue, black, green and red colors, respectively. 16OH2 and
18OH2 and their fragments are omitted for clarity.

122
Table 5.1. List of HRMS peaks and their intensities in the 15-20 Dalton region.
Calculated Observed
Chemical Species
Mass #H #D Mass Rel. %
CHD 15.0219 1 1 15.0216 7.8 CH3 15.0235 3 0 15.0232 2.1 16O 15.9949 15.9949 1.3 CD2 16.0282 0 2 16.0282 9.6 CH2D 16.0298 2 1 16.0299 31.3 CH4 16.0313 4 0 16OH 17.0027 17.0027 2.0 CHD2 17.0360 1 2 17.0361 66.2 CH3D 17.0376 3 1 17.0378 3.1 16OH2 18.0106 18.0106 8.9 CD3 18.0423 0 3 18.0424 37.3 CH2D2 18.0439 2 2 18.0440 100.0 18OH 19.0070 19.0070 3.5 CHD3 19.0501 1 3 19.0501 19.3 18OH2 20.0148 20.0148 13.2 CD4 20.0564 0 4 20.0564 31.5

123
W CRRCH2
RCH2
RCH2
WRCH2
RCH2
CH2R
CD2R
O
W
CH2R
RCH2RCH2
CD2
OR
D2O
E F
W
CH2R
RCH2 CD2R
OR
CH2
D2O
C
CH2R
RCH2
OD
W CDRRCH2
A
G
D
D2O
H
WRCH
RCH2CH2R
CHDR
OD
B
D2O
CD4 CH2D2 CH2D2 CHD3
WRCDHRCH2
CH2R
CHDR
O
W
CH2R
RCDHCHDR
OR
CH2W
CH2R
RCDHRCH2
CHD
OR
+ D2O
4a
+ 12 + 12+ 12-d2 + 12-d2
W
CH2R
RCHD
RCH2
CHD2
OR
ODW
CH2R
RCDH
CH2D
CHDROR
ODW
CH2R
RCH2
CH2D
CD2ROR
ODW
CH2R
RCHD
RCH2
CD3
OR
OD
Scheme 5.2. Reaction of D2O with 4a and the formation of methane
isotopomers.
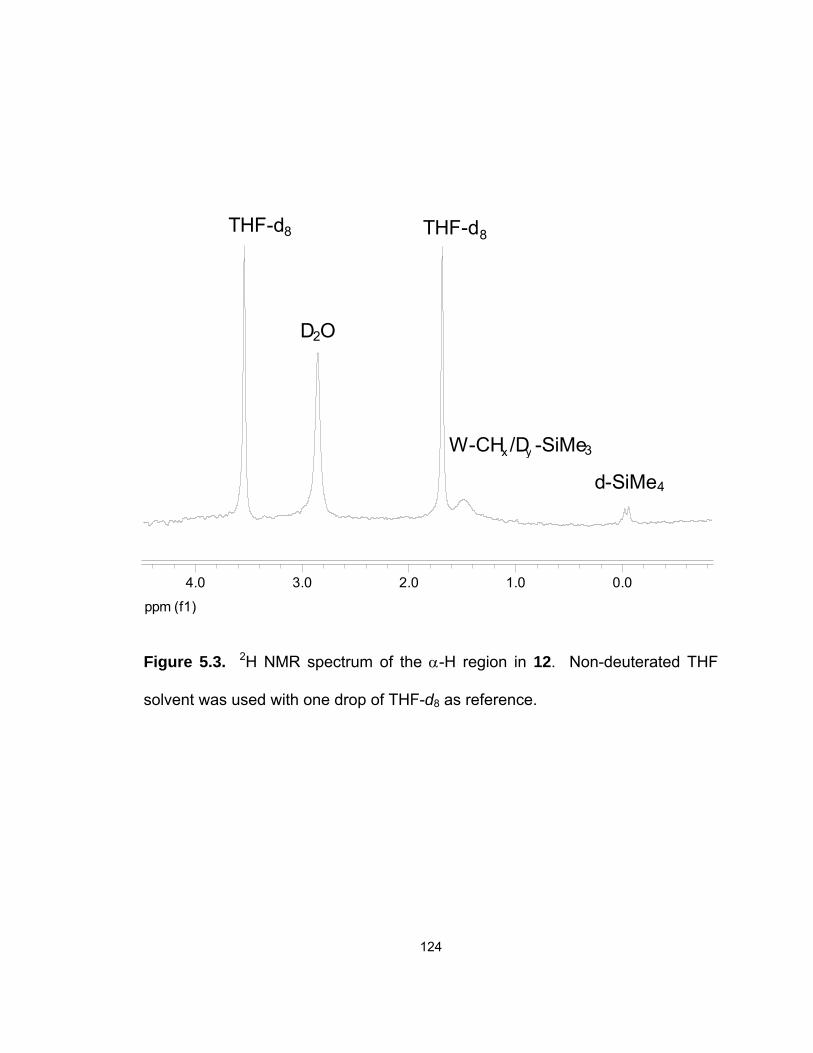
124
ppm (f1)
0.01.02.03.04.0
W-CH /D -SiMe
THF-d THF-d
D O
d-SiMe4
88
2
3x y
Figure 5.3. 2H NMR spectrum of the α-H region in 12. Non-deuterated THF
solvent was used with one drop of THF-d8 as reference.

125
from W−OD to the W=CH− bond yields D. The –SiMe3 group in one of these alkyl
ligands in C and D undergoes the C–Si bond cleavage and migration to the oxo
ligand to give E/F and G/H, respectively, containing a methylene ligand (W=CD2–
in E, W=CH2– in F/G, or W=CHD– in H), which subsequently reacts with D2O to
yield the methane isotopomers.
It is interesting to note that the reactions of H2O with (ButCH2)3W≡CBut (1),
(ButO)3W≡CBut, and (ButCH2)2Mo(=CHBut)(=NBut) give neopentane CMe4 and
oxo complexes W2O3(CH2But)6,43a,44a [NEt4][WO3(CH2But)],44a and
[{Mo(=NBut)(CH2But)3}2-(MoO4)],44b respectively. The alkylidene intermediate
(ButCH2)3W(=CDBut)(OD) in the reaction of 1 with D2O does not undergo α-
hydrogen migration.44a Reactions of H2O/O2 with d4 Cp*M(NO)(CH2SiMe3)2 (M =
Mo, W) afford Cp*M(=O)2(CH2SiMe3),44c and no –SiMe3 migration to the oxo
ligands was observed. It is not clear why the unusual –SiMe3 migration occurs in
the current reaction of 4a with H2O.
5.2.3. Reaction of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with H2O
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) was found to react with H2O to
give O=W(OSiMe3)(CH2SiMe3)3 (12). This reaction is, however, much slower
than that of phosphine oxide-free (Me3SiCH2)3W≡CSiMe3 (4a), the parent
compound of 11, with H2O. The reaction of 4a with H2O is completed in ca. 1
hour. In the reaction of 11 with 8 equiv of H2O, 1H NMR monitoring of the mixture

126
revealed that, after 5 days, only a small amount of 11 had reacted to give only
6% of 12. A large amount of 11 remained in the mixture.
It is not clear how water reacts with the phosphine oxide complex 11. At
least two pathways are possible: (a) direct attack of 11 by a water molecule,
while (Me3SiCH2)3W≡CSiMe3 (4a) is coordinated to the tungsten center; (b)
dissociation of the O=PMe3 from the metal center to give (Me3SiCH2)3W≡CSiMe3
(4a) which, as discussed earlier in this chapter, undergoes hydrolysis to give the
products. Ligands such as phosphine oxides coordinate to metal centers through
2e, dative bonds to give adducts. Such bonding is known to be reversible. In
other words, equilibria among the metal complexes, ligands and their adducts
such as that in Scheme 5.1 for 11, (Me3SiCH2)3W≡CSiMe3 (4a), and the adduct
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) slow the reaction to form complex 12.
If 11 reacts directly with H2O, the slowness of the reaction of 11 with H2O
is perhaps not surprising, given the fact that it is coordinated by a phosphine
oxide ligand. This coordination provides at least two electrons to the tungsten
center so it is less electron deficient and has less drive to coordinate to water.
The coordination also blocks the tungsten atom from attack by the water
molecule to begin the hydrolysis reaction.
A more likely pathway in the hydrolysis of 11 is that the O=PMe3 ligand
dissociates from 11 through the equilibrium in Scheme 5.3 to give 4a, which then
reacts with H2O to give 12. The kinetics of the O=PMe3 dissociation from 11 may
be slow, and this leads to the small overall rate of the reaction of 11 with H2O. In

127
R
CH2RRCH2 RCH2 W
C
2 H2O CH4
O
PMe3
R
CH2RRCH2
RCH2 WC
CH2RRCH2
RCH2 W
O
OSiMe3
11
12
4a
R = SiMe3
+O
PMe3
Scheme 5.3. Formation of 12 from the reaction of 11 with H2O.

128
addition, both O=PMe3 and H2O compete for the small amount of newly formed
4a, further reducing the yield of 12.
5.3. Concluding Remarks
The formation of the novel siloxide 12 demonstrates rich chemistry in the
reactions of d0 complexes with H2O. The presence of β-silicon atoms in 4a and
11 is a critical factor in the observed unique reactivities here. As in the reaction
of 11 with O2, the silyl migration in the reactions of 4a and 11 with H2O is perhaps
driven by the oxophilicity of silicon.
5.4. Experimental Section
5.4.1. General Procedures
All manipulations were performed under a dry nitrogen or argon
atmosphere with the use of either a glovebox or standard Schlenk techniques.
Solvents were purified by distillation from potassium benzophenone ketyl. NMR
solvents were dried and stored over 5 Å molecular sieves. NMR spectra were
recorded on Bruker AC-250 and AMX-400 Fourier transform spectrometers. 1H
and 13C NMR spectra were referenced to solvents (residual protons in the 1H
spectra). 29Si chemical shifts were referenced to SiMe4. (Me3SiCH2)3W≡CSiMe3
(4a) was prepared from WCl3(OMe)3 and ClMgCH2SiMe3 using a procedure
similar to that for the preparation of (Me3CCH2)3W≡CCMe3.15h 11 was prepared
by the procedure in Chapter 4. 13CH4 (99% 13C) was purchased from Cambridge
Isotope Laboratories, Inc. (Andover, Massachusetts) and used as received.

129
5.4.2. Preparation of (Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3
(13) via the Reaction of 4a with H2O.
While a solution of (Me3SiCH2)3W≡CSiMe3 (4a, 183 mg, 0.345 mmol) in
THF (ca. 5–10 mL) at room temperature was stirred, H2O (14 μL, 0.777 mmol)
was added via syringe. An immediate color change from yellow to reddish-
orange was observed upon addition of water. Stirring continued at room
temperature for approximately 1 hour. All volatiles were then removed in vacuo.
In a separate flask, (Me3SiCH2)3W≡CSiMe3 (4a, 183 mg, 0.345 mmol) was added
dropwise to the first Schlenk flask. The mixture was filtered, and the filtrate was
placed in the freezer at −30 °C to give crystals of
(Me3SiCH2)3(Me3SiC≡)W←O=W(OSiMe3)(CH2SiMe3)3 (13, 46.5 mg, 12.5%
yield). When the reaction was conducted in THF-d8 in a Youngs NMR tube, the
yield of 12 was ca. 60% as estimated by NMR. CH4 was observed in the MS
analysis of the gases in the headspace of the Youngs tube.
5.4.3. Quantitative Determination of CH4 Yielded from the Reaction of
(Me3SiCH2)3W≡CSiMe3 (4a) with H2O
In a Youngs NMR tube, (Me3SiCH2)3W≡CSiMe3 (4a, 105 mg, 0.197 mmol)
and 4,4’-dimethylbiphenyl (5.3 mg, 0.029 mmol) were dissolved in THF-d8 (0.4
mL). H2O (ca. 12 mg, 0.67 mmol) was added, and the NMR lid was closed
immediately. The NMR tube was placed in an NMR spectrometer, and the
reaction was monitored by 1H NMR spectroscopy. The reaction was essentially

130
complete within 30 min to give O=W(OSiMe3)(CH2SiMe3)3 (12) (0.118 mmol,
60% yield based on 4a). On a calibrated vacuum line with a volume of 170.7 mL,
0.0893 mmol of 13CH4 (0.0127 atm) was introduced. The Youngs tube containing
the product, attached to the line previously, was opened and allowed to mix with
the 13CH4 gas in the manifold for approximately 15 min. The Youngs tube was
frozen by liquid nitrogen to condense the CH4 gas (13CH4 and CH4) and then
thawed. The tube was then frozen by liquid nitrogen, and was thawed. This
process was repeated for a total of 3 times to allow a good mixing of 13CH4 and
12CH4. The tube was frozen again and the NMR lid was closed. After the tube
was thawed, an HRMS analysis gave a ratio of 13CH4 : 12CH4 was found to be
27.55 : 100.00. Thus the yield of CH4 from the reaction was 0.323 mmol, or ca.
164% yield from 4a. It was found in a separate test that the product 12 reacted
with H2O to yield CH4 as well. In other words, 1 equiv of 4a reacts with excess
H2O to yield at least 2 equiv of CH4. It is reasonable to assume that, in the
current case, the excess H2O (69%) reacted with the product 12, producing
additional CH4 and lowering the yield of 12.
5.4.4. Reaction of (Me3SiCH2)3W≡CSiMe3 (4a) with D2O
4a (30 mg, 0.057 mmol) in THF-d8 (ca. 0.5 mL) at room temperature was
transferred to a Youngs NMR tube. D2O (ca. 5 μL, 0.28 mmol) was added via
syringe in a glove box. An immediate color change from yellow to reddish-
orange was observed upon addition of D2O. 1H NMR spectra of the solution
confirmed formation of O=W(OSiMe3)(CH2-nDnSiMe3)3 (12-dn) in solution. A

131
separate 2D NMR study of this reaction is reported below. Studies with high-
resolution mass spectrometry (HRMS) were conducted shortly after D2O was
added.
5.4.5. 2H NMR Studies of the Reaction of (Me3SiCH2)3W≡CSiMe3 (4a) with
D2O
4a (50 mg, 0.094 mmol) in THF (ca. 0.5 mL) at room temperature was
transferred to a Youngs NMR tube. D2O (ca. 5 μL, 0.28 mmol) was added via
syringe in a glove box. A 2H NMR spectrum at −10 °C showed a broad peak at
1.55 ppm attributable to –CHDSiMe3 and –CD2SiMe3.
5.4.6. The Analysis of Volatile Products by Mass Spectrometry (MS) and
High-Resolution Mass Spectrometry (HRMS)
Electron-ionization mass spectrometry (EI-MS, 70 eV) was conducted on
a VG ZAB-EQ double focusing mass spectrometer for the analysis of CO2 and
CH4. In the HRMS studies, the instrument was tuned to a resolution (M/ΔM ≈
12000) sufficient to discriminate between the various isotopomers of CHxDy (x + y
= 4) and the EI-fragmentation products produced from them (Table 5.1). The
sample gas was introduced through a needle valve to produce a registered
source pressure of ca. 10−6 mbar. Vapor of H2O (ca. 90% 18O, 10% 16O) was
introduced through a separate inlet and used as the mass-calibration standard,
thus allowing assignment of peaks to the CHxDy isotopomers (Table 5.1).

132
5.4.7. Reaction of (Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11) with H2O
H2O (ca. 5 μL, 0.28 mmol) was added via syringe to
(Me3SiCH2)3(Me3SiC≡)W←O=PMe3 (11, 22 mg, 0.035 mmol) in toluene-d8 (ca.
0.5 mL) at room temperature in a glove box. The solution was monitored for 5
days by 1H NMR. A small amount of 11 had reacted to give 6% of the expected
O=W(OSiMe3)(CH2SiMe3)3 (12), and a large amount of unreacted 11 was
observed in solution.
5.4.8. Decomposition of 12 by H2O
12 was found to react with H2O and decompose to CH4 and other
unknown products. H2O (ca. 2 μL, 0.11 mmol) was added via syringe to a J. R.
Youngs NMR tube containing 4a (50 mg, 0.094 mmol) and the reaction mixture
was monitored by 1H NMR spectroscopy. The sample was checked to ensure all
starting material had reacted, and then all volatiles were removed in vacuo.
Another aliquot of H2O (ca. 2 μL, 0.11 mmol) was added to the reaction mixture
dissolved in THF-d8 in a J. R. Youngs NMR tube, and a 1H NMR spectrum was
taken to determine that 12 had fully decomposed to unknown products. The
sample was submitted for MS analysis and CH4 was found to be a product.

133
CHAPTER 6
Future Studies
Several new alkylidyne and bis(alkylidene) complexes have been
prepared and characterized. They were found to undergo unusual exchanges
and reactions with phosphines to give alkyl alkylidene alkylidyne complexes.
Alkylidyne complexes were found to react with O2 or H2O, leading to the
formation of a rare oxo siloxy complex.
Additional studies of the reactions of phosphines with alkylidyne
complexes are needed to understand the nature of the α-hydrogen migration and
abstraction reactions observed in Chapters 2 and 3, and to compare the
differences between –CH2CCMe3 and –CH2SiMe3 ligands. Isolation of
phosphine-oxide analogs of bis-phosphine, alkyl alkylidene alkylidyne complexes
8 and 9 might provide suitable crystals for elemental analysis and for the
determination of the structures of the alkyl alkylidene alkylidyne complexes
through X-ray crystallography. Studies with phosphines other than those used in
this dissertation could provide a more comprehensive picture of the nature of the
reactions reported in Chapters 2 and 3.
High-κ metal oxides have been used as microelectronic materials for
CVD. Additional studies with Group VI metal alkylidene and imido complexes
with oxygen could provide additional mechanistic information. Preliminary
studies revealed that the reaction of 4a or 3 with oxygen and water occurred to

134
give crystalline, trimeric complexes containing W(VI), bridging and terminal oxo
ligands, and alkyl ligands. Isolation and full characterization of these complexes
might provide insight into the nature of the reactions of alkylidyne complexes with
O2. Preliminary studies of the reaction of 4a with oxygen transfer agents, such
as pyridine-oxide and trimethylamine-oxide yield the siloxy complex 12. This
new reaction may provide a rich new area in which to study the nature of oxygen
atom transfer reactions involving alkylidyne complexes. Investigation of the
mechanism and kinetic studies would provide insight into the mechanism of
formation of 12.
An extension of the reactions of water with alkylidyne complexes may
involve their controlled reactions with primary alcohols. Preliminary
investigations revealed that ethanol reacted with 4a to give perhaps an alkylidene
product (based on the 1H NMR spectrum), and this reaction is worth pursuing.
Studies in this area might provide an avenue for precursors for microelectronic
metal oxide materials.

135
REFERENCES

136
1. (a) See, e.g., Metal-Dioxygen Complexes, Special Issue, Chem. Rev. 1994,
94, Issue 3. (b) Holm, R. H. Chem. Rev. 1987, 87, 1401.
2. (a) Fischer, E. O.; Maasbol. A. Angew. Chem., Int. Ed. 1964, 3, 580. (b)
Fischer, H., Kreissl, F. R., Schubert, U., Hofmann, P., Dotz, K. H., Weiss, K.
Transition Metal Carbene Complexes, 1984, VCH Publishers: Weinheim.
3. (a) Schrock, R. R. J. Organomet. Chem. 1986, 300, 249. (b) Schrock, R. R.
J. Am. Chem. Soc. 1974, 96, 6796. (c) Schrock, R. R.; Fellmann, J. D. J.
Am. Chem. Soc. 1978, 100, 3359.
4. (a) Elschenbroich, C.; Salzer, A. Organometallics: A Concise Introduction,
2nd ed, 1992, VCH Publishers: Weinheim, Chapter 14. (b) Crabtree, R. H.
The Organometallic Chemistry of the Transition Metals, 3rd ed.: Wiley, New
York, 2001.
5. See, e.g., (a) Schrock, R. R. Acc. Chem. Res. 1979, 12, 98. (b) Nugent, W.
A.; Mayer, J. M. Metal-Ligand Multiple Bonds; Wiley: New York, 1988. (c)
Schrock, R. R. Acc. Chem. Res. 1986, 19, 342. (d) Fischer H., Kreissl, F.
R., Schrock, R. R., Schubert, U., Hofmann, P., Weiss, K. Carbyne
Complexes, 1988, VCH Publishers: Weinheim.
6. Greenwood, N. N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.,
Butterworth and Heinemann, Oxford, UK, 1997.
7. (a) Simandi, L. I. Catalytic Activation of Dioxygen by Metal Complexes,
Kluwer: Amsterdam, 1992. (b) Sawyer, D. T. Oxygen Chemistry, Oxford
University Press: Oxford, UK, 1991. (c) Martell, A. E.; Sawyer, D. T.
Oxygen Complexes and Oxygen Activation by Transition Metals, Plenum:

137
New York, 1987. (d) Chanon, M.; Julliard, M.; Santamaria, J.; Chanon, F.
New J. Chem. 1992, 16, 171. (e) King, R. B. Encyclopedia of Inorganic
Chemistry, Vol. 2, Wiley: New York, 1994. (f) Pauling, L; Coryell, C. D.
Proc. Natl. Acad. Sci. USA, 1936, 22, 210.
8. (a) Frankland, E. Ann. 1853, 85, 347. (b) Frankland, E. J. Chem. Soc.
1862, 15, 363. (c) Demuth, R., Meyer, V. Ber. 1890, 23, 394.
9. (a) Zucchini, U.,Albizzati, E., Giannini, U. J. Organomet. Chem. 1971, 26,
357. (b) Brindley, P. B., Hodgson, J. C. J. Organomet. Chem. 1974, 65, 57.
10. (a) Blackburn, T. F.; Labinger, J. A.; Schwartz, J. Tetrahedron Lett. 1975,
16, 3041. (b) Labinger, J. A.; Hart, D. W.; Seibert, W. E.; Schwartz, J. J.
Am. Chem. Soc. 1975, 97, 3851. (c) Gibson, T. Organometallics 1987, 6,
918. (d) Brindley, P. B.; Scotton, M. J. J. Chem. Soc. Perkin II 1981, 419.
(e) Van Asselt, A.; Santarsiero, B. D.; Bercaw, J. E. J. Am. Chem. Soc.
1986, 108, 8291. (f) Coughlin, E. B.; Bercaw, J. E. Organometallics 1992,
11, 465. (g) Tilley, T. D. Organometallics 1985, 4, 1452.
11. (a) Lubben, T. V.; Wolczanski, P. T. J. Am. Chem. Soc. 1987, 109, 424. (b)
Lubben, T. V.; Wolczanski, P. T. J. Am. Chem. Soc. 1985, 107, 701. (c)
Wang, R.-J.; Folting, K.; Huffman, J. C.; Chamberlain, L. R.; Rothwell, I. P.
Inorg. Chim. Acta 1986, 120, 81. (d) Gibson, V. C.; Redshaw, C.; Walker,
G. L. P.; Howard, J. A. K.; Hoy, V. J.; Cole, J. M.; Kuzmina, L. G.; De Silva,
D. S. J. Chem. Soc., Dalton Trans. 1999, 161. (e) Brindley, P. B.; Hodgson,
J. C. J. Organomet. Chem. 1974, 65, 57. (f) Kim, S.-J.; Jung, I. N.; Yoo, B.
R.; Cho, S.; Ko, J.; Kim, S. H.; Kang, S. O. Organometallics 2001, 20, 1501.

138
(g) Schaverien, C. J.; Orpen, A. G. Inorg. Chem. 1991, 30, 4968. (h) Adam,
W.; Metz, M.; Prechtl, F.; Renz, M. Synthesis 1994, 563. (i) Adam, W.;
Putterlik, J.; Schuhmann, R. M.; Sundermeyer, J. Organometallics 1996, 15,
4586. See also, (j) Vetter, W. M.; Sen, A. Organometallics 1991, 10, 244.
(k) Van Asselt, A.; Trimmer, M. S.; Henling, L. M.; Bercaw, J. E. J. Am.
Chem. Soc. 1988, 110, 8254. (l) Mansilla, N.; Rheinwald, G.; Lang, H. J.
Organomet. Chem. 2000, 602, 72. (m) Schaverien, C. J. J. Chem. Soc.,
Chem. Comm. 1991, 458.
12. (a) Senzaki, Y.; Hamilton, R. F.; Reid, K. G.; Hobbs, C. C.; Hedge, R. I.;
Tiner, M. J. Mater. Res. Soc. Symp. Proc. 2000, 606, 13. (b) Son, K.-A.;
Mao, A. Y.; Sun, Y.-M.; Kim, B. Y.; Liu, F.; Kamath, A.; White, J. M.; Kwong,
D. L.; Roberts, D. A.; Vrtis, R. N. Appl. Phys. Lett. 1998, 72, 1187. (c) Niimi,
H.; Johnson, R. S.; Lucovsky, G.; Massoud, H. Z. Proc. Electrochem Soc.
2000, 2000-2, 487. (d) Chiu, H.-T.; Wang, C.-N.; Chuang, S.-H. Chem.
Vap. Deposition 2000, 6, 223.
13. Scott, J. F. Annu. Rev. Mater. Sci. 1998, 28, 79.
14. Morton, L. A.; Zhang, X-H.; Wang, R.; Lin, Z.; Wu, Y-D.; Xue, Z-L. J. Am.
Chem. Soc. 2004, 126, 10208.
15. (a) Clark, D. N.; Schrock, R. R. J. Am. Chem. Soc. 1978, 100, 6774. (b)
Schrock, R. R.; Clark, D. N.; Sancho, J.; Wengrovius, J. H.; Rocklage, S.
M.; Pedersen, S. F. Organometallics 1982, 1, 1645. (c) Mayr, A.;
Hoffmeister, H. Adv. Organomet. Chem. 1991, 32, 227. (d) Hua, F.; Mowat,
W.; Skapski, A. C.; Wilkinson, G. J. Chem. Soc., Chem. Commun. 1971,

139
1477. (e) Mowat, W.; Wilkinson, G. J. Chem. Soc., Dalton Trans. 1973,
1120. (f) Kim, H. P.; Angelici, R. J. Adv. Organomet. Chem. 1987, 27, 51.
(g) Andersen, R. A.; Chisholm, M. H.; Gibson, J. F.; Reichert, W. W.;
Rothwell, I. P.; Wilkinson, G. Inorg. Chem, 1981, 20, 3934. (h) Schrock, R.
R.; Sancho, J.; Pederson, S. F. Inorg. Synth. 1989, 26, 45.
16. (a) Schrock, R. R. J. Am. Chem. Soc. 1974, 96, 6796. (b) Schrock, R. R.;
Fellman, J. D. J. Am. Chem. Soc. 1978, 100, 3359. (c) Li, L.; Hung, M.;
Xue, Z. J. Am. Chem. Soc. 1995, 117, 12746. (d) Schrock, R. R. Acc.
Chem. Res. 1979, 12, 98. (e) Schrock, R. R. in Reactions of Coordinated
Ligands, P. S. Braterman, Ed.; Plenum: New York, 1986. (f) Advances in
Metal Carbene Chemistry, Schubert, U. Ed.; NATO ASI Series, Series C,
Vol. 269, 1989. (g) Aguero, A.; Osborn, J. A. New J. Chem. 1988, 12, 111.
(h) Schrock, R. R. Acc. Chem. Res. 1990, 23, 158. (i) Warren, T. H.;
Schrock, R. R.; Davis, W. M. J. Organomet. Chem. 1998, 569, 125. (j)
Feldman, J.; Schrock, R. R. Prog. Inorg. Chem. 1991, 39, 1.
17. (a) Rothwell, I. P. Polyhedron 1985, 4, 177. (b) Comprehensive
Organometallic Chemistry G. Wilkinson, F. G. A. Stone, and E. W. Abel,
Eds.; Pergamon: New York, 1982, Vol. 3, Ch. 25 by J. A. Labinger; Vol. 8,
Ch. 54 by R. H. Grubbs. (c) Comprehensive Organometallic Chemistry II,
E. W. Abel, F. G. A. Stone, and G. Wilkinson Eds.; Pergamon: New York,
1982, Vol. 5, Ch. 2 by D. E. Wigley and S. D. Gray; Ch. 5 by M. J. Winter
and S. Woodward.

140
18. (a) Caulton, K. G.; Chisholm, M. H.; Streib, W. E.; Xue, Z. J. Am. Chem.
Soc. 1991, 113, 6082. (b) Xue, Z.; Caulton, K. G.; Chisholm, M. H. Chem.
Mater. 1991, 3, 384. (c) Xue, Z.; Chuang, S.–H.; Caulton, K. G.; Chisholm,
M. H. Chem. Mater. 1998, 10, 2365.
19. (a) Edwards, D. S.; Biondi, L. V.; Ziller, J. W.; Churchill, M. R.; Schrock, R.
R. Organometallics 1983, 2, 1505. (b) LaPointe, A. M.; Schrock, R. R.
Organometallics 1995, 14, 1875. There is no H/D scrambling among the
neopentyl and neopentylidene ligands in Re(≡CBut)(=CHBut)(CD2But)2 at 80
°C in toluene-d8. (c) LaPointe, A. M.; Schrock, R. R.; Davis, W. M. J. Am.
Chem. Soc. 1995, 117, 4802.
20. (a) Schrock, R. R. Chem. Rev. 2002, 102, 145. (b) Schrock, R. R. Dalton
Trans. 2001, 2541. (c) Feldman, J.; Schrock, R. R. Prog. Inorg. Chem.
1991, 39, 1. (d) Aguero, A.; Osborn, J. A. New J. Chem. 1988, 12, 111. (e)
Mayr, A.; Hoffmeister, H. Adv. Organomet. Chem. 1991, 32, 227. (f) Kim, H.
P.; Angelici, R. J. Adv. Organomet. Chem. 1987, 27, 51. (g) Mowat, W.;
Wilkinson, G. J. Chem. Soc., Dalton Trans. 1973, 1120. (h) Andersen, R.
A.; Chisholm, M. H.; Gibson, J. F.; Reichert, W. W.; Rothwell, I. P.;
Wilkinson, G. Inorg. Chem. 1981, 20, 3934. (i) Li, L.; Hung, M.; Xue, Z. J.
Am. Chem. Soc. 1995, 117, 12746. (j) Xue, Z.; Li, L.; Hoyt, L. K.; Diminnie,
J. B.; Pollitte, J. L. J. Am. Chem. Soc. 1994, 116, 2169.
21. For dn type carbenes and carbynes, see, e.g., Refs. 5b, 5d, 15c and 15f,
and (a) Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18. (b)
Schrock, R. R.; Hoveyda, A. H. Angew. Chem, Inter. Ed. 2003, 42, 4592.

141
(c) Pollagi, T. P.; Manna, J.; Stoner, T. C.; Geib, S. J.; Hopkins, M. D. NATO
ASI Series C 1993, 392, 71. (d) Yong, B. S.; Nolan, S. P. Chemtracts 2003,
16, 205. (e) Doyle, M. P. Pure Appl. Chem. 1998, 70, 1123.
22. (a) Chen, T.; Wu, Z.; Li, L.; Sorasaenee, K. R.; Diminnie, J. B.; Pan, H.;
Guzei, I. A.; Rheingold, A. L.; Xue, Z. J. Am. Chem. Soc. 1998, 120, 13519.
(b) Choi, S.; Lin, Z.; Xue, Z. Organometallics 1999, 18, 5488. (c) Chen, T.-
N.; Zhang, X.-H.; Wang, C.-S.; Wu, Z.-Z.; Li, L.-T.; Sorasaenee, K. R.;
Diminnie, J. B.; Pan, H. J.; Guzei, I. A.; Rheingold, A. L.; Wu, Y.-D.; Xue, Z.-
L. Organometallics 2005, 24, 1214. (d) Wu, Z.; Cai, H.; Yu, Y.-X.; Blanton,
J. R.; Diminnie, J. B.; Pan, H.-J. Xue, Z.-L. Organometallics 2002, 21, 3973.
23. (a) Fellmann, J. D.; Schrock, R. R.; Rupprecht, G. A. J. Am. Chem. Soc.
1981, 103, 5752. (b) Fellmann, J. D.; Rupprecht, G. A.; Wood, C. D.;
Schrock, R. R. J. Am. Chem. Soc. 1978, 100, 5964. (c) Diminnie, J. B.;
Hall, H. D.; Xue, Z. J. Chem. Soc., Chem. Commun. 1996, 2383. (d)
Diminnie, J. B.; Xue, Z. J. Am. Chem. Soc. 1997, 119, 12657. (e) Diminnie,
J. B.; Blanton, J. R.; Cai, H.; Quisenberry, K. T.; Xue, Z. Organometallics
2001, 20, 1504.
24. Churchill, M. R.; Youngs, W. J. Inorg. Chem. 1979, 18, 1930.
25. The liquid sample of 5a-b was shipped overnight in an insulated container
with dry ice for elemental analysis. The sample was analyzed immediately
upon arrival. Repeated attempts to get elemental analysis of 5a-b afforded
the following data: Calcd. C 43.10; H 7.99; P 4.63; Found C 48.47, 48.53; H
7.79, 7.80; P 4.22, 4.18.

142
26. See, e.g., Espenson, J. H. Chemical Kinetics and Reaction Mechanism; 2nd
Ed., McGraw-Hill: New York, 1995, pp. 46-49.
27. Morse, P. M.; Spencer, M. D.; Wilson, S. R.; Girolami, G. S.
Organometallics 1994, 13, 1646.
28. (a) This technique utilizes phase cycling to suppress unwanted signals,
unlike other techniques which apply 13C broad-band decoupling. HMQC
demonstrates the sensitivity advantage of the inverse detection method. (b)
1H-gated-decoupled-13C experiments allow for full NOE-enhancement and
give a fully coupled 13C spectrum. (c) Braun, S.; Kalinowski, H.-O.; Berger,
S. 150 and More Basic NMR Experiments. A Practical Course. 2nd Ed.,
Wiley-VCH: Weinheim, 1998, pp. 384-387.
29. (a) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M.
A.; Cheeseman, J. R.; Montgomery, J. A.; Vreven, Jr., T.; Kudin, K. N.;
Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.;
Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.;
Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.;
Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.;
Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.;
Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.;
Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.;
Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A.
D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.;
Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski,

143
J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin,
R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.;
Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.;
Gonzalez, C.; Pople, J. A. Gaussian 03, Revision B.03, Gaussian, Inc.,
Pittsburgh PA, 2003. (b) Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82,
299. (c) Ehlers, A. W.; Böhme, M.; Dapprich, S.; Gobbi, A.; Höllwarth, A.;
Jonas, V.; Köhler, K. F.; Stegmann, R.; Veldkamp, A.; Frenking, G. Chem.
Phys. Lett. 1993, 208, 111. (d) Andrae, D.; Haeussermann, U.; Dolg, M.;
Stoll, H.; Preuss, H. Theor. Chim. Acta 1990, 77, 123.
30. (a) Murdzek, J. S.; Schrock, R. R. in Ref. 5d, pp. 155-164. (b) Schrock, R.
R.; Sancho, J.; Pederson, S. F. Inorg. Synth. 1989, 26, 44.
31. Diminnie, J. B.; Hall, H. D.; Xue, Z. Chem. Comm. 1996, 2383.
32. (a) Toreki, R.; Schrock, R. R. J. Am. Chem. Soc. 1992, 114, 3367. (b)
Toreki, R.; Vaughan, G. A.; Schrock, R. R.; Davis, W. M. J. Am. Chem. Soc.
1993, 115, 127.
33. Wood, C. D.; McLain, S. J.; Schrock, R. R. J. Am. Chem. Soc. 1979, 101,
3210.
34. (a) The Activation of Dioxygen and Homogeneous Catalytic Oxidation,
Barton, D. H. R., Martell, A. E.; Sawyer, D. T. Eds, Plenum: New York,
1993. (b) Theopold, K. H.; Reinaud, O. M.; Blanchard, S.;
Leelasubeharoen, S.; Hess, A.; Thyagarajan, S. ACS Symp. Ser. 2002, 823,
75. (c) Advances in Catalytic Activation of Dioxygen by Metal Complexes.
Catalysis by Metal Complexes, 2003, 26 (Simándi, L. I. Ed,). (d) Que, L.,

144
Jr.; Tolman, W. B. Angew. Chem. Inter. Ed. 2002, 41, 1114. (e) Metal
Catalyzed Oxidations of Organic Compounds, Sheldon, R. A.; Kochi, J. K.,
Academic Press: New York, 1981.
35. Veige, A. S.; Slaughter, L. M.; Lobkovsky, E. B.; Wolczanski, P. T.;
Matsunaga, N.; Decker, S. A.; Cundari., T. R. Inorg. Chem. 2003, 42, 6204.
36. See, e.g., (a) Feinstein-Jaffe, I.; Gibson, D.; Lippard, S. J.; Schrock, R. R.;
Spool, A. J. Am. Chem. Soc. 1984, 106, 6305. (b) Feng, S. G.; Luan, L.;
White, P.; Brookhart, M. S.; Templeton, J. L.; Young, C. G. Inorg. Chem.
1991, 30, 2582. (c) Blosch, L. L.; Abboud, K.; Boncella, J. M. J. Am. Chem.
Soc. 1991, 113, 7066.
37. The electronegativities of C and Si are 2.55 and 1.90, respectively.
38. Shriver, D.; Atkins, P. Inorganic Chemistry, 3rd ed., W. H. Freeman and Co.:
New York, pp. 296-297.
39. See also reviews on organometallics in aqueous solution: (a) Darensbourg,
D. J. in Aqueous-Phase Organometallic Catalysis, Cornils, B. and
Herrmann, W. A. Eds., 2nd ed.: Wiley-VCH, 2004. (b) Roundhill, D. M.
Adv. Organomet. Chem. 1995, 38, 155.
40. Hillhouse, G. L.; Bercaw, J. E. J. Am. Chem. Soc. 1984, 106, 5412.
41. Rosenthal, U.; Ohff, A.; Michalik, M.; Goerls, H.; Burlakov, V. V.; Shur, V. B.
Organometallics 1993, 12, 5016.
42. van Asselt, A.; Burger, B. J.; Gibson, V. C.; Bercaw, J. E. J. Am. Chem.
Soc. 1986, 108, 5347.
43. See, e.g., (a) Feinstein-Jaffe, I.; Gibson, D.; Lippard, S. J.; Schrock, R. R.;

145
Spool, A. J. Am. Chem. Soc. 1984, 106, 6305. (b) Feng, S. G.; Luan, L.;
White, P.; Brookhart, M. S.; Templeton, J. L.; Young, C. G. Inorg. Chem.
1991, 30, 2582. (c) Blosch, L. L.; Abboud, K.; Boncella, J. M. J. Am. Chem.
Soc. 1991, 113, 7066.
44. (a) Feinstein-Jaffe, I.; Pedersen, S. F.; Schrock, R. R. J. Am. Chem. Soc.
1983, 105, 7176. (b) Schoettel, G.; Kress, J.; Fischer, J.; Osborn, J. A. J.
Chem. Soc. Chem. Comm. 1988, 914. (c) Legzdins, P.; Phillips, E. C.;
Rettig, S. J.; Sanchez, L.; Trotter, J.; Yee, V. C. Organometallics 1988, 7,
1877.
45. See, e.g., (a) Lim, B. S.; Rahtu, A.; Gordon, R. G. Nature Mater. 2003, 2,
749. (b) Hausmann, D. M.; de Rouffignac, P.; Smith, A.; Gordon, R.;
Monsma, D. Thin Solid Films 2003, 443, 1. (c) Gordon, R. G.; Becker, J.;
Hausmann, D.; Suh, S. Chem. Mater. 2001, 13, 2463. (d) Smith, R. C.; Ma,
T.; Campbell, S. A.; Gladfelter, W. L. US Patent Application, 2003,
20030092287. (e) Baum, T. H.; Xu, C.; Hendrix, B. C.; Roeder, J. F. PCT
International Application 2002, WO 2002079211.

146
APPENDIX

147
Table A1. Atomic coordinates (× 104) and equivalent isotropic displacement
parameters (Å2 × 103) for 3b. U(eq) is defined as one third of the trace of the
orthogonalized Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________
W(1) 3333 6667 9210(1) 59(1)
P(1) 3333 6667 11286(4) 63(1)
C(1) 3840(20) 8228(18) 11950(20) 99(8)
C(2) 5036(11) 8174(11) 9337(15) 70(4)
Si(1) 6694(4) 8485(3) 9184(7) 65(1)
C(3) 7440(20) 9410(20) 7903(16) 103(8)
C(4) 7670(20) 9428(19) 10394(17) 105(8)
C(5) 6733(14) 6956(11) 9130(20) 112(6)
C(6) 2560(30) 6480(40) 7679(14) 69(10)
Si(2) 3333 6667 6288(4) 64(1)
C(7) 4730(20) 6450(30) 6460(30) 44(10)
C(8) 3910(40) 8350(20) 5840(50) 310(110)
C(9) 2260(30) 5570(40) 5210(30) 101(18)
________________________________________________________________

148
Table A2. Bond distances (Å) for 3b.
Bond Distance Bond Distance
W(1)-C(2)#1 1.964(12) Si(2)-C(9)#2 1.854(15)
W(1)-C(2) 1.964(12) Si(2)-C(9)#1 1.854(15)
W(1)-C(2)#2 1.964(12) Si(2)-C(9) 1.854(15)
W(1)-C(6)#2 2.041(18) Si(2)-C(6)#1 1.889(19)
W(1)-C(6)#1 2.041(19) Si(2)-C(6)#2 1.889(19)
W(1)-C(6) 2.041(18) Si(2)-C(8)#2 1.874(16)
W(1)-P(1) 2.514(5) Si(2)-C(8) 1.874(16)
P(1)-C(1)#1 1.86(2) Si(2)-C(8)#1 1.874(16)
P(1)-C(1)#2 1.86(2) Si(2)-C(7) 1.858(15)
P(1)-C(1) 1.86(2) Si(2)-C(7)#2 1.858(15)
C(2)-Si(1) 1.860(13) Si(2)-C(7)#1 1.858(15)
Si(1)-C(4) 1.871(13) C(7)-C(8)#1 0.84(7)
Si(1)-C(3) 1.861(13) C(7)-C(9)#2 1.62(6)
Si(1)-C(5) 1.879(10) C(7)-C(6)#2 1.98(4)
C(6)-C(6)#1 1.48(4) C(8)-C(7)#2 0.84(7)
C(6)-C(6)#2 1.48(4) C(8)-C(9)#1 1.06(7)

149
Table A2. Continued
Bond Distance Bond Distance
C(6)-Si(2) 1.889(19) C(9)-C(8)#2 1.06(7)
C(6)-C(7)#1 1.98(4) C(9)-C(7)#1 1.62(6)
Symmetry transformations used to generate equivalent atoms:
#1 -x+y,-x+1,z #2 -y+1,x-y+1,z

150
Table A3. Bond angles (°) in 3b.
Bond Angle Bond Angle
C(2)#1-W(1)-C(2) 119.39(15) C(6)#2-W(1)-C(6) 42.5(11)
C(2)-W(1)-C(2)#2 119.39(14) C(6)#1-W(1)-C(6) 42.5(11)
C(2)#1-W(1)-C(6)#2 71.1(8) C(2)#1-W(1)-P(1) 85.5(5)
C(2)-W(1)-C(6)#2 98.7(14) C(2)-W(1)-P(1) 85.5(5)
C(2)#2-W(1)-C(6)#2 112.7(11) C(2)#2-W(1)-P(1) 85.5(5)
C(2)#1-W(1)-C(6)#1 112.7(11) C(2)#1-W(1)-C(2)#2 119.39(15)
C(2)-W(1)-C(6)#1 71.1(8) C(6)#2-W(1)-P(1) 155.3(6)
C(2)#2-W(1)-C(6)#1 98.7(14) C(6)#1-W(1)-P(1) 155.3(6)
C(6)#2-W(1)-C(6)#1 42.5(11) C(6)-W(1)-P(1) 155.3(6)
C(2)#1-W(1)-C(6) 98.7(14) C(1)#1-P(1)-C(1)#2 102.8(9)
C(2)-W(1)-C(6) 112.7(12) C(1)#1-P(1)-C(1) 102.8(9)
C(2)#2-W(1)-C(6) 71.1(8) C(1)#2-P(1)-C(1) 102.8(9)
C(1)#1-P(1)-W(1) 115.6(7) C(6)#2-C(6)-W(1) 68.8(5)
C(1)#2-P(1)-W(1) 115.6(7) Si(2)-C(6)-W(1) 128.4(12)
C(1)-P(1)-W(1) 115.6(7) C(7)#1-C(6)-W(1) 143(2)
Si(1)-C(2)-W(1) 135.1(7) C(9)#2-Si(2)-C(9)#1 76(2)
C(2)-Si(1)-C(4) 109.5(12) C(9)#2-Si(2)-C(9) 76(2)
C(2)-Si(1)-C(3) 110.8(8) C(9)#1-Si(2)-C(9) 76(2)
C(4)-Si(1)-C(3) 108.5(6) C(9)#2-Si(2)-C(6) 160.5(16)

151
Table A3. Continued
Bond Angle Bond Angle
C(2)-Si(1)-C(5) 111.2(6) C(9)#1-Si(2)-C(6) 122(2)
C(4)-Si(1)-C(5) 108.0(7) C(9)-Si(2)-C(6) 114.5(15)
C(3)-Si(1)-C(5) 108.7(7) C(9)#2-Si(2)-C(6)#1 122(2)
C(6)#1-C(6)-C(6)#2 60.002(17) C(9)#1-Si(2)-C(6)#1 114.5(15)
C(6)#1-C(6)-Si(2) 66.9(6) C(9)-Si(2)-C(6)#1 160.5(16)
C(6)#2-C(6)-Si(2) 66.9(6) C(6)-Si(2)-C(6)#1 46.1(12)
C(6)#1-C(6)-C(7)#1 123.1(13) C(9)#2-Si(2)-C(6)#2 114.5(15)
C(6)#2-C(6)-C(7)#1 87(3) C(9)#1-Si(2)-C(6)#2 160.5(16)
Si(2)-C(6)-C(7)#1 57.4(9) C(9)-Si(2)-C(6)#2 122(2)
C(6)#1-C(6)-W(1) 68.8(5) C(6)-Si(2)-C(6)#2 46.1(11)
C(6)#1-Si(2)-C(6)#2 46.1(11) C(6)#1-Si(2)-C(8)#2 128(2)
C(9)#2-Si(2)-C(8)#2 108.8(10) C(6)#2-Si(2)-C(8)#2 106.1(19)
C(9)#1-Si(2)-C(8)#2 85(3) C(9)#2-Si(2)-C(8) 85(3)
C(9)-Si(2)-C(8)#2 33(2) C(9)#1-Si(2)-C(8) 33(2)
C(6)-Si(2)-C(8)#2 83(2) C(9)-Si(2)-C(8) 108.8(10)
C(6)-Si(2)-C(8) 106.1(19) C(9)#1-Si(2)-C(7)#2 51.7(19)
C(6)#1-Si(2)-C(8) 83(2) C(9)-Si(2)-C(7)#2 120.8(18)
C(6)#2-Si(2)-C(8) 128(2) C(6)-Si(2)-C(7)#2 79.9(18)
C(8)#2-Si(2)-C(8) 112.1(17) C(6)#1-Si(2)-C(7)#2 63.7(16)

152
Table A3. Continued
Bond Angle Bond Angle
C(9)#2-Si(2)-C(8)#1 33(2) C(6)#2-Si(2)-C(7)#2 108.9(12)
C(9)#1-Si(2)-C(8)#1 108.8(10) C(8)#2-Si(2)-C(7)#2 108.4(11)
C(9)-Si(2)-C(8)#1 85(3) C(8)-Si(2)-C(7)#2 26(2)
C(6)-Si(2)-C(8)#1 128(2) C(8)#1-Si(2)-C(7)#2 132.6(11)
C(6)#1-Si(2)-C(8)#1 106.1(19) C(7)-Si(2)-C(7)#2 118.8(4)
C(6)#2-Si(2)-C(8)#1 83(2) C(9)#2-Si(2)-C(7)#1 120.8(18)
C(8)#2-Si(2)-C(8)#1 112.1(18) C(9)#1-Si(2)-C(7)#1 109.9(10)
C(8)-Si(2)-C(8)#1 112.1(18) C(9)-Si(2)-C(7)#1 51.7(19)
C(9)#2-Si(2)-C(7) 51.7(19) C(6)-Si(2)-C(7)#1 63.7(16)
C(9)#1-Si(2)-C(7) 120.8(18) C(6)#1-Si(2)-C(7)#1 108.9(12)
C(9)-Si(2)-C(7) 109.9(10) C(6)#2-Si(2)-C(7)#1 79.9(18)
C(6)-Si(2)-C(7) 108.9(12) C(8)#2-Si(2)-C(7)#1 26(2)
C(6)#1-Si(2)-C(7) 79.9(18) C(8)-Si(2)-C(7)#1 132.6(11)
C(6)#2-Si(2)-C(7) 63.7(16) C(8)#1-Si(2)-C(7)#1 108.4(11)
C(8)#2-Si(2)-C(7) 132.6(11) C(7)-Si(2)-C(7)#1 118.8(4)
C(8)-Si(2)-C(7) 108.4(11) C(7)#2-Si(2)-C(7)#1 118.8(4)
C(8)#1-Si(2)-C(7) 26(2) C(8)#1-C(7)-C(9)#2 36(2)
C(9)#2-Si(2)-C(7)#2 109.9(10) C(8)#1-C(7)-Si(2) 78.0(18)
C(9)#2-C(7)-Si(2) 64.0(12) C(7)#2-C(8)-Si(2) 75.8(16)
153
Table A3. Continued
Bond Angle Bond Angle
C(8)#1-C(7)-C(6)#2 118(2) C(8)#2-C(9)-Si(2) 74.5(15)
C(9)#2-C(7)-C(6)#2 121.9(14) C(9)#1-C(8)-Si(2) 72.4(15)
Si(2)-C(7)-C(6)#2 58.9(11) C(8)#2-C(9)-C(7)#1 28(3)
C(7)#2-C(8)-C(9)#1 116(3) C(7)#1-C(9)-Si(2) 64.2(11)
Symmetry transformations used to generate equivalent atoms:
#1 -x+y,-x+1,z #2 -y+1,x-y+1,z

154
Table A4. Anisotropic displacement parameters (Å2 × 103) for 3b. The
anisotropic displacement factor exponent takes the form: -2π2[h2a*2U11 + ... +
2hka*b*U12].
________________________________________________________________
U11 U22 U33 U23 U13 U12
________________________________________________________________
W(1) 73(1) 73(1) 31(1) 0 0 36(1)
P(1) 78(2) 78(2) 34(2) 0 0 39(1)
C(2) 94(6) 86(7) 53(9) 44(8) 4(6) 62(5)
Si(1) 80(2) 26(1) 76(2) 8(3) -37(3) 16(2)
C(3) 71(11) 170(20) 82(8) 27(13) 5(9) 72(14)
C(4) 113(15) 59(11) 85(9) 1(9) -24(12) 1(11)
C(5) 75(9) 76(9) 196(18) -22(13) 42(19) 46(8)
C(6) 89(19) 120(30) 45(7) 16(19) 6(9) 80(20)
Si(2) 78(2) 78(2) 38(3) 0 0 39(1)
________________________________________________________________

155
Table A5. Hydrogen coordinates (× 104) and isotropic displacement parameters
(Å2 × 103) for 3b.
________________________________________________________________
x y z U(eq)
________________________________________________________________
H(1A) 3666 8107 12742 148
H(1B) 3362 8606 11623 148
H(1C) 4751 8797 11826 148
H(2A) 5003 8789 8821 84
H(2B) 5032 8503 10085 84
H(3A) 7355 10168 7888 154
H(3B) 7012 8876 7257 154
H(3C) 8343 9658 7889 154
H(4A) 7992 10336 10260 157
H(4B) 8382 9278 10498 157
H(4C) 7136 9165 11060 157
H(5A) 6198 6429 8524 168
H(5B) 6410 6497 9832 168
H(5C) 7610 7146 9020 168
H(6A) 2245 7074 7676 83
H(6B) 1811 5654 7676 83
H(7A) 4520 5623 6738 67
________________________________________________________________

156
Table A5. Continued
x y z U(eq)
________________________________________________________________
H(7B) 5135 6573 5749 67
H(7C) 5304 7092 6959 67
H(8A) 4274 8457 5119 469
H(8B) 3157 8413 5805 469
H(8C) 4518 8997 6326 469
H(9A) 2049 4752 5489 152
H(9B) 1499 5626 5140 152
H(9C) 2653 5697 4495 152
________________________________________________________________

157
Table A6. Atomic coordinates (× 104) and equivalent isotropic displacement
parameters (Å2 × 103) for 11. U(eq) is defined as one third of the trace of the
orthogonalized Uij tensor.
X y z U(eq)
C(1) 6791(7) 5740(9) 8323(6) 50(2)
C(2) 7870(4) 7663(6) 6970(4) 48(2)
C(3) 7984(5) 4246(6) 6889(5) 56(2)
C(4) 8409(4) 5930(7) 8145(4) 51(2)
C(5) 6139(6) 3123(9) 7884(5) 89(3)
C(6) 7377(5) 3208(11) 9043(7) 102(4)
C(7) 5852(10) 4203(13) 9481(8) 104(5)
C(8) 8060(14) 9386(19) 8347(13) 263(15)
C(9) 7763(10) 10518(11) 6801(11) 172(8)
C(10) 6532(6) 9112(9) 7591(18) 208(12)
C(11) 8370(10) 5343(18) 5306(7) 172(8)
C(12) 8958(10) 2804(12) 5753(9) 210(11)
C(13) 9593(7) 5070(20) 6514(9) 170(8)
C(14) 9809(5) 7248(10) 8707(6) 96(4)
C(15) 9674(6) 4367(12) 8850(7) 108(4)
C(16) 8732(7) 6051(12) 9883(5) 106(4)
C(17) 5951(11) 4353(13) 5591(8) 107(6)

158
Table A6. Continued
X y z U(eq)
C(18) 6042(6) 7053(12) 5408(7) 121(5)
C(19) 5138(6) 5994(10) 6564(6) 89(3)
O(1) 6657(5) 5842(7) 6637(5) 71(2)
P(1) 6010(1) 5829(2) 6103(1) 58(1)
Si(1) 6558(1) 4098(2) 8669(1) 58(1)
Si(2) 7549(1) 9158(2) 7442(5) 66(1)
Si(3) 8713(1) 4350(2) 6124(1) 64(1)
Si(4) 9152(1) 5915(3) 8886(1) 65(1)
W(1) 7654(1) 5895(1) 7488(1) 35(1)

159
Table A7. Bond distances (Å) for 11.
Bond Distance Bond Distance
C(1)-Si(1) 1.873(10) C(15)-Si(4) 1.878(11)
C(1)-W(1) 2.115(11) C(16)-Si(4) 1.878(11)
C(2)-Si(2) 1.860(8) C(17)-P(1) 1.788(12)
C(2)-W(1) 2.097(7) C(18)-P(1) 1.756(10)
C(3)-Si(3) 1.856(7) C(19)-P(1) 1.760(10)
C(3)-W(1) 2.102(7) O(1)-W(1) 2.307(8)
C(4)-Si(4) 1.842(8) O(1)-P(1) 1.479(8)
C(4)-W(1) 1.763(8) Si(1)-C(1)-W(1) 116.7(5)
C(5)-Si(1) 1.852(9) Si(2)-C(2)-W(1) 120.4(4)
C(6)-Si(1) 1.855(9) Si(3)-C(3)-W(1) 119.8(4)
C(7)-Si(1) 1.886(14) P(1)-O(1)-W(1) 178.7(6)
C(8)-Si(2) 1.820(18) W(1)-C(4)-Si(4) 175.7(5)
C(9)-Si(2) 1.846(13) C(14)-Si(4)-C(4) 109.7(4)
C(10)-Si(2) 1.840(10) C(14)-Si(4)-C(15) 109.5(6)
C(11)-Si(3) 1.856(13) C(2)-W(1)-C(1) 119.5(3)
C(12)-Si(3) 1.801(12) C(3)-W(1)-C(1) 118.3(3)
C(13)-Si(3) 1.871(14) C(4)-W(1)-O(1) 179.5(3)
C(14)-Si(4) 1.856(9)
Symmetry transformations used to generate equivalent atoms: #1 x, -y+1/2, z

160
Table A8. Bond angles (°) for 11.
Bond Angle Bond Angle
C(4)-Si(4)-C(15) 110.1(5) C(11)-Si(3)-C(3) 109.7(5)
C(14)-Si(4)-C(16) 110.3(5) C(12)-Si(3)-C(13) 106.6(9)
C(4)-Si(4)-C(16) 110.0(5) C(11)-Si(3)-C(13) 109.0(9)
C(15)-Si(4)-C(16) 107.2(5) C(3)-Si(3)-C(13) 111.1(5)
C(6)-Si(1)-C(5) 107.0(6) O(1)-P(1)-C(18) 113.0(5)
C(6)-Si(1)-C(1) 113.5(5) O(1)-P(1)-C(19) 114.5(5)
C(5)-Si(1)-C(1) 111.7(5) C(18)-P(1)-C(19) 105.3(6)
C(6)-Si(1)-C(7) 107.6(6) O(1)-P(1)-C(17) 111.1(7)
C(5)-Si(1)-C(7) 107.5(6) C(18)-P(1)-C(17) 107.7(7)
C(1)-Si(1)-C(7) 109.3(5) C(19)-P(1)-C(17) 104.7(7)
C(8)-Si(2)-C(10) 112.4(12) C(4)-W(1)-C(2) 96.4(3)
C(8)-Si(2)-C(9) 107.7(10) C(4)-W(1)-C(3) 96.6(4)
C(10)-Si(2)-C(9) 108.0(8) C(2)-W(1)-C(3) 118.1(3)
C(8)-Si(2)-C(2) 109.2(5) C(4)-W(1)-C(1) 97.3(4)
C(10)-Si(2)-C(2) 110.1(5) C(2)-W(1)-O(1) 84.0(3)
C(9)-Si(2)-C(2) 109.3(7) C(3)-W(1)-O(1) 83.5(3)
C(12)-Si(3)-C(11) 108.7(8) C(1)-W(1)-O(1) 82.2(3)
C(12)-Si(3)-C(3) 111.6(5)
Symmetry transformations used to generate equivalent atoms: #1 x, -y+1/2, z

161
Table A9. Anisotropic displacement parameters (Å2 × 103) for 11. The
anisotropic displacement factor exponent takes the form: -2π2[h2a*2U11 + ... +
2hka*b*U12].
U11 U22 U33 U23 U13 U12
C(1) 58(6) 46(5) 45(5) -8(4) 17(4) -2(4)
C(2) 49(4) 44(4) 49(4) 2(3) 3(3) -5(3)
C(3) 63(5) 43(4) 61(5) 0(3) 22(4) 2(3)
C(4) 44(4) 70(5) 39(4) 8(4) 4(3) -3(4)
C(5) 106(8) 75(6) 87(7) 15(5) 10(6) -32(6)
C(6) 86(7) 97(8) 123(10) 70(7) 22(6) 23(6)
C(7) 107(12) 109(10) 96(9) 1(8) 53(9) 0(8)
C(8) 310(30) 203(19) 280(20) -202(19) -150(20) 144(19)
C(9) 227(18) 41(6) 250(20) 40(9) 99(15) 8(8)
C(10) 72(7) 71(6) 480(40) -46(17) 100(20) 7(5)
C(11) 199(18) 246(19) 70(8) 60(10) 75(10) 82(15)
C(12) 290(20) 102(10) 233(18) -51(10) 218(18) -14(12)
C(13) 71(9) 320(20) 124(12) -40(16) 43(9) -55(13)
C(14) 67(6) 134(10) 86(7) 16(6) -20(5) -36(6)
C(15) 77(7) 163(12) 84(7) 43(8) 6(6) 47(7)
C(16) 114(9) 165(12) 40(5) 16(6) -20(5) -25(8)

162
Table A9. Continued
U11 U22 U33 U23 U13 U12
C(17) 124(14) 112(11) 86(8) -34(8) -32(9) -8(10)
C(18) 88(8) 166(12) 108(9) 77(9) -29(7) -37(8)
C(19) 73(7) 125(9) 68(6) -9(6) -4(5) 11(6)
O(1) 65(5) 85(5) 63(4) 5(4) -21(4) -10(4)
P(1) 50(1) 82(1) 44(1) 1(1) -9(1) -10(1)
Si(1) 54(1) 61(1) 58(1) 14(1) 19(1) -1(1)
Si(2) 70(1) 41(1) 86(2) 6(3) 22(2) 2(1)
Si(3) 71(2) 63(2) 58(1) -6(1) 31(1) 1(1)
Si(4) 50(1) 97(2) 47(1) 14(1) -10(1) -8(1)
W(1) 34(1) 38(1) 33(1) 0(1) 2(1) -1(1)

163
Table A10. Hydrogen coordinates (× 104) and isotropic displacement
parameters (Å2 × 103) for 11.
x y z U(eq)
H(1A) 6930 6247 8772 60
H(1B) 6342 6113 8105 60
H(2A) 8406 7729 6904 57
H(2B) 7654 7642 6452 57
H(3A) 8157 3640 7274 67
H(3B) 7541 3886 6651 67
H(5A) 6480 3075 7454 134
H(5B) 5680 3506 7715 134
H(5C) 6041 2282 8076 134
H(6A) 7216 2403 9245 153
H(6B) 7612 3690 9449 153
H(6C) 7727 3072 8628 153
H(7A) 5760 3369 9686 156
H(7B) 5394 4553 9284 156
H(7C) 6043 4741 9886 156
H(8A) 7714 9408 8774 395
H(8B) 8330 10173 8325 395
H(8C) 8404 8695 8419 395

164
Table A10. Continued
x y z U(eq)
H(9A) 8237 10875 6944 259
H(9B) 7381 11153 6855 259
H(9C) 7783 10234 6271 259
H(10A) 6386 9793 7931 311
H(10B) 6395 8313 7821 311
H(10C) 6283 9205 7100 311
H(11A) 8768 5873 5120 257
H(11B) 7964 5866 5482 257
H(11C) 8199 4802 4892 257
H(12A) 8525 2414 5526 315
H(12B) 9139 2283 6171 315
H(12C) 9341 2891 5366 315
H(13A) 9472 5775 6843 256
H(13B) 9899 5350 6090 256
H(13C) 9859 4439 6811 256
H(14A) 9981 7216 8179 144
H(14B) 10228 7177 9053 144
H(14C) 9558 8041 8798 144
H(15A) 9981 4344 8393 162
165
Table A10. Continued
x y z U(eq)
H(15B) 9324 3675 8836 162
H(15C) 9983 4290 9304 162
H(16A) 9115 6269 10251 159
H(16B) 8511 5253 10027 159
H(16C) 8355 6700 9882 159
H(17A) 6418 4184 5336 161
H(17B) 5560 4401 5209 161
H(17C) 5843 3681 5952 161
H(18A) 6092 7857 5667 181
H(18B) 5590 7047 5107 181
H(18C) 6462 6923 5070 181
H(19A) 5065 5301 6920 133
H(19B) 4749 5985 6180 133
H(19C) 5124 6783 6843 133

166
Table A11. Atomic coordinates (× 104) and equivalent isotropic displacement
parameters (Å2 × 103) for 13. U(eq) is defined as one third of the trace of the
orthogonalized Uij tensor.
________________________________________________________________
x y z U(eq)
________________________________________________________________
C(1) 6667 3333 1544(2) 43(3)
C(2) 6391(5) 4261(5) 1181(1) 63(2)
C(3) 7706(4) 4766(4) 1860(1) 35(1)
C(4) 8866(5) 5961(5) 1514(1) 51(2)
C(5) 9398(5) 4575(5) 1686(1) 58(2)
C(6) 9757(5) 6303(5) 1923(1) 58(2)
C(7) 7283(4) 2455(4) 2447(1) 32(1)
C(8) 9140(4) 3833(5) 2228(1) 49(2)
C(9) 8910(5) 2043(5) 2425(1) 64(2)
C(10) 9180(4) 3733(5) 2671(1) 51(2)
C(11) 7504(4) 4564(4) 3040(1) 48(2)
C(12) 4575(6) 7420(6) 1331(1) 89(3)
C(13) 2262(4) 5223(4) 727(1) 38(1)
C(14) 245(5) 3582(5) 694(1) 90(3)
C(15) 752(4) 5486(5) 508(1) 49(2)
C(16) 643(5) 5286(6) 951(1) 77(2)
________________________________________________________________

167
Table A11. Continued
x y z U(eq)
C(17) 3333 6667 9799(1) 30(2)
C(18) 4110(5) 7908(4) 9430(1) 50(2)
C(19) 2380(4) 5215(4) 10114(1) 34(1)
C(20) 3657(5) 4496(5) 9928(1) 52(2)
C(21) 1709(5) 3975(4) 9752(1) 52(2)
C(22) 1835(5) 3128(5) 10138(1) 64(2)
O(1) 6667 3333 2181(1) 33(2)
O(2) 6667 3333 2716(1) 31(2)
O(3) 3333 6667 464(1) 39(2)
O(4) 3333 6667 997(1) 50(2)
Si(1) 6667 3333 1274(1) 46(1)
Si(2) 8925(1) 5383(1) 1745(1) 37(1)
Si(3) 8637(1) 3032(1) 2443(1) 30(1)
Si(4) 6667 3333 2954(1) 29(1)
Si(5) 3333 6667 1242(1) 61(1)
Si(6) 970(1) 4908(1) 721(1) 45(1)
Si(7) 3333 6667 9528(1) 31(1)
Si(8) 2412(1) 4227(1) 9982(1) 37(1)

168
Table A11. Continued
x y z U(eq)
W(1) 6667 3333 1804(1) 26(1)
W(2) 6667 3333 2434(1) 21(1)
W(3) 3333 6667 717(1) 32(1)
W(4) 3333 6667 10056(1) 24(1)

169
Table A12. Bond distances (Å) for 13.
Bond Distance Bond Distance
C(1)-W(1) 1.775(11) C(1)-Si(1) 1.846(11)
C(2)-Si(1) 1.871(6) C(3)-Si(2) 1.872(6)
C(3)-W(1) 2.100(5) C(4)-Si(2) 1.862(6)
C(5)-Si(2) 1.852(6) C(6)-Si(2) 1.869(6)
C(7)-Si(3) 1.895(5) C(7)-W(2) 2.098(5)
C(8)-Si(3) 1.857(6) C(9)-Si(3) 1.856(6)
C(10)-Si(3) 1.866(6) C(11)-Si(4) 1.851(6)
C(12)-Si(5) 1.848(8) C(13)-Si(6) 1.880(6)
C(13)-W(3) 2.091(5) C(14)-Si(6) 1.862(7)
C(15)-Si(6) 1.853(6) C(16)-Si(6) 1.856(8)
C(17)-W(4) 1.756(9) C(17)-Si(7) 1.859(10)
C(18)-Si(7) 1.873(6) C(19)-Si(8) 1.854(6)
C(19)-W(4) 2.096(5) C(20)-Si(8) 1.866(6)
C(21)-Si(8) 1.863(6) C(22)-Si(8) 1.869(6)
O(1)-W(2) 1.735(6) O(1)-W(1) 2.578(6)
O(2)-Si(4) 1.630(6) O(2)-W(2) 1.924(6)
O(3)-W(3) 1.731(7) O(4)-Si(5) 1.676(8)
O(4)-W(3) 1.913(8) Si(1)-C(2)#1 1.871(6)
Si(1)-C(2)#2 1.871(6) Si(4)-C(11)#1 1.851(6)

170
Table A12. Continued
Bond Distance Bond Distance
Si(4)-C(11)#2 1.851(6) Si(5)-C(12)#3 1.848(8)
Si(5)-C(12)#4 1.848(8) Si(7)-C(18)#3 1.873(6)
Si(7)-C(18)#4 1.873(6) W(1)-C(3)#1 2.100(5)
W(1)-C(3)#2 2.100(5) W(2)-C(7)#2 2.098(5)
W(2)-C(7)#1 2.098(5) W(3)-C(13)#4 2.091(5)
W(3)-C(13)#3 2.091(5) W(4)-C(19)#4 2.096(5)
W(4)-C(19)#3 2.096(5)
Symmetry transformations used to generate equivalent atoms:
#1 -x+y+1,-x+1,z #2 -y+1,x-y,z #3 -y+1,x-y+1,z
#4 -x+y,-x+1,z

171
Table A13. Bond angles (°) for 13.
Bond Angle Bond Angle
W(1)-C(1)-Si(1) 180.0 Si(2)-C(3)-W(1) 123.7(3)
Si(3)-C(7)-W(2) 119.0(3) Si(6)-C(13)-W(3) 119.1(3)
W(4)-C(17)-Si(7) 180.000(1) Si(8)-C(19)-W(4) 123.1(3)
W(2)-O(1)-W(1) 180.0 Si(4)-O(2)-W(2) 180.0
Si(5)-O(4)-W(3) 180.000(1) C(1)-Si(1)-C(2)#1 110.0(2)
C(1)-Si(1)-C(2)#2 110.0(2) C(2)#1-Si(1)-C(2)#2 109.0(2)
C(1)-Si(1)-C(2) 110.0(2) C(2)#1-Si(1)-C(2) 109.0(2)
C(2)#2-Si(1)-C(2) 109.0(2) C(5)-Si(2)-C(4) 107.9(3)
C(5)-Si(2)-C(3) 114.2(3) C(4)-Si(2)-C(3) 108.6(3)
C(5)-Si(2)-C(6) 108.4(3) C(4)-Si(2)-C(6) 110.6(3)
C(3)-Si(2)-C(6) 107.1(3) C(8)-Si(3)-C(9) 109.7(3)
C(8)-Si(3)-C(10) 109.4(3) C(9)-Si(3)-C(10) 109.4(3)
C(8)-Si(3)-C(7) 110.4(3) C(9)-Si(3)-C(7) 106.8(3)
C(10)-Si(3)-C(7) 111.1(3) O(2)-Si(4)-C(11)#1 108.6(2)
O(2)-Si(4)-C(11) 108.6(2) C(11)#1-Si(4)-C(11) 110.3(2)
O(2)-Si(4)-C(11)#2 108.6(2) C(11)#1-Si(4)-C(11)#2 110.3(2)
C(11)-Si(4)-C(11)#2 110.3(2) O(4)-Si(5)-C(12)#3 109.2(3)
O(4)-Si(5)-C(12) 109.2(3) C(12)#3-Si(5)-C(12) 109.7(3)
O(4)-Si(5)-C(12)#4 109.2(3) C(12)#3-Si(5)-C(12)#4 109.7(3)

172
Table A13. Continued
Bond Angle Bond Angle
C(12)-Si(5)-C(12)#4 109.7(3) C(16)-Si(6)-C(15) 110.2(3)
C(16)-Si(6)-C(14) 109.8(4) C(15)-Si(6)-C(14) 109.5(3)
C(16)-Si(6)-C(13) 110.4(3) C(15)-Si(6)-C(13) 110.2(3)
C(14)-Si(6)-C(13) 106.7(3) C(17)-Si(7)-C(18)#3 110.9(2)
C(17)-Si(7)-C(18) 110.9(2) C(18)#3-Si(7)-C(18) 108.0(2)
C(17)-Si(7)-C(18)#4 110.9(2) C(18)#3-Si(7)-C(18)#4 108.0(2)
C(18)-Si(7)-C(18)#4 108.0(2) C(19)-Si(8)-C(21) 108.9(3)
C(19)-Si(8)-C(20) 112.7(3) C(21)-Si(8)-C(20) 110.3(3)
C(19)-Si(8)-C(22) 108.7(3) C(21)-Si(8)-C(22) 108.5(3)
C(20)-Si(8)-C(22) 107.6(3) C(1)-W(1)-C(3)#1 100.45(15)
C(1)-W(1)-C(3) 100.45(15) C(3)#1-W(1)-C(3) 116.79(9)
C(1)-W(1)-C(3)#2 100.45(15) C(3)#1-W(1)-C(3)#2 116.79(9)
C(3)-W(1)-C(3)#2 116.79(9) C(1)-W(1)-O(1) 180.000(1)
C(3)#1-W(1)-O(1) 79.55(15) C(3)-W(1)-O(1) 79.55(15)
C(3)#2-W(1)-O(1) 79.55(15) O(1)-W(2)-O(2) 180.000(1)
O(1)-W(2)-C(7) 92.33(14) O(2)-W(2)-C(7) 87.67(14)
O(1)-W(2)-C(7)#2 92.33(14) O(2)-W(2)-C(7)#2 87.67(14)
C(7)-W(2)-C(7)#2 119.84(2) O(1)-W(2)-C(7)#1 92.33(14)
O(2)-W(2)-C(7)#1 87.67(14) C(7)-W(2)-C(7)#1 119.84(2)

173
Table A13. Continued
Bond Angle Bond Angle
C(7)#2-W(2)-C(7)#1 119.84(2) O(3)-W(3)-O(4) 180.000(1)
O(3)-W(3)-C(13)#4 91.82(17) O(4)-W(3)-C(13)#4 88.18(17)
O(3)-W(3)-C(13) 91.82(17) O(4)-W(3)-C(13) 88.18(17)
C(13)#4-W(3)-C(13) 119.900(18) O(3)-W(3)-C(13)#3 91.82(17)
O(4)-W(3)-C(13)#3 88.18(17) C(13)#4-W(3)-C(13)#3 119.900(19)
C(13)-W(3)-C(13)#3 119.900(19) C(17)-W(4)-C(19)#4 100.96(15)
C(17)-W(4)-C(19) 100.96(15) C(19)#4-W(4)-C(19) 116.47(10)
C(17)-W(4)-C(19)#3 100.96(15) C(19)#4-W(4)-C(19)#3 116.47(10)
C(19)-W(4)-C(19)#3 116.47(9)
Symmetry transformations used to generate equivalent atoms:
#1 -x+y+1,-x+1,z #2 -y+1,x-y,z #3 -y+1,x-y+1,z
#4 -x+y,-x+1,z

174
Table A14. Anisotropic displacement parameters (Å2 × 103) in 13. The
anisotropic displacement factor exponent takes the form: -2π2[h2a*2U11 + ... +
2hka*b*U12].
________________________________________________________________
U11 U22 U33 U23 U13 U12
________________________________________________________________
C(1) 26(4) 26(4) 76(8) 0 0 13(2)
C(2) 81(6) 73(5) 39(4) 18(3) 0(4) 41(5)
C(3) 41(4) 28(3) 37(3) -1(2) 1(3) 18(3)
C(4) 57(5) 49(4) 47(4) 17(3) 15(3) 27(4)
C(5) 51(5) 67(5) 67(5) 16(4) 22(3) 38(4)
C(6) 50(5) 43(4) 70(5) 9(3) -2(3) 15(4)
C(7) 32(3) 29(3) 33(3) -2(2) -2(2) 15(3)
C(8) 40(4) 56(4) 47(4) 11(3) 11(3) 20(4)
C(9) 42(4) 50(5) 109(6) 7(4) 10(4) 30(4)
C(10) 33(4) 63(5) 42(4) -3(3) -6(3) 15(3)
C(11) 48(4) 44(4) 44(4) -5(3) -5(3) 18(3)
C(12) 85(7) 97(7) 80(6) -11(5) -26(5) 41(6)
C(13) 28(3) 27(3) 57(4) 1(3) -3(3) 12(3)
C(14) 36(5) 33(5) 185(9) 15(5) -15(5) 6(4)
C(15) 32(4) 46(4) 68(4) 2(3) -1(3) 19(3)
C(16) 54(5) 96(7) 80(6) 17(5) 16(4) 37(5)

175
Table A14. Continued
U11 U22 U33 U23 U13 U12
C(17) 24(3) 24(3) 41(6) 0 0 12(2)
C(18) 50(4) 45(4) 48(4) 13(3) 8(3) 19(4)
C(19) 28(3) 25(3) 41(3) 4(2) 1(2) 9(3)
C(20) 57(5) 53(4) 60(4) -9(3) -5(3) 39(4)
C(21) 60(5) 36(4) 45(4) -2(3) -9(3) 14(4)
C(22) 91(6) 36(4) 52(4) 4(3) -7(4) 23(4)
O(1) 28(2) 28(2) 41(4) 0 0 14(1)
O(2) 28(2) 28(2) 39(4) 0 0 14(1)
O(3) 26(2) 26(2) 64(5) 0 0 13(1)
O(4) 41(3) 41(3) 68(5) 0 0 20(2)
Si(1) 55(1) 55(1) 28(2) 0 0 27(1)
Si(2) 33(1) 32(1) 42(1) 10(1) 6(1) 14(1)
Si(3) 26(1) 34(1) 34(1) 2(1) 1(1) 17(1)
Si(4) 31(1) 31(1) 27(1) 0 0 15(1)
Si(5) 64(2) 64(2) 55(2) 0 0 32(1)
Si(6) 25(1) 34(1) 70(1) 10(1) 5(1) 10(1)
Si(7) 32(1) 32(1) 30(2) 0 0 16(1)
Si(8) 45(1) 24(1) 36(1) 0(1) -2(1) 14(1)

176
Table A14. Continued
U11 U22 U33 U23 U13 U12
W(1) 25(1) 25(1) 28(1) 0 0 13(1)
W(2) 20(1) 20(1) 24(1) 0 0 10(1)
W(3) 23(1) 23(1) 49(1) 0 0 11(1)
W(4) 20(1) 20(1) 32(1) 0 0 10(1)

177
Table A15. Hydrogen coordinates (× 104) and isotropic displacement
parameters (Å2 × 103) in 13.
x y z U(eq)
H(2A) 5774 4123 1228 95
H(2B) 6387 4253 1041 95
H(2C) 6871 4882 1226 95
H(3A) 7413 5148 1827 42
H(3B) 7808 4818 2000 42
H(4A) 9477 6247 1449 76
H(4B) 8706 6448 1543 76
H(4C) 8385 5488 1430 76
H(5A) 8974 4093 1595 87
H(5B) 9443 4273 1803 87
H(5C) 10022 4939 1628 87
H(6A) 9766 5994 2042 87
H(6B) 9542 6750 1949 87
H(6C) 10391 6638 1869 87
H(7A) 7055 2075 2565 38
H(7B) 7034 2013 2338 38
H(8A) 9819 4076 2221 74

178
Table A15. Continued
x y z U(eq)
H(8B) 9021 4358 2240 74
H(8C) 8842 3479 2111 74
H(9A) 8626 1678 2309 96
H(9B) 8655 1634 2538 96
H(9C) 9592 2307 2421 96
H(10A) 9864 4018 2665 76
H(10B) 8938 3314 2782 76
H(10C) 9016 4227 2683 76
H(11A) 8105 4810 2973 71
H(11B) 7604 4553 3179 71
H(11C) 7236 4969 3014 71
H(12A) 4981 7215 1271 133
H(12B) 4589 7359 1470 133
H(12C) 4799 8078 1298 133
H(13A) 2367 4902 618 46
H(13B) 2357 4950 846 46
H(14A) 378 3283 802 135
H(14B) 407 3392 574 135

179
Table A15. Continued
x y z U(eq)
H(14C) -424 3387 694 135
H(15A) 94 5330 507 74
H(15B) 890 5258 389 74
H(15C) 1160 6168 516 74
H(16A) 1003 5972 961 115
H(16B) 786 5006 1060 115
H(16C) -30 5075 950 115
H(18A) 3822 8291 9458 75
H(18B) 4176 7879 9291 75
H(18C) 4731 8189 9490 75
H(19A) 2446 5119 10252 40
H(19B) 1739 5117 10097 40
H(20A) 3972 5057 9847 78
H(20B) 3635 3962 9860 78
H(20C) 4006 4606 10048 78
H(21A) 1038 3672 9782 77
H(21B) 1825 3558 9670 77
H(21C) 1901 4566 9684 77

180
Table A15. Continued
x y z U(eq)
H(22A) 2172 3256 10260 96
H(22B) 1857 2613 10072 96
H(22C) 1180 2950 10163 96

181
VITA
Laurel Anne Morton (maiden name: Laurel Anne Ray) was born on July
12th, 1977 in Evansville, Indiana, to parents Anne Laurel and John Michael Ray.
Her father was a high school biology and chemistry teacher, football coach, and
later a public health and safety consultant; her mother was a high school
mathematics teacher. She grew up on a small cattle farm in Spring City,
Tennessee, with her three sisters. At age 10, she moved to Chattanooga,
followed four years later by a move to Knoxville, Tennessee, where she attended
Farragut High School and enjoyed her drafting, music and science classes. After
graduation in May 1995, she enrolled at Tennessee Technological University in
Cookeville, Tennessee, the next fall semester. During her first semester at
Tennessee Tech, she met her future husband, colleague and best friend, Samuel
A. Morton III. She studied chemistry at Tennessee Tech, and conducted her
undergraduate research on synthesis of ionic liquids and their use in the
extraction of radioactive metal ions from waste water under the direction of
Professors Dale Ensor and Dan Swartling. She presented this work at the
American Chemical Society (ACS) National Meeting in Anaheim, California, in
March 1999. While at Tennessee Tech, she was awarded the F. U. Foster
Memorial Chemistry Scholarship for her excellence in the ACS curriculum. She
graduated in May 1999 with a Bachelor of science degree in chemistry and
began her Ph.D. studies the next fall in the chemistry department at the
University of Tennessee, Knoxville. On July 7th, 2001, she married Samuel

182
Morton III in Knoxville, Tennessee surrounded by family and friends. She studied
the synthesis and characterization of novel early-transition-metal complexes and
reactions of alkylidyne complexes with oxygen and water under the direction of
Professor Zi-Ling (Ben) Xue. While at the University of Tennessee, she was
elected by the graduate student body to the position of Vice-President of the
Graduate Student Senate. As a fourth year student, Laurel received a
departmental outstanding teaching award. In April 2005, she received the
Chancellor’s Award for Academic Achievement and Chancellor’s Award for
Professional Promise.