trichostatin a increases the levels of plasma gelsolin and amyloid beta-protein in a transgenic...
TRANSCRIPT

Life Sciences 99 (2014) 31–36
Contents lists available at ScienceDirect
Life Sciences
j ourna l homepage: www.e lsev ie r .com/ locate / l i fesc ie
Trichostatin A increases the levels of plasma gelsolin and amyloidbeta-protein in a transgenic mouse model of Alzheimer's disease
Wenzhong Yang, Abha Chauhan, Sangita Mehta, Pankaj Mehta, Feng Gu, Ved Chauhan ⁎NYS Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA
⁎ Corresponding author at: NYS Institute for BasicDisabilities, 1050 Forest Hill Road, Staten Island, NY 1031fax: +1 718 698 7916.
E-mail address: [email protected] (V. Chau
0024-3205/$ – see front matter © 2014 Elsevier Inc. All rihttp://dx.doi.org/10.1016/j.lfs.2014.01.064
a b s t r a c t
a r t i c l e i n f oArticle history:
Received 19 September 2013Accepted 16 January 2014Keywords:Alzheimer's diseaseAmyloid beta-proteinGelsolinHistone deacetylaseTransgenic miceTrichostatin A
Aims: Gelsolin (GSN), a multifunctional protein, binds to amyloid beta-protein (Aβ), inhibits its fibrillization,solubilizes preformed Aβ fibrils, and helps in its clearance from the brain. Trichostatin A (TSA), a histonedeacetylase (HDAC) inhibitor, induces the protein expression of gelsolin. In the present study, we investigatedhow TSA-treatment of APPswe/PS1δE9 transgenic (Tg) mice of Alzheimer's disease (AD) will affect the plasmalevels of gelsolin and Aβ.Mainmethods: TSA (5 mg/kg bodyweight on alternate days for twomonths)was intraperitoneally injected toADTg mice. Gelsolin was measured by Western blotting and Aβ was measured by enzyme-linked immunosorbentassay.Key findings: TSA-treatment significantly increased the levels of plasma gelsolin by 1.79-fold as compared withvehicle-treated control mice (p b 0.01). The levels of Aβ 1–40 and Aβ 1–42 in the plasma were also higher in
TSA-treated mice in comparison with vehicle-treated mice. The treatment of transgenic AD mice with TSA didnot affect the body weight in both male and female groups as compared to vehicle-treated animals. A positivecorrelation was observed between the plasma levels of gelsolin and Aβ 1–40 (r = 0.594, p = 0.042) or Aβ1–42 (r = 0.616, p = 0.033) in AD Tg mice.Significance: These results suggest that TSA increases the levels of plasma gelsolin and Aβ in AD Tg mice, whichmay have implications in gelsolin-mediated clearance of Aβ.© 2014 Elsevier Inc. All rights reserved.
Introduction
Gelsolin (GSN), a multifunctional 90 kDa protein, is a major actin-binding protein. It plays important roles in various diseases such ascancer, Alzheimer's disease (AD), pulmonary disease, cardiac injury,infections, and inflammation as well as in apoptosis (Li et al., 2012). Itis present intracellularly as a cytoplasmic protein, and in the biologicalfluids, i.e., plasma/cerebrospinal fluid (CSF) as a secreted protein(Kwiatkowski et al., 1988). Both forms of gelsolin are products of alter-native splicing of a single 70 kbp-long gene. However, they differ in thelength and the presence of disulfide groups. Plasma gelsolin has a 25amino acid-signal peptide at its amino-terminus (Yin et al., 1984).There are five cysteine (Cys) residues in gelsolin. All five Cys residuesin cytoplasmic gelsolin are free thiols; whereas in plasma gelsolin,three Cys residues are free thiols and the other two are disulfide-linked (Wen et al., 1996).
Extracellular deposition of fibrillar amyloid beta-protein (Aβ) asamyloid plaque, and intracellular formation of neurofibrillary tanglesdue to hyperphosphorylation of tau are two hallmarks of AD (Glenner,
Research in Developmental4, USA. Tel.: +1 718 494 5257;
han).
ghts reserved.
1983; Huang and Jiang, 2009). Aβ is formed by proteolytic cleavage ofamyloid precursor protein (APP) (Pluta et al., 2013). Aβ is normallypresent as a soluble protein. However, in pathological conditions suchas AD, it gets fibrillized and deposited as amyloid plaque in the brain.Fibrillar Aβ 1–40 and Aβ 1–42 are two major constituents of amyloidplaques in AD. Various attempts have been made to prevent thefibrillization of Aβ in both human and transgenic (Tg) animal model ofAD (Antequera et al., 2009; Lahiri et al., 2005; Matsuoka et al., 2003).Since our initial reports on binding of gelsolin to Aβ (Chauhan et al.,1999; Ji et al., 2008, 2010) and inhibition of Aβ fibrillization as well assolubilization of Aβ fibrils by gelsolin (Ray et al., 2000), several studieshave confirmed anti-amyloidogenic role of gelsolin in transgenic animalmodels of AD where it could reduce amyloid load (Antequera et al.,2009; Hirko et al., 2007; Lahiri et al., 2005; Matsuoka et al., 2003).Matsuoka et al. (2003) reported that peripheral administration ofplasma gelsolin could reduce the amyloid load in mutant APP-Tg mice.Similarly, Hirko et al. (2007) showed that transgene expressionof plasmagelsolin in APP/PS1δE9 mice decreased amyloid load. In another study,virally directed expression of gelsolin also reduced the amyloid load indouble-transgenic APP/PS1 mice (Antequera et al., 2009). In a recentstudy, peripheral administration of gelsolin reduced the cerebral amy-loid angiopathy (accumulation of Aβ in the walls of leptomeningealand cortical blood vessels of brains) in Tg2576 mice model of AD(Gregory et al., 2012).

32 W. Yang et al. / Life Sciences 99 (2014) 31–36
The levels of gelsolin can be increased epigenetically by inhibition ofhistone deacetylase. Histone deacetylase inhibitors such as sodiumbutyrate (Kamitani et al., 2002) and trichostatin A (TSA) (Hoshikawaet al., 1994; Kamitani et al., 2002; Yildirim et al., 2008) have beenreported to increase the expression of gelsolin in cell cultures andbrain. However, a relationship between TSA-induced gelsolin and Aβin transgenic AD mice has not been studied. Therefore, we studiedwhether TSA can be used as a potential therapeutic agent in AD forclearance of Aβ by increasing the levels of plasma gelsolin in APPswe/PS1δE9 transgenic mice. We report here that TSA increases the levels ofgelsolin, Aβ 1–40 and Aβ 1–42 in the plasma. The levels of plasmagelsolin correlated with the levels of plasma Aβ 1–40 and Aβ 1–42, sug-gesting that plasma gelsolin probably acts as “peripheral sink protein”to bind Aβ peptides, and may help in Aβ clearance from the brain orother tissues.
Materials and methods
Animal treatment
Twenty six APPswe/PS1δE9 transgenic mice (13 males and 13females) at the age of 4 monthswere purchased from Jackson Laboratory(Bar Harbor, ME) and kept in the animal colony with the food andwaterad libitum. All instructions were followed according to the NationalInstitutes of Health Guidelines for the Humane Treatment of Animals,and the protocol was approved by the Animal Welfare Committee ofNYS Institute for Basic Research in Developmental Disabilities.
The mice were divided randomly into two treatment groups: TSA (7females, 7males) and vehicle control (6 females, 6males). The intraper-itoneal injections of TSA or vehicle to themicewere started at the age of9 months, and continued for 60 days.
Five milligrams of TSA (Selleck Chemicals, Houston, TX) wasdissolved in 0.1 ml of dimethyl sulfoxide (DMSO), and then dilutedwith 9.9ml of phosphate buffered saline (PBS). Themicewere intraper-itoneally injected with TSA at a dose of 0.1 ml/10 g (i.e., 5 mg/kg bodyweight) every alternate day for a total of 30 treatments. The injectionspotwas alternated between the left and right sides to avoid peritonitis.The control mice were injected with the same dose of vehicle (no TSA,i.e., DMSO diluted in PBS). The body weights of mice were monitoredevery week, and the dose of TSA was calculated on the basis of weeklybody weight.
After 30 treatments, the mice were anesthetized with ether andsacrificed. The blood samples were withdrawn and transferred intotubes containing ethylenediaminetetraacetic acid (EDTA) as anti-coagulant. After centrifugation at 1000 g for 10 min at 4 °C, the plasmasamples were collected and stored at−80 °C for further use.
Western blotting for gelsolin
The protein concentrations of plasma samples were measured withthe BCA (bicinchoninic acid) Protein Assay Kit (Thermo Scientific,Rockford, IL). The plasma samples were diluted ten-fold with PBS. Thesamples were mixed with loading buffer, and boiled for 5 min. Thedenatured plasma proteins were separated on a 10% SDS-PAGE gel,and then transferred to a nitrocellulose membrane (0.45 μm; Bio-RadLaboratories, Hercules, CA). After blocking with 5% non-fat milk for 1 hat room temperature (RT), the membrane was incubated overnight at4 °C with 5% non-fat milk containing rabbit monoclonal anti-gelsolinantibody (1:25,000; Abcam, Cambridge, MA). After 3 washes withTris-buffered saline-0.05% Tween-20 (TBST), themembranewas furtherincubated with the horseradish peroxidase-conjugated secondary anti-body (1:5000; Thermo Scientific, Rockford, IL) for 1 h at RT. The mem-brane was again washed 3 times, and the immunoreactive proteinswere visualized using the ECL substrate (Thermo Scientific, Rockford,IL). The densities of the gelsolin bands were measured using ImageJ
software (NIH, Bethesda, MD). The data was normalized with total pro-tein content of the samples.
Measurement of plasma Aβ 1–40 and Aβ 1–42 levels by sandwich enzyme-linked immunosorbent assay [ELISA]
The levels of Aβ 1–40 andAβ 1–42 in theplasma samples of TSA- andvehicle-treated AD Tgmice weremeasured by ELISA as described previ-ously but with minor modifications (Mehta et al., 2000). Briefly, 100 μlof monoclonal Aβ antibody 6E10 (2.5 μg/ml) diluted in carbonate–bicarbonate buffer (pH 9.6) was coated in the wells of microtiter plate,and allowed to incubate overnight at 4 °C. After washing the plateswith PBS containing 0.05% Tween 20 (PBST), the wells were blockedfor 1 h with 200 μl of 10% normal sheep serum in PBS to avoid nonspe-cific binding. The plates were againwashed, and 100 μl of standards (Aβ1–40 and Aβ 1–42; Bachem, Torrance, CA) diluted in PBST with 0.5%bovine serum albumin (BSA) or plasma samples (diluted 1:2) wereadded and incubated for 2 h at RT, followed by overnight incubation at4 °C. After washing, the plates were incubated with rabbit monoclonalantibody (clone 5-139) specific to a peptide corresponding to Aβ 1–40or rabbit monoclonal antibody (clone 1-11-4) specific to a peptide cor-responding to Aβ 1–42 diluted in PBSTwith 0.5% BSA at RT for 2 h. Afterwashing, goat anti-rabbit IgG peroxidase (Invitrogen, Grand Island, NY)diluted in PBST was added to the wells, and the plates were incubatedfor 2 h at RT. The plates were again washed, and QuantaBlu flurogenicperoxidase substrate (Thermoscientific, Rockland, IL) was added. Thereaction was stopped after 30 min by adding QuataBlu stop solution.The relative fluorescence units (RFU) for each well were measuredaccording to the kit instructions. The relationship between RFU and theAβ 1–40 or Aβ 1–42 peptide concentrations was determined using a4-parameter logistic logarithm function.
Statistical analysis
Data is presented asMean± SEM. The unpaired Student's t-test wasused to compare the data between the TSA-treated group and vehicle-treated control group by usingGraphPad prism5.0 (GraphPad Software,Inc., La Jolla, CA). Linear regression analysis was done to study the corre-lation between the levels of plasma gelsolin and Aβ 1–40/Aβ 1–42, andthe Pearson correlation coefficient (r) and p values were obtained. Inaddition, 95% confidence interval (CI) of vehicle-treated control groupwas used as the normal range to evaluate whether the data of theTSA-treated group were out of the normal range. The p values lessthan 0.05 were considered as the significant difference.
Results
Effect of TSA-treatment on the body weight of mice
The body weights of TSA-treated and vehicle-treated control AD Tgmice were measured every week. Percent increases in body weight ofTSA-treated or vehicle-treated control mice (males or females) are shownin Fig. 1. There was no effect of TSA-treatment for two months (9th to11th months of age) on the body weight of either female (Fig. 1A) ormale Tg mice (Fig. 1B) as compared to vehicle-treated control Tg mice.
TSA-treatment increases levels of gelsolin in the plasma of AD Tg mice
Fig. 2A shows theWestern blot of plasma gelsolin of TSA-treated andvehicle-treated AD Tg mice. The density of the gelsolin bands wasnormalized by the total protein content of the samples. TSA-treatmentincreased the plasma concentration of gelsolin by 82% in female(39.98 ± 8.41/μg protein, n = 7) (Fig. 2B) and by 76% in male mice(45.68 ± 5.98/μg protein, n = 7) (Fig. 2C) as compared to vehicle-treated control mice (female group, 21.91 ± 4.24/μg protein, n = 6,andmale group, 26.01± 3.72/μgprotein, n=6). A significantdifference
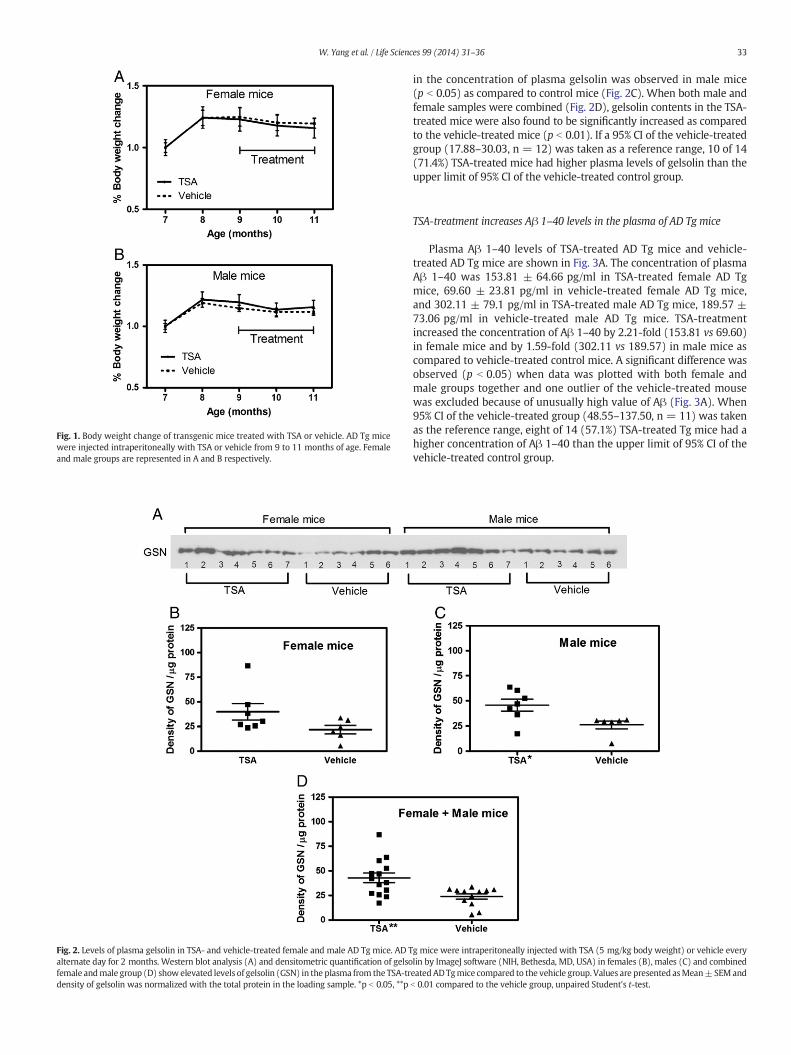
Fig. 1. Body weight change of transgenic mice treated with TSA or vehicle. AD Tg micewere injected intraperitoneally with TSA or vehicle from 9 to 11 months of age. Femaleand male groups are represented in A and B respectively.
Fig. 2. Levels of plasma gelsolin in TSA- and vehicle-treated female and male AD Tg mice. AD Talternate day for 2 months. Western blot analysis (A) and densitometric quantification of gelsofemale andmale group (D) show elevated levels of gelsolin (GSN) in the plasma from the TSA-trdensity of gelsolin was normalized with the total protein in the loading sample. *p b 0.05, **p
33W. Yang et al. / Life Sciences 99 (2014) 31–36
in the concentration of plasma gelsolin was observed in male mice(p b 0.05) as compared to control mice (Fig. 2C). When both male andfemale samples were combined (Fig. 2D), gelsolin contents in the TSA-treated mice were also found to be significantly increased as comparedto the vehicle-treated mice (p b 0.01). If a 95% CI of the vehicle-treatedgroup (17.88–30.03, n = 12) was taken as a reference range, 10 of 14(71.4%) TSA-treated mice had higher plasma levels of gelsolin than theupper limit of 95% CI of the vehicle-treated control group.
TSA-treatment increases Aβ 1–40 levels in the plasma of AD Tg mice
Plasma Aβ 1–40 levels of TSA-treated AD Tg mice and vehicle-treated AD Tg mice are shown in Fig. 3A. The concentration of plasmaAβ 1–40 was 153.81 ± 64.66 pg/ml in TSA-treated female AD Tgmice, 69.60 ± 23.81 pg/ml in vehicle-treated female AD Tg mice,and 302.11 ± 79.1 pg/ml in TSA-treated male AD Tg mice, 189.57 ±73.06 pg/ml in vehicle-treated male AD Tg mice. TSA-treatmentincreased the concentration of Aβ 1–40 by 2.21-fold (153.81 vs 69.60)in female mice and by 1.59-fold (302.11 vs 189.57) in male mice ascompared to vehicle-treated control mice. A significant difference wasobserved (p b 0.05) when data was plotted with both female andmale groups together and one outlier of the vehicle-treated mousewas excluded because of unusually high value of Aβ (Fig. 3A). When95% CI of the vehicle-treated group (48.55–137.50, n = 11) was takenas the reference range, eight of 14 (57.1%) TSA-treated Tg mice had ahigher concentration of Aβ 1–40 than the upper limit of 95% CI of thevehicle-treated control group.
g mice were intraperitoneally injected with TSA (5 mg/kg body weight) or vehicle everylin by ImageJ software (NIH, Bethesda, MD, USA) in females (B), males (C) and combinedeatedADTgmice compared to the vehicle group. Values are presented asMean±SEMandb 0.01 compared to the vehicle group, unpaired Student's t-test.
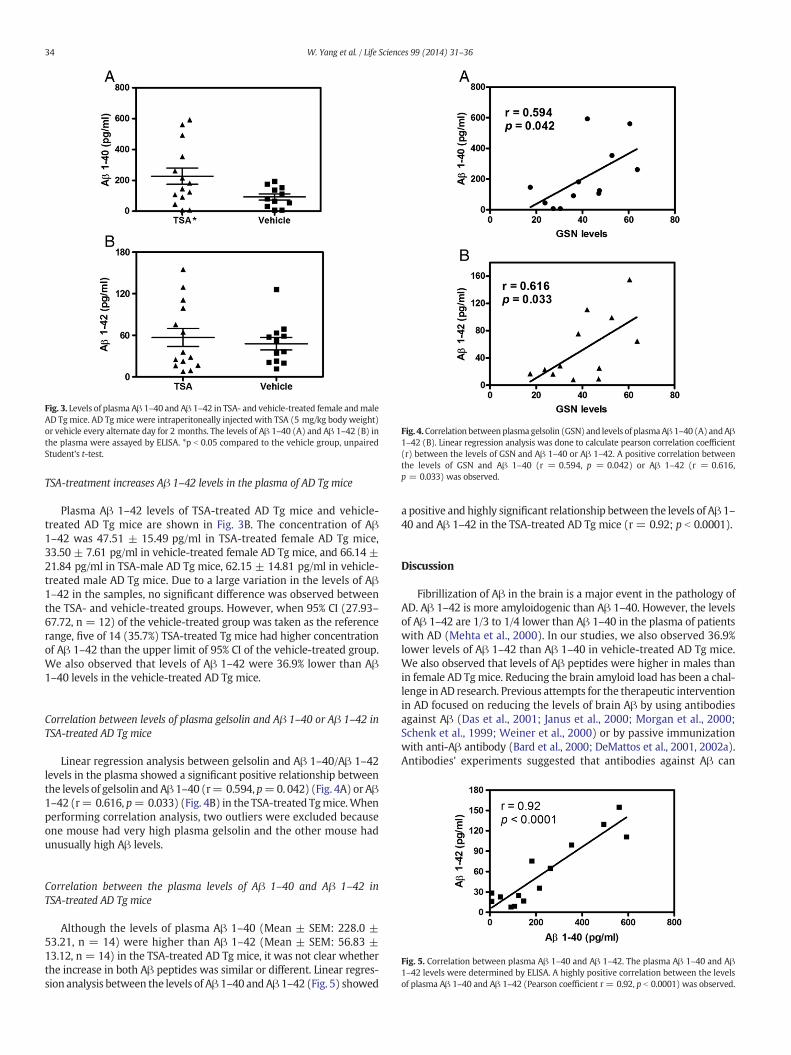
Fig. 3. Levels of plasma Aβ 1–40 and Aβ 1–42 in TSA- and vehicle-treated female andmaleAD Tg mice. AD Tg mice were intraperitoneally injected with TSA (5 mg/kg body weight)or vehicle every alternate day for 2 months. The levels of Aβ 1–40 (A) and Aβ 1–42 (B) inthe plasma were assayed by ELISA. *p b 0.05 compared to the vehicle group, unpairedStudent's t-test.
Fig. 4.Correlation between plasma gelsolin (GSN) and levels of plasmaAβ 1–40 (A) andAβ1–42 (B). Linear regression analysis was done to calculate pearson correlation coefficient(r) between the levels of GSN and Aβ 1–40 or Aβ 1–42. A positive correlation betweenthe levels of GSN and Aβ 1–40 (r = 0.594, p = 0.042) or Aβ 1–42 (r = 0.616,p = 0.033) was observed.
34 W. Yang et al. / Life Sciences 99 (2014) 31–36
TSA-treatment increases Aβ 1–42 levels in the plasma of AD Tg mice
Plasma Aβ 1–42 levels of TSA-treated AD Tg mice and vehicle-treated AD Tg mice are shown in Fig. 3B. The concentration of Aβ1–42 was 47.51 ± 15.49 pg/ml in TSA-treated female AD Tg mice,33.50 ± 7.61 pg/ml in vehicle-treated female AD Tg mice, and 66.14 ±21.84 pg/ml in TSA-male AD Tg mice, 62.15 ± 14.81 pg/ml in vehicle-treated male AD Tg mice. Due to a large variation in the levels of Aβ1–42 in the samples, no significant difference was observed betweenthe TSA- and vehicle-treated groups. However, when 95% CI (27.93–67.72, n = 12) of the vehicle-treated group was taken as the referencerange, five of 14 (35.7%) TSA-treated Tg mice had higher concentrationof Aβ 1–42 than the upper limit of 95% CI of the vehicle-treated group.We also observed that levels of Aβ 1–42 were 36.9% lower than Aβ1–40 levels in the vehicle-treated AD Tg mice.
Correlation between levels of plasma gelsolin and Aβ 1–40 or Aβ 1–42 inTSA-treated AD Tg mice
Linear regression analysis between gelsolin and Aβ 1–40/Aβ 1–42levels in the plasma showed a significant positive relationship betweenthe levels of gelsolin andAβ 1–40 (r= 0.594, p=0. 042) (Fig. 4A) or Aβ1–42 (r= 0.616, p= 0.033) (Fig. 4B) in the TSA-treated Tgmice.Whenperforming correlation analysis, two outliers were excluded becauseone mouse had very high plasma gelsolin and the other mouse hadunusually high Aβ levels.
Fig. 5. Correlation between plasma Aβ 1–40 and Aβ 1–42. The plasma Aβ 1–40 and Aβ1–42 levels were determined by ELISA. A highly positive correlation between the levelsof plasma Aβ 1–40 and Aβ 1–42 (Pearson coefficient r = 0.92, p b 0.0001) was observed.
Correlation between the plasma levels of Aβ 1–40 and Aβ 1–42 inTSA-treated AD Tg mice
Although the levels of plasma Aβ 1–40 (Mean ± SEM: 228.0 ±53.21, n = 14) were higher than Aβ 1–42 (Mean ± SEM: 56.83 ±13.12, n = 14) in the TSA-treated AD Tg mice, it was not clear whetherthe increase in both Aβ peptides was similar or different. Linear regres-sion analysis between the levels of Aβ 1–40 andAβ 1–42 (Fig. 5) showed
a positive and highly significant relationship between the levels of Aβ 1–40 and Aβ 1–42 in the TSA-treated AD Tg mice (r = 0.92; p b 0.0001).
Discussion
Fibrillization of Aβ in the brain is a major event in the pathology ofAD. Aβ 1–42 is more amyloidogenic than Aβ 1–40. However, the levelsof Aβ 1–42 are 1/3 to 1/4 lower than Aβ 1–40 in the plasma of patientswith AD (Mehta et al., 2000). In our studies, we also observed 36.9%lower levels of Aβ 1–42 than Aβ 1–40 in vehicle-treated AD Tg mice.We also observed that levels of Aβ peptides were higher in males thanin female AD Tgmice. Reducing the brain amyloid load has been a chal-lenge in AD research. Previous attempts for the therapeutic interventionin AD focused on reducing the levels of brain Aβ by using antibodiesagainst Aβ (Das et al., 2001; Janus et al., 2000; Morgan et al., 2000;Schenk et al., 1999; Weiner et al., 2000) or by passive immunizationwith anti-Aβ antibody (Bard et al., 2000; DeMattos et al., 2001, 2002a).Antibodies' experiments suggested that antibodies against Aβ can

35W. Yang et al. / Life Sciences 99 (2014) 31–36
modulate the transfer of Aβ between the brain and plasma (DeMattoset al., 2001, 2002a,b). Therefore, compounds that can bind to Aβ in theperiphery without penetrating blood brain barrier may be ideal candi-dates as effective therapeutic agents for AD. One such candidate isgelsolin of 90 kDa protein, which is present as circulating and also intra-cellular protein.
We have previously reported that gelsolin is an anti-amyloidogenicprotein (Chauhan et al., 2008; Ji et al., 2009), which binds to Aβ(Chauhan et al., 1999; Ji et al., 2008, 2010), inhibits Aβ fibrillizationand also promotes its defibrillization (Ray et al., 2000). Interestingly,Guntert et al. (2010) analyzed the plasma protein levels of gelsolin inslow cognitive declining AD patients, rapid cognitive declining ADpatients and non-demented controls using isobaric mass taggingapproach. These authors reported that AD patients had significantlylower plasma gelsolin levels compared to non-demented controls, andgelsolin levels correlated with disease progression rate estimated byMini-Mental Status Examination decline per year. Apart from bindingof gelsolin to Aβ and inhibition of its fibrillization, gelsolin protectscells against apoptosis by blocking cytochrome C release (Koya et al.,2000; Qiao et al., 2005). It also modulates mitochondrial function byincreasing mitochondrial complex IV activity (Antequera et al., 2009),which is decreased in AD patients and animal models of AD (Moraisand De Strooper, 2010; Moran et al., 2012). Antequera et al. (2009)reported that inhibition of gelsolin gene expression inAPP/PS1 transgenicmousemodel of AD resulted in increased amyloid burden and apoptoticresponse.
Many studies have focused on reducing the amyloid load by gelsolin.Anti-amyloidogenic role of gelsolin has been reviewed by us (Chauhanet al., 2008; Ji et al., 2009). Other groups have also confirmed anti-amyloidogenic role of gelsolin (Antequera et al., 2009; Hirko et al.,2007; Matsuoka et al., 2003). Hirko et al. (2007) reported that peripheraltransgene expression of plasma gelsolin reduces amyloid load in trans-genic mouse model of AD. In mutant APP-transgenic mouse model ofAD, intraperitoneal injection of gelsolin resulted in reduced amyloidload in the brain and increased levels of gelsolin in the plasma(Matsuoka et al., 2003). Lemere et al. (2003) also reported increasedserum Aβ levels upon Aβ immunization of transgenic PS/APP mice. Theefflex/clearance of Aβ from brain to plasma is reported by DeMattoset al. (2001, 2002a,b). When cytoplasmic gelsolin was increased byviral-directed overexpressionof gelsolin inAPP/PS1 transgenicmice, a de-crease in the brain Aβ burden was observed (Antequera et al., 2009).From these studies, it can be concluded that increased levels of plasmagelsolin reflect reduced amyloid load in the brain. Although our data of in-creased plasma gelsolin in TSA-treated mice indirectly suggest increasedclearance of Aβ from the brain, it cannot be ruled out that TSA may alsobe affecting the processing of APP which may lead to reduced amyloidload in the brain independent of gelsolin levels. In both scenarios, TSAmay have beneficial effect in reducing the amyloid load in the brain.
In a recent study, Gregory et al. (2012) reported that reducing avail-able soluble Aβ prevents progression of cerebral amyloid angiopathy inTg 2576model of ADmice. However, the present study is the first reportby which we have chemically increased the levels of plasma gelsolin inAD Tg mice, i.e., by using TSA, a class I and II inhibitor of HDACs andstudied its effect on the levels of Aβ. TSA has pleiotropic effects as it pro-motes expression of several genes by inhibition of histone acetylation.One of the genes induced by TSA is gelsolin. Our results indicate thatTSA increases the levels of plasma gelsolin and Aβ 1–40/Aβ 1–42, andthere is a significant positive correlation between the levels of gelsolinand Aβ 1–40/Aβ 1–42 in these mice. A positive correlation betweenthe levels of Aβ 1–40 and Aβ 1–42 suggests that gelsolin sequestersAβ 1–40 and Aβ 1–42 with similar affinity for these peptides. BecauseTSA-treatment of mice for two months did not affect the body weight,TSA-treatment may not have any deleterious effects on health.
Other HDAC inhibitors have also been shown to have beneficialeffects in animal models of AD. Valproic acid (VPA) has been shown todown regulate APP in cancer cells (Venkataramani et al., 2010). VPA
inhibits the production of Aβ peptide in HEK293 cells transfected withSwedish APP751 (Su et al., 2004). VPA is also found to reduce Aβ pro-duction (Qing et al., 2008). Francis et al. (2009) reported that histoneacetylation plays an important role in hippocampal long-term potentia-tion and memory. They observed that levels of hippocampal acetylatedhistone 4 (H4) in APP/PS1 mice were decreased by 50% as compared towild typemice after fear conditioning training. An acute treatmentwithTSA prior to training rescued both acetylated H4 levels and contextualfreezing performance close to wild type animals.
However, there are some differences in the alterations of plasma Aβlevels in various studies, where peripheral Aβ sequestration approacheswere used. We observed increased levels of both Aβ 1–40 and Aβ 1–42in the plasma of mice treated with TSA. On the other hand, Matsuokaet al. reported that plasma levels of Aβ 1–40 and Aβ 1–42 were notaffected in vehicle-treatedmice andmicewithperipherally administratedgelsolin (Matsuoka et al., 2003). In another study, Hirko et al. observedthat levels of plasma Aβ 1–42 were increased but the levels of Aβ1–40were decreased after transgene overexpression of plasma gelsolin(Hirko et al., 2007). At present, it is not clear why different methods ofgelsolin administration affect Aβ levels in differentmanner. In our stud-ies, a positive correlation was also observed between plasma gelsolinand Aβ 1–40/Aβ 1–42 levels, suggesting that increased plasma gelsolinmay be acting as a peripheral sink to reduce brain Aβ. This study furthersuggests that gelsolinmay be acting by binding to Aβ 1–40 andAβ 1–42.It is also supported by our previous in vitro study where we reportedthat plasma gelsolin and cytoplasmic gelsolin bind to Aβ 1–40 and Aβ1–42 (Ji et al., 2008), and inhibit its fibrillization (Ray et al., 2000).While our studies suggest that TSA-induced increased levels of Aβpeptides in the plasma of AD Tg mice come from sequestration ofbrain Aβ peptides by gelsolin, it is also possible that these peptidesmay also arise from increased APP processing in peripheral tissues.
In summary, our results indicate that plasma levels of gelsolin andAβ 1–40 and Aβ 1–42 can be increased by TSA. A correlation betweenthe levels of plasma gelsolin and plasma Aβ 1–40 or Aβ 1–42 suggeststhat TSAmay help in the clearance of Aβ from the brain or other tissues,and may act as a potential therapeutic agent in AD.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Acknowledgments
This workwas supported by funds fromAlzheimer's Association andthe NYS Office for People with Developmental Disabilities.
References
Antequera D, Vargas T, Ugalde C, Spuch C, Molina JA, Ferrer I, et al. Cytoplasmic gelsolinincreases mitochondrial activity and reduces Abeta burden in a mouse model ofAlzheimer's disease. Neurobiol Dis 2009;36:42–50.
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administeredantibodies against amyloid beta-peptide enter the central nervous system and reducepathology in a mouse model of Alzheimer disease. Nat Med 2000;6:916–9.
Chauhan VP, Ray I, Chauhan A,Wisniewski HM. Binding of gelsolin, a secretory protein, toamyloid beta-protein. Biochem Biophys Res Commun 1999;258:241–6.
Chauhan V, Ji L, Chauhan A. Anti-amyloidogenic, anti-oxidant and anti-apoptotic role ofgelsolin in Alzheimer's disease. Biogerontology 2008;9:381–9.
Das P, Murphy MP, Younkin LH, Younkin SG, Golde TE. Reduced effectiveness of Abeta1–42 immunization in APP transgenic mice with significant amyloid deposition.Neurobiol Aging 2001;22:721–7.
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Abeta antibody alters CNS and plasma A beta clearance and decreases brain A betaburden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2001;98:8850–5.
DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer'sdisease. Science 2002a;295:2264–7.
DeMattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, et al. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in amouse model of Alzheimer's disease. J Neurochem 2002b;81:229–36.

36 W. Yang et al. / Life Sciences 99 (2014) 31–36
Francis YI, Fa M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, et al. Dysregulation ofhistone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J AlzheimersDis 2009;18:131–9.
Glenner GG. Alzheimer's disease. The commonest form of amyloidosis. Arch Pathol LabMed 1983;107:281–2.
Gregory JL, PradaCM, Fine SJ, Garcia-AllozaM, BetenskyRA,Arbel-OrnathM, et al. Reducingavailable soluble beta-amyloid prevents progression of cerebral amyloid angiopathy intransgenic mice. J Neuropathol Exp Neurol 2012;71:1009–17.
Guntert A, Campbell J, Saleem M, O'Brien DP, Thompson AJ, Byers HL, et al. Plasmagelsolin is decreased and correlates with rate of decline in Alzheimer's disease.J Alzheimers Dis 2010;21:585–96.
Hirko AC, Meyer EM, King MA, Hughes JA. Peripheral transgene expression of plasmagelsolin reduces amyloid in transgenic mouse models of Alzheimer's disease. MolTher 2007;15:1623–9.
Hoshikawa Y, Kwon HJ, Yoshida M, Horinouchi S, Beppu T. Trichostatin A inducesmorphological changes and gelsolin expression by inhibiting histone deacetylase inhuman carcinoma cell lines. Exp Cell Res 1994;214:189–97.
Huang HC, Jiang ZF. Accumulated amyloid-beta peptide and hyperphosphorylated tauprotein: relationship and links inAlzheimer's disease. J AlzheimersDis 2009;16:15–27.
Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, et al. A beta peptideimmunization reduces behavioural impairment and plaques in amodel of Alzheimer'sdisease. Nature 2000;408:979–82.
Ji L, Chauhan A, Chauhan V. Cytoplasmic gelsolin in pheochromocytoma-12 cells forms acomplex with amyloid beta-protein. Neuroreport 2008;19:463–6.
Ji L, Chauhan A, Chauhan VRole of gelsolin in alzheimer's disease, vol. 4. New York: NovaScience Publisher Inc.; 2009199–218.
Ji L, Chauhan A, Chauhan V. Upregulation of cytoplasmic gelsolin, an amyloid-beta-binding protein, under oxidative stress conditions: involvement of protein kinase C.J Alzheimers Dis 2010;19:829–38.
Kamitani H, Taniura S,Watanabe K, SakamotoM,Watanabe T, Eling T. Histone acetylationmay suppress human glioma cell proliferation when p21 WAF/Cip1 and gelsolin areinduced. Neuro Oncol 2002;4:95–101.
Koya RC, Fujita H, Shimizu S, Ohtsu M, Takimoto M, Tsujimoto Y, et al. Gelsolin inhibitsapoptosis by blocking mitochondrial membrane potential loss and cytochrome crelease. J Biol Chem 2000;275:15343–9.
Kwiatkowski DJ, Mehl R, Yin HL. Genomic organization and biosynthesis of secreted andcytoplasmic forms of gelsolin. J Cell Biol 1988;106:375–84.
Lahiri DK, Chen DM, Lahiri P, Bondy S, Greig NH. Amyloid, cholinesterase, melatonin, andmetals and their roles in aging and neurodegenerative diseases. Ann N Y Acad Sci2005;1056:430–49.
Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, et al. Evidence forperipheral clearance of cerebral Abeta protein following chronic, active Abeta immu-nization in PSAPP mice. Neurobiol Dis 2003;14:10–8.
Li GH, Arora PD, Chen Y, McCulloch CA, Liu P. Multifunctional roles of gelsolin in healthand diseases. Med Res Rev 2012;32:999–1025.
Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, et al. Novel therapeuticapproach for the treatment of Alzheimer's disease by peripheral administration ofagents with an affinity to beta-amyloid. J Neurosci 2003;23:29–33.
Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and cerebro-spinal fluid levels of amyloid beta proteins 1–40 and 1–42 in Alzheimer disease. ArchNeurol 2000;57:100–5.
Morais VA, De Strooper B. Mitochondria dysfunction and neurodegenerative disorders:cause or consequence. J Alzheimers Dis 2010;20(Suppl. 2):S255–63.
MoranM,Moreno-Lastres D,Marin-Buera L, Arenas J, MartinMA, Ugalde C.Mitochondrialrespiratory chain dysfunction: implications in neurodegeneration. Free Radic BiolMed 2012;53:595–609.
Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A beta peptidevaccination prevents memory loss in an animal model of Alzheimer's disease. Nature2000;408:982–5.
Pluta R, Furmaga-Jablonska W, Maciejewski R, Ulamek-Koziol M, Jablonski M. Brainischemia activates beta- and gamma-secretase cleavage of amyloid precursor protein:significance in sporadic Alzheimer's disease. Mol Neurobiol 2013;47:425–34.
Qiao H, Koya RC, Nakagawa K, Tanaka H, Fujita H, Takimoto M, et al. Inhibition ofAlzheimer's amyloid-beta peptide-induced reduction of mitochondrial membranepotential and neurotoxicity by gelsolin. Neurobiol Aging 2005;26:849–55.
Qing H, He G, Ly PT, Fox CJ, Staufenbiel M, Cai F, et al. Valproic acid inhibits Abeta produc-tion, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mousemodels. J Exp Med 2008;205:2781–9.
Ray I, Chauhan A, Wegiel J, Chauhan VP. Gelsolin inhibits the fibrillization of amyloidbeta-protein, and also defibrillizes its preformed fibrils. Brain Res 2000;853:344–51.
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization withamyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse.Nature 1999;400:173–7.
Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P, et al. Lithium, a common drug for bipolardisorder treatment, regulates amyloid-beta precursor proteinprocessing. Biochemistry2004;43:6899–908.
Venkataramani V, Rossner C, Iffland L, Schweyer S, Tamboli IY, Walter J, et al. Histonedeacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the alzheimer amyloid precursor protein. J Biol Chem 2010;285:10678–89.
Weiner HL, Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, et al. Nasal administra-tion of amyloid-beta peptide decreases cerebral amyloid burden in a mouse model ofAlzheimer's disease. Ann Neurol 2000;48:567–79.
Wen D, Corina K, Chow EP, Miller S, Janmey PA, Pepinsky RB. The plasma and cytoplasmicforms of human gelsolin differ in disulfide structure. Biochemistry 1996;35:9700–9.
Yildirim F, Gertz K, Kronenberg G, Harms C, Fink KB, Meisel A, et al. Inhibition of histonedeacetylation protects wildtype but not gelsolin-deficient mice from ischemic braininjury. Exp Neurol 2008;210:531–42.
Yin HL, Kwiatkowski DJ, Mole JE, Cole FS. Structure and biosynthesis of cytoplasmic andsecreted variants of gelsolin. J Biol Chem 1984;259:5271–6.