transmissible spongiform encephalopathies: prion … · normal prion biology. host prpc is a cell...
TRANSCRIPT

TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHIES:
PRION GENETICS, TRANSMISSION BARRIERS, AND DISEASE CONTROL
By
ROBERT DYLAN HARRINGTON
A dissertation submitted in partial fulfillment of the requirements for the degree of
DOCTOR OF PHILOSOPHY in VETERINARY SCIENCE
WASHINGTON STATE UNIVERSITY Department of Veterinary Microbiology and Pathology
AUGUST 2008
© Copyright by ROBERT DYLAN HARRINGTON, 2008 All Rights Reserved

© Copyright by ROBERT DYLAN HARRINGTON, 2008

To the Faculty of Washington State University:
The members of the Committee appointed to examine the dissertation of ROBERT DYLAN HARRINGTON find it satisfactory and recommend that it be accepted.
___________________________________
Chair
___________________________________
___________________________________
___________________________________
___________________________________
ii

ACKNOWLEDGEMENTS
Chapter two is a manuscript originally published in the Journal of General Virology,
Society for General Microbiology, United Kingdom.
I am grateful to John Gorham for many helpful discussions and recommendations;
Janet Alverson for assistance with animal monitoring and necropsies; Tom Truscott,
Huijun Yan, and Charlene Karr-May for histologic procedures; Linda Hamburg, Gina
Kiske, Issana To, Dongyue Zhuang, Liam Broughton, Lowell Kappmeyer, Codie Hanke,
and Marta Henrikkson for expert technical assistance; and Duane Chandler, Pete
Steiner, Amy Hetrick, and Alicia Ewing for animal handling and restraint.
Jean Manson provided prion knockout mice; Margaret Wild and Jenny Powers of Rocky
Mountain National Park provided CWD positive elk and deer brain; Amir Hamir and
Jason Bartz provided TME homogenate; Kurt Vercauteren provided CWD negative deer
brain; Glen Zebarth and the North American Elk Breeders Association provided CWD
negative elk brain; and the staff of the USDA National Sheep Experimental Station,
Dubois, ID, USA provided sheep blood samples.
The work in this dissertation was supported by National Institute of Allergy and
Infectious Disease grant #K08AI060680, USDA-Agricultural Research Service SCA
#58-5348-2-684, and USDA-Agricultural Research Service CRIS #5348-32000-021-
00D.
iii

TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHIES:
PRION GENETICS, TRANSMISSION BARRIERS, AND DISEASE CONTROL
by Robert Dylan Harrington, D.V.M., Ph.D. Washington State University
August 2008
Chair: Donald P. Knowles
Transmissible Spongiform Encephalopathies (TSE) are invariably fatal
neurodegenerative diseases associated with misfolded prion protein. Host prion gene
(PRNP) variation affects TSE transmission barriers within and between species, and
forms the basis of disease control strategies. Reported herein are aspects of PRNP
genetics related to prion transmission, species barriers, and management. A TSE
species barrier in ruminant to carnivore transmission was investigated by the hypothesis
that primary oral challenge with chronic wasting disease (CWD) causes a prion disease
in mink. It was found that while CWD can cause a prion disease when given
intracerebrally to mink, such disease is not characteristic of Transmissible Mink
Encephalopathy (TME) and oral challenge does not result in disease. A novel PRNP
variant at codon 27 variant may affect TSE transmission, possibly by altered membrane
localization of normal prion protein. This study shows that CWD is poorly transmissible
to non-cervid hosts, CWD is an unlikely cause of TME, and mink are an unlikely to be
involved in natural CWD transmission. Thus, Bovine Spongiform Encephalopathy is the
only ruminant TSE orally transmissible to mink suggesting that a previously
unrecognized prion-like disease was a cause of some cases of TME. The effect of
PRNP promoter regions upon TSE transmission was examined by the hypothesis that
transgenic incorporation of the cervid PRNP putative promoter (PP) region and open
reading frame (ORF) renders transgenic mice susceptible to CWD administered by
intracerebral, intraperitoneal, and oral routes. Transgenic insertion of a mule deer
PRNP PP and ORF transgene resulted in stable transcription and translation in mice
without developmental, anatomical, or behavioral abnormalities. Transgenic mice
iv

accumulated disease associated prion protein following challenge with CWD, thus
providing an alternative system for study of peripheral exposure routes in CWD
pathogenesis. To determine adverse affects of PRNP selection for scrapie control a
hypothesis that the sheep PRNP 171 arginine (R) allele is associated with higher
prevalence of ovine progressive pneumonia virus (OPPV) and higher OPPV provirus
levels was tested. Results showed that OPPV presence and provirus levels are
independent of the PRNP 171R allele indicating that PRNP selection will not adversely
affect OPPV within a flock.
v

TABLE OF CONTENTS Page
ACKNOWLEDGEMENTS................................................................................................iii
ABSTRACT..................................................................................................................... iv
LIST OF TABLES............................................................................................................vii
LIST OF FIGURES.........................................................................................................viii
CHAPTERS
1. INTRODUCTION..............................................................................................1
2. A SPECIES BARRIER LIMITS TRANSMISSION OF CHRONIC WASTING
DISEASE TO MINK (Mustela vison)....................................................................15
3. TRANSGENESIS OF A BACTERIAL ARTIFICAL CHROMOSOME RESULTS
IN STABLE TRANSCRIPTION AND TRANSLATION OF MULE DEER PRION
PROTEIN AND REPLICATION OF CWD PATHOGENESIS ………….............. 43
4. OPPV PROVIRUS LEVELS ARE UNAFFECTED BY THE PRNP 171R
ALLELE……..……….……………………………………………………………...….74
5. CONCLUDING REMARKS….…………………………………………….……...88
BIBLIOGRAPHY.............................................................................................................92
APPENDIX
A. ATTRIBUTIONS TO CONTRIBUTING AUTHORS….….…………...............104
B. NOTES ON PRION DISINFECTION……………………………………..…....106
vi

LIST OF TABLES
1. Table 1-1: Examples of prion diseases and major causal link…………….……………2
2. Table 3-1: Animal numbers by treatment group for transgenic mouse challenge…..57
3. Table 3-2: Results of pronuclear microinjection of MD BAC DNA……………………61
4. Table 3-3: Current findings in MD BAC mice challenged with CWD ……...…………67
5. Table 4-1: Distribution of sample set by breed and age………………………………78
6. Table 4-2: Number of OPPV positive or negative sheep among PRNP genotypes…82 7. Table 4-3: Significance level for effect of PRNP genotype upon frequency of OPPV
positive animals. ……………………………………………………………………….…….. 82
8. Table 4-4: Significance level of OPPV proviral load levels between PRNP
genotypes……………………………………………….………………………………….…..84
vii

LIST OF FIGURES
1. Figure 1-1: Diagramatic representation of change in shape of cellular prion protein to
abnormal state…..............................................................................................................5
2. Figure 1-2: Diagram of nucleation between normal prion (PrPc) to abnormal prion
(PrPc) with subsequent fibril formation..............................................................................5
3. Figure 1-3: Prion transmission within a species………………………………………….7
4. Figure 1-4: Prion transmission between species supported by experimental
evidence………………………………………………………………………………………….8
5. Figure 1-5: Prion protein amino acid alignment of mustelids, ruminants, and humans
…………………………………………………………………………………………….……..12
6. Figure 2-1: Immunoreactivity and antigen load in elk brain samples used for
experimental challenge...................................................................................................26
7. Figure 2-2: Photomicrographs illustration vacuoles in TME and CWD positive IC
recipients........................................................................................................................29
8. Figure 2-3: Photomicrographs of PrPd IHC in brain and retina from TME positive IC
and CWD positive IC recipients…..................................................................................30
9. Figure 2-4: Scores of vacuolation and PrPd IHC signal intensity in TME and CWD
positive IC recipients.......................................................................................................31
10. Figure 2-5: Photomicrograph of astrocytes in cerebral cortex and hippocampus....32
11. Figure 2-6: Astrocyte counts by brain region...........................................................32
12. Figure 2-7: Western blot of PK digested brain homogenates from positive IC
recipients………..............................................................................................................33
viii

13. Figure 2-8: Comparative amino acid alignment illustrating positions of disparity
between mustelids and cervids or within mustelids that may effect TSE
susceptibility…………………………………………………………....................................36
14. Figure 3-1: Diagram of serial passage approach to overcome natural murine
resistance to prion disease……………………………………………………………...……45
15. Figure 3-2: Diagram of transgenic approach to overcome natural murine resistance
to prion disease……………………………………………………………………….....…….46
16. Figure 3-3: Diagram of DNA molecule used as construct for pronuclear
microinjection………………............................................................................................48
17. Figure 3-4: General strategy for creation of transgenic founder mice….……………50
18. Figure 3-5: Diagram of backcross breeding to generate Tg mouse of uniform
genetic background or shortcut step breeding…………………………………………......52
19. Figure 3-6: Diagram for breeding transgenic mouse to prion knockout mouse to
eliminate endogenous mouse gene …………………………………………………….…..53
20. Figure 3-7: Representative agarose gel demonstrating positive ORF and PPR of
MD BAC gene in transgenic founder mice……………………………………………….…62
21. Figure 3-8: Representative agarose gel demonstrating PCR products of the wild
type mouse prion gene or the NEO marker cassette of prion deletion……………..……63
22. Figure 3-9: Representative agarose gel confirming MD Tg expression in MD BAC
mouse brain, liver, and spleen by RT-PCR……………………………..………………..…64
23. Figure 3-10: Representative agarose gel confirming MD Tg expression in MD BAC
mouse brain by intron-spanning RT-PCR…………………………………………..…........64
ix

24. Figure 3-11: Representative western blot of PrPc in tissue from MD BAC
mouse………….....……………………………………………………………………….……65
25. Figure 3-12: Immunoreactivity and measurement of antigen load in CWD positive
and CWD negative mule deer brain samples used for experimental challenge………..66
26. Figure 3-13: Western blot of PrPd in brain tissue from MD BAC mouse post
challenge with CWD………………………………………………………………………...…68
27. Figure 3-14: Photomicrographs of the central nervous system from Tg338 and MD
BAC transgenic mice challenged with scrapie and CWD, respectively..........................69
28. Figure 4-1: Number of sheep distributed among PRNP genotypes………………....81
29. Figure 4-2: Odds ratio and 95% confidence interval for effect of PRNP genotype
upon frequency of OPPV positive animals………………………………………………….82
30. Figure 4-3: Provirus levels among PRNP genotypes…………………………….……83
31. Figure 4-4: Adjusted mean log10 provirus levels and 95% confidence interval
among PRNP genotypes used for statistical comparison………………………………....84
x

xi
Dedication
This dissertation is dedicated to my wife Jean for her love, wisdom, and support.

CHAPTER ONE
INTRODUCTION
Transmissible Spongiform Encephalopathies (TSE), also known as prion disorders, are
a group of invariably fatal neurodegenerative diseases affecting humans, domestic
animals, and wildlife. Examples include variant Cruetzfeldt-Jakob Disease (CJD) and
Kuru in humans, scrapie in sheep, chronic wasting disease (CWD) in deer, elk, and
moose, Transmissible Mink Encephalopathy (TME) in mink, and Bovine Spongiform
Encephalopathy (BSE) in cattle (Table 1-1). Scrapie has been known for over 300
years, whereas other forms of TSE are referred to as “new” disease being first reported
in the last 50 years. TSE are characterized by a chronic, invariably fatal, sponge-like
degeneration of the central nervous system (CNS) with accumulation of abnormal
protease resistant prion protein (PrPd), a conformational isoform of the normal protease
sensitive host prion protein (PrPc) [for review see Haywood, 1997; Johnson & Gibbs,
1998; Prusiner, 1998]. Despite recognition and description of these diseases much
remains uncertain regarding prion genetics, the function of PrPc, and mechanisms of
PrPd pathogenesis.
TSE transmissibility is usually limited to hosts of the same species. However,
transmission from one host species to another species has also been documented. The
genetics of the host prion gene (PRNP) determine relative TSE susceptibility within a
species (Prusiner, 1998); how the same genetic factors may affect transmission
between two different species is unclear. Central questions remain regarding the effect
of PRNP genetics on regulation of TSE within a species, effect upon TSE transmission
1

between species, and influence upon other disease processes. The increased
incidence of CWD throughout North America (Joly et al., 2003; Williams & Miller, 2002;
Williams et al., 2002), and increased concern about prion zoonosis (Bonetta, 2002) are
generating additional study of prion transmission within and between species to answer
these questions.
Table 1-1: Examples of prion diseases and major causal link. HGH = human growth
hormone. MBM = meat and bone meal.
HOST DISEASE ACRONYM PATHOGENESIS/TRANSMISSION
Humans Creutzfeldt-Jakob Disease
CJD
new variant vCJD Consumption of BSE infected material familial fCJD Genetic mutation iatrogenic iCJD HGH, tissue graft, instruments sporadic sCJD Spontaneous mutation or PrP conversion
Humans Kuru Cannabalism Humans Fatal Familial Insomnia FFI Genetic mutation Humans Gerstmann-Straussler-
Scheinker syndrome GSS Genetic mutation
Sheep Scrapie Sc Maternal and lateral transmission (placenta, fetal fluids)
Deer, Elk Chronic Wasting Disease
CWD Lateral (Oral). ?blood, urine, feces, other?
Cattle Bovine Spongiform Encephalopathy
BSE Contaminated feedstuffs (MBM)
Mink Transmissible Mink Encephalopathy
TME Contaminated feedstuffs
Cats Feline Spongiform Encephalopathy
FSE Contaminated feedstuffs
Ungulates Exotic Ungulate Encephalopathy
Contaminated feedstuffs (MBM)
Pigs Experimental model Intracardiac, intraperitoneal Hamsters Experimental model Intracranial, intraperitioneal, orally Mice Experimental model Intracranial, intraperitoneal, orally
Normal Prion Biology
Host PrPc is a cell surface glycoprotein that is widely conserved in mammals. It is
expressed from embryogenesis through adulthood. PrPc is ubiquitous in neurons,
2

astrocytes, and glial cells of the central nervous system and in antigen presenting cells.
The prion gene contains three exons, with the entire open reading frame encoded in the
third exon, and produces a protein of 253 amino acid residues (varies slightly by
species). Post translational modifications include glycosylation of two asparigine
residues (codons 181 and 197), formation of a disulphide bridge (cysteines 179 and
214), attachment of a carboxy-terminal glycophosphatidylinositol anchor at codon 231,
and cleavage of an amino-terminal membrane signaling sequence at codon 23 with final
outer membrane localization tethered in lipid rafts of the cell membrane. The protein
has a four to six tandem repeat sequence of eight amino acid residues near the N-
terminus that correspond to copper binding domains (Burns et al., 2002). Copper
binding has been confirmed in vivo (Brown et al., 1997a) and bound copper stimulates
prion endocytosis (Pauly & Harris, 1998) with subsequent cytoplasmic recycling to the
cell surface. Degradation of PrPc is likely mediated by the ubiquitin proteasome system
(Yedidia et al., 2001).
PrPc biology has been implicated in neuronal and immunologic activities (Colling et al.,
1996; Mabbott & Bruce, 2001), however, the critical role of PrPc in daily function
remains controversial [for review see Riesner, 2003; Westergard et al., 2007]. Certainly
PrPc is necessary for TSE pathogenesis since scrapie can not be reproduced in PrPc
deficient mice (Bueler et al., 1992; Sailer et al., 1994); however, what happens in the
absence of TSE infection? Knockout strategies to test normal prion function have had
variable results including no observed phenotype (Bueler et al., 1992) altered circadian
rhythm and behavior (Tobler et al., 1996), or loss of purkinje cells (Sakaguchi et al.,
3

1996). Observations have been made of increased serum copper and decreased
superoxide dismutase activity in PrP knockout mice (Brown et al., 1997a; Brown et al.,
1997b). Others have had contradictory results, indicating no change in copper content
or cuproenzyme activity in mice with varied levels of PrP expression (Waggoner et al.,
2000). In addition to copper homeostasis (Brown et al., 1997a; Prusiner, 1998), some
experiments suggest a role for PrP in synaptic transmission (Collinge et al., 1994),
signal transduction (Mouillet-Richard et al., 2000), or oxidative stress (Brown et al.,
1999; Sorenson, 2001; Wong et al., 2001). The lack of a consistent phenotype may be
due to functionally redundant compensation by other mechanisms or conversely a lack
of sufficient stressors to unmask the phenotype. Additional study of PrPc biology,
particular the role of divalent cations upon protein folding, may provide useful insights to
TSE pathogenesis.
TSE Pathogenesis
Natural TSE pathogenesis can be conceptualized in phases. The first involves entry of
infectious material into the host by oral exposure. The second is a period of
conformational change where PrPc of mostly α-helical structure is converted to β-
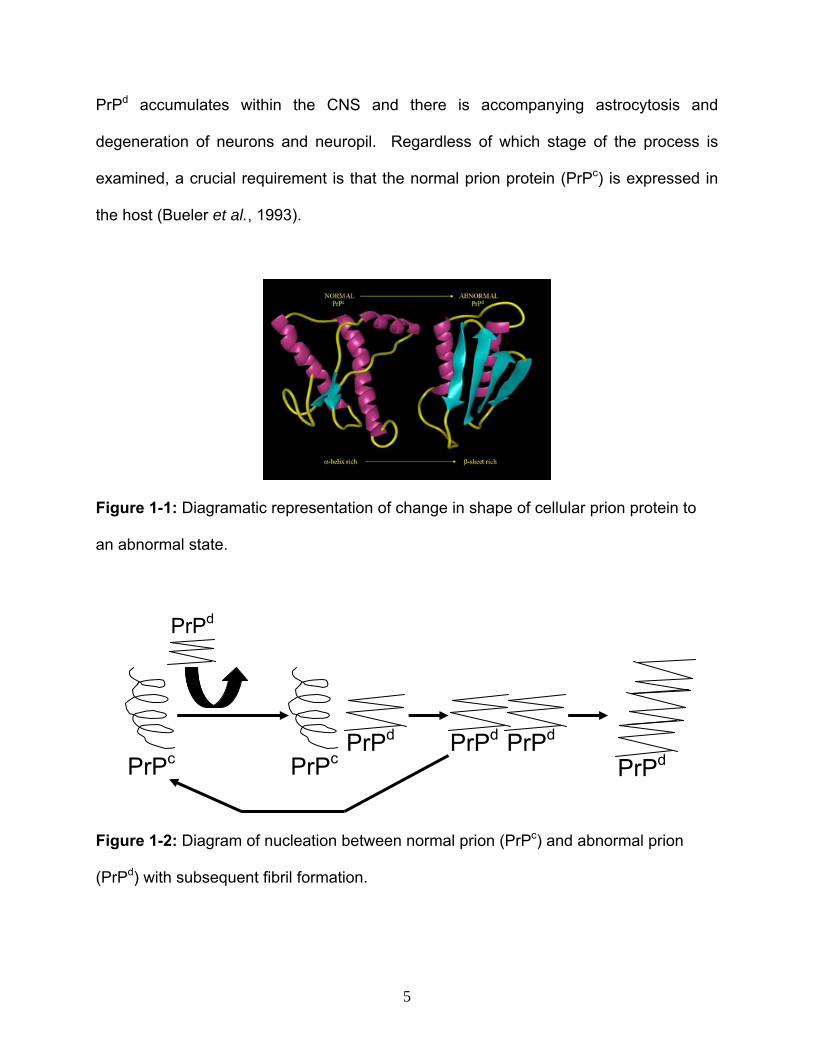
pleated sheet rich PrPd, presumably within lymphoid tissue (Figure 1-1). The
conformational conversion is hypothesized to occur through binding between normal
and abnormal material (Figure 1-2). It is widely accepted that prion protein, while
necessary, is not sufficient for disease and that an unidentified cofactor or molecular
event is required for conversion. Third, there is transport to the central nervous system
(CNS) either hematogenously by lymphoid cells or retrograde along nerves. Finally,
4
PrPd accumulates within the CNS and there is accompanying astrocytosis and
degeneration of neurons and neuropil. Regardless of which stage of the process is
examined, a crucial requirement is that the normal prion protein (PrPc) is expressed in
the host (Bueler et al., 1993).
Figure 1-1: Diagramatic representation of change in shape of cellular prion protein to
an abnormal state.
PrPc PrPd
PrPdPrPcPrPdPrPd
PrPd
Figure 1-2: Diagram of nucleation between normal prion (PrPc) and abnormal prion
(PrPd) with subsequent fibril formation.
5

Biochemical studies of TSE have demonstrated PrPd in lymphoid tissue both local and
distant to the intestine (Andreoletti et al., 2002a; Beekes & McBride, 2000; Bons et al.,
1999; Heggebo et al., 2003; Miller & Williams, 2002; Sigurdson et al., 2002; Sigurdson
et al., 2001; Sigurdson et al., 1999; Terry et al., 2003). PrPd has also been
demonstrated within follicular associated epithelium (FAE) overlaying gut associated
lymphoid tissue (GALT) in studies of scrapie and Bovine Spongiform Encephalopathy,
suggesting a role for M-cells as entry point for infectious material (Beekes et al., 1998;
Bons et al., 1999; Heggebo et al., 2000). Other cells implicated in pathogenesis include
follicular dendritic cells (FDC) (Beekes & McBride, 2000; Herrmann et al., 2003;
Kitamoto et al., 1991; Lezmi et al., 2001; Sigurdson et al., 2002), and B-lymphocytes
(Klein et al., 1997). B cells do not need to express PrPc, rather the B cell role appears
to be via induction of FDC development t(Klein et al., 1998). Macrophages contain
PrPd, but this is likely due to phagocytosis of FDC or B cell components (Sigurdson et
al., 2002). T lymphocytes are generally accepted to be unimportant in prion
pathogenesis (Nicotera, 2001). Despite the identification of PrPd within the
aforementioned cells and the corresponding implication of involvement in pathogenesis,
it is uncertain how the conversion process begins and what role genetic factors play in
promoting or inhibiting the process.
Cross-Species Transmission
Transmissibility of TSE within a species was first documented in studies of scrapie
(Cuille & Chelle, 1936) and Kuru (Gajdusek & Zigas, 1957). Both scrapie and CWD are
readily transmissible within ovid or cervid species, respectively. Conversely,
6

transmssion between cattle, between mink, or between humans affected by other types
of TSE is extremely rare (Figure 1-3).
Figure 1-3: Prion transmission within a species. Top = transmission is common in
sheep, deer, or elk. Bottom = transmission is rare in mink, cattle, or humans.
The potential for cross-species TSE transmission became apparent with evidence that
variant CJD in humans and spongiform encephalopathy in zoo animals originated from
consumption of BSE infected cattle (Bons et al., 1997; Bruce et al., 1997; Collinge,
1999; Ghani, 2002). Outbreaks of TME in mink are likely another example of cross-
species transmission, possibly originating from sheep, cattle, or other sources (Marsh &
Hadlow, 1992). These findings, combined with the spread of CWD in North America,
have raised concern that CWD may cause cross-species infections (Bonetta, 2002).
The host range of CWD could include carnivores (e.g. mink and ferrets), agriculturally
7
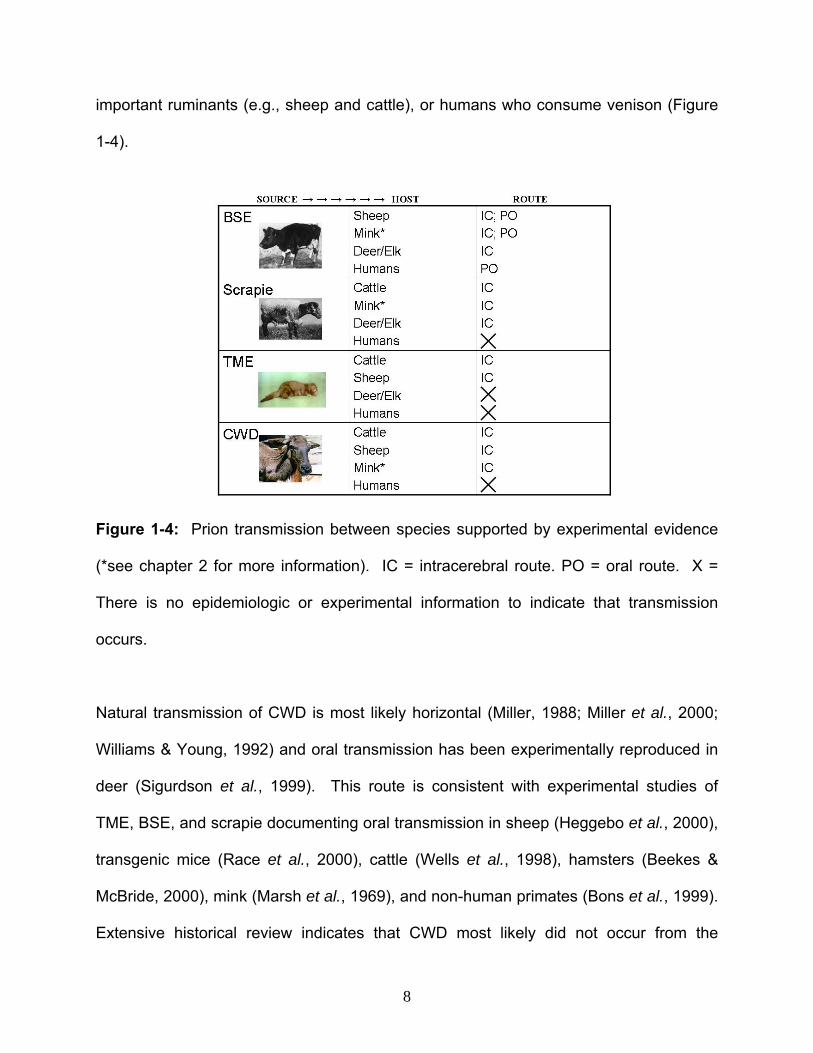
important ruminants (e.g., sheep and cattle), or humans who consume venison (Figure
1-4).
Figure 1-4: Prion transmission between species supported by experimental evidence
(*see chapter 2 for more information). IC = intracerebral route. PO = oral route. X =
There is no epidemiologic or experimental information to indicate that transmission
occurs.
Natural transmission of CWD is most likely horizontal (Miller, 1988; Miller et al., 2000;
Williams & Young, 1992) and oral transmission has been experimentally reproduced in
deer (Sigurdson et al., 1999). This route is consistent with experimental studies of
TME, BSE, and scrapie documenting oral transmission in sheep (Heggebo et al., 2000),
transgenic mice (Race et al., 2000), cattle (Wells et al., 1998), hamsters (Beekes &
McBride, 2000), mink (Marsh et al., 1969), and non-human primates (Bons et al., 1999).
Extensive historical review indicates that CWD most likely did not occur from the
8

feeding of contaminated feedstuffs, unlike some other TSE (Williams & Young, 1992).
Co-housing of sheep and deer that subsequently developed scrapie and CWD,
respectively, has raised concern that scrapie and CWD have a common etiology but this
remains speculative. Alternatively, CWD may have long been present in wild
populations only to be discovered with the advent of increased monitoring and
diagnostic surveillance.
To date, neither CWD nor scrapie has been conclusively linked to human disease. A
report examining three U.S. cases of CJD in young patients who ate venison did not find
a causal link (Belay et al., 2001) and a news report of three hunters contracting vCJD
from deer and elk meat has been determined to be unfounded (CDC, 2003). Cross-
species CWD transmission experiments have been performed by intracerebral (IC)
inoculation of laboratory animals. CWD has little IC infectivity in mice, with only one
study indicating “a very few mice” becoming ill after 500 days of incubation (no further
enumeration was included) (Bruce et al., 2000). IC injected hamsters only develop
disease after CWD is serially passaged through IC injected ferrets (Bartz et al., 1998).
IC injection of CWD has also been studied in cattle, where two of thirteen animals have
developed disease (Hamir et al., 2001); this trial is ongoing. Although these previous
cross-species studies provide data on whether prion conversion can occur in a given
host, they bypass the most probable scenario of oral exposure thus telling us little about
natural CWD transmission. There is a need for additional study of CWD transmission
using the oral route, whether it is in rodents or natural hosts. Mink are a logical choice
to evaluate cross-species CWD transmission by virtue of their susceptibility to oral
9

TSE’s as established for the natural disease TME (Marsh & Hadlow, 1992) and for
experimental infection with BSE (Robinson et al., 1994). If we are to understand natural
cross-species TSE transmission to carnivores and other hosts it is imperative to perform
studies via the oral route.
Disease Susceptibility Polymorphisms
Specific amino acid polymorphisms within the host prion protein have been shown to
influence disease susceptibility within a species for both scrapie and CJD (Prusiner,
1998). The affect of PRNP variation upon traits and diseases other than TSE are
uncertain. Amino acid alignment illustrates that while the majority of residues are
conserved among different species, there are particular codons that may influence
transmission within and between species (Figure 1-5). Susceptibility to scrapie in sheep
is associated with homozygosity for alanine, arginine, and glutamine at codons 136, 154
and 171, respectively (O'Rourke, 2001); such knowledge has been fundamental in
developing scrapie control measures through selective breeding of sheep. The human
prion contains a methionine/valine polymorphism at codon 129 and variant CJD patients
are homozygous for methionine at this position (Zeidler et al., 1997). A
methionine/leucine polymorphism has been identified at codon 132 in elk (O'Rourke et
al., 1999), which positionally corresponds to codon 129 in humans. The elk
polymorphism effects susceptibility and incubation time in elk CWD infection (Hamir et
al., 2006a; O'Rourke et al., 2007).
10

Amino acid polymorphisms may also account for resistance to cross-species infection
(Bartz et al., 1994), however the effect of such variation is undetermined in most cases.
When infectious material from one species affected by a TSE is introduced into a
different species, it typically results in a lengthened incubation period, lowered infection
rates, atypical clinical response, and/or atypical histologic lesions (Pattison, 1966;
Prusiner, 1998). Factors of dose, exposure route, and source of material are certainly
important, but do not fully explain these changes in disease course. Molecular in vitro
conversion assays indicate that cross-species infection can be predicted by examining
the alignment of protein sequence between the endogenous host prion (PrPc) and the
exogenous infectious form (PrPd) (Barron et al., 2001; Race & Chesebro, 1998; Race et
al., 2001; Raymond et al., 2000; Raymond et al., 1997). Comparison of such
alignments to results of experimental cross-species infection may allow correlation of
host genotype to disease susceptibility status, thus identifying amino acid residues that
are important determinants of disease progression. Understanding such
polymorphisms will be important in the control of TSE transmission both within and
between species. Polymorphisms in other genes outside of the prion gene may also
affect disease susceptibility. Many biologic processes and disease are the result of
polygenetic effects. However, it is currently unknown how and if other genetic factors
(e.g. prion pseudogenes, prion regulatory regions, other genes, or immunity haplotypes)
are involved in prion pathogenesis or if PRNP variation effects traits and diseases other
than TSE.
11

Codon 1 60 • • Mink MVKSHIGSWLLVLFVATWSDIGFCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW Ferret MVKSHIGSWLLVLFVATWSDIGFCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW White Tail Deer MVKSHIGSWILVLFVAMWSDVGLCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW Mule Deer MVKSHIGSWILVLFVAMWSDVGLCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW ELK MVKSHIGSWILVLFVAMWSDVGLCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW Sheep MVKSHIGSWILVLFVAMWSDVGLCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW Cattle MVKSHIGSWILVLFVAMWSDVGLCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW Human --MANLGCWMLVLFVATWSDLGLCKKRPKPGG-WNTGGSRYPGQGSPGGNRYPPQGGGGW 61 111 • • Mink GQPHGGGWGQPHGGGWGQPHGG--------GWGQPHGGGGWGQGGGSHGQWGKPSKPKTN Ferret GQPHGGGWGQPHGGGWGQPHGG--------GWGQPHGGGGWGQGGGSHGQWGKPSKPKTN White Tail Deer GQPHGGGWGQPHGGGWGQPHGG--------GWGQPHGGGGWGQSG-THSQWNKPSKPKTN Mule Deer GQPHGGGWGQPHGGGWGQPHGG--------GWGQPHGGGGWGQGG-THSQWNKPSKPKTN ELK GQPHGGGWGQPHGGGWGQPHGG--------GWGQPHGGGGWGQGG-THSQWNKPSKPKTN Sheep GQPHGGGWGQPHGGGWGQPHGG--------GWGQPHGGGGWGQGG-SHSQWNKPSKPKTN Cattle GQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGGWGQGG-THGQWNKPSKPKTN Human GQPHGGGWGQPHGGGWGQPHGG--------GWGQPHG-GGWGQGGGTHSQWNKPSKPKTN 112 132 136 154 171 • • • • • Mink MKHVAGAAAAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYKPVDQ Ferret MKHVAGAAAAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYKPVDQ White Tail Deer MKHVAGAAAAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYRPVDQ Mule Deer MKHVAGAAAAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYRPVDQ ELK MKHVAGAAAAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYRPVDQ Sheep MKHVAGAAAAGAVVGGLGGYMLGSAMSRPFIHFGNDYEDRYYRENMYRYPNQVYYRPVDQ Cattle MKHVAGAAAAGAVVGGLGGYMLGSAMSRPLIHFGSDYEDRYYRENMHRYPNQVYYRPVDQ Human MKHMAGAAAAGAVVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDE • 129 in humans 172 178 223 231 • • • • Mink YSNQNNFVHDCVNITVKQHTVTTTTKGENFTETDMKIMERVVEQMCVTQYQRESEAYYQR Ferret YSNQNNLVHDCVNITVKQHTVTTTTKGENFTETDMKIMERVVEQMCVTQYQQESEAYYQR White Tail Deer YNNQNTFVHDCVNITVKQHTVTTTTKGENFTETDIKMMERVVEQMCITQYQRESQAYYQR Mule Deer YNNQNTFVHDCVNITVKQHTVTTTTKGENFTETDIKMMERVVEQMCITQYQRESQAYYQR ELK YNNQNTFVHDCVNITVKQHTVTTTTKGENFTETDIKMMERVVEQMCITQYQRESEAYYQR Sheep YSNQNNFVHDCVNITVKQHTVTTTTKGENFTETDIKIMERVVEQMCITQYQRESQAYYQR Cattle YSNQNNFVHDCVNITVKEHTVTTTTKGENFTETDIKMMKRVVEQMCITQYQRESQAYYQR Human YSNQNNFVHDCVNITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQR 232 256 • • Mink GASAILFSPPPVILLISLLILLIVG Ferret GASAILFSPPPVILLISLLILLIVG White Tail Deer GASVILFSSPPVILLISFLIFLIVG Mule Deer GASVILFSSPPVILLISFLIFLIVG ELK GASVILFSSPPVILLISFLIFLIVG Sheep GASVILFSSPPVILLISFLIFLIVG Cattle GASVILFSSPPVILLISFLIFLIVG Human GSSMVLFSSPPVILLISFLIFLIVG KEY X= Codon positions where elk and mink agree but differ from sheep, cattle, or humans. X= Codon positions that differ from elk X= Known or suspected codon that effects TSE susceptibility. Putative susceptibility residue is shown. Known alternative alleles are: Leucine at codon 132 of elk; Valine at codon 129 of humans; Valine at codon 132, Histidine at codon 154, and Histidine or Arginine at codon 171 of sheep. X= Additional 8 amino acid repeat found in cattle
Figure 1-5: Prion protein amino acid alignment of mustelids, ruminants, and humans (all codons numbered relative to the elk sequence)
12

Dissertation Subject Items
Information on TSE has increased greatly over the past decade. However the ability for
prion diseases to induce cross-species infections following natural oral exposure is still
unclear and is complicated by information from studies by intracerebral inoculation that
may not translate to natural orally mediated disease transmission. Furthermore, while
some genetic factors that effect transmission within species have been identified the
mechanistic basis for these factors is elusive and it is unclear whether these same
factors apply to transmission between species. In the following chapters I will address
some of the aforementioned issues by documenting research I conducted investigating
prion transmission. These studies focus on the effects of PRNP ORF and putative
promoter regions on prion transmission and possible effects of PRNP variants on other
infectious diseases.
The specific topics are as follows:
1. Ruminant to carnivore prion transmission. I test the hypothesis that primary oral
CWD challenge causes a prion disease in mink; I also performed primary IC CWD
challenge to compare lesions to those of experimental TME. This study examines
PRNP genotype in elk with naturally occurring CWD and a population of mink. It
provides context to species barriers in transmission of prion disease from ruminants to
carnivores.
2. Effect of the cervid putative promoter region on development of prion disease in
mouse models. I tested the hypothesis that transgenic incorporation of the cervid
putative promoter region renders mice susceptible to intracerebral, intraperitoneal, and
13

oral challenge with CWD. This study provides context to the role of PRNP promoter
regions in the development of disease and presents a novel model that may facilitate
future investigations into prion pathogenesis.
3. Effect of PRNP genotype selection upon other infectious diseases. This study
examines whether the predominant economically important infectious disease of sheep,
OPPV, may be affected by selective breeding programs for scrapie control. I tested the
hypotheses that the sheep PRNP 171R allele is associated with 1) the presence of
ovine progressive pneumonia virus (OPPV) provirus and 2) higher OPPV provirus
levels. It provides context to help guide sheep producers when considering PRNP
genetics of herd constituents.
14

CHAPTER TWO
A SPECIES BARRIER LIMITS TRANSMISSION OF
CHRONIC WASTING DISEASE TO MINK (Mustela vison) @
Robert D. Harrington,1,2,3٭ Timothy V. Baszler,1 Katherine I. O’Rourke,1,3 David A.
Schneider, 1,3 Terry R. Spraker,4 H. Denny Liggitt,2 and Donald P. Knowles1,3.
1Department of Veterinary Microbiology and Pathology, Washington State University,
Pullman, WA, 99164-7040, USA
2Department of Comparative Medicine, University of Washington, Seattle, WA 98195-
7190, USA
3Animal Disease Research Unit, Agricultural Research Service, US Department of
Agriculture, Pullman, WA 99164-6630, USA
4Department of Microbiology, Immunology, and Pathology, Colorado State University,
Fort Collins, CO 80523-1619, USA
@Original source published in Journal of General Virology; Volume 89 (4), page 1086-
1096, April 2008.
.Author for correspondence, [email protected]٭
See Appendix A for attributes of contributing authors.
Summary
Transmissible Mink Encephalopathy (TME) occurs as sporadic outbreaks associated
with ingestion of feed presumably contaminated with some type of prion disease. Mink
lack a species barrier to primary oral challenge with Bovine Spongiform
15

Encephalopathy, whereas they have a barrier to such challenge with scrapie. We
investigated whether mink have a species barrier to chronic wasting disease (CWD) by
performing primary intracerebral (IC) and primary oral challenge with CWD positive elk
brain. Primary IC challenge resulted in clinical disease in 2/8 mink at 31 to 33 months
incubation. Affected mink had spongiform vacuolation and astrocytosis within the
central nervous system and immunoreactivity to disease associated prion protein (PrPd)
in brain, retina and lymph node. CWD IC recipients had significantly lower brain
vacuolation and PrPd deposition scores, significantly lower cerebrocortical astrocyte
counts and significantly higher hippocampal astrocyte counts, than TME IC recipients.
Primary oral challenge with CWD positive elk brain (n=22), or CWD negative elk brain
given IC (n=7) or orally (n=23), did not result in clinical or microscopic abnormalities
during 42 months observation. Novel prion gene polymorphisms were identified at
codon 27 (arginine/tryptophan) and codon 232 (arginine/lysine). This study shows that,
while CWD can cause disease when given IC to mink, the lesions are not characteristic
of TME, it is inefficient compared to TME, and oral challenge does not result in disease.
The demonstration of a species barrier in cervid to mustelid prion transmission indicates
mink are unlikely to be involved in natural CWD transmission.
Introduction
Transmissible Mink Encephalopathy (TME) is an uncommon form of prion disease that
has occurred in sporadic outbreaks on commercial mink farms in North America,
Finland, Germany and Russia (Marsh & Hadlow, 1992). Brains from affected mink have
hallmark lesions of Transmissible Spongiform Encephalopathy (TSE) including
16

spongiform vacuolation and astrocytosis that are pronounced throughout the
telencephalon, diencephalon, and mesencephalon (Hadlow & Karstad, 1968; Hartsough
& Burger, 1965). Investigation of TME outbreaks implicated ingestion of ruminant tissue
contaminated with some type of prion as the source of disease (Hartsough & Burger,
1965). However, the ruminant species from which the infected tissue originated is
controversial [for review see Marsh & Bessen, 1993; Marsh & Hadlow, 1992], creating
uncertainty about what role mink may have in natural transmission of prion diseases.
Species barriers in ruminant to mink prion transmission have been evaluated
experimentally (defined for purposes of this report as inefficient primary IC transmission
and lack of primary oral transmission). Primary IC or primary oral challenge with BSE
readily causes a TSE in mink indicating a lack of species barrier from cattle to mink
(Robinson et al., 1994). Conversely, a species barrier exists between sheep and mink
as primary oral challenge with scrapie has not produced disease (Marsh et al., 1991;
Marsh & Hanson, 1979); disease only occurs after IC administration (Hanson et al.,
1971; Marsh & Hanson, 1979). While lesions in the telencephalon, diencephalon, and
mesencephalon of mink challenged IC with TME, BSE or scrapie are similar to natural
TME (Eckroade et al., 1979; Hadlow & Karstad, 1968; Hanson et al., 1971; Hartsough &
Burger, 1965; Marsh & Hadlow, 1992; Marsh & Hanson, 1979; Robinson et al., 1994),
caudal brainstem lesions indicate differences exist among ruminant source species as
these lesions are consistently found in experimental TME or BSE (Eckroade et al.,
1979; Robinson et al., 1994), but are often absent with scrapie challenge (Hanson et al.,
1971).
17

Chronic wasting disease (CWD) is a third ruminant TSE that may be transmissible to
mink, however species barrier characteristics are uncertain as data on primary IC
challenge is unpublished (Williams, 2005) and oral passage has never been performed.
A species barrier has been demonstrated in CWD transmission to ferrets, another
mustelid carnivore similar to mink but without a history of natural TSE. Primary IC
challenge of ferrets causes disease (Bartz et al., 1998) but primary oral challenge does
not; rather CWD material must undergo serial IC passage in ferrets before it will cause
orally mediated disease (Perrott et al., 2004; Sigurdson et al., 2003). Mink and ferrets
differ in susceptibility to experimental TME as the IC incubation period in ferrets is eight
times longer than in mink (Bartz et al., 1994). Whether mink and ferrets also have
differential susceptibility to CWD is undetermined.
We initiated this study of experimental prion transmission from cervids to mink to gain
insight into whether mustelids could be involved in natural CWD transmission. The first
cases of CWD date back to at least 1967 in Colorado and Wyoming (Spraker et al.,
1997; Williams & Young, 1980); as the majority of CWD surveillance programs were
initiated in the past ten years, CWD may have previously gone undetected in North
American wildlife. CWD also occurs in many other areas of North America including
Wisconsin, Minnesota, and Ontario (Williams, 2005). From 1947 to 1985, cases of TME
were documented in the United States and Canada including Wisconsin, Minnesota
(Hartsough & Burger, 1965; Marsh et al., 1991) and Ontario (Hadlow & Karstad, 1968).
Thus, CWD geographically and temporally overlaps with some cases of prion disease in
18

mink. Dietary practices in the mink industry may facilitate food borne prion transmission
as rations are typically prepared by grinding and mixing whole animal carcasses which
are then fed in their entirety. If deer or elk tissue, such as from hunting or road-kills,
were inadvertently included in mink rations then it might be a source of TSE in mink
unbeknown to ranchers, producers, or scientific investigators. If orally administered
CWD were to cause disease in mink, then mink could serve as a disease reservoir in
the wild as these carrion consumers are widely distributed throughout North America.
We performed an oral transmission experiment with elk CWD to test the hypothesis that
primary oral CWD challenge causes a prion disease in mink; we also performed primary
IC CWD challenge to compare lesions to those of experimental TME. This study
provides context to species barriers in transmission of prion disease from ruminants to
carnivores.
Methods
Animals: 60 weanling male and female black mink (e.g. non-Aleutian Disease
phenotype (Marsh et al., 1976) were purchased from a commercial breeder with no
history of TME in the CWD free state of Washington and cared for under guidelines of
the Washington State University Institutional Animal Care and Use and Institutional
Biosafety committees. Animals were given a 4-way vaccine (Distox plus, Schering-
Plough; Kenilwith, NJ) for distemper virus, Pseudomonas aeruginosa, Clostridium
botulinum, and parvoviral enteritis, and dewormed with ivermectin (Merck and
Company; Whitehouse Station, NJ). The breeder used a fish and poultry based wet
feed and kits were adapted to a pelleted ration free of ruminant protein fed ad libidum
19

(MSC; Dundee, IL). Animals were individually housed in stainless steel wire cages with
dedicated nest boxes, located in a secure animal biosafety level-2 facility.
Preparation of inocula: Inocula were prepared from elk brain stored at -20 °C. CWD
positive brains came from elk in Rocky Mountain National Park with naturally occurring
CWD and CWD negative brain came from a normal elk in a closed CWD free herd. Half
brains (including brainstem, cerebellum and cerebrum) were homogenized in sterile
disposable tissue grinders (VWR International; West Chester, PA) and diluted to a final
concentration of 40% (w/v) for feeding and 10% (w/v) for IC injection in sterile saline
(Sigurdson et al., 1999). Bacterial contamination was assessed on 10% sheep blood
agar, and all samples underwent a 3 phase water bath heat treatment cycle of 80 °C for
15 minutes (mins), 37 °C for 60 mins, and 80 °C for an additional 15 mins (bacteria was
found only in CWD positive and TME positive material pretreatment). Gentamycin was
added to IC inocula at 100 µg ml-1. Inocula were stored at -20 °C until use.
Inocula characterization by western and slot blot: PrPd content of elk brain samples
was confirmed by western blot, and antigen load determined by semi-quantitative slot
blot modified from a dot blotting procedure (O'Rourke et al., 2003). Proteinase K (PK)
digest was performed at 50 µg ml-1 at 56 °C for 30 mins, with inactivation at 90 °C for 10
mins. Brain homogenate from scrapie infected sheep or clinically normal elk were used
as positive and negative controls, respectively. Western blot samples were denatured,
run on a 12% bis-tris gel in MOPS SDS running buffer (Invitrogen; Camarillo, CA) at 200
volts for 1 hour (hr), and transferred to methanol soaked PVDF membrane in MOPS
20

transfer buffer (Invitrogen; Camarillo, CA) at 200 mAmps for 1 hr. Slot blot test samples
and a plasmid derived recombinant PrP (rPrP) densitometric reference standard (K.
O’Rourke, USDA-Agricultural Research Service; Pullman, WA) were denatured and
serially diluted 1:2. Duplicate lanes of rPrP ranging from 0.66 to 21.13 ng, one lane of
CWD negative homogenate, and 5 replicate lanes of CWD positive material were
spotted onto nitrocellulose membranes (Sigma-Aldrich; St. Louis, MO) using a slotted
manifold (Biorad Laboratories; Hercules, CA). Western and slot blot membranes were
dried, then blocked for 1 hour in tris-casein buffer (Roche; Palo Alto, CA), with 0.1%
Tween 20. Membrane transfer, blocking and all subsequent steps were done at room
temperature. Membranes were probed for 1 hr with 3.6 µg µl-1 of primary mouse
monoclonal antibody F99/97.6.1 (K. O’Rourke, USDA-ARS; Pullman, WA), that
recognizes prion epitope QYQRES (O'Rourke et al., 2000), followed by biotinylated goat
anti-mouse secondary antibody (Southern Biotech; Birmingham, AL) and enhanced
chemiluminescence (Amersham Biosciences; Piscataway, NJ). Western and slot blot
signal detection was performed with a commercial apparatus (Alphaimager, Alpha
Innotech Corporation; San Leandro, CA). A slot blot standard curve was generated from
densitometric values and known quantity of rPrP and compared to test sample values to
estimate PrPd concentration (ng per mg of wet tissue). Brains from study animals with
TSE underwent western blot and densitometric determination of glycoform ratios with
statistical significance (p ≤ 0.05) determined by the unpaired t-test (GraphPad 5.0; San
Diego, CA).
21

Experimental design and procedures for IC and oral challenge: Male and female
mink were randomly assigned to one of four primary challenge groups, CWD positive
inocula given IC (n=8) and orally (n=22), and CWD negative inocula given IC (n=7) and
orally (n=23). Additional mink were challenged IC with third passage Stetsonville TME
(Marsh et al., 1991) (n=2) or normal mink brain (n=2) for comparison with CWD.
Available Stetsonville TME was used in its entirety for IC challenge. TME and CWD
negative brain samples were administered to control for confounding variables from oral
or IC administration of homologous or heterologous brain tissue. IC injection was
performed using a xylazine-ketamine general anesthetic (Robinson et al., 1994) and
standard surgical site preparation. The skin was incised 2 to 3 cm, and the calvarium
perforated with a 5/16 inch carbide tipped drill bit. 100 µl of 10% (w/v) brain
homogenate was injected into the left cerebral hemisphere at a 1 cm depth. Oral
challenge groups were fed 1 ml of 40% (w/v) brain mixed with 5 grams of canned tuna
fish for five consecutive days and observed to verify consumption of test material
(Diringer et al., 1998; Robinson et al., 1994; Sigurdson et al., 1999).
Clinical observation and necropsy of study animals: Animals were monitored daily
for signs of neurologic disease including ataxia, muscle tremors, head pressing, hind
limb weakness, paresis, or paralysis. Clinical illness was defined as loss of appetite,
lethargy, change in aggressive behavior, decreased awareness of surroundings, or
neurologic symptoms. Animals that could not enter nest boxes or became moribund
were euthanized by intracardiac injection of sodium pentobarbital. Necropsy was
performed at 3, 4, 5, 6, 7, 11, 12, 14, 24, 27, 28, 32 and 38 months with the
22

development of neurologic symptoms or with symptoms related to other organs (e.g.
intercurrent disease). Representative tissue samples from ileum, cecum, colon, heart,
lungs, liver, kidney, spleen, mesenteric and retropharyngeal lymph nodes, cerebrum,
brainstem, and cerebellum were collected in 10% neutral buffered formalin and/or
frozen at -80°C.
Tissue processing and immunohistochemistry: Tissue was formalin fixed for at least
2 days, trimmed, treated with 96% formic acid for 1 hr, processed, paraffin embedded,
sectioned at 5 microns, and placed on glass slides for hematoxylin and eosin (H/E)
staining or immunohistochemistry (IHC). IHC was performed at 37 °C with an
automated immunostainer (Ventana Medical Systems; Tucson, AZ) on samples of
brain, lymph nodes, and/or spleen similar to previously described (Spraker et al., 2002).
Slides for PrPd IHC were blocked with EZ Prep and Cell Conditioner per manufacturers
instructions (Ventana Medical Systems; Tucson, AZ), probed with primary mouse IgG1
monoclonal antibody F99/97.6.1 (provided by K. O’Rourke, USDA-Agricultural Research
Service; Pullman, WA) at 5ug ml-1 for 30 mins, followed by biotinylated secondary goat
anti-mouse IgG antibody for 10 mins, streptavidin-horseradish peroxidase for 10 mins,
and 3-amino-9-ethylcarbazole/H2O2 chromagen (Ventana Medical Systems; Tucson,
AZ). Slides for glial fibrillar acidic protein (GFAP) IHC were blocked with commercial
antibody buffer (Ventana Medical Systems, Tucson, AZ), probed with primary rabbit
polyclonal antibody (CP040C, Biocare Medical; Concord, CA) diluted 1:600 for 12
minutes, followed by a universal secondary antibody/3’,3’-diaminobenzidine chromagen
kit (Ultraview DAB, Ventana Medical Systems; Tucson, AZ) for 8 mins. Positive IHC
control tissues included brain or lymph node from TSE infected elk, deer, sheep or
23

mink. Negative control tissues included tissue from uninoculated, TME negative, or
CWD negative recipient mink. Additional negative antibody controls included omission
of primary antibodies or substitution with unrelated mouse or rabbit primary antibodies
(Ventana Medical Systems; Tucson, AZ).
Tissue examination and definition of disease: Light microscopic examination of
tissue sections was performed blindly on brain ipsilateral and contralateral to the
injection site for vacuolation, PrPd deposition, and astrocytosis. Brain and other
collected tissues were examined for intercurrent disease. Diagnosis of clinical TSE was
based on neurologic signs, and disease was confirmed by detection of spongiform
vacuolation and PrPd immunoreactivity within brain. Brains from asymptomatic animals
were examined by PrPd and GFAP IHC to rule out subclinical disease. Vacuolation and
PrPd IHC scoring was performed on five 1200 x 800 µm fields randomly selected within
the anatomic area of interest. Vacuolation scores were 0=within normal limits,
1=vacuoles confined to white matter, 2=slight vacuolation in grey matter, 3=moderate
vacuolation in grey matter +/- in neurons, 4=Pronounced vacuolation in grey matter +/-
in neurons, 5=Pronounced vacuolation in grey matter and visibly within neuronal
perikaryon [modified from (Bruce et al., 2004)]. PrPd scores were 0=No signal detected
or background only, 1=Slight signal intensity, 2=Moderate signal intensity,
3=Pronounced signal intensity. PrPd IHC was performed on lymph nodes to determine
lymphoreticular distribution. Astrocytes in GFAP IHC sections were manually counted
on five randomly selected grey matter fields within areas of the cerebral cortex,
hippocampus, and thalamus that corresponded to areas of most severe vacuolation in
24

TME positive IC recipients using a 200 by 250 µm grid overlay on commercial imaging
software (Nikon Elements BR, Nikon Corporation; Tokyo, Japan). Statistical
significance (p ≤ 0.05) of scores and counts between treatment groups was determined
using the Mann-Whitney test (GraphPad 5.0; San Diego, CA).
Assessment of mink PRNP genotypes: Frozen pieces of brain or spleen were
homogenized in a DNA lysis buffer (100 mM NaCl, 10 mM Tris-HCl, 25mM EDTA, 0.5%
SDS) with PK (Sigma-Aldrich; St. Louis, MO), incubated overnight at 55°C, and phenol-
chloroform extracted (Sambrook, 1989). PCR amplification was performed using
primers 5’-TGT TTG CAG ATA AGC CAT CAT G-3’ and 5’-ATT TCC CAG GGC CAT
CAG–3’ yielding a 780 base pair amplicon. Sequencing was performed with primers 5’-
GCC ATC ATG GTG AAA AGC CAC-3’, 5’-TCA TCC CAC TAT CAG GAG AAT GAG
C-3’, and 5’-CAT GAT CTT CAT GTC GGT CTC-3’ on automated equipment (Applied
Biosystems; Foster City, CA), and analyzed with commercial software (Vector NTI,
Invitrogen; Carlsbad, CA). Nucleotide polymorphisms were compared with IHC findings
to determine if they were associated with disease. Comparative amino acid alignments
were performed using a public access program (CLUSTAL W, http://bips.u-
strasbg.fr/fr/Documentation/ClustalW/).
Results
Characterization of inocula
We characterized elk brain samples to determine suitability for challenge by assessing
PK resistance, quantity of PrPd antigen, and prion genotype. PrPd immunoreactivity in
25

CWD positive elk brain homogenates was confirmed by western blot of PK digested
samples (Figure 2-1a). Estimated PrPd antigen content for pooled CWD positive
samples was 12.69 +/- 0.21 ng per mg of wet tissue (Figure 2-1b). Total administered
PrPd content was approximately 127 ng for IC, and 25 µg for oral, challenge. Brain from
a CWD negative control elk did not exhibit immunoreactivity by western (Figure 2-1a,
lane 2) or slot blot. Tissue from positive and negative elk had a uniform DNA sequence,
including homozygosity at codon 132, consistent with a reference elk sequence (GEN
BANK #AF016227) (O'Rourke et al., 1999).
a b
Figure 2-1: Immunoreactivity and measurement of antigen load following PK digest in
CWD positive and CWD negative elk brain samples used for experimental challenge. a)
Western blot using 0.5 or 1.0 mg total protein. 0 = Sheep scrapie brain sample reagent
control. 1, 3, 4 = CWD positive elk. 2 = CWD negative elk. b) Graph illustrating
correlation between nanogram quantity of PrP (x-axis) and densitometric values (y-
axis). ■ = Values for recombinant PrP reference standard. ○ = Mean amount of PrPd in
CWD positive elk brain estimated from density values. r2 = 0.9491, dashed line = 95%
confidence interval.
Intracerebral challenge
26

Clinical signs and general histologic observation: IC challenge was performed to
demonstrate pathogenic potential of CWD positive brain homogenates and compare
lesions induced by CWD or TME in mink. CWD positive IC challenge caused
neurologic symptoms in two of eight (25%) mink at 936 and 993 days (mean 964 days,
or 32.1 months). Two of two (100%) TME positive IC recipients developed disease at
173 and 198 days (mean 185 days, or 6.2 months). Six other CWD positive IC
recipients, sampled at 3, 4, 5, 6, 11 and 14 months as serial timepoints or due to
intercurrent disease, did not have spongiform change or PrPd deposits within the central
nervous system and peripheral tissues. Neurologic signs in IC recipients included
lethargy, inappetance, ataxia, hind limb weakness progressing to posterior paresis,
lateral recumbency and inability to return to nest boxes. These signs were similar
between CWD and TME recipients, except for craniodorsal reflection of the tail which
was only observed in the TME cases. Prion disease in CWD and TME IC recipients
with clinical signs was confirmed by detection of spongiform vacuolation and PrPd
immunoreactivity. Vacuolation and PrPd deposition was present in the obex, pons,
thalamus, hypothalamus, hippocampus, and cerebral cortex of CWD positive IC and
TME positive IC recipients, however these lesions were consistently and significantly
more severe in TME IC recipients (Figure 2-2 and 2-3) except for cerebellar
abnormalities which were rare in both groups. Retinal PrPd deposits were present in
both CWD and TME IC recipients (Figure 2-3). In CWD cases, deposits had a distinct
multifocal coarsely globular appearance, whereas retinas of TME recipients had a more
diffuse granular, and rarely globular, presentation. Diffuse PrPd deposits within
germinal centers of mesenteric and retropharyngeal lymph nodes were equivalent in
27

CWD and TME cases (not shown). Neurologic signs and histologic abnormalities were
not present in any control animals receiving CWD negative IC (0/7), or TME negative IC
(0/2).
Vacuolation: TME IC recipients had a high density of 10 to 40 µm round clear vacuoles
within grey matter and neurons throughout the brain, sometimes confluent and
exhibiting a lace-like appearance, particularly in the median layer of the cerebral cortex
(Figure 2-2). Vacuoles in CWD IC recipients tended to be smaller and were much less
frequent, often with only a few present in an examined area (Figure 2-2). Vacuolation
scores were significantly higher in TME IC than in CWD IC recipients, except for the
cerebellum (Figure 2-4).
PrPd IHC: Multifocal PrPd deposits in the brain had a coarse globular appearance in
both groups. Deposits occurred with greater signal intensity and were more uniform in
TME cases. TME cases also had areas of diffuse granular deposits along with globular
signal (Figure 2-3). PrPd deposition scores were significantly higher in TME IC than in
CWD IC recipients, except for the cerebellum (Figure 2-4).
28

Figure 2-2: Photomicrographs illustrating vacuoles in a TME positive IC recipient (top
row; a, b, c), and a CWD positive IC recipient (middle row; d, e, f), but not in a CWD
negative IC recipient (bottom row; g, h, i). Left column= cerebral cortex, middle
column= hippocampus, right column= thalamus. H/E stain. Bar=100 µm.
Figure 2-3 (next page): Photomicrographs of PrPd IHC in brain and retina from TME
positive IC and CWD positive IC recipients. Left column= TME positive IC recipient
mink. Right column= CWD positive IC recipient mink. a, c, e, g= Cerebral cortex,
hippocampus, thalamus and retina, respectively, from TME positive IC recipient. b, d, f,
h= Cerebral cortex, hippocampus, thalamus, and retina, respectively, from CWD
positive IC recipient. Bar=100 µm.
29
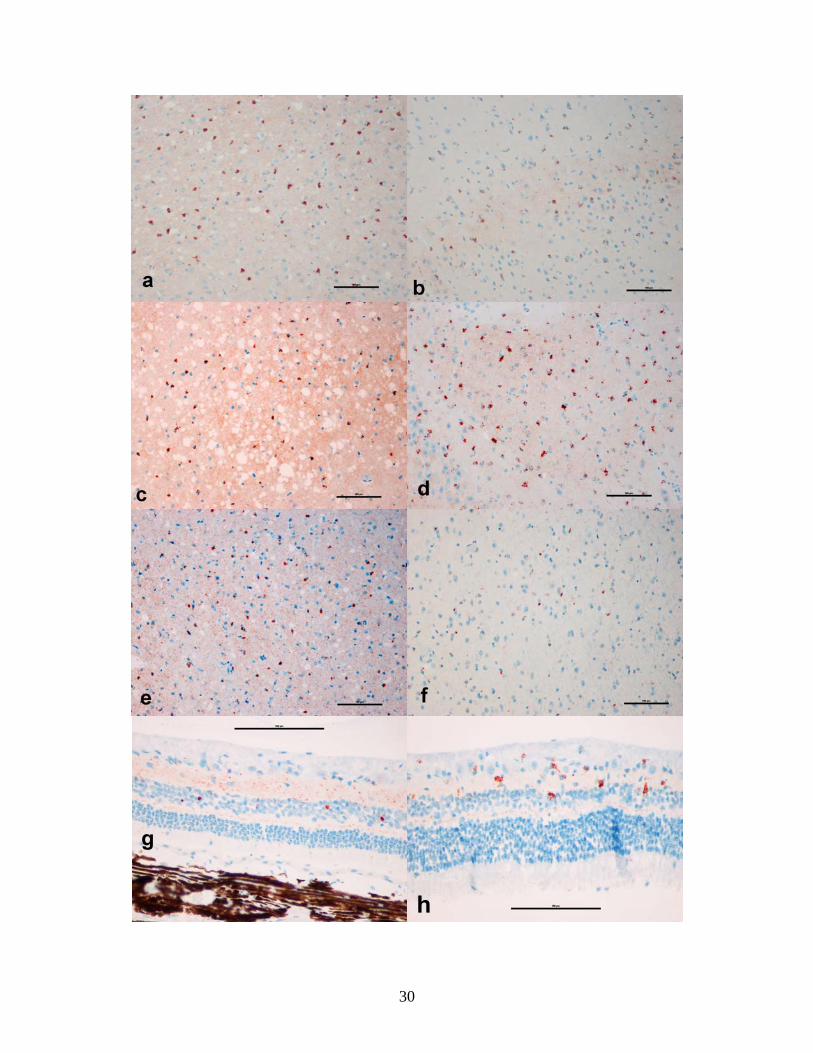
g
h
30

Vacuolation score
0.00
1.00
2.00
3.00
4.00
5.00
6.00
Obex
Pons
Cerebellu
m
Thalamus
Hypoth
alamus
Hippoca
mpus
Cerebral
corte
x
Brain region
Scor
e CWDTME
PrPd IHC score
0.00
0.50
1.00
1.50
2.00
2.50
3.00
3.50
Obex
Pons
Cerebe
llum
Thalam
us
Hypoth
alamus
Hippoc
ampu
s
Cerebra
l Cort
ex
Brain region
Scor
e CWDTME
٭٭٭٭٭٭ ٭ ٭٭٭٭ ٭
Figure 2-4: Scores of vacuolation and PrPd IHC signal intensity in TME positive IC and
CWD positive IC recipients, by brain region (mean and standard error). ٭ = p ≤ 0.05.
Astrocyte quantity: GFAP immunostained sections of the cerebral cortex, hippocampus
and thalamus were examined to determine if astrocytosis differed in the brains of TME
and CWD IC recipients (Figure 2-5 and 2-6). Cerebrocortical astrocyte numbers were
significantly higher in TME recipients than in CWD recipients (p=0.0010), whereas
hippocampal astrocytes were significantly higher in CWD cases than in TME cases
(p=0.0195). Thalamic astrocytes were equivalent between CWD and TME recipients
(p=0.2108). Astrocyte counts in the cerebral cortex, thalamus, and hippocampus were
significantly higher in TSE positive IC recipients than in the negative controls (p=0.0451,
p=0.0019, p=0.0010, respectively).
31
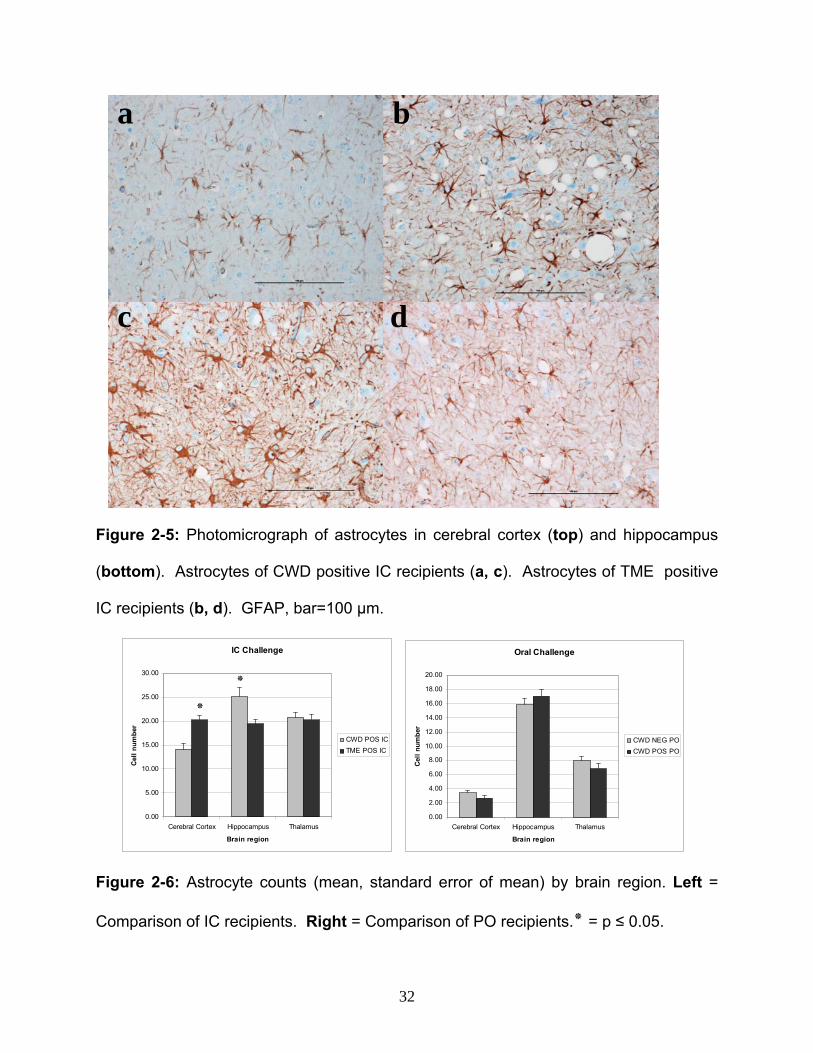
Figure 2-5: Photomicrograph of astrocytes in cerebral cortex (top) and hippocampus
(bottom). Astrocytes of CWD positive IC recipients (a, c). Astrocytes of TME positive
IC recipients (b, d). GFAP, bar=100 µm.
IC Challenge
0.00
5.00
10.00
15.00
20.00
25.00
30.00
Cerebral Cortex Hippocampus Thalamus
Brain region
Cel
l num
ber
CWD POS ICTME POS IC
Oral Challenge
0.00
2.00
4.00
6.00
8.00
10.00
12.00
14.00
16.00
18.00
20.00
Cerebral Cortex Hippocampus Thalamus
Brain region
Cel
l num
ber
CWD NEG POCWD POS PO
a
c
b
d
٭
٭
Figure 2-6: Astrocyte counts (mean, standard error of mean) by brain region. Left =
Comparison of IC recipients. Right = Comparison of PO recipients.٭ = p ≤ 0.05.
32

Western blot migration pattern and band densitometric ratio: Brain extracts from CWD
and TME positive IC recipients were analyzed by PK digestion and western blotting to
determine if glycoform migration patterns differed between the two. Glycoform
migration patterns were identical (Figure 2-7). Densitometric ratio of band intensity was
not significantly different (not shown).
Figure 2-7 (previous page): Western blot of PK digested brain homogenates from
positive IC recipients. Lane 1 = TME IC recipient. Lane 2 = CWD IC recipient. Lane M =
Molecular weight marker (kDa).
Oral challenge
Clinical signs, vacuolation, and PrPd IHC: Mink were orally challenged with CWD brain
homogenates to test the hypothesis that primary oral CWD challenge causes a prion
disease in mink. Oral recipients did not exhibit clinical neurologic symptoms,
vacuolation, or PrPd deposition in neural or lymphoid tissue during the 42 months of
observation. Vacuole and PrPd deposition scores were universally null for all oral
33

recipients including individual animals sampled, with a corresponding positive or
negative control, at 5, 6, 7, 11, 12, 14, 24, 27, 28, 32, and 38 months post challenge as
serial time points or due to intercurrent disease. Causes of intercurrent disease
included oral trauma, pneumonia, interstitial nephritis, intestinal obstruction,
intussusception, colitis, or rectal prolapse.
Astrocyte quantification: GFAP immunostained sections were examined to determine if
astrocytosis was present as an indicator of underlying neurologic damage and
subclinical disease (Figure 2-6). CWD positive PO recipients and CWD negative PO
recipients did not have a significant difference in astrocyte counts for the cerebral
cortex, hippocampus or thalamus indicating a lack of subclinical disease (p=0.1326,
p=0.3499, p=0.2108, respectively).
Prion genotype
The prion gene open reading frame was sequenced to determine if recipient mink had
any codon changes and if such changes correlate with disease status. 56 samples
suitable for genotyping were all homozygous for methionine at codon 133, consistent
with codon 132 of the elk challenge material. Previously unrecognized genetic
changes in mink were detected including an arginine/tryptophan polymorphism at codon
27 (from cgg to tgg, with change at bp 79), and an arginine/lysine polymorphism at
codon 232 (from agg to aag, with change at bp 695) (GEN BANK #EF508270). The
codon 27 polymorphism, found in 7 heterozygous animals, was present in 1 CWD
negative IC recipient, 4 CWD positive PO recipients and 2 CWD negative PO recipients.
34

The codon 232 polymorphism, found in 4 heterozygous animals, was present in two
CWD positive, one CWD negative IC, and one CWD positive PO recipients. Changes in
both codons never occurred within the same animal. Of the two CWD positive IC
recipients with disease, one had the codon 232 change while the other did not. Codon
changes were not present in TME recipients. Silent base pair changes in the population
were noted at bp 69 (c to t, n=5), bp 498, (c to t, n=9), and bp 648 (g to a, n=4);
sequences were otherwise consistent with previously published data (Kretzschmar et
al., 1992). Comparative amino acid alignment identified 23 locations where residues
differ between cervids and mustelids (Figure 2-8).
35

CODON 1 27 60 • • • mink MVKSHIGSWLLVLFVATWSDIGFCKK R PKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW ferret MVKSHIGSWLLVLFVATWSDIGFCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW mule MVKSHIGSWILVLFVAMWSDVGLCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW elk MVKSHIGSWILVLFVAMWSDVGLCKKRPKPGGGWNTGGSRYPGQGSPGGNRYPPQGGGGW *********:******:***:*:************************************* CODON 61 120 • • mink GQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGGWGQGGGSHGQWGKPSKPKTNMKHVAGAA ferret GQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGGWGQGGGSHGQWGKPSKPKTNMKHVAGAA mule GQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGGWGQGG-THSQWNKPSKPKTNMKHVAGAA elk GQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGGWGQGG-THSQWNKPSKPKTNMKHVAGAA ***********************************.*.:*.**.**************** CODON 121 132/133 180 • • • mink AAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYKPVDQYSNQNNFV ferret AAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYKPVDQYSNQNNLV mule AAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYRPVDQYNNQNTFV elk AAGAVVGGLGGYMLGSAMSRPLIHFGNDYEDRYYRENMYRYPNQVYYRPVDQYNNQNTFV ***********************************************:*****.***.:* CODON 181 224 227 232 240 • • • • • mink HDCVNITVKQHTVTTTTKGENFTETDMKIMERVVEQMCVTQYQRESEAYYQ R GASAILFS ferret HDCVNITVKQHTVTTTTKGENFTETDMKIMERVVEQMCVTQYQQESEAYYQRGASAILFS mule HDCVNITVKQHTVTTTTKGENFTETDIKMMERVVEQMCITQYQRESQAYYQRGASVILFS elk HDCVNITVKQHTVTTTTKGENFTETDIKMMERVVEQMCITQYQRESEAYYQRGASVILFS **************************:*:*********:****:**:********.**** CODON 241 257 • • mink PPPVILLISLLILLIVG ferret PPPVILLISLLILLIVG mule SPPVILLISFLIFLIVG elk SPPVILLISFLIFLIVG .********:**:****
Figure 2-8: Comparative amino acid alignment illustrating positions of disparity between
mustelids (e.g. mink, ferrets) and cervids (e.g. mule deer, elk), or mink and ferrets that
may effect TSE susceptibility. Methionine homozygosity at codon 132/133 (elk/mink,
respectively) is conserved. X =Location of new polymorphism identified within mink
(codon 27 R→W, codon 231 R→W). X =Residue difference between mink and ferrets
that may effect TME susceptibility. X=Residue difference between mustelids and
cervids that may effect CWD susceptibility. X=Residue conserved between cervids and
mustelids that is implicated in human TSE susceptibility.
36

Discussion
The results of this study demonstrate a species barrier in transmission of CWD to mink.
Primary oral challenge with CWD infected elk brain did not result in clinical or pathologic
findings of TSE indicating that natural interspecies transmission of CWD to mink is
unlikely to occur on ranches or in wildlife. Furthermore, primary IC challenge of mink
with CWD material was considerably less efficient than IC challenge with TME, as
indicated by prolonged incubation time and different lesion profiles. The lack of orally
mediated disease, despite a total cumulative dose almost 200 fold greater than that
given by the IC route, shows the influence of administration route on TSE pathogenesis.
Species barriers in prion disease are typically defined as increasing attack rate and
decreased incubation time following serial IC passage of infectious material. However
intracerebral injection, while useful for lesion comparison between strains or in the study
of molecular pathogenesis, is an experimental technique that does not occur in nature.
In the context of natural disease transmission from cervids to mustelids, and to
carnivores in general, primary oral transmission is the scenario of consequence.
Therefore, in this study we defined species barriers as inefficient primary IC
transmission and lack of primary oral transmission.
Differences in lesion profile were demonstrated qualitatively by TME IC recipients
having more severe spongiform vacuolation and PrPd deposition than CWD IC
recipients. Quantitatively, TME recipients had significantly higher scores for both
vacuolation and PrPd deposits in all regions of the brain except for the cerebellum.
Different patterns of retinal PrPd IHC further delineated CWD from TME in mink tissue,
37

as the CWD animals had a multifocal globular signal and TME recipients had a
predominately diffuse granular signal. Significant differences in astrocyte quantification
were also informative in both IC and PO recipients. Astrocyte counts were significantly
higher in the hippocampus of CWD IC recipients, whereas cerebrocortical counts were
significantly higher in TME IC recipients. This difference combined with astrocyte
counts that were independent of the degree of vacuolation, shows a clearly different
host response for the two types of challenge inocula. In PO recipients, astrocyte counts
were evaluated as an indicator of subtle neurologic change in the central nervous
system. Counts were not significantly different between CWD positive PO and CWD
negative PO recipients indicating a lack of underlying neural damage or subclinical
disease that may have developed with continued observation. The rare occurrence of
cerebellar lesions in both the CWD and TME IC recipients is consistent with previous
investigations showing minimal cerebellar involvement in mink (Hanson et al., 1971;
Marsh & Hanson, 1979; Robinson et al., 1994). The differences in lesion profile and
extended incubation time for CWD demonstrates that CWD and TME are distinctly
different diseases in the mink host.
This study complements previous ruminant to carnivore investigations where CWD was
administered to ferrets. Results in the ferrets were similar to those in mink as primary
IC administration of deer CWD caused disease (Bartz et al., 1998), whereas primary
oral challenge did not cause disease. In ferrets, serial IC passage is required before
positive PrPd IHC is demonstrable by oral challenge (Perrott et al., 2004; Sigurdson et
al., 2003). CWD infected tissue originated from elk in this study, and from mule deer in
38

the ferret study. It is possible that CWD of deer origin may behave differently in mink
tissue than that of elk, a situation we are currently investigating. Nevertheless, the
cumulative findings demonstrate a species barrier in development of disease in mustelid
carnivores (e.g. mink and ferrets) following primary oral challenge with CWD. By
extension one may speculate that carnivores in general are resistant to consumption of
CWD. Humans may also be resistant to CWD; while nonhuman primates succumb to
IC CWD (Marsh et al., 2005), epidemiologic investigation has not identified a clear link
between CWD and human CJD (Belay et al., 2004; MaWhinney et al., 2006) and
studies in humanized transgenic mice indicate CWD resistance (Kong et al., 2005;
Tamguney et al., 2006). Continued monitoring of human disease and additional oral
transmission studies in animals are needed to confirm or refute primate and carnivore
resistance to orally mediated CWD.
Host prion gene polymorphisms are associated with TSE susceptibility in some species.
We examined prion genetics of both challenge material and recipient mink to identify
residues that might effect interspecies transmission. Source and recipient animals were
universally homozygous for methionine at codon 132/133 (elk and mink, respectively).
Codon 132, the site of a methionine/leucine polymorphism in elk (O'Rourke et al., 1999),
positionally corresponds to human codon 129 where methionine homozygosity is
associated with vCJD (Zeidler et al., 1997). The elk polymorphism segregates with
disease phenotype in CWD, as leucine homozygous elk have a prolonged incubation
period and altered PrPd migration pattern when compared to methionine homozygotes
(Hamir et al., 2006a; O'Rourke et al., 2007). Codon 96 is another site of interest as a
39

glycine/serine polymorphism is associated with relative CWD susceptibility in deer
(O'Rourke et al., 2004). In this study elk and mink had conservation of methionine at
codon 132 and glycine at codon 96 indicating these residues were not limiting factors in
disease transmission. Of the two CWD positive IC recipients with disease, one was
homozygous for arginine at codon 232, the other was a codon 232 arginine/tryptophan
heterozygote. These two animals had similar incubation periods and lesions, thus there
was no obvious effect on disease. The codon 27 polymorphism is intriguing as it is near
the cleavage site of the membrane signaling portion of prion protein (Prusiner, 1998).
As cytosolic accumulation of prion protein has neurotoxic effects (Ma et al., 2002),
signaling sequence variation could influence disease pathogenesis through altered
prion translocation to the cell surface. All diseased animals were homozygous at codon
27, suggesting the polymorphism could modulate relative susceptibility; however, the
small number of affected mink precludes determination of the true effect. Respective
differences between mink and ferrets at codons 179 (phenylalanine/lysine), and 224
(arginine/glutamine) are associated with differential susceptibility to TME (Bartz et al.,
1994). In this study, all mink were homozygous for phenylalanine and arginine and
congruous with challenge material; it is currently unknown if these codon
polymorphisms were a factor in previous CWD-ferret studies. Overall, comparative
amino acid alignment shows 23 divergent residues between cervids and mustelids that
could effect transmission. Additional genetic comparison of cervid challenge material
and recipient mustelids, such as by in vitro conversion assays (Kurt et al., 2007;
Raymond et al., 2000), is needed to further delineate possible roles of these divergent
residues.
40

This and previous studies provide a relative comparison of mustelid susceptibility to
cattle, sheep, or cervid prions. Primary IC or primary oral challenge of mink with BSE
results in clinicopathologic abnormalities at 12 and 15 months of incubation,
respectively, with lesion severity that is independent of challenge route (Robinson et al.,
1994), and occurs close to the estimated 7 to 12 month oral incubation period for
natural TME (Marsh et al., 1991). Conversely, primary oral challenge with scrapie has
not caused disease in mink, despite repeated attempts and observation up to 48
months (Marsh et al., 1991; Marsh & Hanson, 1979); similarly, in this study primary oral
challenge with CWD did not cause disease during 42 months incubation. CWD IC
challenge resulted in minor cerebrocortical involvement while the cerebral cortex is
more extensively involved with scrapie or BSE IC challenge (Hanson et al., 1971; Marsh
& Hanson, 1979; Robinson et al., 1994). IC lesions also vary by source in the caudal
brainstem, including the dorsal motor nucleus of the vagus nerve, as they are of lesser
severity with scrapie or CWD, while severity increases with TME or BSE (Eckroade et
al., 1979; Hanson et al., 1971; Hartsough & Burger, 1965; Marsh & Hadlow, 1992;
Robinson et al., 1994). IC backpassage of TME to cattle causes disease in 14.5
months, similar to TME in mink, and lesions in cattle are similar on both first and second
passage (Hamir et al., 2006b; Robinson et al., 1995). Thus, the overall
clinicopathologic features do not appreciably change between mink and cattle.
Cumulatively, passage of TSE between cattle and mink occurs readily with similar
lesions and incubation times, whereas passage of CWD or scrapie to mink is limited by
route of administration, incubation time, and appearance of lesions when compared to
41

TME. As cattle are the only ruminant without an apparent species barrier in prion
transmission to and from mink, it raises the possibility that in natural settings previously
unrecognized prion, or prion-like, disease in cattle may have been responsible for some
cases of spongiform encephalopathy in mink.
In this study we demonstrated a species barrier between elk CWD and mink as shown
by lack of orally mediated disease and substantive differences in lesions between CWD
and TME IC recipients. While CWD appears to be readily transmissible within cervid
species, this study is additional evidence that cervid prions are poorly transmissible to
non-cervid hosts, and is a strong indication that mink are unlikely to be involved in
natural transmission of CWD among wildlife.
42

CHAPTER THREE
TRANSGENESIS OF A BACTERIAL ARTIFICAL CHROMOSOME RESULTS IN
STABLE TRANSCRIPTION AND TRANSLATION OF MULE DEER PRION PROTEIN
AND REPLICATION OF CWD PATHOGENESIS@
Robert D. Harrington,1,2,3٭ H. Denny Liggitt,2 Katherine I. O’Rourke,1,3 Kelly A. Brayton,
1,3 Donald P. Knowles,1,3 and Carol B. Ware1,4
1Department of Veterinary Microbiology and Pathology, Washington State University,
Pullman, WA, 99164-7040, USA
2Department of Comparative Medicine, University of Washington, Seattle, WA 98195-
7190, USA
3Animal Disease Research Unit, Agricultural Research Service, US Department of
Agriculture, Pullman, WA 99164-6630, USA
4Institute for Stem Cell and Regenerative Medicine, University of Washington, Seattle,
WA 98195- 7750, USA
@ This continuing investigation will be submitted for publication at a future date.
.Author for correspondence, [email protected]٭
See Appendix A for attributions to contributing authors.
Summary
The use of common mouse strains for the study of prion disease has been hampered by
lack of natural susceptibility to prion infection, a short life span relative to long prion
disease incubation period, lack of transgenic (Tg) expression in non-neural tissue, and
43

confounding factors of dominant-negative inhibition by endogenous mouse prion
protein. An alternative transgenic engineering approach was investigated using a
bacterial artificial chromosome (BAC) containing the mule deer (MD) prion gene to drive
prion protein expression for pathobiological study of chronic wasting disease (CWD). A
hypothesis was tested that transgenesis using a bacterial artificial chromosome (BAC)
clone that contained the putative cervid prion promotor region (PP) and the cervid open
reading frame (ORF) would render cervid Tg mice susceptible to CWD administered by
the oral and intraperitoneal routes in addition to the intracerebral route. Pronuclear
microinjection of a 67 kilobase linear or circular BAC molecule resulted in murine in vivo
MD prion transcription as detected by reverse transcriptase PCR and translation of MD
prion protein as detected by western blotting in brain and lymphoreticular tissue. MD
BAC Tg mice were developmentally, anatomically, and behaviorally within normal limits.
MD BAC mice intracerebrally injected with CWD positive material have protease
resistant prion protein evident on western blot and have spongiform change consistent
with TSE. This chapter reports on the ongoing investigation into these mice following
oral, intraperitoneal, and intracerebral challenge with CWD infected brain of MD origin.
Introduction
Laboratory mice are a useful resource for modern biomedical research, however,
standard laboratory mice do not have their own form of natural prion disease and are
resistant to challenge with infectious prion material from cases of CWD. Mice can be
rendered susceptible to a variety of Transmissible Spongiform Encephalopathies (TSE)
by either repeated intracerebral (IC) serial passage (Figure 3-1) or by transgenic
44

insertion of prion coding sequences from the same species as the infectious source of
interest for study (Figure 3-2) (Prusiner, 1998; Telling, 2000). While both methods have
been used, the former approach requires up to 20 serial passages of infectious brain
material over many years and negates study of primary transmission which is a major
scenario of consequence for prion investigation both within and between species.
Creation of transgenic mouse lines using a variety of DNA constructs and insertion
techniques has an advantage over, and eliminates the need for, serial passage. The Tg
approach shortens disease incubation time and renders mice susceptible to TSE
originating from a variety of non-murine species [for review see Telling, 2000; Vilotte &
Laude, 2002; Weissmann & Flechsig, 2003].
Intracerebral injection (IC)
Prion Infectious Material
Remove brain, pass IC
No disease
No
Remove brain, pass IC
Repeat 5 to 20x
disease
No disease
before disease occurs
Figure 3-1: Diagram of serial passage approach to overcome natural murine
resistance to prion disease.
45

WT mouse (expresses mouse prion)
CWD Infectious Material
NO DISEASE
Tg mouse (expresses elk or deer prion)
CWD Infectious Material
SPONGIFORMDISEASE
Dominant-negative
Genetic bckgrnd
Figure 3-2: Diagram of transgenic approach to overcome natural murine resistance to
prion disease. Dashed line = possible confounding variables to pathogenesis in a
genetically engineered mouse.
Several genetic factors must be considered when engineering a prion Tg mouse. First,
the strain of mouse must have a genetically permissive background to prion infection.
Second, the phenomenon of dominant-negative inhibition where expression of the
endogenous mouse gene overrides the ability of Tg expression to produce disease
(Perrier et al., 2002) must be overcome by breeding to a prion “knockout” (PrP KO)
mouse lacking the murine prion gene (Bueler et al., 1993). Third, the elements within
the Tg construct such as the promoter system used to drive transgene expression will
effect how and where the Tg protein is transcribed and translated. TSE transgenic
models have typically relied on over-expression constructs for infectivity bioassays.
However, over-expression resulting from insertion of multiple copies of the gene
increases transcription levels thus raising questions about the relevance to physiologic
levels of gene expression in the natural host. Mouse models of neurodegeneration
typically use neuron specific promoters to drive transgene expression within the central
46

nervous system. While the use of such promoters facilitates central nervous system
prion pathogenesis it has a drawback of minimal or non-existent expression of the
transgene in peripheral tissues such that the Tg mice are only susceptible to disease
following IC injection. As oral transmission is the primary route of natural TSE
transmission additional models that replicate the oral route would be highly useful for
mechanistic studies and would allow primary bioassay to assess infectious potential of
materials otherwise unsuitable for IC injection (e.g., feces, urine, water, soil).
This study was initiated to develop a new Tg mouse model that would express cervid
prion protein in a manner physiologically relevant to the natural host and render Tg mice
susceptible to prion disease regardless of the route of experimental challenge. It was
hypothesized that transgenesis using a bacterial artificial chromosome (BAC) clone that
contained the PP region and the cervid ORF would render cervid Tg mice susceptible to
CWD administered by the oral and intraperitoneal routes in addition to the intracerebral
route. BAC clones can be useful as an alternative method of transgenesis (Yang &
Seed, 2003) and one has been used to generate an ovine Tg mouse for the study of
sheep scrapie (Le Dur et al., 2005). The insertion of a large chromosomal segment
containing the gene of interest may contain the components necessary to closely
replicate how a gene functions and is expressed in the natural host. The BAC facilitates
insertion of up to 150 kilobase long sequences of DNA that can include exons, introns,
promoters, and other regulatory regions pertinent to the gene of interest. This approach
is an intriguing alternative for the study of prion pathogenesis and is particularly
pertinent to CWD as there are few previously established laboratory animal models for
47

CWD study. The approach to generate cervid BAC Tg mice, analysis of transgene
function, and the status of infectious challenge experiments is reported herein.
Methods
DNA molecule for transgenesis: DNA for pronuclear microinjection was derived from
a bacterial artificial chromosome clone, GEN BANK #AY330343 (Figure 3-3) (Brayton et
al., 2004). This clone contains the mule deer prion ORF, all three exons, introns, and
the PP region.
Figure 3-3: Diagram of DNA molecule used as construct for pronuclear microinjection.
XhoI enzyme yields 43.9Kb fragment via two digestion sites. NotI enzyme yields
67.0Kb linearized molecule via a single digestion site.
All cultures were performed at 37°C using LB broth or agar supplemented with
chloramphenicol (20 µg ml-1) and glucose (0.1%). A single colony was picked from
isolation streaks and placed in 5ml broth starter culture for 6 hours then 2 mls were
transferred into 400 mls broth and grown for ~16 hours with rotary incubation at 210
rpm. Bacteria were harvested and BAC DNA extracted using a commercial maxi-prep
kit per the manufacturer’s instructions (Qiagen; Valencia, CA), and the final pellet was
resuspended in 1ml TE, aliquoted, and stored @ -80°C until further use. 20 µl of BAC
48

preparation was digested overnight at 37°C with NotI restriction enzyme (1.6 units µl-1)
to create a linear 67.0 Kb molecule or XhoI restriction enzyme (1.0 units µl-1) to
separate a 43.9 Kb insert from the vector; alternatively BAC DNA was left undigested.
Samples were ran on a sepharose bead column and fractions collected to isolate
regions of interest and remove vector fragments and any residual contaminants.
Collected fractions were run on a 0.3% pulse field gel at 20 volts for 20 hours to verify
and quantify the fraction that contained the DNA of interest. DNA was diluted in a
microinjection buffer composed of 10 mM Tris-HCl, 0.1 mM EDTA, 100 mM NACl and
1x polyamine to a final concentration of approximately 100ng µl-1.
Animals and pronuclear microinjection: Mice were cared for in accordance with the
policies of the University of Washington Institutional Animal Care and Use Committee.
Transgenic mice were created by pronuclear microinjection using B6C3 (C57Bl6 x C3H)
fertilized ova per previously established methods (Nagy, 2003). Pseudopregnant
Swiss-Webster mice were used as recipient females for ova transfer and C57Bl6 mice
used for subsequent backcross breeding (Figure 3-4). Three separate founder lines
were created to confirm that a consistent phenotype developed independent of random
mutation.
PCR screening of transgenic mice: First generation pups born following initial
microinjection were screened by PCR of the MD PRNP ORF and PP region to identify
founder candidates for subsequent breeding. All subsequent offspring were screened
by PCR for the MD ORF, wild-type mouse PRNP (WT), and neomycin resistance
cassette (NEO) to determine suitable genotyped for experimentation. NEO was used
49

as a marker for mouse PRNP deletion as the PrP KO mouse was created by disruption
of the endogenous mouse PRNP ORF by insertion of the NEO cassette (Bueler et al.,
1993).
Figure 3-4: General strategy for creation of transgenic mice by DNA pronuclear
microinjection with PCR screening and subsequent breeding to generate mice of
desired genotype for study (PrPTg/Tg).
Three mm tail biopsy samples for PCR were collected from mouse pups at weaning.
Samples were mixed with a PrP lysis buffer and Proteinase K, incubated overnight at
55°C, phenol-chloroform extracted and ethanol precipitated (Sambrook, 1989). A 716
bp product of the mule deer PRNP ORF was amplified using primers 5'-TGG TGA AAA
50

GCC ACA TAG GCA G-3' and 5'-TGC CCC TCT TTG GTA ATA AGC CTG-3' for 2 mins
at 94°C, 32 cycles of 1min at 94°C, 1 min at 54°C, and 1 min at 72°C then followed by
10 mins @ 72°C and held at 4°C. An 856 bp product of the mule deer PRNP PP region
was amplified using primers 5'-CTG GCC TTT GTG GTC TAA TGG-3' and 5'-AGT AGT
TGT GGA AAG GGA CTT GT-3' by the same conditions as for the ORF. A multiplex
PCR was used for WT PRNP with primers 5' ATG GCG AAC CTT GGC TAC TGG CTG
3' and 5' TCA TCC CAC GAT CAG GAA GAT GAG 3', and for the NEO marker of PrP
KO status using primers 5'-TTG AGC CTG GCG AAC AGT TC-3' and 5'-GAT GGA TTG
CAC GCA GGT TC-3' yielding 764 and 511 bp amplicons, respectively. The cycling
parameters for the multiplex reaction were 3 mins at 94°C, 30 cycles of 30 secs at 94°C,
30 sec at 62°C, and 1 min at 72°C followed by 10 mins @ 72°C and held at 4°C.
Breeding strategy for generation of mice for challenge: Tg mice were bred to yield
a uniform genetic background by backcrossing to C57BL6 mice. PrP KO mice were
also backcrossed to C57Bl6 (PrP KO mice kindly provided by Jean Manson,
Neuropathogenesis Unit, Institute for Animal Health, University of Edinburgh, UK). This
occurred for 5 successive generations resulting in greater than 96% C57BL6 genetic
background (Figure 3-5, center). Subsequently MD BAC Tg mice were bred to PrP KO
mice to eliminate the WT PRNP and supercede dominant-negative inhibition. An initial
cohort was also bred by crossing MDBAC with PRPKO mice, without backcross
breeding, to generate mice on a mixed 129/Ola/C3H/C57BL6 genetic background and
then used for initial challenges (Figure 3-5, far right). In either scenario breeding of MD
51
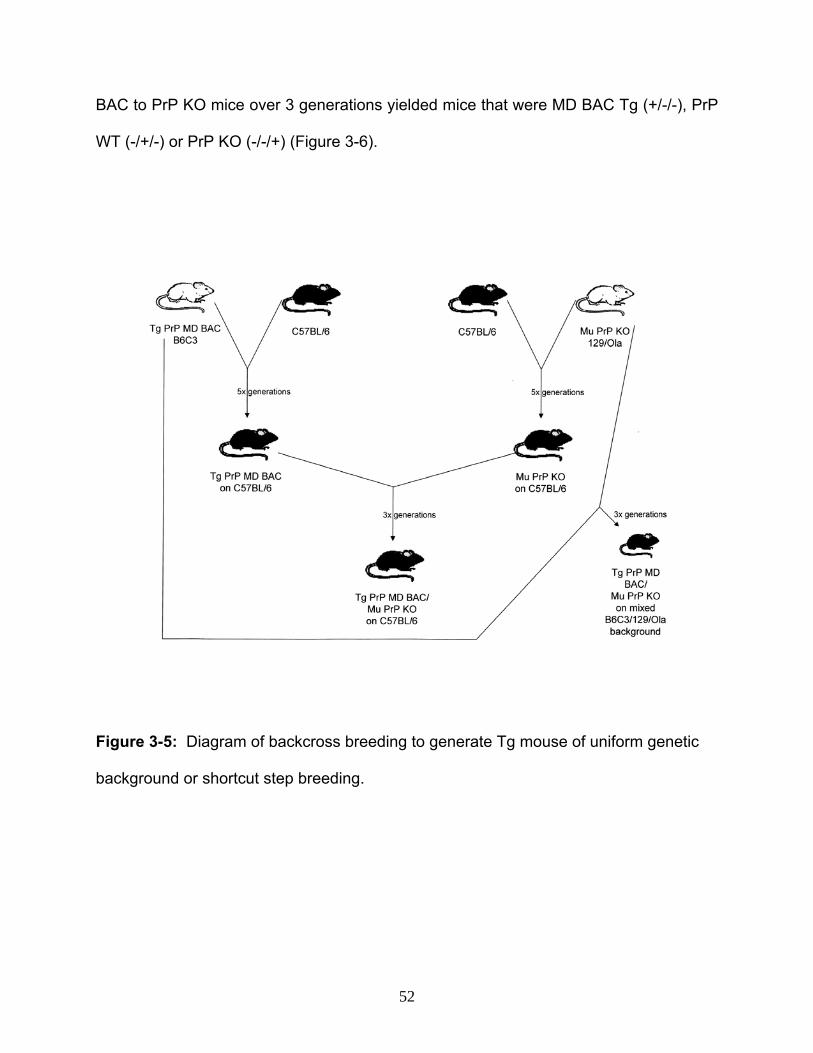
BAC to PrP KO mice over 3 generations yielded mice that were MD BAC Tg (+/-/-), PrP
WT (-/+/-) or PrP KO (-/-/+) (Figure 3-6).
Figure 3-5: Diagram of backcross breeding to generate Tg mouse of uniform genetic
background or shortcut step breeding.
52

Figure 3-6: Diagram for breeding transgenic mice to prion knockout mice to eliminate
endogenous mouse gene from experimental cohort.
Confirmation of transgenic expression in MD BAC x PrP KO mice: RNA was
purified from brain, spleen, and/or liver of MD BAC mice using a commercial kit
following the manufacturer’s instructions (RNAeasy, Qiagen; Valencia, CA). Samples
were treated with DNAse using a commercial kit per manufacturer’s instructions
(TurboDNAse, Ambion; Foster, CA). Reverse transcription was performed using a first-
strand cDNA synthesis kit per manufacturer’s instructions (Protoscript, New England
Biolabs; Ipswich, MA). PCR of cDNA was performed using the MD specific PRNP
53

primers 5'-CTG GCC TTT GTG GTC TAA TGG-3' and 5'-AGT AGT TGT GGA AAG
GGA CTT GT-3' by the same parameters as in the previous section. PCR of cDNA was
also performed using MD PRNP specific intron spanning primer sets 5’- AGC ATC TGT
CTT CAC AGA GAC A -3’ (exon 2) / 5’- GTG GAT AGC GGT TGC CTC CA -3’ (exon 3)
and 5’-TCC GAC TTA AGC TGA ATC ACA G-3’ (exon 2) / 5’- AGC CAC CTC CAT
GAG GTT GG -3’ (exon 3) yielding 221 bp and 261 bp amplicons, respectively. GAPDH
intron spanning primers used as housekeeping controls were 5’- TCC TGC GAC TTC
AAC AGC AAC -3’ (exon 6) and 5’- CTC TCT TGC TCA GTG TCC TTG -3’ (exon 7)
yielding a 206 bp product. Both sets used a cycle of 2 mins at 94°C, 30 cycles with 30
sec at 94°C , 30 sec at TA , 1 min at 72°C, then followed by 10 mins at 72°C and held at
4°C (TA was 52.8°C degrees for MD PRNP and 50.0°C for GAPDH).
Confirmation of transgenic protein production in MD BAC x PrP KO Tg mice by
western blot: Western blotting was used for detection of MD PrPc translation. Tissue
samples were placed in ten times volume of lysis buffer (10mM Tris-HCl ph 7.5, 0.5%
NP-40, 0.5% sodium deoxycholate) and homogenized 45 seconds in a commercial
apparatus (Fastprep, MP biomedicals; Santa Ana, CA) then centrifuged at 1000 g for 5
minutes to pellet cell debris. Samples were denatured, run on a 12% bis-tris gel in
MOPS SDS running buffer (Invitrogen; Camarillo, CA) at 200 volts for 1 hour (hr), and
transferred to methanol soaked PVDF membrane in MOPS transfer buffer (Invitrogen;
Camarillo, CA) at 200 mAmps for 1 hr. Membranes were dried, then blocked for 1 hour
in tris-casein buffer (Roche; Palo Alto, CA), with 0.1% Tween 20. Membrane transfer,
blocking and all subsequent steps were done at room temperature. Membranes were
54

probed for 1 hr with 3.6 µg µl-1 of primary mouse monoclonal antibody F99/97.6.1 (K.
O’Rourke, USDA-ARS; Pullman, WA), that recognizes prion epitope QYQRES
(O'Rourke et al., 2000), followed by biotinylated goat anti-mouse secondary antibody
(Southern Biotech; Birmingham, AL) and enhanced chemiluminescence (Amersham
Biosciences; Piscataway, NJ). Signal detection was performed with a commercial
apparatus (Alphaimager, Alpha Innotech Corporation; San Leandro, CA).
Preparation of inocula for challenge: Inocula were prepared from central nervous
system tissue homogenized in sterile disposable tissue grinders (VWR International;
West Chester, PA) and diluted to a final concentration 10% (w/v) for IC injection in
sterile saline (Sigurdson et al., 1999). Bacterial contamination was assessed on 10%
sheep blood agar, and all samples underwent a 3 phase water bath heat treatment
cycle of 80 °C for 15 mins, 37 °C for 60 mins, and 80 °C for an additional 15 mins.
Gentamycin was added to IC inocula at 100 µg ml-1. Inocula were stored at -20 °C until
use.
Inocula characterization by western and slot blot: PrPd content of mule deer brain
samples was confirmed by western blot, and antigen load determined by semi-
quantitative slot blot modified from a dot blotting procedure (O'Rourke et al., 2003).
Proteinase K (PK) digest was performed at 50 µg ml-1 at 56 °C for 30 mins, with
inactivation at 90 °C for 10 mins. Brain homogenates from clinically normal mule deer
were used as negative controls. Western blot samples were denatured, run on a 12%
bis-tris gel in MOPS SDS running buffer (Invitrogen; Camarillo, CA) at 200 volts for 1
55

hour (hr), and transferred to methanol soaked PVDF membrane in MOPS transfer buffer
(Invitrogen; Camarillo, CA) at 200 mAmps for 1 hr. Slot blot test samples and a plasmid
derived recombinant PrP (rPrP) densitometric reference standard (K. O’Rourke, USDA-
Agricultural Research Service; Pullman, WA) were denatured and serially diluted 1:2.
Duplicate lanes of rPrP ranging from 0.66 to 21.13 ng, one lane of CWD negative
homogenate, and 5 replicate lanes of CWD positive material were spotted onto
nitrocellulose membranes (Sigma-Aldrich; St. Louis, MO) using a slotted manifold
(Biorad Laboratories; Hercules, CA). Western and slot blot membranes were dried,
then blocked for 1 hour in tris-casein buffer (Roche; Palo Alto, CA), with 0.1% Tween
20. Membrane transfer, blocking and all subsequent steps were done at room
temperature. Membranes were probed for 1 hr with 3.6 µg µl-1 of primary mouse
monoclonal antibody F99/97.6.1 (K. O’Rourke, USDA-ARS; Pullman, WA), that
recognizes prion epitope QYQRES (O'Rourke et al., 2000), followed by biotinylated goat
anti-mouse secondary antibody (Southern Biotech; Birmingham, AL) and enhanced
chemiluminescence (Amersham Biosciences; Piscataway, NJ). Western and slot blot
signal detection was performed with a commercial apparatus (Alphaimager, Alpha
Innotech Corporation; San Leandro, CA). A slot blot standard curve was generated from
densitometric values and known quantity of rPrP and compared to test sample values to
estimate PrPd concentration (ng per mg of wet tissue).
Animal sample size determination and experimental design: The sample sizes for
inoculations were determined by computer derived power calculations in consultation
with the Washington State University Statistics department. The rate of infection for this
56

transgenic model is unknown. A paired binomial calculation based on 50% infectivity,
alpha level of 0.05, and power level of 80%, requires a sample size of 14 animals for
each of the groups. Mice in approved biosafety level 2 animal rooms were inoculated
via the oral (PO), intraperitoneal (IP), and intracerebral routes (Table 3-1). MD BAC
mice were used to assess infectivity. PrP KO mice were used as negative controls to
confirm findings are specific to prion disease (Bueler et al., 1993). PRNP WT mice
were used as negative controls to verify that any findings of prion disease in MD BAC
mice were specific to the transgene.
Table 3-1: Animal numbers by treatment group for transgenic mouse challenge. * = A
separate set of 56 animals was used for each of the MDBAC, PrP KO, and PrP WT
genotypes.
Inoculum Number of
animals*
CWD positive brain given IC 14
CWD negative brain given IC 14
CWD positive brain given IP 14
CWD negative brain given IP 14
CWD positive brain given PO 14
CWD negative brain given PO 14
TOTAL 56
57

IC, intraperitoneal, and oral challenge: Oral administration was performed via
gavage of 1ml homogenate. IP administration was used with 100 µl of homogenate
injected into the right caudoventral abdominal quadrant. IC administration was
performed following anesthesia with a xylazine/ketamine mixture (0.008mg/g Xylazine,
0.13mg/g Ketamine) prepared fresh on the day of surgery. Sterile lubricating ointment
was place in the eyes and the skin was disinfected by standard surgical preparation.
Ten to thirty µl of sterile tissue homogenate prepared as above was administered
through the skin and coronal suture approximately 2 mm from the midline with a 27-
gauge needle into the frontal cortex to a depth of 3 mm.
Clinical observation and necropsy: Mice were monitored for clinical signs of
neurologic disease including ataxia, posterior paresis, paralysis, circling, or lethargy.
Animals were observed for up to 2 years (e.g. observed for the duration of a typical
mouse life span) and euthanized upon the development of neurologic symptoms. Mice
that ceased to eat and drink, lost the ability to move about the cage or became
moribund were euthanized, with central nervous tissue collected for analysis of
spongiform change and PrPd deposition. The development of central nervous system
disorders (e.g. circling, head tilt, seizures, recumbency, stupor, ataxia, paresis,
paralysis), served as a basis for euthanasia. Representative tissue samples from ileum,
cecum, colon, heart, lungs, liver, kidney, spleen, mesenteric and retropharyngeal lymph
nodes, cerebrum, brainstem, and cerebellum were collected in 10% neutral buffered
formalin and/or frozen at -80°C.
58

Tissue processing and immunohistochemistry: Tissue was formalin fixed for at least
2 days, trimmed, treated with 96% formic acid for 1 hr, processed, paraffin embedded,
sectioned at 5 µm, and placed on glass slides for hematoxylin and eosin (H/E) staining
or immunohistochemistry (IHC). IHC was performed at 37 °C with an automated
immunostainer (Ventana Medical Systems; Tucson, AZ) on samples of brain, lymph
nodes, and/or spleen similar to previously described (Spraker et al., 2002). Slides for
PrPd IHC were blocked with EZ Prep and Cell Conditioner per manufacturer’s
instructions (Ventana Medical Systems; Tucson, AZ), probed with primary mouse IgG1
monoclonal antibody F99/97.6.1 (provided by K. O’Rourke, USDA-Agricultural Research
Service; Pullman, WA) at 5 ug ml-1 for 30 mins, followed by biotinylated secondary goat
anti-mouse IgG antibody for 10 mins, streptavidin-horseradish peroxidase for 10 mins,
and 3-amino-9-ethylcarbazole/H2O2 chromagen (Ventana Medical Systems; Tucson,
AZ). Slides for glial fibrillar acidic protein (GFAP) IHC were blocked with commercial
antibody buffer (Ventana Medical Systems, Tucson, AZ), probed with primary rabbit
polyclonal antibody (CP040C, Biocare Medical; Concord, CA) diluted 1:600 for 12
minutes, followed by a universal secondary antibody/3’,3’-diaminobenzidine chromagen
kit (Ultraview DAB, Ventana Medical Systems; Tucson, AZ) for 8 mins. Positive IHC
control tissues included brain or lymph node from TSE infected elk, deer, sheep or
mice. Negative controls included tissue from uninoculated or CWD negative recipient
mice. Additional negative antibody controls included omission of primary antibodies or
substitution with unrelated mouse or rabbit primary antibodies (Ventana Medical
Systems; Tucson, AZ).
59

Microscopic examination: Light microscopic examination of tissue sections was
performed blindly on brain ipsilateral and contralateral to the injection site for
vacuolation, PrPd deposition, and astrocytosis. Brain and other collected tissues were
examined for intercurrent disease. Diagnosis of clinical TSE was based on neurologic
signs, and disease was confirmed by detection of spongiform vacuolation and PrPd
immunoreactivity within brain. Brains from asymptomatic animals were examined by
PrPd and GFAP IHC to rule out subclinical disease. Vacuolation and PrPd IHC scoring
was performed on five 1200 x 800 µm fields randomly selected within the anatomic area
of interest. Vacuolation scores were 0=within normal limits, 1=vacuoles confined to
white matter, 2=slight vacuolation in grey matter, 3=moderate vacuolation in grey matter
+/- in neurons, 4=pronounced vacuolation in grey matter +/- in neurons, 5=pronounced
vacuolation in grey matter and visibly within neuronal perikaryon [modified from (Bruce
et al., 2004)]. PrPd scores were 0=no signal detected or background only, 1=slight
signal intensity, 2=moderate signal intensity, 3=pronounced signal intensity. PrPd IHC
was performed on lymph nodes to determine lymphoreticular distribution. Astrocytes in
GFAP IHC sections were manually counted on five randomly selected grey matter fields
within areas of the cerebral cortex, hippocampus, and thalamus that corresponded to
areas of most severe vacuolation in TME positive IC recipients using a 200 by 250 µm
grid overlay on commercial imaging software (Nikon Elements BR, Nikon Corporation;
Tokyo, Japan). Statistical significance (p ≤ 0.05) of scores and counts between
treatment groups was determined using the Mann-Whitney test (GraphPad 5.0; San
Diego, CA).
60

Results
Pronuclear microinjection into fertilized ova and founder screen: 503 ova were
injected and transferred into recipient females using linearized or circular MD BAC DNA
on 7 separate occasions with 99 total pups born for 19.76% injection and transfer
efficiency (Table 3-2). Three pups out of 99 born (3.0% overall efficiency) were positive
by PCR amplification of the putative promoter region (PP) and open reading frame
(ORF) of the mule deer transgene (Figure 3-7). All three of these animals transferred
the PP and ORF to offspring confirming germline transmission of the transgene. Overall
efficiency of founder pup generation from total pups born was 3.0% and from total ova
injected and transferred was 0.6%. Efficiency of founder pups from pups born was
highest with the circular form (5.9%) followed by the 67.0kb form (2.6%) and no pups
were born with the 43.9kb form (0.0%), however these differences were not statistically
significant.
Table 3-2: Results of pronuclear microinjection of MD BAC DNA
# BAC DNA Size
Form injected
Enzymedigest
Ova injected/
Transferred
Pups Born
Founder Pups
Germline Transfer
1 67.0 kb linear NotI 50 0 0 ---
2 43.9 kb linear XhoI 82 33 0 ---
3 67.0 kb linear NotI 58 19 0 ---
4 Circular circular --- 42 9 1 Yes
5 Circular circular --- 98 8 1 Yes
6 67.0 kb linear NotI 124 19 1 Yes
7 43.9 kb linear XhoI 49 11 0 ---
61

PRNP ORF →
PRNP PP →
Figure 3-7: Representative agarose gel demonstrating positive ORF (716 bp) and PP
(856 bp) of MD BAC gene in transgenic founder mice. Far left and right lanes = DNA
Lambda-Hind III and 100 kb molecular weight markers. Lane A/B1 = MD BAC PCR
positive control DNA. Lane A/B6 DNA samples from positive founder mice. Lane
A17/B17 = dH2O negative control.
Genotype analysis of breeding colony: PCR amplification was performed for the MD
BAC transgene, the WT gene, and the NEO cassette (as a marker of the PrP KO), to
confirm genotypes of pups born in the breeding scheme and determine cohorts for
infectious challenge. The assay faithfully represents a single band in WT or KO
homozygous mice, and two bands in a mixed WT-KO heterozygote (Figure 3-8).
62

KO
WT
Figure 3-8: Representative agarose gel demonstrating PCR products of the wild type
mouse prion gene (WT, 764 bp) or the NEO marker cassette of prion deletion (KO, 511
bp). Far left and right lanes = Lambda-Hind III molecular weight markers.
Transcriptional analysis in MD BAC x PrP KO Tg mice: RT-PCR was performed on
tissue from MD BAC mice to determine if the transgene was being expressed. MD BAC
specific primers amplified a 776 bp product from cDNA of brain, liver, and spleen from a
MD BAC mouse (Figure 3-9). Amplification of RNA of the same mice or from cDNA of
WT mice did not yield product ruling out false positive amplification of residual DNA.
Additional RT-PCR performed using two different sets of intron-spanning MD BAC
specific primers yielded 221 and 261bp products confirming transgene transcription
(Figure 3-10). Intron-spanning RT-PCR for GAPDH yielded a 200bp product confirming
integrity of RNA purification and RT steps (data not shown).
63

WT
Mouse
cDN
A
br lv sp
dH2
MD
BA
C 1:10
67Kb
Tg Line R
NA
br lv spbr lv sp
67Kb
Tg Line cD
NAMD
BA
C
Figure 3-9: Representative agarose gel confirming MD Tg expression in MD BAC
mouse brain (br), liver (lv), and spleen (sp) by RT-PCR. Samples (left to right): Lambda
Hind III marker, MD BAC DNA, MD BAC mRNA, MD BAC cDNA, WT mRNA.
Figure 3-10: Representative agarose gel confirming MD Tg expression in MD BAC
mouse brain by intron-spanning RT-PCR. Samples: 100 bp marker, RNA and cDNA
from WT mouse, RNA and cDNA from MD BAC mouse, rat and H20 controls.
300bp →
Wild type MD BAC Tg
dH20
RAT cDNAcDNAcDNARNA RNARNA RNAcDNA cDNA
64

Translational analysis in MD BAC x PrP KO Tg mice: Western blotting was
performed on brain of MD BAC Tg mice to determine if PrPc was being translated.
Western blotting of these mice, on the PrP KO background, produced a typical three
band pattern representing the di-, mono-, and un-glycosylated form of prion protein
migrating at approximately 35, 29, and 25 kDa, respectively, thereby confirming
transgenic production of PrPc (Figure 3-11).
Figure 3-11: Representative western blot of PrPc in tissue from MD BAC mouse.
Lanes: Lane 1 (far left) = Molecular weight marker in kilodaltons, 2 = blank, 3 = spleen,
4 (far right) = brain.
Characterization of inocula
Mule deer brain samples for challenge were characterized to determine suitability for
challenge by assessing PK resistance, quantity of PrPd antigen, and prion genotype.
PrPd immunoreactivity in CWD positive mule deer brain homogenates was confirmed by
western blot of PK digested samples (Figure 3-12, left). Estimated PrPd antigen content
65

for pooled CWD positive samples was 6.61 +/- 2.03 ng per mg of wet tissue (Figure 3-
12, right). Total administered PrPd content was approximately 6.61 ng for IC, 66.1 ng
for IP, and 661.1 for oral recipients. Brain from a CWD negative control mule deer did
not exhibit immunoreactivity by western (Figure 3-12, left) or slot blot. Tissue from
positive and negative mule deer had a uniform DNA sequence consistent with the
PRNP ORF sequence used to generate MD BAC mice.
40-
30-
20-
Figure 3-12: Immunoreactivity and measurement of antigen load following PK digest in
CWD positive and CWD negative mule deer brain samples used for experimental
challenge. Left = Western blot using 50 ug total protein. Lane 1 = kDa molecular weight
marker, Lane 2 = CWD negative homogenate, Lane 3 = CWD positive homogenate.
Right = Graph illustrating correlation between nanogram quantity of PrP (x-axis) and
densitometric values (y-axis). ■ = Values for recombinant PrP reference standard. ○ =
Mean amount of PrPd in CWD positive elk brain estimated from density values. r2 =
0.9749, dashed line = 95% confidence interval.
66

Findings in MD BAC x PrP KO Tg mice post challenge: Mice have been monitored
for signs of neurologic disease and analyzed by histopathology and western blotting for
diagnosis of TSE. Clinical signs observed include ruffled hair coat, lethargy,
dehydration, posterior paresis, and rectal prolapse. Neoplasia and abscesses have
been observed. Necropsy and histopathologic findings are summarized (Table 3-3).
Table 3-3: Current findings in MD BAC mice challenged with CWD. Days = days post
challenge. CNS WNL = Central nervous system within normal limits.
Inocula Route Gender Days Findings CWD + IC Male 140 CNS WNL, obese, hepatic lipidosis CWD - IC Male 156 CNS WNL, lymphoid hyperplasia CWD - IC Male 172 CNS WNL CWD + IC Female 266 CNS WNL, lymphosarcoma CWD + IC Male 375 CNS WNL, hemangiosarcoma CWD + IC Female 438 CNS WB Positive, lymphosarcoma CWD + IC Female 456 CNS ~spongiform change, ocular abscess CWD - IC Male 491 Severe autolysis CWD + IC Female 491 Rectal prolapse. Laboratory data pending CWD + IC Female 499 Laboratory data pending CWD + IC Female 499 Posterior paralysis. Laboratory data pending CWD - IC Male 610 CNS WNL, histocytic sarcoma CWD + IC Male 633 Posterior paralysis. Laboratory data pending
CWD + IP Female 222 CNS WNL CWD - IP Male 402 CNS WNL, lymphosarcoma CWD - IP Female 455 lymphosarcoma
CWD + PO Female 290 CNS WNL
Western blotting of PK digested brain tissue exhibited positive signal in brain of MD
BAC mouse injected with CWD positive material at 438 days post challenge (Figure 3-
13). Histopathology performed on an MD BAC mouse has demonstrated subtle
spongiform change consistent with TSE (Figure 3-14). The mouse in this case was
euthanized due to an ocular abscess; lesions may have become more pronounced with
67
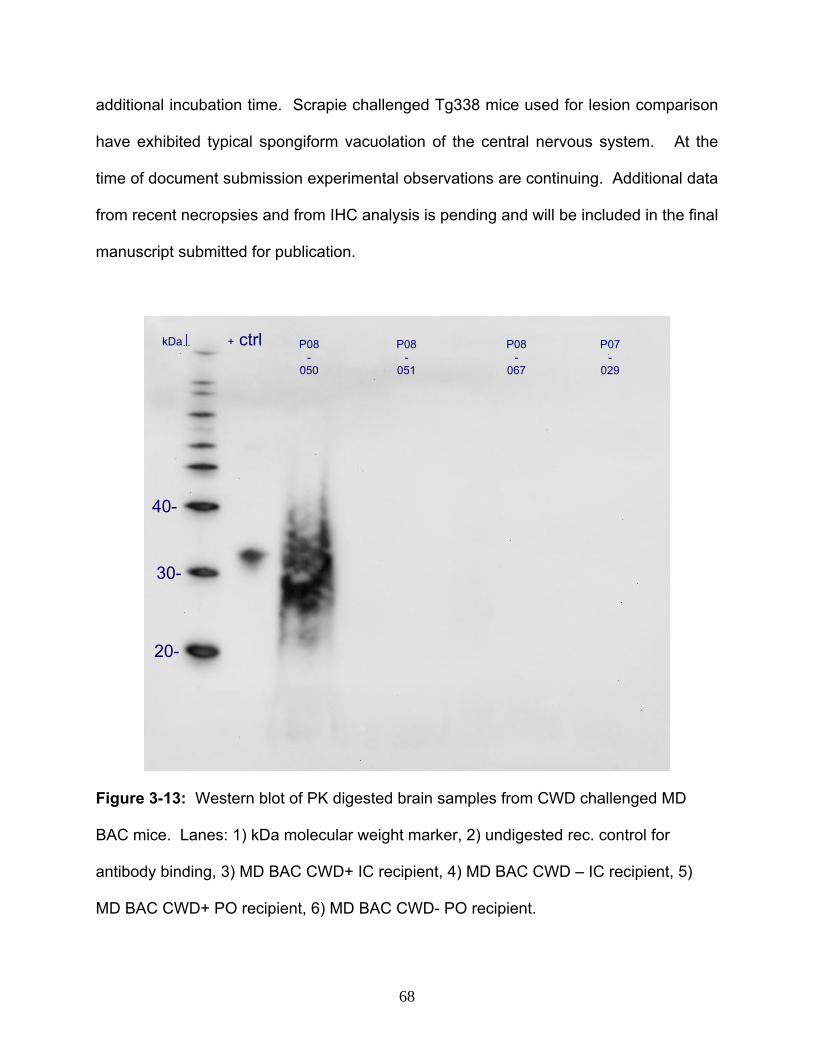
additional incubation time. Scrapie challenged Tg338 mice used for lesion comparison
have exhibited typical spongiform vacuolation of the central nervous system. At the
time of document submission experimental observations are continuing. Additional data
from recent necropsies and from IHC analysis is pending and will be included in the final
manuscript submitted for publication.
P08-
067
P07-
029
P08-
050
+ ctrl kDa↓ P08-
051
20-
30-
40-
Figure 3-13: Western blot of PK digested brain samples from CWD challenged MD
BAC mice. Lanes: 1) kDa molecular weight marker, 2) undigested rec. control for
antibody binding, 3) MD BAC CWD+ IC recipient, 4) MD BAC CWD – IC recipient, 5)
MD BAC CWD+ PO recipient, 6) MD BAC CWD- PO recipient.
68

Figure 3-14: Photomicrographs of the central nervous system showing spongiform
vacuolation in Tg338 and MD BAC transgenic mice following IC challenge with scrapie
or CWD, respectively. Top panel = scrapie challenged Tg338 mouse. Middle panel =
69

CWD+ challenged MD BAC mouse. Bottom panel = MD BAC negative control mouse.
Left column = cerebral cortex. Right column = medulla oblongata.
Discussion
The past few decades have seen widespread use of murine transgenesis for the study
of diseases related to immunity, infectious disease, cancer, congenital abnormalities,
and neurodegeneration among others. These models provide a readily manipulatable
genome, allow high sample size, and require a relatively small commitment of space
and resources compared to other animals. CWD experimentation, as for most studies of
livestock and wildlife, is hampered by the large size of the study animals requiring
significant investments in infrastructure and resources to successfully complete
projects. At the time this project began there were no preexisting mouse models of
CWD. This combined with the previously mentioned limitations of certain mouse
models led to the pursuit of the alternative method of transgenesis using a BAC. The
results of this study show that the transgenic technique results in stable transcription
and translation of mule deer prion protein and replicates disease pathogenesis as
demonstrated by accumulation of PrPd and spongiform change within the central
nervous system.
The usefulness of BAC’s in the generation of Tg mice has been previously
demonstrated (Yang & Seed, 2003) and has been confirmed for the study of scrapie (Le
Dur et al., 2005). The present study demonstrates the use of a BAC molecule to stably
transcribe and translate the mule deer prion gene. The multiple rounds of pronuclear
microinjection, large number of ova transfers, relatively small numbers of pups born,
70

and even fewer founders may make the technique seem daunting. However, the
efficiencies that were observed in this work are similar to what may be expected of more
traditional constructs of much smaller size. These similarities combined with the
advantage of large insert size will likely lead to increased use of BAC to generate
transgenic mice. Initial characterization of this MD BAC mouse confirms that the
necessary components of transgene function are present for prion disease to develop.
The overall low rate of mortality indicates that there is no adverse effect on longevity in
this population and combined with a lack of developmental, anatomical, or behavioral
abnormalities further confirms the feasibility of the use of BAC for transgenesis.
The extra step of breeding onto an endogenous mouse PRNP null background added
approximately two years of time prior to when the mice could be used for infectious
challenge. However, this step was necessary to overcome the complication of
dominant-negative inhibition. Furthermore, an additional two years was added as mice
were backcrossed for five generations to yield greater than 95% uniform C57Bl6 genetic
background. The backcrossing step may provide useful insights into the role of murine
genetic backgrounds on prion disease as both mixed genetic and uniform genetic
background mice have been challenged. If differences are noted between the mixed
and uniform genetic backgrounds in could provide a basis for quantitative trait loci
analysis to identify other genes involved in prion pathogenesis. Some of the elder mice
on the mixed genetic background are approaching two years post challenge and
nearing the end of their normal life span. As a consequence, lesions typical for aged
mice have been observed and are expected to continue. The occurrence of age related
71

lesions will continue to be monitored to determine if it correlates with TSE challenge
status. The pathologic profile will be compared among groups to determine if the
relative heterogeneity or homogeneity of the genetic background affects the TSE
incubation time and/or relative susceptibility.
There are a number of parameters that may modulate the pathobiology as observation
of the MD BAC Tg mouse continues. First, TSE have long incubation periods. The
natural resistance of mice to prion disease has raised the question of whether wild type
mice are truly “immune” to prion infection or if they simply do not live long enough to
manifest disease. Relative level of transgene expression is one facet that may affect
incubation time and is currently being investigated. Second, there may be a component
of the mixed genetic background that alters incubation time or lesion profile. The
uniform genetic background is approximately one year behind the former cohort thus it
will be sometime before conclusions can be reached about the relative contributions of
the genetic background to disease. Third, problems with the brain homogenates used
for challenge, such as low overall prion titer, may lengthen incubation time. The positive
western blots and IHC of challenge material suggest that the mule deer homogenates
are sufficient to induce disease. Nevertheless, alternative samples of CWD material are
currently being administered to evaluate dynamics of incubation time and strain. Fourth,
the biological properties of normal prion protein may play a role in this model.
Specifically, glycosylation could be a modulating factor. The prion surface glycoprotein
has three glycosyslated forms and the relative band intensity and mobility pattern of
these glycosylations varies among prion strains and may be a contributing factor in
72

relative transmissibility within and between species. However, it is mechanistically
uncertain how glycosylation affects TSE pathogenesis. This model may provide a
means to futher explore the role of glycosylation upon both PrPc and PrPd during the
course of TSE pathogenesis.
Time is the element that makes TSE research a daunting task. It is also the essential
factor that will determine the viability of this model. Regardless of the final outcome of
this investigation, it must be remembered that pushing the barriers of current
knowledge, and developing new tools to accomplish that, are not without hurdles.
Perhaps in time the risk associated with long term development of this model will be
rewarded by findings that improve our knowledge of pathogenesis not only in CWD but
for other forms of TSE as well.
73

CHAPTER FOUR
OVINE PROGRESSIVE PNEUMONIA VIRUS PROVIRUS LEVELS ARE
UNAFFECTED BY THE PRION 171R ALLELE IN AN IDAHO SHEEP FLOCK@
Robert D. Harrington,1,2,3٭ Lynn M. Herrmann-Hoesing,1,2 Stephen N. White,1,2,4
Katherine I. O’Rourke,1,2 and Donald P. Knowles1,2
1Animal Disease Research Unit, Agricultural Research Service, US Department of
Agriculture, Pullman, WA 99164-6630, USA
2Department of Veterinary Microbiology and Pathology, Washington State University,
Pullman, WA, 99164-7040, USA
3Department of Comparative Medicine, University of Washington, Seattle, WA 98195-
7190, USA
4Center for Integrated Biotechnology, Washington State University, Pullman, WA 99164,
USA
@This chapter is in submission to the journal Genetics Selection Evolution
.Author for correspondence, [email protected]٭
See Appendix A for attributions to contributing authors.
Summary
Selective breeding of sheep for arginine (R) at prion gene (PRNP) codon 171 confers
resistance to classical scrapie. However, other functional affects of 171R selection are
uncertain. Ovine progressive pneumonia/Maedi-Visna virus (OPPV) may infect more
than half a flock thus any affect of 171R selection upon OPPV susceptibility or disease
74

progression could have major impact upon the sheep industry. Hypotheses that the
PRNP 171R allele is 1) associated with presence of OPPV provirus and 2) associated
with higher provirus levels were tested in an Idaho ewe flock. OPPV provirus was found
in 226 of 358 ewes by quantitative PCR. The frequency of ewes with detectable
provirus did not differ significantly among the 171QQ, 171QR, and 171RR genotypes
(p>0.05). Also, OPPV provirus levels in infected ewes were not significantly different
among codon 171 genotypes (p>0.05). These results show that, in the flock examined,
the presence of OPPV provirus and provirus levels are not related to the PRNP 171R
allele. Therefore, a genetic approach to scrapie control is not expected to increase or
decrease the number of OPPV infected sheep or the progression of disease. This study
provides further support to the adoption of PRNP 171R selection as a scrapie control
measure.
Introduction
Scrapie is the prototypical prion disease and one of several described in animals and
humans. Accumulation of disease associated prion protein (PrPSc), an abnormally
folded form of normal host prion protein (PrPC), is central to disease and expression of
the host prion gene (PRNP) is necessary in pathogenesis (Bueler et al., 1993). PRNP
open reading frame (ORF) variants associate with disease incubation time (Dickinson et
al., 1968) and relative disease susceptibility in sheep (Bossers et al., 2000; Bossers et
al., 1996; Hunter et al., 1996; O'Rourke et al., 1997; Westaway et al., 1994), goats
(Acutis et al., 2006; Papasavva-Stylianou et al., 2007; Vaccari et al., 2006), elk (Hamir
et al., 2006a; Johnson et al., 2003; O'Rourke et al., 1999), deer (Johnson et al., 2003;
75

O'Rourke et al., 2004) and humans (Bishop et al., 2006; Cervenakova et al., 1998;
McCormack et al., 2002; Zeidler et al., 1997).
Polymorphisms in sheep at PRNP codons 136 (Alanine / Valine), 154 (Arginine /
Histidine), and 171 (Glutamine / Arginine) are implicated in scrapie susceptibility [for
review see O'Rourke, 2001]. Codon 171 is an important element of susceptibility in the
United States (US) sheep population (O'Rourke et al., 1997; Westaway et al., 1994).
Sheep homozygous for glutamine at codon 171 (171QQ) are highly susceptible to
Scrapie, whereas sheep that are heterozygous (171QR) or homozygous (171RR) for
arginine are highly resistant to classical strains of US Scrapie.
The PRNP 171Q allele predominates in US sheep whereas the 171R allele and 171RR
genotype are less common (the latter occur in about 37% and 16% of US sheep,
respectively (USDA, 2003)). Selective breeding for the 171R minor allele is sometimes
used as a Scrapie control measure, however the functional consequences of 171R
selection upon other traits is uncertain. Genetic selection may have unexpected
positive or negative effects as individual genes may have multiple biologic roles
(pleiotropy) or may be linked to other genes that impact overall biological functions.
Uncertainty regarding PRNP selection effects (beyond Scrapie resistance) has led to
investigation of multiple ovine traits including reproductive performance (Alexander et
al., 2007; Alexander et al., 2005; De Vries et al., 2005; Sweeney et al., 2007), milk
production (Alvarez et al., 2006; De Vries et al., 2005; Salaris et al., 2007), carcass and
wool quality (Alexander et al., 2005; De Vries et al., 2005; Evoniuk et al., 2007; Vitezica
76

et al., 2007), and genetic diversity (Alfonso et al., 2006). However, PRNP selection
effects on disease susceptibility (besides Scrapie) has only been studied for Salmonella
resistance (Vitezica et al., 2007).
Ovine progressive pneumonia/Maedi-Visna virus (OPPV) is a monocyte/macrophage
tropic lentivirus (a subclass of retrovirus) endemic in many US sheep flocks that causes
pneumonia, mastitis, arthritis, and encephalitis. One of five sheep is infected based on
detection of anti-OPPV serum antibodies and seroprevalence can increase to one in
two sheep in open rangeland environments (Cutlip et al., 1992). Quantitative PCR
(qPCR) is an alternative method for detection of lentivirus that provides both diagnostic
and prognostic information (Arens, 1993; Verhofstede et al., 1994; Vitone et al., 2005).
The qPCR assay measures the presence and amount of virus that has been reverse-
transcribed and integrated into the host genome (provirus). The technique is a useful
indicator of disease progression in the study of OPPV because OPPV provirus levels
are associated with severity of pulmonary lesions (Brodie et al., 1992; Zhang et al.,
2000).
Scrapie is diagnosed in about one of every 500 culled sheep (USDA, 2003) thus OPPV
has greater prevalence. Uncertainty regarding PRNP selection effects can create
producer reluctance to the implementation of 171R selection when OPPV is a more
severe herd-health problem. A prion-retrovirus pathogenic relationship of undetermined
mechanisms has been observed between Scrapie and Murine Leukemia Virus (MuLV)
(Lee et al., 2006), Scrapie and Caprine Arthritis Encephalitis Virus (CAEV) (Stanton,
77

2008), PrPSc accumulation in OPPV associated mastitis (Ligios et al., 2005) and
influence of PrPc on HIV infection (Leblanc et al., 2004). In this study, two hypotheses
were tested in an Idaho ewe flock that 1) the PRNP codon 171R allele is associated
with the presence of OPPV provirus and 2) the PRNP 171R allele is associated with
higher OPPV provirus levels was tested in an Idaho ewe flock. This study will help
guide producer decisions, provides information for future prion-retrovirus co-infection
studies, and advances knowledge of whether PRNP selection affects other infectious
diseases.
Methods
Animals: Three hundred fifty eight ewes were sampled from a flock in southeastern
Idaho in which OPPV is endemic and there are no reported cases of scrapie. Breeding
was performed without prior selection of prion genotype. Approximately equal numbers
of animals were included for each breed and age (Table 4-1).
Table 4-1: Distribution of sample set by breed and age.
Age Columbia Polypay Rambouillet 3yo 39 27 32 4yo 30 31 32 5yo 31 33 36 6yo 17 25 25
Total 117 116 125
Nucleic acid extraction: Peripheral blood leukocytes (PBL) were isolated from whole
blood as described (Herrmann-Hoesing et al., 2007b). Genomic DNA and messenger
RNA were extracted from PBL using commercial kits (Gentra, Minneapolis, Minnesota,
USA and Qiagen Inc., Valencia, CA, respectively). Reverse transcription was
78

performed using a random hexamer first strand system (Invitrogen Corporation,
Carlsbad, CA).
PRNP Genotyping: DNA amplification and sequencing of the ovine PRNP ORF was
performed similar to previous experiments using forward primer 5’-
GGCATTTGATGCTGACACC-3’ and reverse primer 5’-TACAGGGCTGCAGGTAGAC-
3’ (Schneider et al., 2008). Reverse primer 5’-GGTGGTGACTGTGTGTTGCTGA-3’
was used for standard dideoxynucleotide sequencing. All sequencing was performed
at the Laboratory for Biotechnology and Bioanalysis (Washington State University,
Pullman, WA). PRNP genotypes were analyzed using commercial software (Vector
NTI, Invitrogen; Carlsbad, CA or Lasergene Seqman Pro v7.1, DNAstar, Inc, Madison,
WI) and codon variants reported by single letter code (e.g. glutamine, Q; arginine, R;
valine, V; histidine, H; leucine, L; phenylalanine, F).
OPPV quantitative PCR: OPPV provirus level was determined using a previously
described quantitative real-time PCR (qPCR) assay (Herrmann-Hoesing et al., 2007b).
The OPPV qPCR used primers TMENVCONf 5′-TCA TAG TGC TTG CTATCA TGG
CTA-3′ and TMENVCONr 5′-CCG TCC TTG TGT AGG ATT GCT-3′ (Invitrogen
Corporation, Carlsbad, CA) and a Taqman 5′-5′-hexachlorofluorescein-AGC AAC ACC
GAG ACC AGC TCC TGC-3′ Black Hole Quencher-1 probe (Integrated DNA
Technologies, Coralville, IA) targeting the highly conserved transmembrane envelope
region of the North American OPPV strain (Herrmann et al., 2004).
79

Statistical analyses: Two types of genotypic comparison were made using provirus
data and PRNP genotype, with a minimum PRNP allele frequency of 10% required for
analysis. Association between PRNP genotype and presence or absence of OPPV
provirus was tested using logistic regression models from the logistic procedure of SAS
v9.1 (SAS Institute, Cary, NC). Association between PRNP genotype and the level of
logarithm (base 10)-transformed provirus in OPPV positive animals was tested using
the glm procedure in SAS v9.1. In each case the association model included breed as
a categorical predictor, age as a linear covariate, the interaction between breed and
age, and the PRNP genotype of interest. Adjusted odds ratios and 95% confidence
interval were calculated for the pairwise comparison of the frequency of OPPV positive
ewes in each genotype. Adjusted mean log-transformed provirus levels were reported
with 95% confidence intervals. Stepdown Bonferroni p-value correction (Holm, 1979)
was applied separately to each set of analyses.
Results
Distribution of PRNP genotypes
The PRNP genotypes were determined as the first step in testing association with the
presence of OPPV provirus and OPPV provirus levels. ORF coding variants were
identified at codons 101(Q/R), 136(A/V), 141(L/F), 143 (H/R), 154 (R/H), and 171 (Q/R).
Codons 143 and 171 had amino acid substitutions with minor allele frequency of at least
10%. Codons 101, 136, 141, and 154 had less than 10% minor allele frequency and
therefore these four codons were excluded from further association analysis. Of the
358 sheep sampled 100 were 171QQ, 179 were 171QR, and 79 were 171RR, providing
80

representation of all three genotypes (Figure 4-1, left). Examination of the 171R allele
relative to the overall PRNP ORF showed that in all animals of the 171RR genotype
there were no other PRNP codon variants present. Codon changes at other positions
only occurred in animals that had at least one wild type 171Q allele. Of the 358 sheep,
279 were 143HH, 71 were 143HR, and 8 were 143RR (Figure 4-1, right). Due to the
rarity of the 143RR genotype only the 143HH and 143HR genotypes were further
analyzed.
Figure 4-1: Number of sheep distributed among PRNP genotypes. Left = codon 171,
Right = codon 143, y-axis = number of animals.
Frequency of OPP provirus among PRNP genotype
The presence or absence of OPPV provirus was compared among the PRNP 171 and
among the PRNP 143 genotypes to determine if minor alleles within those genotypes
affected the number of sheep infected with OPPV. In the flock, 226 of 358 (63.1%)
sheep had detectable OPPV provirus. Over half of the animals were positive for OPPV
provirus within each PRNP genotype (Table 4-2). The frequency of OPPV positive
animals was not significantly different between the 171QQ, QR, and RR genotypes
81

(Figure 4-2; Table 4-3). Also, the frequency of OPPV positive animals did not differ
significantly between the 143HH and HR genotypes (Table 4-3).
Table 4-2: Number of OPPV positive or negative sheep among PRNP genotypes used
for statistical comparison.
PRNP OPPV Status % OPPV Genotype Negative Positive Positive 171 QQ 36 64 64.0 171 QR 61 118 65.9 171 RR 35 44 55.7 143 HH 103 176 63.1 143 HR 26 45 63.4
OPPV positive vs. negative status
0.00.51.01.52.0
171 QQ vs QR 171 QR vs RR 171 QQ vs RR 143 HH vs RHPRNP genotype
Odd
s rat
io
Figure 4-2: Odds ratio and 95% confidence interval for effect of PRNP genotype upon
frequency of OPPV positive animals.
Table 4-3: Significance level for effect of PRNP genotype upon frequency of OPPV
positive animals. P-values are before (nominal, left) and after (corrected, right)
stepdown Bonferroni multiple test correction.
Genotype OPPV positive vs negative p-value comparison nominal corrected
171 QQ vs QR 0.23 0.90 171 QR vs RR 0.23 0.90 171 QQ vs RR 0.60 1.00 143 HH vs RH 0.78 1.00
82
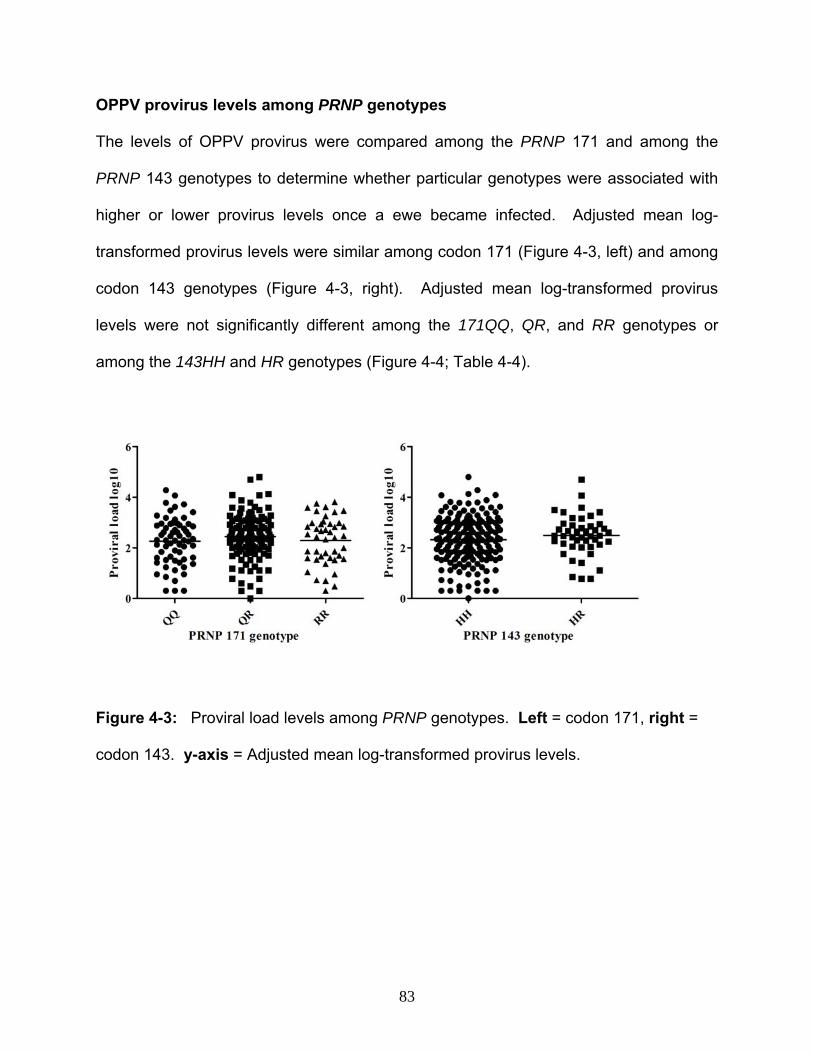
OPPV provirus levels among PRNP genotypes
The levels of OPPV provirus were compared among the PRNP 171 and among the
PRNP 143 genotypes to determine whether particular genotypes were associated with
higher or lower provirus levels once a ewe became infected. Adjusted mean log-
transformed provirus levels were similar among codon 171 (Figure 4-3, left) and among
codon 143 genotypes (Figure 4-3, right). Adjusted mean log-transformed provirus
levels were not significantly different among the 171QQ, QR, and RR genotypes or
among the 143HH and HR genotypes (Figure 4-4; Table 4-4).
Figure 4-3: Proviral load levels among PRNP genotypes. Left = codon 171, right =
codon 143. y-axis = Adjusted mean log-transformed provirus levels.
83
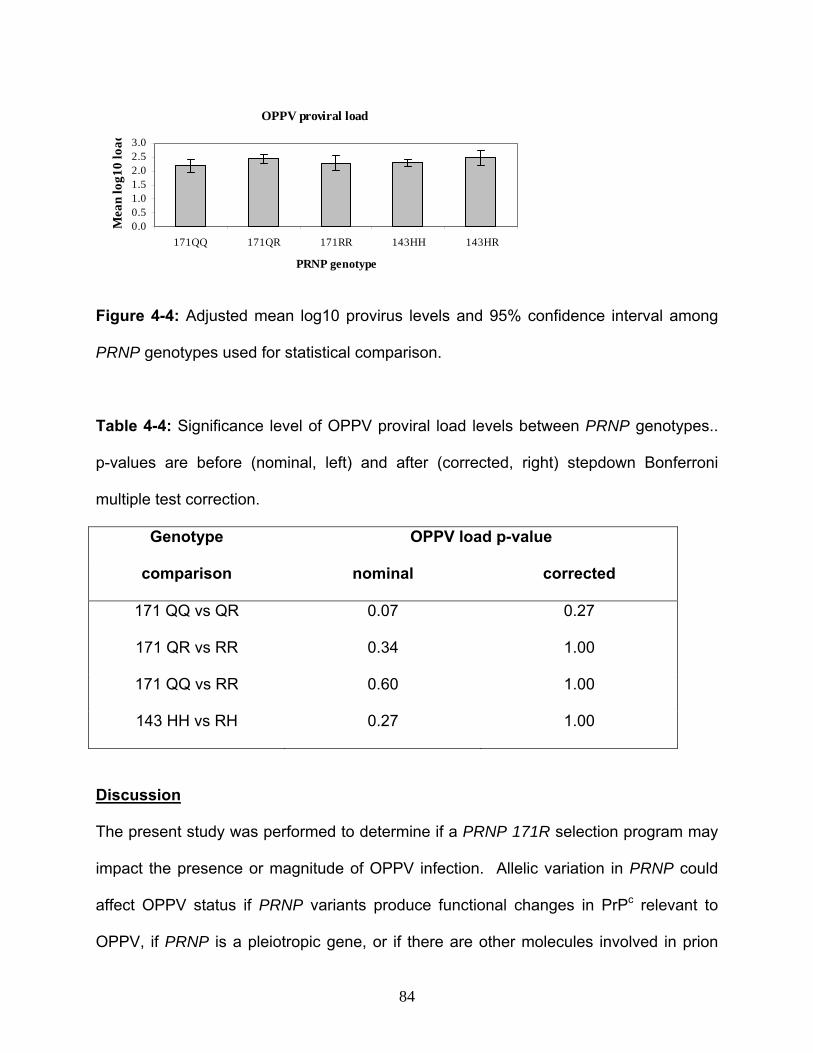
OPPV proviral load
0.00.51.01.52.02.53.0
171QQ 171QR 171RR 143HH 143HR
PRNP genotype
Mea
n lo
g10
load
Figure 4-4: Adjusted mean log10 provirus levels and 95% confidence interval among
PRNP genotypes used for statistical comparison.
Table 4-4: Significance level of OPPV proviral load levels between PRNP genotypes..
p-values are before (nominal, left) and after (corrected, right) stepdown Bonferroni
multiple test correction.
Genotype OPPV load p-value
comparison nominal corrected
171 QQ vs QR 0.07 0.27
171 QR vs RR 0.34 1.00
171 QQ vs RR 0.60 1.00
143 HH vs RH 0.27 1.00
Discussion
The present study was performed to determine if a PRNP 171R selection program may
impact the presence or magnitude of OPPV infection. Allelic variation in PRNP could
affect OPPV status if PRNP variants produce functional changes in PrPc relevant to
OPPV, if PRNP is a pleiotropic gene, or if there are other molecules involved in prion
84

pathogenesis that also affect OPPV pathogenesis. Alternatively, there may be nearby
chromosomal regions affecting OPPV pathogenesis that are in linkage disequilibrium
with certain PRNP alleles. However, the lack of association between PRNP genotype
and OPPV status shows that the presence of a specific PRNP genotype does not
influence the presence or magnitude of OPPV infection in this flock.
The study demonstrated that the frequency of sheep with detectable OPPV provirus
was not related to the PRNP 171R (or 143R) allele in an Idaho ewe flock. This
suggests that it is no more likely that a 171RR or 171QR sheep within a flock would
become infected when compared to a 171QQ sheep. Likewise, the data suggest there
is no difference in frequency of infection between the 143HH and 143HR sheep. Only
ewes were sampled in this study so it is possible that introduction of rams could have a
different affect, however it is unlikely considering that the frequency of OPPV in rams is
equivalent, or perhaps lower than OPPV frequency in ewes (Arsenault et al., 2003;
Cutlip et al., 1992).
Provirus levels in OPPV positive animals were also not related to the PRNP 171R and
143R alleles. Thus, PRNP selection should not affect progression of disease once
animals become infected with OPPV. A shift of flock genetics to a greater frequency of
171QR or 171RR sheep is unlikely to accelerate shedding or transmission of OPPV. In
these sheep there also was no difference in provirus levels between animals of the 143
HH and 143HR genotypes, thus there are no documented cases where PRNP
genotypes impact OPPV infection.
85

Interactions between retrovirus’ and normal or abnormal prion protein have been
previously observed. The current findings do not exclude the possibility that increases
in ovine PrPc or CD230 expression could alter OPPV replication as observed in a
human cell line where over-expression of human PrPc thwarted HIV-1 replication
(Leblanc et al., 2004). OPPV replicates in mammary macrophages and microglia and
transmits via ewe milk (Carrozza et al., 2003; Ebrahimi et al., 2000; Herrmann-Hoesing
et al., 2007a), and PrPSc is found in macrophages of lymphoid follicles and microglia
and transmits via ewe milk (Andreoletti et al., 2002b; Caplazi et al., 2004; Herrmann et
al., 2003; Konold et al., 2008; Ligios et al., 2005) thereby suggesting functional overlap
between host proteins involved in both prion and lentivirus pathogenesis. Additional
links between prion and retrovirus’ are indicated by data showing that caprine arthritis-
encephalitis virus (CAEV) aids PrPd accumulation in immortalized microglia in vitro (J.
Stanton, manuscript in preparation), and that scrapie infection increases MuLV
expression and reciprocally MuLV accelerates scrapie pathogenesis (Lee et al., 2006).
This study is one of many examining PRNP selection effects. The PRNP 171RR
genotype has no apparent effect on reproductive performance (Alexander et al., 2007;
De Vries et al., 2005), ovulation rates and litter sizes (Sweeney et al., 2007), and only
the Suffolk breed has lower lamb weaning weights (Alexander et al., 2005). Milk
production and quality is not effected in Churra (Alvarez et al., 2006), East Friesian milk
sheep (De Vries et al., 2005) or Sardinian sheep, nor are there significant changes in
udder morphology (Salaris et al., 2007). Carcass and wool quality are not impaired (De
86

Vries et al., 2005; Vitezica et al., 2007), and 171R may positively affect average daily
gain (Evoniuk et al., 2007). 171R has no effect on Salmonella resistance (Vitezica et
al., 2007). Finally, pedigree examination in Laxta Black Faced-type Navarra sheep
showed no overall negative effect (Alfonso et al., 2006). It is possible that in future
generations repeated intensive selective pressure may result in loss of low frequency
alleles adjacent to PRNP that affect the above traits. However, it seems a remote
possibility since these nine studies examining reproduction, meat, milk, fiber, and
infectious disease traits in a dozen different breeds found no overt negative effect from
the PRNP 171R allele or 171RR genotype.
The present study and previous investigations indicate that the corresponding affect of
PRNP 171R selection is minimal. This study was restricted to a controlled flock of ewes
in the northwestern US, and the Columbia, Polypay, and Rambouillet breeds. Results
may differ in other breeds, other genders, other environments or management
conditions, with other strains of retrovirus, or with other infectious diseases that remain
unstudied. However, the statistical models used in this study accounted for variation of
age and breed, and previous studies have shown no significant effect of 171R selection
in different breeds and in a wide variety of settings. Unidentified variants of the PRNP
ORF, PRNP promoter regions, or PRNP homologues, or PrPc expression may also
affect future results. Nevertheless, this investigation of a flock with endemic OPPV
shows that prevalence of OPPV infection and level of OPPV provirus loads are not
affected by PRNP 171R selection and supports the implementation of 171R selection as
a component of Scrapie control programs.
87

CHAPTER FIVE
CONCLUDING REMARKS
The past few decades saw great concern about the zoonotic potential of BSE and other
forms of prion disease such as CWD. However, in chapter two it was shown that there
are significant barriers in ruminant to carnivore prion transmission and that CWD likely
has low risk of natural transmission between species. Two new PRNP polymorphisms
were identified with one possibly being involved with relative susceptibility. The
polymorphism identified at codon 27 is near the site of the cell membrane signaling
peptide cleavage site and is intriguing as the codon could theoretically influence
trafficking of PrPc to the cell membrane. As it is known that cytosolic accumulation of
PrPc is neurotoxic it suggests that any alteration of transport to the cell surface could be
a mechanistic factor that may be involved with relative TSE susceptibility. This study
also shows that the biology of CWD and scrapie are significantly different than BSE as
the former two have significant barriers to carnivore transmission whereas the latter
does not. Considering the demonstration of barriers to orally mediated disease despite
the occurrence of IC induced disease, the preponderance of conclusions based on IC
transmission must give us pause when considering the relative risk of prion
transmission between species as compared to transmission within species. Is it true
that transmissibility of prions between species is as dire as predicted and is the concern
warranted? Certainly BSE stands out in its difference from other prion diseases as it (or
some common causal factor) is most likely to affect multiple species, thus caution is
prudent. However, considering that to date there are less than 200 confirmed cases of
variant CJD diagnosed throughout the world we must work to educate the public that
88

while TSE are serious and fatal diseases there are far more imminent threats (influenza
being just one example) with potential for widespread worldwide morbidity and mortality.
Conversely prion transmission within a species occurs readily in sheep, deer, and elk
thus posing a great threat to those species. In the future we may recognize that the
greatest impact of TSE upon humans is not zoonotic but rather economic through
production losses, inhibition of trade, and altered public perception.
TSE transmission dynamics are best studied in the natural host species but as with any
disease process there may be logistical, economic, or ethical obstacles to conducting
research in the natural host. Chapter three reported on the affect of the cervid putative
promoter region on prion biology pertinent to the development of alternative mouse
models that may provide insight into differences between parenteral and non-parenteral
TSE transmission routes. This study provides context to the role of PRNP promoter
regions in the development of disease and presents a novel model that may facilitate
future investigations into prion pathogenesis. The model has promise to be useful in
study of CWD and of prion biology in general. The development of increasingly efficient
models that more closely resemble natural disease processes are needed to further our
scientific knowledge.
Effective TSE control strategies are being developed and, at least for scrapie, may
result in disease eradication. However, as with any management program there may be
questions about unforeseen consequences. In chapter four of this work one possible
detrimental affect of scrapie management was examined. OPPV represents perhaps
89

the most economically important disease of the sheep industry. Thus a producer may
ask whether a Scrapie control program is warranted when, at the least, they have more
pressing disease control issues and, at the most, PRNP selection may have untoward
effects. This investigation demonstrated that Scrapie control measures are unlikely to
affect OPPV and is additional evidence supporting the benefit of adopting PRNP
selection strategies for agricultural management.
Several aspects of prion biology seemed certain when I began my studies but with time
I have discovered discrepancies and inconsistencies in the literature that make TSE
seem more mysterious and enigmatic. As scientists we work to answer one set of
questions but along the way we may encounter others that are increasingly difficult to
answer. Such is the case for TSE. In addition to questions regarding zoonotic potential
there are several other questions about TSE that continue to be debated.
The following is a partial list of examples that are of personal interest:
1. What is the day-to-day function of the normal PrPc?
2. What role, if any, does PrPc play in biologic processes or diseases other than
TSE?
3. What is the role of divalent cations or other ligands in PrPc function?
4. Is PrPd accumulation a function of abnormal turnover, increased production,
impaired degradation, or a combination of all three?
5. Is disease only mediated by post-translational events or is there a transcriptional
component as well?
6. And lastly, what is the true nature of the infectious agent?
90

The prion hypothesis has received international recognition but there is more to the
story. Even the preeminent proponent of the hypothesis suggests an additional factor
(the so-called protein “X” theory) is required for the development of disease (Kaneko et
al., 1997). Others have suggested that the etiologic agent is entirely different, such as a
virion or virus-like particle (Manuelidis, 2007). Another way to consider these diseases
may be as a toxic bioaccumulation rather than as an infectious agent. Protein
misfolding is the key to TSE but it is a phenomenon that also occurs in many other
diseases such as in Alzheimer’s. It is plausible that misfolding of prion protein
represents the end result of a pathologic process rather than the original cause. In
nature proteins are constantly undergoing change in conformational state and there
may be common pathways or initiating factors of misfolding that are not unique to prion
disease. Whatever the ultimate cause and effect may be, the persistent questions
regarding prion diseases warrant continued investigation not only for the diseases
themselves but for greater understanding of protein biology in general.
91

BIBLIOGRAPHY
Acutis, P. L., Bossers, A., Priem, J., Riina, M. V., Peletto, S., Mazza, M., Casalone, C., Forloni, G., Ru, G. & Caramelli, M. (2006). Identification of prion protein gene polymorphisms in goats from Italian scrapie outbreaks. J Gen Virol 87, 1029-1033.
Alexander, B. M., Stobart, R. H. & Moss, G. E. (2007). Scrapie resistance and production traits in Rambouillet rams: Ram performance test 2002-2006. Res Vet Sci.
Alexander, B. M., Stobart, R. H., Russell, W. C., O'Rourke, K. I., Lewis, G. S., Logan, J. R., Duncan, J. V. & Moss, G. E. (2005). The incidence of genotypes at codon 171 of the prion protein gene (PRNP) in five breeds of sheep and production traits of ewes associated with those genotypes. J Anim Sci 83, 455-459.
Alfonso, L., Parada, A., Legarra, A., Ugarte, E. & Arana, A. (2006). The effects of selective breeding against scrapie susceptibility on the genetic variability of the Latxa Black-Faced sheep breed. Genet Sel Evol 38, 495-511.
Alvarez, L., Gutierrez-Gil, B., San Primitivo, F., de la Fuente, L. F. & Arranz, J. J. (2006). Influence of prion protein genotypes on milk production traits in Spanish Churra sheep. J Dairy Sci 89, 1784-1791.
Andreoletti, O., Berthon, P., Levavasseur, E., Marc, D., Lantier, F., Monks, E., Elsen, J. M. & Schelcher, F. (2002a). Phenotyping of protein-prion (PrPsc)-accumulating cells in lymphoid and neural tissues of naturally scrapie-affected sheep by double-labeling immunohistochemistry. J Histochem Cytochem 50, 1357-1370.
Andreoletti, O., Levavasseur, E., Uro-Coste, E., Tabouret, G., Sarradin, P., Delisle, M. B., Berthon, P., Salvayre, R., Schelcher, F. & Negre-Salvayre, A. (2002b). Astrocytes accumulate 4-hydroxynonenal adducts in murine scrapie and human creutzfeldt-jakob disease. Neurobiol Dis 11, 386-393.
Arens, M. (1993). Use of probes and amplification techniques for the diagnosis and prognosis of human immunodeficiency virus (HIV-1) infections. Diagn Microbiol Infect Dis 16, 165-172.
Arsenault, J., Dubreuil, P., Girard, C., Simard, C. & Belanger, D. (2003). Maedi-visna impact on productivity in Quebec sheep flocks (Canada). Prev Vet Med 59, 125-137.
Barron, R. M., Thomson, V., Jamieson, E., Melton, D. W., Ironside, J., Will, R. & Manson, J. C. (2001). Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. Embo J 20, 5070-5078.
Bartz, J. C., Marsh, R. F., McKenzie, D. I. & Aiken, J. M. (1998). The host range of chronic wasting disease is altered on passage in ferrets. Virology 251, 297-301.
Bartz, J. C., McKenzie, D. I., Bessen, R. A., Marsh, R. F. & Aiken, J. M. (1994). Transmissible mink encephalopathy species barrier effect between ferret and mink: PrP gene and protein analysis. J Gen Virol 75, 2947-2953.
92

Beekes, M. & McBride, P. A. (2000). Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci Lett 278, 181-184.
Beekes, M., McBride, P. A. & Baldauf, E. (1998). Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J Gen Virol 79, 601-607.
Belay, E. D., Gambetti, P., Schonberger, L. B., Parchi, P., Lyon, D. R., Capellari, S., McQuiston, J. H., Bradley, K., Dowdle, G., Crutcher, J. M. & Nichols, C. R. (2001). Creutzfeldt-Jakob disease in unusually young patients who consumed venison. Arch Neurol 58, 1673-1678.
Belay, E. D., Maddox, R. A., Williams, E. S., Miller, M. W., Gambetti, P. & Schonberger, L. B. (2004). Chronic wasting disease and potential transmission to humans. Emerg Infect Dis 10, 977-984.
Bishop, M. T., Hart, P., Aitchison, L., Baybutt, H. N., Plinston, C., Thomson, V., Tuzi, N. L., Head, M. W., Ironside, J. W., Will, R. G. & Manson, J. C. (2006). Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet neurology 5, 393-398.
Bonetta, L. (2002). CWD research increases as US concern grows. Nat Med 8, 1338. Bons, N., Mestre-Frances, N., Belli, P., Cathala, F., Gajdusek, D. C. & Brown, P.
(1999). Natural and experimental oral infection of nonhuman primates by bovine spongiform encephalopathy agents. Proceedings of the National Academy of Sciences of the United States of America 96, 4046-4051.
Bons, N., Mestre-Frances, N., Guiraud, I. & Charnay, Y. (1997). Prion immunoreactivity in brain, tonsil, gastrointestinal epithelial cells, and blood and lymph vessels in lemurian zoo primates with spongiform encephalopathy. C R Acad Sci III 320, 971-979.
Bossers, A., de Vries, R. & Smits, M. A. (2000). Susceptibility of sheep for scrapie as assessed by in vitro conversion of nine naturally occurring variants of PrP. Journal of virology 74, 1407-1414.
Bossers, A., Schreuder, B. E., Muileman, I. H., Belt, P. B. & Smits, M. A. (1996). PrP genotype contributes to determining survival times of sheep with natural scrapie. J Gen Virol 77 ( Pt 10), 2669-2673.
Brayton, K. A., O'Rourke, K. I., Lyda, A. K., Miller, M. W. & Knowles, D. P. (2004). A processed pseudogene contributes to apparent mule deer prion gene heterogeneity. Gene 326, 167-173.
Brodie, S. J., Marcom, K. A., Pearson, L. D., Anderson, B. C., de la Concha-Bermejillo, A., Ellis, J. A. & DeMartini, J. C. (1992). Effects of virus load in the pathogenesis of lentivirus-induced lymphoid interstitial pneumonia. J Infect Dis 166, 531-541.
Brown, D. R., Qin, K., Herms, J. W., Madlung, A., Manson, J., Strome, R., Fraser, P. E., Kruck, T., von Bohlen, A., Schulz-Schaeffer, W., Giese, A., Westaway, D. & Kretzschmar, H. (1997a). The cellular prion protein binds copper in vivo. Nature 390, 684-687.
Brown, D. R., Schulz-Schaeffer, W. J., Schmidt, B. & Kretzschmar, H. A. (1997b). Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol 146, 104-112.
93

Brown, D. R., Wong, B. S., Hafiz, F., Clive, C., Haswell, S. J. & Jones, I. M. (1999). Normal prion protein has an activity like that of superoxide dismutase. Biochem J 344 Pt 1, 1-5.
Bruce, M., Boyle, A. & McConnell, I. (2004). TSE strain typing in mice. In Techniques in Prion Research, pp. 132-146. Edited by S. Lehmann & J. Grassi. Switzerland: Birkhauser Verlag Basel.
Bruce, M., Chree, A., Williams, E. S. & Fraser, H. (2000). Perivascular PrP amyloid in the brains of mice infected with chronic wasting disease. Brain Pathol 10, 662-663, Abstract C632-608.
Bruce, M. E., Will, R. G., Ironside, J. W., McConnell, I., Drummond, D., Suttie, A., McCardle, L., Chree, A., Hope, J., Birkett, C., Cousens, S., Fraser, H. & Bostock, C. J. (1997). Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature 389, 498-501.
Bueler, H., Aguzzi, A., Sailer, A., Greiner, R. A., Autenried, P., Aguet, M. & Weissmann, C. (1993). Mice devoid of PrP are resistant to scrapie. Cell 73, 1339-1347.
Bueler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H. P., DeArmond, S. J., Prusiner, S. B., Aguet, M. & Weissmann, C. (1992). Normal development and behaviour of mice lacking the neuronal cell- surface PrP protein. Nature 356, 577-582.
Burns, C. S., Aronoff-Spencer, E., Dunham, C. M., Lario, P., Avdievich, N. I., Antholine, W. E., Olmstead, M. M., Vrielink, A., Gerfen, G. J., Peisach, J., Scott, W. G. & Millhauser, G. L. (2002). Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry 41, 3991-4001.
Caplazi, P., O'Rourke, K., Wolf, C., Shaw, D. & Baszler, T. V. (2004). Biology of PrPsc accumulation in two natural scrapie-infected sheep flocks. J Vet Diagn Invest 16, 489-496.
Carrozza, M. L., Mazzei, M., Bandecchi, P., Arispici, M. & Tolari, F. (2003). In situ PCR-associated immunohistochemistry identifies cell types harbouring the Maedi-Visna virus genome in tissue sections of sheep infected naturally. J Virol Methods 107, 121-127.
CDC (1999). Biosafety in Microbiological and Biomedical Laboratories: Center for Disease Control and National Institute of Health.
CDC (2003). Fatal degenerative neurologic illnesses in men who participated in wild game feasts--Wisconsin, 2002. MMWR Morb Mortal Wkly Rep 52, 125-127.
Cervenakova, L., Goldfarb, L. G., Garruto, R., Lee, H. S., Gajdusek, D. C. & Brown, P. (1998). Phenotype-genotype studies in kuru: implications for new variant Creutzfeldt-Jakob disease. Proceedings of the National Academy of Sciences of the United States of America 95, 13239-13241.
Colling, S. B., Collinge, J. & Jefferys, J. G. (1996). Hippocampal slices from prion protein null mice: disrupted Ca(2+)- activated K+ currents. Neurosci Lett 209, 49-52.
Collinge, J. (1999). Variant Creutzfeldt-Jakob disease. Lancet 354, 317-323.
94

Collinge, J., Whittington, M. A., Sidle, K. C., Smith, C. J., Palmer, M. S., Clarke, A. R. & Jefferys, J. G. (1994). Prion protein is necessary for normal synaptic function. Nature 370, 295-297.
Cuille, J. & Chelle, P.-L. (1936). La maladie dite tremblante du mouton est-elle inoculable? (Is the maladie transmissible by inoculation of sheep). Comptes Rendus Acad Sci 203, 1552-1554.
Cutlip, R. C., Lehmkuhl, H. D., Sacks, J. M. & Weaver, A. L. (1992). Seroprevalence of ovine progressive pneumonia virus in sheep in the United States as assessed by analyses of voluntarily submitted samples. American journal of veterinary research 53, 976-979.
De Vries, F., Hamann, H., Drogemuller, C., Ganter, M. & Distl, O. (2005). Analysis of associations between the prion protein genotypes and production traits in East Friesian milk sheep. J Dairy Sci 88, 392-398.
Dickinson, A. G., Meikle, V. M. & Fraser, H. (1968). Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. J Comp Pathol 78, 293-299.
Diringer, H., Roehmel, J. & Beekes, M. (1998). Effect of repeated oral infection of hamsters with scrapie. J Gen Virol 79 ( Pt 3), 609-612.
Ebrahimi, B., Allsopp, T. E., Fazakerley, J. K. & Harkiss, G. D. (2000). Phenotypic characterisation and infection of ovine microglial cells with Maedi-Visna virus. Journal of neurovirology 6, 320-328.
Eckroade, R., Zu Rhein, G. M. & Hanson, R. P. (1979). Experimental transmissible mink encephalopathy: brain lesions and their sequential development in mink. In Slow Transmissible Diseases of the Nervous System, pp. 409-449: Academic Press, Inc.
Evoniuk, J. M., Berg, P. T., Johnson, M. L., Larson, D. M., Maddock, T. D., Stoltenow, C. L., Schauer, C. S., O'Rourke, K. I. & Redmer, D. A. (2007). Associations between genotypes at codon 171 and 136 of the prion protein gene and production traits in market lambs. American journal of veterinary research 68, 1073-1078.
Fichet, G., Comoy, E., Duval, C., Antloga, K., Dehen, C., Charbonnier, A., McDonnell, G., Brown, P., Lasmezas, C. I. & Deslys, J. P. (2004). Novel methods for disinfection of prion-contaminated medical devices. Lancet 364, 521-526.
Gajdusek, D. C. & Zigas, V. (1957). Degenerative disease of the central nervous system in New Guinea: the endemic occurence of "kuru" in the native population. N Engl J Med 257, 974-978.
Ghani, A. C. (2002). The epidemiology of variant Creutzfeldt-Jakob disease in Europe. Microbes Infect 4, 385-393.
Hadlow, W. J. & Karstad, L. (1968). Transmissible encephalopathy of mink in Ontario. Can Vet J 9, 193-196.
Hamir, A. N., Cutlip, R. C., Miller, J. M., Williams, E. S., Stack, M. J., Miller, M. W., O'Rourke, K. I. & Chaplin, M. J. (2001). Preliminary findings on the experimental transmission of chronic wasting disease agent of mule deer to cattle. J Vet Diagn Invest 13, 91-96.
95

Hamir, A. N., Gidlewski, T., Spraker, T. R., Miller, J. M., Creekmore, L., Crocheck, M., Cline, T. & O'Rourke, K. I. (2006a). Preliminary observations of genetic susceptibility of elk (Cervus elaphus nelsoni) to chronic wasting disease by experimental oral inoculation. J Vet Diagn Invest 18, 110-114.
Hamir, A. N., Kunkle, R. A., Miller, J. M., Bartz, J. C. & Richt, J. A. (2006b). First and second cattle passage of transmissible mink encephalopathy by intracerebral inoculation. Vet Pathol 43, 118-126.
Hanson, R. P., Eckroade, R. J., Marsh, R. F., Zu Rhein, G. M., Kanitz, C. L. & Gustafson, D. P. (1971). Susceptibility of mink to sheep scrapie. Science 172, 859-861.
Hartsough, G. R. & Burger, D. (1965). Encephalopathy of mink. I. Epizootiologic and clinical observations. J Infect Dis 115, 387-392.
Haywood, A. M. (1997). Transmissible spongiform encephalopathies. N Engl J Med 337, 1821-1828.
Heggebo, R., Press, C. M., Gunnes, G., Lie, K. I., Tranulis, M. A., Ulvund, M., Groschup, M. H. & Landsverk, T. (2000). Distribution of prion protein in the ileal Peyer's patch of scrapie-free lambs and lambs naturally and experimentally exposed to the scrapie agent. J Gen Virol 81, 2327-2337.
Heggebo, R., Press, C. M., Gunnes, G., Ulvund, M. J., Tranulis, M. A. & Lsverk, T. (2003). Detection of PrP(Sc) in Lymphoid Tissues of Lambs Experimentally Exposed to the Scrapie Agent. J Comp Pathol 128, 172-181.
Herrmann-Hoesing, L. M., Palmer, G. H. & Knowles, D. P. (2007a). Evidence of proviral clearance following postpartum transmission of an ovine lentivirus. Virology 362, 226-234.
Herrmann-Hoesing, L. M., White, S. N., Lewis, G. S., Mousel, M. R. & Knowles, D. P. (2007b). Development and validation of an ovine progressive pneumonia virus quantitative PCR. Clin Vaccine Immunol 14, 1274-1278.
Herrmann, L. M., Cheevers, W. P., Davis, W. C., Knowles, D. P. & O'Rourke, K. I. (2003). CD21-Positive Follicular Dendritic Cells: A Possible Source of PrP(Sc) in Lymph Node Macrophages of Scrapie-Infected Sheep. Am J Pathol 162, 1075-1081.
Herrmann, L. M., Hotzel, I., Cheevers, W. P., On Top, K. P., Lewis, G. S. & Knowles, D. P. (2004). Seven new ovine progressive pneumonia virus (OPPV) field isolates from Dubois Idaho sheep comprise part of OPPV clade II based on surface envelope glycoprotein (SU) sequences. Virus Res 102, 215-220.
Holm, S. (1979). A simple sequentially rejective bonferroni test procedure. Scandinavian Journal of Statistics 6, 65-70.
Hunter, N., Foster, J. D., Goldmann, W., Stear, M. J., Hope, J. & Bostock, C. (1996). Natural scrapie in a closed flock of Cheviot sheep occurs only in specific PrP genotypes. Arch Virol 141, 809-824.
Johnson, C., Johnson, J., Clayton, M., McKenzie, D. & Aiken, J. (2003). Prion protein gene heterogeneity in free-ranging white-tailed deer within the chronic wasting disease affected region of Wisconsin. J Wildl Dis 39, 576-581.
Johnson, R. T. & Gibbs, C. J., Jr. (1998). Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med 339, 1994-2004.
96

Joly, D. O., Ribic, C. A., Langenberg, J. A., Beheler, K., Batha, C. A., Dhuey, B. J., Rolley, R. E., Bartelt, G., Van Deelen, T. R. & Samuel, M. D. (2003). Chronic wasting disease in free-ranging wisconsin white-tailed deer. Emerg Infect Dis 9, 599-601.
Kaneko, K., Zulianello, L., Scott, M., Cooper, C. M., Wallace, A. C., James, T. L., Cohen, F. E. & Prusiner, S. B. (1997). Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proceedings of the National Academy of Sciences of the United States of America 94, 10069-10074.
Kitamoto, T., Muramoto, T., Mohri, S., Doh-Ura, K. & Tateishi, J. (1991). Abnormal isoform of prion protein accumulates in follicular dendritic cells in mice with Creutzfeldt-Jakob disease. Journal of virology 65, 6292-6295.
Klein, M. A., Frigg, R., Flechsig, E., Raeber, A. J., Kalinke, U., Bluethmann, H., Bootz, F., Suter, M., Zinkernagel, R. M. & Aguzzi, A. (1997). A crucial role for B cells in neuroinvasive scrapie. Nature 390, 687-690.
Klein, M. A., Frigg, R., Raeber, A. J., Flechsig, E., Hegyi, I., Zinkernagel, R. M., Weissmann, C. & Aguzzi, A. (1998). PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat Med 4, 1429-1433.
Kong, Q., Huang, S., Zou, W., Vanegas, D., Wang, M., Wu, D., Yuan, J., Zheng, M., Bai, H., Deng, H., Chen, K., Jenny, A. L., O'Rourke, K., Belay, E. D., Schonberger, L. B., Petersen, R. B., Sy, M. S., Chen, S. G. & Gambetti, P. (2005). Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci 25, 7944-7949.
Konold, T., Moore, S. J., Bellworthy, S. J. & Simmons, H. A. (2008). Evidence of scrapie transmission via milk. BMC veterinary research 4, 14.
Kretzschmar, H. A., Neumann, M., Riethmuller, G. & Prusiner, S. B. (1992). Molecular cloning of a mink prion protein gene. J Gen Virol 73 ( Pt 10), 2757-2761.
Kurt, T. D., Perrott, M. R., Wilusz, C. J., Wilusz, J., Supattapone, S., Telling, G. C., Zabel, M. D. & Hoover, E. A. (2007). Efficient in vitro amplification of chronic wasting disease PrPRES. Journal of virology 81, 9605-9608.
Le Dur, A., Beringue, V., Andreoletti, O., Reine, F., Lai, T. L., Baron, T., Bratberg, B., Vilotte, J. L., Sarradin, P., Benestad, S. L. & Laude, H. (2005). A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proceedings of the National Academy of Sciences of the United States of America 102, 16031-16036.
Leblanc, P., Baas, D. & Darlix, J. L. (2004). Analysis of the interactions between HIV-1 and the cellular prion protein in a human cell line. J Mol Biol 337, 1035-1051.
Lee, K. H., Jeong, B. H., Jin, J. K., Meeker, H. C., Kim, J. I., Carp, R. I. & Kim, Y. S. (2006). Scrapie infection activates the replication of ecotropic, xenotropic, and polytropic murine leukemia virus (MuLV) in brains and spinal cords of senescence-accelerated mice: implication of MuLV in progression of scrapie pathogenesis. Biochem Biophys Res Commun 349, 122-130.
Lezmi, S., Bencsik, A. & Baron, T. (2001). CNA42 monoclonal antibody identifies FDC as PrPsc accumulating cells in the spleen of scrapie affected sheep. Vet Immunol Immunopathol 82, 1-8.
97

Ligios, C., Sigurdson, C. J., Santucciu, C., Carcassola, G., Manco, G., Basagni, M., Maestrale, C., Cancedda, M. G., Madau, L. & Aguzzi, A. (2005). PrPSc in mammary glands of sheep affected by scrapie and mastitis. Nat Med 11, 1137-1138.
Ma, J., Wollmann, R. & Lindquist, S. (2002). Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298, 1781-1785.
Mabbott, N. A. & Bruce, M. E. (2001). The immunobiology of TSE diseases. J Gen Virol 82, 2307-2318.
Manuelidis, L. (2007). A 25 nm virion is the likely cause of transmissible spongiform encephalopathies. Journal of cellular biochemistry 100, 897-915.
Marsh, R. F. & Bessen, R. A. (1993). Epidemiologic and experimental studies on transmissible mink encephalopathy. Dev Biol Stand 80, 111-118.
Marsh, R. F., Bessen, R. A., Lehmann, S. & Hartsough, G. R. (1991). Epidemiological and experimental studies on a new incident of transmissible mink encephalopathy. J Gen Virol 72, 589-594.
Marsh, R. F., Burger, D., Eckroade, R., Zu Rhein, G. M. & Hanson, R. P. (1969). A preliminary report on the experimental host range of the transmissible mink encephalopathy agent. J Infect Dis 120, 713-719.
Marsh, R. F. & Hadlow, W. J. (1992). Transmissible mink encephalopathy. Rev Sci Tech 11, 539-550.
Marsh, R. F. & Hanson, R. P. (1979). On the origin of transmissible mink encephalopathy. In Slow transmissible diseases of the nervous system, pp. 451-460. Edited by S. B. Prusiner & W. J. Hadlow. New York: Academic Press.
Marsh, R. F., Kincaid, A. E., Bessen, R. A. & Bartz, J. C. (2005). Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). Journal of virology 79, 13794-13796.
Marsh, R. F., Sipe, J. C., Morse, S. S. & Hanson, R. P. (1976). Transmissible mink encephalopathy. Reduced spongiform degeneration in aged mink of the Chediak-Higashi genotype. Lab Invest 34, 381-386.
MaWhinney, S., Pape, W. J., Forster, J. E., Anderson, C. A., Bosque, P. J. & Miller, M. W. (2006). Human prion disease and relative risk associated with chronic wasting disease. Emerg Infect Dis 12, 1527-1535.
McCormack, J. E., Baybutt, H. N., Everington, D., Will, R. G., Ironside, J. W. & Manson, J. C. (2002). PRNP contains both intronic and upstream regulatory regions that may influence susceptibility to Creutzfeldt-Jakob Disease. Gene 288, 139-146.
Miller, D. C. (1988). Creutzfeldt-Jakob Disease in Histopathology Technicians. Lancet 318, 853.
Miller, M. W. & Williams, E. S. (2002). Detection of PrP(CWD) in mule deer by immunohistochemistry of lymphoid tissues. The Veterinary record 151, 610-612.
Miller, M. W., Williams, E. S., McCarty, C. W., Spraker, T. R., Kreeger, T. J., Larsen, C. T. & Thorne, E. T. (2000). Epizootiology of chronic wasting disease in free-ranging cervids in Colorado and Wyoming. J Wildl Dis 36, 676-690.
Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J. L., Lehmann, S., Launay, J. M. & Kellermann, O. (2000). Signal transduction through prion protein. Science 289, 1925-1928.
98

Nagy, A. (2003). Manipulating the mouse embryo : a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press.
Nicotera, P. (2001). A route for prion neuroinvasion. Neuron 31, 345-348. O'Rourke, K. I. (2001). Ovine scrapie. New tools for control of an old disease. Vet Clin
North Am Food Anim Pract 17, 283-300, vi. O'Rourke, K. I., Baszler, T. V., Besser, T. E., Miller, J. M., Cutlip, R. C., Wells, G. A.,
Ryder, S. J., Parish, S. M., Hamir, A. N., Cockett, N. E., Jenny, A. & Knowles, D. P. (2000). Preclinical diagnosis of scrapie by immunohistochemistry of third eyelid lymphoid tissue. J Clin Microbiol 38, 3254-3259.
O'Rourke, K. I., Besser, T. E., Miller, M. W., Cline, T. F., Spraker, T. R., Jenny, A. L., Wild, M. A., Zebarth, G. L. & Williams, E. S. (1999). PrP genotypes of captive and free-ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J Gen Virol 80, 2765-2769.
O'Rourke, K. I., Holyoak, G. R., Clark, W. W., Mickelson, J. R., Wang, S., Melco, R. P., Besser, T. E. & Foote, W. C. (1997). PrP genotypes and experimental scrapie in orally inoculated Suffolk sheep in the United States. J Gen Virol 78 ( Pt 4), 975-978.
O'Rourke, K. I., Spraker, T. R., Hamburg, L. K., Besser, T. E., Brayton, K. A. & Knowles, D. P. (2004). Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J Gen Virol 85, 1339-1346.
O'Rourke, K. I., Spraker, T. R., Zhuang, D., Greenlee, J. J., Gidlewski, T. E. & Hamir, A. N. (2007). Elk with a long incubation prion disease phenotype have a unique PrPd profile. Neuroreport 18, 1935-1938.
O'Rourke, K. I., Zhuang, D., Lyda, A., Gomez, G., Williams, E. S., Tuo, W. & Miller, M. W. (2003). Abundant PrP(CWD) in tonsil from mule deer with preclinical chronic wasting disease. J Vet Diagn Invest 15, 320-323.
Papasavva-Stylianou, P., Kleanthous, M., Toumazos, P., Mavrikiou, P. & Loucaides, P. (2007). Novel polymorphisms at codons 146 and 151 in the prion protein gene of Cyprus goats, and their association with natural scrapie. Vet J 173, 459-462.
Pattison, I. H. (1966). The relative susceptibility of sheep, goats and mice to two types of the goat scrapie agent. Res Vet Sci 7, 207-212.
Pauly, P. C. & Harris, D. A. (1998). Copper stimulates endocytosis of the prion protein. J Biol Chem 273, 33107-33110.
Perrier, V., Kaneko, K., Safar, J., Vergara, J., Tremblay, P., DeArmond, S. J., Cohen, F. E., Prusiner, S. B. & Wallace, A. C. (2002). Dominant-negative inhibition of prion replication in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 99, 13079-13084.
Perrott, M. R., Sigurdson, C. J., Mathiason, C. K., Foos, T. L., Eliason, G. A. & Hoover, E. A. (2004). Chronic wasting disease of deer and elk: A ferret model. In Animal Prion Diseases and the Americas, p. 93. Ames, IA, USA.
Prusiner, S. B. (1998). Prions. Proceedings of the National Academy of Sciences of the United States of America 95, 13363-13383.
Race, R. & Chesebro, B. (1998). Scrapie infectivity found in resistant species. Nature 392, 770.
99

Race, R., Oldstone, M. & Chesebro, B. (2000). Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: role of prion protein expression in peripheral nerves and spleen. Journal of virology 74, 828-833.
Race, R., Raines, A., Raymond, G. J., Caughey, B. & Chesebro, B. (2001). Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: analogies to bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease in humans. Journal of virology 75, 10106-10112.
Race, R. E. & Raymond, G. J. (2004). Inactivation of transmissible spongiform encephalopathy (prion) agents by environ LpH. Journal of virology 78, 2164-2165.
Raymond, G. J., Bossers, A., Raymond, L. D., O'Rourke, K. I., McHolland, L. E., Bryant, P. K., 3rd, Miller, M. W., Williams, E. S., Smits, M. & Caughey, B. (2000). Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. Embo J 19, 4425-4430.
Raymond, G. J., Hope, J., Kocisko, D. A., Priola, S. A., Raymond, L. D., Bossers, A., Ironside, J., Will, R. G., Chen, S. G., Petersen, R. B., Gambetti, P., Rubenstein, R., Smits, M. A., Lansbury, P. T., Jr. & Caughey, B. (1997). Molecular assessment of the potential transmissibilities of BSE and scrapie to humans. Nature 388, 285-288.
Riesner, D. (2003). Biochemistry and structure of PrP(C) and PrP(Sc). Br Med Bull 66, 21-33.
Robinson, M. M., Hadlow, W. J., Huff, T. P., Wells, G. A., Dawson, M., Marsh, R. F. & Gorham, J. R. (1994). Experimental infection of mink with bovine spongiform encephalopathy. J Gen Virol 75, 2151-2155.
Robinson, M. M., Hadlow, W. J., Knowles, D. P., Huff, T. P., Lacy, P. A., Marsh, R. F. & Gorham, J. R. (1995). Experimental infection of cattle with the agents of transmissible mink encephalopathy and scrapie. J Comp Pathol 113, 241-251.
Sailer, A., Bueler, H., Fischer, M., Aguzzi, A. & Weissmann, C. (1994). No propagation of prions in mice devoid of PrP. Cell 77, 967-968.
Sakaguchi, S., Katamine, S., Nishida, N., Moriuchi, R., Shigematsu, K., Sugimoto, T., Nakatani, A., Kataoka, Y., Houtani, T., Shirabe, S., Okada, H., Hasegawa, S., Miyamoto, T. & Noda, T. (1996). Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 380, 528-531.
Salaris, S., Casu, S. & Carta, A. (2007). Investigating the relationship between the prion protein locus and udder morphology traits and milk yield in Sardinian sheep. J Anim Sci 85, 2840-2845.
Sambrook, J. F., EF; Maniatis, T (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Schneider, D. A., Yan, H., Fry, L. M., Alverson, J., White, S. N. & O'Rourke K, I. (2008). Myenteric neurons of the ileum that express somatostatin are a target of prion neuroinvasion in an alimentary model of sheep scrapie. in submission
Sigurdson, C. J., Barillas-Mury, C., Miller, M. W., Oesch, B., van Keulen, L. J., Langeveld, J. P. & Hoover, E. A. (2002). PrP(CWD) lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J Gen Virol 83, 2617-2628.
100

Sigurdson, C. J., Mathiason, C. K., Miller, M. W., Perrott, M. R., Eliason, G. A., Spraker, T. R., Bartz, J. C. & Hoover, E. A. (2003). Experimental infection and bioassay of chronic wasting disease (CWD) in the ferret. In Keystone Symposium on the Molecular Aspects of Transmissible Spongiform Encephalopathies. Breckenridge, CO.
Sigurdson, C. J., Spraker, T. R., Miller, M. W., Oesch, B. & Hoover, E. A. (2001). PrP(CWD) in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J Gen Virol 82, 2327-2334.
Sigurdson, C. J., Williams, E. S., Miller, M. W., Spraker, T. R., O'Rourke, K. I. & Hoover, E. A. (1999). Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J Gen Virol 80, 2757-2764.
Sorenson, J. R. (2001). Prion diseases: copper deficiency states associated with impaired nitrogen monoxide or carbon monoxide transduction and translocation. J Inorg Biochem 87, 125-127.
Spraker, T. R., Miller, M. W., Williams, E. S., Getzy, D. M., Adrian, W. J., Schoonveld, G. G., Spowart, R. A., O'Rourke, K. I., Miller, J. M. & Merz, P. A. (1997). Spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus) and Rocky Mountain elk (Cervus elaphus nelsoni) in northcentral Colorado. J Wildl Dis 33, 1-6.
Spraker, T. R., Zink, R. R., Cummings, B. A., Wild, M. A., Miller, M. W. & O'Rourke, K. I. (2002). Comparison of histological lesions and immunohistochemical staining of proteinase-resistant prion protein in a naturally occurring spongiform encephalopathy of free-ranging mule deer (Odocoileus hemionus) with those of chronic wasting disease of captive mule deer. Vet Pathol 39, 110-119.
Stanton, J. (2008). Personal communication. Pullman, WA. Sweeney, T., Hanrahan, J. P. & O'Doherty, E. (2007). Is there a relationship between
prion protein genotype and ovulation rate and litter size in sheep? Anim Reprod Sci 101, 153-157.
Tamguney, G., Giles, K., Bouzamondo-Bernstein, E., Bosque, P. J., Miller, M. W., Safar, J., DeArmond, S. J. & Prusiner, S. B. (2006). Transmission of elk and deer prions to transgenic mice. Journal of virology 80, 9104-9114.
Taylor, D. M. (2000). Inactivation of transmissible degenerative encephalopathy agents: A review. Vet J 159, 10-17.
Telling, G. C. (2000). Prion protein genes and prion diseases: studies in transgenic mice. Neuropathol Appl Neurobiol 26, 209-220.
Terry, L. A., Marsh, S., Ryder, S. J., Hawkins, S. A., Wells, G. A. & Spencer, Y. I. (2003). Detection of disease-specific PrP in the distal ileum of cattle exposed orally to the agent of bovine spongiform encephalopathy. The Veterinary record 152, 387-392.
Tobler, I., Gaus, S. E., Deboer, T., Achermann, P., Fischer, M., Rulicke, T., Moser, M., Oesch, B., McBride, P. A. & Manson, J. C. (1996). Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 380, 639-642.
USDA (2003). Phase II: Scrapie: Ovine Slaughter Surveillance Study 2002-2003. Fort Collins, CO: USDA:APHIS:VS, CEAH, National Animal Health Monitoring System.
101

Vaccari, G., Di Bari, M. A., Morelli, L., Nonno, R., Chiappini, B., Antonucci, G., Marcon, S., Esposito, E., Fazzi, P., Palazzini, N., Troiano, P., Petrella, A., Di Guardo, G. & Agrimi, U. (2006). Identification of an allelic variant of the goat PrP gene associated with resistance to scrapie. J Gen Virol 87, 1395-1402.
Verhofstede, C., Reniers, S., Van Wanzeele, F. & Plum, J. (1994). Evaluation of proviral copy number and plasma RNA level as early indicators of progression in HIV-1 infection: correlation with virological and immunological markers of disease. AIDS 8, 1421-1427.
Vilotte, J. L. & Laude, H. (2002). Transgenesis applied to transmissible spongiform encephalopathies. Transgenic Res 11, 547-564.
Vitezica, Z. G., Moreno, C. R., Lantier, F., Lantier, I., Schibler, L., Roig, A., Francois, D., Bouix, J., Allain, D., Brunel, J. C., Barillet, F. & Elsen, J. M. (2007). Quantitative trait loci linked to PRNP gene controlling health and production traits in INRA 401 sheep. Genet Sel Evol 39, 421-430.
Vitone, F., Gibellini, D., Schiavone, P. & Re, M. C. (2005). Quantitative DNA proviral detection in HIV-1 patients treated with antiretroviral therapy. J Clin Virol 33, 194-200.
Waggoner, D. J., Drisaldi, B., Bartnikas, T. B., Casareno, R. L., Prohaska, J. R., Gitlin, J. D. & Harris, D. A. (2000). Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J Biol Chem 275, 7455-7458.
Weissmann, C. & Flechsig, E. (2003). PrP knock-out and PrP transgenic mice in prion research. Br Med Bull 66, 43-60.
Wells, G. A., Hawkins, S. A., Green, R. B., Austin, A. R., Dexter, I., Spencer, Y. I., Chaplin, M. J., Stack, M. J. & Dawson, M. (1998). Preliminary observations on the pathogenesis of experimental bovine spongiform encephalopathy (BSE): an update. The Veterinary record 142, 103-106.
Westaway, D., Zuliani, V., Cooper, C. M., Da Costa, M., Neuman, S., Jenny, A. L., Detwiler, L. & Prusiner, S. B. (1994). Homozygosity for prion protein alleles encoding glutamine-171 renders sheep susceptible to natural scrapie. Genes Dev 8, 959-969.
Westergard, L., Christensen, H. M. & Harris, D. A. (2007). The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 1772, 629-644.
WHO (1999). World Health Organization Infection Control Guidelines for Transmissible Spongiform Encephalopathies. Geneva, Switzerland.
Williams, E. S. (2005). Chronic wasting disease. Vet Pathol 42, 530-549. Williams, E. S. & Miller, M. W. (2002). Chronic wasting disease in deer and elk in
North America. Rev Sci Tech 21, 305-316. Williams, E. S., Miller, M. W., Kreeger, T. J., Kahn, R. H. & Thorne, E. T. (2002).
Chronic wasting disease of deer and elk: a review with recommendations for management. Journal of Wildlife Management 66, 551-563.
Williams, E. S. & Young, S. (1980). Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 16, 89-98.
Williams, E. S. & Young, S. (1992). Spongiform encephalopathies in Cervidae. Rev Sci Tech 11, 551-567.
102

Wong, B. S., Chen, S. G., Colucci, M., Xie, Z., Pan, T., Liu, T., Li, R., Gambetti, P., Sy, M. S. & Brown, D. R. (2001). Aberrant metal binding by prion protein in human prion disease. J Neurochem 78, 1400-1408.
Yang, Y. & Seed, B. (2003). Site-specific gene targeting in mouse embryonic stem cells with intact bacterial artificial chromosomes. Nat Biotechnol 21, 447-451.
Yedidia, Y., Horonchik, L., Tzaban, S., Yanai, A. & Taraboulos, A. (2001). Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J 20, 5383-5391.
Zeidler, M., Stewart, G., Cousens, S. N., Estibeiro, K. & Will, R. G. (1997). Codon 129 genotype and new variant CJD. Lancet 350, 668.
Zhang, Z., Watt, N. J., Hopkins, J., Harkiss, G. & Woodall, C. J. (2000). Quantitative analysis of maedi-visna virus DNA load in peripheral blood monocytes and alveolar macrophages. J Virol Methods 86, 13-20.
103

APPENDIX A
ATTRIBUTIONS TO CONTRIBUTING AUTHORS
Chapter Two - A species barrier limits transmission of chronic wasting disease to
mink (Mustela vison)
Robert D. Harrington (primary investigator and primary author): Experimental design,
IACUC and EH&S regulatory compliance, experimental protocols, sample
acquisition, inocula preparation and characterization, animal inoculations,
necropsy, histopathology, data analysis, statistical analysis, and manuscript
preparation.
Timothy V. Baszler: Experimental design, data analysis, and editorial comments.
Katherine I. O’Rourke: Experimental design, sample acquisition, experimental protocols,
data analysis, and editorial comments.
David A. Schneider: Animal procedures, statistical analysis, and editorial comments.
Terry R. Spraker: Experimental design, sample acquisition, experimental protocols, and
editorial comments.
H. Denny Liggitt: Experimental design, data analysis, and editorial comments.
Donald P. Knowles (chair): Experimental design, data analysis, and editorial comments.
Chapter Three - Transgenesis of a bacterial artificial chromosome resulsts in
stable transccrition and translation of mule deer prion protein
Robert D. Harrington (primary investigator and primary author): Experimental design,
construct design, IACUC and EH&S regulatory compliance, experimental
protocols, sample acquisition, inocula preparation and characterization, animal
104

inoculations, necropsy, histopathology, data analysis, statistical analysis, and
manuscript preparation.
H. Denny Liggitt: Experimental design, data analysis, and editorial comments.
Katherine I. O’Rourke: Experimental design, sample acquisition, data analysis, and
editorial comments.
Kelly A. Brayton: Experimental design, BAC cloning, and editorial comments.
Donald P. Knowles (chair): Experimental design, data analysis, and editorial comments.
Carol B. Ware: Experimental design, transgenic procedures, data analysis, and editorial
comments.
Chapter Four - OPPV provirus levels are independent of the PRNP 171R allele
Robert D. Harrington (primary investigator and primary author): Experimental design,
experimental protocols, data analysis, statistical analysis, and manuscript
preparation.
Lynn M. Herrmann: Experimental design, sample acquisition, experimental protocols,
data analysis, and editorial comments.
Stephen N. White: Experimental design, statistical analysis, and editorial comments.
Katherine I. O’Rourke: Experimental design, sample acquisition, data analysis, and
editorial comments.
Donald P. Knowles (chair): Experimental design, data analysis, and editorial comments.
105

APPENDIX B:
NOTES ON PRION DISINFECTION
A considerable contribution to concern over prion infection is the widespread belief that
prions cannot be “disinfected” or “killed”. There are actually a number of options for
reducing or eliminating infectious potential [for review see CDC, 1999; Taylor, 2000].
The information below is provided as a general guide to prion biosafety and practices
should be individually tailored to a given situation in consultation with the appropriate
biosafety committees. I take no responsibility for any consequences that may occur
from the use of these strategies whether it arise from prion infection or other
occupational injury. It should further be noted that the information is based on study of
CWD, TME, and scrapie which are considered Biosafety level-2 agents. Work with BSE
or vCJD at Biosafety level-3 and/or 4 will require additional consideration.
The following list includes some strategies that I have found useful:
1. Sodium hypochlorite. Household bleach is an effective method of disinfection when
used at high concentrations. A 40% dilution of bleach (e.g. 4 parts bleach to 6 parts
water) applied to surfaces or for soaking materials for 1 hour is effective (WHO, 1999),
This method may also be used for liquid wastes in which enough bleach is added to the
liquid to reach the 40% concentration.
2. Environ LpH. This phenolic derivative disinfectant has been shown to be effective
for laboratory use at a 5% concentration applied for 30 minutes (Fichet et al., 2004;
Race & Raymond, 2004). This product is potentially caustic but is less irritating than
106

high concentrations of bleach. There are a number of less effective products with the
Environ name therefore its important to use the product with the LpH suffix.
3. Equipment and supplies. Disposable utensils and plastic ware are used whenever
possible and disposed of as biohazardous waste submitted for incineration. Reusable
metal ware and other instruments can be disinfected as listed in items number 1 and 2.
4. Safe laboratory practices and standard universal precautions. In the modern era
older techniques that were standard many decades ago have been replaced by
standards for simple practices that apply to work with any pathogen. A prime example
is pipetting by mouth. It is antiquated, but a good reminder that with the oral exposure
being the primary route of natural TSE transmission the single best preventative
measure is to avoid hand-to-mouth or sample-to-mouth contact. To be blunt - don’t put
it in your mouth! Use of standard personal protective equipment such as gloves and lab
coats further reduces the risk of contamination to personnel. Barrier protection through
the use of biohazard mats or absorbent plastic backed paper provide for easy
containment and cleanup following TSE work.
5. Incineration. This is commonly used for disposal of animal carcasses and other
biohazardous waste. The high temperature involved effectively eliminates infectivity
(CDC, 1999). There is no evidence that incinerator exhaust has any potential to
disseminate infectious material.
107

108
6. Autoclaving. It is correctly reported that standard autoclaving (e.g. 121°C for 30 to
60 minutes) by gravity displacement methods does not eliminate prion infectivity
however it may reduce the infectious titer. Also, there are alternative protocols that are
effective such as increasing gravity displacement autoclave time to 4.5 hours, using
porous load/vacuum displacement autoclaving at 134°C for 1hour, or performing
autoclaving in a bath of sodium hydroxide or other disinfectants (CDC, 1999; Taylor,
2000). Contaminated animal housing equipment used in our experiments is disinfected
by a combination of Environ LpH surface treatment for 1 hour followed by the vacuum
displacement autoclave cycle. It should be noted that a sometimes recommended
method of autoclaving in a sodium hydroxide bath has a high risk of cutaneous or
respiratory caustic burns. It is the opinion of this author that this latter practice
represents a far more serious occupational health hazard than prion infectivity itself.
7. Alkaline hydrolysis. A modification of the techniques in item 6 is the use of large
stainless steel pressurized containers. These may range from a few meters in diameter
to the size of a semi-truck trailer and perform like a giant pressure cooker. High
temperature sodium hydroxide vapor is injected into the chamber and the combination
of heat, pressure, and high pH eliminates infectivity while also significantly reducing the
amount of biomass (reduced to 10 to 20% of the starting volume). The self contained
injection and evacuation system in this apparatus makes it safer than using an alkaline
bath in a standard autoclave. This method also avoids airborne environmental release
of volatile compounds (such as dioxins) that is inherent to incineration.