top 20 observation series # 7 21 cfr 211.42 (subpart c-buildings and facilities – design and...
TRANSCRIPT

21 CFR 211.42(Subpart C-Buildings and Facilities – Design and construction features )
[email protected] Yourself.... ―Know Regulation - No Observation‖

- US FDA inspectional observations - 2014
- List of “Top 20 – CFR parts to know”
- Sec. 21 CFR 211.42
- 483 observations
- Warning Letters
- Other Guidance
- How to avoid observations related to 21 CFR 211.42 ?
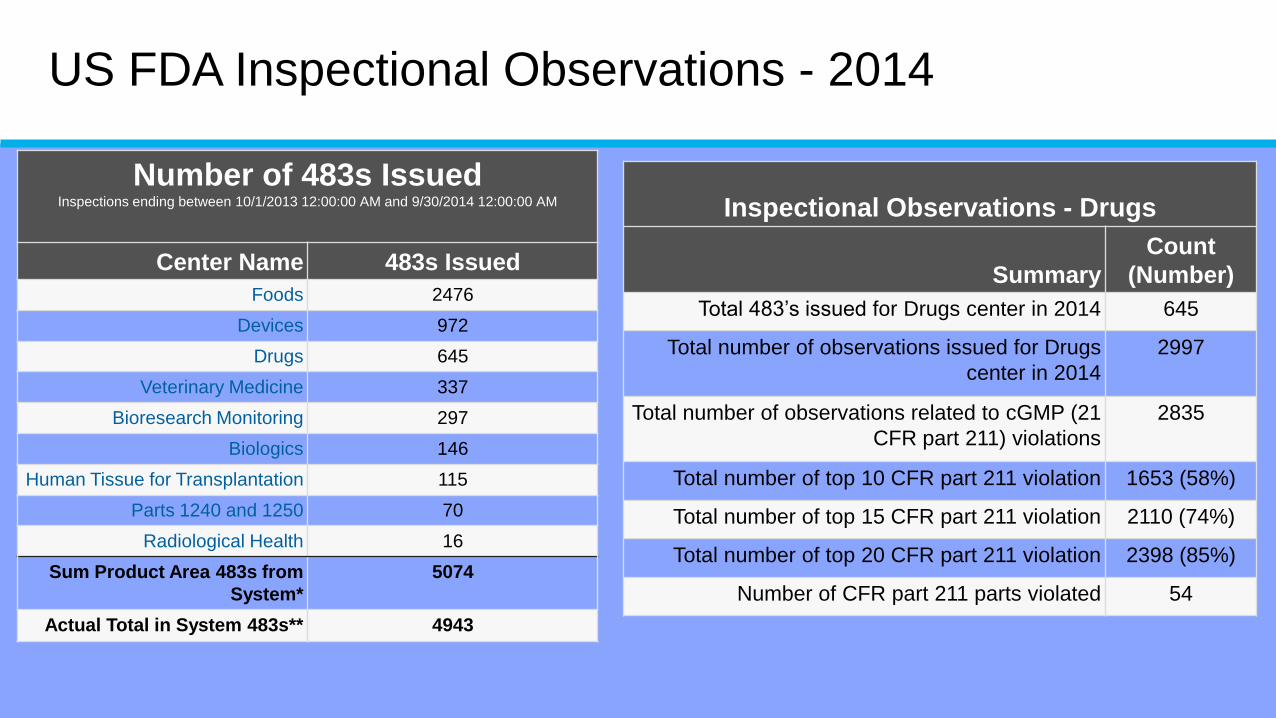
US FDA Inspectional Observations - 2014
Number of 483s IssuedInspections ending between 10/1/2013 12:00:00 AM and 9/30/2014 12:00:00 AM
Center Name 483s Issued
Foods 2476
Devices 972
Drugs 645
Veterinary Medicine 337
Bioresearch Monitoring 297
Biologics 146
Human Tissue for Transplantation 115
Parts 1240 and 1250 70
Radiological Health 16
Sum Product Area 483s from
System*
5074
Actual Total in System 483s** 4943
Inspectional Observations - Drugs
Summary
Count
(Number)
Total 483’s issued for Drugs center in 2014 645
Total number of observations issued for Drugs
center in 2014
2997
Total number of observations related to cGMP (21
CFR part 211) violations
2835
Total number of top 10 CFR part 211 violation 1653 (58%)
Total number of top 15 CFR part 211 violation 2110 (74%)
Total number of top 20 CFR part 211 violation 2398 (85%)
Number of CFR part 211 parts violated 54

S.No. CFR Frequency % S.No. CFR Frequency % S.No. CFR Frequency %
1 21 CFR 211.160 235 8.3 19 21 CFR 211.186 48 1.7 37 21 CFR 211.204 10 0.4
2 21 CFR 211.22 218 7.7 20 21 CFR 211.63 41 1.4 38 21 CFR 211.101 9 0.3
3 21 CFR 211.192 209 7.4 21 21 CFR 211.80 41 1.4 39 21 CFR 211.105 6 0.2
4 21 CFR 211.67 184 6.5 22 21 CFR 211.142 32 1.1 40 21 CFR 211.115 6 0.2
5 21 CFR 211.100 167 5.9 23 21 CFR 211.167 32 1.1 41 21 CFR 211.134 6 0.2
6 21 CFR 211.165 143 5.0 24 21 CFR 211.170 31 1.1 42 21 CFR 211.82 6 0.2
7 21 CFR 211.42 143 5.0 25 21 CFR 211.125 29 1.0 43 21 CFR 211.196 5 0.2
8 21 CFR 211.113 128 4.5 26 21 CFR 211.56 28 1.0 44 21 CFR 211.44 5 0.2
9 21 CFR 211.166 115 4.1 27 21 CFR 211.150 23 0.8 45 21 CFR 211.48 4 0.1
10 21 CFR 211.25 111 3.9 28 21 CFR 211.46 20 0.7 46 21 CFR 211.52 4 0.1
11 21 CFR 211.68 99 3.5 29 21 CFR 211.122 19 0.7 47 21 CFR 211.184 3 0.1
12 21 CFR 211.198 95 3.4 30 21 CFR 211.130 19 0.7 48 21 CFR 211.86 3 0.1
13 21 CFR 211.84 91 3.2 31 21 CFR 211.58 18 0.6 49 21 CFR 211.87 3 0.1
14 21 CFR 211.110 89 3.1 32 21 CFR 211.182 17 0.6 50 21 CFR 211.34 2 0.1
15 21 CFR 211.194 83 2.9 33 21 CFR 211.103 14 0.5 51 21 CFR 211.65 2 0.1
16 21 CFR 211.188 74 2.6 34 21 CFR 211.137 13 0.5 52 21 CFR 211.89 2 0.1
17 21 CFR 211.180 72 2.5 35 21 CFR 211.111 12 0.4 53 21 CFR 211.176 1 0.0
18 21 CFR 211.28 53 1.9 36 21 CFR 211.94 11 0.4 54 21 CFR 211.208 1 0.0
21 CFR 211 Observations – Drugs in 2014
0
50
100
150
200
250
211.2
2
211.2
5
211.2
8
211.3
4
211.4
2
211.4
4
211.4
6
211.4
8
211.5
2
211.5
6
211.5
8
211.6
3
211.6
5
211.6
7
211.6
8
211.8
211.8
2
211.8
4
211.8
6
211.8
7
211.8
9
211.9
4
211.1
211.1
01
211.1
03
211.1
05
211.1
1
211.1
11
211.1
13
211.1
15
211.1
22
211.1
25
211.1
3
211.1
34
211.1
37
211.1
42
211.1
5
211.1
6
211.1
65
211.1
66
211.1
67
211.1
7
211.1
76
211.1
8
211.1
82
211.1
84
211.1
86
211.1
88
211.1
92
211.1
94
211.1
96
211.1
98
211.2
04
211.2
08
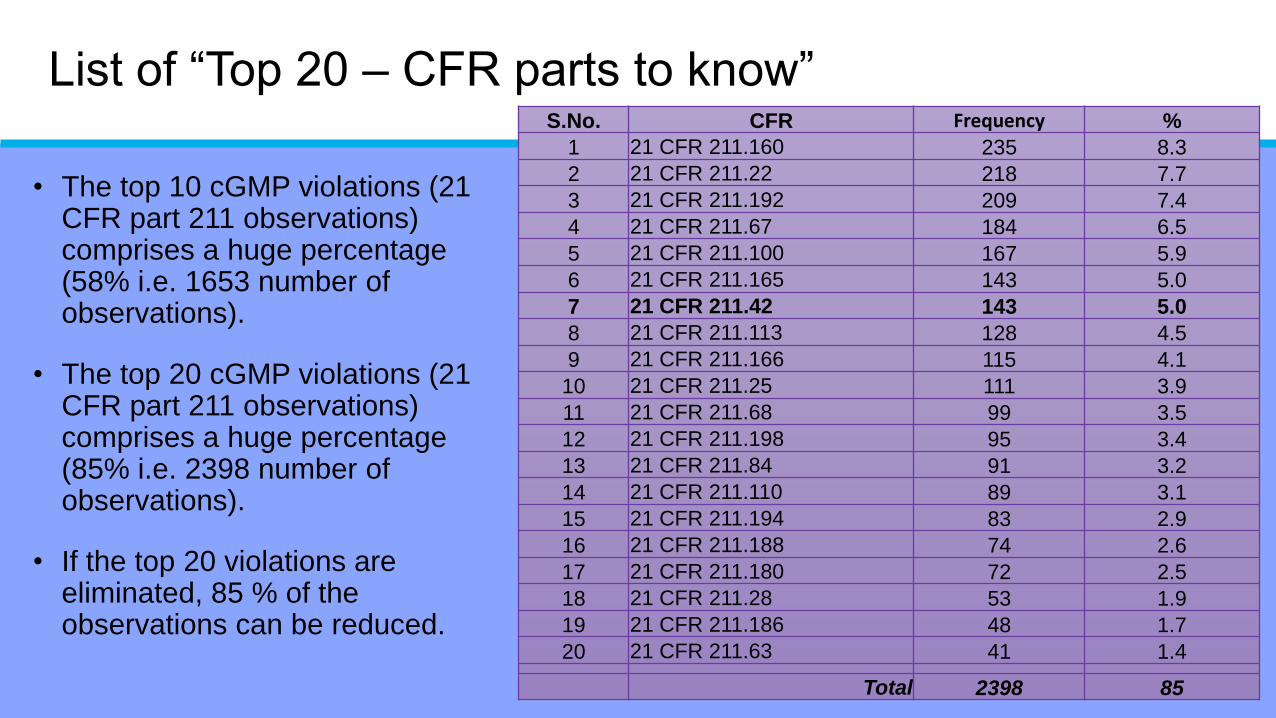
List of “Top 20 – CFR parts to know”S.No. CFR Frequency %
1 21 CFR 211.160 235 8.3
2 21 CFR 211.22 218 7.7
3 21 CFR 211.192 209 7.4
4 21 CFR 211.67 184 6.5
5 21 CFR 211.100 167 5.9
6 21 CFR 211.165 143 5.0
7 21 CFR 211.42 143 5.0
8 21 CFR 211.113 128 4.5
9 21 CFR 211.166 115 4.1
10 21 CFR 211.25 111 3.9
11 21 CFR 211.68 99 3.5
12 21 CFR 211.198 95 3.4
13 21 CFR 211.84 91 3.2
14 21 CFR 211.110 89 3.1
15 21 CFR 211.194 83 2.9
16 21 CFR 211.188 74 2.6
17 21 CFR 211.180 72 2.5
18 21 CFR 211.28 53 1.9
19 21 CFR 211.186 48 1.7
20 21 CFR 211.63 41 1.4
Total 2398 85
• The top 10 cGMP violations (21 CFR part 211 observations) comprises a huge percentage (58% i.e. 1653 number of observations).
• The top 20 cGMP violations (21 CFR part 211 observations) comprises a huge percentage (85% i.e. 2398 number of observations).
• If the top 20 violations are eliminated, 85 % of the observations can be reduced.

Consult Yourself.... ―Know Regulation - No Observation”
Series Regulation Title Internet link
# 1 21 CFR 211.160 Subpart I--Laboratory Controls
Sec. 211.160 General requirements
http://www.slideshare.net/skvemula/top-
20-observation-series-1-21-cfr-211160
# 2 21 CFR 211.22 Subpart B--Organization and Personnel
Sec. 211.22 Responsibilities of quality control
unit.
http://www.slideshare.net/skvemula/top-
20-observation-series-2-21-cfr-21122
# 3 21 CFR 211.192 Subpart J--Records and Reports
Sec. 211.192 Production record review
http://www.slideshare.net/skvemula/top-
20-observation-series-3-21-cfr-211192
# 4 21 CFR 211.67 Subpart D--Equipment
Sec. 211.67 Equipment cleaning and
maintenance
http://www.slideshare.net/skvemula/top-
20-observation-series-4-21-cfr-21167
# 5 21 CFR 211.100 Subpart F- Production and Process Controls
Sec. 211.100 Written procedures; deviations
http://www.slideshare.net/skvemula/top-
20-observation-series-5-21-cfr-211100
# 6 21 CFR 211.165 Subpart I--Laboratory Controls
Sec. 211.165 Testing and release for
distribution
http://www.slideshare.net/skvemula/top-
20-observation-series-6-21-cfr-211165

Subpart C--Buildings and Facilities
Sec. 211.42 Design and construction features
21 CFR 211.42(a)
21 CFR 211.42(b)
21 CFR 211.42(c)
21 CFR 211.42(d)
[email protected] CFR 211.42

483 citations related to 21 CFR 211.42
Year Number
2006 39
2007 43
2008 27
2009 35
2010 55
2011 69
2012 44
2013 118
2014 143
0
20
40
60
80
100
120
140
160
2006 2007 2008 2009 2010 2011 2012 2013 2014
483 Observations


CFR part 211 Regulation - 21 CFR 211.42(a)
Subpart C--Buildings and Facilities
• Sec. 211.42 Design and construction features.
(a) Any building or buildings used in the manufacture, processing, packing, or holding of a
drug product shall be of suitable size, construction and location to facilitate
cleaning, maintenance, and proper operations.

CFR part 211 Regulation - 21 CFR 211.42(b)
Subpart C--Buildings and Facilities
• Sec. 211.42 Design and construction features.
(b) Any such building shall have adequate space for the orderly placement of equipment
and materials to prevent mix-ups between different components, drug product
containers, closures, labeling, in-process materials, or drug products, and to prevent
contamination. The flow of components, drug product containers, closures, labeling, in-
process materials, and drug products through the building or buildings shall be designed
to prevent contamination.

CFR part 211 Regulation - 21 CFR 211.42(c)
Subpart C--Buildings and Facilities
• Sec. 211.42 Design and construction features.
(c) Operations shall be performed within specifically defined areas of adequate size. There shall be
separate or defined areas or such other control systems for the firm's operations as are necessary to
prevent contamination or mixups during the course of the following procedures:
(1) Receipt, identification, storage, and withholding from use of components, drug product containers, closures, and
labeling, pending the appropriate sampling, testing, or examination by the quality control unit before release for
manufacturing or packaging;
(2) Holding rejected components, drug product containers, closures, and labeling before disposition;
(3) Storage of released components, drug product containers, closures, and labeling;
(4) Storage of in-process materials;
(5) Manufacturing and processing operations;
(6) Packaging and labeling operations;
(7) Quarantine storage before release of drug products;
(8) Storage of drug products after release;

CFR part 211 Regulation - 21 CFR 211.42(c)
Subpart C--Buildings and Facilities
• Sec. 211.42 Design and construction features.
(c) Operations shall be performed within specifically defined areas of adequate size. There shall be
separate or defined areas or such other control systems for the firm's operations as are necessary to
prevent contamination or mix-ups during the course of the following procedures:
(9) Control and laboratory operations;
(10) Aseptic processing, which includes as appropriate:
(i) Floors, walls, and ceilings of smooth, hard surfaces that are easily cleanable;
(ii) Temperature and humidity controls;
(iii) An air supply filtered through high-efficiency particulate air filters under positive pressure, regardless of whether
flow is laminar or non-laminar;
(iv) A system for monitoring environmental conditions;
(v) A system for cleaning and disinfecting the room and equipment to produce aseptic conditions;
(vi) A system for maintaining any equipment used to control the aseptic conditions.

CFR part 211 Regulation - 21 CFR 211.42(d)
Subpart C--Buildings and Facilities
• Sec. 211.42 Design and construction features.
(d) Operations relating to the manufacture, processing, and packing of penicillin shall be
performed in facilities separate from those used for other drug products for human use.

[email protected] observations

483 citations related to 21 CFR 211.42(a)
Cite IdReference Number
Short Description Long DescriptionFrequency
2014 2013
1169 21 CFR 211.42(a)Buildings of Suitable Size, Construction, Location
Buildings used in the manufacture, processing, packing, or holding of a drug product do not have the suitable [size] [construction] [location] to facilitate cleaning, maintenance, and proper operations. Specifically, ***
11 11

483 citations related to 21 CFR 211.42(b)
Cite IdReference Number
Short Description Long DescriptionFrequency
2014 2013
1174 21 CFR 211.42(b)Product flow through building is inadequate
The flow of [components] [drug product containers] [closures][labeling] [in-process materials ] [drug products] though the building isnot designed to prevent contamination. Specifically, ***
5 3
4418 21 CFR 211.42(b)Adequate space lacking to prevent mix-ups and contamination
The building lacks adequate space for the orderly placement ofequipment and materials to prevent mix-ups between [differentcomponents] [drug product containers] [closures] [labeling] [in-processmaterials] [drug products] and to prevent contamination. Specifically,***
4 5

483 citations related to 21 CFR 211.42(c)
Cite Id Reference Number Short Description Long DescriptionFrequency
2014 2013
1194 21 CFR 211.42(c)Defined areas of adequate size for operations
The [separate or defined areas][control systems] necessary to prevent contamination or mix-ups are deficient. Specifically, ***
15 15
142121 CFR
211.42(c)(10)Aseptic Processing Area
Separate or defined areas to prevent contamination or mix-ups are deficient regarding operations related to aseptic processing of drug products. Specifically,***
6 7
143021 CFR
211.42(c)(10)(i)Floors, walls, ceiling surfaces
Aseptic processing areas are deficient in that [floors] [walls] [ceilings] are not smooth and/or hard surfaces that are easily cleanable. Specifically,***
1 2
143321 CFR
211.42(c)(10)(iii)Air Supply
Aseptic processing areas are deficient regarding air supply that is filtered through high-efficiency particulate air filters under positive pressure. Specifically, ***
14 12
143421 CFR
211.42(c)(10)(iv)Environmental Monitoring System
Aseptic processing areas are deficient regarding the system for monitoring environmental conditions. Specifically, ***
42 31
143521 CFR
211.42(c)(10)(v)Cleaning System
Aseptic processing areas are deficient regarding the system for cleaning and disinfecting the [room] [equipment] to produce aseptic conditions. Specifically, ***
35 20
143621 CFR
211.42(c)(10)(vi)Equipment to control conditions
Aseptic processing areas are deficient regarding systems for maintaining any equipment used to control the aseptic conditions. Specifically, *** 6 1
1396 21 CFR 211.42(c)(2) Rejected Material AreaSeparate or defined areas to prevent contamination or mix-ups are deficient regarding operations related to the holding of rejected [components] [drug product containers] [closures] [labeling] before disposition. Specifically,***
1 1

483 citations related to 21 CFR 211.42(d)
Cite IdReference Number
Short Description Long DescriptionFrequency
2014 2013
1266 21 CFR 211.42(d)Penicillin processing area not kept separate
The operations relating to the [manufacture] [processing] [packing] of penicillin are not performed in facilities separate from those used for other drug products for human use. Specifically, ***
3 5

483 citations related to 21 CFR 211.42(c) (10)(iv)
Ref: 483 of Millers of Wyckoff Inc. (Jul-2015)

483 citations related to 21 CFR 211.42(c)(10)(iv)
Ref: 483 of Walgreens Home Care (Aug-2015)

483 citations related to 21 CFR 211.42
Ref: 483 of Millers of Wyckoff Inc. (Jul-2015)
21 CFR 211.42(c) (10)(v):
21 CFR 211.42(b);

483 citations related to 21 CFR 211.42(c) (10)(vi)
Ref: 483 of Walgreens Home Care (Aug-2015)

483 citations related to 21 CFR 211.42
Ref: 483 of PharMEDium Services (Jul-2015)
21 CFR 211.42(a):
21 CFR 211.42(c)(10)(iv):

483 citations related to 21 CFR 211.42
Ref: 483 of Unique Pharmaceutical Ltd(Jan-2015)
21 CFR 211.42(c)(10)(iv):
21 CFR 211.42(c)(10)(v):

483 citations related to 21 CFR 211.42(c)(10)(iv)
Ref: 483 of Pyramid Laboratories (Jan-2015)

483 citations related to 21 CFR 211.42(c)(10)(vi)
Ref: 483 of Pyramid Laboratories (Jan-2015)

483 citations related to 21 CFR 211.42(c)(10)(vi)
Ref: 483 of Teva Parenteral Medicines Inc(Jul-2009)

483 citations related to 21 CFR 211.42(c)(10)(iv)
Ref: 483 of Teva Parenteral Medicines Inc(Jul-2009)

483 citations related to 21 CFR 211.42(d)
Ref: 483 of Ameridose, LLC (Nov-2012)

483 citations related to 21 CFR 211.42(a)
Ref: 483 of Ameridose, LLC (Nov-2012)

483 citations related to 21 CFR 211.42(c)(10)(vi)
Ref: 483 of Ameridose, LLC (Nov-2012)

483 citations related to 21 CFR 211.42(c)(10)(vi)
Ref: 483 of Ameridose, LLC (Nov-2012)

483 citations related to 21 CFR 211.42(a)
Ref: 483 of Ameridose, LLC (Nov-2012)

483 citations related to 21 CFR 211.42(a)
Ref: 483 of Ameridose, LLC (Nov-2012)

483 citations related to 21 CFR 211.42(c)(10)(iv)
Ref: 483 of Hospira, Inc (Mar-2013) Continued ..

483 citations related to 21 CFR 211.42(c)(10)(iv)
Ref: 483 of Hospira, Inc (Mar-2013) Continued ..

483 citations related to 21 CFR 211.42(c)(10)(iv)
Ref: 483 of Hospira, Inc (Mar-2013)

483 citations related to 21 CFR 211.42(c)(10)(v)
Ref: 483 of Hospira, Inc (Mar-2013) Continued ..

483 citations related to 21 CFR 211.42(a)
Ref: 483 of Hospira, Inc (Mar-2013)

483 citations related to 21 CFR 211.42(c)(10)(v)
Ref: 483 of Hospira, Inc (Mar-2013)

Warning letter observations - 21 CFR 211.42
Your firm failed to establish an adequate system for monitoring environmental conditions in aseptic
processing areas (21 CFR 211.42(c)(10)(iv)).
a. You do not have a scientific rationale for the environmental monitoring sampling locations in
aseptic filling Suites (b)(4). You did not include factors such as smoke study findings, number and
location of operators, and historical microbial data in your assessment of hazardous points.
For example, we found that settling plates are not appropriately placed in critical areas. Your smoke
study showed that during set-up and filling, air flows toward the front (when (b)(4) open) or back of
the RABS. However, two relevant sampling points were recently eliminated. As a result, these points
of increased risk are not monitored.
b. During our inspection, we noted that you have no justification for two different action levels for
finger dab results. While you have an ISO 5 action level of (b)(4) CFU for set-up personnel, you use
an ISO 6 action level of(b)(4) CFU for operators who do not routinely participate in aseptic
processing operations using the RABS.
Ref: WL: Mylan Laboratories Limited OTL (FEI: 3007512701) - WL: 320-15-14

Warning letter observations - 21 CFR 211.42
• Your firm failed to establish an adequate system for monitoring environmental conditions in aseptic processing areas (21 CFR 211.42(c)(10)(iv)).
• a. Your firm does not have a robust sampling plan as part of its environmental monitoring program.
• No representative non-viable particle (NVP) monitoring data supports your current ISO-5 classification for the product path from the (b)(4) to the (b)(4), which transfers product to the (b)(4) during aseptic processing of finished drug products.
• During our inspection, we documented that your NVP probes are placed (b)(4) surface instead of near the working area. Placing the probe (b)(4) instead of near the working area means you are unable to detect NVPs where sterile drugs are exposed during aseptic processing.
• Additionally, transferring (b)(4) vials from the filling suite to the (b)(4) can take up to (b)(4). This extended exposure time may increase contamination hazards. However, your firm lacks adequate environmental monitoring of this part of the operation. It is essential that your sampling plan include areas where (b)(4) and product are exposed to the environment, and at greater risk of contamination.
Ref: WL: Mylan Laboratories - Agila Specialties Pvt Ltd (SFF) (FEI: 3007648351) - WL: 320-15-14

Warning letter observations - 21 CFR 211.42
• Your firm failed to establish an adequate system for monitoring environmental conditions in aseptic processing areas(21 CFR 211.42(c)(10)(iv)).
• b. In your ISO-5 and ISO-7 environments, the building management system (BMS) monitoring differential pressureand the non-viable particle monitoring system (NVPMS) for non-viable particles appear to be out of control. Forexample:
• We found 456,201 alarmed events registered in the computer system monitoring differential air pressures betweenyour ISO-5, ISO-7, and ISO-8 manufacturing environments from February 14, 2013, through September 26, 2014.
• We also found 16,415 alarmed events registered in NVPMS for Suite (b)(4) ISO-5 areas, and 17,809 forSuite(b)(4), from October 2012 to September 2014.
• You did not conduct a comprehensive evaluation and risk assessment to determine how these frequent eventsaffecting the aseptic processing areas may have compromised product quality.
• c. Your firm failed to identify the source of gram-negative contamination in your ISO 7 area and to implementappropriate corrective actions and preventive actions.
• In your ISO-7 Suite (b)(4), you identified Pseudomonas, sp. during passive air sampling collected from your passageway, in (b)(4) rooms (b)(4) and (b)(4). You did not evaluate the potential routes of contamination.
Ref: WL: Mylan Laboratories - Agila Specialties Pvt Ltd (SFF) (FEI: 3007648351) - WL: 320-15-14

Warning letter observations - 21 CFR 211.42
• Your firm failed to establish an adequate system for monitoring environmental conditions in
aseptic processing areas (21 CFR 211.42(c)(10)(iv)).
• Your firm does not have a scientific justification for alternating the use
of (b)(4) and (b)(4) for sampling by settle plates and swabs on different (b)(4). We are
concerned that you may have underestimated the number and type of bacterial species
that are present on the (b)(4) you use (b)(4) because you have no data to support the
equivalent sensitivity and efficiency of bacterial recovery on the (b)(4) media as
for (b)(4). FDA expects that microbial culture media used for environmental monitoring be
validated as capable of recovering fungi (i.e., yeast and molds), as well as
bacteria. Appropriate trending of environmental monitoring data depends on consistent
methods to provide an indication of the amount and type of microbiological organisms
present.
Ref: WL: Hospira Australia Pty Ltd. 9/26/14 - WL: 320-14-15

Warning letter observations - 21 CFR 211.42
• Your firm failed to perform operations related to the manufacture, processing, and packing
of penicillin in facilities separate from those used for other drug products for human use (21
CFR 211.42(d)).
• Specifically, your firm produces non-penicillin and penicillin containing finished products in
Werk II using shared equipment. As such, all non-penicillin products produced in your
facility are potentially adulterated with penicillin, and constitute a potential serious
allergenic hazard to patients who are sensitive to beta-lactams.
Ref: WL: SANUM-Kehlbeck GmbH & Co. KG 4/11/14 - WL: 320-14-07

Warning letter observations - 21 CFR 211.42
• Your firm failed to establish an adequate system for monitoring environmental conditions in
aseptic processing areas (21 CFR 211.42(c)(10)(iv)).
In your response to the Form FDA 483 dated March 7, 2013, you referenced your
purported compliance with United States Pharmacopeia (USP)-National Formulary (NF)
General Chapter <797>, “Pharmaceutical Compounding - Sterile Preparations.” As noted
above, your firm has manufactured and distributed drugs without valid prescriptions for
individually-identified patients, and the manufacture of such drugs is subject to FDA’s drug
CGMP regulations, 21 CFR Parts 210 and 211.
Ref: WL: Avella of Deer Valley, Inc. 1/17/14 - WL# 10-14

Warning letter observations - 21 CFR 211.42
• Your firm failed to assure an adequate system for cleaning and disinfecting aseptic processing areas and equipment (21 CFR 211.42(c)(10)(v)).
• a) For example, your firm’s SOP 101.74, "General Cleaning, Sanitization and Disinfection ofAseptic and Controlled Areas," lacks provisions to ensure adequate use of sporicidalagents. According to SOP 101.74, sporicidal agents are not required on aseptic filling line stainlesssteel, non-removable components, and the ISO 5 rigid barriers. According to SOP 101.74, (b)(4) willbe used to sanitize/clean stainless steel components in the aseptic or controlled areas.
• Your firm’s SOP 101.74, “General Cleaning, Sanitization and Disinfection of Aseptic and ControlledAreas,” also lacks adequate details on how many times mops and wipes can be used.
• Your response is inadequate because you did not provide scientific data that the corrective actionsimplemented in SOP 101.74, "General Cleaning, Sanitization and Disinfection of Aseptic andControlled Area" are adequate. While this SOP instructs staff to replace mops, wipes, and othersupplies when visually soiled, it is unclear whether this revision will provide for acceptable standardsof sanitization and disinfection in the controlled area.
Ref: WL: Jubilant HollisterStier, LLC 11/27/13

Warning letter observations - 21 CFR 211.42
• Your firm failed to establish an adequate system for monitoring environmental conditions in asepticprocessing (21 CFR 211.42(c)(10)(iv)).
• a. Your firm did not adequately assess contamination risk to determine the worst-case locationsand timing for your active viable air monitoring sites. We noted that you performed a totalof (b)(4) active air sample collections per (b)(4) during idle conditions ((b)(4)), each of which arelocated at the backside of the filling machine and are not representative of the conditions duringproduction. Your sampling was not conducted under dynamic conditions.
• b. Your active air sampling is deficient. The microbiologist sprays the “(b)(4)” of the air samplerwhere (b)(4) with (b)(4), followed by wiping with a cloth. The media plate is loaded onto the airsampler (b)(4)later. There is no assurance that residual (b)(4) does not impact the detection ofcontaminants.
• c. Our inspection found that there is no assurance that personnel monitoring (finger dabs), withperiodic use of the (b)(4) to disinfect the gloves, is conducted at a time that allows accurate recoveryand counts of contaminants.
Ref: WL: Agila Specialties Private Limited 9/9/13 - WL: 320-13-26

Warning letter observations - 21 CFR 211.42
• Your firm failed to establish an adequate system for monitoring environmental conditions in asepticprocessing (21 CFR 211.42(c)(10)(iv)).
• a. Your firm did not adequately assess contamination risk to determine the worst-case locationsand timing for your active viable air monitoring sites. We noted that you performed a totalof (b)(4) active air sample collections per (b)(4) during idle conditions ((b)(4)), each of which arelocated at the backside of the filling machine and are not representative of the conditions duringproduction. Your sampling was not conducted under dynamic conditions.
• b. Your active air sampling is deficient. The microbiologist sprays the “(b)(4)” of the air samplerwhere (b)(4) with (b)(4), followed by wiping with a cloth. The media plate is loaded onto the airsampler (b)(4)later. There is no assurance that residual (b)(4) does not impact the detection ofcontaminants.
• c. Our inspection found that there is no assurance that personnel monitoring (finger dabs), withperiodic use of the (b)(4) to disinfect the gloves, is conducted at a time that allows accurate recoveryand counts of contaminants.
Ref: WL: Agila Specialties Private Limited 9/9/13 - WL: 320-13-26

Warning letter observations - 21 CFR 211.42
• Your firm failed to perform operations within specifically defined areas of adequate size and
to have separate or defined areas of such other control systems necessary to prevent
contamination or mix -ups (21 CFR 211.42(c)).
• a. You do not disinfect the (b)(4) conveyor after storage outside the ISO-5 area; this
conveyor is used to transport filled and partially stoppered vials to the (b)(4).
• b. Our inspection found that the same “mop” is used throughout the production of a batch
and is even stored outside the ISO-5 area before re-use. This “mop” is used to disinfect the
RABS (b)(4) and equipment surfaces inside the RABS during setup and manufacturing
activities. The repeated use of the same “mop” poses a significant risk of cross-
contamination to the open vials with microbial and/or particulate matter from the cloth mop.
Ref: WL: Agila Specialties Private Limited 9/9/13 - WL: 320-13-26

Warning letter observations - 21 CFR 211.42
• Your firm failed to have facilities used in the manufacture, process, packaging and holding of a drug product of appropriate construction to facilitate cleaning, maintenance, and proper operations (21 CFR 211.42(a)).
• a. The FDA investigator observed two holes of approximately 1 cm x 0.5 cm each, between the Class 100,000 (ISO 8) “circulation corridor” and the Class 100 (ISO 5) “component receipt area” of sterile (b)(4) facility.
• b. The entry/exit door into the vial filling/stoppering suite has not been designed in a way that protects the aseptic filling area from disruption of airflow from room entries and exits; for example, there is no mechanism to control the door operation from disrupting the air flow within the filling area when personnel enter and exit the room. This disruption may affect the air flow in this critical area.
• c. The investigator also noted that air was flowing from the circulation corridor into the component receipt area, adjacent to where the aseptic (b)(4) filling of (b)(4) Injection USP (b)(4) mg, lot #(b)(4) was in progress. The circulation corridor, where the holes were observed, is an unclassified area. The component receipt area is directly connected and adjacent to the aseptic vial filling/stoppering suite and is where sterile components are unloaded from the (b)(4).
Ref: WL: Hospira Healthcare India Pvt. Ltd. 5/28/13 - WL: 320-13-18

Warning letter observations - 21 CFR 211.42
• Your firm failed to establish adequate systems for monitoring environmental conditions and for cleaning anddisinfecting the room and equipment in aseptic processing areas (21 CFR 211.42(c)(10)(iv)and (v)).
(a) The aseptic processing environment is not adequately monitored. For example, there is no viable air monitoringinside of the Class 100 (ISO 5) filling barrier on the “(b)(4) Line (b)(4).” This is the critical area where drug product andpre-sterilized components are exposed and it is important that your firm collect air samples that adequately representfilling conditions.
Moreover, outside of the line (b)(4) filling area, the three air samples taken in the Class 100 (ISO 5) area were nottaken under dynamic conditions. These active samples were instead taken after line set-up and before any filling.
We are concerned that the environmental monitoring (EM) program is not adequate to ensure the environment issuitable for aseptic processing of sterile product. The data generated does not sufficiently demonstrate that an ISO 5environment is maintained.
(b) Your firm did not use a sporicidal disinfectant for cleaning inside of the Class 100 (ISO 5) filling areas. Theinspection documented that your firm uses (b)(4) ((b)(4)) alone, which is not effective against spore-formingorganisms such as Bacillus spp. The September 2011 media fill failure investigation for the (b)(4)Line (b)(4) identifiedthe contaminating organism as Bacillus pumilus. Additionally, you did not sufficiently evaluate thedisinfectant (b)(4) on surfaces inside the Class 100 (ISO 5) area including (b)(4).
Ref: WL: Promed Exports Private Limited 8/9/13 - WL: 320-13-24

Warning letter observations - 21 CFR 211.42
• You failed to assure an adequate system for cleaning and disinfecting aseptic processing areas andequipment [21 CFR 211.42(c)(10)(v)]. For example:
• a. Report C017795 entitled “Year 2010 Re-Evaluation of the Approved Disinfectants/SporicidalAgents” (effective date October 8, 2011) used bacterial and mold spores to test the effectiveness ofdisinfectants and sporicidal agents used at your facility, including the BCG aseptic manufacturingareas in Building (b)(4). The effectiveness study is inadequate in that it did not evaluate use of thedisinfectants and sporicidal agents on surfaces other than (b)(4).
• b. Your SOP entitled “Disinfection Program for Equipment and Manufacturing Areas inBuilding (b)(4); BCG Department” is inadequate. Sporicidal disinfection of aseptic manufacturingareas using (b)(4) is only required to be performed (b)(4). There have been no less than 58documented non-conformances relating to the isolation of mold within the BCG aseptic processingareas (Grade (b)(4) areas) of Building (b)(4) since August 2010.
• c. There is no documented evidence that corrective action in followup to non-conformances relatingto the isolation of mold within the BCG aseptic manufacturing areas includes cleaning with asporicidal agent.
Ref: WL: Sanofi, Date Issued: July 12, 2012

Warning letter observations - 21 CFR 211.42
• Your firm has not established separate or defined areas or such other control systems to prevent
contamination during aseptic processing [21 C.F.R. § 211.42(c)]. For example,
• a. There is no documentary evidence of in-situ air pattern analysis (e.g., smoke studies) conducted
at critical areas to demonstrate unidirectional airflow and sweeping action over and away from the
product under dynamic conditions. Your firm failed to demonstrate that the appropriate design and
controls are in place to prevent turbulence and stagnant air in the critical area. It is essential that you
evaluate airflow patterns for turbulence that can act as a channel for air contamination. The studies
should be well documented with written conclusions, and should include an evaluation of the impact
of aseptic manipulations (e.g., interventions) and the equipment design.
• b. Your aseptic processing control systems and operations do not provide assurance that the
production rooms and equipment maintain aseptic conditions. Additionally, your environmental
monitoring practices do not include adequate routine examination of the facilities and equipment to
ensure that possible contaminants can be detected. (continued ..)
Ref: WL: CP Pharmaceuticals, Ltd., Date Issued: October 29, 2010

Warning letter observations - 21 CFR 211.42
• The inspection documented mold contamination in the class 100 production room and poorconditions of a wall in the freeze dryer room, even though maintenance is conducted on the freezedryer every (b)(4) months. An incident report, initiated in November 2009, identifies holes in theceiling and visible light coming from the roof near the ventilation system, bubbling of the vinyland disintegration of the wall under vinyl in the freeze dryer room, visible black mold on the wall, apoor drain system for the freeze dryer steam venting system, and a soft (spongy) wall.
• c. Operators involved in the filling operations for the sterile drug products manufactured at yourfacility do not practice adequate aseptic techniques to prevent product contamination.The environmental monitoring performed at the end of the production run consist of sampling thechest and the hand most frequently used (right or left) of the employee's gown. Also, this procedureis performed by the gowned operator and is not monitored by a second qualified person(e.g., supervisor; quality unit personnel) to ensure the proper techniques are being applied. Thispractice is unacceptable. We expect that all operators who conduct operations within asepticprocessing areas be properly trained and monitored to ensure that proper techniques are utilizedduring all operations, including aseptic filling operations and personnel sampling.
Ref: WL: CP Pharmaceuticals, Ltd., Date Issued: October 29, 2010

Warning letter observations - 21 CFR 211.42
• Controls to prevent contamination in defined (critical and support clean) areas are deficientregarding operations related to aseptic processing of product [21 C.F.R. § 211.42(c)(10)].
• For example, there are no dynamic smoke study evaluations to demonstrate that the personnelactivities during aseptic filling do not compromise the sterile API. The activities conducted duringyour documented smoke studies are not representative of actual operations.
• According to your response, smoke studies were to be completed within the first two weeks ofJanuary 2010. Your response is inadequate because it does not provide an update on all airflowpattern findings and your evaluation of these study results. An in situ air pattern analysis should beconducted at all critical areas, under dynamic conditions, to demonstrate unidirectional airflow andsweeping action at critical work areas. These studies should evaluate the impact of asepticmanipulations (e.g. interventions) and equipment design, and include documentation for theactivities performed with written conclusions. Provide a copy of the smoke study recordings that canbe read using Windows Media Player (as an mpeg file, for example) along with supportingdocumentation. Please also identify the different videos by file name to indicate what is beingpresented in each file.
Ref: WL: Ribbon Pharmaceutical and Chemical Products, Date Issued: May 27, 2010

Warning letter observations - 21 CFR 211.42
• The controls to prevent contamination in defined (critical) areas are deficient regarding
operations related to aseptic processing of products. [21 CFR 211.42(c)(10)]
• A. Smoke studies of Class [(b)(4)] in critical areas were inadequate in that they were not
performed under dynamic conditions and the results were not recorded for subsequent
review. Refer to FDA Form 483, Observation #5.
• You included a CD ROM of the smoke study summary report with your December
response. However, this CD ROM was unable to be opened for review, thus we could not
read the attached documents. Re-submit the supporting documentation including the video
showing the smoke study your firm conducted on November 19, 2008.
Ref: WL: Lupin Limited, Date Issued: May 7, 2009

Warning letter observations - 21 CFR 211.42
• The controls to prevent contamination or mix-ups in defined (critical and supporting clean) areas are deficient regarding operations related to aseptic processing of drug products [21 CFR 211.42(c)(10)].
• A. For parenteral operations, smoke studies were not conducted to demonstrate unidirectional airflow and sweeping action over and away from the product under dynamic conditions during numerous aseptic operations in classified areas of the vial filling facility. For example:
• 1. Various manual operations performed with the [redacted] such as dispensing sterile API and connecting equipment to this [redacted] were not included in smoke studies.
• 2. Other significant manual aseptic activities that can affect airflow, including opening and closing the fill equipment access panels during routine aseptic filling operations, were not evaluated in smoke studies.
• 3. There was no evaluation performed to demonstrate that personnel activities (e.g., manual transfer of material into or out of the ISO [redacted] and ISO [redacted] areas) do not compromise the unidirectional airflow pattern.
• 4. There was no evaluation performed to demonstrate that the horizontal airflow from the [redacted] does not negatively impact upon the vertical airflow within the aseptic Willing areas.
Ref: WL: Ranbaxy Laboratories Limited, Date Issued: September 16, 2008

Warning letter observations - 21 CFR 211.42
• B. For sterile API operations, smoke studies were not representative of actual operations to demonstrate unidirectional airflow and sweeping action over and away from the product under dynamic conditions during numerous aseptic operations in classified areas processing sterile APIs. For example:
• 1. There are no smoke study evaluations to demonstrate that the personnel activities during the [redacted] of sterile API from the [redacted] do not disturb the unidirectional airflow in front of the to prevent compromising the sterile API.
• 2. The smoke study performed for the set up of the [redacted] equipment did not actually reflect the manner with which the equipment and manual aseptic connections are made.
• 3. There are no controls (e.g. physical barrier, curtains) in place to ensure that the [redacted] room's ISO [redacted] unidirectional airflow conditions were not compromised during routine operations performed within the ISO [redacted] area.
• 4. The smoke study performed for the [redacted] steps did not accurately reflect the manner in which routine aseptic connections are made.
Ref: WL: Ranbaxy Laboratories Limited, Date Issued: September 16, 2008

Warning letter observations - 21 CFR 211.42
• C. Failure to conduct aseptic connections of sterile API materials in critical areas (ISO [redacted]) and demonstrateproviding [redacted] unidirectional air flow over the connections. For example, the manual aseptic connections forsterile APIs performed prior to [redacted] were done in an ISO [redacted] (supporting clean) area.
• D. Viewing locations are inadequate to assess processing operations in ISO [redacted] sterile API and drug productoperations. The aseptic processing facility lacks appropriate viewing facilities for aseptic operations in order to assessthe control systems necessary to prevent contamination or mix-ups during the course of aseptic processing. Forexample, the door windows and their locations, used to observe routine operations, precludes the In-Process QualityAssurance (IPQA) and Management from observing all phases of either the [redacted] aseptic API processes or theaseptic finished drug product processes.
• In summary, we are concerned that your aseptic operations are conducted under extensive steps, manualhandling, and inadequate equipment usage as reported above under S.C., D. and E., and 6.C. For example, manualoperations under aseptic conditions should be conducted with minimum operator intervention and no exposed criticalsurfaces and product. Therefore, it is not appropriate to try to overcome major flaws in clean room design andequipment by attempting to validate difficult to perform, intensive manual procedures. These manual practices havethe potential to increase the risk of contamination on critical surfaces and are considered inadequate manufacturingpractices which can not be justified nor validated. Furthermore, design concepts and use of contemporary equipmentand automation technologies should be explored and assessed for suitability to prevent unnecessary activities thatcould increase the potential for introducing contaminants into the aseptic environment. We recommend that youconduct an extensive evaluation of your facilities for opportunities to minimize steps and manual handling.Additionally, appropriate equipment and usage in all related aseptic operations for APIs and finished dosage formsshould be evaluated.
Ref: WL: Ranbaxy Laboratories Limited, Date Issued: September 16, 2008

[email protected] Guidance …

FDA Level 2 Guidance
Ref: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm192869.htm

Comparison between US and EC GMP

Comparison between US and EC GMP

EC GMP Guide
PRINCIPLE : Premises and equipment mustbe located, designed, constructed, adapted andmaintained to suit the operations to be carriedout. Their layout and design must aim to minimisethe risk of errors and permit effective cleaning andmaintenance in order to avoid cross-contamination, build-up of dust or dirt and, ingeneral, any adverse effect on the quality ofproducts.
Ref: http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm

EC GMP Guide
Ref: http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm

EU Non-Compliance Reports
Firm Name Nature of non-compliance
Huzhou Sunflower
Pharmaceutical Co., Ltd.,
China
• The controlled area and the equipment that were used for the final synthesis step in the
manufacture of Povidone Iodinated, namely the complexation reaction of Iodine with Povidone
K30, presented a risk to the patients due to contamination issues with particles and
degradation products
SIMS, Italy • The presence of two unauthorized, not-GMP storage areas (declared as office area) in building
1 illegally used for storage of imported materials from China, expired API, production tails and
for which the company has not provided an inventory list related to origin, status and
destination of each material; an unauthorized, not-GMP repackaging station with craft dust
extraction system was found in the same premises. The company has provided the inspection
team contradictory information about the ownership of the premises and of the materials,
initially attributing them to SIMS Trading and following to SIMS srl.
• Use of above described unauthorized area for the distribution and repackaging of the batch of
lidocaine 170686; the inspection team points out in the report that the conditions for such local
do not ensure the absence of contamination by other products.
Fujian South
Pharmaceutical, China
• In workshop B-03 the room used to perform the first purification of Docetaxel anhydrous by
liquid chromatography was found not suitable for its intended use, as there was a potential risk
of contamination

EU Non-Compliance Reports
Firm Name Nature of non-compliance
ZHUHAI UNITED
LABORATORIES CO.,
LTD, China
• The company was not operating its aseptic manufacture operations in compliance with EU GMP Annex 1.
This was evident from by the high number of observations regarding the aseptic manufacturing facilities
design, equipment, operations, environment monitoring and media fill validation. However, the QA system
of the company failed to notice these problems and therefore was considered weak and inappropriately
implemented.
INTEGRA LIFE
SCIENCES CORP,
United States
• Contamination of Absorbable Collagen Sponges with particulate matter is not under control. ACS sheets
are exposed to an ISO Class 7 environment without protection by Grade A air.
• This is relevant during the loading and unloading of the cross-linking chamber, the cutting of the sponges,
the visual inspections and the (re)packaging stage.
• Embedded particles do occur but are not always removed during visual inspection if seen.
• During visual inspection tweezers are used constantly to remove visible contaminants from the surface of
sponges. This is regarded by the company as a part of routine production.
• A comprehensive analysis of the root cause of contamination is not conducted.
• Packaging material seems to be a source of foreign matter, but other than vacuuming trays before
packaging (which proved to be insufficient) no preventive measures have been taken.
• Static charges contribute to the problem of particulate matter contamination, as was seen by a hair that
was stuck on the outside of a container used to store sponges for a long time.
• The design and functioning of the sponge cutting table as a source of particulate contaminants has not
been considered.

EU Non-Compliance Reports
Firm Name Nature of non-compliance
WOCKHARDT
LIMITED, India
4 major deficiencies were cited , relating to storage, general manufacturing practices, the
performance of tablet coating and qualification and validation of equipment and products.
• Issues seen, included temporary storages areas that had been created immediately prior
to the inspection and were not visible on the site map. Storage areas did not have
adequate temperature control or monitoring and did not protect stored items from
contamination from the area or the environment.

Health Canada – GMP
Premises - Regulation (C.02.004)
• The premises in which a lot or batch of a drug is fabricated, packaged/labelled or stored shall bedesigned, constructed and maintained in a manner that
(a) permits the operations therein to be performed under clean, sanitary and orderly conditions;
(b) permits the effective cleaning of all surfaces therein; and
(c) prevents the contamination of the drug and the addition of extraneous material to the drug.
Equipment - Regulation (C.02.005)
• The equipment with which a lot or batch of a drug is fabricated, packaged/labelled or tested shall bedesigned, constructed, maintained, operated, and arranged in a manner that
(a) permits the effective cleaning of its surfaces;
(b) prevents the contamination of the drug and the addition of extraneous material to the drug; and
(c) permits it to function in accordance with its intended use.

Health Canada – GMP
Sanitation - Regulation (C.02.007)
• (1) Every person who fabricates or packages/labels a drug shall have a written sanitation programthat shall be implemented under the supervision of qualified personnel.
• (2) The sanitation program referred to in subsection (1) shall include:
(a) cleaning procedures for the premises where the drug is fabricated or packaged/labelled and for theequipment used in the fabrication or packaging/labelling of the drug; and
(b) instructions on the sanitary fabrication and packaging/labelling of drugs and the handling of materialsused in the fabrication and packaging/labelling of drugs.
Examples of observations from frequently cited sections of the Food and Drug Regulations - Good Manufacturing Practices Inspections (2013 – 2014)

ICH

[email protected] to avoid 21 CFR 211.42 observations ?

To avoid observations …
To avoid / minimize observations related to 21 CFR 211.42 regulation,
1. Know the Requirements & Regulation
2. Understand the importance of each requirement.
3. Conduct internal audits
4. Identify the gaps
5. Conduct training
6. Implement the change
7. Evaluate periodically

Requirements
GMP regulation 21 CFR 211.42 lists specific
requirements related to
• building and facility• size
• construction
• location
• cleaning
• maintenance
• Manufacturing operations
• aseptic processing
In general, the purpose of this regulation is
to ensure that buildings and facilities are
capable of providing an environment that is
suitable for the manufacture of various
types of drug products.
Building &
Facility
Size
Construction
Location
Cleaning
Maintenance
Air Supply
Environmental Monitoring
System
Manufacturing operations
Aseptic processing
Penicillin processing
area

Regulation at a glance
Regulation Short description
21 CFR 211.42(a) Buildings of Suitable Size, Construction, Location
21 CFR 211.42(b) Adequate product flow through building & Adequate space to prevent mix-ups and contamination
21 CFR 211.42(c) Defined areas of adequate size for operations
21 CFR 211.42(c)(2) Prevent contamination or mix-ups related to the holding of rejected [components] [drug product
containers] [closures] [labeling] before disposition
21 CFR 211.42(c)(10) Aseptic Processing Area –
• Separate or defined areas to prevent contamination or mix-ups regarding operations
• Floors, walls, ceiling surfaces – smooth & easily cleanable
• Air Supply - filtered through high-efficiency particulate air filters under positive pressure
• Environmental Monitoring System - for monitoring environmental conditions.
• Cleaning System - cleaning and disinfecting the [room] [equipment] to produce aseptic conditions
• Equipment to control conditions - for maintaining any equipment used to control the aseptic
condition
21 CFR 211.42(d) Penicillin processing area - separate from those used for other drug products for human use.

Adequate Space & Size – 21 CFR 211.42 (b)
• There are two key points to consider about adequate space & size in the facility.
(1) concerns contamination and mix-ups.
(2) concerned with manufacturing materials and product flow through the steps in the
manufacturing process.
• The layout and design of premises must aim to minimize the risk of errors and permit
effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust
or dirt, and, in general, any adverse effect on the quality of products.

Contamination and Mix-ups - 21 CFR 211.42 (b), (c)
• The placement of equipment and materials is important for preventing contamination and
mix-ups.
• If major pieces of manufacturing equipment are placed too close to each other, it will be
very difficult to perform proper cleaning and maintenance.
• There will almost certainly be hard-to-reach places that accumulate dirt, dust, and residue
that could contaminate materials, equipment, and product. For example, materials that are
stacked against walls prevent anyone from cleaning around them.
• Placement of equipment for packaging and labeling is very important.
• If equipment from two different packaging and labeling operations are placed too close to
each other, the probability of a label or product mix-up is increased.

Materials and Product Flow - 21 CFR 211.42 (b)
• Equipment and materials need to flow in an orderly manner from beginning to completion
through each stage of manufacture.
• Equipment and materials need to be placed, so there is little or no contact of
components, in-process materials, or product as they proceed from one operation to
another. For example, blends that need to be milled should not cross over or be placed in
an area that contains blends that have already been milled.
• Crossovers create the potential for a mix up or contamination.
• A manufacturing process flow that prevents this practice is considered to be compliant with
21 CFR 211.42 (b) requirements for space.

Receipt, Storage, and Withholding - 21 CFR 211.42 (c)(1)
• Compliance with established guidelines regarding adequate space for
receiving, storing, and withholding components, containers, closures, and labeling involves
a separation of at least the receipt and storage areas.
• The receiving area must have adequate space to handle shipments of the various
manufacturing materials.
• Considering that truckloads are involved, this area has to be sufficiently large.
• It is common for a portion of this area to be designated as quarantine, where these
materials are placed pending sampling, testing, or examination by the Quality Unit.
• Take note that this quarantine area has to be large enough to allow for the placement of
materials, so that quality team members are not impeded or restricted from completing
proper sampling procedures.

Rejected Components - 21 CFR 211.42 (c)(2)
• GMP regulations require adequate space for a clearly defined area used for holding
rejected components, containers and closures, and labeling prior to their disposition.
• It is advisable to have enough space to segregate these materials: components in one
section, containers and closures in another section, and labeling in still another.
• This practice facilitates accountability for the disposition of these materials and minimizes
moving them unnecessarily.
• Items in the rejected materials area need to be clearly identified as rejected, with each
individual container being clearly marked.
• The use of a brightly colored label or sticker with the word “rejected” in large type is an
example of clear, visible identification. Such an identification practice will minimize the
chance of a rejected material being mixed with approved or released materials.

Released Components - 21 CFR 211.42 (c)(3)
• The manufacturing facility must have adequate space for the storage of released
components, containers and closures, and labeling to meet another requirement of this
GMP regulation [21 CFR 211.42 (c)].
• A good practice is to keep the released materials segregated and removed from the
quarantine and rejected materials areas.
• This helps minimize the chances that quarantine or rejected material can be mixed with
released materials.
• Visual identification of the materials as released or approved is also helpful.

Storage of In-process Materials - 21 CFR 211.42 (c)(4)
• Adequate space for the storage of in-process materials, such as tablet and capsule
blends, and bulk tablets and filled capsules, is still another requirement of 21 CFR 211.42
(c)(4).
• FDA expects the facility to have adequate space to provide areas where in-process
materials can be segregated to prevent mix-ups.
• A sound practice is keeping tablet blends separate from capsule blends, bulk tablets
separate from filled capsules, and all of these separate from one another.
• Using clear visual identification of contents and stage of manufacture complies with
regulation requirements contained in Subpart F (21 CFR 211.100) of the GMP regulation.

Manufacturing and Process Operations - 21 CFR 211.42 (c)(5)
• There must be defined areas for manufacturing and process operations.
• Adequate space must be available to handle manufacturing and process operations, such as
charge-in of components, blending, liquid manufacture, tableting, and capsule-filling operations.
• There must be space for placement of equipment to provide for proper cleaning and
maintenance, and sufficient separation of equipment to prevent contamination - 21 CFR 211.42 (c).
• Each distinct stage of manufacturing and process operation must be segregated. For
example, blending and milling of different materials or products should not be done in close proximity
to one another.
• This would pose a possible cross contamination issue.
• The best reason for defining areas and providing adequate space for the separation of
manufacturing and process operations is the prevention of contamination.

First Impressions— The Plant Tour
• One of the first significant and critical activities conducted in an audit is the plant
tour.
• The plant tour provides a broad overview of the site operations, both in technical
manufacturing and regulatory compliance.
• The following are two axioms that are particularly applicable to the entire
pharmaceutical plant:
• “A place for everything and everything in its place”
• “Cleanliness is next to godliness.”

Check again …
Building - The following are example primer questions for buildings:
• Do we have adequate space in our work areas to safely and effectively perform our jobs?
• Do we minimize the chance of product contamination, mix-ups, and errors by helping control the internal environment of the workplace?
• Do we quickly report any conditions in our workplace that could be a potential source of product contamination?
Equipment - The following are example primer questions for equipment:
• Do we perform routine maintenance on equipment and do we check to see if any measuring and testing equipment has been properly calibrated?
• Do we keep accurate equipment logs and do we promptly report any maintenance problems to the right people?
• Do we keep equipment and tools clean and store them in the proper manner?

The module Consult Yourself.... ―Know Regulation - No Observation‖ deals with most common (top 20) basic CFR regulations having frequent violations and previous observations, relevant guidance and notes.
The module will be continued with # 8 21 CFR 211.113(Control of microbiological contamination)
For “Pharma Uptoday” free daily newsletter write a mail to [email protected]
for other Pharma Uptoday presentations & Monthly Magazines browse:http://www.slideshare.net/skvemula