((title)) web viewself-assembly of short peptides has been recognized as a powerful strategy to...
TRANSCRIPT

FULL PAPER
Tuning One-Dimensional Nanostructures of Bola-Like Peptide Amphiphiles via Varying the Hydrophilic Amino AcidsYurong Zhao,[a] Li Deng,[a] Wei Yang,[a] Dong Wang,[a] Elias Pambou,[b] Zhiming Lu,[b] Zongyi Li,[b] Jiqian
Wang,[a] Stephen King,[c] Sarah Rogers,[c] Hai Xu,*[a] and Jian R. Lu*[b]
Abstract: By combining experimental measurements and computer simulations, we here show that for the bola-like peptide amphiphiles XI4X, where X=K, R, and H, the hydrophilic amino acid substitutions have little effect on the β-sheet hydrogen bonding between peptide backbones. Whilst all of the peptides self-assemble into one dimensional (1D) nanostructures with completely different morphologies, i.e., nanotubes and helical nanoribbons for KI4K, flat and multilayered nanoribbons for HI4H, and twisted and bilayered nanoribbons for RI4R. These different 1D morphologies can be explained by the distinctive stacking degrees and modes of the three peptide β-sheets along the x-direction (width) and the z-direction (height), which microscopically originate from the hydrogen bonding ability of the sheets to solvent molecules and the pairing of hydrophilic amino acid side chains between β-sheet monolayers via stacking interactions and hydrogen bonding. These different 1D nanostructures have distinctive surface chemistry and functions, with great potential in various applications exploiting the respective properties of these hydrophilic amino acids.
Introduction
Self-assembly of short peptides has been recognized as a powerful strategy to construct supramolecular nanostructures and nanomaterials from the bottom up for a variety of biomedical applications.[1-7] Among various peptide self-assembled aggregates, one-dimensional (1D) nanostructures such as long nanofibers have attracted much more attention due to their ability to entangle into 3D networks to afford hydrogels as well as the subsequent applications of peptide hydrogels in regenerative medicines and drug delivery systems.[8-10] It is known that non-covalent interactions (e.g., hydrogen bonding, hydrophobic and/or aromatic interactions, and electrostatic interactions) are primarily responsible for peptide self-assembly and each of them has a specific role in directing self-assembled
nanostructures. The formation of varied 1D nanostructures including nanofibers,[11-13] nanoribbons,[14-16] and nanotubes[17-21] is correlated to the collaboration or interplay of these non-covalent interactions. As a result, understanding the mechanistic roles of different non-covalent interactions and harnessing their interplay and balance are crucial to unravel the rationales associated with peptide self-assembly and to construct novel nanostructures and nanomaterials for targeted applications.
To this end, many strategies have been applied, and the most widely used ones include variation of solution conditions[14,22-25]
(e.g., pH, ionic strength, solvent polarity, and even ultrasound treatment) and peptide sequence variation.[26-28] In the latter case, Cui et al have shown that four isomeric tetrapeptide amphiphiles with identical composition but different peptide sequences (VEVE, EVEV, VVEE and EEVV, where V=Val and E=Glu) can form drastically different types of 1D nanostructures (nanobelts, twisted nanoribbons, rigid and flexible nanofibers) under the same conditions.[28] For another instance, by changing the position of charged lysine residues in amphiphilic peptides (I4K2, KI4K, and I2K2I2, where I=Ile and K=Lys), we have demonstrated completely different self-assembling ability and self-assembled nanostructures.[26,27] For example, I4K2 formed nanofibers and KI4K formed nanotubes while no well-ordered aggregates were observed in the case of I2K2I2. In these studies, hydrogen bonding between peptide backbones was demonstrated to drive the axial growth of aggregates along the y-direction (length) while the interactions between amino acid side chains and their interplay influenced final 1D morphologies.
Because each amino acid has specific physiochemical properties (e.g., hydrophobicity, aromaticity, and charged state) and propensity for different secondary structures, considerable efforts have been devoted to the effects of amino acid substitutions on peptide self-assembly.[29-32] The amino acids with higher hydrophobicity and/or aromaticity (e.g., Ile, Leu or L, and Phe or F) are generally perceived to favor peptide self-assembly into amyloid-like fibrils. Although isomeric, Ile and Leu have comparable hydrophobicity, we have shown that ultrashort peptide I3K self-assembled into long nanofibers by taking a β-sheet secondary structure while L3K was unable to form well-ordered nanostructures and only adopted a random coil conformation.[29] Such a difference has been ascribed to the stronger propensity of Ile for β-sheet structuring relative to Leu. Nilsson et al have indicated that the hydrophobicity alone did not dictate amyloid potential while aromatic, hydrophobic and steric considerations collectively influence fibril formation. [32] In contrast to hydrophobic amino acid residues that readily cause the association or aggregation of peptides and proteins, hydrophilic ones provide favorable contributions to their solubility. Because hydrophilic amino acids are mostly distributed on the surfaces of peptide assemblies and proteins, they define the aggregated morphology and topography on the microscopic scale through electrostatic interactions and steric effects. Furthermore, hydrophilic amino acid residues are closely related to some key
[a] Dr. Y. Zhao, Dr. L. Deng, W. Yang, Dr. D. Wang, Dr. J. Wang, Prof. H. XuCentre for Bioengineering and BiotechnologyChina University of Petroleum (East China)Changjiang West Road, Qingdao 266580, ChinaE-mail: [email protected]
[b] E. Pambou, Z. Lu, Z. Li, Prof. J. R. LuSchool of Physics and AstronomyThe University of ManchesterOxford Road, Manchester M13 9PL, United KingdomE-mail: [email protected]
[c] Dr. S. King, Dr. S. RogersISIS Pulsed Neutron SourceSTFC Rutherford Appleton LaboratoryDidcot, Oxon OX11 0QX, United Kingdom
Supporting information for this article is given via a link at the end of the document.

FULL PAPERbiological activities such as binding, recognition, signaling, and catalysis. From these perspectives, it is of particular significance to investigate the effects of hydrophilic amino acid substitutions on peptide and protein self-assembly.
Scheme 1. Diagrams of the side chain groups of A) Arg and B) His. The former showing that the planar protonated guanidinium group of an Arg can form 5 hydrogen bonds with water molecules and the latter showing the planar imidazole ring can act both as the hydrogen bonding acceptor and donor. C) Schematic illustration of the y-direction (length) along which the hydrogen bonding between β-strands drives the axial growth of assemblies, the x-direction (width) along which the interaction of hydrophobic amino acid side chains drives the lateral association of β-sheets, and the z-direction (height) along which the pairing of hydrophilic amino acid side chains drives the stacking of β-sheet monolayers. D) Arg-Arg pairing. E) His-His pairing.
KI4K is an ideal model for studying the effects of hydrophilic amino acid substitutions on peptide self-assembly because hydrogen bonding, hydrophobic and electrostatic interactions, and molecular geometry produce a collaborative effect during its self-assembly, leading to significant lateral stacking (association) of β-sheets and finally giving rise to well-ordered nanotubes.[26]
In this work, we replace the two hydrophilic Lys residues with two arginine (Arg or R) or histidine (His or H) residues to get two new cationic amphiphilic peptides, i.e., RI4R and HI4H, respectively, and then investigate their self-assembly by both experimental methods and computer simulations. The side chain guanidinium group of Arg endows the amino acid with very high hydrophilicity and extremely strong ability to form hydrogen bonding with water molecules and other side-chain heteroatoms (Scheme 1A),[33,34] and the side chain imidazole ring of His can act both as the hydrogen bond donor and acceptor (Scheme 1B).[33,35] Furthermore, both guanidinium and imidazole groups are planar, so stacking and other interactions can result in Arg-Arg and His-His pairings (Scheme 1D and 1E), which are often
found in the vicinity of the surfaces of proteins as structural and functional motifs.[36-39] Additionally, Arg is well known to constitute the integrin-binding motif (RGD) for cell adhesion and spreading,[40] and His has strong binding affinity for metal ions such as Cu2+
and Ni2+.[41-44] We show that the hydrophilic amino acid substitutions have little effect on the β-sheet hydrogen bonding between peptide backbones and all the three peptides form 1D nanostructures along the y-direction (length) but with completely different morphologies, i.e., nanotubes for KI4K, flat and multilayered nanoribbons for HI4H, and twisted and bilayered nanoribbons for RI4R. The resulting different 1D morphologies arise from the different stacking degrees and modes of the three peptide β-sheets along the x-direction (width) and the z-direction (height). A schematic illustration on the three directions of assemblies, i.e. the x, y, and z directions was given in Scheme 1C. The work clearly demonstrates the significance of hydrophilic amino acids in tuning self-assembled peptide nanostructures and even their potential applications.
Results and Discussion
The molecular structures of three designed peptides are shown in Figure 1 and both of the C and N terminals were capped during synthesis. The use of four consecutive Ile residues as the hydrophobic core is because of their high hydrophobicity and strong conformational propensity for -sheet structuring. The two identical hydrophilic residues are located at the terminals, making the molecules look like bola-amphiphiles. For all experiments, the peptide concentration was fixed at 16 mM and the solution pH value was carefully adjusted to 3.0 using diluted HCl solution. Under these conditions, the hydrophilic amino acid side chains were 100% protonated and each peptide carried two positive charges at the two terminals.
Figure 1. Molecular structures of KI4K, RI4R, and HI4H.
Secondary structures
Circular dichroism (CD) and FTIR measurements were first performed on the three peptide solutions after incubation for 7 days. As shown in Figure 2A, all three peptides exhibited two characteristic peaks of -sheet conformations, i.e., a negative CD minimum at ~220 nm and a positive maximum at ~200 nm, being indicative of the formation of -sheet hydrogen bonding
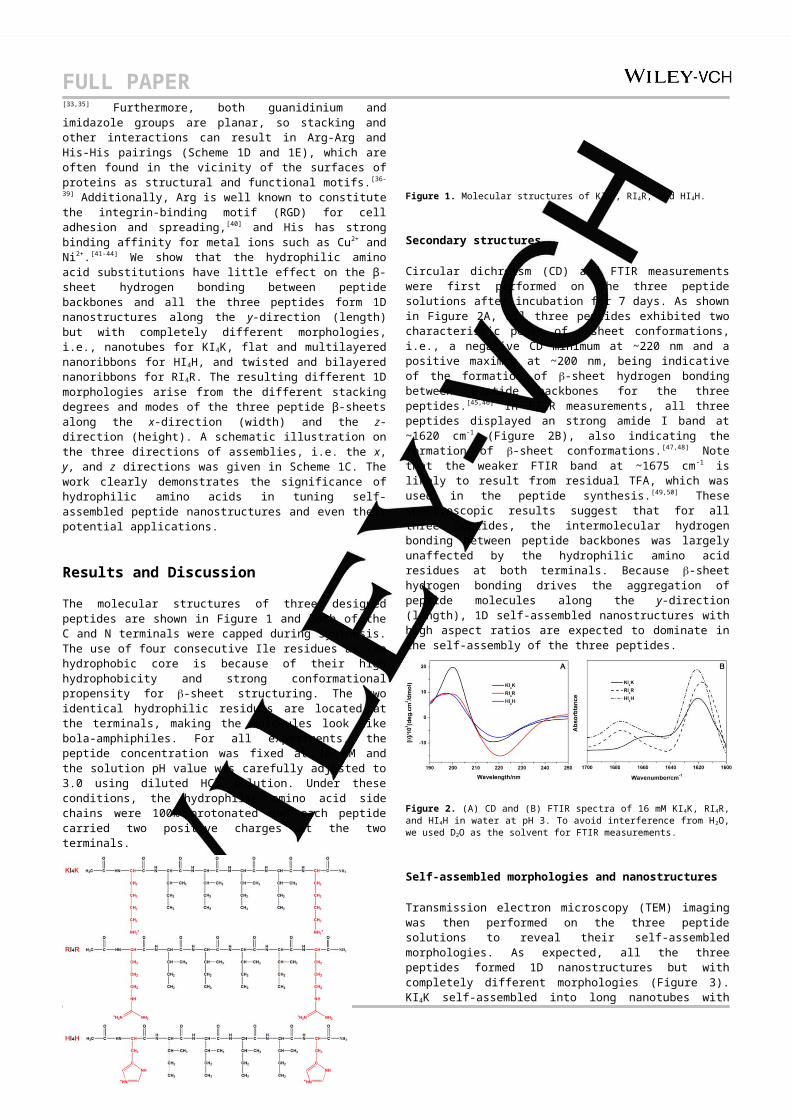
FULL PAPERbetween peptide backbones for the three peptides.[45,46] In FTIR measurements, all three peptides displayed an strong amide I band at ~1620 cm-1 (Figure 2B), also indicating the formation of -sheet conformations.[47,48] Note that the weaker FTIR band at ~1675 cm-1 is likely to result from residual TFA, which was used in the peptide synthesis.[49,50] These spectroscopic results suggest that for all three peptides, the intermolecular hydrogen bonding between peptide backbones was largely unaffected by the hydrophilic amino acid residues at both terminals. Because -sheet hydrogen bonding drives the aggregation of peptide molecules along the y-direction (length), 1D self-assembled nanostructures with high aspect ratios are expected to dominate in the self-assembly of the three peptides.
Figure 2. (A) CD and (B) FTIR spectra of 16 mM KI4K, RI4R, and HI4H in water at pH 3. To avoid interference from H2O, we used D2O as the solvent for FTIR measurements.
Self-assembled morphologies and nanostructures
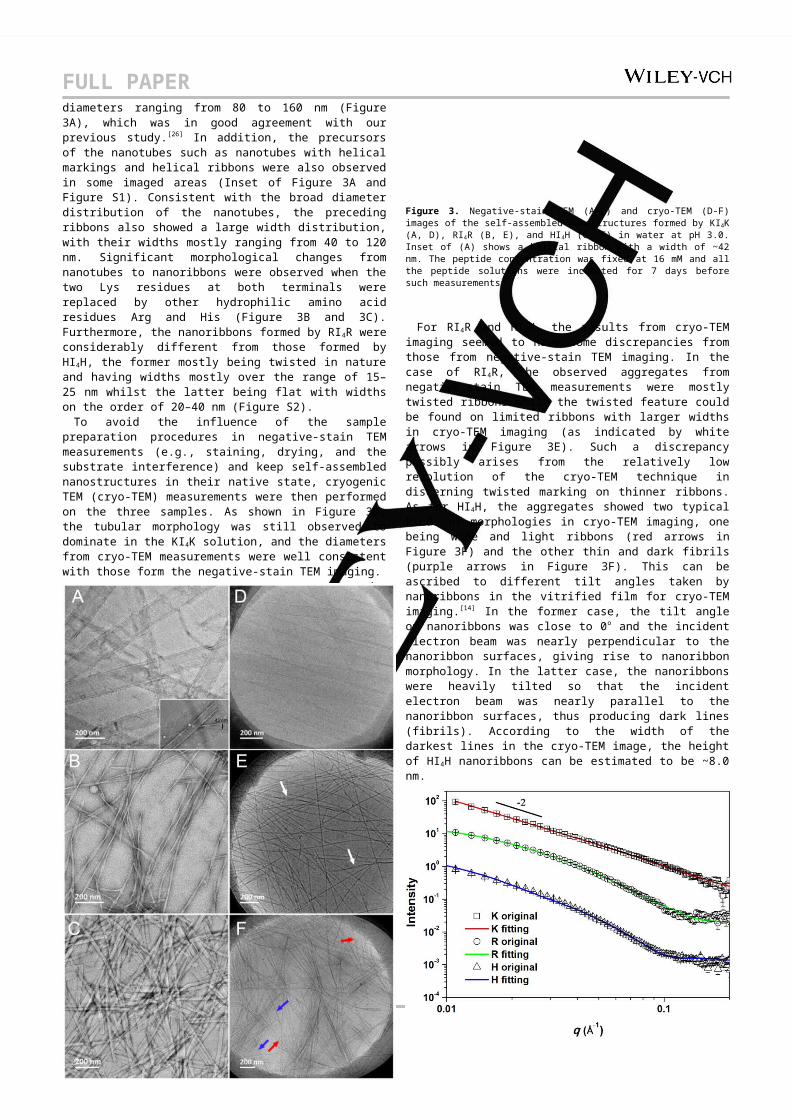
Transmission electron microscopy (TEM) imaging was then performed on the three peptide solutions to reveal their self-assembled morphologies. As expected, all the three peptides formed 1D nanostructures but with completely different morphologies (Figure 3). KI4K self-assembled into long nanotubes with diameters ranging from 80 to 160 nm (Figure 3A), which was in good agreement with our previous study. [26] In addition, the precursors of the nanotubes such as nanotubes with helical markings and helical ribbons were also observed in some imaged areas (Inset of Figure 3A and Figure S1). Consistent with the broad diameter distribution of the nanotubes, the preceding ribbons also showed a large width distribution, with their widths mostly ranging from 40 to 120 nm. Significant morphological changes from nanotubes to nanoribbons were observed when the two Lys residues at both terminals were replaced by other hydrophilic amino acid residues Arg and His (Figure 3B and 3C). Furthermore, the nanoribbons formed by RI4R were considerably different from those formed by HI4H, the former mostly being twisted in nature and having widths mostly over the range of 15–25 nm whilst the latter being flat with widths on the order of 20–40 nm (Figure S2).
To avoid the influence of the sample preparation procedures in negative-stain TEM measurements (e.g., staining, drying, and the substrate interference) and keep self-assembled nanostructures in their native state, cryogenic TEM (cryo-TEM) measurements were then performed on the three samples. As shown in Figure 3D, the tubular morphology was still observed to dominate in the KI4K solution, and the diameters from cryo-TEM measurements were well consistent with those form the negative-stain TEM imaging.
Figure 3. Negative-stain TEM (A-C) and cryo-TEM (D-F) images of the self-assembled nanostructures formed by KI4K (A, D), RI4R (B, E), and HI4H (C, F) in water at pH 3.0. Inset of (A) shows a helical ribbon with a width of ~42 nm. The peptide concentration was fixed at 16 mM and all the peptide solutions were incubated for 7 days before such measurements.
For RI4R and HI4H, the results from cryo-TEM imaging seemed to have some discrepancies from those from negative-stain TEM imaging. In the case of RI4R, the observed aggregates from negative-stain TEM measurements were mostly twisted ribbons while the twisted feature could be found on limited ribbons with larger widths in cryo-TEM imaging (as indicated by white arrows in Figure 3E). Such a discrepancy possibly arises from the relatively low resolution of the cryo-TEM technique in discerning twisted marking on thinner ribbons. As for HI4H, the aggregates showed two typical kinds of morphologies in cryo-TEM imaging, one being wide and light ribbons (red arrows in Figure 3F) and the other thin and dark fibrils (purple arrows in Figure 3F). This can be ascribed to different tilt angles taken by nanoribbons in the vitrified film for cryo-TEM imaging.[14] In the former case, the tilt angle of nanoribbons was close to 0o and the incident electron beam was nearly perpendicular to the nanoribbon surfaces, giving rise to nanoribbon morphology. In the latter case, the nanoribbons were heavily tilted so that the incident electron beam was nearly parallel to the nanoribbon surfaces, thus producing dark lines (fibrils). According to the width of the darkest lines in the cryo-TEM image, the height of HI4H nanoribbons can be estimated to be ~8.0 nm.
FULL PAPER
Figure 4. SANS profiles of 16mM KI4K (□), RI4R (○), and HI4H ( ) in D△ 2O after incubation for 7 days at pH 3.0. The solid lines are the best fits to the models described in the text. For clarity, data for KI4K has been vertically displaced by a factor of ×101, while that of HI4H has been vertically displaced by a factor of ×10-1.5. Error bars have been added to the original data for all the three peptides.
The structural details of the above 1D nanostructures at shorter length scales was further assessed by small angle neutron scattering (SANS) measurements. For all the three peptides, their SANS intensity (I) showed an approximate -2 dependence on wave vector (q) at the low q range (<0.03) (Figure 4), suggesting that the fundamental unit of these peptide assemblies was similar, i.e., all being lamellar structures, [14,26,51,52]
According to the theoretical model of peptide self-assembly proposed by Aggeli and co-workers,[53-55] the basic unit of different 1D peptide nanoaggregates such as ribbons and fibrils,[53-55] and even nanotubes,[26] can be regarded as lamellae. In spite of this structural similarity, the hydrophilic amino acid substitutions were found to have impact on the SANS profiles. At the higher q range (>0.035), the profiles of RI4R and HI4H assumed different shapes from that of KI4K (Figure 4), suggesting their different final self-assembled architectures. Our previous study has indicated that a hollow cylinder model plus a lamellar model fitted the profile of KI4K well, producing an outer diameter of ~90 nm and a wall thickness of ~2.0 nm for the hollow cylinders and a lamellar thickness of ~1.9 nm,[26] as shown in Table 1. Here, a flexible cylinder with elliptical cross-section model fits the SANS profiles of RI4R and HI4H well, producing an average width of ~20 and ~30 nm, respectively,
and an average height of ~4.1 and ~6.1 nm, respectively (Table 1). The fitted architectures and key structural parameters were broadly consistent with those from TEM measurements. Because the lamellar thickness of 1.9 nm from the fitting for KI4K is very close to the extended molecular length of KI4K (~2.0 nm), lamellar structures virtually correspond to β-sheet peptide monolayers with no or just one residue shift.[26,27] Note that there was little difference in the free energy between antiparallel and parallel β-sheet structures due to the symmetric molecular geometry for this kind of bola-like peptide amphiphiles.[27] Driven by the interactions primarily from amino acid side chains of β-sheets, lamellae consisting of β-sheets can undergo higher
ordered self-assembly along the x-direction (width) and/or the z-direction (height) to form different architectures. The self-assembled 1D architectures from the three peptides showed different widths and heights or thicknesses, suggesting they have different aggregation pathways from the initial β-sheets. The larger widths for the KI4K nanotubes indicate significant lateral association of β-sheets during the KI4K self-assembly while the wall thickness of ~2.0 nm suggests no growth along the height direction. The heights of RI4R and HI4H ribbons were nearly integer multiples of 2.0 nm, being indicative of some stacking of β-sheet layers along the height direction, in addition to their lateral association along the width direction.Table 1. Structural Parameters of the Self-Assembled Peptide Nanostructures from TEM, SANS, and AFM Measurements.
The above morphological features and key structural parameters were further confirmed by atomic force microscopy (AFM) measurements (Figure 5). As shown in the height and phase images (Figures 5A and 5G), the tubular feature was evident for the KI4K aggregates, with very clear openings at the two ends of nanotubes. In addition, we also observed the structural transition from helical ribbons to nanotubes or vice versa (green arrows in Figure 5A). Although the contour of the AFM tip readily causes the measured widths to be overestimated, the measured heights are less affected by the
Peptide
Architectures
Width (W) or Diameter (D) (nm)
Height (H) or Wall Thickness (WT) (nm)
Negative-TEM
SANS SANS AFMCryo-TEM
KI4K
Nanotubes & Helical Ribbons
80–160 (D)40–120 (W)
90±10 (D)
2.0±0.1 (WT)1.9±0.1 (H)
2.2±0.1 (WT & H)
/
RI4RTwisted Ribbons
15–25 (W)
20±5 (W)
4.1±0.2 (H)
4.0±0.2 (H)
/
HI4HFlat Ribbons
20–40 (W)
30±5 (W)
6.1±0.2 (H)
(4.2/6.1/8.2)±0.2 (H)
8.0±2.0

FULL PAPER
Figure 5. AFM height images, the corresponding cross-sectional profiles, and the phase images of the assemblies formed by (A, D, G) KI 4K, (B, E, H) RI4R, and (C, F, I) HI4H in water at pH 3.0. The peptide concentration was fixed at 16mM and all the solutions were incubated for 7 days before such measurements.
tip’s convolution with the sample and thus yield more reliable AFM measurements. The AFM cross-sectional profiling revealed a height of ~4.4 nm for the KI4K nanotubes and a height of ~2.2 nm for the KI4K nanoribbons (Figure 5D), indicating a complete flattening of the KI4K nanotubes on the substrate. The wall thickness from AFM was thus consistent with that from SANS, confirming that the tubular wall corresponds to the KI4K β-sheet monolayer, which arises from the lateral association of KI4K β-sheets. As shown in Figures 5B and 5H, the twisted feature of the RI4R nanoribbons was evident. The RI4R nanoribbons are larger in width relative to many narrow twisted nanofibers, so their twisted feature makes them look like “strings of sausages”. Furthermore, the cross-sectional analysis indicated a uniform height of ~4.0 nm nearly for all the nanoribbons (Figure 5E), well consistent with that from SANS, all suggesting that the RI4R nanoribbons correspond to a peptide bilayer, arising from the stacking of RI4R β-sheet monolayers along the z-direction (height). In the case of HI4H, the cross-sectional analysis revealed heights of ~4.2, ~6.1, and ~8.2 nm (Figure 5F), indicating that the HI4H nanoribbons are multilayered (2–4 layered) or multiple β-sheet stacks along the z-direction (height). These heights were very consistent with that (~6.1 nm) from SANS, because only an average height could be deduced from the fitting of SANS data based on the flexible cylinder with elliptical cross-section model. In addition, the phase image showed a terrace structure (as shown by the white arrow in Figure 5I), and the cross-sectional analysis indicated a height difference of ~2.1 nm associated with the terrace (white arrow in Figure 5F).
Microscopic effects of the hydrophilic substitutions on -sheets
The three peptide amphiphiles have an identical hydrophobic core consisting of four consecutive Ile residues, which endows them with a strong propensity for β-sheet structuring and thus favors the self-assembly of 1D nanostructures, as demonstrated by CD, FTIR and TEM measurements. In the following MD
simulations, three -sheets were constructed, each consisting of six strands that were parallel to neighboring strands, and the number of hydrogen bonds between strands within a -sheet and that between the hydrophilic side chains of a -sheet and water molecules were followed. Because of the symmetric molecular geometry for this kind of bola-like peptide amphiphiles, there was little difference in the free energy between antiparallel and parallel β-sheet structures.[27] As shown in Figure 6A, the number of hydrogen bonds between strands remained constant during 10 ns simulations for each -sheet. Furthermore, there was little variation when the two Lys residues were replaced by two Arg or His residues. These microscopic results indicated again that the terminal hydrophilic amino acid substitutions had little effect on the formation of -sheets, well consistent with the above experimental results.
Figure 6. The number of hydrogen bonds between A) strands within a peptide sheet containing 6 strands and B) the hydrophilic side chains of the sheet and water molecules.
In spite of producing little effect on the inter-strand hydrogen bonding, the hydrophilic amino acid substitutions at both terminals are most likely to change the hydrogen bonding with solvent molecules and induce additional interactions and steric hindrance upon aggregation, finally resulting in different self-assembled architectures as observed by experiments. As shown in Figure 6B, the number of hydrogen bonds between the hydrophilic side chains of -sheets and water molecules increased in the order of HI4H < KI4K < RI4R, suggesting an increase in the ability of -sheets to bind water, or in the wetting

FULL PAPERdegree of -sheets. In protein folding, the hydrophobic collapse propensity of surface is negatively related to the wetting degree.[56,57] Thus, an increase in the wetting degree of -sheets disfavors their stacking. Among the three peptide -sheets, the RI4R -sheet was revealed to bind the largest number of water molecules via hydrogen bonding, well consistent with the extremely high ability of Arg to form hydrogen bonding with water molecules.[33,34] As a result, the RI4R -sheet was more easily wetted with water, and the further stacking of RI4R -sheets would be limited, finally leading to the formation of bilayered nanoribbons with thinner widths (15–25 nm), as indicated in Table 1. Because the stacking degree of RI4R -sheets was not large enough to untwist -sheets from their natural states, we observed twisted nanoribbons for the RI4R self-assembly in experiments, as shown in Figures 3B and 3E and Figures 5B and 5H. In contrast, the HI4H -sheet was found to bind the smallest number of water molecules, thus being less wetted with water. Therefore, we expect HI4H -sheets would undergo significant stacking during the subsequent hierarchical self-assembly and we observed multilayered (2–4 layered) nanoribbons with increased widths (20–40 nm). In this case, the stacking might be sufficiently large to untwist -sheets from their natural states, so we observed flat and thick nanoribbons in experiments, as shown in Figures 3C and 3F and Figures 5C, 5F, and 5I. As the precursors of the KI4K nanotubes, the KI4K nanoribbons showed much larger widths but they were monolayered, suggesting -sheet stacking only along the x-direction (width). Hence, these results virtually do not contradict with the intermediate ability of the KI4K -sheet to bind water molecules via hydrogen bonding. The much larger widths cause the KI4K nanoribbons (monolayers) to transform into the helical intermediates leading to the formation of the nanotubes, as shown in Figures 3A and 3D and Figures 5A and 5G.
Figure 7. Fluorescence emission spectra (excitation at 442 nm) of 50 µM ThT in the presence of 16 mM peptides: ThT+KI4K (solid line), ThT+RI4R (dash line), and ThT+HI4H (dot line). Note that there was no fluorescence contribution from ThT alone or the peptide alone in the indicated wavelength range.
ThT binding
Upon binding to -sheet peptide assemblies, ThT usually displays a significant increase in fluorescence emission intensity around 480 nm in aqueous media.[58-60] In the presence of the three peptides, fluorescence emission enhancements of ThT at around 480 nm were observed (Figure 7), indicating the formation of -sheet nanostructures. However, the fluorescent enhancement was the strongest in the presence of HI4H whilst it was the weakest in the presence of KI4K, suggesting different self-assembled nanostructures and different peptide packing modes within the assemblies. Tubular aggregates with -sheet conformations have been widely observed to induce much weaker fluorescence enhancement,[26,61] presumably due to their low molecular packing density relative to other -sheet peptide assemblies such as fibrils and stacked ribbons. Both RI4R and HI4H formed nanoribbons, but the nanoribbons formed by the latter were flat and well-extended with little twisting, thus favoring ThT docking along the -sheet surface.[59]
Proposed self-assembly mechanisms for the three peptides
For the symmetric peptide amphiphiles XI4X, the hydrophilic amino acids at both terminals were observed to produce little

FULL PAPER
Figure 8. Schematic illustration of the effects of the hydrophilic amino acids on the self-assembly processes of A) KI4K, B) RI4R, and C) HI4H. The hydrophilic amino acids at both terminals produce little effect on β-sheet hydrogen bonding between peptide backbones, i.e., all three peptide strands tend to form β-sheets. However, the hydrophilic amino acids have marked impact on the stacking of the β-sheets. KI 4K sheets only undergo lateral association to form wider and monolayered nanoribbons, which finally develop into helical ribbons and nanotubes. The stacking of RI 4R β-sheets is limited and it forms twisted and bilayered nanoribbons, in which Arg-Arg pairings dictate the stacking of RI4R β-sheet monolayers along the z-direction (height). In contrast, the stacking of HI4H -sheets is significant, it forms flat and multilayered nanoribbons, in which His-His pairings are responsible for the stacking of β-sheet monolayers along the z-direction (height). W1, W3, and W2 represent the widths of KI4K, HI4H, and RI4R nanoribbons, respectively, showing a decreasing order. H1, H2, and H3 represent the heights of KI4K, RI4R, and HI4H nanoribbons, respectively, showing an increasing order.
effect on β-sheet hydrogen bonding between peptide backbones and all of them formed 1D nanostructures but with completely different morphologies, i.e., nanotubes for KI4K, flat and multilayered nanoribbons for HI4H, and twisted and bilayered nanoribbons for RI4R. Table 1 summarizes their structural parameters determined from different techniques. Under our experimental conditions, all the hydrophilic residues were protonated. However, owing to the symmetric distribution of the two charged residues along the peptide backbone, the electrostatic repulsion between the charged side chains had little effect on the growth of β-strands along the y-direction (length) and the lateral association of β-sheets along the x-direction (width).[26] Due to the strong β-sheet hydrogen bonding and hydrophobic interactions from four consecutive Ile residues, the growth of aggregates along the two directions were significant for the three peptides. As a result, the 1D nanostructures formed from the three peptides were nanotubes and nanoribbons with relatively large widths, rather than narrower nanofibrils. These extended nanostructures form because growth along the y-direction (length) is much faster than that along the x-direction (width).[14]
Although the hydrophilic amino acids had little impact on the formation of β-sheets and their lateral association only in terms of the electrostatic repulsion, the differences in other properties of the hydrophilic amino acids such as affinities for solvent molecules, interactions and steric considerations may affect the stacking of β-sheets along the x- and even z-directions (width and height), eventually resulting in different 1D architectures. According to our simulation results, the RI4R -sheet could bind the largest number of water molecules via hydrogen bonding, followed by KI4K, and then HI4H (Figure 6B). Thus, RI4R sheets were more easily wetted with water molecules, and their further stacking would be limited, finally leading to the formation of nanoribbons with relatively thin widths. Furthermore, the guanidinium ion C(NH2)3
+ represents a special aromatic system, called “Y-aromaticity”.[62] As a derivative of the guanidinium cation, two protonated Args can form a pairing structure, i.e., Arg-Arg pairing, via cation-π and π-π stacking interactions. [36,37]
Upon lateral association of RI4R -sheets into monolayers, Args are distributed on the surfaces of the monolayers and exposed to aqueous environment. Because the Arg-Arg pairing prefers a polar environment,[36,37] the Arg-Arg pairings between RI4R sheet monolayers are expected to occur and can act as an adhesive force to promote the formation of layered nanoribbons. Also owing to the relatively high wetting degree of RI4R sheets, their
stacking along the z-direction (height) was rather limited and only bilayered nanoribbons were observed in experiments. Because the stacking degree of RI4R -sheets is not large enough to untwist -sheets from their natural states, the observed nanoribbons were twisted (Figures 3B and 3E and Figures 5B and 5H). The schematic illustration of the RI4R self-assembly is given in Figure 8B.
The HI4H -sheet was found to bind the smallest number of water molecules and was thus least wetted with water. Therefore, the stacking of HI4H -sheets was significant. In our experiments, we observed nanoribbons not only with increased widths but also with increased heights (multilayered structures) (Figures 5C, 5F, and 5I). The occurrence of the multilayered structures is also supposed to arise from the residue pairing, i.e., the His-His pairings between HI4H sheet monolayers. Bhattacharyya et al. have indicated that His-His pairing prefers to interact in a face-to-face orientation rather than a T-shaped orientation.[39] In addition, there seem to be more types of hydrogen bonding between His and His in addition to π-π stacking interactions, because the imidazole ring can act both as the hydrogen bond donor and acceptor. Owing to these features including the significant stacking of HI4H -sheets along the x- and z- directions (width and height), the closely packing mode of His-His pairings, and the versatile interactions between His and His, the HI4H nanoribbons were flat with little twisting. The HI4H self-assembly process is illustrated in Figure 8C.
Relative to the rather rigid and planar side chains of Arg and His, the side chain of Lys is flexible. Owing to a lack of π-π stacking interactions, it is unlikely to form the Lys-Lys pairing between KI4K -sheet monolayers in the self-assembly of KI4K. Additionally, the Lys side chain only contains one heteroatom while the His or Arg side chain contains two or three heteroatoms, so the intermolecular hydrogen bonding between Lys side chains is weaker than that between His or Arg side chains, also disfavoring the Lys-Lys pairing. As a result, KI4K β-sheets may only undergo significant lateral association to form monolayered nanoribbons. The KI4K monolayered nanoribbons exhibited much larger widths, although the KI4K -sheet showed a slightly higher ability to bind water than the HI4H sheet. When the widths of nanoribbons are much larger, helical nanoribbons and nanotubes are energetically favored, as indicated by Lynn et al.[63] Therefore, we observed nanotubes and helical nanoribbons for the KI4K self-assembly (Figures 3A and 3D and Figures 5A and 5G), with the schematic illustration of the KI4K self-assembly process given in Figure 8A.

FULL PAPERConclusions
For the bola-like peptide amphiphiles XI4X (X=hydrophilic amino acid residues), we have demonstrated that terminal hydrophilic amino acid substitutions had little effect on the β-sheet hydrogen bonding between peptide backbones and all of them self-assembled into 1D nanostructures. This can be ascribed to the strong intermolecular β-sheet hydrogen bonding and hydrophobic interactions from the hydrophobic core (four consecutive Ile residues) and the symmetric molecular geometry of the peptides. However, hydrophilic amino acid substitutions could significantly affect their self-assembled morphologies. Due to the higher wetting degree of the RI4R -sheet by solvent molecules, RI4R formed twisted and bilayered nanoribbons with widths of 15–25 nm, in which the stacking of RI4R -sheets was limited. The HI4H -sheet was least wetted with water molecules, so HI4H self-assembled into flat and multilayered (2–4) nanoribbons with widths of 20–40 nm, in which the stacking of HI4H -sheets was significant. For the self-assembly of RI4R and HI4H, the residue pairings, i.e., Arg-Arg pairing and His-His pairings were supposed to be responsible for the stacking of -sheet monolayers along the z-direction (height). In addition to stacking interactions, the versatile hydrogen bonding modes between His and His also contributed to the growth of assemblies along the z-direction (height). Owing to a lack of Lys-Lys pairings, there might be only lateral association of -sheets in the self-assembly, which was primarily driven by four hydrophobic amino acid side chains. As a result, KI4K self-assembled into nanoribbons with much larger widths of 40–120 nm, which helically coiled into helical nanoribbons and nanotubes.
Most amino acids have their own unique properties. By taking advantage of this feature, we clearly demonstrated that hydrophilic amino acid substitutions with the bola-like peptide amphiphiles produced different self-assembled 1D architectures, in which hydrophilic amino acids showed different packing modes and densities on the surfaces of the self-assembled nano-objects. Furthermore, different hydrophilic amino acids on the surface can endow the assemblies with distinctive surface chemistry, functions, and even potential applications, provided that these amino acids have their specific functional roles in many essential biological processes.
Experimental Section
Materials
All materials used for peptide synthesis including Fmoc protected amino acids, Rink amide MBHA resin, solvents, and other chemical reagents were obtained from GL Biochem Ltd. (Shanghai, China). The peptides KI4K, RI4R, and HI4H were synthesized on a CEM Liberty microwave synthesizer using the standard Fmoc solid phase synthesis strategy. Their C-termini were amidated using a Rink amide MBHA resin and N-termini were capped using acetic anhydride before cleavage from the resin. The detailed synthesis and purification procedures have been described in our previous work.[29] The high purity (>98%) of the final products was ascertained by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF) and reversed-phase high-performance liquid chromatograph (RP-HPLC) analyses.
Sample Preparation
The three peptides showed high solubility in water. For the sample preparation, the peptide fluffy powder was directly dissolved in Milli-Q water at a concentration of 16 mM. After sonication for ~5 min and further vortexing for ~ 20 sec, homogenous solutions were obtained. After slightly adjusting the solution pH to 3.0 with diluted HCl solution, the peptide solution was incubated for about 7 days at room temperature for further instrumental measurements.
Circular Dichroism (CD)
CD measurements were carried out on a MOS-450 spectrometer (Bio-logic, France) using a 0.1 mm path-length quartz cuvette. The spectra were recorded at room temperature with wavelengths ranging from 190 to 250 nm and a scan speed of 50 nm min -1. A Xe lamp was used as the light source and the bandwidth was set to 0.5 nm. Using the Bio-kine software package (Bio-logic), the solvent background was subtracted and the measured spectra smoothed. The presented CD signals are the average of at least six independent scans and are expressed as [θ] (deg·cm2·dmol-1) versus wavelength.
FTIR
FTIR measurements were performed in absorbance mode on a Nicolet 6700 FT-IR spectrometer equipped with a DGTS detector. A CaF2 cell with 0.1 mm spacer was used. In order to eliminate the interference of H2O to the amide I band, D2O was used as solvent in the sample preparation. For each measurement, the spectra were collected from 4000 to 400 cm-1 with the resolution of 4 cm-1 and 128 scans were performed to improve signal-to-noise. Using the OMINC software (ver. 3.0, Nicolet), the background was subtracted and the measured spectra smoothed.
Cryogenic Transmission Electron Microscope (cryo-TEM)
Samples for cryo-TEM characterizations were prepared in a controlled environment vitrification system (CEVS). The aged peptide solution with a small volume of about 5 μL was dropped onto the TEM copper grid coated with a laced support film and then wicked away with filter paper on both sides for about 3s. The resulting thin film suspending on the mesh holes was quickly plunged into a reservoir of liquid ethane (cooled by liquid nitrogen) at -165 oC. Vitrified samples were stored in liquid nitrogen before transferring to a sample holder (Gatan 626) and examined at about -174 oC on a JEOL JEM-1400 TEM with an accelerating voltage of 120 kV.
Negative-stain TEM
TEM micrographs were recorded on a JEOL JEM-2100 UHR electron microscope with an accelerating voltage of 200 kV. For TEM sample preparation, about 5 μL of the aged peptide solution was dropped onto a TEM copper grid coated with a carbon support film and allowed to adsorb for about 1 min before excess peptide solution was wiped away by filter paper. Then the grid was negatively stained with uranyl acetate (2%, w/v) aqueous solution for about 1 min and excess solution was removed with filter paper.
Atomic force microscopy (AFM)
AFM measurements were performed on a commercial Nanoscope IVa MultiMode AFM (Digital Instruments, Santa Barbara, CA) in tapping mode. Samples for AFM characterization were prepared by dropping a small volume (~10 μL) of the aged peptide solution onto a freshly cleaved mica surface and allowing it adsorb for about 30s before rinsing with water. The resulting surface was gently dried with nitrogen gas, followed by AFM imaging. The scan rate was set to 1.50 Hz with the scan angle at 0°. All images were flattened using a first-order line fit (the ‘flatten’

FULL PAPERfunction in the manufacturers software) to correct for piezo-derived differences between scan lines.
Small angle neutron scattering (SANS)
SANS experiments were performed on the LOQ diffractometer, ISIS Neutron Facility, Rutherford Appleton Laboratory (Oxford, UK). Samples used in this work were all prepared by directly dissolving the peptides in D2O and their pH values slightly adjusted to pD 3.0 using diluted DCl in D2O. After incubation for 7 days under ambient conditions, the resulting homogenous solutions were transferred into 2.0 mm path length disc-shaped fused silica cells (Hellma) for measurements. Incident neutron wavelengths ranging from 2.2 to 10.0 Å at 25 Hz were used. A 64×64 cm2 detector with 5 mm resolution at a distance of 4.05 m from the samples then yielded a wave vector (q) range from 0.008 to 0.24 Å-1. Data were corrected for the wavelength dependence of the measured sample transmission, the incident neutron distribution, and relative detector efficiencies using the Mantid framework[64] before subtracting the solvent background (D2O). Absolute intensity scaling was obtained using the scattering from a partially deuterated polystyrene calibrant. [65] Data fitting were performed using the SansView program (ver. 3.0.0).[66] Three models, including a lamellar model, a hollow cylinder model, and a flexible cylinder with elliptical cross-section model were found to fit the measured SANS profiles well. The details for the three models have been given in our previous studies.[26,67]
ThT binding assay
Fluorescence measurements were performed on a Horiba Jobin Yvon Fluoromax-4 spectrometer at ambient temperature. Thioflavin-T (ThT) was introduced into the aged peptide solutions (16 mM) at a final concentration of 0.05 mM. After mixing, the fluorescence emission spectra of ThT in the presence of peptides were collected from 450 to 550 nm with an excitation wavelength of 442 nm. The excitation and emission slits for all experiments were set to 5 and 2.5 nm, respectively.
Simulation Methods
The REMD simulation in our previous work has demonstrated that the parallel packing mode was the optimal configuration of KI4K during its self-assembly.[27] In this work, we constructed a -sheet consisting of six strands that are parallel to neighbouring strands. All the simulations on the three peptide -sheets were conducted using the GROMACS suite.[68]
The OPLS force field was used to model the peptides, [69-71] and the TIP4P model was used to model water molecules. [72] After the establishment of the initial configuration for the β-sheet of KI4K, the same structure for the hydrophobic core (four consecutive I residues) has been adopted in constructing the initial configuration for the β-sheet of HI4H or RI4R. This would help minimize the effect of initial configuration difference for the three peptides. Because the solution pH was about 3.0 in experiments, the protonated side chains for the two hydrophilic residues were used in our simulations and Cl- was added in simulation for neutralizing the system. The system first underwent an energy minimization process using the steepest decent optimization method. Then the solvent was relaxed for 0.5 ns in the NPT ensemble with peptides restricted. Finally, 10 ns simulation was conducted for each system with peptides released at a constant temperature of 300 K and a constant pressure of 1 atm. The van der Waals and electrostatic interactions were treated with the cutoff method and the particle-mesh-Ewald method, [73,74] respectively, and the cutoff of VDW interactions and electrostatic interactions in real space was set to be 1.2 nm. The time step in simulations was 1 fs and 1000 instantaneous conformations were evenly sampled for equilibrium simulations.
Acknowledgements
The work is supported by the National Science Foundation of China (21503275 and 21373270), the Fundamental Research Funds for the Central Universities (15CX02026A), and the National Science Foundation for Post-doctoral Scientists of China (2012M511555). We thank UK Engineering and Physical Sciences Research Council and Innovative UK for funding support. The ISIS Neutron Source and the STFC are acknowledged for the award of neutron beam time on LOQ. This work benefitted from the SansView software, originally developed by the DANSE project under NSF award DMR-0520547.
Keywords: short peptides • self-assembly • sequence variations • hydrophilic amino acids • nanostructures
[1] X. Zhao, S. Zhang, Chem. Soc. Rev. 2006, 35, 1105–1110.[2] E. Gazit, Chem. Soc. Rev. 2007, 36, 1263–1269.[3] H. Cui, M. J. Webber, S. I. Stupp, Biopolymers 2010, 94, 1–18.[4] I. W. Hamley, Soft Matter 2011, 7, 4122–4138.[5] R.V. Ulijn, A. M. Smith, Chem. Soc. Rev. 2008, 37, 664–675.[6] Y. Loo, S. Zhang, C. A. E. Hauser, Biotechnol. Adv. 2012, 30, 593–603.[7] X. Yan, P. Zhu, J. Li , Chem. Soc. Rev. 2010, 39, 1877−1890.[8] G. Fichman, E. Gazit, Acta Biomater. 2014, 10, 1671–1682.[9] T. C. Holmes, S. de Lacalle, X. Su, G. Liu, A. Rich, S. Zhang, Proc. Natl.
Acad. Sci. U. S. A. 2000, 97, 6728–6733.[10] V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough, R. V.
Ulijn, Adv. Mater. 2006, 18, 611–614.[11] L. C. Serpell, Biochim. Biophys. Acta 2000, 1502, 16–30.[12] J. D. Sipe, A. S. Cohen, J. Struct. Biol. 2000, 130, 88–98.[13] R. Wetzel, Acc. Chem. Res. 2006, 39, 671–679.[14] H. Cui, T. Muraoka, A. G. Cheetham, S. I. Stupp, Nano Lett. 2009, 9,
945–951.[15] Y. Hu, R. Lin, P. Zhang, J. Fern, A. G. Cheetham, K. Patel, R.
Schulman, C. Kan, H. Cui, ACS Nano 2016, 10 , 880–888.[16] P. Zhou, L. Deng, Y. Wang, J. R. Lu, H. Xu, J. Colloid Interf. Sci. 2016,
464, 219–228.[17] I. W. Hamley, Angew. Chem. 2014, 126, 6984–7000; Angew. Chem., Int.
Ed. 2014, 53, 6866–6881.[18] C. C. Cenker, P. H. H. Bomans, H. Friedrich, B. Dedeoglu, V. Aviyente, U.
Olsson, N. A. J. M. Sommerdijk, S. Bucak, Soft Matter 2012, 8, 7463–7470.
[19] S. Bucak, C. Cenker, I. Nasir, U. Olsson, M. Zackrisson, Langmuir 2009, 25, 4262–4265.
[20] M. J. Krysmann, V. Castelletto, J. E. McKendrick, L. A. Clifton, I. W. Hamley, Langmuir 2008, 24, 81588162.
[21] K. Lu, J. Jacob, P. Thiyagarajan, V. P. Conticello, D. G. Lynn, J. Am. Chem. Soc. 2003, 125, 6391–6393.
[22] H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota, J. D. Hartgerink, J. Am. Chem. Soc. 2007, 129, 1246812472.
[23] Q. Li, H. Ma, Y. Jia, J. Li, B. Zhu, Chem. Commun. 2015, 51, 7219–7221.[24] P. Zhu, X. Yan, Y. Su, Y. Yang, J. Li, Chem. Eur. J. 2010, 16, 3176 3183.[25] Y. Kuang, Y. Gao, J. Shi, J. Li, B. Xu, Chem. Commun. 2014, 50, 2772–
2774.[26] Y. Zhao, J. Wang, L. Deng, P. Zhou, S. Wang, Y. Wang, H. Xu, J. R. Lu,
Langmuir 2013, 29, 1345713464.[27] L. Deng, P. Zhou, Y. Zhao, Y. Wang, H. Xu, J. Phys. Chem. B 2014, 118,
1250112510.[28] H. Cui, A. G. Cheetham, E. Thomas Pashuck, S. I. Stupp, J. Am. Chem.
Soc. 2014, 136 , 12461–12468.[29] S. Han, S. Cao, Y. Wang, J. Wang, D. Xia, H. Xu, X. Zhao, J. R. Lu,
Chem. Eur. J. 2011, 17, 1309513102.[30] Y. Liang, S.V. Pingali, A. S. Jogalekar, J. P. Snyder, P. Thiyagarajan, D.
G. Lynn, Biochemistry 2008, 47, 10018–10026.[31] J. A. Lehrman, H. Cui, W.-W. Tsai, T. J. Moyer, S. I. Stupp, Chem.
Commun. 2012, 48, 9711–9713.[32] F. T. Senguen, N. R. Lee, X. Gu, D. M. Ryan, T. M. Doran, E. A.
Anderson, B. L. Nilsson. Mol. BioSyst. 2011, 7, 486–496.

FULL PAPER[33] R. Wolfenden, L. Andersson, P. M. Cullis, C. C. B. Southgate,
Biochemistry 1981, 20, 849–855.[34] C. L. Borders, J. A. Broadwater, P. A. Bekeny, J. E. Salmon, A. S. Lee, A.
M. Eldridge, V. B. Pett, Protein Sci. 1994, 3, 541–548.[35] C. H. Gӧrbitz, Acta Cryst. 1989, B45, 390–395.[36] Z. Zhang, Z. Xu, Z. Yang, Y. Liu, J. Wang, Q. Shao, S. Li, Y. Lu, W. Zhu,
J. Phys. Chem. B 2013, 117, 4827–4835.[37] C. V. Sumowski, B. B. T. Schmitt, S. Schweizer, C. Ochsenfeld, Angew.
Chem. 2010, 122, 10147–10151; Angew. Chem., Int. Ed. 2010, 49, 9951–9955.
[38] S. Marsili, R. Chelli, V. Schettino, P. Procacci, Phys. Chem. Chem. Phys. 2008, 10, 2673–2685.
[39] R. Bhattacharyya, R. P. Saha, U. Samanta, P. Chakrabarti, J. Proteome Res. 2003, 2, 255–263.
[40] S. E. D. Souza, M. H. Ginsberg, E. F. Plow, Trends Biochem. Sci. 1991, 16, 246–250.
[41] T. Miura, K. Suzuki, N. Kohata , H. Takeuchi, Biochemistry 2000, 39, 7024-7031.
[42]J. Dong, J. M. Canfield, A. K. Mehta, J. E. Shokes, B. Tian, W. S. Childers, J. A. Simmons, Z. Mao, R. A.Scott, K. Warncke , D. G. Lynn, Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 13313–13318.
[43]T. Tanaka, T. Mizuno, S. Fukui, H. Hiroaki, J. Oku, K. Kanaori, K. Tajima , M. Shirakawa, J. Am. Chem. Soc. 2004, 126, 14023–14028.
[44]A. Fragoso, P. Lamosa, R. Delgado, O. Iranzo, Chem. Eur. J. 2013, 19, 2076–2088.
[45] S. Brahms, J. Brahms, J. Mol. Biol. 1980, 138, 149–178.[46] N. J. Greenfield, Anal. Biochem. 1996, 235, 1–10.[47] N. Yamada, K. Ariga, M. Naito, K. Matsubara, E. Koyama, J. Am. Chem.
Soc. 1998, 120, 12192–12199.[48] S. Ganesh, S. Prakash, R. Jayakumar, Biopolymers 2003, 70, 346–354.[49] S. Williams, T. P. Causgrove, R. Gilmanshin, K. S. Fang, R. H. Callender,
W. H. Woodruff, R. B. Dyer, Biochemistry 1996, 35, 691–697.[50] J. Madine, H. A. Davies, C. Shaw, I. W. Hamley, D. A. Middleton, Chem.
Commun. 2012, 48, 2976–2978.[51] L. Ziserman, H. Y. Lee, S. R. Raghavan, A. Mor, D. Danino, J. Am. Chem.
Soc. 2011, 133, 2511–2517.[52] M. P. Nieh, N. Kučerka, J. Katsaras, Methods. Enzymol. 2009, 465, 3–20.[53] I. A. Nyrkova, A. N. Semenov, A. Aggeli, N. Boden, Eur. Phys. J. B 2000,
17, 481–497.
[54] I. A. Nyrkova, A. N. Semenov, A. Aggeli, M. Bell, N. Boden, T. C. B. McLeish, Eur. Phys. J. B 2000, 17, 499–513.
[55] A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov, N. Boden, Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 11857–11862.
[56] A. J. Patel, P. Varilly, S. N. Jamadagni, H. Acharya, S. Garde, D. Chandler, Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 17678–17683.
[57] A. J. Patel, P. Varilly, S. N. Jamadagni, M. F. Hagan, D. Chandler, S. Garde, J. Phys. Chem. B 2012, 116, 2498–2503.
[58] H. Levine, Protein Sci. 1993, 2, 404–410.[59] M. Biancalana, K. Makabe, A. Koide, S. Koide, J. Mol. Biol. 2009, 385,
1052–1063.[60] R. Khurana, C. Coleman, C. Ionescu-Zanetti, S. A. Carter, V. Krishna, R.
K. Grover, R. Roy, S. Singh, J. Struct. Biol. 2005, 151, 229–238.[61] H. Shao, J. R. Parquette, Angew. Chem. 2009, 121, 2563–2566; Angew.
Chem., Int. Ed. 2009, 48, 2525–2528.[62] A. M. Sapse, L. J. Massa, J. Org. Chem. 1980, 45, 719–721.[63] W. S. Childers, N. R. Anthony, A. K. Mehta, K. M. Berland, D. G. Lynn,
Langmuir 2012, 28, 6386−6395.[64] http://www.mantidproject.org.[65] G. D. Wignall, F. S. Bates, J. Appl. Cryst. 1987, 20, 28–40.[66] http://www.sasview.org.[67] Y. Zhao, L. Deng, J. Wang, H. Xu, J. R. Lu. Langmuir 2015, 31, 12975–
12983.[68] D. Van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark, H. J. C.
Berendsen, J. Comput. Chem. 2005, 26, 17011718.[69] W. L. Jorgensen, D. S. Maxwell, J. Tirado-Rives, J. Am. Chem. Soc.
1996, 118, 1122511236.[70] W. L. Jorgensen, J. Tirado-Rives, Proc. Natl. Acad. Sci. U. S. A. 2005,
102, 66656670.[71] C. Caleman, P. J. van Maaren, M. Y. Hong, J. S. Hub, L. T. Costa, D. van
der Spoel, J. Chem. Theory Comput. 2012, 8, 6174.[72] W. L. Jorgensen, L. William, J. Chandrasekhar, J. D. Madura, R. W.
Impey, M. L. Klein, J. Chem. Phys. 1983, 79, 926–935.[73] T. Darden, D. York, L. Pedersen, J. Chem. Phys. 1993, 98, 10089–10092.[74] U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee, L. G.
Pedersen, J. Chem. Phys. 1995, 103, 8577–8593.

FULL PAPER
Entry for the Table of Contents
FULL PAPER
Tuning 1D nanostructures: Hydrophilic amino acid residues help define the surface topologies and morphological features of various peptide and protein aggregates. For the bola-like peptide amphiphiles XI4X, the hydrophilic amino acid substitutions have been demonstrated to produce little impact on the β-sheet hydrogen bonding between strands but significant effects on the staking degrees and modes of β-sheets along the width and height directions, finally resulting in completely different 1D nanostructures.
Y. Zhao, L. Deng, W. Yang, D. Wang, E. Pambou, Z. Lu, Z. Li, J. Wang, S. King, S. Rogers, H. Xu,* J. R. Lu*
Page No. – Page No.
Tuning One-Dimensional Nanostructures of Bola-Like Peptide
Amphiphiles via Varying the Hydrophilic Amino Acids