title page anti-inflammatory mechanism of compound k in...
TRANSCRIPT

JPET #189035
1
Title Page
Anti-inflammatory mechanism of compound K in activated microglia
and its neuroprotective effect on experimental stroke in mice
Jin-Sun Park, Jin A. Shin, Ji-Sun Jung, Jin-Won Hyun, Thi Kim Van Le,
Dong-Hyun Kim, Eun-Mi Park, Hee-Sun Kim
Department of Molecular Medicine and Tissue Injury Defense Research Center, Ewha
Womans University Medical School, Seoul, Republic of Korea (J.-S.P., J.-S.J., H.-S.K.),
Department of Pharmacology, Ewha Womans University Medical School, Seoul,
Republic of Korea (J.A.S., E.-M.P.), Department of Biochemistry, College of Medicine,
Cheju National University, Jeju, Republic of Korea (J.-W.H), Department of Life and
Nanopharmaceutical Sciences, College of Pharmacy, Kyung Hee University, Seoul,
Republic of Korea (T.K.V. L, D.-H.K.)
JPET Fast Forward. Published on December 29, 2011 as DOI:10.1124/jpet.111.189035
Copyright 2011 by the American Society for Pharmacology and Experimental Therapeutics.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
2
Running Title Page
Running title: Anti-inflammatory and neuroprotective effects of compound K
Corresponding Author:
Hee-Sun Kim
Department of Molecular Medicine, Ewha Womans University Medical School, Mok-6-
dong 911-1, Yangchun-Ku, Seoul 158-710, Korea
Tel: 82-2-2650-5823
Fax: 82-2-2653-8891
Email: [email protected]
Text pages: 32
Tables: 0
Figures: 7
References: 42
Abstract: 197 words
Introduction: 393 words
Discussion: 1200 words
Abbreviations:
AP-1, activator protein-1; ARE, antioxidant response element; CRE, cyclic-AMP
responsive element; CREB, CRE-binding protein; ERK, extracellular signal-regulated
kinase; GR, glucocorticoid receptor; HO-1, heme oxygenase-1; IL-6, interleukin-6;
iNOS, inducible nitric oxide synthase; JNK, c-Jun N-terminal kinase; LPS,
lipopolysaccharide; MAPK, mitogen-activated protein kinase; MCP, monocyte
chemotactic protein-1; MMP, matrix metalloproteinase; NADPH, nicotinamide adenine
dinucleotide phosphate; NF-κB, nuclear factor-κB; Nrf2, nuclear factor E2-related
factor-2; ROS, reactive oxygen species; TNF, tumor necrosis factor
Section: Neuropharmacology
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
3
Abstract
Microglial activation plays a pivotal role in the pathogenesis of various neurologic
disorders, such as cerebral ischemia, Alzheimer’s disease and Parkinson’s disease. Thus,
controlling microglial activation is a promising therapeutic strategy for such brain
diseases. In the present study, we found that a ginseng saponin metabolite, compound K,
inhibited the expressions of inducible nitric oxide synthase, proinflammatory cytokines,
monocyte chemotactic protein-1, and matrix metalloproteinase-3 and -9 in
lipopolysaccharide (LPS)-stimulated BV2 microglial cells and primary cultured
microglia. Subsequent mechanistic studies revealed that compound K suppressed
microglial activation via inhibiting reactive oxygen species, mitogen-activated protein
kinases, and NF-κB/AP-1 activities with enhancement of HO-1/ARE signaling. To
address the anti-inflammatory effects of compound K in vivo, we used two brain disease
models of mice: sepsis (systemic inflammation) and cerebral ischemia. Compound K
reduced the number of Iba1-positive activated microglia and inhibited the expressions
of tumor necrosis factor-alpha and interleukin-1 beta in the LPS-induced sepsis brain.
Furthermore, compound K reduced the infarct volume of ischemic brain induced by
middle cerebral artery occlusion, and suppressed microglial activation in the ischemic
cortex. The results collectively suggest that compound K is a promising agent for
prevention and/or treatment of cerebral ischemia and other neuroinflammatory disorders.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
4
Introduction
Microglia are major immune cells in the central nervous system, which are readily
activated following brain injury or during neurodegenerative processes, and secrete
growth factors, pro-/anti-inflammatory cytokines, reactive oxygen species (ROS), nitric
oxide (NO), and glutamate (Block and Hong, 2005; Stolp and Dziegielewska, 2009).
While microglial activation is necessary and important for host defense, overactivation
of microglia is neurotoxic. Microglia are also activated after ischemic stroke and
produce cytokines, triggering neuronal death in response to ischemic injury (Wang et al.,
2007). Within 3 days of a stroke, various inflammatory molecules are concomitantly up-
regulated in the brain, cerebrospinal fluid (CSF), and blood, and thus continuous brain
loss is expected during that time. If inflammation can be suppressed, progressive brain
loss following a stroke may be prevented and the clinical outcome improved (Wang et
al., 2007). Thus, development of agents that reduce microglial activation and their
proinflammatory responses are considered to be an important therapeutic strategy for
neuroinflammatory disorders such as cerebral ischemia, Alzheimer’s disease, and
Parkinson’s disease (Block and Hong, 2005; Stolp and Dziegielewska, 2009; Wang et al.,
2007).
Compound K (20-O-D-glucopyranosyl-20(S)-protopanaxadiol) is one of the major
metabolites of ginseng, which are formed by the intestinal bacteria after oral
administration of ginseng extract in humans and rats. The ginseng saponin metabolite,
compound K, is absorbed from the gastrointestinal tract to the blood (Akao et al., 1998).
Compound K has a variety of pharmacological activities, including anti-tumor, anti-
diabetic, anti-allergic and anti-inflammatory effects (Jia et al., 2009; Radad et al., 2010).
Our group has recently reported that compound K suppresses glioma invasion via
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
5
inhibition of MMP-9 expression (Jung et al., 2006). Compound K also suppressed
inflammation in colitic mice via inhibition of interleukin-1 receptor-associated kinase-1
(IRAK-1) activation (Joh et al., 2011).
We have previously reported that ginseng extracts and total saponins exert anti-
inflammatory effects in lipopolysaccharide (LPS)- and/or β-amyloid-stimulated
microglial cells (Park et al., 2009). Among the individual ginsenosides tested,
compound K suppressed LPS-induced NO production. However, the anti-inflammatory
effects of compound K in activated microglia and its’ underlying molecular mechanisms
have not been clearly demonstrated. In the present study, we examined the effects of
compound K on various inflammatory molecules in LPS-stimulated microglial cells and
analyzed the detail molecular mechanisms. Subsequently, we demonstrated the anti-
inflammatory and/or neuroprotective effects of compound K in brain disease models of
mice such as sepsis (systemic inflammation) and cerebral ischemia.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
6
Methods
Reagents
Compound K (20-O-D-glucopyranosyl-20(S)-protopanaxadiol) was prepared as
previously described (Joh et al., 2011). In brief, protopanaxadiol-type ginsenosides were
incubated with Bacteroides JY-6, a human intestinal bacterium in a general anaerobic
medium for 24 h at 37ºC. The incubated medium was extracted with BuOH. The
supernatant was concentrated in vacuum and was processed using silica gel column
chromatography with CHCl3-MeOH-H2O (65:35:10 [v/v]). The isolated compound K
was characterized by mass spectroscopy and 1H-and 13C-nuclear magnetic resonance
(NMR) spectrometry. The chemical structure of compound K is shown in Fig. 1. All
reagents used for cell culture were purchased from Gibco BRL (Grand Island, NY,
USA). Antibodies against MAP kinases or HO-1 were purchased form Cell Signaling
Technology (Beverly, MA, USA). All other chemicals were obtained from Sigma-
Aldrich (St. Louis, MO, USA), unless otherwise stated.
Microglial cell cultures
Immortalized murine BV2 microglial cells (Bocchni et al., 1992) were grown and
maintained in Dulbecco’s modified Eagle’s medium supplemented with 10 % heat-
inactivated FBS, streptomycin (10 μg/ml), and penicillin (10 U/ml) at 37°C. Primary
microglial cells were cultured from the cerebral cortices of 1-2-day-old Sprague-Dawley
rat pups, as described previously (Park et al., 2009). The purity of microglial cultures
was > 95%, which was determined by isolectin B4 staining (data not shown).
Measurement of cytokine, nitrite, and intracellular ROS levels
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
7
Microglial cells (1 x 105 cells per well in a 24-well plate) were pre-treated with
compound K (25, 50, 75 μM) for 30 min and stimulated with LPS (0.1 μg /ml). The
supernatants of the cultured microglia were collected 24 h after LPS stimulation and the
concentrations of TNF-α and IL-1β were measured by an enzyme-linked
immunosorbent assay (ELISA). Accumulated nitrite was measured in the cell
supernatant using the Griess reagent (Promega, Madison, WI, USA). The intracellular
accumulation of ROS was measured with H2DCF-DA (Sigma-Aldrich) by modifying a
previously reported method (Qin et al., 2005).
RT-PCR
BV2 cells (7.5 ×105 cells on a 6-cm dish) and rat primary microglia (7 × 106 cells on
a 6-cm dish) were treated with LPS in the presence of compound K (25, 50, 75 μM) and
total RNA was extracted with TRI reagent (Sigma-Aldrich). For RT-PCR, total RNA (1
μg) was reverse-transcribed in a reaction mixture containing 1 U RNase inhibitor, 500
ng random primers, 3 mM MgCl2, 0.5 mM dNTP, 1 X RT buffer, and 10 U reverse
transcriptase (Promega). The synthesized cDNA was used as a template for the PCR
reaction using GoTaq polymerase (Promega) and primers, as below (Table).
Forward Primer (5’→3’) Reverse Primer (5’→3’) Size
iNOS CCCTTCCGAAGTTTCTGGCAGCAGC GCCTGTCAGAGCCTCGTGGCTTTGG 450 bp
TNF-α CCTATGTCTCAGCCTCTTCT CCTGGTATGAGATAGCAAAT 354 bp
IL-1β GGCAACTGTTCCTGAACTCAACTG CCATTGAGGTGGAGAGCTTTCAGC 447 bp
IL-6 CCACTTCACAAGTCGGAGGCTT CCAGCTTATCTGTTAGGAGA 395 bp
MCP-1 ACTGAAGCCAGCTCTCTCTTCCTC TTCCTTGGGGTCAGCACAGAC 276 bp
MMP-3 ATTCAGTCCCTCTATGGA CTCCAGTATTTGTCCTCTAC 375 bp
MMP-9 GTGATCCCCACTTACTATGGAAAC GAAGCCATACAGTTTATCCTGGTC 352 bp
GAPDH ATGTACGTAGCCATCCAGGC AGGAAGGAAGGCTGGAAGAG 420 bp
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
8
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts from treated microglia were prepared, as described previously
(Woo et al., 2003). The double-stranded DNA oligonucleotides containing the NF-κB,
AP-1, ARE, or CRE consensus sequences (Promega) were end-labeled by [γ-32P]ATP.
EMSA was performed using 30,000 to 50,000 cpm of labeled probe and nuclear
proteins (5 μg) in a final volume of 20 μl of 12.5% glycerol and (in mM): 12.5 HEPES
(pH 7.9), 4 Tris-HCl (pH 7.9), 60 KCl, 1 EDTA, and 1 DTT with 1 μg of poly(dI-dC) as
nonspecific competitor. The reaction was incubated at room temperature for 20 min.
The DNA-protein was resolved on high ionic strength, nondenaturing 6%
polyacrylamide gel followed by autoradiography with an intensifying screen. For the
supershift assay, antibodies to the p65 or p50 subunits of NF-κB (Santa Cruz, CA) were
co-incubated with the nuclear protein in the reaction mixture for 30 min at 4°C prior to
adding the radiolabeled probe.
Transient transfection and luciferase assay
Transfection of the reporter genes into BV2 cells was performed using
GeneporterTM 2 transfection reagent (Gene Therapy Systems, San Diego, CA, USA).
The NF-κB reporter plasmid contains three copies of the κB–binding sequence fused to
the firefly luciferase gene (Clontech, Mountain View, CA, USA). The ARE-luciferase
(ARE-Luc) reporter plasmid contains enhancer 2 (E2) and a minimal promoter sequence
of the mouse HO-1 gene fused to the luciferase gene (So et al., 2006). BV2 cells (2 X
105 cells per well in a 12-well plate) were transfected with 1 μg of the reporter construct
mixed with Geneporter. After 48 h, cells were harvested and luciferase assay was
performed, as previously described (Woo et al., 2003). To determine the effect of
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
9
compound K on reporter gene activity, cells were pre-treated with the agent before
treating with LPS (0.1 μg/ml) and incubated for 6 h prior to harvesting cells.
In vivo administration of compound K
Experiments were performed in male C57BL/6 mice (10-11 weeks old; Orient Bio
Inc., Seongnam, Republic of Korea). All experiments were performed in accordance
with the NIH and Ewha Womans University guidelines for Laboratory Animals Care
and Use, and the study was approved by the Institutional Animal Care and Use
Committee in the Medical School of Ewha Womans University. Compound K was
dissolved in 3% cremophore (Sigma-Aldrich) in normal saline. Mice were randomly
divided into control and treatment groups, and vehicle (3% cremophore in normal
saline) or compound K (30 mg/kg ip) was administered 4 days before LPS (5 mg/kg ip)
treatment or transient MCAO. The doses of LPS and compound K were based on a
previous study (Park et al., 2009).
Transient middle cerebral artery occlusion (MCAO)
Procedures for transient MCAO have been established and published previously
(Shin et al., 2009). Briefly, mice were anesthetized and a fiber optic probe was attached
to the right parietal bone (2 mm posterior and 5 mm lateral to the bregma) and
connected to a laser-Doppler flowmeter (Periflux System 5010; Perimed, Sweden).
Cerebral blood flow (CBF) was continuously recorded during MCAO and reperfusion
periods using a computer-based data acquisition system (Perisoft, Perimed). A 6-0
silicon-coated black monofilament surgical suture (Doccol Cooperation, Redlands, CA,
USA) was inserted into the exposed right external carotid artery, advanced into the
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
10
internal carotid artery, and wedged into the circle of Willis to obstruct the origin of the
MCA. The filament was left in place for 30 min, then withdrawn to re-establish CBF.
Only animals that exhibited a reduction in CBF >85% during MCA occlusion and in
which CBF recovered by >80% after 10 min of reperfusion were included in the study.
Infarct volume measurement
Infarct volume was measured according to procedures described previously (Lin et
al., 1993). Mice were sacrificed 3 days after MCAO, and brains were removed, frozen,
and sectioned (30 µm thick) using a cryostat. Brain sections were collected serially at
600-µm intervals, and stained with cresyl violet. Infarct volume was determined using
an image analyzer (Axiovision LE 4.1; Carl Zeiss, Jena, Germany). Values were
reported after correcting for post-ischemic swelling, as previously described (Lin et al.,
1993).
Immunohistochemistry
Three hour after LPS treatment or 24 h after MCAO, the animals were anesthetized
with sodium pentobarbital (120 mg/kg ip) and perfused transcardially with normal
saline containing heparin (5 U/ml), followed by 4% formaldehyde in 0.1 M sodium
phosphate buffer (pH 7.2). The brains were removed and incubated overnight in
fixatives and stored in a 30% sucrose solution. Serial coronal brain sections (20 µm
thick, at 600-µm intervals) were collected in a cryostat in regions containing the
striatum (between + 1.4 and - 1.0 mm from the bregma). Brain sections were incubated
in TBS containing 0.1% Triton X-100, 5% normal serum, and 1% bovine serum
albumin for 1 h, then subsequently incubated with Iba1 antibody (1:500; Wako Pure
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
11
Chemical Industries, Osaka, Japan). On the following day, the secondary antibody
conjugated to FITC (anti-rabbit, 1:200; Serotec, Raleigh, NC, USA) was applied to
sections for 1 h. TBS was used to wash sections between all steps. The sections were
mounted with Vectashield mounting medium (Vector Laboratories, Inc., Burlingame,
CA, USA), and fluorescence microcopy images were obtained (Axiovert 200; Carl
Zeiss). Densely-stained, round, Iba1-positive cells were quantified using the Axiovison
LE program (Carl Zeiss). Two serial brain sections from each animal were used, and
quantification was performed in three different areas (500 μm2 in size) in the lateral
cortex and striatum of the right hemisphere per brain section. The mean cell number
from six 500 μm2 areas per animal was calculated.
Western blot analysis
Whole cell protein lysates of BV2 cells were prepared in lysis buffer and protein
samples were separated by 12% SDS-PAGE and transferred to nitrocellulose
membranes (Amersham, Piscataway, NJ, USA). The membranes were blocked with 5%
skim milk in 10 mM Tris-HCl containing 150 mM NaCl and 0.5% Tween 20 (TBST),
then incubated with primary antibodies (1:1000) against phospho- or the total form of
MAP kinases, HO-1 (Cell Signaling Technology) or p-p47phox [anti-phospho-(Ser345)-
p47phox Ab, kindly provided by Dr. J.L. Benna (Universite Paris, France)] (Dang et al.,
2006). The cortical tissues from each animal were subjected to simultaneous Western
blot hybridization, as previously described (Shin et al., 2010a), and the protein-
transferred membrane was incubated overnight with antibodies against IL-1β or TNF-α
(1:200; R&D, Minneapolis, MN, USA). After thorough washing with TBST, HRP-
conjugated secondary antibodies (1:3000 dilution in TBST; New England Biolabs,
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
12
Beverly, MA, USA) were applied and the blots were developed using an enhanced
chemiluminescence detection kit (Pierce Biotechnology, Rockford, IL, USA).
Statistical Analysis
The data of in vitro and in vivo studies are expressed as the mean ± S.E.M.
Comparisons of in vivo data between two groups were analyzed with an unpaired
Student’s t-test. Multiple comparisons were evaluated with one-way analysis of variance
(ANOVA), followed by a post-hoc Fisher’s protected least significant difference
(PLSD) test with Statview (Statview version 5, SAS Institute Inc., Cary, NC, USA). For
in vitro data, statistical comparisons between groups were performed using one-way
ANOVA, followed by Student’s t-test. A p-value < 0.05 was considered significant.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
13
Results
Compound K inhibited iNOS, cytokines, MCP-1 and MMP-3/-9 expression in LPS-
stimulated microglial cells
To investigate the anti-inflammatory effects of compound K, BV2 cells and
primary microglia were treated with compound K for 30 min before stimulation with
LPS. As shown in Fig. 2A, compound K inhibited LPS-induced production of NO,
TNF-α, and IL-1β in a dose-dependent manner. Subsequent RT-PCR analysis revealed
that compound K suppressed iNOS, TNF-α, and IL-1β expression at the mRNA level
(Fig. 2B). Compound K also inhibited the expression of IL-6, MCP-1, MMP-3 and
MMP-9, which also play an important role in LPS-induced inflammatory reactions.
Compound K inhibited NF-κB and AP-1 activities, while it increased nuclear
protein binding to CRE
To investigate the anti-inflammatory mechanism of compound K, we examined the
effect of compound K on NF-κB and AP-1, which are important transcription factors
modulating cytokines, MMP/MCP-1, and iNOS gene expression in microglia (Lee and
Kim, 2009; Smale, 2010). As shown in Fig. 3, stimulation of BV2 cells with LPS
resulted in strong NF-κB and AP-1 bindings, which were significantly inhibited by
compound K. In addition, compound K inhibited NF-κB-mediated transcriptional
activity, as shown by the κB-luc reporter gene assay (Fig. 3B). Next, we examined the
effect of compound K on CREB, which is known to be involved in anti-inflammatory
mechanisms of activated microglia (Jung et al., 2010; Woo et al., 2003). As shown in
Fig. 3D, compound K significantly enhanced nuclear protein binding to CRE. The data
collectively suggest that compound K may inhibit the expression of various
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
14
inflammatory molecules by modulating NF-κB, AP-1, and CREB.
Compound K inhibits LPS-induced ROS production, phosphorylation of p47phox of
NADPH oxidase, and three types of MAP kinases
Excessive ROS generation by microglia contributes to the aggravation of neuronal
damage after a stroke (Sorce and Krause, 2009). In addition, ROS are known as second
messengers in inflammatory reactions (Sayre et al., 2008). In the present study,
compound K significantly attenuated LPS-induced ROS production in BV2 cells and
primary microglia (Figs.4A-B). Moreover, compound K suppressed the phosphorylation
of p47phox, a major component of NADPH oxidase (Nox2) responsible for microglial
ROS release (Fig. 4C) (Sorce and Krause, 2009). Thus, the antioxidant effect of
compound K may partly attribute to reduced NADPH oxidase activity. Furthermore,
compound K inhibited the LPS-induced phosphorylation of three types of MAPKs,
which are also important upstream signaling molecules in inflammatory reactions (Fig.
4D).
Compound K up-regulated HO-1 expression via ARE
Next, we examined the effect of compound K on the expression of HO-1, which is
known as a key molecule in the resolution of oxidative stress and inflammation (Min et
al., 2006). RT-PCR and Western blot analyses showed that compound K increased HO-1
expression at the mRNA and protein levels (Figs. 5A & B). Because the AREs on the
HO-1 promoter are critical for HO-1 transcription, we examined the effect of compound
K on nuclear protein binding to ARE. As shown in Fig. 5C, compound K significantly
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
15
increased the ARE-nuclear protein complex. Moreover, compound K increased ARE-
driven luciferase activity (Fig. 5D).
Compound K repressed inflammatory responses in the septic brain of mice
To determine whether compound K also reduced microglial activation in in vivo,
we measured the immunoreactivity of Iba1, which is a marker for activation of
microglia (Ito et al., 2001), in the mouse cortex and striatum 3 h after systemic
administration of LPS. In the brain of LPS-injected mice, the number of Iba1-positive
cells with a densely-stained round shape, indicative of activated cells, was increased
compared to controls. However, pre-treatment with compound K (30 mg/kg) reduced
the number of activated microglia, as seen following the quantification of activated
microglia both in the cortex and the striatum (p < 0.01 compared to vehicle; Fig. 6A). In
addition, compound K significantly reduced the expression of LPS-induced IL-1β and
TNF-α protein in the cortex 6 h after LPS injection (Figs. 6B).
Neuroprotective and anti-inflammatory effects of compound K on ischemic brain
injury in mice
Inflammation plays important roles in ischemic brain damage (Wang et al., 2007).
Based on above results, we further examined the therapeutic potential of compound K
on ischemic stroke induced by transient MCAO in mice. As shown in Fig. 7A,
pretreatment of compound K (30 mg/kg) significantly decreased the total infarct volume
compared to vehicle treatment (46% reduction, p = 0.012). The reduction in infarct
volume was prominent in the cortex (-56% compared to vehicle, p = 0.049), the area of
ischemic penumbra, but not in the striatum, which is the ischemic core. These findings
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
16
are consistent with the previous reports that the penumbra is salvageable area by
neuroprotective therapeutics (Ginsberg, 2003; Shin et al., 2009 and 2010b). Next, we
examined the effects of compound K on microglial activation in the ischemic brain by
using Iba1 antibody. Iba1 expression was assessed by immunofluorescence labeling in
the ipsilateral brain 24 h after MCAO. As shown in Fig. 7B, MCAO led to the
appearance of numerous densely-stained, activated microglial cells in the cortex and
striatum in vehicle-treated mice. Pre-treatment of compound K significantly reduced the
number of activated microglia in the cortex, but not in the striatum (Fig. 7B). The
results are correlated with the data in Fig. 7A, which show a reduction in infarct volume
in the cortex, but not in the striatum. The data suggest that microglial inactivation is at
least partly responsible for the neuroprotective effects of compound K against an
ischemic insult.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
17
Discussion
In the present study, we demonstrate the anti-inflammatory effects of compound K
in activated microglia in vitro and in vivo. Compound K suppressed the expression of
various inflammatory molecules in LPS-stimulated BV2 cells and primary microglia.
The anti-inflammatory effects of compound K were supported by two animal models:
systemic inflammation and cerebral ischemia. Compound K suppressed microglial
activation induced by systemic LPS administration in vivo. In addition, compound K
showed neuroprotective effects with reduction of microglial activation in the ischemic
brains of mice. Detail mechanistic studies by using in vitro cell culture systems revealed
that compound K suppresses inflammatory molecules via modulating ROS, MAPKs,
NF-κB/AP-1 as well as HO-1/ARE signaling pathways.
A number of studies have reported on the beneficial effects of ginsenosides
(ginseng saponins) in the CNS. Ginsenosides potentiate brain functions by promoting
neurogenesis and affecting neurotransmission (Liu et al., 2007; Shen and Zhang, 2007).
Ginsenosides, such as Rh1, Rh2, Rb1, and Rg1, support memory and learning (Wang et
al., 2009; Zhao et al., 2009). Among the ginsenosides, Rb1 is most thoroughly studied
for its neuroprotective effects in cerebral ischemia. Several groups have reported that
Rb1 prevents ischemic neuronal death by up-regulating the expression of anti-apoptotic
Bcl-2, Bcl-XL proteins, and glial-derived neurotrophic factor (GDNF) in rats subjected to
MCAO (Yuan et al., 2007; Zhang et al., 2006). Because Rb1 is metabolized into
compound K by intestinal microflora before absorption in the body (Akao et al., 1998;
Bae et al., 2004), the in vivo anti-ischemic effect of ginsenoside Rb1 might be due to the
metabolites. In support of this notion, the present study demonstrates that compound K
exerts protective effects against ischemic stroke. We found that compound K reduced
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
18
ischemic brain injury in the cortex, but not in the striatum, which is destined to be the
necrotic core (Fig. 7A), indicating that compound K can be a effective therapeutic to
rescue the salvageable penumbra from ischemic insults. In addition, we found that
compound K repressed microglial activation, which contributes to ischemic brain
damage by releasing proinflammatory cytokines and free radicals (Wang et al., 2007).
The number of morphologically activated microglia that were detected prominently at
24 h after MCAO (Shin et al., 2010b) was decreased in the ischemic cortex of mice
pretreated with compound K (Fig. 7B). The repression of microglia is not likely a result
of reduced infarct volume because cortical infarction was not definitely visible at 24 h
after MCAO, the time point that Iba-1 expression was examined. The reason that
compound K repressed microglial activation only in the cortex might be explained by
the different pathophysiology between the core and the penumbra. It has been shown
that molecules released from damaged neurons, such as ATP and glutamate, activate
microglia (Volonte et al, 2003), and the activated microglia can again increase neuronal
death by releasing cytotoxic agents such as cytokines, ROS, proteases and glutamate
(Kato and Kogure, 1999). Therefore, more prominent activation of microglia in the
striatum may indicate more severe cytotoxic responses in the core of infarction, and the
dose or treatment timing of compound K may not be enough to repress microglial
activation in the core area. Although a direct link between microglial activation and
neuroprotection by compound K remains to be further investigated in ischemic stroke
models, we suggest that the reduction of infarct volume by compound K is associated
with repression of microglial activation, and the data support the concept that the
penumbra is salvageable area by suitable protective drugs.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
19
Effects of compound K on microglial activation was further supported by studies
using mice exposed to systemic administration of LPS, an acute model of infection
causing inflammation in global area of the brain (Qin et al., 2007; Shin et al., 2010a).
Compound K reduced microglial activation not only in the cortex but also in the
striatum because there was no regional difference of injury severity between the cortex
and the striatum in this model. Furthermore, compound K also reduced the expression
of proinflammatory cytokines IL-1β and TNF-α (Fig. 6). Collectively, the data support
the view that anti-inflammatory action may be one of the factors contributing to the
neuroprotective effects afforded by compound K.
In BV2 microglial cells and primary cultured microglia, compound K inhibited not
only the expression of pro-inflammatory molecules (iNOS, TNF-α, IL-1β, IL-6, MCP-1,
and MMP-3/-9) but also ROS production. A recent paper reported on in vitro
antioxidant properties of compound K (Lee et al., 2011). Compound K exhibits strong
radical scavenging activities against DPPH (1,1-diphenyl-2-picrylhydrazyl), hydroxyl,
superoxide and ABTS (2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid).
Compound K also inhibited lipid peroxidation. Another paper reported that compound
K regulates zymosan-induced inflammatory signaling through inhibition of ROS
(Cuong et al., 2009). Therefore, the results suggest that compound K may be a useful
antioxidant agent against reactive oxygen species.
In the present study, we found that compound K increased HO-1, which is known
to have antioxidant, anti-apoptotic, and anti-inflammatory functions (Naidu et al., 2009).
Previous studies have shown a link between HO-1 activity and reduction in iNOS
expression (Min et al., 2006). In addition, HO-1 inhibits LPS-induced TNF-α and IL-1β
expression through suppression of NF-κB (Rushworth et al., 2008). Furthermore,
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
20
carbon monoxide (CO), one of the products of HO-1, inhibits NADPH oxidase and
TLR4, which are involved in LPS signaling (Nakahira et al., 2006). Thus, the induction
of HO-1 may provide the basis not only for the anti-inflammatory activity of compound
K, but also the antioxidant activity.
When we examined the effects of compound K on various transcription factors
involved in the regulation of inflammatory molecules, compound K was shown to
inhibit NF-κB and AP-1 activities, while increasing nuclear protein binding to CRE and
ARE in LPS-stimulated microglia. Recent studies indicate that CREB, as well as Nrf2
(ARE binding protein), play a role in the regulation of ROS detoxification (Lee et al.,
2009). In addition, the anti-oxidant enzyme, HO-1, has been shown to be under the
influence of the CREB/CRE transcriptional pathway (Lee et al., 2009; Min et al., 2006).
Therefore, CREB, in concert with HO-1, appears to play an important role in compound
K-mediated anti-inflammation/ anti-oxidant effects in microglia.
A recent study reported that compound K acts as an agonist of glucocorticoid
receptor (GR) and induces tolerance to endotoxin-induced lethal shock (Yang et al.,
2008). They suggested that the therapeutic effect of compound K on lethal sepsis is
mediated through the modulation of TLR4-associated signaling via GR. Thus, the GR
agonist function might be also related with the anti-inflammatory effects of compound
K in our neuroinflammatory model systems. Further studies will be necessary to clarify
this issue.
In conclusion, we report for the first time the anti-inflammatory effects of
compound K in microglial cell culture systems and two in vivo models of brain disease
in which inflammation plays important roles. Furthermore, the neuroprotective effect of
compound K was demonstrated in a mouse model of ischemic brain injury. Detailed
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
21
mechanistic studies indicate that the inhibition of ROS, MAPKs, and NF-κB/AP-1, and
the enhancement of the CREB and Nrf2/HO-1 signaling axis are responsible for strong
anti-inflammatory/antioxidant effects of compound K in activated microglia.
Therefore, compound K is a promising therapeutic agent for prevention and/or
treatment of ischemic brain injuries and other neurodegenerative diseases, which are
accompanied by microglial activation.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
22
Authorship Contributions
Participated in research design: E.M. Park, and H.S. Kim
Conducted experiments: J.S. Park, Shin, Jung, and E.M. Park
Contributed new reagents or analytical tools: Hyun, Le, and D.H. Kim
Performed data analysis: E.M. Park, and H.S. Kim
Wrote or contributed to the writing of the manuscript: J.S. Park, E.M. Park, and H.S.
Kim
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
23
References
Akao T, Kanaoka M, and Kobashi K (1998) Appearance of compound K, a metabolite
of ginsenoside Rb1 by intestinal bacteria, in rat plasma after oral administration-
measurement of compound K by enzyme immunoassay. Biol Pharm Bull 21:245-249.
Bae EA, Hyun YJ, Choo MK, Oh JK, Ryu JH, and Kim DH (2004) Protective effects of
fermented red ginseng on a transient focal ischemic rats. Arch Pharm Res 27:1136-1140.
Block ML, and Hong JS (2005) Microglia and inflammation-mediated
neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol
76:77-98.
Bocchni V, Mazzolla R, Barluzzi R, Blasi E, Sick P, and Kettenmann H (1992) An
immortalized cell line expresses properties of activated microglial cells. J Neurosci Res
6:16-21.
Cuong TT, Yang CS, Yuk JM, Lee HM, Ko SR, Cho BG, and Jo EK (2009)
Glucocorticoid receptor agonist compound K regulates dectin-1-dependent
inflammatory signaling through inhibition of reactive oxygen species. Life Sci 85:625-
633.
Dang PM, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, Hayem G, Jensen
ON, Gougerot-Pocidalo MA, and El-Benna J (2006) A specific p47phox-serine
phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at
inflammatory sites. J Clin Invest 116:2033–2043.
Ginsberg MD (2003) Adventures in the pathophysiology of brain ischemia: penumbra,
gene expression, neuroprotection: the 2002 Thomas Willis Lecture. Stroke 34:214-223.
Ito D, Tanaka K, Suzuki S, Dembo T, and Fukuuchi Y (2001) Enhanced expression of
Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia
in rat brain. Stroke 32:1208-1215.
Jia L, Zhao Y, and Liang XJ (2009) Current evaluation of the millennium
phytomedicine-ginseng (II): collected chemical entities, modern pharmacology, and
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
24
clinical applications emanated from traditional Chinese medicine. Curr Med Chem
22:2924-2942.
Joh EH, Lee IA, Jung IH, and Kim DH (2011) Ginsenoside Rb1 and its metabolite
compound K inhibit IRAK-1 activation-The key step of inflammation. Biochem
Pharmacol 82:278-286
Jung JS, Shin JA, Park EM, Lee JE, Kang YS, Min SW, Kim DH, Hyun JW, Shin CY,
and Kim HS (2010) Anti-inflammatory mechanism of ginsenoside Rh1 in
lipopolysaccharide-stimmulated microglia: critical role of the protein kinase A and
hemeoxygenase-1 expression. J Neurochem 115:1668-1680.
Jung SH, Woo MS, Kim SY, Kim WK, Hyun JW, Kim EJ, Kim DH, and Kim HS
(2006) Ginseng saponin metabolite suppresses phorbol ester-induced matrix
metalloproteinase-9 expression through inhibition of activator protein-1 and mitogen-
activated protein kinase signaling pathways in human astroglioma cells. Int J Cancer
118:490-497.
Kato H, and Kogure K (1999) Biochemical and molecular characteristics of the brain
with developing cerebral infarction. Cell Mol Neurobiol 19:93-108.
Lee B, Cao R, Choi YS, Cho HY, Rhee AD, Hah CK, Hoyt KR, and Obrietan K (2009)
The CREB/CRE transcriptional pathway: protection against oxidative stress-mediated
neuronal cell death. J Neurochem 108:1251-1265.
Lee CH, and Kim SY (2009) NF-κB and therapeutic approach. Biomol Ther 17:219-240.
Lee NJ, Lee JW, Sung JH, Lee YJ, and Kang JK (2011) In vitro antioxidant properties
of a ginseng intestinal metabolite IH-901. Lab Anim Res 27(3):227-234.
Lin TN, He YY, Wu G, Khan M, and Hsu CY (1993) Effect of brain edema on infarct
volume in a focal cerebral ischemia model in rats. Stroke 24:117-121. Liu JW, Tian SJ, de Barry J, and Luu B (2007) Panaxadiol glycosides that induce
neuronal differentiation in neurosphere stem cells. J Nat Prod 70:1329-1334. Min KJ, Yang MS, Kim SU, Jou I, and Joe E (2006) Astrocytes induce hemeoxygenase-
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
25
1 expression in microglia: a feasible mechanism for preventing excessive brain
inflammation. J Neurosci 26:1880-1887.
Naidu S, Vijayan V, Santoso S, Kietzmann T, and Immenschuh S (2009) Inhibition and
genetic deficiency of p38 MAPK up-regulates heme oxygenase-1 gene expression via
Nrf2. J Immunol 11:7048-7057.
Nakahira K, Kim HP, and Geng XH (2006) Carbon monoxide differentially inhibits
TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J
Exp Med 203:2377-2389.
Park JS, Park EM, Kim DH, Jung K, Jung JS, Lee EJ, Hyun JW, Kang JL, and Kim HS
(2009) Anti-inflammatory mechanism of ginseng saponins in activated microglia. J
Neuroimmunol 209:40-49.
Qin L, Li G, Qian X, Liu Y, Wu X, Liu B, Hong JS, and Block ML (2005) Interactive
role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia
activation. Glia 52:78-84.
Radad K, Moldzio R, and Rausch WD (2010) Use Ginsenosides and their CNS targets.
CNS Neurosci Ther 17(6):761-768
Rushworth SA, MacEwan DJ, and O’Connell MA (2008) Lipopolysaccharide-induced
expression of NAD(P)H:quinone oxidoreductase 1 and hemeoxygenase-1 protects
against excessive inflammatory responses in human monocytes. J Immunol 181:6730-
6737.
Sayre LM, Perry G, and Smith MA (2008) Oxidative stress and neurotoxicity. Chem Res
Toxicol 2:172–188.
Shen L, and Zhang J (2007) NMDA receptor and iNOS are involved in the effects of
ginsenoside Rg1 on hippocamcal neurogenesis in ischemic gerbils. Neurol Res 29:270-
273.
Shin JA, Park EM, Choi JS, Seo SM, Kang JL, Lee KE, and Cho S (2009) Ischemic
preconditioning-induced neuroprotection is associated with differential expression of
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
26
IL-1beta and IL-1 receptor antagonist in the ischemic cortex. J Neuroimmunol 217:14-
19.
Shin JA, Lee EJ, Seo SM, Kim HS, Kang JL, and Park EM (2010a) Nanosized titanium
dioxide enhanced inflammatory responses in the septic brain of mouse. Neuroscience
165:445-454.
Shin JA, Lee H, Lim YK, Koh Y, Choi JH, and Park EM (2010b) Therapeutic effects of
resveratrol during acute periods following experimental ischemic stroke. J
Neuroimmunol 227:93-100.
Smale ST (2010) Selective transcription in response to an inflammatory stimulus. Cell
140:833-844.
So HS, Kim HJ, and Lee JH (2006) Flunarizine induces Nrf2-mediated transcriptional
activation of heme oxygenase-1 in protection of auditory cells from cisplatin. Cell
Death Differen 13:1763-1775.
Sorce S, and Krause KH (2009) Nox enzymes in the central nervous system: from
signaling to disease. Antioxid Redox Signal 11:2481-2504.
Stolp HB, and Dziegielewska KM (2009) Review: Role of developmental inflammation
and blood-brain barrier dysfunction in neurodevelopmental and neurodegenerative
diseases. Neuropathol. Appl Neurobiol 35:132-146.
Volonté C, Amadio S, Cavaliere F, D'Ambrosi N, Vacca F, and Bernardi G (2003)
Extracellular ATP and neurodegeneration. Curr Drug Targets CNS Neurol Disord 2:403-
412.
Wang Q, Tang XN, and Yenari MA (2007) The inflammatory response in stroke. J
Neuroimmunol 184:53-68.
Wang YZ, Chen J, Chu SF, Wang YS, Wang XY, Chen NH, and Zhang JT (2009)
Improvement of memory in mice and increase of hippocampal excitability in rats by
ginsenoside Rg1’s metabolites ginsenoside Rh1 and protopanaxatriol. J Pharmacol Sci
109:504-510.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
27
Woo MS, Jang PG, Park JS, Kim WK, Joh TH, and Kim HS (2003) Selective
modulation of lipopolysaccharide-stimulated cytokine expression and mitogen-activated
protein kinase pathways by dibutyryl-cAMP in BV2 microglial cells. Brain Res Mol
Brain Res 113:86-96.
Yang CS, Ko SR, Cho BG, Shin DM, Yuk JM, Li S, Kim JM, Evans RM, Jung JS, Song
DK, and Jo EK (2008) The ginsenoside metabolite compound K, a novel agonist of
glucocorticoid receptor, induces tolerance to endotoxin-induced lethal shock. J Cell Mol
Med 12:1739-1753.
Yuan QL, Yang CX, Xu P, Gao XQ, Deng L, Chen P, Sun, ZL, and Chen QY (2007)
Neuroprotective effects of ginsenoside Rb1 in transient cerebral ischemic in rats. Brain
Res 1167:11-12.
Zhang B, Hata R, Zhu P, Sato K, Wen TC, Yang L, Fujita H, Mitsuda N, Tanaka J,
Samukawa K, Maeda N, and Sakanaka M (2006) Prevention of ischemic neuronal death
by intravenous infusion of a ginseng saponin Rb1 that upregualtes Bcl-XL expression. J
Cereb Blood Flow Metab 26:708-721.
Zhao H, Li Q, Pei X, Zhang Z, Yang R, Wang J, and Li Y (2009) Long-term ginsenoside
administration prevents memory impairment in aged C57BL/6J mice by upregulating
the synaptic plasticity-related proteins in hippocampus. Behav Brain Res 201:311-317.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
28
Footnotes
This work was supported by the National Research Foundation of Korea (NRF) grant
funded by the Korea government [Grant 2010-0029354].
H.S.K. and E.M.P. contributed equally to this work.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
29
Legends for Figures
Fig. 1. The chemical structure of compound K.
Fig. 2. Compound K inhibits the expressions of proinflammatory molecules in
LPS-stimulated BV2 and primary microglial cells. (A) Cells were pre-treated with
compound K for 1 h, followed by treatment with LPS (0.1 μg/ml) for 24 h. The amounts
of nitrite in the supernatants were measured using Griess reagent, and TNF-α and IL-1β
were measured by ELISA. The data are expressed as the mean ± S.E.M. of four
independent experiments. *P < 0.05, compared to the value in cells treated with LPS
alone. (B) Total RNA was isolated from microglial cells treated with LPS in the absence
or presence of compound K for 6 h, and RT-PCR was performed to determine the
mRNA levels of iNOS, TNF-α, IL-1β, IL-6, MCP-1, MMP-3 and MMP-9. The data are
representative of three independent experiments.
Fig. 3. Compound K inhibits DNA binding activities of NF-κB and AP-1 with
increasing nuclear protein binding to CRE. (A) Electrophoretic mobility shift assay
(EMSA) for NF-κB DNA binding activity. The arrow indicates a DNA-protein complex
of NF-κB. ‘F’ indicates free probe. Antibody supershift assay data is shown in the right
panel. Coincubation with p65 or p50 antibody but not with Sp1 antibody diminished
formation of NF-κB complex and/or generated supershift bands as indicated by arrows.
(B) NF-κB reporter gene assay. BV2 cells were transfected with (κB)3-luc plasmid and
pre-treated with compound K for 1 h before addition of LPS. After 6 h of LPS treatment,
cells were harvested and the luciferase assay was performed. Values correspond to the
mean ± S.E.M. of three independent experiments performed in duplicate. *P < 0.05,
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
30
significantly different from the value in cells treated with LPS alone. (C, D) EMSA for
AP-1 and CRE binding activity. The autoradiograms are representative of three
independent experiments.
Fig. 4. Compound K inhibits LPS-induced ROS production and phosphorylation
of NADPH oxidase p47phox and MAP kinases. (A) BV2 and primary microglial cells
were pre-treated with compound K for 1 h, followed by treatment with LPS (0.1 μg/ml)
for 16 h. Then, the intracellular ROS levels were measured by the DCF-DA method as
described in the Materials and Methods section. The data are expressed as the mean ±
S.E.M. of three independent experiments. *P < 0.05, significantly different from LPS-
treated sample. (B) The representative picture of DCF-derived fluorescence in BV2
cells (n = 3). (C) Western blot analysis for phosphorylation of p47phox in BV2 cells and
densitography. Values correspond to the mean ± S.E.M. of three independent
experiments. *P < 0.05, significantly different from LPS-treated samples. (D) Western
blot detection of MAP kinases phosphorylation in BV2 cells. The blots are
representative of four independent experiments.
Fig. 5. Compound K increased HO-1 expression and ARE-mediated
transcriptional activities in LPS-stimulated microglia. (A) Western blot analysis
shows the effect of compound K on HO-1 protein expression. BV2 cells were treated
with compound K with or without LPS for 24 h and cell lysates were obtained. (B) RT-
PCR was performed to determine the HO-1 mRNA expression. Quantification data are
shown in the graph (n = 3). (C) EMSA for nuclear protein binding to ARE element (n =
3). The arrow indicates a DNA-protein complex of ARE. (D) BV2 cells were
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
31
transfected with ARE-luc plasmid and pre-treated with compound K for 1 h before
addition of LPS. After 6 h of LPS treatment, cells were harvested and the luciferase
assay was performed. Data are the mean ± S.E.M. of three independent experiments. *P
< 0.05, significantly different from the control sample. #P < 0.05, significantly different
from the LPS-treated sample.
Fig. 6. Compound K reduced inflammatory responses in the septic brain of mice.
(A) Immunofluorescence labeling of Iba1 and quantification of the number of activated
Iba1-positive cells in the cortex and striatum 3 h after systemic LPS treatment (5 mg/kg).
Representative images were obtained from one set of experiments and three
experiments were done independently. Veh, vehicle; Comp K, compound K. The data
are the mean ± S.E.M. (B) Western blots for IL-1β, TNF-α, and actin in the cortex 6 h
after LPS treatment, and quantification of IL-1β and TNF-α protein levels with
normalization by actin (n = 3 in each group). Values (mean ± S.E.M.) are the ratio vs.
controls (assigned a nominal value of 1). *P < 0.05, significantly different from the
value in control mice. #P < 0.05, significantly different from the value in vehicle-treated
mice.
Fig. 7. Compound K reduced ischemic brain injury of mice and suppressed
microglial activation in the post-ischemic brain. (A) Representative brain sections of
cresyl violet staining and infarct volumes in total brain, cortex, and striatum from mice
pre-treated with vehicle (n = 7) or compound K (n = 8). Dotted lines in brain sections
indicate infracted areas. *P < 0.05, significantly different from vehicle treated mice. The
data are the mean ± S.E.M. (B) Immunofluorescence labeling and quantification of
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

JPET #189035
32
Iba1-positive cells (green) in the cortex and striatum 24 h after MCAO. Representative
images from one of three independent experiments are shown. Comp K, compound K.
The data are the mean ± S.E.M. *P < 0.05, significantly different from the value in
control mice. #P < 0.05, significantly different from the value in vehicle-treated mice.
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from
Fig. 1
OH
Glc-O
HO
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

[BV2 cells] [Primary microglia]
iNOS
TNF-α
IL-1β
(-)
+ LPS
25 50 75 (-)
+ LPS
(μM)25 50 75
MMP-3
IL-6
MCP-1
0 0Comp K
+ LPS + LPS
A [BV2 cells] [Primary microglia] B
Fig. 2
0
500
1000
25 50 75Comp K
TN
F-α
(pg
/ml)
1500
2000
0
500
1000
100
1500
2000
2500
200
300
400
IL-1
β
(pg
/ml)
100
200
300
400
25 50 75
0
20
40
60
80
Nit
rite
( μM
)
0
10
20
30
40
(μM)(-) (-)0 00 0
(μM)
**
*
**
** *
** *
** *
**
*
500500
100 50
2500 3000
GAPDH
MMP-9
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

Fig. 3
A
2
4
6
8
Rel
ativ
e lu
cife
rase
act
ivit
y
Reporter plasmid: (κB)3-Luc
25 50Comp K (μM)(-) 0
+ LPS
B
C D
Comp K (μM)
NF-κB
0 25 50
+ LPS
AP-1
◀ F
◀ F
CREB
◀ F
(-)
**
NF-κB
◀ F
supershift
α p50 α p65 α Sp1Ab :
Comp K (μM)0 25 50
+ LPS
(-) Comp K (μM)0 25 50
+ LPS
(-)
(-)
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from
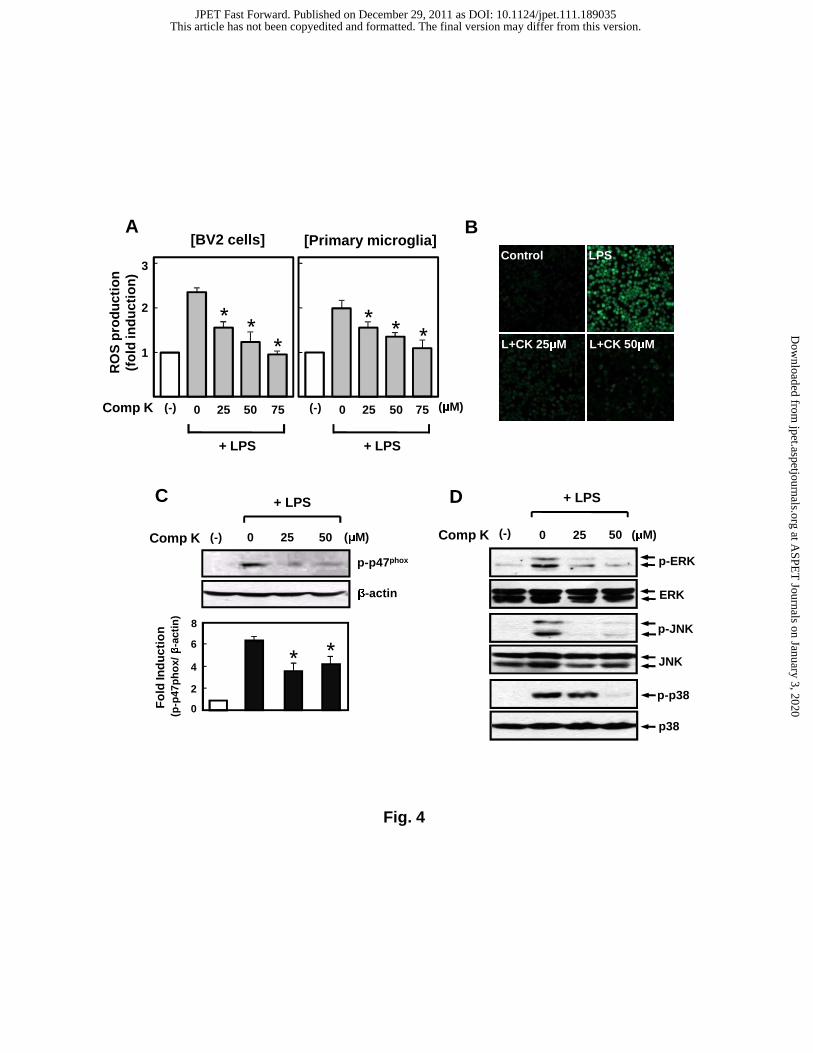
Fig. 4
(-)
p-p47phox
β-actin
(μM)0 25 50
+ LPS
2
6
8
Fo
ld In
du
ctio
n
4
(p-p
47p
ho
x/ β
-act
in)
Comp K
0
C
(-) (μM)0 25 50
+ LPSD
Comp K
p-ERK
ERK
JNK
p-JNK
p38
p-p38
* *
1
2
3Control LPS
L+CK 25μM L+CK 50μM
25 50 75Comp K 25 50 75 (μM)(-) (-)0 0
[BV2 cells] [Primary microglia] A
+ LPS + LPS
B
* **
* * *
RO
S p
rod
uct
ion
(f
old
ind
uct
ion
)
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

Fig. 5
ARE+ protein
complex
Comp K (μM)0 0 25 50
+ LPS
◀ F
C DReporter plasmid: ARE-Luc
Rel
ativ
e lu
cife
rase
act
ivit
y
1
2
3
(μM)25 50Comp K (-) 25 50 0
+ LPS
HO-1
0 25 50Comp K 0 25 50 (μM)
+ LPS
β-actin
A
Fo
ld in
du
ctio
n
2
4
6 # #
(HO
-1/β
-act
in)
**
HO-1
0 25 50Comp K 0 25 50 (μM)
+ LPSB
Fo
ld in
du
ctio
n4
8
12
(HO
-1/G
AP
DH
)
##
* *
**
##
GAPDH
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

Fig. 6
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from

Fig. 7
This article has not been copyedited and formatted. The final version may differ from this version.JPET Fast Forward. Published on December 29, 2011 as DOI: 10.1124/jpet.111.189035
at ASPE
T Journals on January 3, 2020
jpet.aspetjournals.orgD
ownloaded from