tissue tropisms of aav vectors deficient in receptor binding · tissue tropisms of aav vectors...
TRANSCRIPT

TISSUE TROPISMS OF AAV VECTORS DEFICIENT
IN RECEPTOR BINDING
A Thesis
Presented in Partial Fulfillment of the Requirements for Distinction for the
Degree Bachelor of Science in the Division of Medical Technology
in the Allied Medical Professions of The Ohio State University
By
Jennette Kathleen Crumrine
*****
The Ohio State University
2005
Advisors:
Jeffery S. Bartlett, Ph.D., Research Advisor Department of Pediatrics and Department of Molecular Virology, Immunology, and Medical Genetics The Ohio State University
Approved by
Kathy Waller, Ph.D., Honors Advisor Associate Professor, Division of Medical Technology ▬▬▬▬▬▬▬▬▬▬▬▬ School of Allied Medical Professions Advisor The Ohio State University Division of Medical Technology


ABSTRACT
Adeno-associated virus (AAV) is a single stranded DNA virus with an
icosahedral 20-25 nm capsid comprised of three structural proteins, VP1, VP2, and VP3.
AAV is of great interest as a gene therapy vector for the treatment of cancer and genetic
diseases due to its ability to transduce both mitotic and postmitotic tissues, be produced at
high titers, and mediate long-term gene expression without pathogenicity. However, one
concern is that the AAV2 receptor, heparan sulfate proteoglycan (HSPG), is expressed on
a wide variety of human tissues. This can cause unwanted gene transfer and expression
when only a specific cell type needs to be transduced. Changing arginine amino acids
(a.a.) at positions 585 and 588, to alanines (R585A,R588A) has been shown to effectively
eliminate AAV binding to HSPG, while the insertion of small peptide epitopes (<20 aa)
that bind alternative cellular receptors has been shown to effectively retarget AAV
vectors to these new receptors. The amino acid sequence of the major capsid protein VP3
is contained within the two larger and less abundant capsid proteins VP1 and VP2, thus
mutations within VP3 are also present in VP1 and VP2. PCR-based site directed
mutagenesis of pACG2, which expresses all of the AAV capsid proteins, was used to
change the arginines at positions 585 and 588 within the VP1, VP2, and VP3 proteins to
alanines. Similarly, these arginine residues were also changed to alanines in a second
plasmid that expresses only VP1 and VP3 proteins. Different peptides that target the

vasculature endothelial growth factor receptor 2(VEGFR2) were then incorporated into
the R585A, R588A AAV2 capsid constructs. The ability of wild-type AAV2 vectors,
R585A, R588A AAV2 vectors, VEGFR2-targeted AAV2 vectors, and an AAV2 vector
containing the R585A, R588A mutation on only the VP1 and VP3 proteins to bind to
polystyrene beads coated with heparin and to HSPG-expressing Hela C12 cells were
assessed. Our preliminary data suggest that AAV2 vectors comprised of capsids with
R585A, R588A mutations were unable to bind to HSPG. The wild-type AAV2 was the
only virion that successfully bound to the beads and Hela cells. An affinity assay was
performed showing that with a competitor in 100-fold excess, AAV2 did not bind as well
to the Hela C12 cells similar to the heparan mutants. These preliminary findings suggest
that AAV2 does naturally bind to heparan, and that the rAAV2 mutants were deficient in
receptor binding. This can lead to successful elimination of unwanted gene transfer and
expression and the retargeting of the rAAV2 vector to tissues that were once thought
untargetable.

ACKNOWLEDGMENTS
Dr. Kathy Waller- Thank you for giving me the motivation and self-assurance to do the best that I could. Not only have you broaden my horizons in class, but also with your aspects on life. A positive attitude can always get you far in life. Continue you teaching style and never stop helping students find the right path to follow. Michele Suhie – Thank you for starting me on my honor’s curriculum and assisting in choosing a topic. The guidelines and goals that you gave me really helped overcome that wall of problems/stress. You were always there as a counselor to listen to all of my problems and lead me in the right direction. Dr. Jeffrey Bartlett- Thank you for having faith in me and allowing me to be a member of your research project. I am very grateful to have had this opportunity to work with you. I will always cherish the experience you have given me. Matthew Stachler- Thank you for your patience and insightful ideas that guided me in the right direction on numerous occasions in the laboratory. You were always there to help me out of a lot of sticky situations when no one else was. I wish you the best of luck in your future endeavors, for you have the possibility to go far. Sferra’s laboratory- Thank you for your gift of the competitor recombinant virus. I could not have completed my project on time if it wasn’t for the small amount of virus needed. Dr. Wilson - Thank you for your generous gift of paying for all of the Honor students posters. I would also like to thank my family who made all of this possible through their love, encouragement and sacrifices. You have always taught me to work hard and dream big. I owe all of my great accomplishments in my life to you, thank you. Finally I want to give thanks to my husband, Bryce, because he was always there for me regardless of the type of day I was having. When things went wrong, you helped me believe in myself and gave me the reassurance that everything would be all right. One thing you have taught me is that everything happens for a reason. Once you open up you mind, body, and soul you can finally listen to your spiritual side.

TABLE OF CONTENTS
Page
Abstract……………………………………………………………………………………ii
Acknowledgments………………………………………………………………………..iv List of Figures…..… ………..……………………………………………………….….vii
Chapters:
1. Introduction……………………………………………………………………..1-4
1.1 Gene therapy and vectors……………………………………………………...1
1.2 Adeno-associated virus……………………..………………………………1-4
1.3 Background of problem……………………………………………………….4
2. Review of Literature……………………………………………………………5-7
3. Materials and Methods………………………………………………………...8-15
3.1 Construction of AAV capsid mutant plasmid……………………………….8-9
3.2 Ligation and Transformation…………………………………………….……9
3.3 Screening and purifying individual plasmids……………………………...9-10
3.4 In vitro studies…………………………………………………………....11-12
3.4.1 Cell culture…………………………………………………………11
3.4.2 Production of recombinant AAV particles with modified capsid….11
3.4.3 Transfection………………………………………………………..12
3.5 Purification……………………………………………………………...……13
3.6 Virus Titers…………………………………………………………..……....13
3.7 Infectious Titers……………………………………………………………..14
3.8 Heparin-Binding Assay………..………………………………………...14-16

3.8.1 Poros Beads…………………………………………………….14-15
3.8.2 Cell Binding Assay………………………...………………………15
3.8.3 Affinity Binding Assay………………………………………….…16
4. Research Results………………………………………………………...……17-28
5. Conclusion……………………………………………………………….…...29-30
6. Bibliography……………………………………………………………….…31-32

LIST OF FIGURES
Figure Page
1 AAV Capsid………...…………………………………………………………2
2 Outer and inner surface map of AAV…………...…………………………….2
3 Satellitism of AAV with Adeno virus infection………...…………………….3
4 Western Blot of AAV capsid…………………………...……………………..5
5 Visual description of how the three plasmids are placed into the 293 cells and
recombine to produce virus…………………………………………..………12
6 Map of restriction digest for pACG…….……………………………………17
7 Photograph of restriction digest separated on 1.1 % agarose gel……………19
8 Graph of Heparin binding to POROS beads…………………………………22
9 Graph of Heparan Cell Binding experiment 1...……………..…………..…..24
10 Graph of Heparan Cell Binding experiment 2……...……....….………….…25
11 Graph of Hela Cell Binding with competitor………………………………...27

CHAPTER 1
INTRODUCTION
1.1 Gene therapy
Gene therapy is an experimental treatment being investigated to correct defective
genes that are responsible for disease development. Researchers are studying several
approaches of gene therapy to treat cancers and genetic diseases. One of the biggest
challenges in gene therapy is finding a suitable vector to carry DNA into tissues.
Problems for certain vectors in gene therapy include: provocation of immune response,
not invading both quiescent and dividing cells, lack of indefinite expression and
producible high titers (1). Leading candidates for gene therapy vectors are the adenovirus,
retrovirus, and recombinant engineered adeno-associated virus (rAAV). The adeno-
associated virus (AAV) may be a solution to the problems with gene therapy (1, 2).
1.2 AAV
The development of gene transfer vectors from the human parvovirus, adeno-
associated virus (AAV), has provided an efficient and effective way of delivering genes
into mammalian cells AAV genome is encapsulated as a single stranded DNA molecule
of 4.7 KB. The AAV capsid is comprised of three structural proteins VP1, VP2, and VP3
in an approximate ratio of 1:1:18 (3,4,5,6). The non-enveloped virion is icosohedral in

shape and is one of the smallest (20-25 nM diameter)
that has been described. Figure 1 demonstrates the size
and shape of the AAV vector courtesy of
http://www.scripps.edu/mb/goodsell/.
Figure 1: AAV capsid
Even though humans are a natural host for the wild-type AAV virus and at least
80% of the human population is seropositive, no known diseases have emerged from its
infection (7). An electron cryo-microscopy and image reconstruction of adeno-associated
virus type 2 is shown below in figure 2. Picture A is a view of the outer surface of the
capsid and B is the inner surface. The surface maps are colored in such a way to
demonstrate the depth of the surface, higher radii are shown in red and lower radii appear
blue (8).
Figure 2: Outer and inner surface map of AAV(8).
Advantages of AAV as a gene delivery vector include its ability to transduce both
mitotic and postmitotic tissues, be produced at high titers, and mediate long-term gene
expression without pathogenicity (2). AAV is a non-pathogenic defective member of the

human parvovirus family that requires co-infection of a helper virus to propagate itself
into the host genome. Figure 3
demonstrates the human adenovirus with adeno-
associated virus (arrow), a satellite virus.
AAV requires co-infection with a helper virus to
cause a productive infection (9). In the
absence of a helper virus, AAV will integrate
itself into the host, establishing latency
throughout the full life cycle of a cell (10).
Figure 3: Satellitism of AAV (arrow) with Adeno virus infection (9).
AAV is stable in its latent form and can be rescued to enter into a lytic phase upon
infection with a helper virus. In its latent form, AAV based vector is capable of long-
term, high-level gene expression even in competent hosts without showing any cellular
immune response or toxicity (11). This is practical because only a single injection of a
therapeutic gene is required; therefore, no re-administering is necessary. Furthermore, re-
administering the virus will increase the chances of the host to produce antibodies against
the gene vector, rendering it ineffective.
Another aspect of AAV is its ability to transduce both mitotic and postmitotic
tissues such as the lungs, intestines, neurons, muscle, and hematopoietic cells. Even
though AAV vectors seem to be a nearly ideal means of genetic therapy, major
limitations for clinical application still exist. Limitations of the AAV vector include its
inability to target all tissue types, redirecting viral vectors without unwanted transfer of
genes and expression, and incorporation of peptides into the viral capsid. The size of

ligands that can be inserted into the AAV capsid is limited due to its small capsid size of
20-24nm (1,2).
1.3 Background of problem
In gene therapy, adeno-associated virus is used to deliver a transgene into the
body. One of the major concerns with AAV2 as a gene therapy vector is the fact that its
receptor, heparan sulfate proteoglycan (HSPG), is expressed on a wide variety of human
tissues (1, 2, 3). This can cause unwanted gene transfer and expression when only a
specific cell type needs to be transduce. Further clinical development of AAV based gene
therapy will require developing AAV vectors with the ability to target specific tissue
types. This can be accomplished by the elimination of binding to HSPG and the
incorporation of targeting peptides directly into the AAV capsid. By eliminating the virus
from utilizing heparan receptors on its capsid protein through mutations, specific sites
can be targeted without unwanted gene expression. Point mutations and an addition of
small peptides can control how much AAV accumulates in organs of non-interest and
successfully retarget the virus to specific sites (3).

CHAPTER 2
REVIEW OF LITERATURE
The AAV capsid is composed of three structural proteins VP1, VP2, and VP3 in
an approximate ratio of 1:1:18 (3, 4, 5, 6). Figure 4 is a western blot
of the AAV capsids, VP1, VP2 and VP3 showing the concentrations
of each capsid. The capsids were detected by using monoclonal
antibodies that recognizes all three capsid proteins. Certain regions
of the AAV vector capsid protein can tolerate the insertion Figure 4: Western Blot of AAV capsid (8).
of exogenous peptides. One such example has been the insertion of an Arg-Gly-Asp (RGD) peptide following amino acid 588 in the normal
AAV capsid protein sequence. This RGD peptide allows the modified AAV vector to
invade via an interaction with cell surface integrin receptors and without the use of
HSPG. By adding the RGD to the virus, one does not need to worry if HSPG is present
(2). RGD insertion into the wild type capsid only enhances the chances of the virus to
bind to cells. RGD can still bind to heparan, thus in order to redirect the virus, an
elimination of HSPG receptors on the capsid is necessary.
Efforts to improve the packaging capacity of the recombinant AAV vectors are
still being investigated. Currently, only about 4.5Kb of DNA can be efficiently packaged

into an AAV particle. Understanding the primary steps of viral entry will also be
important in defining parameters for efficient gene delivery.
The tropism of AAV can be changed by genetically introducing a ligand peptide
into the viral capsid, redirecting the binding of AAV to other cellular receptors. By
introducing a mutant insertion into the capsid protein of AAV, we can retarget the
tropisms of the virus to cells that are normally resistant to AAV infection (12,13). AAV
has been mutated at numerous capsid protein subunits, one site being arginine 484 (R484)
and another being arginine 585 (R585). These mutations of the AAV capsid have been
shown to drastically reduce heparan binding. Tissue distribution in mice of the double
mutated AAV at R484 and R585 indicated marked infection reduction of the liver,
compared to infection with the wild-type AAV, but showed an increase of infection in the
heart (14). Various mutations sites have been undergone to the AAV capsid, but the
specificity and sensitivity of the mutated virus to bind heparan sulfate proteoglycan
in-vitro and in-vivo has never been demonstrated.
Current research has demonstrated that AAV can utilize different binding sites
other then HSPG to deliver transgenes into the body. Changing arginine amino acids
(a.a.) at positions 585 and 588, to alanines (R585A, R588A) has been shown to
effectively eliminate AAV binding to HSPG (3, 14, 15). The insertion of small peptide
epitopes (<20 a.a.) that bind alternative cellular receptors has been shown to effectively
retarget AAV vectors to these new receptors. The amino acid sequence of the major
capsid protein VP3 is contained within the two larger and less abundant capsid proteins
VP1 and VP2, thus mutations within VP3 are also present in VP1 and VP2. Addition of
small peptides (<20 a.a.) that bind specific receptors after amino acid 588 should

effectively retarget rAAV. Another approach involves the incorporation of larger proteins
(< 30 kDa) into the capsid into the minor capsid protein region VP2. The VP2 capsid
protein region is nonessential and can tolerate large peptide insertions at its N terminus.
This will minimize the disruption of the major structural protein, VP3 (6). What is
apparent is that there is a lack of correlation of heparan binding and infectivity of
some heparan-binding mutants. This means that HSPG might facilitate and enhance
AAV cellular interaction and infectivity, yet appears not to be necessary as a
receptor site (14). Further testing needs to be done to confirm that various point
mutations that reduce the binding and infectivity of the AAV capsid does not bind to
heparan sulfate proteoglycan. Once the tissue tropisms of AAV vectors deficient in
receptor binding have been investigated, the virus can be retargeted to tissues of interest
for gene therapy treatment.

CHAPTER 3
MATERIALS AND METHODS
The effects of various mutants that change receptor binding of AAV particles of
tissues in direct contact with blood and tissue fenestrations large enough for vectors to
enter (liver, lung, spleen, and bone marrow) was monitored. Different mutant viruses
were made with mutations in the AAV capsid that allowed infection independent of
HSPG and with peptide inserts for retargeting to tissues of interest. Dr. Bartlett’s lab has
developed methodologies for production and purification of AAV vectors discussed
below.
3.1 Construction of AAV capsid mutant plasmid
The plasmid pACG2 (16) was used as the template for all mutant constructions.
ACG contains the AAV genome, less the two viral inverted terminal repeats (ITRs), and
an ATG-to-ACG mutation of the Rep 78/68 start codon. This mutation has been shown
to increase recombinant AAV vector yield by attenuating Rep 78/ 68 synthesis (17).
Sites for mutagenesis were chosen on the basis of a computer-generated model of AAV
structure. Point mutations at the VP3 region on 585/588 where an arginine has been
change to an alanine were undertaken. This prevented the binding of AAV2 to heparan
and allows an insertion of new peptides to retarget the vector.

Another type of mutation is the insertion of peptides. Two PCR primers were
designed that contained the sequence of the desired insertion plus a unique
endonuclease restriction site flanked by 15 to 20 homologous base pairs on each side of
the insertion. The PCR products were digested with DpnI endonuclease to eliminate the
parental plasmid template. DpnI digests methylated DNA, which degrades the parent
plasmid. Mutant plasmids that were made in the PCR are unmethylated.
3.2 Ligation and Transformation
The plasmid were ligated together by T4 ligase and transformed into bacteria
(DH5-α), to allow screening of individual plasmids. The PCR purified ligated plasmid
was transformed by adding it to competent bacteria into an electroporation cuvette. The
cuvette is placed into an electroporation unit where two pulses of 1800 volts will be
passed through it. Time constants were monitored to assure recombination. The ligated
bacteria will be grown in liquid media (SOC) and then plated onto a selective media
plate with selection marker (AMP).
3.3 Screening and purifying individual plasmids
Colonies from plates were picked and grown in liquid media (LB or 2X-YT)
over night. The liquid bacteria will then go through a mini-prep procedure as
determined from QIAGEN. The bacteria will be purified and lysed to leave only DNA.
Once the plasmid DNA is purified it will be digested with specific restriction enzyme
endonucleases and ran out on a gel. This allows for screening of the plasmids to assess
if ligation of the peptides of interest were incorporated into the DNA plasmid. This step

can be repeated numerous times until a digestion that looks promising arise or changes
in mutations that should have been added demonstrate an extra band. An agarose gel is
made to the thickness desired of a one percent concentration. 1x TAE is used in
combination of powder agarose to make a gel. The liquid is heated and mixed until the
agarose dissolves and is cooled to about body temperature. Ethidium bromide is added
to the one percent solution in a ratio of 1µl per every 20 ml of mixture. Combs are
added to produce wells for the digested plasmid to enter. Loading dye is added to the
plasmid that will allow us to see the bands once ran out. A base ladder is added to the
sides to assess where the bands are migrating and whether or not the bands are the
correct base pair. Any mini preps whose digestion looks promising are sequenced to
confirm the proper mutant plasmid map. The sequences are read carefully to assure that
the mutant was added in the correct site with no other unwanted mutations in the capsid.
Sequences that were correct were grown in a larger amount of media (500ml)
and a maxi prep procedure from QIAGEN was preformed. The maxi prep procedure is
very similar to the mini prep, but only a larger amount of volume is used. The purified
plasmid will be placed in a spectrophotometer and read at wavelengths of 260 and 280
to find out the quantity and quality of the plasmid prep. 260 λ tells us the concentration
and the ratio of 260 to 280λ tells us the purity of the sample. All plasmids are stored in a
-20˚C freezer until further use. A portion of the liquid media with bacteria/plasmid
growth is mixed 50/50 with glycerol and stored in the -86˚C freezer until more plasmid
is needed. Once all the plasmids that are required for viral preps are prepared and
purified, tissue cultures can be started beginning the in vitro studies of the project.

3.4 In vitro studies
3.4.1 Cell culture
Low passage number Hela C12 cells, Hela cells, and HEK 293 cells (17)
were grown in Dulbecco’s modified Eagles medium (DMEM) supplemented with
10% fetal bovine serum, penicillin (100U/ml), and streptomycin (100U/ml) at 37 ˚C
and 5% CO2. Once the desired confluency was obtained, tests can be preformed or
the cells can be split using trypsin to aid in the breaking of the adhesion of the cells
to the bottom of the plate.
3.4.2 Production of recombinant AAV particles with modified capsids
To produce rAAV with mutant capsid proteins, 293 cells were transfected
with three plasmids. The three plasmids used were as follows: (1) pTR-UF5, which
contains the enhanced green fluorescent protein (eGFP) gene driven by the
cytomegalovirus (CMV) promoter and flanked by the AAV terminal repeats; (2) the
pXX6 plasmid, which supplies the adenovirus helper gene products in trans to allow
rAAV production in an adenovirus-free environment (16); and (3) the modified
pACG plasmid, which supplies the mutant capsid proteins. As a control, rAAV was
also prepared with unmodified pACG plasmid to make the wild type. The plasmids
were mixed at a 1:3:1 molar ratio. Plasmid DNAs used for transfection were
purified with a Quiagen (Valencia, CA) Maxi-prep kit according to the supplier
manual.

3.4.3 Transfection
Transfections were carried out as follows. 293 NC2 cells were split 48 hours
before transfection so that they could reach confluency around 75% at the time of
transfection. Four 10-cm plates were transfected at 37°C, using the calcium
phosphate transfection protocol standardized in Dr. Bartlett’s laboratory, and
incubated at 37°C (19). Forty-eight hours after transfection, cells were harvested by
centrifugation at 1200rpm for 10 minutes, resuspended in TMN (200mM Tris pH 8,
10mM MgCl2, 1.5M NaCl) and freezing and thawing three times released virus.
The crude lysate was clarified by centrifugation at 4200rpm for 20minutes and
treated with Benzonase (Sigma, St. Louis, MO) at 50-U/ml final concentrations at
37°C for 30 minutes. Virus was further purified by iodixanol step gradient (20).
Figure 5 demonstrates the three plasmids that are added to the 293 cells. After 48-72
hours, the virus is harvested out of the cells and then purified.
Figure 5. Visual description of how the three plasmids are placed into the 293 cells and recombine to produce virus.

3.5 Purification Once the virus has been harvested from the cells by a freeze thaw method, it
can be further purified through an iodixanol gradient. Different concentrations of
iodixanol is made and layered from highest concentration to lowest to a centrifuge tube
(Quick-Seal Ultra Clear). 60% iodixanol is placed onto the bottom of the tube, followed
slowly, trying not to mix, by decreasing concentrations (40%, 25%, and 15%). The
clarified lysate/virus is added to the top of the tube and then spun at 350,000 x g for 1
hour at 18˚C with a low brake. The tube is carefully removed and is aspirated with an
18-gauge needle at the 40% layer removing only the 40% solution. The virus will
migrate to the 40% layer due to weight/ density and proteins and salts will stay in the
25% layer.
3.6 Virus titers
The concentration of DNA-containing particles was determined by performing a
real-time (RT) PCR using a Perkin-Elmer (PE) – Applied Biosystems (Foster City,
Calif.) Prism 7700 sequence detector system. Briefly, 1µl of the purified rAAV stock
was diluted in 99µl of digestion buffer (10mM Tris-HCl [pH 8.3], 5 mM MgCl2, and 50
mM KCl ) containing Dnase I (350µg/ml). Samples were incubated at 37°C for 30
minutes, then 95°C for 10 minutes. Proteinase K was added to a final concentration of 1
µg/ml and the samples were incubated at 37°C for 30 minutes. Two and a half
microliters was used for RT-PCR in a Taqman reaction mixture and completed in
compliance as described by Clark (21). The only values that were accepted was within

the linear portion of a standard curve having a coefficient of linearity greater than 0.98.
The average RT-PCR titer was calculated from virus preparations assayed two times.
3.7 Infectious titers
Infectious titers of rAAV containing wild type or mutant capsids were
determined by gene transduction assay. Briefly, Hela C12 cells were seeded in 24-well
plates the day before infection so that they would reach about 75% confluency the next
day. Serial dilutions of wild type and mutant preparations were added to the cells in the
presence of Ad5 at a multiplicity of infection (MOI) of 3. The cells and viruses were
incubated at 37°C for 48 hours. Cells expressing eGFP transgenes were detected by
fluorescence-activated cell sorting (FACS) analysis. Infectious titers were also
determined in the absence of adenovirus with no differences in the relative titers of
mutants.
3.8 Heparin-binding assay
3.8.1 POROS Beads
The ability of mutants to bind heparin sulfate was assessed by precipitation with
heparin sulfate affinity resin (POROS). Briefly, 293 cells that were transfected and
contains wild type and mutant capsids were centrifuged at 4,600 rpm for 3 minutes
and the pellet was resuspended in a 500µl of lysis/binding buffer which contains
TMN as described before with the addition of 0.5% DOC, Benzonase (50 unit/ml),
and 10µl of POROS HE-20 resin. The samples were incubated for 15 minutes at
37°C to ensure complete cell lysis and then gently mixed at 25˚C for 2 hours. The
resin was washed three times with a wash buffer (12.5 mM Tris, pH 8.0; 0.5mM

MgCl2; 100 mM NaCl). The virus was eluted by adding 500µl of elution buffer (20
mM Tris, pH 8.0; 1mM MgCl2; 600 mM NaCl) and then a Taqman/DRP analysis
was performed as described previously. Controls included precipitation of vectors
with wild-type capsids and precipitation of vectors with unconjugated resin.
3.8.2 Cell Binding Assay
The ability of mutants to bind to the cell surface of Hela cells utilizing
heparan sulfate was assessed. Testing the ability of mutants to bind to Hela cells
versus heparin coated plastic polystyrene beads will give an efficient and more
realistic measurement of binding. Hela cells have been proven to express heparan
on their cellular surface in cell culture. The cellular binding assay is testing the
ability of the virus to bind to cells, most likely using HSPG. Hela cells were
harvested and centrifuged at 2,000 x g for 3 minutes and the pellet was washed once
with DMEM without serum and then twice with ice-cold binding buffer (DMEM
with 2mM glucose, 10 mM HEPES [pH 8] and 1% BSA). AAV vector/virus was
added at the highest MOI as possible. The samples were incubated for 60-120
minutes at 4 ˚C occasionally mixing gently. After incubation, the samples were
washed and resuspended in 1x TMN buffer with 0.5% DOC and Benzonase (50
unit/ml). The samples were incubated for 15 minutes at 37˚C to ensure complete cell
lysis and then spun to clarify the lysate. Taqman/DRP analyses on various serial
dilutions were performed.

3.8.3 Affinity binding assay
The ability of mutants to bind heparan sulfate on Hela cells was assessed in
combination with a competitor. Similarly to the cell-binding assay, viruses were
tested to determine if they could bind to the surface of the Hela cells. After cell
lysis, the lysate was analyzed by a Taqman/DRP titer to determine the amount of
virus in the solution. The viral mutants in our laboratory contain a CMV promoter
that drives the enhanced green fluorescent protein and allows the lab to determine
the concentration of virus in Taqman. A CMV probe is used in the Taqman/DRP
analysis procedure that will pick up any amount of virus in a solution tested. The
competitor virus was a gift from Sferra laboratory, which does not contain the CMV
promoter. Thus the Taqman CMV probe will not pick up the virus. The virus
CC119 from Sferra’s lab is a rAAV2 Eflα eGFP at a concentration of 7.6x 1012
DRP/ml. The affinity assay was performed following the cell-binding assay, only
when the viral mutants were added, as was the competitor. The competitor was
added in an excess of 100-fold. The procedure was continued exactly as the cell-
binding assay.
All methodologies used in this study are well established in Dr. Bartlett’s lab at
CRI. The appropriate AAV plasmids, antibodies, and other reagents were all on
hand or commercially available. Dr. Bartlett is an assistant professor in the
Department of Pediatrics and Department of Molecular Virology, Immunology, and
Medical Genetics at The Ohio State University.
CHAPTER 4
RESEARCH RESULTS
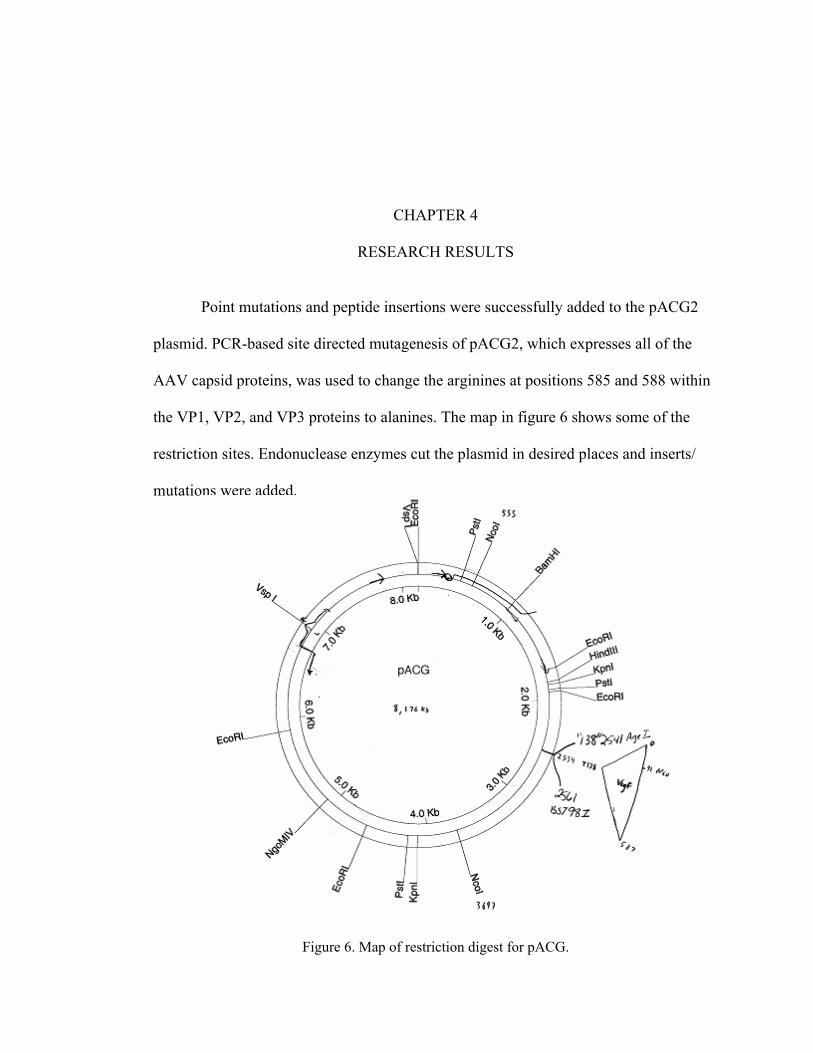
Point mutations and peptide insertions were successfully added to the pACG2
plasmid. PCR-based site directed mutagenesis of pACG2, which expresses all of the
AAV capsid proteins, was used to change the arginines at positions 585 and 588 within
the VP1, VP2, and VP3 proteins to alanines. The map in figure 6 shows some of the
restriction sites. Endonuclease enzymes cut the plasmid in desired places and inserts/
mutations were added.
Figure 6. Map of restriction digest for pACG.

The vector plasmid with a 585/588 point mutation of an arginine to an alanine of
the VP1, VP2, and VP3 capsid was screened, made, and peptide inserts were
successfully added. Peptides inserted that are of interest were a vascular endothelial
growth factor receptor 2 (VEGFR2) peptide. This would retarget the AAV virus to the
vasculature, which is of interest in our laboratory. Wild-type AAV does not transduce
the vasculature very well. Many potential benefits by targeting the vasculature can be
envisioned for gene therapy of cancers and cardiovascular diseases. Peptides include:
VEGFR2-A (VR2-A), VR2-B, VR2-C, VR2-D, and VR2-E. The peptide sequences for
the inserts were added, screened, and made. The inserted peptides will be displayed on
all three viral capsid proteins (VP1, VP2, and VP3). This allows for a large number of
peptides to be displayed, but only a limited size of peptides can be added. Below is the
actual peptides inserted into the 585/588 mutated pACG capsid.
VEGFR 2 A (VR2-A) 5’ CC GGT TGC GCC ACC TGG TTG CCC CCC CGC TGC GGC 3’ A ACG CGG TGG ACC AAC GGG GGG GCG ACG CCG AAT T
VR2-B 5’ CC GGT TGC TTG CCC CCC AAC CCC ACC AAG TGC GGC 3’ A ACG AAC GGG GGG TTG GGG TGG TTC ACG CCG AAT T
VR2-C 5’ CC GGT TGC TAC GCC ATC ATG CCC TTG GTG TGC GGC 3’ A ACG ATG CGG TAG TAC GGG AAC CAC ACG CCG AAT T
VR2-D 5’ CC GGT TGC ATG CAC TCC GAC ATG CAC GCC CCC GTG TCC GAC ATC TGC GGC 3’ A ACG TAC GTG AGC CTG TAC GTG CGG GGG CAC AGG CTG TAG ACG CCG AAT T
VR2-E 5’ CC GGT TGC CAC ACC ATG TAC TAC CAC CAC TAC CAG CAC CAC TTG TGC GGC 3’ A ACG GTG TGG TAC ATG ATG GTG GTG ATG GTC GTG GTG AAC ACG CCG AAT T
The pACG plasmid was cut with enzymes BST 98 I and AGE I for the above
VR2 peptides. The peptides VR2A-E were ordered with matching sticky ends shown

above as the underlined peptides, to match the cut plasmid. The peptides were annealed
into the capsid of the plasmid with the point mutations of the arginine amino acids (a.a.)
at positions 585 and 588, to alanines (R585A, R588A), and then ligated back together in
a PCR unit. The ligations were transformed into dsDNA, grown, and mini prep to look
for clones. The mini preps were digested with the enzymes BST 98I and VSP I, and ran
out on a gel. Figure 7 shows a few of the VR2 samples that were ran out and examined.
Figure 7: Photograph of restriction digest separated on 1.1% agarose gel. The vectors were cut using BST 98 I
and VSPI enzymes in the restriction digest.
The mini preps with arrows are the bands that looked promising based off of the ladder,
and were further sequenced to confirm the mutation.
Mutations of the AAV2 capsid were also undertaken in only the VP1, VP3
portion. VP2 was removed so larger peptides could be inserted into the plasmid, but

only displayed a limited amount of times. One of the pitfalls of the AAV capsid is its
small size, so it cannot withstand large peptide insertions. What is apparent is that the
VP2 capsid is not necessary for viral production. An AAV2 HS- VP1, 3 regions can be
combined with a separate VP2 region and still make virus. The VP2 protein can
withstand large insertions of peptides into its N-terminal region, like the VEGF
receptor. A larger VEGF insertion into the VP2 region has been under investigation, but
is still to be made. This would knock out heparin binding and allow the rAAV vector to
be retargeted to an area of interest. Once we get the VP2 regions with larger peptides to
incorporate properly back into the capsid protein, we can target areas in the body with
AAV2 that has never been targeted before.
HS-RGD plasmids were also made where an insertion of an Arg-Gly-Asp
(RGD) peptide following mutation of R585A, R588A in the normal AAV capsid
protein sequence. This RGD peptide allows the modified AAV vector to interact with
cell surface integrin receptors and without the use of HSPG. Current research has
demonstrated that the RGD peptide can successfully enhance the binding capacity of
rAAV regardless if HSPG was present. Unfortunately the RGD insertion in the
laboratory is not working and is currently under investigation. The HS- RGD AAV does
not bind heparan but it also does not infect cells either.
All of the mutated plasmids were transfected into 293 cells, harvested, purified
and tested for its ability to infect cells. Viruses were DRP titered to determine their
concentrations. A 24 well plate with 90% confluent Hela c12s cells were infected with a
MOI of 1000 of cell lysate virus of wild-type AAV, HS- AAV, and a control with no
virus were tested. Adenovirus was added to half of the wells with an MOI of 10 to show

the increase infectivity when a helper virus is present. The wild-type virus infected the
Hela c12 cells the highest with the helper virus and also without a helper virus. Only a
few cells were infected with the HS- AAV with helper virus and none without the
helper virus. The control was absent of infectivity with and without the helper virus.
The mutated viruses were tested for its ability to bind HSPG.
The ability of wild type AAV2 vectors, and heparan mutants AAV2 vectors to
bind to polystyrene beads coated with heparin was assessed. Every assay was performed
in triplicates to assure no variations between tubes. A wild type AAV2 vector, a heparan
mutant AAV2, and a control were run with every test. Variations in the original
protocol had to be undertaken due to unstable results. The original protocol called for
viral preps as crude lysates and after numerous repetitions of the assay, variations had to
be undertaken. Crude lysates is the viral particles straight out of the harvest cells. The
viral preparations are very “crude” in that cell debris, proteins, and salts are also present
in the lysate, which was a possibility for the unstable results. All of the crude lysate
viral preparations undergone iodixanol gradient purification which gave more reliable
results with lower standard deviations between samples.
Viruses that were tested for heparin binding included: wild-type AAV2, R585A
R588A AAV2 vectors, VEGFR2-targeted AAV2 vectors, AAV2 vector containing the
R585A, R588A mutation on only the VP1 and VP3 proteins, RGD AAV2 vectors, and
R585A R588A RGD AAV2 vectors. The RGD vectors did not behave as envisioned,
bringing on another twist onto why the vector was not infecting cells. RGD AAV2
vectors used to work in the lab by binding to cells whether heparan was present or not.
This would aid the AAV2 vector in binding when heparan sulfate proteoglycan may not

be present in the body. Unfortunately, the RGD vector recently stopped working and
investigation has been undertaken to troubleshoot the dilemma. The entire RGD vector
runs on the heparin resign were ignored due to the inconsistencies. All of the viral
vectors were allowed to bind to the polystyrene beads coated with heparin, washed, and
eluted off. While over 63 percent of the wild-type AAV2 was recovered in the elution,
less than 1 percent of the heparan mutants were found in the elution. Figure 8 is a graph
of the binding for an assay with wild type AAV2, R585A R588A AAV2 VR2B, and
AAV2 R585A R588A VP1, 3 plus VP2.
Heparin Binding
0
10
20
30
40
50
60
70
80
90
AAV2 R585A R588A AAV2VR2B
AAV2 R585A R588AVP1,3+VP2
Perc
en
t V
iru
s Elu
ted
Figure 8: Heparin binding using polystyrene beads coated with heparin and
iodixanol purified virus. Error bars indicate the standard deviation between the triplicate samples of each virus. Wild-type AAV2 bound 63%of the time while the R585A R588A AAV2 mutants bound less than 1%.

The above results demonstrated that I successfully knocked out heparan binding
in the AAV2 vectors. The viruses were next tested for their ability to bind to heparan
sulfate proteoglycan in a more realistic measurement. Cell binding of the viral preps to
Hela cells were examined for heparin binding. First experiments were run with 2.5 x 105
cells, which produced a weak pellet and were barely visible for washing. After the first
run, it was decided to start with 5.0 x 105 cells for a larger, visible pellet. All cells were
spun at 2,000 x g for three minutes and then washed with ice-cold DMEM without
serum one time. Next the cells were washed two times with a Binding Buffer and then
resuspended in a minimal volume. The AAV vectors were added to the appropriate
tubes at the determined MOI. The MOI for the first run with only 2.5 x 105 cells was
10,000 and MOI for second run was 5,000, resulting in 2.5 x 109 total virus added to all
test tubes in both runs. All tubes were run in triplicate alongside a control tube without
virus, consisting of just Hela cells. Due to unwanted background pickup caused by the
Taqman machine noticed in previous experiments with the polystyrene beads heparin
binding, serial dilutions were preformed on the viral samples to determine if any
significant binding occurred.
In the first experiment the viral samples were diluted 1:1, 1:10, and 1:100. All
viral samples that were diluted 1:100 were DRP titer around the same concentration as
the cell control tube. This suggests that the samples must contain greater than 107
amount of virus in order to give accurate results without interference with the CMV
probes or background clutter. Wild type AAV2, AAV2 R585A R588A VP1,3 VP2, and
a control were incubated in the harvested Hela cells at 4˚C for 60 minutes allowing for
binding to occur. The samples were washed three times to remove any unbound virus

and the cells were lysed with a lysis buffer. All of the clarified lysates were
Taqman/DRP analyzed for concentrations. Figure 9 demonstrates a linear chart of the
serial dilutions increasing in concentration from left to right. As the amount of virus
increased, the amount of virus recovered for wild type AAV2 also increased. The
heparan mutants continued to stay close to the cell control concentrations. This
concludes that the heparan mutants did not successfully bind to the Hela cells and
appeared similar to background being picked up by Taqman. The cell control should
have came back with an almost zero concentration due to the fact that no virus was
present and thus no CMV promoters were present to be picked up by the CMV probe.
Heparan Cell Binding 1
0
100000000
200000000
300000000
400000000
500000000
600000000
700000000
25000000 250000000 2500000000
Virus added
Vir
us
Bo
un
d
AAV2
HS-VP1,3+VP2Cell lysatewith no virus
Figure 9: Heparan Cell Binding experiment 1. Serial dilutions of the viral preps were from left to right,
1:100, 1:10, and 1:1. As the concentration increased of the viral preps, the amount of cellular binding that did occur also increased. Wild type AAV2 was the only virus that successfully bound to the Hela cells
and the heparan mutants and cell control can back similar to background pickup.

In the second experiment the viral samples were diluted 1:1, 1:10 and 1:50.
Since all viral samples from the first experiment that was diluted 1:100 came back as
just background pickup, a 1:50 dilution was undertaken to see if improvements could be
made. The amount of Hela cells was also increased from previous concentrations of 2.5
x 105 cells to 5.0 x 105 cells so a larger pellet could be seen to control the amount of
cells that was not aspirated off by accident. The viral samples were treated the same as
the first experiment and results are found in Figure 10 below.
Heparan Cell Binding 2
0
10000000
20000000
30000000
40000000
50000000
60000000
70000000
50000000 250000000 2500000000
Virus Added
Vir
us
Bo
un
d
AAV2
HS-VP1,3 +VP2
Cell lysate withno virus
Figure 10: Heparan Cell Binding experiment 2. Serial dilutions were preformed on the viral samples from left to right as 1:50, 1:10, and 1:1. As concluded from the first experiment as the amount of virus
concentration increases, the amount of background pickup decreases and viruses bound increases. AAV2 again was the only significant Hela cell binding when compared to the heparan mutants.

The ability of mutants to bind heparan sulfate on Hela cells was assessed in an
affinity assay in combination with a competitor. This affinity assay will determine the
amount of specific binding that is occurring to the Hela cells as compared to non-
specific binding. Specific binding of wild-type AAV should occur in the absence of a
competitor. AAV binds to its natural receptor HSPG that is expressed on the surface of
Hela cells. AAV should not demonstrate specific binding in the presence of a
competitor in a 100-fold excess that is competing for the same HSPG binding. The
R585A, R588A AAV should demonstrate non-specific binding since its ability to bind
to its natural receptor, HSPG, has been eliminated.
The protocol was set up similar to the cell binding assay, only in conjunction
with the AAV vectors made in Dr. Bartlett’s laboratory with a CMV promoter, another
AAV vector without the CMV promoter was added. The competitor virus called CC119
was a gift from Sferra’s lab, which was a rAAV2 Eflα eGFP at a concentration of 7.6x
1012 DRP/ml. To analyze the concentration of our viral vectors through the
Taqman/DRP titer, a CMV probe is utilized. The CMV probe picked up any virus that
bound to the Hela cells and a concentration would be given. This value is further
calculated out to determine the active concentration of the virus of every particle (DRP)
per microliters.
The viruses were tested to determine if they could bind to the surface of the Hela
cells in the presence of a competitor in 100-fold excess. The samples were allowed to
bind to the Hela cells at 4˚C for 120 minutes. After cell lysis, the lysates was analyzed
by a Taqman/DRP titer to determine the amount of virus in the solution. AAV2 did not
bind as well to the Hela cells similar to the heparan mutants in the cellular binding assay
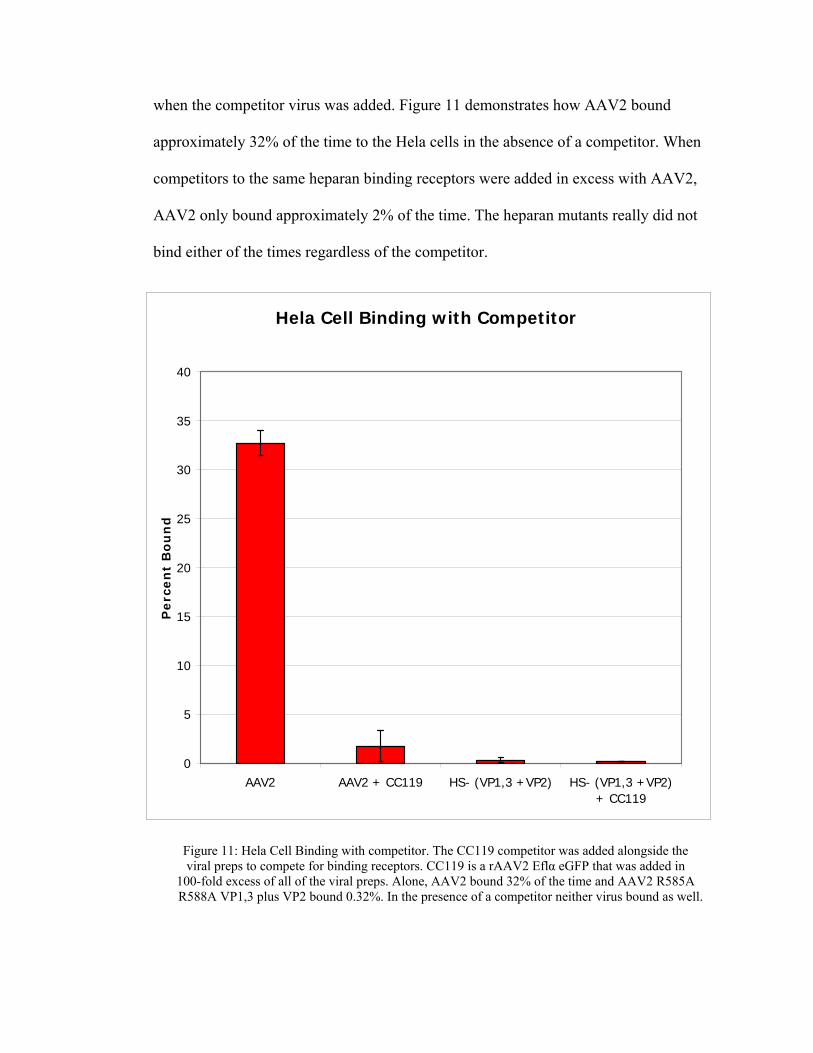
when the competitor virus was added. Figure 11 demonstrates how AAV2 bound
approximately 32% of the time to the Hela cells in the absence of a competitor. When
competitors to the same heparan binding receptors were added in excess with AAV2,
AAV2 only bound approximately 2% of the time. The heparan mutants really did not
bind either of the times regardless of the competitor.
Figure 11: Hela Cell Binding with competitor. The CC119 competitor was added alongside the viral preps to compete for binding receptors. CC119 is a rAAV2 Eflα eGFP that was added in
100-fold excess of all of the viral preps. Alone, AAV2 bound 32% of the time and AAV2 R585A R588A VP1,3 plus VP2 bound 0.32%. In the presence of a competitor neither virus bound as well.
Hela Cell Binding with Competitor
0
5
10
15
20
25
30
35
40
AAV2 AAV2 + CC119 HS- (VP1,3 +VP2) HS- (VP1,3 +VP2)+ CC119
Perc
en
t B
ou
nd

These preliminary findings suggest that AAV2 does bind to heparan, and that
the rAAV2 mutants were deficient in receptor binding. AAV2 demonstrates specific
binding in the absence of a competitor for the same receptor HSPG. In the presence of a
competitor, AAV2 demonstrates non-specific binding to the Hela cells. The heparan
mutants demonstrated non-specific binding to the Hela cells in both incidences when a
competitor was present and without. This truly demonstrates the elimination of HSPG
binding of the AAV vector. Eliminating the heparan binding portion of the AAV2
vector can lead to successful elimination of unwanted gene transfer and expression and
the retargeting of the rAAV2 vector to tissues that were once thought untargetable.

CHAPTER 5
DISCUSSION AND CONCLUSION
Modifying plasmids with R585A R588A point mutations has successfully
eliminated heparan sulfate proteoglycan binding of the AAV2 vector. Eliminating the
binding to natural receptor of AAV2 and introduction of new peptides improves the
gene transfer and expression of the virus to tissues of interest. AAV2 has the ability to
be the solution to all of the problems of gene therapy. Its ability to invade both dividing
and non-dividing cells, long term gene expression, reproducibility of high titers, and
lack of pathogenicity makes AAV2 a suitable vector for gene therapy. Retargeting
AAV2 to tissues of interests can improve the effectiveness and efficiency of gene
therapy for treating cancers and genetic disorders.
In vitro studies are conclusive that AAV2 with mutations at the R585A R588A
positions does not bind heparin on polystyrene beads, heparan expressing cells, and
specific binding has been eliminated in the presence of a competitor. The R585A
R588A AAV2 mutation does not infect cells, which is thought to be or probably due to
the lack of binding to the heparan expressing Hela cells. What is not apparent is whether
the modified viral vectors can bind to cells expressing their new peptide receptors.
Future studies will be tested on tissue culture cells that have been known to express the
various receptor sites on their surface. VEGF receptor binding ligands were added to the

modified AAV2 vectors deficient in receptor binding. Once tissue tropisms of the AAV
vector are deficient in receptor binding and retargeted to new wanted receptors, in vivo
studies can be investigated.
Future studies will examine the in vivo dissemination of the viral vectors
deficient in natural receptor binding developed in our laboratory. The modified vectors
should transduce cells with matching viral receptors. Gene expression of the modified
heparan mutants will have to be monitored and the levels of the vectors that are
expressed determined. The objectives of the original proposal were to test the modified
heparan mutants mainly in vivo. Goals were set high, but were not unobtainable. As
with any research topic, unforeseen obstacles surfaced. Troubleshooting and pondering
the problems that went wrong helped me to understand the reasoning behind what all I
did. If everything went as planned, I could have completed a lot more objectives
originally proposed, but then it would not be research. Albert Einstein already foresaw
the pitfalls in my research. He once said, “If we knew what it was we were doing, than
it would not be called research, would it?” If research was easy to complete in a short
amount of time, than everyone would be able to fix all of the problems and cancers in
the world. I was given a wonderful opportunity to complete something once believed
unimaginable. With all of my new gained knowledge I acquired from my research, I
feel even more lost in understanding it. As the old saying goes, "The more you know,
the more you realize how much you don't know."

CHAPTER 6
BIBLOGRAPHY
1. Bartlett, J.S., Wilcher, R., Samulski, R. “Infectious Entry Pathway of Adeno-Associated Virus and Adeno-Associated Virus Vectors.” Journal of Virology. 74(6): 2777-2785 (2000).
2. Shi, W., Bartlett, J.S. “RGD Inclusion in VP3 Provides Adeno-Associated Virus Type 2 (AAV2)- Based Vectors with a Heparan Sulfate-Independent Cell Entry Mechanism”. American Society for Gene Therapy: Molecular Therapy. 7-4: 515-525 (2003).
3. Opie, S. R., K. W. Warrington, Jr., et al. “Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding.” Journal of Virology. 77(12): 6995-7006 (2003).
4. Buller, R.M., Rose, J.A.. “Characterization of adenovirus-associated virus – induced polypeptides in KB cells.” Journal of Virology. 25:331-338 (1978).
5. Shi,W., et al. “Insertional mutagenesis of the adeno-associated virus type 2 (AAV2) capsid gene and generation of AAV2 vectors targeted to
alternative cell- surface receptors.” Human Gene Therapy 12:1697-1711(2001).
6. Warrington, K., et al. “ Adeno-associated Virus Type 2 VP2 Capsid Protein Is Nonessential and Can Tolerate Large Peptide Insertions at its N Terminus.” Journal of Virology. 78(12): 6595-6609 (2004).
7. Xie, Q., W. Bu, et al. “The atomic structure of adeno-associated virus (AAV2), a vector for human gene therapy.” Proc Natl Acad Sci USA 99(16): 10405-10 (2002).
8. Kronenberg, S, et al. “Electron cryo-microscopy and image reconstruction of adeno associated virus type 2 empty capsids.” EMBO reports 2(11): 997–1002 (2001).
9. Büchen-Osmond, C. “Dependovirus.” 2002. ICTVdB The Universal Virus Database. 18 March 2005. <http://www.ncbi.nlm.nih.gov/ICTVdb/ICTVdB/index.htm>.
10. Rabinowitz, J., Samulski, R.J. “Adeno-associated virus expression systems for gene transfer”. Current Opinion in Biotechnology.9:470-475 (1998).
11. Feuder, E., Alwis, M., Thrasher, A., Ali, R., Fauser, S. “Optimization of recombinant adeno-associated virus production using an herpes simplex

virus amplicon system.” Journal of Virological Methods. 96, 97-105 (2001).
12. Girod, A., M. Ried, et al. “Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2[published erratum appears in Nat Med 1999 Dec; 5(12): 1438].” Nature Medicine 5(9): 1052-6 (1999).
13. Müller, O., et al. “Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors.” Nature Biotechnology. 21(9): 1040-1046 (2003).
14. Kern, A., K., Schmidt, et al. “Identification of a heparin-binding motif on adeno-associated virus type 2 capsids.” Journal of Virology. 77(20): 11072-11081 (2003).
15. Nicklin, S.A., et al. “Efficient and selective AAV2-mediated gene transfer directed to human vascular endothelial cells.” Mol. Ther. 4(3) : 174-181(2001).
16. Xiao, X., Li, J., and Samulski, R. “Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus.” Journal of Virology. 72: 2224-2232 (1998).
17. Li, J., Samulski, R.j., and Xiao, X. “Role for highly regulated rep gene expression in adeno-associated virus vector production.” Journal of Virology.71: 5236-5243 (1997).
18. Clark, K., Voulgaropoulou, F., and Johnson, P. “A stable cell line carrying adenovirus-inducible rep and cap genes allows for infectivity titration of adeno-associated virus vectors.” Gene Therapy. 3:1124-1132. (1996).
19. Jordan, M., et al. “Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation.” Nucleic Acids Res. 24: 596-601(1996).
20. Zolotukhin, S. “Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield.” Gene Therapy 6: 973-985. (1999).
21. Clark, K. R., Liu, X., McGrath, J.P., Johnson, P.R., “Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Human Gene Therapy 10, 1031-1039.