thermodynamics of viscous flow and elasticity of glass forming liquids in the glass transition range
TRANSCRIPT

THE JOURNAL OF CHEMICAL PHYSICS 135, 184501 (2011)
Thermodynamics of viscous flow and elasticity of glass forming liquidsin the glass transition range
T. Rouxela)
LARMAUR, ERL CNRS 6274, Université de Rennes 1, Campus de Beaulieu, 35042 Rennes cedex, France
(Received 14 July 2011; accepted 9 October 2011; published online 8 November 2011)
The elastic moduli of glasses from different chemical systems, including oxide, chalcogenide,oxynitride, and metallic, were investigated through the glass transition (Tg), typically from 0.4 to1.3 Tg. These data were used to interpret the temperature sensitivity of the shear viscosity coefficientobtained on the same materials. The relevant Gibbs free activation energy was estimated from theapparent heat of flow by means of the temperature dependence of the shear elastic modulus. Theactivation entropy associated with the viscous flow was also derived and was found to correlatewith the fragile versus strong character of the glass forming liquids. Finally, the physicochemistryof the flow process was described on the basis of the glass network de-structuration which showsup through the temperature dependence of Poisson’s ratio, and an expression for the shear viscositycoefficient is proposed which is chiefly based on the high temperature elastic behavior. © 2011American Institute of Physics. [doi:10.1063/1.3656695]
I. INTRODUCTION
Viscous flow is a common property of glass forming liq-uids which is essential for processing and shaping glass partsand is thus largely responsible for the immense success of thisclass of materials. However, the understanding of the physicsand the chemistry of this irreversible deformation processis still far from reaching the refinement of the dislocationstheory developed for crystalline materials. In particular, thereare only mainly empirical arguments for the broad range oftemperature sensitivity differences stemming from differentglass compositions. A reason for the description being es-sentially phenomenological lies in the fact that it is very dif-ficult to get a direct insight—or an in situ analysis—of themicroscopic events at the source for the deformation process.This situation opened the field to a large variety of modelsbased on hypothetical structural perturbations. In this study,we start from a thermodynamic description of viscosity con-sidered as a thermally activated process. We especially em-phasize the omnipresence of the elastic characteristics andcautiously avoided adding questionable structural ingredientswhich would narrow the frame of the analysis. In Adam andGibb’s (AG) (Ref. 1) approach of the viscous flow process,the temperature dependence of relaxation phenomena in glassforming liquids is correlated to the temperature dependence ofthe size of the cooperatively rearranging region (z: number ofmolecules or monomeric segments) and to the height (!g)of the potential energy hindering the cooperative rearrange-ment of the elementary structural units involved in the process
τ = τo exp[z!g/(RT)], (1)
where τ is the characteristic relaxation time, τ o is a constant,T is temperature, and R the perfect gas constant.
a)Electronic mail: [email protected]. Tel.: +33-2-23236718.Fax: +33-2-23236111.
Assuming a simple Maxwell relaxation model, this givesthe following expression for the shear viscosity coefficient
η = µτ o exp[z!g/(RT)], (2)
where µ is the shear elastic modulus.In AG model, z is inversely proportional to the configu-
rational entropy Sc and !g is taken as a temperature indepen-dent term. Consequently, Eq. (2) can be rewritten as
η = ηo exp [!G/TSc]. (3)
Subsequent papers on the AG model for viscous flow also as-sumed !g ((or !G) is temperature independent, and mostlyfocused on the expression for Sc, systematically neglectingthe temperature dependence of µ. Starting from the premiseof the AG model, namely kinetics of thermally activated co-operative rearrangements, the purpose of this paper is to shedsome light on the close relationships existing between thetemperature dependence of the elastic moduli and the viscos-ity, based on the following statement
(i) As in the case of plasticity in crystalline materials, theenergy barrier for viscous flow chiefly depends on theshear elastic modulus.2 Therefore µ comes into play bothin the prefactor (Eq. (3)) and implicitly in !G.
(ii) Glassy materials differ from crystalline ones by a rapidsoftening in the supercooled liquid range (i.e., forT > Tg), so that µ is mostly very sensitive to temperature.Consequently, !G depends much on temperature andthis can be analyzed within the classical framework ofthermodynamics, recalling that the entropy change writes
!S = − ∂!G∂T
∣∣∣∣σ
. (4)
(iii) The configurational entropy in Eq. (3) is a temperaturedependent parameter (especially for T > Tg) which iscommonly assimilated to the excess entropy (directly
0021-9606/2011/135(18)/184501/15/$30.00 © 2011 American Institute of Physics135, 184501-1
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-2 T. Rouxel J. Chem. Phys. 135, 184501 (2011)
available from calorimetry). Since both vibrational andconfigurational entropy contribute to the overall entropychange, it is difficult to extract the configurational part. Itis often assumed either that the constant pressure specificheat capacity Cp is temperature independent and that aglass (T < Tg) and a corresponding liquid (T > Tg) havethe same vibrational entropy, so that
Sc(T) = Sc(Tg) +∫ T
Tg
CLiqp − CGlass
p
TdT, (5)
where Sc(Tg) (≈Sc(0)) can be obtained from calorimetricmeasurements.3 Since Cp is related to the elastic moduli(through Debye’s temperature for instance4) and since liquidsmoduli suffer from larger temperature sensitivities, such as-sumptions are discussed in this paper, especially in the caseof weakly cross-linked glass atomic networks, or of fragileliquids.
It becomes clear that the high temperature elastic moduliand the viscosity of glass forming liquids are strongly corre-lated. In order to determine general trends, materials cover-ing a wide range of glass transition temperature, from muchdifferent chemical systems, and with very different networkconnectivity were studied.
II. MATERIALS
The investigations were limited to 18 glass formingliquids, covering a broad spectrum of chemical systems andatomic bonding types (Van der Waals, metallic, ionic, andcovalent) for which both reliable elasticity (especially shearmodulus) and viscosity data were reported as a function oftemperature through the transition range, typically from 0.8to 1.3 Tg. Those include GexSe1−x glasses, with x between0 and 0.3. These glasses were obtained from high purityelements Ge (99.9999%) and Se (99.999%), following acautious procedure described in details in Refs. 5 and 6 inorder to avoid oxygen contamination and to obtain homoge-neous, x-ray amorphous materials. The studied vitreous silicais a commercially available glass (Vitreosil, Saint-Gobain).ZBLAN is a fluoride glass with the following composition:ZrF4(53)BaF2(20)LaF3(4)AlF3(3)NaF3(3)NaF(20),7, 8 wherenumbers in brackets here and below are molar or atomicpercentages. Data reported on amorphous boron oxide(a-B2O3) are also analyzed.9, 10 Unlike cristallized B2O3
or a-SiO2 consisting of a tri-dimensional cross-linked, a-B2O3
has relatively low elastic moduli and Tg because the atomicnetwork is built on corner-sharing BO3 planar triangles whichare weakly bonded together. Some medium range ordershows up in a-B2O3 with the occurrence of boroxol ringscomposed of three corner sharing BO3 triangles forming anequilateral triangle.11 Data reported on three basalt-type alu-minosilicate compositions, namely Ca3Al2Si3O12(grossular),CaAl2Si2O8(anorthite), and CaMgSi2O6(diopside),3, 12 whichare characterized by relatively highly cross-linked networks,and two (Ca, Na) aluminosilicate glasses13—Aluminosilicate1: SiO2(60.06)Al2O3(14.9)CaO(15.8)Na2O(5.64)Fe2O3(3.4)and Aluminosilicate 2: SiO2(57.15)Al2O3(17.32)CaO(15.0)
Na2O(6.92)Fe2O3(3.4)—were analyzed. In silicate glasses,the partial substitution of oxygen for nitrogen results ina significant enhancement of both the refractoriness (Tg
reaches typically 1200 K) and the mechanical properties.14–16
However, interestingly these glasses behave as rela-tively “fragile” liquids (following the concept introducedby Angell),17 with apparent activation energy for vis-cous flow typically reaching 1000 kJ mol−1 at Tg. Twooxynitride glass samples, referred to as Oxynitride 1:Y(12.31)Si(18.46)Al(7.03)O(54.73)N(7.47) and Oxyni-tride 2: Y(10.99)Si(18.46)Al(8.35)O(54.73)N(7.47), wereinvestigated. In equivalent percent charge concentration18
these compositions write Y(28)Si(56)Al(16)O(83)N(17) andY(25)Si(56)Al(19)O(83)N(17), respectively. Two composi-tions of bulk metallic glasses (BMG), namely Zr(55)Cu(30)Al(10)Ni(5) (Refs. 19 and 20) and Pd(42.5)Cu(30)Ni(7.5)P(20) (Refs. 21 and 22) were investigated in this work. Theseglasses are characterized by a very high atomic packingdensity, and thus by relatively high elastic moduli (typicallyabove 80 GPa for Young’s modulus) although their Tg liebelow 700 K. These glasses are known to lack structural unitsof high order of symmetry. It has been suggested that theatomic network of such glasses consists of quasi-equivalentcluster-type units eventually packed with icosahedral-likemedium range order.23, 24 Data reported for a standard win-dow glass—Si(0.25)Na(0.092)Ca(0.035)Mg(0.021)O(0.6)—(Planilux, Saint-Gobain) and glycerol were also includedin this analysis.
It is noteworthy that in most cases, the same glass batcheswere used for both high temperature elasticity and viscositymeasurement.
III. EXPERIMENTAL METHODS
High temperature elasticity data were obtained by meansof three different methods, depending on the glass system andon the temperature range. These methods are, namely: Ultra-sonic echography (USE), direct mechanical vibration (DMV),including mechanical spectroscopy and Brillouin scattering(BS). Owing to the high loading frequencies involved (espe-cially for USE and BS), E, µ, and ν can be understood as E∞,µ∞, and ν∞, i.e., as the frequency-independent “solid-like”moduli (even for the glass forming melts within the tempera-ture range of concern).
USE: Beside the standard measurements using piezoelec-tric transducers and assuming infinite specimen transversedimension,25 a long beam mode set-up is particularly suitablefor high temperature investigations. In this latter cases, theprinciple of the method26, 27 consists in calculating the elasticmoduli from the velocity of an ultrasonic pulse propagating ina long, thin, and refractory wave-guide sealed to a specimen(about 1 × 3 × 40 mm3), using either a longitudinal wave (forE) or a torsional wave (for µ). Magnetostrictive transducers(100–300 kHz) are used and experiments are performedunder argon atmosphere with a heating rate of 5 K min−1
from room temperature and up to 1673 K. In the present case,the longitudinal wave velocity is about 10 km sec−1, and itfollows that the wavelength is about 33 mm, that is muchlarger than the characteristic dimension of the specimen cross
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-3 Viscous flow and elasticity of glass J. Chem. Phys. 135, 184501 (2011)
section. The condition of very long wavelength in comparisonto the scale of the microstructure is thus satisfied, and thematerial response is governed by the effective properties ofthe equivalent homogeneous medium. Furthermore, the longbeam mode approximation holds, so that E can be calculatedfrom the density (ρ) and the longitudinal wave velocity (Vl)according to26
E = ρV21. (6)
As usual µ is given by
µ = ρV2t , (7)
where Vt is the transverse wave velocity.Poisson’s ratio (ν) is then calculated as follows:
ν = E/(2µ) − 1. (8)
The room temperature elastic moduli were first accuratelymeasured by means of piezo-electric transducers (10 MHz)in direct contact with the specimen. Since the density doesnot vary more than a few per cent over the range of these ex-periments, the velocity changes recorded are primarily due tothe effect of temperature on the modulus of elasticity, as wasalready noticed by Ide.28 This technique allows for reliablecontinuous in situ measurements with a better than ±1 GPa.
DMV: Young’s modulus can also be determined atelevated temperature by means of mechanical vibration tech-niques, including torsion pendulum such as in mechanicalspectroscopy and mechanical resonance frequency measure-ments in rectangular bars.29, 30 This latter method allowsto perform experiments at 1 K mn−1 under high vacuum(10−4 Pa) up to 1300 K without any harmful contact, thesample beam being maintained horizontally between steelwires located at the vibration nodes. Furthermore, excitationand detection are insured by an electrostatic device (capaci-tance created between the sample and a single electrode). Theaccuracy of this method is better than 0.5% for conductingbulk materials whatever the rigidity range. Young’s modulus(E) is expressed as29
E = 0.9464ρF2B(t
L4(t))h2
T(h/L, ν), (9)
where FB is the resonance frequency in bending mode, ρ thespecific mass, ν Poisson’s ratio, h and L, the beam thicknessand span length, and T(h/L,ν) a correcting factor close to 1.
The shear modulus of plates (typically 30 mm × 12 mm× 1.5 mm) can be measured in torsion mode30
µ = 4ρ
RL2F2T
, (10)
where R is a shape factor equal to 17.51 in the present case, ρ
is the specific mass, L is the length of the plate-like specimen,and FT is the torsion resonant frequency.
Resonance excitation and detection are obtained elec-trostatically using a capacitance geometry enabling elas-tic or viscoelastic moduli to be recorded by analyzing theresonance.21, 31, 32
BS: In Brillouin scattering measurements12,33–35 the ul-trasound velocities of the longitudinal and transverse modes(Vl,t) are related to the incident light wavelength (λo), to the
scattering angle (φ), to the refractive index of the material(nr), and to the frequency shift !fl,t resulting from the energyconversion according to
Vl,t=!fl,tλo/(2nrsin(φ/2)). (11)
Elastic moduli are then derived by means of the classicalequations for linear elasticity of isotropic media. In mostcases the scattered light is detected at 90◦ to the incident light(φ = 90◦). Note that with a peculiar set-up consisting in twoincident beams intersecting at a right angle and with the spec-imen being placed in the median plane (i.e., with a 45◦ anglebetween the normal to the specimen surface and the incidentbeams), it is possible to estimate the velocity without knowingthe value for nr. High temperature measurements are gener-ally done by suspending the specimen in a small furnace withtwo slits at 90◦ one from the other allowing for the incidentlight beam to enter and for the scattered light to reach the col-limator. Heating is mostly achieved by means of a platinumcoil.
The shear viscosity coefficient (η) was derived eitherfrom a laboratory-made instrumented indentation testing (IIT)dedicated to penetration viscosimetry and consisting in a hotchamber equipped with an alumina tube and a sapphire inden-ter, and allowing for a better than 2 K accuracy along a 10 mmtesting zone,36 or from creep testing (CT) (either in compres-sion, tension (fiber elongation), bending, or torsion (parallel-plate)). In the case of IIT, experiments were performed inthe micro-indentation range, with applied loads between 0.01and 15 N. The whole equipment is situated in a vibration-free and air disturbance-free environment. The load (P) is ap-plied using a piezoelectric actuator and the penetration depthis measured with a capacitive sensor having a resolution of10 nm. The load fluctuation is less than ±12 mN. The max-imum target temperature is 1473 K, with a thermal stabilitywithin 1 K variation up to 1323 K. The shear viscosity coef-ficient (η) was estimated from experiments conducted in airusing a ball indenter (750 µm radius (R)) and is given by36–38
η = 3P
16√
R
(d(u3/2(t))
dt
)−1
, (12)
where u is the penetration depth.In the experimental set-up, the load axis is slightly shifted
from the central axis of symmetry, enabling several indents ona single specimen by rotating the alumina tube by an appro-priate angle. Recall that the viscous flow regime is associatedto the stationary creep regime following the viscoelastictransient one. Consequently, the load was maintained longenough (typically 1 min above Tg to more than 1 h for pointsrecorded below Tg) to insure the occurrence of a stationarycreep regime, as evidenced by a constant slope in the u3/2/Pversus t curves. This problem is particularly critical as soonas measurements are carried out below Tg, i.e., in a rangewhere the characteristic relaxation time increases rapidly andcompares with the experimental duration. The specimen andthe indentation set-up were kept at each testing temperaturefor 4 h before loading to ensure a thermal equilibrium.
CT: In the case of creep experiments, the viscosity isestimated from the strain-rate (dε/dt) associated with the
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-4 T. Rouxel J. Chem. Phys. 135, 184501 (2011)
FIG. 1. Temperature dependence of shear modulus (µ). The curve fitting atT > Tg corresponds to µ/µ(Tg) = (T/Tg)α .
stationary creep regime, following the standard viscosityrelationship
η = σ/[2(1 + ν)dε/dt], (13)
where σ is the normal stress and ν is Poisson’s ratio.The expression for σ depends on the loading configu-
ration. For a compressive experiment or a fiber elongationtest σ = P/So
2 where P is the axial force and So the crosssection area. Under the assumption of an isochoric flow, theformer equation reduces to the classical Trouton’s law: η
= σ /(3dε/dt). Note that this expression is also valid in thecase of strain-rate imposed experiments such as for fiber elon-gation tests.
IV. EXPERIMENTAL RESULTS
A. Temperature dependence of the shear elasticmodulus and Poisson’s ratio
High temperature shear elastic modulus data wereextracted from previously reported high temperatureinvestigations7, 12, 13, 18, 21, 29, 39–46 and are drawn together inFigs. 1 and 2. The corresponding experimental methods areindicated in Table I. In all cases but a-SiO2 the temperaturedependence of the shear modulus exhibits a clear transitionrange between a slow softening rate and a faster one corre-sponding within 15◦ to Tg as obtained by classical meanssuch as DSC and dilatometry. Tg temperatures as measuredfrom µ(T) curves stemming from USE are usually smaller by10–20 K than the ones obtained by dilatometry with the sameheating rate. This is quite surprising since the USE methodsupposes fast displacements of atoms (300 kHz transducersare used) and is thus more dynamic by nature (Tg is expectedto increase with the loading rate or frequency). Although atentative explanation is proposed further, additional investi-gations are required to clarify this point. The elastic moduliexhibit only minor changes between room temperature (RT)and Tg. Their values at Tg are typically more than 80%of their RT values. The glass transition temperatures as
FIG. 2. Temperature dependence of Poisson’s ratio (note that temperatureis normalized to Tg as estimated from the µ(T) data). Data for bitumen areredrawn from Ref. 39.
well as the room temperature values of µ and the softeningrates immediately below and above Tg (dµ/dT(Tg
−) anddµ/dT(Tg
+)), i.e., in the glassy and in the supercooled liquidranges respectively, are reported in Table I. In most casesthe high temperature (T>Tg) shear modulus data could besmoothly fitted by a power law expression (Fig. 1)
µ/µ(Tg) = (Tg/T)α, (14)
where α ranges between 0.07 (a-SiO2) and 10 (a-Se)(Table I).
For a-SiO2, µ changes little (eventually increases athigh T) with temperature above Tg and for T/Tg > 1.05(T > 1530 K) it is likely that some crystals form (such asβ-cristobalite) and alter the behavior. It is noteworthy thatthe temperature at which the curve describing the elastic-ity of the supercooled liquid departs from the experimentaldata upon cooling as the transition range is reached is fewkelvins higher than the Tg temperature mentioned above andis in better agreement with DSC or thermal expansion val-ues. Alternate analytical expressions for µ(T) are discussed inRef. 47 and in Sec. VI D. The temperature dependence ofYoung’s modulus for the investigated glasses was already dis-cussed by the present author in a former paper,39 and for rela-tively stiff glasses with E > 10 GPa and for Tg ≤ T ≤ 1.1Tg,it was found that
E = E(Tg)Tg/T. (15)
High temperature Poisson’s ratio (ν) data (Fig. 2) wereobtained by combining both E(T) and µ(T) results usingEq. (8).
As soon as glasses from radically different chemicalsystems are compared, there is no direct correlation be-tween µ(Tg) (or µ(RT)) and Tg, simply because elastic mod-uli represent volume density of energy as explicitly for-mulated by the 1st Grüneisen rule, K ∼ Uo/Vo (K: bulk
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
184501-5 Viscous flow and elasticity of glass J. Chem. Phys. 135, 184501 (2011)
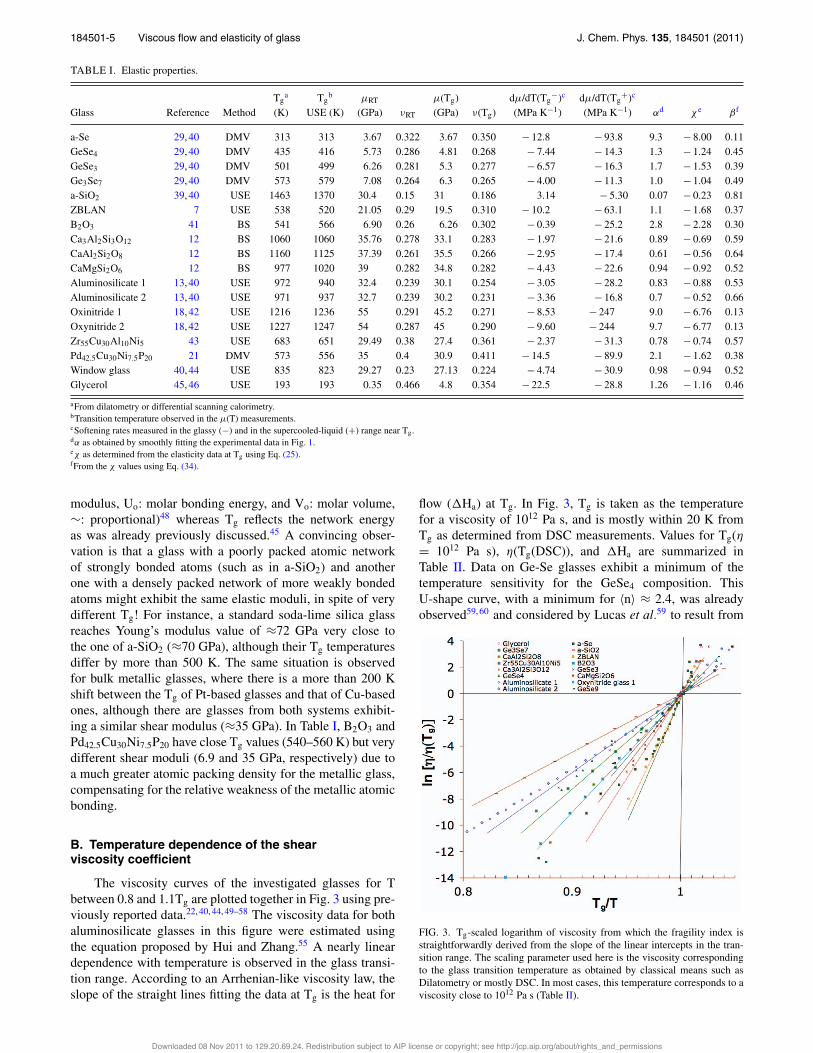
TABLE I. Elastic properties.
Tga Tg
b µRT µ(Tg) dµ/dT(Tg−)c dµ/dT(Tg
+)c
Glass Reference Method (K) USE (K) (GPa) νRT (GPa) ν(Tg) (MPa K−1) (MPa K−1) αd χ e βf
a-Se 29, 40 DMV 313 313 3.67 0.322 3.67 0.350 − 12.8 − 93.8 9.3 − 8.00 0.11GeSe4 29, 40 DMV 435 416 5.73 0.286 4.81 0.268 − 7.44 − 14.3 1.3 − 1.24 0.45GeSe3 29, 40 DMV 501 499 6.26 0.281 5.3 0.277 − 6.57 − 16.3 1.7 − 1.53 0.39Ge3Se7 29, 40 DMV 573 579 7.08 0.264 6.3 0.265 − 4.00 − 11.3 1.0 − 1.04 0.49a-SiO2 39, 40 USE 1463 1370 30.4 0.15 31 0.186 3.14 − 5.30 0.07 − 0.23 0.81ZBLAN 7 USE 538 520 21.05 0.29 19.5 0.310 − 10.2 − 63.1 1.1 − 1.68 0.37B2O3 41 BS 541 566 6.90 0.26 6.26 0.302 − 0.39 − 25.2 2.8 − 2.28 0.30Ca3Al2Si3O12 12 BS 1060 1060 35.76 0.278 33.1 0.283 − 1.97 − 21.6 0.89 − 0.69 0.59CaAl2Si2O8 12 BS 1160 1125 37.39 0.261 35.5 0.266 − 2.95 − 17.4 0.61 − 0.56 0.64CaMgSi2O6 12 BS 977 1020 39 0.282 34.8 0.282 − 4.43 − 22.6 0.94 − 0.92 0.52Aluminosilicate 1 13, 40 USE 972 940 32.4 0.239 30.1 0.254 − 3.05 − 28.2 0.83 − 0.88 0.53Aluminosilicate 2 13, 40 USE 971 937 32.7 0.239 30.2 0.231 − 3.36 − 16.8 0.7 − 0.52 0.66Oxinitride 1 18, 42 USE 1216 1236 55 0.291 45.2 0.271 − 8.53 − 247 9.0 − 6.76 0.13Oxynitride 2 18, 42 USE 1227 1247 54 0.287 45 0.290 − 9.60 − 244 9.7 − 6.77 0.13Zr55Cu30Al10Ni5 43 USE 683 651 29.49 0.38 27.4 0.361 − 2.37 − 31.3 0.78 − 0.74 0.57Pd42.5Cu30Ni7.5P20 21 DMV 573 556 35 0.4 30.9 0.411 − 14.5 − 89.9 2.1 − 1.62 0.38Window glass 40, 44 USE 835 823 29.27 0.23 27.13 0.224 − 4.74 − 30.9 0.98 − 0.94 0.52Glycerol 45, 46 USE 193 193 0.35 0.466 4.8 0.354 − 22.5 − 28.8 1.26 − 1.16 0.46
aFrom dilatometry or differential scanning calorimetry.bTransition temperature observed in the µ(T) measurements.cSoftening rates measured in the glassy (−) and in the supercooled-liquid (+) range near Tg.dα as obtained by smoothly fitting the experimental data in Fig. 1.eχ as determined from the elasticity data at Tg using Eq. (25).fFrom the χ values using Eq. (34).
modulus, Uo: molar bonding energy, and Vo: molar volume,∼: proportional)48 whereas Tg reflects the network energyas was already previously discussed.45 A convincing obser-vation is that a glass with a poorly packed atomic networkof strongly bonded atoms (such as in a-SiO2) and anotherone with a densely packed network of more weakly bondedatoms might exhibit the same elastic moduli, in spite of verydifferent Tg! For instance, a standard soda-lime silica glassreaches Young’s modulus value of ≈72 GPa very close tothe one of a-SiO2 (≈70 GPa), although their Tg temperaturesdiffer by more than 500 K. The same situation is observedfor bulk metallic glasses, where there is a more than 200 Kshift between the Tg of Pt-based glasses and that of Cu-basedones, although there are glasses from both systems exhibit-ing a similar shear modulus (≈35 GPa). In Table I, B2O3 andPd42.5Cu30Ni7.5P20 have close Tg values (540–560 K) but verydifferent shear moduli (6.9 and 35 GPa, respectively) due toa much greater atomic packing density for the metallic glass,compensating for the relative weakness of the metallic atomicbonding.
B. Temperature dependence of the shearviscosity coefficient
The viscosity curves of the investigated glasses for Tbetween 0.8 and 1.1Tg are plotted together in Fig. 3 using pre-viously reported data.22, 40, 44, 49–58 The viscosity data for bothaluminosilicate glasses in this figure were estimated usingthe equation proposed by Hui and Zhang.55 A nearly lineardependence with temperature is observed in the glass transi-tion range. According to an Arrhenian-like viscosity law, theslope of the straight lines fitting the data at Tg is the heat for
flow (!Ha) at Tg. In Fig. 3, Tg is taken as the temperaturefor a viscosity of 1012 Pa s, and is mostly within 20 K fromTg as determined from DSC measurements. Values for Tg(η= 1012 Pa s), η(Tg(DSC)), and !Ha are summarized inTable II. Data on Ge-Se glasses exhibit a minimum of thetemperature sensitivity for the GeSe4 composition. ThisU-shape curve, with a minimum for 〈n〉 ≈ 2.4, was alreadyobserved59, 60 and considered by Lucas et al.59 to result from
FIG. 3. Tg-scaled logarithm of viscosity from which the fragility index isstraightforwardly derived from the slope of the linear intercepts in the tran-sition range. The scaling parameter used here is the viscosity correspondingto the glass transition temperature as obtained by classical means such asDilatometry or mostly DSC. In most cases, this temperature corresponds to aviscosity close to 1012 Pa s (Table II).
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-6 T. Rouxel J. Chem. Phys. 135, 184501 (2011)
TABLE II. Viscous flow properties in the transition range.
!Ha (Tg) !Ga (Tg) !Sa (Tg)Tg
a η (Tg)b (Eq. (26)) (Eq. (28)) (Eq. (27)) !Ga (Tg)d !Sa (Tg)d V*(Tg)d
Glass Reference Method (K) (×1012 Pa s) (kJ mol−1) (kJ mol−1) (J K−1 mol−1) mc (kJ mol−1) (J K−1 mol−1) (×10−6 m3 mol−1)
a-Se 40 IIT 301 0.2 369 41 1090 64 95 910 11.2GeSe9 40 IIT 356 0.2 249 x x 37 112 383 xGeSe4 40 IIT 430 1.3 243 109 312 30 136 249 22.6GeSe3 40 IIT 492 0.5 256 101 315 27 155 204 19.1Ge3Se7 40 IIT 580 3 294 144 258 26 183 191 22.9a-SiO2 49 CT 1463 0.8 510 413 66 18 462 33 13.3ZBLAN 50 CT 538 0.07 781 291 910 76 170 1136 14.9B2O3 50, 93 CT 541 0.4 385 117 495 37 171 396 18.7Ca3Al2Si3O12 51 CT 1060 1 1096 649 422 54 335 718 19.6CaAl2Si2O8 52 CT 1160 1 1014 648 315 46 367 558 18.3CaMgSi2O6 53, 54 CT 1005 1 875 455 418 45 318 555 18.2Aluminosilicate 1 63 x 940 0.25 484 257 241 27 297 199 8.6Aluminosilicate 2 63 x 937 0.25 484 318 177 27 296 201 10.5Oxinitride 1 18 CT 1216 1.9 1115 144 799 48 384 601 3.2Oxynitride 2 18 CT 1227 1.9 1114 143 791 47 388 592 3.2Zr55Cu30Al10Ni5 56 CT 683 8 428 245 267 33 216 311 9.0Pd43Cu27Ni10P20 22 CT 573 1 708 270 769 65 180 928 8.8Window Glass 36, 44 CT-IIT 835 3 571 295 331 36 264 368 10.9Glycerol 57, 58 CT 193 3 253 117 704 68 61 995 24.4
x: non-measured.aGlass transition temperature taken as the temperature at which η = 1012 Pa s.bViscosity at Tg as (mostly) determined by DSC.cAs defined by m = dlog10η/d(T(η = 1012 Pa s)/T) = !Ha/(2.303RTg) (Ref. 17).dFrom the approach proposed by Nemilov,64–66 using Eqs. (29), (30) and (36) and µ(Tg).
the percolation of a rigid phase through a floppy phase,in agreement with a bimodal phase structure consistingof over-constrained GeSe2 domains and under-constrained(floppy) Sen chain- or ring-like units. The relatively strongtemperature sensitivity of the viscosity at large germaniumcontents, with a fragility index remaining as large as ≈27 forboth GeSe3 and Ge3Se7, stems from the fact that the break-down of the cross-linked network structure (disappearance ofthe medium range order) above Tg favors shear in-betweenstructural units, which are no more strongly interconnected.Note that there are high Tg glasses such as basalt andoxynitride glasses (Tg > 1000 K) which show an astonishingtemperature sensitivity, with !Ha over 1000 kJ mol−1,and m values around 50, while there are much less refractoryglasses such as Zr-based BMG’s and Ge-Se glasses withmore than 20% Ge exhibiting heats for flow smaller than500 kJ mol−1 and m values below 35. There is hence nostraightforward relationship between Tg, !Ha, and m.
V. THERMODYNAMICS OF THE VISCOUS FLOWPROCESS
Let’s !Gb be the height of the energy barrier for viscousflow. External forces (work !Wa) may contribute to reducethis height hence favoring flow, so that following the theoryof thermally activated phenomena as introduced by Boltz-mann the relaxation rate (corresponding to the activation rate,i.e., the fraction of successful jumps per unit time) writes
1/τ ∼ exp[− (!Gb − !Wa)
RT
](∼: proportional),
(16)
where τ is a relaxation time, R is the perfect gas constant, and!Wa is the mechanical work.
In a first approximation !Wa can be simply expressed as!Wa = σVa, where σ is a shear stress and Va is an activationvolume. Both temperature (thermal activation !Ga) and stressact to overcome the energy barrier (!Gb = !Ga+ !Wa) sothat considering a simple Maxwell model for the relaxationprocess (τ = η/µ),
η ∼ µ exp[!Ga
RT
], (17)
where !Ga is the free activation enthalpy for flow.It is noteworthy in this expression that the temperature
sensitivity of the exponential term strongly predominates overthe temperature dependence of the shear modulus (recallthat η decreases of orders of magnitude over ten of degreeswhereas µ mostly decreases by less than 50% over the sametemperature interval). Thus the former expression results inthe classical Boltzmann-Arrhenius expression for the shearviscosity coefficient
η = ηo exp[!Ga
RT
], (18)
where ηo is a weakly temperature dependent term often con-sidered as a constant.
It must be emphasized that !Ga is by essence atemperature-dependent parameter (differentiation of the freeenthalpy with respect to T is the negative of the entropy),so that the derivation of Eq. (18) with respect to temperature
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-7 Viscous flow and elasticity of glass J. Chem. Phys. 135, 184501 (2011)
leads to
R∂ ln η
∂(1/T)
∣∣∣∣σ,stru
= !Ga − T∂!Ga
∂T
∣∣∣∣σ,stru
, (19)
where σ is the stress applied on the specimen (see Refs. 2 and61 for further background on this analysis) and is mentionedhere to recall that T and σ are the two independent externalvariables in this problem (“stru” stands for “structure” andimplies that the derivation supposes the invariability of thematerial structure).
But since
− ∂!Ga
∂T
∣∣∣∣σ
= !Sa (dG = − SdT − Vdσ : 2nd principle).
(20)It comes that
R∂ ln η
∂(1/T)
∣∣∣∣σ,stru
= !Ga + T!Sa = !Ha. (21)
By analogy with the formalism introduced in ChemicalKinetics,62 !Sa can be considered as the entropy of activa-tion of the flow process. Hence, the only directly available ex-perimental parameter is the activation enthalpy (heat of flow),!Ha.
Since !Ga = !Gb − !Wa, Eq. (20) gives
!Sa = − ∂!Gb
∂T
∣∣∣∣σ,stru
+ σ∂Va
∂T
∣∣∣∣σ,stru
. (22)
As for crystals and polymers,63 the height of the energy bar-rier can be considered to be to an overwhelming part of elas-ticity origin (!Gb ∼ µ), so that
∂!Gb
∂T
∣∣∣∣σ,stru
= !Gb
µ
∂µ
∂T
∣∣∣∣σ,stru
. (23)
This expression brings to light the two physical origins forthe activation entropy: The first term in the right hand sideof Eq. (22), !Sµ
a = − ∂!Gb∂T
∣∣σ,stru, is the entropy change to
the temperature dependence of the elastic moduli, whereasthe second term, !Sσ
a = σ ∂Va∂T
∣∣σ,stru, reflects stress-induced
structural changes which will not be discussed in the presentpaper.
As the mechanical stress becomes small, !Wa + !Ga,and !Ga can also be considered as proportional to µ. ThenEqs. (22) and (23) lead to
!Sa = −χ
T(!Ga+σVa) + σ
∂Va
∂T, (24)
with : χ = Tµ
∂µ
∂T. (25)
It is noteworthy that stress effects usually do not show up forapplied stresses below GPa order. The viscosity coefficient isnearly stress independent in the usual stress range for viscos-ity measurements. Further considering that σ can be made assmall as desirable and that the activation volume is typicallysmaller than 50 × 10−6 m3 mol−1, it turns out that stress andvolume terms in the right hand side of Eq. (24) are mostlynegligible. (For σ = 10 MPa and Va = 50 cm3 mol−1, σVa
= 500 J mol−1, to be compared with the order of activationenergies, 100–1000 kJ mol−1).
FIG. 4. Temperature dependence of χ , as expressed by Eq. (25) and deter-mined from the experimental µ(T) data.
Finally, the following expressions are derived for theactivation parameters:
Activation enthalpy or heat for flow :
!Ha = R∂ ln η
∂(1/T)
∣∣∣∣σ,stru
, (26)
Activation entropy : !Sa = − χ
(1 − χ ) T!Ha, (27)
Gibbs free enthalpy : !Ga=1
(1 − χ)!Ha. (28)
The temperature dependence of χ , as expressed by Eq. (25)and determined from the experimental µ(T) data is presentedin Fig. 4 and χ (Tg) is reported in Table I. Although a compari-son of Eqs. (14) and (25) results in α = − χ , slightly differentvalues are reported in Table I. This is because α is derivedfrom the modelling of the entire µ(T) curve in the super-cooled liquid range, whereas χ (Tg) is obtained from the slopedµ/dT at Tg. !Ha, as determined from the slope of the linearintercepts in the glass transition range (Fig. 3), !Ga, and !Sa
are reported in Table II. Following the classical theory ofthermally activated flow phenomena, it is possible to estimate!Ga once the temperature dependence of the shear modulusis known (two important assumptions here are: (i) the heightof the energy barrier is proportional to the shear modulusand (ii) the contribution of the mechanical work to overcomethe barrier is small in comparison to that of thermal activa-tion). Keeping in mind that on the contrary to the commonassumption, both !Ha and !Ga are temperature-dependentparameters.
Another approach was proposed by Nemilov,64–66 assum-ing that all viscosity curves meet in the supercooled liquidregion at a given Tg/T ratio corresponding to a viscosity ofabout 10−4.5 Pa s. In this latter approach, both !Ga and
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-8 T. Rouxel J. Chem. Phys. 135, 184501 (2011)
!Sa are solely estimated from the viscosity data on the ba-sis of Eqs. (18) and (21). For instance at Tg one finds (seeAppendix A)
!Sa = −316 + !Ha/Tg (in J mol−1K−1), (29)
!Ga(Tg) = 316Tg (in J mol−1). (30)
A direct correlation can also be obtained between χ and the β
exponent of the stretch-exponential relaxation function: Re-laxation kinetics in slowly relaxing materials, such as glassesin the Tg range, is known to follow a so-called Kohlrausch-Williams-Watt—or stretch-exponential—function
φ(t) = exp[−(t/τ )β], (31)
where τ is the characteristic relaxation time and β is theso-called correlation factor, and ranges between 0 and 1(Debye relaxation). Following Palmer et al.,67 Perez et al.,68
and Donzel et al.,42 this phenomenological expression can beinterpreted on the basis of a hierarchically constrained relax-ation process. The nucleation of shear microdomains occursfirst, with an elementary characteristic time
τ = τ oexp[!Ga
RT
], (32)
where τ o is a temperature-independent time constant.Then these domains expand and promote shear at a con-
tinuously increasing scale until permanent flow of the struc-ture is achieved. This results in a “macroscopic” time constantbeing expressed as τ exp α τ 1/β . The experimentally availableheat for the relaxation process is thus
!Ha =∂lnτ exp
(1/RT), (33)
and therefore (some assumption being made on the tempera-ture dependence of β and !Ga (Ref. 42)) the apparent activa-tion energy is
!Ha = !Ga/β.
Identification with Eq. (28) gives
β = 11 − χ
. (34)
Values for β as calculated as estimated from relaxation exper-iments are usually slightly larger than those deduced from χ
(Eq. (34)) but follow a similar trend, with an increase of β asthe glass forming liquid becomes stronger.39, 69, 70 In a generalreview of non-exponential relaxation process in glass formingliquids (organic and inorganic systems) Böhmer et al.70 pro-posed a linear relationship between m and β, m = 250–320β.Combined with Eq. (34) this would lead to a simple mono-tonic relationship between m and χ , but no such simple trendis observed in the present study where β is estimated fromelasticity measurements as discussed further.
VI. DISCUSSION
A. Composition dependence
A major finding of the present analysis is that thereare supercooled liquids with relatively small softening rates(dµ/dT) above Tg which show up as particularly fragile liq-uids, and refractory glasses (high Tg) which exhibit a dramaticsensitivity to temperature for both elastic moduli and viscos-ity, so that there is no straightforward correlations betweenthe temperature dependence of elastic moduli and viscosity.This is because both quantities are fundamentally differentby essence: whereas elastic moduli reflect the behavior ofthe continuum, regardless of the size and shape of nanoscaleheterogeneities (phase separation, clusters, low-density vs.high density regions, etc.), viscous flow is very sensitive toany spatial distribution and local features (presence of weakchannels, type and size of structural units, etc.). Think of theexpression for the elastic moduli of multiple phase materialswhere only volume fractions mater and conversely of grainboundary sliding where a small amount of lubricant betweenhard grains governs the kinetics and where the grain size is akey parameter. For instance a-SiO2 has a highly cross-linkedand percolating atomic network. Thus both elastic moduliand viscosity weakly change with temperature. As expected,a-SiO2 exhibits an anomalous behavior characterized bya rising shear modulus in the [0.5–0.95] range for T/Tg
(dµ/dT(Tg−) = 3.14 MPa K−1) and a strong resistance to
de-structuration above Tg as evidenced by a remarkably slowsoftening (dµ/dT(Tg
+) = −5.3 MPa K−1) and by the fact thatν remains almost constant from 0.5 to 1.2Tg, suggesting thatthe liquid keeps a strong memory of the atomic structure ofthe glassy solid, with little changes of the average coordina-tion and dimensionality throughout the glass transition range.This well documented anomalous behavior would stem froman amorphous-amorphous transformation (for instance theincrease of E with T).71 This transformation would consistin atomic displacements similar to those associated to the α
to β-cristoballite phase transformation in crystalline silica.72
The β-form is stiffer than the α one, the natural temperatureweakening of the interatomic bonding is compensated.
On the contrary to a-SiO2, oxynitride glasses softenrapidly with rising temperature in spite of a relatively highdegree of interatomic connectivity and high Tg and elasticmoduli. This suggests that the oxynitride glass network con-sists of stiff (likely nitrogen-rich) structural units embeddedin a weaker phase (likely concentrating impurities, and alka-line and alkaline-earth species). Recalling that ν shows up asa mirror of the overall network connectivity, then the largerdecrease of µ(T) (α ∼ 9–10, larger than for glycerol!) withrespect to E(T) and the corresponding rapid increase in ν(T)-–comparing with those of organic liquids such bitumen orglycerol—is an indication of the dramatic softening of the“grain-boundary” phase (case 2 in Fig. 5). Amorphous se-lenium consists of weakly bonded together (Van der Waalsforces) (Se)n chains or rings and thus also adopts an island-like behavior, with m > 50 and α > 4.29, 40 With more than25 at.% germanium the softening rate (in absolute value) be-comes three to four times smaller and m decreases from 64(a-Se) to 26 (Ge3Se7). Nevertheless at larger Ge content a
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
184501-9 Viscous flow and elasticity of glass J. Chem. Phys. 135, 184501 (2011)
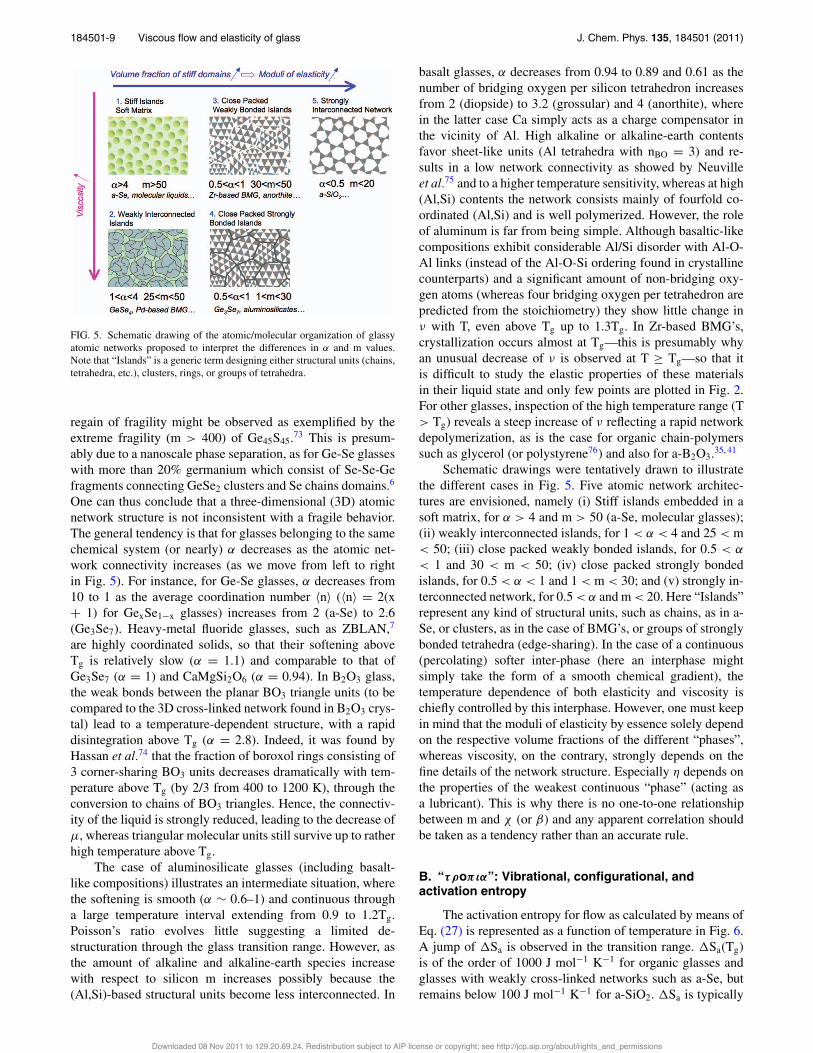
FIG. 5. Schematic drawing of the atomic/molecular organization of glassyatomic networks proposed to interpret the differences in α and m values.Note that “Islands” is a generic term designing either structural units (chains,tetrahedra, etc.), clusters, rings, or groups of tetrahedra.
regain of fragility might be observed as exemplified by theextreme fragility (m > 400) of Ge45S45.73 This is presum-ably due to a nanoscale phase separation, as for Ge-Se glasseswith more than 20% germanium which consist of Se-Se-Gefragments connecting GeSe2 clusters and Se chains domains.6
One can thus conclude that a three-dimensional (3D) atomicnetwork structure is not inconsistent with a fragile behavior.The general tendency is that for glasses belonging to the samechemical system (or nearly) α decreases as the atomic net-work connectivity increases (as we move from left to rightin Fig. 5). For instance, for Ge-Se glasses, α decreases from10 to 1 as the average coordination number 〈n〉 (〈n〉 = 2(x+ 1) for GexSe1−x glasses) increases from 2 (a-Se) to 2.6(Ge3Se7). Heavy-metal fluoride glasses, such as ZBLAN,7
are highly coordinated solids, so that their softening aboveTg is relatively slow (α = 1.1) and comparable to that ofGe3Se7 (α = 1) and CaMgSi2O6 (α = 0.94). In B2O3 glass,the weak bonds between the planar BO3 triangle units (to becompared to the 3D cross-linked network found in B2O3 crys-tal) lead to a temperature-dependent structure, with a rapiddisintegration above Tg (α = 2.8). Indeed, it was found byHassan et al.74 that the fraction of boroxol rings consisting of3 corner-sharing BO3 units decreases dramatically with tem-perature above Tg (by 2/3 from 400 to 1200 K), through theconversion to chains of BO3 triangles. Hence, the connectiv-ity of the liquid is strongly reduced, leading to the decrease ofµ, whereas triangular molecular units still survive up to ratherhigh temperature above Tg.
The case of aluminosilicate glasses (including basalt-like compositions) illustrates an intermediate situation, wherethe softening is smooth (α ∼ 0.6–1) and continuous througha large temperature interval extending from 0.9 to 1.2Tg.Poisson’s ratio evolves little suggesting a limited de-structuration through the glass transition range. However, asthe amount of alkaline and alkaline-earth species increasewith respect to silicon m increases possibly because the(Al,Si)-based structural units become less interconnected. In
basalt glasses, α decreases from 0.94 to 0.89 and 0.61 as thenumber of bridging oxygen per silicon tetrahedron increasesfrom 2 (diopside) to 3.2 (grossular) and 4 (anorthite), wherein the latter case Ca simply acts as a charge compensator inthe vicinity of Al. High alkaline or alkaline-earth contentsfavor sheet-like units (Al tetrahedra with nBO = 3) and re-sults in a low network connectivity as showed by Neuvilleet al.75 and to a higher temperature sensitivity, whereas at high(Al,Si) contents the network consists mainly of fourfold co-ordinated (Al,Si) and is well polymerized. However, the roleof aluminum is far from being simple. Although basaltic-likecompositions exhibit considerable Al/Si disorder with Al-O-Al links (instead of the Al-O-Si ordering found in crystallinecounterparts) and a significant amount of non-bridging oxy-gen atoms (whereas four bridging oxygen per tetrahedron arepredicted from the stoichiometry) they show little change inν with T, even above Tg up to 1.3Tg. In Zr-based BMG’s,crystallization occurs almost at Tg—this is presumably whyan unusual decrease of ν is observed at T ≥ Tg—so that itis difficult to study the elastic properties of these materialsin their liquid state and only few points are plotted in Fig. 2.For other glasses, inspection of the high temperature range (T> Tg) reveals a steep increase of ν reflecting a rapid networkdepolymerization, as is the case for organic chain-polymerssuch as glycerol (or polystyrene76) and also for a-B2O3.35, 41
Schematic drawings were tentatively drawn to illustratethe different cases in Fig. 5. Five atomic network architec-tures are envisioned, namely (i) Stiff islands embedded in asoft matrix, for α > 4 and m > 50 (a-Se, molecular glasses);(ii) weakly interconnected islands, for 1 < α < 4 and 25 < m< 50; (iii) close packed weakly bonded islands, for 0.5 < α
< 1 and 30 < m < 50; (iv) close packed strongly bondedislands, for 0.5 < α < 1 and 1 < m < 30; and (v) strongly in-terconnected network, for 0.5 < α and m < 20. Here “Islands”represent any kind of structural units, such as chains, as in a-Se, or clusters, as in the case of BMG’s, or groups of stronglybonded tetrahedra (edge-sharing). In the case of a continuous(percolating) softer inter-phase (here an interphase mightsimply take the form of a smooth chemical gradient), thetemperature dependence of both elasticity and viscosity ischiefly controlled by this interphase. However, one must keepin mind that the moduli of elasticity by essence solely dependon the respective volume fractions of the different “phases”,whereas viscosity, on the contrary, strongly depends on thefine details of the network structure. Especially η depends onthe properties of the weakest continuous “phase” (acting asa lubricant). This is why there is no one-to-one relationshipbetween m and χ (or β) and any apparent correlation shouldbe taken as a tendency rather than an accurate rule.
B. “τρoπια”: Vibrational, configurational, andactivation entropy
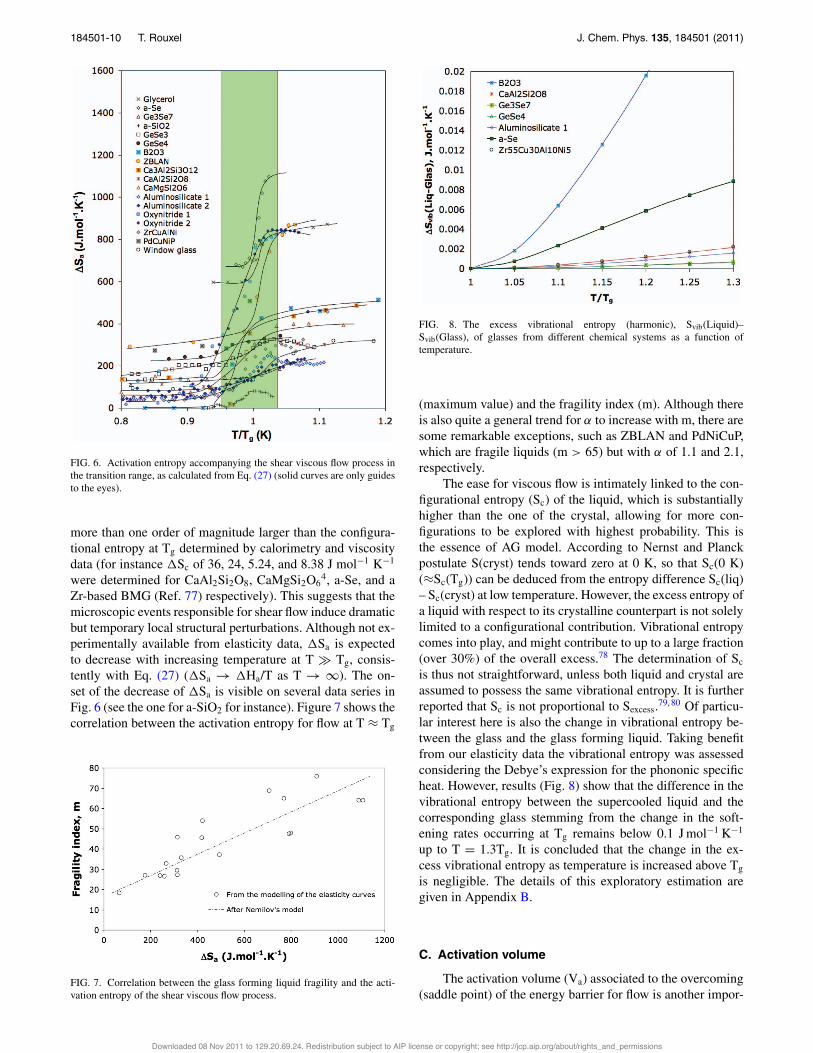
The activation entropy for flow as calculated by means ofEq. (27) is represented as a function of temperature in Fig. 6.A jump of !Sa is observed in the transition range. !Sa(Tg)is of the order of 1000 J mol−1 K−1 for organic glasses andglasses with weakly cross-linked networks such as a-Se, butremains below 100 J mol−1 K−1 for a-SiO2. !Sa is typically
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions
184501-10 T. Rouxel J. Chem. Phys. 135, 184501 (2011)
FIG. 6. Activation entropy accompanying the shear viscous flow process inthe transition range, as calculated from Eq. (27) (solid curves are only guidesto the eyes).
more than one order of magnitude larger than the configura-tional entropy at Tg determined by calorimetry and viscositydata (for instance !Sc of 36, 24, 5.24, and 8.38 J mol−1 K−1
were determined for CaAl2Si2O8, CaMgSi2O64, a-Se, and a
Zr-based BMG (Ref. 77) respectively). This suggests that themicroscopic events responsible for shear flow induce dramaticbut temporary local structural perturbations. Although not ex-perimentally available from elasticity data, !Sa is expectedto decrease with increasing temperature at T - Tg, consis-tently with Eq. (27) (!Sa → !Ha/T as T → ∞). The on-set of the decrease of !Sa is visible on several data series inFig. 6 (see the one for a-SiO2 for instance). Figure 7 shows thecorrelation between the activation entropy for flow at T ≈ Tg
FIG. 7. Correlation between the glass forming liquid fragility and the acti-vation entropy of the shear viscous flow process.
FIG. 8. The excess vibrational entropy (harmonic), Svib(Liquid)–Svib(Glass), of glasses from different chemical systems as a function oftemperature.
(maximum value) and the fragility index (m). Although thereis also quite a general trend for α to increase with m, there aresome remarkable exceptions, such as ZBLAN and PdNiCuP,which are fragile liquids (m > 65) but with α of 1.1 and 2.1,respectively.
The ease for viscous flow is intimately linked to the con-figurational entropy (Sc) of the liquid, which is substantiallyhigher than the one of the crystal, allowing for more con-figurations to be explored with highest probability. This isthe essence of AG model. According to Nernst and Planckpostulate S(cryst) tends toward zero at 0 K, so that Sc(0 K)(≈Sc(Tg)) can be deduced from the entropy difference Sc(liq)– Sc(cryst) at low temperature. However, the excess entropy ofa liquid with respect to its crystalline counterpart is not solelylimited to a configurational contribution. Vibrational entropycomes into play, and might contribute to up to a large fraction(over 30%) of the overall excess.78 The determination of Sc
is thus not straightforward, unless both liquid and crystal areassumed to possess the same vibrational entropy. It is furtherreported that Sc is not proportional to Sexcess.79, 80 Of particu-lar interest here is also the change in vibrational entropy be-tween the glass and the glass forming liquid. Taking benefitfrom our elasticity data the vibrational entropy was assessedconsidering the Debye’s expression for the phononic specificheat. However, results (Fig. 8) show that the difference in thevibrational entropy between the supercooled liquid and thecorresponding glass stemming from the change in the soft-ening rates occurring at Tg remains below 0.1 J mol−1 K−1
up to T = 1.3Tg. It is concluded that the change in the ex-cess vibrational entropy as temperature is increased above Tg
is negligible. The details of this exploratory estimation aregiven in Appendix B.
C. Activation volume
The activation volume (Va) associated to the overcoming(saddle point) of the energy barrier for flow is another impor-
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-11 Viscous flow and elasticity of glass J. Chem. Phys. 135, 184501 (2011)
tant parameter. Since
Va = − ∂!Ga
∂τ
∣∣∣∣T, (35)
it is in principal required to perform temperature jump exper-iments for different values of the applied stress to estimatethe activation volume. However Nemilov64 proposed a simpleestimation for a volume V*, considered to be that of kineticunits overcoming the activation barrier, from the free activa-tion enthalpy for flow and from the shear modulus,
V∗ = !Ga/µ, (36)
and concluded that V*/N (N: Avogadro number) can be writ-ten as ro
3 where ro is in excellent agreement (within 10%)with some interatomic distance (see Ref. 64 for details). It isnoteworthy that expressions similar to Eq. (36) were derivedeither by Argon87 or Dyre.88 The later author used a volumeexpansion model (“shoving” model) and assumed that the ac-tivation energy simply equals the elastic energy stored in thesurroundings of a flow event site (volume V) as a consequenceof local volume expansion (by !V). Then !Ga(T) = µ(T)Vc,with Vc = 2 (!V)2/(3V).Values for V* are summarized inTable II. These values are mostly much smaller than typicalvalues directly obtained mechanical testing. For instance, Va
of the order of 0.1–10 × 10−3 m3 mol−1 and 30–40 × 10−3
m3 mol−1 were determined for Zr-based BMG’s (Refs. 56and 89) and oxynitride glasses,90 respectively. The size of thestructural units responsible for the microscopic shear events isseveral times larger than the one predicted from Eq. (36) andmuch larger than the representative equivalent molecule. Thissuggests a cooperative movement of relatively large units.
D. New insight into the viscous flow process and thenon-Arrhenian behavior
Starting from a classical expression for thermally acti-vated kinetic processes (Eq. (1) and (17)) the non-Arrheniusbehavior can be analyzed by invoking either the temperaturedependence of the critical lower limit size (z in Eq. (1)) forthe cooperative regions involved in the local shear events, orthe temperature dependence of the energy barrier. So far inglass science, most authors followed the first approach as-suming with the AG model a temperature-independent en-ergy barrier and the inverse proportionality between z and thetemperature-dependent configurational entropy. Then, con-sidering the excess entropy in-lieu-of the configurational one,rather good fitting of the experimental data was found inthe few cases for which the calorimetric data were avail-able. Alternatively, referring to classical thermodynamics(Sec. V) and assuming a simple relationship between !Ga
and µ, further accounting for the temperature dependence ofµ (Eq. (14)), results in
!Ga = !Ga(Tg)(Tg/T)α. (37)
Interestingly, replacing this expression for !Ga in Eq. (17)gives an equation for η identical to that proposed by Avramovand Michel (AM) (Ref. 91) considering a temperature inde-pendent shear modulus of (1010 Pa at Tg for all glasses!) andassuming a Poisson’s distribution function for the local energy
FIG. 9. Temperature dependence of the shear viscosity coefficient (η) andcurve fitting using Eq. (38) and optimized α and !Ga parameters.
barriers for flow (note that αAvramov-Milchel = α + 1). Howeverthe temperature exponent is mostly significantly larger in thecase of the AM model (as discussed further). Thus we arriveat the following expression for η(T):
η = ηoexp[!Ga(Tg)(Tg/T)α
RT
], (38)
where, according to Eq. (3) ηo = µ(T)η(Tg)/µ(Tg) and fromEq. (28)
!Ga(Tg) =!Ha(Tg)
(1 − Tg
µ(Tg)∂µ∂T
∣∣∣Tg
) , (39)
and !Ha(Tg) is directly available from the tangent of the vis-cosity curve at Tg (Arrhenius plot) and all other parametersare determined from the high temperature elasticity data. Theability of Eq. (38) to predict viscosity data over a wide tem-perature range is illustrated in Fig. 9 for seven glass composi-tions for which high temperature elasticity and viscosity dataare available, using the above expression for ηo. In this fig-ure, the references for the high temperature viscosity data are:a-Se,92 a-SiO2,
49 B2O3,93 CaMgSi2O6,
53 Oxynitride,94 andGlycerol.57, 58 Note that (i) for the aluminosilicate glass datawere drawn from the empirical equation proposed by Hui55
and (ii) the composition of the oxynitride glass (here a (Ca,Al)silicon oxynitride glass) is slightly different from those ofOxynitride 1,2. Almost similar results can be obtained byconsidering ηo as a constant (ηo ≈ η(Tg)exp[−!Ga(Tg)/R]≈ 1012exp[−!Ga(Tg)/R)] and/or by approximating !Ga(Tg)by 316Tg (Eq. (30))). Values for α and !Ga(Tg) reportedin Tables I and II were found to provide a smooth fit ofthe experimental viscosity data for T/Tg < 1.3, i.e., in thetemperature range where elastic moduli were measured. How-ever, in the case of fragile liquids (such as diopside-likecomposition and glycerol) the best fit of the full data rangerequires slightly higher values for α and smaller values for!Ga(Tg). This discrepancy is likely to originate from the
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-12 T. Rouxel J. Chem. Phys. 135, 184501 (2011)
rapid destructuration (and thus structural changes) of frag-ile liquids at T > 1.3 Tg. In the case of the less frag-ile liquids, owing to the fact that elasticity data and vis-cosity ones were obtained separately (thus from differentspecimens), and that the curve fitting parameters are very sen-sitive to minor changes of Tg as obtained from both tech-niques, a good agreement is observed between the experi-mental data and the theoretical expression. The (α, !Ga(Tg))couples leading to the best curve fitting are as follows: a-Se(9.3, 55 kJ mol−1), a-SiO2(0.07, 413 kJ mol−1), B2O3(3.2,114 kJ mol−1), CaMgSi2O6(3, 246 kJ mol−1), Aluminosili-cate 1(0.83, 245 kJ mol−1), Oxynitride(5.5, 229 kJ mol−1),and Glycerol(3.2, 50 kJ mol−1). Nevertheless, the numbersin brackets are mostly close and almost identical (as for a-SiO2) to those given in Tables I and II. Thus we have reachedan expression for the shear viscosity which is suitable over abroad temperature range above Tg and requires no calibrationprovided accurate high temperature elasticity data are avail-able and that the heat for viscous flow at Tg is known. Thisfinding is in support of the form of the AM equation for theviscosity, notwithstanding the important theoretical differ-ences, and corroborates the shoving model proposed byDyre.95, 96 In particular, it strengthens a major assumptions ofthe later model: The main resistance to overcome the energybarrier for flow is controlled by the shear elastic modulus. Vis-cous flow is thus governed by the elastic properties and theirtemperature dependence! Besides, in the new viscosity equa-tion, η appears to solely depend on temperature, on Tg, andon elastic moduli, so that viscosity which can be viewed asa mirror of internal friction, does not show up as an intrinsicmaterial parameter. It relates to the height of the energy bar-rier for flow (proportional to Tg according to Nemilov64, 65),to the rate of the loading, and to external pressure.
It is noteworthy that different empirical expressions canequally fit the high temperature elasticity data. For instancea good agreement was found between the experimental dataabove Tg and an expression of the form
µ(T) = µ∞ exp (Eµ/T). (40)
Combining this expression with Eq. (18) still assuming that!Ga is proportional to µ leads to an equation for η for-mally identical to the one recently proposed by Mauro et al.(MYEGA equation)93 based on AG’s model and on a specificexpression for the configurational entropy, again suggestingthat the temperature dependence of the shear modulus is thekey parameter. Nevertheless, Eq. (40) introduces two calibra-tion constants (µ∞ and Eµ) and it is not straightforward togive a meaning to µ∞.
The slight discrepancy observed in the case of the mostfragile liquids (a-Se, glycerol), especially in the high tempera-ture range, brings to mind the fact that an important ingredientis missing in Eq. (39). Indeed the relationship between !Ga
and µ implicitly involves a characteristic molar volume (recallthat !Ga is in J mol−1 and µ is in J m−3) such as in Eq. (36).This volume corresponds to the volume swept during an ele-mental flow event at saddle point and is intimately related tothe atomic packing density (Cg). It was shown earlier9, 39 thatCg is a monotonic rising function of ν. Therefore, as dramaticde-structuration occurs (as for fragile liquids) and shows up
through a rapid increase of Poisson’s ratio (Fig. 2) the freeactivation enthalpy for flow is decreased.
VII. CONCLUSIONS AND PERSPECTIVES
The elastic and the viscous behavior of glasses from dif-ferent chemical systems through the glass transition rangewere investigated. Starting from simple thermodynamics con-siderations it is showed that the non-Arrhenian temperaturedependence of the shear viscosity can be directly interpretedin the light of the temperature dependence of the shear elasticmodulus and of Poisson’s ratio. An expression for the viscos-ity has been proposed where all terms were given physicalmeanings and allows for a good fit of the viscosity curvesfor most glass forming liquids but the most fragile ones. Thisexpression corroborates the essential hypothesis of the shov-ing model proposed by Dyre,88 and is strictly identical in itsform to the one of the AM model.91 A form identical to theone of the MYEGA model97 is also reached simply by us-ing a different—but nearly equally satisfactory—constitutivelaw for µ(T). In the case of fragile liquids, accounting for therapid de-structuration occurring at high temperature by incor-porating a function of Poisson’s ratio in the argument of theexponential term in Eq. (39) might bring some refinement.Different atomic network organizations are proposed basedon the values for α and m which describe the temperaturesensitivity of µ and η, respectively. Another perspective of thepresent study lies in the analysis of the second entropy term inEq. (22) (σ (∂Va/∂T)|σ,stru) which is the relevant parameter toinvestigate non-Newtonian flow and hydrostatic pressure ef-fects. Avramov98 found an almost linear relationship betweenα and the specific heat capacity (Cp) and Richet4 obtaineda good fit of the viscosity data of several silicate glasses byimplementing experimental data of excess entropy in the AGmodel. In view of the present work, this suggests that the ex-cess entropy and high temperature elasticity data are stronglycorrelated. For instance, in the cases where viscosity curvescan be modelled equally nicely on the basis of the tempera-ture dependence of the shear modulus, or by substituting Sc
in AG’s model for actual data for the temperature dependentexcess entropy, the excess entropy is supposed to be of thefolllowing form (Eqs. (3) and (39)):
Sexcess = R
(
1 −Tg
µ(Tg)∂µ
∂T
∣∣∣∣Tg
) (TTg
)α
, (41)
where shear modulus, taken as a volume density of energy, ac-counts both for the atomic bonding energy and atomic pack-ing density.
ACKNOWLEDGMENTS
I am particularly grateful to Marc M. Huger (ENSCI,Limoges) and to Pascal Gadaud (ENSMA, Poitiers) for theirhelp and for their long lasting support in the determinationof high temperature elastic moduli of glasses, to Annaik Gen-son (Ambassade de France, Ireland) for her contribution to thecompilation of the viscosity data, and to Hervé Lemercier, HuiJi, Yann Gueguen, Cedric Bernard, and Marie-Laure Vaillant,
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-13 Viscous flow and elasticity of glass J. Chem. Phys. 135, 184501 (2011)
FIG. 10. Myuller-Nemilov approach64–66 to derive the activation entropy(!Sa) for the viscous flow process above Tg.
who provided unique sets of high temperature elasticity andviscosity data during their PhD at ENSCI and at LARMAUR.This work was initiated during a study leave in China, thanksto Professor Gu Hui (SICCAS, Shanghai) for offering aChinese Academy of Science visiting professorship.
APPENDIX A: MYULLER NEMILOV APPROACH
Nemilov65 starts from Eq (18). Taking the logarithmicform of this equation gives lnη = lnηo + !Ga/(RT), so that!Ga = RT(ln(η/ηo)). Since !Ga = !Ha – T!Sa (Eq. (21)),η = ηo exp(!Ha /(RT))exp(!Sa /R). This expression can bewritten
η = ηoe exp (!Ha/(RT)), (A1)
where
ηoe = ηo exp (!Sa/R). (A2)
where ηoe is a temperature dependent coefficient which is ob-tained at T = Tg from the intersection of the tangent to theviscosity curve at Tg (slope !Ha/(RTg), where !Ha is di-rectly experimentally available (Eq. (21))) with the lnη axis(Fig. 10).
At T = Tg, η = 1012 Pa s and Eq. (A1) gives
ηoe = exp (ln1012 − !Ha/(RTg)). (A3)
Then,
!Sa(T) = R(ln(ηo/ηoe)), (A4)
where ηo is taken as the limit of η as T → ∞. FollowingMyuller,66 Nemilov considered ηo to be close to 10−4.5 Pa sin most cases (oxide glasses). Combining Eqs. (A3) and (A4)results in
!Sa(Tg) = −316 + !Ha/(Tg). (A5)
With Eq (21) it comes
!Ga = 316Tg. (A6)
APPENDIX B: ATTEMPT TO ESTIMATE THE EXCESSVIBRATIONAL ENTROPY
The extent to which the vibrational entropy of a liquid(above Tg) might differ from that of an “over-heated” glass,i.e., a glass which would be heated above Tg without experi-encing the Tg transition, Sv(Liq)–Sv(Glass), can be deducedfrom the high temperature elasticity data, considering thesoftening rates on both sides of Tg. The vibrational entropyof the crystal is expected to be smaller (glass being lessstiff than their crystalline counterparts) with the notoriousexception of silica, for which the vibrational entropy is muchless than that of the crystalline polymorphs.81 Changes in thevibrational entropy arise from differences in Cp(vib) throughthe temperature dependence of the Debye temperature (TD).82
Considering in a first approximation the Debye expressionfor Cp (better descriptions of Cp were proposed for peculiarglass compositions by Inaba et al.83 and Richet84)
Cp = 9R(T/TD)3∫ TD/T
0
x4ex
(ex − 1)2dx, (B1)
where R is the perfect gas constant, T is temperature, and TD
is Debye’s temperature defined as
TD= (3/(4π ))1/3(h/k)(N/Ve)1/3VD, (B2)
where k and h are Boltzmann and Planck’s constants, respec-tively, N is Avogadro’s number, Ve is an equivalent atomicvolume (for a AxBy glass, Ve = (xMA + yMB)/ρ where MA
and MB are atomic masses and ρ is the specific mass of theglass) and vD is a mean acoustic velocity defined as
VD =((
1/V3l + 2/V3
t
)/3
)−1/3. (B3)
Therefore, the temperature dependence of the elastic moduli(and chiefly of the shear modulus) has a direct incidenceon the value of Cp. Taking vD ≈ vt in Eq. (B2), replacingvt by (µ/ρ)1/2, and further substituting µ for µ(Tg)(Tg/T)α
(Eq. (14)) allows to calculate TD and Cp at any temperature.Note that although Cp is close to 3R (Dulong-Petit limit) in thetransition range, the full expression for Cp was used to assessthe incidence of the elastic modulus. The vibrational entropyis then obtained by integration of Cp/T and considering thatmost of the excess vibrational entropy above Tg arises fromthe softening rate difference between the glass (extrapolationof the data obtained below Tg) and the liquid at T > Tg,
!SLiq−Glasvib =
∫ T
Tg
CLiqp − CGlas
p
TdT. (B4)
This approach was applied to B2O3 (ρ = 2400 kg m−3, Ve
= 1.132 × 10−6 m3 mol−1), CaAl2Si2O8 (ρ = 2690 kg m−3,Ve = 7.956 × 10−6 m3 mol−1), a-Se (ρ = 4280 kg m−3,Ve = 18.45 × 10−6 m3 mol−1), Ge3Se7 (ρ = 4320 kg m−3,Ve = 1.784 × 10−6 m3 mol−1), GeSe4 (ρ = 4370 kg m−3,Ve = 1.77 × 10−6 m3 mol−1), ZrCuAlNi (ρ = 6772 kgm−3, Ve = 11.06 × 10−6 m3 mol−1), and Aluminosilicate1 (ρ = 2600 kg m−3, Ve = 2.656 × 10−6 m3 mol−1)(Fig. 8). Although Debye’s expression is known to be quiteinaccurate in the transition range (Cp increases more slowlythan predicted by Eq. (B1) and reaches values slightly largerthan the Dulong-Petit limit),85 a good agreement is found
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-14 T. Rouxel J. Chem. Phys. 135, 184501 (2011)
between the vibrational entropy of glass as calculated byimplementing our elasticity data in the above equations andas taken from literature. For instance a value of 43.2 J mol−1
K−1 is reported at Tg for a-Se using the harmonic form of thetemperature dependent density of state with experimentallymeasured frequencies86—we obtained 36 J mol−1 K−1—andvalues of 8.4 and 14 J mol−1 K−1 are reported for GeSe4 atTg and 1.2Tg, respectively77—we found 8.9 and 13.4 J mol−1
K−1, respectively. However, it turns out that the vibrational(phononic) entropy difference depends on the sensitivity ofthe shear modulus to temperature but remains below 0.1 Jmol−1 K−1 in all cases over the temperature range of concern[Tg–1.3Tg], and is thus negligible.
1G. Adam and J. H. Gibbs, J. Chem. Phys. 43, 139 (1965).2G. Schoeck, Phys. Status Solidi B 8, 499 (1965).3P. Richet, J. Non-Cryst. Solids 355, 628 (2009).4P. Debye, Ann. Phys. 39, 789 (1912).5J. Lucas, in Glasses and Amorphous Materials, edited by J. Zarzycki(VCH, New York, 1991), Chap. 8, p. 457.
6B. Bureau, J. Troles, M. Le Floch, P. Guenot, F. Smektala, and J. Lucas, J.Non-Cryst. Solids 319, 145 (2003).
7C. C. Chen, Y. J. Wu, and L. G. Hwa, Mater. Chem. Phys. 65, 306(2000).
8A. Delben, Y. Messaddeq, M. D. Caridade, and M. E. Aegerter, J.Non-Cryst. Solids 161, 165 (1993).
9A. Makishima and J. D. Mackenzie, J. Non-Cryst. Solids 12, 35 (1973).10B. Bridge, N. D. Patel, and D. N. Waters, Phys. Status Solidi A 77, 655
(1983).11P. Hudon and D. R. Baker, J. Non-Cryst. Solids 303, 354 (2002).12V. Askarpour, M. H. Manghnani, and P. Richet, J. Geophys. Res. 98, 17683,
doi:10.1029/93JB01558 (1993).13M. Huger and T. Chotard, Internal Report, ENSCI, Limoges, 2005.14D. R. Messier and A. Broz, J. Am. Ceram. Soc. 65, C-123 (1982).15S. Hampshire, R. A. L. Drew, and K. H. Jack, J. Am. Ceram. Soc. 3, C-46
(1984).16S. Hampshire, E. Nestor, R. Flynn, J. L. Besson, T. Rouxel, H. Lemercier,
P. Goursat, M. Sebai, D. P. Thompson, and K. Liddell, J. Eur. Ceram. Soc.14, 261 (1994).
17C. A. Angell, J. Non-Cryst. Solids 131, 13 (1991).18H. Lemercier, T. Rouxel, D. Fargeot, J. L. Besson, and B. Piriou, J.
Non-Cryst. Solids 201, 128 (1996).19A. Inoue, T. Zhang, and T. Matsumoto, Mater. Trans., JIM 31, 177
(1990).20V. Keryvin, M. L. Vaillant, T. Rouxel, M. Huger, T. Gloriant, and Y. Kawa-
mura, Intermetallics 10, 1289 (2002).21K. Tanaka, T. Ichitsubo, and E. Matsubara, Mater. Sci. Eng. A 442, 278
(2006).22G. J. Fan, H. J. Fecht, and E. J. Lavernia, Appl. Phys. Lett. 84(4), 487
(2004).23D. B. Miracle, Nature Mat. 3, 697 (2004).24H. W. Sheng, W. K. Luo, F. M. Alamgir, J. M. Bai, and E. Ma, Nature
(London) 439, 419 (2006).25H. Bornhöft and R. Brückner, Glasstech. Ber. Glass Sci. Technol. 67, 241
(1994).26E. P. Papadakis, K. A. Fowler, L. C. Lynnworth, A. Robertson, and
E. D. Zysk, J. Appl. Phys. 45, 2409 (1974).27C. Gault, Mater. Res. Soc. Symp. Proc. 142, 263 (1989).28J. M. Ide, J. Geol. 7, 689 (1937).29P. Gadaud, and S. Pautrot, J. Non-Cryst. Solids 316, 146 (2003).30P. Gadaud, X. Milhet, and S. Pautrot, Mater. Sci. Eng. A 521, 303 (2009).31G. Roebben, B. Bollen, A. Brebels, J. Van Humbeeck, and O. Van der Biest,
Rev. Sci. Instrum. 68, 4511 (1997).32A. Migliori, and J. L. Sarrao, Resonant Ultrasound Spectroscopy (Wiley,
New York, 1997).33R. Vacher and L. Boyer, Phys. Rev. B 6, 639 (1972).34Y. Y. Huang, J. L. Hunt, and J. R. Stevens, J. Appl. Phys. 44, 3589 (1973).35J. Kieffer, Phys. Rev. B 50, 17 (1994).36C. Bernard, V. Keryvin, J.-C. Sangleboeuf, and T. Rouxel, Mech. Mater.
42, 196 (2010).
37T. C. T. Ting, J Appl. Mech. 33, 845 (1966).38M. Sakai and T. Shimizu, J. Non-Cryst. Solids 282, 236 (2001).39T. Rouxel, J. Am. Ceram. Soc. 90, 3019 (2007).40Y. Gueguen, T. Rouxel, P. Gadaud, C. Bernard, V. Keryvin, and J.-C.
Sangleboeuf, Phys. Rev. B 82, 195206 (2010).41R. E. Youngman, J. Kieffer, J. D. Bass, and L. Duffrène, J. Non-Cryst.
Solids 222, 190 (1997).42L. Donzel, A. Lakki, and R. Schaller, Philos. Mag. A 76, 933 (1997).43V. Keryvin, T. Rouxel, M. Huger, and L. Charleux, J. Ceram. Soc. Jpn. 116,
851 (2008).44T. Rouxel and J. C. Sangleboeuf, J. Non-Cryst. Solids 271, 224 (2000).45W. M. Slie, A. R. Donfor, and T. A. Litovitz, J. Chem. Phys. 44, 3712
(1966).46F. Scarponi, L. Comez, D. Fioretto, and L. Palmieri, Phys. Rev. B 70,
054203 (2004).47Starting from the theory of shear viscosity proposed by Eyring [H. Eyring,
J. Chem. Phys. 4, 283 (1936)] based on the existence of holes in the liquidand assuming that flow proceeds through thermally activated jumps (ac-tivation energy Eη) of “molecules” (volume Vm) between holes [N. Hi-rai and H. Eyring, J. Appl. Phys. 29, 810 (1958)], Litovitz and Davis[T. A. Litovitz and C. M. Davis, Soc. Phys. Acoust. 2a, 281 (1965)] ar-rived at: µ(T) = (RT/Vm)exp[Eη/(RT)]. However, as was also recognizedby Litovitz and Meister [R. Meister, C. Marhoeffer, R. Schamanda, L. Cot-ter, and T. Litovitz, J. Appl. Phys. 31, 854 (1960)] this expression mostlyprovides a poor prediction of the experimental data. On the one hand, ex-perimental data shows a smaller temperature sensitivity (in many cases a µapproaches a linear dependence on T), and on the other hand unreasonablevalues of Eη are required to fit the data near Tg. For instance, in the case ofAluminosilicate 1, Eη ≈ 151 kJ mol−1, whereas that heat for flow (!Ha) is484 kJ mol−1, and for GeSe3, Eη≈12 kJ mol−1, whereas !Ha≈256 kJ mol−1.
48E. Grüneisen, Handbuch Phys. 10, 1 (1926).49G. Hetherington, K. H. Jack, and J. C. Kennedy, Phys. Chem. Glasses 5,
130 (1964).50C. T. Moynihan, J. Am. Ceram. Soc. 76, 1081 (1993).51A. Sipp, and P. Richet, J. Non-Cryst. Solids 298, 202 (2002).52C. M. Scarfe, D. J. Cronin, J. T. Wenzel, and D. A. Kaufman, Am. Mineral.
68, 1083 (1983).53R. J. Kirkpatrick, Am. J. Sci. 274, 215 (1974).54P. Richet, Geochim. Cosmochim. Acta 50, 1521 (1986).55H. Hui and Y. Zhang, Geochim. Cosmochim. Acta 71, 403 (2007).56M. L. Vaillant, Ph.D. dissertation, University of Rennes 1, 2003.57J. B. Segur and H. E. Oberstar, Ind. Eng. Chem. 43, 2117 (1951).58K. Schröter and E. Dorth, J. Chem. Phys. 113, 9101 (2000).59P. Lucas, E. A. King, O. Gulbiten, J. L. Yarger, E. Soignard, and B. Bureau,
Phys. Rev. B. 80, 214114 (2009).60U. Senapati and A. K. Varshneya, J. Non-Cryst. Solids 197, 210
(1996).61A. G. Evans and R. D. Rawlings, Phys. Status Solidi B 34, 9 (1969).62V. K. La Mer, J. Chem. Phys. 1, 289 (1933).63B. Escaig and J. M. Lefebvre, Rev. Phys. Appl. 13, 285 (1978).64S. V. Nemilov, J. Non-Cryst. Solids 352, 2715 (2006); in Thermodynamics
and Kinetic Aspects of the Vitreous State (CRC, Boca Raton, 1995).65S. Nemilov, J. Non-Cryst. Solids 353, 4613 (2007).66R. L. Myuller, Zh. Prikl. Khim (Russ. J. Appl. Chem.) 28, 363 (1955); 28,
1077 (1955).67R. G. Palmer, D. L. Stein, E. Abraham, and P. W. Anderson, Phys. Rev.
Lett. 53, 958 (1984).68J. Perez, J. Y. Cavaillé, and S. Etienne, and C. Jourdan, Rev. Phys. Appl.
23, 125 (1988).69T. Rouxel, J.-C. Sanglebœuf, M. Huger, C. Gault, and S. Testu, Acta Mater.
50, 1669 (2002).70R. Böhmer, K. L. Ngai, C. A. Angell, and D. J. Plazek, J. Chem. Phys. 99,
4201 (1993).71L. Huang and J. Kieffer, Phys. Rev. B 69, 224203 (2004).72D. J. Lacks, Phys. Rev. Lett. 84, 4629 (2000).73A. Zumailan, Mater. Lett. 57, 94 (2002).74A. K. Hassan, L. M. Torell, L. Börjesson, and H. Doweidar, Phys. Rev. B
45, 12797, (1992).75D. R. Neuville, L. Cormier, A. M. Flank, V. Briois, and D. Massiot, Chem.
Geol. 213, 153 (2004).76R. Kono, J. Phys. Soc. Jpn. 15, 718 (1960).77C. Way, T. Shaw, P. Wadhwa, and R. Busch, J. Alloys Compd. 434, 88
(2007).
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions

184501-15 Viscous flow and elasticity of glass J. Chem. Phys. 135, 184501 (2011)
78P. D. Gujrati and M. Goldstein, J. Phys. Chem. 84, 859 (1980).79G. P. Johari, J. Non-Cryst. Solids 307, 387 (2002).80E. L. Gjersing, S. Sen, and B. G. Aitken, J. Non-Cryst. Solids 355, 748
(2009).81P. Richet, Geochim. Cosmochim. Acta 48, 471 (1984).82P. Debye. Ann. Phys. 39, 789 (1912).83S. Inaba, S. Oda, and K. Morinaga, J. Non-Cryst. Solids 325, 258
(2003).84P. Richet, Chem. Geol. 62, 111 (1987).85L. Ferrari, W. A. Phillips, and G. Russo, Europhys. Lett. 3, 611 (1987).86W. A. Phillips, U. Buchenau, N. Nücker, A.-J. Dianoux, and W. Petry, Phys.
Rev. Lett. 63, 2381 (1989).87A. S. Argon, Acta Metall. 27, 47 (1979).88J. C. Dyre, J. Non-Cryst. Solids 235–237, 142 (1998).
89M. Blétry, P. Guyot, J. J. Blandin, and J. L. Soubeyroux, Acta Mater. 54,1257 (2006).
90T. Rouxel, M. Huger, and J. L. Besson, J. Mater. Sci. 27, 279 (1992).91I. Avramov and A. Milchev, J. Non-Cryst. Solids 104, 253 (1988).92S. Hamada, N. Yoshida, and T. Shirai, Bull. Chem. Soc. Jpn. 42, 1025
(1969).93A. Napolitano, P. B. Macedo, E. G. Hawkins, J. Am. Ceram. Soc. 48, 613
(1965).94G. Urbain, P. Verdier, and J. Lang, Br. Ceram. Trans. J. 83, 12 (1984).95J. C. Dyre, N. B. Olsen, and T. Christensen, Phys. Rev. B 53, 2171 (1996).96J. C. Dyre and N. B. Olsen, Phys. Rev. E 69, 042501 (2004).97J. C. Mauro, Y. Yue, A. J. Ellison, P. K. Gupta, and D. C. Allan, Proc. Natl.
Acad. Sci. U.S.A. 106, 19780 (2009).98I. Avramov, J. Non-Cryst. Solids 351, 3163 (2005).
Downloaded 08 Nov 2011 to 129.20.69.24. Redistribution subject to AIP license or copyright; see http://jcp.aip.org/about/rights_and_permissions