the study of l-electron equivalent oxidation-reduction reactions
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL. CHEMISTRY Vol. 251, No. 14, Issue of July 25, pp. 4336-4345, 1976
Prinkd in U.S.A.
The Study of l-Electron Equivalent Oxidation-Reduction Reactions by Fast Pulse Generation of Reagents
CYTOCHROME c/FERRI-FERROCYANIDE SYSTEM*
(Received for publication, November 3, 1975)
YIGAL ILAN, AVIGDOR SHAFFERMAN, AND GABRIEL STEIN
From the Department of Physical Chemistry, The Hebrew University, Jerusalem, Israel
The method of pulse radiolysis was used to generate reagents in situ in times (500 ns to 1.5 rs) short compared with the rates of the observed biochemical processes. This “instant” mixing technique is compared with rapid stopped flow measurements (limited in rates and concentrations) and T-jump measurements (limited to relaxation in the neighborhood of equilibrium) for the ferro-ferricytochrome c (C(II)-C(III))/ferro-ferricyanide (FCN(II)-FCN(II1)) system. The reagents generated in situ were C(I1) or FCN(II1).
Kinetically indistinguishable binding sites exist on C(I1) and C(II1) for hexacyanide anions. Reductive electron transfer to the protein proceeds within the FCN(I1). C(II1) complex, with a rate of 400 s- I. The binding of FCN(I1) on C(I1) slows down the oxidation of C(I1) by FCN(II1). The sites of interaction on C(I1) or C(II1) with FCN(I1) or FCN(II1) show effective charges of N +2. The association constant per binding site derived from the kinetics of electron transfer is 2 10’ M-’ for FCN(I1) .C(II) and 5 lo* Me’ for FCN(II1) .C(III). Specific clusters of amino acids in the model of cytochrome c are suggested as binding sites. The oxidation-reduction reactions of FCN appear to involve electron equivalent transfer to and from such somewhat remote binding sites on the protein. Anions such as phosphate or sulphate also bind to these, less strongly than hexacyanides. In the presence of perchlorate the kinetics show the resolution of the pK = 9.3 of C(II1) into two parts: (a) optical changes at 695 nm due to ligand interchange on the heme-iron, unaffected by perchlorate and (b), a kinetic change leading to biphasic oxidation of C(II), with pK = 7.4. This is attributed to the effect of perchlorate on water structure in the close environment of the binding sites. The high rate of oxidation of relaxed C(I1) by FCN(III), (2 x lOa M-’ s-’ at p = 0) is not in agreement with an outer sphere Marcus mechanism. Nonrelaxed C(I1) having a structure closer to C(II1) transfers electrons to FCN(II1) even faster (k = 3 x 1OQ M-’ s-’ at p = 0).
Stopped flow (rapid mixing) and T-jump (relaxation) are equilibrium are not produced and therefore not studied. In the among the more common fast kinetic methods used in the regular T-jump apparatus and in most systems studied study of biochemical oxidation-reduction reactions, e.g. of therein, in order to obtain time resolutions of microseconds, cytochrome c (l-14). high ionic strengths are usually required. This shortcoming
Using the stopped flow technique of rapid mixing of reagents does not exist when laser heating-instead of condenser it is possible to observe short-living intermediates in the discharge-is used. This also enables measurements in the reaction mechanism limited by the mixing time to reactions submicrosecond range. occurring at times greater than or equal to a few milliseconds. Oxidation of ferrocytochrome c by ferricyanide has been Accordingly concentrations of reagents must also be below a studied in several laboratories by the stopped flow technique limit set by the relative slowness of the method, thus prevent- (l-5). Fron- these studies reaction rate constants ranging from ing the detection of some intermediates. 6 x lo6 M-’ s-’ to 16 x lo6 M-’ s-’ were obtained for the
In the relaxation (e.g. T-jump) apparatus a system in oxidation process. equilibrium is perturbed by a fast small change (e.g. tempera- T-jump studies on Equilibrium I (6-8): ture). Here one can detect faster reactions, down to mi- croseconds or shorter. However in this method the starting
C(I1) + Fe”’ (CN),? $ C(II1) + Fe” (CN),-3 (I)
point is always an equilibrium state, and only equilibrated agree fairly well with the values given above for the oxidation
species are detected. Short-lived transients which are not in (forward reaction), as well as present data on the reduction of ferricvtochrome c bV ferrocvanide with k = 2.10’ M-’ s-’ (6).
*This work was supported by the United States Energy Research The kinetic data-accumilated in these studies (and similar and Development Administration. studies with other ions, e.g. complexes of iron, cobalt, ru-
4336
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

l-Electron Redox Reactions by Fast Pulse:Cyt cfFe(CN)m3 4337
thenium and other ions (2, 9-12)) indicate the possibility of an outer sphere mechanism, via the exposed edge of the heme (13, 14).
In the present study we have used a third technique-pulse
radiolysis (PR). The application of this method to the study of biologic oxidation-reduction processes and its technical details were reviewed recently (15). In this method the well estab- lished techniques of radiation chemistry are used to produce in situ within the experimental solution; in short (nanosecond) times, l-electron equivalent oxidation-reduction reagents such as hydrated electrons, H atoms, radicals (OH, organic radi- cals). One of the main advantages of PR’ is that l-equivalent oxidation-reduction reactions are easily resolved, since both
reducing and oxidizing agents generated are l-equivalent reagents. Furthermore it enables detection of short-living transients on a time scale of nanoseconds or even picoseconds
(16). While most of the PR studies on biomolecules cited in the
literature involve the reaction of radicals such as e;“, H, OH and organic radicals with the biomolecule, here we present a study on the oxidation and reduction reactions between cytochrome c and ferri-ferrocyanide, where ferri- or ferrocya- nide. or ferrocytochrome c are produced rapidly in situ. For conditions similar to those used in previous studies (stopped flow and T-jump) we have obtained similar results. In addition we could extend the experimental conditions to time resolu- tions and reagent compositions not accessible by other methods. Here the pulse radiolytic method enabled us to obtain rate constants and observe mechanisms and intermedi-
ates which could not be studied by other methods.
EXPERIMENTAL PROCEDURES
Materials-Ferricytochrome c was Sigma type III or type VI and was used without further purification. The results were similar with both types. Its concentration was determined by measuring the absorption at 528 nm, using an extinction coefficient of 11.2 x lOa Mm’ cm-’ (17, 18). It contained less than 7% of reduced enzyme based on the absorption at 550 nm.
Ferrocytochrome c was obtained by reducing the oxidized enzyme with a 5-fold excess of sodium ascorbate (Sigma). Excess reducing agent was removed on Sephadex G-25 (medium, Pharmacia Fine Chemicals) columns. The reduced enzyme was tested for complete reduction by adding solid Na,S,O, (Riedl de Haen, purified) and
checking the spectra before and after this addition. No changes were observed in the region of 520 to 550 nm. The concentration of C(B) was determined by its absorption at 550 nm, using a molar extinction coefficient of 27.6 x 10s M-’ cm-’ (17, 18).
All solutions were prepared less than 15 hours before use, and were stored in glass bottles. All solutions were prepared in water distilled from alkaline permanganate, then from acid dichromate, and finally redistilled. The pH was adjusted by buffer as stated or by titrating with HClO, (Merck) or NaOH-(Riedl de Ha&). Ionic strength was adjusted by Na,SO, (Mallinckrodt), NaClO,.H,O (Fluka Puriss p.a.) or Na,HPO,.H,O + NaH,P0,.7H,O (Mallinckrodt). Potassium fer- rocyanide was obtained from Mallinckrodt, and potassium ferricya- nide from British Drug House.
Argon was supplied by the Israel Oxygen Center, and N,O by Matheson 98% purity (remainder air). Both were freed of oxygen by bubbling through a solution containing V(B) prepared by in situ reduction of Fluka Purum grade NaVO, with zinc amalgam prepared from British Drug House zinc, and Frutarom Analytical grade mer- cury. The results with and without bubbling the gases through the reducing solution were the same within the experimental error. The tert-butanol used (Merck, pro-analysi) contained no impurities detect- able by ultraviolet absorption.
Apparatus-The Varian linear accelerator of the Hebrew University, the cell and the optical and electronic systems were described elsewhere (19). The pulse produces the primary species: e;“, H atoms
‘The abbreviation used is: PR, pulse radiolysis.
and OH radicals. These react further as described below. Spectra of solutions were taken on a Cary 14 spectrophotometer, pH
measurements on a digital (pHM52 of Radiometer) or on an analog pH-meter (El-Hama Instruments, Israel).
The T-jump apparatus was constructed by Messanlagenstudien gesellschaft, m.b.h., Gottingen.
Procedures-Solutions were deaerated and saturated with argon or N,O by sweeping with the gas for at least 20 min, in large glass syringes equipped with capillary taper joints for automatic transfer of solutions to the irradiation cells (-4ml volume) fitted with optical windows. Irradiations were carried out immediately after deaeration. Only one pulse was delivered to a given aliquot of solution. Absorbed dose per pulse was determined routinely using the spectrum of the solvated electron, by pulsing 1 x lo-* M aqeuous ethanol (Merck, pro-analysi) at pH -9.5, taking c118 = 1.06 x 10’ Mm’ cm-‘, and G(e,) = 2.75 (G = molecules formed/100 eV). All measurements on the pulse radiolytic apparatus were performed at 22 + 2”. Doses used ranged from 350 to 3500 rads per pulse (equivalent to lOme to 10e5 M of e&). To assure pseudo-first order conditions the concentration of C(B) or ferricyanide produced by the pulse was -10 times lower than the initially present Fes’(CN),-3 or C(B).
Oscilloscope traces were analyzed by transferring the data to punched cards by means of a magnifying manual trace follower coupled to an analog to digital converter, and processing the cards in a Control Data Corp. Cyber digital computer.
When two stages of a process could be separated the slow stage was analyzed first. The optical density of this stage was extrapolated by the computer program to zero time. The extrapolated value was taken as the optical density of the end of the fast stage.
The solutions for the T-jump measurements were not deaerated. The jump was from 21.8-25”. The relaxation times were derived from the exponential change of the light transmitted by the solution at 550 nm, recorded on a Tektronix 549 storage oscilloscope, and photo- graphed by means of a Polaroid camera.
RESULTS
Production of Fe1s(CN),-3 in Situ-In order to study the oxidation of C(I1) by Fe”‘(CN),-3 we produced Fe”‘(CN),-S in
the presence of C(R) according to the following equations using appropriate concentrations of reagents:
Fe” (CN),-4 + OH + Fe”’ (CN),-s + OH- k = 1.1 x 1O”‘M-Is-’ (20) (1)
e, + N,O + N, + OH- + OH
e, + C(B) + products k = 5.6 x loeM-IS-’ (21) (2)
k = 1.9 x 10”’ M-‘s-’ (22, 23) (3) OH. + C(B) + products (4)
The solutions were saturated with N,O, which scavenges e;” and converts it to OH radicals (Reaction 2). The concentration of N,O was 25 mM, thus more than 99% of e& was scavenged.
For the reaction of C(II1) + OH a value of 1.4 x 10” Mm’ s-’ (24) was obtained. Assuming a similar rate for C(I1) + OH we have less than 5% of OH radicals reacting according to Reaction 4-the rest of the radicals (>95%) react with Fe’i’(CN),-’ (Reaction 1). The production of Fes’(CN),~3 by the latter reaction is accomplished within less than 5 I.CS. Under these conditions we detected a change in absorption due to the oxidation of C(I1) by Fes’(CN),-S. One phase of oxidation is observed at neutral or basic pH (Fig. 1). This process obeys second order kinetics: first order in C(R) and in Fe(CN),-3. Depending on the ionic strength and the concentration of Fe”(CN),-” (which may cause a back reaction) oxidation yields varied from 100% downward. At lower ionic strength when only Fe”(CN),-’ anions are present both reactions of Equilibrium I are significant (see “Discussion”). These results
are plotted in Fig. 1. The rate constants obtained at ionic strength 0.2 are summarized in Table I. Results are given also for the rate measured at p = 0.01 when no salt other than K,Fe”(CN), were present.
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

4338 l-Electron Redox Reactions by Fast Pulse:Cyt clFe(CN),-3
9, 1 TABLE II
6 ,’
_I’
0 I I 1 2 3 4 6 6
[cr] X10’(M)
FIG. 1. Observed rate constants for the oxidation-reduction re- action between C(U) and Fe(CN).a- formed in situ, as a function of C(II) concentration. pH = 7.0, ~1 = 0.01. [K,Fe(CN),] = 1 x lOma M (0); [K,Fe(CN).] = 2 x lo-* M, (A); [K,Fe(CN),] = 4 x lo-’ M (0); mean value (0). (A, 0, 0, mean values of four different measure- ments.) Inset, oscilloscope trace for the reaction mentioned above. Abscissa, 1 ms per division; ordinate; 5 mV per division (p.m. output); V. = 300 mV. [C(U) ] = 4 x lo-* M; p,Fe(CN)s] = 4 x lo-’ M. A = 550 nm.
TABLE I
Observed and second order rate constants for oxidation of C(H) by Fe(CN).3- produced in situ at neutral and at basic pH, at ionic
strength of 0.2
Solution contained 2 x 10ms M K,Fe(CN).. The kinetics were followed at 550 nm, and at 450 nm. The values given are the mean of at least five experiments.
Salt PH
NaH,PO, + Na,HPO, 6.9 Na,HPO, 8.9 NaBO, 6.9 Na,SO, 6.9 NaClO, 6.8 None (p = 0.01) 7.0
k m*
s-1
150 150 147 252
75
k, [Cyt-CII]
loEM-‘so’
5.0 5.0 4.9 4.2 2.5 9.2”
lo-“M
3 3 3 6 3
D Derived from Fig. 1.
The results obtained with the T-jump apparatus at p = 0.2 were very similar to those obtained in the PR apparatus. These are summarized in Table II. k,. (rate constant for oxidation) was calculated in T-jump experiments using the expression (6):
T-’ = Sk,,[coI + k,,,[Fe(CN),-43, The equilibrium concentration of C(II), [C(II)] was calcu-
lated using the initial concentration of C(I1) (which was determined spectroscopically) and a value of 350 for the equilibrium constant of Equilibrium I. ‘T is the relaxation time observed experimentally, and lJ?e”(CN),-‘I,, the initial con- centration of ferrohexacyanide.
Production of C(ZZJ in Situ-C(I1) is produced in situ in a solution containing initially C(II1) and Fe”‘(CN),-S. The following reactions are relevant to this system:
e, + C(II1) + C(U) k > 2 x 10” M-IS-’ (15) (5)
Values of 7, the relaxation time, 7-l and k,,
The solutions contained 8 x 10e8 M K,Fe(CN). The ionic strength was 0.2, and the pH 6.7 to 7.0 in all cases.
Salt T T -’ wII) I. rccir11 ox k
10-3s s-1 M lo’M-‘s-l
Na,SO, + 2.3 430 6.4 x 1O-K 2.1 x 10m5 6.7 phosphate
NaClO, 9.0 111 1.1 x 10-S 7.9 x 10-s 2.9 NaClO, 4.7 215 4.4 x lo-’ 1.6 x 1O-6 3.8 NaClO, 5.9 170 6.4 x lo-’ 2.1 x lo-’ 2.6
e, + Fe”’ (CNjGe3 + Fe” (CN)ce4
H + C(III) - C(I1)
k = (1 to 2) X 10’ M-’ 5-l (25) (6)
k = 1.1 x 10’0~-‘~-’ (22, 23) (7)
H + Fe(CN)6-3 + Fe”(CNL? + H+ k = 5.6 x 10Q~- s-’ (2‘3 (8)
OH + (CH,),COH + H,O+ .CH,C(CH,), (OH)
OH + C(II1) + products
k = 6.3 x lO’w’s-’ (2-O (9)
k = 1.4 x 10” M-‘s-l (24 (10)
More than 99% of the OH radicals were scavenged by t-butanol according to Reaction 9; thus, the contribution of Reaction 10 is negligible. The t-butanol concentration was 10’ times larger than that of C(II1). t-Butanol radicals do not react with C(II1) (22, 23).
In most of the experiments carried out, the concentration of Fe”’ (CN),-” was equal to or smaller than that of C(III), thus more than 95% of e, reacted with C(II1) to produce C(I1) (Reaction 5). Since the majority of H atoms, which are only 15% of the total amount of reducing radicals, react with C(II1) to produce C(II), the amount reacting with Fe”‘(CN),-S accordingly (Reaction 8) is negligible, and for our purpose the effect of Reaction 7 is additive to that of Reaction 5.
A fast change in absorption-an increase at 550 nm or 520 nm and a decrease at 450 nm-due to the formation of C(I1) was observed. This change was nearly complete within 10 c(s. After 100 ps or later a change in absorption in the opposite direction was observed (Fig. 2a, insert). This latter is the reoxidation process at the end of which the initial absorption is regained. One phase of oxidation is observed at neutral pH (6.7 to 7.0), in the absence or presence of salts such as sulfate or phosphate. The process obeys second order kinetics: first order in C(I1) and in Fe”‘(CN),-9. The results are summarized in Table III. The dependence of this process on the ionic strength is given in Fig. 2a, from which a rate of 2 x 10’ M-’ s-’ at zero ionic strength, and an effective charge of +2.0 (Table IV) are obtained for the oxidation process. In spite of the fact that the ionic strength ranged from 0.001 to 0.2 a linear relationship is observed when log k is plotted uersus pCH, as was also observed in the reduction and oxidation of cytochrome c with other inorganic reagents (9, 11).
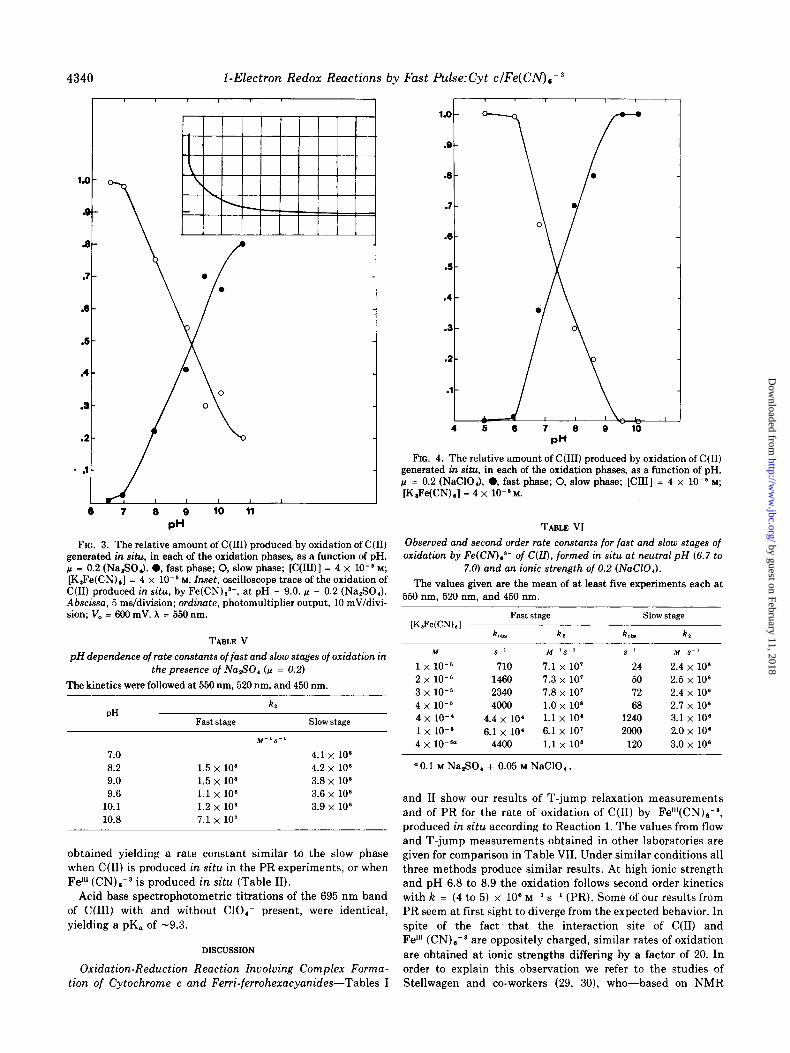
At higher pH two phases of oxidation are observed (Fig. 3, insert). The relative amount of C(II1) produced from these processes is plotted as a function of the pH, and the pK obtained is 9.2 (Fig. 3). Both phases obey second order kinetics: first order in C(I1) and in Fel”(CN),-‘. The rate of the slower phase is equal to that measured at pH 17.0. Rates for each stage at different pH values are given in Table V. The rate of the faster stage, 3 x loo M-’ s-l, at zero ionic strength, as well
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

l-Electron Redox Reactions by Fast Pulse:Cyt clFe(C 4339
1
.
\
.
. .
a , l I l I I .l .2 .3 .4 .5
dr
1 Fm. 2. a, dependence of log k on the square root of the ionic
strength, for the slow phase of oxidation, in the presente of Na,SO,. pH = 6.9. [C<III>] = 4 x lOe6 M [K,Fe(CN),] = 2 x 1O-J M. Inset, oscilloscope trace for the oxidation slow phase at cc = 10m3, and pH = 6.9. Abscissa, 500 pldivision; ordinate, (photomultiplier output) 5
TABLE III TABLE IV
Obseroed and second order rate constants for oxidation by Fe(CN),S- of ferrocytochrome c, produced in situ at neutral (6.7 to 7.0) pH and
ionic strength of 0.2
Effectiue charge of C(H) for oxidation by Fe(CNjSs- in thepresence of NaSO,
The values are the mean of at least five experiments each at 550 nm, 520 nm, and 450 nm.
Effective charge PH
Salt k Ch k, MWCNM
3 ’ 10’hCLS-’ lo-sM
Na,SO, 164 4.1 4 86 4.3 2
Na,HPO, + NaH,PO, 160 4.0 4 90 4.5 2
as the effective charge, +2.1, (Table IV) for this process are obtained from Fig. 2b. At pH > 10 a decrease in rate constant (Table V) and a decrease in effective charge to +0.6 (Table IV and Fig. 26) are observed. To calculate these effective charges we used the value of -2 for the effective charge of ferrocyanide, as derived from spectrophotometric measurements (28) and took the same value as the upper limit for ferricyanide.
Effect of Perchlorate on Oxidation of C(U) by Fe(CIV),-J- When C(I1) is produced in situ in the presente of ClO,-, at neutral pH, two phases of reoxidation are observed, each obeying second order kinetics; first order in C(I1) and in
b , I I 1 I I .l .2 .3 .4 .5
c’r
mV/division; V,, = 200 mV. X = 550 nm. b, dependence of log k on the square root of the ionic strength for the fast phase of oxidation, in the presente of Na$O,. 0, pH = 9.0; 0, pH = 10.5; [C(III)] = 4 x lo-$ M; [K,Fe!CN),] = 2 x 10m5 M.
Stage of oxidation procesa
+2.1 +2.1 +2.0 +0.6
neutral -9 -9
10.5
slow slow fast fast
of these phases as a function of the pH is shown in Fig. 4; a pK, of 7.4 is obtained. The rate constants of both phases are somewhat smaller than in the presente of sulfate or phosphate, (Table VI). The dependence of the logarithm of the rate constants of both phases on the ionic strength is shown in Fig. 5. The dominant effect of ClO,- is the resolution of both phases of oxidation with their characteristic rate constants even when greater concentrations of other anions are present at neutral pH (Table VI).
When Fe1”(CN),-3 is produced in situ and C(I1) is initially present in the solution, only one phase of oxidation is observed in the presente of ClO,-. Again the process is second order: first order in C(I1) and Fe1’1(CN),-3. The rate constant is similar to that of the slower phase of oxidation wh& C(I1) is produced in situ (Table 1).
FeI’1(CN),-3. The relative amount of C(II1) produced by each Using the T-jump apparatus a single relaxation time is
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from
4340 l-Electron Redox Reactions by Fast Pulse:Cyt clFe(Ch96-3
.2-
* .l-
6 7 9 9 10 11
PH
FIG. 3. The relative amount of C(II1) produced by oxidation of C(I1) generated in situ, in each of the oxidation phases, as a function of pH. r.~ = 0.2 (Na,SO,l. 0, fast phase; 0, slow phase; [C(ILll] = 4 x 10e6 M; [I(,Fe(CN),] = 4 x 10ms M. Inset, oscilloscope trace of the oxidation of C(I1) produced in situ, by Fe(CN),a-, at pH = 9.0. p = 0.2 (Na,SO,). Abscissa, 5 ms/division; ordinate, photomultiplier output, 10 mV/divi- sion; V, = 600 mV. A = 550 nm.
TABLE V pH dependence of rate constants of fast and slow stages of oxidation in
the presence of Na,SO, (n = 0.2)
The kinetics were followed at 550 nm, 520 nm, and 450 nm.
PH Fast stage
k,
Slow stage M-ls-l
7.0 4.1 X 106 6.2 1.5 x 108 4.2 x lo8 9.0 1.5 x 108 3.8 x 10” 9.6 1.1 x 106 3.6 x lo6
10.1 1.2 x 108 3.9 x 106 10.8 7.1 x 10’
obtained yielding a rate constant similar to the slow phase when C(R) is produced in situ in the PR experiments, or when Fe”’ (CN).-3 is produced in situ (Table II).
Acid base spectrophotometric titrations of the 695 nm band of C(II1) with and without ClO,- present, were identical, yielding a pK, of -9.3.
DISCUSSION
and II show our results of T-jump relaxation measurements and of PR for the rate of oxidation of C(I1) by Fe1”(CN),-3, produced in situ according to Reaction 1. The values from flow and T-jump measurements obtained in other laboratories are given for comparison in Table VII. Under similar conditions all three methods produce similar results. At high ionic strength and pH 6.8 to 8.9 the oxidation follows second order kinetics with k = (4 to 5) x 10’ M-’ s-’ (PR). Some of our results from PR seem at first sight to diverge from the expected behavior. In spite of the fact that the interaction site of C(R) and Fe”’ (CNIO-’ are oppositely charged, similar rates of oxidation are obtained at ionic strengths differing by a factor of 20. In order to explain this observation we refer to the studies of Oxidation-Reduction Reaction Involving Complex Forma-
tion of Cytochrome c and Ferri-ferrohexacyanides-Tables I Stellwagen and co-workers (29, 30), who-based on NMR
.8 -
FIG. 4. The relative amount of C(W) produced by oxidation of C(U) generated in situ, in each of the oxidation phases, as a function of pH. p = 0.2 (NaC103, 0, fast phase; 0, slow phase; [CIII] = 4 x lo-’ M; [K$e(CN),] = 4 x 10-s~.
TABLE VI
Observed and second order rate constants for fast and slow stages of oxidation by Fe(CN),I- of C(U), f ormed in situ at neutral pH (6.7 to
7.0) and an ionic strength of 0.2 (NaClO,).
The values given are the mean of at least five experiments each at 550 nm, 520 nm, and 450 nm.
Fast stage WWN.1
k ODe k,
M SK’ M-*S.1
1 X 10-s 710 7.1 x 10’ 2 x 10-S 1460 7.3 x 10’ 3 x 10-S 2340 7.8 x 10’ 4 x 10-s 4000 1.0 x 108 4 x lo-’ 4.4 x 10’ 1.1 x 10’ 1 x 10-s 6.1 x 10’ 6.1 x 10’ 4 x 10-50 4400 1.1 X 108
OO.1 M Na,SO, + 0.05 M NaClO,.
Slow stage
k lldo k,
s-1 M-S-’
24 2.4 x 10’ 50 2.5 x 10’ 72 2.4 x lo8 68 2.7 x lOa
1240 3.1 x 106 2000 2.0 x 106
120 3.0 x 106
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

l-Electron Redox Reactions by Fast Pulse:Cyt clFe(CNjGm3 4341
studies-suggested the formation of complexes between cyto- chrome c and ferri-ferrocyanide according to the following equilibria:
C(H) + Fe”’ (CN),-3 - k1 C(II).Fe”’ (CN),-3 -!? k-1 k-z m
C(II1). Fe” (CN),-’ $ C (III) + Fe” (CNjem4 3
They also suggested that complexes such as C(R) .Fe”(CN),-’ and C(II1) . Felll(CN),-S could be formed. Equilibrium dialysis (29, 30) gave association constants for all these complexes in the range of 300 M-’ to 1000 M-I. They suggested that there are
FIG. 5. Dependence of log k on the square root of the ionic strength. in the presence of N&IO,. 0, pH = 6.8, slow phase, - (left scale); 0, pH = 9.4, fast phase, (---) (right scale). [CIII] = 4 x 10mS M; [K$e(CN),] = 2 x lo-lo.
two binding sites of ferro-ferricyanide per one protein, with similar association constants, and that electrostatic interac- tion dominates the binding process (31). In some of our PR measurements, at ionic strength (p) of 0.01 we used high concentrations of Fe” (CN) @-’ (-2 mM), in the presence of C(I1) but no other added salts. Thus, if the complexes suggested above are formed we would have C(I1) molecules bound to Fe”‘(CN),-’ before being oxidized. According to the association
constants per binding site from refs. 29 and 30 (-lo9 M-l),
under these conditions we should have -40% of C(R) in an uncomplexed form. Thus two phases of oxidation should be observed depending on whether free C(I1) or C(II).Fe”(CN),-* reacts with Fe11”(CN),-3. Yet, only one phase of oxidation is observed, slower than expected for uncomplexed C(R). If C(R) molecules are almost entirely bound to Fe”(CN),-‘, the interaction of Fel”‘(CN),-S may be with sites on C(R) already occupied by ferrocyanide, and this can slow down the oxidation in spite of the low ionic strength, as indeed is observed. It follows then that the association constant for equilibrium III should be >_ 10’ M-I.
C(II)Fe” (CN),-* & C(I1) + Fe” (CN) .-’ (III)
In order to confirm this hypothesis we measured the rate of oxidation at the same ionic strength (0.01) but when no complexes such as C(I1) .Fe(CN),-’ could be formed. (This we accomplished by having initially low concentrations of Fel”!(CN),-s and C(II1) and producing C(II) by Reaction 5. The ionic strength was set by phosphate or sulfate.) Under such conditions the rate measured for the oxidation, 7 x 10’ M- ’ s-l, was faster by about 1 order of magnitude than under conditions where C(II) .Fe”(CN),-’ complexes are present (Table VII).
Using a value of 10’ M-’ for the association constant III, one can calculate that under the conditions of the other studies (l-8), the amount of complex formed is negligible. For exam- ple, in T-jump measurements (6-8), when fairly high concen- trations of Fe” (CN),-’ (-8 mM) were used, the concentration of cytochrome c was rather low (0.005 to 0.2 mM), and large amounts of competing anions (29, 30) were present, therefore ferro-ferricyanide complexes are not significant. When the concentration of complexes is negligible, as is the case at high ionic strength, taking kl >> km2 and using steady state for
TABLE VII
Rate constants for oxidation by Fe(CN),s- of C(U)
k, Ir Anion present Method Reference
M.ls-l
1.6 x 10’ 1.2 x 10’ 6.7 x lo6 8.0 x 106 8.1 x 10’ 6.3 x 10’ 1.2 x 10’
4.1 x 10’ 6.7 x 10’ 4.1 x 10’ 2.6 x 10’ 3.1 x 108 9.2 x 106” 7.0 x lo?*
0.10 PO, 0.10 Cl- 0.18 Cl- 0.10 Cl- 0.18 SO,*-, Fe(CN),‘- 0.09 PO, + Cacodylate + Tris 0.17 NO,-
0.2 so,z- 0.2 so,z- + PO, 0.2 PO, 0.2 ClO, 0.2 ClO,- 0.01 Fe(CN),‘- 0.01 so,*-
flOW
flOW
flOW
T-jump T-jump T-jump
PR T-jump PR PR T-jump PR PR
(1) (2) (3) (5) (6) (7) (8)
This work This work This work This work This work This work This work
o C(R) present mainly as C(II)Feii (CN).‘- complex. b No complex present.
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

4342 l-Electron Redox Reactions by Fast Fulse:Cyt cjFe(CN),-3
C(I1). Fe”’ (CN),-S, according to Equilibrium II the following kinetic law can be derived for the oxidation:
V = d[Co= (k,k,)/(k_, + k,) [WI)] [Fe”’ (CN),-3] dt
This is a second order process, with k,, = (k,k,)/(k_, + k,), as is indeed observed under such conditions.
The presentation of k,. in this manner can explain the very low activation energies observed in this process (3, 5).
The higher rate of oxidation obtained in the presence of sulfate or phosphate at p = 0.01 as compared to that at p = 0.01 in excess Fe”(CN),-‘, together with results at higher ionic strength (II = 0.2), indicate that anions such as phosphate and sulfate compete with the ferro-ferricyanide anions for the binding site(s), but have lower affinities. This is in accordance with previous studies from which two binding sites for phos- phate with association constant of 4 x 10’ M-* (31, 32) were determined (it should be pointed out that the association constant for phosphate is for the equilibrium of cytochrome c with two ligands, namely for cytochrome c + 2 L z cytochrome c(L) A.
From the discussion up to this point the following features emerge: (a) Fe”(CN),-’ binds to C(R); (b) the association constant is 210’ Me’ per binding site affecting electron transfer; and (c) other anions such as sulfate and phosphate can bind to the site(s) with lower affinities.
With regard to (b) one should remember that in our studies only binding sites affecting electron transfer can be detected. To account for our observations as to the mechanism of oxidation there have to be at least two different possible routes for electron transfer from the ferroheme in C(I1) to Fe”’ (CN) 6- ‘. I f it is assumed that electron transfer can proceed from the site of binding of the hexacyanide complex, as is the case in the reduction process, the slower rate observed for oxidation of C(II).Fe(CN),-’ as compared to free C(R) is explained by the blocking effect of the Fe”‘(CN),-’ bound on C(I1). Oxidation then proceeds either by an exchange of Fe”’ (CN),-9 with bound Fe”(CN),-’ followed by electron transfer from the ferroheme to Fe”‘(CN),mS, or a chain of electron transfer involving bound Fe”(CN),-‘, bulk Fel”l(CN),-S and ferroheme. Such an explanation will also require that :Fe1”(CN),-3 and Fe”(CN)(-’ share common binding site(s) on cytochrome c. Since electrostatic interaction dominates the binding of the hexacyanide complexes on the protein, the requirement that the similar anions Fe”(CN),-’ and Fe”’ (CN) a-J have identical binding sites is not unreasona- ble. However there is a different mechanism of oxidation by which one can explain the slower rate of oxidation of CII Fe”(CN),-’ as compared to free C(I1). It is possible that the binding of anions (such as Fe”(CN),-‘) induces conforma- tional changes on C(R) which make the electron transfer process less efficient. This will explain the slowness of oxida- tion of C(I1) Fel’;(CN),-S by Fe”’ (CN),-3 without introducing the conditions of identical binding and electron transfer sites for oxidation of C(I1) by Fe”‘(CN),-8 and for reduction of C(II1) by Fe”(CN),-‘.
Whatever the case may be, if indeed C(I1). Feii’(CN),-S is present the oxidation of ferrocytochrome c will produce C(II1). Fe” (CN),-‘. We would have then a back reaction-re- duction. That we have reduction is suggested by the lower yields of oxidation under such conditions. The kinetics of this reduction process should obey true first order law. If the reduction showed pseudo-first order instead of true first order
kinetics we would have kobs = k,.[C(II)] + kred [Fe” (CN),-‘1, .
glvmg an increase of k,,, when Fe(CN),-’ is increased, rather than k,,, = LKNI) I + km,, as observed. Fig. 1 shows that with 1 and 2 mM Fe”,(CN),-‘, the results are identical; with 4 mM kobs slightly decreases, (certainly does not increase). The slight decrease may be due to increased ionic strength. From Fig. 1 we obtain kred = 400 s-’ which is in fair agreement with the value of 208 s-’ obtained for the same reaction from NMR measurements (29, 30).
These observations give further evidence for the existence of C(I1) . Fel’,(CN),-’ complexes and also show that in the reduc- tion of C(II1) electron transfer can proceed from the binding site of the hexacyanide on cytochrome c, and that Fe” (CN),-’ has identical binding sites on C(B) and C(II1). As discussed above we cannot give evidence that oxidation of C(I1) by Fe”’ (CN),-:’ can also proceed from the same site from which reduction occurs. However the oxidation of C(I1) by Fe”‘l(CN),-S is slowed down when Fe”(CN),-’ is bound on C(I1).
Oxidation of Relaxed and Nonrelaxed Ferrocytochrome c by Ferrihexacyanide-The very fast reduction of C(II1) by e, to C(I1) has been studied extensively (19, 33-36) and reviewed recently (15). Of interest in the present study are the following findings: (a) reduction at neutral or basic pH is very rapid with k = 10” to 10” M-’ s-‘; (b) at neutral pH the reduction is followed by two conformational changes of the protein, one in microseconds, the other in milliseconds, at the end of which a relaxed native ferroenzyme is obtained; and (c) at alkaline pH (8 < pH < 10) only one conformational change at 0.1 s is observed. This slow process is also observed when dithionite is used as a reducing agent.
The oxidation of ferrocytochrome c by ferricyanide can be studied by pulse radiolysis in two ways. Starting with C(R) and Fe” (CN),-’ and instantaneously selectively oxidizing Fe” (CN),-’ as in Reaction 1 to produce Fe”’ (CN) o-9, or start- ing with C(II1) and Fe1”(CN),-3 and instantaneously selectively reducing C(II1) as in Reaction 5 to produce C(I1).
We expect that when producing C(I1) in situ (by e,) at neutral pH its oxidation by Fe”’ (CN),-a will be monophasic, since all relaxations of C(I1) to native C(R) are accomplished within a time equal to or shorter than that of the oxidation process. Indeed at neutral pH only one process is observed (Table V and Fig. 2a, insert), obeying second order kinetics with rates similar to those measured by T-jump (Tables II and VII). By T-jump such nonrelaxed protein molecules cannot be produced. Furthermore the rates obtained at neutral pH are identical with those measured under similar conditions but where Fe”‘t(CN)e-S is produced in situ and C(R) is initially present in the native form.
The fact that only one phase of oxidation is observed at all ionic strengths at neutral pH allows an estimation of the upper limit of the association constant for the equilibrium:
C (III) + Fe”’ (CN),+ 8 C (III). Fe”’ (CN),-3 (IV)
I f K,, > 10’ M-l, under our conditions we would have some of the cytochrome c molecules free and some bound to ferricyanide and therefore two phases of oxidation would be observed: thus K,, should be < 10’ M-I.
As originally reported by Theorell, C(II1) has five distinct pH-dependent states,
IA -&,L.,,L. 0.42 2.5 9.35 12.76
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

l-Electron Redox Reactions by Fast Pulse:Cyt clFe(Ch9,-3 4343
pK. = 9.3 is attributed to the replacement of the Met-80 ligand of the iron with a lysine (37). Ferrocytochrome c on the other hand has only two pK values at 4.0 and 12.0. The conforma- tional change at 0.1 s observed in the reduction of C(II1) (4, 36) at alkaline pH was related to the relaxation of C(I1) from the ~(111) conformation to the conformation of relaxed C(I1). Above pH 7.0 we begin to observe two phases of oxidation (Table V, and Fig. 3). A biphasic reduction of C(II1) by Fe”s(CN),-’ was observed at pH = 9.0 (6). Both observations may be connected with the state of binding of the metionine to iron, though the detailed mechanism may differ (see below).
in effective charges found for Co(Phen),+3 (11) and for ferricya- nide.
Each of the oxidation phases obeys simple second order kinetics: first order with respect to C(I1) and to Fe”“(CN)(Im3. As already suggested above, these two reactions may be due to the reaction of Fe”’ (CN),-3 with the two conformations of ferrocytochrome c, namely the relaxed and the nonrelaxed protein which relaxes with z -0.1 s. Support for this assump- tion is given by the titration curve where the relative amount of oxidized enzyme produced by each process is plotted versus the pH (Fig. 3). The pK obtained is 9.2, in agreement with the pK, 9.3 found for ferricytochrome c. Why the fast phase does not yield up to 100% oxidation (Fig. 3) is not fully understood. It is possible that at pH > 10 other conformations of the protein are involved. This is also suggested by the decrease in the rate of the faster process at pH > 10 and the lower effective charge at this pH (Table IV). Changes in reactivity of cytochrome c at pH > 10 were also reported elsewhere (36) and were attributed to structural changes in cytochrome c. The faster reaction is the one between Fe”’ (CN),-” and the nonrelaxed C(B). This is concluded from: (a) the slower phase has rates comparable to those measured with the relaxed ferroenzyme; and (6) the higher the pH the larger the contribution of the faster reaction to the oxidation process. In the faster process Fe”’ (CN),-3 reacts with a nonrelaxed ferrocytochrome c in which the Met-80 ligand of the C(I1) is displaced. Since the rate of the slower phase of the reaction is equal to that measured with native C(II), this rate may give a lower limit for relaxation of the sites involved in oxidation in the ferroenzyme produced in situ, having Met-80 ligated to the iron (19). The effective charges for the two phases of oxidation or that measured when C(I1) is initially present (ionic strength produced by phosphate or sulfate) are identical: +2.0 (Table IV). This high effective charge can be compared to a value of +1.7 found for the reaction of ferricytochrome c with Fe(EDTA)-* (9). It seems thus that the interaction site(s) on C(I1) and C(II1) for oxidizing or reducing anions, respectively, are similar in charge type. According to our observation for Fe”(CN),-’ the binding site(s) and the site(s) from which electron is transferred in C(II1) are identical. We now estimate that the binding site(s) of Fe”:(CN),-’ on C(II1) is * +2 charged. This high effective positive charge suggests an involvement of a cationic cluster on the protein. Note that the effective charges are obtained at high ionic strengths, where the Debye-Huckel equation is not strictly applicable, and are lower limits.
Cationic clusters are known to exist on cytochrome c and are due to lysines and arginines (38). The effect of blocking lysines on oxidation-reduction processes in cytochrome c has been shown in other studies (39-41). The fact that at pH 2 pK (lysine) we have a significant decrease in the effective charge (+0.6) in the oxidation process gives further indication of the involvement of these residues in the mechanism of oxidation. From the model of cytochrome c we observe cationic clusters on the protein: one cluster produced by Lys 88, Lys 86 and Arg 91; the other cluster produced by lysines alone in one of the following configurations: [5,7,8,87]; (13,87,8,5]. According to Stellwagen and Cass (30) lysine 79 is involved in one of the cluster-binding sites for ferricyanide on cytochrome c. How- ever, it is difficult to see how lysines 27, 13, and 79-which are quite far apart from each other-can form a cluster as they suggest (30). Recently arguments were advanced against in- volvement of Lys 79 (42).
An explanation for a faster rate in the reaction of ferricyanide with the nonrelaxed C(B) (having the C(II1) conformation) as compared to native C(B), lies probably in the fact that C(II1) has a much more open conformation as compared to C(R) (43). Oxidation of “partially opened” or denatured ferrocytochrome c is known to proceed more efficiently than that in native C(R) (44). The fact that nonrelaxed enzyme is more effective in the electron transfer process may be relevant to the fast electron transfer in in uiuo conditions (45). Our results give a high rate of oxidation of relaxed C(I1) by Feiii(CN), -3, k = 2 x lOa M-’
s-’ (at zero ionic strength), which is difficult to explain by an outer sphere mechanism using Marcus theory calculations (13). Both self exchange rates of the cytochromes and the hexacyanides decrease through ionic effects in a manner which is not expected to be compensated by the increase of the relevant equilibrium constant. The self exchange rate of ferro- ferricyanide (46) is lo8 to 10’ M-’ s-l, strongly dependent on the cation added but not strongly on the anion or the general ionic strength. The self exchange rate of horse heart cyto- chrome c is lo9 M-‘s-l at 1 = 0.1 (47) reaching 2 x 10’ at high ionic strength when the charges on cytochrome c are screened. At pH = p1 there is no dependence on the ionic strength. Sutin who at first thought that agreement with the Marcus theory is perhaps possible at high ionic strengths (13) later also became doubtful of this (2). Our results are in better agreement with a mechanism in which electron equivalent transfer to and from the iron heme occurs via a somewhat remote binding site on the protein.
The rate of oxidation (Table V) and effective charge (Table IV) on the nonrelaxed protein at pH 210 together with the observed disappearance of binding sites on the protein at pH 210 (29, 30) indicate that binding of the hexacyanide com- plexes is not an obligatory condition for electron transfer; the rate of electron transfer at pH -10.5 in the absence of these binding sites is only by a factor of -2 smaller than at pH <9.
In a study on the oxidation of C(B) (11) an effective “active Oxidation of Relaxed and Nonrelaxed Ferrocytochrome c in site charge” of +0.4 was found, much lower than the value of the Presence of Perchlorate Anion-Results obtained in the +2 (this work) or +1.7 (from Ref. 9) found for reduction of presence of ClO,- are significantly different from those ob- C(II1). However, in the studies on the oxidation the highly tained in the presence of other anions. (a) A pK, of 7.4 (Fig. 4) is positively charged Co(Phen),+3 cation was used (11) and in obtained for the two phases of oxidation of the C(I1) produced reduction studies a negatively charged anion Fe”(EDTA)-’ in situ, instead of 9.3 which is obtained in its absence or in the (9). I f indeed cationic clusters are involved the repulsion of presence of other anions (SO,-2 and HIPO,-). (b) A pK, of 9.3 Co(Phen),+s would be large, which may explain the difference is obtained in the absence, as well as in the presence, of ClO,-
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

l-Electron Redox Reactions by Fast Fulse:Cyt clFe(CNj,-3
for the absorption change at 695 nm, which is connected with the Fe-S bond.
Rate constants of the oxidation of C(I1) produced in situ or of relaxed C(I1) are always smaller in the presence of ClO,- than those obtained in the presence of sulfate or phosphate (Tables I, II, and VI).
The special behavior of cytochrome c in the presence of ClO,- has been reported in other studies. It was found (48) that the spectrum of acid-cytochrome c is affected by ClO,- more than by such other anions as Cl-, Br-, I- and SCN-. It was suggested that perchlorate affects the structure of water, and through this the interaction of the protein with molecules. In a pulse radiolysis study it was also found that reduction of cytochrome c by many organic radicals is slowed down in the presence of ClO,- as compared to other ions at similar ionic strengths. *
Following the reasoning of the previous section, the biphasic oxidation is explained by oxidation of nonrelaxed and relaxed C(I1) by Fe”“(CN),-‘, when C(I1) is produced in situ. That this is indeed the case is shown by T-jump experiments (Table II) and pulse radiolysis (Table I) studies where native relaxed C(I1) is initially present. In these experiments only a single phase of the oxidation process is observed. It obeys second order kinetics. In the previous section (where no ClO,- was present) the biphasic phenomenon showed a pK of 9.2 and could be attributed on the molecular level to the displacement of Met-80 in the newly formed ferroenzyme. In the presence of ClO,-, however, the observed pK is shifted by 2 pH units, to 7.4, there is no kinetically observed pK at 9.2, while the titrations of the 695 mm ‘band, which is related to the S-Fe bond, in the presence or absence of ClO,-, show a pK of 9.3. It seems that the displacement of Met-80 affects indirectly the site(s) in question while ClO,- affects the same site(s) directly, without causing a displacement of the Met-80 ligand. It follows then that this displacement of Met-80 is not an obligatory condition for the observation of the faster phase of oxidation, or to the production of a nonrelaxed ferroenzyme involved in this process. An acid-base equilibrium of a residue with pK 7.4 as well as the dominant effect of ClO,-, even in the presence of excess concentrations of other anions, gives further support for this assumption.
The pK of 7.4 could either be an inherent property of a site or residue of ferricytochrome c being ‘activated’ only when ClO,- is present or a result of the presence of ClO,-, a water structure breaking anion that causes the observed proton equilibrium. Whatever the case may be this residue is not accessible to ClO,- in relaxed C(I1). The more efficient oxidation of non- relaxed C(I1) produced as a result of the presence of ClO,- at neutral pH may be due to the nonrelaxed protein having an open structure, similar to C(II1). Both relaxed and nonrelaxed C(I1) at alkaline pH are oxidized at somewhat slower rates in the presence of ClO,-; the reason for this is not clear. The involvement of a cationic cluster on the protein in the oxidation is shown in Fig. 5 in the presence of ClO,-. At low ionic strength an effective charge greater than or equal to +2.O is again obtained.
There are some general considerations which emerge from the present work. Studies on cytochrome c (49, 50) indicated that binding sites for the large oxidase and reductase com- plexes differ. Studies with some relatively small inorganic reagents (9, 11, 13) indicated that both oxidation and reduction
2A. Shafferman and G. Stein, unpublished results.
of cytochrome c utilize a common site at the heme edge. Our results using FCN are consistent with a common site and indicate that binding involves specific amino acid cluster(s) on the protein.
Nonrelaxed ferrocytochrome c transfers an electron equiva- lent more rapidly (3 x 10’ M-’ s-l at p = 0) than the relaxed ferromolecule. Such a nonrelaxed protein can be produced in aqueous solution by fast reduction of ferricytochrome c having either the Met-80 ligand displaced due to the presence of sufficient concentration of OH- ions (pK = 9.3) or having some amino acid residue(s) on the ferricytochrome affected through the influence on its water environment of the ClO,- ion present, leaving the S-Fe bond intact (pK = 7.4). Neither pK effect is present in C(I1). The residue(s) on ferricytochrome -with which these two ions interact-are no longer accessible for such interactions in the ferroenzyme. The higher efficiency of electron transfer found for a nonrelaxed oxidation-reduction enzyme is a fact to be kept in mind when extrapolations are made from in vitro oxidation-reduction studies to the in uiuo system. What is usually referred to as a relaxed conformation of an enzyme is either related to its properties in aqueous solution or to its conformation obtained from crystal structure. These conformations need not be identical with the “relaxed” conformation of the enzyme in oiuo (e.g. on a membrane). The importance of such a distinction is indicated by the relatively slow rate of 400 s-l for electron transfer from Fe”(CN),-’ bound on C(II1) to the ferriheme, as compared to the rate of 500-1000 s-’ for an electron transfer through the whole mitochondrial oxidative chain and from fast kinetic studies (19, 36) where the final characteristic spectrum of reduced cytochrome c is observed only after slow conformational changes following the fast reduction of C(II1).
The specific effect of ClO,- under such conditions may be due to its presence as noncomplexing gegen-ion assuring electroneutrality in the positively charged active region on the protein. The effect of ClO,- on the water structure near the site may release single water molecules for acid base equilibria of amino acids on the protein revealing the pK of 7.4 which we observed kinetically.
After completion of this manuscript we read the detailed works of Cusanovich and co-workers (51, 52) on the reaction of cytochrome c and Rhodospirillum rubrum cytochrome cZ with iron hexacyanides, studies by stopped flow and T-jump tech- niques. Under similar experimental conditions their results agree with ours. Our critical conclusions reached from studies utilizing the enlarged experimental range of the pulse method apply to their work also.
Acknowledgment-We thank Professor Berta Hayman and Mrs. Ruth Shinar for valuable assistance and advice with the T-jump work.
REFERENCES
1. Sutin, N., and Christman, D. R., (1961) J. Am. Chem. Sot. 83, 1773-1774
2. Creutz, C., and Sutin, N. (1974) J. Biol. Chem. 249, 6788-6795 3. Morton, R. A., Overnell, J., and Harbury, H. A. (1970) J. Biol.
Chem 245, 4653-4657 4. Lambeth, D. O., Campbell, K. L., Zand, R., and Palmer, G. (1973)
J. Biol. Chem. 248, 8130-8136 5. Cassatt. J. C.. and Marini. C. P. (19741 Biochemistry 13,
5323-5328 I \ - - - I
6. Brand& K. G., Parks, P. C., Czerlinski, G. H., and Hess, G. P. (1966) J. Biol. Chem. 241, 4180-4185
7. Zabinski, R. M., Tatti, K., and Czerlinski, G. H. (1974) J. Biol.
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

l-Electron Redox Reactions by Fast Pulse:Cyt clFefCN),-3 4345
Chem. 249, 6125-6129 32. Margalit, R., and Schejter, A. (1973) Eur. J. Biochem. 32,500-505 8. Havsteen, B. H. (1965) Acta Chem. &and. 19, 1227-1231 33. Pecht, I., and Farragi, M. (1972) Proc. N&l. Acad. Sci. U. S. A. 69, 9. Hodges, H. L., Holwerda, R. A., and Gray, H. B. (1974) J. Am. 902-906
Chem. Sot. 96,3132-3137 34. Wilting, J., Nauta, H., and Braams, R. (1971) FEBS Lett. 16, 10. Yandell, J. K., Fay, D. P., and Sutin, N. (1973) J. Am. Chem. Sot. 147-151
95, 1131-1137 35. Land, E. J., and Swallow, A. J. (1971) Arch. Biochem. Biophys. 11. McArdle, J. V., Gray, H. B., Creutz, C., and Sutin, N. (1974) J. 145, 365-372
Am. Chem. Sot. 96, 5737-5741 36. Land, E. J., and Swallow, A. J. (1974) Biochim. Biophys. Acta 368, 12. Ewall, R. X., and Bennett, L. E. (1974) J. Am. Chem. Sot. 96, 86-96
940-942 37. Dickerson, R. E., and Timkovich, R. (1975) in The Enzymes: 13. &tin, N. (1972) Chem. Br. 8, 148-151 Oxidation Reduction (Boyer, P. D., ed) Vol. 11, pp. 397-547, 14. Bennett, L. E. (1973) Progress in Inorganic Chemistry. (Lippard, Academic Press, New York
S. J., ed) Vol. 18, pp. 1-176, John Wiley & Sons, New York 38. Dickerson, R. E., Takano, T., Eisenberg, D., Kallai, 0. B., 15. Shafferman, A., and Stein, G. (1975) Biochim. Biophys. Acta Samson, L. Cooper, A., and Margoliash, E. (1971) J. Biol.
Bioenergetics Reu. 416, 287-317 Chem. 246, 1511-1535 16. Jonah, C. D., Hart, E. J., and Matheson, M. S. (1973) J. Phys. 39. Takemori, S., Wada, K., Ando, K., Hosokawa, M., Sekuzu, I., and
Chem. 77, 1838-1843 Okunuki, K. (1962) J. Biochem. 52, 28-37 17. Margoliash, E., and Frohwirt, N. (1959) Biochem. J. 71, 570-572 40. Wada, K., and Okunuki, K. (1968) J. Biochem. 64, 667-681 18. Massey, V. (1959) Biochim. Biophys. Acta 34,235-236 41. Wada, K., and Okunuki, K. (1969) J. Biochem. 66, 249-262 19. Lichtin, N. N., Shafferman, A. and Stein, G. (1973) Biochim. 42. Power, S. D., Chaucair, A.,
Biophys. Acta 314.117-135 and Palmer, G. (1975) Biochem.
Biophys. Res. Commun. 66, 103-107 20. Rabani, J., and Matheson, M. S. (1969) J. Am. Chem. Sot. 86, 43. Takano, T., Kallai, 0. B., Swanson, R., and Dickerson, R. E.
3175-3176 (1973) J. Biol. Chem. 248, 5234-5255 21. Anbar, M., and Neta, P. (1967) ht. J. App. Rad. Chem. 18, 44. Davison, A. J., Hamilton, R. T., and Kaminsky, L. S. (1971) FEBS
493-523 Lett. 19, 19-21 22. Lichtin, N. N., Shafferman, A., and Stein, G. (1974) Biochim. 45. Chance, B. (1969) in Theoretical Physics and Biology (Morois, M.,
Biophys. Acta 357, 386-398 ed) pp. 156-158, North-Holland Publishing Co., Amsterdam 23. Shafferman, A., and Stein, G. (1974) Science 183, 428-430 46. Campion, R. J., Deck, C. F., King, P., and Wahl, A. C. (1967) 24. Nilsson, K. (1972) Israel J. Chem. 10, 1011-1019 Inorg. Chem. 6, 672-681 25. Gordon, S., Hart, E. J., Matheson, M. S., Rabani, J., and Thomas, 47. Gupta, R. K. (1973) Biochim. Biophys. Acta 292, 291-5
J. K. (1963) J. Am. Chem. Sot. 85, 1375-1377 48. Aviram, I. (1973) J. Biol. Chem. 248, 1894-1896 26. Zehavi, D., and Rabani, J. (1974) J. Phys. Chem. 78, 1368-1373 49. Margoliash, E., Ferguson-Miller, S., Tulloss, J., Hang, C. H., 27. Scholes, G., and Wilson, R. L. (1967) Trans. Faraday Sot. 63, Feinberg, B. A., Brautigan, D. L., and Morrison, M. (1973) Proc.
2983-2993 Natl. Acad. Sci. U. S. A. 70, 3245-3249 28. Shirom, M., and Stein, G. (1969) Israel J. Chem. 7,405412 50. Smith, L., Davies, H. C., Reichlin, M., and Margoliash, E. (1973) 29. Stellwagen, E., and Shulman, R. G. (1973) J. Mol. Biol. 80, J. Biol. Chem. 248, 237-243
559-573 51. Wood, F. E., and Cusanovich, M. A. (1975) Bioinorg. Chem. 4, 30. Stellwagen, E., and Cass, R. D. (1975) J. Biol. Chem. 250, 337-352
2095-2098 52. Miller, W. G., and Cusanovich, M. A. (1975) Biophys. Struct. 31. Nicholls, P. (1974) Biochim. Biophys. Acta 346, 261-310 Mech. 1, 97-111
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from

Y Ilan, A Shafferman and G Steingeneration of reagents. Cytochrome c/ferri-ferrocyanide system.
The study of 1-electron equivalent oxidation-reduction reactions by fast pulse
1976, 251:4336-4345.J. Biol. Chem.
http://www.jbc.org/content/251/14/4336Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/251/14/4336.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on February 11, 2018http://w
ww
.jbc.org/D
ownloaded from