the sacred lotus genome provides insights into the evolution of flowering plants
TRANSCRIPT

Acc
epte
d A
rtic
le
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as an 'Accepted Article', doi: 10.1111/tpj.12313 This article is protected by copyright. All rights reserved.
Received Date : 22-Jan-2013
Revised Date : 04-Aug-2013
Accepted Date : 12-Aug-2013
Article type : Original Article
The sacred lotus genome provides insights into the evolution of
flowering plants
Yun Wang1,9, Guangyi Fan2,3,9, Yiman Liu1,9, Fengming Sun2,3,9, Chengcheng Shi2,3,9, Xin
Liu2, Jing Peng1, Wenbin Chen2, Xinfang Huang1, Shifeng Cheng2, Yuping Liu1, Xinming
Liang2, Honglian Zhu1, Chao Bian2, Lan Zhong1, Tian Lv2, Hongxia Dong1, Weiqing Liu2,
Xiao Zhong2, Jing Chen2, Zhiwu Quan2, Zhihong Wang1, Benzhong Tan4, Chufa Lin4, Feng
Mu3, Xun Xu2, Yi Ding5, An-Yuan Guo6, Jun Wang2,7,8 & Weidong Ke1.
1 Wuhan Vegetable Research Institute, Wuhan 430065, China.
2 BGI-Shenzhen, Shenzhen 518083, China.
3 BGI-Wuhan, Wuhan 430075, China.
4Wuhan Academy of Agricultural Sciences and Technology, Wuhan 430065, China.
5 College of Life Sciences, Wuhan University, Wuhan 430072, China.
6Department of Biomedical Engineering, College of Life Science and Technology, Huazhong University of
Science and Technology, Wuhan 430074, China.
7 Department of Biology, University of Copenhagen, Copenhagen, Denmark.
8 King Abdulaziz University, Jeddah, Saudi Arabia.
9These authors contributed equally to this work.
Correspondence should be addressed to Weidong Ke ([email protected]), Jun Wang
([email protected]), An-Yuan Guo ([email protected]) and Yi Ding ([email protected]).

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
SUMMARY
Sacred lotus (Nelumbo nucifera) is an ornamental plant that is also used for food and
medicine. This basal eudicot species is especially important from an evolutionary perspective,
as it occupies a critical phylogenetic position in flowering plants. Here we report the draft
genome of a wild strain of sacred lotus. The assembled genome is 792 Mb, which is
~85–90% of genome size estimates. We annotated 392 Mb of repeat sequences and 36,385
protein-coding genes within the genome. Using these sequence data, we constructed a
phylogenetic tree and confirmed the basal location of sacred lotus within eudicots.
Importantly, we found evidence for a relatively recent whole-genome duplication event; any
indication of the ancient paleo-hexaploid event was, however, absent. Genomic analysis
revealed evidence of positive selection within 28 embryo-defective genes and one annexin
gene that may be related to the long-term viability of sacred lotus seed. We also identified a
significant expansion of starch synthase genes, which likely elevated starch levels within the
rhizome of sacred lotus. Sequencing this strain of sacred lotus thus provided important
insights into the evolution of flowering plant and revealed genetic mechanisms that influence
seed dormancy and starch synthesis.
INTRODUCTION
Sacred lotus (Nelumbo nucifera) belongs to Nelumbonaceae (Angiosperm Phylogeny
Group, 2009), which is a family of basal eudicot plants that contains only one genus,
Nelumbo. There are only two species within the Nelumbonaceae family, sacred lotus and
American lotus (Nelumbo lutea) (Pan et al., 2010). Sacred lotus is primarily found in East
Asia and Northern Australia, whereas American lotus inhabits eastern portions of North
America and northern regions of South America. In China and other Asian countries, sacred
lotus is an economically important crop and is used for food, medicine, and ornamentation.
Sacred lotus is also important from an evolutionary perspective, as Nelumbonaceae
occupies a key phylogenetic position and may provide critical information concerning the
origin of eudicots (Gandolfo et al., 2004). As a basal eudicot species, sacred lotus may also

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
help to better understand gamma, the ancient genome triplication event that likely contributed
to the early diversification of the core eudicots. Analysis of MADS-box genes suggests that
the gamma event occurred before rapid speciation of the earliest core eudicot lineages
(Vekemans et al., 2012). It would be more informative, however, to estimate the time of this
event using the entire genome sequence of sacred lotus.
To date, studies concerning sacred lotus have focused primarily on its medicinal value
(Kashiwada et al., 2005; Ono et al., 2006; Ohkoshi et al., 2007), the regulation of flowering
by temperature (Seymour, 1998; Watling et al., 2006; Li and Huang, 2009), and the genetic
diversity between varieties (Pan et al., 2007; Pan et al., 2010; Hu et al., 2012). Sacred lotus
also has interesting characteristics concerning seed formation, dormancy, and starch synthesis
that warrant investigation. For example, it blossoms and sets seed during the hot summer, a
process that is likely to involve genetic mechanisms related to a high temperature-response.
In addition, the seeds of sacred lotus can remain dormant for extended periods of time before
germinating (Shen-Miller et al., 2002), and its rhizome is rich in starch (9.25% of fresh
weight) (Mukherjee et al., 2009). These phenotypes make sacred lotus an excellent model for
studying biological processes that control seed formation, seed dormancy, and starch
synthesis and underscore the importance of determining the entire genome of this species.
We have sequenced a wild strain of sacred lotus and obtained a draft genome assembly.
These data confirmed the phylogenetic placement of sacred lotus in eudicots and identified a
recent whole-genome duplication (WGD) event in sacred lotus; however, no evidence for an
ancient whole-genome triplication event was found. We also identified genes under positive
selection that may be involved in seed formation and dormancy and found the expansion of
one gene family that may be important for starch synthesis. In addition, this sequenced
genome provides an out-group for studying the evolution of eudicots and will help to develop
genetic markers to improve breeding practices for the sacred lotus crop.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
RESULTS
Sequencing, assembly, and annotation
We sequenced the genome of a wild strain of sacred lotus from Central China, which
represents a typical subtropical ecotype. Using 67.7 Gb of high-quality data (Table 1), a draft
genome was assembled with 792 Mb in length, including 31,452 contigs and 3,031 scaffolds
(>2 kb). Contig N50 and scaffold N50 (50% of the sequences were longer than this length)
were 39.3 kb and 986.5 kb, respectively (Table 2). Estimations of the sacred lotus genome
size were 879 Mb (based on k-mer analysis, Figure S1 and Table S1) (Li et al., 2010a) and
929 Mb (based on flow cytometry) (Diao Y et al., 2006), which were ~10% and ~15% larger,
respectively, than our current version. To assess the assembly, we isolated RNA from the bud
tissue from one sacred lotus plant, generated 4.6 Gb of sequence data from this sample, and
assembled the data into 77,330 transcript fragments. We were able to map more than 95% of
these transcripts to the assembly (Table 3).
Within the sacred lotus genome we identified 392 Mb (49.48% of the assembly) of
sequences related to transposable elements (TEs). Among the identified TEs, the long
terminal repeat (LTR) was most abundant (~40% of the assembly). Two LTR superfamilies,
Gypsy and Copia, represented 15.98% and 24.59% of the genome, respectively. As such, the
Gypsy-to-Copia ratio was 0.65:1, which is substantially lower than that which has been
observed for other eudicots (Figure S2a and Table S2). Similarly, the En-Spm-to-hAT ratio
of DNA TEs was lower in sacred lotus (0.34:1) than in other flowering plants (3.29:1 in
maize and 1.66:1 in Arabidopsis) (Figure S2b and Table S2). To further characterize the TE
composition of the sacred lotus genome, we estimated insertion times for LTR/Copia and
LTR/Gypsy elements (SanMiguel et al., 1998). Compared with other species, insertion times
were longer for sacred lotus, with greater differences between LTR sequences (Figure S3).
As a result, there are more copies of Copia elements than Gypsy elements in sacred lotus,
which differs from other species.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
We then masked repeat sequences and annotated protein-coding genes throughout the
genome assembly. This identified 40,348 gene models in sacred lotus, including 36,385
protein-coding genes and 3,963 potential transposon-related genes (Table 2 and Table S3).
Many of these gene models were structurally similar to homologs identified in other species
(Figure S4). In addition, 76.97% of these genes could be functionally annotated using
homology approaches (Table S4). Finally, 84.00% of these genes (gene coverage >= 50%)
were also represented in our RNA-Seq data (Figure S5).
Genomic evolution of sacred lotus
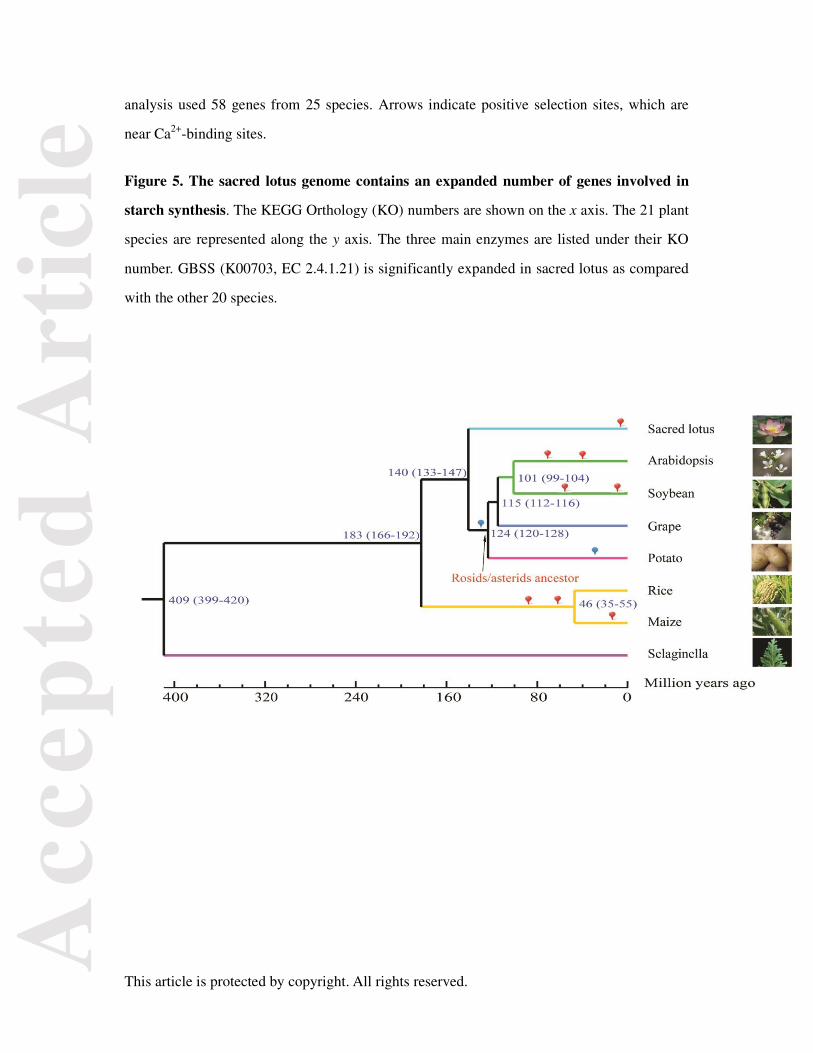
To understand the evolution of sacred lotus, we first identified families of genes by
clustering encoded proteins based on pair-wise similarity. Of 40,348 sacred lotus gene models,
27,562 were classified into 13,834 families. There were 2,075 gene families that were
specific to sacred lotus, and 9,481 families were shared among sacred lotus, Arabidopsis,
grape, and soybean (Figure S6). Then, using gene families that contained a single copy gene
from Selaginella (lycophyte), rice, maize (monocots), Arabidopsis, soybean (rosids), potato
(asterids), and grape, we constructed a phylogenetic tree (Figure 1). Within the phylogenetic
tree, sacred lotus was part of the eudicot cluster but formed an independent basal branch with
respect to other eudicots. This confirmed that sacred lotus is a basal eudicot. The time of
divergence between sacred lotus and other eudicots was estimated to be 140 million years ago
(Mya).
WGD allows for dynamic changes to the genome and accelerates genome evolution (Van
de Peer et al., 2004). To reveal WGD events in sacred lotus, we first characterized the
distribution of four-fold degenerate third-codon transversions (4DTvs) in sacred lotus gene
pairs (Figure 2a and Figure S7). Similar results were obtained when we used SiZer software
(Chaudhuri and Marron, 1999) to analyze the distribution of 4DTvs (Figure S8). There was a
single peak in the 4DTv distribution at lower values (4DTv ≈ 0.17; Ks, ~0.35–0.55),
indicating that there was only one WGD event, which occurred ~18 Mya. This event was
more recent than the alpha WGD in Arabidopsis (~62 Mya; Ks, ~0.47–1.87; 4DTv ≈ 0.25)

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
(Bowers et al., 2003). In fact, among all sequenced plant genomes, only soybean has had a
more recent WGD event (~13 Mya; Ks, ~0.13–0.39; 4DTv ≈ 0.057) (Schmutz et al., 2010).
This recent WGD event is consistent with the sacred lotus genome containing more gene
models than other plant species. According to the fossil record, however, there have been
very few phenotypic changes within the Nelumbonaceae clade, indicating a very slow rate of
evolution during the last 160 million years (Collinson, 1980; Muller, 1981; Cevallos-Ferriz
and Stockey, 1989). There is a discrepancy, therefore, between the fossil record and evidence
of a recent WGD event, which should have resulted in dramatic phenotypic changes. Further
investigations are required to precisely describe the genetic and phenotypic evolution of
sacred lotus.
As we identified only a single recent WGD event in sacred lotus, this genome lacked the
paleo-hexaploid arrangement (gamma WGD) that is common in rosids and asterids. To
confirm this we compared syntenic genes between sacred lotus and grape, as grape has the
paleo-hexaploid arrangement but no other recent genome duplications (Jaillon et al., 2007).
Genome-wide identification of blocks of syntenic genes (Figure 2b) revealed a 2:3
relationship between sacred lotus and grape. This supports a recent WGD event and the
absence of an ancient whole-genome triplication in sacred lotus. We next analyzed scaffolds
that contained duplicated genes (as indicated by the 4DTv distribution). For 307 scaffold
pairs in the 4DTv distribution (representing 59.17% of all scaffolds by length), 77.33% had
three syntenic blocks that corresponded to grape sequences, supporting the 2:3 relationship
(Figure 3a and Figure S9). In addition, when these grape syntenic blocks were compared
with the entire gene set of sacred lotus, 85.84% contained two regions of synteny, which
indicates a 1:2 relationship (Figure 3b and Figure S10). These data also support a WGD
event in sacred lotus but not a whole-genome triplication. Furthermore, among the 40
MADS-box genes in sacred lotus, 16 localized to duplicated regions (Table S5). We also
found a 2:3 syntenic relationship between sacred lotus and grape when we analyzed the AG,
AG32, and SOC1 genes (Figure S11). Taken together, these results strongly support the
occurrence of a recent WGD and the absence of the gamma WGD in sacred lotus. With no
ancient genome triplication in sacred lotus, the paleo-hexaploid arrangement within ancestors

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
of rosids and asterids must have occurred after the split between core eudicots and sacred
lotus. Our results indicate, therefore, that the paleo-hexaploid event probably occurred
124–140 Mya, which represents a more precise estimate concerning the timing of the
triplication event.
Seed formation in sacred lotus
Sacred lotus can flower and set seed during the hot summer, and its seeds retain the
ability to germinate after long periods of dormancy (Shen-Miller et al., 2002). We were
interested to identify, therefore, genetic mechanisms that control embryo development,
dormancy and thermotolerance in sacred lotus. Our whole-genome analysis revealed genes
and gene families that are likely to play specific roles in embryo development and seed
dormancy.
Embryonic development of sacred lotus Genes involved in the embryonic development were
identified from the SeedGenes database (Tzafrir et al., 2003). Clustering these genes with
sacred lotus genes revealed 762 developmental genes within sacred lotus. Twenty-eight of
these genes had sites that were under selection in sacred lotus as compared with other species
(FDR < 0.01, P < 0.01). Homologs of some of these 28 genes affect embryonic development
(Table S6). The homolog of CCG005143.1, for example, is At1g63160, which affects DNA
replication and RNA modification. Eliminating cytosolic translation of At1g63160 results in
100% male and female gametophyte lethality (Berg et al., 2005). Changes made to these
genes within the sacred lotus genome may have affected specific features of embryonic
development.
Dormancy and thermotolerance of the sacred lotus seed Sacred lotus is exceptional in that
its seed can remain dormant for hundreds of years and then germinate when placed into
optimal conditions (Shen-Miller et al., 2002; Chu et al., 2012). Sacred lotus seeds can also
withstand extremely high temperatures, and annexins play important roles in this process
(Ding et al., 2008; Chu et al., 2012). It is thought that the peroxidase activity of annexins
protects membranes against peroxidation (Jami et al., 2008). One annexin gene, NnANN1,

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
has been identified in sacred lotus and regulates seed thermotolerance and germination (Chu
et al., 2012). Here we identified additional annexin genes in sacred lotus by identifying
annexin homologs. Phylogenetic analysis of all annexin homologs within these species
identified five major families (Figure S12). We next examined Ka/Ks values associated with
annexin genes in sacred lotus and discovered that CCG039026.1 (ANNfam5) was under
selection. In addition, Bayes empirical Bayes analysis confirmed two sites of positive
selection within the C-terminal of ANNfam5 (0.916 and 0.948 probability). These sites may
affect binding to Ca2+ (Delmer and Potikha, 1997) and phospholipids (Gerke et al., 2005)
(Figure 4 and Table S7).
Starch-associated genes of sacred lotus
Sacred lotus stores starch in its rhizome. As metabolic pathways associated with starch
and sucrose can affect starch synthesis (Shimada et al., 1993), we examined 35 genes
involved in starch synthesis in 21 plant species with sequenced genomes (Table S8).
Granule-bound starch synthase (GBSS; EC 2.4.1.21) genes were significantly expanded in
sacred lotus (Chi-square test with Bonferroni correction, P = 0.0015) (Figure 5 and Table S9).
GBSS, ADP-glucose pyrophosphorylase (ADPGPPase; EC 2.7.7.27), and starch branching
enzyme (SBE; EC 2.4.1.18) are all important for starch storage (Fisher et al., 1996). GBSS
affects amylose synthesis by catalyzing glucosyl transfer from ADP-glucose to the growing
α-1,4-D-glucan chain (Shimada et al., 1993; Takaha et al., 1993). Phylogenetic and
orthologous analyses of GBSS genes in sacred lotus showed that 70 GBSS genes evolved
recently (Figure S13). Thirty-five of these 70 genes localized to the WGD region, indicating
that the expansion of starch genes was likely caused by the recent WGD. This expansion in
GBSS genes may have resulted in high levels of amylose in sacred lotus. Potatoes also haves
a high starch content in their tubers, but no significant gene expansions within the
starch-synthesis pathway were identified in the potato genome (Table S10). As such,
processes other than gene expansion (e.g., changes in gene expression levels) may play
important roles in starch synthesis in potato (Xu et al., 2011).

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
DISCUSSION
Here we provide a draft genome sequence of sacred lotus, which is an important
agricultural product and a vital tool for understanding plant evolution. The sacred lotus
genome provides solid evidence concerning the divergence of basal eudicots and the ancestor
of rosids/asterids. Because the fossil record indicates a stable phenotype for Nelumbonaceae
plants, it was thought that they retained an ancient genome. Contrary to this hypothesis, we
identified a very recent WGD event (~18 Mya) in sacred lotus. It is clear that the gamma
triplication event is present in Gunnera (NCBI, Vekemans et al., 2012) (Figure S14). As this
triplication event is absent from the sacred lotus genome, we can deduce that all the species
that emerged before the divergence from the ancestor of sacred lotus should not have
experienced the gamma event.
The sacred lotus genome provides interesting resources to study genetic mechanisms that
control agriculturally important features, including seed dormancy and starch synthesis. Seed
dormancy increases the likelihood that a plant can successfully reproduce, even when
confronted with drought conditions. Dormancy increases the ability to survive natural
catastrophes, reduces competition between individuals, and inhibits germination during
inappropriate seasons. Using genetic approaches, researchers have recently identified specific
genes that regulate dormancy in different species (Finkelstein et al., 2008). Comparative
analysis of whole-genome sequences, combined with extensive knowledge concerning
dormancy, will help identify genetic factors associated with this trait that are common to
different species (Finkelstein et al., 2008). Here we identified two sites of positive selection
within the ANNfam5 gene of sacred lotus that may affect seed dormancy. We also identified a
significant expansion of starch-related genes (the GBSS genes) in sacred lotus, which may
explain starch enrichment within rhizome tissue. This genetic expansion may also have
resulted in the crisp taste that is associated with the sacred lotus rhizome. Genomic data
presented here may provide tools and guidance for improving the sacred lotus crop and for
understanding its unique biological features.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Methods
Genome sequencing and assembly
A whole-genome shotgun sequencing strategy was applied using an Illumina Hiseq2000
platform (Illumina, http://www.illumina.com). Approximately 97 Gb of data was generated
from different libraries, with insert sizes of 200 bp, 500 bp, 800 bp, 2 kb, 5 kb, 10 kb, and 20
kb. Libraries were constructed using Illumina reagents. The raw data were filtered before
assembly to remove duplications, adaptor contamination, and sequences with too many
low-quality bases. We used the k-mer (sequences of length k, which we set to 17) depth
distribution to estimate the size of the genome as described (Li et al., 2010a). We randomly
selected 16.4 Gb of filtered data and calculated the frequency of each k-mer. The peak depth
of 17-mers was 15-fold, and a total of 13,186,209,392 17-mers was obtained. By dividing the
total number of 17-mers by the peak depth, we estimated the genome to be 879 Mb. The
genome was assembled using standard steps of the SOAPdenovo software (Li et al., 2010b),
which included contig construction, scaffold linking, and gap closure. Detailed parameters
were as follows: Pregraph -s Lotus.lib, -a 150, -p 12, -K 43, -R, -o Lotus; Contig -g Lotus, -R;
Map -s Lotus.lib, -g Lotus; Scaff -g Lotus -F.
Repeat annotation
DNA TEs were predicted according to a homolog-based search and de novo prediction.
We used RepeatMasker (http://www.repeatmasker.org/), which is based on Repbase (Jurka et
al., 2005), to search for homologs of known repeats. We then used RepeatModeler (RECON
(Bao and Eddy, 2002) and RepeatScout (Price et al., 2005)) to predict repeats de novo. Using
generated de novo repeats as a database, we again applied RepeatMasker to search for these
sequences throughout the genome. Finally, we identified tandem repeats using TRF (Benson
et al., 1999), with the following parameters: Match = 2, Mismatch = 7, Delta = 7, PM = 80,
PI = 10, Minscore = 50, and MaxPeriod = 2000. TE proteins were identified using
RepeatProteinMask in RepeatMasker.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Estimation of LTR divergence
A similar TE annotation process was carried out in Arabidopsis, grape, rice, maize,
sorghum, and sacred lotus. LTR-FINDER (Xu et al., 2007) was used to find complete
LTR/Gypsy and LTR/Copia with 3’- and 5’-LTRs available. All LTR pairs were aligned using
MUSCLE (Edgar et al., 2004), and the distance between them, K, was calculated with the
Kimura two-parameter model using the distmat program implemented within the EMBOSS
package (Hu et al., 2011).
Gene annotation
Homology-based gene prediction For the rough alignment, we aligned protein-coding
sequences from Arabidopsis, strawberry, soybean, potato, and grape to the sacred lotus
genome using TBLASTN (E-value = 1e–5). HSPs were grouped into gene-like structures
using our internal script. For the precise alignment, we first excised target-gene fragments
from the genome by extending 2000 bp from both ends of the aligned regions, including
intronic regions. Parental protein sequences were then aligned to these DNA fragments using
GeneWise (Birney et al., 2004).
De novo gene prediction All TEs within the genome were masked before performing de novo
gene predictions. Two prediction programs were used: Genscan (Salamov et al., 2000) and
Augustus (Stanke et al., 2006). Gene model parameters were trained using Arabidopsis, and
small and partial genes (<150 bp) were filtered before the analysis.
Transcript clustering Data derived from homology-based (five sets from five species) and de
novo predictions (two sets from two programs) were integrated using GLEAN (Elsik et al.,
2007) to generate a consensus set of genes.
Using RNA data to improve GLEAN results RNA-Seq data were obtained from sacred lotus
bud tissue. We used TopHat (Trapnell et al., 2009) to map raw RNA-Seq reads to the sacred
lotus genome, providing information about potential exons. These data were also used to
identify splice donor and acceptor sites. We then used Cufflinks (Trapnell et al., 2010) to

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
assign these potential sites to transcripts and used a fifth-order Markov model to predict open
reading frames. Finally, we integrated these predicted cDNA sequences (based on RNA-Seq
data) with results from GLEAN. If a gene was predicted by both of these methods, the cDNA
sequence as predicted based on the RNA-Seq data was used to represent the gene. Genes
identified only by RNA-Seq were also added to the final gene set.
Functional gene annotation To assess gene function, proteins encoded by predicted sacred
lotus genes were matched (base on BLASTP alignments) to proteins within the Swiss-Prot
(Bairoch et al., 2000) and TrEMBL databases. Protein motifs and domains were determined
using InterProScan (Zdobnov et al., 2001) against the protein databases Pfam, PRINTS,
PROSITE, ProDom, and SMART. Gene Ontology IDs (Ashburner et al., 2000) for each gene
were obtained from corresponding InterPro entries. To assign functional pathways to
predicted genes, gene products were also aligned to proteins within the KEGG database
(Kanehisa et al., 2000).
Annotation of non-coding RNAs The programs tRNAscan-SE (Lowe et al., 1997) and
INFERNAL (Nawrocki et al., 2009) were used to predict non-coding RNAs within the
sacred-lotus genome. Eukaryotic parameters were used with tRNAscan-SE to predict tRNA
genes. To identify ribosomal RNA fragments, we used BLASTN (E-value = 1e–5) to align
potential ribosomal sequences with template sequences of plant ribosomal RNA. Both
microRNA and small nuclear RNA genes were predicted using INFERNAL against the Rfam
database (Griffiths-Jones et al., 2005).
Analysis of sacred-lotus evolution
Ortholog clustering We used OrthoMCL (Li et al., 2003) to define gene families as groups of
genes that descended from a single gene in the last common ancestor of the species under
consideration. First, BLASTP was used to compare all protein sequences with a database that
contained all proteins from all relevant species (E-value = 1e-5). Gene clustering was then
performed using OrthoMCL using the following parameters: OrthoMCL mode = 3;
P-value cut-off = 1e–5; percent identity cut-off = 0; percent match cut-off = 0; MCL inflation

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
= 1.5; maximum weight = 316; BLAST: -p blastp, -e 1e–5, -F F.
Phylogenetic analysis We used single-copy gene families from eight species to construct the
phylogenetic tree. We first used MUSCLE (Edgar et al., 2004) to perform multiple
alignments of protein sequences for each single-copy gene family. Phase-1 sites were
extracted from each family and concatenated into a single super-gene for each species.
Mrbayes (Huelsenbeck et al., 2001) was then used to construct the phylogenetic tree.
Identification of genes under selection CDSs associated with single-copy gene families were
used to investigate non-synonymous to synonymous divergence rates between species.
Branch-specific Ka and Ks values were then estimated using codeml in PAML with the
branch-site model (Yang et al., 2007). The Ka/Ks ratio of each branch allowed us to identify
genes under selection in sacred lotus.
Estimation of divergence time The BRMC approach was used to estimate species divergence
time using MCMCTREE, which is part of the PAML package (Yang et al., 2007). The
“Independent rates molecular clock” and “HKY85” models within the MCMCTREE program
were used to perform these calculations. The MCMC process of the MCMCTREE program
was run 200,000 times, with a sample frequency of 2. This followed a burn-in of 20,000
iterations. “Fine-tune” parameters were set to make acceptance proportions fall within the
interval 0.15–0.70. Other parameters were set to default. Two independent runs were
performed to check convergence. Calibration times of 148 Mya for Arabidopsis-rice
divergence and 109 Mya for soybean-Arabidopsis divergence were acquired from the Time
Tree database (Hedges et al., 2006).
Intergenomic and intragenomic alignments Syntenic blocks between two genomes were
identified using several steps. An initial BLASTP alignment (E-value = 1e–5) was performed
to collect pair-wise information about two proteins. Blast output typically contains multiple
alignments for each protein pair. We selected the alignment with the lowest E-value. Syntenic
blocks (with five or more genes per block) were then constructed using MCscan (Tang et al.,
2008, MCscan: -a, -e 1e–5, -u 1, -s 5; BLAST: -e 1e–5, -p blastp) based on aligned pairs of

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
proteins. Each aligned block represents orthologous pairs of genes that were derived from a
shared ancestor and retained in a particular order. This method was also used to identify
paralogous regions within the sacred lotus genome that arose through genomic duplication.
For each block, a 4DTv value was calculated and revised using the HKY model.
Estimating when the WGD took place To estimate how long ago the sacred lotus genome
was duplicated, we used BLASTP (E-value = 1e–5) to identify paralogous pairs of genes
within the genome. Paralogous gene pairs were then subjected to MCscan, which identified
4,075 syntenic gene pairs. Sequences associated with each pair were aligned using MUSCLE,
and levels of non-synonymous nucleotide substitutions (Ks) were calculated using “yn00”
within the PAML package. The distribution of Ks values revealed a peak at 0.35–0.55,
suggesting a WGD event. This is consistent with the 4DTv distribution. According to the
formula: Time = (Ks peak value)/2γ, where γ is the rate of synonymous substitutions per site
every billion years (γ = 1.5e–8 for dicots), we estimated that this duplication event happened
~18 Mya.
Analysis of seed regulation
The NnANN protein family was downloaded from the NCBI database, and homologous
genes within the sacred lotus genome were identified using BLAST. OrthoMCL (Li et al.,
2003) was used to identify orthologous genes for six species. KEGG annotation was
performed for 25 species using BLASTP. Positive selection sites of selection within these
genes were obtained using Bayes empirical Bayes analysis (Yang et al., 2005).
Identification of MADS-box gene families
Transcription factors including 40 MADS-box genes in sacred lotus and other species
were identified using HMMER 3.0 (http://hmmer.org/).

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
DATA ACCESS
This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank
under the accession APLB00000000. The version described in this paper
is the first version, APLB01000000.
ACKNOWLEDGMENTS
We thank Laurie Goodman (Editor-in-Chief of (Giga)n Science) for assistance in revising the
manuscript. This work was supported by Technology Innovation Projects (YCX201002001,
YCX201101001) supported by Wuhan Academy of Agricultural Sciences and Technology,
the Enterprise Key Laboratory Supported by Shenzhen (CXB201108250096A), National
Public Welfare Sectors (Agriculture) Special Research supported by the Ministry of
Agriculture (200903017), Enterprise Key Laboratory Supported by Guangdong Province
(2011A091000047),The Ministry of Agriculture - 948 program (2010-Z31), National Gene
Bank Project of China and State Key Laboratory of Agricultural Genomics (2012DQ782025).
AUTHOR CONTRBUTION
J. W. and W. K. designed the project. Y. W., G. F., Y. L., F. S and C. S. leaded the
sequencing and analysis. W. L., J. P., Y. D., L. Z. and X. H. did the genome assembly. X. Z.,
H. Z., J. C. and M. W. did the annotation. S. C., C. B., T. L., X. L., Y. L., H. D., Z. Q. and B.
H. did the evolutionary analysis. Z. W., M. B., T. T., B. T., Z. L., C. L. and R. Z. conducted
the TF analysis and starch-related analysis. W. C., X. L., F. M., X. X. and A. Y. G. wrote the
manuscript.
DISCLOSURE DECLARATION
The authors declare no competing financial interests.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
SUPPORTING INFORMATION
Supplemental Data:
Table S1. Genome size estimation by 17-mer analysis.
Table S2. Comparison of TE content in sacred lotus genome and other plant species.
Table S3. General statistics of predicted genes.
Table S4. Functional annotation for sacred lotus genes based on known databases.
Table S5. Statistics for transcriptional factor genes of 10 species.
Table S6. Embryo development related genes in sacred lotus seed under positive selection.
Table S7. Genes of annexin family 5 (ANNfam5) in other 24 species.
Table S8. Statistics of starch-synthesized-pathway related genes in 21 species based on
KEGG database.
Table S9. The result of the Chi-square test for the KEGG term of starch-related genes
between sacred lotus and other 21 species.
Table S10. The result of the Chi-square test for the KEGG term of starch-related genes
between potato and other 21 species.
Figure S1. Depth distribution of 17-mer of sacred lotus genome.
Figure S2. Content distribution of four dominant TE subfamilies in 5 species.
Figure S3. Insertion times statistics of LTR/Copia and LTR/Gypay in five species including
sacred lotus, grape, rice, maize and sorghum.
Figure S4. Comparisons of 4 gene features between sacred lotus and other 3 published
species.
Figure S5. Statistic of gene set coverage by RNA-Seq reads for sacred lotus.
Figure S6. Venn diagram showing shared orthologous groups among genomes of sacred lotus,
arabidopsis, soybean and grape.
Figure S7. Syntenic regions of two duplications within sacred lotus genome.
Figure S8. The 4DTv distribution plotted by SiZer to identify the WGD events. Figure S9.
The copy number of the syntenic gene blocks between the duplicated scaffolds (located in
4TDv region) in sacred lotus and the chromosomes of whole grape genome.
Figure S10. The relation between the syntenic blocks of grape genome (with duplicated
scaffolds of sacred lotus) and the duplicated scaffolds of whole sacred lotus genome.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Figure S11. The copy number of some MADS-box genes in the sacred lotus and grape.
Figure S12. Phylogeny tree of annexin genes in 6 species including sacred lotus, maize, rice,
arabidopsis, soybean and grape.
Figure S13. The phylogeny tree of 70 GBSS genes in sacred lotus.
Figure S14.The phylogenetic tree including sacred lotus by the taxonomy of NCBI.
REFERENCES
Angiosperm Phylogeny Group. (2009) An update of the Angiosperm Phylogeny Group classification for
the orders and families of flowering plants: APG III. Bot. J. Linn. Soc, 161, 105-121.
Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M. et al. (2000) Gene
ontology: tool for the unification of biology. Nat. Genet, 25, 25-29.
Bairoch, A. and Apweiler, R. (2000) The SWISS-PROT protein sequence database and its supplement
TrEMBL in 2000. Nucleic Acids Res, 28, 45-48.
Bao, Z. and Eddy, S.R. (2002) Automated de novo identification of repeat sequence families in sequenced
genomes. Genome Res, 12, 1269-1276.
Benson, G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res, 27,
573-580.
Berg, M., Rogers, R., Muralla, R. and Meinke, D. (2005) Requirement of aminoacyl-tRNA synthetases
for gametogenesis and embryo development in Arabidopsis. Plant J, 44, 866-878.
Birney, E., Clamp, M. and Durbin, R. (2004) GeneWise and Genomewise. Genome Res, 14, 988-995.
Bowers, J.E., Chapman, B.A., Rong, J. and Paterson, A.H. (2003) Unravelling angiosperm genome
evolution by phylogenetic analysis of chromosomal duplication events. Nature, 422, 433-438.
Cevallos-Ferriz, S.R.S. and Stockey, S.A. (1989) Permineralized fruits and seeds from the Princeton
chert (Middle Eocene) of British Columbia: Nymphaeaceae. Bot. Gaz, 150, 207-217.
Chaudhuri, P. and Marron J.S. (1999) SiZer for exploration of structures in curves. J. Am. Sta. Assoc, 94,
807-823.
Chu, P., Chen, H., Zhou, Y., Li, Y., Ding, Y., Jiang, L., Tsang, E.W., Wu, K. and Huang, S. (2012)
Proteomic and functional analyses of Nelumbo nucifera annexins involved in seed thermotolerance
and germination vigor. Planta, 235, 1271-1288.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Collinson, M.E. (1980) Recent and tertiary seeds of the Nymphaeaceae sensu lato with a revision of
Brasenia ovula (Brong.) Reid and Chandler. Ann. Bot, 46, 603-632.
Delmer, D.P. and Potikha, T.S. (1997) Structures and functions of annexins in plants. Cell. Mol. Life Sci,
53, 546-553.
Diao Y., Chen L., Yang G., Zhou M., Song Y., Hu Z. and Lin JY. (2006) Nuclear DNA C-values in 12
species in Nymphaeales. Caryologia, 59, 25-30.
Ding, Y., Cheng, H. and Song, S. (2008) Changes in extreme high-temperature tolerance and activities of
antioxidant enzymes of sacred lotus seeds. Sci. China. Ser. C:Life Sci, 51, 842-853.
Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput.
Nucleic Acids Res, 32, 1792-1797.
Elsik, C.G., Mackey, A.J., Reese, J.T., Milshina, N.V., Roos, D.S. and Weinstock, G.M. (2007) Creating
a honey bee consensus gene set. Genome Biol, 8, R13.
Finkelstein, R., Reeves, W., Ariizumi, T. and Steber, C. (2008) Molecular aspects of seed dormancy.
Annu. Rev. Plant Biol, 59, 387-415.
Fisher, D.K., Gao, M., Kim, K.N., Boyer, C.D. and Guiltinan, M.J. (1996) Allelic analysis of the Maize
amylose-extender locus suggests that independent genes encode starch-branching enzymes IIa and IIb.
Plant physiol, 110, 611-619.
Gandolfo, M.A., Nixon, K.C. and Crepet, W.L. (2004) Cretaceous flowers of Nymphaeaceae and
implications for complex insect entrapment pollination mechanisms in early Angiosperms. Proc. Natl
Acad. Sci. USA, 101, 8056-8060.
Gerke, V., Creutz, C.E. and Moss, S.E. (2005) Annexins: linking Ca2+ signalling to membrane dynamics.
Nat. Rev. Mol. Cell Biol, 6, 449-461.
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S.R. and Bateman, A. (2005) Rfam:
annotating non-coding RNAs in complete genomes. Nucleic Acids Res, 33, D121-124.
Hedges, S.B., Dudley, J. and Kumar, S. (2006) TimeTree: a public knowledge-base of divergence times
among organisms. Bioinformatics, 22, 2971-2972.
Hu, J., Pan, L., Liu, H., Wang, S., Wu, Z., Ke, W. and Ding, Y. (2012) Comparative analysis of genetic
diversity in sacred lotus (Nelumbo nucifera Gaertn.) using AFLP and SSR markers. Mol. Biol. Rep, 39,
3637-3647.
Hu, T.T., Pattyn, P., Bakker, E.G., Cao, J., Cheng, J., Clark, R.M. et al. (2011) The Arabidopsis lyrata

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
genome sequence and the basis of rapid genome size change. Nat. Genet, 43, 476-481.
Huelsenbeck, J.P. and Ronquist, F. (2001) MRBAYES: Bayesian inference of phylogenetic trees.
Bioinformatics, 17, 754-755.
Jaillon, O., Aury, J.M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., Choisne, N. et al. (2007) The
grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature,
449, 463-467.
Jami, S.K., Clark, G.B., Turlapati, S.A., Handley, C., Roux, S.J. and Kirti, P.B. (2008) Ectopic
expression of an annexin from Brassica juncea confers tolerance to abiotic and biotic stress
treatments in transgenic tobacco. Plant Physiol. Biochem, 46, 1019-1030.
Jurka, J., Kapitonov, V.V., Pavlicek, A., Klonowski, P., Kohany, O. and Walichiewicz, J. (2005)
Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res, 110, 462-467.
Kanehisa, M. and Goto, S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res,
28, 27-30.
Kashiwada, Y., Aoshima, A., Ikeshiro, Y., Chen, Y.P., Furukawa, H., Itoigawa, M., Fujioka, T.,
Mihashi, K., Cosentino, L.M., Morris-Natschke, S.L. and Lee, K.H. (2005) Anti-HIV
benzylisoquinoline alkaloids and flavonoids from the leaves of Nelumbo nucifera, and
structure-activity correlations with related alkaloids. Bioorg. Med. Chem, 13, 443-448.
Kurek, I., Chang, T.K., Bertain, S.M., Madrigal, A., Liu, L., Lassner, M.W. and Zhu, G. (2007)
Enhanced thermostability of Arabidopsis Rubisco activase improves photosynthesis and growth rates
under moderate heat stress. Plant cell, 19, 3230-3241.
Laohavisit, A., Brown, A.T., Cicuta, P. and Davies, J.M. (2010) Annexins: components of the calcium
and reactive oxygen signaling network. Plant Physiol, 152, 1824-1829.
Lowe, T.M. and Eddy, S.R. (1997) tRNAscan-SE: a program for improved detection of transfer RNA
genes in genomic sequence. Nucleic Acids Res, 25, 955-964.
Li, J.K. and Huang, S.Q. (2009) Flower thermoregulation facilitates fertilization in Asian sacred lotus.
Ann. Bot, 103, 1159-1163.
Li, L., Stoeckert Jr, C.J. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for
eukaryotic genomes. Genome Res, 13, 2178-2189.
Li, L., Zhang, X., Pan, E., Sun, L., Xie, K., Gu, L. and Cao, B. (2006) Relationship of starch synthesis
with it’s related enzymes’activities during rhizome development of lotus (Nelumbo nucifera Gaertn).

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Sci. Agri. Sin, 39, 2307-2312.
Li, R., Fan, W., Tian, G., Zhu, H., He, L., Cai, J., Huang, Q., Cai, Q., Li, B., Bai, Y., Zhang, Z., Zhang,
Y. et al. (2010a) The sequence and de novo assembly of the giant panda genome. Nature, 463,
311-317.
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., Li, Y., Li, S., Shan, G., Kristiansen, K., Li, S. et
al. (2010b) De novo assembly of human genomes with massively parallel short read sequencing.
Genome Res, 20, 265-272.
Mukherjee, P.K., Mukherjee, D., Maji, A.K., Rai, S. and Heinrich, M. (2009) The sacred lotus
(Nelumbo nucifera) - phytochemical and therapeutic profile. J. Pharm. Pharmacol, 61, 407-422.
Muller, J. (1981) Fossil pollen records of extant angiosperms. Bot. Rev, 47, 1-142.
Nawrocki, E.P., Kolbe, D.L. and Eddy, S.R. (2009) Infernal 1.0: inference of RNA alignments.
Bioinformatics, 25, 1335-1337.
Ohkoshi, E., Miyazaki, H., Shindo, K., Watanabe, H., Yoshida, A. and Yajima, H. (2007) Constituents
from the leaves of Nelumbo nucifera stimulate lipolysis in the white adipose tissue of mice. Planta
medica, 73, 1255-1259.
Ono, Y., Hattori, E., Fukaya, Y., Imai, S. and Ohizumi, Y. (2006) Anti-obesity effect of Nelumbo
nucifera leaves extract in mice and rats. J. Ethnopharmacol, 106, 238-244.
Pan, L., Quan, Z., Li, S., Liu, H., Huang, X., Ke, W. and Ding, Y. (2007) Isolation and
characterization of microsatellite markers in the sacred lotus (Nelumbo nucifera Gaertn.).
Mol. Ecol. Resour, 7, 1054-1056.
Pan, L., Xia, Q., Quan, Z., Liu, H., Ke, W. and Ding, Y. (2010) Development of novel EST-SSRs from
sacred lotus (Nelumbo nucifera Gaertn) and their utilization for the genetic diversity analysis of N.
nucifera. J. Heredity, 101, 71-82.
Price, A.L., Jones, N.C. and Pevzner, P.A. (2005) De novo identification of repeat families in large
genomes. Bioinformatics, 21, i351-358.
Salamov, A.A. and Solovyev, V.V. (2000) Ab initio gene finding in Drosophila genomic DNA. Genome
Res, 10, 516-522.
SanMiguel, P., Gaut, B.S., Tikhonov, A., Nakajima, Y. and Bennetzen, J.L. (1998) The paleontology of
intergene retrotransposons of maize. Nat. Genet, 20, 43-45.
Schmutz, J., Cannon, S.B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., Hyten, D.L. et al. (2010)

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Genome sequence of the palaeopolyploid soybean. Nature, 463, 178-183.
Seymour, R.S. (1998) Physiological temperature regulation by flowers of the sacred lotus. Philos Trans R
Soc Lond B Biol Sci, 353, 935–943.
Shen-Miller, J., Schopf, J.W., Harbottle, G., Cao, R.J., Ouyang, S., Zhou, K.S., Southon, J.R. and Liu,
G.H. (2002) Long-living lotus: germination and soil γ-irradiation of centuries-old fruits, and
cultivation, growth, and phenotypic abnormalities of offspring. Am. J. Bot, 89, 236-247.
Shimada, H., Tada, Y., Kawasaki, T. and Fujimura, T. (1993) Antisense regulation of the rice waxy gene
expression using a PCR-amplified fragment of the rice genome reduce the amylose content in grain
starch. Theor. Appl. Genet, 86, 665-672.
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S. and Morgenstern, B. (2006) AUGUSTUS: ab
initio prediction of alternative transcripts. Nucleic Acids Res, 34, W435-439.
Takaha, T., Yanase, M., Okada, S. and Smith, S.M. (1993) Disproportionating enzyme
(4-alpha-glucanotransferase; EC 2.4.1.25) of potato. Purification, molecular cloning, and potential
role in starch metabolism. J. Biol. Chem, 268, 1391-1396.
Tang, H., Bowers, J.E., Wang, X., Ming, R., Alam, M. and Paterson, A.H. (2008) Synteny and
collinearity in plant genomes. Science, 320, 486-488.
Trapnell, C., Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA-Seq.
Bioinformatics, 25, 1105-1111.
Trapnell, C., Williams, B.A., Pertea, G., Mortazavi, A., Kwan, G. et al. (2010) Transcript assembly and
quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell
differentiation. Nat. Biotechnol, 28, 511-515.
Tzafrir, I., Dickerman, A., Brazhnik, O., Nguyen, Q., McElver, J. et al. (2003) The Arabidopsis
SeedGenes project. Nucleic Acids Res, 31, 90-93.
Van de Peer, Y. (2004) Computational approaches to unveiling ancient genome duplications. Nat. Rev.
Genet, 5, 752-763.
Vekemans, D., Proost, S., Vanneste, K., Coenen, H., Viaene, T., Ruelens, P., Maere, S., Van de Peer, Y.
and Geuten, K. (2012) Gamma paleohexaploidy in the stem lineage of core eudicots: significance for
MADS-Box gene and species diversification. Mol. Biol. Evol, 29, 3793-3806.
Watling, J.R., Robinson, S.A. and Seymour, R.S. (2006) Contribution of the alternative pathway to
respiration during thermogenesis in flowers of the sacred lotus. Plant physiol, 140, 1367-1373.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Xu, X., Pan, S., Cheng, S., Zhang, B., Mu, D., Ni, P., Zhang, G., Yang, S., Li, R. et al. (2011) Genome
sequence and analysis of the tuber crop potato. Nature, 475, 189-195.
Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full-length LTR
retrotransposons. Nucleic Acids Res, 35, W265-268.
Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol, 24, 1586-1591.
Yang, Z., Wong, W.S.W. and Nielsen, R. (2005) Bayes empirical bayes inference of amino acid sites
under positive selection. Mol. Biol. Evol, 22, 1107-1118.
Zdobnov, E.M. and Apweiler, R. (2001) InterProScan-an integration platform for the
signature-recognition methods in InterPro. Bioinformatics, 17, 847-848.
TABLES
Table 1. Statistics of libraries, raw data and filtered data.
Library
insert size
Read
length
(bp)
Raw data Filtered data
Total
data
(Gb)
Sequence
depth
(X)
Physical
depth
(X)
Total
data
(Gb)
Sequence
depth
(X)
Physical
depth
(X)
200bp 100 24.7 27.4 27.4 22.4 24.9 24.9
500bp 100 22.5 25.0 62.5 18.4 20.4 51.0
800bp 100 13.8 15.3 61.2 11.0 12.2 48.8
2Kb 49 12.7 14.1 282.0 9.4 10.4 208.0
5Kb 49 7.0 7.7 385.0 3.6 4.0 200.0
10Kb 49 7.7 8.5 850.0 1.8 1.9 189.0
20Kb 49 8.6 9.5 1900.0 1.1 1.2 236.0
Total --- 97.0 107.7 3568.1 67.7 75.0 957.7
*The estimation of genome size is about 879 Mb.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Table 2. Assembly and annotation statistics of sacred lotus genome
Type Result
Assembly*
Number of scaffolds (>=2kb) 3,031
Total length of assembly 792,334,941 bp
Length of the longest scaffold 4,483,919 bp
Number of contigs (>=2kb) 31,452
Scaffold N50/scaffold N90 986,504 bp/210,888 bp
Contig N50/contig N90 39,303 bp/9,518 bp
GC content 38.7%
Annotation
Number of gene models 40,348
Average length of gene 3431.02 bp
Average length of CDS 908.41 bp
Average exons per gene 3.69
Average length of exon 246.50 bp
Average length of intron 939.43 bp
Number of miRNA genes 273
Number of rRNA fragments 1,327
Number of snRNA genes 806
*The contig is the final contig after filling the gap of intra-scaffold. The contig and scaffolds with length
shorter than 100bp were not included in the statistics.
Table 3. Evaluating assembly by transcripts of RNA-Seq data.
Dataset
(bp) NO.
Total
length (bp)
Covered
by
assembly
(%)
With >90% sequence
in one scaffold
With >50% sequence
in one scaffold
Number Percent
(%) Number
Percent
(%)
All 77,330 32,406,538 95.84 70,461 91.12 72,542 93.81
>200 62,408 29,803,764 95.77 56,633 90.75 57,987 92.92

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Dataset
(bp) NO.
Total
length (bp)
Covered
by
assembly
(%)
With >90% sequence
in one scaffold
With >50% sequence
in one scaffold
Number Percent
(%) Number
Percent
(%)
>500 17,998 16,588,649 98.90 17,466 97.04 17,788 98.83
>1000 5,258 7,852,499 99.32 5,110 97.19 5,223 99.33
FIGURE LEGENDS
Figure 1. Phylogenetic tree including sacred lotus. Sacred lotus is a basal eudicot species
that split from the ancestor of core eudicots ~140 Mya. Blue numbers at nodes are the
estimated divergence time from the present. Red markers indicate a WGD event, and the blue
marker represents a triplication event on that branch. The paleo-hexaploid ancestral genome
rearrangement occurred after divergence from sacred lotus and before the divergence of
rosids and asterids. Mya, million years ago.
Figure 2. WGD events identified in the genome of sacred lotus. a) Distribution of 4DTv
distances between sacred lotus and sacred lotus, and between grape and grape. The horizontal
axis represents the 4DTv distance corrected using the HKY model. The vertical axis
represents the percentage of co-linear gene pairs. b) Whole-genome co-linearity between
sacred lotus and grape. Within this block of synteny, dots represent orthologous gene pair
blocks. The order of sacred lotus scaffolds is based on the order of orthologous genes within
the grape genome.
Figure 3. Syntenic relationships between genes within duplicated scaffolds of the sacred
lotus and grape genomes. a) The 2:3 relationship between duplicated scaffolds of sacred
lotus and grape chromosomes. Sacred lotus scaffolds that are of the same color indicate that
they are syntenic within the peak of 4DTv. b) The 1:2 relationship between the syntenic
block of grape and duplicated scaffolds of sacred lotus.
Figure 4. Annexin genes may regulate thermotolerance of sacred lotus seeds. Two sites of
positive selection were identified within the sacred lotus annexin gene (CCG039026.1). This
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
analysis used 58 genes from 25 species. Arrows indicate positive selection sites, which are
near Ca2+-binding sites.
Figure 5. The sacred lotus genome contains an expanded number of genes involved in
starch synthesis. The KEGG Orthology (KO) numbers are shown on the x axis. The 21 plant
species are represented along the y axis. The three main enzymes are listed under their KO
number. GBSS (K00703, EC 2.4.1.21) is significantly expanded in sacred lotus as compared
with the other 20 species.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.