the interaction of rotation and local mode tunneling in the
TRANSCRIPT

The interaction of rotation and local mode tunneling in the overtone spectra of symmetrical hydrides
Kevin K. Lehmann Department of Chemistry, Princeton University, Princeton, New Jersey 08544
(Received 1 March 1991; accepted 3 May 1991)
In the “local mode limit” where the tunneling time for vibrational energy exchange is long compared to the classical rotational period, one expects that the effective rotational Hamiltonian will reflect the reduced symmetry of the local mode state. Hamiltonians in the local mode basis are given for interaction of rotation and local mode tunneling for molecules of the XH, , XH, , and XH, type. Transformation of these Hamiltonians to a symmetrized basis (which diagonalizes the vibrational problem), produces rotational couplings between the vibrational states. Relations between the spectroscopic constants are derived that are less restrictive than those given earlier by Halonen and Robiette, but reduce to them when the assumptions of their model are met. The present algebraic procedure can be easily extended to include higher order terms. The effect of these couplings is to reduce the size of the pure vibrational splittings. This is due to the fact that in the rovibrational problem, in general, one must reorient the angular momentum vector in the body frame as well as transfer the vibrational action between bonds. This increases the length of the tunneling path and thus decreases the rate of vibrational energy transfer. Model calculations show that a simple semiclassical picture can rationalize the observed trends.
INTRODUCTION
The characterization of highly excited vibrational states has become one of the central goals in chemical physics. Such studies tie spectroscopy to intramolecular dynamics (a connection that is always there but is often only implicit) and reaction dynamics. Often, anharmonic interactions, which in traditional spectroscopic theory are “perturba- tions,” change even the topological character of the motion. Perhaps the best understood of these changes is the transi- tion from normal to local modes of vibration that character- ize the stretching overtone bands of almost all symmetric hydrides.’
Despite the extensive literature on local mode vibra- tions, little is known of the implications of local mode vibra- tional motion on the rovibrational energy levels, and thus the rotational dynamics. The first systematic study on this topic was the work of Halonen and Robiette on XY,, XY,, and XY, molecules.2 By assuming a vibrational potential equal to a sum of uncoupled Morse oscillators, and then making several very restrictive assumptions, Halonen and Robiette derived a set of relations between the resonant terms of the H,, operator.3 This operator contains terms of the form qiqj J, Jb and dominates the vibrational dependence of the rotational structure. An interesting prediction of this model is that the rotational structure of a XH, molecule should resemble a parallel transition of a symmetric top, including the factor of 2 higher statistical weight for the K = 3n levels. This effect has since been observed in the overtone spectra of GeH, ,4 SiH, ,’ and SnH, . 6 An independent derivation of this effect has been given by Michelot er al.’ using an alge- braic approach, and this model has been shown to fit the observed first overtone band of SiH, spectrum with an rms 0.003 cm - ’ with only two adjustable parameters in the effec- tive Hamiltonian.
This symmetric top rotational structure can be under- stood in a simple physical way. In the traditional treatment of the rotational motion, one assumes that the vibrational motion is much faster than the rotation. As a result, the rotational energy level structure is determined by a vibra- tionally averaged effective moment of inertia (the diagonal terms of H,, ), But in the local mode limit, a molecule has nearly degenerate vibrational energy levels.’ These can be viewed as symmetrized combinations of “local-mode” states wherein each X-H stretch has a constant vibrational action unlike normal mode states which have constant vibrational action in each normal mode. The splitting between the dif- ferent symmetry representations of the local-mode states is related to the time scale for tunneling to a wave function for which the excitation has swapped bonds. If this tunneling time is much longer than the rotational period, one would expect that the rotational motion and thus energy level structure will reflect the reduced symmetry of a single local- mode state. The adiabatic separation works in reverse, since rotational motion is now fast compared with tunneling. The local-mode state of XH, with all the excitation in a single bond will look like a slightly prolate symmetric top since, on average, the excited bond will be longer. It should be remem- bered that changes in the vibrationally averaged structure are only part of the vibrational dependence of the rotational constants, but such a simplified model correctly predicts the symmetry of the effective rotational Hamiltonian.
It is clear that the above argument depends only on a separation of time scales, and thus does not depend upon the restrictive assumptions about mass, structure, and bending force constants that Halonen and Robiette made in their work. Halonen and Robiette had given numerical evidence that in fact the effective constants for overtone levels would obey certain of the constraints of their local-mode limit
J. Chem. Phys. 95 (4), 15 August 1991 0021-9606/91 /I 62361-l 0$03.00 @ 1991 American Institute of Physics 2361 Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

2362 Kevin K. Lehmann: Overtone spectra of hydrides
much more than the fundamentals; in particular, that the effective rotational constants for each of the local-mode states tends to become equal for each of the nearly degener- ate local-mode vibrational states and that the effective Cor- iolis coupling of the states vanish. In this paper, we shall examine the spectroscopic consequences of a model that in- vokes an effective rotational Hamiltonian for each local- mode vibrational state plus vibrational tunneling between local-mode states. The relationship of the present approach to that of Halonen and Robiette is similar to the two inde- pendent derivations of the ‘X-K ” relations that allow a lo- cal-mode vibrational Hamiltonian to be expressed in a nor- mal mode basis set. Mills and Robiette’ derived the x-K relationships by starting with a simplified model for the po- tential energy function and then using perturbation theory. My independent derivation” started with the Child and Lawton harmonically coupled, anharmonic oscillator Ham- iltonian, ” expressed as a polynomial in the bond mode rais- ing and lowering operators, and by a symmetry transforma- tion of these operators demonstrated the samex-Krelations. Each method has its own advantages. The perturbation method allows one to use a more general force field and estimate the effect of the neglected terms. General perturba- tion expressions for all possible Darling-Dennison (quar- tic) coupling terms between nondegenerate modes were re- cently published. l2 The algebraic method is much easier to implement, however, and proves an exact equivalence for all eigenvalues, which the perturbation method did not. Della Valle has provided general formulas for the x-K relations for any set of equivalent bonds using the algebraic approach. I3
The present study of vibration-rotation interactions will again start in a local-mode basis with a rotation-inver- sion Hamiltonian operator based upon general physical con- siderations, much as in Child and Lawton’s” treatment of the vibrational coupling. Transformation of this Hamilto- nian to a normal mode basis generates a set of relations between the coefficients of the H22 terms. As with the x-K relationships derived earlier, one does not expect the rela- tionships to hold exactly in real spectra since the starting Hamiltonian is only approximate. But the relationships will be useful in attempts to predict and assign spectra, and as possible constraints to be used in spectral fitting when pa- rameter correlation or limited data sets prevents a free fit of all the possible terms. More importantly, it provides a phys- ical model for interpreting the rotational constants that are derived from a fit of a local-mode spectrum.
An interesting result of the present treatment is that rotation of a molecule can greatly reduce the tunneling rate, stabilizing the molecule for a much longer time with vibra- tional motion localized along a single bond. It will be shown that the qualitative features of this effect can be understood by semiclassical analysis of motion on the rotational energy surface, as used by Harter and Patterson.14 The physical explanation for this effect is that to reach an isoenergetic state, one must not only exchange all of the vibrational ac- tion from one bond to another, but in general, one must also reorient the angular momentum in the body fixed frame. Such a rotational suppression of local-mode tunneling ap- pears to have been observed in the first overtone band of
stannane,‘j but it escaped explicit notice though it was cor- rectly predicted by the fitted constants. The present study provides a physical picture for this effect.
XH, MOLECULES Consider an XH2 molecule in a local-mode state In, )
which means there are n quanta in bond 1. By symmetry the state In,) will be degenerate with energy Go. These two states will be coupled by tunneling, leading to a term in the Hamiltonian R (n, ) (n, (. Here R is the rate at which the IZ quanta of vibrational excitation tunnels from bond 1 to bond 2. It is known from earlier work on local modes that this tunneling rate will decrease exponentially with increasing nk.’ This tunneling term will lead to the vibrational eigen- states being $,,, = (In,) f In,))/+‘% with Es = Go +A and E, = Go - /2. In each local-mode state, the rotational motion is described by an asymmetric top Hamiltonian. But due to the reduced dynamical symmetry ofthe (n, ) state, the principle axes for this state will be rotated in the molecular plane by an angle 19 away from those of the ground state (which are by symmetry the C,, axis and perpendicular to it). We label the axes (X,JJ,Z) as theA,B,Caxes of the equilib- rium structure and thus the C,, axis is along y, and the per- pendicular to the molecular plane is z. To lowest order, the inertial axis rotation comes about because the H22 operator has a term q,q, (J, Jy + J,, J, ). The operator qsq, will have a nonzero expectation value, despite being antisymmetric, be- cause the local-mode state does not have C,, symmetry. Be- cause of the longer average bond length of the excited bond, one expects that the A axis will rotate towards this excited bond. The state In*) by symmetry should have the same rotational constants, but its principal axes will be rotated by an equal but opposite amount compared to (n, ). Thus we write an effective Hamiltonian to describe the rotational- tunneling dynamics of the In, ) and In, ) states as
H = [Go +AJ:, +BJ;, +CJ:,]IMn,I
+ [Go +AJll +BJ;2 +CJ:>]ln,)(n,I +A [IMn,l+ b,>hl]7 (1)
where the principal axes are given by
Jx, = cos t9J, + sin OJ,,,
J,, = - sin OJ, + cos BJ,,,
Jx, = cos OJ, - sin OJ,, (2) (2) Jy2 = sin OJ, + cos BJ,,
Jz, =J+=J*.
If we rewrite this Hamiltonian in terms of projections onto the vibrational eigenstates and rotational operators on axes determined by symmetry we find
H = [Go+~++A,J~+B,J~+C,Jt]Is>(sl
+ [Go -A+A,J: +B,J; +C,J:]I4(al
++-L{J,,J,~[ld(~l + Idbll, (3) where
CJx,J,) = JxJ, + J,Jx,
J. Chem. Phys., Vol. 95, No. 4,15 August 1991 Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

~,=A,=A~os~0+Bsin~t4
B, = B, = A sin’ 0 + B COS’ 0,
c, = c, = c,
d, = (A - B) sin 20. (4)
This is the same form as the Hamiltonian determined by Halonen and Robiette, but the relationship between the pa- rameters is not as restrictive. In absorption from the ground state, one will have x polarized transition amplitude to the I a) rovibrational basis states, and y polarized transition am- plitude to the Is> rovibrational basis states. It is worth point- ing out that, while one is tempted to predict the ratio ofx and y matrix elements from the projections of the bonds on the x and y axes (assuming the transition dipole is along the bond), this is generally a poor approximation.
Several interactions are not present in this Hamiltonian, and their absence needs to be justified. The first is an absence of Coriolis interaction between states 1 n’ ) and 1 n, ) . By sym- metry, there exists a Caxis Coriolis interaction between the s and a stretching fundamentals. However, the Coriolis cou- pling constant is small, since without bend-stretch mixing, it arises only from motion of the central atom,” and thus c T3 is on the order of (mn /m, ). The Coriolis interaction matrix between the symmetrized local-mode basis functions will be 2<f, (nlplO)(Olqln). The product of matrix elements ~~~lplwoJql~) will be approximately given by n!( w/lox] ) ’ - “/n, where w is the bond mode harmonic fre- quency and wx the anharmonicity [see Eqs. (3.17)-( 3.19) in Ref. 11. The ratio of o to wx is typically - 50 for a hydride. Thus the Coriolis interaction will decrease exponentially with increasing n. Halonen16 observed numerically that the Coriolis interaction decreased rapidly as the local-mode lim- it was reached. The lack of Coriolis interaction, even in the fundamentals, was part of the Halonen and Robiette model. We can also consider rotational corrections to 1. But il is already a term of order H2n,0 and rotational corrections would have to be of even higher order and thus smaller still. If we add rotational corrections toil proportional to J’, they would split the rotational constants of the s and a states, but they would not introduce any additional coupling between the symmetrized local-mode states.
When ;I is much less than the rotational separations, only local-mode rovibrational states with the same rota- tional energy will mix significantly. These states will be shift- ed by *;1 times the overlap of their rotational functions. Because both local-mode states share the same C axis, only rotational states that have the same symmetry of Kc, but different symmetry of K, can have nonzero rovibrational overlap. As a result, the Hamiltonian can be factored into four symmetry blocks. If the principle axes rotation is small, then the rotational function overlap will be near unity. But if the XY, molecule is near an accidental symmetric top, as many of the molecules with heavy X are, then this rotation can be large. In the extreme, the A and B axes will switch between the two local-mode states. In this case, the rota- tional overlap will decrease rapidly with increasing K, . This represents a rotational quenching of the local-mode tunnel- ing because in addition to having to transfer the vibrational
action, one must move the direction of the body fixed angu- lar momentum to find a state of the same rovibrational ener- gy-
In order to quantify this discussion, the rotation-tun- neling energy levels have been calculated using parameters that should give a reasonable estimate for the n = 3 overtone band of SeH, . In particular, from the analysis of the interac- tion of the fundamentals by Gillis and Edwards,” it is esti- mated that for the n = 3 levels the spectroscopic constants are A(s) = A(a) = A, - 3~4’ = 7.83, B(s) = B(a) = B, -3af=7.39, C=C, -3&=3.74, d,, =3d,, =0.6,
and /2 = 3A T3/( 8wx2) = - 0.06 cm-‘. These parameters imply inertial axis rotation of & 0.5 tanha;;d_/(As -B,)] = f 27” in then = 3 states.
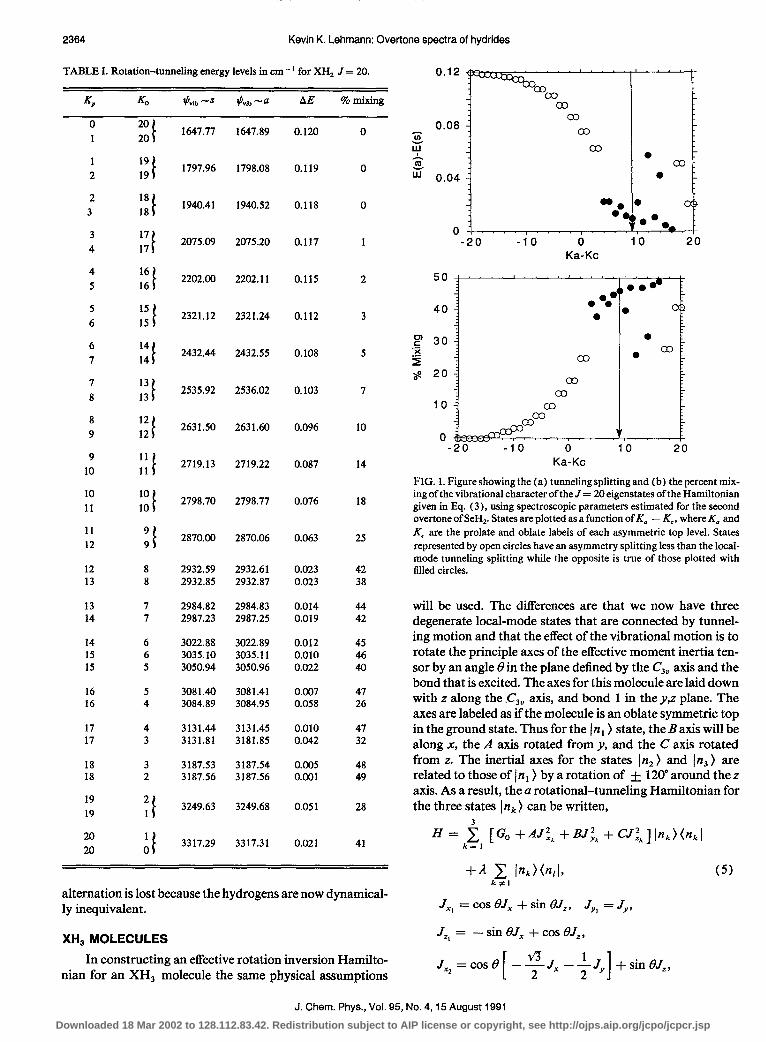
Using the Hamiltonian in Eq. (2) and the parameters just given, the 82 J = 20 levels have been calculated and are given in Table I. In Fig. 1 is plotted both the tunneling splitting and percent mixing of the vibrational character of the eigenstates as a function of K, - Kc. Rotational energy increases mono- tonically from left to right in Fig. 1 This high J was chosen to illustrate the rotational phase space structure of the levels. The lowest energy levels correspond to K, z 0 and are local- ized near the Caxis. The s-u splitting is slightly smaller than 2;1, the value predicted without rotation. We can understand this because these rotational wave functions are almost sin- gle oblate symmetric top functions and thus are the same for both bond modes. With increasing energy, there is an in- crease in the mixing of the symmetric top functions. When the rotational is viewed as motion of the angular momentum vector on the rotational energy surface, the rotational trajec- tories increasingly dip down toward the B axis as K, is in- creased.13 This leads to a slow decrease in the effective tun- neling splitting, down by a factor of two by Kc = 9. This results in the a,s vibrational character being mixed by 25%- 75%. When Kc is decreased further, an interesting effect occurs. The asymmetry splitting, which reflects tunneling between the rotation about the + C and - C axes, grows larger than the tunneling doubling. States for which the asymmetry splitting is larger than the tunneling splitting are marked by solid circles in Fig. 1, where it is obvious that these levels have qualitatively different behavior. Since by symmetry, the s and a vibrational states can only couple across the asymmetry doublet, we see a rapid decrease in the s-u splitting, reaching a value of only 0.0 1 cm - ’ at the level 20’,,6 which is closest to the separatrix separating C- and A- type rotation. The s and a vibrational states are here mixed by 46%-54%. Not until K, reaches 19 does the asymmetry splitting fall below the local mode splitting. The states with K, = 19 and 20 have tunneling splittings of 0.05 1 and 0.02 1 cm-‘, respectively, showing that A-type motion strongly suppresses the tunneling motion. The greater the difference between A and B rotational constants, the greater the volume of rotational phase space that is localized with A axis rota- tion. As a result, a greater fraction of the levels will show suppressed inversion splittings. Figure 1 demonstrates an almost exact inverse relationship between the inversion split- ting and the degree of vibrational mixing. Last, it is noted that when the tunneling splitting is not resolved, the nuclear spin weights of the observed levels will be equal, i.e., the spin
Kevin K. Lehmann: Overtone spectra of hydrides 2363
J. Chem. Phys., Vol. 95, No. 4,15 August 1991 Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
o-,,,.‘,‘,‘,,““,“-‘, or -20 -10 0 IO 20
Ka-Kc
2364 Kevin K.Lehmann:Overtonespectraofhydrides
TABLE I. Rotation-tunneling energy levels in cm - ’ for XH, J = 20. 0.12
Kp KO ?&rib-s 4&b -a AE % mixing
0 0.08 1 205
1647.77 1647.89 0.120 0 3 F
1 19 2 19 t
1797.96 1798.08 0.119 0 c z 0.04
2 18 3 18 1
1940.41 1940.52 0.118 0
3 17 4 17 I
2075.09 2075.20 0.117 1
4 16 5 16 t
2202.00 2202.11 0.115 2
5 15 6 15 t
2321.12 2321.24 0.112 3
6 14 7 14 I
2432.44 2432.55 0.108 5
7 13 8 13 t
2535.92 2536.02 0.103 7
8 12 9 12 1
2631.50 2631.60 0.096 10
9 11 10 11 I
2719.13 2719.22 0.087 14
10 10 11 10 t
2798.70 2798.77 0.076 18
11 9 12 9 I
2870.00 2870.06 0.063 25
12 8 2932.59 2932.61 0.023 42 13 8 2932.85 2932.87 0.023 38
13 7 2984.82 2984.83 0.014 44 14 7 2987.23 2987.25 0.019 42
14 6 3022.88 3022.89 0.012 45 15 6 3035.10 3035.11 0.010 46 15 5 3050.94 3050.96 0.022 40
16 5 3081.40 3081.41 0.007 47 16 4 3084.89 3084.95 0.058 26
17 4 3131.44 3131.45 0.010 47 17 3 3131.81 3181.85 0.042 32
18 3 3187.53 3187.54 0.005 48 18 2 3187.56 3187.56 0.001 49
19 2 19 1 1
3249.63 3249.68 0.051 28
20 1 20 0 I
3317.29 3317.31 0.021 41
FIG. 1. Figure showing the (a) tunneling splitting and (b) the percent mix- ing of the vibrational character of the J = 20 eigenstates of the Hamiltonian given in Eq. (3), using spectroscopic parameters estimated for the second overtone of SeH,. States are plotted as a function ofK, - Kc, where Ka and K, are the prolate and oblate labels of each asymmetric top level. States represented by open circles have an asymmetry splitting less than the local- mode tunneling splitting while the opposite is true of those plotted with filled circles.
will be used. The differences are that we now have three degenerate local-mode states that are connected by tunnel- ing motion and that the effect of the vibrational motion is to rotate the principle axes of the effective moment inertia ten- sor by an angle 8 in the plane defined by the C,, axis and the bond that is excited. The axes for this molecule are laid down with z along the C,, axis, and bond 1 in the y,z plane. The axes are labeled as if the molecule is an oblate symmetric top in the ground state. Thus for the 1 n , ) state, the B axis will be along x, the A axis rotated from y, and the C axis rotated from z. The inertial axes for the states In, ) and In, ) are related to those of 1 n , ) by a rotation of f 120” around the z axis. As a result, the a rotational-tunneling Hamiltonian for the three states Ink) can be written,
Ka-Kc
k#l alternation is lost because the hydrogens are now dynamical- ly inequivalent.
XH, MOLECULES In constructing an effective rotation inversion Hamilto-
nian for an XII, molecule the same physical assumptions
Jx, = cos BJ, + sin t9J,, Jy, = Jy ,
Jz, = - sin 6J, + cos 0J,,
-$Jx --iJy +sinOJ,, 1 Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

Kevin K. Lehmann: Overtone spectra of hydrides 2365
J =aJx -+Jy, Y2 2
Jz, = -sine [
--$Jx -+Jy +coseJ,, 1 A,B, and Care the effective rotational constants in the local- mode state, and 13 is the angle that the C inertial axis is rotat- ed relative to the symmetry axis. Defining symmetrized local mode states by
Jx, = cos e [ $f-Jx -$Jy +sinBJ,, 1
J,, = -$Jx -+Jy,
-+Jy +coseJ,. 1 (6) we can transform the Hamiltonian to a symmetrized vibra- tional basis I
H = [G,,+U+B,(J:+J;) +C,J:]la)(ul+ [G, -A+B,(Jz +J:)+C,J~][le+)(e+ I+k->(e- II
+~[-~rCJ,~J~~+~J2,][l~~~~+I+l~+~~~l+l~+~~~-ll ++[ +idJ,,J+)+qJ’- ][Idb- I + le- )@I +k- )(e+ II, (8)
where & =B, =~[B+AcosZe+Csin2e],
C, =C,=Asin26+Ccos26,
r=(A-C)sin28,
q=Ac0s~8+Csin28-B,
J, =J, -r;l, J- =J, +I;r,. (9) Once again, the form of the terms is the same as Halonen
and Robiette, but the present Hamiltonian is not as restric- tive as to the relationship of the parameters. The relations of Halonen and Robiette are recovered by setting B = C and 0 = 125.26” as required by their model. One obvious feature is that the spectrum should, in the limit of small R, be that of an asymmetric top with A,C hybrid band. This was predicted by Ovchinnikova” who derived a model for the high over- tone bands of NH, that is similar to the present one. Ha- lonen and Robiette found that in their model, the local-mode rotational structure of XH, should be that of a symmetric top. This is due to Halonen and Robiette’s assumed geome- try, which resulted in the molecule being an accidental spherical top in the ground state. As a result, the vibrating bond ensured that one axis would be unique, but the other two retained the same moments of inertia and thus a sym- metric top spectrum resulted. Notice, however, even in this limit the quantization axis is one of the bonds, not the sym- metry axis and thus the rotational levels will not have the 2: 1 nuclear spin weight alternation of the ground vibrational state.
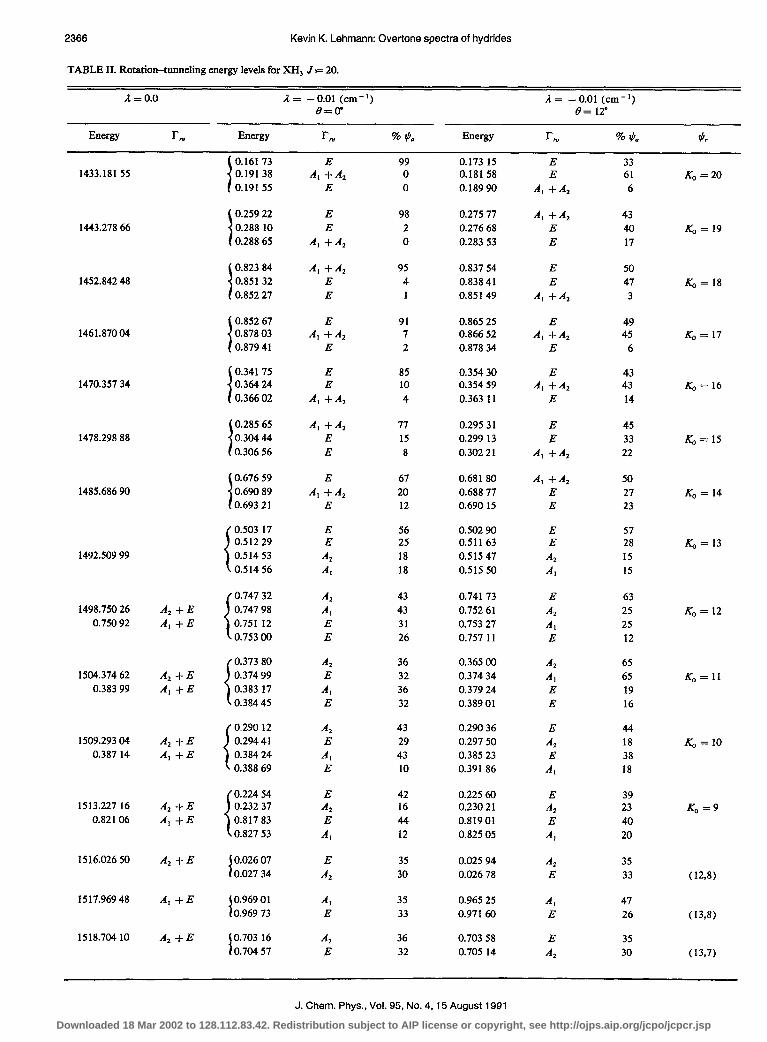
Like before, in the limit of small il, the tunneling between rovibrational levels should be suppressed by a rota- tional overlap factor. Table II contains the J = 20 rota- tional-tunneling energy levels of a XH, molecule calculated with A = 3.72, B = 3.61, C= 3.40, il = - 0.01 cm-‘, and 8 = 0 and 12”. These rotational constants, and the latter an- gle, are approximately those expected for ASH, at n = 3 based upon the analysis of the fundamentals by Olson et aLI9 When 8 = 0, the different local-mode states have their A axes differing by 60” but have the same C axis. As for the XH, case, the states with lowest energy (rotation around the C axis) have essentially an unperturbed tunneling splitting
I and the vibrational states are essentially unmixed. The de- generate E vibrational state is, however, split by a small amount. As the energy rises, there is a decreasing tunneling rate which drops to one-third the rotationless value when K, = 13. When the asymmetry splitting grows to be larger than the tunneling rate, the tunneling rate is strongly sup- pressed further, reaching a rate only 2% as large as the rota- tionless value for the state (2O,,,, ) localized on the separatix between A and C axis motion. When asymmetry splitting once again becomes negligible, at KP = 16, the tunneling rate is back to 25% of the nonrotating value. With increasing KP, the tunneling splitting again decreases, forming a pat- tern of A-E-E-A levels, with a splitting 1:2:1. For K, = J = 20, the total splitting of the pattern is only 6% of the bare vibrational value. The rate of decrease of the tunnel- ing rate for K, -J levels will be larger the more asymmetric the top. This is because, while the distance that the A axes must be rotated to overlap is constant at 60”, the more asym- metric the top, the more localized the highest rotational states will be near the A axis. Again, if the tunneling splitting is unresolved, the observed levels will have identical nuclear spin weights unlike the 2-l alternation expected for a C,, symmetric top. This is expected since the local-mode state has no rotational axis of symmetry.
When we look at the energy levels for 8 = 12”, the most striking change is that the lowest energy levels (K, w J) now have a suppressed tunneling splitting. This is because the rotation of the Caxis means the rotational wave functions of different local-mode states no longer overlap strongly. By the time K, decreases to a value of 13, the tunneling splitting is close to that of the 8 = 0 case. For such values of K,, the rotational functions are sufficiently spread out on the sphere that a 12” rotation has little effect on the overlap. For the state 201,,, , which is localized on the separatix, the C axis rotation increases the overlap, and the tunneling splitting is a factor of 10 larger than was calculated with no rotation of the Caxis (0 = 0). The state with K,, = J has a further reduced splitting. This can be understood since the A axes of the dif- ferent local-mode states are now 64” instead of 60” apart. It is the nature of tunneling that such a small change in rotation angle leads to a factor of 4 reduction in the tunneling rate.
J. Chem. Phys., Vol. 95, No. 4,15 August 1991 Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
2366 Kevin K. Lehmann: Overtone spectra of hydrides
TABLE II. Rotation-tunneling energy levels for XH, J = 20.
R = 0.0 A = -0.01 (cm-‘) A = - 0.01 (cm-‘) 8=(r l9= 12’
Energy rm Energy I-N % *iz Energy l-N % A 95
1433.18155
1443.278 66
1452.842 48
1461.870 04
1470.357 34
1478.298 88
1485.686 90
1492.509 99
1498.750 26 A, +E 0.750 92 A, +E
1504.374 62 A, +E 0.383 99 A, +E
1509.293 04 A, +E 0.387 14 A, -I-E
1513.227 16 A, i-E 0.821 06 A, +E
1516.026 50 A, i-E
1517.969 48 A, -I-E
1518.704 10 A, A-E
0.161 73 E 99 0.173 15 E 33 0.191 38 4 +A, 0 0.181 58 E 61 0.191 55 E 0 0.189 90 A, +A, 6
1
0.259 22 0.288 10 0.288 65
E 98 0.275 77 A, +A, 43 E 2 0.276 68 E 40
A, +A, 0 0.283 53 E 17
1
0.823 84 0.851 32 0.852 27
A, +A, 95 0.837 54 E E 4 0.838 41 E E 1 0.851 49 A, +A,
50 47 3
I
0.852 67 0.878 03 0.879 41
E 91 0.865 25 E 49 A, +A, 7 0.866 52 A, +A, 45
E 2 0.878 34 E 6
i
0.341 75 0.364 24 0.366 02
E 85 0.354 30 E 43 E 10 0.354 59 -4 i-4 43
A, +A, 4 0.363 11 E 14
I
0.285 65 0.30444 0.306 56
A, +A, 77 0.295 31 E E 15 0.299 13 E E 8 0.302 21 A, -i-A,
45 33 22
I
0.676 59 0.690 89 0.693 21
E 67 0.681 80 A, +A, 50 A, +A, 20 0.688 77 E 27
E 12 0.690 15 E 23
i
0.503 17 0.512 29 0.514 53 0.514 56
56 0.502 90 25 0.511 63 18 0.515 47 18 0.515 50
57 28 15 15
t
0.747 32 0.747 98 0.751 12 0.753 00
43 0.741 73 43 0.752 61 31 0.753 27 26 0.757 11
63 25 25 12
1
0.373 80 0.374 99 0.383 17 0.384 45
1
0.290 12 0.294 41 0.384 24 0.388 69
i
0.224 54 0.232 37 0.817 83 0.827 53
E E
4 A,
4 A, E E
4 E
A, E
A* E
A, E
E A2 E
A,
E A2
A, E
‘6 E
36 0.365 CO 32 0.374 34 36 0.379 24 32 0.389 01
65 65 19 16
43 0.290 36 29 0.297 50 43 0.385 23 10 0.391 86
44 18 38 18
42 0.225 60 16 0.230 21 44 0.819 01 12 0.825 05
39 23 40 20
1 0.026 07 0.027 34
35 0.025 94 30 0.026 78
35 33
1 0.969 01 0.969 73
35 0.965 25 33 0.971 60
0.703 16 0.704 57
36 0.703 58 32 0.705 14
E E
4 A,
E A2 A, E
4 A, E E
E 4 E
A,
E 4 E
A,
4 E
A, E
E 4
47 26
35 30
K. = 20
K. = 19
& = 18
K,, = 17
K. = 16
K, = 15
K. = 14
K. = 13
K, = 12
K. = 11
K. = 10
K. =9
(123)
(13,a)
(13,7)
J. Chem. Phys., vol. 95, No. 4.15 August 1991
Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
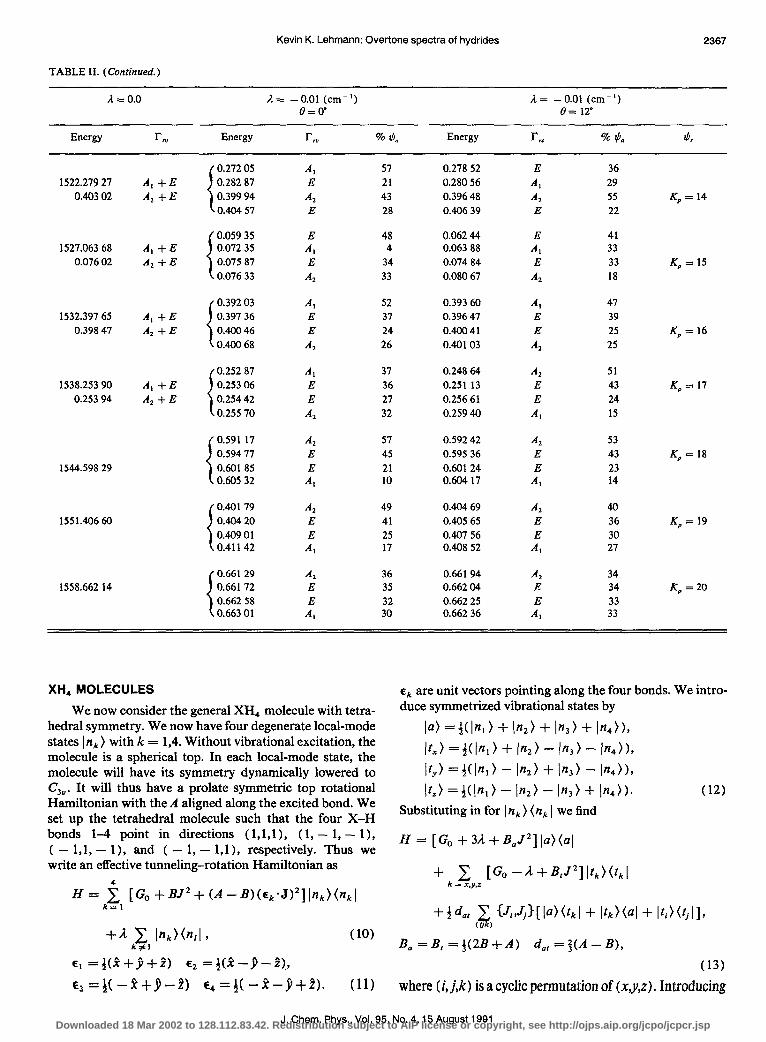
TABLE II. (Continued.)
Kevin K. Lehmann: Overtone spectra of hydrides 2367
A = 0.0 A = - 0.01 (cm-‘) A = - 0.01 (cm-‘) O=o” e= 120
Energy rN Energy r, % *o Energy r, % *a *,
0.272 05 A, 57 0.278 52 E 36 0.282 a7 E 21 0.280 56 A, 29 0.399 94 4 43 0.396 48 A2 55 Kp = 14 0.404 57 E 28 0.406 39 E 22
1522.279 21 A, +E 0.403 02 A, i-E
1527.063 68 A, +E 0.076 02 A, +E
1532.397 65 A, +E 0.398 47 A, +E
1538.253 90 A, -I-E 0.253 94 A, +E
0.059 35 0.072 35 0.075 a7 0.076 33
E Al E
A2
48 0.062 44 E 41 4 0.063 88 A, 33
34 0.074 a4 E 33 33 0.080 67 A2 la
K, = 15
0.392 03 A, 0.397 36 E 0.400 46 E 0.400 68 4
52 0.393 60 A, 37 0.396 47 E 24 0.400 41 E 26 0.40103 A*
47 39 25 K, = 16 25
0.252 87 0.253 06 0.254 42 0.255 70
37 0.248 64 4 51 36 0.251 13 E 43 K, = 17 27 0.256 61 E 24 32 0.259 40 A, 15
Al E E
4
f 0.594 0.605 0.591 0.601 77 32 a5 17 4 57 0.592 42 4 53 E 45 0.595 36 E 43 E 21 0.601 24 E 23
A, 10 0.604 17 A, 14
K,=la 1544.598 29
1551.406 60 0.401 79 4 0.404 20 E 0.409 01 E 0.411 42 A,
49 0.404 69 ‘42 40 41 0.405 65 E 36 K, = 19 25 0.407 56 E 30 17 0.408 52 A, 27
0.661 29 0.661 72 0.662 58 0.663 01
4 E E
A,
36 0.661 94 4 34 35 0.662 04 E 34 K, = 20 32 0.662 25 E 33 30 0.662 36 A, 33
1558.662 14
XH, MOLECULES We now consider the general XH, molecule with tetra-
hedral symmetry. We now have four degenerate local-mode states In,) with k = 1,4. Without vibrational excitation, the molecule is a spherical top. In each local-mode state, the molecule will have its symmetry dynamically lowered to C,,. It will thus have a prolate symmetric top rotational Hamiltonian with the A aligned along the excited bond, We set up the tetrahedral molecule such that the four X-H bonds l-4 point in directions (l,l,l), (1, - 1, - l), ( - l,l, - l), and ( - 1, - 1,l 1, respectively. Thus we write an effective tunneling-rotation Hamiltonian as
ek are unit vectors pointing along the four bonds. We intro- duce symmetrized vibrational states by
Ia) =i(ln,> + In*) + In,> + In,)),
I&) =:(b,) + In*) - 14) - ln4)),
IQ =I(br> - 1%) + I%> - In‘s)), 14) =:(I%) - In*> - I%> + In,)). (12)
Substituting in for Ink) (nk I we find
f~ = [Go + 3A + B,J’] la>(al
H = i [Go +BJ*+ (A-B)(Bk.J)*]Ink)(nkI k=l
(10) +idcZt (& {Ji9Ji)[la>(rkl + Itk)(al + Iti>(tjl],
B, =B, =f(2B+A) da, ={(A--),
(13)
where U,j,k) is a cyclic permutation of (x,y,z). Introducing
J. Chem. Phys., Vol. 95, No. 4,15 August 1991 Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

2366 Kevin K. Lehmann: Overtone spectra of hydrides
TABLE III. Rotation-tunelliug energy levels in cm - ’ XH, J = 20.
4 cl E( rot) % *” AEX 10’ K, r”r E(rot) % 4” AE x 10’
K=O
K=l
K=2
K=3
K=4
K=5
K=6
K=7
K=8
K=9
K= 10
/ A, F,
1 4 4 E
1
E F2 4
4 Fz
t AZ A,
I
E 4 4
1
4 F2 E
F2 A2
f 8 A,
I
E F2 4
1
4 F2 E
i
4 A, 4 A2
i
E 4 F2
839.995 74 36 40.00123 22
40.095 72 36 0.101 29 22 0.104 38 14
40.394 99 37 0.399 18 27 0.404 15 15
40.897 20 32 0.899 11 27 0.902 51 18 0.908 52 4
41.594 86 38 0.600 50 24 0.602 91 17
42.499 03 28 0.499 16 27 0.592 71 la
43.598 66 28 0.599 72 26 OACO 12 25 0.603 98 16
44.894 15 40 0.899 04 27 0.904 a7 13
46.394 85 38 0.402 54 19 0.493 a9 15
48.096 78 33 0.099 3 1 27 0.100 24 24 0.109 67 1
49.994 93 38 50.000 46 24
0.002 a9 18
549
557 309
419 496
191 339 601
564 241
13 355
lo6 40
386
489 583
769 135
253 93
843
553 243
E K= 11 6
F,
K=12
K= 13
K= 14
K=15
K=16
K= 17
K= la
K= 19
K=20
1 4 E
4
1 4 E 4
4 A2
I 4 E Fl
i
4 E 4
A,
i
4 4 4
I
F2 E F*
1
F2 E F1
52.092 02 45 0.100 42 24 0.104 93 13
54.388 80 53 0.399 43 26 0.401 al 20 0.403 75 16
56.898 38 28 0.898 68 28 0.902 43 19
59.596 30 34 0.599 66 26 0.603 97 15
62.494 89 38 0.498 33 29 0.501 71 21 0.505 02 12
65.594 10 40 0.599 99 25 0.605 94 10
68.896 86 33 0.900 03 25 0.903 la 17
72.398 76 28 0.899 60 26 0.400 43 24 0.40127 22
76.099 90 25 0.10001 25 0.100 12 25
ao.ooo oo 25 ao.ooo 01 25 ao.oco 01 25
841 451
63 238 194
30 375
336 431
344 340 331
589 595
317 316
a4 a3 a4
11 11
1 0
the tensor operators of Robiette, Gray, and Birss,” one finds the same relationship for their coefficients as found by Ha- lonen and Robiette. This relationship has also been found to be approximately correct for the XH, overtone spectra that show symmetric top rotational structure.4*6*8
When A ( 2K(A - B) , the energy levels will cluster into a groups of eight quasidegenerate states made up of basis functions Ink,& f K >. These basis functions will be con- nected by tunneling integrals of two different magnitudes A, =Ad;,,(lW) and A, = Ad ;, _ K ( 1090) = Ad & (71”). For K = 3N, the phase factors of the inte-
grals will be equal, and these eight functions will produce a cluster of four energy levels with shifts 3 (A, f A2 > (A, and A,)and - (A, f/2,) (Fr andF,).ForK=3Nf 1,weget three states F, , F2, and E. The order and splitting of these levels depends upon the size of A, and A,. If we look at the J= K levels, then the tunneling rate will fall off rapidly, since d&(109”) = (1/3jJ and d&(71”) = (2/3)J. This will produce the largest rotational quenching of the local- mode tunneling, since the angular momentum vector needs to move the furthest to reach a configuration of equal rota- tional energy.
J. Chem. Phys., Vol. 95, No. 4,15 August 1991
Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

Kevin K. Lehmann: Overtone spectra of hydrides 2369
The rotation-inversion level structure will depend (ex- cept for J dependent scaling and shift) only on the ratio of (A - B) and R. In Table III is given the rotation-inversion energy levels calculated for a XH, top with A = 2.1, B = 2.0, andil = - 0.01 cm - ‘. These constants where cho- sen to emphasize the local-mode limit. For these parameters, the tunneling splitting of the nonrotating molecule is 0.04 cm-’ . Unlike the previous examples, all of the J = 20 levels have a substantial reduction in the tunneling rate, the largest splitting being only about 20% of the rotationless value. It is also shown that the K = J levels have the smallest splitting,
predicted above. But when ?i(J + l/2) = cos( 35”) = 0.82 (which implies K = 16.7 for J = 20), the classical trajectories for K with one bond excited and - K with another bond excited overlap at a tangent. The calculations reveal that the K = 16 is a local maximum in the tunneling splitting. If one looks at K/(J + l/2) = cos(54”) = 0.58(K= 11.8forJ= 20),the classical trajectories for K with one bond excited and K with another bond excited now overlap at a tangent. Again, the splitting of the level K = 11 is a local maximum in the calcu- lated splitting. The slow decrease in the splitting as one goes to K = 0 can be understood by the fact that while the classi- cal trajectories continue to cross, they cross at a steeper an- gle, and the width of the quantum states gets narrower, thus leading to reduced overlap.
It is interesting that in the published figure of the stan- nane overtone,6 the R (6) transition clearly shows a reduc- tion of the tunneling splitting as K increases. This spectrum is only partially in the local-mode limit in that R and A - B are comparable. Since there will be Stark transitions across the tunneling levels (with matrix elements that scale with the size of the vibrationally induced dipole of the local-mode
I
state), some type of double resonance experiment would be ideal for determining the splitting pattern.
EXTENSIONS
This initial work has presented a general procedure for producing rotational-tunneling Hamiltonians for a set of nearly degenerate local mode states. One can clearly extend the work to include quartic and sextic centrifugal distortions terms in the local-mode Hamiltonians. For a quantitative treatment of hydrides, both terms are usually required. The most straightforward way to do a calculation is to use a basis set of unsymmetrized local mode states and use rotational wave functions for each vibrational state aligned with it’s own inertial ellipsoid. Then vibrationally diagonal couplings can be treated by a standard program. The tunneling matrix elements will be izD JK,,K, (#,19,x), where #,19,x are the Euler angles to convert one local-mode principle axis system into another Alternatively, one can use a common set of rota- tional functions, aligned with the symmetry axes, for all the local-mode vibrational functions, and use the rotation matri- ces to transform each of the rotational basis function. This is likely the preferred method, since it allows a convenient cal- culation of transition intensities from the ground state, though the basis functions are not of definite symmetry, and symmetry assignments need to be made by examination of the eigenvectors coefficients of symmetry related basis func- tions.
In order to work in a symmetrized local-mode basis, one needs to convert higher powers of the rotational operators, as give above for the quadratic terms. For the tetrahedral molecule, the terms produced by including DJJ, DJK, and DKK in the local mode, symmetric top, rotational Hamilto- nian are given below:
HD = - D,,J4+DJKJ2(~k.J)2+D~~(~k.J)4](nk)(nk(,
HD = - D,J” -+dJ: + J; + J:)][ lh.4 + 7 \td(b(, -3 D.xJ* 2 CJi,JiI[ la)(f/cl + Ifk)(al + Iti)(rjl] (iik)
1 DKK -- 9 v
SCJf,CJi,JkII + 2CJj,JkI +2CJj,J:I -7CJi,JkI] [ IQ>(fiI + Itt)(al A- Ifj)(fkl]9 (ij )
DLl = D.,., -I- :D.,K -I- Pm. (14)
Only some of these terms appear in the Hamiltonian used by Halonen et al. to fit the spectrum of the first overtone of stannane.6 Such selective inclusion of the terms will destroy the high J,K local-mode energy patterns of the spherical top. In fact, the rovibrational levels predicted by the final fitted constants of Halonen etal. deviate from the expected pattern of falling inversion splitting for the high J = K lines when J become larger than those used in their fit.*l I believe this is due to selective inclusion of terms. Including all of the terms, but imposing the above constraints on the spectroscopic con- stants, has a firmer theoretical basis and may well prove a practical advantage as well. Clearly, the distortion Hamilto-
I
nian is much easier to write (andprogram) in the unsymme- trized basis. As one goes to the sextic distortion terms and beyond, which are often needed for a quantitative fit to the rotational structure of a hyride, the advantage of the unsym- metrized basis only increases.
In fitting the stannane overtone spectrum, Halonen et al. determined a diagonal (in the symmetrized basis) Corio- lis term, but 2Bc3 was only - 0.0042 cm - ‘. Such terms are easily included in the symmetrized local-mode Hamiltonian since they take the same form as the interaction between the fundamentals. In the unsymmetrized basis, one will have a vibrational angular momentum between each pair on local-
J. Chem. Phys., Vol. 95, No. 4,15 August 1991 Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

2370 Kevin K. Lehmann: Overtone spectra of hydrides
mode vibrations, pointing in the direction of the cross prod- uct of the two bonds. One can use Morse oscillator matrix elements to estimate the size of the expected Coriolis term. Halonen ef aL6 also found that a Hz3 higher order Coriolis- type interaction between the A and F, symmetrized local- mode states could also be determined from the spectrum. This term does not show up in the present treatment. It could be generated by adding rotational corrections to the Coriolis coupling between the bond modes, but it is desirable to have a physical model for this interaction so that its magnitude can be estimated from a model force field.
CONCLUSIONS The present paper extends the earlier work of Halonen
and Robiette to provide a systematic treatment of the inter- action of rotation and local-mode tunneling. Effective Ham- iltonians are developed, both in an unsymmetrized and a symmetrized basis. The unsymmetrized basis provides a nat- ural framework for elucidating how rotational motion can reduce the local-mode tunneling rate. At present, it is only applicable when the interaction of the local-mode polyad with other vibrational states can be treated perturbatively, i.e., by an effective rotational Hamiltonian. Cases for Fermi resonance, for example, will require extension but can likely be handled in the same framework. Also, the treatment can be generalized to cases of the local-mode combination bands, with excitation in more than one bond mode, or with a bend- ing mode excited. Note added inproo$ Independently, M. S. Child and Q. Zhu have shown that for an XH, molecule, the normal mode Hamiltonian [ Eq. ( 131 and the Halonen and Robietie con- straints can be transformed to the symmetric top, local mode Hamiltonian [Eq. (lo] thus confirming one aspect of the present work. [ Chem. Phys. Lett. (to be published) I.
ACKNOWLEDGMENTS
I want to thank the Department of Physical Chemistry, University of Helsinki, for their hospitality during the initial stage of this project; Lauri and Marjo Halonen and Quingshi Zhu for helpful discussions on rotational structure in the local-mode limit. This work was supported by the National Science Foundation and the Donors of the Petroleum Re- search Fund, administered by the American Chemical So- ciety.
’ M. S. Child and L. Halonen, Adv. Chem. Phys. 57, 1 ( 1984). 2 L. Halonen and A. G. Robiette, J. Chem. Phys. 84,866l ( 1984). ’ F. W. Birss, Mol. Phys. 31,49 1 ( 1976). 4Q. Zhu, B. A. Thrush, and A. G. Robiette, Chem. Phys. Lett. 150, 181
(1988). “Q. Zhu, B. Zhang, Y. Ma, and H. Qian, Chem. Phys. Lett. 564, 596
(1989). bM. Halonen, L. Halonen, H. Burger, and S. Sommer, J. Chem. Phys. 93,
1607 (1990). ‘F. Michelot, J. Moret-Bailly, and A. DeMarino, Chem. Phys. Lett. 148,
52 (1988). *M. Chevalier, A. DeMartino, and F. Michelot, J. Mol. Spectrosc. 131,382
(1988). 9 I. M. Mills and A. G. Robiette, Mol. Phys. 56, 743 ( 1985). ‘OK. K. Lehman, J. Chem. Phys. 79, 1098 (1983). “M S. Child and R. T. Lawton, Faraday Discuss. Chem. Sot. 71, 273
(lb81). “K. K. Lehmann, Mol. Phys. 66,1129 (1989). “R. G. D. Della Valle, Mol. Phys. 63, 611 ( 1988). I4 W. G. Hatter and C. W. Patterson, J. Chem. Phys. 80,424l (1984). I5 See Eq. ( IO) of Ref. 2. 16L. Halonen, J. Chem. Phys. 86, 588 (1987). “J. R. Gillis and T. H. Edwards, J. Mol. Spectrosc. 85, 74 ( 1981). ‘*M. Ya, Ovchinnikova, Chem. Phys. 120,249 ( 1988). 19W. B. Olson, A. G. Maki, and R. L. Sams, J. Mol. Spectrosc. 55, 252
(1975). “A. G. Robiette, D. L. Gray, andF. W. Birss, Mol. Phys, 32,1591, ( 1976). ” M. Halonen and L. Halonen (private communication).
J. Chem. Phys., Vol. 95, No. 4,15 August 1991
Downloaded 18 Mar 2002 to 128.112.83.42. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp