the hemophilia report
TRANSCRIPT

Differentiating Hemophilia A and B n Genetics of Hemophilia
Prophylactic vs Episodic Therapy n Advances in Clotting Factors
Avoiding Inhibitors n Managing Acute Bleeding
Novel Therapeutics n What’s in the Pipeline?
Steven W. Pipe, MDGuest Editor
Selected Reports from the
65th Annual Meeting of the National Hemophilia Foundation and
55th Annual Meeting of the American Society of Hematology
REPORTHemophilia
CONTINUING EDUC ATION FOR PHYSICIANS AND NURSES: 3.5 CREDITS AVAILABLE
The
This activity is supported by an educational grant from Biogen Idec.
V O L U M E 1 N U M B E R 1 | S P R I N G 2 0 1 4

Guest Editor: Steven W. Pipe, MD
The opinions or views expressed in this publication are those of the authors and do not necessarily reflect the opinions or recommendations of Biogen Idec, the University of Cincinnati, or the publisher, Direct One Communications, Inc. Please consult the full prescribing information before using any medication mentioned in this publication.
This publication was made possible through an educational grant from Biogen Idec.
Copyright © 2014 by Direct One Communications, Inc. All rights reserved. Printed in the USA.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 1
V O L U M E 1 N U M B E R 1 | S P R I N G 2 0 1 4
Selected Reports from the 65th Annual Meeting of the National Hemophilia Foundation and
55th Annual Meeting of the American Society of Hematology
Steven W. Pipe, MD, Guest Editor
HemophiliaThe
REPORT
4 IntroductionSteven W. Pipe, MD, University of Michigan, Ann Arbor, Michigan
6 MeetingtheChallengesofManagingHemophilia: ProphylacticvsEpisodicTherapyandAvoidingInhibitorsDuc Q. Tran, MD, Winship Cancer Institute, Emory University, Atlanta, Georgia
11 AdvancesinClottingFactors: FromBenchtoBedsideAnthony Sung, MD, Duke University Medical Center, Durham, North Carolina
18 HemophiliaAandB: DiseaseDifferencesandtheUseofProphylacticTherapyAnna Chalmers, MD, Rush University Medical Center, Chicago, Illinois
22 GeneticsofHemophilia: TheRoleofGeneticTestingandtheUseofGeneticstoGuideTreatmentAnna Chalmers, MD, Rush University Medical Center, Chicago, Illinois
25 ExpandingTherapeuticOptionsforHemophiliaAandB: ResultsofRecentTrialsandResearchHolleh D. Husseinzadeh, MD, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania
31 TreatmentofHemophilia: What’sinthePipeline?Kerry Hege, MD, Indiana University School of Medicine, Indianapolis, Indiana
37 RecentAdvancesinPreventingBleeding,ReducingInhibitors,andManagingAcuteBleedingNoa Biran, MD, Tisch Cancer Institute, Mount Sinai School of Medicine, New York, New York
42 CME/CNEPostTestandEvaluation

2 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
RATIONALE AND PURPOSEHemophilia, an X-linked recessive bleed-ing disorder, affects more than 400,000 people worldwide. Internal bleeding related to the disorder spans the gamut from spontaneous hemarthroses re-sulting in chronic, disabling joint pain and swelling to deep-muscle, central nervous system, and gastrointestinal bleeding. This inaugural issue of The Hemophilia Report explores the genetic foundation of hemophilia, differentiates between its two major types (A and B), and reviews the current management of this hematologic disease, as well as recent advances in preventing bleed-ing and reducing the development of autoimmune inhibitors to coagulation replacement factor therapy.
Among the many topics discussed by the authors are advances in synthetic blood-coagulation factors that prom-ise to improve patient outcomes with more effective bleeding control and preservation of joint function; reducing the burden of prophylactic replace-ment therapy by extending the half-life of bioengineered coagulation factors; personalizing treatment based on the pharmacokinetics of replacement fac-tors, bleeding phenotypes, and lifestyle; identifying, monitoring, and preventing age-related comorbidities; and poten-tially developing a cure for hemophilia through gene therapy. The articles in this issue are based upon presentations de-livered during the 65th Annual Meeting of the National Hemophilia Foundation, held in Anaheim, California, October 3–5, 2013, and the 55th Annual Meeting of the American Society of Hematology, convened in New Orleans, Louisiana, December 7–10, 2013.
The articles in this issue, written from the academic perspective of physicians-in-training at leading medical institutions, summarize the import of these new find-
About This CME/CNE Activityings and place them into clinical context. This activity has been developed and approved by a planning committee of nationally recognized thought leaders to meet a perceived educational need to provide hematologists, other physi-cians, and nurses with diagnostic and therapeutic strategies to help them perform their clinical roles.
LEARNING OBJECTIVESAfter studying this issue of The Hemo-philia Report, participants in this educa-tional activity should be able to:
• Outline the genetic basis of hemo-philia A and B and differentiate between the two types.
• Review the history of hemophilia management, discuss current best prac-tices in children and adults, and explain the goals of ongoing research to better understand the disease.
• Summarize the developments being made in the laboratory and currently being tested in clinical trials to produce synthetic coagulation factors with longer half-lives than those of present blood products for treating hemophilia or that are less prone to stimulate the production of neutralizing antibodies (inhibitors).
• Discuss the advantages and draw-backs of prophylactic versus on-de-mand therapy, the controversies over therapeutic timing and individualizing therapy for different patients, the factors affecting patient compliance, and the promise of bioengineering and phar-macokinetic strategies to individualize patient management.
TARGET AUDIENCEHematologists, other physicians, and nurses significantly involved in the management of hemophilia should find participating in this educational activity valuable.
ACCREDITATION AND CREDIT DESIGNATIONPhysic ians: Th is activity has been planned and imple-
mented in accordance with the ac-creditation requirements and policies of the Accreditation Council for Con-tinuing Medical Education (ACCME) through the joint providership of the University of Cincinnati and Direct One Communications, Inc. The University of Cincinnati is accredited by the ACCME to provide continuing medical education for physicians.
The University of Cincinnati designates this Enduring Material Activity for a maximum of 3.5 AMA PRA Category 1 Credits ™. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Nurses: A total of 3.5 continuing education contact hours for nurses are approved by the Ohio Board of Nursing through the OBN Approver Unit at the University of Cincinnati College of Nurs-ing, Continuing Education Program (OBN-011-93). Contact hours are valid in most states. Program #140505-1.
CREDIT AVAILABILITYActivity release date: April 30, 2014Expiration date: May 1, 2015 (for CME credit); May 1, 2016 (for CNE credit)
METHOD OF PARTICIPATIONThis Enduring Material Activity is avail-able in print and online at www.Hemo-philiaReport.com and consists of an introduction, seven articles, a postactivity assessment, and an evaluation. Estimated time to complete the activity is 3.5 hours.
To receive credit, participants must read the CME information on these two pages, including the learning objectives and disclosure statements, as well as the full content of this monograph, and then complete the post test and evaluation

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 3
form online at www.HemophiliaReport.com. Upon successful completion of the post test (80% correct) and evaluation form, a CME certificate of participation will be awarded automatically. The cer-tificate may be printed directly from the Web site or e-mailed and printed later.
There are no fees for participating in or receiving credit for this activity.
CME REVIEWERRick Ricer, MD Adjunct Professor of Family Medicine University of Cincinnati Cincinnati, Ohio
CME ACCREDITATIONSusan P. Tyler, MEd, CMP, CCMEP Director, Continuing Medical Education University of Cincinnati Cincinnati, Ohio
FACULTY DISCLOSURESAll faculty members (or anyone else in a position to control content, such as ac-tivity planners) are required to complete a Disclosure of Commercial Interest and Resolution form and to cooperate with identified methods for resolving conflict of interest prior to participating in the activity. The University of Cincinnati requires disclosure to the learners of all relevant financial relationships and adheres strictly to the ACCME Standards for Commercial Support.
Steven W. Pipe, MD, is Professor of Pedi-atrics and Pathology, Division of Pediat-ric Hematology/Oncology, University of Michigan, Ann Arbor, Michigan. He is a consultant to Baxter BioScience, Biogen Idec, CSL Behring, Novo Nordisk, and Pfizer and has received research/grant support from Pfizer.
Duc Q. Tran, MD, a Fellow in Hematol-ogy at Winship Cancer Institute, Emory University, Atlanta, Georgia, has nothing to disclose.
Anthony Sung, MD, a Fellow in Hema-tology/Oncology at Duke University Medical Center, Durham, North Carolina, has nothing to disclose.
Anna Chalmers, MD, a Fellow in He-matology/Oncology at Rush University Medical Center, Chicago, Illinois, has nothing to disclose.
Holleh D. Husseinzadeh, MD, a Fellow in Hematology and Medical Oncology at the University of Pennsylvania, Phila-delphia, Pennsylvania, has nothing to disclose.
Kerry Hege, MD, a Fellow in Pediatric Hematology/Oncology at Riley Hospital for Children, Indiana University Health, In-diana University School of Medicine, India-napolis, Indiana, has nothing to disclose.
Noa Biran, MD, a Fellow in Hematol-ogy/Medical Oncology at Tisch Cancer Institute, Mount Sinai School of Medi-cine, New York, New York, has nothing to disclose.
Rick Ricer, MD, has nothing to disclose.
Susan P. Tyler, MEd, CMP, CCMEP, has nothing to disclose.
Jacqueline Keenan and Edwin Geffner of Direct One Communications, Inc., have nothing to disclose.
DISCLAIMERThis activity is an independent educa-tional activity under the direction of the University of Cincinnati. The activ-ity was planned and implemented in accordance with the accreditation re-quirements and policies of the ACCME, the Ethical Opinions/Guidelines of the American Medical Association, the US Food and Drug Administration (FDA), the Office of Inspector General of the US Department of Health and Human Services, and the Pharmaceutical Re-search and Manufacturers of America Code on Interactions With Healthcare Professionals, thus assuring the highest degree of independence, fair balance, scientific rigor, and objectivity.
However, the planning committee, faculty, University of Cincinnati, Biogen Idec, and Direct One Communications, Inc. shall in no way be liable for the cur-rency of information or for any errors,
omissions, or inaccuracies in this activity. The opinions and recommendations presented herein are those of the faculty and do not necessarily reflect the views of the provider, producer, or grantor. Participants in this activity are encour-aged to refer to primary references or full prescribing information resources.
DISCLOSURE OF UNAPPROVED/OFF-LABEL USEDiscussions concerning drugs, dosages, devices, and procedures may reflect the clinical experience of the planning committee or faculty, may be derived from the professional literature or other sources, or may suggest uses that are in-vestigational and not approved labeling or indications.
In this issue of The Hemophilia Report, Dr. Hege discusses the rationale for develop-ment of new, longer-acting coagulation replacement factors and what’s in the pipeline. Dr. Biran describes a recombi-nant factor VIII crystallizable fragment (Fc) fusion protein currently under FDA review and the off-label use of a four-factor prothrombin complex concentrate (PCC) in patients requiring anticoagulant reversal before undergoing urgent surgi-cal procedures. Dr. Chalmers refers to the potential of PEGylated and fusion protein replacement factors and gene therapy. Drs. Husseinzadeh and Sung discuss recent clinical trials of these and other investigational replacement factors, as well as novel bypassing agents, PCCs, and gene-transfer therapy. Dr. Tran also touches on the potential of gene therapy.
CONTACT INFORMATIONWe would like to hear your comments regarding this or other educational activities produced by Direct One Com-munications, Inc. In addition, sugges-tions for future activities are welcome. Contact us at:
Direct One Communications, Inc. 1424 Ridge Road Syosset, NY 11791 Phone: 516-364-1020 Fax: 516-364-4217 Website: www.CMEdirect.net
AboutThisCME/CNEActivity

4 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
Exploring hemophilia and its ramifications involves a myriad of medical disciplines—hema-tology, immunology, genetics,
infectious disease, radiology, nursing, and pharmacology, to name a few. Dur-ing two recent professional gatherings of hematologists and other health profes-sionals, an international cadre of experts on hemophilia reviewed our current understanding of this difficult disease and shared insights on its management and impact on society.
Some 3,000 healthcare professionals attended or participated in over 40 educa-tional sessions offered during the 65th An-nual Meeting of the National Hemophilia Foundation, held in Anaheim, California, October 3–5, 2013, to better understand this disease and associated issues. During the 55th Annual Meeting of the American Society of Hematology, which took place in New Orleans, Louisiana, December 7–10, 2013, over 18,000 hematologists and other healthcare professionals took part in scientific sessions and presentations, many of which involved the etiology, diag-nosis, and treatment of bleeding disorders. This premiere edition of The Hemophilia
Report delves into subjects pertinent to the practices of hematologists and other healthcare professionals involved in man-aging hemophilia.
n HEMOPHILIA THERAPY
Duc Q. Tran, MD, from Emory Uni-versity in Atlanta, Georgia, provides a historic perspective on the treatment of hemophilia. Among the subjects he cov-ers are the successes and challenges of prophylactic replacement factor therapy in children; in particular, the results of the Joint Outcome Study and the Evalua-tion Study on Prophylaxis: a Randomized Italian Trial (ESPRIT) are compared and contrasted with data collected during other clinical trials of prophylactic regi-mens. Dr. Tran also reviews the major studies on prophylaxis versus episodic, on-demand therapy in adults and the current options for managing the de-velopment of alloimmune inhibitory antibodies to factor VIII (FVIII).
Anthony Sung, MD, from Duke Uni-versity Medical Center in Durham, North Carolina, summarizes the history of hemophilia therapy and a number of discoveries that are advancing treatment of the disease. He writes about strategies toward longer-acting clotting factors through the use of PEGylation, the at-tachment of polyethylene glycol (PEG) molecules to recombinant factor proteins, and fusion protein technologies, among others, that promise to improve the effi-cacy of treatment and reduce the burden of infusions. The latest safety and efficacy data from clinical trials with these novel
agents are provided. Dr. Sung then reviews challenges to reversal and inhibition of bleeding presented by the use of older and newer oral anticoagulants. The efficacy of prothrombin complex concentrates and recombinant activated factor VII for trau-matic or surgical bleeding and reversal of bleeding complications resulting from oral anticoagulation are detailed.
n CHALLENGING DECISIONS
Anne Chalmers, MD, from Rush University Medical Center in Chicago, Illinois, compares hemophilia A with hemophilia B and confronts challenging treatment decisions. When treatment should be started, what the optimal dose of certain therapeutic agents should be, and how long prophylactic therapy should continue remain controversial. A historic view of hemophilia management has led to greater regard for prophylaxis rather than symptomatic treatment, even though optimal prophylactic regimens and sched-ules continue to be explored.
In a separate article, Dr. Chalmers reviews our current understanding of the genetics of hemophilia A and B. She addresses the spectrum of genetic muta-tions that lead to development of each disease, strategies for genetic testing, and how data from such tests could be used to guide treatment.
n NOVEL THERAPEUTICS
Holleh D. Husseinzadeh, MD, from the Hospital of the University of Pennsylvania in Philadelphia, covers recent advances in novel therapeutics for bleeding disorders,
Introduction Selected Reports from the 65th Annual Meeting of the National Hemophilia Foundation and 55th Annual Meeting of the American Society of HematologySteven W. Pipe, MD, Guest Editor
University of Michigan, Ann Arbor, Michigan
Dr. Pipe is Professor of Pediatrics and Pathology, Division of Pediatric Hematology/Oncology, University of Michigan, Ann Arbor, Michigan.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 5
StevenW.Pipe,MD Introduction
including gene-transfer options. Also discussed are new insights into the appli-cation of prophylaxis for hemophilia com-plicated by inhibitors; issues that affect compliance with therapy; and outcomes of total joint replacement in patients with congenital bleeding disorders.
n MEETING THE CHALLENGES AHEAD
Kerry Hege, MD, from the Indiana University School of Medicine in In-dianapolis, takes a closer look at the improved outcomes and remaining chal-lenges with prophylactic therapies for hemophilia and the results of clinical trials with current state-of-the-art agents. Dr. Hege describes efforts to increase patient adherence to prophylactic regimens by
prolonging the half-life of recombinant factor products (and thereby reducing the need for frequent dosing), prevent bleed-ing episodes, and improve patient quality of life. Among the exciting subjects she covers are PEGylation, polysialylation, fu-sion protein technology, and gene therapy.
In recent years, many advances in preventing bleeding, reducing the risk of inhibitor formation, and managing acute bleeds in hemophilia have been reported. Noa Biran, MD, from the Mount Sinai School of Medicine in New York, reports on studies investigating the use of recom-binant FVIII and factor IX crystallizable fragment fusion protein in patients with severe hemophilia A or B, respectively. Dr. Biran also reviews the results of studies comparing early prophylactic measures
with episodic therapy, noting that prophy-laxis has resulted in superior results. He also reports on the usefulness of magnetic resonance imaging to monitor hemophili-ac joint disease, covers strategies to reduce the incidence of inhibitors, and touches upon methods to limit acute bleeding.
By sharing the information imparted by hemophilia experts at these two impor-tant medical meetings, the authors of this report greatly enhance our understanding of many complicated issues involved in hemophilia prophylaxis and manage-ment. Future editions of The Hemophilia Report certainly will add to these insights and further assist busy practitioners and nurses in keeping abreast of novel devel-opments in the optimal management of hemophilia.

6 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
Hemophilia is an X-linked recessive bleeding disorder that results from deficiencies in coagulation factors VIII
(FVIII) and IX (FIX). Decreased or absent levels of these factors lead to hemophilia A and B, respectively. The severity of he-mophilia is classified as mild, moderate, or severe depending on factor activity levels. Mild disease is associated with a factor activity level > 5 IU/dL and < 40 IU/dL; moderate disease with a factor activity level of 1–5 IU/dL; and severe disease with a factor activity level of < 1 IU/dL.
The prevalence of hemophilia A is 1:5,000 male live births and of hemophilia B, 1:30,000 male live births.1 The most common morbidity noted among patients with severe disease is spontaneous joint bleeding (hemarthrosis), which can affect any joint but which primarily affects the ankles, knees, and elbows. In addition to spontaneous hemarthrosis, patients with severe disease also can develop sponta-neous soft-tissue, gastrointestinal, and central nervous system bleeding. Recur-
rent hemarthroses can lead to debilitating joint disease, also known as hemophilic arthropathy. Patients with moderate dis-ease usually bleed in response to some injury but can have spontaneous bleeding; patients with mild disease typically only bleed in response to major insults such as surgery or significant trauma.
n PROPHYLAXIS IN CHILDREN
The cornerstone of hemophilia man-agement is coagulation factor replace-ment.2 Over the past two decades, the focus of therapy has shifted from treat-ment after bleeding has occurred to prevention of bleeding to allow patients to have a more active and fuller lifestyle. Prophylactic factor replacement was first pioneered in Sweden in 1958, when patients with moderate hemophilia were found to be less likely to have chronic debilitating joint disease than patients with severe hemophilia.3
Two recent, randomized, controlled trials that compared prophylaxis with epi-sodic treatment showed definitive benefits
with early prophylaxis. Results of these studies—the Joint Outcome Study4 in the United States and the Evaluation Study on Prophylaxis: a Randomized Italian Trial (ESPRIT)5—are summarized in Table 1. Both evaluated the use of prophylactic factor replacement in young boys who had either no or few joint bleeds.
JointOutcomeStudyThe Joint Outcome Study enrolled 65
male children with a history of minimal or no joint bleeding (Table 1). The subjects, who were < 2.5 years old at study entry, were randomized to receive either prophy-lactic or episodic treatment. Children in the prophylactic treatment arm received 25 IU/kg of recombinant FVIII (rFVIII) every other day; those in the episodic treatment arm received 40 IU/kg of rFVIII during each bleed, followed by a single dose of 20 IU/kg 24 and 72 hours later.
As shown in Figure 1, the frequency of bleeding was significantly less in the prophylactic treatment arm than in the episodic treatment arm. As the children grew older and more active, those in the
Meeting the Challenges of Managing Hemophilia: Prophylactic vs Episodic Therapy and Avoiding InhibitorsDuc Q. Tran, MDWinship Cancer Institute, Emory University, Atlanta, Georgia
Abstract Management of hemophilia has changed over the past few decades. However, the most common complication of hemophilia remains the develop-ment of joint bleeds leading to hemophilic arthropathy. Coagulation factor replacement for children should be started early and given prophylactically to pre-serve joint health and prevent complications. For adults started on prophylaxis, limited studies have reported decreased bleeding episodes, but improvement of joint function and quality of life remains unclear. Inhibitor formation as a com-plication of coagulation factor replacement and its management are reviewed. Future potential therapies for patients with hemophilia are summarized briefly.
Dr. Tran is a Fellow in Hematology at Winship Cancer Institute, Emory University, Atlanta, Georgia.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 7
DucQ.Tran,MD Meeting the Challenges of Managing Hemophilia: Prophylactic vs Episodic Therapy and Avoiding Inhibitors
prophylactic treatment arm continued to have significantly less joint bleeding. Magnetic resonance imaging (MRI) of 56 patients at age 6 years showed that 93% of those in the prophylactic treatment arm did not have joint damage, compared with 55% of those in the episodic treatment arm (Table 2).4
ESPRITThe ESPRIT investigators evaluated 40
older male children (median age, 48–50 months) at study entry and who had no physical or radiographic arthropathy (Table 1). These children were random-ized to receive either prophylactic or episodic treatment. Boys in the prophy-lactic treatment arm received 25 IU/kg of rFVIII three times/week, with doses up to 40 IU/kg to obtain trough levels > 1%. Those in the episodic treatment arm received ≥ 25 IU/kg of rFVIII dur-ing each bleed, with repeated doses every 12–24 hours until the bleeding resolved. As shown in Table 3, children in the prophylactic treatment arm experienced fewer bleeding episodes and had less radiographic evidence of arthropathy than those in the episodic treatment arm.5 Quality-of-life (QOL) measures differed significantly between the two groups because the children in the epi-sodic treatment arm had a greater sense of overprotection.5
In both studies, significantly more re-placement factor was used in the prophy-lactic treatment arm than in the episodic treatment arm. In addition to greater cost due to more factor consumption, there was also a higher rate of complications related to the placement of indwelling catheters to maintain the prescribed pro-phylactic treatment regimen.
OtherStudiesinChildrenOther prophylactic approaches also
have been used. In the Canadian tailored prophylaxis trial conducted by Feldman et al,6 25 boys began primary prophylaxis with 50 IU/kg of rFVIII once weekly, fol-lowed by stepwise increases to 30 IU/kg twice weekly and then 25 IU/kg every other day, depending on the frequency of bleeding episodes. After 5 years, 10 of the
25 children were receiving weekly infu-sions, 8 were being treated twice weekly, and 7 were being treated every other day. At the end of the study, all of the children had normal joints by physical examina-tion and minimal radiographic changes. Only 10 of the 25 children (40%) required indwelling intravenous (IV) catheters, compared with 29 of the 32 children (91%) who were on the prophylactic treatment arm of the Joint Outcome Study.4,6 All of the children in the Canadian study had follow-up MRI examinations at age 9 years, and about 50% of them showed evidence of changes in target joints. This approach resulted in a cost reduction of about 20%–25% over the first 5 years,
as compared with the cost of a standard high-dose prophylactic regimen over that 5-year period.7
In the Netherlands, Fischer and col-leagues8 studied a different approach. Instead of using the standard high-dose prophylactic regimen, they started pro-phylaxis with lower, intermediate doses (usually 15–25 IU/kg 2–3 times/wk) when joint bleeding began. When compared with a Swedish (high-dose) cohort, the Dutch (intermediate-dose) cohort used less than half as many units of FVIII per patient per year.8 Outcomes in the Dutch cohort were good but somewhat inferior to those in the Swedish cohort, since some increase in bleeding and more clinical evi-
TABLE 1Comparison of the Joint Outcome Study and the ESPRIT Study
Parameter JointOutcomeStudy(JOS) ESPRITStudy
Age, years (mean) < 2.5 (1.6) 1–7 (4.1)Prior bleeding ≤ 2 bleeds in index joints ≤ 2 bleeds in a single site (prophylactic therapy arm: 1; (unknown) episodic treatment arm, 0.6)Baseline arthropathy None by physical or x-ray None by physical or x-ray examination examinationTreatment regimen Prophylactic therapy arm: Prophylactic therapy arm: (recombinant factor VIII) 25 IU/kg every other day 25 IU/kg 3 times/wk, up to 40 IU/kg per dose Episodic treatment arm: Episodic treatment arm: 40 IU/kg as needed, then ≥ 25 IU/kg as needed, then 20 IU/kg at 24 and 72 hours every 12–24 h as needed
ESPRIT = Evaluation Study on Prophylaxis: a Randomized Italian Trial Source: Manco-Johnson et al4 (JOS) and Gringeri et al5 (ESPRIT)
FIGURE 1 Frequency of bleeding events in the Joint Outcome Study among boys receiv-ing prophylactic or episodic factor replacement therapy. Adapted, with permission, from Manco-Johnson et al.4
Mean number of hemorrhages per month
1.0
0.8
0.6
1.4
1.6
1.2
1.8
01 2 3
Age (years)4 5
0.4
0.2
Prophylactic therapy: joint hemorrhagesProphylactic therapy: other hemorrhagesEpisodic therapy: joint hemorrhagesEpisodic therapy: other hemorrhages
8 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
DucQ.Tran,MD Meeting the Challenges of Managing Hemophilia: Prophylactic vs Episodic Therapy and Avoiding Inhibitors
dence of arthropathy were noted among the Dutch children (Table 4).8
OtherConsiderationsPatients with hemophilia historically
have been encouraged to be sedentary because of the risk of bleeding. Now, however, with the major societal prob-lem of obesity, treatment centers are encouraging patients with hemophilia to be more active. When compared with inactive patients, active individuals with hemophilia do not develop worse joint disease, although their activity should be coordinated with prophylactic dosing.
No single approach for primary pro-phylaxis is optimal for every child. How-ever, early primary prophylaxis (ie, start-ing before age 2.5–3.0 years) decreases hemophilic arthropathy and improves joint outcomes. Costs and complications
resulting from indwelling IV catheters must also be taken into consideration. Prophylactic dosing using high-dose protocols has the best outcomes, and intermediate-dose protocols have good outcomes.
All of the previously mentioned stud-ies evaluated children with hemophilia A. Because comparative data on patients with hemophilia B receiving prophylactic versus episodic treatment are limited, most treatment centers extrapolate the results from patients with hemophilia A and apply similar principles to maintain FIX activity levels ≥ 1% in children with hemophilia B.
n PROPHYLAXIS IN ADULTS
About one third of young adults who received primary prophylaxis as children choose to switch to episodic treatment.9
One analysis of a self-reported cohort of Danish and Dutch men with severe hemophilia A (median follow-up, 3.6 years) compared outcomes of those who continued on prophylaxis with those of in-dividuals who switched to episodic treat-ment. Although men who chose to switch to episodic treatment experienced more joint bleeding, the incidence (3.2 joint bleeds per year)10 was still considerably lower than the incidence of joint bleeds previously reported in other cohorts of adults with severe hemophilia who were managed with episodic treatment for their entire lives.
The multinational longitudinal Ortho-paedic Outcome Study first supported ini-tiation of prophylaxis in adults because of benefits to joints.11 Patients given full-time prophylactic therapy who were followed for 6 years had less progressive hemophilic arthropathy, as measured by physical and radiographic examinations.
Three recent studies in adults evaluated the efficacy of prophylaxis versus episodic treatment and compared the outcomes of both treatment regimens. Collins et al12 treated 20 adult patients (age, 30–45 years) sequentially with episodic treatment for 6 months and then crossed them over to 6 months of high-dose prophylaxis (20–40 IU/kg of rFVIII three times weekly). A comparison of the two interventions showed that patients with significant joint bleeding histories who were maintained on episodic treatment had no bleeding episodes once switched to prophylaxis. However, because this study was brief, it did not show improvement in joint func-tion or QOL measures.
Valentino et al13 first treated 66 adult patients with 6 months of on-demand
TABLE 2Magnetic Resonance Imaging Findings in the Joint Outcome Study
Prophylactic EnhancedepisodicParameter therapy(n=32) treatment(n=33) Pvalue
Number of patients with primary outcome data 27 29 0.73 Number (%) with joint damage 2 (7) 13 (45) 0.002 Number (%) with no joint damage 25 (93) 16 (55)
Source: Manco-Johnson et al4
TABLE 3ESPRIT Study Outcomes
Episodic Prophylactic Parameter therapy(n=21) therapy(n=19) Pvalue
Months in study, median (range) 81.9 (2–96) 84.4 (13–163) NSAge at study end, months, median (range) 148 (18–193) 154 (27–204) NSHemarthroses/patient/year, median (range) 7 (0–68) 36 (0–117) < 0.01Number of patients with roentgenographic 6 (29%) 14 (74%) < 0.05 evidence of joint damage (%)Number of factor VIII units infused 13,477,251 5,749,085 < 0.01Number of joint bleeding episodes 304,733 701,775 < 0.01
NS = not significantSource: Gringeri et al5
TABLE 4Outcomes of Intermediate- vs High-Dose Prophylaxis for Patients with Severe Hemophilia
Patientsborn1970–1979 Patientsborn1980–1989
Intermediatedose Highdose Intermediatedose Highdose Parameter (n=44) (n=24) Pvalue (n=42) (n=18) Pvalue
Age at evaluation, years (range) 22.7 (20.4–25.3) 17.2 (15.2–20.4) < 0.001 13.5 (10.6–15.7) 9.0 (6.3–13.5) 0.005Joint bleeds/year (range) 2.5 (1–5.7) 0.5 (0.2–1.8) 0.050 3.7 (1.7–5) 0.2 (0–0.3) < 0.001Patients without joint bleeds 5% 25% 0.042 10% 50% 0.001 Clinical score (maximum = 90) 2 (0–5) 0 (0–4) 0.449 0 (0–2) 0 (0–0) < 0.001 Pettersson score (maximum = 78) 10 (3.5–17.5) 4 (0–15) 0.750 0 (0–5) 0 < 0.001Pettersson score = 0 11% 46% 0.560 54% 100% 0.797
Source: Fischer et al8

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 9
DucQ.Tran,MD Meeting the Challenges of Managing Hemophilia: Prophylactic vs Episodic Therapy and Avoiding Inhibitors
treatment and then randomized these individuals to either 12 months of high-dose prophylaxis (20–40 IU/kg of rFVIII every other day) or a pharmacokineti-cally tailored arm in which patients were infused with 20–80 IU/kg of rFVIII every third day to maintain trough levels of > 1%. The annualized bleeding rates for both prophylactic groups were reduced by 99%, but there was no evidence of improvement in joint function.13 QOL measures for pain and physical function were improved at the end of the prophy-lactic treatment period.
The third study, the multinational Trial to Evaluate the Effect of Secondary Prophylaxis with rFVIII Therapy in Severe Hemophilia A Adult and/or Adolescent Subjects Compared to That of Episodic Treatment (SPINART),14 was recently completed. In all, 84 patients were ran-domly assigned to treatment with a high-dose prophylactic regimen or episodic treatment. All patients underwent MRI examination of index joints on study entry and had yearly joint examinations and QOL assessments thereafter. At the end of the 3-year treatment period, the patients underwent another MRI examination. Pa-tients in the prophylaxis arm were treated with 25 IU/kg of rFVIII three times per week (escalation by 5 IU/kg to a maximum of 30 or 35 IU/kg was allowed in patients with bleeding episodes after 1 and 2 years, respectively). For the episodic treatment arm, rFVIII replacement was left to the discretion of the treating physician.
An interim report (median follow-up, 1.4 years) showed a 93% decrease in bleed-ing rate among patients in the prophylaxis arm. There was also an implication that bleeding episodes in the prophylaxis arm were less severe. Data from target joint ex-aminations and MRI and QOL assessments have not yet been reported.
Prophylaxis in adults decreases bleed-ing frequency by > 90% and the severity of bleeding episodes. There is early evi-dence that prophylaxis in adults improves arthropathy and QOL, but this still must be confirmed. Also, if evidence from the pediatric and pharmacokinetic studies is considered, many adults with hemophilia A probably can achieve bleeding control
with less than the standard dose of 20–40 IU/kg of rFVIII three times per week, since the 48-hour trough levels with this dosing regimen have been 3–6 IU/dL in recent studies.12,13 Taking this approach also would help to decrease the discrep-ancy between the amounts of replacement factor used by the prophylaxis treatment groups and the episodic treatment groups. Given that each adult has a different bleeding history, hemophilic arthropathy, and factor activity level, dosing strategies should be individualized to optimize the costs of factor replacement and attain the best outcomes.
n INHIBITORS AND CURRENT MANAGEMENT OPTIONS
Alloimmune inhibitory antibodies to FVIII (inhibitors) are serious complica-
tions of treatment with factor concentrates and develop in about 25%–30% of patients with severe hemophilia A. Inhibitors nor-mally are detected fairly early in treatment after a median of 14–16 treatment days. The most significant risk factor that pre-disposes patients to developing inhibitors is the F8 gene mutation.15 Patients with large multiexon gene deletions have a much higher rate of inhibitor formation than do those with missense mutations, and those with inversions have an inter-mediate risk of inhibitor formation. Race is another risk factor—black males have a higher rate of inhibitor development than do white males,16 although the etiology is unclear.
Treatment-related factors also have been associated with inhibitor develop-ment. For example, early intensive fac-
tor exposure has been associated with increased inhibitor formation, and there may be some protection from inhibitor formation with the use of prophylaxis.17 –19
ImmunogenicityofTherapeuticProducts
Another potential risk factor involves plasma-derived products that are rich in von Willebrand factor (vWF) and less immunogenic than are recombinant fac-tor products. Previous data were based on retrospective studies. The Research of Determinants of Inhibitor Development among Previously Untreated Patients with Haemophilia (RODIN)17 trial is the first study to prospectively observe whether some products are more immunogenic than are others. Investigators collected prospective data on 574 previously un-treated patients with hemophilia and fol-lowed them for 75 exposure days. There was no immunogenic difference between patients treated with plasma-derived products and those treated with recom-binant factor.17 An analysis of individual factor products suggested that a second-generation, full-length recombinant factor might be related to a slightly higher rela-tive risk for inhibitor formation, but the observational nature of this study cannot lead to definitive conclusions.
The ongoing Survey of Inhibitors in Plasma-Product Exposed Toddlers (SIPPET)20 is randomizing previously untreated patients to receive either rFVIII or plasma-derived FVIII product with vWF to determine whether some prod-ucts are more immunogenic than others. Other risk factors for inhibitor formation reported in the literature include poly-morphisms in immune regulatory genes and associations with specific human leu-kocyte antigen (HLA) class II alleles, but the evidence has been inconsistent, and any association with inhibitor formation remains unknown.
ImmuneToleranceInduction(ITI)Eradication of inhibitors uses ITI with
frequent regular infusions of FVIII and works in about 60%–80% of cases (median time to tolerance, approximately 9–12 months). However, the optimal dose of
Dosing strategies should be individualized to optimize the costs of factor replacement and attain the best outcomes.

10 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
DucQ.Tran,MD Meeting the Challenges of Managing Hemophilia: Prophylactic vs Episodic Therapy and Avoiding Inhibitors
factor and optimal product (recombinant or plasma-derived) remain unknown.
The International Immune Tolerance Study looked at the issue of dose and randomized good-risk patients (defined as having a peak historical inhibitor titer of ≤ 200 BU/mL, starting titer level of ≤ 10 BU/mL before randomization, and age < 8 years at the time of randomization) to either a high-dose arm of 200 IU/kg daily of rFVIII or to a low-dose arm of 50 IU/kg three times a week. Inhibitor titers and half-life recovery studies were col-lected throughout the study. There was no difference in the time to tolerance induc-tion and the rate of tolerance induction, although the time to negative inhibitor titer and time to normal recovery were slower in the low-dose arm.21 The trial was stopped early due to significantly more bleeds in the low-dose arm and to futility, since enrollment was not robust enough to determine equivalence between the arms.
Patients who cannot have their inhibi-tor eradicated and have a high inhibitor titer (> 5 BU/mL) are treated with bypass-ing agents, such as activated prothrombin complex concentrate (FEIBA), FVIII inhibitor bypass activity, or recombinant factor VIIa. Prophylactic treatment with either bypassing agent resulted in im-proved bleeding control when compared with episodic treatment.22,23
n POTENTIAL FUTURE TREATMENT OPTIONS
New advances on the horizon include long-lasting factor concentrates soon to be available for both patients with hemo-philia A and those with hemophilia B. Also, three active studies are examining gene therapy for patients with hemophilia B. These studies are employing adeno-associated virus (AAV) serotype 8 vector, which expresses FIX. Only the trial from the University College London/St Jude Children’s Research Hospital has reported data thus far, noting stable FIX levels of 1%–6% among 10 patients 8–40 months after treatment.24 In this study, higher
doses of AAV-8 vector genomes yielded higher plasma FIX levels but were associ-ated with abnormalities in liver function tests and immune responses to the AAV-8 capsids. These patients were managed with short courses of corticosteroids.REFERENCES
1. Tuddenham EGD, Cooper DN. The Molecular Genetics of Haemostasis and Its Inherited Disorders. Oxford Monographs on Medical Genetics. Book 25. Oxford, England: Oxford University Press; 1994.
2. Srivastava A, Brewer AK, Mauser-Bun-schoten EP, et al. Guidelines for the management of hemophilia. Treatment Guidelines Working Group on Behalf of The World Federation of Haemophilia. 2013;19:e1–e47.
3. Nilsson IM, Berntorp E, Löfqvist T, Petters-son H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232:25–32.
4. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–544.
5. Gringeri A, Lundin B, von Mackensen S, Mantovani L, Mannucci PM. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). ESPRIT Study Group. J Thromb Haemost. 2011;9:700–710.
6. Feldman BM, Pai M, Rivgard GE, et al. Tai-lored prophylaxis in severe hemophilia A: interim results from the first 5 years of the Canadian He-mophilia Primary Prophylaxis Study. Association of Hemophilia Clinic Directors of Canada Prophylaxis Study Group. J Thromb Haemost. 2006;4:1228–1236.
7. Risebrough N, Oh P, Blanchette V, Curtin J, Hitzler J, Feldman BM. Cost-utility analysis of Cana-dian tailored prophylaxis, primary prophylaxis and on-demand therapy in young children with severe haemophilia A. Haemophilia. 2008;14:743–752.
8. Fischer K, Astermark J, van der Born JG, et al. Prophylactic treatment for severe haemophilia: comparison of an intermediate-dose to a high-dose regimen. Haemophilia. 2002;8:753–760.
9. Makris M. Prophylaxis in haemophilia should be life-long. Blood Transfus. 2012;10:165–168.
10. van Dijk K, Fischer K, van der Born JG, Scheibel E, Ingersley J, van den Berg HM. Can long-term prophylaxis for severe haemophilia be stopped in adulthood? Results from Denmark and the Netherlands. Br J Haematol. 2005;130:107–112.
11. Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII-deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med. 1994;236:391–399.
12. Collins P, Faradji A, Morfini M, Enriquez MM, Schwartz L. Efficacy and safety of secondary prophylactic vs. on-demand sucrose-formulated
recombinant factor VIII treatment in adults with severe hemophilia A: results from a 13-month crossover study. J Thromb Haemost. 2010;8:83–89.
13. Valentino LA, Mamonov V, Hellmann A, et al. A randomized comparison of two prophylaxis regimens and a paired comparison of on-demand and prophylaxis treatments in hemophilia A management. Prophylaxis Study Group. J Thromb Haemost. 2012;10:359–367.
14. Manco-Johnson MJ, Kempton CL, Reding MT, et al. Randomized, controlled, parallel-group trial of routine prophylaxis vs. on-demand treatment with sucrose-formulated recombinant factor VIII in adults with severe hemophilia A (SPINART). J Thromb Haemost. 2013;11:1119–1127.
15. Gouw SC, van den Berg HM, Oldenberg J, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic re-view and meta-analysis. Blood. 2012;119:2922–2934.
16. Miller CH, Benson J, Ellingsen D, et al. F8 and F9 mutations in US haemophilia patients: cor-relation with history of inhibitor and race/ethnicity. Hemophilia Inhibitor Research Study Investigators. Haemophilia. 2012;18:375–382.
17. Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. PedNet and Research of Deter-minants of INhibitor development (RODIN) Study Group. Blood. 2013;121:4046–4055.
18. Gouw SC, van den Berg JM, le Cessie S, van der Born JG.Treatment characteristics and the risk of inhibitor development: a multicenter cohort study among previously untreated patients with severe hemophilia A. J Thromb Haemost. 2007;5:1383–1390.
19. Astermark J, Altisent C, Batorova A, et al. Non-genetic risk factors and the development of in-hibitors in haemophilia: a comprehensive review and consensus report. Haemophilia. 2010;16:747–766.
20. Mannucci PM, Gringeri A, Peyvandi F, Santagostino E. Factor VIII products and inhibitor development: the SIPPET study (survey of inhibitors in plasma-product exposed toddlers). Haemophilia. 2007.13:65–68.
21. Hay CR, DiMechele DM. The principal re-sults of the International Immune Tolerance Study: a randomized dose comparison. International Im-mune Tolerance Study. Blood. 2012;119:1335–1344.
22. Konkle BA, Ebbesen LS, Erhardtsen E, et al. Randomized, prospective clinical trial of re-combinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007;5:1904–1913.
23. Leissinger C, Gringeri A, Antmen B, et al. Anti-inhibitor coagulant complex prophy-laxis in hemophilia with inhibitors. N Engl J Med. 2011;365:1684–1692.
24. Nathwani AC, Tuddenham EGD, Ranga-rajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 11
Hemophilia is caused by func-tional deficiency of a single coagulation protein. Absence of such proteins—factor VIII
(FVIII) in hemophilia A and factor IX (FIX) in hemophilia B—may lead to spontaneous internal bleeding with joint damage, intracranial hemorrhage, and death. Since the 1840s, transfusion of whole blood has been used to treat hemo-philia-associated bleeding.1 It wasn’t until 1911 that FVIII was detected in plasma2 and 1937 when its role was described in hemostasis and the coagulation cascade3 (Figure 1). These advances led to the de-velopment of plasma transfusions in the 1940s, plasma concentrates in the 1950s, cryoprecipitate in the early 1960s, and freeze-dried FVIII products for storage and home use in the late 1960s.4
However, as use of blood products became more common, concerns began to arise about infectious contamination, highlighted by outbreaks of hepatitis C
virus infection in the 1970s and human immunodeficiency virus (HIV) infection in the 1980s among hemophilia patients receiving pooled blood products. Con-cerns about infected blood products led to the development of recombinant FVIII (rFVIII; approved by the US Food and Drug Administration in 1992) and re-combinant FIX (rFIX; approved in 1997).4
The use of these recombinant coagu-lation proteins has changed hemophilia care and increased the life-expectancy of patients with severe hemophilia from approximately 20 years in the 1970s to an essentially normal lifespan today. Long-term prophylactic factor replace-ment therapy also has reduced morbidity, decreasing the risk of joint damage and intracranial hemorrhage, and improved the quality of life for both children and adults with hemophilia.5–7 Recombinant factors are safe and provide independence from reliance on donor blood products.
However, use of recombinant factors
is not without its challenges. These sub-stances are expensive, costing more than $250,000 a year per adult in the United States.4 Because of their short half-life, they need to be administered every other day or several times a week, representing a significant time commitment and present-ing problems with maintaining adherence. In addition, they require venous access, which often requires placement of a cen-tral venous access device (ie, port) and involves risks of sepsis and thrombosis. Finally, 25%–30% of hemophilia A and 3%–4% of hemophilia B patients develop alloantibodies to these recombinant fac-tors, inhibiting their efficacy and result-ing in renewed problems with bleeding.8 Whereas gene therapy is a potential solu-tion,9 a cure is not yet available.
Thus, there is an urgent need for new, improved clotting factors to treat hemophilia patients and individuals with acquired bleeding problems due to administration of oral anticoagulants ( eg, warfarin) who either have been given a supratherapeutic dose or have developed bleeding in conjunction with trauma and surgery. New oral anticoagulants, such as rivaroxaban and dabigatran, also present challenges to reversal and inhibition of bleeding events. Prothrombin complex concentrates (PCCs), fresh frozen plasma (FFP), anti-inhibitor coagulant complex
Advances in Clotting Factors: From Bench to BedsideAnthony Sung, MDDuke University Medical Center, Durham, North Carolina
Abstract The history of clotting factors is inextricably tied to that of hemophilia. The development of recombinant factor VIII (rFVIII) and factor IX (rFIX) in the 1990s has resulted in hemophilia patients having a virtually normal lifespan and significantly fewer complications, such as joint and intracranial bleeding, following prophylactic infusions. However, these treatments are limited by the short-half lives of rFVIII and rFIX, meaning that patients must endure three- or four-times-weekly infusions. This report summarizes some of the exciting ad-vances in the management of hemophilia that have been reported in recent years. They include the addition of polyethylene glycol (PEGylation) or fusion with another protein, such as immunoglobulin G or albumin, to decrease the frequency of infusions, provide prolonged protection from bleeding, limit the need for central venous access devices, and encourage patients to transition to a prophylaxis regimen. In addition, advances have been made in improving the safety and efficacy of long-lasting rFVIII proteins for hemophilia A and the development of alternative agents to treat hemophilia and anticoagulant-related bleeding (eg, warfarin reversal) and new oral anticoagulants (eg, direct thrombin or factor Xa inhibitors).
Dr. Sung is a Fellow in Hematology/Oncology at Duke University Medical Center, Durham, North Carolina.

12 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
AnthonySung,MD Advances in Clotting Factors: From Bench to Bedside
(factor eight inhibitor bypassing activity; FEIBA), and recombinant activated factor VII (rFVIIa) are being tested in the setting of traumatic or surgical bleeding and to reverse bleeding complications resulting from the use of oral anticoagulants.
n MOLECULAR APPROACHES FOR IMPROVED CLOTTING FACTORS FOR HEMOPHILIA
Based on a presentation by Randal J. Kaufman, PhD, Professor, Del E. Webb Center for Neuroscience, Aging, and Stem Cell Research, and Director, Degenerative Disease Research Program, Sanford-Burnham Medical Research Institute, La Jolla, California.
Although the development of recom-binant factors in the 1990s represented a tremendous advance in the treatment of hemophilia, efforts have been underway to circumvent problems associated with
their short half-life and frequent dosing. Research teams have reported on the struc-ture of FVIII and FIX and their mechanisms of action, allowing engineering of variants with improved half-lives. These methods in-clude conjugation with polyethylene glycol (PEG) in a process known as PEGylation, use of PEGylated liposomes as a mecha-nism for sustained release, fusion with the crystallizable fragment (Fc) of the constant region of immunoglobulin (Ig) or albumin, and novel modifications to clotting factors. Although PEGylated liposomes have failed to demonstrate in vivo efficacy,10–12 other methods have shown considerable promise.
PEGylationPEGylation is achieved by covalently
attaching PEG to residues on target pro-teins such as lysine or N-terminal amines.
However, random PEGylation may reduce the activity of the protein, and product heterogeneity may result in inconsistent effectiveness.
More recently, site-directed PEGylation via attachment of PEG-maleimide to cys-teine residues has improved results with many proteins, including tumor necrosis factor-alpha, monoclonal antibody Fab fragment, vascular endothelial growth factor-aptamer, epoetin β, and interferon alfa.13,14 In this approach, missense muta-tions are introduced at surface residues of FVIII to incorporate cysteine residues for conjugation with PEG-maleimide. This al-lows selective modification at the desired location, so PEGylation does not interfere with protein function and ensures product homogeneity. To date, no long-term safety concerns have arisen with this method, including with PEGylated FVIII products developed by Bayer Healthcare (BAY 94-9027), Baxter (BAX 855), and Novo Nordisk (N8-GP).
Of note, PEGylated products may vary by site of PEGylation and type of PEG used. PEG molecules also may dif-fer in size, with smaller molecules being more rapidly cleared than larger ones. In addition, tissue penetration may vary, with smaller molecules having greater permeability. With PEG molecules > 10 kDa, pinocytotic uptake into macrophages and Kupffer cells is increased; with PEG molecules > 30 kDa, kidney clearance is decreased; and with PEG molecules > 50 kDa, liver clearance is increased.15,16 Therefore, the pharmacokinetics and pharmacodynamics—and safety and ef-ficacy—of different PEG solutions vary.
In addition to improving half-life, PEGylation may reduce immunogenicity. PEGylation of L491C in the A2-domain of FVIII resulted in reduced inhibitory activ-ity of a monoclonal antibody that reacts to this highly immunogenic region of FVIII.17 This finding has been supported by other studies with different PEGylated proteins.15,18 PEGylation also may limit the inhibitory effect of alloantibodies.
FusionProteinsBy covalently fusing coagulation fac-
tors with proteins having a much longer
FXII FXIIa
FXI FXIa
FIX FIXa FVIIa
Intrinsic pathway
FXFX FXa
FII FIIa
Fibrinogen (FI) Fibrin (Ia)
Crosslinked �brin clot
FVIIIaFVIII
FV FVa
FXIIIa FXIII
FVII
Extrinsic pathway
Common pathway
Tissue factor Trauma
Damaged endothelium
Trauma
TFPI
Antithrombin
FIGURE 1 Schematic representation of the coagulation cascade, showing the role and im-portance of the various clotting factors as well as the site of action of tissue factor pathway inhibitor (TFPI) and antithrombin. FXII = factor XII (Hageman factor); FXIIa = activated FXII; FXI = factor XI (deficient in hemophilia C); FXIa = activated FXI; FIX = factor IX (Christmas factor; deficient in hemophilia B); FIXa = activated FIX; FVIII = factor VIII (Brinkhous factor, deficient in hemophilia A); FVIIIa = activated FVIII; FX = factor X (Stuart-Prower factor); FXa = activated FX; FVII = factor VII (stable factor); FVIIa = activated FVII; FV = factor V (proaccelerin); FVa = activated FV; FII = factor II (prothrombin); FIIa = activated factor II (thrombin); FXIII = factor XIII (Laki-Lorand factor); FXIIIa = activated FXIII.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 13
AnthonySung,MD Advances in Clotting Factors: From Bench to Bedside
half-life, such as immunoglobulin G1 (IgG1) or albumin, the half-life of the resulting molecule may be extended. Furthermore, the Fc domain can improve the solubility and stability of the partner molecule, allowing easy and cost-effective purification by protein-A/G affinity chro-matography.19 Support for this concept comes from the results of preclinical and phase 1 studies showing that fusion of the monomeric form of the IgG1 Fc to hu-man FVIII, FIX, and factor VIIa (FVIIa) increases the half-life approximately 1.5- to 4-fold without impairing efficacy. No adverse events have been reported.20,21
Another fusion approach covalently attaches the clotting factor to albumin. Data from both preclinical and phase 1 studies have shown a 1.5- to 3-fold in-crease in plasma half-life and preserved efficacy with rFIX-albumin.22,23 This improvement was not found with recom-binant albumin-bound FVIII, however, possibly because of interactions with von Willebrand factor (vWF).
NovelFVIIIMoleculesAn alternative strategy uses a recom-
binant, single-chain FVIII protein that prevents dissociation of the two chains of FVIII, resulting in higher affinity for vWF. Because vWF protects FVIII from proteolysis and clearance, this strategy improves the stability and half-life of FVIII, as supported by animal studies; this molecule now is being tested in human trials.24 Other strategies focus on reduc-ing the immunogenicity of recombinant FVIII molecules by changing or substitut-ing glycan and sulfyl groups.25
NovelProductsinPreclinicalDevelopment
In addition to modifications of FVIII and FIX and administration of activated FVII (FVIIa), several additional novel approaches are in preclinical develop-ment, including factor Xase mimetics and inhibition of antithrombotic pathways.
FVIII enhances FIXa-mediated cleav-age of FX; factor Xase mimetics accom-plish this same function independent of FVIII. For example, a humanized bispe-cific monoclonal antibody to FIXa and
FX showed both efficacy and a prolonged half-life (2 weeks) in a simian model of ac-quired hemophilia.26 The ultimate goal is to engineer such a product that combines efficacy, a prolonged half-life, and ease of administration via subcutaneous injection or oral delivery.
Whereas one approach of these new agents is to enhance clotting, the desired effect, hemostasis, also may be achieved by inhibiting antithrombotic pathways. For example, tissue factor pathway inhibitor (TFPI) inhibits FVIIa and FXa and helps to foster hemostasis. This concept was demonstrated by a monoclonal antibody to one of the Kunitz domains of TFPI in a rabbit model of hemophilia that resulted in decreased bleeding.27 Other TFPI in-hibitors include nucleic acid aptamers,28 non-anticoagulant sulfated polysaccha-ride,29 and small antagonist peptides.30 Further studies will be needed to evaluate the relative merits of these approaches.
Antithrombin 3 (AT3) is another antithrombotic target. Alnylam Phar-maceuticals has developed a synthetic GalNAc-conjugated RNA interference (ALN-AT3) that suppresses liver pro-duction of AT3 mRNA. Pharmacologic studies have shown dose-dependent and reversible reduction of circulating AT3, which was associated with significantly increased thrombin generation and en-hanced homeostasis in murine models of hemophilia.19 Use in wild-type animals induced thrombotic events and dissemi-nated intravascular coagulation, whereas similar or even higher doses were well tolerated in mice with hemophilia A and B with no evidence of thrombosis.
SummaryConsiderable advances in treating
hemophilia and developing clotting fac-tors have been reported over the past two decades. Current approaches focus on improving the safety, tolerability, and delivery of both existing recombi-nant factors and novel alternatives that target other aspects of the coagulation/antithrombotic pathways. These products are making their way from preclinical development in animal models to clinical trials in humans, offering the promise of
even greater advances for patient care in the years to come.
n LONG-LASTING RECOMBINANT FACTOR VIII PROTEINS FOR HEMOPHILIA A
Based on a presentation by Amy D. Shapiro, MD, Chief Executive Officer and Co-Medical Director, Indiana Hemophilia Treatment Center, Indianapolis, Indiana.
PEGylation and fusion of Ig or albumin to FVIII and FIX are exciting strategies for increasing the half-life of these coagula-tion factors that are now being tested in phase 1–3 clinical trials. This section will review these developments.
ClinicalStudiesofPEG-FVIIIConjugates
Three PEG-FVIII conjugates currently are being tested in clinical trials: B-domain deleted recombinant FVIII (PEG-BDD-rFVIII; BAY 94-9027; www.clinicaltrials.gov ID No. NCT01184820), PEGylated full-length rFVIII (BAX 855; www.clini-caltrials.gov ID No. NCT01736475), and glycol-PEGylated rFVIII (N8-GP; www.clinicaltrials.gov ID No. NCT01480180).
PEGylation can impair a protein’s activity if bound to the wrong site, so it is important that this link is produced in such a way that it maintains the protein’s original function. One method of ensur-ing site-specific PEGylation is site-specific mutagenesis, which introduces cysteine mutations on the surface of B-domain deleted FVIII. Bayer HealthCare used this method in designing BAY 94-9027: PEG was conjugated to surface-exposed cysteines of rFVIII to retain full in vitro activity and vWF binding.17
BAY 94-9027 was evaluated in a phase 1 study of 14 patients with severe hemo-philia A.15 Seven patients were given 25 IU/kg twice weekly, and the other seven received 60 IU/kg once weekly. BAY 94-9027 was well tolerated and effective without causing serious adverse events or immunogenicity. The half-life was 19 hours, representing a 1.6-fold increase over the half-life of standard rFVIII (ap-proximately 12 hours). Phase 2/3 studies are underway.
An alternative strategy was employed by Baxter scientists in the design of BAX

14 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
AnthonySung,MD Advances in Clotting Factors: From Bench to Bedside
855, a 20-kDa PEGylated full-length rFVIII. This conjugation process resulted from the combination of an activated PEG reagent with accessible amino groups on FVIII; it was optimized to target and mod-ify mainly the ε-amino groups of lysine residues.18 BAX 855 reportedly retains all physiologic properties of FVIII except binding to the low-density lipoprotein receptor-related protein clearance recep-tor; preclinical testing revealed normal activity and a prolonged half-life when compared with unmodified rFVIII.
N8-GP is a rFVIII with site-directed glycoPEGylation being developed by Novo Nordisk.31 It is synthesized in a Chinese hamster ovary cell line with a truncated B domain of 21 amino acids; the terminal sialic acid on an O-glycan structure in the truncated B-domain is replaced by a conjugated sialic acid con-taining a branched 40-kDa PEG, resulting in a protein with a single medium-weight PEG attached to the B-domain.32 When the coagulation system is activated, thrombin cleaves the B-domain with the attached PEG, resulting in activated FVIII.
N8-GP was evaluated in a dose-esca-lation study (25, 50, or 75 IU/kg/dose) in 26 previously treated patients with severe hemophilia A.33 It was well tolerated at all dose levels, and no patient developed an inhibitor or binding antibodies to FVIII or N8-GP. N8-GP exhibited a dose-linear pharmacokinetic profile with a mean half-life of 19 hours (range, 11.6–27.3 hours), representing a 1.6-fold increase over that of standard rFVIII.34 Clearance was reduced by 30%, and the volumes of distributions were similar to those of standard products. N8-GP currently is being tested in phase 3 clinical trials. A similar product for hemophilia B, N9-GP (glycoPEGylation of rFIX), also has shown promise in clinical trials.35
FcFusionProteinsrFVIIIFc was developed by Biogen
Idec by fusing a single B-domain deleted rFVIII that is produced from human embryonic kidney cells to the dimeric Fc region of IgG1 without intervening linker sequences.36,37 Studies of rFVIIIFc demon-strated prolonged in vivo activity when
compared with existing rFVIII products and comparable binding to vWF, with ani-mal studies showing an approximate two-fold increase in half-life over rFVIII in murine and canine models of hemophilia. Interestingly, the half-life of rFVIIIFc was comparable to that of rFVIII in neonatal Fc receptor (FcRn) knockout mice, sup-porting the role of the Fc fragment and interaction with FcRn in protecting the fusion protein from degradation.38
A phase 1/2 study of rFVIIIFc in 16 patients with severe hemophilia who were given either 25 or 65 IU/kg rFVIII followed by an equal dose of rFVIIIFc showed a 1.5- to 1.7-fold increase in mean half-life (18.8 hours for both doses) for rFVIIIFc over that of rFVIII (12.2 hours for the lower dose and 11.0 hours for the
higher dose).20 Both products had similar dose-dependent peak plasma concentra-tions. No drug-related adverse events, in-hibitors, or severe bleeding was observed.
A phase 3, multicenter study of rFVIIIFc (A-LONG, www.ClinicalTrials.gov ID No. NCT01181128) was completed recently. Treatment arms included indi-vidualized prophylaxis at 3- to 5-day in-tervals, weekly prophylaxis, and episodic (on-demand) treatment. Preliminary results from this study were presented by Mahlangu and coworkers39 at the 65th Annual Meeting of the National Hemo-philia Foundation in October 2013 and are summarized by Dr. Holleh D. Hus-seinzadeh elsewhere in this edition of The Hemophilia Report. Once the data from
this trial are fully compiled and analyzed, the study should provide valuable infor-mation on the safety and effectiveness of different strategies for prophylaxis of hemophilia A and on-demand treatment of bleeding episodes with rFVIIIFc. A similar study is ongoing in children (Kids A-LONG, www.ClinicalTrials.gov identi-fier NCT01458106).
SummaryPEGylation and Fc fusion are exciting
strategies that may prolong the half-life of rFVIII; extend dosing intervals; and potentially improve compliance, access, and safety. Both PEGylated and Fc fusion products have a half-life of 18–19 hours, whereas the half-life of vWF also is 18 hours.40 Given that vWF is needed to stabilize and protect FVIII, it is possible that the half-life of vWF may represent a new limit to how far the half-life of VIII products may be extended.11,17,38 This remains a significant improvement over conventional products, and the final re-sults of phase 3 studies such as A-LONG are eagerly anticipated.
n THE OLD AND THE NEW: PCCs, rFVIIa, AND LONG-LASTING COAGULATION PROTEINS
Based on a presentation by Margaret V. Ragni, MD, MPH, Professor of Medicine, Division of Hematology/Oncology, University of Pittsburgh, and Director, Hemophilia Center of Western Pennsylvania, Pittsburgh, Pennsylvania.
PCCs contain combinations of clotting factors and proteins C and S. Four-factor PCCs (eg, Beriplex, Octaplex, Kcentra) contain factors II, VII, IX, and X, whereas three-factor PCCs (eg, Bebulin, Profilnine) contain factors II, IX, and X but little VII. These products initially were developed as bypassing agents to treat hemophilia pa-tients with inhibitors to FVIII or FIX. These factors act downstream of FVIII and FIX (Figure 1), bypassing their activity. FEIBA is a formulation of PCCs that has activated clotting factors to enhance hemostasis. PCCs and FEIBA start working within minutes and carry a low risk of infectious transmission due to viral inactivation by filtration, nanofiltration, pasteurization, or solvent detergent treatment.
The substance known as rFVIIa also
PEGylation and Fc fusion are exciting strategies that may prolong the half-life of rFVIII, extend dosing intervals, and potentially improve compliance, access, and safety.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 15
AnthonySung,MD Advances in Clotting Factors: From Bench to Bedside
was developed as a bypassing agent to treat hemophilia-associated bleeding. It may produce a “thrombin burst” via acti-vation of FIX, FX, and FII on the surface of activated platelets. Like PCCs and FEIBA, rFVIIa is expensive, and its use carries a significant risk of thrombosis.
To assess the efficacy of these hemo-static strategies, quantitative and qualita-tive laboratory measures of clot formation are needed. These analytic tools include the thrombin generation assay (TGA), thromboelastography (TEG), and rota-tional thromboelastometry (ROTEM), which provide a more comprehensive assessment of clot formation than do such standard assays as prothrombin time (PT) and activated partial thromboplastin time (aPTT).41 Important parameters of TGA include the lag time (the time to initiation of thrombin generation) and endogenous thrombin potential (area under the curve). Those of TEG include the rate of clot formation and strength and stability of the clot. For ROTEM, impor-tant parameters include the time to clot formation (clotting time), maximum clot firmness, and time to clot lysis.
PCCsandrFVIIainSurgeryandTrauma(WithorWithoutWarfarin)
Preclinical studies in porcine liver lac-eration and spleen injury models provide in vitro and in vivo evidence that PCCs are more effective than rFVIIa in restoring thrombin generation and reducing blood loss.42,43 These results are supported by clinical studies of coagulopathic patients undergoing surgery with excessive bleed-ing requiring PCCs or rFVIIa.
In a study of patients undergoing car-diopulmonary bypass, three-factor PCCs (18.9–30.9 U/kg) reduced transfusion requirements to a greater degree than did rFVIIa (90–120 μg/kg).44 Another comparison study of three-factor PCCs (25 U/kg) with rFVIIa (90 μg/kg) in 85 traumatic brain injury patients revealed significantly greater reductions in red blood cell (RBC) and FFP requirements and a lower mortality among the PCC group.45 In a randomized trial of patients with acute major surgical hemorrhage who also received vitamin K, four-factor
PCCs were superior to FFP, whereas other studies showed that the combination of FFP and PCCs may yield even better results.46,47
At the same time, in coagulopathic trauma patients (half of whom were receiving warfarin), administration of three-factor PCCs (25 U/kg) rapidly cor-rected the international normalized ratio (INR) and reduced the RBC requirement; however, there was no survival benefit, as with most studies of rFVIIa in trauma.48 Controversy remains regarding rFVIIa use, given its high cost, lack of dosing guidelines, and thrombosis risk.
Another challenge of using PCCs and rFVIIa is determining the proper dose. Some studies have used algorithms that adjusted the dose based on INR (eg, three-factor PCC dosed at 25 U/kg for an INR of 2.0–3.9, 35 U/kg for an INR of 4.0–6.0, and 50 U/kg for an INR > 6.0), whereas others have used ROTEM test-ing to guide PCC therapy, and still others have used fixed doses.45,49 The optimal timing and frequency of administration also are unclear.
Of note, four-factor PCCs appear to be more effective than are three-factor PCCs.47 This may be due to consumption of FVII in cases of extensive surgery or trauma, bleeding, or warfarin use and the fact that three-factor PCCs are poor in FVII. When the INR > 6.0, three-factor PCCs may have little efficacy.50,51 Interest-ingly, in cases of warfarin reversal for acute bleeding, administration of 10–90 µg/kg of rFVIIa rapidly corrected the INR; unlike with four-factor PCCs, however, it seemed to do little to reduce bleeding.52 In parallel with these findings, ROTEM clot stability and clot lysis time in warfarin-treated patients appeared to improve more after treatment with PCCs than after use of rFVIIa.53
PCCs,rFVIIa,andtheNewOralAnticoagulants
New oral anticoagulants (thrombin and factor Xa inhibitors) have many advantages over warfarin, including no requirement for monitoring, few drug-drug interactions, and lower bleeding rates. However, reversal of these agents for
life-threatening bleeding is complicated by the absence of an effective antidote. The half-life and duration of action of new oral anticoagulants are short. However, acute bleeding often occurs, and waiting for the drug to wear off is unacceptable.
Unfortunately, there is little evidence of the best approach to stop bleeding in patients on new oral anticoagulants. In preclinical studies, FEIBA, PCCs, and rFVIIa improved parameters such as bleeding time, PT, and aPTT, but there was poor correlation with the amount of blood loss.54,55 The best results appeared to follow the use of high-dose four-factor PCCs (eg, 50 U/kg) and FEIBA, with rFVIIa and FFP having little effect.56,57 Studies in healthy human volunteers ap-peared to support these findings, with reversal of abnormal TEG and ROTEM results seen with FEIBA (20–120 U/kg) and four-factor PCCs (50 U/kg); use of rFVIIa (20–120 µg/kg) was less effective.58 However, few data for these products exist in bleeding patients.
Novel antidotes to reverse new oral anticoagulants are being developed. They include an Xa congener that neutralizes Xa coagulation inhibitor function59 and a dabigatran-specific antidote (aDabi-Fab) that mimics thrombin structure (but not function) and binds to dabigatran with 350-fold greater avidity.60 These agents are still in the exploratory stage.
TreatmentConsiderationsTo reverse warfarin-related bleeding
or surgical and trauma-related bleeding, PCCs appear to be superior to rFVIIa and warfarin.61 The combination of PCC and FFP or PCC and rFVIIa may correct laboratory abnormalities such as INR even more rapidly,46 yet extreme caution must be exercised, given the significant thrombotic risk of each agent, and recommendations regarding combination therapy must await further studies. Furthermore, use of these agents should be avoided in patients with recent (< 3 months) thromboembolism.61 Even in the absence of thromboembolism, dosing should be judicious (eg, 25 U/kg of four-factor PCC for an INR of 2.0–3.9, 35 U/kg for an INR of 4.0–6.0, and 50 U/kg for an INR > 6.0).50,62 The lowest effective

16 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
AnthonySung,MD Advances in Clotting Factors: From Bench to Bedside
dose of rFVIIa has not been established.Regarding new oral anticoagulants,
it is unclear whether PCCs or rFVIIa can reverse their effects. Caution should be exercised, since there apparently is little correlation between correction of laboratory abnormalities and reversal of bleeding, and dosing and monitoring are not established.58,63–65 New antidotes are under development but are experimental at this time. Transfusion support and surgical hemostasis should be provided, if indicated. Of note, dialysis with activated charcoal may be effective for dabigatran if initiated within 2–4 hours of ingestion; however, this may not be effective to re-verse the effects of rivaroxaban, which is highly protein bound.65,66
n CONCLUSION
Tremendous advances have been made in hemostasis over the past sev-eral years. Advances in clotting factors (eg, PEGylation, Ig or albumin fusion with FVIII or FIX) promise to improve hemophilia treatment by increasing fac-tor half-life, decreasing the frequency of infusions, and potentially improving compliance and access while decreasing the risk of bleeding complications. Other novel agents (eg, TFPI, AT3 inhibitors) are also very exciting. Meanwhile, FEIBA, PCCs, and rFVIIa may help decrease life-threatening bleeding for hemophilia patients and those on warfarin or new oral anticoagulants who experience surgical or traumatic bleeding. As with all these agents, caution must be exercised, given the risk of upsetting the balance between hemostasis and thrombosis. Nonetheless, the coming years promise major advances in clotting factor treatments.
REFERENCES
1. Otto JC. An account of an hemorrhagic disposition existing in certain families. Clin Orthop Relat Res. 1996;(328):4–6.
2. Lane S. Haemorrhagic diathesis: successful transduction of blood. Lancet. 1840;1:185–188.
3. Patek AJ, Taylor FH. Hemophilia, pt II: some properties of a substance obtained from normal hu-man plasma effective in accelerating the coagulation of hemophilic blood. J Clin Invest. 1937;16:113–124.
4. Kingdon HS, Lundblad RL. An adventure in biotechnology: the development of haemophilia A therapeutics—from whole-blood transfusion to
recombinant DNA to gene therapy. Biotechnol Appl Biochem. 2002;35:141–148.
5. Nilsson IM, Berntorp E, Löfqvist T, Petters-son H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232:25–32.
6. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–544.
7. Mondorf W, Kalnins W, Klamroth R. Pa-tient-reported outcomes of 182 adults with severe haemophilia in Germany comparing prophylactic vs. on-demand replacement therapy. Haemophilia. 2013;19:558–563.
8. Astermark J. Basic aspects of inhibitors to factors VIII and IX and the influence of non-genetic risk factors. Haemophilia. 2006;12(Suppl 6):8–13.
9. Nathwani AC, Tuddenham EGD, Rangarajan S, et al. Adenovirus-associated virus vector-medi-ated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365.
10. Powell JS. Liposomal approach towards the development of a longer-acting factor VIII. Haemo-philia. 2007;13:23–28.
11. Powell J, Martinowitz U, Windyga J, et al. Efficacy and safety of prophylaxis with once-weekly BAY 79-4980 compared with thrice-weekly rFVIII-FS in haemophilia A patients: a randomised, active-controlled, double-blind study. LipLong Study Investigators. Thromb Haemost. 2012;108:913–922.
12. Powell JS, Nugent DJ, Harrision JA, et al. Safety and pharmacokinetics of a recombinant factor VIII with pegylated liposomes in severe hemophilia A. J Thromb Haemost. 2008;6:277–283.
13. Alconcel SNS, Baas AS, Maynard HD. FDA-approved poly(ethylene glycol)-protein conjugate drugs. Polym Chem. 2011;2:1442–1448.
14. Horton S, Walsh C, Emery P. Certolizumab pegol for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2012;12:235–249.
15. Ivens IA, Baumann A, McDonald TA, Humphries TJ, Michaels LA, Mathew P. PEGylated therapeutic proteins for haemophilia treatment: a review for haemophilia caregivers. Haemophilia. 2013;19:11–20.
16. Tang L, Leong L, Sim D, et al. von Wil-lebrand factor contributes to longer half-life of PEGylated factor VIII in vivo. Haemophilia. 2013;19:539–545.
17. Mei B, Pan , Jiang H, et al. Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood. 2010;116:270–279.
18. Turecek PL, Bossard MJ, Graninger M, et al. BAX 855, a PEGylated rFVIII product with prolonged half-life: development, functional and structural characterisation. Hamostaseologie. 2012;32:S29–S38.
19. Kaufman RJ, Powell JS. Molecular approach-es for improved clotting factors for hemophilia. Blood. 2013;122:3568–3574.
20. Powell JS, Josephson NC, Quon D, et al. Safety and prolonged activity of recombinant fac-tor VIII Fc fusion protein in hemophilia A patients. Blood. 2012;119:3031–3037.
21. Shapiro AD, Ragni MV, Valentino LA, et al.
Recombinant factor IX-Fc fusion protein (rFIXFc) demonstrates safety and prolonged activity in a phase 1/2a study in hemophilia B patients. Blood. 2012;119:666–672.
22. Weimer T, Wormsbächer W, Kronthaler U, Lang W, Liebing U, Schulte S. Prolonged in-vivo half-life of factor VIIa by fusion to albumin. Thromb Haemost. 2008;99:659–667.
23. Santagostinpo, Negrier C, Klamroth R, et al. Safety and pharmacokinetics of a novel recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in hemophilia B patients. Blood. 2012;120:2405–2411.
24. Schulte S. Innovative coagulation factors: albumin fusion technology and recombinant single-chain factor VIII. Thromb Res. 2013;131:S2–S6.
25. Kannicht C, Ramström M, Kohla G, et al. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb Res. 2013;131:78–88.
26. Kitazawa T, Igawa T, Sampei Z, et al. A bi-specific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18:1570–1574.
27. Hilden I, Lauritzen B, Sørensen BB, et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood. 2012;119:5871–5878.
28. Gorczyca ME, Nair SC, Jilma B, et al. Inhibi-tion of tissue factor pathway inhibitor by the aptamer BAX499 improves clotting of hemophilic blood and plasma. J Thromb Haemost. 2012;10:1581–1590.
29. Prasad S, Lillicrap D, Labelle A, et al. Ef-ficacy and safety of a new-class hemostatic drug candidate, AV513, in dogs with hemophilia A. Blood. 2008;111:672–679.
30. Peraramelli S, Thomassen S, Heinzmann A, et al. Direct inhibition of factor VIIa by TFPI and TFPI constructs. J Thromb Haemost. 2013;11:704–714.
31. Agersø H, Stennicke HR, Pelzer H, et al. Pharmacokinetics and pharmacodynamics of turoctocog alfa and N8-GP in haemophilia A dogs. Haemophilia. 2012;18:941–947.
32. Martinowitz U, Bierre J, Brand B, et al. Bio-equivalence between two serum-free recombinant factor VIII preparations (N8 and ADVATE®)—an open-label, sequential dosing pharmacokinetic study in patients with severe haemophilia A. Haemophilia. 2011;17:854–859.
33. Tiede A, Brand B, Fischer R, et al. Enhanc-ing the pharmacokinetic properties of recombinant factor VIII: first-in-human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11:670–678.
34. Møss J, Rosholm A, Laurén A. Safety and pharmacokinetics of a glycoPEGylated recombi-nant activated factor VII derivative: a randomized first human dose trial in healthy subjects. J Thromb Haemost. 2011;9:1368–1374.
35. Negrier C, Knobe K, Tiede A, Giangrande P, Moss J. Enhanced pharmacokinetic properties of a glycoPEGylated recombinant factor IX: a first hu-man dose trial in patients with hemophilia B. Blood. 2011;118:2695–2701.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 17
AnthonySung,MD Advances in Clotting Factors: From Bench to Bedside
36. Peters RT, Low SC, Kamphaus GD, et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood. 2010;115:2057–2064.
37. Peters RT, Toby G, Lu Q, et al. Biochemical and functional characterization of a recombinant monomeric factor VIII-Fc fusion protein. J Thromb Haemost. 2013;11:132–141.
38. Dumont JA, Liu T, Low SC, et al. Prolonged activity of a recombinant factor VIII-Fc fusion protein in hemophilia A mice and dogs. Blood. 2012;119:3024–3030.
39. Mahlangu J, Powell J, Ragni M, et al. A-LONG: Phase 3 study of long-lasting recombinant factor VIII Fc fusion protein (rFVIIIFc) in hemo-philia A. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, California. Abstract CR38.
40. MannuccI PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood. 2013;122:648–657.
41. Tanaka KA, Key NS, Lew JH. Blood coagu-lation: hemostasis and thrombin regulation. Anesth Analg. 2009;108:1433–1446.
42. Mitterlechner T, Innerhofer P, Streif W, et al. Prothrombin complex concentrate and recombinant prothrombin alone or in combination with recombi-nant factor X and FVIIa in dilutional coagulopathy: a porcine model. J Thromb Haemost. 2011;9:729–737.
43. Dickneite G, Dörr B, Kaspereit F, Tanaka KA. Prothrombin complex concentrate versus re-combinant factor VIIa for reversal of hemodilutional coagulopathy in a porcine trauma model. J Trauma. 2010;68:1151–1157.
44. Tanaka KA, Mazzeffi MA, Grube M, Ogawa S, Chen EP. Three-factor prothrombin complex concentrate and hemostasis after high-risk cardio-vascular surgery. Transfusion. 2013;53:920–921.
45. Joseph B, Jadjzacharia P, Aziz H, et al. Pro-thrombin complex concentrate: an effective therapy in reversing the coagulopathy of traumatic brain injury. J Trauma Acute Care Surg. 2013;74:248–253.
46. Holland L, Warkentin TE, Refaai M, Cowther MA, Johnston MA, Sarode R. Subopti-mal effect of a three-factor prothrombin complex concentrate (Profilnine-SD) in correcting supra-therapeutic international normalized ratio due to
warfarin overdose. Transfusion. 2009;49:1171–1177.47. Ogawa S, Szlam F, Ohnishi T, Molinaro
RJ, Hosokawa K, Tanaka KA. A comparative study of prothrombin complex concentrates and fresh-frozen plasma for warfarin reversal under static and flow conditions. Thromb Haemost. 2011;106:1215–1223.
48. Gill R, Herbertson M, Buylsteke A, et al. Safety and efficacy of recombinant activated factor VII: a randomized placebo-controlled trial in the setting of bleeding after cardiac surgery. Circulation. 2009;120:21–27.
49. Weber CF, Görlinger K, Meininger D, et al. Point-of-care testing: a prospective, randomized clinical trial of efficacy in coagulopathic cardiac surgery patients. Anesthesiology. 2012;117:531–547.
50. Pabinger I, Brenner B, Kalina U, Knaumb S, Nagy A, Ostermann H. Prothrombin complex concentrate (Beriplex P/N) for emergency anti-coagulation reversal: a prospective multinational clinical trial. Beriplex P/N Anticoagulation Reversal Study Group. J Thromb Haemost. 2008;6:622–631.
51. Makris M, van Veen JJ. Three or four fac-tor prothrombin complex concentrate for emer-gency anticoagulation reversal? Blood Transfus. 2011;9:117–119.
52. Rosovsky RP, Crowther MA. What is the evi-dence for the off-label use of recombinant factor VIIa (rFVIIa) in the acute reversal of warfarin? Hematol-ogy Am Soc Hematol Educ Program. 2008:36–38.
53. Sniecinski RM, Chen EP, Makadia SS, Kikura M, Bolliger D, Tanaka KA. Changing from aprotinin to tranexamic acid results in increased use of blood products and recombinant factor VIIa for aortic surgery requiring hypothermic arrest. J Cardiothorac Vasc Anesth. 2010;24:959–963.
54. Imberti D, Barillari G, Biasioli C, et al. Emergency reversal of anticoagulation with a three-factor prothrombin complex concentrate in patients with intracranial haemorrhage. Blood Transfus. 2011;9:148–155.
55. Perzborn E, Gruber A, Tinel H, et al. Reversal of rivaroxaban anticoagulation by haemo-static agents in rats and primates. Thromb Haemost. 2013;110:162–172.
56. Zhou W, Schwarting S, Illanes S, et al. Hemostatic therapy in experimental intracerebral hemorrhage associated with the direct thrombin
inhibitor dabigatran. Stroke. 2011;42:3594–3599.57. Pragst I, Zeitler SH, Doerr B, et al. Reversal
of dabigatran anticoagulation by prothrombin com-plex concentrate (Beriplex P/N) in a rabbit model. J Thromb Haemost. 2012;10:1841–1848.
58. Marlu R, Jodaj E, Paris A, Albaladeio P, Cracowski JL, Pernod G. Effect of non-specific reversal agents on anticoagulant activity of dabiga-tran and rivaroxaban: a randomised crossover ex vivo study in healthy volunteers. Thromb Haemost. 2012;108:217–224.
59. Lu G, DeGuzman FR, Hollenbach SJ, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med. 2013;19:446–451.
60. Schiele F, van Ryn J, Canada K, et al. A spe-cific antidote for dabigatran: functional and struc-tural characterization. Blood. 2013;121:3554–3562.
61. Ragni MV. The old and new: PCCs, VIIa, and long-lasting clotting factors for hemophilia and other bleeding disorders. Hematology Am Soc Hematol Educ Program. 2013:44–51.
62. Levy JH, Tanaka KA, Dietrich W. Peri-operative hemostatic management of patients treated with vitamin K antagonists. Anesthesiology. 2008;109:918–926.
63. Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin com-plex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011;124:1573–1579.
64. Weitz JI, Quinlan DJ, Eikelboom JW. Periprocedural management and approach to bleeding in patients taking dabigatran. Circulation. 2012;126:2428–2432.
65. Baglin T, Hillarp A, Tripodi A, Elalamy I, Buller H, Ageno W. Measuring oral direct inhibitors (ODIs) of thrombin and factor Xa: a recommenda-tion from the Subcommittee on Control of Antico-agulation of the Scientific and Standardisation Com-mittee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2013 Jan 24. [Epub ahead of print.]
66. Levi M, Eerenberg E, Kamphuisen PW. Bleeding risk and reversal strategies for old and new anticoagulants and antiplatelet agents. J Thromb Haemost. 2011;9:1705–1712.

18 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
Hemophilia A and B are X-linked, recessive disorders caused by deficiency or ab-sence of coagulation factors
VIII (FVIII) and factor IX (FIX), re-spectively. They are classified into severe (< 1%), moderate (1%–5%), or mild (> 5% to < 40%) categories according to coagula-tion factor activity.
Hemophilia A is four times more common than is hemophilia B. Because they are clinically indistinguishable, they historically were believed to represent the same disease. It was not until 1952 that hemophilia B was considered to be a separate entity. The two types share many similar components (eg, prolongation of activated partial thromboplastin time, recurrent hemarthroses), but they have some distinct differences (eg, frequency of inhibitor development, type of genetic
mutations). For example, hemophilia B is caused by less severe gene mutations than hemophilia A and is characterized by more missense mutations than null mutations. As a result of these differences, only 35% of patients with hemophilia B have severe disease, compared with 45% of those with hemophilia A.1
The subtypes also differ in coagulation factor pharmacokinetics—the half-life of FIX is 18 hours and of FVIII is 11 hours.2 As a result, post-infusion levels after FIX administration are sustained for longer periods, thereby reducing the risk of recurrent bleeding.3 The reduced incidence of recurrent hemarthrosis may contribute to a threefold difference in the need for total joint arthroplasty in people with hemophilia B as compared with those diagnosed with hemophilia A.4 Nonetheless, the treatment approaches are similar—and most clinical research for treatment bundles the two diseases.
During a Baxter Healthcare–spon-sored symposium offered at the 65th An-nual Meeting of the National Hemophilia Foundation, experts described the his-tory of hemophilia B therapy and looked toward its future. The panelists included Amy Shapiro, MD, and Natalie Duncan, MPH, of the Indiana Hemophilia and
Thrombosis Center in Indianapolis; Erik Berntorp, MD, PhD, of the Malmö Centre for Thrombosis and Haemostasis at Lund University in Lund, Sweden; and Marion Koerper, MD, of the University of Cali-fornia, San Francisco School of Medicine in San Francisco, California.
n EVOLUTION OF CURRENT TREATMENT APPROACHES
Multiple factor replacements are avail-able in the United States to treat hemo-philia B (Table 1).5 At one time, the initial management strategy for hemophilia patients was on-demand treatment to provide factor in response to an acute epi-sode of bleeding or trauma. On-demand factor replacement remains the treatment of choice for all patients with mild disease and most of those with moderate disease. In the 1960s, however, clinicians first thought of “converting” individuals with severe disease to moderate manifestations using scheduled prophylactic treatment.
PreventativeMeasuresProphylaxis aims to prevent bleeding
episodes before they result in arthropa-thy and life-threatening hemorrhage. Prophylaxis, however, is expensive and inconvenient, because it requires frequent dosing. Therefore, decades of research were necessary before prophylaxis became the standard of care for children with severe hemophilia.
Some initial evidence to support the prophylactic approach arose from large observational studies. One large study of 156 patients with hemophilia compared Swedish and Norwegian individuals di-
Hemophilia A and B: Disease Differences and the Use of Prophylactic TherapyAnna Chalmers, MDRush University Medical Center, Chicago, Illinois
Abstract The treatment algorithm for hemophilia has evolved substantially since the advent of factor replacement. Prophylaxis is now the standard of care for children with hemophilia A and B. However, unresolved treatment decisions, including the age to start treatment, optimal dosing, and continuation of pro-phylaxis into adulthood, remain. Additionally, exciting new areas of managing hemophilia are on the horizon, including treatment with new recombinant coagu-lation factors that have prolonged half-lives and potentially curative treatments.
Dr. Chalmers is a Fellow in Hematology/Oncology at Rush University Medical Center, Chicago, Illinois.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 19
AnnaChalmers,MD Hemophilia A and B: Disease Differences and the Use of Prophylactic Therapy
FIGURE 1 Approach to primary prophy-laxis of hemophilia B, as practiced at Malmö Hemo philia Center in Sweden. Adapted, with permission, from Berntorp.14
Start via peripheral veinonce per week and increase
frequency of injectionsas soon as possible
Establishaccess
At approximately 1 year ofage, before any joint bleeds
take placeInitiation
Place a Port-A-Cath® ifthe goal for frequency
of injections is notreached
Escalation
TABLE 1Current Factor IX Replacement Products Available in the United States
Tradename Recombinantorplasmaderived Stableatroomtemperature
AlphaNine SD Plasma derived NoBeneFIX Recombinant Yes (up to 6 months)Mononine Plasma derived NoRixubis Recombinant Yes (up to 6 months)
Source: Shapiro5
TABLE 2Definitions of Prophylaxis Used by the European Pediatric Network for Hemophilia Management
Term Definition
Primary prophylaxis A Regular continuous treatment started after the first joint bleed and before the age of 2 yearsPrimary prophylaxis B Regular continuous treatment started before the age of 2 years without previous joint bleedSecondary prophylaxis A Regular continuous (long-term) treatment started after two or more joint bleeds or at an age > 2 yearsSecondary prophylaxis B Intermittent regular (short-term) treatment, because of frequent bleeds
Source: Donadel-Claeyssens12
agnosed with the disease.6 Clinicians in Sweden have long used prophylaxis as the standard of care, whereas those in Norway had embraced an on-demand treatment approach. Patients given on-demand treatment had five times more episodes of bleeding and used more resources outside of the healthcare setting, but the prophylaxis group had significantly higher annual factor-concentrate consumption.
A similar study compared hemophiliac patients in the Netherlands, who were given intermediate-dose prophylaxis, with patients in Sweden, who were given high-dose prophylaxis, and France, who were given on-demand treatment.7 Higher rates of bleeding and arthropathy were noted among the on-demand treatment group; however, there was minimal reported clinical improvement between interme-diate-dose and high-dose prophylaxis. Costs for on-demand and intermediate-dose prophylaxis were similar, whereas high-dose prophylaxis was significantly more expensive.
Despite these and other observational data, authors of a 2006 Cochrane review8 found insufficient evidence to support prophylaxis and called for randomized, controlled trials. Shortly thereafter, a ran-domized, controlled trial showed that 25 IU/kg of prophylactic factor replacement
given every other day decreased bleeding episodes and hemarthroses in patients with hemophilia A.9 The prophylaxis group used twice as much factor annually at a higher financial cost.
Subsequent studies have upheld the positive clinical impact of prophylaxis.10 The latest Cochrane review has recom-mended prophylaxis to preserve joint function.11 According to this review, evidence of benefit from prophylaxis in patients with preexisting joint damage is insufficient. Thus, early initiation of fac-tor replacement is needed for children, although the exact age for treatment initiation is uncertain.
TimingandDosingAreEverythingMultiple methods of prophylaxis have
been defined (Table 2),12 and the best strat-egy to follow has been hotly debated. An early start is important, since it can lessen future degenerative joint disease.13 How-ever, starting treatment in toddlers aged 1–2 years is not without its challenges. For example, complication rates associated with central catheter placement brings venous access into question. The model used at the Malmö Hemophilia Center in Sweden (Figure 1)14 details one approach for primary prophylaxis of hemophilia B.
Optimal dosing is another unresolved
component of prophylaxis. Internation-ally, different treatment approaches have been proposed (Table 3).15 The most expensive scheme, the high-dose Swed-ish prophylactic regimen, previously was considered the best to prevent joint disease and bleeding. More recently, a retrospective analysis of the Dutch intermediate-dose program compared with the much more costly Swedish high-dose regimen concluded that the latter offered minimal added benefit.16 The alternative pharmacokinetic approach remains promising, but there is insuffi-cient evidence that factor level peaks and troughs predict bleeding risk. In clinical practice, prophylactic regimens and dosing usually are tailored to patients’ individual needs.
Lower costs have sparked interest in low-dose regimens, especially when they are used in resource-poor settings. Wu et al17 demonstrated that low-dose prophy-laxis used in China reduced the number of bleeding episodes and improved joint function over 12 weeks. Patients with he-mophilia A were given 10 IU/kg of FVIII twice weekly, and those with hemophilia B were given 20 IU/kg of FIX once weekly. In the Swedish study, however, the aver-age prophylactic dose was 25-40 IU/kg of FVIII given three times a week for patients

20 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
AnnaChalmers,MD Hemophilia A and B: Disease Differences and the Use of Prophylactic Therapy
TABLE 3Dosing Strategies for Long-Term Prophylaxis
Regimen Principle Relativecost
Dutch regimen 15–25 IU/kg 2–3 times/wk, starting early after Moderate (intermediate-dose) the occurrence of joint bleedsTraditional Swedish dosing 25-40 IU/kg 2–3 times/wk, starting before the Most expensive regimen (high-dose) occurrence of joint bleedsAlternative Swedish dosing Individualized to patient needs, starting with Least expensive regimen (pharmacokinetic a high dose and followed by a reduction in dose dosing) and lengthening of the interval between dosesCanadian regimen (dose 50 IU/kg weekly, starting early after the Less expensive escalation) occurrence of joint bleeds and intensified stepwise depending upon bleeding frequency
Source: Berntorp and Shapiro15
with hemophilia A and 25–40 IU/kg of FIX twice a week for those with hemophil-ia B. The Chinese low-dose prophylactic approach reduced costs, but whether it prevented long-term arthropathy-related disability remains unclear, since a moder-ate number of joint bleeds remained.
HowMuchIsTooMuch?Prophylaxis has dramatically improved
quality of life and reduced arthropathy in children with hemophilia. Whether and when to stop prophylaxis remain contro-versial. Presenting data from the HUGS (Hemophilia Utilization Group Study) VB trial, Koerper18 revealed that two thirds of children and one half of adults with hemophilia B at one US hemophilia treatment center were on prophylaxis. Early retrospective data did not show a significant difference in arthropathy scores when on-demand therapy and prophylactic treatment were compared in adults.19 Definitive recommendations for prophylaxis in adults will require long-term, prospective, randomized trials to adequately assess outcomes.
n CHALLENGES IN USING PROPHYLACTIC REGIMENS
One barrier to standard use of the prophylactic regimen is the cost. Prophy-laxis has become less expensive, as it has become the standard of care for children with hemophilia in the United States; however, patient adherence to the regimen remains challenging.
With currently available formulations, patients with hemophilia B require pro-phylaxis two to three times a week. Only
60% of hemophilia patients report infus-ing at least three fourths of the recom-mended factor, most commonly missing doses because of their complexity and the time commitment.20 Paradoxically, the ability of prophylactic factor replace-ment to completely eliminate symptoms decreases adherence.21
This problem, however, is not limited to patients in the United States. A Chinese study examining prophylaxis versus on-demand treatment found that although factor was provided at no cost, adherence rates lagged.22 The primary reason for this phenomenon was that patients and their families did not fully understand the im-portance of therapy. Recent studies by the same group showed significant barriers to prophylaxis in areas without dedicated hemophilia treatment centers.22
ImprovingAdherenceSeveral methods have been developed
to improve adherence rates. Two modi-fiable factors that influence adherence are maintenance of a good relationship between the patient and the healthcare provider and the patient’s positive belief in the necessity of treatment.21–23 With regard to the latter, families reported that expanded health education best facilitated adherence.20 Novel treatment approaches that may help improve adherence rates are being developed.
n NOVEL APPROACHES TO HEMOPHILIA TREATMENT
Current hemophilia treatments have improved the quality of life and longevity of patients with hemophilia dramatically,
yet limitations such as the inconvenience of frequent dosing and its substantial cost remain. Prolonging the half-life of factor replacement may lower costs and de-crease complications and inconvenience related to frequent venous access.
The half-life of therapeutics can be extended by using PEGylation, which involves covalent attachment of poly-ethylene glycol (PEG) polymer chains to liposomes.24 Another technique involves recombinant fusion proteins, in which FIX is attached to the crystallized frag-ment region of immunoglobulin G.25 In these trials, bleeding-free intervals or blood clotting times have been prolonged significantly. Currently, at least three phase 3 clinical trials of long-acting FIX, and many more trials in phase 1 or phase 2, are ongoing.26
An alternative approach is gene therapy. Because a small increase in fac-tor level can significantly improve bleed-ing rates, this method offers a potential cure for hemophilia B. Nathwani et al27 treated six patients with severe hemo-philia using a viral vector that expressed FIX. All patients had less need for factor replacement, and four patients stopped using prophylaxis completely. Additional trials of this extremely promising area of research are ongoing.
REFERENCES
1. Soucie JM, Nuss R, Evatt B, et al. Mortality among males with hemophilia: relations with source of medical care. The Hemophilia Surveillance System Project Investigators. Blood. 2000;96:437–442.
2. Collins PW, Fischer K, Morfini M, Blanchette VS, Björkman S. Implications of coagulation factor VIII and IX pharmacokinetics in the prophylactic treatment of haemophilia. International Prophylaxis Study Group Pharmacokinetics Expert Working Group. Haemophilia. 2011;17:2–10.
3. Björkman S. Prophylactic dosing of factor VIII and factor IX from a clinical pharmacokinetic perspective. Haemophilia. 2003;9:101–108.
4. Tagariello G, Iorio A, Santagostino E, et al. Comparison of the rates of joint arthroplasty in patients with severe factor VIII and IX deficiency: an index of different clinical severity of the 2 coagula-tion disorders. Blood. 2009;114:779–784.
5. Shapiro A. Treatment update on hemophilia B. Presented at the 65th Annual Meeting of the Na-tional Hemophilia Foundation; October 3–5, 2013; Anaheim, California.
6. Steen Carlsson K, Höjgård S, Glomstein A, et al. On-demand vs. prophylactic treatment for severe haemophilia in Norway and Sweden: differences in

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 21
AnnaChalmers,MD Hemophilia A and B: Disease Differences and the Use of Prophylactic Therapy
treatment characteristics and outcome. Haemophilia. 2003;9:555–566.
7. Fischer K, Van Den Berg M. Prophylaxis for severe haemophilia: clinical and economical issues. Haemophilia. 2003;9:376–381.
8. Stobart K, Iorio A, Wu JK. Clotting fac-tor concentrates given to prevent bleeding and bleeding-related complications in people with hemophilia A or B. Cochrane Database Syst Rev. 2006;(2):CD003429.
9. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–544.
10. Gringeri A, Lundin B, von Mackensen S, Mantovani L, Mannucci PM. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). ESPRIT Study Group. J Thromb Haemost. 2011;9:700–710.
11. Iorio A, Marchesini E, Marcucci M, Stobart K, Chan AK. Clotting factor concentrates given to prevent bleeding and bleeding-related complica-tions in people with hemophilia A or B. Cochrane Database Syst Rev. 2011;(9):CD003429.
12. Donadel-Claeyssens S. Current co-ordinat-ed activities of the PEDNET (European Paediatric Network for Haemophilia Management). European Paediatric Network for Haemophilia Management. Haemophilia. 2006;12:124–127.
13. Astermark J, Petrini P, Tengborn L, Schul-
man S, Ljung R, Berntorp E. Primary prophy-laxis in severe haemophilia should be started at an early age but can be individualized. Br J Haematol. 1999;105:1109–1113.
14. Berntorp E. Prophylaxis for hemophilia B: best practices. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, California.
15. Berntorp E, Shapiro AD. Modern haemo-philia care. Lancet. 2012;379:1447–1456.
16. Fischer K, Steen Carlsson K, Petrini P, et al. Intermediate-dose versus high-dose prophylaxis for severe hemophilia: comparing outcome and costs since the 1970s. Blood. 2013;122:1129–1136.
17. Wu R, Luke K-H, Poon M-C, et al. Low dose secondary prophylaxis reduces joint bleeding in severe and moderate haemophilic children: a pilot study in China. Haemophilia. 2011;17:70–74.
18. Koerper M. Burden of illness and quality of life in hemophilia B: HUGS VB study. Presented at the 65th Annual Meeting of the National Hemo-philia Foundation; October 3–5, 2013; Anaheim, California.
19. van Dijk K, Fischer K, van der Bom JG, Scheibel E, Ingerslev J, van den Berg HM. Can long-term prophylaxis for severe haemophilia be stopped in adulthood? Results from Denmark and the Netherlands. Br J Haematol. 2005;130:107–112.
20. Hacker MR, Geraghty S, Manco-Johnson M. Barriers to compliance with prophylaxis therapy in
haemophilia. Haemophilia. 2001;7:392–396.21. De Moerloose P, Urbancik W, van den Berg
HM, Richards M. A survey of adherence to haemo-philia therapy in six European countries: results and recommendations. Haemophilia. 2008;14:931–938.
22. Tang L, Wu R, Sun J, et al. Short-term low-dose secondary prophylaxis for severe/moderate haemophilia A children is beneficial to reduce bleed and improve daily activity, but there are obstacle in its execution: a multi-centre pilot study in China. Haemophilia. 2013;19:27–34.
23. Schrijvers LH, Uitslager N, Schuurmans MJ, Fischer K. Barriers and motivators of adherence to prophylactic treatment in haemophilia: a systematic review. Haemophilia. 2013;19:355–361.
24. Spira J, Plyushch OP, Andreeva TA, An-dreev Y. Prolonged bleeding-free period following prophylactic infusion of recombinant factor VIII reconstituted with pegylated liposomes. Blood. 2006;108:3668–3673.
25. Peters RT, Low SC, Kamphaus GD, et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood. 2010;115:2057–2064.
26. Escobar MA. Advances in the treatment of inherited coagulation disorders. Haemophilia. 2013;19:648–659.
27. Nathwani AC, Tuddenham EGD, Ranga-rajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365.

22 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
Hemophilia A (classic hemo-philia) and B (Christmas disease) are the two main types of inherited bleeding
disorders that hamper the blood-clotting process.1,2 Although they result from mutations in different genes, they have similar clinical courses. Hemophilia A occurs in up to 1:5,000 males around the world, whereas hemophilia B occurs in about 1:20,000 newborn males globally.3
During a Biogen Idec–sponsored symposium offered at the 65th Annual Meeting of the National Hemophilia Foundation, experts discussed the genet-ics of hemophilia A and B and presented talking points that they often share with their patients, including those address-ing family planning and the care of pregnant patients with hemophilia. The panel of speakers included Barbara Konkle, MD, Professor of Medicine at the University of Washington School
of Medicine, Director of Translational Research, and Medical Director of the Hemostasis Reference Laboratory at the Puget Sound Blood Center in Seattle, Washington; Michelle Alabek, MS, CGC, of Norton Cancer Institute Genetic Counseling Services at Norton Suburban Hospital in Louisville, Kentucky; and Jennifer Maahs, PNP, MSN, of the Indi-ana Hemophilia and Thrombosis Center (IHTC) in Indianapolis, Indiana.
n THE GENETIC BASIS OF HEMOPHILIA
Congenital deficiencies in factors VIII (FVIII; hemophilia A) and IX (FIX; hemophilia B) result from mutations of the F8 and F9 genes on the X chromo-some, respectively. Males have only one X chromosome, so a single altered copy of one of these genes in each cell is enough to cause hemophilia. Females, on the other hand, carry two X chromosomes, so women need a mutation in both cop-ies of the gene—one inherited from their mother and the other from their father—to develop the disease. Consequently, hemophilia is exceedingly rare in females.
Many types of mutations of F8 and F9 can lead to hemophilia. Further, the type of mutation can predict disease severity. Deletion and insertion mutations and mutations that cause premature termi-
nation of synthesis usually cause severe disease. Certain missense mutations also can lead to severe hemophilia. More commonly, missense mutations cause mild or moderate disease; in fact, 90% of patients who have mild-to-moderate hemophilia A have missense mutations. The majority of patients with hemo-philia B also have missense mutations.
In hemophilia A, the most com-mon genetic mutation is the intron 22 inversion mutation, which accounts for approximately 45% of all cases.4 This mutation arises from a folding over of the tip of the X chromosome, which leads to a homologous recombination. When the tip unfolds, some exons (nucleotide sequences) are oriented in the opposite direction, or inverted.4 Interestingly, most inversions originate during male meiosis (ie, in the maternal grandfather).5 As a result, almost all mothers of patients with inversion mutations are carriers of hemophilia.
n WHO SHOULD BE TESTED AND HOW?
Understanding the most frequently encountered genetic mutations in pa-tients with hemophilia can help to guide diagnostic testing. Patients with a severe FVIII deficiency are first screened for the intron 22 inversion mutation (Figure 1).5 If they test negative for this mutation, they undergo gene analysis. Patients with mild disease and hemophilia B undergo gene analysis. Currently, the Medical and Scientific Advisory Committee (MASAC) for the National Hemophilia Foundation recommends that all patients
Genetics of Hemophilia: The Role of Genetic Testing and the Use of Genetics to Guide TreatmentAnna Chalmers, MDRush University Medical Center, Chicago, Illinois
Abstract Advances in genetics have led to the isolation and characterization of the genes responsible for hemophilia. The role of hemophilia treatment centers now commonly includes genetic counseling and testing. The ability to individually specify genotype in affected patients may lead to more effective individualized treatment plans.
Dr. Chalmers is a Fellow in Hematology/Oncology at Rush University Medical Center, Chicago, Illinois.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 23
AnnaChalmers,MD Genetics of Hemophilia: The Role of Genetic Testing and the Use of Genetics to Guide Treatment
FIGURE 1 Algorithm for genetic testing in patients with hemophilia A and B. Adapted, with permission, from Pruthi.5
whether they carried null or non-null mutations. Patients with null mutations experienced their first hemarthrosis at a younger age, even though the difference was only 2 months. In the future, genetic characterization may allow individual-ized treatment.
Characteristics of Hemophilia B (Christmas Disease)
• Caused by a mutation in the gene encoding coagulation factor IX (F9) on chromosome Xq27.1• Phenotypically indistinguishable from hemophilia A; on blood testing, however, hemophilia B is
associated with a prolonged activated partial thromboplastin time and a normal prothrombin time.
• A distinction has been made between cross-reactive material (CRM)-negative and CRM-positive hemophilia B mutants based on the detection of F9 antigen in plasma even in the presence of decreased F9 activity. About 90% of hemophilia B patients are CRM-negative.
• Treatment for factor IX deficiency involves replacement of the missing factor by transfusion of plasma from a healthy person or infusion of a recombinant factor IX product.
• A subset of patients develops immunoglobulin G antibodies against normal factor IX, which complicates therapy.
Source: McKusick and Harnosh2
Characteristics of Hemophilia A (Classic Hemophilia)
• Caused by a mutation in the gene encoding coagulation factor VIII (F8) on chromosome Xq28• Clinically heterogeneous, with severity dependent upon plasma levels of factor VIII (FVIII)
— Mild: FVIII levels 6%–30% of normal; excessive bleeding only after trauma or surgery; experienced by 40% of patients
— Moderate: FVIII levels 1%–5% of normal; experienced by 10% of patients— Severe: FVIII levels < 1% of normal; an average of 20–30 episodes/year of spontaneous
and/or excessive bleeding, particularly into the muscles and joints, may occur after minor trauma; experienced by 50% of patients
• Joint involvement causes swelling, pain, decreased function, and degenerative arthritis.• Muscle hemorrhage can cause necrosis, contractures, and neuropathy by entrapment.• Hematuria (occasional) is usually painless. • Intracranial hemorrhage is uncommon but can occur after mild head trauma and lead to severe
complications.• Persistent bleeding from tongue or lip lacerations may occur.
Source: McKusick and Kniffin1
with hemophilia have genotype test-ing. An ongoing initiative is creating a comprehensive de-identified database of these mutations.6
Appropriate family members of hemo-philia patients also can undergo carrier testing. Analysis of data from patients undergoing pretesting counseling found that approximately 30% of cases of hemo-philia are sporadic.5 Despite this finding, most carriers are unaware of their status and require supportive counseling.
Most carriers are asymptomatic. In unusual circumstances, such as cases of Turner syndrome, some carriers may have low enough levels of a blood factor to cause clinical bleeding. For most pa-tients, though, carrier status only affects preconception counseling.
TestingaCarrierTo test a possible carrier, it is best to
target a known mutation. If the mutation is unknown, then the approach is simi-lar to that used for testing patients with hemophilia.
Patients are first screened for intron 22 inversion mutations, the most common overall cause of unspecified hemophilia.5 If the result is negative, then the whole gene is screened. Once carrier status is confirmed, patients can be counseled regarding family planning options, in-cluding preimplantation genetic testing. Patients who are carriers and pregnant with male fetuses need to be followed by a multidisciplinary team of specialists throughout pregnancy to address any possible perinatal testing and ensure a safe delivery.7
n USE OF GENETICS TO GUIDE TREATMENT
Traditionally, analysis of factor levels and clinical presentation have guided the treatment course for hemophilia. However, a small percentage of patients with severe disease, as defined by coagu-lation factor level, have a milder clinical presentation.8 Therefore, there has been some interest in using genetic informa-tion to help guide management of the disease. For example, based on the results of genetic testing, a healthcare provider
may consider inserting a central line ear-lier in some patients or delaying primary prophylaxis in others.
To take a closer look at the value of genetic testing, Carcao and colleagues9 examined 621 previously untreated pa-tients and classified them according to
Test for intron 22inversion mutations Negative
Severe disease Mild/moderatedisease
Gene analysis
Patients with hemophilia A Patients with hemophilia B

24 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
AnnaChalmers,MD Genetics of Hemophilia: The Role of Genetic Testing and the Use of Genetics to Guide Treatment
Internet Resources for Hemophilia Information and Genetic Testing
• GeneReview:HemophiliaAhttp://www.ncbi.nlm.nih.gov/books/NBK1404/
• GeneReview:HemophiliaBhttp://www.ncbi.nlm.nih.gov/books/NBK1495/
• GeneticTestingRegistry:Hemophiliahttp://www.ncbi.nlm.nih.gov/gtr/conditions/C0684275/
• GeneticTestingRegistry:HemophiliaB(M)http://www.ncbi.nlm.nih.gov/gtr/conditions/CN043453/
• GeneticTestingRegistry:HereditaryFactorIXDeficiencyDiseasehttp://www.ncbi.nlm.nih.gov/gtr/conditions/C0008533/
• GeneticTestingRegistry:HereditaryFactorVIIIDeficiencyDiseasehttp://www.ncbi.nlm.nih.gov/gtr/conditions/C0019069/
• LabTestsOnline:BleedingDisordershttp://labtestsonline.org/understanding/conditions/bleeding-disorders
• MedlinePlusEncyclopedia:FactorIXAssayhttp://www.nlm.nih.gov/medlineplus/ency/article/003679.htm
• MedlinePlusEncyclopedia:FactorVIIIAssayhttp://www.nlm.nih.gov/medlineplus/ency/article/003678.htm
• MedlinePlusEncyclopedia:HemophiliaAhttp://www.nlm.nih.gov/medlineplus/ency/article/000538.htm
• MedlinePlusEncyclopediaHemophiliaBhttp://www.nlm.nih.gov/medlineplus/ency/article/000539.htm
• NationalHeart,Lung,andBloodInstitute:HowIsHemophiliaDiagnosed?http://www.nhlbi.nih.gov/health/health-topics/topics/hemophilia/diagnosis.html
• NationalHeart,Lung,andBloodInstitute:HowIsHemophiliaTreated?http://www.nhlbi.nih.gov/health/health-topics/topics/hemophilia/treatment.html
• NationalHemophiliaFoundation:HemophiliaTreatmentCentershttp://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=203&contentid=385
Source: Genetics Home Reference Web site3
Resources for Patient Support
• CanadianHemophiliaSocietyhttp://www.hemophilia.ca
• HemophiliaFederationofAmericahttp://www.hemophiliafed.org
• NationalHemophiliaFoundationhttp://www.hemophilia.org
• NationalOrganizationforRareDisorders(NORD):HemophiliaAhttp://www.rarediseases.org/rare-disease-information/rare-diseases/byID/39/viewFullReport
• NationalOrganizationforRareDisorders(NORD):HemophiliaBhttp://www.rarediseases.org/rare-disease-information/rare-diseases/byID/480/viewFullReport
• ResourcelistfromtheUniversityofKansasMedicalCenterhttp://www.kumc.edu/gec/support/hemophil.html
• WorldFederationofHemophiliahttp://www.wfh.org/en/page.aspx?pid=492
Source: Genetics Home Reference Web site11
DevelopmentofInhibitorsMost patients with the inhibitor phe-
notype have large alterations of the F8 or F9 gene (eg, large deletions, nonsense mutations, frameshift mutations). A small percentage of patients with the inhibitor phenotype can develop anaphylaxis to replacement factor, a life-threatening, difficult-to-manage clinical situation.
Thorland et al10 genotyped eight patients with hemophilia B who had experienced anaphylaxis when exposed to FIX therapy and compared them with patients with severe disease. Those who had complete gene deletions were at the highest risk of developing anaphylaxis.
n SUMMARY
Recognition that patients with hemo-philia are at high risk may impact health-care decisions. For example, patients at high risk of anaphylaxis may need to be
infused in the supervised clinic setting instead of receiving treatment at home. Genetic testing remains expensive, but used judiciously, it has the potential to improve the management of patients with hemophilia.
REFERENCES
1. McKusick VA, Kniffin CL. #306700: Hemo-philia A; HEMA. Online Mendelian Inheritance in Man (OMIM) Web site. March 14, 2013. http://omim.org/entry/306700. Accessed February 20, 2014.
2. McKusick VA, Harnosh H. #306900: Hemo-philia B; HEMB. Online Mendelian Inheritance in Man (OMIM) Web site. February 29, 2012. http://omim.org/entry/306900. Accessed February 20, 2014.
3. Hemophilia. Genetics Home Reference Web site. August 2012. http://ghr.nlm.nih.gov/condition/hemophilia. Accessed Accessed February 20, 2014.
4. Lakich D, Kazazian HH, Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. 1993;5:236–241.
5. Pruthi RK. Hemophilia: a practical approach to
genetic testing. Mayo Clin Proc. 2005;80:1485–1499. 6. MASAC recommendation #214: MASAC
recommendations on the NHF Genotyping Project for persons with hemophilia. National Hemo-philia Foundation Web site. November 11, 2012. http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=57&contentid=1996. Ac-cessed February 20, 2014.
7. MASAC recommendation #192: MASAC guidelines for perinatal management of women with bleeding disorders and carriers of hemo-philia A and B. National Hemophilia Founda-tion Web site. November 1, 2009. http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=57&contentid=1436. Accessed Feb-ruary 20, 2014.
8. Santagostino E, Mancuso ME, Tripodi A, et al. Severe hemophilia with mild bleeding phenotype: molecular characterization and global coagulation profile. J Thromb Haemost. 2010;8:737–743.
9. Carcao MD, van den Berg HM, Ljung R, Mancuso ME. Correlation between phenotype and genotype in a large unselected cohort of children with severe hemophilia A. PedNet and the Rodin Study Group. Blood. 2013;121:3946–3952, S1.
10. Thorland EC, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in haemophilia B patients: complete gene deletions confer the highest risk. Haemophilia. 1999;5:101–105.
11. Hemophilia: Patient support—for patients and families. Genetics Home Reference Web site. August 2012. http://ghr.nlm.nih.gov/condition/hemophilia/show/Patient+support. Accessed Feb-ruary 20, 2014.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 25
During the 2013 Annual Meet-ing of the National Hemo-philia Foundation, research-ers presented the results of
their studies on development of novel therapeutics, gene-transfer therapies, and new technologies for lengthening factor half-lives and battling inhibitors against factor replacement concentrates. In addition, experts in hemophilia and its treatment discussed findings on improv-ing outcomes following total knee and hip replacement in affected patients and the influence of obesity on compliance with prophylactic infusion regimens.
n PRECLINICAL RESEARCH
Phospholipid-BindingAffinityofFactorIX(FIX)
Deficiency of FIX causes hemophilia B. Replacement of FIX with plasma-derived or recombinant factor concentrates is the key step in preventing bleeding episodes but is hampered by the short half-life (approximately 24 hours) of currently available FIX products, requiring frequent infusion. Consequently, researchers are investigating ways to extend their half-life without jeopardizing their safety or efficacy.
FIX is a member of a family of vitamin K–dependent proteins that contain mul-tiple gamma-carboxyglutamic acid (Gla)
residues in homologous amino-terminal Gla domains. Gla allows these proteins to bind to calcium and phospholipids. Of the vitamin K–dependent coagulation factors, FIX has one of the lowest phospholipid-binding affinities, which is thought to shorten its half-life.
Harvey and coworkers1 purposefully mutated three specific amino acids (Y1A, G4Y, and K5L) in the amino-terminal of the FIX protein that were expressed in a human embryonic kidney cell line (K293). Effects of these changes on phospholipid-binding affinity were examined individu-ally and in combination.
The “triple mutant” resulted in a 27-fold increase in phospholipid-binding affinity. The mutation Y1A showed no benefit over wild-type protein, whereas the mutation G4Y resulted in a 2.5-fold increase in binding affinity alone and a 4.3-fold increase when combined with the mutation Y1A. Thus, synergy of the different mutations, G4Y and Y1A, was suggested. The mutation K5L led to an improvement in phospholipid-binding affinity that was not affected by mutations at the other two sites.
The ability to modify the binding af-finity of FIX proteins to phospholipid membranes has attractive implications for the development of novel replacement coagulation factors with a significantly
longer half-life, resulting in less-frequent dosing.
BindingofFactorVIII(FVIII)tovonWillebrandFactor
Binding of FVIII to von WiIlebrand factor plays an important role in the bio-logic activity and clearance of FVIII and influences its presentation to the immune system.
Claar et al2 compared the affinity of a novel, recombinant single-chain factor VIII (rFVIII-SingleChain; CSL627) with that of recombinant full-length FVIII (rFVIII) to purified, plasma-derived von WiIlebrand factor (pdVWF) in vitro. The affinity of CSL627 for pdVWF was significantly higher than that of com-mercially available, full-length rFVIII proteins. Dissociation-rate constants of the two molecules were comparable, which suggested similar bioavailability. Other in vitro characteristics of the novel agent (FVIII enzymatic activity, thrombin generation, and ability to bind to phos-pholipids) were comparable to those of full-length rFVIII.
Systemic availability and mean resi-dence time were higher in hemophilia A mice treated with single doses of CSL627 compared with full-length rFVIII. Addi-
Expanding Therapeutic Options for Hemophilia A and B: Results of Recent Trials and Research Holleh D. Husseinzadeh, MD
Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania
Abstract An overview of current research on bleeding disorders illustrates the vast spectrum of investigative interest. During the 2013 Annual Meeting of the National Hemophilia Foundation, researchers presented the results of a broad variety of studies related to a basic understanding of these diseases in specific patient populations, preclinical experimentation, and the clinical use of current and promising future prophylactic and therapeutic modalities.
Dr. Husseinzadeh is a Fellow in Hematology and Medical Oncology at the University of Pennsylvania, Philadelphia, Pennsylvania.

26 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
HollehD.Husseinzadeh,MD Expanding Therapeutic Options for Hemophilia A and B: Results of Recent Trials and Research
tionally, a decrease in clearance rate and an increase in terminal plasma half-life were observed in mice that had received CSL627 versus full-length rFVIII. The vol-ume of distribution and in vivo recovery of the the two molecules were similar.2
This novel recombinant molecule is a promising new agent being developed to treat hemophilia A. Its higher infinity to pdVWF may delay its elimination from plasma and positively affect its systemic availability.
n CLINICAL TRIALS
BAX 855 is a PEGylated recombinant FVIII product designed to prolong the half-life of commercially available octocog alfa via covalent binding of polyethylene glycol (PEG) moieties. Covalent attach-ment of PEG moieties decreases the systemic clearance of rFVIII, theoretically without affecting its efficacy and safety.
Bevan and others3 assessed the effi-cacy and safety of BAX 855 in previously treated patients with severe hemophilia A and compared the pharmacokinet-ics of this novel compound with those of octocog alfa. In all, 24 patients with hemophilia A who had been exposed to rFVIII for at least 150 days and who had no history of inhibitors were enrolled into this multinational, open-label, phase 1 study. A total of 19 participants completed the study. Patients first received a single dose of octocog alfa and then underwent pharmacokinetic evauation. After a wash-out period, the patients received either 30 IU/kg (cohort 1) or 60 IU/kg (cohort 2) of BAX 855, followed by 7 days of pharmaco-kinetic monitoring. Investigators followed the patients for 4 weeks after they received BAX 855 for safety assessments (adverse events, changes in vital signs, and labo-ratory test results) and immunogenicity (development of FVIII inhibitors; binding antibodies to FVIII, BAX 855, and PEG).
The mean half-life of rFVIIII was 1.4- to 1.5-fold higher for BAX 855 than for oc-tocog alfa in cohorts 1 and 2, respectively. Measurement of thrombin generation showed that mean peak thrombin con-centration was increased above baseline for more than 120 hours after infusion of BAX 855 at 60 IU/kg (cohort 2). Other
pharmacokinetic parameters were similar to, or better than, those of octocog alfa. No serious or treatment-related adverse events were recorded in either cohort. In addition, none of the patients developed FVIII inhibitors or experienced throm-botic or allergic events or significant changes in their vital signs or laboratory measurements.
These results suggested that BAX 855 has a longer half-life than that of octocog alfa, potentially allowing less-frequent dosing while offering similar efficacy and safety. Investigators affiliated with the PROLONG-ATE study currently are enrolling patients for phase 2/3 studies of BAX 855.
Gene-TransferTherapyandBaselineFIXLevels
Reiss and colleagues4 presented interim results from an ongoing phase 1/2 clinical trial in FIX (F9) gene transfer using an adeno-associated virus (AAV) as a vector. A total of 10 adults with severe hemophilia B have received the gene-transfer therapy thus far. The ideal concentration of factor has not yet been determined; two patients received a low dose, two received an in-termediate dose, and six received a high dose of the F9-containing vector.
Post-infusion follow-up currently ranges from 0.5 to 3.3 years. Overall, patients have experienced significant im-provements in disease status. Of the four patients given low- or intermediate-dose vector, baseline FIX levels have remained
between 1% and 3%. This resulted in pro-longed intervals between prophylactic fac-tor infusions in one patient and eliminated the need for prophylactics in two patients. The fourth patient remains on factor pro-phylaxis due to severe hemarthropathy. All six patients given high-dose therapy had postinfusion baseline FIX levels > 3% and an eliminated need for factor prophylaxis.
No serious adverse events were ob-served in the low- or intermediate-dose groups. In the high-dose group, acute and asymptomatic transaminitis was observed 7–9 weeks post infusion. This inflamma-tion likely resulted from capsid-specific T-mediated immunity against hepato-cytes, which preferentially take up the protein vector. In all cases, transaminitis resolved completely after a short course of oral prednisolone, whereas F9 expression was maintained.
The exciting results of this trial repre-sented the first successful gene-transfer therapy in hemophilia resulting in long-term expression of a deficient F9 gene. The trial is reopening and is actively recruit-ing adults (age > 18 years) with severe hemophilia B.
ExtendingFactorHalf-LifewithFusionProteinTechnology
Currently available formulations of rFVIII given to treat hemophilia A have short half-lives (approximately 12 hours). Thus, to prevent and treat bleeding epi-sodes, frequent injections of these agents are needed. Combined use of crystallizable fragment (Fc) fusion protein technology with rFVIII results in an rFVIIIFc fusion protein with an extended half-life (mean, 19 hours). Mahlangu et al5 discussed the A-LONG study, which assessed the ef-ficacy, safety, and pharmacokinetics of rFVIIIFc when given for the prophylaxis and treatment of hemophilia A.
Study participants included 165 previ-ously treated males ≥ 12 years of age who had severe (< 1 IU/dL) hemophilia A. Patients in arm 1 received individualized prophylaxis with pharmacokinetic-driven adjustment of dose and interval, those in arm 2 received weekly prophylaxis at a constant dose, and those in arm 3 received episodic treatment as needed for bleeding
The exciting results of this trial represented the first successful gene-transfer therapy in hemophilia resulting in long-term expression of a deficient F9 gene.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 27
HollehD.Husseinzadeh,MD Expanding Therapeutic Options for Hemophilia A and B: Results of Recent Trials and Research
events. Dosing intervals achieved in arm 1 were ≥ 3.5 days, with 30% of patients achieving a dosing interval of 5 days during the last months of the study. The average number of bleeding episodes was 1.6 in arm 1, 3.6 in arm 2, and 33.6 in arm 3. Administration of just one injection resolved 87.3% of bleeding episodes. None of the participants developed inhibitors to rFVIIIFc. Excellent or good control of bleeding was seen in patients who received rFVIIIFc and underwent major surgery during the study. Adverse events were similar to those expected among the general hemophilia A population; no patient developed inhibitors to rFVIIIFc.
This was the first clinical study of rFVIIIFc fusion protein in patients with hemophilia A. Some prolongation of the dosing interval and other preliminary results suggested that rFVIIIFc offers a safety profile similar to that of existing recombinant factor agents.
ExaminingRecombinantFcFusionProteinTechnologyinHemophiliaB
Available FIX replacement products for patients with hemophilia B have relatively short half-lives. As a result, they require frequent self-injection, which is often disruptive for patients and may lead to decreased compliance and hemophilia-related complications. A novel recom-binant FIX Fc fusion protein (albumin; rFIXFc) with an extended half-life has been developed to help reduce the burden of frequent prophylactic injections.
In the B-LONG study,6 one of the larg-est clinical studies conducted in patients with hemophilia B, investigators assessed the safety, efficacy, and pharmacokinet-ics of rFIXFc given to prevent and treat bleeding episodes. The study involved 123 previously treated males ≥ 12 years of age who were diagnosed with severe he-mophilia B (< 2 IU/dL). Patients received weekly prophylaxis with pharmacokinet-ics-driven rFIXFc dose adjustment (arm 1), individualized interval prophylaxis with pharmacokinetics-driven interval adjustment (arm 2), on-demand admin-istration for acute bleeding episodes (arm 3), or factor management for anticipated surgery (arm 4).
The mean half-life for rFIXFc was 82.1 hours. In arm 2, the dosing interval in all patients was ≥ 7 days, but over one half of these patients were able to be maintained on a dosing interval ≥ 14 days. The median number of bleeding episodes per year was 3.0 in arm 1 and 1.4 in arm 2; these rates were much lower than the median 17.7 episodes per year observed in arm 4. A single dose of rFIXFc was sufficient to control 90.4% of bleeding episodes in arm 3. In arm 4, bleeding control was rated as good or excellent with all major surgeries in study participants. rFIXFc was well tolerated and caused no major side effects or development of inhibitors in study participants.
Thus, the preliminary results from this first clinical study of an rFIXFc fusion protein in hemophilia B patients showed this agent to be well tolerated. Its use led to a markedly increased dosing interval, which may lead to greater compliance, decreased hemophilia-related complica-tions, and improved quality of life.
ProphylaxisinHemophiliawithInhibitors
Factor Eight Inhibitor Bypass Activity Nanofiltered (FEIBA NF; anti-inhibitor coagulant complex) is an activated pro-thrombin complex concentrate used to control spontaneous bleeding episodes or to cover surgical interventions in pa-tients with hemophilia A or B who have developed inhibitors to factor replace-ment concentrates. Antunes and others7 investigated whether prophylactic use of FEIBA NF could be as beneficial and safe as on-demand use.
In this prospective, randomized study, 36 patients with hemophilia A or B who had developed inhibitors against replace-ment factors were randomized to receive either prophylaxis or on-demand regi-mens of FEIBA NF for 12 months. In all, 17 patients were randomized to receive a prophylactic regimen of 85 ± 15 U/kg of FEIBA NF every other day. The remaining 19 patients were given on-demand FEIBA NF as directed by their treating physicians for acute bleeding events.
The results are summarized in Table 1. Patients receiving FEIBA NF prophylacti-cally experienced 196 bleeding episodes, whereas those receiving it on demand had 629 bleeding episodes. The median annualized bleeding rates in the prophy-lactic FEIBA NF arm were significantly lower than those in the on-demand arm (7.9 vs 28.7) in both the intent-to-treat and per-protocol efficacy analysis datasets (P = 0.0003 and P = 0.006, respectively). In addition, the frequency of new target joint bleeds was significantly lower in the prophylactic arm (7 new joint bleeds in 5 of 17 patients), when compared with their frequency in the on-demand arm (23 new joint bleeds in 11 of 19 patients; P = 0.027).
A total of 104 adverse events were reported, including 30 serious and 74 nonserious events. In all, 27 events (26%) were believed to be related to FEIBA NF therapy, including one non-serious hyper-sensitivity reaction. No thromboembolic events occurred. At termination of the study, seven previously negative patients tested positive for hepatitis B surface anti-body (HBsAb), although all were negative for hepatitis B core antibody, hepatitis
TABLE 1Efficacy and Safety of Prophylactic or On-Demand FEIBA NF in Patients with Hemophilia A or Ba
Parameter Prophylaxis(n=17)b On-demand(n=19)c P value
Bleeding episodes 196 629 –Annualized bleeding rate (ABR), median 7.9 28.7 –Occurrence of new target joints 7 in 5/17 patients 23 in 11/19 patients –New target joint ABR, median 0 5.9 0.027
FEIBA NF= Factor Eight Inhibitor Bypass Activity Nanofiltereda Patients (median age, 23 years) with hemophilia A or B had inhibitors against factor replacement concentrates that were refractory to factor VIII or IX.b Received 85 ± 15 U/kg of FEIBA NF every other day for 12 months ± 14 daysc Received FEIBA NF for control of acute bleeding at dosages determined by the treating physicians Source: Antunes et al7

28 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
HollehD.Husseinzadeh,MD Expanding Therapeutic Options for Hemophilia A and B: Results of Recent Trials and Research
B surface antigen, and hepatitis B virus DNA. These findings seemed to result from passive transmission of HBsAb from the donor blood product. One patient in the prophylaxis arm and two patients in the on-demand arm experienced a rise in inhibitor level.
Thus, prophylactic, recurrent admin-istration of FEIBA NF appeared in this study to be as safe as on-demand therapy while significantly reducing all types of bleeds by 72.5% in patients with hemo-philia A or B who had developed inhibi-tors to factor replacement concentrates. Two of the patients who received FEIBA NF prophylactically for 12 months had no bleeding episodes.
PatientAdherencetoFactorInfusionTherapy
The relationship between compliance with factor infusion therapy and specific health outcomes is not well documented. Vietri and colleagues8 assessed the rela-tionship between adherence to prophylac-tic factor infusions in adults and children with moderate-to-severe hemophilia A or B and outcomes, including bleeding events and patient-reported health status.
Adult hemophilia patients or parents of minors with hemophilia were identified through databases at hemophilia treatment centers. A total of 53 adults with moder-ate or severe hemophilia A (n = 43) or B (n = 10) and 56 children with hemophilia receiving prophylaxis participated in the study. Study participants completed online questionnaires, which included an adher-ence assessment, using the Validated He-mophilia Regimen Treatment Adherence Scale–Prophylaxis (VERITAS-Pro), and a self-reported evaluation of health status, using the 12-item Short-Form Health Sur-vey (SF-12v2) for adults and the SF-10 for parents of pediatric patients. Participants in the study were also asked to provide the numbers of breakthrough bleeding events, emergency department visits, hospital admissions, and missed days of work or school they or their children experienced due to bleeding episodes.
In adult patients with hemophilia A or B, lower adherence was associated with an increased number of bleeding episodes
requiring administration of replacement factors (P < 0.001), as well as more days of work or school missed in the past year be-cause of bleeding episodes (P < 0.05). No significant association between adherence and self-reported health status was found among adults (P = 0.91), but increased adherence in children was associated with better physical health (P < 0.01). In pediatric patients, adherence was not significantly associated with the reported number of bleeding episodes (P = 0.95), but it was significantly associated with infection at the injection site (P < 0.05), increased hospital stays for bleeding epi-sodes (P < 0.001), and missed work/school days due to bleeding episodes (P < 0.01).
Although the sample size was limited, these results showed that greater adher-ence to a prophylactic factor replacement regimen by both adults and children with moderate-to-severe hemophilia A or B was associated with significantly better clinical outcomes.
ImprovingOutcomesAfterTotalJointReplacement
Goto et al9 presented a retrospective review of subjective and objective out-comes in patients with congenital bleed-ing disorders after they received total joint arthroplasty (TJA) at a single institution. The goals were assessment of outcomes of TJA for hemophilic arthropathy and the safety and efficacy of standard phar-macologic thromboprophylaxis in this population.
A retrospective chart review of 28 patients with various congenital bleed-ing disorders, including hemophilia A (n = 21), hemophilia B (n = 4), factor-11 deficiency (n = 1), and von Willebrand disease (n = 2), yielded outcomes data from 38 arthroplastic procedures, includ-ing 29 instances of total knee arthroplasty (TKA) and 9 cases of total hip arthroplasty (THA).
Outcomes are summarized in Table 2.9 Objective postoperative clinical outcome data at 2 months were available for 27 of the 28 patients. Seven of the 28 patients (25%) had improvement in range of mo-tion (median, 15°; range, 5°–25°]. At 1.5 years postoperatively, 17 of 29 TKA pa-
tients (59%) experienced improved range of motion (median, 15°; range, 15°–45°), and 100% had decreased knee pain. Two months postoperatively, all nine THA patients had improved range of motion, including internal rotation (89%; median, 45°), external rotation (100%; median, 30°), flexion (56%; median, 35°), exten-sion (78%; median, 15°), and abduction (78%; median, 15°).
Twenty-two of the 28 study patients could be contacted for subjective re-sponses to arthroplasty. All 25 patients undergoing TKA reported significant improvement in pain; 24 of the 25 patients (96%) reported improvement in joint function after joint replacement, and all 25 would elect to repeat the surgery if asked to make the choice again. All six THA patients reported improved joint pain and function, and five of the six (83%) stated they would choose to have surgery if presented with the option again.
Low-molecular-weight heparin (LMWH) was used as thromboprophy-laxis in 29 of 38 procedures (76%), but it was discontinued in three patients for non-joint bleeding or suspected blood loss (two cases of hypotension and anemia). No symptomatic venothrombotic events occurred. Early complications of TJA in-cluded cellulitis in five patients and hem-arthrosis in two patients not on LMWH prophylaxis. Observed late complications of surgery included aseptic loosening of knee arthroplasties requiring repeated TKA, one case of septic arthritis, and development of severe flexion contracture requiring repeated TKA.
LinkingObesitywithDecreasedCompliance
Home infusion of clotting factors is important for proper prophylaxis and treatment of acute bleeding episodes in hemophilia. To be effective, patients need to have both an adequate supply of factor as well as the skill and ability to establish access for intravenous administration. This retrospective analysis, presented by Ullman and others,10 investigated whether an elevated body mass index (BMI) was associated with decreased use of factor home-infusion treatment (HI) and self-

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 29
HollehD.Husseinzadeh,MD Expanding Therapeutic Options for Hemophilia A and B: Results of Recent Trials and Research
infusion (SI) among patients with hemo-philia A or B in the United States.
The investigators analyzed data from 10,814 males aged 6–79 years old. Pa-tients with human immunodeficiency virus (HIV) infection, symptomatic liver disease, or an inhibitor titer > 5 Bethesda units were excluded from the analysis. The prevalence of HI and SI was recorded with demographic and clinical characteristics. Bivariate relationships were assessed using the χ2 test, and independent associations between BMI and HI/SI were evaluated by logistic regression.
Paralleling current trends in the US population, 50% of analyzed males in the study cohort were overweight or obese. In all, 70% of patients studied used HI; 44% of those also used SI. Overweight and obese individuals were less likely to use HI than were those of normal weight (odds ratio [OR] = 0.8; 95% confidence interval [CI] = 0.7–1.0 and OR = 0.7; 95% CI = 0.6–0.8, respectively.) Obese teens and adults also were less likely to participate in HI than were those of normal weight (OR = 0.8; 95% CI = 0.7–0.9). Among all patients, SI use declined after age 40 years, no matter what the patient’s sever-
ity of disease or prophylactic treatment regimen used.
These results suggest that overweight and obese persons with hemophilia A or B may be less likely to use HI or SI—per-haps because of the increased difficulty of performing venipuncture due to ex-cess adiposity—resulting in irregular-ity of prophylaxis, delayed treatment of bleeding episodes, and increased risk of hemophilia-related complications.
ArteriovenousFistulae(AVF)inPatientswithBleedingDisorders
Venous access is a major issue in facilitating factor administration in pa-tients with bleeding disorders. Tapia and others11 reported on long-term follow-up of 17 such patients, including 2 patients with von Willebrand disease, 12 with hemophilia A (3 with inhibitors), and 3 with hemophilia B (1 with an inhibi-tor) who had AVF inserted for venous access. At a mean follow-up of 5 years (range, 1–15 years), 15 patients reported “excellent” results, with continued fis-tula viability. No patients had bleeding complications, AVF-related infection, or difficulty achieving venous access for
administration of factor replacements. Four patients (24%), however, reported dissatisfaction with the appearance of the AVF, resulting in one revision procedure with improvement of appearance.
AVF are a viable option associated with overall satisfaction in patients with bleeding disorders who require long-term venous access. This procedure may be considered in individuals with difficult access or those who have had repeated complications with existing modes of ac-cess. However, AVF are associated with a measurable rate of dissatisfaction in cosmetic appearance.
DeterminingFoodInsecurityinHemophiliaPatients
Ziha et al12 sought to identify the prevalence of food insecurity in families of hemophilia patients at a single treatment center. Patients were screened during an-nual comprehensive visits from May 2012 to January 2013 using a two-question, validated screening tool along with their general health assessments.
Data from 42 male children (age, 0–18 years) were analyzed. The overall preva-lence of food insecurity was 16.7%, which was similar to that of the national aver-ages. Food insecurity was less prevalent among those with mild or moderate dis-ease (5.6%) than among those with severe hemophilia (25.0%; 95% CI = 7.7–42.3). Children who tended to be at increased risk of food insecurity were older, taller, or heavier than the other children; had a higher BMI; or belonged to a minority; however, none of these characteristics had a significant impact (P > 0.05) on food insecurity in this small sample population. Nevertheless, these results highlight the need for food-insecurity screening among hemophilia patients and their families and connection of these families to appropriate community resources.
REFERENCES
1. Harvey S, Kirihara J, Nelsestuen G. Enhanced properties of blood clotting factor IX variants with elevated membrane affinity. Presented at the 65th Annual Meeting of the National Hemophilia Foun-dation; October 3–5, 2013; Anaheim, California. Abstract BCR08.
TABLE 2Outcomes of Total Knee and Hip Arthroplasties for Hemophilic Arthropathya
Totalkneearthroplasty(n=29)Age in years at operation, median 42 (range, 17–74) Results at 2 months postoperatively, n (%) Number of patients 27 Improved range of motion 7 (23%)Results at 1.5 years postoperatively, n (%) Number of patients 29 Improved range of motion 17 (59%) Decreased knee pain 29 (100%)Totalhiparthroplasty(n=9)Age in years at operation, median 45 (range, 18–71) Results at 2 months postoperatively, n (%) Number of patients 9 Improved range of motion 9 (100%) Gains in internal rotation (median, 45°; range, 15°–45°) 8 (89%) Gains in external rotation (median, 30°; range, 15°–45°) 9 (100%) Gains in flexion (median, 35°; range, 27°–55°) 5 (56%) Gains in extension (median, 15°; range, 3°–95°) 7 (78%) Gains in abduction (median, 15°; range, 10°–25°) 7 (78%)
a Procedures were performed in 26 males and 2 females with hemophilia A (n = 21), hemophilia B (n = 4), factor 11 deficiency (n = 1), or von Willebrand disease (n = 2); all patients were treated with hemostatic agents appropriate to their disorders for up to 4–6 weeks postoperatively.Source: Goto et al9

30 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
HollehD.Husseinzadeh,MD Expanding Therapeutic Options for Hemophilia A and B: Results of Recent Trials and Research
2. Claar P, Röder j, Zollner S, Weimer T, Dick-neite G, Schulte S. Characterization of the binding of a novel recombinant single-chain FVIII to von Willebrand factor. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, California. Abstract BCR16.
3. Bevan D, Conlan M, Mant T, et al. A phase 1 study of safety and pharmacokinetics of BAX 855, a longer-acting pegylated full-length recombinant factor VIII in patients with severe hemophilia A. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Ana-heim, California. Abstract CR40.
4. Reiss UM, Nathwani AC, Tuddenham EGD, et al. Prolonged factor IX expression after AAV-mediated gene transfer in adults with severe hemo-philia B. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, California. Abstract CR24.
5. Mahlangu J, Powell J, Ragni M, et al. A-LONG: Phase 3 study of long-lasting recombinant factor VIII Fc fusion protein (rFVIIIFc) in hemo-
philia A. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, California. Abstract CR38.
6. Powell J, Ragni M, Valentino L, et al. B-LONG: Phase 3 study of recombinant factor IX Fc fusion protein (rFIXFc-FP) in hemophilia B. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Ana-heim, California. Abstract CR39.
7. Antunes SV, Tangada S, Stasyshyn O, et al. FEIBA PROOF: A prospective, open-label, random-ized, parallel study with FEIBA NF to evaluate the efficacy and safety of prophylactic versus on-demand treatment in hemophilia A or B subjects with in-hibitors. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, California. Abstract CR41.
8. Vietri J, Furlan R, Krishnan S. Adherence and outcomes in hemophilia. Presented at the 65th Annual Meeting of the National Hemophilia Foun-dation; October 3–5, 2013; Anaheim, California. Abstract SPI56.
9. Goto Y, Holderness B, McKernan L, Bernini
P, Ornstein D. Outcomes of total knee and hip ar-throplasty for hemophilic arthropathy. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, Califor-nia. Abstract OTM51.
10. Ullman M, Zhang QC, Brown D, Soucie JM. Association of obesity with decreased compliance in home factor infusion among males with hemophilia. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Ana-heim, California. Abstract OTM47.
11. Tapia C, Tovar-Herrera M, Urgo M, et al. Long-term follow-up of arteriovenous fistulae in bleeding disorders. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, California. Abstract CR35.
12. Ziha S, Adams E, Black A, Stadler D, Recht M. Prevalence and predictors of food insecurity in children with hemophilia. Presented at the 65th Annual Meeting of the National Hemophilia Foun-dation; October 3–5, 2013; Anaheim, California. Abstract CR25.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 31
Hemophilia A and B are heredi-tary, X chromosome–linked, recessive bleeding disorders caused by mutations in genes
for factor VIII (FVIII; F8) and factor IX (FIX; F9). Both factors are essential components in the coagulation pathway. The prevalence of hemophilia commonly is reported as 1:5,000 live male births for hemophilia A and 1:30,000 for hemo-philia B.1
Hemophilia classification is based on factor activity levels and range from mild (6%–40% activity) and moderate (1%–5% activity) to severe (< 1% activity). The bleeding phenotype generally correlates with the level of factor deficiency. Pa-tients with severe hemophilia experience bleeding episodes involving joints, soft tissues, and muscles. Repetitive bleed-ing into joints can lead to debilitating chronic arthropathy with limited joint mobility, flexion contracture, and muscle wasting.1–5
This report on the present and future of hemophilia management is based on a presentation by Steven W. Pipe, MD, Professor of Pediatrics and Director of the Division of Pediatric Hematology/
Oncology at the University of Michigan Health System in Ann Arbor. Dr. Pipe spoke during a symposium sponsored by Biogen Idec at the 65th Annual Meeting of the National Hemophilia Foundation in Anaheim, California.
n CURRENT THERAPY OF HEMOPHILIA
Current therapy of hemophilia is based on the replacement of missing clotting factors. Factor is derived from either donated human plasma or the more recent and favored recombinant DNA technology.6
Prophylactic factor administration, the current standard of care for severe hemophilia, is started early in life, pref-erably before the onset of hemarthroses.5 Prophylactic FVIII commonly is given at a dosage of 25–50 IU/kg every other day or 3 days/week, assuming an average re-covery of 2 IU/dL for each IU/kg infused. Prophylactic FIX commonly is given at a dosage of 25–40 IU/kg twice weekly, assuming an average recovery of 1 IU/dL for each IU/kg infused.4
Current factor products have limita-tions. First, the short half-lives of 8–12
hours for FVIII and 18–24 hours for FIX require frequent dosing in prophylactic settings and repeat dosing in on-demand settings.4 Second, the development of neutralizing antibodies (inhibitors) renders factor replacement ineffective; these inhibitors develop in up to 30% of patients with hemophilia A and 3%–5% of patients with hemophilia B.2 Third, current therapies require venous access, which may necessitate the placement of central venous access devices in young children. These devices carry the risk for infection and thrombosis.5 Fourth, the cost of current therapies limits their avail-ability to many patients with hemophilia worldwide.6
Results of an investigation of health-care resource use by Valentino et al7 showed that the median annual hemo-philia A–related costs from 2001 to 2007 in the United States were $63,935. This cost increased significantly to a median of $271,357 annually in patients who developed inhibitors. The median annual cost increased significantly from birth to about 15 years of age; thereafter, costs remained stable before dropping again at 21 years of age. The current annual cost of prophylactic factor use is over $150,000.8 Because of this, it is estimated that two thirds of the world do not have proper access to replacement therapies.4,5
Treatment of Hemophilia: What’s in the Pipeline?Kerry Hege, MDIndiana University School of Medicine, Indianapolis, Indiana
Abstract Current management of hemophilia is based on factor-replacement therapy. Because plasma-derived and recombinant factor products have short half-lives, they demand frequent dosing, which, in turn, leads to issues with venous access, inhibitor development, and high financial cost. Bioengineering strategies are bringing new and promising products down the pipeline that may improve patient outcomes via more effective bleeding control and preservation of joint function; reduce the burden of administration by reducing dosing fre-quency and providing more cost-effective therapy; individualize treatment by adapting to individual pharmacokinetics and personalizing treatment regimens based on variability of bleeding phenotype and lifestyle; identify, monitor, and prevent age-related comorbidities; and potentially develop a cure for hemophilia through gene therapy.
Dr. Hege is a Fellow in Pediatric Hematology/Oncology at Riley Hospital for Children, Indiana University Health, Indiana University School of Medicine, Indianapolis, Indiana.

32 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
KerryHege,MD Treatment of Hemophilia: What’s in the Pipeline?
n GOALS OF THE HEMOPHILIA THERAPY PIPELINEThe past four decades have witnessed
great advances in hemophilia therapy (Table 1).2 Goals include improving patient outcomes with more effective bleeding control and preservation of joint function; reducing the burden of factor administration through decreases in dosing frequency and more cost-effective therapy; individualizing therapy by adapt-ing to individual pharmacokinetics and personalizing treatment regimens based on the variability of bleeding phenotype and lifestyle; identifying, monitoring, and preventing age-related comorbidities; and developing a cure for hemophilia through gene therapy. Table 2 provides a list of cur-rent clinical trials of novel recombinant factors in hemophilia A and B.4,9,10
n EXTENDING THE HALF-LIFE OF RECOMBINANT FACTOR
Current factor research has focused on the development of longer-acting
products to decrease the frequency and dosing of factor infusions, improve compliance with prophylactic regimens, prevent bleeding episodes and re-bleeding with bleeding episodes, and improve the overall quality of life for patients with he-mophilia.2,5 Investigations into extending the half-life of current recombinant co-agulation factors have used several strat-egies, including reduction of exposure to clearance receptors through PEGylation, rescue of endocytosed proteins from in-tracellular degradation by crystallizable fragment (Fc) fusion and albumin fusion proteins, and enhanced interactions with von Willebrand factor (vWF).
PEGylationPEGylation improves drug efficacy via
the covalent attachment of polyethylene glycol molecules (PEG) to the protein of interest—in this case, recombinant factor proteins (Figure 1).4 The PEG structures attract water molecules that surround factor proteins. For many therapeutic pro-
teins employing PEGylation, this process effectively increases the size of the protein beyond renal filtration ability. Because factor proteins are already too large for kidney filtration, PEGylation provides other benefits, possibly through disrupted interactions with clearance receptors, and may decrease interactions with immune-mediating cells.4,5
PEGylation of therapeutic proteins, which has been in clinical use since 1990, is considered to be safe and well tolerated.11 Early PEGylation of factor proteins used random introduction of PEG molecules, which often interfered with factor ability to function with re-duced coagulant activity and disrupted vWF-binding in FVIII.4,5 This led to tar-geted, site-specific PEGylation of factor proteins to preserve functional protein-protein interactions.
FactorVIII.A phase 1 study of site-specific PEGylated recombinant FVIIII (rFVIII, BAY 94-9027) demonstrated improved pharmacokinetics with a ter-minal half-life of 19 hours (twice that of B-domain deleted rFVIII).4 BAY 94-9027 was well tolerated; no adverse events were reported, and no inhibitors developed in patients who received it. A phase 2/3 study is ongoing.
A phase 1 study of a similar PEGylated rFVIII, N8-GP replicated these results, with a terminal half-life of 19 hours, no adverse events, and no inhibitor devel-opment.3,4 Testing of a third site-specific PEGylated rFVIII, BAX 855, demonstrat-ed promising preclinical pharmacokinet-
TABLE 1Past, Present, and Future of Hemophilia Treatment
Decade Year Mainevents
1970s: a golden era 1970–1979 Lyophilized factors Home treatment Pioneer prophylaxis programs Comprehensive treatment centers 1977 Desmopressin1980s: many shadows, a few lights 1982 Factor IX gene (F9) cloned Acquired immunodeficiency syndrome (AIDS) recognized 1983 Early virucidal methods (dry-heating) 1984 Factor VIII gene (F8) cloned Human immunodeficiency virus (HIV) isolated 1985 Anti-HIV testing 1987 Safe virus-inactivated plasma factors 1989 Recombinant factor VIII1990s: a new golden era 1994 Immune tolerance 1996 Highly active antiretroviral therapy (HAART) for HIV infection Recombinant factor VIIa 1997 Recombinant factor IX2000s: current hemophilia therapy 2000–2006 First gene-therapy trials 2002 Eradication of hepatitis C virus (HCV) infection by interferon-ribavirinThe next 10 years 2011–2021 More factor concentrate available worldwide Longer-acting recombinant coagulation factors Fusion coagulation factors Cure of hemophilia via gene transfer?
Source: Franchini and Mannucci2
FIGURE 1 Schematic representation of PEGylation. Reproduced, with permission, from Peyvandi et al.4
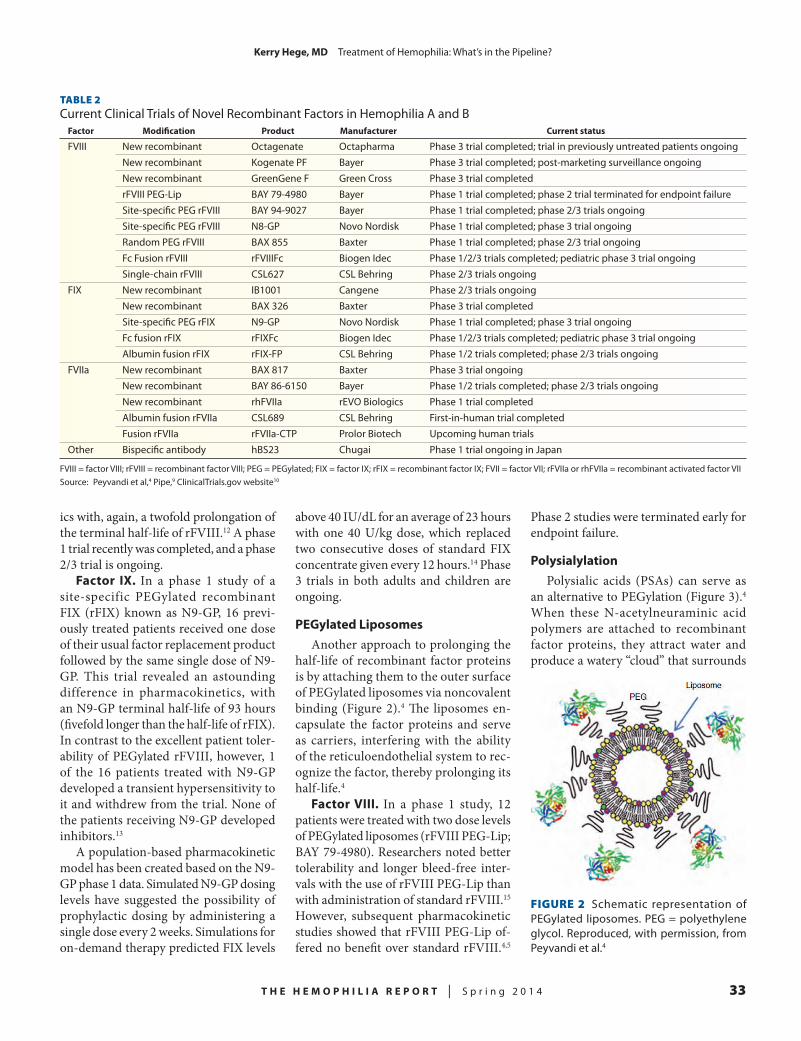
T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 33
KerryHege,MD Treatment of Hemophilia: What’s in the Pipeline?
ics with, again, a twofold prolongation of the terminal half-life of rFVIII.12 A phase 1 trial recently was completed, and a phase 2/3 trial is ongoing.
Factor IX. In a phase 1 study of a site-specific PEGylated recombinant FIX (rFIX) known as N9-GP, 16 previ-ously treated patients received one dose of their usual factor replacement product followed by the same single dose of N9-GP. This trial revealed an astounding difference in pharmacokinetics, with an N9-GP terminal half-life of 93 hours (fivefold longer than the half-life of rFIX). In contrast to the excellent patient toler-ability of PEGylated rFVIII, however, 1 of the 16 patients treated with N9-GP developed a transient hypersensitivity to it and withdrew from the trial. None of the patients receiving N9-GP developed inhibitors.13
A population-based pharmacokinetic model has been created based on the N9-GP phase 1 data. Simulated N9-GP dosing levels have suggested the possibility of prophylactic dosing by administering a single dose every 2 weeks. Simulations for on-demand therapy predicted FIX levels
above 40 IU/dL for an average of 23 hours with one 40 U/kg dose, which replaced two consecutive doses of standard FIX concentrate given every 12 hours.14 Phase 3 trials in both adults and children are ongoing.
PEGylatedLiposomesAnother approach to prolonging the
half-life of recombinant factor proteins is by attaching them to the outer surface of PEGylated liposomes via noncovalent binding (Figure 2).4 The liposomes en-capsulate the factor proteins and serve as carriers, interfering with the ability of the reticuloendothelial system to rec-ognize the factor, thereby prolonging its half-life.4
FactorVIII. In a phase 1 study, 12 patients were treated with two dose levels of PEGylated liposomes (rFVIII PEG-Lip; BAY 79-4980). Researchers noted better tolerability and longer bleed-free inter-vals with the use of rFVIII PEG-Lip than with administration of standard rFVIII.15 However, subsequent pharmacokinetic studies showed that rFVIII PEG-Lip of-fered no benefit over standard rFVIII.4,5
Phase 2 studies were terminated early for endpoint failure.
PolysialylationPolysialic acids (PSAs) can serve as
an alternative to PEGylation (Figure 3).4 When these N-acetylneuraminic acid polymers are attached to recombinant factor proteins, they attract water and produce a watery “cloud” that surrounds
TABLE 2Current Clinical Trials of Novel Recombinant Factors in Hemophilia A and B
Factor Modification Product Manufacturer Currentstatus
FVIII New recombinant Octagenate Octapharma Phase 3 trial completed; trial in previously untreated patients ongoing New recombinant Kogenate PF Bayer Phase 3 trial completed; post-marketing surveillance ongoing New recombinant GreenGene F Green Cross Phase 3 trial completed rFVIII PEG-Lip BAY 79-4980 Bayer Phase 1 trial completed; phase 2 trial terminated for endpoint failure Site-specific PEG rFVIII BAY 94-9027 Bayer Phase 1 trial completed; phase 2/3 trials ongoing Site-specific PEG rFVIII N8-GP Novo Nordisk Phase 1 trial completed; phase 3 trial ongoing Random PEG rFVIII BAX 855 Baxter Phase 1 trial completed; phase 2/3 trial ongoing Fc Fusion rFVIII rFVIIIFc Biogen Idec Phase 1/2/3 trials completed; pediatric phase 3 trial ongoing Single-chain rFVIII CSL627 CSL Behring Phase 2/3 trials ongoingFIX New recombinant IB1001 Cangene Phase 2/3 trials ongoing New recombinant BAX 326 Baxter Phase 3 trial completed Site-specific PEG rFIX N9-GP Novo Nordisk Phase 1 trial completed; phase 3 trial ongoing Fc fusion rFIX rFIXFc Biogen Idec Phase 1/2/3 trials completed; pediatric phase 3 trial ongoing Albumin fusion rFIX rFIX-FP CSL Behring Phase 1/2 trials completed; phase 2/3 trials ongoingFVIIa New recombinant BAX 817 Baxter Phase 3 trial ongoing New recombinant BAY 86-6150 Bayer Phase 1/2 trials completed; phase 2/3 trials ongoing New recombinant rhFVIIa rEVO Biologics Phase 1 trial completed Albumin fusion rFVIIa CSL689 CSL Behring First-in-human trial completed Fusion rFVIIa rFVIIa-CTP Prolor Biotech Upcoming human trialsOther Bispecific antibody hBS23 Chugai Phase 1 trial ongoing in Japan
FVIII = factor VIII; rFVIII = recombinant factor VIII; PEG = PEGylated; FIX = factor IX; rFIX = recombinant factor IX; FVII = factor VII; rFVIIa or rhFVIIa = recombinant activated factor VIISource: Peyvandi et al,4 Pipe,9 ClinicalTrials.gov website10
FIGURE 2 Schematic representation of PEGylated liposomes. PEG = polyethylene glycol. Reproduced, with permission, from Peyvandi et al.4

34 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
KerryHege,MD Treatment of Hemophilia: What’s in the Pipeline?
the protein to protect it from clearance receptors, proteolytic enzymes, and immune-mediating cells.5,8
Unlike PEG, PSAs are naturally occur-ring and biodegradable.5 Polysialylation of rFVIII, FIX, activated factor FVII (FVIIa), and vWF are all currently under study.
FcFusionFusion protein technology links factor
to another naturally occurring protein with a much longer half-life, such as im-munoglobulin G (IgG; Figure 4).4 Fusion of factor to the Fc portion of IgG protects the factor protein from lysosomal degradation via interactions with neonatal Fc receptors when internalized by endothelial cells. Phagocytized fusion proteins are recycled back to the cell surface in a pH-dependent manner and re-released into plasma.5,16,17
FactorVIII. A phase 1/2, first-in-human trial of rFVIII-Fc involved 16 patients who received a single dose of rFVIII followed by an equal dose of rFVIII-Fc. All doses were well tolerated, no adverse events related to study drug
were reported, and none of the patients developed inhibitors. Pharmacokinetic studies demonstrated a terminal half-life 1.6-fold longer than that of rFVIII; clear-ance was 1.5-fold lower.16
A follow-up phase 3 trial in adults was completed recently. Early results demon-strated tolerability in 165 patients, with no reported adverse events or inhibitor development. Pharmacokinetic studies demonstrated a terminal half-life of 19 hours, with a mean interval between doses of 3.5 days to maintain prophylactic factor activity levels; 30% of the patients in the active treatment arm were dosed every 5 days. For on-demand dosing, 87% of bleeding episodes were controlled with one dose of rFVIII-Fc, and 98% of the the patients achieved bleeding control with one or two doses.18 A phase 3 trial in children is ongoing.
FactorIX.In a phase 1/2, dose-escalation trial of rFIX-Fc, 14 patients demonstrated a terminal half-life of 56.7 hours (approxi-mately threefold longer than the half-life of standard rFIX products). rFIX-Fc was well tolerated; no serious adverse events or inhibitor development was reported.19 Pharmacokinetic modeling based on the results of this study suggested that dosing once every 2 weeks at 100 IU/kg was suf-ficient for trough levels 1% above baseline.19
A subsequent phase 3 study in adults was completed recently. Early results demonstrated overall tolerability in 123 patients; one episode of obstructive urop-athy was reported. None of the patients developed inhibitors. Pharmacokinetic studies demonstrated a remarkable termi-nal half-life of 82 hours (two- to threefold longer than with rFIX), suggesting that prophylactic dosing could be given once every 2 weeks. Further, 54% of patients given individualized prophylactic dos-ing achieved dosing intervals ≥14 days. Although 90% of bleeding episodes were controlled with a single on-demand dose of rFIX-Fc, 97% of bleeding episodes were controlled with one or two doses.20 A phase 3 study in children is ongoing.
AlbuminFusionSimilar to Fc fusion, albumin fusion
technology links factor proteins with hu-
man albumin, a natural carrier molecule (Figure 5).4 The resulting albumin-bound molecule has a longer half-life than that of currently available coagulation fac-tors, protects the factor protein from proteolytic degradation, and may prevent exposure to immune-mediating cells, thus prolonging its half-life and decreasing immunogenicity via the same recycling mechanism as described for Fc fusion.4,8
FactorIX.Results of a phase 1, first-in-human, dose-escalation study of the fusion protein linking rFIX with albumin (rFIX-FP) are promising. In 25 patients, rFIX-FP demonstrated a favorable safety profile and a remarkable pharmacoki-netic profile, offering a half-life fivefold longer than that associated with other FIX products.21 The follow-up phase 1/2 study of rFIX-FP echoed these promising results. Preliminary data demonstrated a pharmacokinetic profile suggesting that weekly prophylactic dosing with rFIX-FP provides bleeding prophylaxis and that extended dosing intervals of 10–14 days may be feasible.22 Phase 3 trials in adults and children are ongoing.
Factor VIIa. In a first-in-human study of rFVIIa-FP, 40 healthy subjects were dosed in five consecutive groups. All doses were well tolerated, no serious adverse events were reported, and none of the subjects developed inhibitors. Pharmacokinetic data revealed a mean terminal half-life of 8.5 hours (about 3.5-fold longer than that of commercial rFVIIa products).23
EnhancedInteractionswithvWFThe primary determinant of the half-life
of FVIII is interaction with vWF, which
FIGURE 3 Schematic representation of polysialylation. Reproduced, with permis-sion, from Peyvandi et al.4
FIGURE 4 Schematic representation of crys-tallizable fragment (Fc) fusion. Reproduced, with permission, from Peyvandi et al.4
FIGURE 5 Schematic representation of al-bumin fusion. Reproduced, with permission, from Peyvandi et al.4

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 35
KerryHege,MD Treatment of Hemophilia: What’s in the Pipeline?
naturally protects it from degradation. A unique recombinant, single-chain rFVIII (CSL627) has shown improved stability and higher affinity for vWF when com-pared with other rFVIII formulations.24
In preclinical studies, CSL627 has demonstrated safety and efficacy with equivalent hemostatic activity as com-pared with full-length rFVIII formula-tions. The novel single-chain design provides for higher intrinsic stability and affinity for vWF.25 Whether this translates into a reduced immunogenic potential will be investigated in a recently com-menced phase 1/3 clinical trial.
BispecificAntibodyIn addition to improving factor pro-
teins themselves, a novel approach has been taken to replace FVIII cofactor function by a small molecule.26 This hu-manized bispecific antibody to FIXa and FX (hBS23) binds FIXa with one arm and FX with the other.
Figure 6 illustrates a bispecific an-tibody to mimic FVIIIa activity.27 This molecule brings the two factors together into appropriate positions for hemostatic activity. The most promising hBS23 pre-clinical study revealed a terminal half-life of 14 days and subcutaneous bioavailabil-ity of nearly 100%.27
n GENE THERAPY
Gene therapy involves the transfer of a normal copy of a gene into a person who
harbors a mutation of that gene. Despite early disappointments of gene therapy in hemophilia,28 recent studies of new gene-transfer technology have renewed interest in this potentially curative approach.
Gene therapy trials have used plas-mids, retroviral, and adenoviral vectors directed to autologous fibroblasts, hema-topoietic stem cells, and target cells (eg, skeletal muscle and liver cells). However, investigators have been hampered by low levels of gene expression and failure to achieve long-term gene expression.28 With newer adeno-associated viral-mediated (AAV) models, gene therapy is again under investigation.29 Hemophilia is a good candidate for gene therapy, because it is a monogenetic disease that requires the production of only a small fraction of normal factor activity to ameliorate or cure the bleeding phenotype.
Using AAV-8 delivery of an F9 trans-gene, researchers at University College London documented post–gene transfer FIX levels of 5%–10% and stable FIX levels extending beyond 30 weeks.30 The biggest challenge of gene-transfer therapy is modulating host immune re-sponses to the vector and gene product. Patients in this study experienced mild transaminitis, which likely resulted from a host immune response to the adenoviral vector. This response apparently was suc-cessfully managed with a short course of corticosteroids. No adverse effect on gene transfer or resulting plasma factor levels was noted.30 Thus, gene therapy appar-ently has achieved long-term expression of therapeutic FIX levels,31 but successes with FVIII are lagging behind.
UsingGeneticVariantsInsteadofNormalGenes
Perhaps the two most interesting advancements in gene therapy include the use of lentiviral vectors and the ma-nipulation of genetic variants to produce overexpression of factor as a transgene. Lentiviral vectors can incorporate larger transgenes, which may facilitate FVIII transfer. Lentiviral vectors also avoid pre-existing immunity, which can be a barrier to the use of AAV vectors.31
The idea of using an F9 genetic variant
to achieve overexpression of FIX instead of using the normal F9 gene to achieve nor-mal expression recently was investigated.32 Preclinical mouse models using AAV-8 delivery of a genetically modified F9 trans-gene resulted in over 55% FIX activity. This promising work has resulted in a current human trial (ASKBIO009), which has just enrolled its first two patients.
n AGING AND COMORBIDITIES
With improved availability of quality factor concentrates for factor replace-ment, implementation of prophylactic regimens and advancements in screening, and treatment of blood-borne viral infec-tions, people with hemophilia are now living longer than ever before.2 Improved life expectancy comes with the typical comorbidities seen in an aging popula-
tion, such as cardiovascular and renal diseases and various malignancies.33–35 These issues are being explored, and general aspects of clinical management have been suggested.35,36 These issues will become more prevalent as the hemophilia population ages and will require further attention.
n CONCLUSION
Potential therapies in the pipeline to treat hemophilia focus on prolonging the half-life of recombinant factor products, improving patient outcomes, and develop-ing a cure for hemophilia through gene therapy. Analysis of available data suggests that these early trials appear to be meeting their goals.
At least three different FIX products of three different engineering methods
FIGURE 6 A bispecific antibody capable of mimicking activated factor VIII (FVIIIa) activ-ity. FIXa = activated factor IX; FX = factor X; HC = heavy chain; LC = light chain. Repro-duced, with permission, from Kitazawa et al.27
The biggest challenge of gene-transfer therapy is modulating host immune responses to the vector and gene product.

36 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
KerryHege,MD Treatment of Hemophilia: What’s in the Pipeline?
have shown terminal half-lives as much as fivefold greater than those of standard rFIX products. Prolonging the time that plasma levels can be measured during prophylactic factor use translates into a need for fewer factor infusions and dos-ing intervals lasting as long as 2 weeks. Available data on newly engineered FVIII products have been somewhat less promising. These products currently have terminal half-lives of up to 1.8 times greater than those of standard rFVIII factor. Ultimately, ongoing phase 3 trials will establish the effect this technology will have on individual dosing schedules, overall factor use, patient compliance, inhibitor development, need for venous access, and overall cost of therapy.
Gene therapy is equally as promising, and a new wave of human trials is ongo-ing. The most recent trials have achieved long-term expression of therapeutic fac-tor levels. The field is exploring ways to improve gene delivery, minimize vector immunogenicity, prolong gene expres-sion, and raise factor activity levels.
REFERENCES
1. Mannucci PM, Tuddenham E. The hemophil-ias—from royal genes to gene therapy. N Engl J Med. 2001;344:1773–1779.
2. Franchini M, Mannucci C. Past, present and future of hemophilia: a narrative review. Orphanet J Rare Dis. 2012;7:2–8.
3. Mannucci PM, Mancuso M. Investigational drugs for coagulation disorders. Expert Opin Investig Drugs. 2013;22:945–953.
4. Peyvandi F, Garagiola I, Seregni S. Future of coagulation factor replacement therapy. J Thromb Haemost. 2013;11(suppl 1):84–98.
5. Pipe SW. Hemophilia: new protein therapeu-tics. Hematology Am Soc Hematol Educ Program. 2010;2010:203–209.
6. Lillicrap D. The future of hemostasis man-agement. Pediatr Blood Cancer. 2013;60(suppl 1):S44–S47.
7. Valentino LA, Pipe SW, Tarantino MD, Ye X, Xiong Y, Luo MP. Healthcare resource utilization among haemophilia A patients in the United States. Haemophilia. 2012;18:332–338.
8. Pipe SW. The hope and reality of long-acting hemophilia products. Am J Hematol. 2012;87(suppl
1):S33–S39.9. Pipe SW. What’s in the pipeline? Presented
at the 65th Annual Meeting of the National Hemo-philia Foundation; October 3–5, 2013; Anaheim, California.
10. ClinicalTrials.gov Web site. Bethesda, MD: National Library of Medicine. http://clinicaltrials.gov. Accessed February 20, 2014.
11. Ivens IA, Baumann A, McDonald TA, Humphries TJ, Michaels LA, Mathew P. PEGylated therapeutic proteins for haemophilia treatment: a review for haemophilia caregivers. Haemophilia. 2013;19:11–20.
12. Turecek PL, Bossard MJ, Graninger M, et al. BAX 855, a PEGylated rFVIII product with prolonged half-life: development, functional and structural characterisation. Hamostaseologie. 2012;32(Suppl 1):S29–S38.
13. Negrier C, Knobe K, Tiede A, Giangrande P, Moss J. Enhanced pharmacokinetic properties of a glycoPEGylated recombinant factor IX: a first hu-man dose trial in patients with hemophilia B. Blood. 2011;118:2695–2701.
14. Collins PW, Moss J, Knobe K, Groth A, Colberg T, Watson E. Population pharmacokinetic modeling for dose setting of nonacog beta pegol (N9-GP), a glycoPEGylated recombinant factor IX. J Thromb Haemost. 2012;10:2305–2312.
15. Spira J, Plyushch OP, Andreeva TA, An-dreev Y. Prolonged bleeding-free period following prophylactic infusion of recombinant factor VIII reconstituted with pegylated liposomes. Blood. 2006;108:3668–3673.
16. Powell JS, Josephson NC, Quon D, et al. Safety and prolonged activity of recombinant fac-tor VIII Fc fusion protein in hemophilia A patients. Blood. 2012;119:3031–3037.
17. Shapiro AD. Development of long-acting recombinant FVIII and FIX Fc fusion proteins for the management of hemophilia. Expert Opin Biol Ther. 2013;13:1287–1297.
18. Mahlangu J, Powell JS, Ragni MV, et al. A-LONG: results from a phase 3 study of safety, efficacy, and pharmacokinetics of long-lasting recombinant factor VIII Fc fusion protein (rFVIIIFc). J Thromb Haemost. 2013;11:168–169.
19. Shapiro AD, Ragni MV, Valentino LA, et al. Recombinant factor IX-Fc fusion protein (rFIXFc) demonstrates safety and prolonged activity in a phase 1/2a study in hemophilia B patients. Blood. 2012;119:666–672.
20. Powell JS, Ozelo M, Pasi J, et al. B-LONG: results from a phase 3 study of safety, efficacy, and pharmacokinetics of long-lasting recombinant factor IX Fc fusion protein (rFIXFc). J Thromb Haemost. 2013;11:240.
21. Santagostino E, Negrier C, Klamroth R, et al. Safety and pharmacokinetics of a novel recombinant
fusion protein linking coagulation factor IX with albumin (rIX-FP) in hemophilia B patients. Blood. 2012;120:2405–2411.
22. Martinowitz U, Lubetsky A. Phase I/II, open-label, multicenter, safety, efficacy and PK study of a recombinant coagulation factor IX albumin fu-sion protein (rIX-FP) in subjects with hemophilia B. Thromb Res. 2013;131:S11–S14.
23. Golor G, Bensen-Kennedy D, Haffner S, et al. Safety and pharmacokinetics of a recombinant fusion protein linking coagulation factor VIIa with albumin (rVIIa-FP) in healthy volunteers. J Thromb Haemost. 2013;11:1977–1985.
24. Pabinger-Fasching I, Pipe S. Innovations in coagulation: improved options for treatment of hemophilia A and B. Thromb Res. 2013;131:S1.
25. Zollner SB, Raquet E, Muller-Cohrs J, et al. Preclinical efficacy and safety of rVIII-single chain (CSL627), a novel recombinant single-chain factor VIII. Thromb Res. 2013;132:280–287.
26. Lillicrap D. A complex substitute: antibody therapy for hemophilia. Nat Med. 2012;18:1460–1461.
27. Kitazawa T, Igawa T, Sampei Z, et al. A bi-specific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18:1570–1574.
28. High KA. Gene therapy for haemophilia: a long and winding road. J Thromb Haemost. 2011;9(suppl 1):2–11.
29. High KA. The gene therapy journey for hemo-philia: are we there yet? Blood. 2012;120:4482–4487.
30. Nathwani A, Tuddenhan E, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365.
31. Chuah MK, Evens H, Vandendriessche T. Gene therapy for hemophilia. J Thromb Haemost. 2013;11:99–110.
32. Monahan P. Update on gene therapy clinical research trials. Presented at the 65th Annual Meeting of the National Hemophilia Foundation; October 3–5, 2013; Anaheim, California.
33. Coppola A, Santoro C, Franchini M, et al. Emerging issues on comprehensive hemophilia care: preventing, identifying, and monitoring age-related comorbidities. Semin Thromb Hemost. 2013;39:794–802.
34. Franchini M, Mannucci PM. Co-morbidities and quality of life in elderly persons with haemo-philia. Br J Haematol. 2010;148:522–533.
35. Franchini M, Tagliaferri A, Mannucci PM. The management of hemophilia in elderly patients. Clin Interv Aging. 2007;2:361–368.
36. Mannucci PM, Schutgens RE, Santagostino E, Mauser-Bunschoten EP. How I treat age-related morbidities in elderly persons with hemophilia. Blood. 2009;114:5256–5263.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 37
Hemophilia is caused by a functional or quantitative deficiency of a coagulation protein—factor VIII (FVIII)
in hemophilia A and factor IX (FIX) in hemophilia B—and can lead to a lifelong bleeding disorder. Uncontrolled or unrec-ognized major bleeding events may result in joint damage, intracranial hemorrhage, and death. Complications of hemophilia have included musculoskeletal disease, development of inhibitory antibodies, and transmission of viral bloodborne infections.1 Before the advent of efficient coagulation factor replacement therapy, the life expectancy of a child with severe hemophilia A was about 20 years.2 Cur-rently, the life expectancy of this popula-tion approximates that of the general population.3,4
Recent Advances in Preventing Bleeding, Reducing Inhibitors, and Managing Acute BleedingNoa Biran, MDTisch Cancer Institute, Mount Sinai School of Medicine, New York, New York
Abstract Recent advances in the prevention of bleeding episodes, reduction of inhibitory alloantibodies to factor concentrates (inhibitors), and management of acute bleeding episodes in hemophilia were presented at the 55th Annual Meeting of the American Society of Hematology (ASH). Among the more notable observations were: (1) bleeding tendency is associated with the level of factor VIII (FVIII) activity in patients receiving prophylactic recombinant FVIII crystalliz-able fragment (Fc) fusion protein; (2) prophylactic treatment with long-lasting recombinant factor IX Fc fusion protein is safe and effective in adolescents with hemophilia B; (3) delayed prophylaxis has long-term deleterious effects in patients with acute hemophilia; (4) magnetic resonance imaging is useful for detecting early hemophiliac joint disease; (5) early, low-dose prophylaxis in the absence of immunologic danger signals may reduce inhibitor incidence in patients with hemophilia A; (6) prompt immune tolerance induction at inhibitor diagnosis may increase the success rate in patients with hemophilia A and inhibitors; and (7) four-factor prothrombin complex is superior to plasma administration for achiev-ing hemostasis before urgent surgery. Ongoing studies on prophylaxis, reduction and management of inhibitors, and treatment of acute bleeding are crucial to continuing improvement in the quality of life of patients with hemophilia.
Dr. Biran is a Fellow in Hematology/Medical Oncology at Tisch Cancer Institute, Mount Sinai School of Medicine, New York, New York.
Approval of the first recombinant factor VIII (rFVIII) concentrates in the United States in 19925 improved the product supply, decreased the incidence of viral bloodborne infections to near zero, and advanced the adoption of pro-phylactic treatment regimens. Prophy-lactic treatment regimens achieve a less severe clinical phenotype by maintaining hemostatically protective factor levels, accomplished with regularly scheduled infusions. Specific regimens to achieve optimal bleed suppression and preven-tion vary.6 Current standard prophylactic regimens commonly use infusion therapy given three times weekly, whereas other regimens require administration every other day. Regimens may be individu-ally modified to achieve optimal bleed suppression and tailored to the patient’s
individual needs. Often, patients have difficulty in adhering to demanding therapeutic prophylactic regimens that include frequent morning infusion to achieve adequate hemostatic coverage during periods of highest activity. As chil-dren progress to adolescence, adherence is often further limited,7 making these regimens less effective.
Inhibitors, or inhibitory alloantibod-ies to factor concentrates, develop in approximately 25%–30% of patients with severe hemophilia A and 3%–13% of those with moderate or mild disease.8,9 Inhibitors neutralize infused FVIII and greatly affect a patient’s ability to achieve hemostasis. A bleeding episode in patients with high inhibitor titers requires the use of a bypassing agent; effective use of prophylaxis is greatly diminished in this subset of patients. Patients with inhibitors require considerable management ex-pertise. The use of bypassing agents and immunotolerance regimens that aim to decrease the inhibitory alloantibodies re-main enormously costly. Therefore, much work remains in determining predictors of inhibitor development and in develop-ing regimens to modify the individual’s immune response.

38 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
NoaBiran,MD Recent Advances in Preventing Bleeding, Reducing Inhibitors, and Managing Acute Bleeding
This report will discuss recent advances in prophylaxis and imaging to prevent bleeding episodes, reduction of the in-cidence of inhibitors, and management of acute bleeding episodes in hemophilia patients.
n PREVENTION OF BLEEDING EPISODES: USE OF PROPHYLAXIS AND IMAGING
BleedingTendencyandFVIIIActivityLevel
Joints can be preserved and bleed-ing can be minimized when circulating FVIII levels remain ≥ 1 IU/dL.10 Patients receiving prophylaxis with recombinant FVIII who have had prolonged intervals of circulating FVIII levels < 1 IU/dL ex-perience an increase in bleeding episodes and hemarthroses.11 Thus, prophylactic regimens used in patients with hemophilia A have been designed to maintain the FVIII activity ≥ 1 IU/dL.
Pasi et al12 examined bleeding tendency in relation to predicted FVIII activity levels in patients with severe hemophilia A who were treated with recombinant FVIII crystallizable fragment (Fc) fu-sion protein (rFVIIIFc) in the A-LONG study. A two-compartment population pharmacokinetic model of rFVIIIFc was developed based on activity-time profiles in 180 patients with severe hemophilia A who were 12–65 years of age. Negative binomial regression models were used to correlate the number of bleeding episodes with the annualized time (in days) that the patients’ FVIII activity levels were < 1 IU/dL. Models were adjusted for age, body mass index, baseline human im-munodeficiency virus (HIV) and hepatitis C virus status, baseline von Willebrand factor level, number of bleeding episodes, and time on study.
Multivariable negative binomial re-gression analysis showed that the number of overall bleeding episodes increased the more time that patients experienced a FVIII activity level < 1 IU/dL (P < 0.001). A significant association also was found between the length of time in which pa-tients had a FVIII activity level < 1 IU/dL and different types of bleeding (sponta-neous, traumatic, or joint). In addition
to their increased bleeding tendency, the chance that these patients would be bleed-free was lessened. These findings reinforce the importance of a therapeutic threshold of 1 IU/dL of FVIII activity and contrib-ute to building a stronger foundation for designing effective rFVIIIFc prophylactic dose regimens.
Long-LastingRecombinantFactorIXFcFusionProtein(rFIXFc)
Prophylactic replacement of FIX, which is used to treat people with he-mophilia B, requires up to three weekly intravenous injections. To reduce the frequency of injections, a long-acting recombinant protein (rFIXFc) consisting of one rFIX molecule covalently bonded to the Fc domain of immunoglobulin G1 (IgG1) was developed. Fc fusion uses an endogenous IgG recycling pathway to prolong the half-life of therapeutic proteins. The phase 3 B-LONG study (ClinicalTrials.gov ID No. NCT01027364) examined the pharmacokinetics, safety, and efficacy of rFIXFc given to previously treated subjects with hemophilia B.13 Age may affect the half-life of recombinant coagulation factors.14
A planned subgroup analysis by Sha-piro et al15 examined whether age influ-enced the pharmacokinetics, safety, and efficacy of rFIXFc use. Male subjects with severe hemophilia B (≤ 2 IU/dL) who were at least 12 years of age, who were without inhibitors to FIX, and who had ≥ 100 ex-posure days to FIX were assigned to one of four treatment arms. Arm 1 included those who had weekly prophylaxis, arm 2 included those who had individualized interval prophylaxis, arm 3 included those who had episodic treatment of bleeding episodes, and arm 4 included those who had perioperative management. The study was stopped when prespecified exposure was achieved and the specified number of major surgical procedures were com-pleted. The pharmacokinetic parameters of rFIXFc were estimated in all study par-ticipants by a noncompartmental analysis based on a one-stage clotting assay. A sequential subgroup analysis compared the pharmacokinetic profile of rFIXFc with that of rFIX in arm 1. The primary
endpoints were annualized bleeding rate (ABR), the incidence of adverse events, and inhibitor development. This subgroup analysis compared the outcomes in ado-lescents (12–17 years of age) with those of adults (18–65 years of age). Efficacy of rFIXFc was assessed by comparing the ABR in arms 1 and 2 with that in arm 3 separately for adolescents and adults.
A total of 123 patients were enrolled at 50 study centers in 17 countries. The median treatment duration was 51.4 weeks across all arms. Eleven adolescents were enrolled in arms 1–3 and none in the perioperative management arm (arm 4). All adolescent patients had evaluable rFIXFc pharmacokinetic profiles. The terminal half-life, clearance, incremental recovery, and volume of distribution at steady state were similar between adoles-cents and adults. The ABRs of adolescents also were comparable to those of adults. Both adolescent and adult subgroups in the prophylactic arms (arms 1 and 2) had lower ABRs than did those in the episodic treatment arm (arm 3). No distinctive safety issues were observed in adolescents; their safety profile was similar to that of the adult population.
Pharmacokinetic parameters, safety profiles, and ABRs with rFIXFc prophy-laxis were similar in adults and adoles-cents. This analysis supported the primary result of the B-LONG study, showing that rFIXFc is well tolerated and effective for preventing bleeding episodes in both adults and adolescents with hemophilia B.
DelayedProphylaxisHasLong-TermEffectsinPatientswithSevereHemophilia
The Joint Outcome Study (JOS) was a randomized, controlled clinical trial in boys with severe FVIII deficiency comparing prophylaxis (ie, 25 IU/kg of FVIII given every other day, beginning before age 30 months) with an enhanced episodic regimen used only in response to bleeding.16 At age 6, patients were discon-tinued from the study, and joint outcome by sensitive magnetic resonance imaging (MRI) and physical examination of six index joints (including both ankles, knees, and elbows) were evaluated. The JOS data

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 39
NoaBiran,MD Recent Advances in Preventing Bleeding, Reducing Inhibitors, and Managing Acute Bleeding
demonstrated the superiority of prophy-laxis over episodic therapy (P < 0.05). At the end of the study, all parents were informed of the study results, and patients were switched to prophylaxis.
The JOS Continuation Study (JOSc)17 currently is being performed to determine the results of early prophylaxis on joint development until age 18 years and the impact of delaying initiation of prophy-laxis until age 6 years. All boys in the original JOS trial were eligible to enroll in the continuation study. Data being collected include the cumulative number of index, joint, and total hemorrhages; joint physical examination score of six index joints, using the Colorado Pediatric Joint Assessment Scale18; MRI, soft-tissue, osteochondral, and total scores; and total scores of six index joints using the ex-panded MRI 45 Scale. Additional data are being collected on prophylaxis adherence, activities, surgeries, quality of life, and replacement factor use.
To date, results from 26 (40%) of the original 65 boys, including 16 on early and 10 on delayed prophylaxis, comprising a total of 156 index joints are available for analysis. Following delayed initiation of prophylaxis, adolescents manifested increased numbers of hemarthroses and increased MRI damage affecting particu-larly their bones and cartilage. MRI was more sensitive than was joint physical ex-amination in determining joint outcome.
MRItoDetectHemophiliacJointDisease
Arthropathy can be prevented by routine replacement of FVIII in patients with hemophilia A.16 However, whether prophylaxis retains efficacy if it is initiated after the onset of recurrent hemarthroses or in individuals with established arthrop-athy (secondary or tertiary prophylaxis) is unknown.
At the ASH meeting, Glorioso et al19 presented the results of a meta-analysis that examined the natural history of he-mophiliac arthropathy in patients with hemophilia A who were treated only in response to acute bleeding (on-demand therapy) as a baseline for future work. The researchers sought to estimate the
amelioration of arthropathy attributable to secondary or tertiary prophylaxis.
Data were aggregated from partici-pants given on-demand therapy during three studies—the JOS16 (and JOSc17), baseline results from the SPINART study,20 and the Cross-Sectional MRI Study21—into a single database. To be eligible, patients had to have a FVIII level ≤ 2%; a negative history for inhibitors; a bleeding history of ≥ 12 months before study data acquisition; and assessments of both ankles, knees, and elbows. Joints were assessed using physical examination and T2 gradient echo MRI scored using the World Federation of Hemophilia Gilbert score and the 45-point extended MRI Scales.8,18 MRI data were divided into soft-tissue and osteochondral changes. Bleeding episodes and Gilbert scores for all six joints and MRI data for knees and ankles were analyzed using standard descriptive statistics; regression methods were used to quantify the rate of change in scores of joint damage with age.
Results from the three studies provided data on 275 patients between 1 and 50 years of age. Clinical findings, Gilbert scores, and MRI data were extracted and analyzed from multiple joints in each of 157 individuals given on-demand treatment only. The number of bleeding episodes was approximately constant at 20 per year, regardless of age. Gilbert scores showed a steeper rise in young patients but did not increase as patients passed the late-teenage years. However, when compared with bleeding rates and Gilbert scores, MRI scores showed continued age-related deterioration. Age-related total and osteochondral deterioration on MRI increased steadily with age (on average, 2 MRI points per year of age; P > 0.0001). Soft-tissue scores increased rapidly to approximately 9 by age 20 years and then plateaued at approximately 10 for ages 20–50 years (0.08 MRI points per year; P = 0.28).
The authors of this study concluded that individual patient meta-analysis of bleeding frequency, joint physical ex-amination, and MRI joint structure are useful for determining the natural history of hemophilic arthropathy in patients
receiving on-demand therapy. Joint bone and cartilage abnormalities increased continuously across the age span despite an early leveling in bleeding rate and joint physical function. These data will be criti-cal for determining the quantitative effects of prophylaxis begun at various ages on mitigation of joint deterioration following the onset of joint bleeding.
n REDUCING THE INCIDENCE OF INHIBITORS
EarlyLow-doseProphylaxisintheAbsenceofImmunologicDangerSignals
The development of inhibitors is regulated by patient-specific and treat-ment-related factors associated with the immune response. Of the known risk factors, intensive treatment at an early age has been shown to be significant. Early prophylaxis, defined as first exposure to FVIII in the absence of a bleed in the first year of age, may induce FVIII tolerance, thereby protecting patients from inhibitor development.
The Early Low-Dose Prophylaxis in the Absence of Immunological Danger Signs (EPIC) Study22 prospectively assessed the ability of a once-weekly, low-dose prophy-lactic regimen started before 1 year of age and before the onset of a severe bleeding phenotype (ie, joint bleed) combined with the minimization of immunologic danger signals to reduce the incidence of inhibitor formation in previously untreated pa-tients with severe and moderately severe hemophilia A.
This study enrolled 22 patients, 19 of whom received treatment with FVIII. All patients had severe hemophilia A. Eleven (58%) of these patients were never exposed to factor VIII before, whereas the remaining 42% previously were treated with FVIII concentrates. Eight subjects developed a confirmed inhibitor, and two of these individuals had only borderline positivity at inhibitor testing. Thus, the incidence of an inhibitor titer > 0.6 Bethesda units (BU) in previously untreat-ed patients was 27%. However, 67 major protocol deviations were reported in 15 patients; 44 deviations were reported in 10 patients and were related to the treatment

40 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
NoaBiran,MD Recent Advances in Preventing Bleeding, Reducing Inhibitors, and Managing Acute Bleeding
regimen. These deviations contrasted with the protocol intention, which was to minimize immunologic danger signals and low-dose prophylactic regimens. As a result of the observed inhibitor incidence, the study was terminated based on a futil-ity analysis (the probability to achieve the primary endpoint of inhibitor rate reduc-tion to ≤ 15%).
A significant number of violations in the setting of a demanding study protocol made it difficult to determine whether treatment decisions in the presence of immunologic danger signals correlate with inhibitor formation. Thus, the inves-tigators could not verify or disprove the hypothesis that early low-dose prophy-laxis given in the absence of immunologic danger signals might reduce inhibitor incidence in previously untreated patients with hemophilia A.
PromptImmuneToleranceInductionatInhibitorDiagnosis
Immune tolerance induction (ITI) for patients with hemophilia A with inhibi-tors has an overall reported success rate of 60%–80%, yet it is the only modality known to effectively eradicate inhibitors. One debate concerns the optimal time to start ITI. Recent guidelines recommend delaying ITI until the inhibitor titer is < 10 BU.23
Nakar et al24 performed an analysis to determine the success of ITI relative to time from inhibitor detection to ITI initiation. Data were collected retrospec-tively from two hemophilia centers in the United States. Patients with severe or moderate (≤ 5%) FVIII deficiency under-went ITI; investigators gathered informa-tion on the time interval from inhibitor detection to ITI start, inhibitor titer, and outcome. High-dose ITI was practiced by both centers at ≥ 100 IU/kg per day. Success, partial success, and failure were defined practically, with success involving a negative inhibitor titer and the ability to use FVIII concentrate routinely to treat and prevent bleeding. Partial success in-volved an inhibitor titer < 5 BU with the ability to use FVIII concentrate to treat bleeding episodes. Failure was defined as ongoing ITI > 3 years without achieving
success or partial success or as discontinu-ation of ITI.
Patients were first divided into a low-responding inhibitor (LRI) subgroup and a high-responding inhibitor (HRI) subgroup based on peak inhibitor titer. The HRI subgroup was further subdivided based on the time to ITI initiation; the first subgroup initiated therapy within 1 month, the second from 1 to 6 months, and the third > 6 months from diagnosis of the inhibitor. The HRI subgroup start-ing ITI within 1 month was analyzed based on pre-ITI inhibitor titers.
This analysis included male patients with adequate ITI history documenta-tion. Almost all (95%) had severe FVIII deficiency. Overall, 49 of 58 patients (84%) underwent successful ITI. Among those with LRIs (19 patients [33%]), ITI success
was 100%. Among 39 patients (67%) har-boring HRIs, tolerance was achieved in 30 (77%), and partial success and continued ITI was achieved in 1 individual (3%); therapy failed in 7 (18%).
Of 23 patients who started ITI within 1 month of detection, 91% achieved success. All 13 patients (100%) starting ITI with a pre-ITI inhibitor titer ≥ 10 BU achieved success. Of the 11 patients who started ITI 6 months after detection of inhibitor, only 7 (64%) achieved success; therapy failed in 4 (36%).
These results suggest that the time in-terval from inhibitor detection to the start of ITI may play a critical role in eventual outcome. An inhibitor titer ≥ 10 BU did not influence outcome when ITI was used within 1 month of detection, supporting this approach in contrast to the commonly
accepted practice of delaying the start of ITI until an inhibitor titer < 10 BU is achieved. Patients with hemophilia A may benefit from prompt ITI regardless of the current inhibitor titer and are not subjected to wait periods when bleeding is more likely to occur. Prompt ITI should be considered a worthwhile therapeutic op-tion in newly identified inhibitor patients irrespective of inhibitor titer.
n MANAGEMENT OF ACUTE BLEEDING
Four-FactorProthrombinComplexinAchievingHemostasis
When rapid correction of coagulopa-thy is required, as in cases of intracranial hemorrhage, administration of prothrom-bin complex concentrate (PCC) may offer some advantages over infusion of fresh frozen plasma. Historically, the major drawback of PCCs has been the risk of thrombotic complications.25 However, PCCs have many benefits. For example, they are subjected to at least one viral in-activation step, which reduces the risk of pathogen transmission. Further, they are nanofiltrated, so they are unlikely to pro-voke transfusion-related acute lung injury.
A four-factor prothrombin complex concentrate (4F-PCC) recently was approved by the US Food and Drug Administration for urgent vitamin K antagonist (VKA) reversal in patients with major bleeding. Rapid reversal of VKA-induced anticoagulation often is needed for patients requiring urgent surgical procedures. Plasma currently is the standard of care in the United States for VKA reversal prior to emergency surgery.
A prospective phase 2b, open-label, noninferiority trial compared the effi-cacy and safety of 4F-PCC with those of plasma in patients requiring VKA reversal before undergoing an urgent surgical procedure.26 Patients were randomized to receive a single dose of either 4F-PCC or plasma. Dose was based on baseline international normalized ratio (INR) and weight. There were two primary endpoints: effective hemostasis (preven-tion of excessive perioperative bleeding from the start of study product infusion
Patients with hemophilia A may benefit from prompt immune tolerance induction regardless of the current inhibitor titer.

T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 41
NoaBiran,MD Recent Advances in Preventing Bleeding, Reducing Inhibitors, and Managing Acute Bleeding
until the end of the procedure) and rapid INR reduction (≤ 1.3 at 30 minutes after the infusion ended). Effective hemostasis was defined as the actual blood loss not exceeding the predicted blood loss by 30% and 50 mL, “normal” or “mildly abnor-mal” hemostasis (subjectively assessed), and no administration of non-study coagulation products.
Effective hemostasis was achieved in 89.7% of patients in the 4F-PCC group versus 75.3% of the plasma group, dem-onstrating both noninferiority and su-periority of 4F-PCC over plasma (14.3% difference; 95% confidence interval = 2.8, 25.8). Likewise, rapid INR reduction was achieved in 55.2% of the 4F-PCC group versus 9.9% of the plasma group, demon-strating noninferiority and superiority of 4F-PCC over plasma. Mortality, the fre-quency of serious adverse events, and that of thromboembolic events were similar between the groups. Also, significantly fewer fluid overload events occurred with 4F-PCC than with plasma.
Further phase 3 studies evaluating the safety and efficacy of 4F-PCC should be pursued in patients with hemophilia with and without inhibitors.
n CONCLUSION
Over the past 50 years, the lifespan of an individual affected with severe hemophilia has increased from a mere 20 years to near that of the general unaf-fected population. This improvement is a result of parallel advances in the devel-opment and manufacturing of safe and effective coagulation factor replacement therapies and prophylactic regimens. Furthermore, these strides are products of technologic leaps that allow for rapid control of acute, life-threatening bleeds. Ongoing studies will be crucial in con-tinually improving the quality of life of people with hemophilia and decreasing the cost of their care.
REFERENCES
1. Srivastava A, Brewer AK, Mauser-Bun-schoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–e47.
2. Larsson SA. Life expectancy of Swedish haemo-philiacs, 1831–1980. Br J Haematol. 1985;59:593–602.
3. Darby SC, Kan SW, Spooner RJ, et al. Mor-tality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110:815–825.
4. Plug I, Van Der Bom JG, Peters M, et al. Mortality and causes of death in patients with he-mophilia, 1992–2001: a prospective cohort study. J Thromb Haemost. 2006;4:510–516.
5. Kingdon HS, Lundblad RL. An adventure in biotechnology: the development of haemophilia A therapeutics—from whole-blood transfusion to recombinant DNA to gene therapy. Biotechnol Appl Biochem. 2002;35:141–148.
6. Collins PW, Björkman S, Fischer K, et al. Factor VIII requirement to maintain a target plasma level in the prophylactic treatment of severe hemo-philia A: influences of variance in pharmacokinet-ics and treatment regimens. J Thromb Haemost. 2010;8:269–275.
7. Schrijvers LH, Uitslager N, Schuurmans MJ, Fischer K. Barriers and motivators of adherence to prophylactic treatment in haemophilia: a systematic review. Haemophilia. 2013;19:355–361.
8. Kasper CK. Diagnosis and Management of Inhibitors to Factors VIII and IX: An Introductory Discussion for Physicians. Montreal, QC: World Federation of Hemophilia; 2004. http://www1.wfh.org/publication/files/pdf-1178.pdf. Accessed December 16, 2013.
9. Kempton CL, Allen G, Hord J, et al. Eradi-cation of factor VIII inhibitors in patients with mild and moderate hemophilia A. Am J Hematol. 2012;87:933–936.
10. Nilsson IM, Berntorp E, Lofqvist T, Petters-son H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232:25–32.
11. Blanchette VS. Prophylaxis in the haemo-philia population. Haemophilia. 2010;16:181–188.
12. Pasi J, Potts J, Li S, et al. The bleeding tendency in relation to predicted FVIII activity levels in severe hemophilia A patients treated with recombinant factor VIII Fc fusion protein. Presented at the 55th Annual Meeting of the American Society of Hematology; December 7–10, 2013; New Orleans, Louisiana. Abstract 3590.
13. Fischer K, Kulkarni R, Bradbury M, et al. Pharmacokinetics of recombinant factor IX Fc fusion protein (rFIXFc) in pediatric subjects with hemophilia B: an interim analysis of the kids B-LONG study. Presented at the 55th Annual Meeting of the American Society of Hematology; December 7–10, 2013; New Orleans, Louisiana. Abstract 3599.
14. Björkman S, Oh M, Spotts G, et al. Popula-tion pharmacokinetics of recombinant factor VIII: the relationships of pharmacokinetics to age and body weight. Blood. 2012;119:612–618.
15. Shapiro A, Pasi J, Mahlangu J, et al. Phar-macokinetics, safety, and efficacy of long-lasting recombinant factor IX fc fusion protein (rFIXFc) in adolescent subjects with hemophilia B: a subgroup analysis of the B-LONG study. Presented at the 55th Annual Meeting of the American Society of Hematology; December 7–10, 2013; New Orleans,
Louisiana. Abstract 2350.16. Manco-Johnson MJ, Abshire TC, Shapiro
AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–544.
17. Manco-Johnson MJ, Manco-Johnson ML, Blades TA, et al. Long-term orthopedic effects of delaying prophylaxis in severe hemophilia A until age 6 years: results of the Joint Outcome Study continuation (JOSc). Presented at the 55th Annual Meeting of the American Society of Hematology; December 7–10, 2013; New Orleans, Louisiana. Abstract 210.
18. Manco-Johnson MJ, Nuss R, Funk S, Murphy J. Joint evaluation instruments for chil-dren and adults with haemophilia. Haemophilia. 2000;6:649–657.
19. Glorioso TJ, Kittelson JM, Manco-Johnson ML, et al. Natural history of hemophilic joint dis-ease using sensitive imaging with magnetic reso-nance imaging (MRI). Presented at the 55th Annual Meeting of the American Society of Hematology; December 7–10, 2013; New Orleans, Louisiana. Abstract 209.
20. Manco-Johnson MJ, Kempton CL, Reding MT, et al. Randomized, controlled, parallel-group trial of routine prophylaxis vs. on-demand treatment with sucrose-formulated recombinant factor VIII in adults with severe hemophilia A (SPINART). J Thromb Haemost. 2013;11:1119–1127.
21. Hassan TH, Badr MA, El-Gerby KM. Cor-relation between musculoskeletal function and ra-diological joint scores in haemophilia A adolescents. Haemophilia. 2011;17:920–925.
22. Auerswald G, Kurnik K, Blatney J, Reininger AJ. The EPIC study: a clinical trial to assess whether early low dose prophylaxis in the absence of immu-nological danger signals reduces inhibitor incidence in previously untreated patients with hemophilia A. Presented at the 55th Annual Meeting of the Ameri-can Society of Hematology; December 7–10, 2013; New Orleans, Louisiana. Abstract 576.
23. Dimichele D. The North American Immune Tolerance Registry: contributions to the thirty-year experience with immune tolerance therapy. Haemo-philia. 2009;15:320–328.
24. Nakar CT, Manco-Johnson MJ, Lail A, et al. Prompt immune tolerance induction at inhibitor diagnosis regardless of titer may increase overall success in hemophilia A with inhibitors: experience of two US centers. Presented at the 55th Annual Meeting of the American Society of Hematology; December 7–10, 2013; New Orleans, Louisiana. Abstract 575.
25. Lusher JM. Thrombogenicity associated with factor IX complex concentrates. Semin Hematol. 1991;28:3–5.
26. Refaai MA, Goldstein JN, Milling Jr TJ, et al. Randomized phase IIIb study of rapid vitamin K antagonist reversal in patients requiring an urgent surgical procedure: four-factor prothrombin com-plex concentrate is superior to plasma. Presented at the 55th Annual Meeting of the American Society of Hematology; December 7–10, 2013; New Orleans, Louisiana. Abstract 3588.

42 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
CME/CNE Post TestUsing these two pages as a worksheet, select the best answer to each question based on your reading of the articles in this issue of The Hemophilia Report, then complete the evaluation on page 44 and see the instructions below it to obtain CME or CNE credit.
1. The most common morbidity seen among patients with severe hemophilia A or B is:
a. Splenomegalyb. Spontaneous soft-tissue bleedingc. Hemophilic arthropathyd. Hemarthroses
2. The most significant risk factor predisposing patients with hemophilia A to developing alloimmune inhibitory antibodies to coagulation replacement factor therapy is:
a. Type of F8 gene mutationb. Agec. Caucasian raced. Prolonged use of prophylactic therapy
3. Which of the following statements comparing patients with hemophilia B to those with hemophilia A is true?
a. Patients with hemophilia B have a higher risk of recurrent hemarthrosis and, consequently, undergo more frequent total joint arthroplasty than patients with hemophilia A.
b. Patients with hemophilia B receiving prophylactic factor IX therapy require more frequent doses than patients with hemophilia A receiving prophylactic factor VIII therapy.
c. Patients with hemophilia B are more likely to develop inhibitory antibodies to prophylactic replacement factor therapy than patients with hemophilia A receiving prophylaxis.
d. Patients with hemophilia B have less severe gene mutations than patients with hemophilia A and more missense mutations than null mutations.
4. Analysis of data from patients undergoing counseling prior to genetic testing revealed that approximately ________ of cases of hemophilia are sporadic.
a. 15%b. 30%c. 45%d. 60%
5. Vietri and colleagues reported that in a study of adults and children with moderate-to-severe hemophilia A or B, poorer adherence to therapy was significantly associated with all of the following except:
a. Physical health status in adultsb. Physical health status in childrenc. An increase in bleeding episodes requiring
replacement factor therapy in adultsd. Number of work or school days missed because of
bleeding episodes in adults and children
6. Development of inhibitors renders factor replacement ineffective in up to 30% of patients with hemophilia A and _______ of patients with hemophilia B.
a. 1%–2%b. 3%–5%c. 10%–15%d. 20%–30%
7. Investigators at University College London documented post–gene transfer factor IX (FIX) levels of 5%–10% and stable FIX levels beyond 30 weeks with adeno-associated virus-mediated delivery of an F9 transgene, although patients experienced:
a. Ketoacidosisb. Severe pyrexiac. Mild transaminitisd. Adenovirus infections
8. Patients with hemophilia A receiving prophylactic therapy with recombinant factor VIII (FVIII) who have prolonged intervals of circulating FVIII levels below ______ risk an increase in bleeding episodes and hemarthroses.
a. 1 IU/dLb. 2 IU/dLc. 3 IU/dLd. 4 IU/dL

CME/CNEPostTest
T H E H E M O P H I L I A R E P O R T | S p r i n g 2 0 1 4 43
9. Results of the Joint Outcome Study in children with severe hemophilia A:
a. Proved the superiority of episodic FVIII therapy over prophylactic FVIII therapy
b. Established the superiority of prophylactic FVIII therapy over episodic FVIII therapy
c. Were inconclusive as to whether episodic FVIII therapy or prophylactic FVIII therapy was superior
d. Were limited to physical examination of both ankles, knees, and elbows
10. The only treatment modality known to effectively eradicate immunologic inhibitors in patients with hemophilia A is:
a. Infusion of four-factor prothrombin complex concentrate
b. Early low-dose prophylaxis given in the absence of immunologic danger signals
c. Immunosuppression with systemic corticosteroids and cyclophosphamide
d. Immune tolerance induction
44 T H E H E M O P H I L I A R E P O R T | V o l u m e 1 N u m b e r 1
Instructions for Obtaining CME or CNE CreditTo receive CME or CNE credit for completing this free educational activity and a certificate from the CME/CNE provider:
• Study the educational material presented in this issue of The Hemophilia Report.
• Using pages 42–43 as a worksheet, answer all of the post-test questions based on the content of this issue.
• Visit www.HemophiliaReport.com on the Web by May 1, 2015 (for CME credit) or May 1, 2016 (for CNE credit), click CME/CNE Credit, read the information provided, and then click the appropriate link for physicians or nurses to apply for credit and take the post test and evaluation.
• The full text of each article is available at the HemophiliaReport.com Web site, should you need to refer to it again.
Your candid and thorough completion of this evaluation will help us improve the quality of our CME/CNE activities. Thank you for your participation. Strongly agree Agree Disagree1. As a result of this activity, I am more knowledgeable about the …
a. Genetic basis of hemophilia A and B and the differences between the two ❑ ❑ ❑types of hemophilia.
b. History of hemophilia management, current best practices in children and ❑ ❑ ❑adults, and the goals of ongoing research to better understand the disease.
c. Developments being made in the laboratory and currently being tested in ❑ ❑ ❑clinical trials to produce synthetic coagulation factors with longer half-lives than those of present blood products for treating hemophilia or that are less prone to stimulate the production of neutralizing antibodies (inhibitors).
d. Advantages and drawbacks of prophylactic versus on-demand therapy, the ❑ ❑ ❑controversies over therapeutic timing and individualizing therapy for different patients, the factors affecting patient compliance, and current strategies to individualize patient management.
Strongly agree Agree Disagree2. I found the content of this educational activity …
a. Clearly written and well organized. ❑ ❑ ❑
b. Accurate and timely. ❑ ❑ ❑
c. Related to its overall objectives. ❑ ❑ ❑
d. Free from commercial bias. ❑ ❑ ❑
e. Relevant to my own clinical practice. ❑ ❑ ❑
Yes No Don’t know3. Did the information you received from this CME/CNE activity:
a. Confirm the way you currently manage your patients? ❑ ❑ ❑
b. Suggest new options for managing your patients that you might apply ❑ ❑ ❑in the future?
Patient Board CME/CNE management review credit
4. I used the information in this CME/CNE activity for … (check all that apply) ❑ ❑ ❑
5. Approximately how long (in hours) did it take you to complete this activity, hours including this evaluation?
Evaluation