the growing impact of asymmetric catalysis - aldrichimica acta vol. 40 no. 3
DESCRIPTION
Palladium-Catalyzed Dynamic Kinetic Asymmetric Allylic Alkylation with the dPPBA LigandsDevelopment and Applications of C2-Symmetric, Chiral, Phase-Transfer CatalystsTRANSCRIPT

T h e G r o w i n G i m p a c T o f a s y m m e T r i c c aT a ly s i s
VOL. 40 , NO. 3 • 2007
Palladium-Catalyzed Dynamic Kinetic Asymmetric Allylic Alkylation with the DPPBA Ligands
Development and Applications of C2-Symmetric, Chiral, Phase-Transfer Catalysts

sigma-aldrich.com
DABAL-Me3
Trimethylaluminum is a versatile methylation reagent in organic synthesis. However, because of its pyrophoric nature, it cannot be handled in open air. Developed by the Woodward group (University of Nottingham, U.K.), DABAL-Me3 is a free-flowing solid adduct of trimethylaluminum and DABCO® that can be manipulated without the need for an inert atmosphere.1 This bench-stable reagent has been employed in numerous reactions including methylations of aldehydes and imines,1,2 methylation of aryl and vinyl halides,3 conjugate additions to enones,4 and amide-bond formation.5 In the presence of the appropriate chiral ligand and catalyst, many of these reactions can be performed asymmetrically.
(1) Woodward, S. Synlett 2007, 1490. (2) Mata, Y. et al. J. Org. Chem. 2006, 71, 8159. (3) Cooper, T. et al. Adv. Synth. Catal. 2006, 348, 686. (4) Alexakis, A. et al. Chem. Commun. 2005, 2843. (5) Novak, A. et al. Tetrahedron Lett. 2006, 47, 5767.
Bis(trimethylaluminum)–1,4-diazabicyclo[2.2.2]octane adduct (DaBal-me3)682101
N N AlMe3Me3Al
1 g[137203-34-0]C12H30Al2N2
5 g
FW: 256.34
White Catalyst for Allylic C–H OxidationProfessor Christina White’s group (University of Illinois) recently reported selective allylic C–H oxidation reactions catalyzed by a Pd(II)–bis-sulfoxide system that furnishes branched allylic esters from α-olefins and carboxylic acids.1 These reactions can be performed in an inter- or intramolecular fashion, the latter being capable of yielding highly functionalized, large-ring macrolactone products.2 Finally, the catalyst system allows for a one-pot sequential allylic oxidation–C–H arylation to afford the E arylated allylic ester from the corresponding olefin, carboxylic acid, and arylboronic acid.3
(1) Chen, M. S. et al. J. Am. Chem. Soc. 2005, 127, 6970. (2) Fraunhoffer, K. J. et al. J. Am. Chem. Soc. 2006, 128, 9032. (3) Delcamp, J. H.; White, M. C. J. Am. Chem. Soc. 2006, 128, 15076.
White Catalyst684821
S S PhPhOO
•Pd(OAc)2
250 mg[858971-43-4] C18H20O6PdS2
1 g
FW: 502.90
TarB-NO2 Reducing ReagentsIn conjunction with NaBH4, Singaram’s chiral TarB-NO2 boronic esters rapidly reduce prochiral ketones to optically active secondary alcohols with enantiomeric excesses as high as 99%.1-3 The reagents cleanly reduce aromatic ketones with high enantioselectivity and, in many cases, aliphatic ketones can be reduced with a similar degree of selectivity. Typically, TarB-NO2 reagents perform as well as, or better than, existing hydridic asymmetric reduction methods such as those employing DIP-Chloride™ or the CBS reagents.
(1) Kim, J.; Singaram, B. Tetrahedron Lett. 2006, 47, 3901. (2) Kim, J. et al. Org. Process Res. Dev. 2006, 10, 949. (3) Cordes, D. B. et al. Eur. J. Org. Chem. 2005, 5289.
3-Nitrophenylboronic acid d-tartaric acid ester, 1 M in THF (d-TarB-no2)682748
B
NO2
O
O
CO2H
CO2H
5 mLC10H8BNO8 25 mL
FW: 280.98
3-Nitrophenylboronic acid l-tartaric acid ester, 1 M in THF (l-TarB-no2)682713
B
NO2
O
O
CO2H
CO2H
5 mL[467443-01-2] C10H8BNO8
25 mL
FW: 280.98
N-tert-ButylbenzenesulfenamideIn the presence of NCS, N-tert-butylbenzenesulfenamide catalyzes the selective oxidation of a variety of primary and secondary alcohols to the corresponding aldehydes and ketones in high yield and under mild conditions.1,2 The catalytic oxidation tolerates various functional groups including silyl ethers, epoxides, urethanes, esters, and olefins. The reaction is particularly useful for the preparation of labile or easily epimerized aldehydes.
(1) Mukaiyama, T. Angew. Chem., Int. Ed. 2004, 43, 5590. (2) Matsuo, J.-i. et al. Tetrahedron 2003, 59, 6739.
N-tert-Butylbenzenesulfenamide, 97%681792
SNH
1 g[19117-31-8] C10H15NS
5 g
FW: 181.30
New Products from Aldrich R&DSigma-Aldrich Is Pleased to Offer Cutting-Edge Tools for Organic Synthesis
S S PhPhOO
•Pd(OAc)2
(10–20 mol %)
benzoquinone air, 45 °C
R + R'CO2H
or
R
OC(O)R'
H
CO2H
or
O
O
PMPO
O MeMe
OH
SNH (5 mol %)
NCS (1.1 equiv)PMP
O
O MeMe
CHOK2CO3, 4 Å MS, 0 °C
CH2Cl2, 1.5 h
94%
THF, rt, 30 min
L-TarB-NO2 (1 equiv)NaBH4 (2 equiv)
O OH
84%, 97% ee
DABCO is a registered trademark of Air Products and Chemicals, Inc. DIP-Chloride is a trademark of Sigma-Aldrich Biotechnology, L.P.
N N AlMe3Me3Al
enone
O
Me
cross-coupling
Me
RNH2
R'CO2Et
R' NHR
O
70–99%
79–99%
RCHOR Me
OH
89–95% ee
imine
R Me
HN
75–90%
P
82% eeDABAL-Me3
sigma-aldrich.com

57
VO
L. 4
0, N
O. 3
• 2
007
Aldrich Chemical Co., Inc. Sigma-Aldrich Corporation6000 n. Teutonia ave.milwaukee, wi 53209, Usa
To Place Orders
Telephone 800-325-3010(USA)FAX 800-325-5052(USA) or414-438-2199Mail P.O.Box2060 Milwaukee,WI53201,USA
Customer & Technical Services
CustomerInquiries 800-325-3010TechnicalService 800-231-8327SAFC™ 800-244-1173CustomSynthesis 800-244-1173Flavors&Fragrances 800-227-4563International 414-438-385024-HourEmergency 414-438-3850WebSite sigma-aldrich.comEmail [email protected]
General Correspondence
Editor:SharbilJ.Firsan,Ph.D.P.O.Box355,Milwaukee,WI53201,USA
Subscriptions
TorequestyourFREEsubscriptiontotheAldrichimica Acta,pleasecontactusby:
Phone: 800-325-3010(USA)
Mail: Attn: Mailroom Aldrich Chemical Co., Inc. Sigma-Aldrich Corporation P.O. Box 355 Milwaukee, WI 53201-9358
Email: [email protected]
International customers, please contact your localSigma-Aldrich office. For worldwide contact infor-mation,pleaseseetheinsidebackcover.
The Aldrichimica Acta is also available on theInternetatsigma-aldrich.com.
Aldrich brand products are sold through Sigma-Aldrich, Inc. Sigma-Aldrich, Inc., warrants that itsproducts conform to the information contained inthisandotherSigma-Aldrichpublications.Purchasermustdeterminethesuitabilityoftheproductforitsparticularuse.Seereversesideofinvoiceorpackingslipforadditionaltermsandconditionsofsale.
Aldrichimica Acta(ISSN0002–5100)isapublicationofAldrich.AldrichisamemberoftheSigma-AldrichGroup.©2007Sigma-AldrichCo.
VOL. 40, NO. 3 • 2007
“PLEASE BOTHER US.”
Professor Carsten Bolm of RWTH Aachen University, kindly suggested that we make 2-(trimethylsilyl)ethanesulfonyl chloride (SES-Cl). This reagent is employed to protect an amine in the form of its sulfonamide. In contrast to the harsh conditions sometimes needed to deprotect tosyl-protected amines, the SES group is readily cleaved under mild conditions using a fluoride ion source, regenerating the parent amine along with volatile byproducts. We have also prepared SES-NH2, a useful reagent for the introduction of a protected nitrogen atom into a substrate.1,2
(1) Weinreb. S. M. et al. Tetrahedron Lett. 1986, 27, 2099. (2) Ribière, P. et al. Chem. Rev. 2006, 106, 2249.
SiS
Cl
OOH3C
CH3H3C
SiS
NH2
OOH3C
CH3H3C
681334 2-(Trimethylsilyl)ethanesulfonyl chloride 1 g (SES-Cl) 5 g
681326 2-(Trimethylsilyl)ethanesulfonamide 1 g (SES-NH2)
Naturally, we made these useful reagents. It was no bother at all, just a pleasure to be able to help.
Do you have a compound that you wish Aldrich could list, and that would help you in your research by saving you time and money? If so, please send us your suggestion; we will be delighted to give it careful consideration. You can contact us in any one of the ways shown on this page and on the inside back cover.
TABLE OF CONTENTSPalladium-Catalyzed Dynamic Kinetic Asymmetric Allylic Alkylation with the DPPBA Ligands ..............................................................................................................................................................................................................................................59Barry M. Trost* and Daniel R. Fandrick, Stanford University
Development and Applications of C2-Symmetric, Chiral, Phase-Transfer Catalysts ..........77Takashi Ooi and Keiji Maruoka,* Kyoto University
ABOUT OUR COVEROarsmen at Chatou (oil on canvas, 81.2 × 100.2 cm) was painted in 1879 by the French impressionist painter, Pierre Auguste Renoir (1841–1919), on the river Seine west of Paris. His use of light fresh colors in this painting and throughout his career was the result of his love of paintings from the Rococo period and of his training in a porcelain factory as a young man.
Rowing was the foremost attraction at Chatou. The man in this boat—wearing the typical costume of a short jacket and a straw hat—may be the artist’s brother, Edmond. The man standing on the bank, similarly attired, is probably the painter Gustave Caillebotte, a devoted rowing enthusiast and a friend of Renoir. The woman is most likely Aline Charigot, who was his favorite model and later became his wife.
The painting captures the brilliance of sun and water, summer and youth. In the water, strong blues and white alternate. Their shimmering intensity is enhanced by the equally strong presence of orange in the boat’s reflection and the scarlet accent of Aline’s bow. Renoir has put into practice the principle of simultaneous contrast: colors are perceived stronger when juxtaposed with their opposites—orange with blue, for example, or green with red. The silky texture of Renoir’s feathery brushstrokes mirrors the languid and leisurely scene.
This painting is a gift of Sam A. Lewisohn to the National Gallery of Art, Washington, DC.
Joe porwoll, president aldrich chemical co., inc.
Photograph © Board of Trustees, National Gallery of Art, Washington.
sigma-aldrich.com
Trost Ligands for Asymmetric Allylic Alkylationasymmetric allylic alkylation is a versatile catalytic reaction allowing access to a diversity of chiral molecules. This transformation converts both enantiomers of the substrate into the same enantiomer of the product, allowing theoretical yields of 100% of one enantiomer. professor Trost developed a series of ligands based on diphenylphosphinobenzoic acid (DppBa) and used them with a variety of palladium complexes for the asymmetric allylic alkylation. These ligands perform with a high degree of enantioselectivity and high yields.
O O
MeO2CO
+ NHNs
(R,R)-DACH-PhPd2dba3•CHCl3 (1 mol %)
NEt3, THF O O
NNs
N
OH
OH
H
93% yield99% ee
DACH-Naphthyl Trost Ligands
OCO2Me
+HN NH
O
O O
HN NH
O
O O
(S,S)-DACH-Nap (5 mol %)Pd2dba3•CHCl3 (2.5 mol %)
(n-Bu)4NCl, CH2Cl2
85% yield91% ee
NH HNO O
PPh2 Ph2P
NH HNO O
PPh2 Ph2P
(R,R)-DACH-Phenyl Trost Ligand692808
(S,S)-DACH-Phenyl Trost Ligand692794
DACH-Phenyl Trost Ligands
(R,R)-DACH-Naphthyl Trost Ligand692778
(S,S)-DACH-Naphthyl Trost Ligand692786
For more information, see Professor Trost’s review in this issue.
Sold in collaboration with DowPharmaSM for research purposes only. US Patent 5739396 applies.
DACH-Pyridyl Trost LigandsHNNH
N
O
N
O
(R,R)-DACH-Pyridyl Trost Ligand692751
(S,S)-DACH-Pyridyl Trost Ligand692743
692808
692778
692743
Ovaa, H. et al. Chem. Commun. 2000, 1501.
Trost, B. M.; Schroeder, G. M. J. Org. Chem. 2000, 65, 1569.
Ph OCO2CH3 + NaHC(CO2CH3)2
Ph
HH
H3CO2CCO2CH3
+ PhH
CO2CH3
CO2CH3
(R,R)-DACH-pyridyl (15 mol %)(C2H5CN)3Mo(CO)3 (10 mol %)
THF, rt
4970% yield99% ee (major)
: 1
Trost, B. M.; Hachiya, I. J. Am. Chem. Soc. 1998, 120, 1104.

59
VO
L. 4
0, N
O. 3
• 2
007
Outline1. Introduction2. DYKATthroughConversionofaRacemicSubstrate intoa
MesoIntermediate 2.1. AcyclicSubstrates 2.2. CyclicSubstrates 2.3. ConduritolBSubstrates3. DYKATthroughEnolizationoftheNucleophile 3.1. StabilizedEnolates 3.2. NonstabilizedEnolates 3.3. Azlactones4. DYKAT through Rapid p–s–p Interconversion of
Intermediates 4.1. VinylEpoxidesandAziridinesasSubstrates 4.2. Baylis–HillmanAdductsasSubstrates 4.3. AcyloxyenoatesasSubstrates 4.4. AllenesasSubstrates5. OtherDYKATProcesses6. ConclusionsandOutlook7. Acknowledgment8. References
1. IntroductionThesynthesisofchiralmoleculesisaprominentthemeinorganicchemistry.Thesyntheticcommunityhascomeunder increasedpressuretopreparesyntheticbuildingblocksinanenvironmentallybenignor“green”manner.Tominimizewaste,synthesesshouldbedesignedascatalytictransformationsandshouldtakeplaceinanefficientandatom-economicalfashion.1Asymmetriccatalysishasenabledthecost-effectivepreparationofthesebuildingblocks.Onesuchgeneralmethodisthepalladium-catalyzedasymmetricallylicalkylation(AAA).Themethodologyhasdemonstrateditsabilitytoaffordchiralitythroughnumerousenantiodiscriminatingevents.2AlthoughseveralreviewshavebeenpublishedonAAA,3,4nonehas focusedon themanypalladium-catalyzeddynamickineticasymmetrictransformations(DYKATs)thathavebeendeveloped.Toourknowledge,theonlypalladium-catalyzedDYKATs,whereinasymmetricinductionresultsfromthechiralityofthepalladium
ligand,arethosethattakeplacethroughAAAs.Thisreviewwillfocusonthescopeandsyntheticutilityofthepalladium-catalyzeddynamickineticAAAwithourdiphenylphosphinobenzoicacid(DPPBA)andrelatedfamilyof ligands(Figure 1).Thesebasicligands are constructed with o-diphenylphosphinobenzoic ornaphthoicacidmoieties tetheredbyachiraldiaminebackbone.Themostcommonoftheseligandsarethestandard(LS),naphthyl(LN),stilbene(LST),andanthracene(LA)ones.Theformertwoarecommerciallyavailable.
Thereareseveralgeneralmechanismsforasymmetricinductionin catalyzed transformations. The most common one deriveschiralityfromaprochiralsubstrate,typicallythroughdifferentiationoftheenantiotopicpfaces(Scheme 1).Otherasymmetricprocessesutilizearacemicsubstrate.Inthesecases,thetransformationcanproceedthrougheitherakineticresolutionorDYKAT.5Akineticresolution(KR)resultswhentheenantiomersofaracemicsubstrateareconvertedtothechiralproductsatdifferentrates(Scheme 2).Numerouscatalyticandenzymatic transformationshaveshownhighenantioselectivityforsuchaprocess.Inthebest-casescenario,onlyonesubstrateenantiomerreactsforatheoreticalmaximumyieldof50%,inadditiontothe50%ofrecoveredstartingmaterial.Assuch,thisprocessisnomoreefficientthanaphysicalresolution.Toovercomethislimitation,severalprocessescommonlyknownas dynamic kinetic resolutions (DKRs) have been developedwhereinbothenantiomersofthesubstrateareconvertedintothesameenantiomeroftheproduct.Thisallowsforatheoretical100%yield.Aresolutionimpliesseparationofaracemicsubstrateintoitsenantiomers.Therefore,wepreferthe phrasedynamickineticasymmetrictransformation(DYKAT)6ratherthandynamickineticresolution,sincetheseprocessesarenotresolutionsasthelatterphraseimplies.Currently,therearethreegeneralprocessesforaDYKAT.Inthefirstonethesubstraterapidlyracemizesunderthereactionconditionsandthesubsequenttransformationisselectivefor one substrate enantiomer (Scheme 3).The secondDYKATconvertsthesubstrateintoamesooraprochiralintermediate,andthesubsequentasymmetricinductionresultsfromdifferentiationoftheenantiotopicterminiorfacesofthisintermediate(Scheme 4).TopreventaKR,bothsubstrateenantiomersmustbecompletelyconverted into the meso intermediate. The third DYKAT is
Palladium-Catalyzed Dynamic Kinetic Asymmetric Allylic Alkylation withthe DPPBA Ligands
Barry M. Trost* and Daniel R. FandrickDepartment of ChemistryStanford UniversityStanford, CA 94305-5080, USAEmail: [email protected]
ProfessorBarryM.Trost Dr.DanielR.Fandrick

60
Palla
dium
-Cat
alyz
ed D
ynam
ic K
inet
ic A
sym
met
ric A
llylic
Alk
ylat
ion
with
the
DPP
BA L
igan
dsV
OL.
40,
NO
. 3 •
200
7
Figure 1. The most commonly Used DppBa ligands.
HNNHOO
PPh2Ph2P
HNNHOO
PPh2Ph2P
HNNHOO
PPh2Ph2P
NH N
H
OO
PPh2 Ph2P
(S,S)-LS (S,S)-LN
(S,S)-LST (S,S)-LA
accomplished througha rapid interconversionof intermediates,andasymmetricinductionresultsfromaselectivereactionwithoneintermediate(Scheme 5).Consideringthatenantioselectivityresults from a kinetic reaction of one intermediate, the thirdDYKAT requires a Curtin–Hammett condition be establishedwhereintheenantioselectivityisnotdependent,inprinciple,uponthethermodynamicratiooftheintermediates.
Palladium-catalyzed dynamic kinetic AAAs have beenaccomplishedprimarilybyusingthelattertwoDYKATprocesses.Morespecifically,DYKATshavebeenachievedthroughconversionofthechiralsubstratesintoapseudo-mesoorprochiralintermediate,orthrougharapidp−s−pinterconversionbetweentheenantiotopicfacesofthep-allylPd(II)intermediate.
2. DYKAT through Conversion of a Racemic Substrate into a Meso IntermediateThe most general type of palladium-catalyzed DYKAT proceedsthrough a pseudo-meso-π-allylPd(II) intermediate. In this process, oxidativeadditionofeachenantiomeraffordsdifferentunsymmetricalp-allylPd(II)complexes,whichrequiresequilibrationtoaneffectivelysymmetrical complex for high enantioselectivity. Asymmetricinduction subsequently results from enantiodiscrimination of thetermini(Scheme 6).Duetothechiralcatalyst,thetwoenantiomericsubstrates undergo oxidative addition with palladium at differentrates. Therefore, for a successful DYKAT, complete substrateconversion into the π-allylPd complex is required. Two basic types ofallylicsubstrateshavebeenemployedinthiskindofDYKAT.Theπ-allylPd complexes of acyclic substrates adopt the preferred syn,syn conformation and, due to conformational restrictions, the π-allylPd complexes of cyclic substrates adopt the anti,anti conformation(Figure 2).7Althoughthesecomplexesarestructurallydistinct,theirreaction scopes and efficiencies are similar.3,4AAA of the acyclicsystemspreferentiallygeneratesthetransallylicproductsasaresultoftheformationofthefavoredsyn,synintermediates.
2.1. Acyclic SubstratesThe most common palladium-catalyzed DYKAT involves theasymmetric allylic alkylation of 1,3-diphenyl-3-acetoxypropene(1)(eq 1).8EarlyresultsshowedonlymoderateenantioselectivitieswiththesodiumsaltofthenucleophileandBINAP(R1=Me;81%,50%ee)orBINAPO(R1=Me;75%,68%ee) ligands.However,this reactionhasbecome thestandard test fornew ligands.3,4Asaresult,extensiveresearchhasbeenfocusedonthedevelopmentofalargenumberofdiversechiralligandsforthistransformation.9Highenantiomericexcesseshavebeenobtainedwithmanytypesofchiralligands such as chiraphos (3) (R1=AcNH;98%,86%ee),10P–Nligand4(R1=H;99%,99%ee),11sparteine(5)(R1=H;77%,75%ee),12 isosparteine (6) (R1 =AcNH; 90%, 92% ee),13 and Evans’sP–S ligand7 (R1=H;97%,98%ee).14Auseful extension to thefluorous ligand 8hasenabledhighselectivity(R1=Me;96%,90%ee) for an easily recyclable catalyst.15 However, substrate 1 is theleastsensitiveindeterminingtheasymmetricinductionabilityofthechiralcatalyst.
AlthoughtheDPPBAligandstypicallyaffordlowconversionsandenantioselectivitiesfortheparentsubstrate,1,theseligandshave demonstrated high levels of asymmetric induction withthe more challenging carbonate, 9. This discrepancy has beenrationalized by the DPPBA ligands encountering unfavorablestericinteractionswiththelargersubstrate1.However,duetothisstericallyrestrictivechiralenvironment,DPPBAsaresomeofthemostgeneralligandsforthepalladiumcatalyzedAAA.Forexample,highenantioselectivityandyieldfortheDYKATwithcarbonate9
Scheme 1. Typical asymmetric induction.
S product
R product
k1
k2
k1 >> k2prochiral substrate
Scheme 2. Kinetic resolution (Kr).
S product
R product
k1
k2
k1 >> k2racemic
substrate
Scheme 3. DyKaT through racemization of the substrate.
S product
R product
k1
k2
k1 >> k2
S substrate
R substrate
Scheme 4. DyKaT through conversion to a meso or prochiral intermediate.
S product
R product
k1
k2
k1 >> k2
S substrate
R substrate
meso orprochiral
intermediate
k1'
k2'
Scheme 5. DyKaT through the rapid interconversion of intermediates.
S product
R product
k1 >> k2
S substrate
R substrate
S intermediate
R intermediate
k1
k2
Scheme 6. DyKaT of symmetrical allylic substrates.
R R
LG
R R
LG
R R
Pd(II)Ln
k1'
k2'
k1
k2
R R
Nu
R Rk2k1
R R
NuNu≡

61
Barr
y M
. Tro
st*
and
Dan
iel R
. Fan
dric
kV
OL.
40,
NO
. 3 •
200
7
havebeenachievedbyutilizingthestandarddiaminocyclohexyl(DACH)ligandLS(eq 2).16
These initial results revealed the need for establishing asymmetrical π-allylintermediateforasymmetricinductionoronethatbecomestheequivalentofasymmetricalspeciesbecauseofrapidlyequilibratingnonsymmetricalstructures.Ourgroupalsoobserved that theenantioselectivityof theAAAfor theacyclicsubstrate9 wasdependentuponthesizeofthecountercationofthenucleophile.16Withthesodiumsaltofmalonate,only29%eewasobtained,buttheenantioselectivityincreasedto92%asthesizeofthe cation increased with the use of the cesium salt. The π-allylPd(II) intermediate from the initial oxidative addition is proposed tobe a tight ion pair, which requires relaxation to the necessarysymmetrical intermediate forhighasymmetric induction.17Theexactnatureoftheasymmetrymayderivefromtheconformationsofthemetal-boundligand,althoughotherexplanationshavealsobeenproffered.Reactionswithscalemicsubstratesanddifferentenantiomers of the ligand demonstrated a moderate memoryeffect supporting the requirement forequilibration.Thehigherenantiomericexcessobtainedwithcesiumwassuggestedtoderiveinpartfromaslowerrateofalkylationwithaneffectivelylargernucleophile that allowed for sufficient relaxation of the π-allylPd(II) intermediate.Additionally,thecesiumnucleophilemayalsoafforda less tight ion pair and lead to a faster equilibration. FurthersupportforthisrequirementforrelaxationwasobtainedwhenaderivativeoftheTrostligand withdipodalarms,13,furnishedhighenantioselectivityandreactivityintheAAAwiththepreviouslysluggishsodiumsaltofthemalonatenucleophile(eq 3).18Apossibleexplanationrelatestoafasterequilibrationbycoordinationofthecationtothepolyethersidechainoftheligand.AsimilarbaseandcountercationeffectontheenantioselectivitywasobservedwithBINAP-basedligands.19
Othercarbonnucleophiles,inanalogytomalonate,undergothistypeofDYKATwithhighasymmetricinduction.Thestandardligand,LS,alsoaffordshighdiastereoselectivityfortheadditionofnitroethane(eq 4).20Thisexampledemonstratestheabilityofthe catalyst to simultaneously discriminate between both theenantiotopicterminioftheallylligandandtheenantiotopicfacesoftheenolizednucleophile.
BarbituratesarealsoeffectiveandusefulsoftcarbonnucleophilesforDYKAT.21Utilizationofthestandardligand,LS,andafluorideadditive to slightly improve the ee (vide infra), led to goodenantioselectivityintheAAA(Scheme 7).Simplehydrogenationoftheinitialproductcompletedtheconcisesynthesisofpentobarbital,asedativeandhypnoticagent.
SimilartootherAAAs,numeroussoftheteroatomnucleophilescanbeemployedforDYKAT.InanextensionoftheGabrielaminesynthesis,highenantioselectivitywasachievedfortheasymmetricallylicalkylationwiththestandardligand,LS,andphthalimideasthenucleophile(Scheme 8).16,22Inthisexample,DYKATtoleratedtheunprotectedalcoholfunctionality,andtheproductprovidedausefulbuildingblockforthepreparationofpolyoxamicacid,thenovelaminoacidinseveralantifungalagents.16
2.2. Cyclic SubstratesSimilartoacyclicelectrophiles,palladium-catalyzeddynamickineticAAAsofcyclicsubstratesaffordexcellentenantioselectivitiesforabroad rangeof softnucleophiles. In thebasicalkylation,excellentenantioselectivities were achieved with malonate and phthalimidenucleophilesfor5-,6-,and7-memberedsubstrates(Scheme 9).23Asinthecaseoftheacyclicsubstrates,thenatureofthecountercationandmalonatenucleophilehadadramaticeffectontheenantioselectivity,which again emphasizes the importance of equilibration to the
Figure 2. coordination Geometries for acyclic and cyclic
p-allylpd complexes.
R R
Pd+
acyclicsyn,syn
conformation
R RPd+
cyclicanti,anti
conformation
eq 1
Ph Ph
OAc
Ph Ph
R2O2C CO2R2R1R2O
O
OR2
OM
Pd(0), ligand*
rac-1 2
PPh2 N
O
PhN N
N N
Ph2PPPh2
PO
i-Prt-BuS
Ar
Ar NPPh2MeO
N OR
RO
3chiraphos
4 5sparteine
6isosparteine
7Ar = 1-naphthyl
8R = CO(CF2)10CF3
R1
Ref. 8,10–15
eq 2
OCO2MeMeO2C CO2Me
[η3-C3H5PdCl]2 (2.5 mol %)CH2(CO2Me)2
Cs2CO3, CH2Cl2rac-9
(R,R)-LS (7.5 mol %)
1098%, 92% ee
Ref. 16
eq 3
BnO2C CO2Bn
[η3-C3H5PdCl]2 (0.2 mol %)CH2Cl2
BnO
O
OBn
ONa
Ar = O OO
HNNHO O
PAr2 Ar2P
rac-9 +
11 1268%, 90% ee
(R,R)-13 (0.6 mol %)
(R,R)-13
Ref. 18
eq 4
OCO2Me NO2
Pd2dba3•CHCl3 (0.25 mol %)BSA, CH2Cl2, (n-Bu)4NCl
NO2H
MeH
(S,S)-LS (0.75 mol %)
14 15 1671%, 11:1 dr, 97% ee
+
BSA = N,O-bis(trimethylsilyl)acetamide
Ref. 20
Scheme 7. DyKaT with Barbiturates and application to the
synthesis of pentobarbital.
Pd2dba3•CHCl3 (2.5 mol %)CH2Cl2, TBAT
(R,R)-LS (5 mol %)HN NH
O O
O
HN NH
O O
O
H2, Pd/C
HN NH
O O
O
rac-9 +
17 1896%, 72% ee
pentobarbital99%
MeOH
TBAT = tetra-n-butylammonium triphenyldifluorosilicate
Ref. 7
Ref. 21

62
Palla
dium
-Cat
alyz
ed D
ynam
ic K
inet
ic A
sym
met
ric A
llylic
Alk
ylat
ion
with
the
DPP
BA L
igan
dsV
OL.
40,
NO
. 3 •
200
7equivalent of a symmetrical meso-π-allylPd(II) intermediate. The additionoftetrahexylammoniumbromide(THABr)increasedtheeefrom38%to82%inTHF.Afurtherincreaseintheenantioselectivityto98%wasobtainedbyutilizingmethylenechlorideasthesolvent.Reetzetal.observedthattetra-n-butylammoniummalonateexistsasadimerinpolarsolvents,24andattributedtheeffectsoftheadditiveandsolventtovariationsinthenatureofthenucleophileandsubstrateion pairs in solution. Therefore, tetraalkylammonium malonateis effectively larger than the sodium counterpart and, by analogyto the acyclic substrates, allows the equilibration of the initiallyformed π-allylPd(II) intermediate to the effectively symmetrical complex.17,18
The DYKAT of cyclic substrates with nitromethane25 andbarbiturates21affordsexcellentenantioselectivitieswiththestandardandnaphthylligands(Scheme 10).Inthelatterexample,theAAAprovidedaconciseandefficientsynthesisofcyclopentobarbital,asedativeandhypnoticagent.
The dynamic kinetic asymmetric addition of oxygennucleophilestoracemicsubstratesisoneofthemoresyntheticallyusefulDYKATsforthesynthesisofcomplexnaturalproducts.ThesimplestreactionistheformalderacemizationofallylicalcoholsbythedynamickineticAAAwithcarboxylatenucleophiles.26Inorder toobtainhighenantioselectivities,both thematchedandmismatchedoxidativeadditionof thesubstratemusteffectivelycompetewithionizationoftheproduct,otherwisetheproductwillequilibratetotheracemate.Theutilizationofthecarbonateleavinggroupandacarboxylatenucleophilehasprovedeffective,providinghighenantioselectivitiesforthetypicalcyclicsubstrate32withavarietyofcarboxylicacidnucleophiles(eq 5).Themethodologywasappliedsuccessfullytotheracemicallyliccarbonate35,whichfurnishedpivalate37inhighyieldandeetoconstituteaformalsynthesisoftheantitumoragentphyllanthocin(Scheme 11).27
Extendingouruseofcarbonateandbicarbonatenucleophiles,28Gais and co-workersdevelopedanotherpracticalmethod for asimilarderacemizationofallyliccarbonates.29Inthisprocedure,thereactionproceedsinhighenantioselectivitythroughalkylationwithbicarbonateandsubsequent in situdecarboxylation to thechiralallylicalcohol(eq 6).AnattractivefeatureofthisAAAisthathydrolysisoftheionizedcarbonateleavinggroupinsitugeneratesthebicarbonatenucleophile.Thereactionisgeneralforbothacyclicandcyclicsubstratesandrequirestheuseofallyliccarbonates.
One of the most synthetically useful alkylations is with2-halophenols.AfterAAAwiththesenucleophiles,asubsequentintramolecularHeckreactioncanconstructthedihydrobenzofurancoreofnumerousbiologicallysignificantnaturalproducts.Inthepresenceofthestilbeneligand,LST,carbonate40providedefficientlyand highly enantioselectively 41, an intermediate in the totalsynthesisof(–)-galanthamine30and(–)-morphine31(Scheme 12).TheDYKATbetween39and40 illustratesthetolerancebythecatalystofarylbromidesandfunctionalityinthe2positionoftheelectrophile.
Similarly, AAA with sulfonamide nucleophiles furnishedsynthetically valuable protected amines.32 An interestingexampleofthisalkylationisthehighlyenantioselectivereactionof cyclopentene 42 (Scheme 13).33 Due to the inversion in theoxidativeaddition,34 thepalladiumcatalyst ispositionedon thesamefaceofthecyclopenteneastheacetonidesubstituentwhich,by this example, did not hinder the AAA. Ring-closing–ring-openingmetathesisandsubsequenttransformationsoftheDYKATproduct43quicklyfurnishedausefulentryintothesynthesisofindolizidinealkaloids.
Additionally, modification of the standard ligand, LS, wasrequired toobtainhighenantioselectivity in the intramolecular
Scheme 8. DyKaT with phthalimide and application to the
synthesis of polyoxamic acid.
NPd2dba3•CHCl3 (2.5 mol %) Cs2CO3 (10 mol %), THF
OH HN
O
OHO OH
O O
+
HO
O
OHNH2
OH
OH19 20 21
87%, 82% ee
polyoxamic acid
(R,R)-LS (7.5 mol %)O
Ref. 16
Scheme 9. Dynamic Kinetic aaa of cyclic substrates with
malonate and phthalimide nucleophiles.
[η3-C3H5PdCl]2 (2.5 mol %)Hex4NBr, CH2Cl2, 0 oC
OAc
n
OMe
ONa
MeO
O
+
n = 1; 81%, 98% een = 2; 86%, 96% een = 3; 99%, 93% ee
NK
O
O
+
n
NO
O
n = 1; 87%, 94% een = 2; 95%, 97% een = 3; 84%, 98% ee
OCO2Me
n
n
CO2MeMeO2C
(R,R)-LS (7.5 mol %)
22 23 24
25 26 27
[η3-C3H5PdCl]2 (2.5 mol %)Hex4NBr, CH2Cl2, 0 oC
(R,R)-LS (7.5 mol %)
Ref. 23
Scheme 10. DyKaT of cyclic substrates with nitromethane and
Barbiturates.
HN NH
O O
O
HN NH
O O
OOCO2Me
+
O
O(S,S)-LS (6 mol %)
Pd2dba3•CHCl3 (2 mol %)
(n-Bu)4NCl, BSA, CH2Cl2then CH2N2
CO2Me
NO2+ MeNO2
rac-28 2974%, 99% ee
rac-30 31 cyclopentobarbital85%, 91% ee
(S,S)-LN (5 mol %) Pd2dba3•CHCl3 (2.5 mol %)
(n-Bu)4NCl, CH2Cl2
Ref. 21,25
eq 5
[η3-C3H5PdCl]2 (2.5 mol %)Hex4NBr, CH2Cl2
OCO2Me O
O(R,R)-LS (7.5 mol %)
rac-32 33 3491%, 98% ee
+ EtCO2Na
Ref. 26
Scheme 11. application of the Deracemization of allylic
carbonates to the formal synthesis of phyllanthocin.
CO2Me
OCO2Me
CO2Me
O t-Bu
O
O CO2Me
H
H
O
O
rac-35 36
3798%, 93% ee(+)-phyllanthocin
+ t-BuCO2Na[η3-C3H5PdCl]2 (2.5 mol %)
Hex4NBr, CH2Cl2
(R,R)-LS (7.5 mol %)
O
PhO
Ref. 27

63
Barr
y M
. Tro
st*
and
Dan
iel R
. Fan
dric
kV
OL.
40,
NO
. 3 •
200
7
eq 6
Pd2dba3•CHCl3 (2 mol %)CH2Cl2, H2O
OCO2Me OH(R,R)-LS (8 mol %)
rac-32 3894%, 99% ee
Ref. 29
Scheme 12. DyKaT with phenols and its synthetic applications.
OHMeO Br
CHOCO2Me
TrocOCO2Me
OBr
OHC
OMe
O
OHH
N
OH
[η3-C3H5PdCl]2 (1 mol %) Et3N, CH2Cl2
(S,S)-LST (3 mol %)
O
OMe
OH
N
39 40 4172%, 88% ee
(–)-galanthamine (–)-morphine
+
Ref. 30,31
cyclizationleadingtotheazabicyclo[4.2.1]noneneDYKATproduct,47(Scheme 14).35Standardtransformationsof47furnishedthe“veryfastdeathfactor”anatoxin-a.TwoexplanationsarepossibleforthedifferentresultsobtainedwithLSand45.Asdiscussedpreviously,for high asymmetric induction to occur, the initial p-allylPdintermediatemustequilibrateinordertofunctionasameso-likeintermediatepriortoalkylation.Previousexamplesdemonstratedthat the larger nucleophiles afford a slower alkylation, whichallowsthenecessaryequilibrationtotakeplace.Inthisexample,andbecausethealkylationoccursintramolecularly,thecyclizationisfastandcompeteswiththeequilibration.Thiseffect is likelyoccurringwiththestandardligand,LS.Duetocoordinationtothepyridinefragmentofthemodifiedligand45, theelectrophilicityof the p-allylPd(II) complex is decreased thereby slowing thealkylationandallowingtherequiredequilibrationtotakeplace.Analternativeexplanationisthatabackgroundreactionmaycompetewiththemetal-catalyzedprocess.Usingastericallylesshinderedandamoreelectron-richPd(0)complexthatwouldformwiththepyridylligand45,afasteroxidativeadditionmaythenallowthemetal-catalyzedprocesstoout-competethebackgroundreaction.
Heterocycles are also effective nucleophiles in DYKAT.Applicationofthetypicalconditionswiththestandardligand,LS, andCs2CO3allowedthepreparationofindolocarbazoleproaglyconswithhighenantioselectivity(eq 7).36Additionally,themoreacidicindolewasselectivelyalkylated.
BurgerandTungereportedaninterestingexample,whereintheallylicalkylationwasperformedwithaketoneenolateforaformalasymmetric Claisen reaction.37 In this case, decarboxylation38oftheinitiallyformedβ-ketocarboxylatep-allylPd(II)complexafforded the reactive enolate nucleophile, 53 (eq 8). Good-to-excellentenantioselectivitieswereachieved forbothcyclicandacyclicsubstrates.Interestingly,thereactionproceededthroughcoordinationofthecarboxylatetothepalladium(II)intermediateor, namely, through a covalently bonded “ion pair” which hassufficientabilitytoequilibrate.Nocrossoverwasobservedinatestreaction,suggestingalackofsignificantdissociationoftheionpairpriortoalkylation.However,theasymmetricinductionobservedisconsistentwithalkylationoccurringonthefaceoftheallylligandoppositethepalladium.
In summary, the current technology has achieved highenantioselectivities in thedynamickineticAAAofacyclicandcyclicsubstratesthataffordasymmetricalallylicintermediate.Forasymmetricinductiontooccur,andinadditiontousingachiralcatalyst,conditionsmustbeemployedthatallowrelaxationofthesubstratestotheeffectivemesop-allylintermediate.
2.3. Conduritol B SubstratesA valuable cyclic substrate for the dynamic kineticAAA is tetra-acylatedconduritolB,55.Forthissystem,oxidativeadditionoftheracemicsubstratewiththePdcatalystfurnishesamesointermediate,56,inwhichasymmetricinductionoccursbytheselectivealkylationof one terminus (Scheme 15).39 For a successful DYKAT, bothenantiomers of 55 must completely ionize to the symmetricalintermediate, albeit at different rates.Dialkylationof the substratecanalsooccurbyionizationoftheinitialproduct,57,followedbyanother regio- and enantioselective alkylation. In both cases, fourstereocenters are established in one asymmetric transformationthroughaDYKATofracemicconduritolB.
In the AAA of tetraacetate 62 with a pivalate nucleophile,a kinetic resolution was observed with good regio- andenantioselectivity (eq 9).40 This result demonstrates the differentrates of the oxidative addition. Utilization of the more activatedtetracarbonatesubstrate66andsodiumbenzoateasthenucleophile
Scheme 13. DyKaT with carbonate 42.
Pd2dba3•CHCl3(1 mol %)Et3N, THF
O O
MeO2CO
NHNsO O
NNs
NH
OH
OH
(R,R)-LS
rac-42 4393%, 99% ee
44
+
Ref. 33
Scheme 14. intramolecular asymmetric cyclization of racemic carbonate 46.
TsHN
OCO2Me
CO2Me
NTs
MeO2CHN
O
HNNHO O
NPPh2
Pd2dba3•CHCl3 (2.5 mol %) CH2Cl2
45 (7.5 mol %)
rac-46 4790%, 88% ee
anatoxin-a
Ref. 35
eq 7
Pd2dba3•CHCl3 (1 mol %) Cs2CO3, THFN
HNH
HN
OO
NNH
HN
OO
H
OAc
+
rac-48 49 5099%, 79% ee
(S,S)-LN (3 mol %)
Ref. 36

64
Palla
dium
-Cat
alyz
ed D
ynam
ic K
inet
ic A
sym
met
ric A
llylic
Alk
ylat
ion
with
the
DPP
BA L
igan
dsV
OL.
40,
NO
. 3 •
200
7
eq 8
Pd2dba3•CHCl3 (0.2 mol %)benzene, reflux
O
O
O
Pd(II)O
OO Pd(II)
O
O
51 52 53
5475%, 94% ee
(R,R)-LS (0.4 mol %) – CO2
Ref. 37
Scheme 15. DyKaT mechanism with conduritol B substrates.
OCOR
OCOROCOR
OCOR
OCOR
OCOROCOR
OCOR
OCOR
OCOR
OCOR
LnPd(II)OCOR
OCORNu
OCOR
OCOR
OCOROCOR
Nu
OCOR
OCORNu
Ln(II)Pd
OCOR
OCOR
Nu
Ln(II)Pd
OCOR
OCORNu
Nu
OCOR
OCORNu
Nu OCOR
OCORNu
NuOCOR
OCOR
NuNu
matched ionization
mismatchedionization
matchedattack
mismatchedattack
matched ionization
mismatchedionization
mismatchedattack
matchedattack
matchedattack
55 ent-55
56
57 ent-57
58 59
60 61 ent-60 ent-61
Ref. 39
enabled complete consumption of the mismatched enantiomerand ultimately good enantioselectivity for the dynamic kineticasymmetric di(allylic substitution) (Scheme 16).39 The fourstereocenters in aminocyclohexitol 68 of hygromycin A wereefficientlyestablishedbyuseofthisDYKAT.41
Thechoiceofnucleophileofferscontrolforeithermono-orpolyalkylation.SoftnucleophilessuchasMeldrum’sacid,(phen-ylsulfonyl)nitromethane,andphthalimideaffordmonoalkylationwith good enantioselectivity. Notably, these monoalkylationsdemonstrate how, under the appropriate conditions, themismatchedionizationofthesubstratecansuccessfullycompetewiththematchedionizationofthemonoalkylatedproduct.Thesulfonylnitromethane DYKAT product has been applied tothe efficient synthesis of the HIV inhibitor (–)-cyclophellitol(Scheme 17).42
3. DYKAT through Enolization of the NucleophilePalladium-catalyzedAAAshavedemonstratedauniqueability tonot only afford enantiodiscrimination of the π-allyl electrophile, but also to effectively differentiate the enantiotopic faces of anucleophile. This property opened a new avenue for palladium-catalyzedDYKATs(Scheme 18).For this typeof transformation,the racemic nucleophile is enolized into the active achiralenolate, wherein asymmetric induction results from the catalystdiscriminatingbetweentheenantiotopicfacesoftheenolate.Duetotheconversionoftheracemicsubstrateintotheprochiralnucleophileoccurringwithoutinvolvementofthechiralcatalyst,therateoftheenolizationforbothenantiomersshouldbeidenticalandcircumventany possible memory effect. Asymmetric alkylations of racemicenolizable nucleophiles are typically not considered DYKATs.However,aDYKAToccurswhenbothenantiomersofthesubstrateareconvertedintooneenantiomeroftheproductwithatheoreticalyieldof100%.Ifoneconsiderstheoverallalkylationofaracemicnucleophile, reactionswherein thenucleophile isconverted intoaprochiralenolateorintermediateeveninaprevioustransformationaretechnicallyDYKATs.
3.1. Stabilized EnolatesThe most common palladium-catalyzed AAA wherein chiralityis established at the nucleophile is with substrates that afford astabilizedprochiralenolate.Becauseofthisstabilization,onlymildconditionsarenecessarytogeneratetheactivenucleophile.Inthepalladium-catalyzedAAA,thechiralligandispositionedonthesideofthemetaloppositetheallylligandinasquare-planargeometry.The low-to-moderate enantioselectivities observed with typicalchiralbidentateligands(suchasDIOPandtheP–Noxazolidinone)43havebeenattributedtothedistantchiralenvironmentnoteffectivelydifferentiatingbetweentheenantiotopicfacesofthenucleophile.Inanefforttoextendthechiralenvironmenttothenucleophile,severalgroups developed a series of chiral ferrocenylphosphine ligands,whichincorporateatetheredfunctionalgroup,tointeractwiththenucleophile and enhance the interaction between the nucleophileandligand.44Incontrast,theDPPBAligandshaveshownexcellentasymmetricinductionforthecreationofchiralityatthenucleophilewithout the requirement of an appendant functional group. Forexample, the asymmetric allylation of β-keto ester 71 proceededinhighyieldandenantioselectivitywithuseofthenon-ionicbaseN,N,N’,N’-tetramethylguanidine(TMG)(Scheme 19).45Theutilityofthisalkylationwasdemonstratedinthesynthesisofnitramine,abiologicallyactivespiroalkaloid.Witharacemicelectrophile,thereactionachievedexcellentdiastereoselectivity,againdemonstratingthecatalyst’sabilitytosimultaneouslydiscriminatetheenantiotopicfacesofthenucleophileandterminioftheelectrophile(eq 10).45
eq 9
OAc
OAcOAc
OAc
[η3-C3H5PdCl]2 (1 mol %) (n-Bu)4NBr, CH2Cl2, H2O
OAc
OAc
OAc
OAc
OAc
OAcOAc
O
t-Bu
O
OAc
OAcO
O
t-Bu
O
t-Bu
O
t-BuCO2Na(R,R)-LS (3 mol %)
rac-62 6350%, 83% ee
(unreacted ent-62)
6444%, 97% ee
651%
++
Ref. 40
Scheme 16. DyKaT of conduritol B Tetracarbonate (66).
OTroc
OTrocOTroc
OTroc
[η3-C3H5PdCl]2 ( 2.5 mol %) Hex4NBr, CH2Cl2, H2O
OTroc
OTrocOCOPh
OCOPhNH2
OHOCOPh
OH
O
O
PhCO2Na(S,S)-LS (7.5 mol %)
rac-66 6790%, >99% ee
68
Ref. 39,41
Scheme 17. DyKaT of conduritol B with sulfonylnitromethane.
OTroc
OTrocOTroc
OTroc
(R,R)-LS (7.5 mol %)Pd2dba3•CHCl3 ( 2.5 mol %)
Cs2CO3, THF
OTroc
OTrocOTroc
NPhO2SONa
O–
+
PhO2S NO2
OH
OHOH
OH
O
rac-69 7081%, 88% ee (–)-cyclophellitol
Ref. 42

65
Barr
y M
. Tro
st*
and
Dan
iel R
. Fan
dric
kV
OL.
40,
NO
. 3 •
200
7
Thechiral3-substitutedindolineand3H-indolestructuralmotifsarepresentinnumerousbiologicallyactivecompounds.Asymmetricallylic alkylations of racemic oxindoles provide a valuable andefficiententryintothepreparationoftheseimportantheterocycles.Becauseofthearomaticstabilizationobtainedthroughenolizationofoxindoles,onlymildconditionsarenecessaryto generatetherequirednucleophileinsitu. TheDYKATof3-aryloxindolesproceededinhighenantioselectivityfor thepreparationofaquaternarystereocenterwithout the addition of a base (eq 11).46 Since the catalyst mustdiscriminatebetweentheenantiotopicfacesofthenucleophile,theenantioselectivityshowedamoderatedependenceuponthesubstratesubstitution.Thehighestenantioselectivity,97%,wasachievedwitha3-(ortho-substituted)arylgroup.
3.2. Nonstabilized EnolatesTheasymmetricallylicalkylationsofnonstabilizedenolateshavealsobeen successful.These examples demonstrate how high asymmetricinduction can also be achieved by stoichiometrically converting theketone into the enolate or enol ether prior to alkylation. Using thestandardligand,lithiumenolate,andatinadditive,theallylicalkylationof 2-methyl-1-tetralone proceeded with high enantioselectivity(eq 12).47 The extent of the nucleophile aggregation showed asignificant effect upon the enantioselectivity, and optimal results were obtainedwithtwoequivalentsoftheamidebase.48Whileadditionofa trialkyltin chloride gave the highest ee, only a very small loss (afewpercent)ineeoccurredinitsabsence.Arelatedenolate-structureeffectonbothdiastereoselectivityandenantioselectivitywasobservedby Braun and co-workers with BINAP ligands.47b–d The asymmetricinductionobservedwith thesehardernucleophiles is consistentwithanintermolecularalkylation,inwhichalkylationoccursonthefaceoftheallylmoietyopposite themetal, inanalogy toAAAwith typicalsoftnucleophiles.Theproductsof thismethodologyhavehadbroadsynthetic applications. One particular example involves the AAAof cyclopentanone 81, which efficiently establishes the absolute stereochemistryforthesynthesesofhamigeranB48andallocyathinB2
(Scheme 20).49
Modifying the arms of the standard ligand with ferrocenylcomplexeshasalsoenabledhighenantioselectivity,95%ee,intheAAAofthetetralonesubstrate78(eq 13).50
One of the main limitations of the above methodologies isenolateequilibration.Accordingly,theaboveexamplesutilizeketonesubstratesthataffordonlyonepossibleenolateintermediate.Aneffectivesolutionistheregio-andenantioselectiveallylicalkylationofunsymmetrically substitutedketonesbyuseof their allyl enolcarbonate derivatives. The reaction proceeds after ionization ofthe allylic ester throughapalladium-promoteddecarboxylation38to theenolatenucleophile.Useof theanthracene ligand,LA,hasenabledhighenantioselectivityfortheformalDYKATofracemic2-methylcyclohexanone (eq 14).51 Due to the neutral conditionsemployedinthereaction,thealkylationefficientlyestablishestertiarystereocentersinbothcyclicandacyclicsubstrateswithoutracemizationoftheproduct(Scheme 21).52ThesyntheticutilityoftheprocesswasdemonstratedbyapplicationtotheAAA/Stork–Danheiseradditionsequencefortheformationofchiralγ,γ-disubstitutedcycloalkenones(Scheme 22).53
3.3. AzlactonesThe asymmetric allylic alkylation of azlactones offers an efficient processforthepreparationofquaternaryaminoacids,astructuralmoietypresent in numerous biologically significant molecules. The azlactones provide sufficient stabilization so that enolization can be conducted in situ.Asymmetricprenylationofazlactone94withthestandardligand,LS, proceeded in moderate yield and excellent enantiomeric excess
Scheme 18. DyKaT through enolization of the nucleophile.
OR1
H
O
R1H
OMR1
Pd(II)Ln
OR1
O
R1
enolization
k1
k2
Scheme 19. asymmetric allylic alkylation of β-Keto esters with the standard ligand.
[η3-C3H5PdCl]2 (0.5 mol %) TMG, PhMe
O
OEt
O O
OEt
O
HN
OH
allyl acetate(R,R)-LS (1.2 mol %)
71 7286%, 86% ee
nitramine
TMG = N,N,N',N'-tetra- methylguanidine
Ref. 45
eq 10
O
OBn
O
OAc
+
CO2BnO
H
73 rac-48 7487%, 99:1 dr, 96% ee
[η3-C3H5PdCl]2 (0.4 mol %) TMG, PhMe
(R,R)-LS (1.2 mol %)
eq 11
NO
OMe
NO
OMe
75 7772%, 97% ee
allyl acetate (76)(R,R)-LA (5 mol %)
[η3-C3H5PdCl]2 (2.5 mol %)t-BuOH, PhMe, 4 oC
Ref. 46
eq 12
O O
78 8099%, 88% ee
76, LDA, then(S,S)-LS (5 mol %)
[η3-C3H5PdCl]2 (2.5 mol %)Me3SnCl (79), DME
Ref. 47
Scheme 20. application of the Dynamic Kinetic aaa of nonstabilized enolates to the Total synthesis of hamigeran B and allocyathin B2.
t-BuO
O
t-BuO
O
OH OO
H
OH
OH
81 8277%, 93% ee
hamigeran Ballocyathin B2
76, LDA, thenLS (1 mol %)
[η3-C3H5PdCl]2 (0.5 mol %)79, t-BuOH, DME
Br
Ref. 48,49
Ref. 45

66
Palla
dium
-Cat
alyz
ed D
ynam
ic K
inet
ic A
sym
met
ric A
llylic
Alk
ylat
ion
with
the
DPP
BA L
igan
dsV
OL.
40,
NO
. 3 •
200
7(Scheme 23).54The chiral product, 96, served as a useful substratefor the preparation of α-methylaspartic acid (97).Asdemonstratedinseveralpreviousexamples,thechiralcatalystcansimultaneouslydiscriminatebetweentheenantiotopicfacesofthenucleophileandenantiotopic termini of the racemic electrophile.This asymmetrictransformationaffordedgooddiastereoselectivityandhigheewiththeracemicacetate99(eq 15).55Thepalladium-catalyzedAAAhasalso efficiently discriminated between enantiotopic geminal leaving groups.56Extensionofthemethodologywithazlactonestogeminalacetate101providedausefulprocessfor thepreparationofchiralvicinal amino alcohols and an efficient entry into the total synthesis ofsphingofunginsEandF(Scheme 24).57
4. DYKAT through Rapid p−s−p Interconversion of IntermediatesAnothereffectiveprocessforapalladium-catalyzeddynamickineticAAA relies on the rapid interfacial exchange of the allyl ligandthrough a π−σ−π interconversion. Oxidative addition with inversion of each substrate enantiomer initially forms two diastereomeric π-allylPd(II) intermediates (Scheme 25).2,3,4Withachiralcatalyst,aratedifferenceintheoxidativeadditionisexpected,andaDYKAToccurs with complete consumption of the mismatched substrate.Asymmetric induction results from the preferential alkylationof one diastereomeric intermediate over the other. Accordingly,highenantioselectivityisachievedwhen,inadditiontoaselectivealkylation (k1 >> k2), a Curtin–Hammett condition is establishedwherein interconversion is rapid and successfully competes withnucleophilicaddition.AnotherrequirementforthistypeofDYKATistheexistenceofidenticalgeminalsubstituentsononesideoftheallyl ligand. Ifone terminusof theallyl ligand is substitutedwithdifferent geminal groups, then the π−σ−π interconversion will result in a geometrical isomerization of the allyl ligand. Theseπ-allylPd(II) intermediates cannot “racemize” through a π−σ−π mechanism(Scheme 26).Furthercomplicatingthealkylationwithunsymmetricalsubstratesisalkylationatthedifferenttermini,whichleadstoregioisomers(Scheme 27).RegioselectivityintheAAAhasbeenachievedbybothsubstrateandcatalystcontrol.Althoughthechiral catalyst provides a significant preference for a regioselective alkylation of one diastereomeric intermediate, optimization ofthe reaction conditions is often necessary to establish the Curtin–Hammettsituationforasymmetricinduction.
4.1. Vinyl Epoxides and Aziridines as SubstratesA versatile substrate for the palladium-catalyzed dynamic kineticAAAisvinylepoxide,which,duetotheringstrain,promotestheoxidativeadditionandconsumptionofthemismatchedenantiomerrequired for a DYKAT. Suitable vinyl epoxides have geminalhydrogens or other identical geminal substituents on the olefin terminus, enabling a Curtin–Hammett condition to be establishedthrough a rapid π−σ−π interconversion. In the π-allylPd(II) intermediates, thealcoholoralkoxidecandirect thealkylationforthebranchedproducttypicallythroughhydrogenbondingorothercovalentinteractionwiththeincomingnucleophile(eq 16).
AlthoughBINAP-basedligandshavebeenexaminedfortheDYKAT of vinyl epoxides,58 high enantioselectivities for theintermolecularadditionofnucleophilestovinylepoxidestypicallyrequiredtheuseoftheDPPBAligands.Thesereactionsallowedtheuseofabroadrangeofnucleophilesandenabledapplicationofthisapproachtonumeroustotalsyntheses.TheAAAwithphthalimide59provided the corresponding vinylglycinol derivative in highenantio-andregioselectivity(Scheme 28).60Ourinitialproposalwasthatahydrogen-bondinginteractionbetweenthealkoxideofthep-allylPd(II) intermediateandthenucleophilewoulddirect
eq 13
HNNHO O
Fe
Fe
83
O O
78 8093%, 95% ee
allyl ethyl carbonateLDA, then
83•2H2O (7.5 mol %)
[η3-C3H5PdCl]2 (2.5 mol %)THF
PPh2 Ph2P
Ref. 50
eq 14
O O
O
O
Pd2dba3•CHCl3 (2.5 mol %)PhMe
(R,R)-LA (5.5 mol %)
84 8588%, 85% ee
Ref. 51
Scheme 21. formation of Tertiary stereocenters by the Dynamic Kinetic aaa of allyl enol carbonates.
O O
O
MeO
O
MeO
Ph
O
O
O Ph
O
8690%
8790%, 99% ee
8991%
9094%, 94% ee
O
MeO
Ph
O
n-Pr
88
Pd2dba3•CHCl3(2.5 mol %)
dioxane
(R,R)-LA(5.5 mol %)
Pd2dba3•CHCl3(2.5 mol %)
PhMe
(R,R)-LA(5.5 mol %)
O Cl
O
then
NaHMDSTMEDA
THF, –78 oC
O Cl
O
then
NaHMDSTHF, –78 oC
Ref. 51,52
Scheme 22. The stork–Danheiser application of DyKaT with allyl enol carbonates.
PhS
O O
O
PhS
O
O
91 9275%, 99% ee
93
Pd2dba3•CHCl3(2.5 mol %)
dioxane
(R,R)-LA(5.5 mol %)
Ref. 53
Scheme 23. asymmetric allylic alkylation (aaa) with azlactones.
+OAc
NO
O
Ph
NO
O
Ph
OH
OHO
O NH3Cl
94 95 9653%, 98% ee
97
[η3-C3H5PdCl]2(2.5 mol %)Et3N, PhMe
(R,R)-LS(7.5 mol %)
Ref. 54
eq 15
+N
O
Oi-Bu
Ph
NO
O
Ph
OAci-Bu
H
98 rac-99 10077%, 13:1 dr, 99% ee
[η3-C3H5PdCl]2(2.5 mol %)Et3N, MeCN
(R,R)-LS(7.5 mol %)
Ref. 55

67
Barr
y M
. Tro
st*
and
Dan
iel R
. Fan
dric
kV
OL.
40,
NO
. 3 •
200
7
thealkylation.Reactionswith triphenylphosphine still favoredthebranchedproductwithaslightlylowerregioselectivity(4:1B/L).Withoutdirectingeffects,thelinearproductisfavoredduetoalkylationattheleaststericallyhinderedposition.Therefore,boththesubstrateandcatalystcontributetothehighregioselectivityobservedin theDYKAT.Thevinylglycinolderivativeobtainedbythismethodologyprovidedavaluablesyntheticbuildingblockforthepreparationofseveralbiologicallysignificantcompoundsincluding ethambutol, vigabatrin,61 DMDP, bulgecinine, andbroussonetineG.62
Alcoholsaretypicallypoornucleophilesforthealkylationofp-allylPd(II)complexesand,accordingly,requireactivationforreactivity.Ausefulstrategy toactivate thealcoholnucleophileanddirectthealkylationistoemployaboraneco-catalystforthedynamickineticasymmetricadditionstovinylepoxides.63InthisAAA,thealkoxideofthep-allylPd(II)intermediatecoordinatestotheborontoforman“ate”complex,therebyactivatingthealcoholforanintramolecularalkylation.Theprocessgivestheglycolinhighyieldwithexcellentenantio-andregioselectivity(eq 17).63ThismethodologyisoneofthemostsyntheticallyusefuloftheDYKATs,andhasbeenappliedto theasymmetricsynthesisofnucleosides,64malyngolide,65tipranavir,66andintermediate111intheformalsynthesisofLY333531(Scheme 29).67
Carbonatesarealsoeffectivenucleophileswithvinylepoxides,providinganadditionalefficientsynthesisofchiralvinylglycidols.Underbiphasicconditions,thereactionofisoprenemonoepoxideandbicarbonateaffordsthedioxolanonein88%yieldand93%ee(Scheme 30).28Thegoodyieldandregioselectivityobtainedareattributedtoanintramolecularalkylationstep.Thealkoxideoftheinitialp-allylPd(II)intermediateisproposedtoattacktheinsitugeneratedcarbondioxidetoform113,whichsubsequentlycyclizesto the dioxolanone. The high enantioselectivity results from arapidp−s−pequilibrationoccurringeitherpriortotheadditiontocarbondioxideand/orpriortothecyclization.ComplementingthisDYKAT, theuseofaboronco-catalyst (Et3B)andsodiumcarbonateasnucleophileallowsforadirectalkylationwiththecarbonatenucleophilewithoutcyclizationtothedioxolanone.Inthiscase,theintermediatecarbonate,115,undergoesafacileinsitu decarboxylationtovinylglycidol116inhighyieldandee.
Theasymmetricalkylationwithstabilizedcarbonnucleophileshas shown high regio- and enantioselectivity in the DYKATwith isoprene monoepoxide. Under optimized conditions, thedynamic kinetic AAA of isoprene monoepoxide with β-ketoestersaffordsgoodregioselectivity for thebranchedalkylationproduct and furnishes the corresponding tetrahydrofuran withhighenantiomericexcess(eq 18).68Theregioselectivityislowerin theabsenceof thefluorideadditive, tetra-n-butylammoniumtriphenyldifluorosilicate(TBAT).Thiseffect isattributedtoanintermolecular alkylation, and formation of the linear productis due to the alkylation competing with the necessary p−s−pinterconversion. The asymmetric induction obtained in theallylicalkylationswiththeDPPBAligandsisrationalizedbythepreferentialionizationandalkylationoccurringunderaflapinthe“nun’shat”model.69Thematchedalkylationofthemismatchedintermediatewould favor the linearproduct, and thematched-intermediatematchedalkylationwouldfavorthebranchedproduct(Scheme 31).Halideadditivesincreasetherateofthenecessaryinterconversion,70andimprovetheregioselectivitybypromotingthenecessaryCurtin–Hammettcondition,thusallowingforthepreferredmatchedalkylationof thematched intermediate.Theutilityofthemethodologyhasbeendemonstratedbyapplicationtothesynthesisofthehighlysubstitutedcyclopentylcore,124,ofviridenomycin(Scheme 32).71
Scheme 24. aaa of allylic Geminal acetates with azlactones.
TBDPSOOAc
OAcTBDPSO
OAc
NO
O
Ph
n-C6H13
O
OH
OH
OHCO2
–
NH3+
94 +
101 10270%, 11:1 dr, 89% ee
X = OH, sphingofungin EX = H, sphingofungin F
[η3-C3H5PdCl]2(0.5 mol %)NaH, THF
(R,R)-LS(1.5 mol %)
X
Ref. 57
Scheme 25. DyKaT through a p−s−p interconversion.
R
LG
R
LG
R
Pd(II)L*
R
Pd(II)L*
R Pd(II)L*
Pd(0)L*
k1'
k2'
Pd(0)L*
+
+
+
Nu–
k1
k2
R
Nu
R
Nu
π–σ
π–σ
Nu–
Ref. 2–4
Scheme 26. p−s−p interconversion of 1,3-Disubstituted allylpd(ii) complexes.
R1
Pd(II)L*
R1 Pd(II)L*
+
+R2
R2
R1
L*Pd(II) R2+
Scheme 27. regioselective alkylation of Unsymmetrical allyl complexes.
R1
+Pd(II)L*
R2
Nu–
R1 R2 R1 R2
Nu Nu
Nu–
eq 16
OXNu
Pd(II)
OHNu
Ref. 55
Scheme 28. Dynamic Kinetic aaa of Vinyl epoxide with phthalimide.
O NH
O
O
+
HO
N
O
ONH3Cl
CO2H
OH
NH
NHOH
HN
OHHO
OH
O
O
OH
LN (1.2 mol %)[η3-C3H5PdCl]2 (0.4 mol %)Na2CO3, CH2Cl2
103 104
10598%, 96% ee vigabatrinethambutol
(+)-broussonetine G
HN
OHHO
OHOH
DMDP
HN
HO
CO2H
OH
(–)-bulgecinine
Ref. 60–62

68
Palla
dium
-Cat
alyz
ed D
ynam
ic K
inet
ic A
sym
met
ric A
llylic
Alk
ylat
ion
with
the
DPP
BA L
igan
dsV
OL.
40,
NO
. 3 •
200
72-Vinylaziridinesarealsocompetentsubstratesforthedynamic
kinetic asymmetric cycloaddition with isocyanates to furnishsynthetically useful chiral imidazolidinones (Scheme 33).72 Inthisexample,theuseofaceticacidasaco-catalystsignificantlyimproves the enantioselectivity. This effect is rationalized byprotonationofthenitrogentetheredtothep-allylPd(II)intermediateandslowingoftheacylationbytheisocyanate,therebyallowingthenecessaryp−s−pinterconversiontoeffectivelycompetewithproductformation.Thechiralvicinaldiaminemoietyispresentinnumerousbiologicallyimportantnaturalproducts,73andtheutilityofthismethodologyhasbeendemonstratedbyapplicationtotheconcisetotalsynthesisofpseudodistominD.74
4.2. Baylis–Hillman Adducts as Substrates Asyntheticallyuseful substrate for thedynamickineticAAA is aBaylis–Hillmanadduct.Similar to theDYKATofvinyl epoxides,asymmetricinductionresultsinthiscasefromthekineticalkylationof one diastereomeric π-allylPd(II) intermediate in a mixture of rapidly interconverting complexes through a π−σ−π mechanism (Scheme 34).75InatypicalAAA,thesynpathwayisnormallystronglypreferred.7However,intheDYKATwithBaylis–Hillmanadducts,the presence of a substituent at the 2 position of the allyl ligandincreasestheimportanceoftheantipathway.Forhighasymmetricinductionandregioselectivity,thecatalystmustdiscriminatebetweentheterminioftheallylligandandaffordaselectivealkylationofaspecific geometrical isomer of the intermediate.
ThedynamickineticAAAofBaylis–Hillmanadductswithalcohol nucleophiles provides a useful strategy for the formalderacemizationofthereadilyavailablesubstrates.Underoptimizedconditions,highenantioselectiviesareobtainedintheDYKATofboth2-cyano-and2-carboethoxy-substitutedadducts(Scheme 35).76Anexaminationoftheminor,linearproductsprovidesanindicationofthepreferredallylgeometryoftheintermediates.ExclusiveZdouble-bondgeometryoftheminor,linearproductwasobservedfrom the cyano substrate 127, and exclusive E geometry wasobtainedforthelinearproductfromtheestersubstrate131.Thischangeinallylconformationwasrationalizedbystericinteractionswithinthep-allylPd(II)intermediates.Forthecyanosubstrate,thepreferredallylcomplexesaresynduetominimizationofthetypicalA1,3 strain associated with allyl ligands. In the ester substrate,the larger ester group increases the unfavorable 1,2 repulsionandoverridestheA1,3straintofavortheantiallylintermediates.Additionally,similareffectshavebeenobservedwiththerespectivelinearachiralsubstratestosupporttheconclusionthatthestrongpreferenceforeitherthesynorantipathwayisdependentonthesubstituentinthe2positionoftheallylintermediate.Accordingtotheabovemechanism,intermediatesAandD(seeScheme34)should favor different product enantiomers, contrary to theasymmetricinductionobserved.However,furtherstereochemicalanalysiswithothersubstrateshasrevealedthattheesterandcyanosubstratespreferdifferentcantsofthep-allylPd(II)intermediates.Oppositecantsorallylgeometriesofthep-allylPd(II)intermediateinvert the sense of asymmetric induction to generate oppositeenantiomersoftheproduct.Theestersubstratefavorstheanti allylcomplexwithaforwardcant,andthecyanosubstratefurnishesthesynallylintermediatewiththetypicalbackwardscant.Bothof these intermediates, therefore, favor thesameenantiomerofthe product. In addition to establishing the necessary Curtin–Hammettcondition, theDYKATwithBaylis–Hillmanadductsaffordsremarkableselectivityforspecificconformationsof thep-allylPd(II)intermediates,andresultsinhighenantioselectivitiesforthealkylation.Overall,onlypreliminarystudiesonthesubstrateandnucleophileshavebeenreported.
eq 17
O Pd2dba3•CHCl3 (1 mol %)(s-C4H9)3B (1 mol %), CH2Cl2
OHOMeH
Pd(II)L*
OBO
RR
+
103 106 10782%, 89% ee
MeOH(R,R)-LN (1.2 mol %)
–
Ref. 63
Scheme 29. synthetic applications of the asymmetric addition of alcohols to Vinyl epoxides.
OOH
OR2R1
OHON
N
N
N
Cl
OH
n-C9H19
HO
O
R1
O O
OH
Ph
HNS
N
OO
CF3
HN OO
N N
OOH
R2OH, LN
108 109
110
(–)-malyngolide tipranavir
111
Pd2dba3•CHCl3(s-C4H9)3B, CH2Cl2
Ref. 64–67
Scheme 30. Dynamic Kinetic aaa of isoprene monoepoxide with Bicarbonate.
O
+ Pd(II)L*
O–O
O
OO
O
OH
OHOH
OCO2–Pd2dba3•CHCl3
(1 mol %)Et3B (1 mol %)H2O, CH2Cl2
112
112
113 11488%, 93% ee
115 11691%, 97% ee
NaHCO3(S,S)-LS (1.5 mol %)
NaHCO3 (S,S)-LS (3 mol %)
Pd2dba3•CHCl3(0.5 mol %)
H2O, CH2Cl2
Ref. 28
eq 18
Pd2dba3•CHCl3(1 mol %)
TBAT (1 mol %)PhH
70%, 79:21 B/L, 96% ee
O
OHEtO
O
CO2Et
OOH
n
OCO2Et
112(S,S)-LST (3 mol %)
117 118branched
119linear
n = 1,2
+
Ref. 68
Scheme 31. rationalization of the regioselectivity in the alkylation with the DppBa ligands.
O
O
π–σ–π
OROO
matched acetoacetate
k1
OH
CO2R
O
OH
(R)-112
(S)-112
120branched
121linear
ionization
mismatched
ionization
acetoacetate
k2
Pd
+
OH
Nu
Pd
+
HO
Nu
Ref. 68b,69

69
Barr
y M
. Tro
st*
and
Dan
iel R
. Fan
dric
kV
OL.
40,
NO
. 3 •
200
7
The synthetic utility of the DYKAT with Baylis–HillmanadductswasdemonstratedbyapplicationtothetotalsynthesesoffuraquinocinE77andhippospongicacidA(Scheme 36).76Withthe long chainpresent in thehippospongic acid substrate, fullconversionwasinhibited.Thus,theobservedeeevolvesfromacombination of a kinetic resolution and a DYKAT associatedwith the largechain inhibiting the ionization.Accordingly, theDYKATwiththesmallersubstrate136proceededinhighyieldandenantioselectivity.
4.3. Acyloxyenoates as SubstratesFor typical DYKATs with a π−σ−π mechanism, one terminus of the allylintermediatemustbesubstitutedwithidenticalgroups.However,inacyloxyenoatesthatdonotabidebytheaboverequirement,highenantioselectivities have been achieved through an alternativeequilibration process. In this case, asymmetric induction is due toa rapid π−σ−π interconversion, wherein equilibration between the enantiotopicfacesoccursthroughanachiralO-palladium(II)enolate(Scheme 37).78Highenantiomericexcesswasachievedwithphenol-basednucleophiles(eq 19), and a halide additive showed a significant effectontheasymmetricinduction.DuringoptimizationstudieswithCs2CO3, an ee of 24% was obtained without tetrabutylammoniumchloride(TBACl),andincreasedto75%with30mol%ofTBACl.Thiseffectwasattributedtothehalideadditiveincreasingtherateofthe π−σ−π interconversion to promote the necessary Curtin–Hammett condition.3,4,70Slowingthealkylationratebyremovingthebasefurtherincreased the ee to 84% (74% yield). The methodology efficiently providedtheabsolutestereochemistryforthetotalsynthesesof(+)-aflatoxin B1
79and(+)-brefeldinA(Scheme 38).80Asdemonstratedbytheseexamples,theeefortheDYKATsurpassed95%byutilizinganaphtholorhighlysubstitutedphenolnucleophile.
In addition to the cyclic γ-butenolide substrates, highenantioselectivitieshavealsobeenachievedinanefficientAAAthat results in thederacemizationofacyclicacyloxyenoatesandrelated electrophiles (eq 20).29Asymmetric induction for theseacyclic substratespresumably results fromananalogousp−s−pinterconversionwithaprochiralPd(II)intermediate.
4.4. Allenes as SubstratesInadditiontotheprevioustransformationswhereinastereogeniccenteris created, palladium-catalyzed dynamic kinetic AAAs of racemicalleneshaveshownhighasymmetricinductionfortheestablishmentofaxialchirality.Inthismechanism,theCurtin–Hammettconditionresults from a rapid π−σ−π interconversion through a vinylPd(II) intermediate (Scheme 39).81 Using the standard ligand, LS, highenantioselectivitiesandyieldswereobtainedforthedynamickineticasymmetric addition of malonates and amines to racemic 2,3-alkadienyl acetates (Scheme 40).82 Similarly to the addition ofmalonatenucleophiles tocyclicandacyclic substrates (vida supra),thecountercationofthenucleophileorbasehadapronouncedeffectontheasymmetricinductionforbothtypesofnucleophiles.However,theobservedpattern,whereindifferentcountercationswerenecessaryforoptimalenantioselectivity,lithiumwithmalonatesandcesiumwithamines,isnotconsistentwiththepreviouslyobservedcountercationeffects(videsupra).Adetailedrationalizationforthisdiscrepancyhasyet to be formulated.The malonate products with a tethered dienefunctionalitywereappliedtoaRh(I)-catalyzed[4+2]cycloaddition,in which the axial chirality was efficiently transferred to multiple stereogenic centers and exocyclic olefin geometry.
5. Other DYKAT ProcessesSeveral DYKATs have been reported wherein the asymmetricinduction cannot be rationalized by the previously described
Scheme 32. application of the DyKaT of isoprene mono-epoxide with β-Keto esters to the synthesis of the cyclopentyl core of Viridenomycin.
Pd2dba3•CHCl3(1 mol %)CH2Cl2
O
OHEtO
O
OCO2Et
PhS
SPh
MeO
TBSOOTBS
CO2Et
OMe
112(S,S)-LS (3 mol %)
122 12371%, 94% ee
124
Ref. 71
Scheme 33. Dynamic Kinetic asymmetric cycloaddition of 2-Vinylaziridines with isocyanates.
NDMB
NN
DMB
DMB
O
(S,S)-LN (6 mol %)[η3-C3H5PdCl]2 (2 mol %)
DMB-NCO10% AcOH, CH2Cl2
NH
OHH2N
125 12680%, 94% ee
(+)-pseudodistomin D
DMB = 2,4-dimethoxybenzyl
Ref. 72,74
Scheme 34. p−s−p interconversion of Baylis–hillman adducts.
EWG
R
X
EWGR
X
EWG
R Pd(II)+
EWG
R Pd(II)+
EWG
Pd(II)+
R
EWG
Pd(II)+
R
EWG
R
Nu
EWGR
Nu
EWGR
Nu
EWG
R
Nu
R
EWGNu
EWGNuR
M M MM MM
M
MM
MMMMMM
M
MMπ–σ–π π–σ–π
M = matched; MM = mismatched
A B
C D
Anti Pathway Syn Pathway
Ref. 75
Scheme 35. DyKaT of Baylis–hillman adducts.
n-PrCN
OCO2Me
n-PrCO2Et
OCO2Me
Pd2dba3•CHCl3(1 mol %)CH2Cl2
n-PrCN
OPMPHCN
n-Pr
OPMP
CO2Et
OPMPn-Pr
CO2EtOPMPH
n-Pr
PMP-OH (128)(R,R)-LST (3 mol %)
rac-127 12971%, 93% ee
13015%
rac-131 13264%, 92% ee
13318%
Pd2dba3•CHCl3(1 mol %)CH2Cl2
+
+
PMP-OH (128)(R,R)-LST (3 mol %)
PMP = p-methoxyphenyl
Ref. 76

70
Palla
dium
-Cat
alyz
ed D
ynam
ic K
inet
ic A
sym
met
ric A
llylic
Alk
ylat
ion
with
the
DPP
BA L
igan
dsV
OL.
40,
NO
. 3 •
200
7mechanisms.Furthermore,themechanismsthataffordtheobservedenantioselectivitiesforthesesubstratesmayalsobeoperatinginthepreviously described reactions and contributing to the previouslyobservedhighlevelsofenantioselectivityinthesecasestoo.Hobergandco-workers83andGaisandco-workers29reportedhighee’sfortheDYKATofunsymmetricalacyclicsubstrateswiththestandardLS and BINAP ligands (Scheme 41). Due to the unsymmetricalnatureoftheelectrophile,whichaffordsanallylintermediatewithdifferentgeminalgroupsonbothterminioftheligand,theDYKATcannotproceedthroughthepreviouslydescribedmesointermediateor π−σ−π mechanism. Other processes such as interfacial exchange throughantiadditionviaasecondequivalentofthePd(0)catalyst84andracemizationofthesubstratemayaccountfortheasymmetricinduction. Another possibility is that either the ionization of thecarbonateornucleophilicattackproceedswithretentionintheso-calledmismatchedsituationforanoverall inversionmechanism.85Interestingly,onlythecarbonatesubstrateshaveaffordedaDYKAT,whileacetatesubstrateshavefurnishedaselectiveKR.
6. Conclusions and OutlookInconclusion,thepalladium-catalyzeddynamickineticasymmetricallylic alkylation with the DPPBA ligands is a versatile and
Scheme 36. synthetic applications of the DyKaT of Baylis–hillman adducts.
CNOCO2Me
Pd2dba3•CHCl3(1 mol %)CH2Cl2
I
O
OCN
CN
O
OH
O
O
OHMeO
MeO2COAc
HO ORO2C
[η3-C3H5PdCl]2 (2 mol %)Hex4NCl (30 mol %)
dioxane55% conv, 50% y, 91% ee
100% conv, 65% ee R = Me 135
R = H(+)-hippospongic acid A
rac-134
rac-136
(R,R)-LST (6 mol %)
2-I-1,3-(OH)2C6H3 (137)(R,R)-LST (2.7 mol %)
13897%, 92:8 dr furaquinocin E
LiOH98%
Ref. 76,77
Scheme 37. DyKaT mechanism for γ-Butenolides.
OO
LG
OO
LG
OO
+Pd(II)
OO
+Pd(II)
OO
+Pd(II)
π-σ
π-σ
OO
Nu
OO
Nu
k1
k2
Ref. 78
eq 19
OO
BocO OO
OH
MeO
Pd2dba3•CHCl3 (1 mol %)(n-Bu)4NCl (30 mol %), CH2Cl2
(R,R)-LS (3 mol %)
rac-139 14074%, 84% ee
4-MeOC6H4OH
Ref. 78
Scheme 38. synthetic applications of the DyKaT with γ-Butenolides.
OO
OH
Pd2dba3•CHCl3(2.5 mol %)
(n-Bu)4NCl (30 mol %)CH2Cl2
HO HO
H
H
H
O
OH
HO
OEtO2C
O
MeO OH
I
OO
HO
OEtO2C
O
MeO
IO
O
MeO
O
O
O
H
H
141 14284%, 96% ee
143 14489%, >95% ee
(+)-brefeldin A
(+)-aflatoxin B1
rac-139(R,R)-LS (7.5 mol %)
Pd2dba3•CHCl3(2.5 mol %)
(n-Bu)4NCl (30 mol %)CH2Cl2
rac-139(R,R)-LS (7.5 mol %)
Ref. 79,80
eq 20
R
OCO2Me
R
OH
Pd2dba3•CHCl3 (2 mol %)KHCO3, H2O, CH2Cl2racemic
(R,R)-LS (4 mol %)
SM
145147149151
R
EtO2CPhO2S
CN(MeO)2(O)P
Prd
146148150152
Yield
87%87%87%83%
ee
99%93%61%69%
Ref. 29b
Scheme 39. DyKaT mechanism for allenes.
LnPd(0)
Nu Nu–k1 k2
R
HH
RLG
H
HLG
Pd(II)LnR
H
Pd(II)LnRH
Pd(II)Ln
R +++
RH Nu
R
H NuH H
k1 > > k2
LnPd(0)
–
Ref. 81
Scheme 40. DyKaT of allenes.
H CO2Me
CO2MeH
Pd2dba3•CHCl3 (2.5 mol %)Cs2CO3, THF
Hex4NCl (5 mol %), rt
BnNHMe(S,S)-LS (7.5 mol %)
BnO HO
Ac
BnO
HO
Ac
H
H
+
CO2Me
CO2Me
BnO
HN
Bn
H
BnO
[(C10H8)Rh(cod)]SbF6
CH2Cl2, rt, 0.5 hCO2Me
CO2MeH
H
BnO
153
ent-153 15497%, 90% ee
15589%, 91% ee
15698%, 95% ee
rac-153
Pd2dba3•CHCl3 (2.5 mol %)LiHMDS (1.1 equiv), THF
Hex4NCl (5 mol %), rt
(S,S)-LS (7.5 mol %)
Ref. 82
Scheme 41. DyKaT of Unsymmetrical acyclic substrates.
Ph
OCO2Et
OTBS Pd2dba3•CHCl3 (3 mol %)Cs2CO3, THF
PhOH(S)-BINAP (8 mol %)
Ph
OPh
OTBS
Ph
OCO2Me
Pd2dba3•CHCl3 (2 mol %)KHCO3, H2O, CH2Cl2
Ph
OH
rac-157 15894%, 92% ee
rac-159 16085%, 85% ee
(R,R)-LS (4 mol %)
Ref. 29b,83

71
Barr
y M
. Tro
st*
and
Dan
iel R
. Fan
dric
kV
OL.
40,
NO
. 3 •
200
7
synthetically useful technology. Currently, the predominantDYKATprocesses for asymmetric inductionare (i)discriminationof enantiotopic termini of a π-allylpalladium intermediate, (ii)discrimination of enantiotopic faces of a meso or prochiralintermediate, and (iii) kinetic alkylation of one diastereomericintermediate of rapidly interconverting π-allylPd(II) complexes. Additionally,DYKAThasbeenaccomplishedwithseveralsubstrateswherein the reaction proceeds through alternative processes.These alternative mechanisms may also be operating in the otherDYKATs and contribute to the high enantioselectivities observed.Asdemonstrated,awidevarietyofsubstratesandnucleophilesaretoleratedinDYKAT,andhaveprovidedchiralbuildingblocksforthesynthesisofnumerouscomplexnatural compounds,validating theversatility and flexibility of the methodology. Further development is necessary to broaden the nucleophile and substrate scopes.Furthermore, mechanistic studies are required to further elucidatethesenseofasymmetricinductionobservedinmostreactionsand,accordingly,unravelthefullpotentialofthissyntheticallyenablingmethodology.
7. AcknowledgmentWe thank the National Institutes of Health (GM-13598 and GM-33049) and the National Science Foundation (CHE-0455354) fortheirgeneroussupportofourprograms.
8. References(1) (a)Trost,B.M.Science1991,254,1471.(b)Trost,B.M.Acc. Chem. Res.
2002,35,695.(2) Trost,B.M.Chem. Pharm. Bull. 2002,50,1.(3) (a)Trost,B.M.;vanVranken,D.L.Chem. Rev. 1996,96,395.(b)Trost,
B.M.Acc. Chem. Res.1996,29,355.(4) Trost,B.M.J. Org. Chem.2004,69,5813.(5) ForreviewsonDKR,see:(a)Ward,R.S.Tetrahedron: Asymmetry 1995,
6,1475.(b)Cook,G.R.Curr. Org. Chem. 2000,4,869.(c)Pellissier,H.Tetrahedron 2003,59,8291andreferencestherein.
(6) AgeneraltermforaDKRiskineticasymmetrictransformation:Eliel,E.L.Stereochemistry of Carbon Compounds;McGraw-Hill:NewYork,1962;Chapter4.
(7) (a)Faller,J.W.;Thomsen,M.E.;Mattina,M.J.J. Am. Chem. Soc. 1971,93,2642.(b)Faller,J.W.;Tully,M.T.J. Am. Chem. Soc. 1972,94,2676.
(8) Trost,B.M.;Murphy,D.J.Organometallics 1985,4,1143.(9) Forrecentdevelopmentsofchiralligands,see:(a)Jansat,S.;Gomez,M.;
Philippot,K.;Muller,G.;Guiu,E.;Claver,C.;Castillon,S.;Chaudret,B.J. Am. Chem. Soc. 2004,126,1592.(b)Tokuda,R.;Matsunaga,H.;Ishizuka,T.;Nakajima,M.;Kunieda,T.Heterocycles 2005,66,135.(c)Nemoto,T.;Masuda,T.;Matsumoto,T.;Hamada,Y.J. Org. Chem. 2005,70,7172.(d)Braga,A.L.;Paixão,M.W.;Marin,G.Synlett 2005,1675.(e)Okuyama,Y.;Nakano,H.;Saito,Y.;Takahashi,K.;Hongo,H.Tetrahedron: Asymmetry 2005,16,2551.(f)Laurent,R.;Caminade,A.-M.;Majoral,J.-P.Tetrahedron Lett. 2005,46,6503.(g)Jin,M.-J.;Takale,V.B.;Sarkar,M.S.;Kim,Y.-M.Chem. Commun. 2006,663.(h)Mikhel,I.S.;Bernardinelli,G.;Alexakis,A.Inorg. Chim. Acta 2006,359,1826.(i)Kloetzing,R.J.;Knochel,P.Tetrahedron: Asymmetry 2006,17,116.
(10) Yamaguchi,M.;Shima,T.;Yamagishi,T.;Hida,M.Tetrahedron Lett. 1990,31,5049.
(11) (a)VonMatt,P.;Pfaltz,A.Angew. Chem., Int. Ed. Engl. 1993,32, 566.(b)Sprinz,J.;Kiefer,M.;Helmchen,G.;Reggelin,M.;Huttner,G.;Walter,O.;Zsolnai,L.Tetrahedron Lett. 1994,35,1523.(c)Constantine,R.N.;Kim,N.;Bunt,R.C.Org. Lett. 2003,5,2279.
(12) Togni,A.Tetrahedron: Asymmetry 1991,2,683.(13) Kang, J.;Cho,W.O.;Cho,H.G.Tetrahedron: Asymmetry 1994,5,
1347.
(14) Evans,D.A.;Campos,K.R.;Tedrow,J.S.;Michael,F.E.;Gagne,M.R.J. Org. Chem. 1999,64,2994.
(15) Mino,T.;Sato,Y.;Saito,A.;Tanaka,Y.;Saotome,H.;Sakamoto,M.;Fujita,T.J. Org. Chem. 2005,70,7979.
(16) Trost,B.M.;Krueger,A.C.;Bunt,R.C.;Zambrano,J.J. Am. Chem. Soc. 1996,118,6520andreferencestherein.
(17) Trost,B.M.;Bunt,R.C.J. Am. Chem. Soc. 1996,118,235.(18) Trost,B.M.;Radinov,R.J. Am. Chem. Soc. 1997,119,5962.(19) Kinoshita,N.;Kawabata,T.;Tsubaki,K.;Bando,M.;Fuji,K.Tetrahedron
2006,62,1756.(20) Trost,B.M.;Surivet,J.-P.J. Am. Chem. Soc. 2000,122,6291.(21) Trost,B.M.;Schroeder,G.M.J. Org. Chem. 2000,65,1569.(22) Powell,M.T.;Porte,A.M.;Reibenspies,J.;Burgess,K.Tetrahedron
2001,57,5027.(23) Trost,B.M.;Bunt,R.C.J. Am. Chem. Soc. 1994,116,4089.(24) Reetz,M.T.;Huette,S.;Goddard,R.J. Am. Chem. Soc. 1993,115,
9339.(25) Trost,B.M.;Surivet,J.-P.Angew. Chem., Int. Ed. 2000,39,3122.(26) Trost,B.M.;Organ,M.G.J. Am. Chem. Soc. 1994,116,10320.(27) Trost,B.M.;Kondo,Y.Tetrahedron Lett. 1991,32,1613.(28) Trost,B.M.;McEachern,E.J.J. Am. Chem. Soc. 1999,121,8649.(29) (a)Lüssem,B.J.;Gais,H.-J.J. Am. Chem. Soc. 2003,125,6066.(b)Gais,
H.-J.;Bondarev,O.;Hetzer,R.Tetrahedron Lett. 2005,46,6279.(30) Trost,B.M.;Toste,F.D.J. Am. Chem. Soc. 2000,122,11262.(31) Trost,B.M.;Tang,W.;Toste,F.D.J. Am. Chem. Soc. 2005,127,14785.(32) Mori,M.;Kuroda,S.;Zhang,C.-S.;Sato,Y.J. Org. Chem. 1997,62,
3263.(33) (a) Ovaa, H.; Stragies, R.; van der Marel, G. A.; van Boom, J. H.;
Blechert,S.Chem. Commun. 2000,1501.(b)Trost,B.M.;Sorum,M.T.Org. Process Res. Dev. 2003,7,432.
(34) (a)Trost,B.M.;Verhoeven,T.R.J. Am. Chem. Soc.1976,98,630.(b)Trost,B.M.;Verhoeven,T.R.J. Am. Chem. Soc. 1980,102,4730.(c)Hayashi,T.;Hagihara,T.;Konishi,M.;Kumada,M.J. Am. Chem. Soc. 1983,105,7767.
(35) Trost,B.M.;Oslob,J.D.J. Am. Chem. Soc. 1999,121,3057.(36) Trost,B.M.;Krische,M.J.;Berl,V.;Grenzer,E.M.Org. Lett. 2002,4,
2005.(37) Burger,E.C.;Tunge,J.A.Org. Lett. 2004,6,4113.(38) (a)Tsuda,T.;Chujo,Y.;Nishi,S.;Tawara,K.;Saegusa,T.J. Am. Chem.
Soc. 1980,102,6381.(b)Tsuji,J.Pure Appl. Chem. 1982,54,197.(c)Tsuji,J.;Yamada,T.;Minami,I.;Yuhara,M.;Nisar,M.;Shimizu,I.J. Org. Chem. 1987,52,2988.
(39) Trost,B.M.;Patterson,D.E.;Hembre,E.J.J. Am. Chem. Soc. 1999,121,10834.
(40) Trost,B.M.;Hembre,E.J.Tetrahedron Lett. 1999,40,219.(41) Trost,B.M.;Dudash, J., Jr.;Hembre,E. J.Chem.—Eur. J. 2001,7,
1619.(42) Trost,B.M.;Patterson,D.E.;Hembre,E.J.Chem.—Eur. J. 2001,7,
3768.(43) (a)Fiaud,J.C.;deGournay,A.H.;Larcheveque,M.;Kagan,H.B.J.
Organomet. Chem. 1978,154,175.(b)Genet,J.P.;Ferroud,D.;Juge,S.;Montes,J.R.Tetrahedron Lett. 1986, 27,4573.(c)Genet,J.-P.;Juge,S.;Achi,S.;Mallart,S.;Montes,J.R.;Levif,G.Tetrahedron 1988,44,5263.(d)Genet,J.-P.;Juge,S.;Montes,J.R.;Gaudin,J.-M.J. Chem. Soc., Chem. Commun. 1988,718.(e)Baldwin,I.C.;Williams,J.M.J.Tetrahedron: Asymmetry 1995,6,679.
(44) (a) Ito, Y.; Sawamura, M.; Matsuoka, M.; Matsumoto, Y.; Hayashi,T. Tetrahedron Lett. 1987, 28, 4849. (b) Hayashi, T.; Kanehira, K.;Hagihara,T.;Kumada,M.J. Org. Chem. 1988,53,113.(c)Sawamura,M.;Nagata,H.;Sakamoto,H.;Ito,Y.J. Am. Chem. Soc. 1992,114,2586.(d)Sawamura,M.;Nakayama,Y.;Tang,W.-M.;Ito,Y.J. Org. Chem. 1996, 61,9090.(e)Kaneko,S.;Yoshino,T.;Katoh,T.;Terashima,S.Tetrahedron: Asymmetry 1997,8,829.(f)He,X.-C.;Wang,B.;Bai,D.

72
Palla
dium
-Cat
alyz
ed D
ynam
ic K
inet
ic A
sym
met
ric A
llylic
Alk
ylat
ion
with
the
DPP
BA L
igan
dsV
OL.
40,
NO
. 3 •
200
7Tetrahedron Lett. 1998,39,411.(g)He,X.-C.;Wang,B.;Yu,G.;Bai,D.Tetrahedron: Asymmetry 2001,12,3213.
(45) Trost,B.M.;Radinov,R.;Grenzer,E.M.J. Am. Chem. Soc. 1997,119,7879.
(46) Trost,B.M.;Frederiksen,M.U.Angew. Chem., Int. Ed. 2005,44,308.(47) (a)Trost,B.M.;Schroeder,G.M.J. Am. Chem. Soc. 1999,121,6759.
(b)Braun,M.;Laicher,F.;Meier,T.Angew. Chem., Int. Ed. 2000,39,3494.(c)Braun,M.;Meier,T.Synlett2005,2968.(d)Braun,M.;Meier,T.Angew. Chem., Int. Ed. 2006,45,6952.
(48) Trost,B.M.;Pissot-Soldermann,C.;Chen,I.Chem.—Eur. J. 2005,11,951.
(49) (a)Trost,B.M.;Dong,L.;Schroeder,G.M.J. Am. Chem. Soc. 2005,127,2844.(b)Trost,B.M.;Dong,L.;Schroeder,G.M.J. Am. Chem. Soc. 2005,127,10259.
(50) You,S.-L.;Hou,X.-L.;Dai,L.-X.;Zhu,X.-Z.Org. Lett. 2001,3,149.(51) Trost,B.M.;Xu,J.J. Am. Chem. Soc. 2005,127,2846.(52) Trost,B.M.;Xu,J.J. Am. Chem. Soc. 2005,127,17180.(53) Trost,B.M.;Bream,R.N.;Xu, J.Angew. Chem., Int. Ed. 2006,45,
3109.(54) Trost,B.M.;Ariza,X.J. Am. Chem. Soc. 1999,121,10727.(55) Trost,B.M.;Ariza,X.Angew. Chem., Int. Ed. Engl. 1997,36,2635.(56) (a)Trost,B.M.;Lee,C.B.J. Am. Chem. Soc. 2001,123,3671.(b)Trost,
B.M.;Lee,C.B.J. Am. Chem. Soc. 2001,123,3687.(57) (a)Trost,B.M.;Lee,C.B.J. Am. Chem. Soc. 1998,120,6818.(b)Trost,
B.M.;Lee,C.J. Am. Chem. Soc. 2001,123,12191.(58) (a)Hayashi,T.;Yamamoto,A.;Ito,Y.Tetrahedron Lett. 1988,29,99.(b)
Larksarp,C.;Alper,H.J. Am. Chem. Soc. 1997,119,3709.(59) Trost,B.M.;Bunt,R.C.Angew. Chem., Int. Ed. Engl. 1996,35,99.(60) Trost,B.M.;Bunt,R.C.;Lemoine,R.C.;Calkins,T.L.J. Am. Chem.
Soc. 2000,122,5968.(61) Trost,B.M.;Lemoine,R.C.Tetrahedron Lett. 1996,37,9161.(62) (a)Trost,B.M.;Horne,D.B.;Woltering.M.J.Angew. Chem., Int. Ed.
2003,42,5987.(b)Trost,B.M.;Horne,D.B.;Woltering.M.J.Chem.—Eur. J.2006,12,6607.
(63) Trost,B.M.;McEachern,E.J.;Toste,F.D.J. Am. Chem. Soc. 1998,120,12702.
(64) Trost,B.M.;Brown,B.S.;McEachern,E.J.;Kuhn,O.Chem.—Eur. J. 2003,9,4442.
(65) Trost,B.M.;Tang,W.;Schulte,J.L.Org. Lett. 2000,2,4013.(66) Trost,B.M.;Andersen,N.G.J. Am. Chem. Soc. 2002,124,14320.(67) Trost,B.M.;Tang,W.Org. Lett. 2001,3,3409.(68) (a)Trost,B.M.;Jiang,C.J. Am. Chem. Soc. 2001,123,12907.(b)Jiang,
C.Ph.D.Dissertation,StanfordUniversity,Stanford,CA,2005.(69) Trost,B.M.;Toste,F.D.J. Am. Chem. Soc. 1999,121,4545.(70) Burckhardt,U.;Baumann,M.;Togni,A.Tetrahedron: Asymmetry 1997,
8,155.(71) Trost,B.M.;Jiang,C.Org. Lett. 2003,5,1563.(72) Trost,B.M.;Fandrick,D.R.J. Am. Chem. Soc. 2003,125,11836.(73) Lucet,D.;LeGall,T.;Mioskowski,C.Angew. Chem., Int. Ed. 1998,37,
2580.(74) Trost,B.M.;Fandrick,D.R.Org. Lett. 2005,7,823.(75) Trost,B.M.;Tsui,H.-C.;Toste,F.D.J. Am. Chem. Soc. 2000,122,
3534.(76) Trost,B.M.;Machacek,M.R.;Tsui,H.C.J. Am. Chem. Soc. 2005,127,
7014.(77) (a)Trost,B.M.;Thiel,O.R.;Tsui,H.-C.J. Am. Chem. Soc. 2002,124,
11616.(b)Trost,B.M.;Thiel,O.R.;Tsui,H.-C.J. Am. Chem. Soc. 2003,125,13155.
(78) Trost,B.M.;Toste,F.D.J. Am. Chem. Soc. 1999,121,3543.(79) Trost,B.M.;Toste,F.D.J. Am. Chem. Soc. 2003,125,3090.(80) (a)Trost,B.M.;Crawley,M.L.J. Am. Chem. Soc. 2002,124,9328.(b)
Trost,B.M.;Crawley,M.L.Chem.—Eur. J. 2004,10,2237.(81) (a)Ogasawara,M.;Ikeda,H.;Hayashi,T.Angew. Chem., Int. Ed. 2000,39,
1042.(b)Ogasawara,M.;Ikeda,H.;Nagano,T.;Hayashi,T.J. Am. Chem. Soc.2001,123,2089.(c)Imada,Y.;Ueno,K.;Kutsuwa,K.;Murahashi,S.-I.Chem. Lett.2002,140.(d)Ogasawara,M.;Ueyama,K.;Nagano,T.;Mizuhata,Y.;Hayashi,T.Org. Lett.2003,5,217.(e)Ogasawara,M.;Nagano,T.;Hayashi,T.J. Org. Chem. 2005,70,5764.
(82) Trost,B.M.;Fandrick,D.R.;Dinh,D.C.J. Am. Chem. Soc. 2005,127,14186.
(83) Dong,Y.;Teesdale-Spittle,P.;Hoberg,J.O.Tetrahedron Lett. 2005,46,353.
(84) ForevidenceofaPd–Pddisplacementmechanism,seeGranberg,K.L.;Bäckvall,J.-E.J. Am. Chem. Soc. 1992,114,6858.
(85) Forevidenceofa reductiveeliminationofanallylPd(II)acetate, seeLofstedt,J.;Franzen,J.;Bäckvall,J.-E.J. Org. Chem. 2001,66,8015andreferencestherein.
About the AuthorsBarry M. Trost wasbornin1941inPhiladelphia,Pennsylvania,wherehebeganhisuniversitytrainingattheUniversityofPennsylvania(B.A.,1962).HeobtainedaPh.D.degreeinchemistryjustthreeyearslaterattheMassachusettsInstituteofTechnology(1965).HethenmovedtotheUniversityofWisconsin-Madison,wherehewaspromotedtoProfessorofChemistryin1969and,in1982,becametheVilasResearchProfessor.In1987,he joined the faculty atStanfordUniversity asProfessorofChemistry,andbecametheTamakiProfessorofHumanitiesandSciencesin 1990. In addition, he has beenVisiting Professor of Chemistry inGermany(UniversitiesofMarburg,Hamburg,andMunich),Denmark(UniversityofCopenhagen),France(UniversitiesofParisVIandParis-Sud),Italy(UniversityofPisa),andSpain(UniversityofBarcelona).In1994,hewaspresentedwithaDocteur Honoris CausaoftheUniversitéClaude-Bernard (LyonI,France)and, in1997,aDoctor Scientiarum Honoris CausaoftheTechnion,Haifa,Israel.In2006,hewasappointedHonoraryProfessoroftheShanghaiInstituteofOrganicChemistry.
ProfessorTrost’sworkhasbeencharacterizedbyaveryhighorderofimagination,innovation,andscholarship.Hehasrangedovertheentirefield of organic synthesis, particularly emphasizing extraordinarily novel methodologies. In recognition of his many contributions, ProfessorTrosthas receivedanumberofawards, including theACSAward inPureChemistry(1977),theACSAwardforCreativeWorkinSyntheticOrganicChemistry(1981),theBaekelandAward(1981),theArthurC.CopeScholarAward(1989),theGuentherAwardintheChemistryofEssentialOilsandRelatedProducts(1990),theDr.PaulJanssenPrize(1990),theASSUGraduateTeachingAward(1991),theBingTeachingAward(1993), theACSRogerAdamsAward(1995), thePresidentialGreenChemistryChallengeAward(1998),theHerbertC.BrownAwardforCreativeResearchinSyntheticMethods(1999),theBelgianOrganicSynthesisSymposiumElsevierAward(2000),theNicholsMedal(2000),theYamadaPrize(2001),theACSNobelLaureateSignatureAwardforGraduateEducationinChemistry(2002),theACSCopeAward(2004),andtheJohnScottAwardofthecityofPhiladelphia(2004).ProfessorTrosthasbeenelectedafellowoftheAmericanAcademyofSciences(1992)andamemberoftheNationalAcademyofSciences(1990).Hehas published two books and over 790 scientific articles.
Daniel R. Fandrick received his B.S. degree with a major inchemistryin2001fromtheUniversityofCalifornia,SanDiego.Duringhisundergraduatestudiesunder theguidanceofProfessor JosephM.O’Connor,hecontributedtothesynthesisofastrainedcyclicferrocenylenediyne complex. In 2006, he received his Ph.D. degree in organicchemistry at Stanford University under the supervision of ProfessorBarry M. Trost. His graduate studies focused on the developmentof several palladium-catalyzed dynamic kinetic asymmetric allylicalkylations and their applications in total synthesis.After graduation,hejoinedthechemicaldevelopmentgroupatBoehringerIngelheiminRidgefield, Connecticut.^
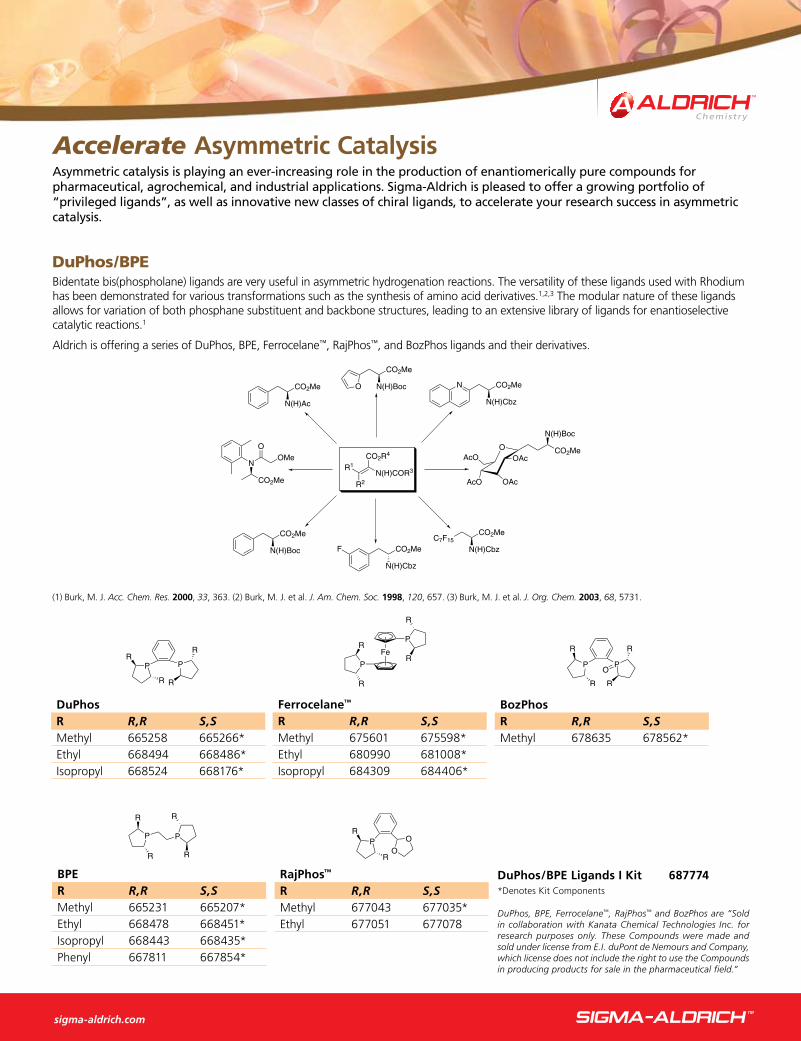
sigma-aldrich.com
Accelerate Asymmetric Catalysisasymmetric catalysis is playing an ever-increasing role in the production of enantiomerically pure compounds for pharmaceutical, agrochemical, and industrial applications. sigma-aldrich is pleased to offer a growing portfolio of “privileged ligands”, as well as innovative new classes of chiral ligands, to accelerate your research success in asymmetric catalysis.
DuPhos/BPE
DuPhosR R,R S,SMethyl 665258 665266*Ethyl 668494 668486*Isopropyl 668524 668176*
BPER R,R S,SMethyl 665231 665207*Ethyl 668478 668451*Isopropyl 668443 668435*Phenyl 667811 667854*
Ferrocelane™
R R,R S,SMethyl 675601 675598*Ethyl 680990 681008*Isopropyl 684309 684406*
RajPhos™
R R,R S,SMethyl 677043 677035*Ethyl 677051 677078
BozPhosR R,R S,SMethyl 678635 678562*
DuPhos/BPE Ligands I Kit 687774*Denotes Kit Components
DuPhos, BPE, Ferrocelane™, RajPhos™ and BozPhos are “Sold in collaboration with Kanata Chemical Technologies Inc. for research purposes only. These Compounds were made and sold under license from E.I. duPont de Nemours and Company, which license does not include the right to use the Compounds in producing products for sale in the pharmaceutical field.”
Bidentate bis(phospholane) ligands are very useful in asymmetric hydrogenation reactions. The versatility of these ligands used with Rhodium has been demonstrated for various transformations such as the synthesis of amino acid derivatives.1,2,3 The modular nature of these ligands allows for variation of both phosphane substituent and backbone structures, leading to an extensive library of ligands for enantioselective catalytic reactions.1
Aldrich is offering a series of DuPhos, BPE, Ferrocelane™, RajPhos™, and BozPhos ligands and their derivatives.
R1
R2N(H)COR3
CO2R4
CO2Me
N(H)Ac
CO2Me
O N(H)Boc CO2MeN
N(H)Cbz
CO2Me
O
AcO OAc
OAcAcOCO2Me
N(H)Boc
NOMe
O
C7F15CO2Me
N(H)CbzCO2Me
N(H)Cbz
F
CO2Me
N(H)Boc
(1) Burk, M. J. Acc. Chem. Res. 2000, 33, 363. (2) Burk, M. J. et al. J. Am. Chem. Soc. 1998, 120, 657. (3) Burk, M. J. et al. J. Org. Chem. 2003, 68, 5731.
P PR
RR
R
PP
R R
R R
Fe
P
P
R
R
R
R
PPO
R
RR
R
PO
OR
R
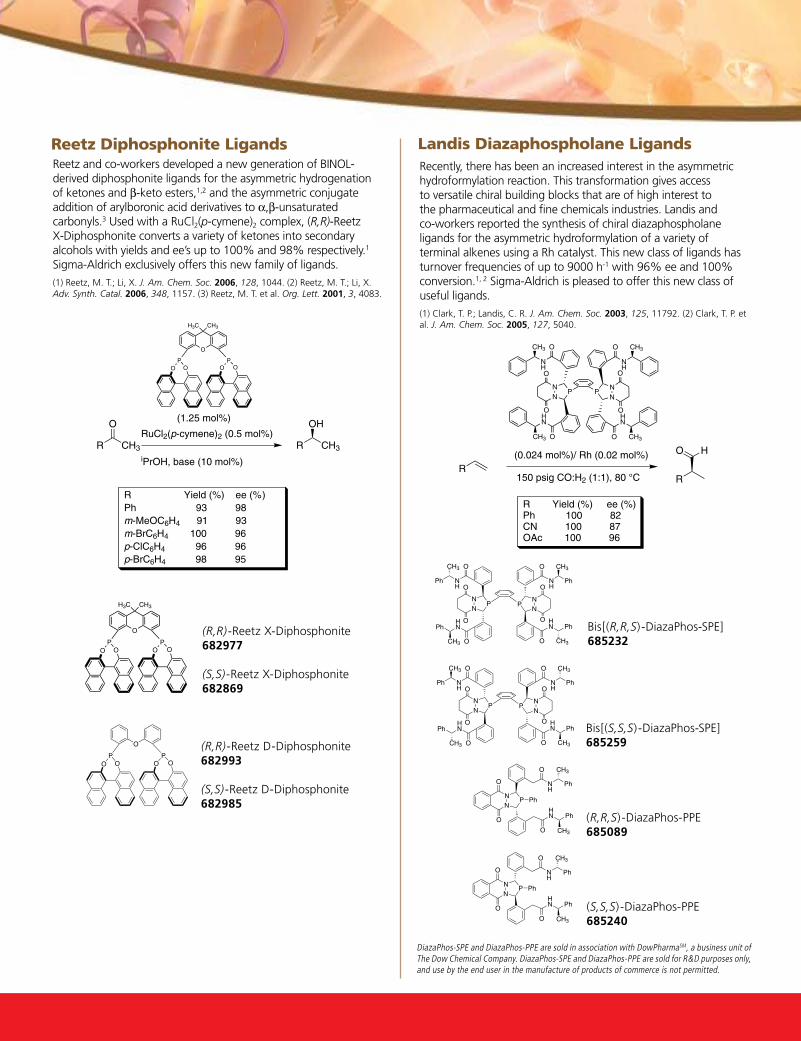
Landis Diazaphospholane LigandsRecently, there has been an increased interest in the asymmetric hydroformylation reaction. This transformation gives access to versatile chiral building blocks that are of high interest to the pharmaceutical and fine chemicals industries. Landis and co-workers reported the synthesis of chiral diazaphospholane ligands for the asymmetric hydroformylation of a variety of terminal alkenes using a Rh catalyst. This new class of ligands has turnover frequencies of up to 9000 h-1 with 96% ee and 100% conversion.1, 2 Sigma-Aldrich is pleased to offer this new class of useful ligands.(1) Clark, T. P.; Landis, C. R. J. Am. Chem. Soc. 2003, 125, 11792. (2) Clark, T. P. et al. J. Am. Chem. Soc. 2005, 127, 5040.
RR
O H
P PNNN
N
NH
NH
HN
HN
OO
OO
CH3CH3
CH3 CH3
OO
OO
R Yield (%) ee (%) Ph 100 82CN 100 87OAc 100 96
(0.024 mol%)/ Rh (0.02 mol%)
150 psig CO:H2 (1:1), 80 °C
Reetz Diphosphonite LigandsReetz and co-workers developed a new generation of BINOL-derived diphosphonite ligands for the asymmetric hydrogenation of ketones and β-keto esters,1,2 and the asymmetric conjugate addition of arylboronic acid derivatives to α,β-unsaturated carbonyls.3 Used with a RuCl2(p-cymene)2 complex, (R,R)-Reetz X-Diphosphonite converts a variety of ketones into secondary alcohols with yields and ee’s up to 100% and 98% respectively.1 Sigma-Aldrich exclusively offers this new family of ligands.(1) Reetz, M. T.; Li, X. J. Am. Chem. Soc. 2006, 128, 1044. (2) Reetz, M. T.; Li, X. Adv. Synth. Catal. 2006, 348, 1157. (3) Reetz, M. T. et al. Org. Lett. 2001, 3, 4083.
O
O OP
O OP
H3C CH3
O
O OP
O OP
(R,R)-Reetz X-Diphosphonite 682977
(S,S)-Reetz X-Diphosphonite 682869
(R,R)-Reetz D-Diphosphonite 682993
(S,S)-Reetz D-Diphosphonite 682985
NN
P
O
O
NN
P
O
O
NH
O
Ph
CH3
HN
O
Ph
CH3
NH
O
Ph
CH3
HN
O
Ph
CH3
NN
P
O
O
NN
P
O
O
NH
O
Ph
CH3
HN
O
Ph
CH3
NH
O
Ph
CH3
HN
O
Ph
CH3
Bis[(R,R,S)-DiazaPhos-SPE] 685232
Bis[(S,S,S)-DiazaPhos-SPE] 685259
N
O
N
O
P Ph
O
NH
CH3
Ph
O
HN
CH3
Ph
N
O
N
O
P Ph
O
NH
CH3
Ph
O
HN
CH3
Ph
(R,R,S)-DiazaPhos-PPE 685089
(S,S,S)-DiazaPhos-PPE 685240
DiazaPhos-SPE and DiazaPhos-PPE are sold in association with DowPharmaSM, a business unit of The Dow Chemical Company. DiazaPhos-SPE and DiazaPhos-PPE are sold for R&D purposes only, and use by the end user in the manufacture of products of commerce is not permitted.
O
O OP
O OP
H3C CH3
R
O
CH3 R CH3
OHRuCl2(p-cymene)2 (0.5 mol%)
iPrOH, base (10 mol%)
(1.25 mol%)
R Yield (%) ee (%)Ph 93 98m-MeOC6H4 91 93m-BrC6H4 100 96p-ClC6H4 96 96p-BrC6H4 98 95

sigma-aldrich.com
FeSulPhos Ligand for the Enantioselective 1,3-Dipolar Cycloaddition
Reference: Cabrera, S. et al. J. Am. Chem. Soc. 2005, 127, 16394.
Ph N CO2Me N
O
O
CH3+
(R)-FeSulPhos ( 3 mol %)Cu(CH3CN)4ClO4 (3 mol %)
NEt3 (18 mol %)CH2Cl2, rt
N
NH
CO2MePh
OO
Me
97% yield>99% ee
(RP)-FeSulPhos 687561
Sulfoximine Ligands for Asymmetric Aldol Reactions
Reference: Okamura, H.; Bolm, C. Chem. Lett. 2004, 33, 482.
OSiMe3Me
OMeO
O
OMeO Me OH
O
sulfoximine ligand (10 mol %)Cu(OTf)2 (10 mol %)
THF, rt
+
99% yield93% ee
(R)-S-Methyl-S-phenyl-N-[2-(2,4,6-triisopropylbenzylamino)-phenyl]sulfoximine669857
(S)-S-Methyl-S-phenyl-N-[2-(2,4,6-triisopropylbenzylamino)-phenyl]sulfoximine669970
Fe
S-t-Bu
PPh2
The asymmetric 1,3-dipolar cycloaddition reaction is of the utmost importance for the enantioselective synthesis of five-membered-ring heterocycles. Cabrera et al. introduced a new family of ligands consisting of a planar-chiral P,S-ligand, named FeSulPhos, for the 1,3-dipolar cycloaddition of azomethine ylides. The catalytic reaction is carried out with the FeSulPhos ligand, a copper salt, and triethylamine in methylene chloride. This new catalytic system demonstrated complete enantiocontrol (ee >99%) with conversions up to 97%. Sigma-Aldrich is pleased to offer this new ligand for asymmetric synthesis.
Chiral sulfoximine ligands have been studied for the past 15 years for use in catalytic asymmetric reactions. Bolm’s group developed a new class of sulfoximine used with copper salts for asymmetric aldol reactions. Using these bidentate ligands, Bolm and co-workers reported up to 93% ee’s and 99% yields for the Mukaiyama-type aldol reaction of 1-phenyl-1-(trimethylsilyloxy)ethane and methyl pyruvate. This new class of ligands is offered exclusively by Sigma-Aldrich.
N HNSO
H3C
H3C
H3C
H3CCH3
H3CCH3
Sigma-Aldrich’s ChemBlogs
An Industry First for Open Scientific Discussion
Stay on Topof the Latest Chemistry Innovations
from Sigma-Aldrich
Got ChemNews?
Monthly Chemistry E-Newsletter
Visit sigma-aldrich.com/chemnews.Visit chemblogs.com.

sigma-aldrich.com
PPh2
PPh2
FePh2P
N
PPh2
CH3
CH3
Boc-l-Ala Ligand for the Enantioselective Reduction of Ketones
O OH
90% yield96% ee
Boc-l-Ala (1.1 mol %)RuCl2(p-cymene)2 (0.5 mol %)NaOH (5 mol %)
2-propanol, rt
Reference: Bøgevig, A. et al. Chem.—Eur. J. 2004, 10, 294.
H3CNH
OCH3
HN OHBoc
Boc-l-alanine (2S)-2-hydroxypropylamide 684414
8 Chiral Quest Ligands for Asymmetric Hydrogenation
BoPhoz and PhanePhos* for Asymmetric Hydrogenation
(S)-Me-f-KetalPhos 685674
(R)-PhanePhos 682144
(S)-PhanePhos 682136
* Sold in collaboration with Johnson Matthey for research purposes only. US5874629 and any patents arising therefrom apply.
Chemists extensively use the enantioselective reduction of ketones to secondary alcohols. This reaction gives access to important functionalities for the synthesis of natural products. Adolfsson and co-workers reported a novel class of ligands, based on pseudo-dipeptides, for the efficient reduction of ketones. The ligand is used with RuCl2(p-cymene)2 in the presence of NaOH in 2-propanol. Yields of up to 90% with 96% ee have been reported. This new ligand is now part of the Sigma-Aldrich ligand library for asymmetric transformations.
Fe
PP
H3C
H3C
CH3
CH3O
O
CH3
CH3O
OH3CH3C
(S,S)-f-Binaphane 685925
P
P
Fe
P
P
H3C CH3
CH3
CH3CH3
CH3
CH3H3C
(R)-Methyl-BoPhoz 682322
(S)-Methyl-BoPhoz 682314
(R)-Xylyl-PhanePhos 682306
(S)-Xylyl-PhanePhos 682292
Chiral Quest Ligands Kit*
Chiral Quest ligands are some of the most potent for asymmetric hydrogenation. This new kit includes 7 ligands with 100 mg of each for rapid screening of chiral catalysts. The Chiral Quest ligands Kit I includes (R)-C3-TunePhos, (R)-Binaphane, (S,S’,R,R’)-TangPhos, (1R,1’R,2S,2’S)-Duan-Phos, (S)-Binapine, (S)-Me-f-KetalPhos, and (S,S)-f-Binaphane.
* Sold in collaboration with Chiral Quest for research purposes only. U.S. Patent: 6,828,271; 6,525,210; and additional patents pending.

77
VO
L. 4
0, N
O. 3
• 2
007
Outline1. Introduction2. Alkylation 2.1. Asymmetric Synthesis of α-Amino Acids and Their
Derivatives 2.1.1. Monoalkylation of Schiff Bases Derived from
Glycine 2.1.2. Dialkylation of Schiff Bases Derived from
α-Alkyl-α-aminoAcids 2.1.3. AlkylationofPeptidesActivatedbyaSchiffBase 2.2. OtherAlkylations3. TheMichaelAddition4. TheAldolandRelatedReactions5. TheDarzensCondensation6. TheNeberRearrangement7. Epoxidation8. Cyanation9. Conclusions10.Acknowledgements11.ReferencesandNotes
1. IntroductionTheevolutionofphase-transfercatalysis(PTC)wasledmainlybythedemandfromindustryinthemid-1960sforatrulyeffectiveprocedurefor transferringhydrophilicanionstoorganicmedia.Withitssimpleexperimentaloperations,mildreactionconditions,inexpensiveandenvironmentallybenignreagentsandsolvents,andthepossibilityofconductinglarge-scalepreparations,PTChassincebeenrecognizedasaversatilemethodologyfororganicsynthesisinbothindustrialandacademiclaboratories.1AsymmetricPTC,thatisbasedontheuseofstructurallywell-definedchiral,nonracemiccatalysts,hasbecomeatopicofgreatscientificinterestinthepasttwodecades.Recent,enormouseffortshaveresulted innotableachievements,makingitfeasibletoperformvariousbond-formingreactionsundermildphase-transfer-catalyzedconditions.2Thisreviewwillfocusonrecentadvancesinasymmetricreactions—whichareenabledbyC2-symmetric,chiral,phase-transfercatalysts
and reported between 2000 and 2006—and will showcase thevariationsintheirdesignsandapplications.OtherasymmetricPTCs,withcinchona-alkaloid-derived,chiralquaternaryammoniumsaltsandchiralcrownetherslackingC2-symmetry,arenotcoveredduetospacelimitation,andtheirrelevantreferencesarecitedonlyinconjunctionwith related reactions.Other, excellent reviewsonasymmetricphase-transfercatalysishavealsobeenpublished.2
2. Alkylation2.1. Asymmetric Synthesis of α-Amino Acids and Their Derivatives2.1.1. monoalkylation of schiff Bases Derived from GlycineIn1989,theresearchgroupledbyMartinO'Donnellsuccessfullyutilized chiral quaternary ammonium salts, prepared fromnaturallyoccurringalkaloids,for theasymmetricsynthesisofα-amino acids by using glycinate Schiff base 1asakeysubstrate(eq 1).3Theasymmetricalkylationof1proceededsmoothlyundermild phase-transfer conditions with N-(benzyl)cinchoniniumchloride (3a)ascatalyst togive thealkylationproduct (R)-2ain good yield and moderate enantioselectivity. This practicalasymmetric alkylation procedure has been strengthened intoanevenmorevaluableprotocol through thedevelopmentofanew class of cinchona-alkaloid-derived catalysts bearing anN-anthracenylmethylfunction.In1997,Lygo’sgroupdesignedN-anthracenylmethylammonium salts 3b and 4a and appliedthem to the asymmetric phase-transfer alkylation of 1 tosynthesize α-amino acids with much higher enantioselectivities.4At thesame time,Coreyandco-workerspreparedO-allyl-N-anthracenylmethylcinchonidiniumbromide(4b),andachievedhighasymmetricinductionintheenantioselectivealkylationof1bythecombineduseofsolidCsOH•H2Oatverylowtemperature.5These reports helped generate a great deal of interest inasymmetric phase-transfer catalysis, and the enantioselectivefunctionalizationof1,particularlyalkylation,hasbeenextensivelyutilizedasabenchmarkreactiontoevaluatetheefficienciesofnewlydevisedcatalystsincludingC2-symmetricones.
Development and Applications of C2-Symmetric, Chiral, Phase-Transfer Catalysts
Takashi Ooi† and Keiji Maruoka*Department of ChemistryGraduate School of ScienceKyoto UniversitySakyo, Kyoto 606-8502, JapanEmail: [email protected]
ProfessorTakashiOoi ProfessorKeijiMaruoka

78
VO
L. 4
0, N
O. 3
• 2
007
Dev
elop
men
t an
d A
pplic
atio
ns o
f C
2-Sy
mm
etric
, Chi
ral,
Phas
e-Tr
ansf
er C
atal
ysts
N+
Ar
Ar
Br–
N+
Ar
Ar
N+
R1
R1
R2
R2
N+
Ar
Ar
Ar
Ar
(S,S)-5a, Ar = 3,4,5-F3C6H2c, Ar = 3,5-(3,5-(t-Bu)2C6H3)2C6H3d, Ar = 3,5-(CF3)2C6H3e, Ar = 3,5-(3,5-(CF3)2C6H3)2C6H3f, Ar = 3,5-(3,4,5-F3C6H2)2C6H3g, Ar = 6-CF3-naphthalen-2-ylh, Ar = 4-CF3C6H4
(R,R)-5b, Ar = 3,5-Ph2C6H3
N+
Ar
Ar
Ar
Ar Ar
Ar
Ar
Ar
(S)-9R1 = 3,5-Ph2C6H3, R2 = Ph
(S,S)-6Ar = 3,5-Ph2C6H3
(S,S)-7Ar = 3,5-Ph2C6H3
N+
R'R'
R' R'
R'R'
R'R'
(R,R)-8R' = SiMe2(CH2CH2C8F17)
Br–
Br– Br–
Br– Br–
eq 1
Ph2C NO
Ot-Bu + BnBr Ph2C NO
Ot-BuH Bn
N
N
HX–
OR
2a
N
H R
Cl–OH
N
+
+
*
1
3a, R = Bn3b, R = (9-anthracenyl)CH2
4a; X = Cl, R = H 4b; X = Br, R = allyl
Cat.a
3a3b4a4b
Base
50% NaOH (aq)50% KOH (aq)50% KOH (aq)
CsOH•H2O
a Catalyst loading = 10 mol %.
Solvent
CH2Cl2PhMePhMe
CH2Cl2
Temp
20 oC20 oC20 oC
–78 oC
Time
9 h18 h18 h23 h
Yield
75%63%68%87%
ee
66% (R)89% (R)91% (S)94% (S)
Ref.
3a4a4b5
eq 2
Ph2C NO
Ot-Bu + RX Ph2C NO
Ot-BuH R
1
catalyst(mol %)
*
2
basesolvent, temp
Figure 1. chiral, c2-symmetric, phase-Transfer alkylation catalysts.
In 1999, our group reported the structurally rigid, chiralquaternaryammoniumsaltsoftype5a—derivedfromcommerciallyavailable (S)-or (R)-1,1’-bi-2-naphthol—asnewC2-symmetric,chiral,phase-transfercatalysts,whichweresuccessfullyappliedtothehighlyefficient,catalytic,andenantioselectivealkylationof1undermildphase-transferconditions (eq 2, Figure s 1–2,Table 1).6Thearomaticsubstituents(Ar)atthe3and3'positionsofonebinaphthylsubunitofthecatalysthadasignificanteffectontheenantiocontrollingabilityofthecatalyst,and5awasthecatalystofchoiceforthepreparationofavarietyofessentiallyenantiopureα-amino acids by this transformation.
To fully exploit the potential catalytic activity of chiralammoniumsalts suchas5b, binaryphase-transfer catalysis—usinganappropriateachiralco-catalyst—hasbeendeveloped.Forinstance,thephase-transfer-catalyzedbenzylationof1undertheinfluenceof (R,R)-5b (0.1mol%)and18-crown-6(0.1mol%)proceededsmoothlytofurnish(S)-2a in98%yieldand98%ee[4%yield(92%ee)without18-crown-6asco-catalyst].7
Withthecriticalroleofthe3,3’-diarylsubstituentsof5inmind,ourgroupalsoexaminedtheeffectofthe4,4’and6,6’substituentsofonebinaphthylsubunitonthestereoselectivityofthealkylationof1 throughthepreparationof(S,S)-6.8Wealsoassembledthesymmetrical phase-transfer catalyst 7, which exhibited highcatalyticandchiralefficiencies.9Thesymmetricalstructuralmotifin7ledustothedevelopmentoffluorous,chiral,phase-transfercatalyst8.Afterthealkylationreaction,8waseasilyrecoveredbysimpleextractionwithFC-72(perfluorohexanes)asafluoroussolventandwasusedforthenextrunwithoutanylossofreactivityorselectivity.10
Althoughtheconformationallyrigid,N-spirostructurecreatedbytwochiralbinaphthylsubunitsrepresentsacharacteristicfeatureof5andrelatedcatalysts(suchas6),italsoimposeslimitationsoncatalystdesignduetotheimperativeuseofthetwodifferentchiral binaphthyl moieties. Accordingly, our group developedthe C2-symmetric chiral quaternary ammonium bromide 9,incorporatingaconformationallyflexibleyeteasilymodifiableachiral biphenyl subunit, which exerted chiral efficiencies ashighas thoseof a seriesof conformationally rigidhomochiralcatalysts.11
Our group also undertook efforts to substantially enhancethereactivityofN-spiro,chiral,quaternaryammoniumsaltsandsimplify their structures for thepurposeofdevelopinga trulypractical method for the asymmetric synthesis of α-amino acids and theirderivatives.Ourinitialattemptwastodesignpolyamine-basedchiralphase-transfercatalystswiththeexpectationofamultipliereffectoftheattachedchiralauxiliaries.Gratifyingly,catalyst(S)-10,bearinga3,4,5-trifluorophenylgroupatthe3and3’positionsofthechiralbinaphthylmoieties,gaveriseto95%ee.12Thisobservationledtothediscoverythatchiralquaternaryammoniumbromide(S)-11,possessingflexiblestraight-chainalkylgroupsinsteadofrigidbinaphthylmoieties,functionsasanunusuallyactivechiralphase-transfercatalyst.13Thereactionof1withvariousalkylhalidesproceededsmoothlyandwithexcellentenantioselectivitiesundermildconditionsinthepresenceofonly0.01–0.05mol%of(S)-11.Furthermore,ourgroupsucceededinassemblingahighlyreactivecatalyst, (S)-12, from the readily available, gallic acidderived(S)-4,4’,5,5’,6,6’-hexamethoxybiphenyldicarboxylicacid.14
The usefulness of other chiral sources for the moleculardesignofC2-symmetricphase-transfercatalystshasrecentlybeendemonstratedinquiteanattractivemanner(Figure 3, Table 1).Inconnectionwiththeintensiveinvestigationoftheabilityofchiralmetal–salencomplexesaschiralphase-transfercatalysts in thesynthesis of α,α-dialkyl-α-amino acids from α-substituted α-amino
NNN+
N+Br–
Br–
+Nn-Bu
n-Bu
Ar
ArOMe
MeO
MeO
MeO
MeO
OMe
+N
Ar
Ar
n-Bun-Bu
(S)-10, Ar = 3,4,5-F3C6H2
(S)-12, Ar = 3,4,5-F3C6H2(S)-11, Ar = 3,4,5-F3C6H2
Ar
Ar
Ar
ArAr
Ar
Ar
Ar
Br– Br–
Figure 2. chiral, c2-symmetric, phase-Transfer alkylation catalysts.

79
Taka
shi O
oi a
nd K
eiji
Mar
uoka
*V
OL.
40,
NO
. 3 •
200
7
+N N+
2 Br–+N
i-Pr
OH
HO
OH
i-Pr
i-Pr
Ph
TfO–
N
NNO O
HHMe MeBF4
–NH
N
NH OO
OMe
H
Me
MeO
H
MeCl–
NCu
N
O O
N+
N+O
O
Ar
Me Ar
ArMe
ArR
R'2 X–
18
O O
n-Bu
20
n-Bu
15
N+
+
N+Me
+
Me2 TfO–
1413
(S,S)-16a; R = Me, R' = t-Bu, Ar = p-An, X = Ib; R = Me, R' = t-Bu, Ar = p-An, X = BF4c; R = R' = n-Pr, Ar = p-Tol, X = Id; R = R' = n-Pr, Ar = p-Tol, X = BF4e; R = R' = p-FC6H4CH2CH2, Ar = p-Tol, X = BF4
O
O
OO
O
O
17
(S)-19
Figure 3. chiral, c2- and c3-symmetric, phase-Transfer alkylation catalysts.
N+
N
H
O
Br–
Q Q
Br–
HQ HQF
N+
N
H
OHQ =
HQ HQ
Q
Q
21 22 24
25(PF6
–)
Q
Q
Q
23
N
N
N
N
Q26
Q
Q =
Figure 4. chiral, c2- and c3-symmetric, phase-Transfer alkylation catalysts.
acids,Belokon’sgroupreportedontheeffectivenessof13 intheasymmetricmonoalkylationof1.15
Nagasawaandco-workersreportedaC2-symmetricchiralcyclicguanidine of type 14 for the asymmetric alkylation of 1.16 Thestructurallyrelated15wasalsoevaluatedasachiralphase-transfercatalystbyMurphyandco-workers.17
Shibasaki’sgroupdesigneda tartrate-derivedbis(ammoniumsalt),16,basedontheconceptoftwo-centerasymmetriccatalysis,andsystematicallyoptimizedthereactionparametersforachievinghighenantioselectivity.18 Bycombiningatartratederivativeand2,5-dimethylpyrroline,MacFarland’sgroupprepareddiastereomericbis(ammoniumsalts)17,andtestedthemaschiralphase-transfercatalysts.19
The structurally unique, spiro-type bis(ammonium salt) 18wassynthesizedandsuccessfullyapplied tosimilarasymmetricalkylations of 1 by Sasai and co-workers.20 His group alsopreparedthechiralcrownether(S)-19,whichgaverisetomoderateenantioselectivityinthebenzylationof1inthepresenceofKOH.21
TheC3-symmetric,amine-based,chiralphase-transfercatalyst20hasbeendevelopedbyTakabe’sgroup.22Thehydroxylgroupsareexpectedtoplayanimportantroleashydrogen-bonddonorsintheformationofchiralionpairs.
ThedevelopmentofC2-andC3-symmetriccatalystsbyusingnaturallyoccurringalkaloidsaschiralunitshasalsobeenpursuedbyseveralresearchgroups(Figure 4, Table 1).ThegroupofJewandParkdesigneddimericandtrimericcinchona-alkaloid-derivedcatalysts 21,23 22,24 and 23,25 which substantially enhanced theenantioselectivityofthealkylationof1andexpandedthescopeofusablealkylhalideswhencomparedtotheirmonomericcounterparts.Moreover,thesameworkersinvestigatedtheidealaromaticspacerforoptimaldimericcatalystsandfoundthatcatalyst24,derivedfrom2,7-bis(bromomethyl)naphthaleneandtwocinchonaalkaloidunits, exhibited remarkable catalytic and chiral efficiencies.26Nájera’sgroupalsopreparedadimericsalt,25,whichincorporatesadimethylanthracenylbridgeasaspacer.27Inaddition,SivaandMuruganutilizedacyclictetraamineasaspacerfortheassemblyof26,whichexhibitedanextremelyhighperformanceaschiralphase-transfercatalyst.28
Thesedevelopments,togetherwiththeemergenceofotherchiralphase-transfercatalysts,29haveledtoimportant,enantiomericallyenriched α-amino acids and their derivatives being readily prepared by the asymmetric alkylation (Figure 5).30–35 These α-amino acidsandderivativeshavebeenemployedinthetotalsynthesisofbiologicallyactivecompounds.
2.1.2. Dialkylation of schiff Bases Derived from α-alkyl-α-amino acidsNonproteinogenic, chiral α,α-dialkyl-α-amino acids possessing stereochemically stable quaternary carbon centers have beensignificantsynthetictargets,notonlybecausetheyoftenareeffectiveenzymeinhibitors,butalsobecausetheyareindispensablefortheelucidation of enzymatic mechanisms. Accordingly, numerousstudieshavebeenconductedtodeveloptrulyefficientmethodsfortheirpreparation,36andphase-transfercatalysishasmadeuniquecontributions.
OnthebasisofO’Donnell’spioneeringstudyoftheasymmetricalkylationofthealdimineSchiffbasederivedfromalanineunderphase-transferconditions,37,38Belokonetal.demonstratedthat(R,R)-TADDOL(28)39 and thecopper(II)–salencomplex,13,15,40wereemployablefortheenantioselectivealkylationofalanine-derivedimines27and29(Scheme 1).
Ourgroupdevelopedaone-pot,highlyenantioselectivedoublealkylation of glycine-derived aldimine 30 by utilizing chiral
quaternaryammoniumbromide(S,S)-5a(Scheme 2).41Thisprovidesanattractiveandpowerfulstrategyfortheasymmetricsynthesisofstructurally diverse α,α-dialkyl-α-amino acids.
Sincethestereochemistryofthenewlycreatedquaternarycarboncenterwasapparentlydeterminedinthesecondalkylationprocess,thecoreofthismethodshouldbeapplicabletotheasymmetricalkylationofaldimineSchiffbase32 derived from the corresponding α-alkyl-α-amino acids. This approach was pursued by our group,41aswellasShibasaki’s18bandMaeda’s,42byusingC2-symmetricquaternaryammoniumsaltsascatalysts(Scheme 3).dl-Alanine-,phenylalanine-,leucine-,andphenylglycine-derivedimines32a–dwerealkylatedsmoothlywith (S,S)-5a and (S,S)-16b under similar conditions,affordingthedesirednoncodeddialkylaminoacidesters31withexcellent asymmetric induction. This powerful quaternizationmethod has also allowed the catalytic asymmetric synthesis ofquaternaryisoquinolinederivatives30and4-hydroxy-2-phenylprolinederivatives42from32c.
The efficient phase-transfer-catalyzed alkylation strategythatutilizes(S,S)-5awassuccessfullyappliedbyJewandPark’sgroup to the asymmetric synthesis of α-alkylserines starting with phenyloxazolinederivative33a.Thereactionisgeneralandpractical,and leads to a variety of optically active α-alkylserines after acidic hydrolysis(Scheme 4).43
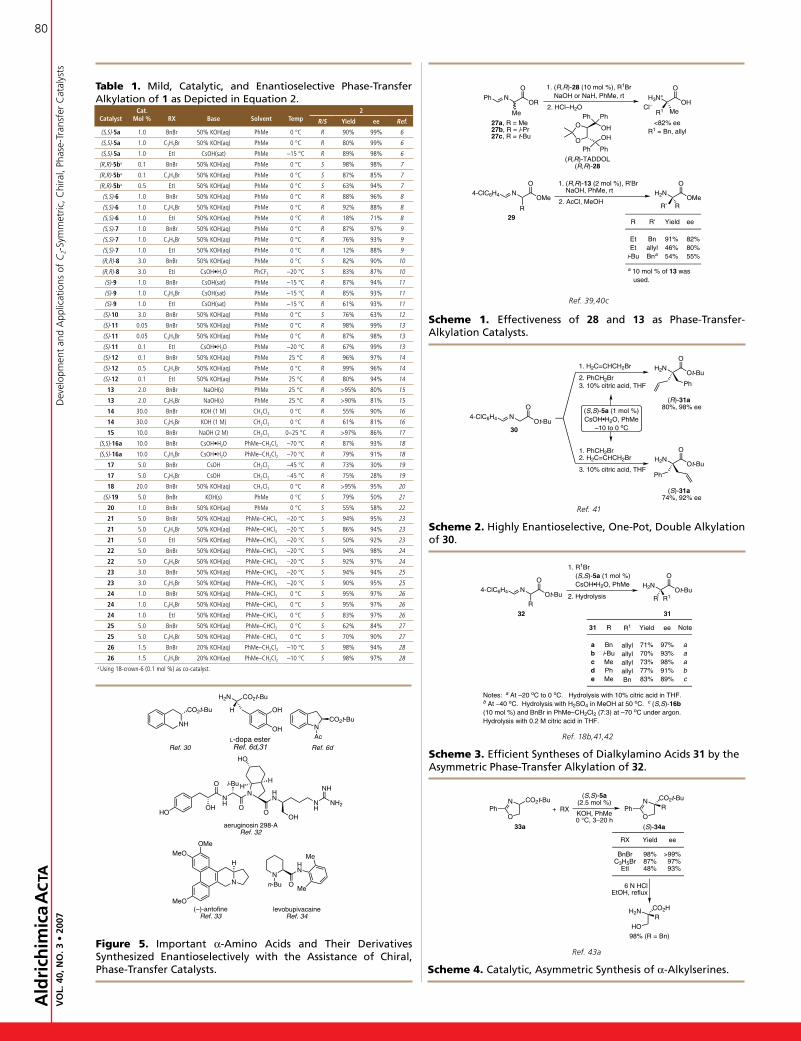
80
VO
L. 4
0, N
O. 3
• 2
007
Dev
elop
men
t an
d A
pplic
atio
ns o
f C
2-Sy
mm
etric
, Chi
ral,
Phas
e-Tr
ansf
er C
atal
ysts
NO
ORPh
Me
H3N+O
OH
O
O OHOH
Ph Ph
PhPh
NO
OMe4-ClC6H4
R2. AcCl, MeOH
1. (R,R)-28 (10 mol %), R1Br NaOH or NaH, PhMe, rt
H2NO
OMe
27a, R = Me27b, R = i-Pr27c, R = t-Bu
<82% eeR1 = Bn, allyl
(R,R)-TADDOL(R,R)-28
1. (R,R)-13 (2 mol %), R'Br NaOH, PhMe, rt
29
R1 Me
R’ R
2. HCl–H2O Cl–
R
EtEt
i-Bu
R'
BnallylBna
Yield
91%46%54%
ee
82%80%55%
a 10 mol % of 13 was used.
Scheme 1. effectiveness of 28 and 13 as phase-Transfer-alkylation catalysts.
Ref. 39,40c
4-ClC6H4 NO
Ot-Bu
H2NO
Ot-Bu
H2NO
Ot-Bu
30
(S)-31a 74%, 92% ee
(R)-31a 80%, 98% ee
Ph
Ph
1. H2C=CHCH2Br
3. 10% citric acid, THF
2. PhCH2Br3. 10% citric acid, THF
(S,S)-5a (1 mol %)CsOH•H2O, PhMe
–10 to 0 oC
1. PhCH2Br2. H2C=CHCH2Br
Scheme 2. highly enantioselective, one-pot, Double alkylation of 30.
Ref. 41
4-ClC6H4 NO
Ot-BuR
H2NO
Ot-BuR1R
31
abcde
R
Bni-BuMePhMe
R1
allylallylallylallylBn
Yield
71%70%73%77%83%
ee
97%93%98%91%89%
3132
1. R1Br (S,S)-5a (1 mol %) CsOH•H2O, PhMe
2. Hydrolysis
Note
aaabc
Notes: a At –20 oC to 0 oC. Hydrolysis with 10% citric acid in THF. b At –40 oC. Hydrolysis with H2SO4 in MeOH at 50 oC. c (S,S)-16b (10 mol %) and BnBr in PhMe–CH2Cl2 (7:3) at –70 oC under argon. Hydrolysis with 0.2 M citric acid in THF.
Scheme 3. efficient syntheses of Dialkylamino acids 31 by the asymmetric phase-Transfer alkylation of 32.
Ref. 18b,41,42
+ RXO
NPh
CO2t-Bu
O
NPh
CO2t-BuR
33a
(S,S)-5a(2.5 mol %)
(S)-34a
HO
H2N CO2HR
98% (R = Bn)
KOH, PhMe0 °C, 3–20 h
Yield
98%87%48%
RX
BnBrC3H5Br
EtI
ee
>99% 97% 93%
6 N HClEtOH, reflux
Scheme 4. catalytic, asymmetric synthesis of α-alkylserines.
Ref. 43a
NH
OH
i-BuN
O
HN
NH
OH
HO
HHO
NH2
NH
HOO
NO
HN
Me
Men-Bu
OMeMeO
MeO
N
H
NH
CO2t-Bu
NCO2t-Bu
aeruginosin 298-ARef. 32
H2N CO2t-Bu
H
OH
OH
levobupivacaineRef. 34
(–)-antofineRef. 33
L-dopa esterRef. 6d,31Ref. 30 Ref. 6d
Ac
Figure 5. important α-amino acids and Their Derivatives synthesized enantioselectively with the assistance of chiral, phase-Transfer catalysts.
Table 1. mild, catalytic, and enantioselective phase-Transfer alkylation of 1 as Depicted in equation 2.
Catalyst
Cat. Mol %
RX
Base
Solvent
Temp
2
R/S Yield ee Ref.
(S,S)-5a 1.0 BnBr 50% KOH(aq) PhMe 0 °C R 90% 99% 6
(S,S)-5a 1.0 C3H5Br 50% KOH(aq) PhMe 0 °C R 80% 99% 6
(S,S)-5a 1.0 EtI CsOH(sat) PhMe –15 °C R 89% 98% 6
(R,R)-5ba 0.1 BnBr 50% KOH(aq) PhMe 0 °C S 98% 98% 7
(R,R)-5ba 0.1 C3H5Br 50% KOH(aq) PhMe 0 °C S 87% 85% 7
(R,R)-5ba 0.5 EtI 50% KOH(aq) PhMe 0 °C S 63% 94% 7
(S,S)-6 1.0 BnBr 50% KOH(aq) PhMe 0 °C R 88% 96% 8
(S,S)-6 1.0 C3H5Br 50% KOH(aq) PhMe 0 °C R 92% 88% 8
(S,S)-6 1.0 EtI 50% KOH(aq) PhMe 0 °C R 18% 71% 8
(S,S)-7 1.0 BnBr 50% KOH(aq) PhMe 0 °C R 87% 97% 9
(S,S)-7 1.0 C3H5Br 50% KOH(aq) PhMe 0 °C R 76% 93% 9
(S,S)-7 1.0 EtI 50% KOH(aq) PhMe 0 °C R 12% 88% 9
(R,R)-8 3.0 BnBr 50% KOH(aq) PhMe 0 °C S 82% 90% 10
(R,R)-8 3.0 EtI CsOH•H2O PhCF3 –20 °C S 83% 87% 10
(S)-9 1.0 BnBr CsOH(sat) PhMe –15 °C R 87% 94% 11
(S)-9 1.0 C3H5Br CsOH(sat) PhMe –15 °C R 85% 93% 11
(S)-9 1.0 EtI CsOH(sat) PhMe –15 °C R 61% 93% 11
(S)-10 3.0 BnBr 50% KOH(aq) PhMe 0 °C S 76% 63% 12
(S)-11 0.05 BnBr 50% KOH(aq) PhMe 0 °C R 98% 99% 13
(S)-11 0.05 C3H5Br 50% KOH(aq) PhMe 0 °C R 87% 98% 13
(S)-11 0.1 EtI CsOH•H2O PhMe –20 °C R 67% 99% 13
(S)-12 0.1 BnBr 50% KOH(aq) PhMe 25 °C R 96% 97% 14
(S)-12 0.5 C3H5Br 50% KOH(aq) PhMe 0 °C R 99% 96% 14
(S)-12 0.1 EtI 50% KOH(aq) PhMe 25 °C R 80% 94% 14
13 2.0 BnBr NaOH(s) PhMe 25 °C R >95% 80% 15
13 2.0 C3H5Br NaOH(s) PhMe 25 °C R >90% 81% 15
14 30.0 BnBr KOH (1 M) CH2Cl2 0 °C R 55% 90% 16
14 30.0 C3H5Br KOH (1 M) CH2Cl2 0 °C R 61% 81% 16
15 10.0 BnBr NaOH (2 M) CH2Cl2 0–25 °C R >97% 86% 17
(S,S)-16a 10.0 BnBr CsOH•H2O PhMe–CH2Cl2 –70 °C R 87% 93% 18
(S,S)-16a 10.0 C3H5Br CsOH•H2O PhMe–CH2Cl2 –70 °C R 79% 91% 18
17 5.0 BnBr CsOH CH2Cl2 –45 °C R 73% 30% 19
17 5.0 C3H5Br CsOH CH2Cl2 –45 °C R 75% 28% 19
18 20.0 BnBr 50% KOH(aq) CH2Cl2 0 °C R >95% 95% 20
(S)-19 5.0 BnBr KOH(s) PhMe 0 °C S 79% 50% 21
20 1.0 BnBr 50% KOH(aq) PhMe 0 °C S 55% 58% 22
21 5.0 BnBr 50% KOH(aq) PhMe–CHCl3 –20 °C S 94% 95% 23
21 5.0 C3H5Br 50% KOH(aq) PhMe–CHCl3 –20 °C S 86% 94% 23
21 5.0 EtI 50% KOH(aq) PhMe–CHCl3 –20 °C S 50% 92% 23
22 5.0 BnBr 50% KOH(aq) PhMe–CHCl3 –20 °C S 94% 98% 24
22 5.0 C3H5Br 50% KOH(aq) PhMe–CHCl3 –20 °C S 92% 97% 24
23 3.0 BnBr 50% KOH(aq) PhMe–CHCl3 –20 °C S 94% 94% 25
23 3.0 C3H5Br 50% KOH(aq) PhMe–CHCl3 –20 °C S 90% 95% 25
24 1.0 BnBr 50% KOH(aq) PhMe–CHCl3 0 °C S 95% 97% 26
24 1.0 C3H5Br 50% KOH(aq) PhMe–CHCl3 0 °C S 95% 97% 26
24 1.0 EtI 50% KOH(aq) PhMe–CHCl3 0 °C S 83% 97% 26
25 5.0 BnBr 50% KOH(aq) PhMe–CHCl3 0 °C S 62% 84% 27
25 5.0 C3H5Br 50% KOH(aq) PhMe–CHCl3 0 °C S 70% 90% 27
26 1.5 BnBr 20% KOH(aq) PhMe–CH2Cl2 –10 °C S 98% 94% 28
26 1.5 C3H5Br 20% KOH(aq) PhMe–CH2Cl2 –10 °C S 98% 97% 28a Using 18-crown-6 (0.1 mol %) as co-catalyst.

81
Taka
shi O
oi a
nd K
eiji
Mar
uoka
*V
OL.
40,
NO
. 3 •
200
7
eq 3
Ph2C NO
NH
O
Ot-BuBn
Ph2C NO
NH
O
Ot-BuBn
Bn
Ph2C NO
NH
O
Ot-BuBn
Bn
DL-36
(S,S)-5c (2 mol %)PhCH2Br (1.1 equiv)
PhMe–50% KOH(aq)0 °C, 6 h
97%, 97% de35
LL-36
>>
Ref. 44
O
CO2t-Bu
O
R
CO2t-Bu(S,S)-5d (1 mol %)
RBr (1.2 equiv)
PhMe, CsOH•H2O–40 to –60 °C, 2.5–9 h
NHBn
R
CO2t-BuOH
R
CO2t-Bu
37
3890% (dr = 86:14)(R = Bn, 97% ee)
L-Selectride®
THF, –78 °C
3998% (dr = 84:16)(R = Bn, 97% ee)
R = Bn; 94%, 97% eeR = PhCH=CHCH2; 80%, 92% ee
BnNH2NaBH3CN, AcOH
4 Å MS, MeOH65 °C
Scheme 5. asymmetric phase-Transfer alkylation of β-Keto esters for the construction of Quaternary stereocenters, and the stereoselective conversion of the intermediates into β-hydroxy and β-amino esters.
Ref. 46
N
O
CO2t-Bu
1-NpN
O
CO2t-Bu
1-Np
CO2Et
CO2H
+ 1-NpCO2H
H2N
OHCO2H
6 N HCl
(S,S)-5a (2.5 mol %)H2C=CHCO2Et
93%, 97% ee
95% 98%
33b1-Np = 1-naphthyl
BEMP (1.25 equiv)CH2Cl2, –60 °C, 20 h
Scheme 6. asymmetric synthesis of (2s)-α-(hydroxymethyl)-glutamic acid.
Ref. 52
eq 4
OO
O
OOO
OO
OO
OO
OO
Ph2C NO
Ot-Bu
O
Y
N+
R
R R
RBr–
Ph2C NO
Ot-Bu
H (CH2)2COY
+
1
40R = 4-CF3C6H4CH2O 42
41R = 4-CF3C6H4CH2OR' = CH2N+Et3 Br–
Cat.
16c16d40414242
Cat.Mol %
101010 12020
Y
OEtOBn
Ot-BuMeEta
OEta
Base
Cs2CO3
Cs2CO3
CsOH•H2OCs2CO3
t-BuOKt-BuOK
Solvent
PhClPhCl
t-BuOMePhCl
CH2Cl2CH2Cl2
Temp
–30 oC–30 oC–60 oC–30 oC–78 oC–78 oC
Time
26 h 10 h 26 h114 h 0.3 h 24 h
Yield
71% 84% 73%100% 65% 76%
ee
82%81%77%75%96%87%
Ref.
18b18b49505151
R
R'
R'
R
a Ph2C=NCH2CO2Et was used instead of 1.
2.1.3. alkylation of peptides activated by a schiff BaseOurgrouphasfoundthatPTCwithC2-symmetricchiralquaternaryammonium salts of type 5 can be successfully applied to thestereoselectiveN-terminalalkylationofsmallpeptidessuchasGly-l-Phederivative35.Forinstance,thebenzylationof35with(S,S)-5c—possessingstericallyhinderedaromaticsubstituentsatthe3and3’positionsofthebinaphthylmoiety—underbiphasicconditions proceeded with almost complete diastereocontrol(eq 3).44Thismethodcanbeextendedto thediastereoselectivealkylationofSchiffbaseactivatedtripeptidesandtetrapeptides.
2.2. Other AlkylationsDue to the relatively high acidity of the α-methine proton, α-substituted β-keto esters are considered to be suitable substrates foralkylationunderphase-transferconditions.45Highefficienciesandenantioselectivitieshavebeenattainedintheconstructionofquaternary stereocenters on β-keto esters by such alkylation in the presenceofthesuitablymodifiedchiralquaternaryammoniumbromide5d.Thisreactionsystemhasabroadscopewithrespectto the β-keto esters and alkyl halides that can be used. The resulting alkylationproducts37 canbe readily converted intothe corresponding β-hydroxy esters 38 and β-amino esters 39(Scheme 5).46,47
3. The Michael Addition The asymmetric Michael addition of active methylene ormethine compounds to electron-deficient olefins, particularlyα,β-unsaturated carbonyl compounds, represents a fundamental approachforconstructingfunctionalizedcarbonframeworks.Thecombination of glycinate Schiff bases with α,β-unsaturated esters and ketones as electrophiles offers a practical route to various α-aminoacidshavinganadditionalcarbonylfunctionality.48
In this regard, the researchgroupsofShibasaki,18AraiandNishida,49,50 and Akiyama51 have carried out the asymmetricMichaeladditionofglycinederivative1 toacrylatesandvinylketones in thepresenceofC2-symmetric chiral phase-transfercatalystssuchaschiralquaternaryammoniumsalts16,40,and41,andachiralcrownether,42(eq 4).
Jew,Park,andco-workersachievedthehighlyenantioselectivesynthesis of (2S)-α-(hydroxymethyl)glutamic acid, a potent metabotropic receptor ligand, through theMichael additionof2-(naphthalen-1-yl)oxazoline-4-carboxylic acid tert-butyl ester(33b)toethylacrylateinthepresenceof(S,S)-5aascatalystandBEMPasbase(Scheme 6).52
Nitroalkanesarevaluableactivemethylenecompounds,53andourgroupdevelopedadiastereo-andenantioselectiveconjugateaddition of nitroalkanes to alkylidenemalonates54 and cyclicα,β-unsaturated ketones55undermildphase-transferconditions.In this transformation, the nature of the 3 and 3’ aromaticsubstituentsofthecatalystwascriticalforattainingahighlevelofstereoselectivitywitheachelectrophile(Scheme 7).
The structurally related chiral phase-transfer catalyst 5denables the enantioselective Michael addition of β-keto esters to α,β-unsaturated aldehydes and ketones, leading to the construction ofquaternarystereocentershavingthreedifferentfunctionalitiesofcarbonylorigin(Scheme 8).46Itisworthmentioningthattheuseofthefluorenylestergreatlyimprovedtheenantioselectivityofthereaction.
In conjunction with our research effort to design effectivecatalysts for the asymmetric epoxidation of α,β-unsaturated ketones(seeScheme11),ourgrouphasaddressedtheimportanceofdual-functioningchiralphase-transfercatalystssuchas46afor

82
VO
L. 4
0, N
O. 3
• 2
007
Dev
elop
men
t an
d A
pplic
atio
ns o
f C
2-Sy
mm
etric
, Chi
ral,
Phas
e-Tr
ansf
er C
atal
ysts
n-PrNO2
PhCO2i-Pr
CO2i-Pr
O2N *
*
99% (anti/syn = 86:14)97% ee (anti isomer)
O
NO2
* *
99% (anti/syn = 4:96)91% ee (syn isomer)
PhCH=C(CO2i-Pr)2(S,S)-5e (1 mol %)
PhMe, Cs2CO30 °C, 2.5 h
(S,S)-5f (1 mol %)PhMe, Cs2CO3
–20 °C, 6 h
O
Scheme 7. michael addition of nitroalkanes to alkylidene-malonates and α,β-Unsaturated Ketones.
Ref. 54,55
O
CO2R
O O
OCO2R
O
OCO2R
O
O
43
45 (R = 9-fluorenyl)>99%, 97% ee
(S,S)-5d (2 mol %)K2CO3 (10 mol %)
cumene
44a (R = t-Bu)84%, 79% ee
44b (R = 9-fluorenyl)92%, 90% ee
H2C=CHC(O)Me (2 equiv)–78 °C, 5 min
then –40 °C, 8 h
1. H2C=CHCHO (2 equiv) –78 oC, 5 min then –40 to –35 oC
2. p-TsOH (cat.), rt
Scheme 8. asymmetric michael addition of β-Keto esters to acrolein and methyl Vinyl Ketone.
Ref. 46
eq 5
Ph
O
Ph Ph
O Ph
X
X
N+
R
R
OH
OH
Ar Ar
Ar Ar
Br–
(S)-46a (3 mol %)
*
X = CO2Et (–20 °C)99%, 90% ee (R)
X = CN (–50 °C)98%, 81% ee (R)
(S)-46a, Ar = R = 3,5-Ph2C6H3b, Ar = 3,5-Ph2C6H3, R = H
K2CO3 (10 mol %)PhMe
+ CH2X2
Ref. 56
NO
Ot-BuPh2C
O
Ot-BuRNH2
OH
O
OEtHN
t-BuONH2
O4-An
1
47anti/syn = 96:4
4-An
NHBocCO2t-Bu
NCPh2
1. (R,R)-5e (2 mol %) RCHO (2–5 equiv)
1% NaOH(aq) (15 mol %) NH4Cl (10 mol %) PhMe, 0 °C2. 1 N HCl, THF
48, 88%syn/anti = 82:18
91% ee (syn)
49, 95%syn/anti = 95:582% ee (syn)
R
Ph(CH2)2
Me(CH2)4
(i-Pr)3SiOCH2
MeCya
Yield
82%79%73%54%83%
ee
98%97%98%99%98%
a CPME used as solvent.
1. (R,R)-5a (2 mol %) 4-AnN=CHCO2Et
17% NaOH(aq) MesH, –20 °C, 6 h2. 1 N HCl, THF
(S,S)-16e (10 mol %)4-AnC=NBoc
Cs2CO3 (2 equiv)PhF, –45 °C, 48 h
Scheme 9. highly Diastereo- and enantioselective Direct aldol and mannich reactions of a Glycine Derivative.
Ref. 58b,60,61
achievingahighlyenantioselectiveMichaeladditionofmalonatesormalononitriletochalconederivatives(eq 5).56,57
4. The Aldol and Related ReactionsAlthoughthephase-transfer-catalyzed,enantioselectivedirectaldolreactionofaglycinedonorwithaldehydeacceptorscouldprovide an ideal method for the simultaneous constructionof the primary structure and stereochemical integrity ofβ-hydroxy-α-amino acids—extremely important chiral units forpharmaceuticalchemistry—theexamples reported todateareverylimited.Accordingly,ourgrouprecentlydevelopedanefficientandhighlydiastereo-andenantioselectivedirectaldolreactionofglycinateSchiffbase1withawiderangeofaliphaticaldehydesundermildphase-transferconditionsemployingchiralquaternaryammoniumsalt5easakeycatalyst(Scheme 9).58,59The highly enantioselective phase-transfer-catalyzed, directMannich reactionof1with imineswasaccomplishedbyourgroup60 and the group of Ohshima and Shibasaki61using thestructurally related chiral ammonium bromide (R,R)-5a andthetartrate-derivedbis(ammoniumsalt)(S,S)-16eascatalysts,respectively(Scheme 9).
5. The Darzens CondensationTheDarzensreactionrepresentsoneofthemostpowerfulmethodsfor the synthesis of α,β-epoxy carbonyl and related compounds. Arai’sgroupsynthesizedanewquaternarybis(ammoniumsalt),50,from(S)-1,1’-bi-2-naphthol,andutilizeditforthepreparationof optically active α,β-epoxy amides as a mixture of cis and transisomers,52and53,throughreactionofhaloamides51 withaldehydes(eq 6).62
6. The Neber RearrangementThe Neber rearrangement of oxime sulfonates into α-amino ketones proceeds via a nitrene or an anion pathway. If thelattermechanism isoperating, theuseof a chiral base couldresult in the discrimination of two enantiotopic α protons to furnish optically active α-amino ketones. Verification of this hypothesiswasprovidedby thesuccessfulasymmetricNeberrearrangementofsimpleoximesulfonate55,generatedinsitufromtheparentoxime(Z)-54.Underphase-transferconditions,andusingC2-symmetricchiralquaternaryammoniumbromide5gor5h as catalyst, the corresponding protected α-amino ketone 56wasisolatedinhighyieldandmoderateenantiomericexcess(Scheme 10).63
7. EpoxidationSince the first reportbyWynberg’sgroupon theasymmetricepoxidationofelectron-deficientolefinsunderphase-transferconditions,64anumberofusefulcatalyst–oxidantcombinationshave been elaborated particularly for the epoxidation ofchalconederivatives.65Alongthisline,Murphyandco-workerspreparedtetracyclicC2-symmetricguanidiumsaltsof type15 from(S)-malicacid,andapplied themto theenantioselectiveepoxidationofchalconederivatives(eq 7).17
Ourgroupdesignedanew,dual-function,andhighlyefficientchiral quaternary ammonium salt, 46, for the asymmetricepoxidationofvariousenonesubstrates (Scheme 11).66 In theX-ray structure of the PF6 salt of 46a, the exceedingly highasymmetricinductionisascribabletothemolecularrecognitionabilityofthecatalyst towardenonesubstratesbyvirtueoftheappropriatelyalignedhydroxylfunctionalityaswellasthechiralmolecularcavity.Indeed,theobservedenantioselectivityhighlydependsonthesizeandtheelectronicpropertiesofArandRin

83
Taka
shi O
oi a
nd K
eiji
Mar
uoka
*V
OL.
40,
NO
. 3 •
200
7
eq 6
OPh
H
CONPh2
HX
NPh2
O OH
Ph
CONPh2
H
N
N
2 Br
52
PhCHO50 (2 mol %)
51 53
+
+
+
_
50
X
ClBr
Yield
81%81%
52:53
3.5:12.3:1
52
52%58%
53
51%63%
ee
RbOH•H2OCH2Cl2, rt
Ref. 62
Bn
N
Ar
OH
Ph
N
Ar
OTs
HH
Ph
N
ArH
PhO
ArNHBz
(Z)-54Ar = 4-FC6H4
p-TsCl (1.2 equiv)5 (5 mol %), MeOH (10 equiv)
PhMe–50% KOH(aq)0 °C
56(S,S)-5g; 95%, 50% ee(S,S)-5h; 90%, 63% ee
B*_
55
1. BzCl, Py CH2Cl2
2. 6 N HCl
Scheme 10. The asymmetric neber rearrangement of oxime sulfonate 55.
Ref. 63
eq 7
Ph
O
R Ph R
OO
N
NNO O
HHMe MeBF4
–
(R,R)-15 (5 mol %)
8% NaOCl, PhMe0–25 °C R = Ph, 93% ee
R = C6H13, 91% ee
(R,R)-15
+
Ref. 17
R1
O
R2 R1 R2
OO
Ph
O
Ph
OO
(S)-46 (3 mol %)
13% NaOClPhMe, 0 °C
91%, 99% ee
(S)-46a (3 mol %)
13% NaOClPhMe, 0 °C
R1
Pht-But-Bu
R2
PhPhCy
Cat.
(S)-46a(S)-46b(S)-46b
Yield
99%99%80%
ee
96%92%96%
Scheme 11. Dual-functioning catalyst, 46, for asymmetric epoxidations.
Ref. 66
46.ThegroupofJewandParkdemonstratedthatthecombineduse of a surfactant such as Span® 20 and dimeric cinchona-alkaloid-derivedphase-transfercatalyst57enabled thehighlyefficientandenantioselectiveepoxidationofchalconederivativesusing30%aqueoushydrogenperoxideasoxidant(eq 8).67
8. CyanationThe phase-transfer-catalyzed and highly enantioselectivecyanationofaldiminederivatives58withaqueousKCNhasbeenrealizedbyourgroupbasedonthechiralquaternaryammoniumiodide(R,R,R)-60,whichpossessesastereochemicallydefinedtetranaphthylbackbone.Awide rangeof aliphatic aldiminesincluding those having α-tert-alkylsubstituentsaretoleratedbythissystem(Scheme 12).68 The use of α-amide sulfones 59 asprecursorsofthereactiveimines58wasfoundtoenhanceboththechemicalyieldsandtheenantioselectivitiesinthepresenceofonlyaslightexcessofKCN(1.05equiv).69,70ThisstudyrepresentsanessentiallynewapproachtowardtheasymmetricStrecker-typereactions.Itharnessesthedistinctsyntheticadvantagesofchiralphase-transfercatalysistoprovideatrulypracticalrouteto various unusual, optically pure α-amino acids.
9. ConclusionsThe development of C2-symmetric, chiral, phase-transfercatalystslargelyreliesonthemoleculardesignofpurelysyntheticchiralquaternaryammoniumsalts.Thesesaltsoftendelivernotonly a higher reactivity and stereoselectivity but also create
eq 8
N+
N
H
OH
F
57
OMe
N+
HON
OMeH
R
O
Ph R Ph
OO
30% H2O2 (10 equiv)50% KOH (1 equiv)
i-Pr2O, rt, 4 h
57 (1 mol %)Span® 20 (1 mol %)
R
Ph4-FC6H4
4-MeC6H4
Yield
95%94%96%
ee
>99% 98% 97%
2 Br–
Ref. 67
R H
NSO2Mes
R CN
HNSO2Mes
NMe
Me
Ar
Ar
Ar
Ar
I–
R SO2p-Tol
HNSO2Mes
(R,R,R)-60 (1 mol %)KCN(aq) (1.5 equiv)
R
(R,R,R)-60Ar = 4-CF3C6H4
H
NSO2Mes
59
58
+
From
5858585959
R
Ph(CH2)2Cy
PhMe2CPh(CH2)2
Cy
Yield
81%89%95%99%99%
ee
90%95%98%94%97%
PhMe, 0 °C2–8 h
(R,R,R)-60 (1 mol %)KCN(aq) (1.05 equiv)
PhMe, 0 °C, 2 h
Scheme 12. The phase-Transfer-catalyzed asymmetric strecker reaction of aldimines and α-amide sulfones with (r,r,r)-60.
Ref. 68,69
newsyntheticopportunities,thusexpandingtheapplicabilityofasymmetricphase-transfercatalysisinmodernorganicsynthesis.Efforts need to continue to be made toward understandingthe relationship between catalyst structure, activity, andstereocontrollingability.Thesystematicaccumulationofsuchknowledgewould allowus to conduct an evenmore rationalcatalystdesignforpursuingselectivechemicalsynthesis inareliableandpracticalmanner.
10. AcknowledgementsWethankourcolleaguesatHokkaidoandKyotoUniversities,whosenamesappearinthecitedreferences,fortheirpersonalandscientificcollaborations.Withouttheirenthusiasmfororganicchemistry,ourresearchonthedevelopmentandapplicationofC2-symmetric,chiral,phase-transfercatalystswouldnothavebeenachieved.

84
VO
L. 4
0, N
O. 3
• 2
007
Dev
elop
men
t an
d A
pplic
atio
ns o
f C
2-Sy
mm
etric
, Chi
ral,
Phas
e-Tr
ansf
er C
atal
ysts 11. References and Notes
(†) Current address: Department of Applied Chemistry, GraduateSchoolofEngineering,NagoyaUniversity,Chikusa,Nagoya464-8603,Japan.
(1) (a)Dehmlow,E.V.;Dehmlow,S.S.Phase Transfer Catalysis,3rded.;Wiley-VCH:Weinheim,1993.(b)Starks,C.M.;Liotta,C. L.; Halpern, M. Phase-Transfer Catalysis: Fundamentals, Applications, and Industrial Perspectives;Chapman&Hall:NewYork,1994.(c)Handbook of Phase Transfer Catalysis;Sasson,Y.,Neumann,R.,Eds.;Chapman&Hall:NewYork,1997.(d)Phase-Transfer Catalysis: Mechanisms and Syntheses;Halpern,M.E.,Ed.;ACSSymposiumSeries659;AmericanChemicalSociety:Washington,DC,1997.
(2) (a)Shioiri,T.InHandbook of Phase Transfer Catalysis;Sasson,Y.,Neumann,R.,Eds.;Chapman&Hall:NewYork,1997;Chapter14.(b)O'Donnell,M.J.Phases–The Sachem Phase Transfer Catalysis Review1998,Issue4,5.(c)O'Donnell,M.J.Phases–The Sachem Phase Transfer Catalysis Review 1999, Issue 5, 5. (d) Nelson,A.Angew. Chem., Int. Ed. 1999,38,1583. (e)Shioiri,T.;Arai,S. InStimulating Concepts in Chemistry;Vogtle,F., Stoddart,J.F.,Shibasaki,M.,Eds.;Wiley-VCH:Weinheim,2000;p123.(f)O’Donnell,M.J.InCatalytic Asymmetric Syntheses,2nded.;Ojima, I., Ed.; Wiley-VCH: New York, 2000; Chapter 10. (g)O'Donnell,M.J.Aldrichimica Acta2001,34,3.(h)Maruoka,K.;Ooi,T.Chem. Rev.2003,103,3013.(i)O'Donnell,M.J.Acc. Chem. Res.2004,37,506.(j)Lygo,B.;Andrews,B.I.Acc. Chem. Res.2004,37,518.(k)Vachon,J.;Lacour,J.Chimia2006,60,266.
(3) (a)O'Donnell,M.J.;Bennett,W.D.;Wu,S.J. Am. Chem. Soc.1989,111,2353.Seealso: (b)Lipkowitz,K.B.;Cavanaugh,M.W.;Baker,B.;O'Donnell,M.J.J. Org. Chem.1991,56,5181.(c)Esikova,I.A.;Nahreini,T.S.;O'Donnell,M.J.InPhase-Transfer Catalysis: Mechanisms and Syntheses;Halpern,M.E.,Ed.;ACSSymposiumSeries659;AmericanChemicalSociety:Washington,DC,1997;Chapter 7. (d)O'Donnell,M. J.;Esikova, I.A.;Mi,A.; Shullenberger, D. F.; Wu, S. In Phase-Transfer Catalysis: Mechanisms and Syntheses;Halpern,M.E.,Ed.;ACSSymposiumSeries659;AmericanChemicalSociety:Washington,DC,1997;Chapter10.
(4) (a)Lygo,B.;Wainwright,P.G.Tetrahedron Lett.1997,38, 8595.(b)Lygo,B.;Crosby,J.;Lowdon,T.R.;Wainwright,P.G.Tetrahedron2001,57, 2391.(c)Lygo,B.;Crosby,J.;Lowdon,T.R.;Peterson,J.A.;Wainwright,P.G.Tetrahedron2001,57, 2403.
(5) Corey, E. J.; Xu, F.; Noe, M. C. J. Am. Chem. Soc. 1997, 119, 12414.
(6) (a)Ooi,T.;Kameda,M.;Maruoka,K.J. Am. Chem. Soc.1999,121,6519.(b)Maruoka,K.J. Fluorine Chem. 2001,112,95.(c)Ooi,T.;Uematsu,Y.;Maruoka,K.Adv. Synth. Catal.2002,344,288.(d)Ooi,T.;Kameda,M.;Maruoka,K.J. Am. Chem. Soc.2003,125,5139.
(7) Shirakawa,S.;Yamamoto,K.;Kitamura,M.;Ooi,T.;Maruoka,K.Angew. Chem., Int. Ed.2005,44, 625.
(8) Hashimoto,T.;Maruoka,K.Tetrahedron Lett.2003,44, 3313.(9) Hashimoto,T.;Tanaka,Y.;Maruoka,K.Tetrahedron: Asymmetry
2003,14, 1599.(10) Shirakawa,S.;Tanaka,Y.;Maruoka,K.Org. Lett.2004,6,1429.(11) Ooi,T.;Uematsu,Y.;Kameda,M.;Maruoka,K.Angew. Chem.,
Int. Ed.2002,41,1551.(12) (a) Kano, T.; Konishi, S.; Shirakawa, S.; Maruoka, K. Kyoto
University,Kyoto,Japan.Unpublishedwork,2006.(b)Kano,T.;Konishi,S.;Shirakawa,S.;Maruoka,K.Tetrahedron: Asymmetry2004,15,1243.
(13) Kitamura,M.;Shirakawa,S.;Maruoka,K.Angew. Chem., Int. Ed.2005,44, 1549.
(14) Han,Z.;Yamaguchi,Y.;Kitamura,M.;Maruoka,K.Tetrahedron Lett.2005,46,8555.
(15) Belokon,Y.N.;North,M.;Churkina,T.D.;Ikonnikov,N.S.;Maleev,V.I.Tetrahedron2001,57,2491.
(16) Kita,T.;Georgieva,A.;Hashimoto,Y.;Nakata,T.;Nagasawa,K.Angew. Chem., Int. Ed.2002,41,2832.
(17) Allingham,M.T.;Howard-Jones,A.;Murphy,P.J.;Thomas,D.A.;Caulkett,P.W.R.Tetrahedron Lett.2003,44,8677.
(18) (a)Shibuguchi,T.;Fukuta,Y.;Akachi,Y.;Sekine,A.;Ohshima,T.;Shibasaki,M.Tetrahedron Lett.2002,43,9539. (b)Ohshima,T.;Shibuguchi,T.;Fukuta,Y.;Shibasaki,M.Tetrahedron2004,60,7743.
(19) Kowtoniuk,W.E.;MacFarland,D.K.;Grover,G.N.Tetrahedron Lett.2005,46,5703.
(20) Sasai,H.Jpn.KokaiTokkyoKohoJP2003335780,2003.(21) Yonezawa,K.;Patil,M.L.;Sasai,H.;Takizawa,S.Heterocycles
2005,66,639.(22) (a)Mase,N.;Ohno,T.;Hoshikawa,N.;Ohishi,K.;Morimoto,H.;
Yoda,H.;Takabe,K.Tetrahedron Lett.2003,44,4073.(b)Seealso:Mase,N.;Ohno,T.;Morimoto,H.;Nitta,F.;Yoda,H.;Takabe,K.Tetrahedron Lett.2005,46,3213.
(23) Jew, S.-s.; Jeong, B.-S.; Yoo, M.-S.; Huh, H.; Park, H.-g. Chem. Commun.2001,1244.
(24) Park,H.-g.;Jeong,B.-S.;Yoo, M.-S.; Lee,J.-H.;Park,B.-s.;Kim,M.G.;Jew,S.-s.Tetrahedron Lett.2003,44, 3497.
(25) (a)Park,H.-g.;Jeong,B.-S.;Yoo,M.-S.;Park,M.-k.;Huh,H.;Jew,S.-s.Tetrahedron Lett.2001,42, 4645.(b)Forarelatedstructure,seeSiva,A.;Murugan,E.Synthesis2005,2927.
(26) Park,H.-g.;Jeong,B.-S.;Yoo,M.-S.;Lee,J.-H.;Park,M.-k.;Lee,Y.-J.;Kim,M.-J.;Jew,S.-s.Angew. Chem., Int. Ed.2002,41, 3036.
(27) Chinchilla,R.;Mazón,P.;Nájera,C.;Ortega, F. J.Tetrahedron: Asymmetry2004,15, 2603.
(28) Siva,A.;Murugan,E.J. Mol. Catal. A: Chem.2005,241,111.(29) For recent representative contributions with non-C2-symmetric
catalysts: (a)Belokon,Y.N.;Kochetkov,K.A.;Churkina,T.D.;Ikonnikov,N.S.;Larionov,O.V.;Harutyunyan,S.R.;Vyskocil,S.;North,M.;Kagan,H.B.Angew. Chem., Int. Ed.2001,40,1948.(b)Nakoji,M.;Kanayama,T.;Okino,T.;Takemoto,Y.Org. Lett.2001,3, 3329.(c)Chinchilla,R.;Mazón,P.;Nájera, C.Tetrahedron: Asymmetry2002,13, 927.(d)Mazón,P.;Chinchilla,R.;Nájera,C.;Guillena,G.;Kreiter,R.;KleinGebbink,R.J.M.;vanKoten,G.Tetrahedron: Asymmetry2002,13, 2181.(e)Jew,S.-s.;Yoo,M.-S.;Jeong,B.-S.;Park,I.Y.;Park,H.-g.Org. Lett. 2002, 4,4245.(f)Nakoji,M.;Kanayama,T.;Okino,T.;Takemoto,Y.J.Org. Chem.2002,67, 7418.(g)Guillena,G.;Kreiter,R.;vandeCoevering,R.;KleinGebbink,R.J.M.;vanKoten,G.;Mazón,P.;Chinchilla,R.;Nájera,C.Tetrahedron: Asymmetry2003,14, 3705.(h)Lygo,B.;Allbutt,B.;James,S.R.Tetrahedron Lett.2003,44,5629.(i)Lygo,B.;Allbutt,B.Synlett2004,326.(j)Yoo,M.-S.;Jeong,B.-S.;Lee,J.-H.;Park,H.-g.;Jew,S.-s.Org. Lett. 2005, 7,1129.(k)Elango,S.;Venugopal,M.;SureshandEni,P.S.Tetrahedron2005,61,1443.(l)Kumar,S.;Sobhia,M.E.;Ramachandran,U.Tetrahedron: Asymmetry2005,16,2599.(m)Andrus,M.B.;Ye,Z.;Zhang,J.Tetrahedron Lett. 2005,46,3839.(n)Kumar,S.;Ramachandran,U.Tetrahedron 2005,61,4141.(o)Kumar,S.;Ramachandran,U.Tetrahedron2005,61,7022.
(30) Ooi,T.;Takeuchi,M.;Maruoka,K.Synthesis2001,1716.(31) Ooi,T.;Kameda,M.;Tannai,H.;Maruoka,K.Tetrahedron Lett.
2000,41,8339.(32) (a) Ohshima, T.; Gnanadesikan, V.; Shibuguchi, T.; Fukuta, Y.;
Nemoto,T.;Shibasaki,M.J. Am. Chem. Soc.2003,125,11206.(b)Fukuta,Y.;Ohshima,T.;Gnanadesikan,V.;Shibuguchi,T.;Nemoto,T.;Kisugi,T.;Okino,T.;Shibasaki,M.Proc. Natl. Acad. Sci. U.S.A. 2004,101,5433.

85
Taka
shi O
oi a
nd K
eiji
Mar
uoka
*V
OL.
40,
NO
. 3 •
200
7
(33) Kim,S.;Lee,J.;Lee,T.;Park,H.-g.;Kim,D.Org. Lett.2003,5,2703.
(34) Kumar,S.;Ramachandran,U.Tetrahedron Lett.2005,46,19.(35) For other representative examples: (a) Lygo, B.; Crosby, J.;
Peterson,J.A.Tetrahedron2001,57,6447.(b)Nitz,M.;Mezo,A.R.;Ali,M.H.;Imperiali,B.Chem. Commun.2002,1912.(c)Boeckman,R.K.,Jr.;Clark,T.J.;Shook,B.C.Org. Lett.2002,4,2109.(d)Lygo,B.;Humphreys,L.D.Tetrahedron Lett.2002,43,6677.(e)Castle,S.L.;Srikanth,G.S.C.Org. Lett.2003,5,3611.(f)Lygo,B.;Andrews,B.I.Tetrahedron Lett.2003,44,4499.(g)Lygo,B.;Andrews,B.I.;Slack,D.Tetrahedron Lett.2003,44,9039. (h)Lemaire,C.;Gillet,S.;Guillouet,S.;Plenevaux,A.;Aerts,J.;Luxen,A.Eur. J. Org. Chem.2004,2899.
(36) (a)Cativiela,C.;Diaz-de-Villegas,M.D.Tetrahedron: Asymmetry1998,9, 3517. (b)Schöllkopf,U.Top. Curr. Chem.1983,109,65.
(37) O’Donnell,M.J.;Wu,S.Tetrahedron: Asymmetry1992,3,591.(38) For related contr ibutions with cinchona-alkaloid-derived
catalysts:(a)Jew,S.-s.;Jeong,B.-S.;Lee,J.-H.;Yoo,M.-S.;Lee,Y.-J.;Park,B.-s.;Kim,M.G.;Park,H.-g.J. Org. Chem.2003,68,4514.(b)Seealsoreference29f.
(39) Belokon,Y.N.;Kochetkov,K.A.;Churkina,T.D.;Ikonnikov,N.S.;Chesnokov,A.A.;Larionov,O.V.;Singh,I.;Parmar,V.S.;Vyskočil, Š.; Kagan, H. B. J. Org. Chem.2000,65,7041.
(40) (a)Belokon,Y.N.;Davies,R.G.;North,M.Tetrahedron Lett.2000, 41, 7245. (b) Belokon, Y. N.; Davies, R. G.; Fuentes, J.A.;North,M.;Parsons,T.Tetrahedron Lett.2001,42,8093.(c)Belokon,Y.N.;Bhave,D.;D’Addario,D.;Groaz,E.;North,M.;Tagliazucca,V.Tetrahedron2004,60,1849.(d)Belokon,Y.N.;Fuentes,J.;North,M.;Steed,J.W.Tetrahedron2004,60,3191.
(41) Ooi,T.;Takeuchi,M.;Kameda,M.;Maruoka,K.J. Am. Chem. Soc.2000,122,5228.
(42) Maeda, K.; Miller, R. A., Jr.; Szumigala, R. H.; Shafiee, A.;Karady, S.; Armstrong, J. D., III Tetrahedron Lett. 2005, 46,1545.
(43) (a)Jew,S.-s.;Lee,Y.-J.;Lee,J.;Kang,M.J.;Jeong,B.-S.;Lee,J.-H.;Yoo,M.-S.;Kim,M.-J.;Choi,S.-h.;Ku,J.-M.;Park,H.-g.Angew. Chem., Int. Ed.2004,43, 2382. (b)With cinchona-alkaloid-derived catalyst, see: Lee, Y.-J.; Lee, J.; Kim, M.-J.;Kim,T.-S.;Park,H.-g.;Jew,S.-s.Org. Lett.2005,7,1557.
(44) (a) Ooi, T.; Tayama, E.; Maruoka, K. Angew. Chem., Int. Ed.2003,42,579.(b)Maruoka,K.;Tayama,E.;Ooi,T.Proc. Natl. Acad. Sci. U.S.A.2004,101,5824.
(45) For recent exampleswithcinchona-alkaloid-derivedcatalysts,see:(a)Dehmlow,E.V.;Düttmann,S.;Neumann,B.;Stammler,H.-G.Eur. J. Org. Chem.2002,2087. (b)Park,E.J.;Kim,M.H.; Kim, D. Y. J. Org. Chem. 2004, 69, 6897. (c) Bella, M.;Kobbelgaard,S.;Jørgensen,K.A.J. Am. Chem. Soc.2005,127,3670.
(46) (a)Ooi,T.;Miki,T.;Taniguchi,M.;Shiraishi,M.;Takeuchi,M.;Maruoka,K.Angew. Chem., Int. Ed.2003,42,3796. (b)Forasimilar enantioselective alkylation of cyclic α-amino-β-ketoesters, see Ooi, T.; Miki, T.; Maruoka, K. Org. Lett. 2005, 7,191.
(47) For recent examples of other alkylations using C2-symmetriccatalysts, see: (a)Kumar,S.;Ramachandran,U.Tetrahedron: Asymmetry 2005,16,647.(b)Ooi,T.;Fukumoto,K.;Maruoka,K.Angew. Chem., Int. Ed.2006,45,3839.
(48) For recent examples: (a) Belokon, Y. N.; Bespalova, N. B.;Churkina,T.D.;Cisarová,I.;Ezernitskaya,M.G.;Harutyunyan,S.R.;Hrdina,R.;Kagan,H.B.;Kocovsky,P.;Kochetkov,K.A.;Larionov,O.V.;Lyssenko,K.A.;North,M.;Polásek,M.;Peregudov, A. S.; Prisyazhnyuk, V. V.; Vyskočil, Š. J.Am. Chem.
Soc.2003,125,12860.(b)Siebum,A.H.G.;Tsang,R.K.F.;vanderSteen,R.;Raap,J.;Lugtenburg,J.Eur. J. Org. Chem. 2004,4391.(c)Lygo,B.;Allbutt,B.;Kirton,E.H.M.Tetrahedron Lett.2005,46,4461.(d)Ramachandran,P.V.;Madhi,S.;Bland-Berry,L.;RamReddy,M.V.;O’Donnell,M.J.J. Am. Chem. Soc.2005,127,13450.
(49) Arai,S.;Tsuji,R.;Nishida,A.Tetrahedron Lett.2002,43,9535.(50) Arai,S.;Tokumaru,K.;Aoyama,T.Chem. Parm. Bull.2004,52,
646.(51) Akiyama,T.;Hara,M.;Fuchibe,K.;Sakamoto,S.;Yamaguchi,
K.Chem. Commun.2003,1734.(52) Lee,Y.-J.;Lee,J.;Kim,M.-J.;Jeong,B.-S.;Lee,J.-H.;Kim,T.-
S.;Lee,J.;Ku,J.-M.;Jew,S.-s.;Park,H.-g.Org. Lett.2005,7,3207.
(53) For examples of conjugate addition under phase-transferconditionswithcrown-ether-derivedcatalysts,see:(a)Novák,T.;Tatai,J.;Bakó,P.;Czugler,M.;Keglevich,G.;Tõke,L.Synlett2001, 424. (b) Novák, T.; Bakó, P.; Keglevich, G.; Dobó, A.;Vékey,K.;Tõke,L.J. Inclusion Phenom. Macrocyclic Chem.2001,40,207.(c)Bakó,T.;Bakó,P.;Szöllõsy,Á.;Czugler,M.;Keglevich,G.;Tõke,L.Tetrahedron: Asymmetry2002,13,203.(d)With cinchona-alkaloid-derived catalyst, seeZhang,F.-Y.;Corey,E.J.Org. Lett.2004,6,3397.
(54) Ooi,T.;Fujioka,S.;Maruoka,K.J. Am. Chem. Soc.2004,126,11790.
(55) Ooi,T.;Takada,S.;Fujioka,S.;Maruoka,K.Org. Lett.2005,7,5143.
(56) Ooi,T.;Ohara,D.;Fukumoto,K.;Maruoka,K.Org. Lett.2005,7,3195.
(57) For relatedreactionswithnon-C2-symmetriccatalysts,see: (a)Zhang,F.-Y.;Corey,E.J.Org. Lett.2000,2,1097.(b)Perrard,T.;Plaquevent,J.-C.;Desmurs,J.-R.;Hébrault,D.Org. Lett.2000,2, 2959. (c)O’Donnell,M. J.;Delgado,F.;Domínguez,E.; deBlas,J.;Scott,W.L.Tetrahedron: Asymmetry2001,12,821.(d)Kim,D.Y.;Huh,S.C.;Kim,S.M.Tetrahedron Lett.2001,42,6299. (e)Dere,R.T.;Pal,R.R.;Patil,P.S.;Salunkhe,M.M.Tetrahedron Lett.2003,44,5351.(f)Donnoli,M.I.;Scafato,P.;Nardiello,M.;Casarini,D.;Giorgio,E.;Rosini,C.Tetrahedron2004,60,4975.
(58) (a)Ooi,T.;Taniguchi,M.;Kameda,M.;Maruoka,K.Angew. Chem., Int. Ed. 2002, 41, 4542. (b) Ooi, T.; Kameda, M.;Taniguchi,M.;Maruoka,K.J. Am. Chem. Soc.2004,126,9685.
(59) For the additionofdiazoacetate to aldehydesusing cinchona-alkaloid-derivedcatalysts,seeArai,S.;Hasegawa,K.;Nishida,A.Tetrahedron Lett.2004,45,1023.
(60) Ooi,T.;Kameda,M.;Fujii,J.-i;Maruoka,K.Org. Lett.2004,6,2397.
(61) Okada,A.;Shibuguchi,T.;Ohshima,T.;Masu,H.;Yamaguchi,K.;Shibasaki,M.Angew. Chem., Int. Ed.2005,44,4564.
(62) Arai,S.;Tokumaru,K.;Aoyama,T.Tetrahedron Lett.2004,45,1845.
(63) Ooi,T.;Takahashi,M.;Doda,K.;Maruoka,K.J. Am. Chem. Soc.2002,124,7640.
(64) Helder,R.;Hummelen,J.C.;Laane,R.W.P.M.;Wiering,J.S.;Wynberg,H.Tetrahedron Lett.1976,17,1831.
(65) For recent examples with non-C2-symmetric catalysts, see:(a) Adam, W.; Rao, P. B.; Degen, H.-G.; Saha-Möller, C. R.Tetrahedron: Asymmetry2001,12,121. (b)Lygo,B.;To,D.C.M.Tetrahedron Lett.2001,42,1343.(c)Adam,W.;Rao,P.B.;Degen,H.-G.;Levai,A.;Patonay,T.;Saha-Möller,C.R.J. Org. Chem.2002,67,259.(d)Arai,S.;Tsuge,H.;Oku,M.;Miura,M.;Shioiri,T.Tetrahedron2002,58,1623.(e)Lygo,B.;To,D.C.M.Chem. Commun. 2002,2360.(f)Ye,J.;Wang,Y.;Liu,R.;Zhang,

86
VO
L. 4
0, N
O. 3
• 2
007
Dev
elop
men
t an
d A
pplic
atio
ns o
f C
2-Sy
mm
etric
, Chi
ral,
Phas
e-Tr
ansf
er C
atal
ysts G.;Zhang,Q.;Chen,J.;Liang,X.Chem. Commun. 2003,2714.
(g)Ye,J.;Wang,Y.;Chen,J.;Liang,X.Adv. Synth. Catal. 2004,346,691. (h)Bakó,P.;Bakó,T.;Mészáros,A.;Keglevich,G.;Szöllõsy,A.;Bodor,S.;Makó,A.;Tõke,L.Synlett2004,643.(i)Bakó,T.;Bakó,P.;Keglevich,G.;Bombicz,P.;Kubinyi,M.;Pál,K.;Bodor,S.;Makó,A.;Tõke,L.Tetrahedron: Asymmetry2004,15,1589.(j)Geller,T.;Gerlach,A.;Krüger,C.M.;Militzer,H.-C.Tetrahedron Lett.2004,45,5065.(k)Geller,T.;Krüger,C.M.;Militzer,H.-C.Tetrahedron Lett.2004,45,5069.
(66) Ooi,T.;Ohara,D.;Tamura,M.;Maruoka,K.J. Am. Chem. Soc.2004,126,6844.
(67) Jew,S.-s.;Lee,J.-H.;Jeong,B.-S.;Yoo,M.-S.;Kim,M.-J.;Lee,Y.-J.;Lee,J.;Choi,S.-h.;Lee,K.;Lah,M.S.;Park,H.-g.Angew. Chem., Int. Ed. 2005,44,1383.
(68) Ooi,T.;Uematsu,Y.;Maruoka,K.J. Am. Chem. Soc.2006,128,2548.
(69) Ooi,T.;Uematsu,Y.;Fujimoto,J.;Fukumoto,K.;Maruoka,K.Tetrahedron Lett.2007,48,1337.
(70) For a similar asymmetricStrecker synthesiswith a cinchona-alkaloid-derived catalyst, see Herrera, R. P.; Sgarzani, V.;Bernardi, L.; Fini, F.; Pettersen, D.; Ricci, A. J. Org. Chem.2006,71,9869.
l-Selectride is a registered trademark of Sigma-AldrichBiotechnology, L.P. Span is a registered trademark of ICIAmericas,Inc.
About the AuthorsTakashi Ooi receivedhisPh.D.degree in1994fromNagoyaUniversityunderthedirectionofProfessorHisashiYamamoto,and then joined the group of Professor Julius Rebek, Jr., atMITasapostdoctoralfellow(1994–1995).HewasappointedassistantprofessoratHokkaidoUniversityin1995andpromotedtolecturerin1998.In2001,hemovedtoKyotoUniversityasanassociateprofessor,andbecameafullprofessoratNagoyaUniversity in 2006. He was awarded the Chugai Award inSynthetic Organic Chemistry (Japan, 1997), the ChemicalSocietyofJapanAward forYoungChemists (1999),and theThiemeJournalAward(2006).Hiscurrentresearch interestsare focusedon thedevelopmentofnewandusefulsyntheticmethodologies by designing organic molecular catalystsincludingC2-symmetric,chiral,quaternaryammoniumsalts.
Keiji Maruoka received his Ph.D. degree in 1980 fromthe University of Hawaii with Prof. Hisashi Yamamoto. Hewas appointed assistant professor at Nagoya University in1980,andpromotedtoassociateprofessorin1990.HemovedtoHokkaidoUniversity as a fullprofessor in1995, andhasbeenaprofessoratKyotoUniversitysince2000.HisresearchinterestsarefocusedonorganicsynthesiswithbidentateLewisacidsanddesignerchiralorganocatalysts.Hisawardsincludethe Japan Synthetic Organic Chemistry Award (2003), theNagoyaSilverMedal(2004), theGSCAward(2006),andtheChemicalSocietyofJapanAward(2006).^
Continuing the Tradition of Excellencesigma-aldrich’s highly acclaimed chemistry review journal, Aldrichimica Acta, has again been ranked #1 (out of 56 similar journals) by impact factor in the field of organic chemistry. in 2006, the latest year for which such rankings are available, the Acta achieved an impact factor of 10.692. The Acta was also ranked #1 in 2005 and 2004. These rankings appear annually in the science edition of Journal Citation Reports® (JCR), which is published by Thomson isi®. JCR, science edition, tracks close to 5,900 international science journals annually and offers quantifiable statistical data, which allows the systematic and objective ranking of journals by relative importance within their subject categories.
The Aldrichimica Acta publishes in-depth reviews on topics of current interest to chemists. for 40 years, it has been an international forum for the frontiers of chemical research. articles, written by chemists from around the world, cover a variety of topics usually based on a synthetic theme involving organic, organometallic, bio-organic, or inorganic chemistry.
To get your free subscription to the Aldrichimica Acta, please visit our award-winning web site at sigma-aldrich.com/acta, call toll-free 800-325-3010 (U.s.a.), or contact your local sigma-aldrich office.
isi and Journal Citation Reports are registered trademarks of The Thomson corporation.
6.3337.077
8.833
9.91710.692
0.000
2.000
4.000
6.000
8.000
10.000
12.000
2002 2003 2004 2005 2006
Year
Imp
act
Fact
or
Aldrichimica ActaGrowing Impact over the Past Five Years

sigma-aldrich.com
Asymmetric Alkylation Phase-Transfer Catalystschiral quaternary ammonium salts derived from c2-symmetric amines or cinchona alkaloids are powerful phase-transfer catalysts for the preparation of optically active molecules. Both types of phase-transfer catalysts allow for the stereocontrolled monoalkylation of glycine-derived schiff bases with alkyl halides to afford protected α-alkyl-α-amino acids. catalysts developed by maruoka effect these reactions with a high degree of enantioselectivity at exceptionally low catalyst loadings (<0.1 mol %).
Ot-Bu
ON
Ph
Ph
R X, toluene–50% aq. KOH, 0 °COt-Bu
ON
Ph
Ph
R
677086 (0.01–0.05 mol %)
Ot-Bu
ON
Ph
Ph
Bn
98%, 99% ee
Ot-Bu
ON
Ph
Ph
87%, 98% ee
Ot-Bu
ON
Ph
Ph
87%, 98% ee
C2-Symmetric Maruoka Catalysts
N
CH3
CH3
Br
F
FF
F
FF
N
CH3
CH3
Br
F
FF
F
FF
677086 687596
For further application information, see Professor Maruoka’s review article
in this issue.
Ot-Bu
ON
Ph
Ph
Bn Br, solvent, baseOt-Bu
ON
Ph
Ph
Bn
Cinchona alkaloid catalyst (10 mol %)
up to 84%, 94% ee
*
N
HCl
HN
HON
HCl
H
NOH
N
HBr
H
NOR
R = allyl
499617 366188515701
Corey, E. J. et al. J. Am. Chem. Soc. 1997, 119, 12414.
O'Donnell, M. J. et al. J. Am. Chem. Soc. 1989, 111, 2353.
Lygo, B.; Wainwright, P.G. Tetrahedron Lett. 1997, 38, 8595.
Cinchona Alkaloid Catalysts
For more information, please visit
sigma-aldrich.com/cinchona.

sigma-aldrich.com
ChemDose® is a novel technology allowing for the use of chemical reagents and catalysts in the form
of tablets. Co-developed with Reaxa Ltd., the ~5 millimeter tablets are composed of a chemically inert
magnesium aluminosilicate matrix, together with an absorbed reagent or catalyst. Upon exposure to solvents,
the reagent or catalyst readily dissolves out, leaving behind an easily removed insoluble tablet.1
Fast, Accurate, and Convenient Dosing of Catalysts and Reagents
For more information, visit sigma-aldrich.com/chemdose.
Convenient handling and dispensing of reagents and catalysts.
Eliminates the tedious weighing process for milli- and micromolar chemical quantities.
Simple reaction workup: inert tablet easily removed upon completion of reaction.
Consistent chemical loadings of tablets, controlled release rates, microwave compatible.
Virtually no “learning curve” when using ChemDose®.
•
•
•
•
•
0
25
50
75
100
0 1 2 3 4 5 Time (h)
Co
nve
rsio
n (
%)
Conventional
ChemDose®
ChemDose®
repeat run
Reference: (1) Ruhland, T. et al. J. Comb. Chem. 2007, 9, 301.
Br
CH3
+
B(OH)2
H3C
Pd(dppf)Cl2•CH2Cl2 (2 mol%)
K2CO3, water–i-PrOH, 80 °C
X-Phos Pd(OAc)2 Pd(dppf)Cl2•CH2Cl2 PEPPSI™-IPr PdCl2(PPh3)2 Pd2(dba)3 HATU
Discovery Chemistry with ChemDose®
PEPPSI is a trademark of Total Synthesis Ltd. (Toronto, Canada). ChemDose is a registered trademark of Reaxa Ltd.
Simplify

sigma-aldrich.com
Sigma-Aldrich Worldwide Locations
ArgentinaSIGMA-ALDRICHDEARGENTINAS.A.FreeTel:08108887446Tel:(+54)1145561472Fax:(+54)1145521698
AustraliaSIGMA-ALDRICHPTYLTD.FreeTel:1800800097FreeFax:1800800096Tel:(+61)298410555Fax:(+61)298410500
AustriaSIGMA-ALDRICHHANDELSGmbHTel:(+43)16058110Fax:(+43)16058120
BelgiumSIGMA-ALDRICHNV/SA.FreeTel:080014747FreeFax:080014745Tel:(+32)38991301Fax:(+32)38991311
BrazilSIGMA-ALDRICHBRASILLTDA.FreeTel:08007017425Tel:(+55)1137323100Fax:(+55)1155229895
CanadaSIGMA-ALDRICHCANADALTD.FreeTel:18005651400FreeFax:18002653858Tel:(+1)9058299500Fax:(+1)9058299292
ChinaSIGMA-ALDRICH(SHANGHAI)TRADINGCO.LTD.FreeTel:8008193336Tel:(+86)2161415566Fax:(+86)2161415567
Czech RepublicSIGMA-ALDRICHS.R.O.Tel:(+420)246003200Fax:(+420)246003291
DenmarkSIGMA-ALDRICHDENMARKA/STel:(+45)43565910Fax:(+45)43565905
FinlandSIGMA-ALDRICHFINLANDOYTel:(+358)93509250Fax:(+358)935092555
FranceSIGMA-ALDRICHCHIMIES.à.r.l.FreeTel:0800211408FreeFax:0800031052Tel:(+33)474822800Fax:(+33)474956808
GermanySIGMA-ALDRICHCHEMIEGmbHFreeTel:08005155000FreeFax:08006490000Tel:(+49)8965130Fax:(+49)8965131160
GreeceSIGMA-ALDRICH(O.M.)LTD.Tel:(+30)2109948010Fax:(+30)2109943831
HungarySIGMA-ALDRICHKftIngyeneszöldtelefon:0680355355Ingyeneszöldfax:0680344344Tel:(+36)12359055Fax:(+36)12359050
IndiaSIGMA-ALDRICHCHEMICALSPRIVATELIMITEDTelephoneBangalore:(+91)8066219600NewDelhi:(+91)1141654255Mumbai:(+91)2225702364Hyderabad:(+91)4066845488FaxBangalore:(+91)8066219650NewDelhi:(+91)1141654266Mumbai:(+91)2225797589Hyderabad:(+91)4066845466
IrelandSIGMA-ALDRICHIRELANDLTD.FreeTel:1800200888FreeFax:1800600222Tel:(+353)14041900Fax:(+353)14041910
IsraelSIGMA-ALDRICHISRAELLTD.FreeTel:1800702222Tel:(+972)89484100Fax:(+972)89484200
ItalySIGMA-ALDRICHS.r.l.NumeroVerde:800827018Tel:(+39)0233417310Fax:(+39)0238010737
JapanSIGMA-ALDRICHJAPANK.K.TokyoTel:(+81)357967300TokyoFax:(+81)357967315
KoreaSIGMA-ALDRICHKOREAFreeTel:(+82)800237111FreeFax:(+82)800238111Tel:(+82)313299000Fax:(+82)313299090
MalaysiaSIGMA-ALDRICH(M)SDN.BHDTel:(+60)356353321Fax:(+60)356354116
MexicoSIGMA-ALDRICHQUÍMICA,S.A.deC.V.FreeTel:018000075300FreeFax:018007129920Tel:527222761600Fax:527222761601
The NetherlandsSIGMA-ALDRICHCHEMIEBVFreeTel:08000229088FreeFax:08000229089Tel:(+31)786205411Fax:(+31)786205421
New Zealand
SIGMA-ALDRICHNEWZEALANDLTD.
FreeTel:0800936666
FreeFax:0800937777
Tel:(+61)298410555
Fax:(+61)298410500
Norway
SIGMA-ALDRICHNORWAYAS
Tel:(+47)23176060
Fax:(+47)23176050
Poland
SIGMA-ALDRICHSp.zo.o.
Tel:(+48)618290100
Fax:(+48)618290120
Portugal
SIGMA-ALDRICHQUÍMICA,S.A.
FreeTel:800202180
FreeFax:800202178
Tel:(+351)219242555
Fax:(+351)219242610
Russia
SIGMA-ALDRICHRUS,LLC
Tel:+7(495)6216037
Fax:+7(495)6215923
Singapore
SIGMA-ALDRICHPTE.LTD.
Tel:(+65)67791200
Fax:(+65)67791822
South Africa
SIGMA-ALDRICH
SOUTHAFRICA(PTY)LTD.
FreeTel:0800110075
FreeFax:0800110079
Tel:(+27)119791188
Fax:(+27)119791119
Spain
SIGMA-ALDRICHQUÍMICA,S.A.
FreeTel:900101376
FreeFax:900102028
Tel:(+34)916619977
Fax:(+34)916619642
Sweden
SIGMA-ALDRICHSWEDENAB
Tel:(+46)87424200
Fax:(+46)87424243
Switzerland
SIGMA-ALDRICHCHEMIEGmbH
FreeTel:0800800080
FreeFax:0800800081
Tel:(+41)817552828
Fax:(+41)817552815
United Kingdom
SIGMA-ALDRICHCOMPANYLTD.
FreeTel:0800717181
FreeFax:0800378785
Tel:(+44)1747833000
Fax:(+44)1747833313
SAFC(UK)FreeTel:0800717117
United States
SIGMA-ALDRICH
P.O.Box14508
St.Louis,Missouri63178
Toll-Free:8003253010
Toll-FreeFax:8003255052
CallCollect:(+1)3147715750
Tel:(+1)3147715765
Fax:(+1)3147715757
Internet
sigma-aldrich.com
As a leading Life Science and High Technology company, we are always looking for talented individuals to join our team. At Sigma-Aldrich we value the contributions of our employees, and recognize
the impact they have on our success. We strive to foster creativity and innovation, and encourage professional development.
Our biochemical and organic chemical products and kits are used in scientific and genomic research, biotechnology, pharmaceutical development, the diagnosis of disease, and as key components in pharmaceutical and other high technology manufacturing. We have customers in life science companies, university and government institutions, hospitals, and in industry.
Learn more about our career opportunities by visiting our award-winning Web site at sigma-aldrich.com/careers.
Sigma-Aldrich Career Opportunities
Sigma-Aldrich Corporation is an equal opportunity employer.
UNLEASH YOUR TALENTS

JRB02472-503200
0087
P.O. Box 14508St. Louis, MO 63178USA
sigma-aldrich.com
View table of contents, search, browse, or order from our entire library at sigma-aldrich.com/books.
Chemical Synthesis
The Sigma-Aldrich Library is your guide to finding new and
best-selling chemistry books.whether your interest includes Drug Discovery,
chemical synthesis, materials science, or a wide range of other areas of interest, we can help you find the right book.
Z730203Palladium in Heterocyclic Chemistry A Guide for the Synthetic Chemist, 2nd EditionJ. J. Li and G. W. Gribble, Elsevier, 2007, 658 pp. Softcover.
Z704113Asymmetric Organocatalysis – From Biomimetic Concepts to Applications in Asymmetric SynthesisA. Berkessel and H. Gröger, Wiley-VCH, 2005, 454 pp. Hardcover.
Z703354Side Reactions in Organic Synthesis: A Guide to Successful Synthesis DesignF. Z. Dörwald, Wiley-VCH, 2005, 389 pp. Softcover.
Z705284Handbook of Chiral Chemicals, 2nd EditionD. Ager, Ed., CRC Press, 2006, 664 pp. Hardcover.
Z706043Chiral AnalysisK. W. Busch and M. A. Busch, Eds., Elsevier, 2006, 720 pp. Hardcover.
Z730181The Pilot Plant Real Book, 2nd EditionF. X. McConville, FXM Engineering, 2006, 320 pp. Softcover.
New for 2007
SciBookSelect is a trademark of Sigma-Aldrich Biotechnology, L.P.