the blood - dm5migu4zj3pb.cloudfront.net · the gaps in the anamnesis result from the fact that...
TRANSCRIPT

THEBLOODFLOWANDOXYGENCONSUMPTIONOFTHEHUMANBRAIN IN DIABETIC ACIDOSIS ANDCOMA'
By SEYMOURS. KETY, B. DAVID POLIS, CARL S. NADLER,AND CARL F. SCHMIDT
(From the Department of Pharmacology, University of Pennsylvania, and the Metabolic Service,Philadelphia General Hospital, Philadelphia)(Received for publication December 4, 1947)
Despite recent gratifying advances in our knowl-edge of carbohydrate metabolism, diabetic comaremains an important medical emergency and ourunderstanding of its fundamental pathogenesis andrational treatment is far from complete. The se-quence of events leading from disturbed carbohy-drate utilization through acidosis, coma and deathhas been. variously defined and important roleshave been assigned at one time or another toketosis, acidosis, dehydration, circulatory failure,and cerebral anoxia.
In an effort better to define these fundamentalderangements and their inter-relationships, thebrain was chosen as an important object of studysince its integrity is so intimately associated withsurvival. Clinicians who have studied diabetic aci-dosis are agreed that cerebral function as reflectedin mental state is closely correlated with the sever-ity of the disease and its prognosis (1-6). Thecentral nervous system, apparently dependent oncarbohydrate for its normal source of energy (7 to9), would be expected significantly to reflect dis-turbances in the metabolism of this foodstuff.For many reasons recently recapitulated by Soskinand Levine (10), the results of extensive in vitrostudies may not rigorously be applied to the metab-olism of tissues in the living organism so that in-ferences drawn from work with the Warburg ap-paratus require validation by studies on living or-gans in situ. Such studies are comparatively rarebecause suitable methods have been developed onlyin the past few years.
The recently devised nitrous oxide method forthe quantitative measurement of cerebral bloodflow in man (11, 12) makes possible calculation ofthe utilization or production by the brain of any
1 This work was supported by grants from the Com-mittee on Research in Dementia Precox, founded by theSupreme Council, 330 Scottish Rite, Northern MasonicJurisdiction, U. S. A., and from the Life Insurance Medi-cal Research Fund.
substance susceptible of accurate analysis in ar-terial and cerebral venous blood. In applyingthis method to diabetic acidosis it was decided firstto obtain an overall estimate of cerebral metabo-lism by determination of the oxygen consumptionof the brain and, if possible, an indication of thefactors which may influence it in this condition.If this approach appeared feasible, it was hopedthat analytical techniques could be refined or de-veloped to permit examination of more specificphases of cerebral metabolism and the aberrationsassociated with diabetic acidosis. The present re-port concerns itself largely, therefore, with cere-bral oxygen utilization.
METHODS
Members of the investigating team were notified assoon as a diagnosis of diabetic coma or severe acidosiswas made in the receiving ward and confirmed by bloodsugar and CO2 determinations. Studies were not at-tempted where too great a delay in treatment would haveresulted, where the acidosis was not very severe, or ifthe patient was moribund. There was no difference be-tween the mortalities in this series and in comparablepatients on the same services before this investigationwas undertaken.
Respiratory minute volume was measured in a Tissotspirometer attached to a well-fitted face mask equippedwith expiratory and inspiratory valves. Mean arterialblood pressure was read from a damped mercury manom-eter communicating with a needle in the femoral artery.The nitrous oxide method (12) employing a gas mixtureof 15% N20; 21%o 02, 64% N2, was used for measuringcerebral blood flow.
Blood samples were analyzed for oxygen and carbondioxide (13), hemoglobin (14), glucose (15), total ke-tones (16), protein (17), chlorides (18), total base (19)and urea nitrogen (20). Hydrogen ion concentrationwas measured potentiometrically at 37° under anaerobicconditions using a glass electrode. Values for CO2 ten-sion were calculated from CO2 content and pH by meansof nomograms (13).
Immediately after the required blood samples weretaken, treatment was begun. In seven instances thestudies above were repeated at some time (varying fromtwo to 48 hours) after the initiation of treatment.
500
CEREBRALMETABOLISM IN DIABETIC ACIDOSIS
RESULTS
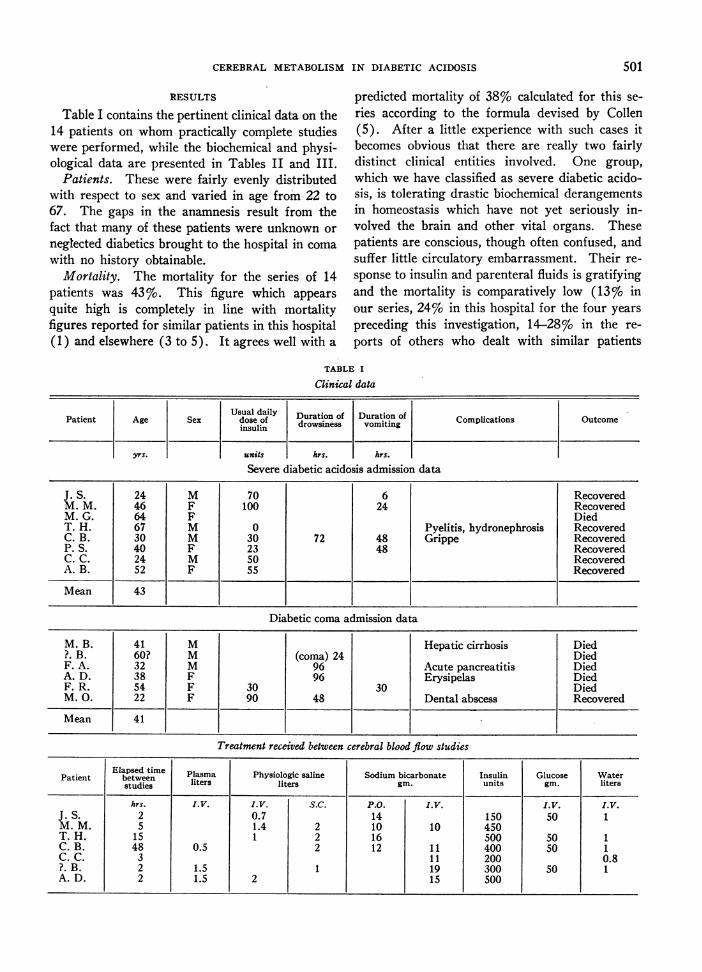
Table I contains the pertinent clinical data on the14 patients on whom practically complete studieswere performed, while the biochemical and physi-ological data are presented in Tables II and III.
Patients. These were fairly evenly distributedwith respect to sex and varied in age from 22 to67. The gaps in the anamnesis result from thefact that many of these patients were unknown or
neglected diabetics brought to the hospital in coma
with no history obtainable.Mortality. The mortality for the series of 14
patients was 43%o. This figure which appears
quite high is completely in line with mortalityfigures reported for similar patients in this hospital(1) and elsewhere (3 to 5). It agrees well with a
predicted mortality of 38% calculated for this se-ries according to the formula devised by Collen(5). After a little experience with such cases itbecomes obvious that there are really two fairlydistinct clinical entities involved. One group,which we have classified as severe diabetic acido-sis, is tolerating drastic biochemical derangementsin homeostasis which have not yet seriously in-volved the brain and other vital organs. Thesepatients are conscious, though often confused, andsuffer little circulatory embarrassment. Their re-
sponse to insulin and parenteral fluids is gratifyingand the mortality is comparatively low (13%o inour series, 24%o in this hospital for the four yearspreceding this investigation, 14-28%o in the re-
ports of others who dealt with similar patients
TABLE I
Clinical data
Usual daily Duration of Duration ofCopiaon
dose of drowsiness vomiting Complications Outcomeinsulin
yrs. units hrs. hrs.
Severe diabetic acidosis admission data
J. S. 24 M 70 6 RecoveredM. M. 46 F 100 24 RecoveredM. G. 64 F DiedT. H. 67 M 0 Pyelitis, hydronephrosis RecoveredC. B. 30 M 30 72 48 Grippe RecoveredP. S. 40 F 23 48 RecoveredC. C. 24 M 50 RecoveredA. B. 52 F 55 Recovered
Mean 43
Diabetic coma admission data
M. B. 41 M Hepatic cirrhosis Died?. B. 60? M (coma) 24 DiedF. A. 32 M 96 Acute pancreatitis DiedA. D. 38 F 96 Erysipelas DiedF. R. 54 F 30 30 DiedM. 0. 22 F 90 48 Dental abscess Recovered
Mean 41
Treatment received between cerebral blood flow studies
Elapsed time Plasma Physiologic saline Sodium bicarbonate Insulin Glucose WaterPatient between liters liters gin. units gin. litersstudies
hrs. I.V. I.V. S.C. P.O. I.V. I.V. I.V.J. S. 2 0.7 14 150 50 1M. M. 5 1.4 2 10 10 450T. H. 15 1 2 16 500 so 1C. B. 48 0.5 2 12 11 400 50 1C. C. 3 11 200 0.8?. B. 2 1.5 1 19 300 50 1A. D. 2 1.5 2 15 500
501

S. S. KETY, B. D. POLIS, C. S. NADLER, AND C. F. SCHMIDT
C 0 +0 m ON -
tr rz Z- -.: b6 Z.:t:t r .
0 - Ce9 t - - C.q
-4 --4 -4 U- C-
00
0
1-4 -4 -4 -4 -4
0%N00 0C.-c U'0o - 11
1- '0m )C9-' U)oI
C-4 m r --'-U'0-.4000%0C.OCqcq-
tC~--~ U4-:dt:r~ C-z
0u)C-'tr_ 4 00M -I -
M C1% - ' 0 V
- - -
. * * * * *.
C14m00% - C--\0U6 vic~4 6 o:~6 6C-4 C4-4-- C.'1- C.- -4 -
0 0 '0 C o0% 00 U'\00S 0%
t- C-- C-C C.1 0 I-\ \ c-C U) U) C.iI.. -e " 1-t -4 -. -4
00CC t- a, o tr t-C
- - - - -
'o 14 C-I t- C- -
000000% 0000% 0%
U)t - - tU 14 4j
*0 . . . . ..U1-4
on e)
r- ofi r- \o r- Co _- oo
m aw I o21 oo I-d \0
t- 00 00 0-C-I C--4000%'"03 cUm 0% 00Ct- NO-'O0-'0o e'
,,, 4m0C_ _ * * * -* Q
CU
CU
-
CU
rQ
0C.)
~0
00 -0 C~l4 eU) 0)N - 00 N00s
C- ''4-A00 t- eC.-4 C14 eU) C1 CNU C
(14o 0 U-) C"! Io6o o m o oo
C---
C; c; e
0.- ". -. .-
mCNU) Cen
0 n4 C.4 '0 '0 00 -- 0% 00 00tozt-zX oo~6IzN
U) U) ") me'1 00
C4 4 C4 -4
C-.1-4 1_4-
o~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~C4 0%'O0- --
_4 rZ 6 U) oo oO
- U)
0 )00% 0% ONCA
u) r- 00 +4 00 +4C-. c*i *) C-i 4
4 _- -4 -
C,(4-U 00
U) U)oq-4 -4 4 1-
- C-
-0000%0 00C-
iU) - 000%
e U) U)Ueci
0 'o0000 C- 4
16% 160
U) U)i0 0 t-c-i 0a _b4
000 Ck - Id4
C14 0 C14 ON03 000% C0%'o mUt) -14 m to %0 %0
04 0 ,Uai 4 C
0mm,:~4 ~ 4
00%
'0
0C"
00000%'0
0%.
00
0
o6
0ooo
~4
0
'At00C0
Ul)
cd
4-J
'04)
0a4
00eq 0% 00C1en 0
) ) tU ) +Cr- *- .- *- *- *
C-
'tCNUl)to I1f)eq U)
~o (01 %o 11~o-l;4e~Ci6 U~
Co60o> o
00o 4 U)
4 U40t N
10 \U)0 0 0
o6'0~'0c)ei o6
* 0**
U')0U)- 0\0 I- m \0 C\ m OlI 0
6o-i 00
o1 It~ C14() ~'O00% 0% 0NC00 00 CS
u Cseto en Lo
.d \. .d . \. . .14 C0 0N n- O60e 0)0%6e06 if;U.
_%_%0\0%0 0
u-400-O0U)O0 U)
-%0C+0U)e40 U)
0000U)o%000'- 0
- c e- -- N4~
CU E-5~.3 (~P~dC
Cd
CUc*R Y 4)w0 0O. 0 c
502
CU
bO
c
0-
0
a0
1,
cd
Co~0
cd
U)
0
-0
C.
04 *
I0
00
0
000
a..I
0s
4i
000
0
0 a
0
C)
I.
0
24
~bo
*C I
I
00d
0
4l-
0Ci
I A
NI-
-
.
12

CEREBRALMETABOLISM IN DIABETIC ACIDOSIS
TABLE III
Cerebral and other physiological data
Cerebral Respiratory
A-V CB CMR Minute Tidal Rate Mean Pulse RectalPatient 0v BF 02 CVR RQ volume volume per arterial rate temp.
IMental min. B. P. per min OF.state
cc./100 cc./100 cc./100vol. % B.gmin. g./min. g./min. liters CC. mm. Hg
Severe diabetic acidosis admission data
J. S. Confused 9.9 41 4.1 1.8 0.84 7.5 500 15 74 130 95M. M. Confused 4.2 58 2.4 1.4 0.98 33 920 36 79 128 100M. G. Confused 4.7 55 2.6 1.1 0.87 35 880 40 58 120 102T. H. Confused 5.6 32 1.8 3.8 0.98 21 1100 19 120 110 99C. B. Confused 6.9 36 2.5 2.4 0.84 26 1300 20 86 116 100P. S. Confused 5.6 50 2.8 2.1 0.96 31 1200 25 107 94 99C. C. Confused 6.4 40 2;6 2.1 0.99 10.7 400 26 84 110 99A. B. Confused 6.9 44 3.0 1.8 0.88 16.9 770 22 80 110 98
Mean 6.3 45 2.7 2.1 0.92 23 880 25 86 115 99
Diabetic coma admission data
M. B. Uncon. 1.2 63 0.8 1.3 0.70 80 160 96?. B Uncon. 4.3 35 1.5 1.8 0.98 28.4 64 120 96F. A. Uncon. 5.7 35 2.0 1.0 0.83 23.9 34 105 101A. D. Uncon. 2.4 80 1.9 0.8 0.96 65 110 98F. R. Uncon. 1.8 101 1.8 0.8 0.91 20 850 24 82 106 95M. 0. Uncon. 2.8 78 2.1 0.9 0.81 25 780 32 72 135 97
Mean 3.0 65 1.7 1.1 0.87 66 123 97
Post treatment data
J.S. Alert 4.9 63 3.1 1.4 1.25 7.7 550 14 90M. M. Alert 5.8 54 3.1 1.1 0.93 18.7 750 25 62 112T. H. Alert 5.2 42 2.2 2.0 0.87 10 560 18 86C. B. Alert 8.1 40 3.2 2.3 0.90 5 560 9 92 88C. C. Confused 7.2 45 3.2 1.9 1.02 6.8 340 20 84 100?. B. Uncon. 3.5 48 1.7 1.6 0.92 26.6 76A. D. Confused 2.0 120 2.4 0.7 0.90 83
Normal 6.3 54 3.3 1.6 0.99 8 500 16 86
valuesIIIII[1 to 5]). The other type is characterized byunconsciousness, frequently associated with cir-culatory depression. These alone we have feltjustified in classifying as diabetic coma. Satisfac-tory correction of the chemical disturbances in theblood of these patients has little effect on the courseof the disease and the mortality is almost alwayshigh (83%o in our series, 80%o in the four yearspreceding this investigation, 31%, 58%, 70%,73%o, 75%o and 81% in other series [1 to 6]).The overall mortality of any series of cases ofdiabetic acidosis and coma depends far more onthe relative frequency of these two groups inthe series than on the minor differences which
may occur in a treatment so universally acceptedand used. It should be pointed out that a diag-nosis of coma on the basis of the plasma CO2 com-bining power, rather than on mental state, doesnot differentiate between these two groups andmay be misleading from the point of view of prog-nosis and therapeutic results. A case in point isthe overall mortality of 11% reported by onegroup (6) in a series of 525 cases of "diabeticcoma" based on the plasma CO2, only 18%o ofwhom were unconscious. The mortality in these93 unconscious patients, however, was only 31%,an enviable record in itself. It is worthy of notethat all the deaths in our series were in patients
503

S. S. KETY, B. D. POLIS, C. S. NADLER, AND C. F. SCHMIDT
who had never attended the Metabolic Clinic ofthe hospital, and in five of the six deaths the pres-ence of diabetes had been unknown.
Acid-base balance. There was a marked reduc-tion in the arterial carbon dioxide content, tensionand pH, a picture of uncompensated metabolicacidosis. The lowest value for pH (6.80) was ob-tained in a patient who subsequently recovered.There is surprisingly little difference between the"acidosis" and "coma" patients on the basis ofthese three functions. In all cases studied duringor after treatment, which included moderateamounts of sodium bicarbonate, there was a strik-ing recovery of pH to normal or nearly normallevels even though the plasma bicarbonate laggedfar behind. The reason for this is seen in theslow return of the CO2 tension to normal indica-tive of only a gradual reduction of hyperventila-tion in response to the improved pH. The prac-tical implications of this with respect to the ques-tion of alkali administration are important. Thereis some controversy at present on the advisability,amount and route of administration of bicarbonatein the treatment of diabetic acidosis. Unfortu-nately the criterion of acidosis and response totreatment has been the plasma CO2 combiningpower, at best a very rough guide. Actually it isthe ratio of bicarbonate to CO2 tension (pCO2)which is important and this can best be determinedby measurement of the pH which represents theresultant of all the blood buffer systems. Sincethe pCO2 does not rise parn pass with the ad-ministered bicarbonate the pH will be brought tonormal much sooner than will the blood bicar-bonate concentration. Thus, any attempt to ad-minister alkali rapidly in quantities calculatedto restore the CO2 combining power to a normallevel can only result in a severe alkaline shift ofthe pH until the relatively sluggish respiratoryadjustments are made. Our results demonstratethat it is possible to bring the arterial pH withinnormal limits quickly by means of the intravenousadministration of amounts of bicarbonate smallenough to raise the blood CO2 content only 10 or15 vol. %'. For example, patients ? B., M. M. andC. C. showed a normal arterial pH with an alkalireserve less than half the normal after two to fivehours of treatment. In the case of A. D. the ar-terial pH rose from 6.96 to 7.24 with a change inBHCO3of from 5.1 to only 8.4 m.Eq./1. In pa-
tient J. S. who was not severely acidotic, pulmon-ary ventilation was never above normal. He wastherefore able to retain C02, hence the pH andalkali reserve rose concomitantly. Similarly inpatients T. H. and C. B., where sufficient time hadelapsed between the two studies for adequate ad-justment, both the pH and BHCO3 had beenbrought to normal values. It is clear that de-termination of arterial or, less exactly, venous pHconstitutes a more reliable and accurate pictureof the acidosis and, by techniques now available,a more conveniently estimated measure than theusual plasma CO2 combining power. With thissafeguard, much of the objection which has beenraised against the intravenous use of alkalies inthe treatment of diabetic acidosis may be met.If it be conceded that acidosis in itself is not de-sirable there is little argument against the rapidcorrection of pH toward normal limits.
Arterial oxygen content and saturation. Manyof these patients exhibited on admission highervalues for arterial hemoglobin and oxygen con-tent than is usual for hospital patients. This ismost likely a reflection of associated dehydrationand hemo-concentration. Even those patientswhose admission values for blood hemoglobin con-centration were not high were probably both de-hydrated and anemic since determinations madeafter treatment invariably showed greater hemo-dilution. The figures for arterial oxygen satura-tion are approximations since oxygen capacity wasnot directly determined but was derived from thehemoglobin concentration and the factor 1.34 rep-resenting the oxygen capacity of one gram ofhemoglobin. They indicate on the whole only avery slight reduction in arterial blood oxygen satu-ration (mean = 94%) which, in view of the ex-perimental errors involved plus the generally se-vere acidosis which would be expected to depressthe oxygen capacity of hemoglobin slightly, indi-cates the absence of significant interference withpulmonary -gas exchange.
Pulmonary ventilation. The respiration was ap-preciably stimulated in practically all patients and,in many, was of the Kussmaul type. In an effortto ascertain the mechanisms possibly involved inthis hyperpnea, the respiratory minute volume wastested for possible relationship with mean arterialblood pressure, arterial CO2 tension, pH andketone concentration, and rectal temperature.
504

CEREBRALMETABOLISM IN DIABETIC ACIDOSIS
There was little correlation except in tarterial pH, a graph of which is shown1. This includes some data on patientswise studied. The relationship between Iventilation and pH is somewhat complexis evidence of a threshold at about pHpiration being relatively unaffected donpoint. Below pH 7.20 respiratory volsharply to a maximum of 35 liters/mregion of pH 7.0. With increasing a
ventilation appears gradually to fallinitial threshold is similar to that of tbodies to acidosis demonstrated by CcSchmidt (21 ), and suggests that the mec&this hyperpnea may be a chemorecepiThat this stimulation should eventuallyto a depression as the acidosis increases ipossibly through depression of the medtters, is not unreasonable. It is interestithat in these patients, respiratory stimt
RESP. MIN.VOL.40i LIT./ MIN.
30
x
N
/-I * \
/0
8,/
20/ x
I o
\\*\N
6.8 7.0ARTERIAL pH
7.2
FIG. 1. THE RELATIONSHIP BETWEENREsMINUTE VOLUMEAND ARTERIAL PI
The circles represent patients who ultimatelythe crosses those who succumbed.
.he case of curs in the face of a profound fall in arterial CO2in Figure tension, indicating that at least in this conditionnot other- the important regulatory function of carbon di-pulmonary oxide has been superseded by another agent, quite~x. There possibly hydrogen ion concentration. Our data
7.20, res- for pulmonary ventilation and arterial pH afterwn to that the acidosis has been corrected, although incom-lume rises plete, suggest that the respiratory response lagsfin. in the considerably behind the response of pH to treat-Lcidity the ment, so that the hyperpnea may continue for aoff. The time after the arterial pH is close to normal.
.he carotid Should this observation be confirmed it would indi-)mroe and cate either that the hydrogen ion is not the realhanism for factor concerned in the hyperpnea, or that sometor reflex. time must elapse before a change in the pH of
give way arterial blood is reflected in the hydrogen ion con-
in severity, centration of the cells responsible for the respira-ullary cen- tory stimulation.nIg to note Blood glucose and electrolytes. Values forelation oc- blood glucose were elevated in all patients and
indiscriminately with respect to the presence orabsence of coma. This has also been the findingof others (1, 3, 5). The mean values for plasmaelectrolytes are shown graphically in Figure 2.Such studies have been made in some detail byothers (22 to 24). These electrolyte patterns, inalmost complete numerical agreement with thoseof Peters and associates (23), show a reductionin total base in diabetic acidosis, a considerablereduction in bicarbonate ion, and a progressiveincrease in the undetermined anion fraction (X)associated with, but by no means entirely ex-plained by, an increase in total ketone bodies.These substances are abnormally elevated in the"acidosis" group and even more so in the patientswith coma although the difference falls short ofstatistical significance. Thus, from the degree ofacidosis, of hyperglycemia, of ketosis, and of dis-turbance in the blood electrolyte pattern there islittle to explain the marked difference in mentalstate and prognosis between patients in diabeticcoma and those with severe diabetic acidosis.
Mean arterial blood pressure. The average74 value for mean arterial blood pressure is definitely
lower for the comatose patients (66 mm. Hg) thanthe normal figure of 86 mm. found in those who
SPIRATORY were acidotic but conscious, although this differ-ence is short of statistical significance. This ob-
recovered, servation is explained by the well-established ele-ment of circulatory failure in diabetic coma most
505

S. S. KETY, B. D. POLIS, C. S. NADLER, AND C. F. SCHMIDT
160
140
120
100
60
40
2C
NORMAL
\
B+ Cl
z_
or
DIABETICACIDOSIS COMA
(CVR) (1.1 resistance units) while this functionwas slightly increased in those patients in acidosiswithout coma (2.1 units). The normal value forcerebrovascular resistance is 1.6 mm.Hg/cc./100 g./min. (12).
Some explanation for the surprising observa-tion of an actual increase in cerebral blood flowin diabetic coma is suggested if the blood flow beplotted against the pH of arterial blood (Figure3). There is a fairly good correlation (r =-0.70,p = 0.01 ) and the shape of the curve is similar tothat found in normal subjects (26) where pH isaltered by changing the pCO2 of the blood. In thecase of the diabetic acidosis group, however, thecurve is shifted to the left so that for comparablelevels of pH the blood flow is lower in these pa-tients than in normal subjects breathing 5-7%ocarbon dioxide. This may be due to the fact thatin the case of normal subjects breathing carbon di-oxide both pCO2 and hydrogen ion concentrationare increased and their individual effects are likelyto be summated, while in diabetic acidosis the in-
CBFCc/IOOG/MIN
I 001 X
90
FIG. 2. THE BLOODELECTROLYTEPATTERN IN DIABETICAcIDosIS AND COMA
The anion fraction (X) represents phosphate, sulphate,and organic acids.
recently enunciated by Schecter, Wiesel and Cohn(25).
Cerebral blood flow and metabolism. In spiteof the circulatory depression found in the comatosepatients, only two showed a reduction in cerebralblood flow (CBF). The others yielded figuresfor this function somewhat in excess of the normaland the mean value for the group was 65 as com-
pared with a normal figure of 54 cc./100 g./min.(12). The patients with acidosis displayed a
greater consistency in cerebral blood flow whichwas slightly reduced on the average (45 cc./100 g./min.). This difference between the two groups
was largely due to a difference in tone of cerebralvessels, patients in coma showing a fairly con-
sistent decrease in cerebrovascular resistance
r = 0.70
p =<O.O I
80j
70
60
50
40
30
\
X\XO
6.8 7.0
ARTERIAL pH7.2 7.4 7.6
FIG. 3. THE RELATIONSHIP BETWEENCEREBRAL BLOODFLOWAND ARTERIAL PH
The circles represent patients who ultimately recovered,the crosses those who succumbed,
MEO/L
506

CEREBRALMETABOLISMIN DIABETIC ACIDOSIS
crease in hydrogen ion concentration occurs in theface of a marked fall in pCO2. It is not yet clearin the case of CO2 inhalation whether the cere-brovascular dilation is due to the CO2 itself or tothe concomitant pH shift or to both; it is never-theless evident that at least in the severe acidosisof diabetes, cerebral vessels may dilate eventhough the CO2 tension be remarkably low.
Of all the studies performed, measurement ofcerebral metabolic rate in terms of oxygen con-sumed (CMRo2) yielded by far the most sig-nificant difference between the comatose and non-comatose patients. As compared with a consump-tion of 3.3 (a = ± 0.4) cc. of oxygen per 100 g.of brain per minute found in mentally alert normalsubjects (12), the comatose patients yielded anaverage of only 1.7 (oa = + 0.4), a reduction of48%o and highly significant statistically. Thosepatients who were acidotic and confused but notunconscious on admission exhibited an averagefigure for cerebral 02 consumption of 2.7 (a =+- 0.4) cc./100 g./min., moderately lower thanthe normal. In fact there seems to be a criticallevel for cerebral oxygen utilization of 2.1 cc./100 g./min. at or below which consciousness disap-pears.
The cerebral respiratory quotient was consist-ently below unity, averaging 0.87 and 0.92 in thecomatose and non-comatose groups, respectively.At the present state of our knowledge there islittle justification for speculation on the basis forthis slight but significant deviation from the normalof 0.99 (12).
It was hoped that by determination of cerebralarteriovenous glucose difference the utilization ofthis substance by the brain could be measured inthis condition as has already been done in hy-poglycemia (8). At the high blood sugar levelsencountered, however, the arteriovenous differ-ence was well within the error of the glucose de-terminations so that such an estimation is notpossible until blood glucose methods of muchhigher precision are available.
DISCUSSION
It has been a discouraging finding repeatedlyconfirmed that the usual biochemical analyses inpatients with severe diabetic acidosis fail to showany significant difference between those who are
conscious, whose metabolic derangements are read-ily reversible by present therapy, and those whoare in true coma with a grave prognosis, and afrequently. irreversible pattern of deterioration.The present studies indicate that cerebral oxygenutilization is at least one biochemical functionwhich serves to differentiate these two groups.In all of the patients who were in coma on admis-sion the cerebral oxygen consumption was 2.1,cc./100 g./min. or less while seven of the eightpatients who still retained consciousness yieldeda value for this function of 2.4 cc./100 g./min. ormore. This measurement was also the only one ofthe many performed which had definite prognosticsignificance. With but one exception in eachgroup, a cerebral oxygen consumption below 2.1cc./100 g./min. was incompatible with survival,whereas in those with values above that figure re-sponse to treatment was satisfactory and recoverytook place. This depression in oxygen utilizationis not limited to the brain. Schecter, Wiesel andCohn have demonstrated a decrease in oxygenconsumption in the extremities of patients in di-abetic acidosis (25). In the brain, however, a de-fect in metabolism is of grave and immediate sig-nificance. The interesting correlation betweenmental state and utilization of oxygen by the brainis shown in Figure 4. The reduction in cerebralmetabolism is accompanied by and undoubtedly
CEREBRAL METABOLISM IN, DIABETIC ACIDOSISCMs
3
2-
C.C. 02PER100 G. BRAIN PERMIN.NORMALDIABETIC ACIDOSIS
CONFUSED
UNCONSCIOUS
CMR
3
2 * e
r O.52
Oal1l0 | O v ~~~~~~~p<O.02MENTAL STATE
0 6.8 7.0 7.2 7.4ARTERIAL pH
CMR4 * 4 0
r. 0.19 ra-0.663 00 0 0 p>>O. 0 3 000 0 p<O.OOI
*0 .0 00
*2 00 * 2 no-*
0 040 60 OD0100 1001 60 120 80OCEREBRAL. BLOOD FLOWccsoowO BLOODKETONES aAoomo.FIG. 4. CORRELATIONSOF CEREBRALOXYGENCONSUMP-
TION (CMR) WITH MENTAL STATE, CEREBRAL BLOODFLOW, ARTERIAL PH AND ARTERIAL KETONE CONCEN-TRATION
The closed circles represent admission data, the opencircles observations made in the course of therapy.
507
0 0 o

S. S. KETY, B. D. POLIS, C. S. NADLER, AND C. F. SCHMIDT
responsible for the progressive deterioration ofmental function which occurs in the course of un-corrected diabetic acidosis. We have observeda strikingly similar phenomenon in the hypogly-cemia and coma induced by insulin (8), the stageof severe hypoglycemia and confusion being as-sociated with a CMRo2 of 2.6 and the state ofdeep coma yielding an average of 1.9 cc. 02/100 g./min. Whereas in insulin hypoglycemia the de-pression of cerebral metabolism may reasonably beattributed to the profound drop in glucose avail-able for utilization by the brain, it is by no meansas easy to identify the process responsible for thedecreased cerebral oxygen utilization in diabeticcoma. There is evidence to indicate that insulinfrom the pancreas maybe dispensable in the utili-zation of glucose by the brain (9), a very cogentobservation being that in untreated human di-abetics and depancreatized animals mental func-tion is interfered with only late in the progress ofthe acidosis and other biochemical derangements.In a search for the possible factors directly re-sponsible for this depressed utilization of oxygenby the brain, it may be pertinent to determine thecorrelation between CMRo2and each of the bio-chemical and physiological disturbances known tooccur in diabetic coma. On the basis of the cir-culatory failure usually found in this condition thereasonable thesis has been proposed that the comais due to a deficiency in cerebral circulation (27).Our findings demonstrate that this is not so(Table III), and a correlation between CMRo2and CBF (Figure 4) indicates that the cerebraloxygen utilization in this condition is almost com-pletely unrelated to the cerebral blood flow. Onthe basis of this fact and the relatively normalarterial oxygen saturations observed, it may beconcluded that the fault in cerebral oxygen utili-zation does not lie in the supply of oxygen to thebrain. The possibility that the mechanisms for re-lease of oxygen from the blood to cerebral tissuemay be disturbed is now under investigation by astudy of the oxyhemoglobin dissociation curveand the oxygen tensions in cerebral venous blood.With the reservation that a significant defect maybe found there, it is probable that the fundamentalderangement is in the cellular biochemical proc-esses responsible for the normal utilization ofoxygen. If these processes are at all dependenton the environmental pH, and this can hardly be
doubted, the acidosis itself might be expected tocontribute to the depression in cerebral metabo-lism. There is a fair correlation (Figure 4) be-tween CMRo2 and arterial pH which tends tojustify this supposition. Of course the pH of ar-terial blood only indirectly affects the hydrogen ionconcentration inside the cell and it is possible thatthis correlation would be considerably improvedwere the latter quantity measurable.
In 1914 Hurtley (28) suggested that aceto-acetic acid was a noxious agent in diabetic acidosisand despite conflicting evidence (29), recent workhas corroborated the toxic properties of this sub-stance. Schneider and Droller (30) found thatslow intravenous infusion of acetoacetic acid inrabbits regularly produced coma where even agreater acidosis resulting from administration ofhydrochloric or beta hydroxybutyric acids hadno such effect. In fact coma was consistently ob-tained even with sodium aceto-acetate where noacidosis accompanied the administration. Thusat least one of the ketone bodies is capable of pro-ducing coma although- the blood concentrationsnecessary are not known and may well be higherthan those found in the coma of diabetic acidosis.Our own observations are compatible with thethesis that ketosis is an important factor contrib-uting to'this type of coma. Figure 4 shows a fairlygood correlation between CMRo2 and blood ke-tone concentrations. Cerebral oxygen utilizationfell as the blood ketone level rose. Such a cor-relation is open to a number of interpretations.It may mean that one or more of the ketone sub-stances, acting as a histotoxic agent, is respon-sible for the depression in cerebral metabolism.An equally good possibility is that the blood ke-tone level is simply an index of less defined butmore fundamental aberrations just as the bloodurea concentration reflects, but is hardly respon-sible for, the disturbances in uremia. There ismore to be said for the causal efficacy of the ke-tones, however, in that at least one of their numberhas been shown to be capable of producing comain itself.
The results reported here are compatible with,but by no means demand, the following sequenceof events. The glycosuria plus the loss of sodiumfrom the body lead to the well-recognized but in-adequately verified contraction in extracellularfluid space of which the blood volume is an im-
508

CEREBRALMETABOLISM IN DIABETIC ACIDOSIS
portant component (31, 32). This can only re-sult in a decrease in cardiac output compensatedby a restriction in peripheral blood flow (25) andquite probably a marked decrease in renal bloodflow. The work of McCance and Lawrence (33)as well as that of Peters, Kydd, Eisenman andHald (23) stresses the importance of renal regu-latory mechanisms in the excretion of keto-acids,while the experiments of Stadie, Zapp and Lukens(34) demonstrate the major role which periph-eral tissues play in the utilization of ketone bodies.Thus this diversion of the decreased cardiac out-put from the kidneys and muscles, although neces-sary for the maintenance of blood flow throughmore immediately vital centers, sharply restrictsthe available mechanisms for the utilization andexcretion of ketone bodies produced in this con-dition in excessive amounts (35). The impor-tant renal adjustment of the body hydrogen ionconcentration is also disorganized (33). The re-sultant acidosis and ketosis, not to mention a num-ber of poorly defined but possibly more importantbiochemical disturbances, produce serious derange-ments in cellular oxidations throughout the body.In the heart these derangements may lead to fur-ther circulatory failure, now on the basis of myo-cardial inefficiency in addition to the decreasedblood volume. These disturbances in metabolismin the brain are probably responsible for the devel-opment of coma and eventual death.
The nature of the "irreversibility" of severediabetic coma is somewhat indicated by the presentstudies but by no means clearly defined. It is onething to establish a critical level of cerebral oxygenconsumption below which death occurs in spiteof therapy, but quite another to explain the natureof the process which cannot be reversed. One canonly hope that further study will lead to a deeperinsight into these processes for certainly the ir-reversible stage in this disease is relative only toour ability to comprehend and correct the bio-chemical and physiological aberrations which com-prise it.
SUMMARY
1. Studies of blood gases, electrolytes, acid-basebalance, respiration, blood pressure, cerebral bloodflow and cerebral oxygen consumption are reportedon 14 patients in severe diabetic acidosis, six ofwhom were in deep coma.
2. Respiratory minute volume in these patientswas well correlated with arterial pH.
3. Coma was associated with and probably theresult of a 40%o reduction in cerebral utilization ofoxygen which occurred in spite of a generally aug-mented cerebral blood flow and a normal arterialoxygen saturation.
4. The depression in cerebral oxygen con-sumption is partly related to the acidosis and moresignificantly to the ketosis in this condition al-though other factors as yet poorly defined areundoubtedly operating.
5. The results establish the feasibility of apply-ing these techniques in diabetic coma and open thepossibility of further definition of the biochemicalderangements in the living human brain by thestudy of more specific metabolic components.
ACKNOWLEDGMENT
The authors wish to acknowledge the cooperation ofDoctors Edward S. Dillon and Anthony Sindoni, Jr.,chiefs of service in the Metabolic Division; the splendidassistance of Miss Edith Erikson and the Metabolic nurs-ing staff, and the painstaking analytical work of MissesMaxine Sortwell and Ruth Spear.
BIBLIOGRAPHY
1. Dillon, E. S., and Dyer, W. W., Factors influencingthe prognosis in diabetic coma. Ann. Int. Med.,1937, 11, 602.
2. Baker, T. W., A clinical survey of 108 consecutivecases of diabetic coma. Arch. Int. Med., 1936, 58,373.
3. Owens, L. B., and Rockwern, S. S., Prognosis indiabetic coma: basic importance of mental state.Am. J. M. Sc., 1939, 198, 252.
4. Rabinowitch, I. M., Fowler, A. F., and Bensley, E. H.,Diabetic coma (an investigation of mortalities andreports of a severity index for comparative stud-ies). Ann. Int. Med., 1939, 12, 1403.
5. Collen, M. F., Mortality in diabetic coma. Arch.Int. Med., 1942, 70, 347.
6. Joslin, E. P., Root, H. F., White, P., and Marble,A., Diabetic coma. J. A. M. A., 1942, 119, 1160.
7. Mulder, A. G., and Crandall, L. A., Cerebral metab-olism in fat fed dogs. Am. J. Physiol., 1942, 137,436.
8. Kety, S. S., Woodford, R. B., Harmel, M. H., Frey-han, F. A., Appel, K. E., and Schmidt, C. F.,Cerebral blood flow and metabolism in schizo-phrenia. The effects of barbiturate semi-narcosis,insulin coma and electroshock. Am. J. Psychiat.,1948, In press.
509

S. S. KETY, B. D. POLIS, C. S. NADLER, AND C. F. SCHMIDT
9. Himwich, H. E., and Nahum, L. H., The respiratoryquotient of the brain. Am. J. Physiol., 1932, 101,446.
10. Soskin, S., and Levine, R., Carbohydrate Metabolism.University of Chicago Press, Chicago, 1946.
11. Kety, S. S., and Schmidt, C. F., The determinationof cerebral blood flow in man by the use of nitrousoxide in low concentrations. Am. J. Physiol.,1945, 143, 53.
12. Kety, S. S., and Schmidt, C. F., The nitrous oxidemethod for the quantitative determination of cere-
bral blood flow in man; theory, procedure andnormal values. J. Clin. Invest., 1948, 27, 476.
13. Peters, J. A., and Van Slyke, D. D., QuantitativeClinical Chemistry. Williams & Wilkins, Balti-more, 1931.
14. Evelyn, K. A., and Malloy, H. T., Microdetermina-tion of oxyhemoglobin, methemoglobin, and sulf-hemoglobin in a single sample of blood. J. Biol.Chem., 1938, 126, 655.
15. Polis, B. D., and Sortwell, M., Rapid photocolori-metric micro procedure for blood sugar using cop-per reduction with perchloric acid deproteinizedfiltrates. Arch. Biochem., 1946, 11, 229.
16. Greenberg, L. A., and Lester, D., A micromethod forthe determination of acetone and ketone bodies.J. Biol. Chem., 1944, 154, 177.
-17. Kingsley, G. R., The determination of serum totalprotein, albumin, and globulin by the biuret reac-
tion. J. Biol. Chem., 1939, 131, 197.18. Sendroy, J., Jr., Microdetermination of chloride in
biological fluids, with solid silver iodate. III.Colorimetric analysis. J. Biol. Chem., 1937, 120,419.
19. Polis, B. D., and Reinhold, J. G., The determinationof total base of serum by ion exchange reactionsof synthetic resins. J. Biol. Chem., 1944, 156, 231.
20. Karr, W. G., A method for the determination of bloodurea nitrogen. J. Lab. & Clin. Med., 1924, 9, 329.
21. Comroe, J. H., Jr., and Schmidt, C. F., The partplayed by reflexes from the carotid body in thechemical regulation of respiration in the dog. Am.J. Physiol., 1938, 121, 75.
22. Atchley, D. W., Loeb, R. F., Richards, D. W., Jr.,Benedict, E. M., and Driscoll, M. E., On diabeticacidosis; a detailed study of electrolyte balances
following the withdrawal and reestablishment ofinsulin therapy. J. Clin. Invest., 1933, 12, 297.
23. Peters, J. P., Kydd, D. M., Eisenman, A. J., andHald, P. M., The nature of diabetic acidosis. J.Clin. Invest., 1933, 12, 377.
24. Hartmann, A. F., and Darrow, D. C., Chemicalchanges occurring in the body as the result of cer-tain diseases. III. The composition of the plasmain severe diabetic acidosis and the changes takingplace during recovery. J. Clin. Invest., 1928, 6,257.
25. Schecter, A. E., Wiesel, B. H., and Cohn, C., Periph-eral circulatory failure in diabetic acidosis and itsrelation to treatment. Am. J. M. Sc., 1941, 202,364.
26. Kety, S. S., and Schmidt, C. F., Effects of alteredarterial tensions of carbon dioxide and oxygen oncerebral blood flow and cerebral oxygen consump-tion of normal young men. J. Clin. Invest., 1948,27, 484.
27. Dillon, E. S., Riggs, H. E., and Dyer, W. W., Cere-bral lesions in uncomplicated fatal diabetic acidosis.Am. J. M. Sc., 1936, 192, 360.
28. Hurtley, W. H., The four carbon atom acids of dia-betic urine. Quart. J. Med., 1915, 9, 301.
29. Dodds, E. C., and Robertson, J. D., The relation ofaceto-acetic acid to diabetic coma and the cause ofdeath. Lancet, 1930, 218, 852.
30. Schneider, R., and Droller, H., Relative importanceof ketosis and acidosis in production of diabeticcoma. Quart. J. Exper. Physiol., 1938, 28, 323.
31. Chang, H. C., Harrop, G. A., Jr., and Schaub, B. M.,The circulating blood volume in diabetic acidosis.J. Clin. Invest., 1928, 5, 407.
32. Jacobson, S. D., and Lyons, R. H., The changesin the blood volume produced by diabetic acidosis.J. Lab. & Clin. Med., 1942, 27, 1169.
33. McCance, R. A., and Lawrence, R. D., The secretionof urine in diabetic coma. Quart. J. Med., 1935,4, 53.
34. Stadie, W. C., Zapp, J. A., Jr., and Lukens, F. D. W.,Effect of insulin upon ketone metabolism of nor-mal and diabetic cats. J. Biol. Chem., 1940, 132,423.
35. Stadie, W. C., Fat metabolism in diabetes mellitus.J. Clin. Invest., 1940, 19, 843.
510