the active role of vitamin c in mammalian iron metabolism: much more than just enhanced iron...
TRANSCRIPT

Review Article
The active role of vitamin C in mammalian iron metabolism:Much more than just enhanced iron absorption!
Darius J.R. Lane n, Des R. Richardson n
Molecular Pharmacology and Pathology Program, Department of Pathology and Bosch Institute, University of Sydney, Sydney, NSW 2006, Australia
a r t i c l e i n f o
Article history:Received 13 May 2014Received in revised form4 July 2014Accepted 8 July 2014Available online 15 July 2014
Keywords:AscorbateVitamin CIronTransferrinFerritinDcytbIRPHIFFree radicals
a b s t r a c t
Ascorbate is a cofactor in numerous metabolic reactions. Humans cannot synthesize ascorbate owing toinactivation of the gene encoding the enzyme L-gulono-γ-lactone oxidase, which is essential for ascorbatesynthesis. Accumulating evidence strongly suggests that in addition to the known ability of dietaryascorbate to enhance nonheme iron absorption in the gut, ascorbate within mammalian systems canregulate cellular iron uptake and metabolism. Ascorbate modulates iron metabolism by stimulating ferritinsynthesis, inhibiting lysosomal ferritin degradation, and decreasing cellular iron efflux. Furthermore,ascorbate cycling across the plasma membrane is responsible for ascorbate-stimulated iron uptake fromlow-molecular-weight iron–citrate complexes, which are prominent in the plasma of individuals with iron-overload disorders. Importantly, this iron-uptake pathway is of particular relevance to astrocyte brain ironmetabolism and tissue iron loading in disorders such as hereditary hemochromatosis and β-thalassemia.Recent evidence also indicates that ascorbate is a novel modulator of the classical transferrin–iron uptakepathway, which provides almost all iron for cellular demands and erythropoiesis under physiologicalconditions. Ascorbate acts to stimulate transferrin-dependent iron uptake by an intracellular reductivemechanism, strongly suggesting that it may act to stimulate iron mobilization from the endosome. Theability of ascorbate to regulate transferrin iron uptake could help explain the metabolic defect thatcontributes to ascorbate-deficiency-induced anemia.
& 2014 Elsevier Inc. All rights reserved.
Contents
Ascorbate biochemistry in mammals . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70Cellular ascorbate uptake . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70Sodium-dependent vitamin C transporters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71Facilitative glucose transporters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
Iron uptake, storage, and efflux: new roles for ascorbate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72Non-transferrin-bound iron uptake and ascorbate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72
NTBI and ferrireductases: Dcytb and the cytochromes b561 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72Dcytb as a ubiquitous oxidoreductase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73Ascorbate efflux and ferrireduction: a novel paradigm. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73
Transferrin iron uptake and ascorbate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73Ascorbate stimulates iron uptake by an intracellular reductive mechanism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74Ascorbate may act at the level of transferrin-cycle endosomes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
Cellular iron storage, efflux, and homeostasis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75Ferritins and iron storage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
Contents lists available at ScienceDirect
journal homepage: www.elsevier.com/locate/freeradbiomed
Free Radical Biology and Medicine
http://dx.doi.org/10.1016/j.freeradbiomed.2014.07.0070891-5849/& 2014 Elsevier Inc. All rights reserved.
Abbreviations: Asc, L--ascorbate; CGcytb, chromaffin granule cytochrome b561; Dcytb, duodenal cytochrome b561; DHA, dehydroascorbate; DMT1, divalent metaltransporter isoform 1; FBXL5, F-box and leucine-rich repeat protein 5; FIH, factor inhibiting HIF; FPN1, ferroportin 1; FTH1, H-ferritin; FTL, L-ferritin; GULO, L-gulono-γ-lactone oxidase; GLUT, facilitative glucose transporter; HIF, hypoxia-inducible factor; holo-Tf, mono- or diferric Tf; IRE, iron-responsive element; IRP, iron-regulatory protein;ISC, iron–sulfur cluster; Lcytb, lysosomal cytochrome b561; NTBI, non-Tf-bound iron; PHD, prolyl-4-hydroxylase domain-containing iron-dependent prolyl hydroxylase;SDR2, stromal cell-derived receptor 2; Steap3, six-transmembrane epithelial antigen of the prostate-3; SVCT, sodium–vitamin C cotransporter; Tf, transferrin; TfR, Tfreceptor; UTR, untranslated region
n Corresponding authors. Fax: þ61 2 9351 3429.E-mail addresses: [email protected] (D.J.R. Lane), [email protected] (D.R. Richardson).
Free Radical Biology and Medicine 75 (2014) 69–83

Systemic iron homeostasis: cellular iron efflux, hepcidin,and ferroportin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75Iron homeostasis: the iron-regulatory protein (IRP)–iron-responsive element (IRE) system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75Roles for ascorbate in regulating iron storage, efflux, and the IRP–IRE system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76Iron homeostasis: the HIF system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77Roles for ascorbate in regulating the HIF system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78Some important caveats: cells in culture versus cells in vivo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
Ascorbate biochemistry in mammals
It is well known that L-ascorbate (Asc)1 is a physiologicalreductant and enzyme cofactor that contributes to numerous well-defined enzymatic reactions involving collagen hydroxylation, carni-tine and norepinephrine biosynthesis, tyrosine metabolism, andpeptide hormone amidation [1–4]. Ascorbate also contributes sig-nificantly to cellular antioxidation as a water-soluble chain-breakingradical scavenger [5–7] and to the recycling of plasma membraneα-tocopherol (vitamin E) by reducing the α-tocopheroxyl radical[3,8–10]. Ascorbate also recycles tetrahydrobiopterin [11,12], which isimportant for nitric oxide synthase and tyrosine hydroxylase activ-ities. The recycling of vitamin E by Asc is important for protectionagainst lipid peroxidation in membranes [8–10,13]. Most mammalsare capable of de novo hepatic synthesis of Asc from glucose, througha biosynthetic pathway that employs the enzyme L-gulono-γ-lactone
oxidase (GULO) for the terminal oxidation reaction [14–17]. However,higher primates (including humans), guinea pigs, and some bats areobligatorily dependent on dietary sources of the vitamin [18,19]. Thisrequirement is due to an inactivation of the GULO gene [14,15,20].After absorption from the intestinal lumen in humans, Asc istransported in the blood and is typically found at millimolarconcentrations intracellularly (nucleated cells) and at micromolarconcentrations in extracellular fluids and erythrocytes [21–23].
Under physiological conditions, Asc typically undergoes a reversibleone-electron oxidation to the ascorbyl radical (also known as semi-dehydroascorbate, monodehydroascorbate, or the ascorbate free radi-cal; Fig. 1). The ascorbyl radical is relatively stable [6] and can beenzymatically recycled to Asc (see below). If they are not rapidlyreduced, two ascorbyl radicals can dismutate to one Asc and onedehydroascorbate (DHA) molecule [8,17,20] (Fig. 1). The dismutation oftwo ascorbyl radicals leads to Asc being able to effectively donate tworeducing equivalents in distinct monoelectronic cellular redox reac-tions [5]. Within cells, DHA is rapidly reduced back to Asc byglutathione (GSH)- and NAD(P)H-dependent enzymatic and none-nzymatic reactions [17]. The mitochondrial electron transport chain isalso an important site of intracellular DHA reduction in nucleated cells[24–29], with reduction taking place at an apically exposed section ofcomplex III in mammals [26]. The reduction of DHA appears to occuras a two-electron step that bypasses the intermediate production ofthe ascorbyl radical [8]. Intracellular Asc regeneration from either theascorbyl radical or DHA has been reviewed by Linster and VanSchaftingen [17] and Du et al. [30] and is not described in detail herein.
Importantly, DHA is structurally unstable, with a half-life ofseveral minutes under physiological conditions [31–33]. The rapiddegradation of DHA by ring opening to diketogulonic acid isirreversible in mammalian systems [31–33] (Fig. 1). This propertyhas probably been an evolutionary driving force in biochemical andcellular adaptations to Asc auxotrophy in GULO-deficient species.Indeed, under physiological conditions, vitamin C is maintainedpredominantly in its fully reduced form in both intra- and extra-cellular biological fluids. As already alluded to, mammalian cellspossess a variety of conservative reductive mechanisms for main-taining both intra- and extracellular Asc [8,17,20,34,35]. Evencultured cells, which are chronically Asc-deficient owing to the lackof supplementation and/or biosynthesis under standard cultureconditions [23,36,37], still maintain a high capacity for Asc regen-eration. The net effect of this regeneration is the maintenance ofphysiological Asc concentrations within biological fluids (e.g.,plasma, interstitial and cerebrospinal fluid) at the expense of other,predominantly intracellular, reductants (e.g., GSH and NAD(P)H).
Cellular ascorbate uptake
The majority of mammalian cells, with the notable exception ofhuman erythrocytes [21], maintain intracellular Asc concentrationsthat are higher (e.g., up to 30-fold in some cases) than those in theextracellular fluid [8,20,22]. For instance, lymphocytes accumulate
Fig. 1. Biological redox reactions of ascorbate. The Asc monoanion is the pre-dominant species of vitamin C present under physiological conditions. Asc under-goes a thermodynamically favorable and reversible one-electron oxidation to theascorbyl radical (AR; Reaction (1)). The AR is stabilized by resonant distribution ofthe resultant unpaired electron over the ring structure and can be enzymaticallyrecycled back to Asc. If not rapidly reduced, two ARs can dismutate to form onemolecule each of the two-electron oxidized form, dehydroascorbate (DHA; Reac-tion (2)) and Asc. DHA can be reduced within the cell interior back to Asc in a two-electron reduction, which bypasses the AR (Reaction (3)). DHA is very unstable and,if not rapidly reduced back to Asc, undergoes an essentially irreversible hydrolyticring opening to 2,3-diketogulonic acid with a half-life of several minutes underphysiological conditions (Reaction (4)). Adapted from Lane and Lawen, [78].
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–8370
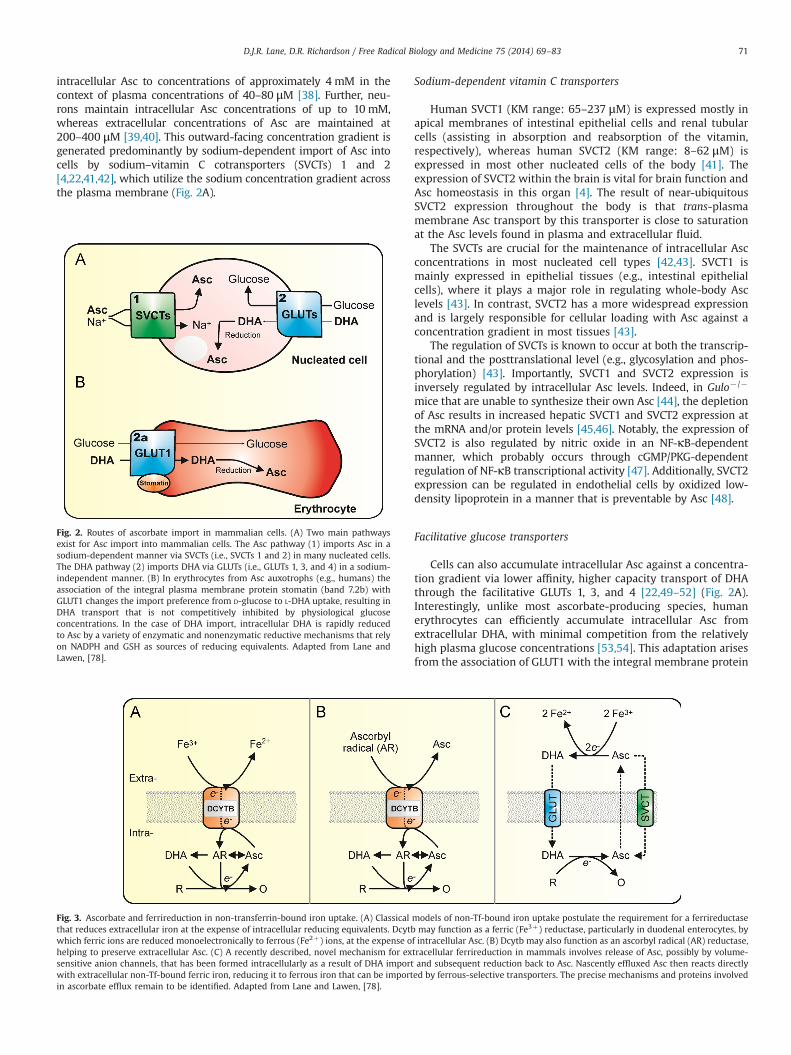
intracellular Asc to concentrations of approximately 4 mM in thecontext of plasma concentrations of 40–80 μM [38]. Further, neu-rons maintain intracellular Asc concentrations of up to 10 mM,whereas extracellular concentrations of Asc are maintained at200–400 μM [39,40]. This outward-facing concentration gradient isgenerated predominantly by sodium-dependent import of Asc intocells by sodium–vitamin C cotransporters (SVCTs) 1 and 2[4,22,41,42], which utilize the sodium concentration gradient acrossthe plasma membrane (Fig. 2A).
Sodium-dependent vitamin C transporters
Human SVCT1 (KM range: 65–237 μM) is expressed mostly inapical membranes of intestinal epithelial cells and renal tubularcells (assisting in absorption and reabsorption of the vitamin,respectively), whereas human SVCT2 (KM range: 8–62 μM) isexpressed in most other nucleated cells of the body [41]. Theexpression of SVCT2 within the brain is vital for brain function andAsc homeostasis in this organ [4]. The result of near-ubiquitousSVCT2 expression throughout the body is that trans-plasmamembrane Asc transport by this transporter is close to saturationat the Asc levels found in plasma and extracellular fluid.
The SVCTs are crucial for the maintenance of intracellular Ascconcentrations in most nucleated cell types [42,43]. SVCT1 ismainly expressed in epithelial tissues (e.g., intestinal epithelialcells), where it plays a major role in regulating whole-body Asclevels [43]. In contrast, SVCT2 has a more widespread expressionand is largely responsible for cellular loading with Asc against aconcentration gradient in most tissues [43].
The regulation of SVCTs is known to occur at both the transcrip-tional and the posttranslational level (e.g., glycosylation and phos-phorylation) [43]. Importantly, SVCT1 and SVCT2 expression isinversely regulated by intracellular Asc levels. Indeed, in Gulo�/�
mice that are unable to synthesize their own Asc [44], the depletionof Asc results in increased hepatic SVCT1 and SVCT2 expression atthe mRNA and/or protein levels [45,46]. Notably, the expression ofSVCT2 is also regulated by nitric oxide in an NF-κB-dependentmanner, which probably occurs through cGMP/PKG-dependentregulation of NF-κB transcriptional activity [47]. Additionally, SVCT2expression can be regulated in endothelial cells by oxidized low-density lipoprotein in a manner that is preventable by Asc [48].
Facilitative glucose transporters
Cells can also accumulate intracellular Asc against a concentra-tion gradient via lower affinity, higher capacity transport of DHAthrough the facilitative GLUTs 1, 3, and 4 [22,49–52] (Fig. 2A).Interestingly, unlike most ascorbate-producing species, humanerythrocytes can efficiently accumulate intracellular Asc fromextracellular DHA, with minimal competition from the relativelyhigh plasma glucose concentrations [53,54]. This adaptation arisesfrom the association of GLUT1 with the integral membrane protein
Fig. 3. Ascorbate and ferrireduction in non-transferrin-bound iron uptake. (A) Classical models of non-Tf-bound iron uptake postulate the requirement for a ferrireductasethat reduces extracellular iron at the expense of intracellular reducing equivalents. Dcytb may function as a ferric (Fe3þ) reductase, particularly in duodenal enterocytes, bywhich ferric ions are reduced monoelectronically to ferrous (Fe2þ) ions, at the expense of intracellular Asc. (B) Dcytb may also function as an ascorbyl radical (AR) reductase,helping to preserve extracellular Asc. (C) A recently described, novel mechanism for extracellular ferrireduction in mammals involves release of Asc, possibly by volume-sensitive anion channels, that has been formed intracellularly as a result of DHA import and subsequent reduction back to Asc. Nascently effluxed Asc then reacts directlywith extracellular non-Tf-bound ferric iron, reducing it to ferrous iron that can be imported by ferrous-selective transporters. The precise mechanisms and proteins involvedin ascorbate efflux remain to be identified. Adapted from Lane and Lawen, [78].
Fig. 2. Routes of ascorbate import in mammalian cells. (A) Two main pathwaysexist for Asc import into mammalian cells. The Asc pathway (1) imports Asc in asodium-dependent manner via SVCTs (i.e., SVCTs 1 and 2) in many nucleated cells.The DHA pathway (2) imports DHA via GLUTs (i.e., GLUTs 1, 3, and 4) in a sodium-independent manner. (B) In erythrocytes from Asc auxotrophs (e.g., humans) theassociation of the integral plasma membrane protein stomatin (band 7.2b) withGLUT1 changes the import preference from D-glucose to L-DHA uptake, resulting inDHA transport that is not competitively inhibited by physiological glucoseconcentrations. In the case of DHA import, intracellular DHA is rapidly reducedto Asc by a variety of enzymatic and nonenzymatic reductive mechanisms that relyon NADPH and GSH as sources of reducing equivalents. Adapted from Lane andLawen, [78].
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–83 71

stomatin (band 7.2b) [55] and appears to be a compensatoryregenerative mechanism for the lack of endogenous Asc produc-tion [55–59] (Fig. 2B). With respect to DHA uptake by cells, aninward-facing DHA gradient is maintained by the rapid reductionof imported DHA back to Asc, which largely occurs in an NADPH-and GSH-dependent manner [22,53,60].
Iron uptake, storage, and efflux: new roles for ascorbate
Iron is vital for cellular survival, as evidenced by the onset of celldeath after excessive iron depletion [61,62]. Almost all organismsrequire iron for metabolism and it plays a multitude of roles inbacteria, plants, and animals. Adult humans have 3–5 g of iron in thebody [63], 480% of which is found in the hemoglobin of erythro-cytes and 10–15% in muscle myoglobin and other iron-containingproteins and enzymes. Typically, only 0.1% circulates in the plasmabound to the major plasma iron-binding protein, transferrin (Tf)[61]. Cellular iron storage predominantly occurs within proteinnanocages created by ferritin [64]. Whereas most iron in mamma-lian systems is contained within the oxygen-binding proteinshemoglobin and myoglobin, many other cellular proteins also relyon iron for their function. These include other heme-containingproteins and enzymes (hemoproteins; e.g., the cytochromes) as wellas iron–sulfur cluster (ISC)-containing proteins (e.g., succinate dehy-drogenase) [61] and nonheme, non-ISC, iron-containing proteins(e.g., iron- and 2-oxoglutarate-dependent dioxygenases).
Iron that is improperly sequestered, however, can be highly toxic tocells. This occurs predominantly through its ability to act as a pro-oxidant. Indeed, redox-active iron can catalyze the production ofreactive oxygen species through the Fenton [65] and Haber–Weiss[66] reactions, resulting in the formation of highly damaging hydroxylradicals [61]. As too much or too little iron is detrimental, cellular ironhomeostasis must be tightly controlled through the regulation ofimport, storage, and efflux [61,67,68]. Far from the classical view thatAsc is merely a passive dietary factor that enhances nonheme dietaryiron absorption, a growing body of evidence indicates Asc is an activemolecular participant in cellular, and perhaps systemic, iron metabo-lism. These findings are reviewed below.
Non-transferrin-bound iron uptake and ascorbate
In its physiological form, extracellular iron is typically com-plexed by low-molecular-weight (Mr) ligands or iron-bindingproteins. The most important iron complexants in the extracellularfluids of mammalian systems are Tf (see below) and citrate [61,68].Although the basic mechanisms involved in the uptake of iron arequite well understood, the mechanisms of ferrireduction in bothTf- and non-Tf-bound iron (NTBI) uptake by mammalian cellsremain unclear [61].
Virtually all iron in plasma is bound to Tf under physiologicalconditions [61] (see below for further discussion). However, indiseases resulting in iron overload (e.g., hereditary hemochroma-tosis and β-thalassemia), Tf can become saturated with iron, suchthat excess plasma iron occurs in the circulation as NTBI [69].Although the precise biochemical nature of NTBI is ill defined [70],the term typically refers to a putative low-Mr pool of iron bound tosmall organic complexants, such as citrate and ATP [70,71]. Thispool of NTBI is present at very low concentrations in extracellularbiological fluids such as plasma and interstitial fluid [72,73].However, it becomes significant in the context of iron-overloaddiseases such as hereditary hemochromatosis, hypotransferrine-mia, and hemolytic anemias (e.g., β-thalassemia), in which plasmairon levels increase and may exceed Tf-binding capacity [72,73].Low levels (o1 μM) of NTBI have been documented in healthy
individuals, but may rise up to 10–20 μM under conditions ofsevere iron overload [70].
NTBI and ferrireductases: Dcytb and the cytochromes b561
In most instances, iron uptake from NTBI can be blocked byextracellular iron(II) chelators [74–76], which supports the notionthat extracellular iron(III) must first be reduced to iron(II) beforeuptake. In fact, it was originally hypothesized that a plasmamembrane ferrireductase was responsible for NTBI reductionbefore uptake by mammalian cells [77,78] (Fig. 3A). However,identification of the enzyme(s) responsible for cellular ferrireduc-tion before iron uptake has been the subject of much controversy.
Dcytb (also known as CYBRD1) is an iron-regulated ferrireduc-tase in the striated border membrane of duodenal enterocytes,which has been suggested to be responsible for nonheme ironreduction during dietary iron absorption [79]. This hypothesis wassubsequently challenged by the observation that Cybrd1� /� (i.e.,Dcytb-knockout) mice did not develop iron deficiency on astandard lab diet, or greater iron deficiency on an iron-deficientdiet, compared to wild-type mice [80]. These results suggest thatDcytb's activity may be supplemented by the action of otherferrireductases or ferrireductants. As the expression of Dcytb isiron regulated [81–84], and as the expression of this proteinclearly stimulates iron uptake in vitro [85], Dcytb is likely to playsome role (albeit probably a conditionally redundant one), innonheme iron absorption.
Dcytb is a member of the cytochrome b561 family, which exists inall eukaryotic kingdoms [86]. Cytochrome b561 (also known aschromaffin granule cytochrome b561; CGcytb/CYB561/CYB561A1)catalyzes transmembranous electron transfer from cytosolic Asc tointravesicular ascorbyl radicals in neuroendocrine secretory gran-ules [87].
CGcytb was initially recognized as a redox-active component ofcatecholamine storage granules in the early 1960s [88]. Flatmark andothers demonstrated that this activity was the result of a uniqueheme-containing cytochrome located in the membranes of thesevesicles [89–91]. In the 1980s, spectroscopic and EPR studies demon-strated that this cytochrome was responsible for equilibrating theascorbate–ascorbyl radical pair inside the granule lumen with thisredox pair in the cytosol [92–94]. Indeed, CGcytb is canonicallyinvolved in transmembranous electron transfer from cytosolic Asc tothe intravesicular ascorbyl radical in neuroendocrine secretory gran-ules [87], which occurs by a series of concerted electron/protontransfers across the membrane [95,96]. The resulting Asc is subse-quently oxidized by copper-containing dopamine β-hydroxylase andpeptidylglycine α-amidating monooxygenase [97]. The CGcytb-catalyzed reaction involves a histidine cycling mechanism of coupledproton/electron transfer between Asc and ascorbyl radical [98]. Addi-tional members of the cytochrome b561 family of enzymes inmammals include lysosomal cytochrome b561 (Lcytb/CYB561A3)[86,99], stromal cell-derived receptor 2 (SDR2/FRRS1) [100,101], andthe putative tumor suppressor 101F6 (CYB561D2/TSP10) [102,103].Notably, Dcytb, Lcytb, CGcytb, 101F6, and SDR2 are all capable ofstimulating cellular ferrireduction and are expressed at the plasmamembrane as well as at intracellular sites [79,86,100,104,105].
Although the bulk of evidence suggests that Asc is the majorelectron donor for these proteins [106], a recent report suggests thatdihydrolipoic acid can donate reducing equivalents to cytochromeb561 proteins [107]. Although the cellular levels of dihydrolipoic acidare relatively low compared to Asc, this observation may help explainthe apparent Asc-independent ferrireductase activity of Dcytb thathas been reported [105]. With the exception of Dcytb, there is still apaucity of direct evidence directly linking any of these proteins toendogenous extracellular or intracellular ferrireduction reactionsin vitro or in vivo. Although much evidence suggests likely roles
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–8372

for some of the cytochrome b561 family members in mammalianferrireduction, the precise cellular functions of these proteins remainunclear.
Dcytb as a ubiquitous oxidoreductase
In addition to being implicated as a ferrireductase at thestriated border membrane of duodenal enterocytes [79,108], Dcytbhas also been suggested to function as an oxidoreductase in avariety of tissue and cell types including human erythrocytes[109], lung epithelial cells [105], K562 cells [110,111], HepG2 cells[111], Caco-2 cells [111], and astrocytes [112,113]. This observationsuggests a more general function for Dcytb than one of justenhancing dietary nonheme iron absorption. In support of thisnotion, a growing number of studies [79,82,104,105,114–116] haveimplicated Dcytb as a ferrireductase in the cellular reduction ofNTBI. Importantly, as Dcytb demonstrates partial conservation ofthe canonical Asc-binding motif originally identified in CGcytb[86,97], Asc is a likely proximal electron donor for this activity,although dihydrolipoic acid may also be involved [107]. Accord-ingly, several studies in which Dcytb-expressing cells have beensupplemented with Asc or DHA provide support for this conclu-sion [82,86,104,116].
Ascorbate efflux and ferrireduction: a novel paradigm
The ferrireductase-dependence hypothesis for extracellularferrireduction has been challenged by studies showing that NTBIreduction can occur owing to efflux of reductants such as super-oxide [117], dihydrolipoic acid [118], and Asc [76,119–121]. We andothers have demonstrated that extracellular Asc oxidase, whichrapidly degrades Asc to DHA, abolished the reduction of iron(III)citrate by Asc-loaded cells, as well as greatly inhibiting Asc-loading-dependent iron uptake [76,119,121]. This sensitivityclearly indicates a requirement for effluxed Asc in the reductionof NTBI before uptake. Importantly, our data indicate that Asc isexported from the cells and directly reduces low-Mr iron(III)citrate complexes to iron(II) for cellular uptake, with the latteroccurring via an iron(II)-selective transporter such as divalentmetal transporter isoform 1 (DMT1) [119,122–125].
Regarding the mechanisms of ascorbate efflux from mammaliancells (for reviews see [22,77,78,126]), several plausible candidates fora release pathway have been proposed [22]. These include(i) exocytosis of ascorbate [22,127], (ii) connexin hemichannels[128], and (iii) plasma membrane (volume-sensitive) anion channels(VSOACs) [22,129–131]. The VSOAC hypothesis for ascorbate efflux isperhaps the best supported by experimental data [22]. Thesefunctionally defined channels are thought to be plasma membraneanion channels that are involved in regulatory volume decrease ofcells after cellular swelling caused by hypotonic shock [132] orglutamate-transporter-dependent [133] glutamate/aspartate uptakeby astrocytes [130,134]. Electrophysiological evidence demonstratesthat VSOACs are permeable to ascorbate [132], although the mole-cular identities of many of these channels remain uncertain. More-over, the observation that both VSOAC permeability [135] andascorbate efflux from cells [22,76,129] can often be blocked bynonspecific, sulfonate anion channel inhibitors, such as 4,40-dii-sothiocyanatostilbene-2,20-disulfonic acid and 4-acetamido-40-iso-thiocyanostilbene-2,20-disulfonic acid, suggests that a significantproportion of ascorbate release occurs via these channels. Ascorbateefflux, which is highly sensitive to sulfonate anion channel inhibitors,occurs in a variety of cell types, including astrocytes [130,133,136],hepatocyte-like HepG2 cells [137], SH-SY5Y neuroblastoma cells[131], coronary artery endothelial cells [138], and K562 cells [76],but not in endothelial cells [139]. Importantly, not all anion channelsare sensitive to sulfonate inhibitors [140]. In the case of endothelial
cells, the release of ascorbate is largely insensitive to such inhibitors[138,139], but appears to be stimulated by Ca2þ and ATP andinhibited by gluconate [138,141]. Taken together, these resultssuggest that ascorbate efflux in endothelial cells involves Ca2þ-dependent anion channels that are regulated by the G-protein-coupled purinergic P2Y2 receptors [138,141]. However, as previouslysuggested, these pharmacological properties may also suggest aCa2þ-dependent, P2Y2-regulated exocytotic mechanism [78]. In thecase of VSOAC involvement in ascorbate efflux from astrocytes andother cell types, a plasma membrane isoform of the voltage-dependent anion channel 1 [142,143] is a plausible candidate thatshould be seriously considered. Clearly, further molecular studies areneeded to resolve the involvement of these pathways.
We have proposed a model of ascorbate-dependent NTBIreduction and uptake by mammalian cells in which iron uptakeis preceded by iron reduction by extracellular Asc, which issubsequently regenerated from DHA by trans-plasma membraneAsc cycling [76–78,119] (Fig. 3C). Notably, a recent study hasidentified an analogous Asc efflux mechanism as a novel strategyfor iron reduction and transport in plants [144]. Indeed, Pisumsativum and Arabidopsis embryos exhibit ferrireduction activitythat is not catalyzed by the typically attributed membrane ferrir-eductase activity, but is instead due to the efflux of high levels ofAsc that directly reduce iron(III) present in extracellular iron–citrate–malate complexes [144].
Transferrin iron uptake and ascorbate
Under normal physiological conditions, virtually all plasma iron istightly bound to Tf [61]. Iron that is taken up by enterocytes isreleased into the bloodstream and oxidized to the ferric statepotentially by the transmembrane multicopper ferroxidase, hephaes-tin, which occurs predominantly in the basolateral membrane ofenterocytes [145]. The homologous soluble multicopper ferroxidase,ceruloplasmin, which is abundant in plasma, is likely also to playsome role in this activity [146]. The iron(III) that is formed by theaction of these ferroxidases is specifically bound to serum Tf, an 80-kDa glycoprotein that is mainly synthesized by the liver [147]. Each Tfmolecule can bind one or two iron(III) ions [148,149] to form onemolecule of mono- or diferric Tf (holo-Tf), respectively.
The binding of iron(III) to Tf occurs with high affinity in a pH-dependent manner, with maximal binding occurring at pH 7.4(affinity constant at atmospheric pCO2 for each binding site of�1020 M�1) [150]. Holo-Tf binds to the integral membrane proteinTf receptor 1 (TfR1) to donate iron to cells [61,68,147] (Fig. 4A).TfR1 is a glycoprotein of about 90-95 kDa, which forms a homo-dimer of approximately 180-190 kDa that is linked by disulfidebonds [151] and is capable of binding two molecules of holo-Tf[152]. TfR1 is expressed by most cells, with the exception ofmature erythrocytes [153] and possibly oligodendrocytes, micro-glia, and astrocytes in vivo [154].
The affinity of TfR1 for diferric Tf at pH 7.4 is about 2000-foldhigher than for apo-Tf [149,155] and about 20-fold higher than foreither of the monoferric Tf forms [156]. The entire holo-Tf–TfR1complex is then endocytosed via receptor-mediated endocytosis inclathrin-coated pits [157]. The pH of the endosome decreases afterinternalization, owing to the activity of an ATP-dependent protonpump, the vacuolar-type Hþ-ATPase, in the endosomal membrane[158,159]. Owing to the resulting acidic environment (i.e., a lumenpH of 5.3–5.6 [160]), the iron(III) dissociates from Tf, whereas apo-Tf remains tightly bound to TfR1 [158,161].
Before the transport of iron from the endosome to the cytoplasmby DMT1 [162,163], iron(III) that is released from Tf must first bereduced to iron(II) [164,165]. The only ferrireductase identified to beinvolved in this process is the six-transmembrane epithelial antigen
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–83 73

of the prostate-3 (Steap3) [166,167], but this activity may berestricted to erythroid precursors. It should also be noted that otheriron(II) transporters have been proposed to contribute to ironmobilization from Tf-cycle endosomes, including ZIP14 [168],although their relative contributions in different cell types remainto be established. Finally, the complex of apo-Tf and TfR1 is thenrecycled back to the plasma membrane and the apo-Tf dissociatesfrom TfR1 at the slightly alkaline pH of the extracellular space [169](Fig. 4A).
Ascorbate stimulates iron uptake by an intracellular reductivemechanism
We recently demonstrated that Asc regulates iron uptake fromTf in a variety of human cell types [120]. This observation isintriguing, as Asc is a ubiquitous and normally abundant cellularreductant in vivo, yet it is absent under standard cell cultureconditions [170]. Significantly, typical physiological plasma Ascconcentrations enhance iron uptake by Asc-derived cells by up to100% from Tf, which is accompanied by a corresponding increasein cellular ferritin expression and ferritin-iron loading [120].Although Asc can increase ferritin either by promoting de novosynthesis [171] or by inhibiting ferritin autophagy [172] (see belowfor further discussion), we demonstrated that none of theseprocesses were responsible for Asc's ability to stimulateTf-dependent iron uptake [120]. Indeed, these results supportthe general notion that ferritin does not contribute to Tf-dependent iron uptake per se, although it serves as the majoriron storage depot for nascently internalized iron.
Ascorbate may act at the level of transferrin-cycle endosomes
To gain insight into Asc's mechanism of action in stimulating Tf-dependent iron uptake, we initially adopted a comparative approachto examine key differences between the Asc-stimulated uptake fromiron–citrate complexes and holo-Tf [120]. We determined that, unlike
iron–citrate [76,119,121], the uptake of iron from Tf was largelyindependent of: (i) the reductive action of extracellular Asc and (ii)the extracellular labilization of iron from Tf [120]. In fact, the results ofour experiments with membrane-impermeative and membrane-permeative Asc-oxidizing reagents strongly suggest that Asc actsintracellularly to enhance Tf-dependent iron uptake [120].
Consistent with these findings, and with the fact that mostknown biological activities of Asc are mediated by the molecule'sreducing activity [5,17], our results demonstrated that the redu-cing ene-diol moiety of Asc was required for stimulation of ironuptake [120]. Importantly, we also showed that this was notdue to a general increase in cellular reducing capacity in thepresence of Asc or to a specific increase in NADPH. In fact,incubation of SK-Mel-28 cells with a physiological level of Asc(50 μM) resulted in depletion of cellular NADPH, but not totalNADP [120]. This finding is consistent with the involvement ofNADPH and thioredoxin reductase [35,173] during intracellularAsc recycling reactions. Taken together, our results suggest thatAsc enhances Tf-dependent iron uptake in a manner dependentspecifically on Asc's reducing activity and an intracellular pool ofthe vitamin.
Of importance to the mechanism by which Asc acts to enhanceTf-dependent iron uptake, Asc is at least as effective as NADHin vitro at mobilizing iron from isolated Tf-containing endosomes[164,165]. We also demonstrated that, although Asc did not affectthe kinetics of Tf cycling, the expression of human TfR1 or TfR2was necessary for the stimulatory effect of Asc on iron uptake[120]. As an aside, our observation that Asc stimulated iron uptakein a TfR2-dependent manner lends support to the hypothesis that,in addition to regulating hepcidin production and systemic ironhomeostasis [174], TfR2 contributes to iron acquisition from Tf[175,176]. However, any direct role for TfR2 in Tf-dependent ironuptake is likely to be minor in comparison to that of TfR1[177,178].
Finally, using an array of well-characterized inhibitors of endocy-tosis or endosome acidification, we demonstrated that endocytosis
Fig. 4. Model of ascorbate-dependent stimulation of transferrin–iron uptake. (A) In Asc-depleted cells, holo-Tf binds to cell surface Tf receptors (TfR) and is internalized byreceptor-mediated endocytosis. The endosomes that form become acidified via the proton-pumping vacuolar-type Hþ-ATPase (V-ATPase). Acidification leads to release ofiron from Tf, whereas iron-free Tf (apo-Tf) remains bound to the TfR. The intraendosomal Fe3þ is then reduced to Fe2þ and transported across the endosomal membrane bythe divalent metal transporter 1 (DMT1) and/or Zip14. The apo-Tf–TfR complex returns to the cell surface, where apo-Tf dissociates at neutral pH. After transport across theendosomal membrane, iron initially enters the labile iron pool (LIP) and can be stored in ferritin. (B) In contrast, intracellular ascorbate enhances: (i) Tf-dependent ironuptake, (ii) LIP size, (iii) ferritin synthesis, and (iv) iron deposition in ferritin. The last two effects on ferritin may be secondary to the increase in the LIP and Tf-dependent Feuptake. Asc appears to act intracellularly via a reductive mechanism, either by direct ferrireduction or via a transmembrane reductase. This process probably (as indicated bydotted lines) involves direct import of Asc into the Tf cycle endosome and direct or chemical ferrireduction and/or provision of electrons to an endosomal ferrireductase toenhance Tf-dependent delivery of iron to cells. Adapted from Lane et al. [120].
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–8374

and intracellular vesicle acidification were critical for Asc-enhancedTf-dependent iron uptake [120]. Specifically, endocytosis was neces-sary for stimulation to occur, whereas blockade of intracellularvesicle acidification inhibited both control and Asc-stimulated ironuptake similarly, although the relative stimulation provided by Ascwas not affected. Together, our recent findings demonstrate that:(i) endocytosis is essential for Asc to stimulate Tf-dependent ironuptake and (ii) intracellular vesicle acidification facilitates bothcontrol and Asc-stimulated iron uptake from holo-Tf equally [120].These observations show that Asc enhances Tf-dependent ironuptake downstream of an endocytosis event, which is furtherenhanced by intracellular vesicle acidification. Thus, Asc acts intra-cellularly via a reductive mechanism to enhance Tf-dependent irondelivery of iron to cells, and this appears to occur subsequent to theTfR-dependent endocytosis of holo-Tf (Fig. 4B).
As mentioned above, our results are consistent with previousobservations that Asc efficiently promotes iron mobilization fromisolated and acidified Tf-containing endosomes [164,165]. Consid-ering these data collectively, we have proposed a model for Asc'sactivity in enhancing intraendosomal ferrireduction and ironmobilization from the Tf-containing endosome (Fig. 4B). As sug-gested by studies on isolated endosomes containing Tf, this couldoccur by Asc uptake into the endosomal vesicle followed bychemical ferrireduction [179] and/or by supply of reducing equiva-lents from Asc to an endosomal ferrireductase [164,165].
Importantly, the only confirmed ferrireductase of the Tf cycle isSteap3, which appears to be most active in this role in erythroidcells [166]. It is unlikely that Asc contributes electrons to Steap3,which instead appears to utilize NAD(P)H [166,167], for thefollowing reasons. First, we observed that NADPH levels weredecreased and not increased by incubation with Asc, probably dueto NADPH-dependent intracellular ascorbate recycling reactions[120]. Second, we also observed that Steap3 expression wasunaffected by the vitamin [120]. Although Asc may act to stimulateSteap3 activity indirectly, the possibility that other yet-to-be-identified endosomal Asc-dependent ferrireductases contributeto intraendosomal ferrireduction must now be considered.
The finding that Asc stimulates Tf-dependent iron uptake [120] issignificant for the following reasons: (i) the majority of in vitrostudies on Tf-dependent iron uptake have been with performed withAsc-depleted cells and (ii) severe Asc deficiency in humans(i.e., scurvy), as well as experimentally induced Asc deficiency inguinea pigs, causes an “anemia of scurvy” [180,181]. The observationthat Asc enhances iron uptake from Tf in all human cells examinedsuggests that the many previous studies on cellular iron metabolismand Tf-dependent iron uptake may have overlooked the involvementof this important cellular reductant. This is probably due to the factthat most cell culture studies are performed in the absence of Asc.Moreover, the ability of Asc to enhance iron uptake from Tf [120],which is the major donor of iron to the erythropoietic compartment[62], could assist in explaining the metabolic defect that contributesto Asc-deficiency-induced anemia.
Cellular iron storage, efflux, and homeostasis
Ferritins and iron storage
In nonerythroid cells, iron that has been nascently internalizedfrom holo-Tf or NTBI initially enters the poorly characterized labileiron pool, after which the majority (70–80%) is incorporated intoferritin [182,183]. Ferritin is a typically cytoplasmic heteromericprotein complex composed of 24 subunits that form a hollowsphere that can store up to 4500 atoms of iron as a mineralizedferric, phosphate, and hydroxide core [183,184]. In mammals, thereare two ferritin subunits, H-ferritin (heavy; also known as FTH1)
and L-ferritin (light; also known as FTL), which heteropolymerize toform a wide range of iso-ferritins (i.e., comprising different ratios ofFTH1 to FTL) with tissue-specific distributions [184]. The iron(II)entering ferritin is readily oxidized by the intrinsic ferroxidaseactivity of H-ferritin in an oxygen-dependent manner to iron(III),followed by nucleation and mineralization of the iron center byL-ferritin, which is devoid of ferroxidase activity [183,184]. Thisform of storage is vital for cells as it avoids adventitious Fenton- andHaber–Weiss-type redox reactions from occurring.
To be an effective store of iron, intracellular ferritins must also beable to release their iron. The major mechanism of iron release fromferritin under in vivo conditions is autolysosomal proteolysis, althoughproteasomal degradation of the protein can also occur [185–188].Lysosomal degradation of ferritin requires the action of the autophagicapparatus [189]. Interestingly, in vitro studies with isolated ferritinsindicate that reductive mobilization reactions, which can be mediatedby Asc [190,191], are involved in iron release from ferritin. The in vivorelevance of this protein-degradation-independent release of ironfrom ferritin, which occurs through the eight hydrophilic channels inthe ferritin protein [183,192,193], is unclear.
Systemic iron homeostasis: cellular iron efflux, hepcidin,and ferroportin
Systemic iron homeostasis is controlled primarily at the level ofiron efflux by the only known iron exporter, ferroportin 1 (FPN1/SLC40A1), from key cell types into the circulation: (i) dietary ironabsorption by duodenal enterocytes (i.e., via reduced iron effluxinto the portal circulation from duodenal enterocytes), (ii) ironrecycling by splenic macrophages (i.e., via reduced iron efflux intothe plasma after the phagocytic turnover of effete and/or damagederythrocytes), and (iii) iron release from iron stored withinhepatocytes (Fig. 5).
Iron homeostasis: the iron-regulatory protein (IRP)–iron-responsiveelement (IRE) system
Cellular iron homeostasis is predominantly regulated at thelevel of the translation of key iron metabolism proteins involved iniron uptake, storage, and release [194,195]. The IRP–IRE system isresponsible for this regulation [196] and allows for rapid altera-tions in the synthesis of key iron metabolism proteins in responseto intracellular iron levels [36,171]. This system depends on themRNA-binding proteins IRPs 1 and 2 [197,198], which posttran-scriptionally control the expression of proteins whose mRNApossesses an IRE [67,68] (Fig. 6). IRPs bind to IREs in the 50- or30-untranslated regions (UTRs) of key mRNAs involved in ironmetabolism with high affinity in iron-depleted cells, either sup-pressing the translation of the mRNA (i.e., mRNAs in which the IREis located in the 50-UTR, e.g., FTH1 or FTL) or enhancing mRNAstability against nuclease attack (i.e., mRNAs in which the IRE islocated in the 30-UTR, e.g., TfR1 and DMT1-I) [61,62].
The two IRPs are homologous to each other and possess a highdegree of amino acid sequence identity (i.e., 64% in humans[194,196]). IRPs 1 and 2 are members of the aconitase gene familyand are probably derived from gene duplication events. Despitetheir homology, the molecular responses of IRPs 1 and 2 to ironlevels are significantly different. IRP1 is an intriguing bifunctionalprotein that responds to intracellular iron primarily through an ISC“IRP1/aconitase switch” mechanism [199]. Under conditions ofincreased cellular iron, which can be potentiated by Asc [171], IRP1loses its IRE-binding activity by acquiring a cubane ISC (4Fe–4Scluster) [195]. The acquisition of this 4Fe–4S cluster converts IRP1 intoa cytosolic aconitase: an enzyme capable of catalyzing the stereo-specific isomerization of citrate to isocitrate via cis-aconitate [200].
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–83 75

Whereas acquisition of an ISC seems to be the predominant formof iron-mediated regulation of IRP1, iron-regulated proteasomaldegradation is the major regulatory mechanism for IRP2 [194].IRP2, which is unable to acquire an ISC in response to increasedcellular iron, is regulated at the level of protein abundance by theubiquitin–proteasome system [194]. The major mechanism by whichthis occurs is the IRP2-targeting E3 ubiquitin ligase complex contain-ing F-box and leucine-rich repeat protein 5 (FBXL5), which itself isregulated at the level of protein stability by iron and oxygen [201–204]. Rather than being a substrate for iron- and oxygen-dependenthydroxylases, as with other iron- and oxygen-regulated proteins (seebelow), FBXL5 contains a hemerythrin-like domain that directlyincorporates iron in a di-iron center coordinated by histidine andglutamate residues, leading to stabilization (i.e., decreased degrada-tion) of the protein [201–204]. In turn, this leads to increasedassembly of the FBXL5 ubiquitin E3 ligase complex and increasedrates of IRP2 degradation [201–204].
Roles for ascorbate in regulating iron storage, efflux, and the IRP–IREsystem
Ascorbate has been shown to regulate ferritin expression bymultiple mechanisms [36,37,83,120,171,172,205–207]. Incubationof cells with physiological levels of Asc has been shown to inhibitautophagic degradation of ferritin, leading to increased steady-state levels of the protein [172]. Unfortunately, the mechanism ofthis inhibition of autophagy remains unknown, but may relate toAsc-dependent regulation of the multiple intersecting intracellularsignaling cascades that regulate this process.
Ascorbate has also been described to stimulate ferritin synthesis,which appears to be related to an increase in the IRP1/aconitase“switch” mechanism [36] (Fig. 6). Indeed, Asc increases the propor-tion of ISC-containing IRP1 that functions as a cytosolic aconitaseand, consequently, decreases the pool of the mRNA-binding form ofIRP1 [171]. It is important to note that although the combination ofAsc and holo-Tf increases both FTH1 and FTL levels, as well asdecreasing TfR1 levels, this effect does not appear to be responsiblefor the ability of Asc to increase iron uptake from holo-Tf [120]. This
is based on the following observations: (i) blockade of translationdoes not consistently affect Asc-stimulated iron uptake from holo-Tf, and the effect of Asc on ferritin appears to require a source ofiron, suggesting that an increase in ferritin synthesis is downstreamof increased iron uptake; (ii) Asc causes iron-dependent TfR1downregulation, not upregulation, which occurs downstream ofincreased iron uptake; and (iii) blockade of cytosolic aconitaseactivity with the aconitase inhibitor oxalomalate does not affectAsc-stimulated iron uptake from holo-Tf [120].
In addition to decreasing the IRE-binding activity of IRP1, Asc canalso promote the proteasomal degradation of IRP2 [208,209]. Theactivity of prolyl hydroxylases, such as those involved in thehydroxylation of HIFα proteins (see below), has been implicated inthe proteasomal degradation of IRP2 [208,209]. In contrast, a recentreport indicates that physiological levels of Asc cause an increase inIRP2 in Caco-2 cells [206]. The question of whether Asc affects theregulation of IRP2 abundance by modulating the FBXL5-dependentmechanism [201,202] or by an apparently distinct mechanisminvolving the activity of ascorbate-stimulated iron-dependent prolylhydroxylases [208], remains to be determined (see below). Interest-ingly, the regulation of IRP2 degradation by ascorbate could berelated to the well-described antioxidant activity of ascorbate[6,7,30]. It is known that IRP2 can be stabilized in the presence ofiron by pro-oxidants such as H2O2, and this can be antagonized byantioxidants such as ascorbate, α-tocopherol, and N-acetylcysteine[208,209]. These findings do not discriminate between the ascorbate-mediated redox control of the di-iron center in FBXL5 [201,202], theapparent prolyl-hydroxylase-dependent mechanism [208], or analternative cysteine-dependent S-nitrosylation-regulated pathwayfor IRP2 degradation [210].
In summary, by modulating IRP1 and/or IRP2, cellular Ascstatus can alter the expression of key IRP–IRE-regulated proteinsand is consequently a likely modulator of cellular iron home-ostasis. Further studies to explore this hypothesis and its ramifica-tions for mammalian iron metabolism are essential.
Ascorbate may also be able to modulate FPN1 [206] and/orcellular iron efflux activity [206,211]. We have shown that Asc candecrease iron release from various cell types [120,211], although
Fig. 5. Systemic iron homeostasis and the major cell types involved. Systemic iron homeostasis is primarily regulated by the hepcidin–ferroportin (FPN1) axis. The major celltypes known to be involved in regulating/consuming iron at the systemic level are shown. Duodenal enterocytes import dietary iron from several sources including nonhemeiron, which typically must be reduced at the level of the apical membrane by chemical reductants such as Asc or by plasma membrane oxidoreductases (e.g., Dcytb). It is thensubsequently imported by ferrous iron transporters such as divalent metal transporter 1 (DMT1). Both DMT1 and Dcytb can be regulated by a recently described IRP1–HIF2αaxis of control. Nascently imported iron then enters a common intracellular pool of iron within the enterocyte. (A) Under conditions of low iron, this iron can be readilyreleased into the circulation by the iron efflux protein, FPN1, that is localized to the basolateral membrane. This release is coupled to reoxidation of iron by the membrane-bound ferroxidase, hephaestin (HEPH), and iron loading of Tf. Additionally, the consumption of senescent/damaged erythrocytes by specialized macrophages (e.g., splenicmacrophages) leads to the release of iron within the macrophage followed by cellular efflux by FPN1 coupled to a plasma-membrane-bound variant of the ferroxidaseceruloplasmin (CP). Oxidized iron is then bound by circulating Tf to form holo-Tf. The Tf-bound iron is the major source of iron for virtually all cells in the body and isprimarily consumed by erythroid progenitors during erythropoiesis. (B) Under conditions of iron overload, hepatocytes “sense” the level of Tf saturation and levels of ironstores and consequently upregulate hepcidin expression. Hepcidin is then released into the plasma where it can then bind to FPN1 (e.g., at the level of duodenal enterocytes,splenic macrophages, and hepatocytes), thereby triggering its internalization and degradation. Adapted from Lawen and Lane, [61].
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–8376

whether this iron efflux is FPN1-dependent is unclear. Consideringthis, it is notable that physiological Asc levels caused an increase inFPN1 expression in intestinal epithelial-like human Caco-2 cells,which was associated with an increase in IRP2 and HIF2α [206].Although the mechanism was not identified in that study, theincrease in FPN1 may be due to the Asc-dependent IRP1/aconitaseswitch mechanism, despite the observed increase in IRP2 levels.Further studies on the regulation of FPN1 and iron efflux activityby Asc are clearly needed.
Iron homeostasis: the HIF system
Cellular iron homeostasis is also controlled at the level of thetranscription of iron metabolism genes. A major regulator of thesechanges in transcription is the HIF system, which includes the oxygen-and iron-regulated proteins HIF1α and HIF2α [212,213] (Fig. 7).Low oxygen tensions (i.e., hypoxia), as well as low intracellular iron
concentrations, activate HIF1- and HIF2-regulated transcription by theincreased formation of heterodimers of HIF1α or HIF2α and theconstitutively expressed HIF1β subunit (also known as the arylhydrocarbon receptor nuclear translocator) [212]. HIF1α is ubiqui-tously expressed, whereas HIF2α has a more restricted tissue distribu-tion [214]. The HIFα/β heterodimers form transcription factors thatregulate a wide range of genes encoding proteins that are importantfor cellular oxygen homeostasis and the response to hypoxia [61,213].
Both HIF1α and HIF2α are posttranslationally regulated atthe level of protein degradation in an oxygen-dependent andiron-dependent manner [212]. This occurs by a specific class of2-oxoglutarate-dependent dioxygenases: the prolyl-4-hydroxylasedomain-containing iron-dependent prolyl hydroxylases (PHDs) 1–3and the asparaginyl hydroxylase, “factor inhibiting HIF” (FIH) [213].The PHD-type hydroxylases are fully active under conditions of 21%oxygen, found under standard cell culture conditions (see below),and iron repletion and they hydroxylate HIFα proteins at specificproline residues [215,216]. Importantly, it is the strict dependence ofthese hydroxylases on iron that is presumed to be largely respon-sible for the ability of cellular iron levels to modulate HIF-regulatedgene expression [213,217,218]. Hence, low levels of cellular iron willlead to less iron for PHD metallation, reducing PHD activity and
Fig. 6. Ascorbate and the iron-regulatory protein (IRP)–iron-responsive element(IRE) system: posttranscriptional control of iron homeostasis. The IRP–IRE system is amajor regulator of iron metabolism at the cellular level. It is also the most rapidresponder to perturbations in intracellular iron levels. This system provides a meansof regulating the expression of proteins involved in iron storage (H-ferritin (FTH1)and L-ferritin (FTL)), iron export (FPN1), iron uptake (TfR1, DMT1, MRCKα), themitochondrial citric acid cycle (ACO2), mitochondrial hemoglobinization (eALAS),oxygen sensing (HIF2α), and cell cycle control (CDC14A). Under conditions of lowiron, the IRPs are in their IRE-binding forms. Under conditions of high iron, the IRE-binding activity of the IRPs is decreased. IRP1 acquires an iron–sulfur cluster (ISC),which converts the protein into a cytosolic aconitase that is incapable of bindingIREs, whereas IRP2 is targeted for degradation by the proteasome. Ascorbate canpotentiate the loss of IRE-binding activity by promoting IRP1/cytosolic aconitaseswitching and IRP2 degradation. Under conditions of high iron, the binding of IRP1 orIRP2 to cis-regulatory motifs known as IREs in the 50-UTR of select mRNAs (i.e., thoseencoding FTH1, FTL, eALAS, FPN1, HIF2α, and ACO2) serves to inhibit translation (↓)of the mRNA by preventing ribosomal docking (translational control), whereas thebinding of IRPs to IREs in the 30-UTRs of select mRNAs (i.e., those encoding TfR1,DMT1, MRCKα, and CDC14A) protects the transcripts against nuclease-mediateddegradation (mRNA stability control), leading to increased protein expression (↑). Inthe absence of IRPs binding to IREs, the translation of mRNAs possessing 50 IREs canproceed (↑),whereas mRNAs possessing 30 IREs are degraded, leading to decreasedprotein expression (↓). In both cases, the IRE–IRP system effectively modulates therate at which IRE-containing mRNAs are translated into their corresponding proteins.Adapted from Lawen and Lane, [61].
Fig. 7. Ascorbate and the HIF system: transcriptional control of iron homeostasis.Cellular iron metabolism is also regulated at the transcriptional level. A majortranscriptional control mechanism involved in the regulation of certain genesinvolved in iron metabolism is the hypoxia-inducible factor (HIF) system. Thissystem is the primary homeostatic responder to changes in oxygen (O2) tensionand is also sensitive to changes in intracellular iron and Asc levels. Underconditions of high iron, O2, and Asc concentrations, HIF1α and HIF2α are hydro-xylated at specific proline residues by a class of prolyl hydroxylase domain proteins(PHDs 1–3) whose activities vary directly with the intracellular concentrations ofO2, iron, and Asc. Prolyl hydroxylation targets HIF1/2α proteins for ubiquitinationby the E3 ubiquitin ligase, von Hippel-Lindau protein (VHL), which subsequentlymarks them for proteasomal degradation. Under conditions of low iron, O2, and Ascconcentrations, HIF1/2α proteins are stabilized and form heterodimers with theconstitutively expressed HIFβ protein. These heterodimers then translocate to thenucleus and activate the transcription of specific genes that contain hypoxia-response elements (HREs; e.g., genes encoding Tf (TF), TfR1 (TFRC), DMT1(SLC11A2), FPN1 (SLC40A1), Cp (CP), Dcytb (CYBRD1), HO1 (HMOX1), and EPO(EPO)). Additionally, high iron, O2, and Asc levels increase hydroxylation activity ofthe asparaginyl hydroxylase known as factor inhibiting HIF1 (FIH-1). Notably, FIHinhibits the transcriptional activity of the HIF complex by inhibiting the interactionof the C-terminal transactivation domain of HIF1α with the transcriptionalcoactivator CBP/p300. Adapted from Lawen and Lane, [61].
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–83 77

limiting the hydroxylation of HIF1α, which subsequently increasesHIF1α protein levels and its transcriptional activity. Indeed, suchiron limitation would be expected to limit the activity of other 2-oxoglutarate oxidases in an analogous manner. The hydroxylated αsubunits of HIF are then targeted for ubiquitination by the E3ubiquitin ligase von Hippel-Lindau tumor suppressor protein, whichdirects the proteins to be degraded by the proteasome [212,217].
Roles for ascorbate in regulating the HIF system
In addition to its effects on the IRP–IRE system, Asc probably alsoregulates the expression of iron metabolism proteins by affecting HIFsignaling [219,220]. In fact, Asc is known to inhibit HIF1- and 2-dependent gene expression [220] by downregulating the 1 or 2αsubunit of HIF [219–222]. This seems to occur because Asc is requiredas a cofactor for the full activity of the PHDs that hydroxylate [215]and target HIF1α for proteasomal degradation [220,223] (Fig. 7).
In addition, it has recently been shown that in addition topromoting HIF1α degradation, Asc also prevented HIF1α-dependenttranscriptional activity, even under conditions that allowed for HIF1αstabilization [221]. Intriguingly, both effects appeared to be mediatedby Asc's effects on the 2-oxoglutarate- and iron-dependent dioxy-genases, including PHDs 1–3 and the asparaginyl hydroxylase FIH.Importantly, ascorbate acts as a cofactor for these hydroxylases bymaintaining the catalytic iron center in its functional ferrous state[215]. As such, Asc promotes HIF1α prolyl hydroxylation, leading todegradation by the proteasome.
Ascorbate also promotes asparginyl hydroxylation, resulting indecreased binding of the transcriptional coactivator p300 andinactivation of HIF1α-dependent transcriptional activity, respec-tively [221]. These findings strongly suggest that cellular Ascdeficiency is a likely enhancer of the HIF-dependent transcrip-tional response that is known to be activated under conditions ofiron deficiency, hypoxia, metabolic disturbance, and oxidativestress [224].
Interestingly, although Asc is required in vitro for maximalhydroxylation activity of the PHDs, a recent study using Asc-depleted Gulo�/� mice showed normal HIF-dependent gene expres-sion and normal hypoxic HIF1α induction [225]. Although thesefindings suggest that Asc may be dispensable in vivo for HIFα-dependent oxygen (and perhaps iron) sensing, they should beinterpreted with caution. This is because Asc-depleted Gulo�/� miceare likely to retain intracellular Asc levels [44,220], which may besufficient to stimulate hydroxylase activity. The role of Asc inregulating the HIF system and its involvement in intracellular ironsensing and iron homeostasis require further investigation.
Some important caveats: cells in culture versus cells in vivo
It is important to note that although the vast majority of cellculture studies are performed in incubators with an oxygentension equivalent to that of atmospheric oxygen levels (21%), thisis much greater than the oxygen levels experienced by most cellsin most adult tissues in vivo (e.g., �3–5% O2) [226]. Under therelatively “hyperoxic” conditions that cultured cells typicallyexperience, there are marked differences in iron metabolism thatare relevant to the role of ascorbate in regulating these processes.
First, the IRPs, which posttranscriptionally regulate expression ofvarious proteins, including TfR1 and ferritin, are differentiallyregulated by tissue oxygen levels in vivo [227]. IRP2� /� micedemonstrate global dysregulation of iron metabolism and developneurodegeneration, whereas IRP1� /� mice are largely spared [228].This has led to the conclusion that IRP2 predominates over IRP1 inthe posttranscriptional regulation of iron metabolism. However,whereas IRP2� /� cells demonstrate severely dysregulated ironmetabolism at mildly hypoxic in vivo oxygen concentrations, no
apparent dysregulation is observed at 21% oxygen [228]. Intrigu-ingly, under the latter condition, the latent RNA-binding activity ofthe relatively large IRP1 pool [229] in cells is activated, allowing it tosubstitute for the loss of IRP2 [228]. Thus, although IRP2 is thedominant IRP involved in regulation of cellular iron homeostasisunder in vivo oxygen concentrations, in contrast, IRP1 becomesmore important under hyperoxic conditions (i.e., 21% oxygen) and isbetter able to respond to changes in cellular iron status [228].
This oxygen-dependent behavior of the IRPs may be explained, atleast partially, as follows. The cubane ISC of IRP1, which converts theprotein from an RNA-binding protein to a cytosolic aconitase, isoxidation-sensitive and susceptible to degradation by oxidantsincluding oxygen, superoxide, hydrogen peroxide, nitric oxide, andperoxynitrite [230–234]. Thus, such conditions may lead to anincrease in the IRE-binding pool of IRP1. With respect to the role ofascorbate in modulating cellular iron metabolism, ascorbate may beexpected to play a more pronounced role in enhancing the IRP1/aconitase switch mechanism described above, owing to the increasedlevels of the RNA-binding form of the protein, under the higheroxygen levels found under standard cell culture conditions [171].
In the case of IRP2, the high oxygen concentrations experiencedunder typical cell culture conditions are thought to stabilize thedi-iron center in the hemerythrin domain of the FBXL5 andthereby downregulate IRP2 levels by the activity of the FBXL5-containing ubiquitin E3 ligase complex [201–204]. In contrast, thedegradation of IRP2 in response to iron proceeds efficiently undermildly hypoxic conditions typical of the in vivo environment (viz.3% oxygen), but this is compromised under conditions of severehypoxia (viz. 0.1% oxygen) [209]. Additionally, IRP2 is stabilized byexposure to H2O2 [209]. Under such highly oxidizing conditions,H2O2-dependent oxidation of Fe(II) to Fe(III) could occur, decreas-ing the formation of the di-iron center of FBXL5 [209]. Intriguingly,the protective effect of H2O2 on IRP2 protein stability can beabolished by ascorbate [209], which is consistent with earlierstudies demonstrating that ascorbate promotes IRP2 degradation[208] (see above). Taken together, these results may suggest thatunder 21% oxygen, in which levels of oxidants such as H2O2 aretypically higher, ascorbate may play a proportionally greater rolein promoting protection and/or formation of the di-iron center ofFBXL5, leading to increased levels of FBXL5 and subsequentdownregulation of IRP2. Clearly, further studies are needed toelucidate the various possibilities under different oxygen levels.
Second, HIF-dependent regulation of iron metabolism is alsoknown to be different in vivo compared to that found in cell culture[219–221]. This differential regulation could be due to the effectsthat hyperoxia has on PHD activity [215]. That is, PHD activity hasbeen shown to be increased under such conditions, leading to farlower HIF1 levels than those typically observed under in vivooxygen levels [219–221]. Considering the possible difference ineffects of ascorbate on iron metabolism under 21% oxygen andmore hypoxic conditions, it may be expected that the effect ofascorbate on downregulating HIF in a PHD-dependent mannerwould be more pronounced at lower oxygen concentrations, whichare typical of the in vivo situation. Indeed, this is precisely what isobserved in experimental studies [219–221].
Such potential differences in the role of ascorbate in regulatingiron metabolism under hyperoxic cell culture conditions andmildly hypoxic in vivo conditions should be taken into considera-tion in further studies in this area.
Conclusion
In this review, we have explored the emerging hypothesis that Asccontributes to cellular physiology, in part by functioning as a mod-ulator of cellular iron metabolism. Indeed, accumulating evidence
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–8378

strongly suggests that in addition to the known ability of dietary Ascto enhance nonheme iron absorption in the gut, Asc can regulatecellular iron uptake and downstream cellular metabolism. Vitamin Cregulates iron metabolism by increasing Tf-dependent and non-Tf ironuptake, the latter of which occurs by a novel transplasma membraneascorbate-cycling mechanism, stimulating ferritin synthesis, inhibitinglysosomal ferritin degradation via autophagy, and decreasing cellulariron efflux.
Recent evidence indicates that Asc stimulates iron uptake bythe classical Tf–iron uptake pathway, which provides almost alliron for cellular demands and erythropoiesis under physiologicalconditions. Ascorbate acts to enhance Tf-dependent iron uptake byan intracellular reductive mechanism, strongly suggesting that itmay act to stimulate iron mobilization from the endosome into thecell. The ability of Asc to regulate Tf–iron uptake could helpexplain the metabolic defect that contributes to Asc-deficiency-induced anemia.
Additionally, Asc appears to be capable of regulating both theIRP–IRE and the HIF systems, which are integrally involved incellular and systemic iron homeostasis, although some crucialmechanistic details remain the subject of controversy. However,there is little doubt that Asc plays an active role in regulating theseprocesses, and the case for the multifaceted involvement ofascorbate in iron metabolism is strong. As such, future studiesshould aim to directly assess the mechanisms of involvement ofAsc in all tiers of mammalian iron homeostasis.
Acknowledgments
D.J.R.L. thanks the Cancer Institute New South Wales for anEarly Career Fellowship (10/ECF/2-18) and the National Health andMedical Research Council (NHMRC) of Australia for an Early CareerPostdoctoral Fellowship (1013810). D.R.R. thanks the NHMRC for aSenior Principal Research Fellowship and project grants.
References
[1] Padayatty, S. J.; Levine, M. New insights into the physiology and pharmacol-ogy of vitamin C. Can. Med. Assoc. J. 164:353–355; 2001.
[2] May, J. M. Vitamin C transport and its role in the central nervous system.Subcell. Biochem. 56:85–103; 2012.
[3] Aguirre, R.; May, J. M. Inflammation in the vascular bed: importance ofvitamin C. Pharmacol. Ther. 119:96–103; 2008.
[4] Harrison, F. E.; May, J. M. Vitamin C function in the brain: vital role of theascorbate transporter SVCT2. Free Radic. Biol. Med. 46:719–730; 2009.
[5] Asard, H. Ascorbate. In: Banerjee, R., Becker, D. F., Dickman, M. B., Gladyshev,V. N., Ragsdale, S. W., editors. Redox Biochemistry. Hoboken: Wiley; 2007.p. 22–37.
[6] Buettner, G. R. The pecking order of free radicals and antioxidants: lipidperoxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 300:535–-543; 1993.
[7] Buettner, G. R.; Jurkiewicz, B. A. Catalytic metals, ascorbate and free radicals:combinations to avoid. Radiat. Res. 145:532–541; 1996.
[8] May, J. M. Is ascorbic acid an antioxidant for the plasma membrane? FASEB J.13:995–1006; 1999.
[9] Li, X.; Huang, J.; May, J. M. Ascorbic acid spares α-tocopherol and decreaseslipid peroxidation in neuronal cells. Biochem. Biophys. Res. Commun.305:656–661; 2003.
[10] Huang, J.; May, J. M. Ascorbic acid spares α-tocopherol and prevents lipidperoxidation in cultured H4IIE liver cells. Mol. Cell. Biochem. 247:171–176;2003.
[11] Huang, A.; Vita, J. A.; Venema, R. C.; Keaney Jr. J. F. Ascorbic acid enhancesendothelial nitric-oxide synthase activity by increasing intracellular tetra-hydrobiopterin. J. Biol. Chem. 275:17399–17406; 2000.
[12] Heller, R.; Unbehaun, A.; Schellenberg, B.; Mayer, B.; Werner-Felmayer, G.;Werner, E. R. L-ascorbic acid potentiates endothelial nitric oxide synthesisvia a chemical stabilization of tetrahydrobiopterin. J. Biol. Chem. 276:40–47;2001.
[13] May, J. M.; Qu, Z. -c.; Mendiratta, S. Protection and recycling of α-tocopherolin human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys.349:281–289; 1998.
[14] Challem, J. J.; Taylor, E. W. Retroviruses, ascorbate, and mutations, in theevolution of Homo sapiens. Free Radic. Biol. Med. 25:130–132; 1998.
[15] Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning andchromosomal mapping of the human nonfunctional gene for L-gulono-γ-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man.J. Biol. Chem. 269:13685–13688; 1994.
[16] Nishikimi, M.; Yagi, K. Molecular basis for the deficiency in humans ofgulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am. J.Clin. Nutr. 54:1203S–1208S; 1991.
[17] Linster, C. L.; Van Schaftingen, E. Vitamin C: biosynthesis, recycling anddegradation in mammals. FEBS J. 274:1–22; 2007.
[18] Chatterjee, I. B.; Majumder, A. K.; Nandi, B. K.; Subramanian, N. Synthesis andsome major functions of vitamin C in animals. Ann. N. Y. Acad. Sci. 258:24–47;1975.
[19] Michels, A. J.; Hagen, T. M.; Frei, B. Human genetic variation influencesvitamin C homeostasis by altering vitamin C transport and antioxidantenzyme function. Annu. Rev. Nutr. 33:45–70; 2013.
[20] Rumsey, S. C.; Levine, M. Absorption, transport and disposition of ascorbicacid in humans. J. Nutr. Biochem. 9:116–130; 1998.
[21] May, J. M.; Qu, Z. -c.; Qiao, H.; Koury, M. J. Maturational loss of the vitamin Ctransporter in erythrocytes. Biochem. Biophys. Res. Commun. 360:295–298;2007.
[22] Wilson, J. X. Regulation of vitamin C transport. Annu. Rev. Nutr. 25:105–125;2005.
[23] Michels, A. J.; Frei, B. Myths, artifacts, and fatal flaws: identifying limitationsand opportunities in vitamin C research. Nutrients 5:5161–5192; 2013.
[24] Li, X.; Cobb, C. E.; Hill, K. E.; Burk, R. F.; May, J. M. Mitochondrial uptake andrecycling of ascorbic acid. Arch. Biochem. Biophys. 387:143–153; 2001.
[25] May, J. M.; Li, L.; Qu, Z. -c.; Cobb, C. E. Mitochondrial recycling of ascorbicacid as a mechanism for regenerating cellular ascorbate. Biofactors 30:35–48;2007.
[26] Li, X.; Cobb, C. E.; May, J. M. Mitochondrial recycling of ascorbic acid fromdehydroascorbic acid: dependence on the electron transport chain. Arch.Biochem. Biophys. 403:103–110; 2002.
[27] Szarka, A.; Horemans, N.; Kovács, Z.; Gróf, P.; Mayer, M.; Bánhegyi, G.Dehydroascorbate reduction in plant mitochondria is coupled to the respira-tory electron transfer chain. Physiol. Plant. 129:225–232; 2007.
[28] Xu, D. P.; Wells, W. W. α-Lipoic acid dependent regeneration of ascorbic acidfrom dehydroascorbic acid in rat liver mitochondria. J. Bioenerg. Biomembr.28:77–85; 1996.
[29] Sagun, K. C.; Carcámo, J. M.; Golde, D. W. Vitamin C enters mitochondria viafacilitative glucose transporter 1 (Glut1) and confers mitochondrial protec-tion against oxidative injury. FASEB J. 19:1657–1667; 2005.
[30] Du, J.; Cullen, J. J.; Buettner, G. R. Ascorbic acid: chemistry, biology and thetreatment of cancer. Biochim. Biophys. Acta 1826:443–457; 2012.
[31] Jung, C. -H.; Wells, W. W. Spontaneous conversion of L-dehydroascorbic acidto L-ascorbic acid and L-erythroascorbic acid. Arch. Biochem. Biophys.355:9–14; 1998.
[32] Koshiishi, I.; Mamura, Y.; Imanari, T. Bicarbonate promotes a cleavage oflactone ring of dehydroascorbate. Biochim. Biophys. Acta 1379:257–263;1998.
[33] Koshiishi, I.; Mamura, Y.; Liu, J.; Imanari, T. Degradation of dehydroascorbateto 2,3-diketogulonate in blood circulation. Biochim. Biophys. Acta 1425:209–214; 1998.
[34] May, J. M.; Qu, Z. -c.; Whitesell, R. R. Ascorbic acid recycling enhances theantioxidant reserve of human erythrocytes. Biochemistry 34:12721–12728;1995.
[35] May, J. M.; Qu, Z. -c.; Cobb, C. E. Human erythrocyte recycling of ascorbicacid: relative contributions from the ascorbate free radical and dehydroas-corbic acid. J. Biol. Chem. 279:14975–14982; 2004.
[36] Toth, I.; Rogers, J. T.; McPhee, J. A.; Elliott, S. M.; Abramson, S. L.; Bridges, K. R.Ascorbic acid enhances iron-induced ferritin translation in human leukemiaand hepatoma cells. J. Biol. Chem. 270:2846–2852; 1995.
[37] Hoffman, K. E.; Yanelli, K.; Bridges, K. R. Ascorbic acid and iron metabolism:alterations in lysosomal function. Am. J. Clin. Nutr. 54:1188S–1192S; 1991.
[38] Levine, M.; Wang, Y.; Padayatty, S. J.; Morrow, J. A new recommended dietaryallowance of vitamin C for healthy young women. Proc. Natl. Acad. Sci. USA98:9842–9846; 2001.
[39] Rice, M. E.; Russo-Menna, I. Differential compartmentalization of brainascorbate and glutathione between neurons and glia. Neuroscience82:1213–1223; 1998.
[40] Rice, M. E. Ascorbate regulation and its neuroprotective role in the brain.Trends Neurosci. 23:209–216; 2000.
[41] Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M. V. SVCT1 and SVCT2:key proteins for vitamin C uptake. Amino Acids 34:347–355; 2007.
[42] May, J. M. The SLC23 family of ascorbate transporters: ensuring that you getand keep your daily dose of vitamin C. Br. J. Pharmacol. 164:1793–1801; 2011.
[43] Burzle, M.; Suzuki, Y.; Ackermann, D.; Miyazaki, H.; Maeda, N.; Clemencon,B.; Burrier, R.; Hediger, M. A. The sodium-dependent ascorbic acid trans-porter family SLC23. Mol. Aspects Med. 34:436–454; 2013.
[44] Maeda, N.; Hagihara, H.; Nakata, Y.; Hiller, S.; Wilder, J.; Reddick, R. Aorticwall damage in mice unable to synthesize ascorbic acid. Proc. Natl. Acad. Sci.USA 97:841–846; 2000.
[45] Amano, A.; Aigaki, T.; Maruyama, N.; Ishigami, A. Ascorbic acid depletionenhances expression of the sodium-dependent vitamin C transporters,SVCT1 and SVCT2, and uptake of ascorbic acid in livers of SMP30/GNLknockout mice. Arch. Biochem. Biophys. 496:38–44; 2010.
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–83 79

[46] Meredith, M. E.; Harrison, F. E.; May, J. M. Differential regulation of theascorbic acid transporter SVCT2 during development and in response toascorbic acid depletion. Biochem. Biophys. Res. Commun. 414:737–742; 2011.
[47] Portugal, C. C.; da Encarnacao, T. G.; Socodato, R.; Moreira, S. R.; Brudzewsky,D.; Ambrosio, A. F.; Paes-de-Carvalho, R. Nitric oxide modulates sodiumvitamin C transporter 2 (SVCT-2) protein expression via protein kinase G(PKG) and nuclear factor-kappaB (NF-kappaB). J. Biol. Chem. 287:3860–3872;2012.
[48] May, J. M.; Li, L.; Qu, Z. C. Oxidized LDL up-regulates the ascorbic acidtransporter SVCT2 in endothelial cells. Mol. Cell. Biochem. 343:217–222; 2010.
[49] Astuya, A.; Caprile, T.; Castro, M.; Salazar, K.; García, M. d.1. A; Reinicke, K.;Rodríguez, F.; Vera, J. C.; Millán, C.; Ulloa, V.; Low, M.; Martínez, F.; Nualart, F.Vitamin C uptake and recycling among normal and tumor cells from thecentral nervous system. J. Neurosci. Res. 79:146–156; 2005.
[50] Guaiquil, V. H.; Farber, C. M.; Golde, D. W.; Vera, J. C. Efficient transport andaccumulation of vitamin C in HL-60 cells depleted of glutathione. J. Biol.Chem. 272:9915–9921; 1997.
[51] Nualart, F. J.; Rivas, C. I.; Montecinos, V. P.; Godoy, A. S.; Guaiquil, V. H.; Golde,D. W.; Vera, J. C. Recycling of vitamin C by a bystander effect. J. Biol. Chem.278:10128–10133; 2003.
[52] Vera, J. C.; Rivas, C. I.; Zhang, R. H.; Farber, C. M.; Golde, D. W. Human HL-60myeloid leukemia cells transport dehydroascorbic acid via the glucosetransporters and accumulate reduced ascorbic acid. Blood 84:1628–1634;1994.
[53] Himmelreich, U.; Drew, K. N.; Serianni, A. S.; Kuchel, P. W. 13C NMR studies ofvitamin C transport and its redox cycling in human erythrocytes. Biochem-istry 37:7578–7588; 1998.
[54] Mendiratta, S.; Qu, Z. -c.; May, J. M. Erythrocyte ascorbate recycling:antioxidant effects in blood. Free Radic. Biol. Med. 24:789–797; 1998.
[55] Montel-Hagen, A.; Kinet, S.; Manel, N.; Mongellaz, C.; Prohaska, R.;Battini, J. -L.; Delaunay, J.; Sitbon, M.; Taylor, N. Erythrocyte Glut1 triggersdehydroascorbic acid uptake in mammals unable to synthesize vitamin C.Cell 132:1039–1048; 2008.
[56] Troadec, M. -B.; Kaplan, J. Some vertebrates go with the GLO. Cell 132:921–-922; 2008.
[57] Carruthers, A.; Naftalin, R. J. Altered GLUT1 substrate selectivity in humanerythropoiesis? Cell 137:200–201; 2009.
[58] Montel-Hagen, A.; Kinet, S.; Manel, N.; Mongellaz, C.; Prohaska, R.; Battini, J.-L.; Delaunay, J.; Sitbon, M.; Taylor, N. Species diversity in GLUT expressionand function. Cell 137:201–202; 2009.
[59] Montel-Hagen, A.; Sitbon, M.; Taylor, N. Erythroid glucose transporters. Curr.Opin. Hematol. 16:165–172; 2009.
[60] Rumsey, S. C.; Kwon, O.; Xu, G. W.; Burant, C. F.; Simpson, I.; Levine, M.Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbicacid. J. Biol. Chem. 272:18982–18989; 1997.
[61] Lawen, A.; Lane, D. J. R. Mammalian iron homeostasis in health and disease:uptake, storage, transport, and molecular mechanisms of action. Antioxid.Redox Signaling 18:2473–2507; 2013.
[62] Richardson, D. R.; Lane, D. J. R.; Becker, E. M.; Huang, M. L.; Whitnall, M.;Rahmanto, Y. S.; Sheftel, A. D.; Ponka, P. Mitochondrial iron trafficking andthe integration of iron metabolism between the mitochondrion and cytosol.Proc. Natl. Acad. Sci. USA 107:10775–10782; 2010.
[63] Zhang, A. S.; Enns, C. A. Iron homeostasis: recently identified proteinsprovide insight into novel control mechanisms. J. Biol. Chem. 284:711–715;2009.
[64] Andrews, S. C. The ferritin-like superfamily: evolution of the biological ironstoreman from a rubrerythrin-like ancestor. Biochim. Biophys. Acta1800:691–705; 2010.
[65] Fenton, H. J. H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc.Trans. 65:899–910; 1894.
[66] Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide by ironsalts. Proc. R. Soc. Ser. A 147:332–351; 1934.
[67] Hentze, M. W.; Kuhn, L. C. Molecular control of vertebrate iron metabolism:mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidativestress. Proc. Natl. Acad. Sci. USA 93:8175–8182; 1996.
[68] Richardson, D. R.; Ponka, P. The molecular mechanisms of the metabolismand transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta1331:1–40; 1997.
[69] Hershko, C.; Graham, G.; Bates, G. W.; Rachmilewitz, E. A. Non-specific serumiron in thalassaemia: an abnormal serum iron fraction of potential toxicity.Br. J. Haematol. 40:255–263; 1978.
[70] Breuer, W.; Hershko, C.; Cabantchik, Z. I. The importance of non-transferrinbound iron in disorders of iron metabolism. Transfus. Sci. 23:185–192; 2000.
[71] Grootveld, M.; Bell, J. D.; Halliwell, B.; Aruoma, O. I.; Bomford, A.; Sadler, P. J.Non-transferrin-bound iron in plasma or serum from patients with idio-pathic hemochromatosis: characterization by high performance liquid chro-matography and nuclear magnetic resonance spectroscopy. J. Biol. Chem.264:4417–4422; 1989.
[72] Anderson, G. J. Ironing out disease: inherited disorders of iron homeostasis.IUBMB Life 51:11–17; 2001.
[73] De Silva, D. M.; Askwith, C. C.; Kaplan, J. Molecular mechanisms of ironuptake in eukaryotes. Physiol. Rev. 76:31–47; 1996.
[74] Inman, R. S.; Wessling-Resnick, M. Characterization of transferrin-independent iron transport in K562 cells: unique properties provide evi-dence for multiple pathways of iron uptake. J. Biol. Chem. 268:8521–8528;1993.
[75] Jordan, I.; Kaplan, J. The mammalian transferrin-independent iron transportsystem may involve a surface ferrireductase activity. Biochem. J. 302:875–879; 1994.
[76] Lane, D. J. R.; Lawen, A. Non-transferrin iron reduction and uptake areregulated by transmembrane ascorbate cycling in K562 cells. J. Biol. Chem.283:12701–12708; 2008.
[77] Lane, D. J. R.; Lawen, A. Transplasma membrane electron transport comes intwo flavors. Biofactors 34:191–200; 2009.
[78] Lane, D. J. R.; Lawen, A. Ascorbate and plasma membrane electron transport—enzymes vs efflux. Free Radic. Biol. Med. 47:485–495; 2009.
[79] McKie, A. T.; Barrow, D.; Latunde-Dada, G. O.; Rolfs, A.; Sager, G.; Mudaly, E.;Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; Peters, T. J.; Raja, K. B.;Shirali, S.; Hediger, M. A.; Farzaneh, F.; Simpson, R. J. An iron-regulated ferricreductase associated with the absorption of dietary iron. Science 291:1755–1759; 2001.
[80] Gunshin, H.; Starr, C. N.; Direnzo, C.; Fleming, M. D.; Jin, J.; Greer, E. L.;Sellers, V. M.; Galica, S. M.; Andrews, N. C. Cybrd1 (duodenal cytochrome b)is not necessary for dietary iron absorption in mice. Blood 106:2879–2883;2005.
[81] Jacolot, S.; Ferec, C.; Mura, C. Iron responses in hepatic, intestinal andmacrophage/monocyte cell lines under different culture conditions. BloodCells Mol. Dis. 41:100–108; 2008.
[82] Latunde-Dada, G. O.; Van der Westhuizen, J.; Vulpe, C. D.; Anderson, G. J.;Simpson, R. J.; McKie, A. T. Molecular and functional roles of duodenalcytochrome b (Dcytb) in iron metabolism. Blood Cells Mol. Dis. 29:356–360;2002.
[83] Scheers, N. M.; Sandberg, A. S. Ascorbic acid uptake affects ferritin, Dcytb andNramp2 expression in Caco-2 cells. Eur. J. Nutr. 47:401–408; 2008.
[84] Shah, Y. M.; Matsubara, T.; Ito, S.; Yim, S. H.; Gonzalez, F. J. Intestinal hypoxia-inducible transcription factors are essential for iron absorption followingiron deficiency. Cell Metab. 9:152–164; 2009.
[85] Latunde-Dada, G. O.; Simpson, R. J.; McKie, A. T. Duodenal cytochrome bexpression stimulates iron uptake by human intestinal epithelial cells. J. Nutr.138:991–995; 2008.
[86] Su, D.; Asard, H. Three mammalian cytochromes b561 are ascorbate-dependent ferrireductases. FEBS J. 273:3722–3734; 2006.
[87] Fleming, P. J.; Kent, U. M. Cytochrome b561, ascorbic acid, and transmem-brane electron transfer. Am. J. Clin. Nutr. 54:1173S–1178S; 1991.
[88] Spiro, M. J.; Ball, E. G. Studies on the respiratory enzymes of the adrenalgland. I. The medulla. J. Biol. Chem. 236:225–230; 1961.
[89] Flatmark, T.; Terland, O. Cytochrome b-561 of the bovine adrenal chromaffingranules: a high potential b-type cytochrome. Biochim. Biophys. Acta253:487–491; 1971.
[90] Terland, O.; Flatmark, T. Oxidoreductase activities of chromaffin granuleghosts isolated from the bovine adrenal medulla. Biochim. Biophys. Acta597:318–330; 1980.
[91] Ichikawa, Y.; Yamano, T. Cytochrome 559 in the microsomes of the adrenalmedulla. Biochem. Biophys. Res. Commun. 20:263–268; 1965.
[92] Njus, D.; Knoth, J.; Cook, C.; Kelly, P. M. Electron transfer across thechromaffin granule membrane. J. Biol. Chem. 258:27–30; 1983.
[93] Kelley, P. M.; Njus, D. Cytochrome b561 spectral changes associated withelectron transfer in chromaffin-vesicle ghosts. J. Biol. Chem. 261:6429–6432;1986.
[94] Wakefield, L. M.; Cass, A. E.; Radda, G. K. Electron transfer across thechromaffin granule membrane: use of EPR to demonstrate reduction ofintravesicular ascorbate radical by the extravesicular mitochondrial NADH:ascorbate radical oxidoreductase. J. Biol. Chem. 261:9746–9752; 1986.
[95] Njus, D.; Kelley, P. M. The secretory-vesicle ascorbate-regenerating system: achain of concerted Hþ/e�-transfer reactions. Biochim. Biophys. Acta1144:235–248; 1993.
[96] Jalukar, V.; Kelley, P. M.; Njus, D. Reaction of ascorbic acid with cytochromeb561: concerted electron and proton transfer. J. Biol. Chem. 266:6878–6882;1991.
[97] Tsubaki, M.; Takeuchi, F.; Nakanishi, N. Cytochrome b561 protein family:expanding roles and versatile transmembrane electron transfer abilities aspredicted by a new classification system and protein sequence motifanalyses. Biochim. Biophys. Acta 1753:174–190; 2005.
[98] Nakanishi, N.; Takeuchi, F.; Tsubaki, M. Histidine cycle mechanism for theconcerted proton/electron transfer from ascorbate to the cytosolic haem bcentre of cytochrome b561: a unique machinery for the biological trans-membrane electron transfer. J. Biochem. 142:553–560; 2007.
[99] Zhang, D. -l.; Su, D.; Bérczi, A.; Vargas, A.; Asard, H. An ascorbate-reduciblecytochrome b561 is localized in macrophage lysosomes. Biochim. Biophys.Acta 1760:1903–1913; 2006.
[100] Vargas, J. D.; Herpers, B.; McKie, A. T.; Gledhill, S.; McDonnell, J.; van denHeuvel, M.; Davies, K. E.; Ponting, C. P. Stromal cell-derived receptor 2 andcytochrome b561 are functional ferric reductases. Biochim. Biophys. Acta1651:116–123; 2003.
[101] Picco, C.; Scholz-Starke, J.; Naso, A.; Preger, V.; Sparla, F.; Trost, P.; Carpaneto,A. How are cytochrome b561 electron currents controlled by membranevoltage and substrate availability? Antioxid. Redox Signaling 21:384–391;2014.
[102] Mizutani, A.; Sanuki, R.; Kakimoto, K.; Kojo, S.; Taketani, S. Involvement of101F6, a homologue of cytochrome b561, in the reduction of ferric ions. J.Biochem. 142:699–705; 2007.
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–8380

[103] Recuenco, M. C.; Rahman, M. M.; Takeuchi, F.; Kobayashi, K.; Tsubaki, M.Electron transfer reactions of candidate tumor suppressor 101F6 protein, acytochrome b561 homologue, with ascorbate and monodehydroascorbateradical. Biochemistry 52:3660–3668; 2013.
[104] Wyman, S.; Simpson, R. J.; McKie, A. T.; Sharp, P. A. Dcytb (Cybrd1) functionsas both a ferric and a cupric reductase in vitro. FEBS Lett. 582:1901–1906;2008.
[105] Turi, J. L.; Wang, X.; McKie, A. T.; Nozik-Grayck, E.; Mamo, L. B.; Crissman, K.;Piantadosi, C. A.; Ghio, A. J. Duodenal cytochrome b: a novel ferrireductase inairway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 291:L272–L280;2006.
[106] Asard, H.; Barbaro, R.; Trost, P.; Berczi, A. Cytochromes b561: ascorbate-mediated trans-membrane electron transport. Antioxid. Redox Signaling19:1026–1035; 2013.
[107] Berczi, A.; Zimanyi, L.; Asard, H. Dihydrolipoic acid reduces cytochrome b561proteins. Eur. Biophys. J. 42:159–168; 2013.
[108] McKie, A. T. The role of Dcytb in iron metabolism: an update. Biochem. Soc.Trans. 36:1239–1241; 2008.
[109] Su, D.; May, J. M.; Koury, M. J.; Asard, H. Human erythrocyte membranescontain a cytochrome b561 that may be involved in extracellular ascorbaterecycling. J. Biol. Chem. 281:39852–39859; 2006.
[110] Kovar, J.; Neubauerova, J.; Cimburova, M.; Truksa, J.; Balusikova, K.; Horak, J.Stimulation of non-transferrin iron uptake by iron deprivation in K562 cells.Blood Cells Mol. Dis. 37:95–99; 2006.
[111] Balusikova, K.; Neubauerova, J.; Dostalikova-Cimburova, M.; Horak, J.; Kovar,J. Differing expression of genes involved in non-transferrin iron transportacross plasma membrane in various cell types under iron deficiency andexcess. Mol. Cell. Biochem. 321:123–133; 2009.
[112] Jeong, S. Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin isrequired for iron efflux from cells in the central nervous system. J. Biol. Chem.278:27144–27148; 2003.
[113] Tulpule, K.; Robinson, S. R.; Bishop, G. M.; Dringen, R. Uptake of ferrous ironby cultured rat astrocytes. J. Neurosci. Res. 88:563–571; 2010.
[114] Atanasova, B.; Mudway, I. S.; Laftah, A. H.; Latunde-Dada, G. O.; McKie, A. T.;Peters, T. J.; Tzatchev, K. N.; Simpson, R. J. Duodenal ascorbate levels arechanged in mice with altered iron metabolism. J. Nutr. 134:501–505; 2004.
[115] Brookes, M. J.; Hughes, S.; Turner, F. E.; Reynolds, G.; Sharma, N.; Ismail, T.;Berx, G.; McKie, A. T.; Hotchin, N.; Anderson, G. J.; Iqbal, T.; Tselepis, C.Modulation of iron transport proteins in human colorectal carcinogenesis.Gut 55:1449–1460; 2006.
[116] Oakhill, J. S.; Marritt, S. J.; Gareta, E. G.; Cammack, R.; McKie, A. T. Functionalcharacterization of human duodenal cytochrome b (Cybrd1): redox proper-ties in relation to iron and ascorbate metabolism. Biochim. Biophys. Acta1777:260–268; 2008.
[117] Ghio, A. J.; Nozik-Grayck, E.; Turi, J.; Jaspers, I.; Mercatante, D. R.; Kole, R.;Piantadosi, C. A. Superoxide-dependent iron uptake: a new role for anionexchange protein 2. Am. J. Respir. Cell Mol. Biol. 29:653–660; 2003.
[118] Jones, W.; Li, X.; Qu, Z. -c.; Perriott, L.; Whitesell, R. R.; May, J. M. Uptake,recycling, and antioxidant actions of α-lipoic acid in endothelial cells. FreeRadic. Biol. Med. 33:83–93; 2002.
[119] Lane, D. J. R.; Robinson, S. R.; Czerwinska, H.; Bishop, G. M.; Lawen, A. Tworoutes of iron accumulation in astrocytes: ascorbate-dependent ferrous ironuptake via the divalent metal transporter (DMT1) plus an independent routefor ferric iron. Biochem. J. 432:123–132; 2010.
[120] Lane, D. J. R.; Chikhani, S.; Richardson, V.; Richardson, D. R. Transferrin ironuptake is stimulated by ascorbate via an intracellular reductive mechanism.Biochim. Biophys. Acta 1833:1527–1541; 2013.
[121] May, J. M.; Qu, Z. -c.; Mendiratta, S. Role of ascorbic acid in transferrin-independent reduction and uptake of iron by U-937 cells. Biochem. Pharma-col. 57:1275–1282; 1999.
[122] Fleming, M. D.; Trenor 3rd C. C.; Su, M. A.; Foernzler, D.; Beier, D. R.; Dietrich,W. F.; Andrews, N. C. Microcytic anaemia mice have a mutation in Nramp2, acandidate iron transporter gene. Nat. Genet. 16:383–386; 1997.
[123] Illing, A. C.; Shawki, A.; Cunningham, C. L.; Mackenzie, B. Substrate profileand metal-ion selectivity of human divalent metal-ion transporter-1. J. Biol.Chem. 287:30485–30496; 2012.
[124] Mackenzie, B.; Takanaga, H.; Hubert, N.; Rolfs, A.; Hediger, M. A. Functionalproperties of multiple isoforms of human divalent metal-ion transporter 1(DMT1). Biochem. J. 403:59–69; 2007.
[125] Mackenzie, B.; Ujwal, M. L.; Chang, M. -H.; Romero, M. F.; Hediger, M. A.Divalent metal-ion transporter DMT1 mediates both Hþ-coupled Fe2þ
transport and uncoupled fluxes. Pflugers Arch. Eur. J. Physiol. 451:544–558;2006.
[126] Corti, A.; Casini, A. F.; Pompella, A. Cellular pathways for transport and effluxof ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 500:107–115;2010.
[127] von Zastrow, M.; Tritton, T. R.; Castle, J. D. Exocrine secretion granulescontain peptide amidation activity. Proc. Natl. Acad. Sci. USA 83:3297–3301;1986.
[128] Ahmad, S.; Evans, W. H. Post-translational integration and oligomerization ofconnexin 26 in plasma membranes and evidence of formation of membranepores: implications for the assembly of gap junctions. Biochem. J. 365:693–699; 2002.
[129] Kónya, C.; Ferdinandy, P. Vitamin C: new role of the old vitamin in thecardiovascular system? Br. J. Pharmacol. 147:125–127; 2006.
[130] Wilson, J. X.; Peters, C. E.; Sitar, S. M.; Daoust, P.; Gelb, A. W. Glutamatestimulates ascorbate transport by astrocytes. Brain Res. 858:61–66; 2000.
[131] May, J. M.; Li, L.; Hayslett, K.; Qu, Z. -c. Ascorbate transport and recycling bySH-SY5Y neuroblastoma cells: response to glutamate toxicity. Neurochem.Res. 31:785–794; 2006.
[132] Fürst, J.; Gschwentner, M.; Ritter, M.; Bottà, G.; Jakab, M.; Mayer, M.;Garavaglia, L.; Bazzini, C.; Rodighiero, S.; Meyer, G.; Eichmüller, S.; Wöll, E.;Paulmichl, M. Molecular and functional aspects of anionic channels activatedduring regulatory volume decrease in mammalian cells. Pflügers Arch.444:1–25; 2002.
[133] Lane, D. J. R.; Lawen, A. The glutamate aspartate transporter (GLAST)mediates L-glutamate-stimulated ascorbate-release via swelling-activatedanion channels in cultured neonatal rodent astrocytes. Cell. Biochem. Biophys.65:107–119; 2012.
[134] Wilson, J. X.; Dragan, M. Sepsis inhibits recycling and glutamate-stimulatedexport of ascorbate by astrocytes. Free Radic. Biol. Med. 39:990–998; 2005.
[135] Sánchez-Olea, R.; Peña, C.; Morán, J.; Pasantes-Morales, H. Inhibition ofvolume regulation and efflux of osmoregulatory amino acids by blockers ofCl� transport in cultured astrocytes. Neurosci. Lett. 156:141–144; 1993.
[136] Siushansian, R.; Dixon, S. J.; Wilson, J. X. Osmotic swelling stimulatesascorbate efflux from cerebral astrocytes. J. Neurochem. 66:1227–1233; 1996.
[137] Upston, J. M.; Karjalainen, A.; Bygrave, F. L.; Stocker, R. Efflux of hepaticascorbate: a potential contributor to the maintenance of plasma vitamin C.Biochem. J. 342:49–56; 1999.
[138] Davis, K. A.; Samson, S. E.; Best, K.; Mallhi, K. K.; Szewczyk, M.; Wilson, J. X.;Kwan, C. -Y.; Grover, A. K. Ca2þ-mediated ascorbate release from coronaryartery endothelial cells. Br. J. Pharmacol. 147:131–139; 2006.
[139] May, J. M.; Qu, Z. -c. Ascorbic acid efflux and re-uptake in endothelial cells:maintenance of intracellular ascorbate. Mol. Cell. Biochem. 325:79–88; 2009.
[140] Nilius, B.; Droogmans, G. Amazing chloride channels: an overview. ActaPhysiol. Scand. 177:119–147; 2003.
[141] Davis, K. A.; Samson, S. E.; Wilson, J. X.; Grover, A. K. Hypotonic shockstimulates ascorbate release from coronary artery endothelial cells by aCa2þ-independent pathway. Eur. J. Pharmacol. 548:36–44; 2006.
[142] De Pinto, V.; Messina, A.; Lane, D. J. R.; Lawen, A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett. 584:1793–-1799; 2010.
[143] Lawen, A.; Ly, J. D.; Lane, D. J. R.; Zarschler, K.; Messina, A.; De Pinto, V.Voltage-dependent anion-selective channel 1 (VDAC1)—a mitochondrialprotein, rediscovered as a novel enzyme in the plasma membrane. Int. J.Biochem. Cell Biol. 37:277–282; 2005.
[144] Grillet, L.; Ouerdane, L.; Flis, P.; Hoang, M. T.; Isaure, M. P.; Lobinski, R.; Curie, C.;Mari, S. Ascorbate efflux as a new strategy for iron reduction and transport inplants. J. Biol. Chem. 289:2515–2525; 2014.
[145] Vulpe, C. D.; Kuo, Y. M.; Murphy, T. L.; Cowley, L.; Askwith, C.; Libina, N.;Gitschier, J.; Anderson, G. J. Hephaestin, a ceruloplasmin homologue impli-cated in intestinal iron transport, is defective in the sla mouse. Nat. Genet.21:195–199; 1999.
[146] Eid, C.; Hemadi, M.; Ha-Duong, N. T.; El Hage Chahine, J. M. Iron uptake andtransfer from ceruloplasmin to transferrin. Biochim. Biophys. Acta1840:1771–1781; 2014.
[147] Morgan, E. H. Transferrin, biochemistry, physiology and clinical significance.Mol. Aspects Med. 4:1–123; 1981.
[148] Bailey, S.; Evans, R. W.; Garratt, R. C.; Gorinsky, B.; Hasnain, S.; Horsburgh, C.;Jhoti, H.; Lindley, P. F.; Mydin, A.; Sarra, R.; et al. Molecular structure ofserum transferrin at 3.3-A resolution. Biochemistry 27:5804–5812; 1988.
[149] Wally, J.; Halbrooks, P. J.; Vonrhein, C.; Rould, M. A.; Everse, S. J.; Mason, A. B.;Buchanan, S. K. The crystal structure of iron-free human serum transferrinprovides insight into inter-lobe communication and receptor binding. J. Biol.Chem. 281:24934–24944; 2006.
[150] Aisen, P.; Listowsky, I. Iron transport and storage proteins. Annu. Rev.Biochem. 49:357–393; 1980.
[151] Jing, S. Q.; Trowbridge, I. S. Identification of the intermolecular disulfidebonds of the human transferrin receptor and its lipid-attachment site. EMBOJ. 6:327–331; 1987.
[152] Lawrence, C. M.; Ray, S.; Babyonyshev, M.; Galluser, R.; Borhani, D. W.;Harrison, S. C. Crystal structure of the ectodomain of human transferrinreceptor. Science 286:779–782; 1999.
[153] Enns, C. A.; Suomalainen, H. A.; Gebhardt, J. E.; Schroder, J.; Sussman, H. H.Human transferrin receptor: expression of the receptor is assigned tochromosome 3. Proc. Natl. Acad. Sci. USA 79:3241–3245; 1982.
[154] Moos, T. Immunohistochemical localization of intraneuronal transferrinreceptor immunoreactivity in the adult mouse central nervous system.J. Comp. Neurol. 375:675–692; 1996.
[155] Tsunoo, H.; Sussman, H. H. Characterization of transferrin binding andspecificity of the placental transferrin receptor. Arch. Biochem. Biophys.225:42–54; 1983.
[156] Huebers, H. A.; Csiba, E.; Huebers, E.; Finch, C. A. Competitive advantage ofdiferric transferrin in delivering iron to reticulocytes. Proc. Natl. Acad. Sci. USA80:300–304; 1983.
[157] Grant, B. D.; Donaldson, J. G. Pathways and mechanisms of endocyticrecycling. Nat. Rev. Mol. Cell Biol. 10:597–608; 2009.
[158] Mayle, K. M.; Le, A. M.; Kamei, D. T. The intracellular trafficking pathway oftransferrin. Biochim. Biophys. Acta 1820:264–281; 2012.
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–83 81

[159] Presley, J. F.; Mayor, S.; McGraw, T. E.; Dunn, K. W.; Maxfield, F. R. BafilomycinA1 treatment retards transferrin receptor recycling more than bulk mem-brane recycling. J. Biol. Chem. 272:13929–13936; 1997.
[160] Marshansky, V.; Futai, M. The V-type Hþ-ATPase in vesicular trafficking:targeting, regulation and function. Curr. Opin. Cell Biol. 20:415–426; 2008.
[161] Karin, M.; Mintz, B. Receptor-mediated endocytosis of transferrin in devel-opmentally totipotent mouse teratocarcinoma stem cells. J. Biol. Chem.256:3245–3252; 1981.
[162] Gunshin, H.; Mackenzie, B.; Berger, U. V.; Gunshin, Y.; Romero, M. F.; Boron,W. F.; Nussberger, S.; Gollan, J. L.; Hediger, M. A. Cloning and characterizationof a mammalian proton-coupled metal-ion transporter. Nature 388:482–488;1997.
[163] Su, M. A.; Trenor, C. C.; Fleming, J. C.; Fleming, M. D.; Andrews, N. C. TheG185R mutation disrupts function of the iron transporter Nramp2. Blood92:2157–2163; 1998.
[164] Nunez, M. -T.; Gaete, V.; Watkins, J. A.; Glass, J. Mobilization of iron fromendocytic vesicles: the effects of acidification and reduction. J. Biol. Chem.265:6688–6692; 1990.
[165] Watkins, J. A.; Altazan, J. D.; Elder, P.; Li, C. -Y.; Nunez, M. -T.; Cui, X. -X.; Glass,J. Kinetic characterization of reductant dependent processes of iron mobili-zation from endocytic vesicles. Biochemistry 31:5820–5830; 1992.
[166] Ohgami, R. S.; Campagna, D. R.; Greer, E. L.; Antiochos, B.; McDonald, A.;Chen, J.; Sharp, J. J.; Fujiwara, Y.; Barker, J. E.; Fleming, M. D. Identification of aferrireductase required for efficient transferrin-dependent iron uptake inerythroid cells. Nat. Genet. 37:1264–1269; 2005.
[167] Ohgami, R. S.; Campagna, D. R.; McDonald, A.; Fleming, M. D. The Steapproteins are metalloreductases. Blood 108:1388–1394; 2006.
[168] Zhao, N.; Gao, J.; Enns, C. A.; Knutson, M. D. ZRT/IRT-like protein 14 (ZIP14)promotes the cellular assimilation of iron from transferrin. J. Biol. Chem.285:32141–33250; 2010.
[169] Bomford, A.; Young, S. P.; Williams, R. Release of iron from the two iron-binding sites of transferrin by cultured human cells: modulation by methy-lamine. Biochemistry 24:3472–3478; 1985.
[170] Frikke-Schmidt, H.; Lykkesfeldt, J. Keeping the intracellular vitamin C at aphysiologically relevant level in endothelial cell culture. Anal. Biochem.397:135–137; 2010.
[171] Toth, I.; Bridges, K. R. Ascorbic acid enhances ferritin mRNA translation by anIRP/aconitase switch. J. Biol. Chem. 270:19540–19544; 1995.
[172] Bridges, K. R. Ascorbic acid inhibits lysosomal autophagy of ferritin. J. Biol.Chem. 262:14773–14778; 1987.
[173] May, J. M.; Qu, Z. C.; Neel, D. R.; Li, X. Recycling of vitamin C from its oxidizedforms by human endothelial cells. Biochim. Biophys. Acta 1640:153–161;2003.
[174] Gao, J.; Chen, J.; Kramer, M.; Tsukamoto, H.; Zhang, A. S.; Enns, C. A.Interaction of the hereditary hemochromatosis protein HFE with transferrinreceptor 2 is required for transferrin-induced hepcidin expression. Cell Metab.9:217–227; 2009.
[175] Graham, R. M.; Reutens, G. M.; Herbison, C. E.; Delima, R. D.; Chua, A. C.;Olynyk, J. K.; Trinder, D. Transferrin receptor 2 mediates uptake oftransferrin-bound and non-transferrin-bound iron. J. Hepatol. 48:327–334;2008.
[176] Johnson, M. B.; Chen, J.; Murchison, N.; Green, F. A.; Enns, C. A. Transferrinreceptor 2: evidence for ligand-induced stabilization and redirection to arecycling pathway. Mol. Biol. Cell 18:743–754; 2007.
[177] Herbison, C. E.; Thorstensen, K.; Chua, A. C.; Graham, R. M.; Leedman, P.;Olynyk, J. K.; Trinder, D. The role of transferrin receptor 1 and 2 intransferrin-bound iron uptake in human hepatoma cells. Am. J. Physiol. CellPhysiol. 297:C1567–C1575; 2009.
[178] Chua, A. C.; Delima, R. D.; Morgan, E. H.; Herbison, C. E.; Tirnitz-Parker, J. E.;Graham, R. M.; Fleming, R. E.; Britton, R. S.; Bacon, B. R.; Olynyk, J. K.; Trinder,D. Iron uptake from plasma transferrin by a transferrin receptor 2 mutantmouse model of haemochromatosis. J. Hepatol. 52:425–431; 2010.
[179] Escobar, A.; Gaete, V.; Nunez, M. T. Effect of ascorbate in the reduction oftransferrin-associated iron in endocytic vesicles. J. Bioenerg. Biomembr.24:227–233; 1992.
[180] Mettier, S. R.; Chew, W. B. The anemia of scurvy: effect of vitamin C diet onblood formation in experimental scurvy of guinea pigs. J. Exp. Med. 55:971–980; 1932.
[181] Cox, E. V.; Meynell, M. J.; Northam, B. E.; Cooke, W. T. The anaemia of scurvy.Am. J. Med. 42:220–227; 1967.
[182] Young, S. P.; Roberts, S.; Bomford, A. Intracellular processing of transferrinand iron by isolated rat hepatocytes. Biochem. J. 232:819–823; 1985.
[183] Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation ofcellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta1800:783–792; 2010.
[184] Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: a family of molecules for ironstorage, antioxidation and more. Biochim. Biophys. Acta 1790:589–599; 2009.
[185] Zhang, Y.; Mikhael, M.; Xu, D.; Li, Y.; Soe-Lin, S.; Ning, B.; Li, W.; Nie, G.; Zhao,Y.; Ponka, P. Lysosomal proteolysis is the primary degradation pathway forcytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit.Antioxid. Redox Signaling 13:999–1009; 2010.
[186] Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai,K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted andiron-replete cells. Mol. Cell. Biol. 31:2040–2052; 2011.
[187] De Domenico, I.; Ward, D. M.; Kaplan, J. Specific iron chelators determine theroute of ferritin degradation. Blood 114:4546–4551; 2009.
[188] De Domenico, I.; Vaughn, M. B.; Li, L.; Bagley, D.; Musci, G.; Ward, D. M.;Kaplan, J. Ferroportin-mediated mobilization of ferritin iron precedes ferritindegradation by the proteasome. EMBO J. 25:5396–5404; 2006.
[189] Kurz, T.; Eaton, J. W.; Brunk, U. T. The role of lysosomes in iron metabolismand recycling. Int. J. Biochem. Cell Biol. 43:1686–1697; 2011.
[190] Sakurai, K.; Nabeyama, A.; Fujimoto, Y. Ascorbate-mediated iron release fromferritin in the presence of alloxan. Biometals 19:323–333; 2006.
[191] Boyer, R. F.; McCleary, C. J. Superoxide ion as a primary reductant inascorbate-mediated ferritin iron release. Free Radic. Biol. Med. 3:389–395;1987.
[192] Takagi, H.; Shi, D.; Ha, Y.; Allewell, N. M.; Theil, E. C. Localized unfolding atthe junction of three ferritin subunits: a mechanism for iron release? J. Biol.Chem. 273:18685–18688; 1998.
[193] Theil, E. C. Ferritin protein nanocages use ion channels, catalytic sites, andnucleation channels to manage iron/oxygen chemistry. Curr. Opin. Chem. Biol.15:304–311; 2011.
[194] Anderson, C. P.; Shen, M.; Eisenstein, R. S.; Leibold, E. A. Mammalian ironmetabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta1823:1468–1483; 2012.
[195] Rouault, T. A. The role of iron regulatory proteins in mammalian ironhomeostasis and disease. Nat. Chem. Biol. 2:406–414; 2006.
[196] Recalcati, S.; Minotti, G.; Cairo, G. Iron regulatory proteins: from molecularmechanisms to drug development. Antioxid. Redox Signaling 13:1593–1616;2010.
[197] Dunn, L. L.; Rahmanto, Y. S.; Richardson, D. R. Iron uptake and metabolism inthe new millennium. Trends Cell Biol. 17:93–100; 2007.
[198] Hentze, M. W.; Muckenthaler, M. U.; Galy, B.; Camaschella, C. Two to tango:regulation of mammalian iron metabolism. Cell 142:24–38; 2010.
[199] Volz, K. The functional duality of iron regulatory protein 1. Curr. Opin. Struct.Biol. 18:106–111; 2008.
[200] Pitula, J. S.; Deck, K. M.; Clarke, S. L.; Anderson, S. A.; Vasanthakumar, A.;Eisenstein, R. S. Selective inhibition of the citrate-to-isocitrate reaction ofcytosolic aconitase by phosphomimetic mutation of serine-711. Proc. Natl.Acad. Sci. USA 101:10907–10912; 2004.
[201] Salahudeen, A. A.; Thompson, J. W.; Ruiz, J. C.; Ma, H. W.; Kinch, L. N.; Li, Q.;Grishin, N. V.; Bruick, R. K. An E3 ligase possessing an iron-responsivehemerythrin domain is a regulator of iron homeostasis. Science 326:722–726; 2009.
[202] Vashisht, A. A.; Zumbrennen, K. B.; Huang, X.; Powers, D. N.; Durazo, A.; Sun,D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; Spruck, C.; Leibold, E.A.; Wohlschlegel, J. A. Control of iron homeostasis by an iron-regulatedubiquitin ligase. Science 326:718–721; 2009.
[203] Chollangi, S.; Thompson, J. W.; Ruiz, J. C.; Gardner, K. H.; Bruick, R. K.Hemerythrin-like domain within F-box and leucine-rich repeat protein 5(FBXL5) communicates cellular iron and oxygen availability by distinctmechanisms. J. Biol. Chem. 287:23710–23717; 2012.
[204] Thompson, J. W.; Salahudeen, A. A.; Chollangi, S.; Ruiz, J. C.; Brautigam, C. A.;Makris, T. M.; Lipscomb, J. D.; Tomchick, D. R.; Bruick, R. K. Structural andmolecular characterization of iron-sensing hemerythrin-like domain withinF-box and leucine-rich repeat protein 5 (FBXL5). J. Biol. Chem. 287:7357–7365; 2012.
[205] Bridges, K. R.; Hoffman, K. E. The effects of ascorbic acid on the intracellularmetabolism of iron and ferritin. J. Biol. Chem. 261:14273–14277; 1986.
[206] Scheers, N.; Sandberg, A. S. Iron transport through ferroportin is induced byintracellular ascorbate and involves IRP2 and HIF2a. Nutrients 6:249–260;2014.
[207] Goralska, M.; Harned, J.; Fleisher, L. N.; McGahan, M. C. The effect of ascorbicacid and ferric ammonium citrate on iron uptake and storage in lensepithelial cells. Exp. Eye Res. 66:687–697; 1998.
[208] Wang, J.; Chen, G.; Muckenthaler, M.; Galy, B.; Hentze, M. W.; Pantopoulos, K.Iron-mediated degradation of IRP2, an unexpected pathway involving a2-oxoglutarate-dependent oxygenase activity. Mol. Cell. Biol. 24:954–965;2004.
[209] Hausmann, A.; Lee, J.; Pantopoulos, K. Redox control of iron regulatoryprotein 2 stability. FEBS Lett. 585:687–692; 2011.
[210] Kim, S.; Wing, S. S.; Ponka, P. S-nitrosylation of IRP2 regulates its stability viathe ubiquitin–proteasome pathway. Mol. Cell. Biol. 24:330–337; 2004.
[211] Richardson, D. R. Role of ceruloplasmin and ascorbate in cellular iron release.J. Lab. Clin. Med. 134:454–465; 1999.
[212] Cassavaugh, J.; Lounsbury, K. M. Hypoxia-mediated biological control. J. Cell.Biochem. 112:735–744; 2011.
[213] Mole, D. R. Iron homeostasis and its interaction with prolyl hydroxylases.Antioxid. Redox Signaling 12:445–458; 2010.
[214] Wiesener, M. S.; Jurgensen, J. S.; Rosenberger, C.; Scholze, C. K.; Horstrup, J. H.;Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U. A.; Pugh, C. W.; Ratcliffe, P. J.;Bachmann, S.; Maxwell, P. H.; Eckardt, K. U. Widespread hypoxia-inducibleexpression of HIF-2a in distinct cell populations of different organs. FASEB J.17:271–273; 2003.
[215] Flashman, E.; Davies, S. L.; Yeoh, K. K.; Schofield, C. J. Investigating thedependence of the hypoxia-inducible factor hydroxylases (factor inhibitingHIF and prolyl hydroxylase domain 2) on ascorbate and other reducingagents. Biochem. J. 427:135–142; 2010.
[216] Pappalardi, M. B.; McNulty, D. E.; Martin, J. D.; Fisher, K. E.; Jiang, Y.; Burns, M.C.; Zhao, H.; Ho, T.; Sweitzer, S.; Schwartz, B.; Annan, R. S.; Copeland, R. A.;Tummino, L.; Luo, L. Biochemical characterization of human HIF hydroxylases
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–8382

using HIF protein substrates that contain all three hydroxylation sites.Biochem. J. 436:363–369; 2011.
[217] Chepelev, N. L.; Willmore, W. G. Regulation of iron pathways in response tohypoxia. Free Radic. Biol. Med. 50:645–666; 2011.
[218] Haase, V. H. Hypoxic regulation of erythropoiesis and iron metabolism. Am. J.Physiol. Renal Physiol. 299:F1–F13; 2010.
[219] Kuiper, C.; Molenaar, I. G. M.; Dachs, G. U.; Currie, M. J.; Sykes, P. H.; Vissers,M. C. M. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endome-trial cancer. Cancer Res. 70:5749–5758; 2010.
[220] Vissers, M. C.; Gunningham, S. P.; Morrison, M. J.; Dachs, G. U.; Currie, M. J.Modulation of hypoxia-inducible factor-1α in cultured primary cells byintracellular ascorbate. Free Radic. Biol. Med. 42:765–772; 2007.
[221] Kuiper, C.; Dachs, G. U.; Currie, M. J.; Vissers, M. C. M. Intracellular ascorbateenhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferen-tially suppresses the HIF-1 transcriptional response. Free Radic. Biol. Med.69:308–317; 2014.
[222] Page, E. L.; Chan, D. A.; Giaccia, A. J.; Levine, M.; Richard, D. E. Hypoxia-inducible factor-1alpha stabilization in nonhypoxic conditions: role ofoxidation and intracellular ascorbate depletion. Mol. Biol. Cell 19:86–94;2008.
[223] Nytko, K. J.; Spielmann, P.; Camenisch, G.; Wenger, R. H.; Stiehl, D. P.Regulated function of the prolyl-4-hydroxylase domain (PHD) oxygen sensorproteins. Antioxid. Redox Signaling 9:1329–1338; 2007.
[224] Ozer, A.; Bruick, R. K. Non-heme dioxygenases: cellular sensors and regula-tors jelly rolled into one? Nat. Chem. Biol. 3:144–153; 2007.
[225] Nytko, K. J.; Maeda, N.; Schlafli, P.; Spielmann, P.; Wenger, R. H.; Stiehl, D. P.Vitamin C is dispensable for oxygen sensing in vivo. Blood 117:5485–5493;2011.
[226] Semenza, G. L. Hypoxia-inducible factors in physiology and medicine. Cell148:399–408; 2012.
[227] Meyron-Holtz, E. G.; Ghosh, M. C.; Rouault, T. A. Mammalian tissue oxygenlevels modulate iron-regulatory protein activities in vivo. Science 306:2087–2090; 2004.
[228] Meyron-Holtz, E. G.; Ghosh, M. C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.;Berger, U. V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; Rouault,T. A. Genetic ablations of iron regulatory proteins 1 and 2 reveal why ironregulatory protein 2 dominates iron homeostasis. EMBO J. 23:386–395; 2004.
[229] Wang, W.; Di, X.; D'Agostino Jr R. B.; Torti, S. V.; Torti, F. M. Excess capacity ofthe iron regulatory protein system. J. Biol. Chem. 282:24650–24659; 2007.
[230] Brazzolotto, X.; Gaillard, J.; Pantopoulos, K.; Hentze, M. W.; Moulis, J. M.Human cytoplasmic aconitase (iron regulatory protein 1) is converted into its[3Fe–4S] form by hydrogen peroxide in vitro but is not activated for iron-responsive element binding. J. Biol. Chem. 274:21625–21630; 1999.
[231] Pantopoulos, K.; Mueller, S.; Atzberger, A.; Ansorge, W.; Stremmel, W.;Hentze, M. W. Differences in the regulation of iron regulatory protein-1(IRP-1) by extra- and intracellular oxidative stress. J. Biol. Chem. 272:9802–9808; 1997.
[232] Hanson, E. S.; Leibold, E. A. Regulation of the iron regulatory proteins byreactive nitrogen and oxygen species. Gene Expression 7:367–376; 1999.
[233] Bouton, C.; Drapier, J. C. Iron regulatory proteins as NO signal transducers.Sci. STKE 2003:pe17; 2003.
[234] Styś, A.; Galy, B.; Starzyński, R. R.; Smuda, E.; Drapier, J. C.; Lipiński, P.;Bouton, C. Iron regulatory protein 1 outcompetes iron regulatory protein 2 inregulating cellular iron homeostasis in response to nitric oxide. J. Biol. Chem.286:22846–22854; 2011.
D.J.R. Lane, D.R. Richardson / Free Radical Biology and Medicine 75 (2014) 69–83 83