targeting suppressive myeloid cells potentiates checkpoint...
TRANSCRIPT

Cancer Therapy: Preclinical
Targeting Suppressive Myeloid Cells PotentiatesCheckpoint Inhibitors to Control SpontaneousNeuroblastomaYumeng Mao1, Nina Eissler2, Katarina Le Blanc3, John Inge Johnsen2, Per Kogner2,and Rolf Kiessling1
Abstract
Purpose: Neuroblastoma is the most common extracranialsolid cancer type in childhood, and high-risk patients have poorprognosis despite aggressive multimodal treatment. Neuroblas-toma-driven inflammation contributes to the induction of sup-pressive myeloid cells that hamper efficient antitumor immuneresponses. Therefore, we sought to enhance antitumor immunityby removing immunosuppression mediated by myeloid cells.
Experimental Design: The prognostic values of myeloid cellsare demonstrated by analyzing genomic datasets of neuroblasto-mapatients. The impact of tumor-derived factors onmyelopoiesisand local induction of suppressive myeloid cells is dissected by invitro culturemodels using freshly isolatedhumanCD34þhemato-poietic stem cells, primary human monocytes, and murine bonemarrow cells. To test the therapeutic efficacy of BLZ945 as amonotherapy or in combination with checkpoint inhibitors, weused a transgenic murine model (TH-MYCN) that developsaggressive spontaneous neuroblastoma.
Results: We report that infiltrating CSF-1Rþ myeloid cellspredict poor clinical outcome in patients with neuroblastoma.In vitro, neuroblastoma-derived factors interfere with earlydevelopment of myeloid cells and enable suppressive func-tions on human monocytes through M-CSF/CSF-1R interac-tion. In a transgenic mouse model (TH-MYCN) resemblinghigh-risk human neuroblastoma, antagonizing CSF-1R with aselective inhibitor (BLZ945) modulates the induction ofhuman and murine suppressive myeloid cells and efficientlylimit tumor progression. While checkpoint inhibitors areinsufficient in controlling tumor growth, combining BLZ945with PD-1/PD-L1 blocking antibodies results in superiortumor control.
Conclusions:Our results demonstrate the essential role ofCSF-1R signaling during the induction of suppressive myeloid cellsand emphasize its clinical potential as an immunotherapy forhuman cancers. Clin Cancer Res; 22(15); 3849–59. �2016 AACR.
IntroductionNeuroblastoma is a solid pediatric tumor of the developing
sympathetic nervous system (1). High-risk metastatic tumorsfrequently show amplified MYCN proto-oncogene, and overallpatient survival is less than 50% (1, 2). Progression of neuroblas-toma is associated with expression of inflammatory factors (3),and intratumoral myeloid cells predict poor clinical outcome(4, 5). A transgenic murine model resembling the aggressivegrowth pattern of high-risk human neuroblastoma has beencharacterized, where the human MYCN oncogene (TH-MYCN)
drives the establishment of spontaneous tumors (6, 7). We andothers have reported prolonged survival of these animals usingnovel treatments (8–10). However, once the tumors are estab-lished, it is difficult to achieve tolerable therapeutic effects, due tothe known short treatment window and rapid disease progression(7, 11, 12).
Recently, eliciting antitumor immunity has been proposed tobe a promising option for treating high-risk childhood neuro-blastoma (13, 14). However, tumor-induced inflammation limitsefficient antitumor immune responses through recruitment ofvarious suppressive immune cell types (15), including myeloid-derived suppressor cells (MDSC) and "M2-biased" tumor-asso-ciated macrophages (TAM) (16–18). These cells demonstrateoverlapping phenotypic markers as well as inhibitory mechan-isms (19, 20) and predict poor clinical outcome in patients withcancer (21–24).
Macrophage colony-stimulating factor (M-CSF or CSF-1) isknown to be essential for the differentiation and survival ofmyeloid cells (25). In malignant conditions, tumor-derived M-CSF is associated with poor survival in various human cancers(26). Thus, targeting strategies against its receptor (CSF-1R) havebeen extensively explored (27–29). When included in combina-tional approaches, blockade of CSF-1R signaling exhibits syner-gistic effects of various anticancer therapies (30–34).
Here, we show that infiltration of CSF-1Rþ myeloid cellspredicts poor survival in patients with neuroblastoma. Further,
1Department of Oncology-Pathology, Cancer Center Karolinska, Kar-olinska Institutet, Stockholm, Sweden. 2Childhood Cancer ResearchUnit, Department of Women's and Children's Health, Karolinska Insti-tutet, Astrid Lindgren Children's Hospital, Stockholm, Sweden.3Department of Medicine, Karolinska Institutet, Hematology Centre,Karolinska University Hospital, Stockholm, Sweden.
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
Y. Mao and N. Eissler contributed equally to this article.
Corresponding Authors: Rolf Kiessling, Karolinska Institutet, CCK R8:01, KSRingen, R8:01, Stockholm, S-17176, Sweden. Phone: 46851776857; Fax:468309195; E-mail: [email protected] and Yumeng Mao, [email protected]
doi: 10.1158/1078-0432.CCR-15-1912
�2016 American Association for Cancer Research.
ClinicalCancerResearch
www.aacrjournals.org 3849
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912

we reveal the mechanistic insight of the M-CSF/CSF-1R axisduring the induction of suppressiveMDSCs and TAMs in humansand mice. Finally, combining a highly selective CSF-1R inhibitor,BLZ945, with PD-1/PD-L1–blocking agents, elicits robust anti-tumor effects against established aggressive tumors in theTH-MYCN murine neuroblastoma model.
Materials and MethodsTumor cell lines
The NHO2A murine neuroblastoma cell line was maintainedin RPMI1640 medium (Life Technologies) supplemented with10% heat-inactivated FBS (Life Technologies), 1% antibiotics(penicillin and streptomycin), and 2 mmol/L L-glutamine (LifeTechnologies). Tumor-conditioned medium (TCM) was har-vested and used on the same day when the cells reached 90%confluence.
Human neuroblastoma cell lines, including SK-N-BE(2),SK-N-AS, and SK-N-FI, were routinely maintained in IMDMmedium (Life Technologies) supplemented with 10% heat-inactivated FBS. Cell lines were routinely tested for mycoplasmacontamination (MycoAlert, Lonza) and authenticated by an STRidentifier kit (Supplementary Table S2; Applied Biosystems) Forobtaining TCM, cells were cultured in IMDM medium supple-mented with 10% heat-inactivated, pooled human AB serumfrom six donors (referred to as "culture medium" later), andmedia were harvested and stored in aliquots at �80 �C whencells reached 90% confluence.
Flow cytometryDetailed information of fluorochrome-conjugated antibodies
utilized in this studywas summarized in Supplementary Table S3.Briefly, 1�105 to 2�105 cells were placed in a 96 v-bottom plateand washed twice in 200 mL FACS buffer (PBS with 1% humanserum albumin, HSA). Next, cells were resuspended in 20 mL PBScontaining the appropriate antibody cocktails for extracellularantigens and incubated at 4�C for 25 minutes. For samplesrequiring intracellular staining, cellswerefixed andpermeabilizedin BD cytofix/cytoperm buffer for 15 minutes and stained for 40minutes at 4�C. All cells were washed and acquired in a BD LSRII
instrument, and data were further analyzed with FlowJo software(Treestar Inc.).
TH-MYCN murine neuroblastoma modelTH-MYCN animals were obtained from the Mouse Model of
Human Cancer Consortium Repository as an N16 backcross tothe 129�1/SvJ background and have been kept as a continuousinbreeding. All experimental protocols were reviewed andapproved by the regional ethical committee (ethical permitN42/14). Heterozygous mice received abdominal palpations 3times weekly to follow tumor development and were moni-tored closely for signs of discomfort. At the time tumors werepalpable (day 0), mice were treated with BLZ945 by daily oralgavage as described above for 10 consecutive days. Alternative-ly, blocking antibodies for PD-1 (clone: RMP1-14) and PD-L1(clone: 10F.9G2, both purchased from BioXcell) were injectedintraperitoneally at 12.5 mg/kg on days 0, 3, and 6 or concur-rently with BLZ945. The combination was well tolerated, andneither weight loss nor toxicity of the internal organs wasobserved. Spleens and tumors were collected and homoge-nized, erythrocytes were lysed by BD Pharm Lyse Buffer (BDBiosciences), and immune cells were analyzed by flow cyto-metry. For tumors smaller than 0.5 g, an additional staining fortumor antigen GD2 was performed in order to confirm thepresence of neuroblastoma cells.
Isolation of primary human cellsTo obtain humanprimarymonocytes, peripheral bloodmono-
nuclear cells (PBMC) were first isolated from buffy coats by ficollgradient centrifugation (GE Healthcare) and washed three timesin PBS. Next, monocytes were labeled with CD14þ selectionmicrobeads and purified using an LS column (Miltenyi Biotech).CD34þ hematopoietic progenitor cells were purified from umbil-ical cord blood provided by the KarolinskaUniversityHospital. Inbrief, T cells were first removed during gradient centrifugationusing a RossetteSep CD3þ depletion kit (StemCells), followed byCD34þ positive selection using microbeads and MS columns(Miltenyi Biotech).
CSF-1R inhibitorsBLZ945, a highly selective small-molecule inhibitor for tyro-
sine kinase of CSF-1R (>3,200-fold more than other tyrosinekinases; ref. 27), was kindly provided by Novartis. For in vitro–blocking experiments, stock solutions were prepared by dissol-ving BLZ945 or GW2580 (Selleck Chemicals) in DMSO at 10mmol/L and 1 mmol/L, respectively. For in vivo treatment,BLZ945 was dissolved in 20% Captisol at 16 mg/mL and deliv-ered by daily oral gavage at the dose of 200mg/kg, according to aprevious study (29).
Differentiation of CD34þ hematopoietic progenitor cellsMaturation of CD34þ cells was performed using 900 mL culture
medium containing 50 ng/mL GM-CSF and 5 ng/mL TNFa (bothwere from PeproTech) in a 24-well plate. Alternatively, super-natants harvested from the three above-mentioned human neu-roblastoma cell lines were added in addition to the cytokines at a2:1 ratio to theprogenitor cells. CD34þ cellsmaintained in culturemedium were used as controls. To block CSF-1R signaling,BLZ945 (500 nmol/L) was added to cells matured with cytokinesor the combination of cytokines and SK-N-BE(2) supernatant.
Translational Relevance
Tumor-driven induction of suppressive myeloid cells ham-pers sufficient antitumor immune responses. We show herethat combining a highly selective CSF-1R inhibitor (BLZ945)with blocking antibodies against the PD-1/L1 axis providesstriking tumor control in a transgenic murine model thatresembles the aggressive growth pattern of high-risk humanneuroblastoma. Further, we demonstrate the prognostic valueof CSF-1R–expressing myeloid cells in patients with neuro-blastoma and reveal themechanistic insight that the inductionof suppressivemyeloid cells is governedbyCSF-1R signaling inhumans and mice. Given that the therapeutic efficacy of PD-1blockade and the prognostic values of suppressive myeloidcells are being validated in clinical trials of various humancancers, we believe that thesefindings have broad implicationsin designing novel combinational immunotherapies forpatients with cancer.
Mao et al.
Clin Cancer Res; 22(15) August 1, 2016 Clinical Cancer Research3850
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912

After 7 days, all cells were harvested by washing and gentlyscraping, and phenotypes and functions of the cells were evalu-ated by flow cytometry or in CFSE-based T-cell proliferationassays.
Human monocyte–tumor coculturePrimary human monocytes were cocultured with human
neuroblastoma cell lines according to our published protocol(35). In brief, monocytes were cocultured with 4�105 SK-N-BE(2), SK-N-AS, or 6�105 SK-N-FI neuroblastoma cells in 3-mLculture medium in a 6-well plate. Monocytes cultured withouttumor cells were used as controls. After 64 hours, cells wereharvested by vigorously washing, followed by gently scrapingof the plates. Phenotypic changes of the cells were evaluated byflow cytometry and HLA-DRþ cells were sorted with microbe-ads and MS columns (Miltenyi Biotech). To investigate the roleof CSF-1R, 500 nmol/L BLZ945 (Novartis) or 1 mmol/LGW2580 (Selleck Chemicals) was added to monocytes or thecocultures.
Differentiation of murine bone marrow cellsSuppressive myeloid cells were induced from bone marrow
cells harvested from negatively genotyped TH-MYCN mice, inaccordance with a previously described protocol (36). In brief,1�106 isolated bone marrow cells were cultured in 6-well platesin the presence of NHO2A tumor-conditioned medium (TCM,1:1 dilution to fresh medium). As controls, cells were cultured infresh medium or with M-CSF (20 ng/mL, Biolegend). To blockCSF-1R signaling, BLZ945 or GW2580 were added at 1 mmol/L tothe cultures andDMSOwas included as control. After 4 days, cellswere harvested by collecting floating cells and carefully scrapingthe adherent cells of the wells, and flow cytometric analysis or T-cell suppression assays were conducted subsequently.
T-cell suppression assaysPrimary human T cells were isolated from PBMCs by CD3þ
positive selection andMS columns (Miltenyi Biotech) and labeledwith 1.5 mmol/L 5(6)-CFDA/CFSE tracing dye (Invitrogen). Forthe tumor-educatedmonocytes, 1�105 T cells were added togeth-er at 2:1 or 4:1 with HLA-DR–sorted cells and activated bymicrobeads coated with anti-CD3/CD28mAb (Invitrogen). After4 days, cells were harvested and proliferations of T cells wereevaluated by flow cytometry. In mixed lymphocyte reactions,CFSE-labeled T cells from PBMCs were mixed at 4:1, 8:1, or16:1 with differentiated CD34þ cells, and proliferation of T cellswas analyzed after 6 days. In some experiments, IFNg concentra-tions in the culturemediumat the endof proliferation assayswerealso measured by ELISA (Mabtech).
In order to evaluate murine T-cell functions, 1�106 CFSE-labeled splenocytes from na€�ve mice were preincubated for 24hours at different ratios with Gr1þ cells isolated from spleens oftumor-bearing mice or bone marrow–derived myeloid cells dif-ferentiated in vitro in a 96-well plate. Next, T cells were activatedwith 1 mLmicrobeads coated with anti-CD3/CD28mAb (Invitro-gen) and proliferation and expression of CD69 on T cells weremeasured by flow cytometry after 4 days. To investigate thesuppressive mechanisms, pharmacologic inhibitors for IDO(1-DL-MT, 250 mmol/L, Sigma-Aldrich), JAK/STATs (AG490,10 mmol/L, Sigma-Aldrich), arginase (nor-NOHA, 200 IU/mL,Calbiochem), or iNOS (1400W, 200 mmol/L, Sigma-Aldrich)were added to the wells during the T-cell activation.
Cytokine analysisCytokine contents in culture medium or supernatants har-
vested from SK-N-BE(2), SK-N-AS, or SK-N-FI neuroblastomatumor cell lines were analyzed by a 27-parameter Luminexmultiplex assay (R&D Systems) in the core facility at KarolinskaUniversity Hospital. Concentrations of human or murineM-CSF (CSF-1) in the TCM were determined using ELISA(R&D Systems).
R2 databaseMultiple parameters were searched and correlated in patient
datasets available in the public "R2: microarray analysis andvisualization platform" (http://r2.amc.nl). For comparisonsbetween benign and malignant tumors, three datasets, includingVersteeg (cohort 1, n ¼ 88), Hiyama (cohort 2, n ¼ 51), andLastowska (cohort 3, n ¼ 30), were compared with the Millerneurofibromadataset (n¼ 86)usingone-wayANOVA. For survivalof patients, Kaplan–Meier analysis were performed on two previ-ously reported datasets of patients with untreated neuroblastomatumor biopsies, including Versteeg (cohort 1, n ¼ 88; ref. 37) andSeeger (cohort 4, n ¼ 102; ref. 5), and the cutoff expression levelswere calculatedwithin theR2database. Inaddition, a largerdataset,Kocak (cohort 5, n ¼ 649; ref. 38), was included to study thecorrelations between expression of CSF-1R and other markers.
Statistical analysisUnless otherwise stated, all results were collected frommultiple
experiments, and figures were prepared in the Prism software(GraphPad). Normal distribution and equal variant of the data-sets were first tested, thereafter appropriate Student t tests ornonparametric, Mann–Whitney U tests were applied (as statedin the figure legends). All results were presented as means � SD,and representative histograms or pictures were selected based onthe average values.
ResultsInfiltrating CSF-1Rþ myeloid cells predicted poor survival inpatients with neuroblastoma
To investigate the immune signatures in human neuroblasto-ma, we analyzed the expression patterns in different patientdatasets (`R2: microarray analysis and visualization platform',http://r2.amc.nl).
In comparison to benign neurofibroma, neuroblastoma fromthree patient cohorts (refs. 37, 39; P values are summarized inSupplementary Table S1) all showed significantly increasedexpression of CD14 and CD68 monocyte/macrophage markers(Fig. 1A). In contrast, granulocytes (CD66b), regulatory T cells(FoxP3) or dendritic cell (DC) markers CD11c and CD83remained unchanged (Supplementary Fig. S1A). Moreover,CSF-1R showed enhanced expression in tumor tissues (Fig.1B) and correlated strongly with expression levels of CD14and CD68 (R ¼ 0.801 and 0.752, respectively; Fig. 1C). Accord-ingly, the myeloid differentiation factor M-CSF (Fig. 1D), butnot GM-CSF, G-CSF, or another CSF-1R ligand, IL34 (Supple-mentary Fig. S1B), was expressed at significantly higher levels inhuman neuroblastoma.
Furthermore, we validated the prognostic significance ofM-CSFand CSF-1R in two previously published datasets. We restrictedthese analyses to data collected from biopsies of untreatedpatients (5, 37), because myeloid cells are sensitive to treatment,
Block CSF-1R to Improve Checkpoint Blockade in Neuroblastoma
www.aacrjournals.org Clin Cancer Res; 22(15) August 1, 2016 3851
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912
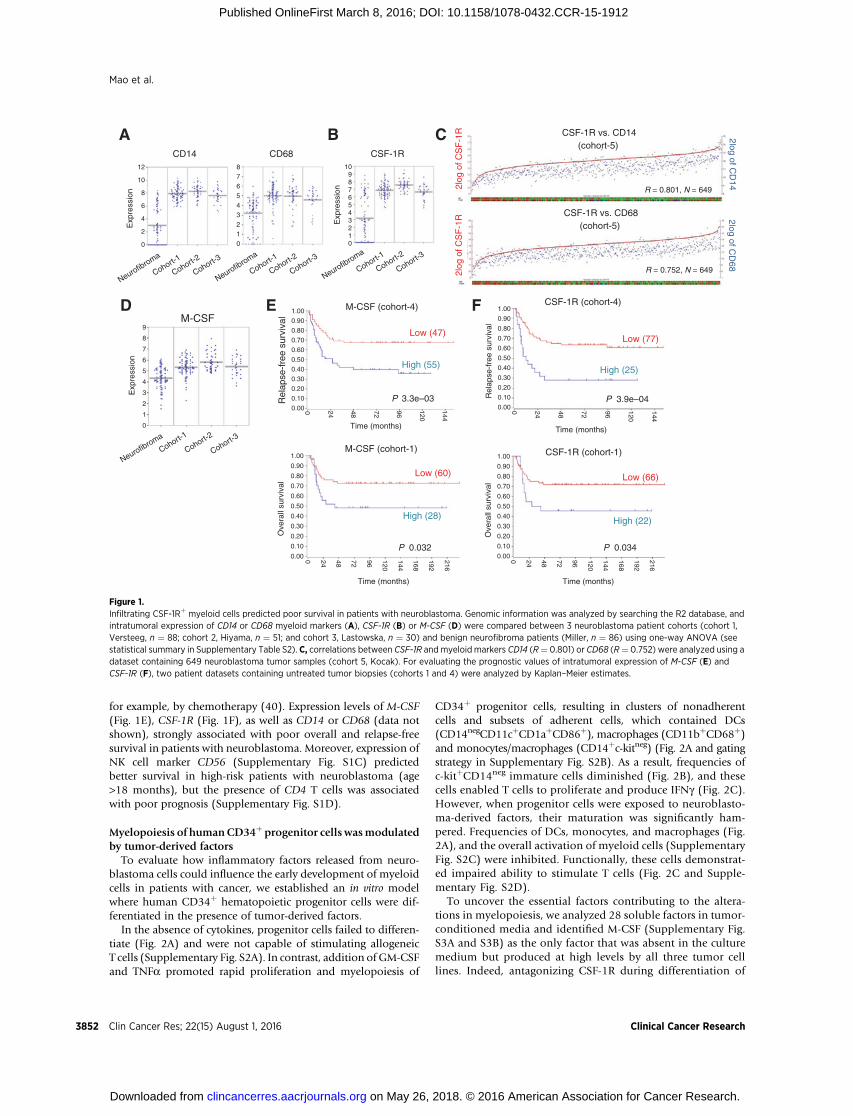
for example, by chemotherapy (40). Expression levels of M-CSF(Fig. 1E), CSF-1R (Fig. 1F), as well as CD14 or CD68 (data notshown), strongly associated with poor overall and relapse-freesurvival in patients with neuroblastoma. Moreover, expression ofNK cell marker CD56 (Supplementary Fig. S1C) predictedbetter survival in high-risk patients with neuroblastoma (age>18 months), but the presence of CD4 T cells was associatedwith poor prognosis (Supplementary Fig. S1D).
Myelopoiesis of humanCD34þ progenitor cells wasmodulatedby tumor-derived factors
To evaluate how inflammatory factors released from neuro-blastoma cells could influence the early development of myeloidcells in patients with cancer, we established an in vitro modelwhere human CD34þ hematopoietic progenitor cells were dif-ferentiated in the presence of tumor-derived factors.
In the absence of cytokines, progenitor cells failed to differen-tiate (Fig. 2A) and were not capable of stimulating allogeneicT cells (Supplementary Fig. S2A). In contrast, addition of GM-CSFand TNFa promoted rapid proliferation and myelopoiesis of
CD34þ progenitor cells, resulting in clusters of nonadherentcells and subsets of adherent cells, which contained DCs(CD14negCD11cþCD1aþCD86þ), macrophages (CD11bþCD68þ)and monocytes/macrophages (CD14þc-kitneg) (Fig. 2A and gatingstrategy in Supplementary Fig. S2B). As a result, frequencies ofc-kitþCD14neg immature cells diminished (Fig. 2B), and thesecells enabled T cells to proliferate and produce IFNg (Fig. 2C).However, when progenitor cells were exposed to neuroblasto-ma-derived factors, their maturation was significantly ham-pered. Frequencies of DCs, monocytes, and macrophages (Fig.2A), and the overall activation of myeloid cells (SupplementaryFig. S2C) were inhibited. Functionally, these cells demonstrat-ed impaired ability to stimulate T cells (Fig. 2C and Supple-mentary Fig. S2D).
To uncover the essential factors contributing to the altera-tions in myelopoiesis, we analyzed 28 soluble factors in tumor-conditioned media and identified M-CSF (Supplementary Fig.S3A and S3B) as the only factor that was absent in the culturemedium but produced at high levels by all three tumor celllines. Indeed, antagonizing CSF-1R during differentiation of
ACD14
Exp
ress
ion
CD68
BCSF-1R
Exp
ress
ion R = 0.801, N = 649
CSF-1R vs. CD14(cohort-5)
2log
of C
SF
-1R 2log of C
D14
CSF-1R vs. CD68(cohort-5)
R = 0.752, N = 649
2log of CD
68
2log
of C
SF
-1R
M-CSF
Exp
ress
ion
D E
High (25)
Low (77)
P 3.9e–04
CSF-1R (cohort-4)
Time (months)
Rel
apse
-fre
e su
rviv
al
High (22)
Low (66)
P 0.034P 0.032
CSF-1R (cohort-1)
Time (months)
Ove
rall
surv
ival
F
High (55)
Low (47)
P 3.3e–03
M-CSF (cohort-4)
Time (months)
Rel
apse
-fre
e su
rviv
al
C
12
10
8
6
4
2
0
109876543210
876543210
9
8
7
6
5
4
3
2
1
0
1.00
0.90
0.80
0.70
0.60
0.50
0.40
0.30
0.20
0.10
0.00
1.00
0.90
0.80
0.70
0.60
0.50
0.40
0.30
0.20
0.10
0.00
1.00
0.90
0.80
0.70
0.60
0.50
0.40
0.30
0.20
0.10
0.00
1.00
0.90
0.80
0.70
0.60
0.50
0.40
0.30
0.20
0.10
0.000 24 48 72 96
120
144
0 24 48 72 96 120
144
168
192
216
0 24 48 72 96 120
144
168
192
216
0 24 48 72 96 120
144
M-CSF (cohort-1)
Time (months)
Ove
rall
surv
ival
Neurofibroma
Neurofibroma
Neurofibroma
Neurofibroma
Cohort-1
Cohort-1
Cohort-2
Cohort-3
Cohort-1
Cohort-1
Cohort-2
Cohort-2
Cohort-2
Cohort-3
Cohort-3
Cohort-3
Low (60)
High (28)
Figure 1.Infiltrating CSF-1Rþ myeloid cells predicted poor survival in patients with neuroblastoma. Genomic information was analyzed by searching the R2 database, andintratumoral expression of CD14 or CD68 myeloid markers (A), CSF-1R (B) or M-CSF (D) were compared between 3 neuroblastoma patient cohorts (cohort 1,Versteeg, n ¼ 88; cohort 2, Hiyama, n ¼ 51; and cohort 3, Lastowska, n ¼ 30) and benign neurofibroma patients (Miller, n ¼ 86) using one-way ANOVA (seestatistical summary in Supplementary Table S2). C, correlations between CSF-1R andmyeloid markers CD14 (R¼ 0.801) or CD68 (R¼ 0.752) were analyzed using adataset containing 649 neuroblastoma tumor samples (cohort 5, Kocak). For evaluating the prognostic values of intratumoral expression of M-CSF (E) andCSF-1R (F), two patient datasets containing untreated tumor biopsies (cohorts 1 and 4) were analyzed by Kaplan–Meier estimates.
Mao et al.
Clin Cancer Res; 22(15) August 1, 2016 Clinical Cancer Research3852
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912
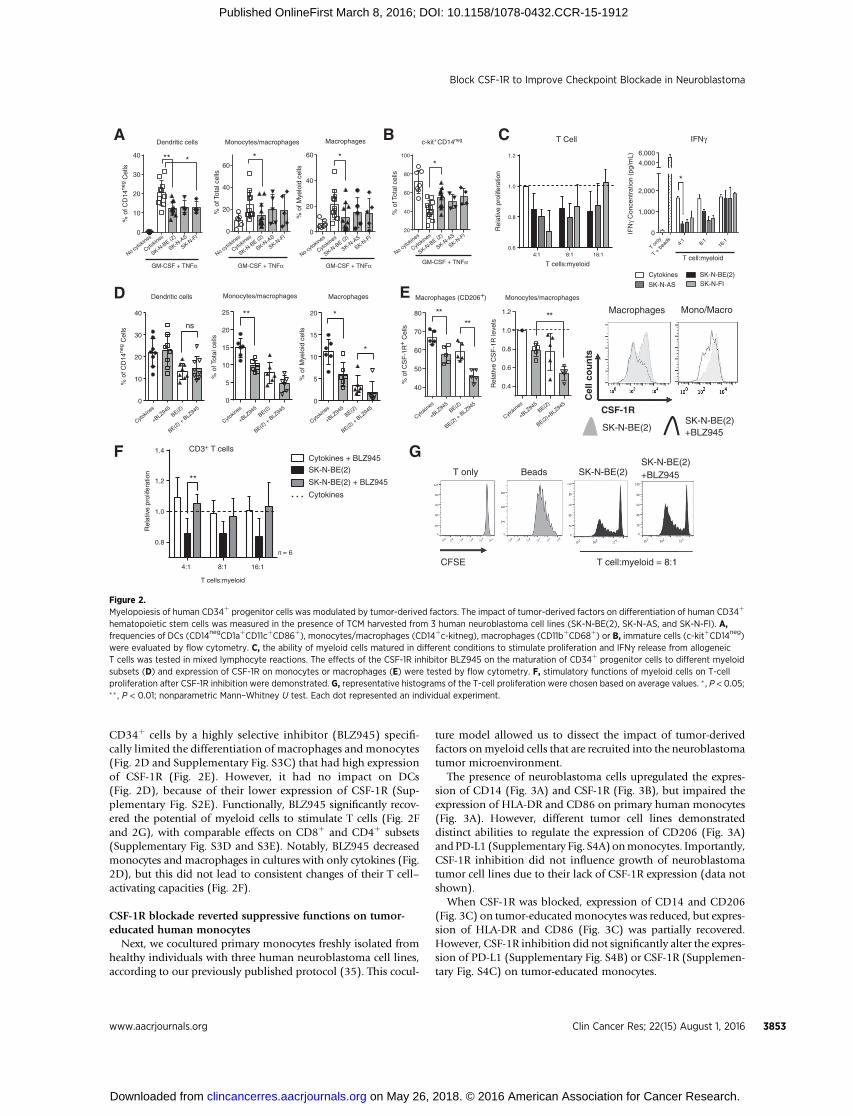
CD34þ cells by a highly selective inhibitor (BLZ945) specifi-cally limited the differentiation of macrophages and monocytes(Fig. 2D and Supplementary Fig. S3C) that had high expressionof CSF-1R (Fig. 2E). However, it had no impact on DCs(Fig. 2D), because of their lower expression of CSF-1R (Sup-plementary Fig. S2E). Functionally, BLZ945 significantly recov-ered the potential of myeloid cells to stimulate T cells (Fig. 2Fand 2G), with comparable effects on CD8þ and CD4þ subsets(Supplementary Fig. S3D and S3E). Notably, BLZ945 decreasedmonocytes and macrophages in cultures with only cytokines (Fig.2D), but this did not lead to consistent changes of their T cell–activating capacities (Fig. 2F).
CSF-1R blockade reverted suppressive functions on tumor-educated human monocytes
Next, we cocultured primary monocytes freshly isolated fromhealthy individuals with three human neuroblastoma cell lines,according to our previously published protocol (35). This cocul-
ture model allowed us to dissect the impact of tumor-derivedfactors onmyeloid cells that are recruited into the neuroblastomatumor microenvironment.
The presence of neuroblastoma cells upregulated the expres-sion of CD14 (Fig. 3A) and CSF-1R (Fig. 3B), but impaired theexpression of HLA-DR and CD86 on primary human monocytes(Fig. 3A). However, different tumor cell lines demonstrateddistinct abilities to regulate the expression of CD206 (Fig. 3A)andPD-L1 (Supplementary Fig. S4A) onmonocytes. Importantly,CSF-1R inhibition did not influence growth of neuroblastomatumor cell lines due to their lack of CSF-1R expression (data notshown).
When CSF-1R was blocked, expression of CD14 and CD206(Fig. 3C) on tumor-educated monocytes was reduced, but expres-sion of HLA-DR and CD86 (Fig. 3C) was partially recovered.However, CSF-1R inhibition did not significantly alter the expres-sion of PD-L1 (Supplementary Fig. S4B) or CSF-1R (Supplemen-tary Fig. S4C) on tumor-educated monocytes.
16:18:14:10.6
0.8
1.0
1.2
T CellDendritic cells Monocytes/macrophages Macrophages
T cells:myeloid
T cells:myeloid
T cell:myeloid
T only
T + b
eads 4:
18:
116
:1
IFN
γ C
once
ntra
tion
(pg/
mL)
A
No cytokin
es
Cytokin
es
SK-N-B
E (2)
SK-N-F
I
SK-N-A
S
No cytokin
es
No cytokin
es
No cytokin
es
Cytokin
es
Cytokin
es
Cytokin
es
Cytokin
es
Cytokin
es
Cytokin
es
Cytokin
es
Cytokin
es
SK-N-B
E (2)
SK-N-B
E (2)
SK-N-B
E (2)
SK-N-F
I
SK-N-F
I
SK-N-F
I
SK-N-A
S
SK-N-A
S
SK-N-A
S
20
40
60
80
100
c-kit+CD14neg
GM-CSF + TNFαGM-CSF + TNFα GM-CSF + TNFα GM-CSF + TNFα
B C
0
5
10
15
20
25
Monocytes/macrophages Monocytes/macrophages
+BLZ945
+BLZ945
+BLZ945
+BLZ945
+BLZ945
BE(2)BE(2)
BE(2)BE(2)
BE(2)
BE(2) + B
LZ945
BE(2) + B
LZ945
BE(2) + B
LZ945
BE(2) + B
LZ945
BE(2)+BLZ9450
10
20
30
40
Dendritic cells
ns
0
5
10
15
20
Macrophages
*
*
D
40
50
60
70
80
Macrophages (CD206+)
% o
f CS
F-1
R+
Cel
ls
** ****
Rel
ativ
e C
SF
-1R
leve
ls
E
F
CSF-1R
Cel
l co
un
ts
Macrophages Mono/Macro
SK-N-BE(2)SK-N-BE(2)+BLZ945
SK-N-BE(2)BeadsT onlySK-N-BE(2)+BLZ945
T cell:myeloid = 8:1CFSE
GCD3+ T cells
IFNγ
40
30
20
10
0
60
40
20
0
60
40
20
0
% o
f CD
14ne
g C
ells
% o
f CD
14ne
g C
ells
% o
f Tot
al c
ells
% o
f Tot
al c
ells
% o
f Tot
al c
ells
Rel
ativ
e pr
olife
ratio
n
Rel
ativ
e pr
olife
ratio
n
% o
f Mye
loid
cel
ls%
of M
yelo
id c
ells
1.2
1.0
0.8
0.6
0.4
6,000
4,000
2,000
1,000
0
Cytokines
SK-N-AS SK-N-FISK-N-BE(2)
1.4
1.2
1.0
0.8
4:1 8:1 16:1
n = 6
Cytokines + BLZ945
SK-N-BE(2)
SK-N-BE(2) + BLZ945
Cytokines
**
**
**
* * **
*
Figure 2.Myelopoiesis of human CD34þ progenitor cells was modulated by tumor-derived factors. The impact of tumor-derived factors on differentiation of human CD34þ
hematopoietic stem cells was measured in the presence of TCM harvested from 3 human neuroblastoma cell lines (SK-N-BE(2), SK-N-AS, and SK-N-FI). A,frequencies of DCs (CD14negCD1aþCD11cþCD86þ), monocytes/macrophages (CD14þc-kitneg), macrophages (CD11bþCD68þ) or B, immature cells (c-kitþCD14neg)were evaluated by flow cytometry. C, the ability of myeloid cells matured in different conditions to stimulate proliferation and IFNg release from allogeneicT cells was tested in mixed lymphocyte reactions. The effects of the CSF-1R inhibitor BLZ945 on the maturation of CD34þ progenitor cells to different myeloidsubsets (D) and expression of CSF-1R on monocytes or macrophages (E) were tested by flow cytometry. F, stimulatory functions of myeloid cells on T-cellproliferation after CSF-1R inhibition were demonstrated. G, representative histograms of the T-cell proliferation were chosen based on average values. � , P < 0.05;�� , P < 0.01; nonparametric Mann–Whitney U test. Each dot represented an individual experiment.
Block CSF-1R to Improve Checkpoint Blockade in Neuroblastoma
www.aacrjournals.org Clin Cancer Res; 22(15) August 1, 2016 3853
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912
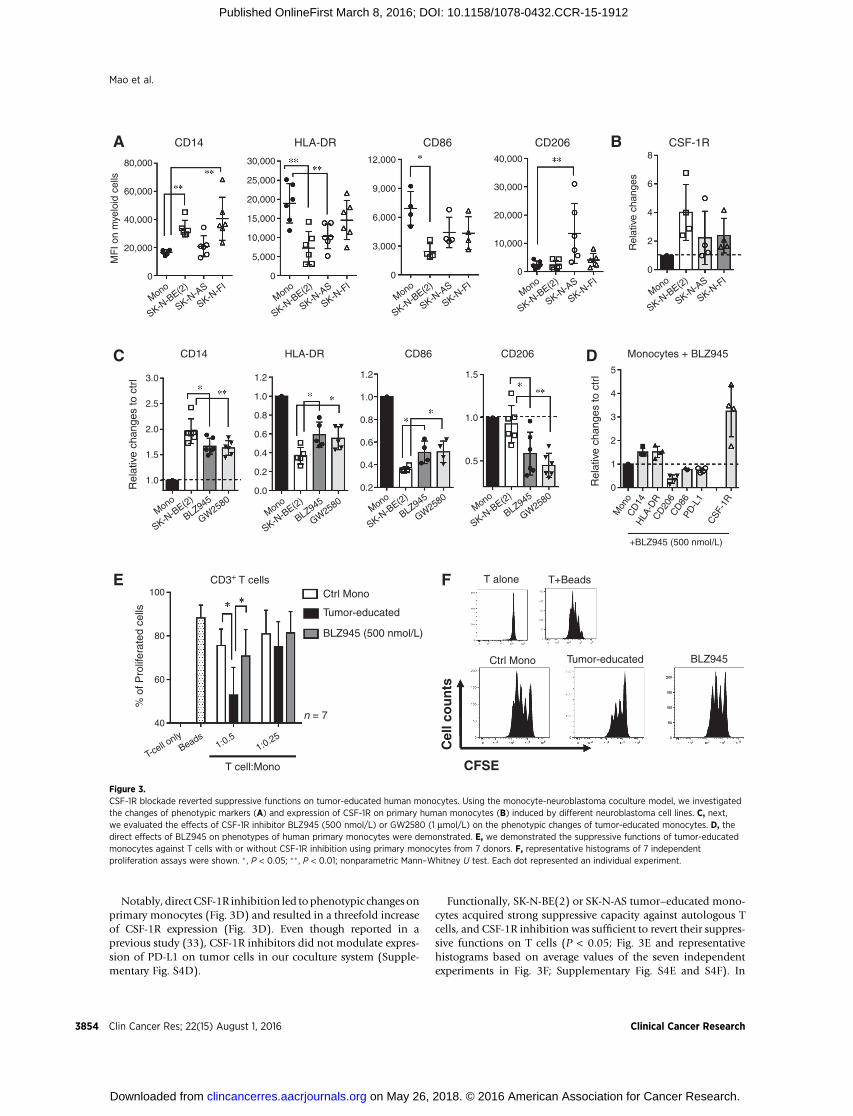
Notably, direct CSF-1R inhibition led tophenotypic changes onprimary monocytes (Fig. 3D) and resulted in a threefold increaseof CSF-1R expression (Fig. 3D). Even though reported in aprevious study (33), CSF-1R inhibitors did not modulate expres-sion of PD-L1 on tumor cells in our coculture system (Supple-mentary Fig. S4D).
Functionally, SK-N-BE(2) or SK-N-AS tumor–educated mono-cytes acquired strong suppressive capacity against autologous Tcells, and CSF-1R inhibition was sufficient to revert their suppres-sive functions on T cells (P < 0.05; Fig. 3E and representativehistograms based on average values of the seven independentexperiments in Fig. 3F; Supplementary Fig. S4E and S4F). In
80,000
60,000
40,000
20,000
0
30,000
25,000
20,000
15,000
10,000
5,000
0
12,000
9,000
6,000
3,000
0
40,000
30,000
20,000
10,000
0
8
6
4
2
0
MF
I on
mye
loid
cel
ls
Rel
ativ
e ch
ange
s
CD14 HLA-DR CD86 CD206 CSF-1R
MonoMono
MonoMono
Mono
SK-N-B
E(2)
SK-N-B
E(2)
SK-N-B
E(2)
SK-N-B
E(2)
SK-N-B
E(2)
SK-N-A
S
SK-N-A
S
SK-N-A
S
SK-N-A
S
SK-N-A
S
SK-N-F
I
MonoMono
MonoMono
Mon
o
CD206
CD86PD
-L1
CSF
-1R
CD
14H
LA-D
R
SK-N-B
E(2)
SK-N-B
E(2)
SK-N-B
E(2)
SK-N-B
E(2)
BLZ945
BLZ945BLZ945
BLZ945
GW2580
GW2580
GW2580
GW2580
SK-N-F
I
SK-N-F
I
SK-N-F
I
SK-N-F
I
+BLZ945 (500 nmol/L)
3.0
2.5
2.0
1.5
1.0
1.2
1.0
0.8
0.6
0.4
0.2
0.0
1.2
1.0
0.8
0.6
0.4
0.2
1.5
1.0
0.5
5
4
3
2
1
0
CD14
CD3+ T cells
HLA-DR CD86 CD206 Monocytes + BLZ945
Rel
ativ
e ch
ange
s to
ctr
l
Rel
ativ
e ch
ange
s to
ctr
l100
80
60
40
% o
f Pro
lifer
ated
cel
ls
Ctrl Mono
Tumor-educated
BLZ945 (500 nmol/L)
n = 7
T-cell o
nlyBeads
1:0.51:0.25
T cell:Mono
T alone T+Beads
Ctrl Mono BLZ945Tumor-educated
A
C
E F
D
B
CFSE
Cel
l co
un
ts
Figure 3.CSF-1R blockade reverted suppressive functions on tumor-educated human monocytes. Using the monocyte-neuroblastoma coculture model, we investigatedthe changes of phenotypic markers (A) and expression of CSF-1R on primary human monocytes (B) induced by different neuroblastoma cell lines. C, next,we evaluated the effects of CSF-1R inhibitor BLZ945 (500 nmol/L) or GW2580 (1 mmol/L) on the phenotypic changes of tumor-educated monocytes. D, thedirect effects of BLZ945 on phenotypes of human primary monocytes were demonstrated. E, we demonstrated the suppressive functions of tumor-educatedmonocytes against T cells with or without CSF-1R inhibition using primary monocytes from 7 donors. F, representative histograms of 7 independentproliferation assays were shown. � , P < 0.05; ��, P < 0.01; nonparametric Mann–Whitney U test. Each dot represented an individual experiment.
Mao et al.
Clin Cancer Res; 22(15) August 1, 2016 Clinical Cancer Research3854
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912
contrast, treatment of healthy monocytes with BLZ945 did notshow changes in their functions toward T-cell proliferation (Sup-plementary Fig. S4G).
BLZ945 elicited therapeutic effects through modulatingsuppressive myeloid cells
To confirm that the M-CSF/CSF-1R interaction in mice isinvolved in the development of suppressive myeloid cells, weestablished an in vitro model based on a previous study (36).Na€�ve bone marrow cells were differentiated in the presence ofNHO2A neuroblastoma tumor-conditioned medium (TCM) orrecombinant murine M-CSF. Similar to the results using primaryhuman myeloid cells, TCM and M-CSF induced expansion ofMDSCs and TAMs (Supplementary Fig. S5A), and the resultedcells inhibited proliferation of autologous T cells (SupplementaryFig. S5B). Blocking CSF-1R signaling efficiently abolished theinduction of CSF-1R–expressing suppressive myeloid cells (Sup-plementary Fig. S5C and S5D) and recovered functions of T cells(Supplementary Fig. S5E).
Motivated by our in vitro findings, we sought to evaluate thetherapeutic potential of BLZ945 in vivo in a transgenic murine
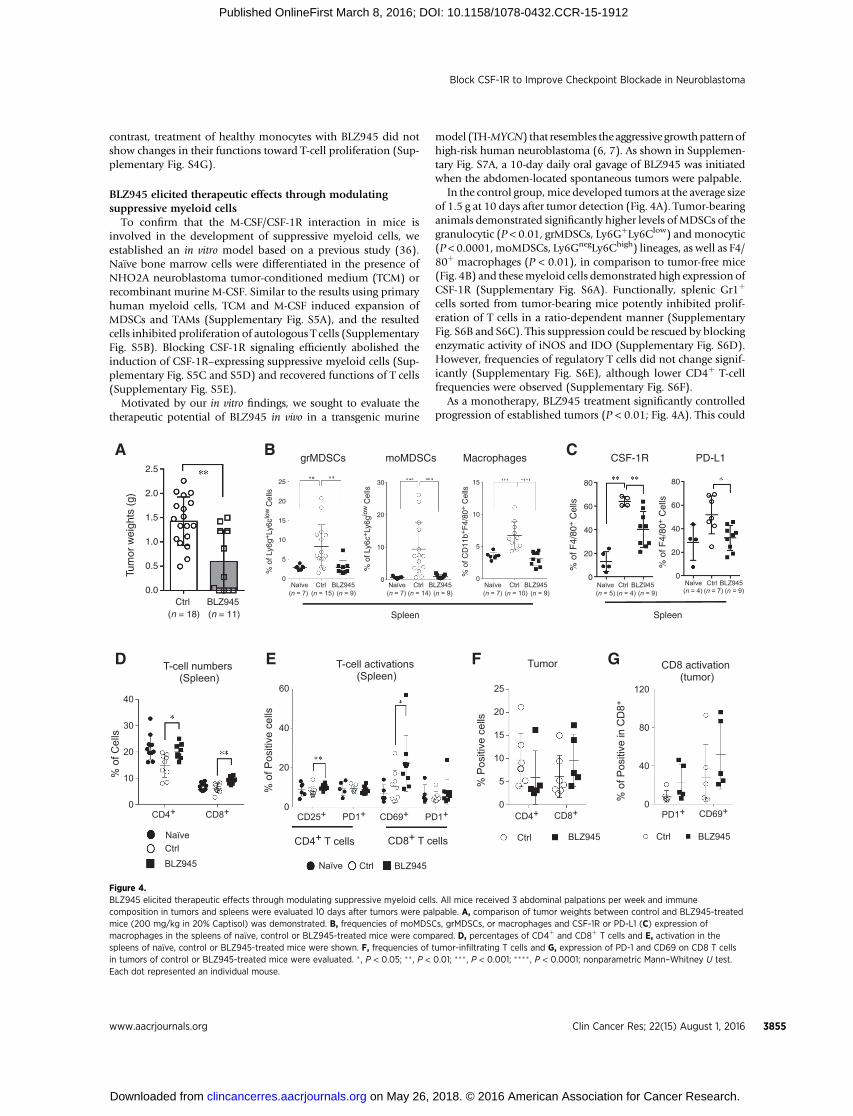
model (TH-MYCN) that resembles the aggressive growthpatternofhigh-risk human neuroblastoma (6, 7). As shown in Supplemen-tary Fig. S7A, a 10-day daily oral gavage of BLZ945 was initiatedwhen the abdomen-located spontaneous tumors were palpable.
In the control group,mice developed tumors at the average sizeof 1.5 g at 10 days after tumor detection (Fig. 4A). Tumor-bearinganimals demonstrated significantly higher levels of MDSCs of thegranulocytic (P < 0.01, grMDSCs, Ly6GþLy6Clow) andmonocytic(P < 0.0001,moMDSCs, Ly6GnegLy6Chigh) lineages, as well as F4/80þ macrophages (P < 0.01), in comparison to tumor-free mice(Fig. 4B) and thesemyeloid cells demonstrated high expression ofCSF-1R (Supplementary Fig. S6A). Functionally, splenic Gr1þ
cells sorted from tumor-bearing mice potently inhibited prolif-eration of T cells in a ratio-dependent manner (SupplementaryFig. S6B and S6C). This suppression could be rescued by blockingenzymatic activity of iNOS and IDO (Supplementary Fig. S6D).However, frequencies of regulatory T cells did not change signif-icantly (Supplementary Fig. S6E), although lower CD4þ T-cellfrequencies were observed (Supplementary Fig. S6F).
As a monotherapy, BLZ945 treatment significantly controlledprogression of established tumors (P < 0.01; Fig. 4A). This could
2.5
2.0
1.5
1.0
0.5
0.0
Tum
or w
eigh
ts (
g)
grMDSCs moMDSCs Macrophages CSF-1R PD-L1
Ctrl BLZ945(n = 18) (n = 11) Spleen Spleen
25
20
15
10
5
0
30
20
10
0
15
10
5
0
80
60
40
20
0
80
60
40
20
0
% o
f Ly6
g+Ly
6clo
w C
ells
% o
f Ly6
c+Ly
6glo
w C
ells
% o
f CD
11b+
F4/
80+ C
ells
% o
f F4/
80+ C
ells
% o
f F4/
80+ C
ells
Naïve
Naïve
Naïve
Naïve Naïve NaïveCtrl Ctrl
Ctrl
Ctrl
Ctrl CtrlBLZ945 BLZ945
BLZ945 BLZ945
BLZ945 BLZ945 Naïve Ctrl BLZ945(n = 15) (n = 14) (n = 10) (n = 4) (n = 9)(n = 7) (n = 7) (n = 7) (n = 5) (n = 4) (n = 7) (n = 9)(n = 9) (n = 9) (n = 9)
40
30
20
10
0
60
40
20
0
25
20
15
10
5
0
120
80
40
0
% o
f Cel
ls
% o
f Pos
itive
cel
ls
% o
f Pos
itive
in C
D8+
% P
ositi
ve c
ells
CD4+ CD25+ CD69+ CD4+
CD4+ T cells CD8+ T cells
CD8+ CD69+PD1+ PD1+ PD1+CD8+
T-cell numbers (Spleen)
T-cell activations (Spleen)
CD8 activation (tumor)
Tumor
Ctrl CtrlBLZ945 BLZ945
A
D E F G
B C
Figure 4.BLZ945 elicited therapeutic effects through modulating suppressive myeloid cells. All mice received 3 abdominal palpations per week and immunecomposition in tumors and spleens were evaluated 10 days after tumors were palpable. A, comparison of tumor weights between control and BLZ945-treatedmice (200 mg/kg in 20% Captisol) was demonstrated. B, frequencies of moMDSCs, grMDSCs, or macrophages and CSF-1R or PD-L1 (C) expression ofmacrophages in the spleens of na€�ve, control or BLZ945-treated mice were compared. D, percentages of CD4þ and CD8þ T cells and E, activation in thespleens of na€�ve, control or BLZ945-treated mice were shown. F, frequencies of tumor-infiltrating T cells and G, expression of PD-1 and CD69 on CD8 T cellsin tumors of control or BLZ945-treated mice were evaluated. � , P < 0.05; �� , P < 0.01; ��� , P < 0.001; ���� , P < 0.0001; nonparametric Mann–Whitney U test.Each dot represented an individual mouse.
Block CSF-1R to Improve Checkpoint Blockade in Neuroblastoma
www.aacrjournals.org Clin Cancer Res; 22(15) August 1, 2016 3855
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912

be explained by the selective effects on CSF-1R–expressing mye-loid cells, because numbers of CD11bþ cells (P < 0.01; Supple-mentary Fig. S7B), including grMDSCs (P < 0.01), moMDSCs(P < 0.001), and macrophages (P < 0.0001), were significantlyreduced in spleens of the treated mice (Fig. 4B). Further, BLZ945treatment strongly limited the expression of CSF-1R (P < 0.01)and PD-L1 (P < 0.01) on macrophages and MDSCs (Fig. 4C andSupplementary Fig. S7C).
In addition, treatment with BLZ945 led to a significant recoveryof CD4þ (P < 0.05) and CD8þ (P < 0.01) T cells in spleens(Fig. 4D). Activation of CD4þ and CD8þ T cells were improved(Fig. 4E), demonstrated by significantly higher expression ofCD25 (P < 0.05) and CD69 (P < 0.01). However, the CD4/CD8ratio remained unchanged in the treated mice when comparedwith the controls (Supplementary Fig. S7D).
In tumors of BLZ945-treated mice, neither did we observechanges in CD11bþ cell numbers (Supplementary Fig. S7E), northe frequencies of grMDSCs, moMDSCs, and macrophages (Sup-plementary Fig. S7F). However, lower numbers of infiltratingCD4þ T cells but increased CD8þ T cells were observed (Fig.4F), leading to decreased CD4/CD8 ratios (SupplementaryFig. S7G). Moreover, tumor-infiltrating CD8þ (Fig. 4G) andCD4þ T cells (Supplementary Fig. S7H) were more activated inBLZ945-treated mice.
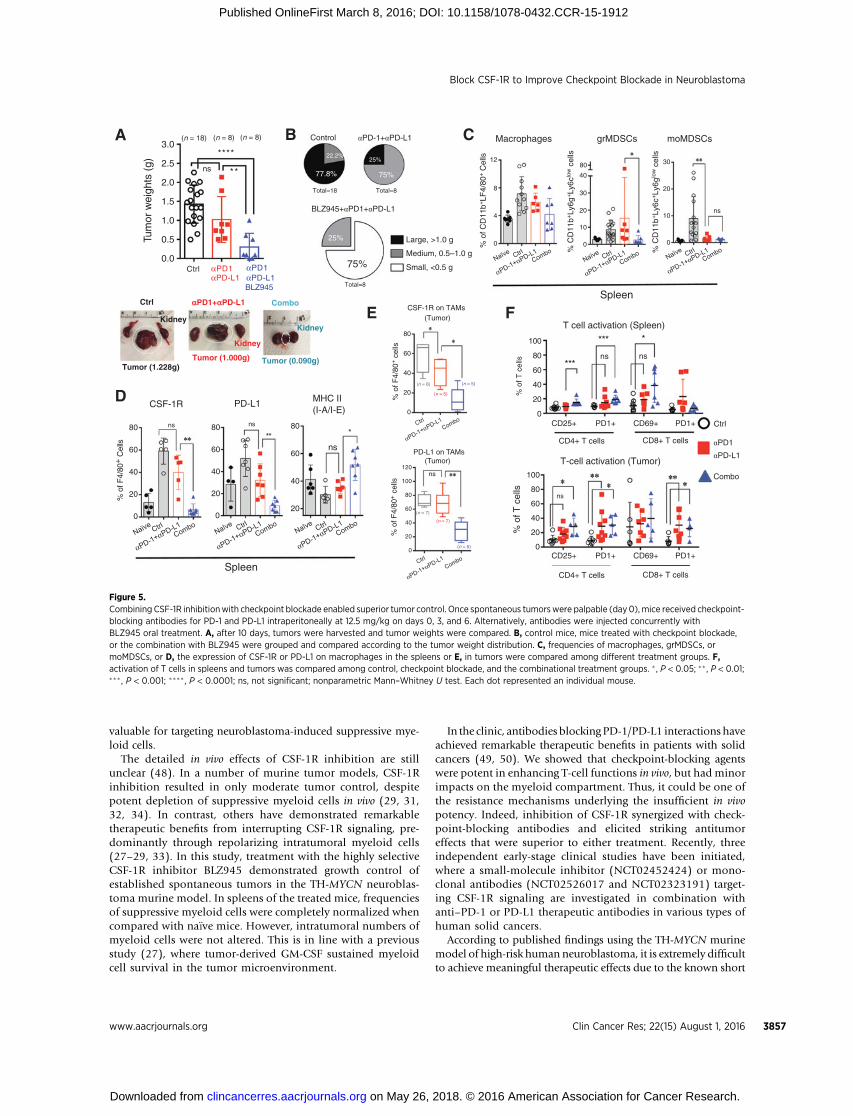
Combining CSF-1R inhibition with checkpoint blockadeenabled superior tumor control
Given that suppressive myeloid cells expressed PD-L1 at highlevels and T cells demonstrated elevated PD-1 expression afterCSF-1R inhibition, we asked whether checkpoint blockade couldfurther improve the therapeutic effects of BLZ945 treatment.Given that PD-1 has multiple ligands (41) and PD-L1 couldinhibit functions of PD-1–deficient T cells (42), we treatedtumor-bearing mice with blocking antibodies against both PD-1 and PD-L1 to maximize the blockade of this pathway, asmonotherapy or combined with BLZ945 (Supplementary Fig.S8A). This therapeutic scheme was motivated by the observationthat both PD-L1 and PD-L2 were positively correlated with CSF-1R expression in human neuroblastoma (R ¼ 0.542 and 0.512,respectively; Supplementary Fig. S8B).
To our surprise, antagonizing the PD-1/PD-L1 axis (Fig. 5A) orthe isotype controls (data not shown) did not delay progression ofestablished spontaneous tumors in the TH-MYCN mice. In con-trast, the addition of BLZ945 to the checkpoint-blocking agentsresulted in potent synergistic antitumor effects (P < 0.05; Fig. 5A).In6of8 (75%) treatedmice (Fig. 5B),weobserved complete tumorregression or small tumors (<0.5 g). Strikingly, the combinationprevented the progressive growth of large tumors (>1.0 g; Fig. 5B).
In comparison to checkpoint blockade, the combination effi-ciently normalized the numbers of CD11bþ cells (P < 0.05;Supplementary Fig. S8C), including macrophages and grMDSCs(P < 0.05; Fig. 5C), and all treatments could diminish moMDSCs(Fig. 5C) in the spleens. Strikingly, the combined treatmentreactivated macrophages in spleens, demonstrated by decreasednumbers of CSF-1Rþ (P < 0.01) or PD-L1þ (P < 0.01) macro-phages, but increased expression of MHC class II (P < 0.05) onmacrophages (Fig. 5D). Although intratumoral frequencies ofCD11bþ cells (Supplementary Fig. S8D) or macrophages (Sup-plementary Fig. S8E) were not significantly modulated, weobserved similar effects on macrophages in tumor tissues(Fig. 5E). Despite the low total numbers, there was also a marked
reduction of grMDSCs (P < 0.05) andmoMDSCs in the tumors ofmice received combination therapy (Supplementary Fig. S8E).
When checkpoint-blocking agents were administered, weobserved enhanced infiltration (Supplementary Fig. S8G) andactivation (Fig. 5F) of T cells in tumor tissues. It was morepronounced on CD4þ T cells, leading to increased CD4/CD8ratios (Supplementary Fig. S8F and S8G). Removal of suppressivemyeloid cells by CSF-1R inhibition did not further elevate T-cellnumbers in spleens (Supplementary Fig. S8F) or tumors (Sup-plementary Fig. S8G), but boosted activation of T cells (Fig. 5F).
DiscussionStimulated by the finding that CSF-1Rþmyeloid cells in tumors
of patients with neuroblastoma predict poor overall and disease-free survival, we investigated the functional and therapeutic sig-nificance of targeting CSF-1R signaling in this study. The essentialrole of the tumor-drivenM-CSF/CSF-1R axis on the differentiationand activation of suppressive myeloid cells is dissected by in vitromodels, reflecting myelopoiesis in the bone marrow or activationof myeloid cells in the tumor microenvironment. As therapeuticapproaches, the highly selective CSF-1R inhibitor BLZ945 elicitsstrong antitumor effects against established aggressive spontane-ous tumors in vivo and could exert superior tumor control whencombined with PD-1/PD-L1–blocking antibodies.
Emerging evidence has demonstrated that tumor-derivedinflammatory mediators support circulating hematopoietic pro-genitor cells to engraft in the spleen, leading to splenic productionof immature suppressive myeloid cells (43, 44). Previous studieshave confirmed the importance of GM-CSF, G-CSF, and IL6 (36,45) in driving the differentiation of suppressive myeloid cellsfrommurine bonemarrow.However, the detailedmechanisms ofthis effect in humans remain elusive. In a recent study, Wu andcolleagues confirmed the role of GM-CSF and IL6 during theexpansion of MDSCs from human CD34þ hematopoietic pro-genitor cells (46).
Utilizing murine bone marrow cells, human CD34þ hemato-poietic progenitor cells and a human monocyte-neuroblastomacoculture model, we provided insightful results emphasizing thekey contribution of the M-CSF/CSF-1R pathway during the induc-tion of MDSCs and TAMs. Notably, CSF-1R inhibition did notrescue frequencies of DCs after in vitro differentiation of CD34þ
progenitor cells. This indicated that other tumor-derived factors,such as IL10 or VEGF, might be responsible for the suppression.Given that long-term in vitro culture could result in growth ofselected tumor cell subsets, it is important to validate thesefindingswith primary or short-passage human neuroblastoma cells (47).
It is well documented that MDSCs and TAMs are associatedwith poor clinical outcome in patientswith solid tumors (22–24).In human neuroblastoma, tumor-infiltrating inflammatory mye-loid cells were analyzed by immunohistochemistry stainings in133metastatic tumors anddemonstrated independentprognosticvalues (4). However, due to the extremely heterogeneous natureof suppressive myeloid cells, it remains challenging to specificallytarget these cells. Our analyses revealed that human neuroblas-toma produced M-CSF at high levels and recruited substantialnumbers of CSF-1Rþmyeloid cells. These findings were validatedin vitro utilizing human neuroblastoma cell lines and primarymonocytes. Indeed, M-CSF levels as well as numbers of CSF-1Rþ
myeloid cells in human tumors predicted worse patient outcome.Collectively, we proposed that theM-CSF/CSF-1R axis is clinically
Mao et al.
Clin Cancer Res; 22(15) August 1, 2016 Clinical Cancer Research3856
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912
valuable for targeting neuroblastoma-induced suppressive mye-loid cells.
The detailed in vivo effects of CSF-1R inhibition are stillunclear (48). In a number of murine tumor models, CSF-1Rinhibition resulted in only moderate tumor control, despitepotent depletion of suppressive myeloid cells in vivo (29, 31,32, 34). In contrast, others have demonstrated remarkabletherapeutic benefits from interrupting CSF-1R signaling, pre-dominantly through repolarizing intratumoral myeloid cells(27–29, 33). In this study, treatment with the highly selectiveCSF-1R inhibitor BLZ945 demonstrated growth control ofestablished spontaneous tumors in the TH-MYCN neuroblas-toma murine model. In spleens of the treated mice, frequenciesof suppressive myeloid cells were completely normalized whencompared with na€�ve mice. However, intratumoral numbers ofmyeloid cells were not altered. This is in line with a previousstudy (27), where tumor-derived GM-CSF sustained myeloidcell survival in the tumor microenvironment.
In the clinic, antibodies blocking PD-1/PD-L1 interactions haveachieved remarkable therapeutic benefits in patients with solidcancers (49, 50). We showed that checkpoint-blocking agentswere potent in enhancing T-cell functions in vivo, but had minorimpacts on the myeloid compartment. Thus, it could be one ofthe resistance mechanisms underlying the insufficient in vivopotency. Indeed, inhibition of CSF-1R synergized with check-point-blocking antibodies and elicited striking antitumoreffects that were superior to either treatment. Recently, threeindependent early-stage clinical studies have been initiated,where a small-molecule inhibitor (NCT02452424) or mono-clonal antibodies (NCT02526017 and NCT02323191) target-ing CSF-1R signaling are investigated in combination withanti–PD-1 or PD-L1 therapeutic antibodies in various types ofhuman solid cancers.
According to published findings using the TH-MYCN murinemodel of high-risk human neuroblastoma, it is extremely difficultto achieve meaningful therapeutic effects due to the known short
0.0
0.5
1.0
1.5
2.0
2.5
3.0(n = 18) (n = 8)
ns **
(n = 8)
Ctrl αPD1αPD-L1
αPD1αPD-L1BLZ945
****
A B C Macrophages
% C
D11
b+Ly
6g+Ly
6clo
w c
ells
% C
D11
b+Ly
6c+Ly
6glo
w c
ells
% o
f CD
11b+
LF4/
80+ C
ells
grMDSCs moMDSCs
Spleen
D
PD-L1 on TAMs(Tumor)
% o
f F4/
80+ c
ells
% o
f F4/
80+ c
ells
% o
f T c
ells
CSF-1R on TAMs (Tumor) FE
PD1+CD69+PD1+CD25+
PD1+CD69+PD1+CD25+
0
20
40
60
80
100
***
***
ns ns
ns
*
CD4+ T cells CD8+ T cells
CD4+ T cells CD8+ T cells
T cell activation (Spleen)
% o
f T c
ells
T-cell activation (Tumor)
Ctrl
αPD1
αPD-L1
Combo
Ctrl
Kidney
Tumor (1.228g)
aPD1+aPD-L1
Kidney
Tumor (1.000g)
Combo
Kidney
Tumor (0.090g)
Spleen
% o
f F4/
80+
Cel
ls
CSF-1R
0
20
40
60
80 ns
**
PD-L1
20
40
60
80
ns
ns
ns
*
MHC II(I-A/I-E)
Large, >1.0 g
Medium, 0.5–1.0 g
Small, <0.5 g
Control
Total=18
77.8%
22.2%
αPD-1+αPD-L1
Total=8
75%
25%
BLZ945+αPD1+αPD-L1
Total=8
75%
25%Tum
or w
eigh
ts (
g)
ns80
60
40
20
0
Naïve Ctrl Ctrl CtrlNaïve
NaïveCombo
ComboCombo
αPD-1+αPD-L1
NaïveNaïve Naïve
Ctrl
Ctrl
Ctrl CtrlCombo
Combo
ComboCombo
αPD-1+αPD-L1
αPD-1+αPD-L1
CtrlCombo
αPD-1+αPD-L1
αPD-1+αPD-L1
αPD-1+αPD-L1
αPD-1+αPD-L1
αPD-1+αPD-L1
12
8
4
0
80
40
30
20
10
0
30
20
10
0
80
60
40
20
0
100
80
60
40
20
0
120
100
80
60
40
20
0
(n = 6)
(n = 7)
(n = 5)
(n = 7)
(n = 5)
(n = 5)
Figure 5.Combining CSF-1R inhibitionwith checkpoint blockade enabled superior tumor control. Once spontaneous tumorswere palpable (day0), mice received checkpoint-blocking antibodies for PD-1 and PD-L1 intraperitoneally at 12.5 mg/kg on days 0, 3, and 6. Alternatively, antibodies were injected concurrently withBLZ945 oral treatment. A, after 10 days, tumors were harvested and tumor weights were compared. B, control mice, mice treated with checkpoint blockade,or the combination with BLZ945 were grouped and compared according to the tumor weight distribution. C, frequencies of macrophages, grMDSCs, ormoMDSCs, or D, the expression of CSF-1R or PD-L1 on macrophages in the spleens or E, in tumors were compared among different treatment groups. F,activation of T cells in spleens and tumors was compared among control, checkpoint blockade, and the combinational treatment groups. � , P < 0.05; �� , P < 0.01;��� , P < 0.001; ���� , P < 0.0001; ns, not significant; nonparametric Mann–Whitney U test. Each dot represented an individual mouse.
Block CSF-1R to Improve Checkpoint Blockade in Neuroblastoma
www.aacrjournals.org Clin Cancer Res; 22(15) August 1, 2016 3857
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912

treatment window and rapid tumor progression once the tumorsare established andpalpable (7, 12).Novel therapeuticmodalitiesof these animals developed by us and others (9, 10) resulted inprolonged survival, but in contrast to current findings, not a singleanimal was cured or even showed shrinkage of tumors. However,due to abdominally located tumors in the TH-MYCN mice, it ischallenging to determine an objective endpoint to monitor thelongitudinal effects of the treatment. Facilitated by imaging tools,several well-establishedmurinemodels of neuroblastoma shouldbe used, in order to further explore the mechanistic insights andlongitudinal impact of combination immunotherapy (51–54).
Taken together, we conclude that tumor-driven CSF-1R signal-ing regulates the induction of suppressive myeloid cells, whichhampers antitumor effects of checkpoint inhibitors. Given thatthe therapeutic efficacy of PD-1 blockade and prognostic values ofsuppressive myeloid cells are being validated in clinical trials, ourresults support CSF-1R inhibition as a novel treatment option forimmunotherapy in human cancers, including high-risk child-hood neuroblastoma.
Disclosure of Potential Conflicts of InterestP. Kogner is a consultant/advisory board member for Roche/Genentech.
No potential conflicts of interest were disclosed by the other authors.
Authors' ContributionsConception and design: Y. Mao, N. Eissler, J.I. Johnsen, P. Kogner, R. KiesslingDevelopment of methodology: Y. Mao, N. Eissler, K. Le Blanc, P. Kogner,R. KiesslingAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): Y. Mao, N. Eissler, P. Kogner, R. KiesslingAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): Y. Mao, N. Eissler, P. Kogner
Writing, review, and/or revision of the manuscript: Y. Mao, N. Eissler,K. Le Blanc, J.I. Johnsen, P. Kogner, R. KiesslingAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): P. KognerStudy supervision: J.I. Johnsen, P. Kogner, R. Kiessling
AcknowledgmentsThe authors appreciate the excellent technical support from colleagues Ms.
Lotta Elfman, Anna Kock (doctoral candidate), and Andrew Sheppard at theKarolinska Institutet. They are grateful for the help fromMs. Sandra Olsson andMichelle Gustafsson from the AKM animal facility at the Karolinska Institutetduring animal experiments. They thank Dr. Cecilia G€otherstr€om, Ulrica Askel€of(doctoral candidate), and Ms. Malin Hjertqvist at the Karolinska UniversityHospital (Huddinge) for coordinating the cord bloodmaterials. They also thankNovartis for providing the BLZ945 compound and the assistance received fromDr. James Sutton (Novartis) and Dr. Dylan Daniel during the process.
Grant SupportThe study was generously supported by grants received from the Swedish
Cancer Society, the Swedish Medical Research Council, the Cancer Society ofStockholm, the ALF-project grant from Stockholm City Council, the Knut andAlice Wallenberg's Foundation, the Karolinska-sponsored Center for ImmuneModulatory Therapies for Autoimmunity and Cancer (IMTAC), the SwedishChildhood Cancer Foundation, and the Neuroblastoma and CNS TumorNetwork of Sweden (NBCNS). In addition, N. Eissler is supported by apostdoctoral fellowship from the Swedish Childhood Cancer Foundation andY. Mao received an individual grant from the Robert Lundberg MemorialFoundation.
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received August 11, 2015; revised February 9, 2016; accepted February 23,2016; published OnlineFirst March 8, 2016.
References1. Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and ther-
apy. Annu Rev Med 2015;66:49–63.2. Johnsen JI, Kogner P, Albihn A, Henriksson MA. Embryonal neural
tumours and cell death. Apoptosis 2009;14:424–38.3. Song L, Asgharzadeh S, Salo J, Engell K, WuHW, Sposto R, et al. Valpha24-
invariant NKT cells mediate antitumor activity via killing of tumor-asso-ciated macrophages. J Clin Invest 2009;119:1524–36.
4. Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, Berthold F, et al.Clinical significance of tumor-associated inflammatory cells in metastaticneuroblastoma. J Clin Oncol 2012;30:3525–32.
5. Asgharzadeh S, Pique-Regi R, Sposto R, Wang H, Yang Y, Shimada H, et al.Prognostic significance of gene expression profiles of metastatic neuro-blastomas lacking MYCN gene amplification. J Natl Cancer Inst 2006;98:1193–203.
6. Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targetedexpression of MYCN causes neuroblastoma in transgenic mice. EMBO J1997;16:2985–95.
7. Rasmuson A, Segerstrom L, Nethander M, Finnman J, Elfman LH, Javan-mardi N, et al. Tumor development, growth characteristics and spectrumofgenetic aberrations in the TH-MYCNmousemodel of neuroblastoma. PloSOne 2012;7:e51297.
8. Carlson LM, Rasmuson A, Idborg H, Segerstrom L, Jakobsson PJ, Sveinb-jornsson B, et al. Low-dose aspirin delays an inflammatory tumor pro-gression in vivo in a transgenic mouse model of neuroblastoma. Carcino-genesis 2013;34:1081–8.
9. ZirathH,FrenzelA,OliynykG,SegerstromL,WestermarkUK,LarssonK, etal.MYC inhibition induces metabolic changes leading to accumulation of lipiddroplets in tumor cells. Proc Natl Acad Sci USA 2013;110:10258–63.
10. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al.Targeting MYCN in neuroblastoma by BET bromodomain inhibition.Cancer Discov 2013;3:308–23.
11. Chesler L, Goldenberg DD, Seales IT, Satchi-Fainaro R, Grimmer M,Collins R, et al. Malignant progression and blockade of angiogenesis ina murine transgenic model of neuroblastoma. Cancer Res 2007;67:9435–42.
12. Chesler L, Weiss WA. Genetically engineeredmurinemodels–contributionto our understanding of the genetics, molecular pathology and therapeutictargeting of neuroblastoma. Sem Cancer Biol 2011;21:245–55.
13. Mackall CL, Merchant MS, Fry TJ. Immune-based therapies for childhoodcancer. Nat Rev Clin Oncol 2014;11:693–703.
14. Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancergenomics and immunotherapy. Nat Rev Cancer 2013;13:397–411.
15. Mao Y, Poschke I, Kiessling R. Tumour-induced immune suppression: roleof inflammatory mediators released by myelomonocytic cells. J Int Med2014;276:154–70.
16. MaoY, SarhanD, StevenA, Seliger B, Kiessling R, Lundqvist A. Inhibition oftumor-derived prostaglandin-e2 blocks the induction of myeloid-derivedsuppressor cells and recovers natural killer cell activity. Clin Cancer Res2014;20:4096–106.
17. GabrilovichDI,Nagaraj S.Myeloid-derived suppressor cells as regulators ofthe immune system. Nat Rev Immunol 2009;9:162–74.
18. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance,tolerance, and diversity. Curr Opin Immunol 2010;22:231–7.
19. Peranzoni E, Zilio S,Marigo I, Dolcetti L, Zanovello P,Mandruzzato S, et al.Myeloid-derived suppressor cell heterogeneity and subset definition. CurrOpin Immunol 2010;22:238–44.
20. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S.Cross-talk between myeloid-derived suppressor cells and macrophagessubverts tumor immunity toward a type 2 response. J Immunol 2007;179:977–83.
21. Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM,et al. Myeloid-derived suppressor cells predict survival of patients with
Mao et al.
Clin Cancer Res; 22(15) August 1, 2016 Clinical Cancer Research3858
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912

advancedmelanoma: comparisonwith regulatory T cells andNY-ESO-1- ormelan-A-specific T cells. Clin Cancer Res 2014;20:1601–9.
22. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ,Montero AJ. Increased circulating myeloid-derived suppressor cells corre-late with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother 2009;58:49–59.
23. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophagesin tumour progression: implications for new anticancer therapies. J Pathol2002;196:254–65.
24. Huang A, Zhang B, Wang B, Zhang F, Fan KX, Guo YJ. Increased CD14(þ)HLA-DR (-/low)myeloid-derived suppressor cells correlate with extrathor-acic metastasis and poor response to chemotherapy in non–small cell lungcancer patients. Cancer Immunol Immunother 2013;62:1439–51.
25. Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflam-mation. Curr Opin Immunol 2006;18:39–48.
26. Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growthfactor CSF-1 in mammary gland development and tumor progression. JMammary Gland Biol Neoplasia 2002;7:147–62.
27. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, QuailDF, et al. CSF-1R inhibition alters macrophage polarization and blocksglioma progression. Nat Med 2013;19:1264–72.
28. Ries CH, CannarileMA,Hoves S, Benz J,Wartha K, Runza V, et al. Targetingtumor-associated macrophages with anti-CSF-1R antibody reveals a strat-egy for cancer therapy. Cancer Cell 2014;25:846–59.
29. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al.CSF1R inhibition delays cervical and mammary tumor growth inmurine models by attenuating the turnover of tumor-associated macro-phages and enhancing infiltration by CD8 T cells. Oncoimmunology2013;2:e26968.
30. DeNardoDG, BrennanDJ, Rexhepaj E, Ruffell B, Shiao SL,Madden SF, et al.Leukocyte complexity predicts breast cancer survival and functionallyregulates response to chemotherapy. Cancer Discov 2011;1:54–67.
31. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1Rsignaling blockade stanches tumor-infiltrating myeloid cells andimproves the efficacy of radiotherapy in prostate cancer. Cancer Res2013;73:2782–94.
32. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1receptor improves the antitumor efficacy of adoptive cell transfer immu-notherapy. Cancer Res 2014;74:153–61.
33. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1Rblockade reprograms tumor-infiltratingmacrophages and improvesresponse to T-cell checkpoint immunotherapy inpancreatic cancermodels.Cancer Res 2014;74:5057–69.
34. Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX,Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells byinhibiting CSF-1 receptor: combating tumor evasion of antiangiogenictherapy. Blood 2010;115:1461–71.
35. Mao Y, Poschke I, Wennerberg E, Pico de Coana Y, Egyhazi Brage S, SchultzI, et al. Melanoma-educated CD14þ cells acquire a myeloid-derivedsuppressor cell phenotype through COX-2-dependent mechanisms.Cancer Res 2013;73:3877–87.
36. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates mye-loid inflammation and T cell immunity in pancreatic cancer. Cancer Cell2012;21:822–35.
37. Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van derPloeg I, et al. Sequencing of neuroblastoma identifies chromothripsis anddefects in neuritogenesis genes. Nature 2012;483:589–93.
38. Hero B, Simon T, Spitz R, Ernestus K, Gnekow AK, Scheel-Walter HG, et al.Localized infant neuroblastomas often show spontaneous regression:results of the prospective trials NB95-S and NB97. J Clin Oncol 2008;26:1504–10.
39. LastowskaM, Viprey V, Santibanez-KorefM,Wappler I, Peters H, CullinaneC, et al. Identification of candidate genes involved in neuroblastomaprogression by combining genomic and expression microarrays withsurvival data. Oncogene 2007;26:7432–44.
40. De Palma M, Lewis CE. Macrophage regulation of tumor responses toanticancer therapies. Cancer Cell 2013;23:277–86.
41. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present,and future. J Clin Invest 2015;125:3384–91.
42. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmeddeath-1 ligand 1 interacts specificallywith the B7-1 costimulatorymoleculeto inhibit T cell responses. Immunity 2007;27:111–22.
43. Bronte V, Pittet MJ. The spleen in local and systemic regulation of immu-nity. Immunity 2013;39:806–18.
44. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A,Berger C, et al. Origins of tumor-associated macrophages and neutrophils.Proc Natl Acad Sci U S A 2012;109:2491–6.
45. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbetatranscription factor. Immunity 2010;32:790–802.
46. Wu WC, Sun HW, Chen HT, Liang J, Yu XJ, Wu C, et al. Circulatinghematopoietic stem and progenitor cells are myeloid-biased in cancerpatients. Proc Natl Acad Sci U S A 2014;111:4221–6.
47. Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, Biedler JL, et al.Characteristics of stem cells from human neuroblastoma cell lines and intumors. Neoplasia 2004;6:838–45.
48. Bronte V, Murray PJ. Understanding local macrophage phenotypes indisease: modulating macrophage function to treat cancer. Nat Med 2015;21:117–9.
49. Hamid O, Robert C, Daud A, Hodi FS, HwuWJ, Kefford R, et al. Safety andtumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl JMed 2013;369:134–44.
50. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A(anti-PD-L1) treatment leads to clinical activity in metastatic bladdercancer. Nature 2014;515:558–62.
51. Kroesen M, Brok IC, Reijnen D, van Hout-Kuijer MA, Zeelenberg IS, DenBrok MH, et al. Intra-adrenal murine TH-MYCN neuroblastoma tumorsgrow more aggressive and exhibit a distinct tumor microenvironmentrelative to their subcutaneous equivalents. Cancer Immunol Immunother2015;64:563–72.
52. Kroesen M, Nierkens S, Ansems M, Wassink M, Orentas RJ, Boon L, et al. Atransplantable TH-MYCN transgenic tumor model in C57Bl/6 mice forpreclinical immunological studies in neuroblastoma. Int J Cancer 2014;134:1335–45.
53. Althoff K, Beckers A, Bell E, Nortmeyer M, Thor T, Sprussel A, et al. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improvedtool for preclinical studies. Oncogene 2015;34:3357–68.
54. Heukamp LC, Thor T, SchrammA, De Preter K, Kumps C, DeWilde B, et al.Targeted expression of mutated ALK induces neuroblastoma in transgenicmice. Sci Translat Med 2012;4:141ra91.
www.aacrjournals.org Clin Cancer Res; 22(15) August 1, 2016 3859
Block CSF-1R to Improve Checkpoint Blockade in Neuroblastoma
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912

2016;22:3849-3859. Published OnlineFirst March 8, 2016.Clin Cancer Res Yumeng Mao, Nina Eissler, Katarina Le Blanc, et al. Inhibitors to Control Spontaneous NeuroblastomaTargeting Suppressive Myeloid Cells Potentiates Checkpoint
Updated version
10.1158/1078-0432.CCR-15-1912doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2016/05/20/1078-0432.CCR-15-1912.DC2
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/22/15/3849.full#ref-list-1
This article cites 54 articles, 17 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/22/15/3849.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/22/15/3849To request permission to re-use all or part of this article, use this link
on May 26, 2018. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 8, 2016; DOI: 10.1158/1078-0432.CCR-15-1912