tailored ruthenium polypyridyl complexes as molecular...
TRANSCRIPT

Indian Juurnal of Che mi stry Vol. 42A. August 2003. pp. 1797-18 14
Advances in Contemporary Research
Tailored ruthenium polypyridyl complexes as molecular electronic materials t
L Mishra* & A K Yadaw Department ofChemi5try, Banaras Hindu University. Varanasi 221005, India
and
G Govil
Department of Chemical Physics, TIFR, Mumbai, Indi a
Received II February 2003; revised 26 JUlle 2003
In the present review, salient features of ruthenium polypyridyl complexes tailored for molecular electronic properties ar.:: presented. Attempts are also made to explore these newly synthesized material s in the construction of molecular wires. molecular rods, luminescence sw itches as well as sensors.
Dr. Lallan Mishra is currently Professor of Chemistry , Banams Hindu Uni versity. Varanasi. Ind ia. His research interest lies in the design and development of metal based bioactive molecular and supramolecIar complexes. Prof. Mi shra is a recipient of Prof. P. Ray Memori al Lecture award of Indian Che mical Society, Kolkata for the year J 997. During 1999 he was invited to
preselll hi s work in Gordon Research Conferencc on Supramolecu lar Chemistry held in Henniker, USA. He was a visi ting Professor in the University of Sevi ll a, Spain (May 2000-Nov 2000) . Recently Prof. Mishra was honoured with CHEMITO 200 1 award .
Prof. Girjesh Govil is a Senior Professor a t Tata Insti tute of Fundamental Research, Mumbai . Hi s current interests are in the field o f molecular biophysics. He has coauthored/co-edited four books. published over 200 papers in international journuls. and has written a number o f scientitic articles of general interest. He wurkcd for his Ph .D. under the supervision o f (late) Prof. S S
Dharmatli at TIFR, Mumbai . He has held visiting posit ions at the National Physical Laboratory, UK. National Research Counci l, Canada, Na ti onal Instiwte of Heal th, USA and Kobe University, Japan. Awards and honurs rece ived by Prof. Govil inc lude Shanti Swarup Bhatnagar Award, Acharya J C Ghose Memorial Award, Acharya PC Ray Memurial Award, Vikram Sarabhai Gold Medal and ihe Fourth lawaharlal Nehru Bi rth Centenary Visiting Fe lluwship of INSA. He is Fellow of Third World Academy of Sciences. Indian National Science Academy, Indi an Academy of Scien(:es and Natiollul Academy o f Sc iences.
t in the memory of Late Dr (Mrs) Ratna S Phadke, TIFR , Mumbal, InJia
Dr. Aj ay K Yadaw did hi s Ph.D. ulluer the supervIsion o f Pro f. Lallan Mi ,hra . Department of Chemistry. Banaras Hindu University. Varanasi. Indi a. Hi s an:a or research is studies on ru thenium po lypyridyl complexes. Currently Dr. Yadaw i, a research scienti st in C HEMB IOT El\. Research Internati onal Pvt. Ltd. , Kolkala . Dr Yadaw has publi shed several research pape rs
including review artic les.
Multidisciplinary scientific researches have provided technology for the fabrication of devices wi th the purpose of signal' and information receiving2, storing
l
and processing. Development of such devices on a molecular level fall s under the purview of molecular electronics . However, the term electronics is more generic in nature and does not imply utili sation of motion of electrons for information processing. For example the concept of machine can be extended to the molecular level. A molecular level machine could be defined as an assembl y of a discrete numher of molecular components designed to perform mechanical movements (output) as a consequence or appropriate external stimuli4 (input ). It is very young field of science initiated in Jate 1970.
The investigation of molecular machines and motors is an exciting area of contemporary research which lies at the interface of chemistry , biology. physics and material science.
Molecular level machines operate5 via elect ronic and molecular rearrangements i.e. bringing some kind of chemical reactions. Like macroscopic machil1e~ ,
they are characterized by (i) energy input to make them work, (ii ) movement performed by their

1798 INDIAN J CHEM, SEC A, AUGUST 2003
components, (iii) the way in which their operation can be contro ll ed and monitored, (iv) the possibility to repeat the operation at will, (v) time scale needed to complete a cycle of operation and (vi) the function performed.
Since a machine has to work by repeating cycles, an important requirement is that any chemical reaction in a molecular device has to be reversible. Currently there is an increasing interest in the development of photon- and electronlhole-powered molecular-level machines by taking advantage of the outstanding progress made in supramolecular photochemistry and electrochemistry. In the case of photoexci tation, the most suitable candidates are photoisomerisation and photoinduced redox reactions. Photochemical and electrochemical energy inputs offer other advantages compared to alternati ve chemical energy inputs. For example, they can be switched on and off easily and rapidly.
Thus, the development of concepts for the construction of the machines on the molecular level is very important not only from basic research point of view but also from the growth6
.7 of nanoscience and
nanotechnology. The idea of constructing artificial molecular-level machines was introduced2 for the first time in 1980 by F L Carter, Naval Research Laboratory, USA and Richard P Feynman, who got Nobel pri ze in Physics.
Microelectronics which is based on bulk properties of inorganic substances, has so far succeeded in producing cost effective, high speed, low energy consuming, small sized, low weight, long lasting and hi ghly reliable devices . However, when the size of individual elements reaches less than tenth of a micron, thermal and quantum size effects will make the electronic components unreliable8
.9
. Hence, the need to look for molecular devices. Molecular electronics should not be anticipated to emerge as a revolutionary technology, threatening to replace conventional solid state electronics but could be considered as a complementary technology. One may explore possibilities of novel applications which may be beyond present day electronics. A perfect marri age between man-made microelectronics and nature evolved systems is envisaged to produce more intelligent and high performance devices. Construction of molecular-based devices is ex panding. especially, in the design and development of nanomachines. Bottom-up construction of miniaturized components capable of performing specific functions are important challenges before
2+
PHOTOPHYSICS PHOTOCHEMISTRY
Fig. I-Left: Photophysics of Ru(lI )LJ complexes. Ri ghi : Photoelectron transfer from the excited (triplet) stale of the Ru(II)L3 complexes.
modern chemists. Progress in this fi eld requires the availability of molecular components (building blocks) having well-defined structures and properti es.
In this context, attention has been focussed on systems containing M(N-Nh2+ (M = Ru" or OSII ; N-N
= 2,2'-bipyridine or 1, lO-phenanthro line) units as building blocks. The choice of such blocks is justified by their favorable excited-state 10, redox propert ies 11.1 2
as well as chemical stabilityl3. The general behaviour of Ru(II) L3 complexes used in the constructi on of molecular devices is depicted 14 in Fig. 1.
Synthetic Ru-based Materials The development of any device needs the
corresponding materials synthesized by chemi sts. Therefore, it is of worth to look on the intelligent design and development of ruthenium-based materials, specially, from molecular electronic viewpoint.
In general, mononuclear and dinuclear (homo/hetero) ruthenium complexes are sy nthes ized l
(,
following Scheme 1.
Mononuclear RuL2Ch + BL ~[RuL2 (BL)f+ [RuLCI3] + 2BL ~[RuL (BLhf+ RuCb + 3BL ~[Ru (BLhf+
BL = Differen t bridging li gands like
Pyrazine 4,4'-bipyridinium etc. Binuclear complexes RuL2Ch+[RuL2(BL)f+ ~[L2Ru (fl-BL)RuL2r l+
Scheme 1
A detailed account of such sy nthes is and characteri zation techniques have been publ ished
. I 17- 18 prevIOus y . To obtain dinuclear heterometallic complexes. the
usual approach involves two steps. First, a mononuclear complex-l igand is prepared by reac ting

MISHRA et at.: TAILORED RUTHENIUM POL YPYRIDYL COMPLEXES 1799
a metal precursor with an excess of the ligand. During this step, dinuclear homo metallic by-products are unavoidably formed which may be separated from desired mononuclear complexes using column chromatography. In the following step, the second metal is added so that only the dinuclear homometallic species are obtained.
The complexes incorporating the Ru(bpyh 2+ moiety are of interest since they construct efficient intramolecular electron or energy transfer systems. Several multicomponent systems (often called supramolecular species) have been designed and studied in terms of their photo and/or redox properties 19-20. In fact these properties have spurred the development of newer Ru(1I) polypyridyl complexes either by incorporating the desired groups within the bipyridine or phenanthroline moiety, or by usi ng other donor sites along with the [Ru(bpy/phenh]2+ core to form mixed ligand trischelate to modulate the photoredox activities of this class of complexes.
Complexes where bpy/phen-based photoactive centres are linked with the metal ions have shown problems21 in photoinduced processes due to the possible existence of different stereoisomers. To overcome such problems tridentate, terpyridine (tpy) type of ligands have emerged as a better choice22. Such complexes are in fact achiral and the introduction of a single substituent in the 4'-position of each terpyridine ligand overcomes isomeric problems and offers the possibility of designing triads in which the two additional components are in opposi te directions with respect to the photosensi tizer22.
However, such replacement of bpy to tpy-type ligand in the construction of Ru(lI) complexes resulted into shorter excited-state lifetime and a much weaker luminescence intensity in solutions at room temperature. A systematic investigation of Ru(tpyh 2+ type complexes has recently shown that this drawback can be overcome partially by appropriate substituents (e.g. MeS02) on the tpy ligand frame.
In the process of such development, Constable 's group in U.K. along with his collaborator in France successfully synthesized doubly cyclometallated diruthenium complex with strongly coupled metal centres23
. These complexes could easily be oxidized giving strongly coupled mixed Ru(III)-Ru(fl) species. Thus, during the investigation of photoinduced energy and electron-transfer in geometrically well-defined polynuclear Ru-based systems, electronic and nuclear factors governing these processes could be studi ed24
.
In this connection, the emission properties of a new tpy based Ru(1I) complex25 in which a 2.S-thiophenedyl spacer connecting [Ru(tpyh ]-chromophore modified the properties such that di- and tri- nuc lear complexes luminesce like Ru(bpyh 2+ have been reported. The excited states responsi ble for the luminescence were formally triplet Ru-tpy charge transfer. Luminescence maxima were found consistent with these sequential stabilization and passing from mono- to di- and tri-nuclear complexes. Barrigelleti et al?1 also prepared rigid rod-like dinuclear Ru(II)/Os(Il) terpyridine complexes as shown in Fig. 2. These complexes were also studied for their luminescence and electronic energy tran sfer through phenylene bridges.
Thus, while assembling these units in the construction of supramolecular species, the selection of terminal as well as bringing ligand is very important and the distance between two metal centres bridging efficient bond coupling can be tuned by the bridging ligands. Supramolecular species consisting of oligo metal complexes having capability for use in photochemical molecular devices have been cited by Balzani et al.25
,.
Keeping these points in view, Haga el al. ~(, developed novel bridging ligand based on benzimidazole unit. This unit has strong IT-donor property and hence stabilizes Ru(III) state better than pyridine or pyrazing units . In the benzimidazole bridged dinuclear complexes (Fig. 3), when excited photochemically, negative charge on the bridging ligand enhanced hole-type super exchange interaction between the metal centres.
l6+ eM,
.-1,2
Fig. 2-Heterotrinuclear complex having phenyl bridged exteded conjugation

1800 INDIAN J CHEM, SEC A, AUGUST 2003
L = 2,2'-bipyridinel I,IO-phenanthroline Fig. 3-Homometallic dinuclear Ru(II) polypyridyl complexes
Fig. 4- Crown ether bridged dinuclear Ru(Il) complexes
Later, crown ether-bridged dinuclear Ru(lI) polypyridyl complexes (Fig. 4) were synthesized by Beer e l al.27 which show anion binding properties.
Recently our group has reported28.32 a series of
homodinuclear Ru(lI) polypyridyl complexes (Fig. 5) having extended conjugated bridging ligands. We have also studied their redox and luminescent properties. Extensive luminescence quenching observed from dinuclear Ru(lI) complexes as compared to their mononuclear analogues suggested photoinduced intramolecular electron and/or energytransfer between the two metal centres.
Heterodinuclear complexes containing Ru(lI) polypyridyl component as one of the core has been h b· f . 31·34 I' h t e su ~ect 0 great Importance ' lor t e
intramolecular electron/energy transfer processes which have vital role in designing the photochemical molecular devices.
Komatsuzaki el al. 35 reported a series of dinuclear heterometallic ruthenium-cobalt and ruthenium-nickel complexes which are shown in Fig. 6.
The lowest energy charge-transfer bands, attributable to the Ru~bpy transition, were little affected by the intramolecularly attached Co(Il[) or Ni(lI) moiety , and indicate that Ru(1I) and Co(III) or Ni CH) have no electronic interaction . Their luminescence behaviour had also been studied. [Co(bpyh ]3+ and [Ni(bpYh]2+ did not show emission since these compounds do not absorb light in this wavelength region. Other complexes containing the
when N-N = 2,2'·bipyridine· = O' and n = 2, when N-N = 1,1 O-phenanthroline • = OH and n = 4
N-N = 2,2'-bipyridine II ,1 O-phenanthro line
l4 +
.4 PF,,:
where N-N = 2,2' -bipyridine II, IO·phenanthroline
Fig. 5-Dinuclear Ru(lI) complexes having d ifferent conjugated bridging ligands.
[Ru(bpyh]2+ moiety emitted at -610 nm though the relative luminescence intensity from the dinuc lear complexes was found to be 30-47% less th an analogous mononuclear complexes.
Though the excited-state of [Ru(bpYh f + moiety in 3 (Fig. 6) should have been quenched intramolecularly by the attachment of [Co(bpY)2f + and [Ni(bpY)2]2+ moieties as observed by several research groups, these dinuclear complexes did not
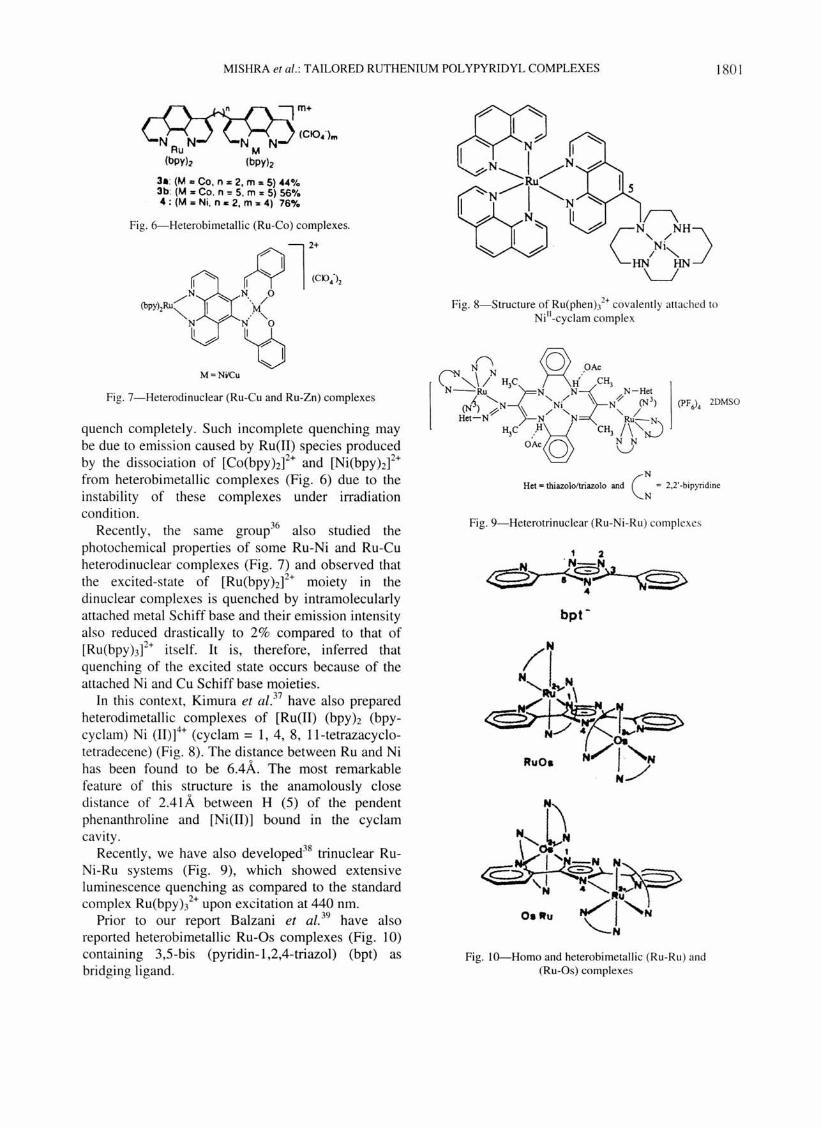
MISHRA et al.: TAILORED RUTHENIUM POL YPYRIDYL COMPLEXES 180 1
~m+
~N~N-> <-." N(-<" . .J (CIO.)m Au M N
(bPY)2 (bPY)2
31: (M .. Co, n .. 2, m .. S) .<40;. 3b: (M '" Co. n = 5. m '" 5) 56% 4 ; (M .. Ni. n c 2, m", 4) 76,..
Fig. 6-Heterobimetallic (Ru-Co) complexes.
Fig. 7-Heterodi nuclear (Ru-Cu and Ru-Zn) complexes
quench completely. Such incomplete quenching may be due to emission caused by Ru(lI) species produced by the dissociation of [Co(bpYhf+ and [Ni(bpYhf+ from heterobimetallic complexes (Fig. 6) due to the instability of these complexes under irradiation condition .
Recently , the same group36 also studied the photochemical properties of some Ru-Ni and Ru-Cu heterodinuclear complexes (Fig. 7) and observed that the excited-state of [Ru(bpyh]2+ moiety in the dinuclear complexes is quenched by intramolecularly attached metal Schiff base and their emission intensity also reduced drastically to 2% compared to that of [Ru(bpyhf+ itself. It is, therefore, inferred that quenching of the excited state occurs because of the attached Ni and Cu Schiff base moieties.
In this context, Kimura et al. 37 have also prepared heterodimetallic complexes of [Ru(lI) (bpyh (bpycyclam) Ni (11)]4+ (cyclam = 1, 4, 8, Il-tetrazacyclotetradecene) (Fig. 8). The distance between Ru and Ni has been found to be 6.4,A.. The most remarkable feature of this structure is the anamolously close distance of 2.41 A between H (5) of the pendent phenanthroline and [Ni(II)] bound in the cyclam cavity.
Recently , we have also developed38 trinuclear RuNi-Ru systems (Fig. 9) , which showed extensive luminescence quenching as compared to the standard complex Ru(bpyh2+ upon excitation at 440 nm.
Prior to our report Balzani et al. 39 have also reported heterobimetallic Ru-Os complexes (Fig. lO) containing 3,5-bis (pyridin-l,2,4-triazol) (bpt) as bridging ligand .
Fig. 8-Structure of Ru(phenh2+ covalently attached to
Ni"-cyclam complex
N Het = thiazolo/triazolo and eN = 2,2··bipyridine
Fig. 9-Heterotrinuclear (Ru-Ni-Ru) complexes
Fig. lO--Homo and heterobimetallic (Ru-Ru ) and (Ru-Os) complexes

1802 INDIAN J CHEM, SEC A, AUGUST 2003
® ® ® \Ru-bpy) I(Ru-bpy) I(Ru_bpy) -
1(0. -bpy)
3(Ru_bpy) 3(Ru_bpy) 3(Ru_bpy)
I
" \ . I 3(0._bpy)
I I I I I I Ru11_0.'I1l
h~, I h~2 h~, h~3 I h"'~ hv, I I I I I I hilS
t I t ~
Fig. II-Schematic energy level diagrams (a) mononuclear [Ru(bpyhbptt; (b) RuOs and OsRu species; (c) one-electron Ru"OS I
I and OslllRu" forms
Luminescence and redox properties of these Ru-Os complexes [(bpy)z (Ru) (bpt) Os (bpy)zf+ and [(bpY)2 Os (bpt) (Ru (tpyhh3+ have been compared with those of the corresponding dinuclear homometallic Ru(II)Ru(J1) and Os(Il)-Os(IJ) complexes. Excitation in the Ru-based component is fo llowed by :: 100% efficient energy transfer to Os-based component. One electron ox idation products (which makes Os in the +3 oxidation state) are not luminescent because of the presence of a low-energy intravalence charge transfer band which is likely to deactivate the luminescent level. It is observed that the bpt bridge is not involved in the lowest energy excited states, which allows energy transfer to occur very rapidly from Ru(bpy)/ + component to Os(bpYh2+ or Os(bpY)23+ components in the dimetall ic species. A schematic presentation34 of such processes is depicted in Fig. 11.
There has been great interest in chiral complexes4o
.4 1
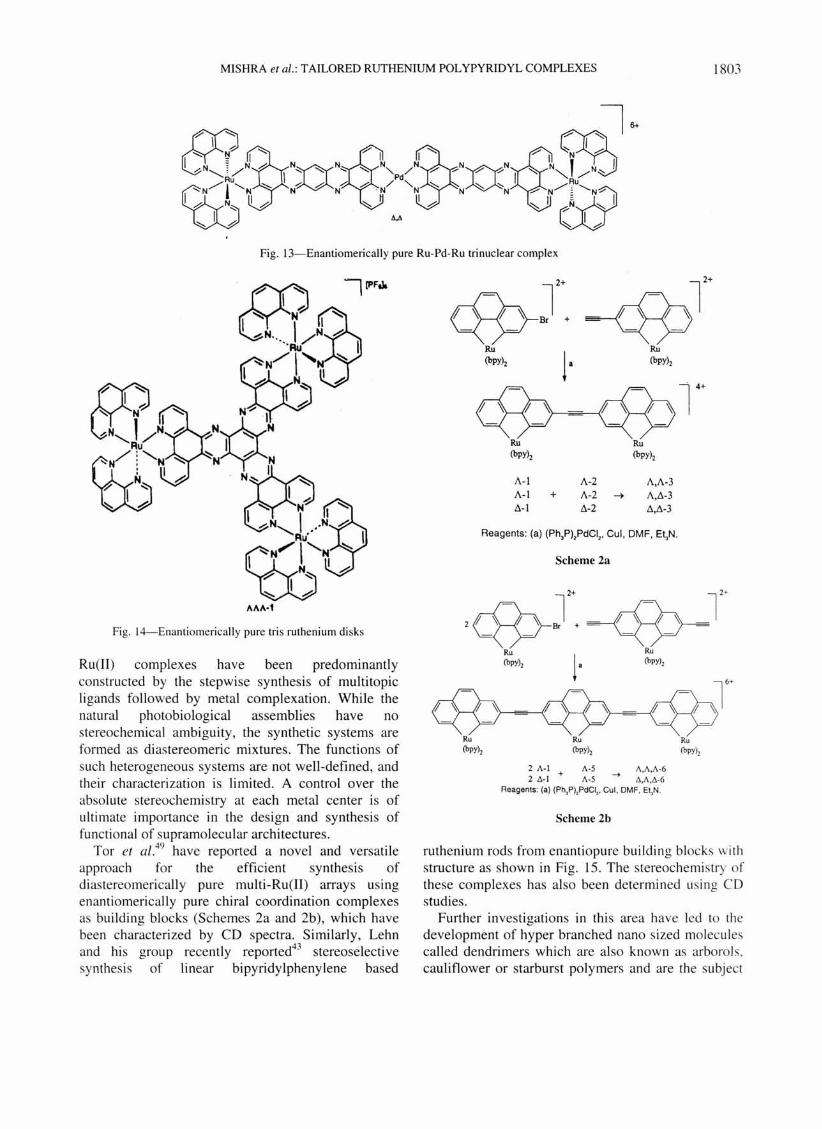
. The report by Tzalis et al.42 abou t the synthesis of an interesting rod like arrangement (Fig. 12) could be probably the first example of controlled sy nthesis of stereochemically defined multinuclear complexes based on Ru(II) centres.
Adopting similar syn thetic strategy, a mixed metal Ru(II)-Pd(II) trinuclear complex43 based on enantiomerically pure Ru-centres (Fig. 13) is made which is another interesting rod like structure (nm level).
As compared to Ru(II)-tpy systems, Ru(II)-bpy and phen complexes generally have a longer excited-state lifetime and are found suitable for the construction of photosensitizers44
-45 and light harvesting devices46
.
Fig. 12-Enantiomerically pure trinuclear Ru(1I1 complex
However, the latter systems give ri se to diastereoisomers when more than one octahedral metal tris-(bidentate ligand) centre is incorporated. Development of diastereomerically pure compounds is , therefore, important as diastereoisomers differ in their physical properties such as rates of energy or electron transfer. The use of inorganic enantiomerically pure building blocks allows the cons truction of diastereomerically pure polymetallic archi tectures without subsequent separati on of diastereoisomers which avoids complicat ions due to its exponential increase 2N (N = the number of incorporated metal centres) in the number of stereoisomers .
In this connection, the work of Warn mark ef al. is worth mentioning as they have reported43 the synthesis of extended enantio and dias tereomericall y pure tris-ruthenium disks from pure building bloc ks (Fig. 14).
Multi metallic assemblies containi ng Ru(ll ) polypyridyl complexes are important due to their chemical stabi li ty, favourable redox properties as well as photophysical characterist ics47
.48. These multinuc lear
MISHRA et al.: TAILORED RUTHENIUM POL YPYRIDYL COMPLEXES 1803
Fig. 13-Enantiomerically pure Ru-Pd-Ru trinuclear complex
Fig. 14-Enanliomerically pure tri s ruthenium di sks
Ru(II) complexes have been predominantly constructed by the stepwise synthesis of multitopic ligands followed by metal complexation. While the natural photobiological assemblies have no stereochemical ambiguity, the synthetic systems are formed as diastereomeric mixtures. The function s of such heterogeneous systems are not well-defined, and their characterization is limited. A control over the absolute stereochemistry at each metal center is of ultimate importance in the design and synthesis of functional of supramolecular architectures.
Tor et al.49 have reported a novel and versatile approach for the efficient synthesis of diastereomerically pure multi-Ru(ll) arrays usi ng enantiomerically pure chiral coordination complexes as building blocks (Schemes 2a and 2b), which have been characterized by CD spectra. Similarly, Lehn and his group recently reported43 stereoselective synthesis of linear bipyridylphenylene based
~r
Ru (bPY),
A-\ A-2 A-I + A-2 6.- 1 6.-2
~
Ru
Ru (bPY),
(bPY),
A,A-3 A,6.-3 6.,A-3
Reagents: (a) (Ph,P),PdCI" Cui, DMF, Et,N.
Scheme 2a
'c8=>-r~r Ru Ru
Ru (bPY),
(bpy), 1- (bpy),
Ru (bpy),
22 ~~ Il + A-5
5 ~ 1\,1\.,1\-6
u II· 6,11,6-6 Reagents: (a) (Ph,P),PdCI,. CuI. DMF. Et,N.
Scheme 2b
Ru (bpy),
ruthenium rods from enantiopure building blocks with structure as shown in Fig. 15. The stereochemistry of these complexes has also been determined using CD studies.
Further investigations in thi s area have led to the development of hyper branched nano sized molecu les called dendrimers which are also known as arborols. cauliflower or starburst polymers and are the subject

1804 INDIAN] CHEM, SEC A, AUGUST 2003
of much interest because of their unique structure and properties. These aesthetically appealing synthetic macromolecules distinguish themselves from normal polymers in several ways. First, they are constructed
n=O.1
t.,I:.
Fig. l5-Enantiomerically pure ruthenium based rod like structure
from ABn monomers (n usually 2 or 3) rather than the standard AB monomers which produce linear polymers. Secondly, they are synthesized in an iterative fashion. The combination of these two features leads to a non-linear, stepwise synthetic growth wherein the number of monomer units incorporated in each successive iteration rough ly doubles (AB2) or triples (AB3) to that in the previous cycle.
Strategies for synthesizing dendrimers which have emerged during the past decade can be classified into divergent and convergent approaches . In the divergent strategy, dendrimers are built from the central core out to the periphery whereas the convergent strategy builds the dendrimers from the periphery toward the central core. Metal based dendrimers50
.52 have
received significant attention in recent years because they contain multiple coordination sites or
~: o
00
o ,-r'OR
~OR
Fig. l6--A metallodendrimer reported by Newkome, Constable and co-workers53

MISHRA el at.: TAILORED RUTHENIUM POL YPYRIDYL COMPLEX ES 1805
multielectron transfer redox centres and hence have the potential for wider applications like catalysts, molecular electronics, photochemical devices for information storage and switching as well as energy transfer and conversion devices.
A variety of Ru-based dendrimers have recently been synthesized. A representative example is the ruthenodendrimer reported by Newkome and coworkers53 (Fig. 16).
Recently , much attention has been focussed on the incorporation of luminescent, redox-active transition metal complexes, such as ruthenium polypyridine species, into the dendrimer structure as the potential of such systems for applications in light harvesting and energy storage devices has been realized. Bodige et al. 54 reported stereoselective synthesis of rigid Dr symmetric tetranuclear ruthenium complex (Scheme 3).
Scheme 3--Tetranuclear ruthenium(lI) complex with D) sy mmetry
Applications
Molecular wires The origin of molecular wires can be traced to
soliton theory55 of electronic conduction in DNA. The widespread interes t in the studies of energy or electron transport schemes has resulted in the construction of molecular materials holding photoactive components usually at the termini of the system as well as in the storage and utilization of li ghtenergy. As a consequence of specific interactions with light or after undergoing electrochemical process, such active un its can be the initial and final sites for the temporary storage of energy. Other components,
a (Booge)
Fig. 17-Structure of the molecular wire containing Ru(lI ) complex as donor and (j bridging ligand as acceptor
the linkers or bridge playa structural role and provide electronic connection between the active units.
A molecular wire thus possesses following criteria: (i) Contains an electron conducting chain ; (ii ) possesses terminal electroactive and pl anar groups for reversible electron exchange; and (iii) it should be long enough to span a typical molecul ar supporting element such a monolayer or bilayer membrane.
Molecular wires can be represented as D - (J - A or D+ - 1t - A- where D = donor, A = acceptor, and (J
and 1t are bridging or spacer groups56 as shown in Fig. 17.
Examples of other Ru(lI) complexes which may ac t as donor and electron acceptors are shown in Fig. 18.
A favourable factor which allows better performance includes a good energy match ing between the donor and bridge components . The molecular wires incorporating ruthenium polypyridy l complexes are amenable to studies of directi onal energy and electron transfer processes. The ruthenium based units usually exhibit luminescence and playas donor (D) or acceptor (A) units.
A bridging ligand provides both the structural and electronic connectivity between D and A and D-B-A unit could be either flexible or rigid depending upon the nature of spacer included within the bridge. The ruthenium polypyridyl complexes can frequently undergo well-defined oxidation and reduction process. In the presence of identical metal (homodimetallic), the conduction properties of the bridge can be evaluated via intervalence studies. There is a great interest57-58 in dinuclear Ru polypyridine complexes for understanding the electronic interaction between the metal ions and photoinduced intramolecular electron/energy transfer processes.
Energy-transfer process takes place either by coulombic (Forster) or exchange (Dexter) mechani sm. In the former case, the main contribution to the rate

1806 INDIAN J CHEM, SEC A, AUGUST 2003
2+
2+
Donon
2+
t.~. R-N~N-R
R = m:thyt, propyt. pentyl
Accepton
Fig. IS- Structure of the e lectron donor Ru(lI) complexes (Top) and electron acceptor complexes (bottom) employed in electron transfer research.
constant comes from the dipole-dipole interaction between donor and acceptor. The rate constant according to thi s mechanism can be calculated from spectroscopic and structural parameters uSing59 Eqs (l and 2).
ken= litO (RoIr)6 .. . (1 )
Ro6=5 .87x I0-25<1>°/11 4 f F(V) £('1)'1-4 dv ... (2)
where Ro is critical radius i.e. the distance at which the energy transfer rate and the intrinsic deactivation of the donor are equal, V is the frequency (cm-I
) and
11 , <1>0 and r are the refractive index of the solvent, the luminescence quantum yield of the donor and the donor-acceptor distance (A.) respectively.
Dexter-type energy transfer mechanism is described as a double exchange of electrons between donor and acceptor and is related to direct or superexchange mediated electronic interaction between the two partners. The rate constant of energy transfer can be calculated60 using Eqs (3 - 5).
ken = Ven exp (-flG""/RD ... (3)
... (4)
. .. (5)
H = intercomponent electronic interaction.
Scheme 4
The free energy change flGo can be ex pressed as the difference between the spectroscopic energies of the donor (D) and acceptor (A) and reorganisat ional energy 0.) can be estimated from the spectroscopic Stokes shift of the acceptor.
In context of ruthenium system, these reactions could be understood in terms of Scheme 4 proposed by Haga et al. 26
•
In the electron type superexchange mechanism, the mixing between the LUMO n* orbitals and Ru(lI) dn orbital is the major contribution for metal-metal interaction . On the other hand, the hole type superexchange mechanism is attributable to electronic interactions between the HOMO n-orbitals of the bridged ligand and Ru(III) dn orbitals.

MISHRA et at.: TAILORED RUTHENIUM POL YPYRIDYL COMPLEXES 1807
In terms of distance, the rate constant for electrontransfer processes in bis-porphyrin donor/acceptor compounds with one, two and three phenylene spacers is expressed59 as:
ken = An exp (-Br) ... (6)
where B = 0.4 A and r = donor-acceptor distance Since intramolecular electron and energy transfer
rates in donor-acceptor (D-A) linked by spacer are dependent on several molecular and environmental parameters, it is often difficult to design D-A systems where a single parameter can be studied independently . In view of this, Larson et al. 60 have studied the rates of intra-molecular energy transfer from the MLCT excited state of a tris (bipyridine) Ru (lI) donor to a ground state tris (bipyridine) iron (II) acceptor within a series of heterodinuclear (Ru-Fe) complexes (Fig. 19).
Due to identical metal-metal separation as well as relative ligand orientations, these complexes provide a unique opportunity to investigate the role of D-A connectivity in an energy transfer process while maintaining other parameters virtually constant.
Complex dMM , (A) kgf (ns· l)
I 7,61 2.440 II 7,57 2.870 III 7.50 2.710 IV 7.50 2,940 V 8.70 0.370 VI 9.23 0.075 VlI 9.89 0,029
dMM = metal-metal separation; kq = energy transfer rate constant.
The data shown above indicate that for J-VII, the quenching rate falls exponentially as metal-metal separation increases, After many supporting experiments, they concluded that the MLCT state of the RuL] is quenched by an exchange energy-transfer mechanism. It is remarkable that the rate is independent of the nature of the bridges and appears to depend only on the donor-acceptor separation.
For the design of an efficient molecular wire, the role of the spacer is significant which could be easily understood with the following examples61
.62 (Fig, 20),
Metal centres (A) Rull /Osll (B) Ru" /OS" (C) Ru" /OS"
L7 4,5 250.0
13.0 12.8 13.8
dMM = metal-metal separation; ken = electron transfer rate constant
3
l. X=(C~}z
II. X = (CHz}J
III . X = CHz.S.CHz IV. X = c~.oCHz
v. X = (CHz).
VI. X=(C~)5
VlI . X=~ Fig. 19-Heterodinuclear Ru(ll) -Fe(l l) complexes ha ving
di fferent type of spacer
Fig. 20-Heterodinuc1ear Ru-Os based complexes having different type of spacers
Thus, the above rate constant data indicate that the alkane chain does not provide a good electronic interaction between 0 and A and D-A interaction is of dipole-dipole interaction type. On comparing the above three cases, we find that in complex C, the energetic and spatial parameters are simi lar to those of A and B. However, Ru -7 Os energy step is faster by ~everal orders of magnitude, and the alkyne bridge provides an efficient electronic interact ion between D and A centres .
An unique case of energy traping has been identified by looking at the homodimetallic complex (Fig. 21) reported by Mishra et a163
. In this case the anthraquinone unit collects the excitation energy from either of the two identical Ru centres.

1808 INDIAN J CHEM. SEC A. AUGUST 2003
Fig. 2 1- Homodimeta lli c Ru(Il ) complex
2+
4' 4'
Fig. 22- Structu re of mononuclear Ru(I1 ) terpyridyl complex
As mentioned earlie r in the Ru-bipyrid ine system, there is always a poss ibility of co-exi stence of d ifferent stereoisomers which may cause specific d iffic ulti es in the study of photo induced processes. In thi s regard , a be tte r a lternati ve fo r the ligand coordinati on is ensured by the use of 2,2':6',2 /1-terpyridine building block via the possible use of 4'position of tpy fo r developing wires (Fig. 22).
In view of above, a se ries of Ru ( lI) - Rh (JII ) dyads of the general formula64 [ttpy Ru-tpy- (ph)n-tpy-Rh (ttpy]5+ with n = 0 , I , 2 (ttpy = 4'-p-tolyl-2,2' : 6,2/1-terpyridine; tpy-(ph)n- tpy = bridgi ng ligand where two 2.2':6,2/1-terpyrid ine units are connected at the 4' pos itio n th rough a variable number of p-phenylene spacers) have been studied for their photo induced electron-transfer p rocesses . When n = 1, excitation of the Ru(ll )-based molecular component is followed by efficient intramolecular quenching by e lec tron transfer to the Rh(I1I ) centre (Fi g. 23). The rate
constant k ~ 3 X 109 s· ' is high despite the re latively small driv ing force of the process (ca . 0.1 eV). When n = 2. with the same dri ving force as above, no intramo lecular electron transfer quenching is observed (k < 5 x 108 s" ). T he decrease in e lectron transfer rate is go ing fro m n = I to n = 2 is in line with the behav iour of other systems containing po ly-pphenylene spacers.
Similarly, rod like dinuclear ruthen ium terpy ri dy l complexes have been developed65,66 by coupling at 4'position of the terpyridine ring .
Sau vage el al. 67 synthes ized a series of rod li ke Ru B-Os heterodinuclear complexes (Fig. 24) for the study of e lectronic energy transfer and observed that the te mperature independent Ru ~ Os electroni c energy transfer process occurs over the 200-90 K interval and energy transfer is neither affec ted by temperature changes nor by the state of the solvent. which is fluid at T > 110 K and froze n below that temperature.
The study of the excited state properti es of chi r;)1 transition metal complexes appear~ to be ve ry promising. At present, however, ex tensio n to cases o f' mi xed metal enantio meri cally pure compl exes does not seem to be easy. Of course, these comp lexes would be suitable for energy/electron transfer stud ies o f the type treated abo ve and seem to be appropriate for further development. Some ruthen ium po lypy ri dy l based rod like structures68
.70 are shown in Scheme 5.
Molecular redox switches f h . 7 ' -7" · I Last ew years ave seen a great Interest - In t le
fie ld of mo lecular switches in the developmen t or molecul ar e lectronics and photonics . Severa l examples of the latter are based on supramolecular devices whose optical properties (pho tochromislll. luminescence, optical no n-linearity) can be sw itched by external stimuli such as chemical, e lectrochemica l and light inputs. Attentio n has been paid to molecu lar switches whose emiss io n properties are in fl uenced by redox potenti a l data as they could find appli cati o n in conjuncti on with imaging techniques 73 , in b i o log/~ and biochemistry, to detect synaptic acti vity and in general to perfo rm membrane potential measurements in organelles (e.g . mitochondri a) and ce ll s.
Design of fluorescence redox swi tches consist. of assembling a redox acti ve subunit (contro l unit). which can exist in two di fferent ox ida ti on states of comparable stability (i .e. bi stable sys tem) and a

MISHRA el at.: TAILORED RUTHENIUM POL YPYRIDYL COMPLEXES 1809
Fig. 23-Structure of phenyl bridged hetero dinuclear Ru(II )-Rh(II1) complex
[ttp Ru (tpy-ph-bCOph-tpy) Os(ttp)t+
Fig. 24-Structure of heterodimetallic Ru-Os complex
,.
(3)
(') A = n-hexyl
Scheme 5- Rulhenium based rod like structures (1-4) hav ing di fferent types of bridging ligands
luminescent fragment (the active unit ) whose emission depends on the oxidation state of the contro l unit. It is the different extent of interacti on between the control subunit and the active subunit that generates the function which can be turned onlo ff through the external parameter. Thus the control subunit behaves as a switch .
Transition metal complexes are found to be good candidates for molecular switches due to fo ll owi ng properties.
1. They can form couples of consecutive ox idati on states which are easily interconvertable th rough a fast and reversible one electron redox change. The relative stability of the two states can also be modulated by the variation of co-ordination environment around the metal centre.
2. In most cases the one electron change significantly modifies the properties of the metal centre.
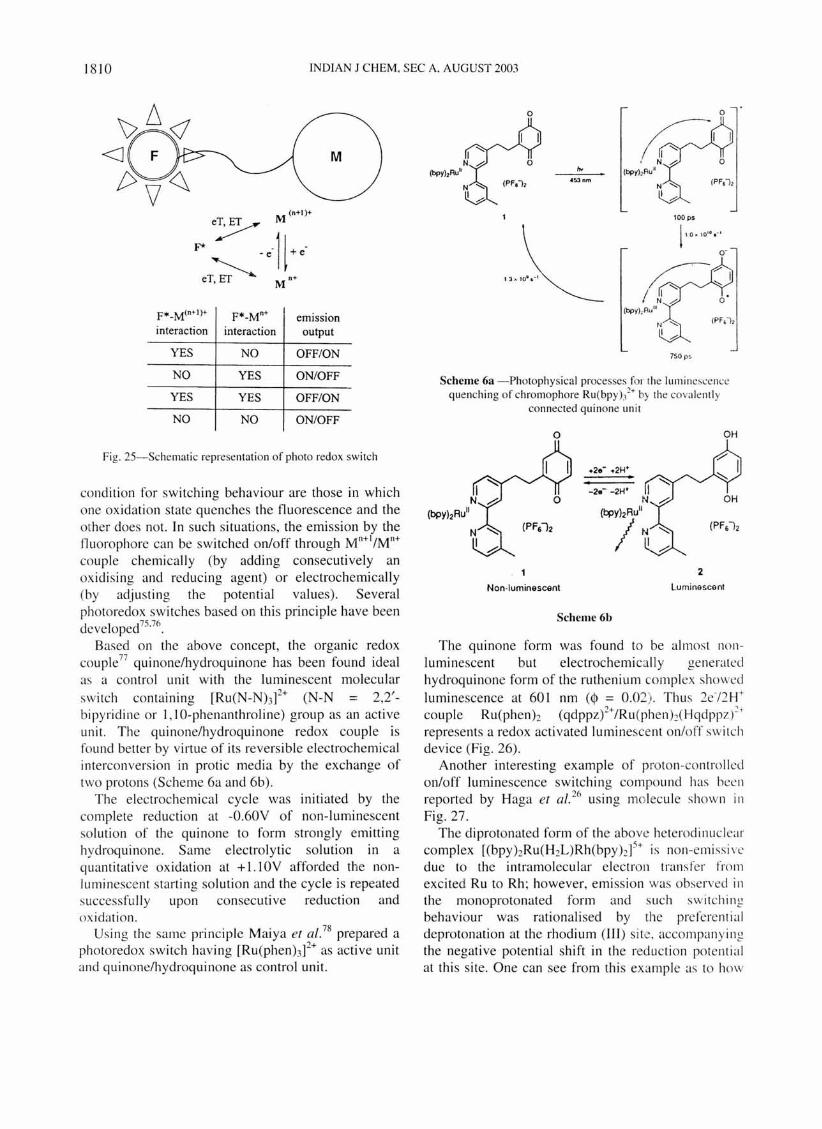
A pictorial presentation of such behaviour is show n as depicted in Fig. 25.
An organic f1uorophore F is linked covalently to a multidentate receptor which hosts a metal centre M. The metal complex part is redox acti ve through the two oxidation states M"+1IM n+ which are connected in a fast and reversible manner. Each state mayor may not interact with proximate photoexcited f1uorosphere through either an electron transfer or on energy transfer process . Interaction results in the quenching of light emission by the f1uorosphere. Favourable
1810 INDIAN J CHEM, SEC A, AUGUST 2003
M
F*_M(n+I)+ F*_Mn+ emission interaction interaction output
YES NO OFF/ON
NO YES ON/OFF
YES YES OFF/ON
NO NO ON/OFF
Fig. 2S-Schematic representation of photo redox switch
condition for sw itching behaviour are those in which one oxidation state quenches the fluorescence and the mher does not. In such situations, the emission by the tluorophore can be switched on/off through M"+I/M"+ couple chemically (by adding consecutively an ox idising and reducing agent) or electrochemjcally (by adjusting the potential values) . Several photoredox sw itches based on this principle have been developed75.7().
Based on the above concept, the organic redox couple77 quinone/hydroquinone has been found ideal as a control unit with the lumjnescent molecular
switch contall1l11g [Ru(N-Nh ]2+ (N-N 2,2'bipyridine or I, IO-phenanthroline) group as an active unit. The quinone/hydroquinone redox couple is fo und better by virtue of its reversible electrochemical interconversion in protic media by the exchange of two protons (Scheme 6a and 6b).
The electrochemical cycle was initiated by the complete reduction at -0.60Y of no n-luminescent solution of the quinone to form strongly emitting hydroquinone. Same electrolytic solution in a quantitative oxidation at + I. lOY afforded the nonluminescent starting solution and the cycle is repeated successfully upon consecutive reduction and (lx idati on.
Using the same principle Maiya et al.78 prepared a photoredox sw itch having [Ru(phenh ]2+ as active unit and quinone/hydroquinone as control unit.
o
N '" I ~
100 p,
N '" I ~
I' 0. '0" ,-'
o·
750 ps
Scheme 6a -Photophysical processes for the lumi nescence quenching of chromophore Ru(bpy)/+ by the covalently
connected quinone un it
o OH
2
Non·luminescent Luminescent
Scheme 6b
The quinone form was found to be a lmost 11 0 nluminescent but electrochemically generated hydroquinone form of the ruthen ium complex showed
luminescence at 601 nm (<I> = 0.02) . Thus 2d2 H+ couple Ru(phenh (qdppz)2+/Ru(phen)c(Hqdppz)c+ represents a redox activated luminesce nt on/off sw itch device (Fig. 26).
Another interesting example of proton-controll ed on/off luminescence switching compollnd has bee n reported by Haga et al. 26 using mo lecule shown in Fig. 27.
The diprotonated form of the above heterodinucl ear complex [(bpyh Ru(H2L)Rh(bpyh]5+ is non-emissi ve due to the intramolecular e lectron trans fe r from excited Ru to Rh ; however, emiss ion was observed ill the monoprotonated form and such sw itching beh av iour was rationalised by the pre ferential deprotonation at the rhodium (Ill) site , acco mpany ing the negative potential shift in the reducti on potential at this site. One can see from thi s exa mpl e as to how

MISHRA et at. : TAILORED RUTHENIUM POL YPYRlDYL COMPLEXES 1811
l2+ ~ ~ # I 0 I
N " -:7 ""
~ k,J/gx' ~ I 0
"N/~~ ~ ~N ""
.0 N .0
I " ""
[Ru(phenM qdppz) )2+
Fig. 26-(a) Luminescence spectra (CH3CNI 5%H20 0.1 M TBAPF6 Am = 440 nm of [Ru(phenh(qdppz))2+ (I) and [Ru(phen h(hqdppz)f+ (2) as obtained by exhaustive electrolyses at the indicated potential in each case. The arrows refer to the reversible changes observed upon electrochemical interconverat ion of these complexes
Fig. 27- Heterod inuclear Ru(I1)-Rh(lll) complex
the introduction of asymmetry can provide not only a potential difference but a preferential protonation site in dinuclear complexes.
With the above examples in view, a schematic representation of such behaviour is shown in Fig. 28.
Fluorescent PET "on/off' switches are the opposite of the "off/on" switches with regard to the nature of the phenomenon and their popularity.
Metal based redox systems has also been found amenable to signalling schemes. In the heterodinuclear complex6s
.79 Ru-BL-Os, no
luminescence was detected when the two metal centres (Ru, Os) are bridged with azo groups which act as a trap for the excitation based on the two terminal chromophores. However, reduction of azo group allows the occurrence of Ru ~ Os energy transfer. Thus, the complex (Fig. 29) acts as a redox responsive molecular switch.
SCI/sor
The assembly of a specific receptor with a subunit capable of signall ing a receptor-substrate interaction
Fluorophore Spacer Recepto r 0
Fluorophore Spacer Recepto r
Fig. 28-Fluorescent PET "on-off sw itch respondin g to a redox-active guest
Fig. 29-Azo bridged heterodinuclear (Ru-Os) compl ex showing molecul ar sw itching property
Signal Receptor
Fig . 30--Assembly of the constituents of the sensor
constitutes a sensor70. It involves basical ly three units
(i) receptor, (ii) signalling, and (iii) spacer as depicted in Fig. 30.
The receptor unit plays the role of recognizing and reversibly binding a given target substrate. The s ignalling unit must be capable of either producing a signal or significantly altering its intensity followin g the binding of the substrate, preferably withoul changing other chemical properties. Finally, the spacer links binding and signalling unit controlling their mutual separation and geometric arrangements.

1812 INDIAN J CHEM, SEC A. AUGUST 2003
The efficiency of a sensor is related to a comparable extent (i) to the selectivity of the binding and (ii) to the simplicity of detecting and measuring the displaying system.
A number of fluorescent sensors for metal ions developed 1.80 by several groups during the past decades contain anthracene as a fluorescent subunit because of its strong and well-characterized emission and chemical stability. The anthracene fluorescence is quenched via an electron-transfer process from the fa irly reducing tertiary amine group of macrocyclic ring to the photo-excited f1uorophore . Lowering in oxidation potential of the amine group occurs as a result of metal coordination and consequently the electron transfer is prevented on thermodynamic grounds. As a consequence, fluorescence is restored. Therefore, the interaction between the receptor and K+ ion is communicated to the outside by the appearance of the intensive emission spectrum of anthracene.
Sensors which work through a photo-induced electron transfer mechanism (PET) contain81 besides the receptor and the f1uorophore, an internal device providing the electron release (Fig. 31).
Ruthenium based species have been frequently used in the construction at various sensors due to unique combination of their luminescence characteristics, chemical stability and redox
. 10- 1:1 propertIes -. Recently a new class of chemosensor82 containing a
polyamine unit connected to a Ru(II) metal centre has been sy nthesized (Fig. 32) and studied for its cation [Cu(TI) and Zn(IJ)] and anion binding properties.
In a similar way a ruthenium based complex (Fig. 33) containing free bipyridine site has been synthesi zed83 and used fo r the study of cation bindings.
The luminescence properties undergo changes in the presence of the guest and thi s may be an useful approach towards molecular based devices for chemical sensing.
The molecular recognition of anionic guest species of biochemical and environmental importance by positively charged or neutral electron-deficient abiotic receptor molecules is an area of ever increasi ng research acti vity. Anions play various fundamental roles in biological and chemical processes . In this contex t. Beer ef a/.27 synthesized different ruthenium based azaCl'own ether type macrocyclic system (Fig, 34) having amide linkages for the study of group I metal cation and halide amon complexing properties.
·~·~·· ...... hv_ ~~.~. .... ~"-
. .....
Fig. 3 I- Design of a PET se nsor for a tran sition metal. The redox ac tive metal ion itself prov ides the electron release to the excited fluorophore
Fig. 32- Aza crown bridged Ru-based receptor
Fig. 33-Ru(lI ) homobimetallic complex sensing metal ions
Fig. 34-Ruthenium based aza crown bridged receptors for alkali metal

MISHRA et al.: TAILORED RUTHENfUM POL YPYRlDYL COMPLEXES 1813
400.-------------------,
~N
DPPN= 0000 NO N
300
200
100
WAVELENGTH (nm)
Fig. 35-Steady-state emission spectrum of Ru(phenh(DPPN)2+ in the absence and in the presence of sonictaed calf-thymus DNA
Chloride anion coordination studies indicate that receptors containing secondary amide can complex and electrochemically sense the halide anions through mutual electrostatic attraction and favourable amide CONH anion hydrogen binding interaction27
.
Ru(II) polypyridal complexes also provide sensitive luminescent probes84
-89 for double helical DNA. The
mixed ligand complexes (Fig. 35) Ru(phenh DPPN2+
(phen = 1,1O-phenanthroline and Ru(bpyh DPPN2+
(bpy 2,2' -bipyridine) causes molecular light switching for DNA. No detected emission was observed in aqueous solution due to quenching by hydrogen bonding between water and the phenazine nitrogens of the dppz ligand . Binding to DNA, however, protects the phenazine nitrogens from water th rough preferential intercalation of the dppz ligand and leads to intense photoluminescence.
Conclusion Ruthenium polypyridyl based systems serve as
promising candidates in the fi eld of molecular electronics due to their unique combination of photophysical, photochemical and redox properties. It is expected that these systems have bright future in development of molecular devices.
Acknowledgement Authors gratefully thank Department of Atomic
Energy (DAE) Mumbai for a financial support through BRNS .
References De Silva A P. G unartne H Q N, Gunnlaugsson T, Huxley A J M. McCoy C P, Rade macher J T & Rice T E, Chem Rev, 97 ( 1997) 1515 .
2 Phadke R S. III dian J p ure applied Physics , 33 (1995) 583 and references therein.
3 Di Casa M. Fabbrizzi L. Licchelli M, Poggi A. Sacchi 0 & Zema M, J chem Soc, Dallon Trans , (200 I) 167 1.
4 Fabbrizzi L & Poggi A, Gem Soc Rev, (1995) 197. 5 Ballardini R, Balzani V, Credi A, Gandolfi M T & Venturi
M. Ace chem Res, 34 (200 1) 445.
6 Balzani V, Gomez-Lopez M & Stoddart J F. Ace ehem Res. 31 (1998) 405 .
7 Sauvage J-P, Acc chem Res , 3 1 ( 1998) 6 11 . 8 Chiabrera A, Zitu E Di , Costa F & Bisio G M. J PhI'S D. App
Phy, 22 (1989) 1571 . 9 Lehn J M, Allgew Chem Internal Ed En81. 27 ( 1988) 89.
10 Kalyansundaram K, Coord Chem Rev, 46 ( 1982) 159. II Ghosh B K & Chakravorty A. Coord Gem Rev. 95 ( 19X9)
239. 12 Lahiri G K, Bhattacharya S , Mukherjee M, Mukhe lj ee A K
& Chakravorty A, Illorg Gem, 26 ( 1987) 3359. 13 Kalyansundram K, Gratzel M & Pe li ze tti E. Coord Ch l'lI/
Rev, 69 (1986) 57. 14 Keerthi K D, Santra B K & Lahiri G K. Polyhedroll . 17
( 1998) 1387. 15 Coe B J, Friessen D A, Thompson 0 W & Meyer T J. Illorg
G em , 35 (1996) 4575. 16 Denti G, Campagna S, Sabatino L, Serroni S. C iano M &
Balzani V in Photochemical cOll versioll alld SlO ra f.:e or solar energy; (Kluwer Academic Press: Netherlands) 1991.
17 Yadaw A K in Studies all some Ilew Rulli/II polrpvridrl complexes (Ph .D. Thesis, Banaras Hindu Uni versity: Varanasi) 2001.
18 Sinha R in Swdies all mo/w lluclear alld pO/\'IIUc/l'I II' ruthellium polypyridyl complexes cOllta illillg al'l)II 1(11ic alld heterocyclic compounds as co-ligands (Ph .D. Thes i ~ .
Banaras Hindu Uni vers ity: Varanasi) 200 I. 19 Balzani V, Zuris A, Venturi M, Compag na S & Serroni S.
Chem Rev, 96 ( 1996) 759. 20 Chakravarty A R & Chakravorty A, J chem Soc, Daholl
Trans, (1982) 1765. 2 1 Barige lletti F, Flamigni L, Balzani V, Collin J P. Sau vage J
P, Sour A, Co nstab le E C & Thompson A M W C. J Am chem Soc, 116 (1994) 7692.
22 Collin J-P, Guillevez S & Sauvage J-P. J che/II SoC, Ch('ll/ COl1lmun, (1989) 776.
23 Constable E C, Thompson A M W C & Greu lich S. J chl'lI/ Soc, Chem Commun , (1993) 1444.
24 Lehn J-M, Angew G em lilt Ed Engl, 29 ( 1990) 1304. 25 Constable E C. Housecroft C E. Scho field E R. Encinas S.
Armaro li N, Barigelle tti F, Flamigni L. Figgeme ier E & Vos J G, J chem Soc. Chem COml11UlI , ( 1999) 869 .
25a Balzani V, Moggi L & Scando la F in Supramolecular photochemistry (Reide l Dord recht : Ne therl ands) 1987. I.
26 Haga M, Ali M. Koseki S, Yoshimura A. Nozaki K & Ohnn T , 1110rg Chil1l Acta, 226 ( 1994) 17.
27 Beer P D, Dent S W, Fletcher N C & Wear T J. PO/I'hl'dmll. 15 (1996) 2983.
28 Mishra L, Yadaw A K, Cho i C S & Araki K. Ill r/ilill J Chl'lII. 38A (1999) 339.
29 Mi shra L, Yadaw A K. Srivastava S & Pale l A B. NI'II ' .I Chelll , 24 (2000) 505 .
30 Mi shra L, Yadaw A K, Phadke R S. C ho i C S & Araki K. Metal Based Drugs. 8 (200 I) 65 .
31 Mi shra L & Sinha R, Indiall J Chem (A), 39 (2000) 1295. 32 Mi shra L & Si nha R, Illdiall J Chem (A). 39 (2000) 11 3 1. 33 Kimura E, Wada S. Shionoya M, Takanasi T & Litaka Y. J
chem Soc Chem Commwl , ( 1990) 397. 34 Haga M, Ano T . Ishi zaki T. Kano K. Wazak i K & Ohllo T. J
chem Soc Daltoll TrailS. ( 1994) 263.
35 Komatsuzaki N, Himeda Y, Hi rose T. Sugihara H & Kasliga K, Bull chem Soc Japal/ , 72 ( 1999) 725.

1814 INDIAN J CHEM, SEC A, AUGUST 2003
36 Komatsuzak i N, Himeda y, Goto M, Kasuga K, Sugihara H & Arakawa H. Gem Lell, (1999) 327.
37 Kimura E. Bu X, Shionoya M, Wada S & Maruyama S, Inurg Gem. 31 ( 1992) 4542.
38 Mishra L & Yadaw A K. J trans met Chelll, 25 (2000) 579. 39 Cola L D, Barigelleni F & Balzani V, Chelllical Phy Lell.
178 ( 1991 ) 491. -10 Keene FR. Gelll Soc Rev, 27 ( 1998) 185. 40a Zkgler M & Zelewsky A V, Coord C/WIl Rev, 177 (1998)
257. 4 1 Knopt U & Zelewsky A V, Angew Chelll lilt Ed Engl , 38
(1999) 302. 42 Tzali s D & Tor Y. J Alii chelll Soc, 119 (1997) 852. 43 Warn mark K. Baxter P N W & Lehn J-M. J chelll Soc Chem
Comllllln , (1998) 993. 44 Kimura E, Bu X. Shionoya M, Wada S & Maruyama S,
Illorg Gem. 3 1 ( 1992) 4542. 45 Hawec ker J, Lehn J-M & Ziessel R, J chem Soc Chem
CO II IIIIUlI . ( 1983) 536. 46 Denti G, Campagna S. Serroni S, Ciano M & Balwni V. J
Alii che/II Soc. 114 ( 1992) 2944. 47 Kalyansundram K & Nazeeruddin Md K, /llorg Chil1l Acta.
226 (1994) 213. -18 Harriman A & Sauvage J-P, Chem Soc Rev, (1996) 41. 49 Connors Jr P J, Tzalis J D, Dunnik A L & Tor Y, /llorg
Gem, 37 (1998) 1121 . 50 Tomali a D A, Naylor A M & Goddard W A, Angew Chem
11/1 Ed Engl. 29 ( 1990) 138. 51 Frechet J-M, Science, 263 ( 1994) 1710. 52 Zeng F & Zimmerman S C. Chelll Rev, 97 (1997) 1681. 53 Newkome G R, Cardullo F. Constable E C, Moorefield C N
& Thompson A M W C, J chem Soc Chem Comlllun, (1993) 925 .
54 Bodige S, Torres A S, Maloney D J, Tate D, Kinsel G R, Walker A K & Macdonnell F D. J Alii chelll Soc, 119 (1997) 10364.
55 Winkler J R & Gray H B, Gem Rev, 92 (1992) 369 and references therein .
56 Shreder K, Harriman A & Iverson B L, J Alii chelll Soc, 11 7 ( 1995 ) 2673.
57 Ward M D, Chelll Rev, ( 1995) 121 and references therein. 58 Curti s J C. Bernstein J S & Meyer T J, /norg Chelll , 24
( 1985) 385. 59 Barigelleui F. Flamigni L, Balzani V, Collin J-P, Sauvage J
P. Sour A. Constable E C & Thompson A M W C, J Alii chem Soc. 116 (1994) 7692.
60 Larson S L, Hedrickson S M, Ferrere S, Derr D L & Elliott C M, J Alii chelll Soc, 117 (1995) 5881.
6 1 Furue M, Yoshidzumi T, Kinoshita S, Kushida T, Nozakura S & Kamachi M, Bull chelll Soc Japan , 64 (199 1) 1632.
62 Harriman A & Ziessel R, J chem Soc Chem Commul1, (1996) 1707.
63 Mishra L, Choi C S & Arak i K, Chelll Lell, (1997) 447.
64 lndelli M T. Scando la F. Flam igni L, Co llin J-P. Sauvag.: J-P & Sour A, Inorg Gem, 36 (1997) 4247.
65 Barigelletti F & Flamigni L. Chem Soc Re I'. 29 (2000) I. 66 Indelli M T. Bignozzi C A. Harriman A. Schoonover J R &
Scandola F, JAm chem Soc, 116 ( 1994) 3768. 67 Hammarstrom L, Barigelleni F. Flamigni L. Armaroli N.
Sour A. Collin J P & Sauvage J P. J Alii che/II Soc. I I X (1996) 11972.
68 Sauvage J P, Collin J 1', Cham broil J C. Guillerez S. Coudr':l C, Balzani V. Barigelletti F. Cola L De & Flailligni L. Chelll Rev, 94 (1994) 993.
69 Schicke B, Belser 1', Co la L De, Sabbioni E & Balz:lIli \' . .I Am chem Soc, 121 (1999) 4207.
70 Rastogi R I' & Mishra L, Indian J Chelli . 37A ( 199X) 377 and references therein.
7 1 Cosa M D, Fabbrizzi L. Licchelli M. Poggi A. Sacchi D & Zema M, J chelll Soc Dalton Trans. (200 I) 1671 .
71 Fabbrizzi L, Licchelli M & Pallavicini P. Ace chell/ Res . 32 (1999) 846.
73 Rohr S & Salzborg B M, J Biophys, 67 ( 1994) 1301. 74 Toth PT, Bindokas V 1', Bleakman D, Colmers W F & Miler
R .1, Nature (London), 364 (1993) 635 . 75 Santis G De. Fabbrizzi L, Licchelli M. Sardone N & V.:ld.:r,
A H, Gem EliI' J, 2 ( 1996) 1243. 76 Fabbrizzi L, Licche lli M, Pallavicini P. Perotti A. Tagli ': lli A
& Sacchi D, Chem EliI' J , 2 (1996) 75. 77 Goudie V, Harriman A & Lehn J-M . J che/II Soc Che/II
CO/WIIWI , (1993) 1034. 78 Ambroise A & Maiya B G. Ill org Chell/. 39 (2000) 4256. 79 Otsuki J, Tsujino M, Izaki T, Araki K, Seno M. Takatera K
& Watanabe T, JAm chelll Soc. 11 9 ( 1997) 7895. 80 Bissel R A, de Sil va A 1', Gunaratne H Q N. Lynch D L M.
Maguire GEM & Sundanayake K R A S, Chelll Soc R('l '. (1992) 187.
81 Fabbrizzi L, Licchelli M. Pallav icini P. Perott i A & Sacchi D, Allgew Chem Int Ed Engl. 33 (1994) 1975.
82 Lodeiro C, Pina F. Parola A J. Bencini A. Bianchi A. Bazzicalupi C, Ciattini S. Giorgi C. Masotti G A. VaitaIlcll li B & Demelo J S, /norg Chem. 40 (200 I) 68 13.
83 Hi ssler M, EI-ghayoury A. Harriman A & Ziessel R. Al/gclI '
G elll In t Ed ElIgl, 37 (1998) 1717. 84 Holmlin R E & Barton J K, Illorg Chem , 34 ( 1995) 7. 85 Kumar C V, Barton J K & Turro N J, J Alii che/II Soc . 107
( 1985) 5518. 86 Turro N J, Barton J K & Tomalia D A. Ace chell/ Res . 2-1
(1991) 332. 87 Barton J K. Go ldberg J M. Kumar C V & Turro N J. J Alii
chelll Soc. 108 ( 1986) 2081 . 88 Pyle A M, Rehmann J 1', Meshoyrer R. Kumar C V. BartoIl J
K & Turro N J, JAm chem Soc, 107 ( 19,)5) 5S 18.
89 Murphy C J & Barton J K, Methods £1I~VlII()I . 226 ( llJ93) 576.