systemic exposure to the metabolites of lesogaberan in
TRANSCRIPT

DMD #56614
1
Systemic Exposure to the Metabolites of Lesogaberan in Humans
and Animals – a Case Study of Metabolites in Safety Testing
Ann Aurell Holmberg, Anja Ekdahl, and Lars Weidolf
AstraZeneca R&D, Mölndal, Sweden
DMD Fast Forward. Published on March 21, 2014 as doi:10.1124/dmd.113.056614
Copyright 2014 by the American Society for Pharmacology and Experimental Therapeutics.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
2
Running title: Lesogaberan Metabolites in Safety Testing
Address correspondence to: Ann Aurell Holmberg, AstraZeneca R&D, S-431 83, Mölndal,
Sweden.
Phone: +46 (31) 776 10 00
Fax: +46 (31) 776 37 60
E-mail: [email protected]
Number of text pages: 15
Number of tables: 3
Number of figures: 2
Number of references: 25
Number of words in Abstract: 234
Number of words in Introduction: 642
Number of words in Discussion: 1201
ABBREVIATIONS: ADME, absorption, distribution, metabolism and excretion; AUC, area
under the plasma concentration–time curve; Ct, concentration at the time of the last plasma
sample; DRM, drug-related material; FDA, US Food and Drug Administration; GERD,
gastroesophageal reflux disease; HILIC, hydrophilic interaction liquid chromatography; ICH,
International Conference on Harmonisation of Technical Requirements for Registration of
Pharmaceuticals for Human Use; k, apparent terminal rate constant; LC-MS, liquid
chromatography with mass spectrometry detection; LLOQ, lower limit of quantification; MIST,
Metabolites in Safety Testing; PPI, proton pump inhibitor.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
3
Abstract
During preclinical and early-phase clinical studies of drug candidates, exposure to metabolites
should be monitored to determine whether safety conclusions drawn from studies in animals can
be extrapolated to humans. Metabolites accounting for more than 10% of total exposure to drug-
related material (DRM) in humans are of regulatory concern, and for any such metabolites
adequate exposure should be demonstrated in animals before large-scale phase 3 clinical trials
are conducted. We have previously identified six metabolites, M1–M6, of the gastroesophageal
reflux inhibitor lesogaberan. Here, we measure exposure in humans, rats and beagle dogs to
lesogaberan and these metabolites. Plasma samples were taken at various time points after
lesogaberan dosing in two clinical and three preclinical studies. Concentrations of lesogaberan
and its metabolites were measured, and exposures during a single dosing interval were
calculated. The parent compound and metabolites M1, M2, M4 and M5 were together shown to
comprise all significant exposure to DRM in humans. Only M4 and M5 were present at levels of
regulatory concern (10.6% and 18.9% of total exposure to DRM, respectively, at steady state).
Absolute exposure to M5 was greater in rats during toxicology studies than the highest absolute
exposure observed in humans at steady state (117.0 µmol×h/l versus 52.2 µmol×h/l). In contrast,
exposure to M4 in rats was less than 50% of the highest absolute exposure observed in humans.
Further safety testing of this metabolite may therefore be required.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
4
Introduction
In 2008, the US Food and Drug Administration (FDA) published guidance on monitoring
exposure in humans and animals to the metabolites of novel drug candidates during preclinical
and early-phase clinical safety studies (Metabolites in Safety Testing [MIST]) (FDA, 2008).
According to this guidance, any metabolite for which the total exposure in humans accounts for
more than 10% of exposure to the parent compound at steady state is of regulatory concern. For
such metabolites, the absolute level of exposure in at least one animal species used in general
toxicology studies should equal or exceed that observed in humans. If this requirement is not
met for any metabolite of regulatory concern, further safety testing is required. Two strategies
are recommended: the identification of and safety evaluation in an alternative animal species
that produces the metabolite in sufficient quantities, or dosing of synthetic metabolite to a
species already tested. Regulatory guidance does not distinguish between metabolite
characteristics; stable, reactive and pharmacologically inert or active metabolites are treated the
same.
The FDA MIST guidance has been superseded by guidance from the International Conference
on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human
Use (ICH) (ICH, 2009; ICH, 2012). The major difference between the FDA and ICH guidance
lies in the definitions of metabolites of regulatory concern; according to the ICH, such
metabolites are those for which exposure in humans accounts for more than 10% of the total
exposure to drug-related material (DRM), not parent compound alone. The ICH guidance also
considers adequate exposure in animals to metabolites of regulatory concern to be anything
greater than 50% of the maximum absolute exposure observed in humans, unless the metabolite
constitutes the majority of the total human exposure to DRM (ICH, 2012). In this case, exposure
in animals should exceed the maximum absolute exposure observed in humans. The FDA
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
5
adopted the ICH guidance in 2010 (Yu et al., 2010), but has not officially changed or withdrawn
its original guidance.
Although there is much published literature focusing on MIST strategies, perspectives and
methodology, as well as reviews of the topic (Ma et al., 2010; Yu et al., 2010; Gao and Obach,
2011; Luffer-Atlas, 2012), there are very few published case studies demonstrating the
application of regulatory guidance (Luffer-Atlas, 2008; Nedderman et al., 2011). Here, we
report the results of MIST studies carried out during the preclinical and clinical development of
the GABAB receptor agonist lesogaberan ([R]-[3-amino-2-fluoropropyl]phosphinic acid).
Lesogaberan has been developed as a reflux inhibitor for the treatment of patients with
gastroesophageal reflux disease (GERD) who have a partial response to proton pump inhibitor
(PPI) therapy (Cioffi et al., 1999; Dent et al., 2005; Boeckxstaens et al., 2010a; Boeckxstaens et
al., 2010b; El-Serag et al., 2010; Boeckxstaens et al., 2011). The parent compound was shown to
be stable in human and animal hepatocytes in vitro, but to be extensively metabolised in humans
in vivo (Niazi et al., 2011). Levels of metabolism in animals in vivo were considerably lower
(data on file). This finding was communicated to the FDA, and further experiments were
undertaken to identify lesogaberan metabolites and to develop methods for their quantification
(Dunér et al., 2013; Ekdahl et al., 2013).
The metabolite profile of lesogaberan in rats was found to be similar to that in humans, despite
lower overall levels of metabolism of the parent compound in rats (data on file). Six metabolites
were identified in rat urine, the primary route of drug excretion (Ekdahl et al., 2013). These were
designated M1–M6, and were shown by comparison with synthetic reference standards of their
behaviour in liquid chromatography and mass spectrometry (LC-MS) to be: M1, ([2R]-3-
acetamido-2-fluoropropyl)phosphinic acid; M2, 3-hydroxypropylphosphinic acid; M3, [2R]-2-
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
6
fluoro-3-hydroxyphosphonoylpropanoic acid; M4, ([2R]-2-fluoro-3-guanidinopropyl)phosphinic
acid; M5, 3-hydroxyphosphonoylpropanoic acid; and M6, ([2R]-3-amino-2-
fluoropropyl)phosphonic acid (Fig. 1) (Ekdahl et al., 2013). Qualified methods were
subsequently developed to determine their plasma concentrations (Dunér et al., 2013). Here, we
report the results of investigations conducted to determine exposure to lesogaberan and these six
metabolites in humans, rats and beagle dogs.
Materials and Methods
Plasma samples and determination of metabolite concentrations
Animal and human plasma samples were obtained at various time points after lesogaberan
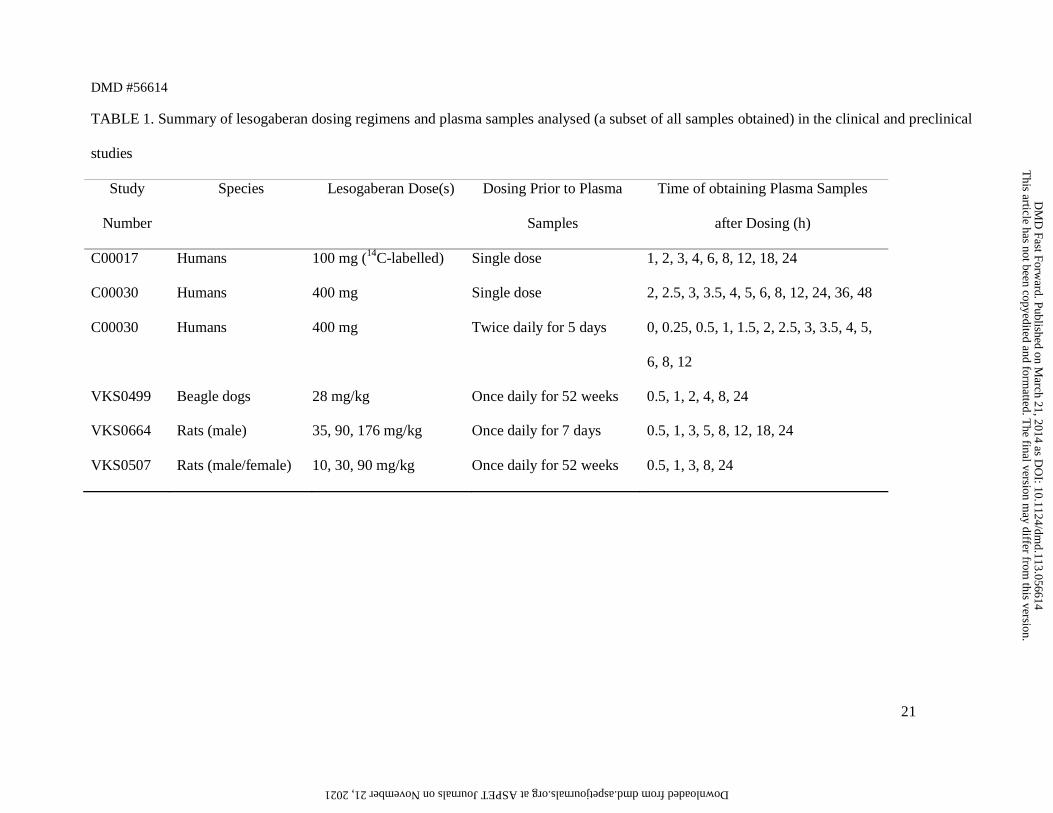
dosing in five clinical and preclinical studies, as outlined in Table 1. All analyses were carried
out in accordance with the methods and ethical standards outlined in the relevant study protocol.
Metabolite concentrations in each plasma sample were measured using LC-MS, as described
previously (Dunér et al., 2013). In the human studies, concentrations of the parent compound
were measured by an accredited laboratory (PRA International-Bioanalytical Laboratory B.V.,
Assen, Netherlands) using LC-MS, and in the animal studies, as described previously (Fakt et
al., 2003). In the human study in which 14C-labelled lesogaberan was administered, total
radioactivity was measured by Covance Laboratories Ltd (Harrogate, UK) using liquid
scintillation counting.
Pharmacokinetic calculations
The pharmacokinetic parameters of lesogaberan and its metabolites were calculated by non-
compartmental analysis using WinNonlin Enterprise (Pharsight Corporation, Mountain View,
CA). At steady state, the area under the plasma concentration–time curve for the parent
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
7
compound and each metabolite during a single dosing interval (AUCτ) was calculated using the
linear trapezoidal method. For determination of AUC0–∞ in the human study in which a single
400 mg dose of lesogaberan was administered, AUC0–48 h was calculated and extrapolated to
infinity by adding Ct/k, where Ct is the concentration at the time of the last plasma sample and k
is the apparent terminal rate constant, obtained by linear least-squares regression analysis of the
logarithm of the last three plasma concentrations versus time. For the determination of 24-hour
exposure in the human study in which twice-daily 400 mg doses were given, AUCτ (12-hour
exposure) was calculated and multiplied by 2.
Results
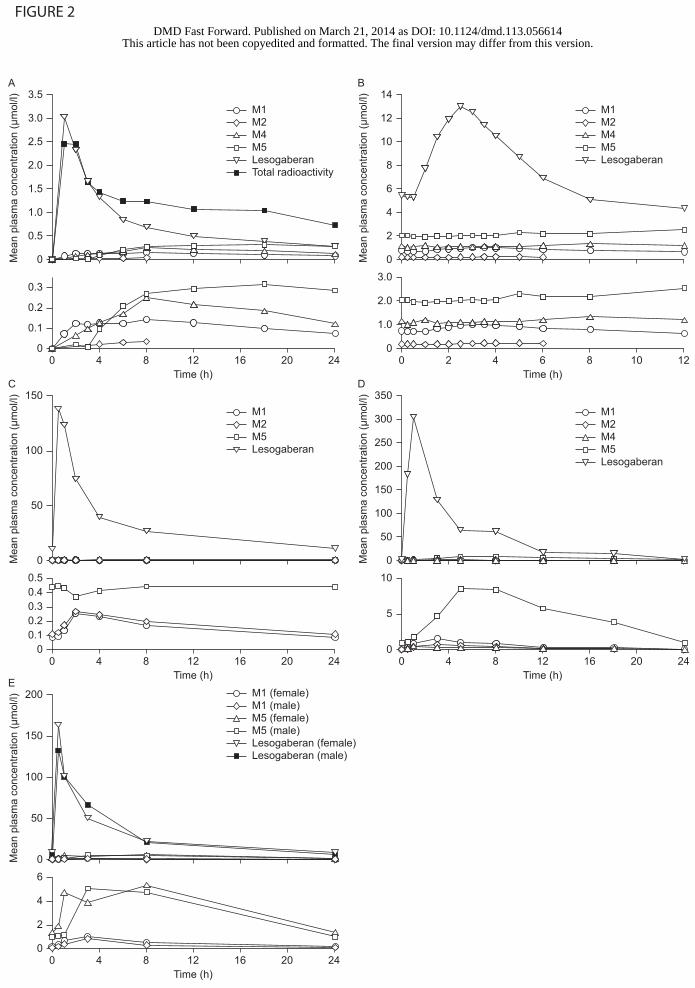
Exposure to lesogaberan and its metabolites in humans. Exposure to lesogaberan and its
metabolites was measured in humans in three experiments carried out on plasma samples taken
from two clinical studies (Table 1; Fig. 2, A and B). Two of these experiments assessed
exposure after single lesogaberan doses (100 mg and 400 mg), and one assessed exposure during
a 24-hour period at steady state during twice-daily 400 mg dosing. In the study assessing both
single and repeated twice-daily 400 mg dosing, metabolites M3 and M6 were not present at
levels above the lower limit of quantification, 0.1 µmol/l, at any of the time points analysed
(Table 2). In this study, a single 400 mg lesogaberan dose was administered, after which plasma
samples were taken for 48 hours. After a further 1-day washout period, a 5-day period of
400 mg twice-daily dosing was started, and plasma samples were obtained for 12 hours after the
final dose. Because 24-hour exposure to either M3 or M6 at 0.1 µmol/l would represent a
theoretical maximum AUC of 2.4 µmol×h/l (i.e. less than 10% of exposure to total DRM), these
metabolites were concluded not to be of regulatory concern and were omitted from any further
analysis (including in the 100 mg single-dose radiolabelled study).
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
8
In the study in which a single 100 mg dose was given, 14C-labelled lesogaberan was
administered, allowing exposure to total radioactivity to be measured (Niazi et al., 2011). In this
experiment, all radioactivity was accounted for by the parent compound and the four measured
metabolites: the AUC0–24 h values for lesogaberan, M1, M2, M4 and M5 summed to 105.4% of
the AUC0–24 h for total radioactivity (Table 2). Thus, it was concluded that no major metabolites
remain to be identified and quantified.
At steady state, after 5 days of twice daily 400 mg dosing, exposure to M1 accounted for 7.1%
of exposure to total DRM, exposure to M2 accounted for 1.7%, exposure to M4 accounted for
10.6% and exposure to M5 accounted for 18.9% (Table 2). Similar results were seen in the
single-dose experiments. Thus, only M4 and M5 were shown to be metabolites of regulatory
concern, according to the ICH guidance (ICH, 2009; ICH, 2012).
Exposure to lesogaberan and its metabolites in animals. Exposure to parent compound and
metabolites was measured in rats and beagle dogs during three preclinical studies (Table 1;
Table 3; Fig. 2, C–E). All calculations were for exposure during a single 24-hour dosing interval
at steady state after repeated once-daily lesogaberan administration and, in all studies, no
adverse effects were observed at the highest dose tested. Exposure to M2 was not measured in
rats in the 52-week study, as this metabolite was not of regulatory concern in humans. Exposure
to M4 in rats could only be measured in the 7-day study, owing to lack of availability of plasma
samples in the 52-week study at the time that M4 was identified and the quantification
methodology developed. Exposure to M4 was not measured in beagle dogs, as it was expected
from the results of early dog absorption, distribution, metabolism and excretion (ADME) studies
to be present only at very low levels (data on file).
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
9
Of the two metabolites of regulatory concern, the highest absolute AUC observed in humans for
M5, 52.2 µmol×h/l (AUCτ after twice-daily 400 mg dosing), was exceeded in rats after 7 days of
once-daily 176 mg/kg dosing (117.0 µmol×h/l). Thus, extrapolation to humans of safety
conclusions from preclinical studies performed in rats can be considered valid with respect to
this metabolite. In contrast, the highest AUC for M4 in animals, which was only measured in the
7-day study in rats, did not exceed 50% of the highest AUC observed in humans (i.e. was below
the level considered adequate in the ICH MIST guidance for a metabolite that does not
constitute the majority of exposure to total DRM in humans) (ICH, 2012). Further toxicology
testing may therefore be necessary for this metabolite. However, the development program for
lesogaberan has been halted, as phase 2b study results did not meet criteria for progression into
phase 3 clinical trials (Shaheen et al., 2012).
Discussion
Although the current literature on MIST includes numerous methodology, perspective and
strategic papers, and literature reviews (Ma et al., 2010; Yu et al., 2010; Gao and Obach, 2011;
Luffer-Atlas, 2012), there are very few case studies of the application of regulatory guidance
(Luffer-Atlas, 2008; Nedderman et al., 2011). Here, we have presented the results of MIST
experiments carried out during the clinical development of lesogaberan, which therefore
represent an important addition to available reports. The experiments were initially undertaken
after communication to the FDA of results showing that, despite the high in vitro stability of
lesogaberan in hepatocytes from various species, it is highly metabolised in humans in vivo. In
the human phase 1 ADME study, in which 14C-labelled lesogaberan was administered orally and
intravenously to healthy individuals, 84% of the dose was excreted in urine (based on recovery
of radioactivity after both oral and intravenous dosing), but renal clearance of unchanged parent
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
10
compound accounted for only approximately 22% of total clearance (Niazi et al., 2011). Thus,
the majority of the parent compound was metabolised. After oral dosing, total plasma
radioactivity levels were clearly higher than the concentration of the parent compound at time
points later than 1 hour after dosing, indicating circulating metabolites. In contrast, parent
compound accounted for approximately 65% of total DRM excreted in urine and faeces in rats,
and approximately 74% in dogs (data on file). Experiments were therefore conducted to identify
the metabolites of lesogaberan in humans, and to develop methods to quantify them in plasma.
FDA and ICH guidance recommends that any metabolite for which systemic exposure in
humans at steady state accounts for 10% or more of total exposure to DRM is of regulatory
concern (FDA, 2008; ICH, 2009; ICH, 2012). For such metabolites, the absolute level of
exposure observed in at least one animal species used in general toxicology studies should be
50% or more of the maximum exposure observed in humans, in order to conclude that the
contribution of the metabolite to the toxicity of the drug has been established (ICH, 2012). An
exception to this is in cases in which the metabolite constitutes the majority of human total
DRM exposure; for any such metabolite, exposure in animals should be shown to exceed the
maximum level observed in humans. For any metabolites of regulatory concern that do not meet
these requirements, further toxicology studies are recommended. Any potential concerns
regarding metabolite toxicity should be resolved before beginning large-scale phase 3 clinical
trials (ICH, 2009).
The high in vitro stability of lesogaberan presented initial challenges for metabolite
identification, exacerbated by the highly polar and zwitterionic nature of the parent compound
and its metabolites, which resulted in poor LC retention and MS response. Consequently, the
elegant in vivo non-radiolabelled cross-species systemic metabolite exposure comparison that
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
11
has been proposed in the literature could not be applied (Ma et al., 2010; Gao and Obach, 2011).
Our standard approach to the early assessment of metabolite exposures under steady-state
conditions would compare human plasma samples obtained after dosing to the highest level
expected to be used in the clinic, with samples obtained from animal species at the highest ‘no
observed adverse effect’ level. For lesogaberan, however, because of the very low metabolic
turnover in vitro in combination with the poor MS response of formed metabolites, the
metabolite profile could not be assessed using samples from our early studies. It was therefore
not possible to follow this relatively simple protocol for early exposure comparisons.
These difficulties were overcome by the use of hydrophilic interaction liquid chromatography
(HILIC) to separate metabolites excreted in rat urine after administration of a high dose of
lesogaberan (rats had previously been shown to have a similar lesogaberan metabolite profile to
humans, despite overall lower levels of metabolism of the parent compound), coupled with
detection using linear trap quadrupole orbitrap MS (Ekdahl et al., 2013). In HILIC, a mixture of
an organic solvent and water is used in the mobile phase, together with a hydrophilic silica or
modified silica stationary phase; this offers better retention, separation, sensitivity and efficacy
than traditional reversed-phase LC in the separation of small and highly polar compounds
(Ikegami et al., 2008; Chirita et al., 2010; Hsieh, 2010; Jian et al., 2011). HILIC offers the
further benefit of being favorable for electrospray MS owing to the high organic content of the
eluent.
Six unique compounds (M1–M6; Fig. 1) in addition to the parent compound were detected in rat
urine, and their identities were confirmed by comparison of their LC-MS properties with those
of synthesised reference compounds (Ekdahl et al., 2013), followed by unambiguous structural
elucidation by nuclear magnetic resonance spectroscopy (data on file). Lesogaberan was found
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
12
to be metabolised via oxidative pathways, including by deamination and subsequent oxidation to
the corresponding carboxylic acid, oxidation to the phosphonic acid, and conjugation to an N-
acetylated species. The routes to formation of the more surprising defluorinated and guanidino
metabolites have previously been discussed (Ekdahl et al., 2013). Bioanalytic methods were
subsequently developed and qualified for the determination of metabolite concentration in
human and animal plasma samples (Dunér et al., 2013). This is in accordance with the “tiered
approach” recommended by the European Bioanalytical Forum (Timmerman et al., 2010):
preliminary screening to detect metabolites, followed by the development and use of qualified
and/or validated bioanalytic methods to determine absolute parent compound and metabolite
exposures.
In the experiments described in the current report, we have analysed samples taken from human
ADME (single 100 mg dose) and dose escalation (single and repeat 400 mg dosing) studies, and
animal studies carried out in rats and beagle dogs. For beagle dogs, plasma samples were
remainder from a 12-month toxicology study, while in rats, the 7-days study was carried out in
order to mimic longer term safety studies in rats, from which no sample was left to analyse and
for which the maximum dose was 176 mg/kg. The 52-week rat samples were remainder from a
carcinogenesis study, which confirmed the results from the 7-day study. In the human dose
escalation study, use of plasma samples for exploratory metabolite work was specified in the
protocol. We have shown that, in addition to parent compound, four of the six metabolites (M1,
M2, M4 and M5) identified in rats account for all significant exposure to DRM in humans. Of
these, only two (M4 and M5) are present at levels of regulatory concern. M5 was particularly
prevalent, representing 18.9% of total exposure to DRM at steady state, while metabolite M4
was closer to the 10% threshold, at 10.6% of total exposure to DRM at steady state. Overall
exposure to each metabolite was similar in humans after single doses and at steady state.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
13
For M5, absolute exposures observed in rats at the highest doses tested in both the 7-day and 52-
week studies exceeded the greatest absolute exposure seen in humans. Adequate exposure to this
metabolite could be achieved in animals despite overall lower levels of metabolism than in
humans, as high lesogaberan doses could be administered without apparent safety concerns. The
contribution of M5 to the toxicology of lesogaberan can therefore be considered to have been
established. This was not the case, however, for M4, the concentration of which was only
measured in the 7-day rat study. For this metabolite further safety testing may be required
(although it should be noted that 400 mg twice daily is a very high lesogaberan dose in humans,
and was considerably higher than the highest dose administered during the phase 2b study
[240 mg twice daily] (Shaheen et al., 2012)). Possibilities for such experiments include testing
an alternative animal species that generates M4 in sufficient quantities, or dosing animals
directly with synthetic M4. However, because the phase 2b study results did not meet the criteria
for progression into phase 3 clinical trials, the development program for lesogaberan has been
halted (Shaheen et al., 2012). Further toxicology experiments are therefore likely to be delayed
until the future developmental process for this compound becomes clear.
In conclusion, we have described how MIST regulatory guidance was followed during the
development of the drug candidate lesogaberan. Despite challenges such as the high polarity,
low molecular weight and low MS response of lesogaberan and its metabolites, robust
quantification was achieved that allowed comparisons of metabolite exposure in humans and
animals to be made with confidence. Our data indicate that further studies would be necessary to
ensure adequate exposure of at least one animal species to metabolite M4 before large-scale
phase 3 trials could be conducted. If the clinical development program for lesogaberan is
continued, we are well-equipped to address the safety assessment of this remaining metabolite.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
14
Acknowledgments
Medical writing support was provided by Dr. Stephen Sweet of Oxford PharmaGenesis™ Ltd.,
and was funded by AstraZeneca R&D, Mölndal, Sweden.
Authorship Contributions
Participated in research design: Holmberg, Ekdahl and Weidolf.
Conducted experiments: None of the authors.
Contributed new reagents or analytic tools: Ekdahl.
Performed data analysis: Holmberg.
Wrote or contributed to the writing of the manuscript: Holmberg, Ekdahl and Weidolf.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
15
References
Boeckxstaens GE, Beaumont H, Hatlebakk JG, Silberg DG, Bjorck K, Karlsson M, and
Denison H (2011) A novel reflux inhibitor lesogaberan (AZD3355) as add-on
treatment in patients with GORD with persistent reflux symptoms despite proton
pump inhibitor therapy: a randomised placebo-controlled trial. Gut 60:1182–1188.
Boeckxstaens GE, Beaumont H, Mertens V, Denison H, Ruth M, Adler J, Silberg DG, and
Sifrim D (2010a) Effects of lesogaberan on reflux and lower esophageal sphincter
function in patients with gastroesophageal reflux disease. Gastroenterology 139:409–
417.
Boeckxstaens GE, Rydholm H, Lei A, Adler J, and Ruth M (2010b) Effect of lesogaberan, a
novel GABA(B)-receptor agonist, on transient lower oesophageal sphincter
relaxations in male subjects. Aliment Pharmacol Ther 31:1208–1217.
Chirita RI, West C, Finaru AL, and Elfakir C (2010) Approach to hydrophilic interaction
chromatography column selection: application to neurotransmitters analysis. J
Chromatogr A 1217:3091-3104.
Cioffi U, Rosso L, and De Simone M (1999) Gastroesophageal reflux disease. Pathogenesis,
symptoms and complications. Minerva Gastroenterol Dietol 45:43–49.
Dent J, El-Serag HB, Wallander MA, and Johansson S (2005) Epidemiology of gastro-
oesophageal reflux disease: a systematic review. Gut 54:710–717.
Dunér K, Bottner P, and Norlén A (2013) Development of analytical methods for the
quantification of metabolites of lesogaberan in a MIST investigation. Biomed
Chromatogr doi: 10.1002/bmc.3029.
Ekdahl A, Aurell-Holmberg A, and Castagnoli N, Jr. (2013) Identification of the metabolites
of lesogaberan using linear trap quadrupole orbitrap mass spectrometry and
hydrophilic interaction liquid chromatography. Xenobiotica 43:461–467.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
16
El-Serag H, Becher A, and Jones R (2010) Systematic review: persistent reflux symptoms on
proton pump inhibitor therapy in primary care and community studies. Aliment
Pharmacol Ther 32:720–737.
Fakt C, Jacobson B, Leandersson S, Olsson B, and Persson B (2003) Determination of a
small, highly polar aminopropylphosphinic acid as racemate in plasma and urine and
as separated enantiomers in plasma by liquid chromatography and tandem mass
spectrometry. Anal Chim Acta 492:261–269.
FDA (2008) Safety testing of drug metabolites. Available from:
http://www.fda.gov/OHRMS/DOCKETS/98fr/FDA-2008-D-0065-GDL.pdf (accessed
13 November 2013)
Gao H and Obach RS (2011) Addressing MIST (Metabolites in Safety Testing): bioanalytical
approaches to address metabolite exposures in humans and animals. Curr Drug Metab
12:578-586.
Hsieh Y (2010) Hydrophilic interaction liquid chromatography-tandem mass spectrometry
for drug development. Curr Drug Discov Technol 7:223-231.
ICH (2009) Topic M3(R2): non-clinical safety studies for the conduct of human clinical trials
and marketing authorization for pharmaceuticals. Available from:
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/
09/WC500002720.pdf (accessed 07 November 2012)
ICH (2012) M3(R2) Implementation Working Group, M3(R2) guideline: guidance on
nonclinical safety studies for the conduct of human clinical trials and marketing
authorization for pharmaceuticals questions & answers (R2). Available from:
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidiscip
linary/M3_R2/Q_As/M3_R2_Q_A_R2_Step4.pdf (accessed 07 November 2012)
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
17
Ikegami T, Tomomatsu K, Takubo H, Horie K, and Tanaka N (2008) Separation efficiencies
in hydrophilic interaction chromatography. J Chromatogr A 1184:474-503.
Jian W, Xu Y, Edom RW, and Weng N (2011) Analysis of polar metabolites by hydrophilic
interaction chromatography–MS/MS. Bioanalysis 3:899-912.
Luffer-Atlas D (2008) Unique/major human metabolites: why, how, and when to test for
safety in animals. Drug Metab Rev 40:447-463.
Luffer-Atlas D (2012) The early estimation of circulating drug metabolites in humans. Expert
Opin Drug Metab Toxicol 8:985-997.
Ma S, Li Z, Lee KJ, and Chowdhury SK (2010) Determination of exposure multiples of
human metabolites for MIST assessment in preclinical safety species without using
reference standards or radiolabeled compounds. Chem Res Toxicol 23:1871-1873.
Nedderman AN, Dear GJ, North S, Obach RS, and Higton D (2011) From definition to
implementation: a cross-industry perspective of past, current and future MIST
strategies. Xenobiotica 41:605-622.
Niazi M, Skrtic S, Ruth M, and Holmberg AA (2011) Pharmacokinetic profile of lesogaberan
(AZD3355) in healthy subjects: a novel GABA(B)-receptor agonist reflux inhibitor.
Drugs R D 11:77–83.
Shaheen NJ, Denison H, Björck K, Karlsson M, and Silberg DG (2012) Efficacy and safety
of lesogaberan in gastroesophageal reflux disease: a randomized controlled trial. Gut
doi: gutjnl-2012-302737.
Timmerman P, Anders Kall M, Gordon B, Laakso S, Freisleben A, and Hucker R (2010) Best
practices in a tiered approach to metabolite quantification: views and
recommendations of the European Bioanalysis Forum. Bioanalysis 2:1185-1194.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
18
Yu H, Bischoff D, and Tweedie D (2010) Challenges and solutions to metabolites in safety
testing: impact of the International Conference on Harmonization M3(R2) guidance.
Expert Opin Drug Metab Toxicol 6:1539-1549.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
19
Footnotes
The study was funded by AstraZeneca R&D, Mölndal, Sweden.
Ann Aurell Holmberg, Anja Ekdahl and Lars Weidolf are employees of AstraZeneca R&D,
Mölndal, Sweden.
Presented as a poster at the International Society for the Study of Xenobiotics (ISSX) annual
meeting, 2013. Abstract published in ISSX Online Abstracts, Supplement 8, No. 2, 2013.
(http://issx.confex.com/issx/intl10/webprogrampreliminary/Paper29458.html)
Address for reprint requests: Ann Aurell Holmberg, AstraZeneca R&D, S-431 83, Mölndal, Sweden (email: [email protected]).
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from

DMD #56614
23617551-file00 20
Figures and Tables
Fig. 1. Chemical structures of lesogaberan and metabolites M1–M6. Lesogaberan, [2R]-(3-amino-
2-fluoropropyl)phosphinic acid; M1, [2R]-3-acetamido-2-fluoropropyl]phosphinic acid; M2, 3-
hydroxypropylphosphinic acid; M3, [2R]-2-fluoro-3-hydroxyphosphonoylpropanoic acid; M4,
([2R]-2-fluoro-3-guanidinopropyl)phosphinic acid; M5, 3-hydroxyphosphonoylpropanoic acid; M6,
([2R]-3-amino-2-fluoropropyl)phosphonic acid.
Fig. 2. Lesogaberan and metabolite concentrations plotted against time after the final lesogaberan
dose in clinical and preclinical studies. (A) Humans, single 100 mg dose (14C-labelled); (B)
humans, 400 mg twice daily for 5 days; (C) beagle dogs, 28 mg/kg once daily for 52 weeks; (D)
male rats, 176 mg/kg once daily for 7 days; (E) male and female rats, 90 mg/kg once daily for 52
weeks. The lower graphs in each panel are at an increased magnification to show the concentrations
of metabolites present at low levels.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from
DMD #56614
21
TABLE 1. Summary of lesogaberan dosing regimens and plasma samples analysed (a subset of all samples obtained) in the clinical and preclinical
studies
Study
Number
Species Lesogaberan Dose(s) Dosing Prior to Plasma
Samples
Time of obtaining Plasma Samples
after Dosing (h)
C00017 Humans 100 mg (14C-labelled) Single dose 1, 2, 3, 4, 6, 8, 12, 18, 24
C00030 Humans 400 mg Single dose 2, 2.5, 3, 3.5, 4, 5, 6, 8, 12, 24, 36, 48
C00030 Humans 400 mg Twice daily for 5 days 0, 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5,
6, 8, 12
VKS0499 Beagle dogs 28 mg/kg Once daily for 52 weeks 0.5, 1, 2, 4, 8, 24
VKS0664 Rats (male) 35, 90, 176 mg/kg Once daily for 7 days 0.5, 1, 3, 5, 8, 12, 18, 24
VKS0507 Rats (male/female) 10, 30, 90 mg/kg Once daily for 52 weeks 0.5, 1, 3, 8, 24
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
DM
D Fast Forw
ard. Published on March 21, 2014 as D
OI: 10.1124/dm
d.113.056614 at ASPET Journals on November 21, 2021 dmd.aspetjournals.org Downloaded from

DMD #56614
22
TABLE 2. Mean AUC values for lesogaberan and its metabolites in human studies
AUC
value
measured
Lesogaberan
Dose
Dosing
Regimen
Total
Radioactivity
AUC,
µmol×h/l
AUC, µmol×h/l
(% Parent; % Total DRM)
Lesogaberan M1 M2 M3 M4 M5 M6
AUC0–24 h 100 mg Single dose 28.100 17.7
(100.0; 59.7)
2.56
(14.5; 8.6)
0.145
(0.8; 0.5)
– 3.66
(20.7; 12.4)
5.56
(31.4; 18.8)
–
AUC0–∞ 400 mg Single dose – 87.9
(100.0; 52.8)
10.5
(11.9; 6.3)
3.41
(3.9; 2.0)
< 2.40 17.8
(20.3; 10.7)
47.0
(53.5; 28.2)
< 2.40
AUCτ × 2 400 mg Twice daily
for 5 days
– 171
(100.0; 61.8)
19.6
(11.5; 7.1)
4.66
(2.7; 1.7)
< 2.40 29.2
(17.1; 10.6)
52.2
(30.5; 18.9)
< 2.40
–, not measured; AUC, area under curve; DRM, drug-related material (expressed as the sum of the AUCs for parent compound and measured metabolites).
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
DM
D Fast Forw
ard. Published on March 21, 2014 as D
OI: 10.1124/dm
d.113.056614 at ASPET Journals on November 21, 2021 dmd.aspetjournals.org Downloaded from

DMD #56614
23
TABLE 3. Mean AUCτ values for lesogaberan and its metabolites in animal studies
Species Lesogaberan
Dose
Dosing Regimen AUCτ, µmol×h/l
Lesogaberan M1 M2 M4 M5
Beagle dogs 28 mg/kg Once daily for 52 weeks 712 3.25 3.85 – 9.11
Rats (male) 35 mg/kg Once daily for 7 days 322 3.24 1.54 1.31 27.6
90 mg/kg 784 7.59 3.41 2.76 55.5
176 mg/kg 1280 12.2 5.75 4.71 117
Rats (male) 10 mg/kg Once daily for 52 weeks 107 0.790 – – 15.8
30 mg/kg 253 1.90 – – 35.1
90 mg/kg 632 6.59 – – 77.0
Rats (female) 10 mg/kg Once daily for 52 weeks 90.5 0.809 – – 11.6
30 mg/kg 277 5.06 – – 43.7
90 mg/kg 648 10.4 – – 87.3
–, not measured; AUC, area under curve.
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
DM
D Fast Forw
ard. Published on March 21, 2014 as D
OI: 10.1124/dm
d.113.056614 at ASPET Journals on November 21, 2021 dmd.aspetjournals.org Downloaded from

Lesogaberan (AZD3355)
H2N
F O
P OH
H
M2: C3H9O3P
HO
O
P
H
OH
M1: C5H11NO3PF
F O
O
P
H
NHH3COH
M4: C4H11O2N3PF
F O
NH
P
H
NHH2NOH
M3: C3H6O4PF
OH
F O
O P
H
OH
H2N
F O
P OH
OH
M6: C3H9O3NPFM5: C3H7O4P
HO
O
O P
H
OH
FIGURE 1
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from
E
Mea
n pl
asm
a co
ncen
tratio
n (µ
mol
/l)
A
Mea
n pl
asm
a co
ncen
tratio
n (µ
mol
/l)
B
Mea
n pl
asm
a co
ncen
tratio
n (µ
mol
/l)
C
Mea
n pl
asm
a co
ncen
tratio
n (µ
mol
/l)
D
Mea
n pl
asm
a co
ncen
tratio
n (µ
mol
/l)
200
100
50
0
150
14
12
10
8
6
4
2
0
150
100
50
0
350
300
250
200
150
100
50
0
3.5
3.0
2.5
2.0
1.5
1.0
0.5
0
6
0
4
3.0
2.0
0
0.50.4
0
10
5
0
0 4 8 12Time (h)
16 20 24
2
0.3
00 4 8 12
Time (h)16 20 24
0.2
0.1
0 2 4 6Time (h)
8 10 12
1.0
0 4 8 12Time (h)
16 20 24
0.30.20.1
0 4 8 12Time (h)
16 20 24
M1 (female)M1 (male)M5 (female)M5 (male)Lesogaberan (female)Lesogaberan (male)
M1M2M4
LesogaberanM5
M1M2M5Lesogaberan
M1M2M4
LesogaberanTotal radioactivity
M5
M1M2M4
LesogaberanM5
FIGURE 2
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on March 21, 2014 as DOI: 10.1124/dmd.113.056614
at ASPE
T Journals on N
ovember 21, 2021
dmd.aspetjournals.org
Dow
nloaded from