synthetic peptide ligand mimetics and tumor...
TRANSCRIPT

Synthetic Peptide Ligand Mimetics and Tumor Cell Motility
Item Type text; Electronic Dissertation
Authors Sroka, Thomas Charles
Publisher The University of Arizona.
Rights Copyright © is held by the author. Digital access to this materialis made possible by the University Libraries, University of Arizona.Further transmission, reproduction or presentation (such aspublic display or performance) of protected items is prohibitedexcept with permission of the author.
Download date 09/06/2018 10:56:08
Link to Item http://hdl.handle.net/10150/194831

SYNTHETIC PEPTIDE LIGAND MIMETICS AND TUMOR CELL MOTILITY
by
Thomas Charles Sroka
_______________________________
A Dissertation Submitted to the Faculty of the
GRADUATE INTERDISCIPLINARY PROGRAM IN CANCER BIOLOGY
In Partial Fulfillment of the Requirements For the Degree of
DOCTOR OF PHILOSOPHY
In the Graduate College
THE UNIVERISTY OF ARIZONA
2005

2
THE UNIVERSITY OF ARIZONA GRADUATE COLLEGE
As members of the Dissertation Committee, we certify that we have read the dissertation prepared by Thomas Charles Sroka entitled Synthetic Peptide Ligand Mimetics and Tumor Cell Motility and recommend that it be accepted as fulfilling the dissertation requirement for the Degree of Doctor of Philosophy _______________________________________________________________________ Date: (10/21/05)Anne E. Cress, Ph.D. _______________________________________________________________________ Date: (10/21/05)Roger L. Miesfeld, Ph.D. _______________________________________________________________________ Date: (10/21/05)Scott E. Klewer, M.D.
Final approval and acceptance of this dissertation is contingent upon the candidate’s submission of the final copies of the dissertation to the Graduate College. I hereby certify that I have read this dissertation prepared under my direction and recommend that it be accepted as fulfilling the dissertation requirement.
________________________________________________ Date: (10/21/05)Dissertation Director: Anne E. Cress, Ph.D.

3
STATEMENT BY AUTHOR
This dissertation has been submitted in partial fulfillment of requirements for an advanced degree at The University of Arizona and is deposited in the University Library to be made available to borrowers under rules of the Library.
Brief quotations from this dissertation are allowable without special permission, provided that accurate acknowledgement of source is made. Requests for permission for extended quotation from or reproduction of this manuscript in whole or in part may be granted by the head of the major department of the Dean of the Graduate College when in his or her judgment the proposed use of the material is in the interests of scholarship. In all other instances, however, permission must be obtained from the author.
SIGNED: Thomas Charles Sroka

4
ACKNOWLEDGEMENTS
In working toward this dissertation the most valuable knowledge gained was an
understanding that research is a collaborative effort. The work presented here would not
be possible without many minds and helping hands. I am eternally grateful to members
of the Cress laboratory, fellow students in the Cancer Biology Interdisciplinary Program,
members of Core Services, and my dissertation committee members.
To my mentor, Anne Cress, thank you. Thank you for your patience, excitement,
and encouragement that have enriched my development as a scientist. With your
guidance I now feel prepared to meet the challenges of Science and challenge my goals.
I am grateful for your honest, critical review of my work and cherish our discussions,
both science and non-science related. I feel lucky to have been a part of your laboratory.
Finally I would like to thank my family, my friends, and my Isis. Thank you for
your support and patience as I travel through an extended career-training path. None of
this work would have been possible without you.

5
DEDICATION
I dedicate this work to anyone that has been a victim, witness, or survivor of
cancer. May it serve as a step toward new therapies, new hope.

6
TABLE OF CONTENTS LIST OF FIGURES………………………………………………………………….9 LIST OF TABLES…………………………………………………………………..12 ABSTRACT…………………………………………………………………………. 13
I. INTRODUCTION……………………………………………………………….15 Cancer Metastasis……………………………………………………………………..15 Prostate Cancer………………………………………………………………………..16 Integrins and Cell Motility…………………………………………………………….19 Protrusive and Contractile Contacts…………………………………………………...21 Focal Adhesion Proteins: The Role of Focal Adhesion Kinase and the Extracellular Signal-Regulated Protein Kinase/Mitogen-Activated Protein Kinase Pathway in Cell Migration………………………………………………………………………24 Isolation of Tumor Cell Adhesion Peptides Using a One-Bead-One-Compound Combinatorial Library……………………………………………………………...27 II. MATERIALS AND METHODS…………………………………………...31 Affinity Precipitation with Peptides…………………………………………………..31 Antibodies and Reagents……………………………………………………………...32 Biotinylation of Cell Surface Proteins………………………………………………...33 Cell Lines and Culture Conditions……………………………………………………33 Cell Motility Assays…………………………………………………………………..34 Cell Adhesion Assay and Peptide Inhibition of Adhesion…………………………….37 Cell Lysis and Western-Blot…………………………………………………………..37 Immunoprecipitation…………………………………………………………………..38 Epithelial and Stromal Cell Adhesion Assay………………………………………….39 FACS Analysis and Confocal Microscopy……………………………………………40 In Vivo Actin Distribution Assay……………………………………………………...40 Immunofluorescence…………………………………………………………………..41 Synthetic Peptides, Fibronectin, Laminin-1, Laminin-5, and Collagen IV…………...42 Laminin-5 Coating Conditions for Motility and Signaling Experiments……………..43 III. IDENTIFICATION AND CHARACTERIZATION OF TUMOR CELL ADHESION PEPTIDES………………………………………………44 Introduction……………………………………………………………………………44 Results…………………………………………………………………………………46 RZ-3, HYD-1 and AG-73 Bind to Prostate Tumor Cell Surfaces………………….46 DU-H Prostate Tumor Cells Adhere to Immobilized Peptide and ECM Proteins…49

7
TABLE OF CONTENTS – CONTINUED-
Tumor Cell Adhesion to Immobilized Peptides is Integrin Dependent…………….51 Inhibition of DU-H Cell Attachment to ECM Proteins…………………………….53 Inhibition of SCC-25 and HFF Co-Culture Interaction by HYD-1 Peptide………..56 Adhesion to Immobilized HYD1 Prevents Cell Spreading and Adhesion Dependent Phosphorylation……………………………………………………...56 Discussion……………………………………………………………………………..61
IV. SYNTHETIC D-AMINO ACID PEPTIDE INHIBITS TUMOR CELL MOTILITY ON LAMININ-5………………………………………...64 Introduction……………………………………………………………………………64 Results…………………………………………………………………………………66 HYD1 Blocks Laminin-5 Dependent Migration and Invasion……………………..66 HYD1 Induces Actin Cytoskeletal Remodeling and Cortactin Mediated Actin Dynamics at the Cell Membrane…………………………………………………72 HYD1 interacts with α6 and α3 Integrins and Activates Integrin-Associated Signaling…………………………………………………………………………75 HYD1 Enhances Signaling on Laminin-5 Resulting in Transient Activation of ERK………………………………………………………………………………77 Discussion……………………………………………………………………………..81
V. IDENTIFICATION OF THE MINIMAL ACTIVE ELEMENT OF THE SYNTHETIC D-AMINO ACID PEPTIDE HYD1 (kikmviswkg)………………………………………………………………………85 Introduction……………………………………………………………………………85 Results…………………………………………………………………………………86 The Minimal Element that Mediates Cell Ahesion to Immobilized HYD1 is Xkmvixw…………………………………………………………………………86 Analysis of the Minimal Element of HYD1 Necessary for Activation of ERK…...91 Analysis of the Minimal Element of HYD1 Necessary to Block Cell Migration on Laminin-5……………………………………………………………………..94 Discussion……………………………………………………………………………..97

8
TABLE OF CONTENTS – CONTINUED –
VI. PRELIMINARY DATA: EFFECT OF THE SYNTHETIC D-AMINO ACID PEPTIDE, HYD1 (kikmviswkg), ON INTEGRIN LATERAL-ASSOCIATIONS……………………………....100 Introduction……………………………………………………………………….....100 Results…………………………………………………………………………….....102 HYD1 Interacts with CD151/Integrin Complexes………………………………..102 HYD1 Increases CD81 Association with the α6 Integrin………………………...102 HYD1 Alters the Lateral-Associations of α6 and α3-Integrins and the Tetraspanin CD81……………………………………………………………………………105 Discussion……………………………………………………………………………105
VII. CONCLUDING STATEMENTS………………………………………..111
REFERENCES……………………………………………………………………..120

9
LIST OF FIGURES Figure 1: Schematic of two-dimensional cell migration……………………………….22
Figure 2: Schematic of integrin-activated focal adhesion signaling……………………25
Figure 3: Isolation of tumor cell adhesion peptides using a one-bead-one-compound
combinatorial library……………………………………………………………………..30
Figure 4: Fluorescence-activated cell sorting analysis of biotinylated peptides bound to
DU-H prostate carcinoma cells…………………………………………………………..47
Figure 5: Confocal microscopy of the peptides RZ-3 and HYD1 binding to the surface
of human prostate tumor cells……………………………………………………………48
Figure 6: Comparison of human prostate tumor cell adhesion to immobilized peptides or
ECM proteins…………………………………………………………………………….50
Figure 7: Effect of antibodies and reagents on DU-H cell adhesion to peptides and
extracellular matrix proteins……………………………………………………………..52
Figure 8: Inhibition of DU145 cell adhesion to immobilized β1 integrin antibody by
peptides…………………………………………………………………………………..54
Figure 9: Inhibition of DU-H cell adhesion to extracellular matrix proteins
by peptides……………………………………………………………………………….55
Figure 10: Inhibition of SCC-25 cell adhesion to HFF monolayers by peptides………57
Figure 11: Adhesion to immobilized HYD1 prevents cell spreading…………………..59
Figure 12: Suppression of adhesion dependent phosphorylation by immobilized
HYD1…………………………………………………………………………………….60
Figure 13: HYD1 blocks cell migration on laminin-5………………………………….67

10 LIST OF FIGURES – CONTINUED – Figure 14: Soluble HYD1 does not effect cell spreading on immobilized laminin-5…70
Figure 15: HYD1 inhibits laminin-5 dependent cellular invasion……………………..71
Figure 16: HYD1 induces cytoskeletal reorganization on laminin-5…………………..73
Figure 17: HYD1 induces phosphorylation of cortactin and alters membrane actin
dynamics…………………………………………………………………………………74
Figure 18: HYD1 interacts with α6 and α3 integrins…………………………………..76
Figure 19: HYD1 activates integrin-associated signaling……………………………...78
Figure 20: HYD1 temporally enhances activation of FAK and phosphorylation of MEK
on laminin-5……………………………………………………………………………...79
Figure 21: HYD1 enhances Laminin-5 signaling and transiently activates ERK……...80
Figure 22: Cell adhesion to immobilized HYD1 is mediated by N- and C-terminal
regions of the peptide…………………………………………………………………….87
Figure 23: The minimal sequence mediating cell adhesion to immobilized HYD1 is
ikmviswk…………………………………………………………………………………89
Figure 24: The minimal element mediating cell adhesion to immobilized HYD1 is
xkmvixw…………………………………………………………………………………90
Figure 25: xikmviswkx is the minimal element of HYD1 required to activate ERK…..92
Figure 26: HYD1 induced ERK activation is mediated by N- and C-terminal regions of
the peptide………………………………………………………………………………..93
Figure 27: xikmviswxx is the minimal element necessary to block cell migration on
laminin-5…………………………………………………………………………………95

11 LIST OF FIGURES – CONTINUED – Figure 28: HYD1 inhibition of cell migration on laminin-5 is mediated by N- and C-
terminal regions of the peptide…………………………………………………………..96
Figure 29: HYD1 interacts with CD151/Integrin complexes…………………………103
Figure 30: HYD1 does not change the amount of CD151 associated with α6 or α3-
containing integrins……………………………………………………………………..104
Figure 31: HYD1 increases the amount of CD81 associated with α6-integrins and
decreases the amount of CD81 associated with α3-integrins…………………………..106
Figure 32: HYD1 induces α6 integrin clustering……………………………………..107
Figure 33: HYD1 alters the lateral-associations of α6 and α3-containing integrins and
the tetraspanin CD81……………………………………………………………………108
Figure 34: Potential mechanisms for the anti-migratory activity of HYD1…………..116

12 LIST OF TABLES Table 1: Alanine-substituted and N- and C-terminal truncated peptides of HYD1 with
their cell adhesion, cell signaling, and anti-migratory activity…………………………..98

13 ABSTRACT
Human tumor cell progression and metastasis is partially dependent on the ability
of tumor cells to adhere to the proteins of the extracellular matrix and migrate to distant
locations. Using a combinatorial screening approach, six novel D-amino acid containing
peptides were identified and analyzed for their ability to adhere to human prostate tumor
cells, support tumor cell adhesion and inhibit tumor cell adhesion to ECM proteins. Two
peptides, RZ-3 (kmviywkag) and HYD1 (kikmviswkg) bound to tumor cell surfaces. A
scrambled peptide derivative of HYD1, HYDS (wiksmkivkg) is not active. As
immobilized ligands, RZ-3 and HYD1 can support prostate tumor cell adhesion. Prostate
tumor cell adhesion to immobilized RZ-3 and HYD1 is integrin dependent. Soluble RZ-3
and HYD1 inhibits tumor cell adhesion to extracellular matrix proteins in a concentration
dependent manner. These results indicate that RZ-3 and HYD1 are biologically active D-
amino acid containing peptides that can support tumor cell adhesion and can inhibit
tumor cell adhesion to immobilized extracellular matrix proteins.
Cell migration is dependent on adhesive interactions with the extracelluar matrix.
These interactions induce signaling and cytoskeletal responses necessary for migration.
HYD1 completely blocks random haptotactic migration and inhibits invasion of prostate
carcinoma cells on laminin-5. This effect is adhesion independent and reversible. The
inhibition of migration by HYD1 involves a dramatic remodeling of the actin
cytoskeleton resulting in increased stress fiber formation and actin colocalization with
cortactin at the cell membrane. HYD1 interacts with α6 and α3 integrin subunits and
elevates laminin-5 dependent intracellular signals including focal adhesion kinase,

14 mitogen activated protein kinase kinase, and extracellular signal-regulated kinase. The
scrambled derivative of HYD1, HYDS, does not interact with the α6 or α3 integrin
subunits and is not biologically active. The minimal element for bioactivity of HYD1
was determined using alanine-substituted analogs of HYD1 and N- and C-terminal
deletion mutants of HYD1. The minimal element necessary to block cell migration on
laminin-5 and activate cell signaling through ERK is xikmviswxx. Taken together, these
results indicate that HYD1 is a biologically active integrin-targeting peptide that
reversibly inhibits tumor cell migration on laminin-5 and uncouples phosphotyrosine
signaling from cytoskeletal dependent migration.

15 I. INTRODUCTION
Cancer Metastasis
Metastasis is the result of the spread of cancer from the primary tumor site to a
distant site or organ. The process of metastasis consists of several steps in order for a
tumor to achieve successful growth at a distant site (Reviewed in [1-5]). First, a tumor
must be able to support its metabolic needs during progressive growth through
angiogenesis [6]. After vascularization, tumor cells may escape into the blood supply
through these new vessels or by impeding into vessels in the vicinity. In order to escape
into the vasculature or lymphatics, tumor cells must also invade through normal
surrounding tissue and vascular endothelium. Once in the blood supply they must
survive in the circulation and arrest in capillary beds at distant organs. Extravasation into
a distant site requires proper adhesive interactions as well as the ability to invade into
normal tissue. Finally, these tumor cells must be adaptive to progressively grow in a new
environment. The multiple events necessary for the development of metastasis
foreshadow its molecular complexity.
Considering cancer mortality, most of the deaths are a direct cause of the
consequences of metastatic tumors rather than the primary tumor itself [1,7]. There is an
intense need for new therapeutics to target the processes involved in metastasis given that
cancer diagnosis often occurs at a relatively late stage in cancer progression. In order to
develop novel therapeutics, further research is needed to identify the critical molecular
components involved in metastasis. Although metastasis is a complicated in vivo

16 process, it is valuable to initially examine the molecular players at the cellular level in
vitro. In general, study of the machinery responsible for cellular motility can lead to the
important discovery of key molecular players that regulate cell migration and/or invasion.
Some of these molecules may prove to be appropriate anti-metastatic targets.
Prostate Cancer
Adenocarcinoma of the prostate is the second most common cause of cancer
deaths in the United States with a projection of 30,350 deaths in 2005 [8,9]. New
diagnoses in 2005 will reach 232,090, making this type of cancer the most common non-
cutaneous malignancy in American men [8,9]. The problem of prostate cancer also exists
outside America. According to the World Health Organization, there were 679,023 new
diagnoses and 221,002 deaths due to prostate cancer worldwide in 2002 [10].
Undoubtedly, the high incidence of prostate cancer is directly related to improved
prostate cancer screening such as the use of prostate specific antigen (PSA) as a
molecular marker for cancer progression and recurrence [11]. In addition, improved
prostate screening and detection may have had a direct effect on the mortality rates given
that in the early 1990s there were approximately 40,000 deaths per year; this figure
dropped to 29,000 in 2004 [8]. Nevertheless, a staggering figure from the American
Cancer Society reveals that the five-year survival rate for localized or regionally detected
prostate cancer is nearly 100%. However, the five-year survival rate drops to 33.5% for
prostate cancer at a distant stage at the time of diagnosis [8]. Distant stage is defined as
cancer that has spread to parts of the body remote from the primary tumor, either by

17 direct extension or by metastasis to distant organs. Therefore, it is clear that this type of
cancer is in need of new therapies to target and ablate its ability to metastasize.
The clinical history of prostate cancer progression indicates that it is an indolent
disease. Without initial treatment, prostate cancer diagnosed at an early clinical stage
will cause relatively few deaths within the first 10-15 years following diagnosis [12-14].
This is most likely related to a small rate of progression to metatstatic disease during that
period. However, a study has reported that additional follow-up at 15-20 years post-
diagnosis significantly changes disease-specific survival [15]. This particular cohort
reports a 3-fold increase in the prostate specific mortality rate compared with the first 15
years of follow-up. Therefore, early stage prostate cancer has a large period of latency
before metastatic conversion.
The indolent nature of prostate cancer has important implications regarding
therapy. First, it is not a highly proliferative cancer and therefore will not be sensitive to
cytotoxic agents used in other proliferative tumors. Second, as previously stated, survival
rates are linked to a loss of organ-confined disease and development of distant metastasis.
Therefore, non-cytotoxic agents that modify the mechanisms of cell invasion and motility
can be useful at preventing the spread of localized disease. Identifying key molecules
involved in prostate cancer invasion and metastasis should be a priority for novel
therapeutics.
Much work has been focused on characterization of both genetic and protein
changes that take place during prostate cancer progression. Chromosomal instability is a
hallmark of cancer and accordingly many genetic alterations have been discovered and

18 described for prostate cancer. All of these changes are out of the scope of this
dissertation. Although not exhaustive, the major findings can be found in several reviews
[16-19]. Alterations at the genetic and protein level act as molecular clues to the etiology
of prostate cancer as well as providing a foundation to understand its progression to
metastatic disease.
Integrins are cell adhesion molecules that play a crucial role in not only cell
adhesion, but also dictate multiple aspects of cell behavior including cell motility and
cellular metastasis [20-22]. Changes that occur in integrin expression during prostate
cancer progression seem to be early events [16,23,24]. Complete loss of specific alpha
and beta subunits have been described and include α5, α4, αv, β1C, and β4 [23,25,26].
Coupled with these findings, conservation of the integrins α6β1 and α3β1 have been
documented [23,24], making these laminin receptors critical molecular components and
targets encompassing prostate cancer metastasis. The role of alpha-6 integrins has been
solidified by the findings that α6β1 maintains expression in prostate micrometastases
[27]. Teleologically, the retention of these receptors correlates with the finding that
prostate tumors often invade out of the capsule along nerve-endings and exit into the
laminin-rich perineural space [28-31]. Taken together, these findings warrant further
study of the biology and functional role of α6 and α3-containing integrins in prostate
cancer progression. Specifically, how do these integrins functionally regulate tumor cell
invasion and metastasis?

19 Integrins and Cell Motility
Integrins are evolutionarily conserved heterodimeric cell surface molecules [32-
34]. Structurally, they contain an alpha and beta subunit, each consisting of a large
extracellular domain, a single-pass transmembrane domain, and a short cytoplasmic tail
with the exception of the β4 subunit, which has a large cytoplasmic domain [32,35]. To
date there are 18 distinct α and 8 distinct β subunits that pair in a restrictive manner to
give about 24 different integrins that have individual ligand binding specificities and
nonredundant functions given their phenotypes in knockout mice [32,35,36]. The main
function of integrins is to mediate and regulate cell adhesion to extracellular matrix
proteins. In addition to cell adhesion, integrins affect many aspects of cell behavior
including cell survival, proliferation, motility, and cytoskeletal reorganization. These
roles in cell biology mediate the impact of integrins on biological processes including
hemostasis, immunity, inflammation, and cancer metastasis [32-34]. The ability to act as
signal transducers is central to their multitude of functions. Their effect on cell signaling
is directly related to the ability to form mechanical links from the extracellular matrix
(ECM) to the cytoskeleton. Through these links integrins support the formation of
membrane proximal adhesion complexes that contain a vast array of different proteins
that are involved in cytoskeletal structure and signaling [32,37,38].
Given the complexities of their function, integrin activation must be tightly
regulated and much work has focused on uncovering the mechanisms of integrin
activation. Integrin adhesion and activation is rapidly regulated by changes in affinity for
extracellular matrix ligands [33]. Mutational studies and the crystal structure of the αvβ3

20 [39] suggests that affinity modulation is linked through a conformational change that
spans the integrin protein. In support of this, it has been demonstrated that a conserved
GFFKR sequence in the cytoplasmic domain of the α subunit is a negative regulator of
activation [40]. A critical affinity-independent mechanism of activation is integrin
clustering [33,34]. When integrins bind to the ECM they become clustered in the plane
of the membrane and recruit cytoskeletal and signaling elements that promote the
formation and reorganization of actin filaments. This process is thought to be dependent
on the action of the Rho family of GTPases and lateral associating proteins that interact
with integrins such as caveolin-1 or tetraspanin molecules [34,41,42]. Importantly, this
process of activation can be induced by binding to ECM proteins, termed “outside-in”
signaling or by response to soluble or internal processes, termed “inside-out” signaling.
Both result in the adhesive and signaling responses that elicit integrin function [32]. Cell migration is a complex process coordinating the actions of many different
cellular players. The adhesive complexes formed from integrin ligation and activation
serve as signaling regulators of migratory processes as well as traction points over which
the body of the cell moves [43]. Work has shown both in vitro and in vivo that integrins
influence the ability of a malignant cell to invade and metastasize [44]. As an example,
use of a anti-α6 antibody co-injected into nude mice with invasive human melanoma
cells will reduce metastasis of the xenograft [45].

21 Protrusive and Contractile Contacts
The process of migration is complicated, requiring the dynamics of many
processes. In general, cell migration is a reiterative process that requires extensive
cortical actin reorganization including lamellipodia and filopodia extension [43,46,47],
integrin dependent adhesion leading to the formation of focal complexes, and maturation
of focal complexes into focal adhesions [46,48]. These adhesions then form a platform
for traction over which the cell body can move [43]. With the description just given, two
types of contacts can be described that regulate this process, protrusive and contractile.
Protrusive contacts are membrane extensions formed by cells to examine the surrounding
environment. Cellular protrusions at the leading-edge of the cell form adhesive contacts
with the extracellular matrix and polarize the cell for migration [37]. These contacts can
be any type of extension ranging from very defined, localized extensions such as
filopodia or microspikes, to very broad protrusions such as lamellipodia or membrane
ruffles. Contractile contacts are those involved in stable, prolonged attachments such as
those displayed by focal adhesions [37]. These types of contacts apply tension to the cell,
showing that the adhesions formed are stable and worthy of use as a migration surface
[49]. Figure 1 is an overview of the important events necessary for two-dimensional cell
migration. Dysregulation of the migration machinery during cancer progression may
create hypermotile tumor cells that have an increased capacity to invade and metastasize.

22
Figure 1. Schematic of two-dimensional cell migration. This figure illustrates important components at both the leading edge and the retracting end of a migrating cell on a 2-dimensional, rigid surface.

23
Assembly of protrusive contacts are influenced by two members of the Rho
family of GTPases. Specifically, Rac1 and Cdc42 act as signal transducers to form
lamellipodia and filopodia respectively [43,47,48,50]. Activation of these GTPases can
take place from “outside-in” or “inside-out” signaling and are reliant upon guanine
nucleotide exchange factors for their activation [47]. There is accumulating evidence
suggesting that tips of lamellipodia and filopodia serve as docking sites to localize factors
that contribute to actin polymerization required for cell motility. A good example is
highlighted by a study illustrating that the amount of vasodilator-stimulated
phosophoprotein (VASP) in lamellipodium tips increased directly with protrusion rate
[51]. The molecular players involved in initiating actin polymerization are starting to be
characterized and the Arp 2/3 complex has been strongly implicated in nucleating and
structuring actin networks [52,53]. In addition, it has been shown that a member of the
Wiskott-Aldrich syndrome protein family, Scar/Wave, is integral in activating Arp 2/3 in
lamellipodium formation [54]. Further work is warranted to determine the additional
players that mediate actin polymerization at the tips of protrusive contacts. The small GTPase Rho is a major player in contractile contacts through its role in
regulating the organization of actin stress fibers. Additionally, Rho activation directly
correlates to the formation of focal adhesions [43,48]. As described above, contractile
contacts are important in cell motility by providing traction over which the cell moves.
The molecular mechanism in how Rho regulates this process is well characterized. Rho
can activate Rho kinase, which phosphorylates and inhibits myosin light chain
phosphatase [55]. This allows myosin light chain to be in a highly phosphorylated, pro-

24 contractile state, contributing to integrin clustering and stress fiber formation. This is one
characterized mechanism, however, given the multiplicity of Rho targets, there are
undoubtedly many more molecular players regulating this process.
Focal Adhesion Proteins: The Role of Focal Adhesion Kinase and the Extracellular
Signal-Regulated Protein Kinase/Mitogen-Activated Protein Kinase Pathway in Cell
Migration
As previously mentioned, integrin activation results in recruitment of many
structural and signaling proteins to the cell membrane at the site of integrin/ECM contact
[56]. These protein complexes organize into focal adhesions that physically link
integrins to the actin cytoskeleton and are capable of integrating external cues into
cellular signals that regulate cell behavior. The dynamic regulation in the formation and
disassembly of focal adhesions are crucial to cell motility, [56] and often the components
of the adhesion complex directly regulate its dynamic. Because of the abundance of
proteins involved, this section will focus on the role of focal adhesion kinase (FAK) and
the mitogen-activated protein kinase pathway in cell migration events. Figure 2 is a
schematic that displays a few important cytoskeletal and signaling proteins incorporated
into focal adhesions and their downstream effects on motility. A prominent biochemical change that occurs with integrin clustering is tyrosine
phosphorylation of several proteins, including FAK [57]. FAK is a 125 kD protein that is
a member of non-receptor protein kinases. Integrin clustering leads to rapid recruitment

25
Figure 2. Schematic of integrin-acitvated focal adhesion signaling. Upon interaction with the extracellular matrix (ECM), integrins become activated and recruit multiple structural and signaling proteins to the cell membrane. These proteins compose the structural integrity of the adhesion site and serve as signal-transducers to modulate many aspects of cell behavior including motility. This figure displays a few of the proteins linked to cell migration discussed in the introduction. PP2 and Y-27632 are chemical inhibitors of Src and ROCK respectively that were used in this study.

26 of FAK to focal contact sites [46]. FAK has several phosphorylation sites. In particular
phosphorylation on tyrosine 397, an autophosphorylation site, correlates with increased
kinase activity [58], thus providing a marker for FAK activation. FAK is implicated in
migratory processes by several lines of evidence. First, FAK deficient fibroblasts migrate
poorly in response to haptotactic or chemotactic cues while forming prominent focal
adhesions by vinculin staining [46,59,60]. In addition, FAK overexpression increases
fibronectin stimulated motility only if tyrosine 397 site is kept intact [61]. Finally, FAK
tyrosine phosphorylation has been shown to be critical for cell spreading and migration of
endothelial cells on fibronectin [62]. These data indicate that FAK has both signaling
and focal adhesion remodeling functions that are crucial for cell migratory processes.
Like FAK, the extracellular signal-regulated kinase/mitogen-activated protein
kinase pathway (ERK/MAPK) has a role in cell migration through both signaling and
focal adhesion remodeling activities [56]. It has been shown that the ERK/MAPK
activity stimulates myosin light chain kinase (MLCK) activation, which phosphorylates
myosin light chain (MLC). Phosphorylation of MLCs initiates myosin dimerization and
engagement with the actin cytoskeleton producing cellular contraction [63,64]. In
addition, activation of the ERK/MAPK pathway, in cooperation with Src, may negatively
regulate the Rho-ROCK-LIMK pathway and thus alters actin dynamics [65]. The focal
adhesion remodeling activities are thought to be directed by interplay with the protease
calpain 2 [56]. Specifically, chemotactic-induced detachment from the matrix during cell
motility required calpain 2 at the cell membrane. The proper localization of this protease
was dependent on ERK/MAPK signaling [66], and furthermore, ERK can directly

27 regulate calpain activity through phosphorylation [67]. Finally, calpain activity is
attenuated by inhibition of kinase components of the MAPK pathway [68]. Taken
together, the ERK/MAPK pathway influences cell motility through its ability to regulate
cytoskeletal dynamics and focal adhesion proteolysis.
Isolation of Tumor Cell Adhesion Peptides Using a One-Bead-One-Compound
Combinatorial Library
The retention of α6β1 and α3β1 integrins in prostate cancer progression [23,24]
foreshadows their importance in prostate cancer metastasis, marking them as potential
therapeutic targets. The field of combinatorial peptide chemistry has enabled scientists to
create peptide ligand mimetics to multiple protein targets including kinases and cell
surface receptors [reviewed in [69]]. Two particularly useful methods to isolate
candidate peptide ligands for cell surface receptors are phage-display and the one-bead-
one-compound (OBOC) approach. Isolation and characterization of these peptides not
only creates potential inhibitors, it also fosters increased understanding of protein biology
and function. This section will briefly compare the phage-display method to the OBOC
approach and highlight the methodology behind the one-bead-one-compound
combinatorial library for the development of tumor cell adhesion peptides.
Combinatorial peptide chemistry allows the screening for peptide ligands among
billions of random peptides created through a variety of approaches. The one-bead-one-
compound combinatorial library method [70,71] and the phage-display peptide library
approach [72-75] have been successfully used to identify peptide ligands for cell surface

28 molecules. Specifically, the phage display method and the OBOC approach has
identified peptides that interact with the surface idiotypes of B-cell lymphoma and human
chronic lymphocytic lymphoma [76-78]. Both of these approaches have also been
efficient at identifying ligands that interact with integrins [79-84]. Although each
technique is effective, they each have specific advantages and disadvantages.
Phage-display is a biological library method that manipulates filamentous phage
to express peptides on specific surface proteins such as the viral pIII coat [85]. Random
deoxyoligonuleotides dictating peptide size and orientation are inserted directly into the
pIII gene creating a distinct peptide on the surface of individual phage [85]. Panning for
peptides with specificity to a cell surface protein can be performed using purified cell
surface receptors [86] or live cells [87,88]. This technique is advantageous because up to
109 peptides can be screened per library while there is no restriction on the size of peptide
displayed.
The one-bead-one-compound combinatorial method uses a solid phase technique
and a “split-mix” synthesis approach to create a peptide library [70,89,90]. In essence,
individual peptides are synthesized on polyethylene glycol-grafted polystyrene beads that
yield approximately 1013 copies of the same peptide per bead [70,71]. Like the phage
display method, OBOC libraries can identify cell surface ligands using purified receptors
[71] or intact cells [81]. Some specific advantages of using the OBOC approach
compared to phage display are that peptides from the OBOC method are spatially
separable, meaning that multiple motifs can easily be identified from isolation and
characterization of specific beads. Most importantly, the OBOC method is not limited to

29 the use of L-amino acids as in phage display. D-amino acids, unnatural amino acids or
even nonamino acid moieties can be used in OBOC libraries. Peptides created for
example, from D-amino acids are much more resistant to proteolysis in vivo [91,92] and
are therefore more desirable for therapeutic purposes.
We have used the OBOC peptide library method to identify peptide sequences
that support tumor cell adhesion via the alpha-6 integrin [81,82]. The method of
selection was based on functional cell adhesion of live tumor cells to peptide-containing
beads. Briefly, DU-145H cells, a prostate tumor cell line that expresses high levels of α6
[93], was applied to OBOC libraries and analyzed under a dissecting microscope. Beads
with cells attached were retrieved, and those beads were rescreened with the same tumor
cell line in the presence of a functional-blocking antibody to α6, GoH3. The beads that
no longer bound to tumor cells in this condition contained candidate α6-specific adhesion
peptides. Those beads were isolated, retested for adhesion without the antibody, and
sequenced. Figure 3 displays the strategy for the selection of a cell adhesion peptide
using the OBOC method. This method is particularly powerful because it does not
require purification of the receptor and thus allows the appropriate post-translational
modifications. In addition, by using live cells in this scheme, peptides can be identified
that interact with conformationally sensitive surface molecules. In principle, this method
allows the identification of specific cell adhesion peptides for any cell surface receptor
for which a function-blocking reagent is available.

30
Figure 3. Isolation of tumor cell adhesion peptides using a one-bead-one-compound combinatorial library. This selection scheme was used to isolate and characterize peptides that interact with α6 integrins. The function-blocking reagent was GoH3, a function-blocking antibody to α6.

31 II. MATERIALS AND METHODS
Affinity Precipitation with Peptides
1.2 x108 DU-145H cells were harvested with 2mM EDTA in PBS. Cells were
washed in AP buffer containing 25 mM HEPES pH 8.0, 100 mM KCl, 1 mM MgCL, 1
mM CaCl, 20% glycerol, 1 mM PMSF, 1 µg/ml leupeptin and aprotinin. Cells were lysed
using a dounce homogenizer then centrifuged for 5 minutes at 15,000 g at 4oC in an
Eppendorf microcentrifuge. Supernatants were further centrifuged for 15 minutes at
96,000 g at 4oC in a Sorvall RC M150GX ultracentrifuge. Membrane fraction pellets
were suspended in AP buffer containing 0.2% NP-40 and analyzed for protein
concentration using the BCA protein assay kit (Pierce, Rockford, IL). 500 µg of
biotinylated peptide was preloaded on to 30 µl of UltraLink Immobilized NeutrAvidin
Plus beads (Pierce, Rockford, IL) in a buffer containing 0.5mM KCl, 0.3mM KH2PO4,
27.6mM NaCl, and 1.6mM Na2HPO4 pH 7.4 for 1 hour at room temperature. The
resulting beads were washed twice with AP buffer and incubated with 30µg of the
membrane fraction adjusted to a total volume of 500 µl for 18 hours at 4oC. NeutrAvidin
beads were washed 3 times with AP buffer containing 0.2% NP-40. All samples were
suspended in 2x laemmli buffer and analyzed by SDS-PAGE electrophoresis.

32 Antibodies and Reagents
GoH3, a rat anti-α6 functional-blocking antibody was purchased from Serotech
Inc. (Raleigh, NC). The anti-α3 functional-blocking antibody, P1B5, was purchased
from Chemicon (Temecula, CA). AIIB2, a rat anti-β1 functional-blocking antibody, was
purified from its hybridoma (Developmental Studies Hybridoma Bank, University of
Iowa). Integrin antibodies used for western blotting: Anti-α6, AA6A rabbit polyclonal,
was generated and purified by Bethyl Laboratories Inc. (Montgomery, TX). Anti-α3,
AB1920, was purchased from Chemicon (Temecula, CA.). Antibodies to FAK, Tyr-397
and 4.47, in addition to phospho-Mek (serine 298) and cortactin (clone 4F11) were
purchased from Upstate (Lake Placid, NY). MEK1 antibody was obtained from BD
Transduction Laboratories (San Diego, CA). Map Kinase antibodies, phospho-p44/42
(Thr202/Tyr204) and total p44/42, were purchased from Cell Signaling Technology Inc.
(Beverly, MA). CD151 monoclonal antibody, 5C11, was a gift from Dr. Martin Hemler
(Dana-Farber Cancer Institute and Department of Pathology, Harvard Medical School,
Boston, MA.). CD81 monoclonal antibody, JS64, was purchased from Immunotech
(France). Inhibitors for Src (PP2) and ROCK (Y-27632) were purchased from
Calbiochem (San Diego, CA). Anti-mouse and anti-rabbit secondary antibodies
conjugated to horseradish peroxidase for immunoblotting were obtained from Chemicon
(Temecula, CA). Anti-mouse secondary antibody conjugated to Alexa Fluor 568 and
phalloidin conjugated to Alexa Fluor 488, used in immunofluorescence, were obtained
through Molecular Probes (Eugene, OR).

33
The mouse monoclonal antibodies used for the adhesion blocking studies were as
follows: P4C10, anti-β1 integrin; P1E6, anti-α2 integrin; P1B5, anti-α3 integrin; and
P1D6, anti-α5 integrin (all from Life Technologies Inc.). Mouse preimmune IgG was
purchased from ICN Biomedicals Inc. (Aurora, OH). The β1 antibody used for in the
adhesion assay was TS2/16 obtained from the American Type Culture Collection.
Biotinylation of Cell Surface Proteins
Cells were adhered to laminin-5 coated tissue culture plates for 1 hour prior to
biotinylation. Cells were washed twice in HBSM (20mM Hepes, pH 7.5, 150mM NaCl,
and 5mM MgCl2 ) followed by biotinylation with 0.2 mg/ml sulfo-NHS-LC biotin
(Pierce Chemical Co) in HBSM for 1 hour at room temperature. Cells were rinsed three
times with HBSM prior to lysis.
Cell Lines and Culture Conditions
All cell lines were incubated at 37o C in a humidified atmosphere of 95% air and
5% CO2. The human prostate carcinoma cell lines, PC3N and DU-145H, were grown in
Iscove’s Modified Dulbecco’s Medium (Gibco BRL, Gaithersburg, MD.) plus 10% fetal
bovine serum (Gibco BRL). All medium was supplemented with penicillin/streptomycin,
100 units/ml (Gibco BRL). Serum-Free medium was supplemented with 0.1% Bovine
Serum Albumin (Sigma, St. Louis, MO.). PC3N cells are a variant of the human PC3
prostate carcinoma cell line [94] . DU-145H (DU-H) cells are DU-145 cells, a prostate
carcinoma cell line originating from a brain metastasis, selected for high expression of α6

34 integrin [93]. DU-145 and PC3 cell lines are available from ATCC (Rockville, MD).
HaCaT cells [95] were obtained from Dr. Norbert E. Fusenig (German Cancer Research
Center, University of Heidelberg, Heidelberg, Germany). The human oral squamous cell
carcinoma line, SCC-25, was obtained from American Type Culture Collection
(Rockville, MD). Human foreskin fibroblasts (HFF) were obtained as previously
described [96]. SCC-25 and HFF cells were maintained in Ham's F:12/DMEM
purchased from Gibco BRL (Grand Island, NY) supplemented with 10% fetal bovine
serum from Gemini-Bio-Products (Calabasas, CA), 0.1% hydrocortisone and penicillin
(100units/ml)/streptomycin (100units/ml).
Cell Motility Assays
Both time-lapse video microscopy and modified Boyden chamber transwell
assays were performed. Time-lapse video microscopy is offered as a core service
through the Southwest Environmental Health Services Center Cellular Imaging Core.
Time-lapse video microscopy was used to analyze random haptotaxis of PC3N cells on
laminin-5. The effect of the peptides or pharmacological inhibitors was observed with
differential interference contrast optics on an inverted Olympus IMT2 microscope
(Olympus America, Melville NY). The microscope was equipped with a BiopTechs
Delta T live Cell system (BiopTechs, Butler PA) with a humidified 5/95% CO2/Air
atmosphere and was maintained at 37oC. Images were obtained with a grayscale CCD
camera (ORCA-100, Hamamatsu, Japan) and analyzed using Compix imaging software
(SimplePCI 4.0, Compix Imaging, Cranberry Township PA). 70,000 cells were plated

35 for 1 hour in serum-free conditions on laminin-5 coated Delta T dishes (0.15 mm,
BiopTechs, Butler PA). Coating was done as described above. Soluble peptides or
chemical inhibitors in serum-free media at the stated concentrations were added to the
adhered cells and video was started at approximately 30 min post peptide or inhibitor
treatment. For peptide reversibility experiments, cells were adhered and treated as above
but after 30 minutes of treatment with peptide the cells were washed 1 time with PBS and
serum-free media or soluble laminin-5 was added back. Videos were four hours in length
with a frame capture every 5 minutes. The speed of each cell was calculated manually
with the Compix software by measuring the distance traveled by the cell nuclei over the 4
hour time period. The average rate of migration (15 cells/experiment) in microns was
calculated. Each experiment was performed at least three times.
Analysis of invasion was performed using a modified Boyden chamber assay.
Falcon polyethylene terephthalate (PET) track-etched membrane 8µm pore inserts
(Becton Dickinson Labware, Franklin Lakes NJ) were coated on the underside with
laminin-5 (HaCaT conditioned media) for two hours at room tempurature and were
washed 1 time with HEPES buffer (20mM HEPES, 130mM NaCl, 5mM KCl, 0.8mM
MgCl2, 1mM CaCl2, pH 7.4). Membranes were blocked with 1% BSA in PBS for 30
minutes at room temperature and were washed 1 time with HEPES buffer. 50,000 cells
were seeded in the upper chamber in serum free media and allowed to attach for 1 hour
followed by the indicated treatments with peptide or chemical inhibitor. Peptides and
inhibitors were in serum-free media and in both upper and lower chambers. For
experiments with integrin blocking antibodies, cells were pretreated in suspension with

36 the indicated antibody(s) for 15 min before attachment. Cells were allowed to invade for
18 hours at 37o C and cells remaining in the upper well were removed with a cotton swab
while cells on the lower surface were fixed with methanol and stained with crystal violet.
The number of cells on the lower surface of the insert was quantified by solubilizing the
dye in 0.1 M sodium citrate and reading the absorbance at 562 nm. Triplicate
determinations were done with each treatment and each experiment was performed at
least 3 times.
The scratch assay was performed with PC3N cells grown to confluency on
laminin-5 coated square coverslips in IMDM (Gibco BRL, MD, USA) plus 10% fetal
bovine serum (FBS). The source of laminin-5 was serum-free conditioned medium from
HaCaT cells. A scratch was made diagonally across the square coverslip with a
plastic cell scraper (Fisherbrand Cat. # 08-773-2). The coverslips were then
rinsed in medium and placed in fresh medium containing 75 µg/ml peptide and 1% FBS
for 12 hours at 37oC. The cells were fixed with chilled methanol for 10 min
followed by 10 dips in chilled acetone. On drying, the cells were then stained
with 0.5 µg/ml DAPI for 10 min. The coverslips were washed in PBS, post-fixed
in Ethanol for 4 min and mounted using Prolong Antifade (Molecular Probes, Or,
USA). The slides were visualized on a Zeiss Axiovert microscope. Pictures were
taken using Axiocam camera at 10 X magnification. Quantification was done using
Scion Image.

37 Cell Adhesion Assay and Peptide Inhibition of Adhesion.
Ten µg of ligand or 20 µg of neuralite avidin (Molecular Probes, Inc.) was
dissolved in 1 ml of distilled water and 100 µl of solution was added to each well of a
tissue culture 96 multiwell plate (Falcon, Franklin Lakes, NJ). The solution of either
ECM protein or neuralite avidin was allowed to dry overnight. Use of neuralite avidin
ensures maximum binding of the biotinylated peptide to the surface, avoiding the
variability of peptide coating. In the β1 antibody adhesion assay, the TS2/16 antibody
(2µg/ml) was coated for 18 hours in the HEPES buffer at 4C. The wells for all substrates
were then blocked with 100 µl of 1% bovine serum albumin (BSA) for 1 hour. The wells
were washed with HEPES buffer and varying amounts of peptide added per well. After
one hour of peptide incubation, wells were washed with IDMEM without serum and
suspended DU-H cells (5 x 104) in serum free IDMEM were added in each well. The
cells were allowed to adhere for 60 minutes at 37C. The wells were washed three times
with HEPES buffer and fixed with 2.5% formaldehyde in PBS. The cells were then
stained with 0.5% crystal violet in 20% (v/v) methanol/water and viewed under a
microscope. The amount of bound cells was estimated by solubilizing the dye using
0.1M sodium citrate and reading the absorbance at 570nm. Triplicate determinations
were done at each data point.
Cell Lysis and Western-Blot
For cell signaling experiments involving FAK, MEK and ERK, cells were
incubated in serum-free media overnight and harvested with 5mM EDTA in PBS for 10

38 minutes. Cells were washed in serum-free media and added to laminin-5 coated plates or
BSA coated plates for 1 hour at 37o C. Cells were then treated with peptide in serum-free
media for stated times at 37o C. Two minutes before lysis, 0.5mM sodium orthovanadate
was added into the media. Cells were lysed for 5 minutes in a modified RIPA lysis buffer
containing 40mM Tris, pH 7.5, 150mM NaCl, 1% Triton X-100, 6mM EDTA, 100mM
NaF, 1mM sodium orthovanadate, 10mM sodium pyrophosphate, 1mM PMSF, and
1µg/ml leupeptin and aprotinin. Lysates were processed for SDS-PAGE after adjusting
for equal loading by using the BCA protein assay kit (Rockford IL). Proteins resolved in
the gel were electrotransferred to Millipore Immobilon-P polyvinylidene fluoride (PVDF)
membrane (Millipore, Bedford MA). Blots were developed using chemiluminescence
(ECL Western Blotting Detection System, Amersham, Arlington Heights, IL) and band
densities were analyzed by densitometry using NIH Image software (Scion). For
stripping, membranes were incubated in stripping buffer (62.5mM Tris, pH 6.75, 2%
SDS, and 100mM beta-mercaptoethanol) at 50o C for 30 minutes followed by washing 5
times in TBS.
Immunoprecipitation
All immunoprecipitations, unless specified, contained 200 µg total protein lysate.
Immunoprecipitation of cortactin per reaction contained, 200µg total protein lysate, 35 µl
of protein G sepharose and 3 µg of antibody. The final volume was adjusted to 400 µl of
RIPA lysis buffer described above. The mixture was rotated at 4o C for 18 hours and the
complexes were washed 3 times with cold lysis buffer. For co-immunoprecipitation

39 studies, cells were lysed in HBSM ( 20mM Hepes, pH 7.5, 150mM NaCl, and 5mM
MgCl2 ) supplemented with 1% Brij 96 (or 97). These complexes were washed 4 times
with cold lysis buffer following rotation at 4o C for 18 hours. Immunoprecipitation of
CD151 was performed on the supernatants from affinity precipitations described above.
Briefly, the supernatant was added to an immunoprecipitation reaction containing 1 µl of
5C11 antibody and 30 µl of protein G sepharose. The mixture was rotated for 18 hours at
4oC. Additional 5C11 immunoprecipitations were carried out with 1 µl of 5C11, 20 µg
membrane fraction and 30 µl protein G beads adjusted to a total volume of 500 µl with
the standard rotated for 18 hours at 4oC. Protein G sepharose beads were washed 3 times
with RIPA buffer. All samples were suspended in 2x laemmli buffer and analyzed by
SDS-PAGE electrophoresis. When western blot analysis was for α6, non-reducing
laemmli buffer was used.
Epithelial and Stromal Cell Adhesion Assay.
HFF cells were used at 3 x 105 cells/well in 6-well plates in serum free medium
and allowed to attach at 37C. After 24 hours the medium was removed, the well was
washed once with PBS and 3 x 105 SCC-25 cells were placed on top of the HFF
monolayer. The SCC-25 cells were added using 1ml of serum free medium containing
either 40ug/ml HYD-1, 40ug/ml HYDS-1 or no treatment. The number of SCC-25
unable to attach were recovered by a PBS wash and counted every hour for three hours
using a hemocytometer. The determinations were done in triplicate.

40 FACS Analysis and Confocal Microscopy.
The peptides were synthesized with a biotin at the amino terminus. The peptides
were then allowed to bind to neuralite avidin conjugated with Bodipy (Molecular Probes,
Eugene, OR) and the tetrameric peptide-avidin complexes were incubated with the cells.
The non-specific binding was determined using the neuralite avidin-Bodipy alone
incubated with the cells. Suspended DU-H cells (1x106) in 1 ml of IDMEM without
serum were incubated with 10 µg of peptide and 10 µg Bodipy conjugated to neuralite
avidin at 4oC in the dark for 30 minutes. The cells were washed several times with
IDMEM without serum and then analyzed for fluorescence on a FACStarPlus (Becton
Dickenson) or examined using a Laser Scanning Confocal Microscope (LSM 10, Zeiss ).
Digital images were collected on each Z-series using identical contrast and brightness
settings.
In Vivo Actin Distribution Assay
The distribution of globular actin and filamentous actin was analyzed using an F-
actin/G-actin in vivo assay kit (Cytoskeleton Inc., Denver CO). Briefly, after peptide
treatment, cells were lysed in a cell lysis and F-actin stabilization buffer and
homogenized using 27 gauge needles. In order to isolate cellular filamentous actin, cell
lysates were centrifuged at 100,000 g for 60 minutes at 37o C and the supernatants (G-
actin) were immediately separated from the pellets (F-actin). The pellets were
resuspended in the same volume of dH2O as the supernatants and were incubated on ice
for 60 minutes. Cytochalasin D (2µM) was added to the resuspended pellets to promote

41 dissociation of filamentous-actin in the pellets into actin monomers. The pellet was then
further fractioned to insoluble and soluble forms by centrifugation at 14,000 rpm for 1
minute. The insoluble portion containing the cell membrane was solubilized in RIPA
buffer. Equal amounts (2µg of protein for each sample) of each fraction were subjected
to SDS-PAGE and analyzed by Western Blot with an anti-actin antibody provided in the
kit.
Immunofluorescence
Coverslips were coated with laminin-5 as stated in materials and methods and
washed with PBS before plating the cells. Cells were plated subconfluently and allowed
to adhere for one hour before treatments. Peptide was added in serum-free media for 30
minutes before fixation. Coverslips were dipped in PBS and fixed in PBS containing
3.2% formaldehyde (Ted Pella Inc. Irvine CA) for 5 minutes. The coverslips were rinsed
in water before incubation with 50 mM NH4Cl in PBS for 5 minutes. Following a wash
in PBS for 5 minutes cells were treated with 0.2% Triton X-100 in PBS for 5 minutes.
Fixation and washing were performed at room temperature. Cells were washed again in
PBS and blocked with 1% BSA for 30 min at room temperature before incubating with
the primary antibody (cortactin 1:100) and phallodin (1:40) for 1 hour at room
temperature. Staining for alpha-6 (J1B5 1:50) and CD81 (JS64 1:50) followed the same
protocol. Secondary antibody was diluted at 1:200 for 30 minutes at room temperature.
The coverslips were mounted with Anti-Fade (Molecular Probes, Eugene OR) and
observed at 40X with an inverted Zeiss Axiophot (Carl Zeiss, Gottingen, Germany)

42 equipped with an Axiocam camera. Captured images were analyzed with Photoshop 7.0
software (Adobe Systems Inc., San Jose CA).
Synthetic Peptides, Fibronectin, Laminin-1, Laminin-5, and Collagen IV
The RZ series of peptides was found using the “one-bead one-compound”
combinatorial library method and a D-isomer Octyl-mer peptide library as previously
described by us [81]. The amino acid sequences of the D-amino acid peptides are RZ-3,
kmviywkag; RZ-4, kggrhykfg; RZ-6, arkfkglig; RZ-12, yiknrkhhg. The HYD-1 peptide
(kikmviswkg) was generated by overlapping the positive adhesion peptides and
postulating a hybrid sequence. The HYDS-1 scrambled peptide (wiksmkivkg) was
generated by random ordering of the HYD-1 peptide sequence. AG73
(RKRLQVQLSIRT) is a previously characterized L-amino acid peptide from the G-
domain of the laminin α1 chain [97]. The six peptides were synthesized with a biotin at
the amino terminus and purified by Molecular Resources Core Facility, Colorado State
University, Dept. of Biochemistry, Ft. Collins, Colorado. Peptides were also synthesized
and purified by High-Performance-Liquid-Chromatography to greater than 90% by
Global Peptide Service (Fort Collins CO). The human fibronectin and mouse collagen
IV was obtained from Gibco BRL, Inc. The laminin-1 was obtained from Becton
Dickinson Corp., Franklin Lakes, NJ. and is derived from the Engelbreth-Holm-Swarm
(EHS) mouse tumor. The laminin-5 was obtained from the matrix of HaCaT cells[95]
(Dr. Norbert Fusenig, University of Heidelberg).

43
Laminin-5 Coating Conditions for Motility and Signaling Experiments
Laminin-5 was obtained from conditioned media of HaCaT cells. Briefly, sub-
confluent HaCaT cells were incubated with serum-free media overnight. The media was
collected and filtered through a 0.2µm acetate filter. For laminin-5 coating of tissue
culture plastic, coverslips, and time-lapse video plates, conditioned media was added to
the surface for 2 hours at room temperature and washed 1 time with PBS (2.7mM KCl,
1.5mM KH2PO4, 138mM NaCl, 8.1mM Na2HPO4, pH 7.4) before use. For signaling
experiments, plates were blocked with 1% BSA for 30 minutes at room temperature.

44 III. IDENTIFICATION AND CHARACTERIZATION OF TUMOR
CELL ADHESION PEPTIDES
Introduction
Tumor cell adhesion to the extracellular matrix (ECM) within tissues greatly
influences a malignant cell’s ability to invade and metastasize to outlying tissues,
reviewed in [98]. Further, the survival of the metastasized tumor cell depends in part
upon the activity of the ECM receptors [99-104]. Cell adhesion and the accompanying
intermediate filaments in tumor cells are also an important factor in resistance to the
killing effects of several chemotherapeutic drugs. Fibronectin mediated adhesion of
myeloma cells confers a survival advantage in a phenomenon known as cell adhesion
mediated drug resistance (CAM-DR) [105-108]. The pivotal role of adhesion
modulation in both tumor cell metastasis and their survival prompted us to characterize
tumor cell adhesion peptides for their ability to alter cell adhesion to the ECM.
The proteins of the ECM consist of type I and IV collagens, laminins, heparin
sulfate proteoglycan, fibronectin, and other noncollagenous glycoproteins [109]. Cell
adhesion to the laminins, fibronectin, and collagens is mediated in part by a group of
heterodimeric transmembrane proteins called integrins, which are composed of a non-
covalently associated α- and β- subunit that define the integrin ligand specificity [20].
The integrins α6β1, α3β1, and α6β4 are laminin receptors [110,111]. These integrins in
particular are associated with epithelial tumor progression in prostate, breast, colon,
pancreatic carcinomas, head and neck tumors, and melanoma [23,112-115]. Prostate

45 micrometastases continue to express the α6 integrin whereas other cell adhesion
molecule expression is suppressed [27].
Previous attempts to block adhesion of cells to the ECM (i.e. laminin-1, 5 or
collagen) have been accomplished using integrin specific, function blocking monoclonal
antibodies or peptide mimics of the natural ECM protein [116-118]. For example, human
melanoma cells co-injected into nude mice with an anti-α6 antibody have a lowered
ability to metastasize [119]. Similarly, the α1β1 and α2β1 integrins are collagen
receptors and antibodies to these integrins reduced invasion of basement membranes
[120]. Biologically active adhesion peptides derived from specific regions of the ECM
protein laminin-1 have had reported effects on a variety of biological processes including
invasion, metastasis, migration and matrix metalloproteinase expression [97,121-131].
Our previous work identified several L-amino acid containing peptide candidates
as anti-adhesive agents by selecting peptides based upon the ability of the prostate tumor
cells to bind to the immobilized peptide [81]. Further screening with a D-amino acid
library yielded several new peptide candidates. Adhesion and biological properties of
these peptides are analyzed here, a hybrid peptide proposed and further tested. The
identification of D-amino acid peptides for the interruption of cell adhesion to ECM
proteins or dermal fibroblasts should be useful in the increased killing of distant
metastases, as well as providing insight into regulation of integrin-ligand interactions.

46 Results
RZ-3, HYD-1 and AG-73 Bind to Prostate Tumor Cell Surfaces
The RZ-3, RZ-4, RZ-6, RZ-12, HYD-1 peptides and the previously reported AG-
73 peptide [97] were tested for their ability to bind to DU-H cells by FACS analysis. In
Figure 4 (top panel), the mean peak of the fluorescence (MPF) for each peptide is shown
and indicates that Bodipy neuralite avidin alone resulted in a barely detectable amount of
non-specific binding. The RZ-4, RZ-6, and RZ-12 peptides all demonstrated slight
activity above the background of Bodipy neuralite avidin alone. The RZ-3, HYD-1 and
AG-73 peptides exhibited significantly higher amounts of cell binding as compared to
Bodipy neuralite avidin alone. Use of the RZ-3 peptide resulted in a mean peak of
fluorescence (MPF) of 350; HYD-1 resulted in a MPF of 1200. The previously reported
AG-73 peptide resulted in a MPF of 500. The scrambled peptide, HYDS-1, had a MPF
similar to the non-specific binding detected by the bodipy neuralite avidin alone
indicating that the scrambled peptide did not bind to the cells. The fluorescence
histogram for each binding peptide is shown in the remaining panels of Figure 4. All
three peptides result in a normal distribution pattern of cell binding, indicating that there
are populations of cells which are low and high binders of the peptides. The AG-73
peptide had the broadest distribution pattern, followed by RZ-3 and HYD-1 respectively.
Of the three peptides, the HYD-1 peptide displays the most uniform distribution. The
distribution of both RZ-3 and HYD-1 peptides was also examined in live DU-H prostate
tumor cells by confocal microscopy. The data in Figure 5 indicate that the peptides
remained on the surface of the cells during the 30-minute incubation period.

47
Figure 4. Fluorescence-activated cell sorting (FACS) analysis of biotinylated peptides bound to DU-H prostate carcinoma cells. The cells were incubated with BODIPY-peptide complexes for 30 minutes and the amount of peptide bound was estimated using FACS analysis. The mean peak of fluorescence (MPF) for each peptide from the resulting histograms are shown in the top panel. The representative histograms for the AG-73, HYD-1, HYDS-1 and the RZ-3 peptides are shown in the remaining panels.

48
Figure 5. Confocal microscopy of the peptides RZ-3 and HYD1 binding to surface of human prostate tumor cells. The DU-H cells were incubated with BODIPY-RZ-3 (top panel,) or BODIPY-HYD-1 middle panel) peptide or BODIPY alone (bottom panel) for 30 minutes and the peptide (green) was observed using confocal microscopy on the live cells. The nuclei are stained with propidium iodide (red). The bar is 25 microns.

49 The confocal image also indicates that all of the cells were decorated with the peptide.
DU-H Prostate Tumor Cells Adhere to Immobilized Peptide and ECM Proteins
Using the three peptides that displayed the most cell-binding activity, RZ-3,
HYD-1 and AG-73, the biological activities of the peptides were further tested. The
ability of DU-H cells to adhere to the peptides immobilized on a plastic surface was
compared to the ability of the cells to adhere to several immobilized natural ECM
ligands, such as fibronectin, laminin-1 and collagen-1 (Figure 6).
All three peptides supported cell adhesion within the 60-minute incubation period
(Figure 6). The HYD-1 peptide promoted the most adhesion, with concentrations as low
as 2µg/well supporting cell attachment. Maximal cell adhesion to HYD-1 occurred at
10µg/well. The scrambled peptide, HYDS-1, did not support cell adhesion at any
concentration tested (i.e.10-100µg/well). The RZ-3 peptide required 10-50µg/well for
tumor cell adhesion. A comparison of the peptides indicates that 50% cell binding
occurred using 1.5µg, 20µg or 30µg per well for HYD-1, AG-73 and RZ-3 respectively.
Mixtures of the peptides were not synergistic for promoting adhesion (data not shown).
All three ECM proteins are sufficient ligands for DU-H adhesion, and can support
adhesion in a similar manner at concentrations as low as 0.1µg/well. A comparison of
the ECM proteins indicates that 50% cell binding occurred using 0.1µg, 0.6µg and 1.5µg
of fibronectin, laminin-1 and collagen IV respectively. Taken together, a comparison of
the ability of the peptides and the ECM proteins to support adhesion indicates that the

50
Figure 6. Comparison of human prostate tumor cell adhesion to immobilized peptides or ECM proteins. The DU-H cells were allowed to attach to HYD1 (open circles), AG73 (open triangle), RZ3 (closed triangle), or HYDS (open square) peptide coated 96-well microtiter plates (top panel) or fibronectin (closed square), laminin-1 (open circle), or collagen IV (closed triangle) protein coated 96-well microtiter plates (bottom panel). The number of cells attached after 60 minutes was determined by absorbance at 570 nm. Data are the mean triplicates. Error bars = SD.

51
HYD-1 peptide compares favorably to the natural ligands.
Tumor Cell Adhesion to Immobilized Peptides is Integrin Dependent
Next, we tested if cell adhesion to the peptides or the natural ligands was
dependent upon cations or whether the interaction could be blocked by integrin function
blocking antibodies. Cell attachment to laminin-1 and fibronectin was inhibited by 3mM
EDTA as expected (Figure 7), indicating that these interactions are conducted through an
interaction that requires cations [132]. In contrast, cell attachment to RZ-3, and HYD-1
was not affected by 3mM EDTA, suggesting that the adhesive properties occur through
an integrin interaction which does not require cations. This is consistent with a peptide
interaction occurring independent of the calcium binding motif. The calcium binding
motifs of the α integrin subunit lie on a surface of the integrin far from the ligand contact
sites [132]. Cell attachment to laminin-1 and fibronectin was not affected by the
addition of 10 ug/ml heparin suggesting that the cell adhesion to the immobilized matrix
proteins was not due to the positive charge interactions with the cell surface. Similarly,
attachment to RZ-3, and HYD-1 was unaffected by adding heparin (data not shown).
Cell attachment to fibronectin was inhibited by the anti-α5 and the anti-β1 antibody,
consistent with its known function as a ligand for the α5β1 integrin. Cell attachment to
laminin-1 was inhibited by anti-α3, 6 and anti-β1 integrin antibodies, consistent with the
fact that adhesion to laminin-1 is mediated by both the α3, 6 and β1 integrins. Cell
attachment to laminin-5 was partially inhibited by the α3, 6 antibodies and unaffected by

52
Figure 7. Effect of antibodies and reagents on DU-H cell adhesion to peptides and extracellular matrix proteins. The DU-H cells were allowed to attach to Fibronectin (1µg/well), Laminin-1 (1µg/well), Collagen IV (1µg/well), HYD-1 (10µg/well), or RZ-3 (50µg/well) coated plates in the presence of 10µg/ml IgG, 3mM EDTA; anti-α 2; anti-α5; anti α3 and 6; anti α2 and 5 integrin antibodies or anti-β1 integrin antibody, as indicated. After 60 minutes of incubation at 37oC, the number of attached cells was determined by absorbance at 570nm. Data are expressed as a mean of triplicate results; error bars are the standard deviation. The percent of control is the absorbance value of cells with the function-blocking antibody divided by the absorbance value of cells without the function-blocking antibody X 100.

53 the β1 blocking antibody. A combination of the α2, 3, 5, and 6 integrin blocking
antibodies partially inhibited adhesion to immobilized HYD-1 or RZ-3 peptides.
Although cell adhesion to HYD-1 was not inhibited by β1 blocking antibodies, HYD-1
peptide can inhibit cell adhesion to the β1 integrin specific activating antibody TS2/16
(Figure 8). Taken together, the data suggest that cell adhesion to the immobilized RZ-3
and HYD-1 are integrin dependent.
Inhibition of DU-H Cell Attachment to ECM Proteins
The next test of the biological properties of the peptides was to determine
whether the soluble peptides could inhibit DU-H adhesion to the immobilized ECM
proteins. The DU-H cell attachment to immobilized laminin-1 and fibronectin was
decreased in a concentration-dependent manner when HYD-1 or RZ-3 was added to the
wells (Figure 9). The RZ-3 peptide was the most effective peptide for inhibiting
attachment to all ECM proteins, including laminin-5, as judged by comparing the
concentration of peptide required to inhibit 50% of the cell adhesion (IC50) to the
respective ECM protein. The previously reported AG-73 peptide inhibited cell adhesion
to fibronectin, requiring approximately 9ug/well and did not inhibit cell adhesion to
laminin-1 or collagen IV. The HYD-1 peptide inhibited cell adhesion to laminin-1,
fibronectin and collagen IV in a similar manner. The HYD-1 peptide inhibition of cell
adhesion showed a threshold response in that greater than 2µg/well of peptide was
required before an inhibition of cell adhesion was detected. The scrambled HYDS-1
peptide was not able to inhibit adhesion

54
Figure 8. Inhibition of DU145 cell adhesion to immobilized ββββ1 integrin antibody by peptides. The cells were allowed to attach to the immobilized β1 integrin antibody called TS2/16 (2µg/well) in the presence of increasing concentrations of soluble peptide (HYD-1, RZ-3, HYDS-1 or AG-73) using serum free medium. After one hour at 37oC, the number of attached cells was determined by absorbance at 570nm. Data are expressed as a mean of triplicate results; error bars are the standard deviation.

55
Figure 9. Inhibition of DU-H cell adhesion to extracellular matrix proteins by peptides. The cells were allowed to attach to laminin-1 (closed triangle, 1µg/well), fibronectin (open circle, 1µg/well), laminin-5 (closed circle, 1µg/well) or collagen IV (open triangle, 1µg/well) coated 96 well microtiter plates in the presence of various amounts the RZ-3, HYD-1 or HYDS-1 peptides using serum free media. After a 60 min incubation at 37oC, the number of attached cells was determined by absorbance at 570nm. Data are expressed as a mean of triplicate results. The percent of control is the absorbance value of cells with the peptide divided by the absorbance value of the cells without the peptide X 100.

56 to any ECM protein. A comparison of the IC50 values of the peptides reveals that the
most potent inhibitor for attachment to all three ECM proteins is RZ-3.
Inhibition of SCC-25 and HFF Co-Culture Interaction by HYD-1 Peptide
The biological relevance of the adhesion blocking ability of the HYD-1 peptide
was tested using an epithelial-fibroblast co-culture model system. Epithelial stromal
interactions are increasingly recognized as determinants of tumor progression. The
adhesion of SCC-25 cells to a HFF monolayer is a time dependent process that was
maximal within one hour of incubation at 37oC (Figure 10). The HYD-1 peptide
inhibited approximately half of the SCC-25 cells from attaching to the HFF monolayer.
The inhibition of SCC-25 adhesion was maintained over the three-hour period of the
assay. The scrambled form of the peptide, HYDS-1, was ineffective in altering adhesion.
Adhesion to Immobilized HYD1 Prevents Cell Spreading and Adhesion Dependent
Phosphorylation
Cell spreading is a critical event that normally follows cell adhesion to the
extracellular matrix. Cell adhesion is coupled to integrin activation, initiating multiple
signaling pathways [133,134]. In general, cell spreading occurs from extensive actin
cytoskeletal remodeling through actions of the Rho Family of small GTPases [133].
Several focal adhesion proteins play a regulatory role in cell spreading events such as
focal adhesion kinase (FAK), paxillin, and p130CAS [135]. In addition, phosphorylation

57
Figure 10. Inhibition of SCC-25 cell adhesion to HFF monolayers by peptides. Approximately 3 x 105 SCC-25 cells were plated onto HFF monolayers in the absence (circle) or presence of the HYD-1 (square), HYDS-1 (triangle) peptides. After the indicated times of incubation, the number of SCC-25 cells unable to attach was determined using a hemocytometer. The number of HFF cells unable to attach was also determined in the presence of the HYD-1 peptide (diamond). A minimum of three determinations were done at each time point. The percent adhesion is the number of SCC-25 cells adhering to the HFF monolayer divided by the number of SCC-25 cells added to the wells X 100.

58 of mitogen activated protein kinase kinase (MEK) at a specific residue, serine 298, is an
adhesion dependent phosphorylation regulated by p21-activated kinase (PAK) [136].
Using immobilized peptide as a ligand, we found that HYD1 is an adhesion
agonist by mediating integrin-dependent tumor cell adhesion (Figures 6-8). To determine
if HYD1 is a sufficient ligand to initiate cell spreading, we quantified the amount of cell
spreading and average area occupied by the cells after adhesion to immobilized HYD1 or
laminin-5 after 1 hour. As expected, adhesion to laminin-5 initiates cell spreading in
nearly 100% of the cells within a microscopic field (Figure 11A). Interestingly, adhesion
to HYD1 prevented cell spreading even in the presence of EGF (10ng/ml) (Figure 11B)
suggesting that adhesion to immobilized HYD1 uncouples integrin-dependent adhesion
from cell spreading.
Since cell spreading events are tightly regulated by intracellular signals [133,135],
we examined two integrin-proximal cellular signals that occurred during adhesion to
HYD1. Adhesion to HYD1 induced phosphorylation of FAK on tyrosine 397 and MEK
on serine 298 after thirty minutes of attachment (Figure 12A and B). The magnitude of
the response was quite different when compared to cells attached to laminin-5 over the
same time course (Figure 12A and B). Laminin-5 induced more activation of FAK at
both 10 and 30 minutes post-adhesion while inducing significantly more phosphorylation
of MEK at 10 minutes post-adhesion. Adhesion to HYDS was not able to induce a
comparable response with either signal. Taken together these data suggest that adhesion
to immobilized HYD1 uncouples cellular adhesion from cell spreading in part by
suppressing adhesion dependent phosphorylation events.

59
Figure 11. Adhesion to immobilized HYD1 prevents cell spreading. Spreading of PC3N cells was determined using differential interference contrast optics on an inverted Olympus IMT2 microscope. A) PC3N cell adhesion on a native ECM ligand, laminin-5 (HaCaT conditioned media), after 1 hour. B) PC3N cell adhesion on HYD1 (150µg) + EGF 10ng/ml after 1 hour. C) Cell spreading was quantified with Compix Imaging software. Error bars are standard error from the mean area spread of 15 cells/experiment.

60
Figure 12. Suppression of adhesion dependent phosphorylation by immobilized HYD1. Phosphorylation of focal adhesion kinase (FAK) on tyrosine 397 (A) and the mitogen-activated protein kinase MEK1 on serine 298 (B) was analyzed by western blot using phosphospecific antibodies. Densitometry analysis of percent phosphorylation is shown. PC3N cells were serum-starved overnight and attached in serum-free conditions to the indicated ligands for 10 and 30 minutes. Peptides were coated at a concentration of 150µg/plate. HYDS is a scrambled peptide derivative of HYD1.

61 Discussion
Using a previously described method to isolate candidate peptides for biological
activity with the α6 integrin, we report the existence of two bioactive D-amino acid
containing peptides called RZ-3, and HYD-1. These peptides attach to the surfaces of live
tumor cells, can themselves support cell attachment, can inhibit attachment to ECM
proteins such as fibronectin, laminin-1, laminin-5 and collagen IV and can inhibit
epithelial stromal adhesion interactions.
The inhibition of tumor cell adhesion to a number of ECM proteins was observed
with both peptides. In addition, the integrin blocking antibodies only partially inhibited
the adhesion of the cells to the peptides. Taken together, these data suggest that the
peptides can affect adhesion processes that are both integrin dependent and independent.
This fact may have significance for both understanding cross-talking pathways of cell
adhesion to the ECM and interrupting adhesion dependent biological events. The broad
nature of the inhibition could prove useful in disabling the varied adhesion events that are
activated within human tumor cell populations. It should be noted that these peptides do
not show homology to any known proteins in the SwissProt database from a BLASTP
search (data not shown).
The potential use of the peptides could include the prevention of tumor cells
adhesion in vivo. Preliminary experiments utilizing a mouse human tumor xenograft
model system [137] suggests that the peptides themselves are not toxic to animals (data
not shown). The alteration of tumor cell adhesion could be of benefit for preventing
metastasis as well as increasing the killing of tumor cells that are refractory to treatment.

62
The peptides may be particularly useful in human tumors with a wild type p53
function since clustering of integrin in tumor cells containing wt p53 can trigger
apoptosis [104]. Our preliminary experiments indicate that the altering of attachment can
sensitize the tumor cells to the killing effects of ionizing radiation (data not shown). We
are currently testing both the efficacy of the peptides to inhibit tumor cell adhesion in
vivo and the potential for synergistic lethality with DNA damaging agents in vivo.
It has been demonstrated that fibronectin-mediated adhesion confers a survival
advantage for myeloma cells acutely exposed to cytotoxic drugs in a phenomenon known
as cell adhesion mediated drug resistance (CAM-DR)[108,138]. Myeloma cells within
the bone marrow are adherent to fibronectin and are thought to serve as a reservoir of
tumor cells that are difficult to eradicate. The anti-adhesive peptides HYD-1 and RZ-3
may be lead candidates to overcome CAM-DR.
The role of the ECM as a cell survival factor has been demonstrated in both
normal cell types and fibroblasts. The ECM is critical for normal survival signals and the
loss of the ECM will result in normal cell death in a process termed anoikis [139-142].
However, during transformation, normal cells down regulate many of their adhesive
interactions and display anchorage independent cell growth. Although the majority of the
adhesion events are suppressed during transformation, selected adhesion events are
preserved. For example, in prostate cancer progression, the majority of the integrin ECM
receptors and the E-cadherin class of adhesion molecules are suppressed [143] while the
laminin receptors and N-cadherin remain expressed [23,94]. The preserved adhesion

63 events may be important targets for inactivating the known survival advantage of
invasive or metastatic tumor cells.
Finally, the cell adhesion peptides characterized in this study have both agonist
and antagonistic properties when compared to native extracellular matrix ligands. Both
HYD1 and RZ3 were active as adhesion mimetics by both supporting tumor cell adhesion
when immobilized and inhibiting tumor cell adhesion to immobilized extracellular matrix
proteins. However, adhesion to HYD1 was not sufficient to produce cell spreading. In
addition, compared to laminin-5, adhesion to HYD1 did not initiate similar adhesion-
dependent signals. Therefore, HYD1 uncouples cell adhesion and cell spreading in part
by preventing the appropriate adhesion-dependent phosphorylation events. The ability of
HYD1 to interrupt the normal response following cell adhesion may prove useful in
developing agents to block cell invasion and metastasis.

64 IV. SYNTHETIC D-AMINO ACID PEPTIDE INHIBITS TUMOR
CELL MOTILITY ON LAMININ-5
Introduction
Cell migration is a complex process integral to normal biological events such as
wound healing and inflammatory responses as well as the pathological circumstances of
tumor invasion and metastasis. The motile nature of all cell types depends upon the
actions of many different molecular components [144]. Central to this process are the
signaling and cytoskeletal responses elicited by the interactions of integrins with the
extracellular matrix (ECM). The adhesive complexes formed from integrin ligation and
activation regulate intracellular signaling events that dictate the cytoskeletal
reorganization necessary for cell movement [56]. Several signaling pathways have been
shown to be important for cell movement and specific pathways may have crucial roles
depending on the extracellular environment [56,145-147]. In addition, tumors associated
with an invasive and migratory phenotype may favor a certain integrin repertoire
[93,148,149], displaying a pivotal role of specific integrin/ECM interactions that favor
tumor metastasis.
It is well established that the ECM can induce integrin dependent cell spreading
and migration by activation of specific signaling programs that regulate focal adhesion
and cytoskeletal dynamics [56,144]. Interestingly, specific integrin/ECM pairs have been
shown to differentially modulate the activities of these programs [147,150] suggesting
that integrin and ECM composition will dictate the signaling response and phenotype.

65 Laminin-5 dependent cell spreading and migratory activities, for example, has been
linked to the activities of focal adhesion kinase (FAK), phosphoinositide 3-OH kinase
(PI3-K), p21-activated kinase, and the mitogen-activated protein kinase pathway
[147,151,152]. Inhibition of these pathways using small molecules can ultimately block
a migratory phenotype. However, given the multitude of components in these pathways
and their redundancy in function, targeted dysregulation of integrin/ligand activity may
prove to be a more potent method to inhibit motility.
Integrins are evolutionarily conserved heterodimeric cell surface molecules. To
date there are 18 distinct α and 8 distinct β subunits that pair in a restrictive manner to
give about 24 different integrins that have individual ligand binding specificities [134].
The primary ligands for integrins are proteins of the ECM that consist of type I and IV
collagens, fibronectin, laminins, heparin sulfate proteoglycan, and other noncollagenous
glycoproteins [109]. The integrins α6β1, α6β4, and α3β1 are laminin receptors,
[110,111] and these integrin pairs are associated with the progression of many epithelial
tumors [23,112,115]. In particular, the α6 subunit is continually expressed during
prostate cancer progression and found in micrometastases [23,27,149]. Previous studies
have shown that biologically active peptides developed from defined regions within
laminin chains can have profound effects on biological events including cell migration
and metastasis [97,123,125,127,153-156]. These findings prompted us to develop α6-
binding cell adhesion peptides.
Our previous work [81,82] has identified human tumor cell adhesion peptides
capable of binding prostate carcinoma cells expressing the α6 integrin. We characterized

66 two D-amino acid peptides, HYD1 (kikmviswkg) and RZ3 (kmviywkag), as cell
adhesion peptides based on their ability to both support tumor cell adhesion themselves
and inhibit tumor cell adhesion to immobilized ECM proteins [82]. In the present study,
we examine the effect of these peptides on laminin-5 dependent haptotaxis. HYD1
causes dramatic cytoskeletal reorganization in prostate tumor cells adhered to laminin-5,
resulting in a loss of cell migration. HYD1 interacts with both α6 and α3 integrin
complexes and induces signaling through FAK, MEK, and ERK. These data show that
HYD1 is a novel synthetic peptide that disconnects pro-migration phosphorylation
signals from cytoskeletal dependent migration.
Results
HYD1 Blocks Laminin-5 Dependent Migration and Invasion
Effect of the peptides, HYD1 and RZ3, on laminin-5 mediated cell migration and
invasion was investigated. Video microscopy was used to measure random haptotaxis
and a modified Boyden chamber assay was used to assess the effects on invasion toward
laminin-5. In order to analyze the effect of the peptides independent of disrupting
adhesive contacts with the ligand, cells were allowed to adhere for 1 hour prior to
treatment with peptides in serum-free media. Treatment with HYD1 completely blocked
random haptotaxis on laminin-5 that was quantified for a four-hour period (Figure 13A
and B). A scrambled variant of HYD1, HYDS, was inactive. The activity of HYD1 was
a post-ligand-occupancy event; there was no loss of adhesion and the cells remain spread
over the course of the video (4 hours). In addition, membrane surfaces were observed to

67
Figure 13. HYD1 blocks cell migration on laminin-5. (A) PC3N cells were placed on laminin-5 for 1 hour followed by the addition of serum-free media or serum-free media containing HYD1 (75µg/ml). Videos were started approximately 30 minutes post-treatment. Time-lapse images were taken at start of video T=0, T=15 (15 minutes), and T=30 (30 minutes). White arrowhead in T=0 frame marks the cell followed during analysis at T=30. Scale bar = 100µm.

68
Figure 13 Continued. (B) Cells were added as described in A and treated with 75µg/ml of the peptides HYD1, RZ3, AG73, or HYDS in serum-free media. The Src inhibitor, PP2, was used at a concentration of 10µM. Videos were started 30 minutes post-treatment and images were taken every 5 minutes for 4 hours. (C) The effect of HYD1 is reversible. Cells were added as described in A. Cells were treated with HYD1 for the entire course of the video (4 hours). Alternatively, cells were treated for 30 minutes, the peptide was washed-out, and the cells were exposed to serum-free media alone or serum-free media containing soluble laminin-5 and allowed to recover over the 4-hour period. Migration rates were quantified as described in Experimental Procedures. Error bars are standard error from the mean migration rate of 15 cells/experiment.

69 remain active after peptide treatment and cell division occurred normally. Cells remained
non-motile after cell division and reattachment. The effect of HYD1 was reversible
following peptide wash-out (Figure 13C). Adding soluble laminin-5 after peptide wash-
out enabled a faster recovery of migration over a four-hour period as compared to adding
serum-free media, indicating that the cells retain the capacity to respond to a pro-
migratory stimulus. The inhibitory activity for HYD1 was comparable quantitatively to
PP2, a Src family kinase inhibitor previously published to inhibit migration of prostate
carcinoma cell lines [157] (Figure 13B). The morphology of cells treated with HYD1 as
compared to PP2 suggest a distinct mechanism of action. HYD1 treated cells remain
spread and have active membrane surfaces compared to the non-spread, rounded
morphology of cells treated with PP2 (Figure 14). RZ3 reduced the rate of random
haptotaxis (Figure 13B), however its inability to remain soluble may contribute to the
activity since many cells were trapped in an insoluble peptide web (data not shown). In
addition, AG73, a cell adhesion peptide derived from the alpha globular domain of
laminin-1 [97] had no migration-blocking activity in this assay.
HYD1 also was functional in inhibiting invasion toward immobilized laminin-5.
The activity of HYD1 to inhibit invasion was concentration dependent with
approximately 60% inhibition at a concentration of 250µg/ml (Figure 15A). The
scrambled variant, HYDS, was inactive at all concentrations tested. The HYD1 peptide
was able to inhibit invasion with similar potency as other known inhibitors of invasion
such as the Rho-kinase inhibitor, Y-27632 [158], as well as integrin-function blocking

70
Figure 14. Soluble HYD1 does not effect cell spreading on immobilized laminin-5 Spreading of PC3N cells was determined using differential interference contrast optics on an inverted Olympus IMT2 microscope. Following 1 hour adhesion to laminin-5 in serum-free conditions cells were treated with serum-free media alone (SFM), HYD1 (75mg/ml), a Src inhibitor, PP2 (10mM), or a Rho-kinase inhibitor, Y-27632 (25mM) in serum-free media.

71
Figure 15. HYD1 inhibits laminin-5 dependent cellular invasion. (A) Concentration-dependent inhibition of PC3N cell invasion toward laminin-5. Cells were added into the upper chamber of laminin-5 coated transwells for 1hour prior to treatment with 75, 100, 250, or 500 (µg/ml) HYD1 or HYDS in serum-free media as described in Experimental Procedures. (B) Comparison of HYD1 activity to other known inhibitors of invasion. Cells were added as in A and followed by treatment with 250µg/ml HYD1 or HYDS in serum-free media. The Rho-kinase inhibitor, Y-27632, was used at a concentration of 25µM in serum-free media. Integrin function-blocking antibodies P1B5 (α3), GoH3 (α6), and AIIB2 (β1) were used at a concentration of 10µg/ml and were incubated with cells for 15 minutes at room temperature prior to adding the cells into transwells. Error bars are standard deviation from the mean.

72 antibodies to α6, α3, and β1 (Figure 15B). These data indicate that HYD1 can block
random haptotaxis on laminin-5 and inhibit cellular invasion toward laminin-5.
HYD1 Induces Actin Cytoskeletal Remodeling and Cortactin Mediated Actin
Dynamics at the Cell Membrane
A critical component of cell motility dynamics is the regulation of the actin
cytoskeleton [159,160]. Given the immediate loss of motility after HYD1 treatment we
investigated the concurrent changes in actin organization. Cells treated for 30 minutes
with HYD1 displayed increased filamentous actin and an accumulation of actin at the cell
membrane with associated microspikes (Figure 16A and B-asterisk). This robust change
in the cytoskeletal architecture is not seen with treatment of the cells with scrambled
peptide, HYDS. Interestingly, although HYD1 generated the formation of stress fibers
(Figure 16B-arrows), the peptide did not increase Rho activity (data not shown)
suggesting Rho-independent signaling events are responsible for the phenotype.
Cortactin is a cortical actin binding protein that localizes at sites of dynamic
membrane activity [161]. Given the accumulation of actin and actin microspikes at the
cell membrane following HYD1 treatment, we hypothesized that this phenotype may be
mediated through cortactin. We show that cortactin is involved in HYD1 induced
accumulation of filamentous actin at the cell membrane by its colocalization with actin at
the cell membrane in cells treated with HYD1 for 30 minutes (Figure 16B Inset). In
addition, HYD1 induced an increase in tyrosine phosphorylation of cortactin over time
(Figure 17A). This is consistent with a concomitant increase in filamentous actin in the

73
Figure 16. HYD1 induces cytoskeletal reorganization on laminin-5. (A) HYD1 treatment causes global actin reorganization. PC3N cells were placed on laminin-5 coated coverslips for 1 hour followed by addition of HYD1 or HYDS (75µg/ml) in serum-free media for 30 minutes. Coverslips were fixed and stained for actin (green) and cortactin (red) as described in Materials and Methods. Asterisks designate points of microspike formation at the cell membrane. Closed arrowhead marks the cell enlarged in B. (B) HYD1 induces colocalization of cortactin with actin at the cell membrane. The cells marked in A were enlarged using Photoshop 7.0 software. The insets were created by using the auto level function in Photoshop on the boxed region of the cell membrane. Asterisks designate points of microspike formation at the cell membrane. Closed arrows designate areas containing stress fibers.

74
Figure 17. HYD1 induces phosphorylation of cortactin and alters membrane actin dynamics. (A) HYD1 induces phosphorylation of cortactin. PC3N cells were placed on laminin-5 for 1 hour followed by treatment with HYD1 or HYDS (75µg/ml) in serum-free media for 5, 10, 15, or 30 minutes before lysis. The phosphorylation state of cortactin was analyzed after immunoprecipitation with the phosphotyrosine antibody 4G10. (B) HYD1 induces F-actin assembly at the cell membrane. PC3N cells were added as described in A and treated with HYD1 or HYDS for 30 minutes. The distribution of F-actin in the membrane compared to the globular pool of actin was analyzed as described in Experimental Procedures. (C) Desitometry plot of results shown in B.

75 cell membrane fraction following treatment with HYD1 for 30 minutes (Figure 17B, C).
These data show that HYD1 stimulates cytoskeletal reorganization mediated in part by
signaling through cortactin.
HYD1 Interacts with αααα6 and αααα3 Integrins and Activates Integrin-Associated
Signaling
Given that cell migration on laminin-5 is mediated by α6 and α3 integrins
[152,162-165], we determined if either peptide interacted with α6 or α3 integrin
subunits. To define receptor specificity, each peptide was incubated with membrane
preparations from DU-145H prostate carcinoma cells and the resulting precipitate was
analyzed for integrin content. HYD1 interacted with complexes containing full-length
α6 and α3 integrins (Fig. 18A and B). RZ3 interacted with α3 integrin to a greater extent
than α6 integrin as judged by the amount of integrin retrieved. HYD1 was superior to
RZ3 in retrieving the α6 and α3 subunits. The scrambled derivative, HYDS, does not
interact with either alpha integrin subunit (Fig. 18A and B).
Upon engagement with the ECM, integrins are signal transduction receptors with
the capability of activating many intracellular protein kinases and influencing a wide
array of cellular processes [134]. One kinase linked to integrin activation and cell
motility is focal adhesion kinase (FAK), and phosphorylation of FAK on tyrosine 397 is
indicative of FAK activation [58,166]. Phosphorylation of mitogen activated protein
kinase kinase (MEK) on serine 298 is an adhesion dependent phosphorylation regulated
by p21-activated kinase (PAK) [136]. Using these two signals as biochemical markers

76
Figure 18. HYD1 interacts with αααα6 and αααα3 integrins. Affinity precipitation reactions were performed with HYD1, RZ3, or HYDS peptides on DU-145H cell membrane fractions as described in Experimental Procedures. (A) The precipitates were analyzed for α6 or (B) α3 integrin by SDS-PAGE electrophoresis followed by a western blot with AA6A antibody (α6 specific) or Antibody 1920 (α3 specific). ML (membrane lysate) was used to indicate the starting material.

77 for integrin activation, we determined if HYD1 or RZ3 could act as signaling ligands to
PC3N cells attached to BSA-coated plates. After 30 minutes of exposure, HYD1 induced
both activation of FAK and phosphorylation of MEK on serine 298 (Figure 19A Lane 2
and B Lane 2). RZ3 and the scrambled derivative, HYDS, and AG73 showed no activity
compared to cells treated with serum-free media alone.
HYD1 Enhances Signaling on Laminin-5 Resulting in Transient Activation of ERK
Cellular haptotaxis involves the dynamic regulation of signaling and cytoskeletal
components. Ultimately, signals elicited from integrin interaction with the ECM drive
the dynamic adhesive and cytoskeletal remodeling necessary for movement [56]. The
anti-migratory activity of HYD1 coupled with its ability to reorganize the cytoskeleton
prompted us to analyze the immediate effect on laminin-5 dependent pro-migratory
signals. As mentioned previously, cell migration on laminin-5 is coupled to the activity
of several kinases including FAK and p21-activated kinase (PAK) [147,152]. After 5
minutes of treatment, HYD1 increased the amount of activated FAK and elevated
phosphorylation of MEK on serine 298, which is mediated by PAK [136], compared to
baseline levels generated by haptotaxis on laminin-5 (Figure 20A and B Lanes 1 and 2).
Both of these signals had the greatest difference above baseline at 15 minutes post-
treatment (Figure 20A and B Lanes 3 and 4). At this time-point RZ3 had similar activity,
albeit less than HYD1, while AG73 did not show an effect (Figure 21A and B). Given
that PAK phosphorylation of MEK on serine 298 primes the MAPK pathway for
activation, [136,167] we investigated the activation of ERK in response to the peptides.

78
Figure 19. HYD1 activates integrin-associated signaling. PC3N cells were attached to BSA coated plates in the presence of serum-free media for 1 hour followed by the addition of peptides (75µg/ml) in serum-free media or serum-free media (SFM) alone for 30 minutes. Cell lysates were analyzed for phosphorylation of FAK Tyr-397 (A) and phosphorylation of MEK Serine 298 (B). Blots were stripped and reprobed for total FAK and MEK and the amount of activity was normalized by densitometry.
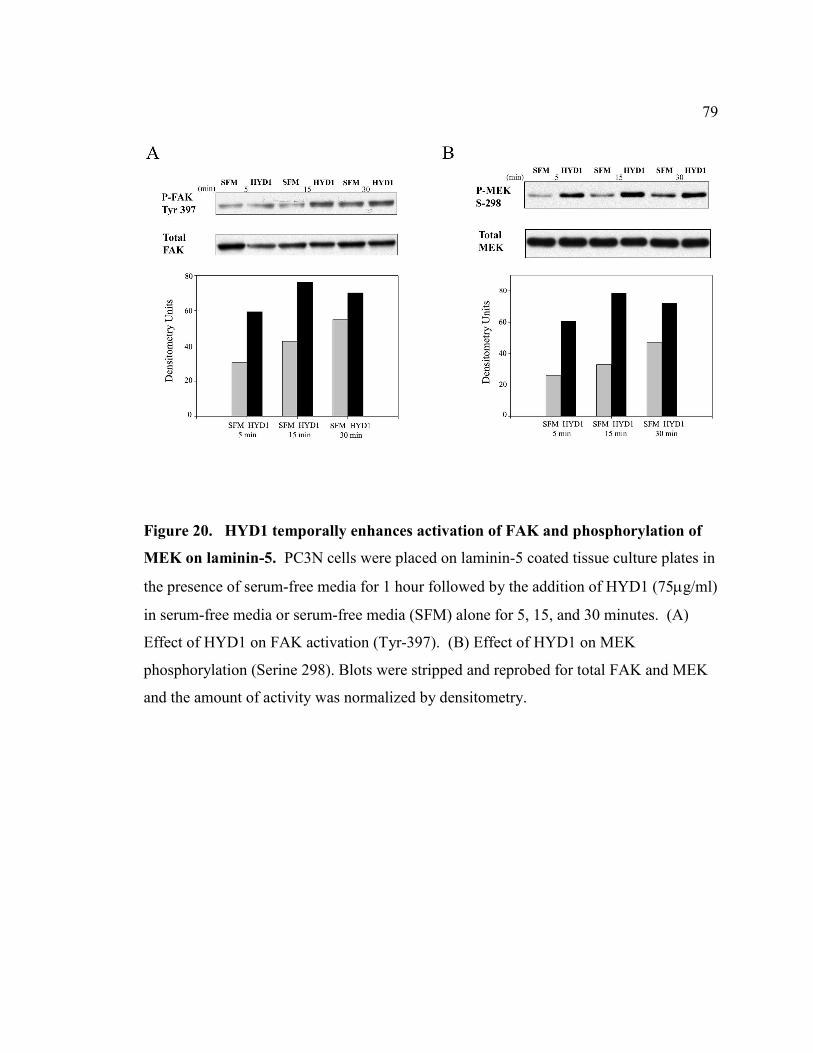
79
Figure 20. HYD1 temporally enhances activation of FAK and phosphorylation of MEK on laminin-5. PC3N cells were placed on laminin-5 coated tissue culture plates in the presence of serum-free media for 1 hour followed by the addition of HYD1 (75µg/ml) in serum-free media or serum-free media (SFM) alone for 5, 15, and 30 minutes. (A) Effect of HYD1 on FAK activation (Tyr-397). (B) Effect of HYD1 on MEK phosphorylation (Serine 298). Blots were stripped and reprobed for total FAK and MEK and the amount of activity was normalized by densitometry.
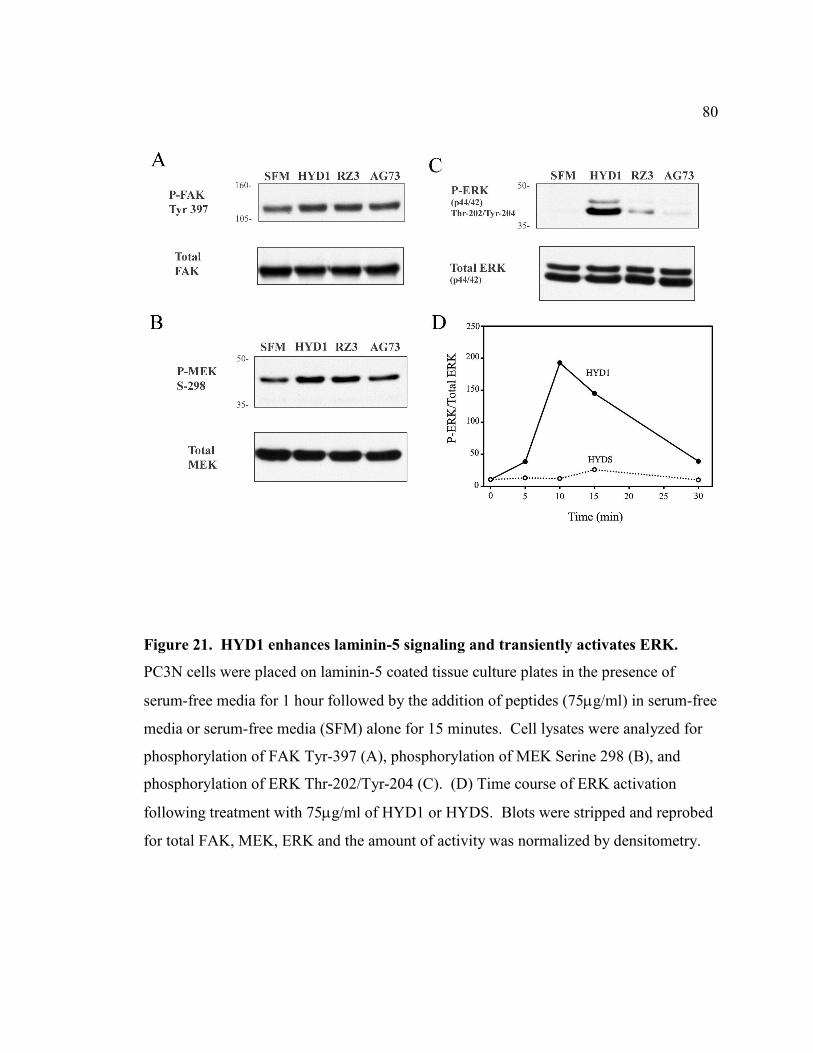
80
Figure 21. HYD1 enhances laminin-5 signaling and transiently activates ERK. PC3N cells were placed on laminin-5 coated tissue culture plates in the presence of serum-free media for 1 hour followed by the addition of peptides (75µg/ml) in serum-free media or serum-free media (SFM) alone for 15 minutes. Cell lysates were analyzed for phosphorylation of FAK Tyr-397 (A), phosphorylation of MEK Serine 298 (B), and phosphorylation of ERK Thr-202/Tyr-204 (C). (D) Time course of ERK activation following treatment with 75µg/ml of HYD1 or HYDS. Blots were stripped and reprobed for total FAK, MEK, ERK and the amount of activity was normalized by densitometry.

81 HYD1 induced activation of ERK (Figure 21C) that was maximal at 10 min post-
treatment and returned to baseline levels after 30 minutes (Figure 21D). Taken together
these data demonstrate that HYD1 disrupts the normal pro-migratory signaling dynamic
on laminin-5 and results in a transient activation of ERK.
Discussion
We have identified a D-amino acid peptide, HYD1 (kikmviswkg), which inhibits
tumor cell migration on laminin-5. HYD1 interacts with both α6 and α3-containing
integrin complexes. Thus, HYD1 interacts with the integrins α6β1, α6β4, and α3β1.
These are all receptors for laminin-5 [168] and although α3β1 has been documented as
the primary integrin receptor involved in mediating migration on laminin-5 by use of
function-blocking antibodies to α3 [152,162], evidence also suggests that α6 integrins
play a major regulatory role in motility on laminin-5 [163-165]. The receptor specificity
mediating the direct effects on motility, signaling, and the cytoskeleton will require
further investigation.
It is well known that integrin function-blocking antibodies or RGD-based
peptiomimetics can inhibit cell invasion by blockage of integrin/ECM interaction. HYD1
is novel in that its activity is not dependent on interrupting integrin/laminin-5
interactions. This suggests that the effects are induced through binding to available
integrin complexes on the surface rather than integrin complexes pre-engaged with the
ECM. Alternatively, our previous study [82] suggested that HYD1 interaction was
independent of the calcium-binding motif. Therefore, the peptide may effect both non-

82 engaged and pre-engaged integrin complexes. The post-ligand occupancy activity of
HYD1 is reinforced by the fact that cells remained attached and spread on laminin-5 after
peptide treatment. The inhibitory effect of the peptide was reversible by removing the
peptide and adding soluble laminin-5. This suggests that the migration-blocking activity
is not a lethal event and can be rescued by the normal ligand.
Several peptide sequences that bind α6β1 have been isolated from the laminin
α1 chain [97,169] as well as a peptide from CCN1, [170] a protein that is involved in
angiogenesis. Screening of a phage display library [171] has also isolated α6β1 peptide
ligand mimetics. These peptides do not reveal a consensus binding motif and do not
overlap with the sequence of HYD1. In addition, since HYD1 is a D-amino acid peptide
it might be structurally distinct from other L-amino acid ligand mimetics and therefore
not a good candidate for comparative purposes. The diversity of the ligand sequences for
α6β1 may allude to the fact that integrins are involved in a multitude of biological
processes and specific ligands could interact with distinct binding sites on integrins to
give the appropriate biological response.
Studies have shown that migration rates of cells are dependent on the organization
of the actin cytoskeleton and the presence of long, centrally distributed stress fibers
correlates with reduced migration rates [150,151,172]. HYD1 generates this type of
cytoskeletal morphology within 30 minutes, producing a global increase in filamentous
actin both at the membrane and throughout the cytoplasm. Cortactin, a regulator of
membrane actin dynamics, is also shown to be tyrosine phosphorylated in response to the
peptide and colocalized at the cell membrane with filamentous actin. Although the full

83 significance of cortactin phosphorylation has yet to be determined, tyrosine
phosphorylation is partially mediated by Src in response to cell matrix interactions [173].
This suggests that the cytoskeletal remodeling events at the cell membrane produced by
HYD1 are activated in part by integrin mediated signaling through cortactin.
Interestingly, HYD1 induced cytoskeletal reorganization occurred independent of
increased RhoA activity (data not shown), however, RhoA effectors can be modulated by
other signaling pathways such as the MAPK pathway [56,64]. Therefore, HYD1 may
elicit changes in cytoskeletal architecture directly through RhoA effectors rather than
increasing RhoA activity. Alternatively, it has been shown in keratinocytes that ablation
of α3β1 created a cytoskeletal reorganization event that resulted in enhanced stress fiber
formation [174]. The interaction of HYD1 with α6 and α3 integrins is consistent with
the idea that the cytoskeletal effects could be a consequence of inhibiting integrin
function independent of modulating actin dynamics through cellular signals.
Laminin-5 dependent motility depends on the activity of multiple cell signaling
molecules including FAK, PAK, PI3K, and the MAP-kinase pathway, and studies have
shown that inhibition of these mediators can reduce migration [151,152,157]. It is
somewhat surprising then that HYD1 could block migration yet stimulate signaling
through these pathways. However, studies with disintegrins have displayed similar
phenomena. For example, it has been shown that contortrostatin, a snake venom
disintegrin that inhibits cell motility, induces the tyrosine phosphorylation of FAK,
p130CAS, and activation of ERK2 [175-178]. Disintegrins have also generated actin
cytoskeletal reorganization that was dependent on tyrosine kinase activity [179].

84 Although HYD1 does not contain a conserved disintegrin domain, the similarity of
activity is remarkable.
In summary, we have identified an integrin-targeting peptide, HYD1, that blocks
tumor cell migration on laminin-5. Loss of cell migration involved intense cytoskeletal
remodeling events mediated in part by cortactin. HYD1 interacts with full-length α6 and
α3 integrins and disrupts the pro-migratory signaling dynamic induced from laminin-5
haptotaxis. Therefore HYD1 uncouples the induction of pro-migratory signals with the
appropriate cytoskeletal response necessary for motility. The dramatic effect of HYD1
on cell migration and invasion prompts for further study to determine if it can efficiently
block invasion and metastasis in vivo. In addition, we are determining the minimal
element for activity in this peptide by deletion analysis. The minimal element of HYD1
may provide insight into novel compounds for drug development. HYD1 is novel
because it does not have to compete with the native ligand for activity, suggesting that
integrin functions can be regulated independently from adhesion to the extracellular
matrix. This study may provide new insights into development of antagonists of specific
integrin functions to target cancer metastasis.

85 V. IDENTIFICATION OF THE MINIMAL ACTIVE ELEMENT OF
THE SYNTHETIC D-AMINO ACID PEPTIDE HYD1 (kikmviswkg)
Introduction
Multiple studies have isolated biologically active peptides from defined regions
within laminin chains and documented profound effects on biological events including
cell migration and metastasis [97,123,125,127,153-156]. The discovery of RGD, the
tripeptide sequence found in many adhesive proteins such as fibronectin and vitronectin
[180,181], has led to several studies showing that this cell adhesion peptide has anti-
invasive and anti-metastatic effects both in vitro and in vivo [182,183]. Peptides derived
from the laminin β1 chain, YIGSR, and α5 chain, RLVSYNGIIFFLK, have also been
effective at blocking experimental metastasis [127,153,184]. HYD1, kikmviswkg, has
shown potent anti-migratory effects in vitro and warrants further study in experimental
models of metastasis. HYD1 is a linear peptide consisting of ten D-amino acids.
Because HYD1 is relatively large in size compared to other bioactive cell adhesion
peptides used in clinical studies, it is important to determine if the amino acid motif
contains a minimal element that mediates the biological activity of the peptide before
testing in mouse models. This will help in developing strategies to increase peptide
bioavailability and provide insight into the synthesis of novel compounds to target tumor
cell migration and metastasis.
Previous groups that isolated cell adhesion peptides from the laminin α1 chain
successfully determined the minimum active sequences of these peptides by using N-

86 terminal and C-terminal deletion mutants of the peptides [124]. To determine the
minimal element of HYD1, five N-terminal and five C-terminal deletion mutants were
synthesized. In addition, alanine substitution was performed throughout the sequence of
HYD1 to identify amino acids that are critical for activity. Three tests of bioactivity were
applied to determine the minimal element of HYD1: 1) Tumor cell adhesion. 2) ERK
activation. 3) Inhibition of tumor cell migration on laminin-5.
Results
The Minimal Element that Mediates Cell Adhesion to Immobilized HYD1 is
xkmvixw
Previously we demonstrated that HYD1, when immobilized, promoted cell
adhesion at concentrations as low as 2µg/well. Maximal cell adhesion to HYD1 occurred
at 10µg/well [82]. Since mutants of HYD1 have different molecular weights, equal
molar concentrations of each mutant was used to determine if adhesive activity was
retained. Cell adhesion to 50µM of immobilized HYD1, HYDS, N- and C-terminal
mutants, and alanine-substituted HYD1 was determined (Figure 22). Adhesion activity
of HYD1 was reduced following deletion of the second N-terminal lysine (Figure 22
Mutant 14). Additional deletion from the N-terminus did not entirely eliminate the
adhesive capacity of HYD1 suggesting that the five C-terminal amino acids retained
some adhesion-promoting activity. Deletion from the C-terminus reduced activity starting
with the loss of the C-terminal lysine (Figure 22 Mutant 18). Loss of additional C-
terminal residues (Figure 22 Mutants 19, 20, 21) completely abolished the adhesive

87
Figure 22. Cell adhesion to immobilized HYD1 is mediated by N- and C-terminal regions of the peptide. The DU-H cells were allowed to attach to HYD1, HYDS, deletion mutants (12-21), and alanine-substituted (2-11) peptides coated in 96-well microtiter plates. Each peptide was used at a concentration of 50µM. The number of cells attached after 90 minutes was determined by absorbance at 570 nm. Data are the mean triplicates. Error bars = SD.

88 activity of HYD1. The alanine-substituted mutants of HYD1 did not alter the adhesive
capacity of the peptide (Figure 22 Mutants 2-11).
Data from the deletion mutants suggested that both N- and C-terminal regions of
the peptide were bioactive. This initial screen indicated a minimal element of kmviswk.
To further explore crucial residues within this peptide, four additional truncated mutants
of HYD1 were synthesized. Adhesion activity of the peptides, ikmviswk- ikmvisw-
kmviswk- kmvisw, were determined over a concentration range of zero to four-hundred
micromolar (Figure 23). Two of these peptides, ikmviswk and ikmvisw, supported tumor
cell adhesion in a concentration dependent manner with maximal adhesion occurring at a
concentration of 100µM. The peptides kmviswk and kmvisw did not have adhesive
activity over the concentration range tested. These data suggest that ikmvisw is the
active adhesive sequence within HYD1 (kikmviswkg).
Although alanine-substitution mutants of full-length HYD1 did not reveal critical
amino acids (Figure 22), we postulated that alanine substitution with the smaller peptide
would be informative. Alanine-substitution mutants of ikmvisw were synthesized. Cell
adhesion to these mutant peptides revealed that the serine and N-terminal isoleucine were
not critical elements for adhesion to immobilized ikmvisw (Figure 24A). However,
substitution of alanine for any remaining amino acid within ikmvisw resulted in loss of
peptide activity. Interestingly, this minimal sequence is also a conserved element within
the sequence of RZ3 (Figure 24B). These data show that the minimal element of HYD1
that mediates cell adhesion is xkmvixw.

89
Figure 23. The minimal sequence mediating cell adhesion to immobilized HYD1 is ikmvisw. The DU-H cells were allowed to attach to peptides coated in 96-well microtiter plates. Four peptides, ikmviswk (closed circle)- ikmvisw (open circle)- kmviswk (closed triangle)- kmvisw (open triangle), were used over a concentration range of 0 to 400µM. The number of cells attached after 90 minutes was determined by absorbance at 570 nm. Data are the mean triplicates. Error bars = SD.

90
Figure 24. The minimal element mediating cell adhesion to immobilized HYD1 is xkmvixw. (A) The DU-H cells were allowed to attach to 100µM of peptide coated in 96-well microtiter plates. Alanine-substitution peptides were generated from the parent peptide (ikmvisw) that was derived from HYD1 (kikmviswkg). The number of cells attached after 90 minutes was determined by absorbance at 570 nm. Data are the mean triplicates. Error bars = SD. (B) Table comparing HYD1 sequence and type of amino acids with other cell adhesion peptides obtained with a combinatorial screening approach [82].

91 Analysis of the Minimal Element of HYD1 Necessary for Activation of ERK
HYD1, when introduced as a soluble ligand, completely blocks random hapotaxis
on laminin-5 (Figure 13). Enhanced cellular signaling was coupled to the loss of cell
motility (Figure 20 and 21). HYD1 induces activation of ERK that is maximal at 10 min
post-treatment (Figure 21D). Using this signal as an indicator for bioactivity of HYD1,
we tested the truncated and alanine-substituted mutants of HYD1 for their ability to
activate ERK. Each peptide was used at a concentration of 50µM. Activation of ERK at
10 min post-treatment was conserved with alanine-substitution only at the N-terminal
lysine and C-terminal glycine (Figure 25). All other alanine-substitution resulted in
partial or complete loss of ERK activation, suggesting that the interior residues,
ikmviswk, are critical.
Deletion analysis of HYD1 revealed that both N- and C-terminal regions are
involved in activation of ERK (Figure 26). Specifically, the N-terminal 5 amino acid
residues, kikvm, and the C-terminal 5 amino acid residues, iswkg, alone induced partial
activation of ERK. The N-terminal region, kikvm, induced a stronger signal than iswkg.
In addition, some of the truncated mutants were able to partially activate ERK (Figure
26), however the amount of activation was substantially reduced in comparison to the
full-length peptide. In addition, mixing the two terminal regions of HYD1, kikmv and
iswkg, enhanced ERK activation compared to the response of these two peptides alone
(data not shown). Taken together, these data show that both the N- and C-terminal
regions of HYD1 contain an element that can induce ERK activation while alanine-
substitution

92
Figure 25. xikmviswkx is the minimal element of HYD1 required to activate ERK. PC3N cells were placed on laminin-5 coated tissue culture plates in the presence of serum-free media for 1 hour followed by the addition of HYD1 (kikmviswkg), alanine-substitution mutants of HYD1, or HYDS (wiksmkivkg) at a concentration of 50µM in serum-free media for 10 minutes. Cell lysates were analyzed for phosphorylation of ERK Thr-202/Tyr-204. Blots were stripped and reprobed for total ERK.

93
Figure 26. HYD1 induced ERK activation is mediated by N- and C-terminal regions of the peptide. PC3N cells were placed on laminin-5 coated tissue culture plates in the presence of serum-free media for 1 hour followed by the addition of HYD1 (kikmviswkg), truncated mutants of HYD1, or HYDS (wiksmkivkg) at a concentration of 50µM in serum-free media for 10 minutes. Cell lysates were analyzed for phosphorylation of ERK Thr-202/Tyr-204. Blots were stripped and reprobed for total ERK.

94 analysis of HYD1 suggests that the critical amino acid residues mediating this response
are xikmviswkx.
Analysis of the Minimal Element of HYD1 Necessary to Block Cell Migration on
Laminin-5
HYD1 completely blocks random haptotaxis on laminin-5 (Figure 13). This
response to the peptide is independent of disrupting adhesion to laminin-5 since the cells
were adhered to the ligand before treatment with the peptide. A wound-healing assay
was used to investigate the minimal element of HYD1 necessary to inhibit cell migration.
Alanine-substitution analysis of HYD1 revealed that the N-terminal lysine and the final
two C-terminal residues, a lysine and glycine, were not critical amino acids (Figure 27).
All other alanine-substitution mutants resulted in loss of peptide activity. These data
suggest that the active sequence for blocking cell migration is xikmviswxx.
Analysis using the truncated mutants of HYD1 showed that minimal deletion
from either end of the peptide resulted in at least partial loss of activity (Figure 28).
Using the control peptide, HYDS, to indicate 100% cell migration into the scratch,
several peptides including, ikmviswkg- kmviswkg- kikmviswk, were effective at
blocking cell migration by approximately 50%. These data are consistent with the results
found in the adhesion and signaling assays in that both the N- and C-terminal regions of
HYD1 are responsible for its bioactivity. Taken together, these data show that the
minimal element necessary to block cell migration on laminin-5 is xikmviswxx while

95
Figure 27. xikmviswxx is the minimal element necessary to block cell migration on laminin-5. PC3N cells were grown to a monolayer on laminin-5. Cells were treated with HYD1 (kikmviswkg), alanine-substitution mutants of HYD1, or HYDS (wiksmkivkg) at a concentration of 50µM in the presence of 1% FBS for 12 hours following the scratch. Data represent the percent of cells in a microscopic field migrating into the scratch. Three fields were analyzed per sample. Error bars = SD.

96
Figure 28. HYD1 inhibition of cell migration on laminin-5 is mediated by N- and C-terminal regions of the peptide. PC3N cells were grown to a monolayer on laminin-5. Cells were treated with HYD1 (kikmviswkg), truncated mutants of HYD1, or HYDS (wiksmkivkg) at a concentration of 50µM in the presence of 1% FBS for 12 hours following the scratch. Data represent the percent of cells in a microscopic field migrating into the scratch. Three fields were analyzed per sample. Error bars = SD.

97 loss of amino acids from either end will attenuate activity.
Discussion
Our previous data has characterized the cell adhesion peptide, HYD1
(kikmviswkg), as a bioactive peptide. Specifically, when immobilized, HYD1 acts as an
adhesion agonist by supporting tumor cell adhesion [82]. In addition, when introduced in
soluble form to prostate tumor cells adhered to laminin-5, HYD1 completely blocked
random haptotaxis (Figure 13). Coupled to the loss of cell motility, HYD1 enhanced cell
signaling on laminin-5 (Figure 20) resulting in transient activation of ERK (Figure 21).
The objective of this study was to determine the minimal element within the linear 10
amino acid structure of HYD1 that mediates these biological activities. We utilized both
N- and C-terminal deletion mutants of HYD1 in addition to alanine substitution analysis
of the peptide. Table 1 summarizes the adhesion, signaling, and anti-migratory effects of
these peptides.
Analysis of the peptide mutants suggests the minimal element necessary to
support prostate tumor cell adhesion was ikmvisw, however, we found that deletion of
amino acids from both the N- and C-terminal regions of the peptide severely attenuated
its ability to activate ERK and block cell migration on laminin-5. Alanine-substitution
revealed that xikmviswxx was the minimal element necessary to induce ERK activation
and block cell migration. This is consistent with the idea that the bioactive regions of
HYD1 could be sensitive to structural changes produced with amino acid deletion when

98
Table 1. Alanine-substituted and N- and C-terminal truncated peptides of HYD1 with their cell adhesion, cell signaling, and anti-migratory activity. Biological activity was scored on the following subjective scale: + +, activity comparable to HYD1; +, activity apparent but weaker; -, no activity.

99 HYD1 is used in soluble form, such as with the signaling and migration assays.
Nevertheless, the interior motif ikmvisw was critical in each biological assay tested.
A challenge to the use of peptide-based drugs is their poor bioavailability
[185,186]. Modifications to increase bioavailability and potency have been applied to the
cell adhesion peptides RGD and YIGSR with some success. These modifications include
peptide-cyclization, D-amino acid substitution, and conjugation to water-soluble
polymeric modifiers [187-189]. Other groups have made chemical modifications or used
the peptide backbone as template to create pseudo-peptide analogs with increased
stability and activity [190,191]. HYD1 is a linear peptide consisting of ten D-amino
acids. These data suggest that HYD1 may have a specific secondary structure in solution
given that its bioactivity was lost following minimal deletions from both the N- and C-
terminal ends of the peptide. Techniques such as circular dichroism and NMR can be
used to study the conformation and secondary structure of peptides [192-195]; these
techniques would be useful in determining if HYD1 has a structure/activity relationship.
In summary, the minimal element for bioactivity of the cell adhesion peptide
HYD1 (kikmviswkg) was determined. When immobilized as an adhesive substrate the
minimal element is xkmvixw. In solution, the minimal element necessary to block cell
migration and activate cell signaling through ERK is xikmviswxx. Our current focus is
testing the bioavailability and activity of HYD1 in mouse models of metastasis. HYD1
structural information in combination with these data could provide a template for the
creation of a small molecule mimetic of HYD1 that may prove useful in the clinical
arena.

100 VI. PRELIMINARY DATA: EFFECT OF THE SYNTHETIC D-
AMINO ACID PEPTIDE, HYD1 (kikmviswkg), ON INTEGRIN
LATERAL-ASSOCIATIONS
Introduction
A particularly remarkable activity of HYD1 is the ability to block cell migration
on a native extracellular matrix protein, laminin-5. Data presented has shown that the
inhibition of migration by HYD1 involves a dramatic remodeling of the actin
cytoskeleton resulting in increased stress fiber formation and actin colocalization with
cortactin at the cell membrane. In addition, HYD1 causes an altered signaling response
on laminin-5 resulting in transient activation of ERK. In Chapter III it is suggested that
HYD1 acts through a post-ligand occupancy event on the integrin that initiates the
cascade of events leading to loss of cell motility. It is possible that HYD1 interacts with
a common binding partner of α6 and α3 on the cell surface and initiates a change in
integrin function indirectly by altering the assocation of this protein with α6 or α3.
Integrins interact directly with other transmembrane proteins such as tetraspanins and
members of the immunoglobulin superfamily of proteins [196]. Integrin lateral-
association with these proteins can dramatically influence integrin function including
adhesion, signaling, and motility [197-209]. This chapter will discuss preliminary data
exploring if HYD1 alters α6 and α3 integrin lateral-associations on the cell surface.

101
Several studies have shown functional lateral-associations with the tetraspanin
CD151 and integrins [202-204,207,208]. CD151 forms stable associations with α3β1
and α6-containing integrins [206,210,211]. Specifically, these studies have shown that
association of CD151 with these integrins contributes to cell motility events in vitro
[203,204] and this association also correlates to increased tumor cell motility in vivo
[202]. Although the exact mechanism of tetraspanin/integrin regulation of cell motility
remains unclear, it seems likely that tetraspanin/integrin association is important in
integrin-dependent signaling events that lead to cell migration [206,212].
Another potential tetraspanin important in integrin-mediated cell migration is
CD81. Like CD151, CD81 associates with α3 and α6-containing integrins [211] and has
been shown to contribute to integrin-dependent migration events in vitro [203,213]. In
addition, a member of the immunoglobulin superfamily of proteins, EWI2, associates
with the tetraspanins CD81 and CD9 [214,215] and can regulate α3β1 dependent
functions on laminin-5 [198]. These data show that integrin function can be regulated
through primary or secondary interactions on the cell surface.
Preliminary data has shown that HYD1 interacts with specific integrin/tetraspanin
complexes on the cell surface. PC3N cells on laminin-5 treated with HYD1 had an
increase in the amount of CD81 associated with α6, however it had no effect on the
amount of CD151 associated. Biotinylation studies revealed that HYD1 induces the
association of a 60kD protein with α6 and α3-containing integrins as well as the
tetraspanin CD81. These data suggest that HYD1 changes the cell surface associations
with α6 and α3-containing integrins.

102 Results
HYD1 Interacts with CD151/Integrin Complexes
Our previous data showed that HYD1 interacted with α6 and α3 integrin subunits
(Figure 29A and B). To define receptor specificity, the peptides were incubated with
membrane preparations from DU-145H prostate carcinoma cells and the resulting
precipitate was analyzed for integrin content. Since α6 and α3-containing integrins form
stable associations with the tetraspanin CD151, [206,210,211] we wanted to determine if
the peptide was interacting with integrin/CD151 complexes. HYD1 depleted membrane
preparations of CD151 associated with α3 and α6 integrins, (Figure 29C and D)
suggesting that HYD1 targets integrin/tetraspainin complexes.
HYD1 Increases CD81 Association with the αααα6 Integrin
As mentioned above, integrin association with specific tetraspanin proteins can
effect multiple aspects of integrin function including adhesion, signaling, and motility
[197-209]. Association with the tetraspanins CD151 and CD81 have specifically been
shown to influence cell motility [203,213]. Given that HYD1 interacts with
integrin/tetraspanin complexes, we hypothesized that HYD1 may alter
integrin/tetraspanin association and thus provide an initiating event to the blockade of cell
motility on laminin-5. PC3N cells on laminin-5 were treated with HYD1 or the
scrambled derivative, HYDS, for 10 and 30 minutes before cell lysis. The samples were
immunoprecipitated for CD151 and CD81 and analyzed for α6 and α3 integrin content.

103
Figure 29. HYD1 interacts with CD151/Integrin Complexes. Affinity precipitation reactions were performed with HYD1, RZ3, or HYDS peptides on DU-145H cell membrane fractions as described in Experimental Procedures. (A) The precipitates were analyzed for α6 or (B) α3 integrin by SDS-PAGE electrophoresis and western blot. ML (membrane lysate) was loaded as a control. Supernatants from the affinity precipitation reactions in A and B were then used in an immunoprecipitation reaction for the tetraspanin CD151 (C) and (D) and blotted for α6 and α3 respectively. ML (membrane lysate) was used for the control immunoprecipitation reactions.

104
Figure 30. HYD1 does not change the amount of CD151 associated with αααα6 or αααα3-containing integrins. PC3N cells were placed on laminin-5 coated tissue culture plates in the presence of serum-free media for 1 hour followed by the addition of the peptides, HYD1 or HYDS (75µg/ml), in serum-free media for 10 and 30 minutes. Samples were immunoprecipitated (Brij 96) for α6 (J1B5) and CD151 (5C11) overnight at 4oC. The precipitates were analyzed for α6 by western blot. The blot was stripped and reprobed for α3.

105
HYD1 did not change the amount of CD151 associated with either α6 or α3 compared to
the control peptide HYDS (Figure 30). However, after 30 minutes of treatment, HYD1
caused an increase in the amount of CD81 associated with α6 and decreased the amount
associated with α3 (Figure 31). HYD1 induced clustering of α6 on laminin-5 (Figure
32), however, this was only seen in 2-3 cells/microscopic field. In addition, α6 did not
colocalize with CD81 at these adhesion sites.
HYD1 Alters the Lateral-Associations of αααα6 and αααα3-Integrins and the Tetraspanin
CD81
To determine if HYD1 disrupted or induced other changes in integrin associations
on the cell surface, we biotinylated PC3N cells on laminin-5 that were treated with
HYD1 or the scrambled derivative, HYDS, for 30 minutes. The samples were
immunoprecipitated for α6 and α3 integrins in addition to the tetraspanin CD81. HYD1
treatment induced a protein at approximately 60kD to co-immunoprecipitate with α6 and
α3 integrins, as well as CD81 (Figure 33). These data suggest that HYD1 changes the
cell-surface protein associations of α6 and α3-containing integrins and the tetraspanin
CD81.
Discussion
Integrin lateral-associations with tetraspanins and other cell surface molecules can
influence integrin function. Specifically, the tetraspanins CD151 and CD81 have been

106
Figure 31. HYD1 increases the amount of CD81 associated with αααα6-integrins and decreases the amount of CD81 associated with αααα3-integrins. PC3N cells were placed on laminin-5 coated tissue culture plates in the presence of serum-free media for 1 hour followed by the addition of the peptides, HYD1 or HYDS (75µg/ml), in serum-free media or serum-free media alone for 30 minutes. Samples were immunoprecipitated (Brij 96) for CD81 (JS64) overnight at 4oC. The precipitates were analyzed for α6 and α3 by western blot. WCL = whole cell lysate.

107
Figure 32. HYD1 induces αααα6 integrin clustering. PC3N cells were placed on laminin-5 coated coverslips for 1 hour followed by addition of HYD1 or HYDS (75µg/ml) in serum-free media for 30 minutes. Coverslips were fixed and stained for CD81 (JS64) (green) and Alpha-6 Integrin (J1B5) (red) as described in Experimental Procedures.

108
Figure 33. HYD1 alters the lateral-associations of αααα6 and αααα3-containing integrins and the tetraspanin CD81. PC3N cells were adhered to laminin-5 for 1 hour followed by addition of HYD1 or HYDS (75µg/ml) in serum-free media for 30 minutes. Cells were biotinylated before immunoprecipitation for α6 (J1B5), α3 (P1B5), and CD81 (JS64) as described in materials and methods. HYD1 induced a protein at approximately 60kD to co-immunoprecipitate with α6, and α3 integrins as well as CD81.

109 shown to have crucial roles in cell motility through interaction with integrins
[203,204,213]. Here we explored if the anti-migratory effect of HYD1 on laminin-5 was
dependent on disrupting or altering α6 or α3 associations with these tetraspanins. HYD1
was shown to interact with integrin complexes containing CD151, however HYD1 did
not change the amount of CD151 associated with α6 or α3-containing integrins.
Interestingly, HYD1 did alter integrin association with the tetraspanin CD81. Peptide
treatment for 30 minutes induced increased CD81 association with α6 while reducing the
amount associated with α3. It is tempting to speculate that a shift in the amount of CD81
interacting with α6 or α3 could be an initiating step in the loss of cell motility induced by
HYD1.
There are several possibilities that could explain how a change in
integrin/tetraspainin interaction could have such a profound effect on cell motility. First,
tetraspanins have been shown to modulate integrin-dependent signaling events such as
focal adhesion kinase activation that lead to cell migration [206,212]. In addition,
integrin/tetraspanin association has been shown to recruit signaling molecules such as
phosphatidylinositol 4-kinase [204] and protein kinase C [209]. It is possible that an
increase in CD81 association with α6 recruits a signaling complex that initiates a
signaling cascade that functionally blocks cell motility. Although most data show that
CD81/integrin interaction is a pro-migratory association [203,213], it is possible that a
pro-migratory signaling complex is recruited to α3β1 while an inhibitory complex is
recruited to α6-containing integrins after association with CD81. This is consistent with

110 the data that HYD1 reduces association of CD81 with α3-containing integrins while
increasing its association with α6.
A second possibility is that HYD1 induces a change in the protein content of the
tetraspanin protein microdomain (tetraspanin-web) associated with the integrin. This
could, as stated above, recruit specific signaling complex that could alter integrin
function. A group recently reported that EWI2, a cell surface protein of the
immunoglobulin superfamily and a major CD81 and CD9 partner [214], can regulate
α3β1 functions on laminin-5 [198]. In this study, overexpression of EWI2 suppressed
cell migration on laminin-5. The association of EWI2 with α3β1 was mediated through
the tetraspanins CD81 and CD9. This is particularly intriguing due to the fact that HYD1
induced an additional protein of about 60kD to associate with α6, α3, and CD81. The
approximate molecular weight of EWI2 is 55-70kD. These data suggest that HYD1
induces a change in the tetraspanin-web associated with α6 and α3-containing integrins.
Whether the new associative protein is EWI2 and if this new association is coupled to the
loss of motility is to be determined.

111 VII. CONCLUDING STATEMENTS
Cancer is a national health concern for men and women worldwide. Although
methods for diagnosis and treatment have improved with technological innovations,
cancer mortality rates remain significant. Metastases, rather than the primary tumor,
accounts for most cancer related deaths [1,7]. There is an intense need for new
therapeutics that target the processes involved in metastasis given that cancer diagnosis
often occurs at a relatively late stage in the disease. In order to develop novel
therapeutics, research must focus on determining essential molecules involved in
metastasis. This dissertation focused on the development and biological properties of
syntheic cell adhesion peptides. Using a combinatorial method, peptides were selected to
target α6 integrins. These peptide ligand mimetics were used to explore integrin biology
of cell adhesion and migration. This study has produced a novel integrin-targeting
peptide, HYD1 (kikmviswkg) that is capable of blocking both tumor cell adhesion to
extracellular matrices and cell migration of tumor cells adhered to laminin-5. These in
vitro data provide rationale to determine the activity of this peptide in animal models of
metastasis.
The biological activity of HYD1 characterizes it as having both agonist and
antagonist effects on integrin functions. Since the combinatorial screen was based on
functional cell ahesion, it is logical that the peptide works as an adhesion agonist. Two
peptides, RZ3 and HYD1, could both support tumor cell adhesion and inhibit adhesion to
extracellular matrix proteins. In addition, as an integrin adhesion ligand, a true agonist

112 would promote other integrin-mediated functions such as signal transduction, leading to
cell spreading and motility. However, HYD1 uncoupled integrin-mediated cell adhesion
from cell spreading and migratory events. The most likely cause is due to the fact that
the peptide did not initiate the identical signaling program as the native extracellular
matrix. Therefore, when immobilized, HYD1 is not sufficient as agonist for all integrin
functions. One explanation could be that the immobilized peptide lacks the proper
avidity for integrin activation. Alternatively, HYD1 could initiate a signaling response
that antagonizes particular integrin functions. It would be interesting to test if cell
spreading and migration would occur with cell adhesion to multivalent forms of HYD1.
HYD1 had antagonistic effects when introduced in soluble form to cells adhered
to laminin-5. Specifically, HYD1 blocked tumor cell random haptotaxis and invasion.
The initiating event for this response remains unknown, however the effects on
cytoskeletal reorganization and cell signaling mirrors what has been described for
disintegin blockade of cell migration [175-179]. This suggests that HYD1 has a direct
effect on integrins. Further study is needed to solidify the mechanism for HYD1
inhibition of cell motility.
A couple of observations provide insight into the mechanism of action for HYD1.
First, the activity of HYD1 is a post-ligand occupancy event; the loss of cell motility
induced by HYD1 is independent of disrupting cell adhesion to the native ligand. Indeed,
following treatment with HYD1, cells remain adhered and retain a “spread” morphology.
This is consistent with the idea that the peptide does not compete for the ligand binding
sites with laminin-5, suggesting that the peptide interacts with integrins at a distinct site.

113 It is reinforced by the fact that, unlike adhesion to native ECM proteins such as laminin-5
and Fibronectin, tumor cell adhesion to immobized HYD1 is not affected by the presence
of EDTA.
Integrin function is intimately linked to the ability to modulate receptor structure
[33,216]. It has been documented that integrin affinity states are modulated whether the
receptor is in a ligand-bound, primed, or inactive confirmation [32,33,216-218]. A major
tool to study the link between receptor shape and activity is conformational-dependent
monoclonal antibodies. Antibodies have been identified that modulate the ability of
integrins to bind ligand or recognize epitopes that represent active confirmations of
integrins [219-221]. The mechanism of the former is thought to occur through
stabilization of ligand-bound or unbound integrin confirmations following antibody
exposure. It is tempting to speculate that HYD1 interaction with integrins causes a
conformational change that is integrated into an anti-migratory confirmation.
Conformational-dependent antibodies may prove useful to identify if HYD1 acitivty is
linked to integrin conformational shifts.
Chapter V. introduced the importance of integrin lateral-associations in mediating
integrin functions including adhesion, signaling, and motility [197-209]. An alternative
hypothesis should include that HYD1 disrupts or changes the protein milleu in the
integrin-associated tetraspanin web. Any such change could maintain integrin adhesion
while blocking cell motility through initiation of an inhibitory signal. This hypothesis
might also explain potential promiscuity of HYD1 in blocking cell migration on different
matrices. Preliminary data has shown that HYD1 is equally effective in blocking cell

114 motility of HT-1080 cells on Fibronectin (data not shown). This would seem confusing
since the peptides were screened for specificity to Laminin-receptors, however it is
possible that HYD1 interacts at a conserved site on alpha-subunits that is crucial in
mediating integrin lateral-associations. In this case, HYD1 would be expected to have
anti-migratory activity on a wide array of matrix proteins. This phenomenon could also
be explained by integrin cross-talk, meaning that HYD1 ligation of Laminin receptors
functionally regulates other integrin receptors through “inside-out” signaling.
A second observation is that although random haptotaxis is completely blocked,
cell surfaces remain active after treatment with HYD1. This is consistent with the data
that HYD1 induces actin cytoskeletal reorganization and cell signaling through focal
adhesion kinase and the extracellular signal-regulated protein kinase pathway. Focal
adhesion and actin dynamics are crucial regulators of cell motility [56], with integrins
serving as the molecular “feet” of a cell and the site of formation for adhesion complexes
[222]. Since membrane ruffling occurs yet existing adhesions seem to remain static,
HYD1 could influence the formation and/or disassembly of focal adhesions.
Focal adhesion dymanics is an essential part of cell locomotion by a reiterative
process involving the formation of new matrix adhesions at the leading edge coupled
with focal adhesion disassembly and retraction at the trailing end [56,222]. Observation
of cells treated with HYD1 suggests two possibilities. First, tumor cells treated with
HYD1 have active membrane surfaces, including membrane ruffling, yet the cells do not
seem to create viable adhesion sites from these protrusive events. This suggests a defect
in focal complex assembly at the leading edge or an inability of focal complexes to

115 mature into focal adhesions. Using live-cell imaging and flourescently labeled focal
adhesion components, a recent study has determined the rate constants for focal adhesion
disassembly [223]. Application of this technique would offer valuable insight as to
whether HYD1-treated cells disrupted the proper kinetics of adhesion assembly at the
leading edge. In addition, another group has characterized the adhesion protein zyxin as
a definitive marker for a mature focal adhesion [224]. Analysis of zyxin incorporation to
adhesions proximal to the leading edge would help determine if the defect was linked to
adhesion maturation.
Tumor cells treated with HYD1 do not seem to reorganize existing adhesions that
tether the cell body to the matrix. This suggests a loss in focal adhesion turnover
resulting in static adhesion sites. Focal ashesion disassembly is linked to the activity of
many signaling proteins and proteases [56]. The most direct approach to study the
regulation of mature focal adhesions is by applying the technology described above to
observe the kinetics of adhesion proteins with live-cell imaging. A significant change in
the half-life of mature adhesions, that is those distinct from the leading edge, would
suggest that HYD1 induces a defect in adhesion remodeling. Figure 34 is a schematic
representing the possible mechanisms for the antimigratory activity of HYD1.
Cancer therapeutics has entered an era of molecular targeting. New agents are
being developed to target specific molecular components important to tumor progression
and survival. Signal transduction pathways such as the extracellular signal-regulated
protein kinase/mitogein-activated protein kinase (ERK/MAPK) pathway have shown to

116
Figure 34. Potential mechanisms for the anti-migratory activity of HYD1. This model illustrates the hypotheses that HYD1 may influence integrin lateral-associations and/or focal adhesion dynamics to prevent cell migration.

117 be important for the pathogenesis of several cancers and inhibitors to specific molecules
in this pathway have been developed and are in clinical trials [225,226]. Other agents
have been developed for specific abnormalities. STI-571, an inhibitor of BCR-ABL
tyrosine kinase, is used to treat chronic myeloid leukemia [227,228]. Monoclonal
antibody therapy has also been employed with some success. Rituxan, an anti-CD-20
monoclonal antibody, has been approved for the treatment of B-cell lymphoma
[229,230]. Herceptin, a monoclonal antibody to the HER2-neu receptor has been
effective in treating certain types of breast cancer [231,232]. It is conceivable to imagine
that cancer therapy will evolve into individualized therapy, treatment regimens will be
designed based on the molecular characteristics of individual tumors.
Integrins are attractive as a therapeutic target because they are sentinel molecules
for a plethora of cellular functions critical to cancer progression [22,32]. This
characteristic is particularly appealing in regard to cancer metastasis because integrins
serve directly as a mediator for the adhesion events necessary in metastasis while also
integrating information from the extracellular environment into cellular signals that are
required for cell motility and survival at distant sites in the body. There are currently
several integrin inhibitors under investigation for cancer therapy. Antagonists to αvβ3
and αvβ5 have been developed as antibody and peptide-based inhibitors to target
angiogenesis and metastasis [233,234]. Vitaxin, a humanized αvβ3 antibody, is currently
in Phase II trials [235-238]. Cilengitide, a peptide inhibitor of αvβ3/αvβ5, is in Phase II
trial for advanced solid tumors and has shown promise for use in combination therapy
[239-242]. In addition, other αvβ3 and α5β1-blocking peptides have been documented

118 but have not yet entered clinical trials [243,244]. A humanized antibody has also been
developed for α5β1 integrin to target angiogenesis and is entering a Phase I trial
(www.pdl.com). Although the current integrin-based therapeutics are promising, they are
focused on αv and α5 integrin pairs. Antagonists to other integrins, specifically the
laminin receptors, may prove to be equally beneficial.
The integrins α6β1, α3β1, and α6β4 are laminin receptors [110,111]. These
integrins are associated with epithelial tumor progression in prostate, breast, colon,
pancreatic carcinomas, head and neck tumors, and melanoma [23,112-115,245].
Combinatorial screening, such as the one-bead-one-compound approach, has been
successful in identifying and characterizing cell adhesion peptides [84] with potential
antagonistic effects on the targeted cell surface receptor. An argument against the use of
peptide-based drugs is their poor bioavailability [185,186,234]. However, modifications
to increase bioavailability and potency have been applied to the cell adhesion peptides
RGD and YIGSR with some success [187-189]. In addition, using the peptide backbone
as a template, chemical modifications can create pseudo-peptide analogs with increased
stability and activity [190,191]. The studies shown in chapter IV should aid in the
modifcation of HYD1 into a small molecule mimetic, increasing its potential use as a
novel drug.
The work presented in this dissertation supports the use of synthetic cell adhesion
peptides as a means to study cell biology and develop novel therapeutics. The priority for
further development of HYD1 is determining in vivo activity. Both spontaneous and
experimental mouse models of metastasis should be employed to identify if the peptide

119 has preferential activity to early or late stages in metastasis [1,246]. After determining
therapeutic potential by these measures, studies of bioavailability will help clarify if
peptide modification is necessary before testing in the clinical arena.

120 REFERENCES
1. Chambers, A.F., Groom, A.C. and MacDonald, I.C. (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer, 2, 563-72.
2. Fidler, I.J. (2003) The pathogenesis of cancer metastasis: the 'seed and soil'
hypothesis revisited. Nat Rev Cancer, 3, 453-8. 3. Fidler, I.J. (1999) Critical determinants of cancer metastasis: rationale for therapy.
Cancer Chemother Pharmacol, 43 Suppl, S3-10. 4. Chambers, A.F., Naumov, G.N., Varghese, H.J., Nadkarni, K.V., MacDonald, I.C.
and Groom, A.C. (2001) Critical steps in hematogenous metastasis: an overview. Surg Oncol Clin N Am, 10, 243-55, vii.
5. Woodhouse, E.C., Chuaqui, R.F. and Liotta, L.A. (1997) General mechanisms of
metastasis. Cancer, 80, 1529-37. 6. Folkman, J. (1992) The role of angiogenesis in tumor growth. Semin Cancer Biol,
3, 65-71. 7. Chambers, A.F., MacDonald, I.C., Schmidt, E.E., Morris, V.L. and Groom, A.C.
(2000) Clinical targets for anti-metastasis therapy. Advances in Cancer Research,79, 91-121.
8. Society, A.C. (2005) Cancer Facts and Figures 2005. American Cancer Society
Atlanta, Ga.
9. Routh, J.C. and Leibovich, B.C. (2005) Adenocarcinoma of the prostate: epidemiological trends, screening, diagnosis, and surgical management of
localized disease. Mayo Clin Proc, 80, 899-907. 10. Parkin, D.M., Bray, F., Ferlay, J. and Pisani, P. (2005) Global cancer statistics,
2002. CA Cancer J Clin, 55, 74-108. 11. Hankey, B.F., Feuer, E.J., Clegg, L.X., Hayes, R.B., Legler, J.M., Prorok, P.C.,
Ries, L.A., Merrill, R.M. and Kaplan, R.S. (1999) Cancer surveillance series: interpreting trends in prostate cancer--part I: Evidence of the effects of screening in recent prostate cancer incidence, mortality, and survival rates. J Natl Cancer Inst, 91, 1017-24.

121 12. Johansson, J.E., Holmberg, L., Johansson, S., Bergstrom, R. and Adami, H.O.
(1997) Fifteen-year survival in prostate cancer. A prospective, population-based study in Sweden. Jama, 277, 467-71.
13. Chodak, G.W., Thisted, R.A., Gerber, G.S., Johansson, J.E., Adolfsson, J., Jones,
G.W., Chisholm, G.D., Moskovitz, B., Livne, P.M. and Warner, J. (1994) Results of conservative management of clinically localized prostate cancer. N Engl J Med,330, 242-8.
14. Adolfsson, J., Steineck, G. and Hedlund, P.O. (1997) Deferred treatment of
clinically localized low-grade prostate cancer: actual 10-year and projected 15-year follow-up of the Karolinska series. Urology, 50, 722-6.
15. Johansson, J.E., Andren, O., Andersson, S.O., Dickman, P.W., Holmberg, L.,
Magnuson, A. and Adami, H.O. (2004) Natural history of early, localized prostate cancer. Jama, 291, 2713-9.
16. Banyard, J. and Zetter, B.R. (1998) The role of cell motility in prostate cancer.
Cancer Metastasis Rev, 17, 449-58. 17. Ozen, M. and Pathak, S. (2000) Genetic alterations in human prostate cancer: a
review of current literature. Anticancer Res, 20, 1905-12. 18. Pilat, M.J., Kamradt, J.M. and Pienta, K.J. (1998) Hormone resistance in prostate
cancer. Cancer Metastasis Rev, 17, 373-81. 19. Dong, J.T., Isaacs, W.B. and Isaacs, J.T. (1997) Molecular advances in prostate
cancer. Curr Opin Oncol, 9, 101-7. 20. Hynes, R.O. (1992) Integrins: versatility, modulation, and signaling in cell
adhesion. Cell, 69, 11-25. 21. Hood, J.D. and Cheresh, D.A. (2002) Role of integrins in cell invasion and
migration. Nat Rev Cancer, 2, 91-100. 22. Jin, H. and Varner, J. (2004) Integrins: roles in cancer development and as
treatment targets. Br J Cancer, 90, 561-5. 23. Cress, A.E., Rabinovitz, I., Zhu, W. and Nagle, R.B. (1995) The alpha 6 beta 1
and alpha 6 beta 4 integrins in human prostate cancer progression. Cancer Metastasis Rev, 14, 219-28.

122 24. Nagle, R.B., Hao, J., Knox, J.D., Dalkin, B.L., Clark, V. and Cress, A.E. (1995)
Expression of hemidesmosomal and extracellular matrix proteins by normal and malignant human prostate tissue. Am J Pathol, 146, 1498-507.
25. Fornaro, M., Tallini, G., Bofetiado, C.J., Bosari, S. and Languino, L.R. (1996)
Down-regulation of beta 1C integrin, an inhibitor of cell proliferation, in prostate carcinoma. Am J Pathol, 149, 765-73.
26. Perlino, E., Lovecchio, M., Vacca, R.A., Fornaro, M., Moro, L., Ditonno, P.,
Battaglia, M., Selvaggi, F.P., Mastropasqua, M.G., Bufo, P. and Languino, L.R. (2000) Regulation of mRNA and protein levels of beta1 integrin variants in human prostate carcinoma. Am J Pathol, 157, 1727-34.
27. Putz, E., Witter, K., Offner, S., Stosiek, P., Zippelius, A., Johnson, J., Zahn, R.,
Riethmuller, G. and Pantel, K. (1999) Phenotypic characteristics of cell lines derived from disseminated cancer cells in bone marrow of patients with solid epithelial tumors: establishment of working models for human micrometastases. Cancer Res, 59, 241-8.
28. Hassan, M.O. and Maksem, J. (1980) The prostatic perineural space and its
relation to tumor spread: an ultrastructural study. Am J Surg Pathol, 4, 143-8. 29. Villers, A., McNeal, J.E., Redwine, E.A., Freiha, F.S. and Stamey, T.A. (1989)
The role of perineural space invasion in the local spread of prostatic adenocarcinoma. J Urol, 142, 763-8.
30. Stephens, H., Bendayan, M. and Gisiger, V. (1985) Simultaneous labelling of
basal lamina components and acetylcholinesterase at the neuromuscular junction. Histochem J, 17, 1203-20.
31. Haraida, S., Nerlich, A.G., Bise, K., Wiest, I. and Schleicher, E. (1992)
Comparison of various basement membrane components in benign and malignant peripheral nerve tumours. Virchows Arch A Pathol Anat Histopathol, 421, 331-8.
32. Hynes, R. (2002) Integrins: bidirectional, allosteric signaling machines. Cell, 110,
673-87. 33. Liddington, R.C. (2002) Integrin Activation Takes Shape. Journal of Cell
Biology, 158, 833-839. 34. Giancotti, F.G. and Ruoslahti, E. (1999) Integrin signaling. Science, 285, 1028-
32.

123 35. Green, L.J., Mould, A.P. and Humphries, M.J. (1998) The integrin beta subunit.
International Journal of Biochemistry & Cell Biology, 30, 179-84. 36. Hemler, M. (1999) Integrin Guidebook. Oxford Publishing. 37. Adams, J.C. (2002) Regulation of protrusive and contractile cell-matrix contacts.
Journal of Cell Science, 115, 257-65. 38. Wehrle-Haller, B. (2002) The Inner Lives of Focal Adhesions. Trends in Cell
Biology, 12, 382-389. 39. Xiong, J.P., Stehle, T., Diefenbach, B., Zhang, R., Dunker, R., Scott, D.L.,
Joachimiak, A., Goodman, S.L. and Arnaout, M.A. (2001) Crystal structure of the extracellular segment of integrin alpha Vbeta3. [see comments.]. Science, 294,339-45.
40. O'Toole, T.E., Mandelman, D., Forsyth, J., Shattil, S.J., Plow, E.F. and Ginsberg,
M.H. (1991) Modulation of the affinity of integrin alpha IIb beta 3 (GPIIb-IIIa) by the cytoplasmic domain of alpha IIb. Science, 254, 845-7.
41. Hemler, M.E. (1998) Integrin associated proteins. Current Opinion in Cell
Biology, 10, 578-85. 42. Schwartz, M.A. (2001) Integrin signaling revisited. Trends in Cell Biology, 11,
466-70. 43. Horwitz, A.R. and Parsons, J.T. (1999) Cell migration--movin' on. [letter;
comment.]. Science, 286, 1102-3. 44. Boudreau, N. and Bissell, M.J. (1998) Extracellular matrix signaling: integration
of form and function in normal and malignant cells. Current Opinion in Cell Biology, 10, 640-6.
45. Ruiz, P., Dunon, D., Sonnenberg, A. and Imhof, B.A. (1993) Suppression of
mouse melanoma metastasis by EA-1, a monoclonal antibody specific for alpha 6 integrins. [erratum appears in Cell Adhes Commun 1993 Sep;1(2):following 190.]. Cell Adhesion & Communication, 1, 67-81.
46. Parsons, J.T., Martin, K.H., Slack, J.K., Taylor, J.M. and Weed, S.A. (2000) Focal
adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene, 19, 5606-13.
47. Small, J.V. (2002) The lamellipodium: where motility begins. Trends in Cell
Biology, 12, 112-120.

124 48. Ridley, A.J. (2001) Rho family proteins: coordinating cell responses. Trends in
Cell Biology, 11, 471-7. 49. Zamir, E., Katz, M., Posen, Y., Erez, N., Yamada, K.M., Katz, B.Z., Lin, S., Lin,
D.C., Bershadsky, A., Kam, Z. and Geiger, B. (2000) Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nature Cell Biology, 2, 191-6.
50. Nobes, C.D. and Hall, A. (1995) Rho, rac, and cdc42 GTPases regulate the
assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell, 81, 53-62.
51. Rottner, K., Behrendt, B., Small, J.V. and Wehland, J. (1999) VASP dynamics
during lamellipodia protrusion. Nature Cell Biology, 1, 321-2. 52. Pantaloni, D., Boujemaa, R., Didry, D., Gounon, P. and Carlier, M.F. (2000) The
Arp2/3 complex branches filament barbed ends: functional antagonism with capping proteins. Nature Cell Biology, 2, 385-91.
53. Blanchoin, L., Amann, K.J., Higgs, H.N., Marchand, J.B., Kaiser, D.A. and
Pollard, T.D. (2000) Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature, 404, 1007-11.
54. Miki, H., Suetsugu, S. and Takenawa, T. (1998) WAVE, a novel WASP-family
protein involved in actin reorganization induced by Rac. EMBO Journal, 17,6932-41.
55. Kimura, K. (1996) Regulation of myosin phosphatase by Rho and Rho-associated
kinase (Rho-kinase). [see comments.]. Science, 273, 245-8. 56. Carragher, N.O. and Frame, M.C. (2004) Focal adhesion and actin dynamics: a
place where kinases and proteases meet to promote invasion. Trends Cell Biol, 14,241-9.
57. Guan, J.L., Trevithick, J.E. and Hynes, R.O. (1991) Fibronectin/integrin
interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell Regulation, 2, 951-64.
58. Calalb, M.B., Polte, T.R. and Hanks, S.K. (1995) Tyrosine phosphorylation of
focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Molecular & Cellular Biology, 15, 954-63.
59. Schlaepfer, D.D., Hauck, C.R. and Sieg, D.J. (1999) Signaling through focal
adhesion kinase. Progress in Biophysics & Molecular Biology, 71, 435-78.

125 60. Sieg, D.J., Hauck, C.R. and Schlaepfer, D.D. (1999) Required role of focal
adhesion kinase (FAK) for integrin-stimulated cell migration. Journal of Cell Science, 112, 2677-91.
61. Cary, L.A., Chang, J.F. and Guan, J.L. (1996) Stimulation of cell migration by
overexpression of focal adhesion kinase and its association with Src and Fyn. Journal of Cell Science, 109, 1787-94.
62. Gilmore, A.P. and Romer, L.H. (1996) Inhibition of focal adhesion kinase (FAK)
signaling in focal adhesions decreases cell motility and proliferation. Molecular Biology of the Cell, 7, 1209-24.
63. Klemke, R.L., Cai, S., Giannini, A.L., Gallagher, P.J., de Lanerolle, P. and
Cheresh, D.A. (1997) Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol, 137, 481-92.
64. Fincham, V.J., James, M., Frame, M.C. and Winder, S.J. (2000) Active
ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. Embo J, 19, 2911-23.
65. Pawlak, G. and Helfman, D.M. (2002) MEK mediates v-Src-induced disruption of
the actin cytoskeleton via inactivation of the Rho-ROCK-LIM kinase pathway. JBiol Chem, 277, 26927-33.
66. Glading, A., Chang, P., Lauffenburger, D.A. and Wells, A. (2000) Epidermal
growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J Biol Chem, 275, 2390-8.
67. Glading, A., Bodnar, R.J., Reynolds, I.J., Shiraha, H., Satish, L., Potter, D.A.,
Blair, H.C. and Wells, A. (2004) Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol, 24, 2499-512.
68. Cuevas, B.D., Abell, A.N., Witowsky, J.A., Yujiri, T., Johnson, N.L., Kesavan,
K., Ware, M., Jones, P.L., Weed, S.A., DeBiasi, R.L., Oka, Y., Tyler, K.L. and Johnson, G.L. (2003) MEKK1 regulates calpain-dependent proteolysis of focal adhesion proteins for rear-end detachment of migrating fibroblasts. Embo J, 22,3346-55.
69. Liu, R., Enstrom, A.M. and Lam, K.S. (2003) Combinatorial peptide library
methods for immunobiology research. Exp Hematol, 31, 11-30.

126 70. Lam, K.S., Salmon, S.E., Hersh, E.M., Hruby, V.J., Kazmierski, W.M. and
Knapp, R.J. (1991) A new type of synthetic peptide library for identifying ligand-binding activity. Nature, 354, 82-4.
71. Lam, K.S., Lebl, M. and Krchnak, V. (1997) The "One-Bead-One-Compound"
Combinatorial Library Method. Chem Rev, 97, 411-448. 72. Cwirla, S.E., Peters, E.A., Barrett, R.W. and Dower, W.J. (1990) Peptides on
phage: a vast library of peptides for identifying ligands. Proc Natl Acad Sci U S A, 87, 6378-82.
73. Devlin, J.J., Panganiban, L.C. and Devlin, P.E. (1990) Random peptide libraries:
a source of specific protein binding molecules. Science, 249, 404-6. 74. Rodi, D.J., Makowski, L. and Kay, B.K. (2002) One from column A and two
from column B: the benefits of phage display in molecular-recognition studies. Curr Opin Chem Biol, 6, 92-6.
75. Trepel, M., Arap, W. and Pasqualini, R. (2002) In vivo phage display and
vascular heterogeneity: implications for targeted medicine. Curr Opin Chem Biol,6, 399-404.
76. Buhl, L., Szecsi, P.B., Gisselo, G.G. and Schafer-Nielsen, C. (2002) Surface
immunoglobulin on B lymphocytes as a potential target for specific peptide ligands in chronic lymphocytic leukaemia. Br J Haematol, 116, 549-54.
77. Renschler, M.F., Bhatt, R.R., Dower, W.J. and Levy, R. (1994) Synthetic peptide
ligands of the antigen binding receptor induce programmed cell death in a human B-cell lymphoma. Proc Natl Acad Sci U S A, 91, 3623-7.
78. Lam, K.S., Lou, Q., Zhao, Z.G., Smith, J., Chen, M.L., Pleshko, E. and Salmon,
S.E. (1995) Idiotype specific peptides bind to the surface immunoglobulins of two murine B-cell lymphoma lines, inducing signal transduction. Biomed Pept Proteins Nucleic Acids, 1, 205-10.
79. Assa-Munt, N., Jia, X., Laakkonen, P. and Ruoslahti, E. (2001) Solution
structures and integrin binding activities of an RGD peptide with two isomers. Biochemistry, 40, 2373-8.
80. Koivunen, E., Wang, B. and Ruoslahti, E. (1995) Phage libraries displaying cyclic
peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Biotechnology (N Y), 13, 265-70.

127 81. Pennington, M.E., Lam, K.S. and Cress, A.E. (1996) The use of a combinatorial
library method to isolate human tumor cell adhesion peptides. Mol Divers, 2, 19-28.
82. DeRoock, I.B., Pennington, M.E., Sroka, T.C., Lam, K.S., Bowden, G.T., Bair,
E.L. and Cress, A.E. (2001) Synthetic peptides inhibit adhesion of human tumor cells to extracellular matrix proteins. Cancer Res, 61, 3308-13.
83. Lau, d.h., Guo, l.l, Liu, r.w., Song, a.m., Shao, c.k., Lam, k.s. (2002) Identifying
peptide ligands for cell surface receptors using cell-growth-on-bead-assay and one-bead-one-compound combinatorial library. Biotechnology Letters, 24, 497-500.
84. Aina, O.H., Marik, J., Liu, R., Lau, D.H. and Lam, K.S. (2005) Identification of
novel targeting peptides for human ovarian cancer cells using "one-bead one-compound" combinatorial libraries. Mol Cancer Ther, 4, 806-13.
85. Parmley, S.F. and Smith, G.P. (1988) Antibody-selectable filamentous fd phage
vectors: affinity purification of target genes. Gene, 73, 305-18. 86. Fukuda, M.N., Ohyama, C., Lowitz, K., Matsuo, O., Pasqualini, R., Ruoslahti, E.
and Fukuda, M. (2000) A peptide mimic of E-selectin ligand inhibits sialyl Lewis X-dependent lung colonization of tumor cells. Cancer Res, 60, 450-6.
87. Romanov, V.I., Durand, D.B. and Petrenko, V.A. (2001) Phage display selection
of peptides that affect prostate carcinoma cells attachment and invasion. Prostate,47, 239-51.
88. Zhang, J., Spring, H. and Schwab, M. (2001) Neuroblastoma tumor cell-binding
peptides identified through random peptide phage display. Cancer Lett, 171, 153-64.
89. Houghten, R.A., Pinilla, C., Blondelle, S.E., Appel, J.R., Dooley, C.T. and
Cuervo, J.H. (1991) Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature, 354, 84-6.
90. Furka, A., Sebestyen, F., Asgedom, M. and Dibo, G. (1991) General method for
rapid synthesis of multicomponent peptide mixtures. Int J Pept Protein Res, 37,487-93.
91. Hamamoto, K., Kida, Y., Zhang, Y., Shimizu, T. and Kuwano, K. (2002)
Antimicrobial activity and stability to proteolysis of small linear cationic peptides with D-amino acid substitutions. Microbiol Immunol, 46, 741-9.

128 92. Van Regenmortel, M.H. and Muller, S. (1998) D-peptides as immunogens and
diagnostic reagents. Curr Opin Biotechnol, 9, 377-82. 93. Rabinovitz, I., Nagle, R.B. and Cress, A.E. (1995) Integrin alpha 6 expression in
human prostate carcinoma cells is associated with a migratory and invasive phenotype in vitro and in vivo. Clin Exp Metastasis, 13, 481-91.
94. Tran, N.L., Nagle, R.B., Cress, A.E. and Heimark, R.L. (1999) N-Cadherin
expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion withStromal cells. Am J Pathol, 155, 787-98.
95. Breitkreutz, D., Schoop, V.M., Mirancea, N., Baur, M., Stark, H.J. and Fusenig,
N.E. (1998) Epidermal differentiation and basement membrane formation by HaCaT cells in surface transplants. Eur J Cell Biol, 75, 273-86.
96. Borchers, A.H., Sanders, L.A., Powell, M.B. and Bowden, G.T. (1997)
Melanocyte mediated paracrine induction of extracellular matrix degrading proteases in squamous cell carcinoma cells. Exp Cell Res, 231, 61-5.
97. Nomizu, M., Kim, W.H., Yamamura, K., Utani, A., Song, S.Y., Otaka, A., Roller,
P.P., Kleinman, H.K. and Yamada, Y. (1995) Identification of cell binding sites in the laminin alpha 1 chain carboxyl-terminal globular domain by systematic screening of synthetic peptides. J Biol Chem, 270, 20583-90.
98. Boudreau, N. and Bissell, M.J. (1998) Extracellular matrix signaling: integration
of form and function in normal and malignant cells. Curr Opin Cell Biol, 10, 640-6.
99. Wewer, U.M., Shaw, L.M., Albrechtsen, R. and Mercurio, A.M. (1997) The
integrin alpha 6 beta 1 promotes the survival of metastatic human breast carcinoma cells in mice. Am J Pathol, 151, 1191-8.
100. Meerschaert, J., Vrtis, R.F., Shikama, Y., Sedgwick, J.B., Busse, W.W. and
Mosher, D.F. (1999) Engagement of alpha4beta7 integrins by monoclonal antibodies or ligands enhances survival of human eosinophils in vitro. J Immunol,163, 6217-27.
101. Sethi, T., Rintoul, R.C., Moore, S.M., MacKinnon, A.C., Salter, D., Choo, C.,
Chilvers, E.R., Dransfield, I., Donnelly, S.C., Strieter, R. and Haslett, C. (1999) Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med, 5, 662-8.

129 102. Streuli, C.H. and Gilmore, A.P. (1999) Adhesion-mediated signaling in the
regulation of mammary epithelial cell survival. J Mammary Gland Biol Neoplasia, 4, 183-91.
103. Brizzi, M.F., Defilippi, P., Rosso, A., Venturino, M., Garbarino, G., Miyajima,
A., Silengo, L., Tarone, G. and Pegoraro, L. (1999) Integrin-mediated adhesion of endothelial cells induces JAK2 and STAT5A activation: role in the control of c-fos gene expression. Mol Biol Cell, 10, 3463-71.
104. Bachelder, R.E., Ribick, M.J., Marchetti, A., Falcioni, R., Soddu, S., Davis, K.R.
and Mercurio, A.M. (1999) p53 inhibits alpha 6 beta 4 integrin survival signaling by promoting the caspase 3-dependent cleavage of AKT/PKB. J Cell Biol, 147,1063-72.
105. Cress, A.E. and Dalton, W.S. (1996) Multiple drug resistance and intermediate
filaments. Cancer Metastasis Rev, 15, 499-506. 106. St Croix, B., Man, S. and Kerbel, R.S. (1998) Reversal of intrinsic and acquired
forms of drug resistance by hyaluronidase treatment of solid tumors. Cancer Lett,131, 35-44.
107. Kerbel, R.S., Rak, J., Kobayashi, H., Man, M.S., St Croix, B. and Graham, C.H.
(1994) Multicellular resistance: a new paradigm to explain aspects of acquired drug resistance of solid tumors. Cold Spring Harb Symp Quant Biol, 59, 661-72.
108. Damiano, J.S., Cress, A.E., Hazlehurst, L.A., Shtil, A.A. and Dalton, W.S. (1999)
Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood, 93, 1658-67.
109. Hay, E.D. (1991) Cell Biology of the Extracelluar Matrix. Plenum Press, New
York. 110. Lee, E.C., Lotz, M.M., Steele, G.D., Jr. and Mercurio, A.M. (1992) The integrin
alpha 6 beta 4 is a laminin receptor. J Cell Biol, 117, 671-8. 111. Mercurio, A.M. and Shaw, L.M. (1991) Laminin binding proteins. Bioessays, 13,
469-73. 112. Friedrichs, K., Ruiz, P., Franke, F., Gille, I., Terpe, H.J. and Imhof, B.A. (1995)
High expression level of alpha 6 integrin in human breast carcinoma is correlated with reduced survival. Cancer Res, 55, 901-6.
113. Rabinovitz, I. and Mercurio, A.M. (1996) The integrin alpha 6 beta 4 and the
biology of carcinoma. Biochem Cell Biol, 74, 811-21.

130 114. Rosendahl, A., Neumann, K., Chaloupka, B., Rothmund, M. and Weinel, R.J.
(1993) Expression and distribution of VLA receptors in the pancreas: an immunohistochemical study. Pancreas, 8, 711-8.
115. Carey, T.E., Laurikainen, L., Ptok, A., Reinke, T., Linder, K., Nair, T.S. and
Marcelo, C. (1992) Culture conditions affect expression of the alpha 6 beta 4 integrin associated with aggressive behavior in head and neck cancer. Adv Exp Med Biol, 320, 69-79.
116. Weinel, R.J., Rosendahl, A., Neumann, K., Chaloupka, B., Erb, D., Rothmund,
M. and Santoso, S. (1992) Expression and function of VLA-alpha 2, -alpha 3, -alpha 5 and -alpha 6-integrin receptors in pancreatic carcinoma. Int J Cancer, 52,827-33.
117. Niessen, C.M., Hogervorst, F., Jaspars, L.H., de Melker, A.A., Delwel, G.O.,
Hulsman, E.H., Kuikman, I. and Sonnenberg, A. (1994) The alpha 6 beta 4 integrin is a receptor for both laminin and kalinin. Exp Cell Res, 211, 360-7.
118. Brown, J.C. and Goodman, S.L. (1991) Different cellular receptors for human
placental laminin and murine EHS laminin. FEBS Lett, 282, 5-8. 119. Ruiz, P., Dunon, D., Sonnenberg, A. and Imhof, B.A. (1993) Suppression of
mouse melanoma metastasis by EA-1, a monoclonal antibody specific for alpha 6 integrins. Cell Adhes Commun, 1, 67-81.
120. Lochter, A., Navre, M., Werb, Z. and Bissell, M.J. (1999) alpha1 and alpha2
integrins mediate invasive activity of mouse mammary carcinoma cells through regulation of stromelysin-1 expression. Mol Biol Cell, 10, 271-82.
121. Kibbey, M.C., Grant, D.S. and Kleinman, H.K. (1992) Role of the SIKVAV site
of laminin in promotion of angiogenesis and tumor growth: an in vivo Matrigel model. J Natl Cancer Inst, 84, 1633-8.
122. Grant, D.S., Kinsella, J.L., Fridman, R., Auerbach, R., Piasecki, B.A., Yamada,
Y., Zain, M. and Kleinman, H.K. (1992) Interaction of endothelial cells with a laminin A chain peptide (SIKVAV) in vitro and induction of angiogenic behavior in vivo. J Cell Physiol, 153, 614-25.
123. Bresalier, R.S., Schwartz, B., Kim, Y.S., Duh, Q.Y., Kleinman, H.K. and Sullam,
P.M. (1995) The laminin alpha 1 chain Ile-Lys-Val-Ala-Val (IKVAV)-containing peptide promotes liver colonization by human colon cancer cells. Cancer Res, 55,2476-80.

131 124. Nomizu, M., Kuratomi, Y., Song, S.Y., Ponce, M.L., Hoffman, M.P., Powell,
S.K., Miyoshi, K., Otaka, A., Kleinman, H.K. and Yamada, Y. (1997) Identification of cell binding sequences in mouse laminin gamma1 chain by systematic peptide screening. J Biol Chem, 272, 32198-205.
125. Skubitz, A.P., Letourneau, P.C., Wayner, E. and Furcht, L.T. (1991) Synthetic
peptides from the carboxy-terminal globular domain of the A chain of laminin: their ability to promote cell adhesion and neurite outgrowth, and interact with heparin and the beta 1 integrin subunit. J Cell Biol, 115, 1137-48.
126. Graf, J., Ogle, R.C., Robey, F.A., Sasaki, M., Martin, G.R., Yamada, Y. and
Kleinman, H.K. (1987) A pentapeptide from the laminin B1 chain mediates cell adhesion and binds the 67,000 laminin receptor. Biochemistry, 26, 6896-900.
127. Iwamoto, Y., Robey, F.A., Graf, J., Sasaki, M., Kleinman, H.K., Yamada, Y. and
Martin, G.R. (1987) YIGSR, a synthetic laminin pentapeptide, inhibits experimental metastasis formation. Science, 238, 1132-4.
128. Sakamoto, N., Iwahana, M., Tanaka, N.G. and Osada, Y. (1991) Inhibition of
angiogenesis and tumor growth by a synthetic laminin peptide, CDPGYIGSR-NH2. Cancer Res, 51, 903-6.
129. Skubitz, A.P., McCarthy, J.B., Zhao, Q., Yi, X.Y. and Furcht, L.T. (1990)
Definition of a sequence, RYVVLPR, within laminin peptide F-9 that mediates metastatic fibrosarcoma cell adhesion and spreading. Cancer Res, 50, 7612-22.
130. Kleinman, H.K., Graf, J., Iwamoto, Y., Sasaki, M., Schasteen, C.S., Yamada, Y.,
Martin, G.R. and Robey, F.A. (1989) Identification of a second active site in laminin for promotion of cell adhesion and migration and inhibition of in vivo melanoma lung colonization. Arch Biochem Biophys, 272, 39-45.
131. Charonis, A.S., Skubitz, A.P., Koliakos, G.G., Reger, L.A., Dege, J., Vogel,
A.M., Wohlhueter, R. and Furcht, L.T. (1988) A novel synthetic peptide from the B1 chain of laminin with heparin-binding and cell adhesion-promoting activities. J Cell Biol, 107, 1253-60.
132. Loftus, J.C. and Liddington, R.C. (1997) Cell adhesion in vascular biology. New
insights into integrin-ligand interaction. J Clin Invest, 99, 2302-6. 133. Arthur, W.T., Noren, N.K. and Burridge, K. (2002) Regulation of Rho family
GTPases by cell-cell and cell-matrix adhesion. Biol Res, 35, 239-46. 134. Hynes, R.O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell,
110, 673-87.

132 135. Panetti, T.S. (2002) Tyrosine phosphorylation of paxillin, FAK, and p130CAS:
effects on cell spreading and migration. Front Biosci, 7, d143-50. 136. Slack-Davis, J.K., Eblen, S.T., Zecevic, M., Boerner, S.A., Tarcsafalvi, A., Diaz,
H.B., Marshall, M.S., Weber, M.J., Parsons, J.T. and Catling, A.D. (2003) PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. JCell Biol, 162, 281-91.
137. McCandless, J.R., Cress, A.E., Rabinovitz, I., Payne, C.M., Bowden G.T., Knox,
J.D., and Nagle R.B. (1997) A human xenograft model for testing early events of epithelial neoplastic invasion. Int J Oncol., 10, 279-285.
138. Hazlehurst, L.A., Foley, N.E., Gleason-Guzman, M.C., Hacker, M.P., Cress, A.E.,
Greenberger, L.W., De Jong, M.C. and Dalton, W.S. (1999) Multiple mechanisms confer drug resistance to mitoxantrone in the human 8226 myeloma cell line. Cancer Res, 59, 1021-8.
139. Ilic, D., Almeida, E.A., Schlaepfer, D.D., Dazin, P., Aizawa, S. and Damsky,
C.H. (1998) Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol, 143, 547-60.
140. Frisch, S.M. and Ruoslahti, E. (1997) Integrins and anoikis. Curr Opin Cell Biol,
9, 701-6. 141. Schwartz, M.A. (1997) Integrins, oncogenes, and anchorage independence. J Cell
Biol, 139, 575-8. 142. Ruoslahti, E. and Reed, J.C. (1994) Anchorage dependence, integrins, and
apoptosis. Cell, 77, 477-8. 143. MacGrogan, D. and Bookstein, R. (1997) Tumour suppressor genes in prostate
cancer. Semin Cancer Biol, 8, 11-9. 144. Lauffenburger, D.A. and Horwitz, A.F. (1996) Cell migration: a physically
integrated molecular process. Cell, 84, 359-69. 145. Sahai, E. and Marshall, C.J. (2003) Differing modes of tumour cell invasion have
distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol, 5, 711-9.
146. Friedl, P. and Wolf, K. (2003) Tumour-cell invasion and migration: diversity and
escape mechanisms. Nat Rev Cancer, 3, 362-74.

133 147. Zhou, H. and Kramer, R.H. (2005) Integrin engagement differentially modulates
epithelial cell motility by RhoA/ROCK and PAK1. J Biol Chem, 280, 10624-35. 148. Guo, W. and Giancotti, F.G. (2004) Integrin signalling during tumour
progression. Nat Rev Mol Cell Biol, 5, 816-26. 149. Schmelz, M., Cress, A.E., Scott, K.M., Burger, F., Cui, H., Sallam, K., McDaniel,
K.M., Dalkin, B.L. and Nagle, R.B. (2002) Different phenotypes in human prostate cancer: alpha6 or alpha3 integrin in cell-extracellular adhesion sites. Neoplasia, 4, 243-54.
150. Gu, J., Sumida, Y., Sanzen, N. and Sekiguchi, K. (2001) Laminin-10/11 and
fibronectin differentially regulate integrin-dependent Rho and Rac activation via p130(Cas)-CrkII-DOCK180 pathway. J Biol Chem, 276, 27090-7.
151. Kariya, Y. and Miyazaki, K. (2004) The basement membrane protein laminin-5
acts as a soluble cell motility factor. Exp Cell Res, 297, 508-20. 152. Shang, M., Koshikawa, N., Schenk, S. and Quaranta, V. (2001) The LG3 module
of laminin-5 harbors a binding site for integrin alpha3beta1 that promotes cell adhesion, spreading, and migration. J Biol Chem, 276, 33045-53.
153. Hibino, S., Shibuya, M., Engbring, J.A., Mochizuki, M., Nomizu, M. and
Kleinman, H.K. (2004) Identification of an active site on the laminin alpha5 chain globular domain that binds to CD44 and inhibits malignancy. Cancer Res, 64,4810-6.
154. Ponce, M.L., Hibino, S., Lebioda, A.M., Mochizuki, M., Nomizu, M. and
Kleinman, H.K. (2003) Identification of a potent peptide antagonist to an active laminin-1 sequence that blocks angiogenesis and tumor growth. Cancer Res, 63,5060-4.
155. Nakahara, H., Mueller, S.C., Nomizu, M., Yamada, Y., Yeh, Y. and Chen, W.T.
(1998) Activation of beta1 integrin signaling stimulates tyrosine phosphorylation of p190RhoGAP and membrane-protrusive activities at invadopodia. J Biol Chem, 273, 9-12.
156. Song, S.Y., Nomizu, M., Yamada, Y. and Kleinman, H.K. (1997) Liver
metastasis formation by laminin-1 peptide (LQVQLSIR)-adhesion selected B16-F10 melanoma cells. Int J Cancer, 71, 436-41.
157. Slack, J.K., Adams, R.B., Rovin, J.D., Bissonette, E.A., Stoker, C.E. and Parsons,
J.T. (2001) Alterations in the focal adhesion kinase/Src signal transduction

134
pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene, 20, 1152-63.
158. Somlyo, A.V., Bradshaw, D., Ramos, S., Murphy, C., Myers, C.E. and Somlyo,
A.P. (2000) Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem Biophys Res Commun, 269, 652-9.
159. Mitchison, T.J. and Cramer, L.P. (1996) Actin-based cell motility and cell
locomotion. Cell, 84, 371-9. 160. Pollard, T.D. and Borisy, G.G. (2003) Cellular motility driven by assembly and
disassembly of actin filaments. Cell, 112, 453-65. 161. Weed, S.A. and Parsons, J.T. (2001) Cortactin: coupling membrane dynamics to
cortical actin assembly. Oncogene, 20, 6418-34. 162. Plopper, G.E., Domanico, S.Z., Cirulli, V., Kiosses, W.B. and Quaranta, V.
(1998) Migration of breast epithelial cells on Laminin-5: differential role of integrins in normal and transformed cell types. Breast Cancer Res Treat, 51, 57-69.
163. Li, X., Ma, Z., Zhu, J., Li, K., Wang, S., Bie, P. and Dong, J. (2004) Laminin-5
promotes cell motility by regulating the function of the integrin alpha6beta1 in pancreatic cancer. Carcinogenesis, 25, 2283.
164. Mercurio, A.M., Rabinovitz, I. and Shaw, L.M. (2001) The alpha 6 beta 4 integrin
and epithelial cell migration. Curr Opin Cell Biol, 13, 541-5. 165. Russell, A.J., Fincher, E.F., Millman, L., Smith, R., Vela, V., Waterman, E.A.,
Dey, C.N., Guide, S., Weaver, V.M. and Marinkovich, M.P. (2003) Alpha 6 beta 4 integrin regulates keratinocyte chemotaxis through differential GTPase activation and antagonism of alpha 3 beta 1 integrin. J Cell Sci, 116, 3543-56.
166. Parsons, J.T. (2003) Focal adhesion kinase: the first ten years. J Cell Sci, 116,
1409-16. 167. Slack-Davis, J.K. and Parsons, J.T. (2004) Emerging views of integrin signaling:
implications for prostate cancer. J Cell Biochem, 91, 41-6. 168. Colognato, H. and Yurchenco, P.D. (2000) Form and function: the laminin family
of heterotrimers. Dev Dyn, 218, 213-34.

135 169. Nakahara, H., Nomizu, M., Akiyama, S.K., Yamada, Y., Yeh, Y. and Chen, W.T.
(1996) A mechanism for regulation of melanoma invasion. Ligation of alpha6beta1 integrin by laminin G peptides. J Biol Chem, 271, 27221-4.
170. Leu, S.J., Liu, Y., Chen, N., Chen, C.C., Lam, S.C. and Lau, L.F. (2003)
Identification of a novel integrin alpha 6 beta 1 binding site in the angiogenic inducer CCN1 (CYR61). J Biol Chem, 278, 33801-8.
171. Murayama, O., Nishida, H. and Sekiguchi, K. (1996) Novel peptide ligands for
integrin alpha 6 beta 1 selected from a phage display library. J Biochem (Tokyo),120, 445-51.
172. Huttenlocher, A., Ginsberg, M.H. and Horwitz, A.F. (1996) Modulation of cell
migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol, 134, 1551-62.
173. Vuori, K. and Ruoslahti, E. (1995) Tyrosine phosphorylation of p130Cas and
cortactin accompanies integrin-mediated cell adhesion to extracellular matrix. JBiol Chem, 270, 22259-62.
174. Hodivala-Dilke, K.M., DiPersio, C.M., Kreidberg, J.A. and Hynes, R.O. (1998)
Novel roles for alpha3beta1 integrin as a regulator of cytoskeletal assembly and as a trans-dominant inhibitor of integrin receptor function in mouse keratinocytes. JCell Biol, 142, 1357-69.
175. Ritter, M.R., Zhou, Q. and Markland, F.S., Jr. (2001) Contortrostatin, a
homodimeric disintegrin, actively disrupts focal adhesion and cytoskeletal structure and inhibits cell motility through a novel mechanism. Cell Commun Adhes, 8, 71-86.
176. Ritter, M.R. and Markland, F.S., Jr. (2000) Contortrostatin activates ERK2 and
tyrosine phosphorylation events via distinct pathways. Biochem Biophys Res Commun, 274, 142-8.
177. Schmitmeier, S., Markland, F.S., Ritter, M.R., Sawcer, D.E. and Chen, T.C.
(2003) Functional effect of contortrostatin, a snake venom disintegrin, on human glioma cell invasion in vitro. Cell Commun Adhes, 10, 1-16.
178. Ritter, M.R., Zhou, Q. and Markland, F.S., Jr. (2000) Contortrostatin, a snake
venom disintegrin, induces alphavbeta3-mediated tyrosine phosphorylation of CAS and FAK in tumor cells. J Cell Biochem, 79, 28-37.
179. Coelho, A.L., De Freitas, M.S., Mariano-Oliveira, A., Rapozo, D.C., Pinto, L.F.,
Niewiarowski, S., Zingali, R.B., Marcinkiewicz, C. and Barja-Fidalgo, C. (2004)

136
RGD- and MLD-disintegrins, jarastatin and EC3, activate integrin-mediated signaling modulating the human neutrophils chemotaxis, apoptosis and IL-8 gene expression. Exp Cell Res, 292, 371-84.
180. Pierschbacher, M.D. and Ruoslahti, E. (1984) Cell attachment activity of
fibronectin can be duplicated by small synthetic fragments of the molecule. Nature, 309, 30-3.
181. Ruoslahti, E. and Pierschbacher, M.D. (1987) New perspectives in cell adhesion:
RGD and integrins. Science, 238, 491-7. 182. Humphries, M.J., Olden, K. and Yamada, K.M. (1986) A synthetic peptide from
fibronectin inhibits experimental metastasis of murine melanoma cells. Science,233, 467-70.
183. Gehlsen, K.R., Argraves, W.S., Pierschbacher, M.D. and Ruoslahti, E. (1988)
Inhibition of in vitro tumor cell invasion by Arg-Gly-Asp-containing synthetic peptides. J Cell Biol, 106, 925-30.
184. Engbring, J.A. and Kleinman, H.K. (2003) The basement membrane matrix in
malignancy. J Pathol, 200, 465-70. 185. Mahato, R.I., Narang, A.S., Thoma, L. and Miller, D.D. (2003) Emerging trends
in oral delivery of peptide and protein drugs. Crit Rev Ther Drug Carrier Syst, 20,153-214.
186. Hamman, J.H., Enslin, G.M. and Kotze, A.F. (2005) Oral delivery of Peptide
drugs : barriers and developments. BioDrugs, 19, 165-77. 187. Kumagai, H., Tajima, M., Ueno, Y., Giga-Hama, Y. and Ohba, M. (1991) Effect
of cyclic RGD peptide on cell adhesion and tumor metastasis. Biochem Biophys Res Commun, 177, 74-82.
188. Kaneda, Y., Yamamoto, Y., Okada, N., Tsutsuml, Y., Nakagawa, S., Kakiuch,
M., Maeda, M., Kawasaki, K. and Mayumi, T. (1997) Antimetastatic effect of synthetic Glu-Ile-Leu-Asp-Val peptide derivatives containing D-amino acids. Anticancer Drugs, 8, 702-7.
189. Yamamoto, Y., Tsutsumi, Y. and Mayumi, T. (2002) Molecular design of
bioconjugated cell adhesion peptide with a water-soluble polymeric modifier for enhancement of antimetastatic effect. Curr Drug Targets, 3, 123-30.
190. Fujii, H., Nishikawa, N., Komazawa, H., Suzuki, M., Kojima, M., Itoh, I., Obata,
A., Ayukawa, K., Azuma, I. and Saiki, I. (1998) A new pseudo-peptide of Arg-

137
Gly-Asp (RGD) with inhibitory effect on tumor metastasis and enzymatic degradation of extracellular matrix. Clin Exp Metastasis, 16, 94-104.
191. Dechantsreiter, M.A., Planker, E., Matha, B., Lohof, E., Holzemann, G., Jonczyk,
A., Goodman, S.L. and Kessler, H. (1999) N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem, 42,3033-40.
192. Greenfield, N.J. (1996) Methods to estimate the conformation of proteins and
polypeptides from circular dichroism data. Anal Biochem, 235, 1-10. 193. Kelly, S.M. and Price, N.C. (2000) The use of circular dichroism in the
investigation of protein structure and function. Curr Protein Pept Sci, 1, 349-84. 194. Klassen, R.B. and Opella, S.J. (1997) NMR studies of peptides and proteins
associated with membranes. Methods Mol Biol, 60, 271-97. 195. Reid, D.G., MacLachlan, L.K., Edwards, A.J., Hubbard, J.A. and Sweeney, P.J.
(1997) Introduction to the NMR of proteins. Methods Mol Biol, 60, 1-28. 196. Hemler, M.E. (1998) Integrin associated proteins. Curr Opin Cell Biol, 10, 578-
85. 197. Stipp, C.S., Kolesnikova, T.V. and Hemler, M.E. (2003) Functional domains in
tetraspanin proteins. Trends Biochem Sci, 28, 106-12. 198. Stipp, C.S., Kolesnikova, T.V. and Hemler, M.E. (2003) EWI-2 regulates
alpha3beta1 integrin-dependent cell functions on laminin-5. J Cell Biol, 163,1167-77.
199. Kolesnikova, T.V., Stipp, C.S., Rao, R.M., Lane, W.S., Luscinskas, F.W. and
Hemler, M.E. (2004) EWI-2 modulates lymphocyte integrin alpha4beta1 functions. Blood, 103, 3013-9.
200. Zhang, X.A., Bontrager, A.L., Stipp, C.S., Kraeft, S.K., Bazzoni, G., Chen, L.B.
and Hemler, M.E. (2001) Phosphorylation of a conserved integrin alpha 3 QPSXXE motif regulates signaling, motility, and cytoskeletal engagement. Mol Biol Cell, 12, 351-65.
201. Zhang, X.A., Lane, W.S., Charrin, S., Rubinstein, E. and Liu, L. (2003)
EWI2/PGRL associates with the metastasis suppressor KAI1/CD82 and inhibits the migration of prostate cancer cells. Cancer Res, 63, 2665-74.

138 202. Gesierich, S., Paret, C., Hildebrand, D., Weitz, J., Zgraggen, K., Schmitz-
Winnenthal, F.H., Horejsi, V., Yoshie, O., Herlyn, D., Ashman, L.K. and Zoller, M. (2005) Colocalization of the tetraspanins, CO-029 and CD151, with integrins in human pancreatic adenocarcinoma: impact on cell motility. Clin Cancer Res,11, 2840-52.
203. Stipp, C.S. and Hemler, M.E. (2000) Transmembrane-4-superfamily proteins
CD151 and CD81 associate with alpha 3 beta 1 integrin, and selectively contribute to alpha 3 beta 1-dependent neurite outgrowth. J Cell Sci, 113 ( Pt 11),1871-82.
204. Yauch, R.L., Berditchevski, F., Harler, M.B., Reichner, J. and Hemler, M.E.
(1998) Highly stoichiometric, stable, and specific association of integrin alpha3beta1 with CD151 provides a major link to phosphatidylinositol 4-kinase, and may regulate cell migration. Mol Biol Cell, 9, 2751-65.
205. Mazzocca, A., Carloni, V., Sciammetta, S., Cordella, C., Pantaleo, P., Caldini, A.,
Gentilini, P. and Pinzani, M. (2002) Expression of transmembrane 4 superfamily (TM4SF) proteins and their role in hepatic stellate cell motility and wound healing migration. J Hepatol, 37, 322-30.
206. Hemler, M.E. (2001) Specific tetraspanin functions. J Cell Biol, 155, 1103-7. 207. Lammerding, J., Kazarov, A.R., Huang, H., Lee, R.T. and Hemler, M.E. (2003)
Tetraspanin CD151 regulates alpha6beta1 integrin adhesion strengthening. Proc Natl Acad Sci U S A, 100, 7616-21.
208. Kazarov, A.R., Yang, X., Stipp, C.S., Sehgal, B. and Hemler, M.E. (2002) An
extracellular site on tetraspanin CD151 determines alpha 3 and alpha 6 integrin-dependent cellular morphology. J Cell Biol, 158, 1299-309.
209. Zhang, X.A., Bontrager, A.L. and Hemler, M.E. (2001) Transmembrane-4
superfamily proteins associate with activated protein kinase C (PKC) and link PKC to specific beta(1) integrins. J Biol Chem, 276, 25005-13.
210. Serru, V., Le Naour, F., Billard, M., Azorsa, D.O., Lanza, F., Boucheix, C. and
Rubinstein, E. (1999) Selective tetraspan-integrin complexes (CD81/alpha4beta1, CD151/alpha3beta1, CD151/alpha6beta1) under conditions disrupting tetraspan interactions. Biochem J, 340 ( Pt 1), 103-11.
211. Berditchevski, F., Zutter, M.M. and Hemler, M.E. (1996) Characterization of
novel complexes on the cell surface between integrins and proteins with 4 transmembrane domains (TM4 proteins). Mol Biol Cell, 7, 193-207.

139 212. Berditchevski, F. and Odintsova, E. (1999) Characterization of integrin-
tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. J Cell Biol, 146, 477-92.
213. Domanico, S.Z., Pelletier, A.J., Havran, W.L. and Quaranta, V. (1997) Integrin
alpha 6A beta 1 induces CD81-dependent cell motility without engaging the extracellular matrix migration substrate. Mol Biol Cell, 8, 2253-65.
214. Stipp, C.S., Kolesnikova, T.V. and Hemler, M.E. (2001) EWI-2 is a major CD9
and CD81 partner and member of a novel Ig protein subfamily. J Biol Chem, 276,40545-54.
215. Clark, K.L., Zeng, Z., Langford, A.L., Bowen, S.M. and Todd, S.C. (2001) PGRL
is a major CD81-associated protein on lymphocytes and distinguishes a new family of cell surface proteins. J Immunol, 167, 5115-21.
216. Humphries, M.J. (2000) Integrin structure. Biochem Soc Trans, 28, 311-39. 217. Mould, A.P. (1996) Getting integrins into shape: recent insights into how integrin
activity is regulated by conformational changes. J Cell Sci, 109 ( Pt 11), 2613-8. 218. Shimaoka, M., Takagi, J. and Springer, T.A. (2002) Conformational regulation of
integrin structure and function. Annu Rev Biophys Biomol Struct, 31, 485-516. 219. Mould, A.P., Akiyama, S.K. and Humphries, M.J. (1996) The inhibitory anti-
beta1 integrin monoclonal antibody 13 recognizes an epitope that is attenuated by ligand occupancy. Evidence for allosteric inhibition of integrin function. J Biol Chem, 271, 20365-74.
220. Mould, A.P., Garratt, A.N., Askari, J.A., Akiyama, S.K. and Humphries, M.J.
(1995) Identification of a novel anti-integrin monoclonal antibody that recognises a ligand-induced binding site epitope on the beta 1 subunit. FEBS Lett, 363, 118-22.
221. Bazzoni, G., Shih, D.T., Buck, C.A. and Hemler, M.E. (1995) Monoclonal
antibody 9EG7 defines a novel beta 1 integrin epitope induced by soluble ligand and manganese, but inhibited by calcium. J Biol Chem, 270, 25570-7.
222. Ridley, A.J., Schwartz, M.A., Burridge, K., Firtel, R.A., Ginsberg, M.H., Borisy,
G., Parsons, J.T. and Horwitz, A.R. (2003) Cell migration: integrating signals from front to back. Science, 302, 1704-9.

140 223. Webb, D.J., Donais, K., Whitmore, L.A., Thomas, S.M., Turner, C.E., Parsons,
J.T. and Horwitz, A.F. (2004) FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol, 6, 154-61.
224. Zaidel-Bar, R., Ballestrem, C., Kam, Z. and Geiger, B. (2003) Early molecular
events in the assembly of matrix adhesions at the leading edge of migrating cells. J Cell Sci, 116, 4605-13.
225. Rinehart, J., Adjei, A.A., Lorusso, P.M., Waterhouse, D., Hecht, J.R., Natale,
R.B., Hamid, O., Varterasian, M., Asbury, P., Kaldjian, E.P., Gulyas, S., Mitchell, D.Y., Herrera, R., Sebolt-Leopold, J.S. and Meyer, M.B. (2004) Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol, 22, 4456-62.
226. Allen, L.F., Sebolt-Leopold, J. and Meyer, M.B. (2003) CI-1040 (PD184352), a
targeted signal transduction inhibitor of MEK (MAPKK). Semin Oncol, 30, 105-16.
227. Druker, B.J., Talpaz, M., Resta, D.J., Peng, B., Buchdunger, E., Ford, J.M.,
Lydon, N.B., Kantarjian, H., Capdeville, R., Ohno-Jones, S. and Sawyers, C.L. (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med, 344, 1031-7.
228. Mauro, M.J. and Druker, B.J. (2001) STI571: targeting BCR-ABL as therapy for
CML. Oncologist, 6, 233-8. 229. White, C.A., Larocca, A. and Grillo-Lopez, A.J. (1999) Anti-CD20 monoclonal
antibodies as novel treatments for non-Hodgkin's lymphoma. Pharm. Sci. Technol. Today, 2, 95-101.
230. Grillo-Lopez, A.J., White, C.A., Varns, C., Shen, D., Wei, A., McClure, A. and
Dallaire, B.K. (1999) Overview of the clinical development of rituximab: first monoclonal antibody approved for the treatment of lymphoma. Semin Oncol, 26,66-73.
231. Dillman, R.O. (1999) Perceptions of Herceptin: a monoclonal antibody for the
treatment of breast cancer. Cancer Biother Radiopharm, 14, 5-10. 232. Graziano, C. (1998) HER-2 breast assay, linked to Herceptin, wins FDA's okay.
CAP Today, 12, 1, 14-6. 233. Kerr, J.S., Slee, A.M. and Mousa, S.A. (2002) The alpha v integrin antagonists as
novel anticancer agents: an update. Expert Opin Investig Drugs, 11, 1765-74.

141 234. Tucker, G.C. (2003) Alpha v integrin inhibitors and cancer therapy. Curr Opin
Investig Drugs, 4, 722-31. 235. Gutheil, J.C., Campbell, T.N., Pierce, P.R., Watkins, J.D., Huse, W.D., Bodkin,
D.J. and Cheresh, D.A. (2000) Targeted antiangiogenic therapy for cancer using Vitaxin: a humanized monoclonal antibody to the integrin alphavbeta3. Clin Cancer Res, 6, 3056-61.
236. Mikecz, K. (2000) Vitaxin applied molecular evolution. Curr Opin Investig
Drugs, 1, 199-203. 237. Patel, S.R., Jenkins, J., Papadopolous, N., Burgess, M.A., Plager, C., Gutterman,
J. and Benjamin, R.S. (2001) Pilot study of vitaxin--an angiogenesis inhibitor-in patients with advanced leiomyosarcomas. Cancer, 92, 1347-8.
238. Posey, J.A., Khazaeli, M.B., DelGrosso, A., Saleh, M.N., Lin, C.Y., Huse, W. and
LoBuglio, A.F. (2001) A pilot trial of Vitaxin, a humanized anti-vitronectin receptor (anti alpha v beta 3) antibody in patients with metastatic cancer. Cancer Biother Radiopharm, 16, 125-32.
239. Smith, J.W. (2003) Cilengitide Merck. Curr Opin Investig Drugs, 4, 741-5. 240. Eskens, F.A., Dumez, H., Hoekstra, R., Perschl, A., Brindley, C., Bottcher, S.,
Wynendaele, W., Drevs, J., Verweij, J. and van Oosterom, A.T. (2003) Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur J Cancer, 39, 917-26.
241. Burke, P.A., DeNardo, S.J., Miers, L.A., Lamborn, K.R., Matzku, S. and
DeNardo, G.L. (2002) Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res, 62, 4263-72.
242. Raguse, J.D., Gath, H.J., Bier, J., Riess, H. and Oettle, H. (2004) Cilengitide
(EMD 121974) arrests the growth of a heavily pretreated highly vascularised head and neck tumour. Oral Oncol, 40, 228-30.
243. Reinmuth, N., Liu, W., Ahmad, S.A., Fan, F., Stoeltzing, O., Parikh, A.A.,
Bucana, C.D., Gallick, G.E., Nickols, M.A., Westlin, W.F. and Ellis, L.M. (2003) Alphavbeta3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res, 63, 2079-87.
244. Stoeltzing, O., Liu, W., Reinmuth, N., Fan, F., Parry, G.C., Parikh, A.A.,
McCarty, M.F., Bucana, C.D., Mazar, A.P. and Ellis, L.M. (2003) Inhibition of

142
integrin alpha5beta1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int J Cancer, 104, 496-503.
245. Weinel, R.J., Rosendahl, A., Pinschmidt, E., Kisker, O., Simon, B. and Santoso,
S. (1995) The alpha 6-integrin receptor in pancreatic carcinoma. Gastroenterology, 108, 523-32.
246. Welch, D.R. (1997) Technical considerations for studying cancer metastasis in
vivo. Clin Exp Metastasis, 15, 272-306.