synthesis of caseinolytic protease agonists towards … · synthesis of caseinolytic protease...
TRANSCRIPT

Synthesis of Caseinolytic Protease Agonists Towards the Synthesis of the Natural
Acyldepsipeptides
by
Michele Cossette
A thesis submitted in conformity with the requirements for the degree of Master’s of Science
Department of Chemistry University of Toronto
© Copyright by Michele Cossette 2010

ii
Synthesis of Caseinolytic Protease Agonists Towards the Synthesis of the Natural Acyldepsipeptides
Michele Cossette
Masterʼs of Science
Department of Chemistry University of Toronto
2010
Abstract Caseinolytic protease (ClpP) is a cylindrical protease forming the core of
protein degradation machinery in eubacteria. ClpP is tightly regulated and is non-
functional without a member of the Clp-ATPases. A new class of antibiotics,
termed ADEPs, bind to ClpP and allow for activation without the Clp-ATPases;
leading to cell death.
A more efficient synthetic route to the ADEPs utilizing solid-phase peptide
synthesis was investigated. A linear peptide was synthesized, however attempts
to close the depsipeptidic macrocycle via macrolactonization failed. Further
attempts of assembling a branched depsipeptide for ring closure via a
macrolactamization resulted in products that were not stable to cleavage
conditions.
A group of molecules termed Activators of Self-Compartmentalizing Proteases
(ACP) were identified through a screen for activity towards ClpP. Compound
ACP1 was synthesized along with twelve analogs and their activity towards ClpP
evaluated. The project resulted in a compound with a higher activity than its
natural product counterpart.

iii
Acknowledgements I would first like to thank my research supervisor Prof. Robert Batey. With
his support and guidance I have gained knowledge in the fundamentals of total
synthesis and many problem-solving skills required from any synthetic chemist.
His approach encourages students to be creative, resourceful chemists, and
therefore I would like to thank Rob most.
I would like to thank the NMR staff, Dr. Tim Burrows, and Dr. Alex Young
for help with spectral analysis.
Next I would like to acknowledge the current and past members of the
Batey group. Their good company and enthusiasm towards chemistry made for a
nurturing environment to learn. Pete, Tabitha, Rivka, and John were particularly
helpful when I required some advice in synthesis. I would especially like to thank
Xiong Zhao and Jordan Goodreid for their contributions to the research project.
I would also like to thank my family and friends for their support these past
two years. My parents have been supportive in everything I have done, and
therefore I would like to dedicate my thesis to them. Most importantly I want to
thank Meldon, your love and support has made all the hard work worth it. I can
only hope you know how much it has meant to me.

iv
Table of Contents
Abstract ................................................................................................................ii Acknowledgements............................................................................................iii List of Tables .........................................................................................................v
List of Figures .......................................................................................................vi List of Abbreviations ............................................................................................vii 1.0 Introduction .................................................................................................1
1.1 The Growing Demand for New Antibiotics...................................................... 1 1.2 Cyclic Depsipeptide Antibiotics ....................................................................... 1 1.3 The Acyldepsipeptide Antibiotics .................................................................... 2 1.4 The Target of ADEPs is Caseinolytic Protease............................................... 4 1.5 New Antibacterial Target ClpP ......................................................................... 5 1.6 Previous Synthesis of ADEPs .......................................................................... 8
1.6.1 Structure Activity Relationship Studies of the ADEPs ................................. 11 1.7 Proposed Synthetic Approach ....................................................................... 13
1.7.1 Solid Phase Peptide Synthesis ................................................................... 13 1.7.2 Macrolactonization Approach...................................................................... 16 1.7.3 Macrolactamization Approach..................................................................... 17
1.8 Small Molecule Agonists of ClpP................................................................... 19
2.0 Results and Discussion............................................................................20 2.1 Towards the synthesis of ADEPs................................................................... 20
2.1.1 Linear approach .......................................................................................... 20 2.2 Macrolactamization Approach to the ADEPs ................................................ 24
2.2.1 Synthesis of the Depsipeptide Unit ............................................................. 24 2.2.2 Towards the Solution Phase Synthesis of the ADEPs ................................ 29
2.3 Synthesis of ACP1 and analogs ..................................................................... 31 2.4 Discussion of SAR studies ............................................................................. 35 2.5 Conclusions...................................................................................................... 39
3.0 Experimental..............................................................................................40 3.1 General Experimental...................................................................................... 40
Synthetic Preparations......................................................................................41 3.1.1 Linear Peptide Synthesis ............................................................................. 41 Synthesis of the Lipophilic Side Chain .................................................................. 43 3.1.2 Synthesis of Depsipeptide........................................................................... 44
3.2 Branched Depsipeptide Synthesis ................................................................. 49 3.4 Synthesis of ACP1 Analogs............................................................................ 54
Appendix I ..........................................................................................................68 Spectra of Selected Compounds ............................................................................ 68
Appendix II .........................................................................................................91 Endeavor 90 Peptide Synthesis Protocols............................................................. 91

v
List of Tables Table 1: Optimization of Horner-Wadsworth-Emmons Reaction .........................22
Table 2: Amide Coupling Conditions for ACP1 Synthesis ...................................33
Table 3: Structures of ACP1 and “eastern” modification analogs ........................34
Table 4: Structures of ACP1 “western” modification analogs ..............................35
Table 5: RD25 Values of the Natural ADEPs, ACP1 and Analogs........................37

vi
List of Figures Figure 1: Structures of Therapeutic Cyclic Depsipeptides .....................................2
Figure 2: Structure of Natural Enopeptin Acyldepsipeptide Antibiotics ..................3
Figure 3: Structure of B. subtilis ClpP15 .................................................................5
Figure 4: Model of ClpP mechanism14 ...................................................................6
Figure 5: Conformational changes of ADEP-ClpP complex15 ................................7
Figure 6: Structure of Optimized ADEP 2 ............................................................11
Figure 7: Binding site of ADEP 1A in BsClpP15....................................................12
Figure 8: Process of solid-phase peptide synthesis ............................................14
Figure 9: Structure of ACP1.................................................................................19
Figure 10: O,N-Acyl shift for the depsipeptide methodology................................26
Figure 11: Structures of ACP compounds ...........................................................31
Figure 12: in vitro assay of ACP1 analogs...........................................................36

vii
List of Abbreviations ADEP acyldepsipeptide
BnBr benzyl bromide
Boc tert-butyl carbonyl
BsClpP Bacillus subtilis caseinolytic protease 13C NMR carbon-13 nuclear magnetic resonance spectroscopy
Casein-FITC fluorescein isothiocyanate labeled casein
CBz carboxybenzyl
ClpA caseinolytic protease A
ClpC caseinolytic protease C
ClpE caseinolytic protease E
ClpP caseinolytic protease
ClpX caseinolytic protease X
2-Cl-Trt 2-chlorotrityl
DBU 1,8-diazabicyclo[5.4.0]undec-7-ene
DCU dicyclohexylurea
DIC diisopropylcarbodiimide
DIEA diisopropylethylamine
DMAP N,N-dimethylpyridine
DME dimethoxyethane
DMF dimethylformamide
DMSO dimethylsulfoxide
EDC 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
ESI MS electrospray ionization mass spectroscopy
EtOAc ethyl acetate
Fmoc fluorenyl-9-methyloxycarbonyl
FmocOSu Fmoc-O-succinimide
FT-IR Fourier transform infrared spectroscopy
HATU 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
HBTU 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate
HF hydrogen fluoride

viii
1H NMR proton nuclear magnetic resonance spectroscopy
HOAc acetic acid
HPLC high pressure liquid chromatography
HRMS high resolution mass spectroscopy
MBHA 4-methylbenzylhydrylamine
mCPBA meta-chloroperoxybenzoic acid
mp melting point
MS mass spectroscopy
MTBE methoxy-tert-butyl ether
NMI N-methylimidazole
NMP N-methylpyrrolidone
Pac phenacyl
Pfp pentafluorophenol
PivOCl pivoyl chloride
PyBOP benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
RD25 relative degradation at 25 µM
Rf retention factor
SAR structure activity relationship
SPPS solid phase peptide synthesis tBu tert-butyl
TFA trifluoroacetic acid
THF tetrahydrofuran
TIS triisopropylsilane
TLC thin layer chromatography
Trt trityl

1
1.0 Introduction 1.1 The Growing Demand for New Antibiotics
The increasing prevalence of drug resistant bacteria is a growing concern and
greatly impairs the treatment of life-threatening infections.1,2 The evolution of antibiotic
resistance among Gram-positive bacteria such as staphylococci, streptococci, and
enterococci are of particular concern because of their prevalence in hospitals and
community settings.3,4 The development of new resistance free antibiotic agents with
novel mechanisms of action is an important need.
1.2 Cyclic Depsipeptide Antibiotics
Cyclic depsipeptides belong to a large class of naturally occurring compounds
with a variety of biological activities. Depsipeptides belong to the heterodetic class as
they contain ester bonds in their peptide backbone. Both cyclic peptides and
depsipeptides have gained much interest due to the challenges they pose from a
synthetic point of view, as well as their potential in the pharmaceutical industry.5 In
comparison to their open chain analogs, cyclic peptides have demonstrated increased
metabolic stability, improved receptor selectivity, and more controlled bioavailability.5
One of the characteristic features of cyclic depsipeptides is their conformational rigidity
(cyclic vs. open chain) and stability in vivo (amide vs. ester hydrolysis) which may
contribute to their therapeutic potential.6 Several of these compounds have entered
clinical trials for antitumor, antiviral, and antimicrobial therapeutics. Figure 1 shows
Didemnin B, Daptomycin 1, and FR 901228, three cyclic depsipeptides in phase I and II
clinical trials for the treatment of cancer.7-9

2
NHO
HNO O
ON
O
OH
O
NH
ON
N
OMe
O
O
O
Didemnin B
ON
OHO
NHNH O
HN
O O
NH
HN
HO
NH
NH
HNHOOC
O
+H3N
HOOC
O
O
NH
O
NH2
O
OHO
OO
O
NH O
OHN
COOHO
NH
CONH2O
NH
OH3C 8
NH
Daptomycin 1
NHNH
NHO
NH
OO
O
O
O
S
S
H
FR 901228
Figure 1: Structures of Therapeutic Cyclic Depsipeptides
1.3 The Acyldepsipeptide Antibiotics
A mixture of eight acyldepsipeptide (ADEP) factors (A-H) were first isolated form
a strain of Streptomyces hawaiiensis NRRL 15010 and described as the ʻA54556
complexʼ in a patent in 1985.10 The structure ADEP 1A (also known as A54556A),
shown below in figure 2 was determined to be the main component of the A54556
complex.11 The ADEP structure consists of a macrolactone core, composed of five S-
configured amino acids and a lipophilic acyl side chain attached to the serine
nitrogen.10,12 Later, in 1991, the depsipeptide antibiotics, enopeptin A and B, were
isolated from a culture broth of Streptomyces sp. RK-1051, found in a soil sample in
Tsuruoka city, Japan.13 The enopeptins are related through their cyclic depsipeptidic

3
core and differ only in the lipophilic side chain, where the serine nitrogen is connected to
an aminocyclopentanedione through a dodecapentaenedioic acid residue (Figure 2).
N
OO
O
ONHO
N NO N
H
O
HN
O
R
NH
OHO
O
N
OO
O
ONHO
NN
O NH
O
HN
O
R
R: CH3 enopeptin AR: H enopeptin B
R: CH3 A 54556 AR: H A 54556 B
Figure 2: Structure of Natural Enopeptin Acyldepsipeptide Antibiotics
These ADEPs all have been shown to exhibit in vitro antibacterial activity against
Gram-positive bacteria, including multidrug-resistant strains such as penicillin-resistant
Streptococcus pneumonia (a community acquired respiratory-tract pathogen) and
methicillian-resistant Staphylococcus aureus (a major cause of hospital-acquired
infections).11 Brotz-Oesterhelt et al., from Bayer Healthcare, was able to demonstrate
the efficacy of several of the ADEPs for the treatment of mice with a lethal systemic
infection of E. faecalis and S. aureus. This resulted in 100 % survival of mice infected
with E. faecalis, and 80 % rescue of the latter. Also, the ADEPs showed superior
treatment of rats infected with S. pneumoniae over the Pfizer marketed antibiotic

4
linezolid (Zyvoxam).11 Thus, the ADEPs show promising efficacy in two rodent models
against three known pathogens among Gram-positive bacteria.
1.4 The Target of ADEPs is Caseinolytic Protease
In 2005, the target of the ADEP antibiotics was determined as caseinolytic
protease (ClpP). Using reverse genomics, Brotz-Oesterhelt et al. demonstrated that B.
subtilis, S. aureus, E. faecalis, and S. pneumoniae strains all carrying a point mutation
rendering ClpP inactive, were resistant to the antibiotic activity of the ADEPs (MIC ≥ 100 µg/mL).11 In another experiment microscope examination of B. subtilis after treatment
with 1.6 µg/mL of ADEP 1A showed filament formation, suggesting the mechanism of
action involves inhibition of cell division.11 This novel mechanism shows promise for
further development of ClpP as an antibacterial target.

5
1.5 New Antibacterial Target ClpP
Proteases play an essential role in maintaining cell homeostasis. They participate
in the general degradation of misfolded or damaged proteins as well as targeted
proteolysis of specific substrates including, but not limited to, transcriptional factors and
other key regulatory proteins.11,14 They usually consist of two components: the
proteolytic machinery for degradation and an energy-consuming ATPase, which is
responsible for substrate selection, unfolding and translocation. ClpP is the proteolytic
core that makes up the core unit of a major bacterial-protease complex.11,15
Figure 3: Structure of B. subtilis ClpP15
ClpP is a cylindrical tetradecamer serine protease that is organized into two
stacked heptameric rings. The proteolytic chamber is located within the cylinder and is
accessible via small entrance pores.14 Therefore by itself it has limited degradative
activity. ClpP works in conjunction with ClpX or ClpA (in Escherichia coli) or ClpX, ClpC
or ClpE (in Bacillus subtilis) the chaperone proteins.14 These ATPases denature the
native proteins and thread them through the axial pores into the proteolytic chamber of
ClpP where it is cleaved into peptides 7-8 residues in length.14 The peptide fragments
are then released from the chamber. A model of the proteolytic mechanism of ClpP is
summarized below in Figure 4.

6
Figure 4: Model of ClpP mechanism14
A detailed proteolytic activation mechanism remains elusive due to the lack of a
ClpP-ClpATPase complex structure. The ADEPs bind to ClpP, and serve as a model for
the activation mechanism by Clp-ATPases.11,16 Lee and co-workers were able to solve
the structure of B. subtilis ClpP (BsClpP) complexed with ADEP 1A, in its activated
state, gaining insight into the mechanism.15 Each BsClpP tetradecamer binds to 14
ADEPs in a 1:1 stoichiometry at the apical and distal surfaces. The ADEP
macrolactone core binds in the cavities formed between two adjacent subunits where
BsClpP interacts with its physiological activators; subsequently blocking its interaction
with Clp-ATPases.15 Binding of the ADEPs has also shown to trigger subunit assembly
to form a functional ClpP tetradecamer through stabilizing intersubunit interactions.

7
Complex formation with the ADEPs induces conformational changes in ClpP,
which reveals the activation mechanism. ADEPs binding triggers concerted movement
of all the subunits laterally to the periphery, opening the axial pores, allowing larger
unfolded peptides to be degraded (Figure 5).15
Figure 5: Conformational changes of ADEP-ClpP complex15
In order to investigate the effect of ADEPs as antibiotics and more explicitly their
effect on ClpP, synthetic methods were developed to obtain sufficient material for these
studies.

8
1.6 Previous Synthesis of ADEPs
Synthetic methods have been developed to obtain such amounts of the ADEPs
needed for their investigation as antibacterial agents. The first synthesis of Enopeptin B
was reported by Schmidt et al. in 1997 (Schemes 1 and 2).12 A macrolactamization
approach was employed for the synthesis of the depsipeptide macrocycle 3. First the
linear depsipeptide was assembled, then closure of the macrocycle was achieved via
amide bond formation between proline and a pentafluorophenyl ester. Subsequent side-
chain attachment led to the formation of 4.
NN
NHO
O
O
N
OONHCBz
O OPAc
Boc1. Zn, HOAc, r.t., 4 h
2. CH2Cl2, EDC, pentafluorophenol,-20 ˚C - r.t., 20 h
1. HCl, dioxane, r.t., 2 h
NO
OO
NHO
NH
ON
ONHCBz
2. CHCl3, NaHCO3, H2O,r.t., 6 h 68 % 4 steps
1. MeOH, HCl, Pd/C/H2, r.t., 6 h2. BocPheOH, HATU,DIEA, 0 ˚C, 12 h
NO
OO
NHO
NH
ON
ONH
O
NH23. HBr, HOAc, r.t., 30 min, 84 % 3 steps
NN
NHO
O
O
N
OONHCBz
O OPfp
Boc
1 2
3 4 Scheme 1: Synthesis of macrolactone core by Schmidt et al.12

9
The lipophilic side-chain 8 was then synthesized from dialdehyde 4 by a Wadsworth-
Horner-Emmons reaction sequence. Coupling of the macrolactone 4 with side-chain
acid 8 using HATU, was followed by t-butyl ester deprotection, activation with pivaloyl
chloride as the mixed anhydride, and coupling with amine 2-aminocyclopentane-1,3-
dione gave Enopeptin B in overall 0.6 % yield (Scheme 2).
HH
O
OTHF, NaH, r.t., 3 h,75%
EtOPO
OEtO
OSi(tBu)Ph2
H
OOSi(tBu)Ph2
OTHF, NaH, r.t., 3 h,91%
EtOPO
OEtOtBu
O
OSi(tBu)Ph2
OO
OtBu
THF, HF, H2O,CH3CN, 1 h, 95% OH
OO
OtBu
1. DMF, HATU, DIEA,0 ˚C, 12 h2. CH2Cl2, TFA, r.t., 2 h
3. THF, PivOCl, NMM, r.t., 2 h
H2N
O
O
HCl
DMF, NMM, r.t., 3h6 % 3 steps
NO
OO
NHO
NH
ON
ONH
O
HN
ONH
O
O
HO
5 6
7 8
Enopeptin B
Scheme 2: Synthesis of lipophilic side chain and assembly of Enopeptin B

10
As interest in their antibacterial activity grew, the natural acyldepsipeptides A
54556 A and A 54556 B (ADEP 1A and 1B respectively) were investigated through a
structure activity relationship (SAR) medicinal chemistry approach, eventually leading to
optimized ADEP 2 (Scheme 3).16 The synthetic approach followed a similar route to that
employed by Schmidt et al., utilizing their method of macrolactonization with a
pentafluorophenyl ester.12 By using a convergent synthesis, several analogs were
prepared for SAR studies allowing for pharmacophore identification.
OH
ONH
N
ONBoc
O
NH
O
O
NHCBzO
O
PhO
+
1) CH2Cl2, HOBt, TBTU,DIEA, 0 ˚C - r.t., 62 % O
NH
N
ONBoc
O
N
O
O
NHCBzO
OH2) AcOH, H2O, Zn, 2h,r.t., 67 %
1) CH2Cl2, C6H5OH, EDC0 ˚C - r.t., 18 h2) 4 N HCl in dioxane, 1h
3) CH2Cl2, H2O, NaHCO3,r.t. 62 % 3 steps
NO
OO
NHO
N
ON
ONHCBz
1) MeOH, aq. HCl, H2,Pd/C, 92 %2) 3,5-difluor-N-Boc-Phe,HATU, DIEA, r.t., 87 %
3) CH2Cl2, TFA, H2O, 45 min,r.t., quant.
NO
OO
NHO
N
ON
ONH
O
NH2
F F
2-hexenecarboxylic acid,DMF, HATU, DIEA, 88 %
NO
OO
NHO
N
ON
ONH
O
HN
O
F F
910 11
12
13 ADEP 2 Scheme 3: Synthesis of optimized ADEP 21

11
1.6.1 Structure Activity Relationship Studies of the ADEPs
N
OO
O
ONHO
NN
O NH
O
HN
O
FF
Figure 6: Structure of Optimized ADEP 2
Modifications of the northern trans-4-methylproline residue were not well
tolerated. In addition to the synthetic challenges posed, substitution of the alkyl group to
an ethyl or methoxy group, and changing the position (4- to 3-) resulted in a loss of
antibacterial activity.16 Hinzen et al. found that substitution of the N-Me-alanine reside
with pipecolic acid resulted in an increase in potency by rigidification of the macrocyclic
scaffold.16 SAR studies of the side chain phenylalanine showed that incorporating
fluorine at the 3- and 5- position of the aryl ring increased potency, while introducing a
fluorine at the 4- position resulted in a loss of antibacterial activity. Replacing the phenyl
ring with an aliphatic (cyclohexyl) or heteroaromatic (pyridine) groups were not tolerated. Analog synthesis identified that the lipophilic side chain only required simple α,β-trans
unsaturation, allowing for increased chemical stability compared to the natural products
without compromising activity towards ClpP. The other double bonds were not essential
for biological activity.16 Moreover the length of the alkyl chain was important for potency.
It was found that heptenoic acid derivatives were superior to their shorter or longer alkyl
side chains.16

12
Figure 7: Binding site of ADEP 1A in BsClpP15
A crystal structure of ADEP 1A in the binding site of BsClpP shows several
interactions. Ser60 and Tyr62 are involved in hydrogen bonding interactions with the
side chain as well as the depsipeptide core.15 The octatrienoyl tail lies inside a
hydrophobic pocket created by Leu, Ile, Arg residues as well as the Ala52ʼ and Phe49ʼ
residues from the adjacent subunit. This supports the observation made earlier that
ADEP binding gives rise to subunit assembly and increased stability of the
tetradecamer.15 The phenyl ring also lies in a hydrophobic pocket surrounded by the
Tyr62, Leu189, Phe82ʼ, and Leu144 residues. In contrast the pentapeptidic core seems
rather solvent accessible, also supporting the SAR studies showing that modifications to
the linking phenylalanine and lipophilic side chain showed the most increase in
potency.15,16

13
1.7 Proposed Synthetic Approach
1.7.1 Solid Phase Peptide Synthesis
The previously reported synthesis of the ADEPs was performed in solution
phase. Unfortunately solution phase peptide synthesis can be labor intensive, requiring
isolation and purification of each intermediate along the peptide chain. Robert Bruce
Merrifield was awarded the Nobel Prize in Chemistry in 1984 for the development of his
solid phase peptide synthesis (SPPS) methodology, which has provided a major
breakthrough in peptide chemistry.17 The concept involves heterogeneous reactions on
an insoluble polymer. The peptide chain is assembled starting from the C-terminus,
where the first amino acid in the sequence is anchored to an insoluble polymer (Figure
8). This allows for easy separation of the excess reagents and side products from the
growing peptide chain through a simple filtration. Although SPPS does pose some
limitations in terms of scale, it is appropriate for an approach where only small quantities
of compounds are required. Therefore when synthesis is carried out on a polymeric
support, this eliminates the need for time-consuming isolation and purification of the
intermediates.

14
linkerO
OH2N
R1
coupling agent,base
linkerO
OHN
R1
NH
R2
OH
O
Y
ONH
R2
Y
piperidine (Fmoc synthesis)or TFA (Boc synthesis)
linkerO
OHN
R1OH2N
R2
linkerO
OHN
R1ONH
R2OHN
R3ONH
Rn
Cleavage from Resin
OH2N
Rn+1
n
OH
OHN
R1ONH
R2OHN
R3ONH
RnOH2N
Rn+1
n
coupling agent,baseN
H
R3
OH
O
Y
linkerO
OHN
R1ONH
R2O
R3
HN
Y
1. Coupling of the first amino acid
2. Deprotection of amine
3. Coupling of the next amino acid
4. Elongation of peptide repeat deprotection and coupling
Figure 8: Process of solid-phase peptide synthesis
The standard Merrifield system is based on a Boc-protecting group for the N-
terminal amine, and relies on a selective deprotection of the temporary protecting group
(Boc) using trifluoroacetic acid (TFA).17 Typically these peptides are attached to the
resin using 4-methylbenzyhydrylamine (MBHA) linkers.18 MBHA linkers require
treatment with liquid hydrogen fluoride (HF) for final cleavage of the peptide from the
solid support. In this case stability of the peptide during these deblocking and final
cleavage steps is essential. More specialized methods have been developed since the
introduction of Merrifieldʼs invention.

15
The Sheppard tactic is a widely applied alternative to the Boc scheme.19 This
approach utilizes the base liability of the fluorenyl-9-methyloxycarbonyl (Fmoc) group,
as a temporary protecting group for the N-terminal amine. The Fmoc is cleaved under
base-catalyzed conditions using piperidine, or 1,8-diazabicyclo[5.4.0]undec-7-ene
(DBU).19 The peptides are linked to their solid support using 2-chlorotrityl linkers (2-Cl-
Trt), which are cleaved under mildly acidic conditions. Typically the Fmoc approach is
preferred due to the associated hazards of working with TFA and anhydrous HF in the
Boc approach.
Our goal was to develop a rapid solid phase synthetic route to provide a
synthesis of similar ADEPs in order to further study their effect on ClpP. By using
conventional solid phase peptide synthesis, we proposed two routes to the ADEPs.

16
1.7.2 Macrolactonization Approach
The first proposed approach is based on a macrolactonization for formation of the
depsipeptide macrocycle (Scheme 4). This would consist of synthesizing the linear
chain on a solid support, then closing the ring using the serine hydroxyl and the free
carboxylate of the northern proline.
NO
OO
NHO
N
ON
ONH
O
HN
O
NO
NH
ON
O
N
O
NH
OH
O
HN
O
O OHADEP 1B 14
SPPS Scheme 4: Retrosynthesis of ADEPs, macrolactonization approach
A resin-bound synthesis would allow for quick assembly of the linear chain,
removing several purification steps. Also, a solid phase approach makes it much easier,
and faster to prepare several analogs, based on a combinatorial approach. Several
challenges face this proposed route, most notably the key step of forming the
macrocycle. Kahalalide B, a cyclic depsipeptide formed from six amino acids was
synthesized by a similar approach in 28 % yield.20 Cyclic depsipeptides are usually
synthesized utilizing an amide bond formation (macrolactamization) rather than through
an ester bond formation (macrolactonization), due to the greater nucleophilicity of an
amine over a hydroxyl group. Despite this challenge the proposed strategy would
provide a fast route to the formation of ADEPs.

17
1.7.3 Macrolactamization Approach
The second synthetic route proposed for the ADEPs also involves solid phase
peptide synthesis followed by a macrolactamization. Syntheses of several cyclic
depsipeptides take a macrolactamization approach including, emericellamide A,
largazole, and FK901228.9,21,22 In these three examples, macrolactonization was
attempted but failed due to the decreased nucleophlicity of the hydroxyl group for ester
formation. For this route the ester bond of the depsipeptide would be synthesized at an
earlier stage, and then the branched depsipeptide assembled on the resin. Subsequent
cleavage, deprotection, and the key macrolactamization step between the northern
proline and alanine residues of 15 would give ADEP 1B (Scheme 5). The branched
depsipeptide 15 can be assembled by solid phase peptide synthesis. Further
retrosynthetic analysis reveals three fragments: tripeptide 22, the depsipeptide unit 19,
and the lipophilic side chain 16. The side chain can be synthesized from the coupling of
phenylalanine with (E,E,E)-2,4,6-octatrienoic acid 17. The depsipeptide 19 can be
formed from the coupling of proline to an appropriately protected serine, and lastly the
tripeptide 22 can be prepared using Fmoc-chemistry for SPPS. This route benefits from
the solid support in much the same way as the macrolactonization route, in that the
branched depsipeptide 15 would be assembled without the need for purification of the
elongating depsipeptide. The exception being that a more feasible macrolactamizaion is
used for the ring-closure in comparison to the macrolactonization route.

18
NO
OO
NHO
N
ON
ONH
O
HN
OO
NH
ON
O
N
O
NH
O
O
HN
O
ON
O
NH
OH
O
O
Fmoc
ONBoc HPro-NMeAla-AlaO
O
HN
OOH
NBoc
OHO
+NH O
OBn
OH
PGOH
O
+
+ +
H2N
Ph
OPG
O
SPPS
ADEP 1B 15
16 19
22
17 18 20 21
Boc
Scheme 5: Retrosynthesis of ADEPs via a macrolactamization approach

19
1.8 Small Molecule Agonists of ClpP
While research was pursued in the area of the ADEPs as agonists of ClpP, Prof.
Walid Houry from University of Toronto, Department of Biochemistry, led a team
investigating compound libraries to find alternative compounds for the activation of ClpP.
By screening the Maybridge collection (50,000 compounds) and Chembridge collection
(10,000 compounds), several compounds were found to activate ClpP towards the
degradation of fluorescein isothiocyanate labelled casein (casein-FITC).
These compounds were later termed Activators of Self-Compartmentalizing
Proteases (ACP). One compound from the Maybridge collection, BTB09142 termed
ʻACP1ʼ stood out for itʼs high biological activity in the assay (Figure 9). Other
compounds tested in the screen also showed ClpP activation properties, but the “drug-
like” character of ACP1 and the ease of synthesis, encouraged us to choose ACP1 as a
lead structure for analog studies.
N
F
F F
SHN
OS
O
O
Figure 9: Structure of ACP1

20
2.0 Results and Discussion
2.1 Towards the synthesis of ADEPs
2.1.1 Linear approach
Model peptide 22 was prepared by SPPS in order to determine the optimal
coupling and cleavage conditions. Several changes were made from the natural ADEP
1B system. The N-Me-alanine residue was replaced with proline, in an attempt to
conserve the number of N-H bonds in the cyclic depsipeptide. The octatrienoic acid
fragment was replaced with commercially available sorbic acid in the model system. The
peptide synthesis proceeded with 78 % yield (mass gain on resin) on the 2-Cl-Trt resin
using standard Fmoc chemistry (Scheme 6).23 Utilizing the standard TFA + scavenger
cleavage system for the 2-Cl-Trt resin, the peptide was recovered in low yield (17 %) by
washing with diethyl ether then extraction with chloroform.24 Due to the highly lipophilic
nature of the peptide, it could not be recovered from the crude TFA cleavage by
conventional methods such as precipitating in methoxy-tert-butyl ether (MTBE). An
attempt to close the macrocycle using PyBOP/DIEA (3:6 eq) in DMF (10-3 M) for 24
hours, a previous method described for Kahalalide B, was unsuccessful with this system
and lead to decomposition.20

21
NH
ON
ON
ONH
TrtOO
NH
O
O
N
O O-
TFA/TIS/H2O
NH
ON
ON
ONH
HOO
NH
O
O
N
O OH
PyBOP, DIEA,DMF, r.t., 24 h
NO
OO
NHO
N
ON
ONH
O
HN
O
17 %SPPS
FmocAA, HBTU,iPr2EtN, NMP
78 %
22
23 24 Scheme 6: Synthesis of model peptide 24
In order to synthesize the natural ADEP 1B, the octatrienoic acid side chain 26
was prepared using a Horner-Wadsworth Emmons approach with sorbic aldehyde
(Scheme7).25-27 Variation of the base and solvent in the Horner-Wadsworth-Emmons
reaction did not significantly alter the yield or stereoselectivity of the product.
H
O (EtO)2POCH2CO2Et,base, solvent, time
O
O
25 26 Scheme 7: Synthesis of (2E,4E,6E)-ethyl octa-2,4,6-trienoate 26

22
Table 1: Optimization of Horner-Wadsworth-Emmons Reaction
Entry Base Solvent Time (h) Conversion (%) E/Z
1 LiOH⋅H2O THF 8 63 94:6
2 LiOH⋅H2O THF 48 73 92:8
3 LiOH⋅H2O THF 16 82 85:15
4 LiOH⋅H2O CH2Cl2 16 55 82:18
5 LiOH⋅H2O DME 16 94 83:17
6 nBuLi THF 16 98 85:15
7 nBuLi DME 16 95 86:14
8 NaH DME 16 81 89:11
The use of sodium hydride (NaH) in DME afforded the optimal conditions for a
gram scale reaction, typically yielding the ester 26 in 70–80 % yield. Hydrolysis of the
ester gave (2E,4E,6E)-octa-2,4,6-trienoic acid 17 in 56% yield.
H
O (EtO)2POCH2CO2Et,NaH, THF, r.t.
O
O NaOH, MeOH/H2O,reflux, 45 min
OH
O
81 %25 26
17
56 %
Scheme 8: Synthesis of lipophilic side chain
Synthesis of the linear peptide 28 gave the precursor to an ADEP 1B analog,
differing only in the length of the lipophilic side chain. The peptide was prepared by
SPPS in 81 % yield (by gained mass on resin). Monitoring the peptide coupling using
the appropriate colorimetric test (Kaiser, isatin, p-chloranil) revealed that the standard
ʻdouble couplingʼ method was insufficient for coupling N-Me-alanine to the proline
carboxylate and required the use of a more active coupling reagent, HATU. Cleavage
from the resin yielded 53 % of the linear peptide 14. Attempts to close the macrocycle 14 using EDC⋅HCl, PyBOP, diisopropylcarbodiimide (DIC) and N-methylimidazole (NMI)
were unsuccessful and resulted in either decomposition, or no reaction.

23
Macrolactonization was not found to be a widely utilized route to other known cyclic
depsipeptide natural products, and therefore was abandoned as a short route to the
ADEPs.6,28-31
NO
NH
ON
O
N
O
NH
OTrt
O
HN
O
O O
SPPS
TFA/TIS/H2O(92.5:5:2.5)
N
OOH
NHO
O
NN
O
O NH
OHO
HN
O
macrolactonization
NO
OO
NHO
N
ON
ONH
O
HN
O
FmocAA, HBTU,iPr2EtN, NMP
53 %
81 %
27
14 ADEP 1B - analog Scheme 9: Linear Approach to ADEP 1B

24
2.2 Macrolactamization Approach to the ADEPs
2.2.1 Synthesis of the Depsipeptide Unit
The macrolactamization route to the ADEPs required depsipeptide unit 19 for
assembly of the branched depsipeptide 15. The synthesis of 19 (Scheme 8) was
adapted from a previous report on the depsipeptide methodology for SPPS.32 The
protection of the serine carboxylate was achieved with benzyl chloride in 48 % yield.
This step was improved by using benzyl bromide, giving typical yields of 90 % or greater
on a gram scale. Protection of the orthogonal hydroxyl group was not found to be necessary. Coupling of the serine hydroxyl group with excess BocProOH and EDC⋅HCl
in CH2Cl2 gave the protected depsipeptide in 83 % yield. Next hydrogenolysis of the
CBz and benzyl ester protecting groups gave the unprotected depsipeptide 31 in
quantitative yield. In order for this depsipeptide unit to be compatible with the SPPS
approach, an Fmoc protecting group needed to be installed on the serine amine.
Attempts were made to install this protecting group without success. Coupling of the
amino group with Fmoc-O-succinimide (FmocOSu) in acetonitrile and water under basic
conditions (Et3N or NaHCO3) gave a crude product, which was not soluble in methanol,
chloroform, dichloromethane, dimethylsulfoxide (DMSO), acetone, ethyl acetate,
hexanes, or water. Possibly the use of a stronger base (K2CO3) would have afforded the
desired product.

25
NH
OH
OO
OHO
NH
O
O
CBz
OH
NH
O
O
O
O
CBzBn
NBoc
H2NOH
O
O
ON
NH
OH
O
ONBoc
1) Cs2CO3, MeOH2) BnBr, DMF
BocProOH, EDC⋅HCl, DMAP, CH2Cl2
H2, Pd-C, THF FmocOSu, base, CH3CN/H2O (1:1)
O
Fmoc
Boc
90 % 96 %
quant.
28 29
30 31 19 Scheme 10: Synthesis of Depsipeptide 19 from CBzSerOH
In this case an alternate approach was taken to the synthesis of the depsipeptide
unit. It was previously reported that the benzyl ester could be removed by
hydrogenolysis with minimal cleavage of the Fmoc protecting group.32 Therefore the
synthesis was carried out with FmocSerOH, outlined in Scheme 11.
NH
OH
O
OH
NH
O
O
Fmoc
OH
NH
O
O
O
O
FmocBn
NBoc
NH
OH
O
O
ON
1) Cs2CO3, MeOH2) BnBr, DMF
BocProOH, EDC⋅HCl, DMAP, CH2Cl2
H2, Pd-C, THF Boc
Fmoc
Fmoc72 % 77 %
90 %
32 33
34 19 Scheme 11: Synthesis of Depsipeptide 19 from FmocSerOH
The synthesis was carried out with an overall yield of 50 % on a gram scale. The
final hydrogenolysis was achieved in 90 % yield to give depsipeptide 19, which was
readily soluble in several organic solvents. This result implies that the product obtained
from the route shown in Scheme 10 was not the desired depsipeptide 19. The next
steps involved incorporating this unit into a branched depsipeptide using SPPS.

26
Typically the reported ʻdepsipeptide methodʼ developed by Irene Coin, is used for
the synthesis of difficult sequences when synthetic difficulty arises due to aggregation of
the elongating peptide.32 When using this methodology, depsipeptide bonds are
incorporated in the elongating peptide during SPPS, but are isomerized in an O,N-acyl
shift after cleavage from the solid support. The resulting peptide does not actually
incorporate the depsipeptide bond, which is only used to prevent aggregation (by
increasing the peptideʼs solubility) during the synthesis of the peptide on the resin.29,32-34
We attempted to apply the ʻdepsipeptide methodʼ with the absence of the O,N-acyl shift
step to the synthesis of the ADEPs.
NH
C-TermO
H2N
O
OHN
RN-Term
N-TermHN
NH
O
R
HN
OH
OC-Term
pH > 7
Figure 10: O,N-Acyl shift for the depsipeptide methodology

27
Depsipeptides have been incorporated in SPPS to synthesize natural cyclic
depsipeptides such as cotransin, which used an anchored lactic acid to install the
depsipeptide bond with N-Me-alanine.29 It was demonstrated that the depsipeptide
bonds were stable to standard Fmoc SPPS in the synthesis of cotransin, and other
depsipeptides.20,29,32
Fmoc-Leu-MeLeu-Leu-MePhe-Leu-MeAla-(-)-lac
O
H-[linear cotransin]-OH
NHN
O O
OO
N ONH
ON
O
O
HN
cotransin
OHO
O
OO
ON
Fmoc-MeAla-OHDIC/NMI
Fmoc
SPPS
HATU/DIEA,CH2Cl2 (0.5 mM)
acidic deprotection
Scheme 12: Solid-phase synthesis of cotransin a cyclic depsipeptide29
The branched depsipeptide 37 was then assembled as outlined in scheme 13. In
an attempt to increase stability of the resulting depsipeptide, it was cleaved from the
resin after addition of the depsipeptide fragment 19 under previous TFA cleavage
conditions. The depsipeptide was isolated by precipitation in cold diethyl ether to give a
brown sticky residue in low yield (14 %). 1H NMR and ESI MS of the crude product
suggested that the Boc protecting group was also successfully cleaved from the proline
residue under these conditions.
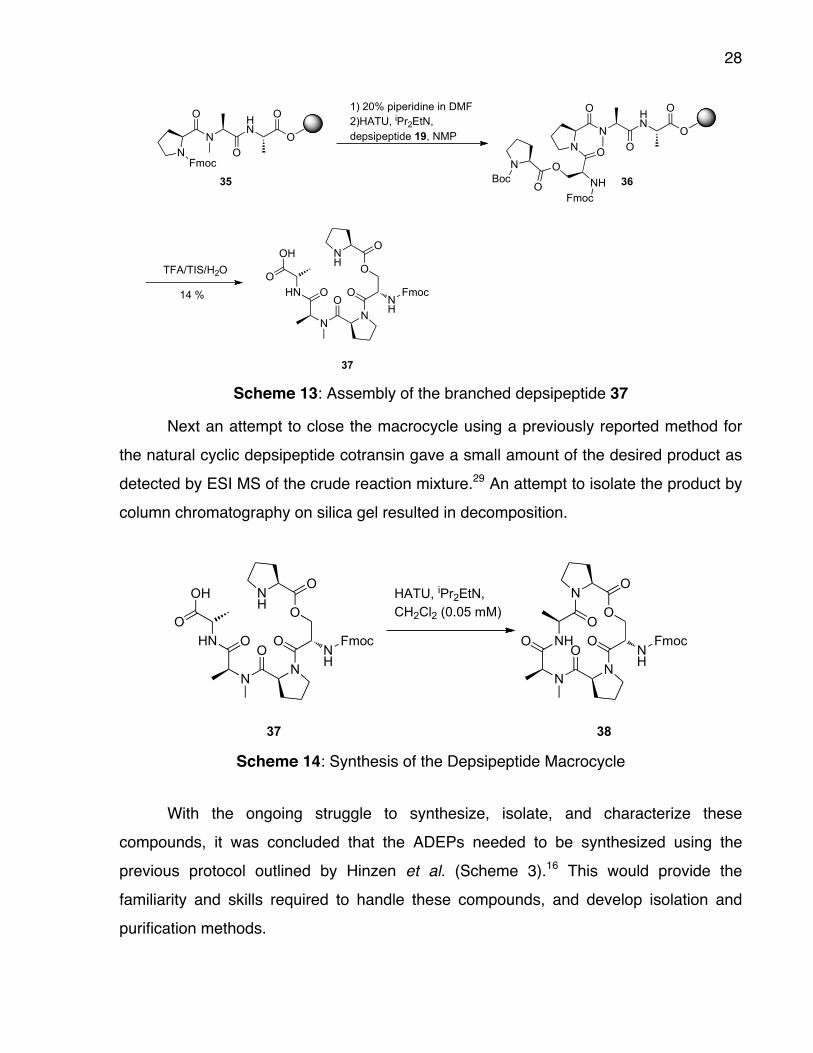
28
O
OHN
ON
O
NFmoc
1) 20% piperidine in DMF2)HATU, iPr2EtN, depsipeptide 19, NMP O
OHN
ON
O
N O
NHO
O
NBoc
Fmoc
TFA/TIS/H2ONH
O
O
N
ON
ONH
FmocOHNO
OH
14 %
35 36
37 Scheme 13: Assembly of the branched depsipeptide 37
Next an attempt to close the macrocycle using a previously reported method for
the natural cyclic depsipeptide cotransin gave a small amount of the desired product as
detected by ESI MS of the crude reaction mixture.29 An attempt to isolate the product by
column chromatography on silica gel resulted in decomposition.
NO
OO
NHO
N
ON
ONH
Fmoc
HATU, iPr2EtN,CH2Cl2 (0.05 mM)
NH
O
O
N
ON
ONH
FmocOHNO
OH
37 38 Scheme 14: Synthesis of the Depsipeptide Macrocycle
With the ongoing struggle to synthesize, isolate, and characterize these
compounds, it was concluded that the ADEPs needed to be synthesized using the
previous protocol outlined by Hinzen et al. (Scheme 3).16 This would provide the
familiarity and skills required to handle these compounds, and develop isolation and
purification methods.
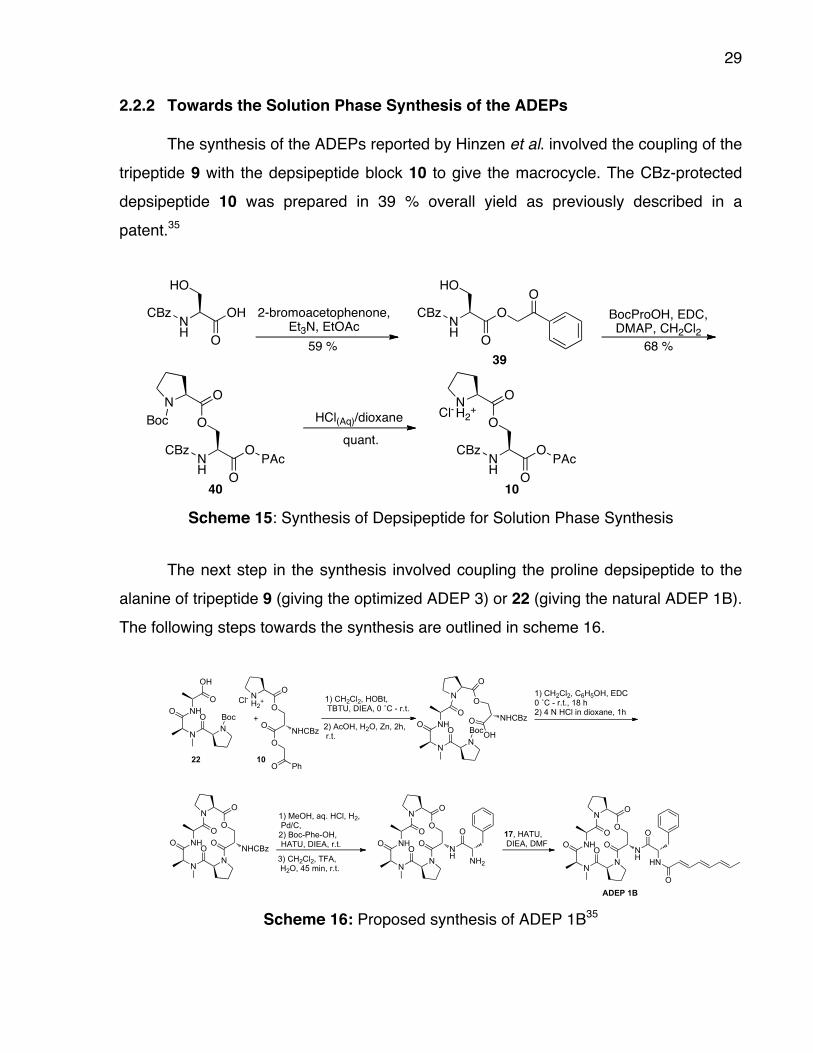
29
2.2.2 Towards the Solution Phase Synthesis of the ADEPs The synthesis of the ADEPs reported by Hinzen et al. involved the coupling of the
tripeptide 9 with the depsipeptide block 10 to give the macrocycle. The CBz-protected
depsipeptide 10 was prepared in 39 % overall yield as previously described in a
patent.35
NH
HO
OH
O
2-bromoacetophenone,Et3N, EtOAc N
HO
O
OBocProOH, EDC, DMAP, CH2Cl2
NH
O
OPAc
O
ONBoc HCl(Aq)/dioxane
quant.
CBz CBz
CBz
HO
59 % 68 %
NH
O
OPAc
O
ONH2
+
CBz
Cl-
39
40 10 Scheme 15: Synthesis of Depsipeptide for Solution Phase Synthesis
The next step in the synthesis involved coupling the proline depsipeptide to the
alanine of tripeptide 9 (giving the optimized ADEP 3) or 22 (giving the natural ADEP 1B).
The following steps towards the synthesis are outlined in scheme 16.
OH
ONH
N
ONBoc
O
NH2
+
O
O
NHCBzO
O
PhO
+
1) CH2Cl2, HOBt, TBTU, DIEA, 0 ˚C - r.t. O
NH
N
ONBoc
O
N
O
O
NHCBzO
OH2) AcOH, H2O, Zn, 2h, r.t.
1) CH2Cl2, C6H5OH, EDC0 ˚C - r.t., 18 h2) 4 N HCl in dioxane, 1h
NO
OO
NHO
N
ON
ONHCBz
1) MeOH, aq. HCl, H2, Pd/C,2) Boc-Phe-OH, HATU, DIEA, r.t.
3) CH2Cl2, TFA, H2O, 45 min, r.t.
NO
OO
NHO
N
ON
ONH
O
NH2
17, HATU, DIEA, DMF
NO
OO
NHO
N
ON
ONH
O
HN
O
22 10
Cl-
ADEP 1B Scheme 16: Proposed synthesis of ADEP 1B35

30
Due to time restraints and the discovery of new compounds that displayed
biological activity for ClpP activation the synthesis of the ADEPs was delayed. This
allowed for the investigation of a new compound ACP1 that displayed promising activity
towards ClpP, including the synthesis and preliminary SAR studies of a number of
analogs of ACP1.

31
2.3 Synthesis of ACP1 and analogs
In collaboration with Professor Walid H. Houry and Elisa Leung at the University
of Toronto, Biochemistry department, several small molecules were identified with
activity in a high throughput screen with ClpP and Fluorescein isothiocyanate labelled
casein (Casein-FITC). The compounds that displayed the highest in vitro activity to
induce ClpP protease in the absence of ClpATPases are shown below in figure 11.
N
F
FF
SHN
OS
O
ONH
Fmoc
O O
tBu
HN
OStBu
NH
OO
tBu
OH
O
N
N
ClN
O
HO
O
O
O
ClCl
O2N
HO
O
O
O
ClCl
Br
ACP2ACP1
ACP3 ACP4
ACP5 Figure 11: Structures of ACP compounds
ACP1 was picked as a lead from the five compounds for a number of reasons.
ACP2 is simple a tBu-protected tripeptide, which likely would not survive enzymatic
degradation in vivo. ACP 4, and 5 present synthetic challenges, particularly for analog

32
synthesis and therefore were not investigated. ACP1 displays several characteristics of
a lead compound according to Lipinskiʼs rule of five36: it has less than five hydrogen
bond donors, and less than ten acceptors, and the molecular weight is less than 500.
ACP3 also displays these desirable characteristics and like ACP1 appears to be “drug-
like”, since the activity of ACP1 was higher than ACP3, ACP1 was chosen as the lead
structure for further investigation.
The synthesis of ACP1 was adapted from a recent patent,37 as shown in scheme 17. By coupling 2-mercapto-5-(trifluoromethyl)pyridine with ethyl-α-bromoisobutyrate,
ester 43 could be formed under basic conditions in 96 % yield. Oxidation of the thioether
to the sulfone was first performed using Oxone© monopersulfate, but resulted in low
yields and a complex mixture of products that was difficult to separate using column
chromatography. It was found that oxidation using mCPBA gave comparable yields but
the resulting sulfone was relatively pure after aqueous workup. Hydrolysis of the ester
provided the acid 45 in 46 % overall yield. The final step in the synthesis of the ACP1
analogs was the coupling of 45 with an amine. Ultimately, a PyBOP based coupling
procedure was adopted.
N
F
FF
SH
Br O
O+
LiOH⋅H2O, EtOHreflux, 16 h
N
F
F F
S O
O
mCPBA, NaHCO3,CH2Cl2, 0 °C - r.t.
N
F
FF
SO
OO
O
LiOH⋅H2O,THF/H2O (4:1)
N
F
FF
SOH
OO
O
PyBOP, iPr2EtN,RNH2, DMF
N
F
FF
SHN
OO
OR
96 %
60 % 81 %
30 - 80 %
41 42 43
44 45
Scheme 17: Synthesis of ACP1 Analogs

33
The first target structure was ACP1 for which coupling with amine 46 was
necessary. Amine 46 was prepared through a nucleophilic substitution of 2-
bromoethylamine hydrobromide with thiophenol (Scheme 18).
HS+Br
H2NHBrK2CO3, EtOH
89 % SH2N
46 Scheme 18: Synthesis of 2-(Phenylthio)ethanamine
The reported reaction conditions for formation of amides from acid 45 involved
the use of an acyl chloride thionyl chloride. Attempted formation of the acyl chloride
using oxalyl chloride and DMF led to decomposition of the starting material. EDC
coupling conditions only returned unreacted starting materials, likely due to the steric
bulk of the gem-dimethyl substitution adjacent to the carboxylic acid. DCC coupling gave
poor yields of ACP1, and isolation of the product required purification by column
chromatography twice to remove the dicyclohexylurea (DCU) byproduct. The use of
uronium (HATU) and phosphonium (PyBOP) based coupling reagents gave ACP1 in
moderate yields with the latter giving slightly better yields.
N
F
F F
SOH
OO
O H2NS
N
F
F F
SHN
OS
O
O+ conditions
45 46 ACP1 Scheme 19: Synthesis of ACP1
Table 2: Amide Coupling Conditions for ACP1 Synthesis
Entry Conditions Yield (%) Comments
1 1. (COCl)2, DMF, Et2O 2. 46, Et3N, CH2Cl2
28 Complex mixture
2 EDC⋅HCl, DMAP, DMF, 48 h 0 S.M. recovered
3 DCC, DIEA, DMF, 18 h 14 Contaminated with DCU
4 PyBOP, DIEA, DMF, 1 h 56
5 HATU, DIEA, DMF, 1 h 46

34
The PyBOP conditions were then applied for the coupling of various amines to
acid 45 to prepare a set of analogs in which the “eastern” side of the molecule was
varied relative to ACP1. The compounds shown below in table 3 were synthesized from
coupling the corresponding amine to acid 45 in the presence of PyBOP and DIEA (1 eq:
3 eq) in peptide grade DMF and isolated in moderate to good yields by column
chromatography. Table 3: Structures of ACP1 and “eastern” modification analogs
Compound Structure Yield (%) Compound Structure Yield (%)
ACP1
56 54
51
47
88 55
31
48
42 56
26
49
Quant. 57
65
50
48

35
Several other analogs were synthesized to investigate different aspects of the
“western” half of the molecule, i.e., the sulfonylpyridine moiety. Analogs with deletions of
the trifluoromethyl group, gem-dimethyl substitution, and oxidation state of the sulphur
attached to the pyridine ring, were all synthesized by varying the starting materials in the
synthesis of the analogs as outlined in Scheme 17. The modified sulfonylpyridine moiety
was coupled to amine 46 in the presence of PyBOP and DIEA (1 eq: 3 eq) in peptide
grade DMF. The compounds synthesized are outlined in table 4 and were isolated by
column chromatography in low to moderate yields.
Table 4: Structures of ACP1 “western” modification analogs
Compound Structure Yield (%)
51 N
F
FF
SHN
OS
31
52 N SHN
OS
O
O
62
53 N
F
FF
SHN
OS
O
O
68
2.4 Discussion of SAR studies
These analogs were then evaluated in an in vitro assay performed by Elisa
Leung at the University of Toronto, Biochemistry Department. The assay involved
incubating E. coli ClpP with the ACP1 analog and observing the degradation of Casein-
FITC after 6 hours, with varying concentrations, in relation to the control system (ClpP
incubated with the chaperone ClpA). This relationship was presented as the relative
degradation of Casein-FITC at 25 μM concentration of the compound (RD25). Therefore,
a higher RD25 value implies a more activating compound.

36
FF
F
F
F F F F
F
F
F
F
FF
FF
FF
F
ProteolyticDigestion
F
F
FF
F
F
F
F
F
F
Fluorescent Signal:Ex. 485 nmEm. 538 nm
Casein-FITC
ClpP + Compound(25 µM)
+
Figure 12: in vitro assay of ACP1 analogs
The structures of the ACP1 analogs as well as ADEP 1 and 2 and their
respective RD25 values are presented below in Table 2. The ADEPs 1A and 1B were
included in the assay as a reference. The activation of ClpP by the natural ADEPs 1A
and 1B was confirmed, and the ranking of the activators was ADEP 1A > ADEP 1B >
ACP1 in the initial assay. The target of the medicinal chemistry optimization of ACP1
was to synthesize a compound with comparable in vitro activity as ADEP 1A. There
were several interesting results involving the variations on the ʻwesternʼ sideʼ of ACP1
(left of the amide bond). It was found that the key structural elements found in the
ʻwesternʼ fragment of ACP1 were required for activity towards ClpP. Deletion of the
trifluoromethyl group or gem-dimethyl groups as for 56 and 57 resulted in a loss of
activity. Also analog 55 having a fully reduced sulphur adjacent to the pyridine did not
show any activity in the screen. Therefore, the modifications of the ʼwesternʼ side of
ACP1 were poorly tolerated, and hence these three groups were important for biological
activity of ACP1.
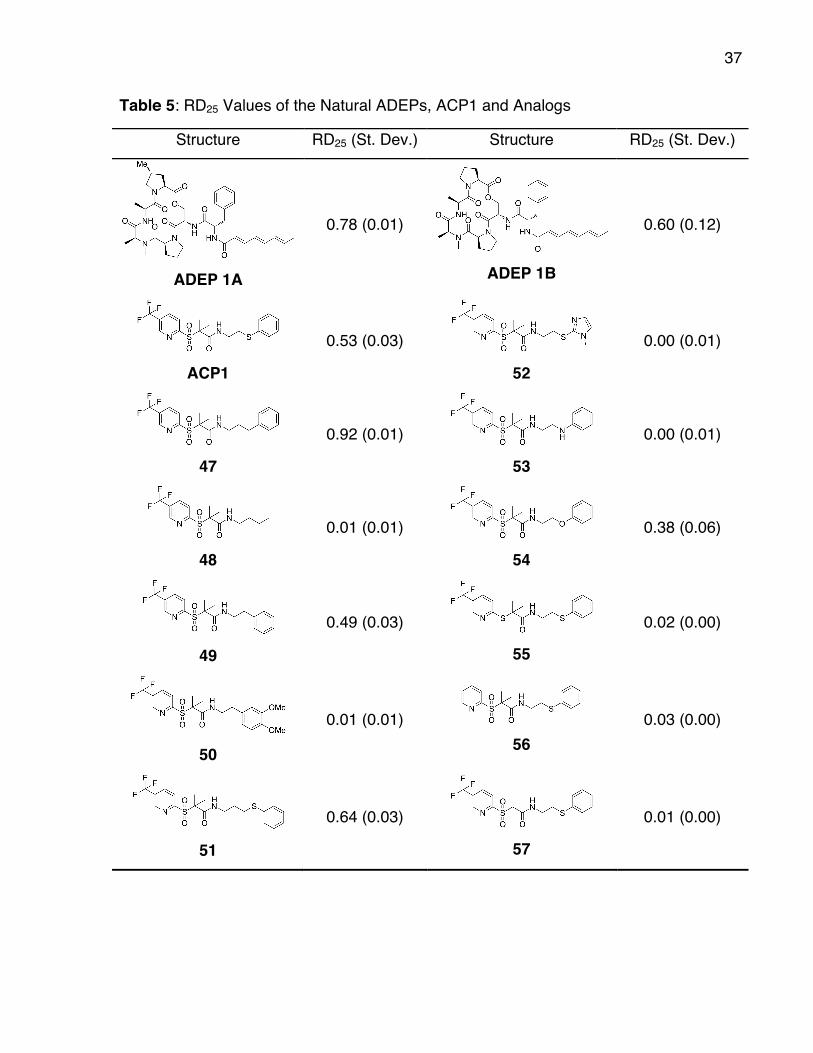
37
Table 5: RD25 Values of the Natural ADEPs, ACP1 and Analogs
Structure RD25 (St. Dev.) Structure RD25 (St. Dev.)
ADEP 1A
0.78 (0.01)
ADEP 1B
0.60 (0.12)
ACP1
0.53 (0.03) 52
0.00 (0.01)
47
0.92 (0.01) 53
0.00 (0.01)
48
0.01 (0.01) 54
0.38 (0.06)
49
0.49 (0.03) 55
0.02 (0.00)
50
0.01 (0.01) 56
0.03 (0.00)
51
0.64 (0.03) 57
0.01 (0.00)

38
Modifications to the ʻeasternʼ fragment of ACP1 (right of the amide bond) were
also interesting and led to a more activating compound 47. Deletion of the sulfide and
replacement with a methylene group (analog 47) gave a large increase in activity
relative to ACP1, and even having improved activity over ADEP 1A. Analogs 50, 53, and
54, with more electron-rich aryl rings displayed either reduced or no detectable activity.
In analogs 53 and 54 the sulfur was replaced by the more electronegative heteroatoms,
nitrogen and oxygen respectively. The oxygen substituted analog 54 showed reduced
activity, while the nitrogen substituted analog 53 displayed no activity. The latter result
could be attributed to the fact that a hydrogen bond donor may not be tolerated at this
site, and the analog may not bind to ClpP. Synthesis of an N-methylated analog may
gave insight to this observation.
Next the length between the amide bond and the benzene ring was studied.
Increasing the space by one methylene group (analog 51) gave a slight increase in
activity. Deletion of the sulfur and truncation of the space between the benzene and
amide bond (analog 49) gave a slight decrease in activity. Removal of the aromatic ring
to give a simple alkyl group for the ʻeasternʼ side of the molecule in analog 48 led to no
activity towards ClpP indicating that the aromatic ring is required for activity. Analog 52
in which substitution of the aryl ring for a heteroaromatic group has occurred, also gave
no activity, though it is not know how less drastic substitutions would affect activity.
These analogs have allowed us to gain insight into the important
pharmacophores present in the lead compound ACP1. Further studies are required to
understand the mechanism by which the ACP1 analogs function, and whether the mode
of action is the same as for the ADEPs. Analog synthesis yielded a compound (analog
47) which had improved activity towards ClpP over the lead ACP1 and the natural ADEP
1A. An x-ray crystal structure of a ClpP-ACP1 or ClpP-47 would provide invaluable
information for the guided synthesis of new ACP1 analogs.

39
2.5 Conclusions
ClpP has presented itself as a promising target for the development of novel
antibiotics. New antibiotics that function by a novel mechanism are of great need in a
world where antibiotic resistance is prevalent. The ADEP natural products have
demonstrated activity towards ClpP turning it from tightly controlled to an unregulated
protease. Previous work by Hinzen et al. investigated the SAR of the ADEPs leading to
a medicinal chemistry optimized ADEP.16 Our goal of developing a faster, more efficient
synthetic route to the ADEPs is still in progress, while an interesting compound ACP1
was presented that displayed similar in vitro activity. ACP1 was synthesized along with
12 compounds, one of which displayed increased activity towards ClpP relative to ACP1
and even ADEP 1A. This compound, 47 should be further investigated in ex vivo studies
with similar bacteria that displayed sensitivity to the ADEPs, B. subtilis, S. aureus, E.
faecalis, and S. pneumoniae. Compound 47 should also be further studied to determine
the binding affinity and the site of binding, preferably gaining an x-ray crystal structure of
the ClpP-47 complex. This would guide future analog synthesis to one-day lead to the
development of a new antibiotic.

40
3.0 Experimental 3.1 General Experimental THF was distilled from sodium metal/benzophenone ketyl under nitrogen. CH2Cl2 was
distilled from CaH2 under argon. All other solvents were used as obtained except 1,4-
dioxane, which was distilled prior to use. All commercial reagents were used as
received (Aldrich, Fischer Scientific Ltd., Aapptec, Nova Biochem etc.). Solvents DMF,
and NMP were peptide grade and used as received (Caledon Laboratory Chemicals).
Solid-phase peptide synthesis was performed using the Aapptec Endeavor 90 Peptide
Synthesizer. All glassware was oven dried and cooled under a stream of nitrogen, or
flame dried under a stream of dry nitrogen with the exception of the reaction vessels for
the Endeavor 90 peptide synthesizer, which were dried under vacuum. All FT-IR spectra
were obtained on a Shimadzu FT-IR 8400S spectrometer, with samples loaded as neat
films on NaCl plates. 1H NMR and 13C NMR spectra were obtained on Varian Unity 300
or 400 spectrometers as solutions in CDCl3, CD3OD, or DMSO-d6. Chemical shifts are expressed in δ (ppm) values. Spectra are referenced 7.24 ppm, 3.31 ppm, and 2.50
ppm for CDCl3, CD3OD, and DMSO-d6 respectively for proton chemical shifts, and
77.00 ppm and 49.00 ppm for CDCl3, and CD3OD respectively for carbon chemical
shifts. The following abbreviations are used: s = singlet, d = doublet, t = triplet, q =
quartet, m = multiplet, dd = doublet of doublets, td = triplet of doublets, and br = broad.
Low resolution mass spectra (MS) were recorded on an Hewlett Packard 1100 HPLC
with mass spectrometer detector, high resolution mass spectra (HRMS) were recorded
on an AB/Sciex QStar mass spectrometer. Flash chromatography on silica gel (60 Å,
230 – 400 mesh, obtained from Silicycle® Inc.). Analytical thin layer chromatography
(TLC) was performed on precoated silica gel plates, (SiliaPlate™Aluminium with F245
indicator purchased form Silicycle® Inc.), visualized with UV254 lamp (Entela, long/short
wave assembly 115 V, 60 Hz, 0.16 Amps) and stained with either 20 %
phosphomolybdic acid in ethanol or ninhydrin. Solvent systems used to determine Rf
values and for chromatrography are reported as v/v ratios.

41
Synthetic Preparations
3.1.1 Linear Peptide Synthesis (2E,4E)-Hexa-2,4,-dienoyl-Phe-Ser-Pro-N-MeAla-Ala-ProOH (14):
N
OOH
NHOO
NN
O
O NH
OHO
HN
O Standard solid phase peptide synthesis was performed on the Aapptec Endeavor 90
Peptide Synthesizer using a standard Fmoc protocol (0.5 M). A preloaded H-Pro-2ClTrt
resin (1.00 g, 100 – 200 mesh, 0.71 mmol/g) was used as the solid support. Couplings
were completed using 3.0 equivalents of Fmoc-protected amino acid, 3.0 equivalents of
HBTU, and 6.0 equivalents of diisopropylethylamine. The preactivated amino acid
solution was added to the resin and mechanically mixed under N2 atmosphere for 15
minutes. Each cycle consisted for double couplings with the exception of Fmoc-NMe-
AlaOH and FmocProOH, which were performed using 1.5 equivalents of Fmoc-
protected amino acid, 1.5 equivalents of HATU, and 3.0 equivalents of
diisopropylethylamine. These activated amino acid solutions were manually added to
the resin and mechanically mixed under N2 atmosphere for 20 minutes. These cycles
(Fmoc-NMe-AlaOH, and FmocProOH) consisted of single couplings. Coupling reactions
were monitored using the appropriate colorimetric test of the 0.5 – 1 mg of resin beads
(Kaiser test for primary amines38, Isatin test for proline39, and p-Chloranil test for
secondary amines40). Fmoc removal was performed using 20 % (v/v) piperidine in DMF
(1 x 3 min then 1 x 15 min). The resin-supported product was obtained with 81 % yield
(1.352 g, resin mass).
The peptide was cleaved from the solid support by treatment with a mixture of TFA
(9.25 mL), triisopropylsilane (0.5 mL), and distilled H2O (0.25 mL). The reaction mixture

42
was cooled in an ice bath with occasional swirling for 3 h. The resin was filtered off by
passing the mixture through a plug of glass wool in a Pasteur pipette and the resin was
rinsed with additional TFA (3 x 0.5 mL). The combined filtrates were collected and
reduced under N2 flow. The crude yellow residue was suspended in H2O (10 mL) and
extracted with Et2O (2 x 10 mL), followed by CHCl3 (3 x 10 mL). The CHCl3 extract was
washed with brine (5 mL), dried over MgSO4, filtered and concentrated to give a white
powder (101 mg, 15 %). The Et2O extract was washed with brine (5 mL), dried over
MgSO4, filtered and concentrated. The residue from the Et2O was suspended in H2O (5
mL) and extracted with CHCl3 (3 x 5 mL). The second CHCl3 extract was washed with
brine (5 mL), dried over MgSO4, filtered, and concentrated to recover and additional 69
mg (10 %) of white powder; mp = 133 – 136 °C; 1H NMR (rotamers, 400 MHz, CDCl3) δ
9.05 (1H, br s), 8.70 (1H, br s), 8.35 (1H, br s), 7.59–7.58 (1H, m), 7.29–7.16 (5H, m),
6.96 (1H, d, J = 6.4 Hz), 6.74 (0.7H, d, J = 6.4 Hz), 6.58–6.56 (0.3H, d, J = 6.4 Hz),
6.17–6.07 (2H, m), 5.77 (1H, d, J = 14.8 Hz), 5.18 (1H, d, J = 8.5 Hz), 4.99–4.97 (2H,
m), 4.91–4.80 (2H, m), 4.74–4.71 (1H, m), 4.66 (1H, q, J = 6.4 Hz), 4.55 (1H, dd, J =
4.2, 8.5 Hz), 4.50 (1H, dd, J = 5.2, 8.5 Hz), 4.02 (1H, m), 3.88–3.81 (1H, m), 3.74–3.55
(4H, m), 3.26 (1.5H, d, J = 4.0 Hz), 3.21 (0.5H, d, J = 5.2 Hz), 3.17 (1H, s), 2.77 (1H, s),
2.30–2.25 (2H, m), 2.21–2.08 (2H, m), 2.03-2.00 (3H, m), 1.84 (3H, d, J = 6.0 Hz), 1.43
(2H, d, J = 7.2 Hz), 1.39 (1H, d, J = 7.2 Hz), 1.35-1.33 (2H, m), 1.25 (2H, s), 1.05 (1H, s); 13C NMR (100 MHz, CDCl3) δ 173.5, 173.1, 172.3, 171.8, 170.7, 166.6, 143.1, 139.2,
136.2, 129.9, 129.6, 128.1, 126.9, 120.6, 59.2, 57.4, 53.8, 52.6, 48.2, 47.7, 47.3, 38.9,
31.0, 29.9, 29.1, 28.9, 28.3, 25.0, 23.9, 18.8, 18.6, 17.9, 15.8; MS (ESI) m/z 735
(C35H48N6O9K+, 8),719 (C35H48N6O9Na+, 40), 697 (C35H49N6O9+, 53), 582 (7), 551 (10),
529 (10), 511 (15), 456 (21), 423 (7), 388 (19), 376 (97), 368 (100), 337 (11), 288 (7);
HRMS (ESI) m/z (C35H49N6O9+) calcd. 697.3555, found 697.3529.

43
Synthesis of the Lipophilic Side Chain (2E,4E,6E)-Ethyl octa-2,4,6-trienoate (25)26,41:
H
O (EtO)2POCH2CO2Et,NaH, THF, r.t.
O
O
A suspension of NaH (132 mg, 3.3 mmol, 60 % dispersion in mineral oil) in DME was
treated drop wise with triethylphosphonoacetate (0.65 mL, 3.3 mmol). The reaction
mixture was stirred at room temperature until H2 gas evolution ceased, then sorbic
aldehyde (0.33 mL, 3.0 mmol) was added drop wise giving a brown solution. The
reaction mixture was stirred overnight (18 h) at room temperature. The mixture was
quenched with H2O (50 mL) and extracted with CH2Cl2 (3 x 50 mL). The combined
organic fractions were washed with brine (25 mL), dried over MgSO4, filtered and
concentrated to give a yellow oil. The crude product was purified by column
chromatography on silica gel (1:4, EtOAc: hexanes) to give a yellow oil (406 mg, 81 %, mixture of E:Z isomers, 87:13); 1H NMR (400 MHz, DMSO-d6) δ 7.24 (1H, dd, J = 11.3,
15.4 Hz), 6.70 (1H, ddd, J = 0.5, 10.7, 14.8 Hz), 6.30 (1H, ddd, J = 0.5, 11.3, 14.8 Hz),
6.20–6.11 (1H, m), 5.99–5.95 (1H, m), 5.91 (1H, d, J = 15.4 Hz), 4.12 (2H, q, J = 7.1
Hz), 1.80 (3H, d, J = 6.8 Hz), 1.21 (3H, t, J = 7.1 Hz).
(2E,4E,6E)-Octa-2,4,6-trienoic acid (17)41:
O
O NaOH, MeOH/H2O,reflux, 45 min
OH
O
A solution of 25 (226 mg, 1.35 mmol) in MeOH/H2O (12 mL/ 24 mL) was treated with
solid NaOH pellets (3.26 g, 81.5 mmol) in one portion. The reaction mixture was heated
to reflux (heating mantle) for 45 min. The reaction mixture was cooled to room
temperature then diluted with brine (20 mL), and extracted with Et2O (2 x 20 mL). The
aqueous layer was collected and acidified (pH ~ 2) with 10 % (v/v) HCl giving a brown
suspension. The crude product was extracted with EtOAc (3 x 25 mL) and the combined

44
organic fractions were washed with H2O (30 mL), brine (30 mL), dried over MgSO4,
filtered and concentrated to give a brown solid. The crude product was purified by
column chromatography to give a light orange powder (105 mg, 56 %); mp = 155 – 160 °C, lit. mp = 185 – 187 ºC; 1H NMR (400 MHz, DMSO-d6) δ 12.13 (1H, s), 7.19 (1H, dd,
J = 11.4, 15.2 Hz), 6.65 (1H, dd, J = 10.8, 15.2 Hz), 6.31 (1H, dd, J = 11.4, 15.2 Hz),
6.20 (1H, ddd, J = 1.2, 10.8, 14.8 Hz), 5.99–5.94 (1H, m), 5.83 (1H, d, J = 15.2 Hz),
1.80 (3H, d, J = 6.8 Hz).
3.1.2 Synthesis of Depsipeptide
(S)-Benzyl 2-(((benzyloxy)carbonyl)amino)-3-hydroxypropanoate (29)32:
NH
HO
CBz
O
OH
1. Cs2CO3, MeOH2. BnBr, DMF
NH
HO
CBz
O
O Bn
A solution of CBz-SerOH (1.60 g, 6.69 mmol) in anhydrous MeOH (10 mL) was treated
with Cs2CO3 (1.31 g, 4.02 mmol) to give a clear solution. The MeOH was removed
under vacuum to give a white solid, which was dissolved in anhydrous DMF (30 mL)
and treated with benzyl bromide (0.870 mL, 7.36 mmol). The reaction mixture was
stirred at room temperature overnight (18 h) and the DMF removed under vacuum. The
residue was suspended in 10% NaHCO3 (30 mL) and extracted with CH2Cl2 (3 x 30
mL). The combined organic extracts were washed with H2O (50 mL), brine (50 mL),
dried over MgSO4, filtered and concentrated to give a crude yellow oil. The crude
product was purified by column chromatography on silica gel (2:3 EtOAc: hexanes) to give a white solid (1.97 g, 90%); 1H NMR (400 MHz, CDCl3) δ 7.36–7.31 (10H, m), 5.74
(1H, d, J = 6.4 Hz), 5.20 (2H, s), 5.11 (2H, s), 4.48 (1H, dd, J = 3.6, 3.6 Hz), 4.00 (1H, d,
J = 10.6 Hz), 3.92 (1H, d, J = 10.6 Hz), 2.22 (1H, s).
(S)-2-((S)-3-(Benzyloxy)-2-(((benzyloxy)carbonyl)amino)-3-oxopropyl) 1-tert-butyl pyrrolidine-1,2-dicarboxylate (30)32:

45
NH
HO
CBz
O
O Bn
BocProOH, EDC⋅HCl,DMAP, CH2Cl2, 0 °C - r.t.
NH
O
CBz
O
O Bn
ONBoc
A solution of 29 (1.88 g, 5.71 mmol), Boc-ProOH (2.46 g, 11.41 mmol), and DMAP (209 mg, 1.71 mmol) in anhydrous CH2Cl2 (60 mL) was cooled to 0 °C (ice bath) and treated
with a solution of EDC⋅HCl (3.28 g, 17.11 mmol) in anhydrous CH2Cl2 (10 mL). The
reaction mixture was allowed to come to room temperature and stirred for an additional
5 hours. The reaction was diluted with EtOAc (200 mL) and washed with 10 % citric
acid(aq) (150 mL), followed by saturated NaHCO3(aq) (150 mL) and brine (150 mL). The
organic fraction was dried over MgSO4, filtered and concentrated to give an orange oil.
The crude product was purified by column chromatography on silica gel (1:1, EtOAc: hexanes) to give a clear oil (2.86 g, 96 %); 1H NMR (rotamers, 400 MHz, CDCl3) δ 7.34–
7.31 (10H, m), 5.93 (0.5H, d, J = 8.4 Hz), 5.51 (0.5 H, d, J = 8.4 Hz), 5.23–5.07 (4H, m),
4.68–4.45 (3H, m), 4.24 (0.5H, dd, J = 8.6, 3.8 Hz), 4.13 (0.5H, dd, J = 8.6, 3.8 Hz),
3.46–3.37 (2H, m), 2.12–2.08 (1H, m), 1.87–1.77 (3H, m), 1.41 (4.5H, s), 1.37 (4.5H, s).
(S)-2-Amino-3-(((S)-1-(tert-butoxycarbonyl)pyrrolidine-2-carbonyl)oxy)propanoic acid (31)32:
NH
O
O
CBz O
OBn
NBoc Pd-C/H2, EtOH
H2N
O
O
OH
O
NBoc
A solution of 30 (466 mg, 0.886 mmol) in absolute EtOH (40 mL) was treated with 10 %
palladium on carbon (46 mg, 0.04 mmol). The flask was stoppered and purged with H2
gas three times. The reaction mixture was stirred at room temperature under H2
atmosphere (balloon pressure) overnight (16 h). The reaction mixture was filtered
through a pad of celite and washed with MeOH (6 x 5 mL). The filtrate was concentrated

46
to give a grey foam (283 mg, quant. + 6 % EtOH). 1H NMR (rotamers, 400 MHz, DMSO-d6) δ 7.85 (2H, br s), 4.51–4.45 (1H, m), 4.19–4.06 (2H, m), 3.46–3.43 (1H, m), 3.41–
3.38 (1H, m), 3.29–3.25 (1H, m), 2.20–2.11 (1H, m), 2.02–1.98 (1H, m), 1.82–1.77 (2H,
m), 1.39 (4H, s), 1.32 (5H, s); MS (ESI) m/z 325 (9, C13H22N2O6Na+), 303 (100,
C13H23N2O6+), 247 (46).
(S)-Benzyl-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-hydroxypropanoate
(33)45:
NH
HO
Fmoc
O
OH
1. Cs2CO3, MeOH2. BnBr, DMF
NH
HO
Fmoc
O
O Bn
A solution of Fmoc-SerOH (2.00 g, 6.110 mmol) in anhydrous MeOH (10 mL) was
treated with Cs2CO3 (1.19 g, 3.67 mmol) to give a clear solution. The MeOH was
removed under vacuum to give a white solid, which was dissolved in anhydrous DMF
(10 mL) and treated with benzyl bromide (1.09 mL, mmol). The reaction mixture was
stirred at room temperature overnight (18 h) and the DMF removed under vacuum. The
residue was suspended in 10% NaHCO3 (30 mL) and extracted with CH2Cl2 (3 x 30
mL). The combined organic extracts were washed with H2O (50 mL), brine (50 mL),
dried over MgSO4, filtered and concentrated to give crude yellow oil. The crude product
was purified by column chromatography on silica gel (3:7 – 1:1 EtOAc: hexanes) to give a white solid (1.83 g, 72%); mp = 96-97 °C; 1H NMR (400 MHz, CDCl3) δ 7.75 (2H, d, J
= 7.5 Hz), 7.58 (2H, d, J = 7.5 Hz), 7.39 (2H, dd, J = 7.5, 7.5 Hz), 7.33 (5H, s), 7.27 (2H,
dd, J = 7.5, 7.5 Hz), 5.80 (1H, d, J = 7.0 Hz), 4.47–4.39 (3H, m), 4.20 (1H, t, J = 7.0 Hz),
4.03–3.99 (1H, m), 3.93–3.90 (1H, m), 2.40 (1H, br s), 1.70 (1H, s).

47
2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(benzyloxy)-3-oxopropyl) 1-tert-butyl pyrrolidine-1,2-dicarboxylate (34)32:
A solution of 33 (1.757 g, 4.209 mmol), Boc-ProOH (1.812 g, 8.419 mmol), and DMAP (155 mg, 1.263 mmol) in anhydrous CH2Cl2 (40 mL) was cooled to 0 °C (ice bath) and
treated with a solution of EDC⋅HCl (2.420 g, 12.627 mmol) in anhydrous CH2Cl2 (10
mL). The reaction mixture was allowed to warm to room temperature while stirring
overnight (18 h). The reaction was quenched with H2O (50 mL) and extracted into
CH2Cl2 (3 x 50 mL). The combined organic fractions were washed with brine (70 mL),
dried over Mg2SO4, filtered and concentrated to give a yellow oil. The crude product
was purified by column chromatography on silica gel (2:3, EtOAc: hexanes) to give a clear oil (2.004 g, 77 %); 1H NMR (400 MHz, CDCl3) δ 7.75 (2H, d, J = 7.3 Hz), 7.66–
7.57 (2H, m), 7.41–7.38 (2H, m), 7.34–7.32 (5H, m), 7.27–7.24 (2H, m), 6.12 (0.5H, d, J
= 8.0 Hz), 5.64 (0.5H, d, J = 8.0 Hz), 5.28–5.15 (2H, m), 4.72–4.66 (1H, m), 4.62–4.24
(4H, m), 4.21–4.12 (1H, m), 3.50–3.38 (2H, m), 2.18–2.11 (1H, m), 1.87–1.80 (4H, m),
1.47 (5H, s), 1.39 (4H, s).

48
2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(benzyloxy)-3-oxopropyl) 1-tert-butyl pyrrolidine-1,2-dicarboxylate (19):
A solution of 34 (490 mg, 0.797 mmol) in anhydrous THF (20 mL) was treated with solid
10 % Pd on activated carbon (50 mg, 0.05 mmol) and the flask purged with H2 gas (3
times). The reaction mixture was stirred under H2 atmosphere (balloon pressure)
overnight (16 h). The reaction mixture was then filtered through a pad of celite, and the
celite washed with methanol (12 x 5 mL). The filtrate was concentrated to give a white foam (374 mg, 90 %); 1H NMR (400 MHz, CDCl3) δ 10.11 (1H, br s), 7.73 (2H, d, J = 7.2
Hz), 7.61–7.54 (2H, m), 7.37 (2H, dd, J = 7.2, 7.2 Hz), 7.28 (2H, dd, J = 7.2, 7.2 Hz),
6.21 (0.5 H, d, J = 7.4 Hz), 6.02 (0.5 H, d, J = 7.4 Hz), 4.68–4.47 (3H, m), 4.35–4.34
(2H, m), 4.21–4.17 (1H, m), 3.49–3.35 (2H, m), 2.16 (1H, br s), 1.97–1.86 (4H, m), 1.45 (5H, s), 1.38 (4H, s); 13C NMR (100 MHz, CDCl3) δ 172.6, 156.5, 155.2, 154.6, 144.0,
141.5, 127.9, 127.2, 125.5, 120.2, 81.0, 67.7, 64.7, 59.0, 53.7, 47.3, 46.5, 30.6, 28.6,
24.1; MS (ESI) m/z 579 (6), 563 (10, C28H32N2O8K+), 547 (38, C28H32N2O8Na+), 525 (6,
C28H33N2O8+), 425 (100, C28H33N2O8
+-Boc); HRMS (ESI) m/z (C28H32N2O8Na+) calcd.
547.2035, found 547.2050.

49
3.2 Branched Depsipeptide Synthesis FmocSer(Pro)-Pro-N-Me-Ala-AlaOH (37):
NH
O
O
N
ON
ONHFmocOHN
O
OH
Standard solid phase peptide synthesis was performed on an Aapptec Endeavor 90
Peptide Synthesizer using a standard Fmoc protocol (0.5 M). A preloaded H-Pro-2ClTrt
resin (500 mg, 100 – 200 mesh, 0.71 mmol/g) was used as the solid support. Couplings
were completed using 3.0 equivalents of Fmoc-protected amino acid, 3.0 equivalents of
HBTU, and 6.0 equivalents of diisopropylethylamine. The preactivated amino acid
solution was added to the resin and mechanically mixed under N2 atmosphere for 15
minutes. Each cycle consisted for double couplings with the exception of Fmoc-NMe-
AlaOH and Fmoc-Ser-(BocPro)OH, which were performed using 1.5 equivalents of
Fmoc-protected amino acid, 1.5 equivalents of HATU, and 3.0 equivalents of
diisopropylethylamine. These activated amino acid solutions were manually added to
the resin and mechanically mixed under N2 atmosphere for 20 minutes. These cycles
(Fmoc-NMe-AlaOH, and Fmoc-Ser-(BocPro)OH) consisted of single couplings. Coupling
reactions were monitored using the appropriate colorimetric test of the 0.5 – 1 mg of
resin beads (Kaiser test for primary amines38, Isatin test for proline39, and p-Chloranil
test for secondary amines40). Fmoc removal was performed using 20 % (v/v) piperidine
in DMF (1 x 3 min then 1 x 15 min). The resin-supported peptide was obtained with 78
% yield (546 mg, resin mass).
The peptide 37 was cleaved from the solid support by treatment with a mixture of
TFA (9.25 mL), triisopropylsilane (0.5 mL), and distilled H2O (0.25 mL). The reaction
mixture was cooled in an ice bath with occasional swirling for 3 h. The resin was filtered
off by passing the mixture through a plug of glass wool in a Pasteur pipette and the
resin was rinsed with additional TFA (3 x 0.5 mL). The combined filtrates were collected

50
and reduced under N2 flow. The crude filtrate was added to Et2O at 0 °C to give a
precipitate. The suspension was stored at 0 °C overnight (16 h) to allow the precipitate
to settle. The Et2O was decanted off and the product dried under reduced pressure to give a brown sticky solid (47 mg, 14 %). 1H NMR (rotamers, 400 MHz, CDCl3) δ 10.81
(1H, br s), 7.93 (0.5 H, br s), 7.87 (1H, br s), 7.76 (2H, d, J = 6.8 Hz), 7.60–7.58 (2H, m),
7.40 (2H, dd, J = 6.8, 6.8 Hz), 7.31 (2H, dd, J = 6.8, 6.8 Hz), 6.82 (0.5H, br s), 6.34–
6.15(1H, m), 5.19–5.00 (6H, m), 4.85–4.68 (3H, m), 4.50–4.40 (4H, m), 4.22–4.21 (1H,
m), 3.78–3.68 (1H, m), 3.10 (1H, br s), 2.99 (1H, br s), 2.78 (1H, br s), 2.47–2.40 (1H,
m), 2.18–2.00 (4H, m), 1.43–1.39 (6H, m), 1.05 (3H, s); MS (ESI) m/z 678
(C35H44N5O9+, 42), 581 (C35H43N5O9-Pro+, 100).

51
3.3 Synthesis of Depsipeptide 10
(S)-2-Oxo-2-phenylethyl2-(((benzyloxy)carbonyl)amino)-3-hydroxypropanoate (39)35:
A solution of CBz-SerOH (500 mg, 2.09 mmol) and Et3N (582 µL, 4.18 mmol) in EtOAc
(5.5 mL) was treated with 2-bromoacetophenone (457 mg, 2.30 mmol) in one portion at
room temperature. The reaction mixture was stirred at room temperature for 24 h then
diluted with EtOAc (4.5 mL). The reaction mixture was washed with 2 N NaOH (10 mL)
and extracted with additional EtOAc (2 x 10 mL). The combined organic fractions were
washed with saturated NaHCO3 (10 mL), dried over MgSO4, filtered and concentrated to
give a yellow solid. The crude product was purified by column chromatography on silica
gel (1:1, EtOAc: hexanes) to give a white solid (438 mg, 59 %); mp = 104-105 °C (lit. mp
= 113-144.5 °C46); 1H NMR (400 MHz, CDCl3) δ 7.91 (2H, d, J = 7.4 Hz), 7.65 (1H, tt, J
= 7.4, 1.4 Hz), 7.51 (2H, dd, J = 7.4, 7.4 Hz), 7.36–7.25 (5H, m), 5.79 (1H, d, J = 8.5
Hz), 5.72 (1H, d, J = 16.4 Hz), 5.32 (1H, d, J = 16.4 Hz), 5.14 (2H, s), 4.63 (1H, dt, J =
8.5, 2.8 Hz), 4.34–4.31 (1H, m), 3.92–3.87 (1H, m), 3.63 (1H, dd, J = 9.6, 4.6 Hz).

52
(S)-2-((S)-2-(((Benzyloxy)carbonyl)amino)-3-oxo-3-(2-oxo-2-phenylethoxy)propyl) 1-tert-butyl pyrrolidine-1,2-dicarboxylate (40)35:
A solution of 39 (413 mg, 1.16 mmol) and BocProOH (497 mg, 2.31 mmol) in anhydrous CH2Cl2 (10 mL) was cooled to 0 °C (ice bath) and treated with EDC⋅HCl (664 mg, 3.47
mmol) and DMAP (42 mg, 0.347 mmol). The reaction mixture was stirred at 0 °C for 1 h
then warmed to room temperature and stirred for 48 h. The reaction was quenched with
H2O (25 mL) and extracted with CH2Cl2 (2 x 25 mL). The combined organic fractions
were washed with 1 N HCl (2 x 25 mL), saturated NaCl(aq) (25 mL), dried over MgSO4,
filtered and concentrated. The residue was purified by column chromatography (1:3 –
1:1, EtOAc: hexanes) to give a colourless oil (438 mg, 68 %). 1H NMR (rotamers, 400 MHz, CDCl3) δ 7.88 (2H, dd, J = 7.4, 2.4 Hz), 7.64–7.60 (1H, m), 7.51–7.47 (2H, m),
7.35–7.29 (5H, m), 6.00 (0.5 H, d, J = 8.1), 5.60 (0.5 H, d, J = 8.1 Hz), 5.49–5.45 (1H,
m), 5.38 (0.5H, d, J = 4.5 Hz), 5.35 (0.5H, d, J = 4.5 Hz), 5.14–5.12 (2H, m), 4.87–4.82
(1H, m), 4.69–4.54 (2H, m), 4.33–4.25 (1H, m), 3.56–3.33 (2H, m), 2.25–2.13 (1H, m),
2.06–2.01 (1H, m), 1.96–1.80 (2H, m), 1.41 (4.5H, s), 1.38 (4.5H, s).

53
(S)-2-(((S)-2-(((Benzyloxy)carbonyl)amino)-3-oxo-3-(2-oxo-2-phenylethoxy)propoxy)carbonyl)pyrrolidin-1-ium chloride (10)35:
A solution of 40 (184.5 mg, 0.336 mmol) in 4 N HCl in dioxane (1.0 mL) was stirred
overnight under N2 atmosphere at room temperature. The reaction mixture was
concentrated, taken up once with EtOAc (5 mL), and taken up with CH2Cl2 (2 x 5 mL).
The product was dried under reduced pressure to give a yellow foam (184.5 mg, 12 % dioxane present). 1H NMR (400 MHz, CD3OD) δ 11.49 (0.5H, br s), 10.43 (0.5H, br s),
9.58 (1H, d, J = 8.5 Hz), 9.45 (2H, d, J = 7.5 Hz), 9.19 (1H, dd, J = 7.5, 7.5 Hz), 9.05
(2H, dd, J = 7.5, 7.5 Hz), 8.88–8.78 (5H, m), 7.15 (1H, d, J = 17.0 Hz), 7.04 (1H, d, J =
17.0 Hz), 6.56 (2H, s), 6.24–6.19 (1H, m), 6.03 (1H, dd, J = 4.1, 11.0 Hz), 5.98 (1H, dd,
J = 6.5, 11.0 Hz), 5.89 (1H, dd, J = 6.5, 8.4 Hz), 4.70 (2H, t, J = 7.3 Hz), 3.75–3.65 (1H,
m), 3.59–3.51 (1H, m), 3.36 (2H, tt, J = 7.3, 7.3 Hz).

54
3.4 Synthesis of ACP1 Analogs Ethyl 2-methyl-2-((5-(trifluoromethyl)pyridin-2-yl)thio)propanoate (43)37:
A solution of 2-mercapto-5-(trifluoromethyl)pyridine (5.00 g, 27.9 mmol) and LiOH⋅H2O
(1.171 g, 27.9 mmol) in EtOH (30 mL) was heated to reflux (~80 °C, heating mantle) and
treated drop wise with ethyl-α-bromoisobutyrate (4.14 mL, 27.9 mmol). The resulting
yellow reaction mixture was stirred at reflux for 16 h. The reaction mixture was cooled,
the EtOH removed in vacuo and the residue suspended in H2O (50 mL). The product
was extracted into EtOAc (3 x 50 mL). The combined organic fractions were washed
with brine (100 mL), dried over MgSO4, filtered and concentrated to give a yellow liquid.
The crude product was purified by column chromatography on silica gel (1:9 EtOAc: hexanes) to give a colourless oil (8.04 g, 98 %). 1H NMR (400 MHz, CDCl3) δ 8.52 (1H,
s), 7.60 (1H, dd, J = 8.4, 2.3 Hz), 7.17 (1H, d, J = 8.4 Hz), 4.10 (2H, q, J = 7.1 Hz), 1.63
(6H, s), 1.11 (3H, t, J = 7.1 Hz).

55
Ethyl 2-methyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanoate (44)37:
A solution of 43 (1.78 g, 6.08 mmol) in anhydrous CH2Cl2 (60 mL) was cooled to 0 °C
(ice bath) and treated with NaHCO3 (3.57 g, 42.6 mmol) followed by mCPBA (2.73 g,
12.2 mmol) portion wise. The reaction mixture was allowed to warm to room
temperature and stirred vigorously for 2.5 h, upon which a white precipitate formed. The
reaction mixture was quenched with sat. Na2SO3 solution (40 mL) and the mixture
filtered through a celite pad. The pad was rinsed with excess CH2Cl2 (5 x 15 mL). The
mixture was transferred to a 500 mL separatory funnel and the aqueous layer removed.
The organic fraction was washed with additional sat. Na2SO3 (50 mL), followed by sat.
NaHCO3 (2 x 50 mL), and brine (50 mL). The organic fraction was then dried over
MgSO4, filtered and concentrated to give a white solid. The crude product was purified
by column chromatography on silica gel twice (5:95, EtOAc: hexanes, 1:9, EtOAc: hexanes) to give a clear oil (594 mg, 30 %). 1H NMR (400 MHz, CDCl3) δ 9.00 (1H, s),
8.22 (2H, d, J = 1.6 Hz), 4.14 (2H, q, J = 7.2 Hz), 1.77 (6H, s), 1.19 (3H, t, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 168.9, 159.5, 146.9, 135.4, 130.0 (q, J = 36.1 Hz), 125.1,
122.7 (q, J = 270.7 Hz), 69.8, 62.7, 20.5, 13.9; MS (ESI) m/z 348 (C12H14F3NO4SNa+,
8), 326 (C12H15F3NO4S+, 100).

56
Ethyl 2-methyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanoate (44)37:
N
F
FF
S O
ON
F
FF
S O
OO
OOxone, dioxane/H2O
(5:1)
A solution of 43 (4.00 g, 13.6 mmol) in dioxane/H2O (160 mL / 40 mL) was treated with
solid Oxone© monopersulfate (16.76 g, 27.27 mmol) portion wise. The reaction mixture
was allowed to stir at room temperature for 16 h. The precipitate was filtered off through
a fritted glass funnel and washed with additional dioxane (50 mL). The filtrate was
concentrated and the residue was suspended in additional H2O (50 mL). The crude
product was extracted with CH2Cl2 (3 x 100 mL). The combined organic fractions were
then washed with brine (100 mL), dried over MgSO4, filtered and concentrated to give a
crude oil (3.63 g). The crude product was purified by column chromatography on silica
gel (1:9, EtOAc: hexanes) to give the product as a clear oil (955 mg, 21 %). 1H NMR (400 MHz, CDCl3) δ 9.00 (1H, s), 8.22 (2H, d, J = 1.6 Hz), 4.14 (2H, q, J = 7.2 Hz), 1.77
(6H, s), 1.19 (3H, t, J = 7.2 Hz); MS (ESI) m/z 364 (C12H14F3NO4SK+, 2), 348
(C12H14F3NO4SNa+, 37), 326 (C12H15F3NO4+, 100), 280 (70); HRMS (ESI) m/z
(C12H15NF3O4S+) calcd. 326.0668, found 326.0655.

57
2-Methyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanoic acid (45)37:
A solution of 44 (552 mg, 1.70 mmol) in THF (24 mL) and H2O (6 mL) was treated with LiOH⋅H2O (142 mg, 3.39 mmol) in one portion. The reaction mixture was stirred at room
temperature overnight. The reaction mixture was concentrated and the residue was
suspended in H2O (20 mL). The aqueous layer was washed with CH2Cl2 (3 x 20 mL)
and then acidified to ~pH 2 with concentrated HCl resulting in the formation of a white
precipitate. The product was extracted from the aqueous fraction with CHCl3 (3 x 20
mL). The combined organic fractions were washed with brine (40 mL), dried over
MgSO4, filtered and concentrated to give a white crystalline solid (383 mg, 76 %); mp = 189-192 °C; 1H NMR (400 MHz, CD3OD) δ 9.07 (1H, s), 8.46 (1H, dd, J = 8.2, 2.9 Hz),
8.27 (1H, d, J = 8.2 Hz), 1.70 (6H, s); MS (ESI) m/z 336 (C10H10F3NO4SK+, 7), 320
(C10H10F3NO4SNa+, 53) 298 (C10H10F3NO4SH+, 81), 280 (100); HRMS (ESI) m/z
(C10H11F3NO4S+) calcd. 298.0355, found 298.0344.

58
2-Methyl-2-((5-(trifluoromethyl)pyridin-2-yl)thio)propanoic acid (45-A)37:
N
F
FF
S O
O
LiOH⋅H2O, THF/H2O,(4:1), r.t.
N
F
FF
S OH
O
A solution of 43 (160 mg, 0.545 mmol) in THF (16 mL) was treated with a solution of LiOH⋅H2O (45 mg, 1.09 mmol) in H2O (4 mL). The resulting yellow solution was stirred at
room temperature for 18 h. The THF was removed in vacuo, and the residue suspended
in H2O (10 mL) and extracted into CH2Cl2. The aqueous phase was acidified (pH ~ 2)
with concentrated HCl and the resulting cloudy suspension was extracted with CH2Cl2 (3
x 10 mL). The combined organic extracts were washed with brine (10 mL), dried over
MgSO4, filtered and concentrated to give a yellow waxy solid (98 mg, 68 %). 1H NMR (400 MHz, CDCl3) δ 8.70 (1H, s), 8.67 (1H, dd, J = 2.0, 8.4 Hz), 7.48 (1H, d, J = 8.4 Hz),
1.71 (6H, s); MS (ESI) m/z 288 (C10H10F3NO2SNa+, 40), 266 (C10H11F3NO2S, 100).
2-(Phenylthio)ethanamine (46)40:
HSBrH2N +HBr
K2CO3, EtOHSH2N
A suspension of 2-bromoethylamine hydrobromide (3.00 g, 14.6 mmol) and K2CO3 (3.03
g, 25.0 mmol) in EtOH (30 mL) was treated drop wise with thiophenol (0.75 mL, 7.32
mmol). The reaction mixture was stirred at room temperature for 3 d. The reaction
mixture was concentrated and the residue dissolved in H2O (40 mL) and extracted into
CHCl3 (3 x 40 mL). The combined organic extracts were washed with brine (40 mL),
dried over MgSO4, filtered and concentrated to give a light yellow oil (1.00 g, 89 %). 1H
NMR (400 MHz, CD3OD) δ 7.37–7.33 (2H, m), 7.28–7.24 (2H, m), 7.19–7.16 (1H, m),
2.99 (2H, t, J = 6.2 Hz), 2.89 (2H, t, J = 6.2 Hz), 1.44 (2H, s).

59
3-(Phenylthio)propan-1-amine48:
HSH2N Br +HBr
K2CO3, CH2Cl2SH2N
A suspension of 3-brompropylamine hydrobromide (4.28 g, 19.5 mmol) and K2CO3 (4.05
g, 29.3 mmol) in CH2Cl2 (30 mL) was treated drop wise with thiophenol (0.75 mL, 7.32
mmol). The reaction mixture was stirred at room temperature for 16 h. The reaction
mixture was concentrated and the residue dissolved in H2O (40 mL) and extracted into
CHCl3 (3 x 40 mL). The combined organic extracts were washed with brine (40 mL),
dried over MgSO4, filtered and concentrated to give a light yellow oil. The crude was
purified by column chromatography on silica gel (10:89:1, MeOH: CH2Cl2: Et3N), to give a waxy solid (808 mg, 49 %). 1H NMR (400 MHz, CD3OD) δ 7.39–7.36 (2H, m), 7.32–
7.27 (2H, m), 7.19 (1H, tt, J = 7.4, 1.2 Hz), 3.02 (2H, t, J = 7.2 Hz), 2.94 (2H, t, J = 7.2
Hz), 1.89 (2H, tt, J = 7.2, 7.2 Hz).
2-((1-Methyl-1H-pyrrol-2-yl)thio)ethanamine:
K2CO3, EtOHN
NHSBrH2NHBr +
N
NSH2N
A suspension of 2-bromoethylamine hydrobromide (500 mg, 2.44 mmol) and K2CO3
(506 mg, 3.66 mmol) in EtOH (10 mL) was 2-mercapto-1-methylimidazole (139 mg, 1.22
mmol) in one portion. The reaction mixture was stirred at room temperature for 2 d. The
reaction mixture was concentrated and the residue suspended in 2 N NaOH (25 mL)
and extracted into CH2Cl2 (3 x 25 mL). The combined organic extracts were washed
with brine (25 mL), dried over MgSO4, filtered and concentrated to give a brown oil (73 mg, 38 %); 1H NMR (400 MHz, CD3OD) δ 7.22 (1H, s), 7.03 (1H, s), 3.75 (3H, s), 3.02
(2H, t, J = 6.4 Hz), 2.81 (2H, t, J = 6.4 Hz).

60
N-(2-Phenoxyethyl)propionamide44:
HOO
N neat, 160 °C+ O
HN
O
A mixture of phenol (508 mg, 5.39 mmol) and 2-ethyl-2-oxazoline (0.545 mL, 5.39
mmol) in a 25 mL round bottom flask equipped with an N2 inlet and reflux condenser was heated to 160 °C (oil bath) for 6 h. The reaction mixture was cooled and quenched
with H2O (10 mL). The mixture was extracted with CH2Cl2 (2 x 15 mL). The combined
organic extracts were dried over MgSO4, filtered and concentrated. The crude product
was purified by column chromatography on silica gel (1:1, EtOAc: hexanes) to give a white solid (260 mg, 25 %); mp = 64–65 °C; Rf = 0.14 (1:1, EtOAc: hexanes); 1H NMR
(400 MHz, CDCl3) δ 7.29–7.24 (2H,m), 6.96 (1H, tt, J = 1.2, 7.5 Hz), 6.90–6.88 (2H, m),
6.02 (1H, br s), 4.03 (2H, t, J = 5.2 Hz), 3.66 (2H, q, J = 5.2 Hz), 2.22 (2H, q, J = 7.6
Hz), 1.16 (3H, t, J = 7.6 Hz).
2-Phenoxyethanamine44, 47:
OHN
O
H3PO4, H2O,reflux, 8 h
OH2N
A solution of N-(2-phenoxyethyl)propionamide (200 mg, 1.03 mmol) in H3PO4 (3 g, 85 %
in H2O) and H2O (1 mL) in a 10 mL round bottom flask equipped with a N2 inlet and
reflux condenser was heated to reflux (heating mantle) for 16 h. The reaction mixture
was cooled to room temperature and neutralized (pH ~7) with 2 N NaOH. The mixture
was extracted into toluene (3 x 10 mL). The combined organic fractions were washed
with brine (15 mL), dried over MgSO4 and concentrated to give a grey solid (84 mg, 59 %). 1H NMR (400 MHz, CDCl3) δ 7.29–7.24 (2H,m), 6.94 (1H, tt, J = 1.2, 7.2 Hz), 6.91–
6.88 (2H, m), 3.95 (2H, t, J = 5.2 Hz), 3.05 (2H, t, J = 5.2 Hz), 1.56 (2H, br s).

61
Synthesis of ACP1 and Analogs
2-Methyl-N-(2-(phenylthio)ethyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (ACP1):
N
F
FF
S OH
OO
O H2NS
PyBOP, iPr2EtN,DMF
N
F
FF
SHN
OS
O
O+
To a solution of 45 (20 mg, 0.067 mmol) in anhydrous DMF (2 mL) was added a solution
of PyBOP (38 mg, 0.074 mmol) in anhydrous DMF (0.5 mL) followed by DIEA (35 µL,
0.20 mmol). A solution of 46 (10 mg, 0.067 mmol) in anhydrous DMF (0.5 mL) was
added drop wise. The resulting yellow solution was stirred at room temperature for 1 h.
The DMF was removed in vacuo and the crude product was absorbed onto silica gel.
The product was purified by column chromatography (20–40 % EtOAc/hexanes) to give a clear oil (16 mg, 56 %). 1H NMR (400 MHz, CDCl3) δ 8.91 (1H, dd, J = 1.6, 0.8 Hz),
8.20 (1H, d, J = 8.4 Hz), 8.15 (1H, ddd, J = 0.4, 2.0, 8.4 Hz), 7.41–7.38 (2H, m), 7.32–
7.29 (2H, m), 7.22 (1H, tt, J = 7.3, 1.3 Hz), 3.51 (2H, q, J = 6.4 Hz), 3.12 (2H, t, J = 6.4 Hz), 1.63 (6H, s); 13C NMR (100 MHz, CDCl3) δ 167.9, 158.3, 147.3, 135.8, 135.7,
135.0, 130.2, 130.1 (q, J = 34.4 Hz), 129.4, 126.8, 124.6, 67.9, 39.7, 33.2, 20.6; IR (film) ν 3390, 3063, 2993, 2939, 1662, 1527 cm-1; MS (ESI) m/z 460 (C18H19F3N2O3S2K+, 7),
455 (C18H19F3N2O3SNa+, 56) 433 (C18H20F3N2O3S2+, 100), 280 (38); HRMS (ESI) m/z
(C18H20N2O3F3S2) calcd. 433.0861, found 433.0868.

62
2-Methyl-N-(3-phenylpropyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (47):
Obtained in 88 % yield. White powder, mp 83-86 °C; 1H NMR (400 MHz, CDCl3) δ 8.90
(1H, s), 8.18–8.17 (2H, m), 7.30 (2H, t, J = 7.7 Hz), 7.22–7.18 (3H, m), 6.99 (1H, br),
3.33 (2H, q, J = 6.6 Hz), 2.71 (2H, t, J = 7.6 Hz), 1.92 (2H, tt, J = 7.3, 7.3 Hz), 1.62 (6H, s); 13C NMR (100 MHz, CDCl3) δ 167.5, 158.4, 147.4, 141.5, 135.7, 130.3 (q, J = 33.9
Hz), 128.7, 128.5, 126.3, 124.7, 122.5 (q, J = 273.5 Hz), 67.9, 40.2, 33.3, 30.7, 20.6; IR (film) ν 3394, 3063, 3028, 2939, 2862, 2360, 1670, 1531 cm-1; MS (ESI) m/z 453
(C19H21F3N2O3SK+, 7), 437 (C19H21F3N2O3SNa+, 100), 415 (C19H22F3N2O3S+, 71); HRMS
(ESI) m/z (C19H22N2O3F3S+) calcd. 415.1297, found 415.1280.
N-Butyl-2-methyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (48):
Obtained in 42 % yield. Grey solid, mp = 63–65 °C; Rf = 0.36 (2:3, EtOAc: hexanes); 1H NMR (400 MHz, CDCl3) δ 8.98 (1H, s), 8.22–8.18 (2H, m), 6.95 (1H, br s), 3.30 (2H, q, J
= 7.2 Hz), 1.65 (6H, s), 1.55 (2H, tt, J = 7.2, 7.2 Hz), 1.38 (2H, qt, J = 7.2, 7.2 Hz), 0.94 (3H, t, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 167.4, 158.5, 147.4, 135.7, 130.4 (q, J
= 33.8 Hz), 124.7, 122.6 (q, J = 273.6 Hz), 68.0, 40.2, 31.3, 20.8, 20.3, 13.9; IR (film) ν
3356, 2958, 2935, 1662, 1531, 1315 cm-1; MS (ESI) m/z 375 (C14H19F3N2O3SNa+, 41),

63
353 (C14H20F3N2O3S+, 100), 280 (67); HRMS (ESI) m/z (C14H20N2O3F3S+) calcd.
353.1141, found 353.1127.
2-Methyl-N-phenethyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (49):
N
F
FF
SHN
OO
O
Obtained in quantitative yield. Grey solid; mp = 82–83 °C; Rf = 0.30 (2:3, EtOAc: hexanes); 1H NMR (400 MHz, CDCl3) δ 8.91 (1H, s), 8.17–8.14 (1H, m), 8.08–8.06 (1H,
m), 7.34–7.30 (2H, m), 7.25–7.22 (3H, m), 7.22 (1H, br s), 3.55 (2H, q, J = 7.2 Hz), 2.89 (2H, t, J = 7.2 Hz), 1.61 (6H, s); 13C NMR (100 MHz, CDCl3) δ 167.5, 158.4, 147.3,
138.8, 129.0, 128.9, 126.9, 124.7, 68.0, 41.9, 35.4, 20.7; MS (ESI) m/z 439
(C18H19F3N2O3SK+, 6), 423 (C18H19F3N2O3SNa+, 100). 401 (C18H20F3N2O3S+, 70), 280
(36); HRMS (ESI) m/z (C18H20F3N2O3S+) calcd.401.1141, found 401.1153.
N-(3,4-Dimethoxyphenethyl)-2-methyl-2-((5-(trifluoromethyl)pyridin-2-
yl)sulfonyl)propanamide (50):
Obtained in 48 % yield. Clear oil; Rf = 0.37 (2:3, EtOAc: hexanes); 1H NMR (400 MHz, CDCl3) δ 8.75 (1H, d, J = 2.4 Hz), 7.88 (1H, dd, J = 8.6, 2.4 Hz), 7.22 (1H, d, J = 8.6
Hz), 6.75 (1H, d, J = 8.0 Hz), 6.70–6.67 (2H, m), 4.13–4.09 (2H, m), 3.84 (6H, s), 2.90–2.86 (2H, m), 1.2 (3H, s), 1.11 (3H, s); 13C NMR (100 MHz, CDCl3) δ 178.1, 158.7,
149.2, 147.9, 146.2, 135.2, 131.4, 124.2 (q, J = 33.3 Hz), 123.5 (q, J = 272.1 Hz), 121.0,

64
120.7, 112.4, 111.5, 56.1, 49.9, 34.7, 32.7, 19.9; IR (film) ν 3059, 2966, 2935, 2874,
2835, 1670, 1604, 1516 cm-1; MS (ESI) m/z 461 (C20H24F3N2O5S+, 100), 397 (26), 353
(63).
2-Methyl-N-(3-(phenylthio)propyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (51):
N
F
FF
SHN
OO
OS
Obtained in 51 % yield. White solid; mp = 74–78 °C Rf = 0.22 (2:3, EtOAc: hexanes); 1H
NMR (400 MHz, CDCl3) δ 8.92 (1H, s), 8.18 (2H, s), 7.36–7.34 (2H, m), 7.33–7.26 (2H,
s), 7.17 (1H, tt, J = 1.3, 7.2 Hz), 7.08 (1H, br s), 3.44 (2H, q, J = 6.8 Hz), 3.03 (2H, t, J =
6.8 Hz), 1.93 (2H, tt, J = 6.8, 6.8 Hz), 1.62 (6H, s); MS (ESI) m/z 485
(C19H21F3N2O3S2K+, 10), 469 (C19H21F3N2O3S2Na+, 100), 447 (C19H22F3N2O3S2+, 87);
HRMS (ESI) m/z (C19H22F3N2O3S2+) calcd. 447.1018, found 447.1008.
2-Methyl-N-(2-((1-methyl-1H-imidazol-2-yl)thio)ethyl)-2-((5-(trifluoromethyl)pyridin-
2-yl)sulfonyl)propanamide (52):
N
F
FF
SHN
OS
O
ON
N
Obtained in 31 % yield. Yellow oil; Rf = 0.31 (4:1, EtOAc: hexanes); 1H NMR (400 MHz, CDCl3) δ 8.64 (1H, s), 7.91 (1H, dd, J = 8.6, 2.6 Hz), 7.58 (1H, d, J = 8.6 Hz), 7.01 (1H,
d, J = 0.8 Hz), 6.91 (1H, d, J = 0.8 Hz), 4.17 (2H, t, J = 7.2 Hz), 3.56 (3H, s), 3.30 (2H, t,
J = 7.2 Hz), 1.12 (6H, d, J = 6.4 Hz).

65
2-Methyl-N-(2-(phenylamino)ethyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (53):
Obtained in 26 % yield. Clear oil; Rf = 0.25 (2:3, EtOAc: hexanes); 1H NMR (400 MHz, CDCl3) δ 8.30 (1H, s), 7.53 (1H, dd, J = 8.8, 2.7 Hz), 7.45–7.35 (3H, m), 7.17–7.14 (2H,
m), 6.47 (1H, d, J = 8.8 Hz), 5.79 (1H, s), 3.96 (2H, t, J = 5.5 Hz), 3.55 (2H, q, J = 5.5 Hz), 2.47–2.40 (1H, m), 1.02 (3H, s), 1.00 (3H, s); 13C NMR (100 MHz, CDCl3) δ 179.5,
160.5, 146.1, 142.6, 134.2, 130.2, 128.5, 128.2, 107.5, 48.9, 41.3, 31.6, 19.9; IR (film) ν
3333, 3163, 3063, 3093, 2978, 2935, 2874, 1597, 1531, 1496, 1207 cm-1.
2-Methyl-N-(2-phenoxyethyl)-2-((5-(trifluoromethyl)pyridin-2-
yl)sulfonyl)propanamide (54):
Obtained in 65 % yield. White crystalline solid, mp 108-109 °C; Rf = 0.25 (2:3, EtOAc: hexanes); 1H NMR (400 MHz, CDCl3) δ 8.76–8.75 (1H, m), 8.11 (1H, d, J = 8.2 Hz),
7.81 (1H, ddd, J = 8.2, 2.2, 0.7 Hz), 7.53–7.51 (1H, br m), 7.31–7.27 (2H, m), 6.97 (1H,
tt, J = 7.3, 1.0 Hz), 6.89–6.87 (2H, m), 4.02 (2H, t, J = 5.3 Hz), 3.69 (2H, q, J = 5.3 Hz), 1.67 (6H, s); 13C NMR (100 MHz, CDCl3) δ 168.0, 158.6, 147.3, 144.9, 135.6, 130.2 (q,
J = 34 Hz) 129.9, 123.9, 122.5 (q, J = 273 Hz), 121.1, 114.7, 68.0, 66.2, 40.2, 20.8; IR (film) ν 3394, 2924, 2854, 1670, 1597, 1531 cm-1; m/z (ESI, %) 455 (3, C18H19F3N2O4S
K+), 439 (33, C18H19F3N2O4S Na+), 417 (100, C18H20F3N2O4S+), 323 (5), 280 (43); HRMS
(ESI) m/z (C18H20F3N2O4S+) calcd. 417.1096, found 417.1090.

66
2-Methyl-N-(2-(phenylthio)ethyl)-2-((5-(trifluoromethyl)pyridin-2-yl)thio)propanamide (55):
N
F
FF
SHN
OS
Obtained in 31 % yield. Yellow oil; Rf = 0.43 (2:3, EtOAc: hexanes); 1H NMR (400 MHz, CDCl3) δ 8.64 (1H, s), 7.72–7.68 (2H, m), 7.29–7.22 (4H, m), 7.16–7.14 (1H, m), 3.44
(2H, q, J = 6.3 Hz), 2.99 (2H, t, J = 6.3 Hz), 1.68 (6H, s); 13C NMR (100 MHz, CDCl3) δ
174.4, 162.4, 146.2, 135.2, 133.2, 129.6, 129.3, 126.6, 123.7 (q, J = 272.0 Hz), 123.4
(q, J = 33.3 Hz), 122.9, 52.8, 39.0, 33.5, 26.7; m/z (ESI, %) 401 (C18H20F3N2OS2+, 100),
365 (90).
2-Methyl-N-(2-(phenylthio)ethyl)-2-(pyridin-2-ylsulfonyl)propanamide (56):
Obtained in 62 % yield. Grey solid; mp = 65–67 °C; Rf = 0.39 (3:4, EtOAc: hexanes); 1H
NMR (400 MHz, CDCl3) δ 8.69 (1H, d, J = 3.6 Hz), 8.06 (1H, d, J = 7.5 Hz), 7.92 (1H, td,
J = 7.5, 1.5 Hz), 7.56–7.51 (2H, m), 7.40 (2H, dd, J = 7.5, 1.5 Hz), 7.30 (2H, t, J = 7.5
Hz), 7.21 (1H, t, J = 7.5 Hz), 3.50 (2H, q, J = 6.6 Hz), 3.12 (2H, t, J = 6.6 Hz), 1.61 (6H, s); 13C NMR (100 MHz, CDCl3) δ 168.3, 155.0, 150.4, 138.2, 135.3, 129.8, 129.4, 128.0,
126.7, 124.9, 67.6, 39.8, 32.9, 20.8; IR (film) ν 3387, 3055, 2993, 2939, 1662, 1577,
1519, 1473 cm-1; m/z (ESI, %) 403 (C17H20N2O3S2K+, 6) 387 (C17H20N2O3S2Na+, 100),
365 (C17H21N2O3S2+, 49); HRMS (ESI) m/z (C17H21N2O3S2
+) calcd. 365.0988, found
365.0991

67
N-(2-(Phenylthio)ethyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)acetamide (57):
Obtained in 68 % yield. Waxy solid; Rf = 0.58 (7:3, EtOAc: hexanes); 1H NMR (400 MHz, CDCl3) δ 8.99 (1H, s), 8.25–8.20 (2H, m), 7.38–7.35 (2H, m), 7.33–7.29 (2H, m),
7.23 (1H, tt, J = 4.9, 1.5 Hz), 4.29 (2H, s), 3.47 (2H, q, J = 6.2 Hz), 3.02 (2H, t, J = 6.2 Hz); 13C NMR (100 MHz, CDCl3) δ 160.4, 147.6, 144.9, 136.2, 134.6, 130.4, 129.4,
127.1, 122.3, 57.8, 39.3, 33.6; IR (film) ν 3321, 3055, 2928, 2360, 2341, 1651, 1550,
1330 cm-1; m/z (ESI, %) 427 (C16H15F3N2O3S2Na+, 100), 405 (C16H16F3N2O3S2+, 99);
HRMS (ESI) m/z (C16H16F3N2O3S2+) calcd. 405.0548, found 405.0539.

68
Appendix I Spectra of Selected Compounds

69
4

70
25
17
O
O
HO
O

71
29
30
NH
HO
CBz O
OBn
NH
O
CBz O
OBn
ONBoc

72
31
33
H2N
O
OH
O
ONBoc
NH
HO
O
O
Fmoc Bn

73
34
NH
O
O
O
ONBoc
Fmoc Bn

74
19
NH
OH
O
ONBoc
O
Fmoc

75
37
39
NH
O
O
OCBz
HO
NH
O
O
N
ON
ONHFmocOHN
O
OH

76
40
NH
O
OPAc
O
ONBoc
CBz
77
10
43
NH
CBz O
OPAc
O
ONH2+Cl-
N
F
FF
S O
O

78
44
45
N
F
FF
S O
OO
O
N
F
FF
S OH
OO
O

79
45-A
46
H2NS
N
F
FF
S OH
O

80
ACP1
N
F
FF
SHN
OS
O
O

81
47
N
F
F F
SHN
OO
O

82
48
N
F
F F
SHN
OO
O

83
49
N
F
FF
SHN
OO
O

84
50
N
F
F F
SHN
OO
OOMe
OMe

85
51
52
N
F
FF
SHN
OO
OS
N
F
FF
SHN
OS
O
ON
N

86
53
N
F
FF
SHN
ONHO
O

87
54
N
F
FF
SHN
OO
O
O

88
55
N
F
FF
SHN
OS

89
56
N SHN
OS
O
O

90
57
N
F
FF
SHN
OS
O
O

91
Appendix II Endeavor 90 Peptide Synthesis Protocols

92
Peptide Synthesizer Protocol for (2E,4E)-Hexa-2,4,-dienoyl-Phe-Ser-Pro-N-MeAla-Ala-ProOH (14): File Name: MC01-60 1g scale Created By: General Created On: 1/30/2009 1. ********SWELL******** 2. Wash RV1 with 30.00 mL from DMF 3Times. 3. Fill RV1 with 16.00 mL from DMF. 4. Mix RV1 via Mechanical Mix for 2.00 minutes. 5. Empty RV1. 6. Repeat from step 2, 1 time. (1) 7. Pause 8. Blank 9. ********Wash******* 10. Fill RV1 with 16.00 mL from DMF. 11. Mix RV1 via Mechanical Mix for 5.00 minutes. 12. Empty RV1. 13. Repeat from step 10, 1 time. (1) 14. Fill RV1 with 16.00 mL from DMF. 15. Wash RV1 with 45.00 mL from DMF 3Times. 16. Fill RV1 with 16.00 mL from NMP. 17. Mix RV1 via Mechanical Mix for 5.00 minutes. 18. Empty RV1. 19. Repeat from step 16, 1 time. (1) 20. Blank 21. ********Coupling Ala******* 22. Dissolve AA1 with 4.40 mL from HBTU for 0.50 minutes. 23. Dissolve AA1 with 2.20 mL from DIEA for 0.20 minutes. 24. Fill RV1 with 25.00 mL from AA1. 25. Clean AA1 with 20.00 mL from DMF. 26. Repeat from step 25, 1 time. (1) 27. Mix RV1 via Mechanical Mix for 15.00 minutes. 28. Empty RV1. 29. Blank 30. Blank 31. Dissolve AA2 with 4.40 mL from HBTU for 0.50 minutes. 32. Dissolve AA2 with 2.20 mL from DIEA for 0.20 minutes. 33. Fill RV1 with 25.00 mL from AA2. 34. Clean AA2 with 20.00 mL from DMF. 35. Repeat from step 34, 1 time. (1) 36. Mix RV1 via Mechanical Mix for 15.00 minutes. 37. Empty RV1. 38. Pause 39. Blank 40. ********Deprotection Cycle 1******* 41. Fill RV1 with 16.00 mL from Piperidine. 42. Mix RV1 via Mechanical Mix for 3.00 minutes. 43. Empty RV1. 44. Fill RV1 with 16.00 mL from Piperidine. 45. Mix RV1 via Mechanical Mix for 10.00 minutes. 46. Empty RV1. 47. Clean Lines with 15.00 mL from DMF. 48. Clean RV1 Vent with 15.00 mL from DMF. 49. ********Wash******* 50. Fill RV1 with 16.00 mL from DMF. 51. Mix RV1 via Mechanical Mix for 0.50 minutes. 52. Empty RV1.

93
53. Repeat from step 50, 1 time. (1) 54. Fill RV1 with 16.00 mL from DMF. 55. Wash RV1 with 45.00 mL from DMF 3Times. 56. Fill RV1 with 16.00 mL from NMP. 57. Mix RV1 via Mechanical Mix for 0.50 minutes. 58. Empty RV1. 59. Repeat from step 56, 1 time. (1) 60. Blank 61. ********Coupling N-Me-Ala******* 62. Pause 63. Fill RV1 with 10.00 mL from NMP. 64. Pause 65. **Addition of N-MeAla and HATU** 66. Mix RV1 via Mechanical Mix for 20.00 minutes. 67. Empty RV1. 68. Blank 69. Pause 70. Blank 71. ********Deprotection Cycle 3******* 72. Fill RV1 with 16.00 mL from Piperidine. 73. Mix RV1 via Mechanical Mix for 3.00 minutes. 74. Empty RV1. 75. Fill RV1 with 16.00 mL from Piperidine. 76. Mix RV1 via Mechanical Mix for 10.00 minutes. 77. Empty RV1. 78. Clean Lines with 15.00 mL from DMF. 79. Clean RV1 Vent with 15.00 mL from DMF. 80. ********Wash******* 81. Fill RV1 with 16.00 mL from DMF. 82. Mix RV1 via Mechanical Mix for 0.50 minutes. 83. Empty RV1. 84. Repeat from step 81, 1 time. (1) 85. Fill RV1 with 16.00 mL from DMF. 86. Wash RV1 with 45.00 mL from DMF 3Times. 87. Fill RV1 with 16.00 mL from NMP. 88. Mix RV1 via Mechanical Mix for 0.50 minutes. 89. Empty RV1. 90. Repeat from step 87, 1 time. (1) 91. Blank 92. ********Coupling Pro******* 93. Pause 94. Fill RV1 with 10.00 mL from NMP. 95. Pause 96. **Addition of Pro and HATU** 97. Mix RV1 via Mechanical Mix for 20.00 minutes. 98. Empty RV1. 99. Blank 100. Pause 101. **2nd coupling of FmocPro** 102. Pause 103. Fill RV1 with 10.00 mL from NMP. 104. Pause 105. **Addition of Pro and HATU** 106. Mix RV1 via Mechanical Mix for 20.00 minutes. 107. Empty RV1. 108. Blank 109. Pause 110. ********Deprotection Cycle 4*******

94
111. Fill RV1 with 16.00 mL from Piperidine. 112. Mix RV1 via Mechanical Mix for 3.00 minutes. 113. Empty RV1. 114. Fill RV1 with 16.00 mL from Piperidine. 115. Mix RV1 via Mechanical Mix for 10.00 minutes. 116. Empty RV1. 117. Clean Lines with 15.00 mL from DMF. 118. Clean RV1 Vent with 15.00 mL from DMF. 119. ********Wash******* 120. Fill RV1 with 16.00 mL from DMF. 121. Mix RV1 via Mechanical Mix for 0.50 minutes. 122. Empty RV1. 123. Repeat from step 120, 1 time. (1) 124. Fill RV1 with 16.00 mL from DMF. 125. Wash RV1 with 45.00 mL from DMF 3Times. 126. Fill RV1 with 16.00 mL from NMP. 127. Mix RV1 via Mechanical Mix for 0.50 minutes. 128. Empty RV1. 129. Repeat from step 126, 1 time. (1) 130. Blank 131. ********Coupling Ser(Trt)******* 132. Dissolve AA3 with 4.40 mL from HBTU for 0.50 minutes. 133. Dissolve AA3 with 2.20 mL from DIEA for 0.20 minutes. 134. Fill RV1 with 25.00 mL from AA3. 135. Clean AA3 with 20.00 mL from DMF. 136. Repeat from step 135, 1 time. (1) 137. Mix RV1 via Mechanical Mix for 15.00 minutes. 138. Empty RV1. 139. Blank 140. Blank 141. Dissolve AA4 with 4.40 mL from HBTU for 0.50 minutes. 142. Dissolve AA4 with 2.20 mL from DIEA for 0.20 minutes. 143. Fill RV1 with 25.00 mL from AA4. 144. Clean AA4 with 20.00 mL from DMF. 145. Repeat from step 144, 1 time. (1) 146. Mix RV1 via Mechanical Mix for 15.00 minutes. 147. Empty RV1. 148. Pause 149. Blank 150. ********Deprotection Cycle 5******* 151. Fill RV1 with 16.00 mL from Piperidine. 152. Mix RV1 via Mechanical Mix for 3.00 minutes. 153. Empty RV1. 154. Fill RV1 with 16.00 mL from Piperidine. 155. Mix RV1 via Mechanical Mix for 10.00 minutes. 156. Empty RV1. 157. Clean Lines with 15.00 mL from DMF. 158. Clean RV1 Vent with 15.00 mL from DMF. 159. ********Wash******* 160. Fill RV1 with 16.00 mL from DMF. 161. Mix RV1 via Mechanical Mix for 0.50 minutes. 162. Empty RV1. 163. Repeat from step 160, 1 time. (1) 164. Fill RV1 with 16.00 mL from DMF. 165. Wash RV1 with 45.00 mL from DMF 3Times. 166. Fill RV1 with 16.00 mL from NMP. 167. Mix RV1 via Mechanical Mix for 0.50 minutes. 168. Empty RV1.

95
169. Repeat from step 166, 1 time. (1) 170. Blank 171. ********Coupling Phe******* 172. Dissolve AA1 with 4.40 mL from HBTU for 0.50 173. Dissolve AA1 with 2.20 mL from DIEA for 0.20 174. Fill RV1 with 25.00 mL from AA1. 175. Clean AA1 with 20.00 mL from DMF. 176. Repeat from step 175, 1 time. (1) 177. Mix RV1 via Mechanical Mix for 15.00 minutes. 178. Empty RV1. 179. Blank 180. Blank 181. Dissolve AA2 with 4.40 mL from HBTU for 0.50 minutes. 182. Dissolve AA2 with 2.20 mL from DIEA for 0.20 minutes. 183. Fill RV1 with 25.00 mL from AA2. 184. Clean AA2 with 20.00 mL from DMF. 185. Repeat from step 184, 1 time. (1) 186. Mix RV1 via Mechanical Mix for 15.00 minutes. 187. Empty RV1. 188. Pause 189. Blank 190. ********Deprotection Cycle 6******* 191. Fill RV1 with 16.00 mL from Piperidine. 192. Mix RV1 via Mechanical Mix for 3.00 minutes. 193. Empty RV1. 194. Fill RV1 with 16.00 mL from Piperidine. 195. Mix RV1 via Mechanical Mix for 10.00 minutes. 196. Empty RV1. 197. Clean Lines with 15.00 mL from DMF. 198. Clean RV1 Vent with 15.00 mL from DMF. 199. ********Wash******* 200. Fill RV1 with 16.00 mL from DMF. 201. Mix RV1 via Mechanical Mix for 0.50 minutes. 202. Empty RV1. 203. Repeat from step 200, 1 time. (1) 204. Fill RV1 with 16.00 mL from DMF. 205. Wash RV1 with 45.00 mL from DMF 3Times. 206. Fill RV1 with 16.00 mL from NMP. 207. Mix RV1 via Mechanical Mix for 0.50 208. Empty RV1. 209. Repeat from step 206, 1 time. (1) 210. Blank 211. ********Coupling Sorbic acid******* 212. Dissolve AA3 with 4.40 mL from HBTU 213. Dissolve AA3 with 2.20 mL from DIEA 214. Fill RV1 with 25.00 mL from AA3. 215. Clean AA3 with 20.00 mL from DMF. 216. Repeat from step 215, 1 time. (1) 217. Mix RV1 via Mechanical Mix for 15.00 minutes. 218. Empty RV1. 219. Blank 220. Blank 221. Dissolve AA4 with 4.40 mL from HBTU for 0.50 minutes. 222. Dissolve AA4 with 2.20 mL from DIEA for 0.20 minutes. 223. Fill RV1 with 25.00 mL from AA4. 224. Clean AA4 with 20.00 mL from DMF. 225. Repeat from step 224, 1 time. (1) 226. Mix RV1 via Mechanical Mix for 15.00 minutes.

96
227. Empty RV1. 228. Pause 229. Blank 230. ********Final Wash******* 231. Fill RV1 with 16.00 mL from DMF. 232. Mix RV1 via Mechanical Mix for 0.50 minutes. 233. Empty RV1. 234. Repeat from step 231, 2 times. (2) 235. Wash RV1 with 70.00 mL from DMF 3Times. 236. Fill RV1 with 16.00 mL from MeOH. 237. Mix RV1 via Mechanical Mix for 5.00 minutes. 238. Empty RV1. 239. Wash RV1 with 80.00 mL from MeOH 3Times. 240. Clean Lines with 15.00 mL from DMF. 241. Clean RV1 Vent with 15.00 mL from DMF. 242. Blank

97
Peptide Synthesizer Protocol for HPro-NMeAla-Ala-2-Cl-Trt resin: File Name: MC01-134 HPro-NMeAla-Ala-OH_500mg Sept 9 2009 Created By: General Created On: 9/17/2009 1. ********SWELL******** 2. Wash RV1 with 15.00 mL from DMF 3Times. 3. Fill RV1 with 8.00 mL from DMF. 4. Mix RV1 via Mechanical Mix for 15.00 minutes. 5. Empty RV1. 6. Repeat from step 2, 1 time. (1) 7. Fill RV1 with 8.00 mL from NMP. 8. Mix RV1 via Mechanical Mix for 0.50 minutes. 9. Empty RV1. 10. Repeat from step 7, 1 time. (1) 11. Pause 12. Blank 13. ********Coupling NMeAla******* 14. Pause 15. **addition of active ester directly to flask** 16. Mix RV1 via Mechanical Mix for 20.00 minutes. 17. Empty RV1. 18. Pause 19. **kaiser test** 20. ********Deprotection Cycle 2******* 21. Fill RV1 with 8.00 mL from Piperidine. 22. Mix RV1 via Mechanical Mix for 3.00 minutes. 23. Empty RV1. 24. Fill RV1 with 8.00 mL from Piperidine. 25. Mix RV1 via Mechanical Mix for 10.00 minutes. 26. Empty RV1. 27. Clean Lines with 15.00 mL from DMF. 28. Clean RV1 Vent with 15.00 mL from DMF. 29. ********Wash******* 30. Fill RV1 with 8.00 mL from DMF. 31. Mix RV1 via Mechanical Mix for 0.50 minutes. 32. Empty RV1. 33. Repeat from step 30, 1 time. (1) 34. Fill RV1 with 8.00 mL from DMF. 35. Wash RV1 with 45.00 mL from DMF 3Times. 36. Fill RV1 with 8.00 mL from NMP. 37. Mix RV1 via Mechanical Mix for 0.50 minutes. 38. Empty RV1. 39. Repeat from step 36, 1 time. (1) 40. Blank 41. ********Coupling Pro******* 42. Dissolve AA1 with 2.20 mL from HBTU for 0.50 minutes. 43. Dissolve AA1 with 1.10 mL from DIEA for 0.20 minutes. 44. Fill RV1 with 15.00 mL from AA1. 45. Clean AA1 with 15.00 mL from DMF. 46. Repeat from step 45, 1 time. (1) 47. Mix RV1 via Mechanical Mix for 15.00 minutes. 48. Empty RV1. 49. Blank 50. Blank 51. Dissolve AA2 with 2.20 mL from HBTU for 0.50 minutes. 52. Dissolve AA2 with 1.10 mL from DIEA for 0.20 minutes.

98
53. Fill RV1 with 15.00 mL from AA2. 54. Clean AA2 with 15.00 mL from DMF. 55. Repeat from step 54, 1 time. (1) 56. Mix RV1 via Mechanical Mix for 15.00 minutes. 57. Empty RV1. 58. Pause 59. Blank 60. ********Deprotection Cycle 3******** 61. Fill RV1 with 8.00 mL from Piperidine. 62. Mix RV1 via Mechanical Mix for 3.00 minutes. 63. Empty RV1. 64. Fill RV1 with 8.00 mL from Piperidine. 65. Mix RV1 via Mechanical Mix for 10.00 minutes. 66. Empty 67. Clean 68. Clean 69. Blank 70. ********Wash******* 71. Fill RV1 with 8.00 mL from DMF. 72. Mix RV1 via Mechanical Mix for 0.50 minutes. 73. Empty RV1. 74. Repeat from step 71, 1 time. (1) 75. Fill RV1 with 8.00 mL from DMF. 76. Wash RV1 with 45.00 mL from DMF 3Times. 77. Fill RV1 with 8.00 mL from NMP. 78. Mix RV1 via Mechanical Mix for 0.50 minutes. 79. Empty RV1. 80. Repeat from step 77, 1 time. (1) 81. Blank 82. ********Complete Synthesis******* 83. Blank 84. ********Final Wash******* 85. Fill RV1 with 8.00 mL from DMF. 86. Mix RV1 via Mechanical Mix for 0.50 minutes. 87. Empty RV1. 88. Repeat from step 85, 2 times. (2) 89. Wash RV1 with 55.00 mL from DMF 4Times. 90. Fill RV1 with 8.00 mL from MeOH. 91. Mix RV1 via Mechanical Mix for 0.50 minutes. 92. Empty RV1. 93. Wash RV1 with 55.00 mL from MeOH 4Times. 94. Clean Lines with 15.00 mL from DMF. 95. Blank

99
Peptide Synthesizer Protocol for FmocSer(Pro)-Pro-N-Me-Ala-AlaOH (37): File Name: MC01-136 addition of depsipeptide to tripeptide Created By: General Created On: 9/18/2009 1. ********SWELL******** 2. Wash RV1 with 15.00 mL from DMF 3Times. 3. Fill RV1 with 8.00 mL from DMF. 4. Mix RV1 via Mechanical Mix for 15.00 minutes. 5. Empty RV1. 6. Repeat from step 2, 1 time. (1) 7. Blank 8. ********Coupling FmocSer(BocPro) OH******* 9. Pause 10. **addition of active ester directly to flask** 11. Mix RV1 via Mechanical Mix for 20.00 minutes. 12. Empty RV1. 13. Pause 14. **isatin test** 15. ********Complete Synthesis******* 16. Blank 17. ********Final Wash******* 18. Fill RV1 with 8.00 mL from DMF. 19. Mix RV1 via Mechanical Mix for 0.50 minutes. 20. Empty RV1. 21. Repeat from step 18, 2 times. (2) 22. Wash RV1 with 55.00 mL from DMF 4Times. 23. Fill RV1 with 8.00 mL from MeOH. 24. Mix RV1 via Mechanical Mix for 0.50 minutes. 25. Empty RV1. 26. Wash RV1 with 55.00 mL from MeOH 4Times. 27. Clean Lines with 15.00 mL from DMF. 28. Blank

100
References (1) Nussbaum, F. v.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Angew.
Chemie. Int. Ed. 2006, 45, 5072–5129.
(2) Levy, S. B.; Marshall, B. Nat. Med. 2004, 10, S122.
(3) Andersson, D. I. Curr. Opin. Microbiol. 2003, 6, 452–456.
(4) Weigel, L. M.; Clewell, D. B.; Gill, S. R.; Clark, N. C.; McDougal, L. K.;
Flannagan, S. E.; Kolonay, J. F.; Shetty, J.; Killgore, G. E.; Tenover, F. C.
Science 2003, 302, 1569–1571.
(5) Anonymous In Peptide characterization and application protocols; Fields, G.
B., Ed.; Humana: Totowa, N.J., 2007.
(6) Hamada, Y.; Shioiri, T. Chem. Rev. 2005, 105, 4441–4482.
(7) Schmidt, U.; Kroner, M.; Griesser, H. Tetrahedron Lett. 1988, 29, 4407–4408.
(8) Perez, E.; Hillman, D.; Fishkin, P.; Krook, J.; Tan, W.; Kuriakose, P.; Alberts,
S.; Dakhil, S. Invest. New Drugs 2005, 23, 257–261.
(9) Nakajima, H.; Kim, Y. B.; Terano, H.; Yoshida, M.; Horinouchi, S. Exp. Cell
Res. 1998, 241, 126–133.
(10) Koshino, H.; Osada, H.; Yano, T.; Uzawa, J.; Isono, K. Tetrahedron Lett.,
1991, 32, 7707–7710.
(11) Brötz-Oesterhelt, H.; Beyer, D.; Kroll, H.; Endermann, R.; Ladel, C.;
Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; Bandow,
J. E.; Sahl, H.; Labischinski, H. Nat. Med. 2005, 11, 1082–1087.
(12) Schmidt, U.; Neumann, K.; Schumacher, A.; Weinbrenner, S. Angew. Chem.
Int. Ed. Engl. 1997, 36, 1110–1112.
(13) Osada, H.; Yano, T.; Koshino, K.; Isono, J. J. Antibiotics 1991, 44, 1463–
1466.
(14) Yu, A. Y. H.; Houry, W. A. FEBS Letters 2007, 581, 3749–3757.
(15) Lee, B.; Park, E. Y.; Lee, K.; Jeon, H.; Sung, K. H.; Paulsen, H.; Rubsamen-
Schaeff, H.; Brotz-Oesterhelt, H.; Song, H. K. Nat. Struct. Mol. Biol. 2010, 17,
471–478.

101
(16) Hinzen, B.; Raddatz, S.; Paulsen, H.; Lampe, T.; Schumacher, A.; Häbich,
D.; Hellwig, V.; Benet-Buchholz, J.; Endermann, R.; Labischinski, H.; Brötz-
Oesterhelt, H. ChemMedChem 2006, 1, 689–693.
(17) Merrifield, R. B. J. Am. Chem. Soc. 1963, 85, 2149–2154.
(18) Sewald, N. In Peptides: Chemistry and Biology; Jakubke, H., Ed.; Wiley-
VCH: Weinheim, 2002.
(19) Atherton, E. In Solid Phase Peptide Synthesis: A Practical Approach;
Sheppard, R. C., Ed.; IRL Press: New York, 1989.
(20) López-Macià, À.; Jiménez, J. C.; Royo, M.; Giralt, E.; Albericio, F.
Tetrahedron Lett. 2000, 41, 9765–9769.
(21) Ghosh, S.; Pradhan, T. K. Tetrahedron Lett. 2008, 49, 3697–3700.
(22) Bowers, A.; West, N.; Taunton, J.; Schreiber, S. L.; Bradner, J. E.; Williams,
R. M. J. Am. Chem. Soc. 2008, 130, 11219–11222.
(23) Chan, W.C. In Fmoc Solid Phase Peptide Synthesis: A Practical Approach;
White, P.D., Ed.; Oxford University Press: New York, 2000.
(24) Coin, I.; Beyermann, M.; Bienert, M. Nat. Protocols 2007, 2, 3247–3256.
(25) Takacs, J. M.; Jaber, M. R.; Clement, F.; Walters, C. J. Org. Chem. 1998,
63, 6757–6760.
(26) Thompson, S. K.; Heathcock, C. H. J. Org. Chem. 1990, 55, 3386–3388.
(27) Bonadies, F.; Cardilli, A.; Lattanzi, A.; Orelli, L. R.; Scettri, A. Tetrahedron
Lett. 1994, 35, 3383–3386.
(28) Parenty, A.; Moreau, X.; Campagne, J. -M. Chem. Rev. 2006, 106, 911–939.
(29) Coin, I.; Beerbaum, M.; Schmieder, P.; Bienert, M.; Beyermann, M. Org. Lett.
2008, 10, 3857–3860.
(30) Cook, A.; Hodge, P.; Manzini, B.; Ruddick, C. L. Tetrahedron Lett. 2007, 48,
6496–6499.
(31) Yokokawa, F.; Inaizumi, A.; Shioiri, T. Tetrahedron 2005, 61, 1459–1480.
(32) Coin, I.; Dolling, R.; Krause, E.; Bienert, M.; Beyermann, M.; Sferdean, C. D.;
Carpino, L. A. J. Org. Chem. 2006, 71, 6171–6177.

102
(33) Bergmann, M.; Brand, E.; Weinmann, F. Hoppe-Seyler´s Zeitschrift für
physiologische Chemie, 1923, 131, 1–17.
(34) Spengler, J.; Koksch, B.; Albericio, F. Peptide Science 2007, 88, 823–828.
(35) Hinzen, Berthold; Brötz-oesterhelt, Heike; Endermann, Rainer; Henninger,
Kerstin; Paulsen, Holger; Raddatz, Siegfried; Lampe, Thomas; Hellwig,
Veronika; Schumacher, Andreas. Antibacterial Macrocycles. U.S. Patent
7,405,201, 2008.
(36) Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Adv. Drug Deliv.
Rev. 2001, 46, 3–26.
(37) Berry, A.; Cirillo, P. F.; Hickey, E. R.; Riether, D.; Thomson, D. S.; Ermann,
M.; Jenkins, J. E.; Mushi, I.; Taylor, M.; Chowdhury, C.; Palmer, C. F.;
Blumire, N. Compounds Which Modulate The CB2 Receptor. U.S. Patent
Application 20080039464, March 2008.
(38) Kaiser, E.; Colescott, R. L.; Bossinge, C. D.; Cook, P. I. Anal. Biochem.
1970, 34, 595–598.
(39) Kaiser, E.; Boddinger, C. D.; Colescott, R. L.; Olsen, D. B. Anal. Chem. Acta.
1980, 118, 149–151.
(40) T. Vojkovsky, Pept. Res. 1995, 8, 236–237.
(41) Karagiozov, S. K.; Abbott, F. S. Synthetic Commun. 2004, 34, 871–888.
(42) Alonso, D. A.; Nájera, C.; Varea, M. Helv. Chim. Acta 2002, 85, 4287–4305.
(43) Katritzky, A. R.; Xu, Y.; He, H.; Mehta, S. J. Org. Chem. 2001, 66, 5590–
5594.
(44) Su, Wei-yang; Speranza, George P. Preparation of phenoxyethanamines.
U.S. Patent 5,276,192, 1994.
(45) Pianowski, Z. L.; Winssinger, N. Chem. Commun. 2007, 3820-3822.
(46) Boggs III, N. T; Goldsmith, B.; Gawley, R. E.; Koehler, K. A.; Hiskey, R. G. J.
Org. Chem. 1979, 44, 2262-2269.
(47) Brasili, L.; Sorbi, C.; Franchini, S.; Manicardi, M.; Angeli, P.; Marucci, G.;
Leonardi, A.; Poggesi, E. J. Med. Chem. 2003, 48, 1504-1511.

103
(48) Merck and Co. Inc. Pyrazinone thrombin inhibitors. U.S. Patent 6,387,911,
2002.