synthesis, experimental and theoretical investigation of …tasiopo/89.pdf · synthesis,...
TRANSCRIPT

Polyhedron 62 (2013) 208–217
Contents lists available at SciVerse ScienceDirect
Polyhedron
journal homepage: www.elsevier .com/locate /poly
Synthesis, experimental and theoretical investigation of a new typenickel dithiolene complex
0277-5387/$ - see front matter � 2013 Elsevier Ltd. All rights reserved.http://dx.doi.org/10.1016/j.poly.2013.06.033
⇑ Corresponding author. Tel.: +30 210 7273838.E-mail address: [email protected] (G.A. Mousdis).
G. Soras a, N. Psaroudakis b, M.J. Manos c, A.J. Tasiopoulos c, D.G. Liakos d, G.A. Mousdis a,⇑a Theoretical & Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vass. Constantinou Aven., 116 35 Athens, Greeceb University. of Athens Chemistry Department, University Campus, Athens GR 157-71, Greecec Department of Chemistry, University of Cyprus, 1678 Nicosia, Cyprusd Max-Planck Institut für Bioanorganische Chemie Stiftstr. 34-36, D-45470 Mülheim an der Ruhr, Germany
a r t i c l e i n f o a b s t r a c t
Article history:Received 25 April 2013Accepted 10 June 2013Available online 1 July 2013
Keywords:Dithiolene complexesUV–Vis-NIR spectraIR and Raman spectraDFT calculationsCrystal structureCyclic voltammetry
A new nickel complex with an extended multisulfur dithiolene ligand, [Ni(dmeodddt)2] (dmeodddt = 5,6-dimethoxy-5,6-dihydro-1,4-dithiine-2,3-dithiolate), has been synthesized and characterized by IR,Raman, UV–Vis and NMR spectroscopy. Its crystal structure has been determined by X-ray crystallogra-phy, showing that the Ni atom is tetra-coordinated and has a square planar geometry with the methoxygroups placed above and below the metal dithiolene core, due to stereochemical hindrance. Electrochem-ical measurements showed that the complex exhibits four 1e� reversible redox waves. The results of the-oretical calculations showed a good agreement with the experimental findings and gave answers aboutits electronic structure.
� 2013 Elsevier Ltd. All rights reserved.
1. Introduction
Metal 1,2-dithiolenes have been widely studied during the pastfew decades. The intense interest in dithiolene complexes is the re-sult of both their intrinsic interest and their applications as molec-ular materials with conducting [1,2], magnetic [3], optical [4–6]and other properties due to their unique electronic structures [7–11]. The electronic, optical and magnetic properties of these mate-rials can be tuned through modification of the organic dithioleneligands [12]. In addition to such tuning at the molecular level,the bulk properties can also be tuned via crystal engineering asthe strong intermolecular interactions arising from the large num-ber of sulfur and other heavy atoms in these complexes allows awide variety of 3-D crystalline structures [13].
Complexes of [Ni(dmit)2]n�, [(dmit) = 1,3-dithiol-2-thione-4,5-dithiolate] were found to be molecular conductors and supercon-ductors [14]. This prompted many laboratories to explore newcomplexes in this area [15]. One of the most widely studied dithio-lene ligands for the preparation of metal complexes has been dddt[dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate]. This ligand is theinorganic structural analog to fragments of BEDT-TTF (bis ethyl-enedithio tetrathiafulvalene), that gave many conducting radicalsalts, some of which with superconducting properies. A detailed
analysis of a number of metal complexes with this ligand,[M(dddt)2] (M = Ni, Pd, Pt, Cu, Au, Co), has been reported [16].
One approach to maximizing electron delocalization in the or-ganic ligand is through the fusion of organic rings onto the squareplanar metal dithiolene core [17], thus increasing the size andenhancing orbital overlap, giving them peculiar molecular proper-ties such as high thermal and photochemical stabilities and intensevis-near-IR absorption [17]. This approach should allow elaboratesystems to be developed with highly tuneable electronic structuresand solid state arrangements. It is difficult to predict which struc-tural changes to a ligand produce significant stereo-electronic dif-ferences in the metal complexes. For that reason, a number ofmetal-bis-1,2-dithiolene complexes with SR groups attached atthe periphery were synthesized and studied [9].
In our previous work we synthesized and studied dddtanalogues by extending the outer rings, either with an ethylenethioxo- group, leading to [Ni(etodddt)2] [etodddt = 2,3,4a,8a-tetrahydro-[1,4]dithiino[2,3-b][1,4]oxathiine-6,7-dithiolate] [18],or with an ethylene dioxo- group, leading to [Ni(edodddt)2] [edo-dddt = 2,3,4a,8a-tetrahydro-[1,4]dithiino[2,3-b][1,4]dioxine-6,7-dithiolate] [4]. In both works the role of the external ligand unit onthe molecular structure and crystal packing was discussed.
In continuation of this work and for an investigation into theelectronic and structural effects of substitution on the external li-gand unit of dddt, a new metal 1,2-dithiolene compound, contain-ing two methoxy groups as an extension, was synthesized, namely

S
S
S
S
S
S
SNi
S
O
O
S
S
S
S
O
O
S
S
SNi
S
S
O
S
S
S
S
O
S
S
S
SNi
S
O
O
S
S
S
S
O
O
S
S
SNi
S
[Ni(dddt)2] [Ni(etodddt)2]
[Ni(edodddt)2] [Ni(dmeodddt)2]
Scheme 1. Structural formulas of [Ni(dmeodddt)2], [Ni(edodddt)2], [Ni(etodddt)2]and [Ni(dddt)2].
G. Soras et al. / Polyhedron 62 (2013) 208–217 209
[Ni(dmeodddt)2]. Electrochemical and X-ray measurements andspectral investigations have been made. These measurements werealso compared to those of the compounds [Ni(etodddt)2] and[Ni(edodddt)2]. DFT studies of the new compound’s structural,electronic, spectral and vibronic properties are also included.
The reason for choosing this group was because[Ni(dmeodddt)2] [dmeodddt = 5,6-dimethoxy-5,6-dihydro-1,4-dithiine-2,3-dithiolate] looks similar to the compounds [Ni(edo-dddt)2] and [Ni(etodddt)2] (Scheme 1), but the MeO groups arenot bonded together to form a ring, so there are more potentialarrangements. Due to this we expect better solubility and maybea different crystal packing.
2. Experimental
2.1. Materials and methods
(Bu4N)2[Zn(dmit)2] was prepared according to the literature[19]. All starting materials were of analytical grade, obtained com-mercially and used as received, except for methanol, which wasdehydrated according to the literature [20]. Scheme 2 summerizesthe synthetic procedure leading to [Ni(dmeodddt)2].
2.2. Synthesis of 1,2-dichloro-1,2-dimethoxy ethane (1) [21]
9.60 g of glyoxal trimeric dihydrate 45.7 mmol) were dissolvedin a mixture of 12.8 mL of methanol and 24.0 mL of carbon tetra-chloride, with the addition of 1.0 mL of thionylchloride. The solu-tion was cooled to 0 �C and 23.0 mL of thionylchloride wereadded dropwise under stirring. After 30% of the SOCl2 had beenadded, a vigorous gaseous evolution took place and the reactionbecame endothermic; the solution was then left to reach roomtemperature. The SOCI2 was then added faster than before to keepthe reaction temperature at 20–25 �C, and if necessary the mixturewas heated. The addition took about 6 hr. and the mixture wasthen stirred overnight at room temp. The solvent and excess SOCI2
were evaporated under vacuum, and the residue distilled at 19–
MeO
MeO
Cl
Cl
S
S
S
SS
CCl4
SOCl2 (Bu4N)2Zn(dmit)2
NiCl2
I2
Me4NOHO
O
S
S
S
S
O
O
S
S
SNi
SMe4N
O
O
S
S
S
S
O
O
S
S
SNi
S
O
O
1 2
2
3
3
4
O
O
O
O
OH
OHHO
HO
Scheme 2. Synthetic route to the [Ni(dmeodddt)2].
20 Torr, to isolate the desired product in the liquid form at 79 �C.It consists mainly of the rac and meso isomers, and when cooledto 0–4 �C crystals of the meso form were precipitated as a whitesolid.
Yield: 4.50 g (28.3 mmol), (20.6%), mp: 72 �C (meso form); 1HNMR (300 MHz, CDCl3) d (ppm): 5.49 and 5.56 (s, 2H meso andrac), 3.60 and 3.59 (s, 6H meso and rac); 13C NMR (300 MHz, CDCl3)d (ppm): 97.36 and 97.20 (s, C–C meso and rac), 58.33 and 58.13 (s,CH3 meso and rac);
2.3. Synthesis of 5,6-dimethoxy-5,6-dihydro-[1,3]dithiolo[4,5-b][1,4]dithiine-2-thione (2)
1,2-Dichloro-1,2-dimethoxy ethane (1) (1.59 g, 10.0 mmol) and4.7 g (5.0 mmol) of (Bu4N)2[Zn(dmit)2] were dissolved in 100 mLacetone. The solution was refluxed for 48 h and the color changedfrom bright red to yellow-brown. The solvent was evaporated un-der vacuum and the resulting dark-brown sticky residue was dis-solved in 200 mL of dichloromethane and washed twice with200 mL of water. The organic layer was dried with magnesium sul-fate and condensed under vacuum to 10 mL. Purification of the fi-nal product was made by column chromatography in silica gelusing CH2Cl2 as the eluent. Yellow-orange crystals were produced.Depending on the starting material, the product can consists of themeso isomer or a mixture of the meso and rac isomers. Yield: 1.4 g,(49%); mp: 130 �C (meso); UV–Vis (CH2Cl2, nm): 405, 272; 1H NMR(300 MHz, CDCl3) d (ppm): 5.33 (s, 1H), 3.58 (s, 3H) for the mesoform and 4.98 (s, 1H), 3.55 (s, 3H) for the rac form; IR data (m,cm�1): 2996, 2948, 2921, 2890, 2875, 2826, 1483, 1445, 1435,1321, 1247, 1230, 1184, 1106, 1064, 1045, 1023, 968, 937, 887,827, 724, 616.
2.4. Bis[5,6-dimethoxy-5,6-dihydro-1,4-dithiine-2,3-dithiolate] nickeltetramethylammonium [(Me4N)Ni(dmeodddt)2] (3)
In a dispersion of thione 2 (200 mg, 0.70 mmol) (meso form) andnickel chloride hexahydrate (50 mg, 0.35 mmol) in 20 mL metha-nol under N2, 1 mL of hydroxyl tetramethylammonium, 25% inmethanol was added. The mixture was stirred for 12 h, under anitrogen atmosphere. The solid was removed by filtration in a vac-uum and washed repeatedly with methanol to give 3 as a brownpowder.
Yield: 165 mg, (77%) of pure product; mp: >290 �C; UV–Vis(CH2Cl2, nm (e, dm3 mol�1 cm�1)): 1144 (25493), 603 (5259), 389(18165), 317 (9114), 250 (95557); IR data (m, cm�1): 2957, 2929,2871, 2824, 1479, 1457, 1366, 1320, 1223, 1185, 1151, 1106,1092, 1027, 933, 856, 839, 737, 629; Elemental Anal. Calc. forC16H28NNiO4S8: C, 31.32; H, 4.60. Found: C, 31.65; H, 4.88%.
2.5. Bis[5,6-dimethoxy-5,6-dihydro-1,4-dithiine-2,3-dithiolate] nickel[Ni(dmeodddt)2] (4)
Compound 3 (65 mg 0.11 mmol) was dissolved in acetonitrile(30 mL), heated and filtered. A solution of iodine (30 mg,0.12 mmol) in acetonitrile (30 mL) was added to the filtrate. Themixture stirred for 40 min and left to stand at room temperaturefor 24 h to give dark green crystalline powder that was filteredand washed with acetonitrile.
Yield: 33 mg, (56%); mp: >290 �C; UV–Vis (CH2Cl2, nm (e, dm3 -mol�1 cm�1)): 986 (69650), 653 (2820), 366 (17872), 364 (18044),311 (92133), 268 (66033); IR data (m, cm�1): 2993, 2956, 2927,2894, 2874, 2852, 2831, 1450, 1344, 1315, 1259, 1239, 1207,1182, 1112, 1072, 1022, 965, 941, 894, 817, 736, 721, 626; RamanShifts (cm�1): 150, 235, 275, 348, 368, 380, 414, 480, 542, 626, 911,1023, 1244, 1342, 1355, 2833, 2933; dH (300 MHz, CDCl3) d (ppm):5.18 (s, 2H), 3.59 (s, 6H); 13C NMR (300 MHz, CDCl3) d (ppm):
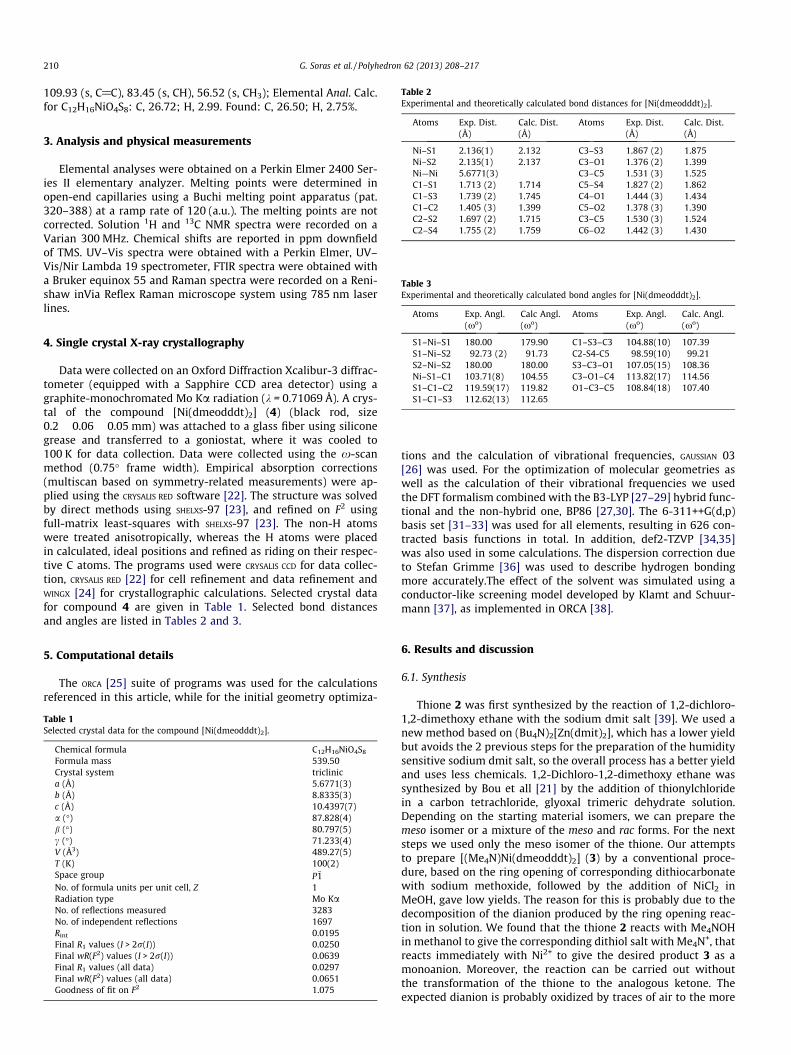
Table 2Experimental and theoretically calculated bond distances for [Ni(dmeodddt)2].
210 G. Soras et al. / Polyhedron 62 (2013) 208–217
109.93 (s, C@C), 83.45 (s, CH), 56.52 (s, CH3); Elemental Anal. Calc.for C12H16NiO4S8: C, 26.72; H, 2.99. Found: C, 26.50; H, 2.75%.
Atoms Exp. Dist.(Å)
Calc. Dist.(Å)
Atoms Exp. Dist.(Å)
Calc. Dist.(Å)
Ni–S1 2.136(1) 2.132 C3–S3 1.867 (2) 1.875Ni–S2 2.135(1) 2.137 C3–O1 1.376 (2) 1.399Ni—Ni 5.6771(3) C3–C5 1.531 (3) 1.525C1–S1 1.713 (2) 1.714 C5–S4 1.827 (2) 1.862C1–S3 1.739 (2) 1.745 C4–O1 1.444 (3) 1.434C1–C2 1.405 (3) 1.399 C5–O2 1.378 (3) 1.390C2–S2 1.697 (2) 1.715 C3–C5 1.530 (3) 1.524C2–S4 1.755 (2) 1.759 C6–O2 1.442 (3) 1.430
Table 3Experimental and theoretically calculated bond angles for [Ni(dmeodddt)2].
Atoms Exp. Angl. Calc Angl. Atoms Exp. Angl. Calc. Angl.
3. Analysis and physical measurements
Elemental analyses were obtained on a Perkin Elmer 2400 Ser-ies II elementary analyzer. Melting points were determined inopen-end capillaries using a Buchi melting point apparatus (pat.320–388) at a ramp rate of 120 (a.u.). The melting points are notcorrected. Solution 1H and 13C NMR spectra were recorded on aVarian 300 MHz. Chemical shifts are reported in ppm downfieldof TMS. UV–Vis spectra were obtained with a Perkin Elmer, UV–Vis/Nir Lambda 19 spectrometer, FTIR spectra were obtained witha Bruker equinox 55 and Raman spectra were recorded on a Reni-shaw inVia Reflex Raman microscope system using 785 nm laserlines.
(xo) (xo) (xo) (xo)
S1–Ni–S1 180.00 179.90 C1–S3–C3 104.88(10) 107.39S1–Ni–S2 92.73 (2) 91.73 C2-S4-C5 98.59(10) 99.21S2–Ni–S2 180.00 180.00 S3–C3–O1 107.05(15) 108.36Ni–S1–C1 103.71(8) 104.55 C3–O1–C4 113.82(17) 114.56S1–C1–C2 119.59(17) 119.82 O1–C3–C5 108.84(18) 107.40S1–C1–S3 112.62(13) 112.65
4. Single crystal X-ray crystallography
Data were collected on an Oxford Diffraction Xcalibur-3 diffrac-tometer (equipped with a Sapphire CCD area detector) using agraphite-monochromated Mo Ka radiation (k = 0.71069 Å). A crys-tal of the compound [Ni(dmeodddt)2] (4) (black rod, size0.2 � 0.06 � 0.05 mm) was attached to a glass fiber using siliconegrease and transferred to a goniostat, where it was cooled to100 K for data collection. Data were collected using the x-scanmethod (0.75� frame width). Empirical absorption corrections(multiscan based on symmetry-related measurements) were ap-plied using the CRYSALIS RED software [22]. The structure was solvedby direct methods using SHELXS-97 [23], and refined on F2 usingfull-matrix least-squares with SHELXS-97 [23]. The non-H atomswere treated anisotropically, whereas the H atoms were placedin calculated, ideal positions and refined as riding on their respec-tive C atoms. The programs used were CRYSALIS CCD for data collec-tion, CRYSALIS RED [22] for cell refinement and data refinement andWINGX [24] for crystallographic calculations. Selected crystal datafor compound 4 are given in Table 1. Selected bond distancesand angles are listed in Tables 2 and 3.
5. Computational details
The ORCA [25] suite of programs was used for the calculationsreferenced in this article, while for the initial geometry optimiza-
Table 1Selected crystal data for the compound [Ni(dmeodddt)2].
Chemical formula C12H16NiO4S8
Formula mass 539.50Crystal system triclinica (Å) 5.6771(3)b (Å) 8.8335(3)c (Å) 10.4397(7)a (�) 87.828(4)b (�) 80.797(5)c (�) 71.233(4)V (Å3) 489.27(5)T (K) 100(2)Space group P�1No. of formula units per unit cell, Z 1Radiation type Mo KaNo. of reflections measured 3283No. of independent reflections 1697Rint 0.0195Final R1 values (I > 2r(I)) 0.0250Final wR(F2) values (I > 2r(I)) 0.0639Final R1 values (all data) 0.0297Final wR(F2) values (all data) 0.0651Goodness of fit on F2 1.075
tions and the calculation of vibrational frequencies, GAUSSIAN 03[26] was used. For the optimization of molecular geometries aswell as the calculation of their vibrational frequencies we usedthe DFT formalism combined with the B3-LYP [27–29] hybrid func-tional and the non-hybrid one, BP86 [27,30]. The 6-311++G(d,p)basis set [31–33] was used for all elements, resulting in 626 con-tracted basis functions in total. In addition, def2-TZVP [34,35]was also used in some calculations. The dispersion correction dueto Stefan Grimme [36] was used to describe hydrogen bondingmore accurately.The effect of the solvent was simulated using aconductor-like screening model developed by Klamt and Schuur-mann [37], as implemented in ORCA [38].
6. Results and discussion
6.1. Synthesis
Thione 2 was first synthesized by the reaction of 1,2-dichloro-1,2-dimethoxy ethane with the sodium dmit salt [39]. We used anew method based on (Bu4N)2[Zn(dmit)2], which has a lower yieldbut avoids the 2 previous steps for the preparation of the humiditysensitive sodium dmit salt, so the overall process has a better yieldand uses less chemicals. 1,2-Dichloro-1,2-dimethoxy ethane wassynthesized by Bou et all [21] by the addition of thionylchloridein a carbon tetrachloride, glyoxal trimeric dehydrate solution.Depending on the starting material isomers, we can prepare themeso isomer or a mixture of the meso and rac forms. For the nextsteps we used only the meso isomer of the thione. Our attemptsto prepare [(Me4N)Ni(dmeodddt)2] (3) by a conventional proce-dure, based on the ring opening of corresponding dithiocarbonatewith sodium methoxide, followed by the addition of NiCl2 inMeOH, gave low yields. The reason for this is probably due to thedecomposition of the dianion produced by the ring opening reac-tion in solution. We found that the thione 2 reacts with Me4NOHin methanol to give the corresponding dithiol salt with Me4N+, thatreacts immediately with Ni2+ to give the desired product 3 as amonoanion. Moreover, the reaction can be carried out withoutthe transformation of the thione to the analogous ketone. Theexpected dianion is probably oxidized by traces of air to the more

Fig. 1. Labeled ball and stick representation of the structure of [Ni(dmeodddt)2]. Hatoms are omitted for clarity. Selected crystal structure bond distances are alsogiven, the calculated ones are in parenthesis.
G. Soras et al. / Polyhedron 62 (2013) 208–217 211
stable monoanion. The neutral complex 4 can be obtained by oxi-dation with an excess of I2. In an attempt to obtain good crystals forX-ray analysis, an ACN solution of 3 in a test tube was left to oxi-dize slowly in an iodine chamber; after 24 h, crystals of 4 wereformed on the walls of the tube.
6.2. Crystal structure of bis[5,6-dimethoxy-5,6-dihydro-1,4-dithiine-2,3-dithiolate] nickel
[Ni(dmeodddt)2] crystallizes in the monoclinic space groupP21/n. A labeled ball and stick representation of the compound isgiven in Fig. 1. Selected bond lengths and angles, from the crystalstructure determination and calculated values from DFT studies,are shown in Tables 2 and 3.
The compound [Ni(dmeodddt)2] contains one Ni2+ ion linked tothe two vicinal exocyclic sulfur atoms of the two chelating ligands.The Ni atom is tetra-coordinate and has a square planar geometry,as in both the [Ni(edodddt)2] and [Ni(etodddt)2] compounds. Thetwo methoxy groups are cis to each other at one end of the mole-cule and they are placed in different manner, due to stereochemi-cal hindrance, above and below the metal dithiolene core, adoptingan envelope-like conformation. By adopting this conformation, astacking formation of the molecules inside the crystal is prohibited,as in the cases of the thioxo- or dioxo-rings analogues [4,18]. Thepacking motif of the crystal down the b axis, shown in the Fig. 2,reveals a parallel alignment of the [Ni(dmeodddt)2] molecules.
Fig. 2. Packing of [Ni(dmeodddt)2] molecules, viewed down the b-axis. Color code: N
A close examination of the packing revealed the existence ofintermolecular hydrogen-bonding interactions involving the meth-oxy groups (C� � �O separations = 3.274–3.473 Å) and also the C andS atoms of the ligands (C� � �S separations = 3.553–3.779 Å). Theclosest Ni–Ni distance is 5.6771(3) Å, which is the length of the aaxis. In fact this distance is larger than in [Ni(etodddt)2](5.0051 Å) or [Ni(edodddt)2] (4.5870 Å). This observation indicatesa stronger repulsion for a single group in the stacking formationthan for a closed ring.
The methoxy group, due to its free rotation across the single C–O bond, enhances the stereochemical hindrance. The Ni–S bondlengths are 2.135(1) [Ni–S1] and 2.137(1) [Ni–S2] Å, whereas theS–C, C@C and C–O bond distances are 1.697(2)–1.867(2),1.405(3) and 1.376(2)–1.444(3) Å respectively. These bond dis-tances of the [Ni(dmeodddt)2] compound lie within the range ob-served for other neutral dithiolenes [5,16,19,40]. In fact thesedistances are very similar to the distances that have been mea-sured in both [Ni(etodddt)2] [18] and [Ni(edodddt)2] [4], sincethe substituents are not coplanar with the nickel-dithiolene core.Furthermore, as has been reported for similar compounds, [41]the C2–S2 and C1–S1 distances are shorter than the C2–S4 andCl–S3 distances, respectively, which can be explained by the reso-nance structures in the radical form that allow for partial C1–S1and C2–S2 double bonds (Scheme 3).
6.3. IR and Raman spectra
The Raman spectrum of [Ni(dmeodddt)2] was acquired using a785 nm laser line. It shows the characteristic vibrations of S–Ni–Sand Ni–S at 414 and 480 cm�1, and the characteristic vibrationsof C–O–C and C–O at 348 and 380 cm�1. The peaks at 542 and911 cm�1 correspond to the S–C–S bending and S–C stretchingvibrations. The C@C stretching vibrations appear at 1023 and1355 cm�1. The intensities at 2833 and 2933 cm�1 correspond toC-H vibrations.
[Ni(dmeodddt)2] also displays a very rich IR spectrum withcharacteristic peaks. The stretching vibrations of the O–C bondsare observed at 965, 1072 1112 and 1182 cm�1. The S–C stretchingvibrations are also present in the IR spectrum at 736 cm�1, whilethe C@C ones appear at 1259 and 1022 cm�1. Furthermore, thereare also characteristic bands in the region between 2800 and3000 cm�1 related to C–H stretching vibrations.
i, blue; O, red; S, yellow; C, black. H atoms are omitted for clarity. (Color online.)

O
O
S
S
SNi
S
O
O
S
S
S
S2+
O
O
S
S
SNi
S
O
O
S
S
S
S2+
O
O
S
S
SNi
S
O
O
S
S
S
S2+
-
--
-
Scheme 3. Resonance forms of [Ni(dmeodddt)2].
200 300 400 500 600 700 800 900 1000 1100 1200 13000
20000
40000
60000
80000
100000
120000
140000
ε (d
m3 m
ol-1cm
-1)
λ (nm)
3
4
Fig. 3. UV–Vis-NIR spectra of 4 (black) and 3 (red) in CH2Cl2. (Color online.)
400 600 800 1000 120
O
O
S
S
S
S
O
O
S
S
S
Ni
S
986
1012
1027
Abs
orba
nce
(a.u
)
λ (nm)
DMSO CS2 CH2Cl2
Fig. 4. UV–Vis-NIR spectra of [Ni(dmeodddt)2] in three different solvents.
212 G. Soras et al. / Polyhedron 62 (2013) 208–217
The theoretical IR and Raman spectra of this compound werecalculated; a comparison between these results and the experi-mental ones will be discussed later.
6.4. UV–Vis-NIR spectra
The UV–Vis-NIR spectra of complexes 3 and 4 are shown inFig. 3. Complex 3 in dichloromethane shows a strong broad absorp-tion at 1144 nm (e = 25493 dm3 mol�1 cm�1) which is assigned to ap–p⁄ transition (2b1u ? 2b2g) [42]. A characteristic long wave tran-sition is also found in complex 4 at 986 nm (e = 69650 dm3 mol�1 -cm�1). The molar extinction coefficient of 4 is higher than the evalue in [Ni(etodddt)2] (e = 21175 dm3 mol�1 cm�1 in CH2Cl2) or[Ni(edodddt)2] (e = 21175 dm3 mol�1 cm�1 in CH2Cl2). This factsuggests that this compound could be a better candidate as a NIRdye than the other two compounds.
Moreover, a low intensity peak at 603 nm for 3 and 653 nm for4 are considered to be due to the n ? p transition between thenon-bonding electron pairs at the sulfur atoms and the 2 b2g MO.There is also a band at 389 nm for 3 and 366 nm for 4 that is notassigned, but mainly n=s ? p⁄ and metal-ligand charge-transferbands are to be expected in this region. The high energy peaks at317 and 311 nm for 3 and 4 respectively, are due to inter-p transi-tions, based on their high intensity [42]. A relatively low absorp-tion appears between 400 and 800 nm providing a transparentwindow, which is important for optical limiting applications.
The UV–Vis absorption spectra of compound 4 in solvents of dif-ferent polarities is plotted in Fig. 4. One can see that as the polarityincreases, there is a bathochromic shift. The solvent dependence onthe wavelength of the light absorption is related to the rearrange-ment of the solvent and the complex electrons. This rearrangementresults in stabilization of the ground and excited states by intermo-lecular forces, which can affect the transition energies [43]. Severalproperties of the solvent (depending on Coulomb, directional,inductive, dispersion, charge-transfer, hydrogen-bonding forces)contribute to the solvation behavior of a solvent, thus it is not
possible to correlate solvatochromism by a single property. Solva-tochromism is common to metal dithiolene complexes [44].
6.5. Cyclic voltammetry
In the class of square-planar d8 metal-dithiolene complexes, themembers of the series, which may spread from the dianionic to thedicationic form [45], can be connected through reversible one-elec-tron redox steps.
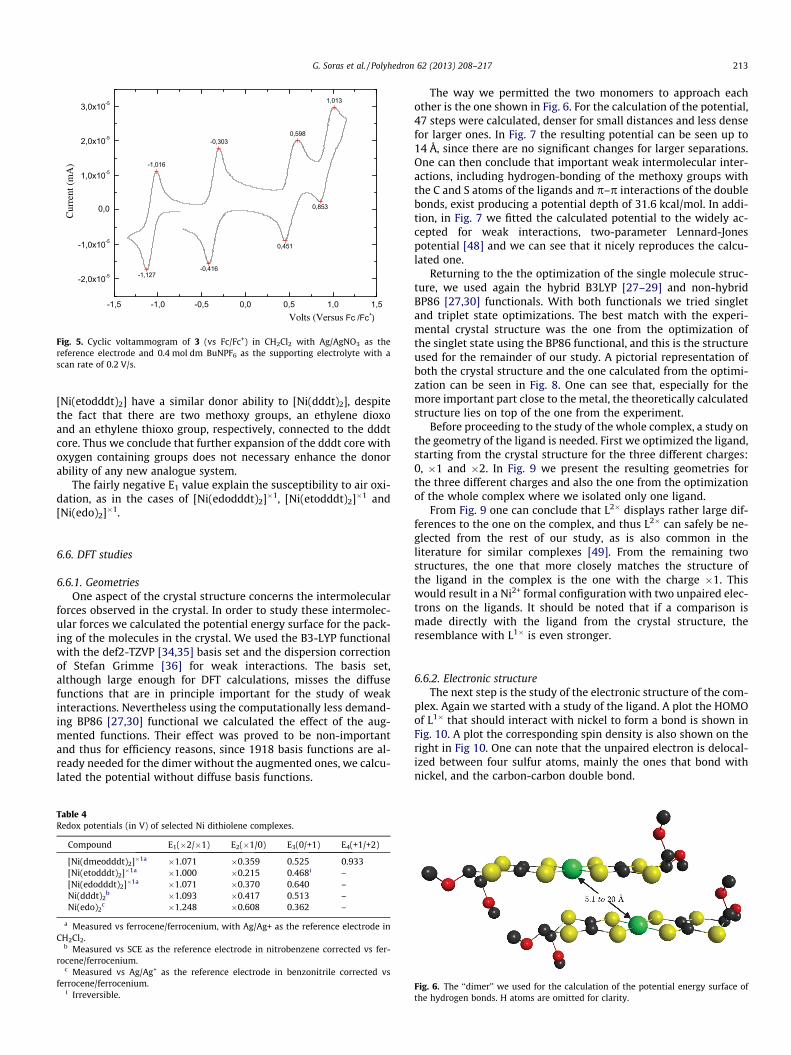
The electrochemistry of complex 3 was investigated using cyclicvoltammetry in CH2Cl2 (versus the ferrocene/ferrocenium couplewith Ag/AgNO3 as a reference electrode and Pt as a working elec-trode) (Fig. 5).
The cyclovoltammogram exhibits four 1e� reversible redoxwaves at E1/2 = �1.071, �0.359, 0.525 and 0.933 V (versus ferro-cene/ferrocenium). These oxidations correspond to the equilibria:
Table 4 shows the voltammetric data, not only of this new com-pound but also of the compounds [Ni(edodddt)2][4], [Ni(etodddt)2][18], [Ni(dddt)2] [46] and [Ni(edo)2] [47], (corrected versus ferro-cene) for a comparison. It is clear that [Ni(edo)2] is the strongest do-nor from all of examples due to the electron donating properties ofthe two oxygen atoms. [Ni(dmeodddt)2], [Ni(edodddt)2] and
-1,5 -1,0 -0,5 0,0 0,5 1,0 1,5
-2,0x10-5
-1,0x10-5
0,0
1,0x10-5
2,0x10-5
3,0x10-5
-1,016
-0,3030,598
1,013
-1,127
0,853
0,451
-0,416
Cur
rent
(m
A)
Volts (Versus Fc /Fc+)
Fig. 5. Cyclic voltammogram of 3 (vs Fc/Fc+) in CH2Cl2 with Ag/AgNO3 as thereference electrode and 0.4 mol dm BuNPF6 as the supporting electrolyte with ascan rate of 0.2 V/s.
G. Soras et al. / Polyhedron 62 (2013) 208–217 213
[Ni(etodddt)2] have a similar donor ability to [Ni(dddt)2], despitethe fact that there are two methoxy groups, an ethylene dioxoand an ethylene thioxo group, respectively, connected to the dddtcore. Thus we conclude that further expansion of the dddt core withoxygen containing groups does not necessary enhance the donorability of any new analogue system.
The fairly negative E1 value explain the susceptibility to air oxi-dation, as in the cases of [Ni(edodddt)2]�1, [Ni(etodddt)2]�1 and[Ni(edo)2]�1.
6.6. DFT studies
6.6.1. GeometriesOne aspect of the crystal structure concerns the intermolecular
forces observed in the crystal. In order to study these intermolec-ular forces we calculated the potential energy surface for the pack-ing of the molecules in the crystal. We used the B3-LYP functionalwith the def2-TZVP [34,35] basis set and the dispersion correctionof Stefan Grimme [36] for weak interactions. The basis set,although large enough for DFT calculations, misses the diffusefunctions that are in principle important for the study of weakinteractions. Nevertheless using the computationally less demand-ing BP86 [27,30] functional we calculated the effect of the aug-mented functions. Their effect was proved to be non-importantand thus for efficiency reasons, since 1918 basis functions are al-ready needed for the dimer without the augmented ones, we calcu-lated the potential without diffuse basis functions.
Table 4Redox potentials (in V) of selected Ni dithiolene complexes.
Compound E1(�2/�1) E2(�1/0) E3(0/+1) E4(+1/+2)
[Ni(dmeodddt)2]�1a �1.071 �0.359 0.525 0.933[Ni(etodddt)2]�1a �1.000 �0.215 0.468i –[Ni(edodddt)2]�1a �1.071 �0.370 0.640 –Ni(dddt)2
b �1.093 �0.417 0.513 –Ni(edo)2
c �1.248 �0.608 0.362 –
a Measured vs ferrocene/ferrocenium, with Ag/Ag+ as the reference electrode inCH2Cl2.
b Measured vs SCE as the reference electrode in nitrobenzene corrected vs fer-rocene/ferrocenium.
c Measured vs Ag/Ag+ as the reference electrode in benzonitrile corrected vsferrocene/ferrocenium.
i Irreversible.
The way we permitted the two monomers to approach eachother is the one shown in Fig. 6. For the calculation of the potential,47 steps were calculated, denser for small distances and less densefor larger ones. In Fig. 7 the resulting potential can be seen up to14 Å, since there are no significant changes for larger separations.One can then conclude that important weak intermolecular inter-actions, including hydrogen-bonding of the methoxy groups withthe C and S atoms of the ligands and p–p interactions of the doublebonds, exist producing a potential depth of 31.6 kcal/mol. In addi-tion, in Fig. 7 we fitted the calculated potential to the widely ac-cepted for weak interactions, two-parameter Lennard-Jonespotential [48] and we can see that it nicely reproduces the calcu-lated one.
Returning to the the optimization of the single molecule struc-ture, we used again the hybrid B3LYP [27–29] and non-hybridBP86 [27,30] functionals. With both functionals we tried singletand triplet state optimizations. The best match with the experi-mental crystal structure was the one from the optimization ofthe singlet state using the BP86 functional, and this is the structureused for the remainder of our study. A pictorial representation ofboth the crystal structure and the one calculated from the optimi-zation can be seen in Fig. 8. One can see that, especially for themore important part close to the metal, the theoretically calculatedstructure lies on top of the one from the experiment.
Before proceeding to the study of the whole complex, a study onthe geometry of the ligand is needed. First we optimized the ligand,starting from the crystal structure for the three different charges:0, �1 and �2. In Fig. 9 we present the resulting geometries forthe three different charges and also the one from the optimizationof the whole complex where we isolated only one ligand.
From Fig. 9 one can conclude that L2� displays rather large dif-ferences to the one on the complex, and thus L2� can safely be ne-glected from the rest of our study, as is also common in theliterature for similar complexes [49]. From the remaining twostructures, the one that more closely matches the structure ofthe ligand in the complex is the one with the charge �1. Thiswould result in a Ni2+ formal configuration with two unpaired elec-trons on the ligands. It should be noted that if a comparison ismade directly with the ligand from the crystal structure, theresemblance with L1� is even stronger.
6.6.2. Electronic structureThe next step is the study of the electronic structure of the com-
plex. Again we started with a study of the ligand. A plot the HOMOof L1� that should interact with nickel to form a bond is shown inFig. 10. A plot the corresponding spin density is also shown on theright in Fig 10. One can note that the unpaired electron is delocal-ized between four sulfur atoms, mainly the ones that bond withnickel, and the carbon-carbon double bond.
Fig. 6. The ‘‘dimer’’ we used for the calculation of the potential energy surface ofthe hydrogen bonds. H atoms are omitted for clarity.

Fig. 7. The calculated and Lennard-Jones fitted potential for the approach of two ‘‘monomers’’.
Fig. 8. Pictorial representation of the crystal structure (blue) and the one from thegeometry optimization (pink). (Color online.)
214 G. Soras et al. / Polyhedron 62 (2013) 208–217
In the actual complex, if nickel is in the formal oxidation state2+ and the ligand is L�1, there are two possibilities. The first oneis for the two orbitals to be coupled ferromagnetically, resultingin a paramagnetic S = 1 ground state. The second possibility is forthe two electrons to be coupled anti-ferromagnetically, resultingin an S = 0 ground state. From a computational point of view, thesetwo solutions present a major difference. While for the S = 1 case, asingle slater determinant is in principle capable of describing thesystem, for the S = 0 case more than one determinant is needed.If using DFT methods, the S = 0 case presents a problem since in
Fig. 9. Optimized structures for the ligand, with different ele
DFT only a single determinant is used. For these scenarios, the wellknown broken symmetry formalism [50–53] can be used.
We first checked to see if a broken symmetry solution exists forour system. Using the BP86 functional, one ends up with a singletsolution. The triplet solution lies 13.8 kcal/mol above this, and ifone tries a broken symmetry solution it converges back to the re-stricted singlet one. Nevertheless, using the B3LYP there is indeed abroken symmetry solution which rests 1.0 kcal/mol below the sin-glet one and 5.82 kcal/mol below the triplet. This result agreeswith reports for similar complexes [54].
The corresponding orbitals [55] that should show the existenceof a broken symmetry solution are plotted in Fig. 11. The overlapbetween these two orbitals is 0.70 and it is this significant overlapthat stabilizes the antiferromagnetically coupled broken symmetrysolution with respect to the triplet one.
The spin density for the broken symmetry solution and the trip-let one is plotted in Fig. 12. For the broken symmetry solution, thespin density is known to be an artifact of the formalism, but it canstill give a hint of the wavefunction. The similarities between thetwo densities suggest that the unpaired electrons are localizedon the ligands, in one case ferromagnetically and in the other anti-feromagnetically coupled.
ctron charges, and the one from the optimized complex.

Fig. 10. HOMO orbital of L�1 (left) and its spin density (right).
Fig. 11. The magnetic corresponding orbitals for the spin up, alpha, electron (left) and the spin down, beta, electron (right).
Fig. 12. Spin densities for the complex, for the broken symmetry singlet solution (left) and the triplet one (right).
500 1000 1500 2000 2500 3000
29332833
1355
1023911542
480
380
414348
O
O
S
S
S
S
O
O
S
S
SNi
S
Calculated
Experimental Excitation: 785 nm laser line
Scat
teri
ng I
nten
sity
[a.
u.]
Raman shifts (cm-1)
Fig. 13. Experimental (black) and theoretical (red) Raman spectra. (Color online.)
3500 3000 2500 2000 1500 1000 5003500 3000 2500 2000 1500 1000 500
2993 29572928
145013441315
1259
12071180
1113
1072
1022
941
895
818
737627
O
O
S
S
S
NiS O
O
S
S
S
S
Abs
orba
nce
(a.u
.)
Wavenumber (cm-1)
Experimental spectra
Calculated spectra
Fig. 14. Experimental (black) and theoretical (red) IR spectra. (Color online.)
G. Soras et al. / Polyhedron 62 (2013) 208–217 215
What is also important to notice in Fig. 12 is the similarity be-tween the spin densities on the two complexes and the spin den-sity of the L1� free ligand. This again supports the description ofthe complex as having nickel with a formal oxidation number of+2 and the ligands as radicals with a charge of �1, antiferromag-netically coupled.
Finally the molecular orbitals of the complex were analyzed.Both the HOMO and LUMO molecular orbitals seem not to haveany important metal character. They are constructed mainly fromthe pz orbitals of the four sulfur atoms and the pz orbitals of thecarbon atoms of the double bond. For the HOMO molecular orbital,
the orbitals of the two ligands are in-phase, while in the LUMOthey are out of phase, resulting in a little higher energy. In theSupporting information Fig. S1, the most important orbitals ofthe complex are plotted. The analysis reveals what is expectedfor this kind of complex [49,56].
The nickel dx2�y2 orbital is empty and contributes to a sigmaanti-bonding molecular orbital. The dz2 orbital is the HOMO-4molecular orbital and is non-bonding. The HOMO-1 and HOMO-2orbitals have important contributions from the dyz and dxz orbitalsof nickel.

Fig. 15. The experimental and calculated electronic absorption spectrum of 4.
216 G. Soras et al. / Polyhedron 62 (2013) 208–217
No contribution from the methoxy groups can be noticed forboth MOs, probably because they are out of plane with respect tothe rest of the pz atomic orbitals, as is demonstrated in Fig. S1 ofthe Supporting information.
6.6.3. IR-RamanThe theoretical and experimental Raman spectra of 4 are dis-
played in Fig. 13. The experimental Raman spectrum shows char-acteristic S–Ni–S and Ni–S vibrations at 368 (sh) 380 and480 cm�1. In the theoretical spectrum, those vibrational peaks ap-pear at 356 (str), 364 (b) and 484 cm�1. The intensity at 348 cm�1
corresponds to the C–S–C bending vibration, similar to the calcu-lated one (348 cm�1) and the peak at 911 cm�1 corresponds tothe S–C–S bending vibration (calc. 906 cm�1) The C@C stretchingvibrations appear in the experimental spectrum at 1023 cm�1 to-gether with a strong peak at 1355 cm�1, compared to the theoret-ical spectrum with peaks at 1028 and 1380 cm�1. In general, theintensity of these peaks seems to be underestimated in our calcu-lations. The C–H stretching vibrations appear at 2833 and2933 cm�1 in the experimental spectrum. In the calculated spec-trum these peaks were shifted, at 2996 and 3004 cm�1, asexpected.
The experimental and the calculated infrared spectra of 4 aredisplayed in Fig. 14. The Ni–S symmetric and asymmetric stretchesappeared in the theoretically calculated spectrum at 379, 409 and486 cm�1, with low intensities, and this area is out of our instru-ment detection limit. The S–C stretching and S–C–S bending vibra-tions appeared in the region between 540 and 920 cm�1, whereasthe region between 1180 and 1500 cm�1 consists mainly of C@Cand CH3 vibrations. The characteristic peaks at 2800–3000 cm�1,related to the C–H stretching vibrations in the experimental spec-trum, are shifted in the theoretical one to higher wavenumbers.Overall it seems that there is a good agreement between the theo-retically calculated and the experimentally recorded spectra of theinvestigated complex. It was very important to propose both Ra-man and IR band assignments for the investigated compound.These data (Table S1 in the Supporting information) should be use-ful for the design of new dithiolene complexes with desired phys-ical properties.
6.6.4. UV–VisIn order to reproduce the UV–Vis spectrum, the TD-DFT formal-
ism was used. It is possible to use more accurate multi-referencemethods, but this was outside the scope of the current study.The calculated spectrum seems to nicely agree with the experi-mental one. We should emphasize that we did not tried to fit thecalculated spectrum to the experimental one, but in order to make
the pictorial representation more clear for the figures that follow,the following adjustments were made. Two general half-widthswere used, one of 1000 cm�1 up to 15000 cm�1 and one of2500 cm�1 from there on. This produces the thin spectrum ofFig. 15. The calculated spectrum was then shifted by 0.414 eVand scaled by 0.752 so that the left intense peak fits the experi-mental one at 10.141 cm�1. This is the spectrum tagged as ‘‘calcu-lated’’ on Fig. 15.
In Fig. S2 of the Supporting information, the difference densityfor the main peaks is plotted. The prominent peak A has 74% per-cent of the HOMO to LUMO transition and is, as expected, the p–p⁄ transition to the b2g orbital. In the region from 15000 to30000 three peaks were located. The first of them, B1, correspondsto a LMCT excitation. The next two, B2 and C2, are mainly interli-gand bands, with the first one having the main contribution froman electron transfer to the b2g HOMO orbital. The high intensityD and E peaks are ligand to metal charge transfer transitions andalso intraligand ones.
7. Conclusion
The synthesis of the new metal donor Ni(dmeodddt)2 wasmade. Its crystal structure was resolved with X-ray crystallogra-phy. Structure and spectral investigations were made both experi-mentally and theoretically. A comparison of the results in bothcases showed a very good agreement, helping us to understand thisnew complex.
Theoretical calculations confirmed that nickel is in a Ni2+ formaloxidation state, with both ligands in anionic states, antiferromag-netically coupled. The effect of weak interactions in the formationof the crystal was calculated and found to have an interaction en-ergy of around 31 kcal/mol for a dimer. The vibrational spectrumwas calculated and found to agree well with the experimentalone. Finally the absorption spectrum was calculated throughTDDFT and analyzed based on the molecular orbitals found, show-ing the classical behavior expected for this kind of complexes.
Appendix A. Supplementary data
CCDC 918380 contains the supplementary crystallographic datafor [Ni(dmeodddt)2]. These data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/conts/retrieving.html, or from theCambridge Crystallographic Data Centre, 12 Union Road, Cam-bridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: [email protected]. Supplementary data associated with thisarticle can be found, in the online version, at http://dx.doi.org/10.1016/j.poly.2013.06.033.

G. Soras et al. / Polyhedron 62 (2013) 208–217 217
References
[1] C. Faulmann, P. Cassoux, Dithiolene Chemistry, John Wiley & Sons, Inc., 2004.pp. 399.
[2] T.D. Anthopoulos, S. Setayesh, E. Smits, M. Cölle, E. Cantatore, B. de Boer,P.W.M. Blom, D.M. de Leeuw, Adv. Mater. 18 (2006) 1900.
[3] A.T. Coomber, D. Beljonne, R.H. Friend, J.L. Bredas, A. Charlton, N. Robertson,A.E. Underbill, M. Kurmoo, P. Day, Nature 380 (1996) 144.
[4] G. Soras, N. Psaroudakis, M.J. Manos, A.J. Tasiopoulos, D.G. Liakos, G.A. Mousdis,Polyhedron 28 (2009) 3340.
[5] D. Espa, L. Pilia, Inorg. Chem. 50 (2011) 2058.[6] D. Qing, C.X. Feng, C. Hong, G. Xing, Z.X. Ping, C. Zhusheng, Supramol. Sci. 5
(1998) 531.[7] L. Qu, Y. Guo, H. Luo, C. Zhong, G. Yu, Y. Liu, J. Qin, Chem. Commun. 48 (2012)
9965.[8] F. Alary, J.-L. Heully, A. Scemama, B. Garreau-de Bonneval, K. Chane-Ching, M.
Caffarel, Theor. Chem. Acc. 126 (2010) 243.[9] B. Garreau-de Bonneval, K.I. Moineau-Chane Ching, F. Alary, T.-T. Bui, L. Valade,
Coord. Chem. Rev. 254 (2010) 1457.[10] S. Sproules, K. Wieghardt, Coord. Chem. Rev. 255 (2011) 837.[11] R. Eisenberg, H.B. Gray, Inorg. Chem. 50 (2011) 9741.[12] P.G. de Gennes, J. Prost, The Physics of Liquid Crystals International Series of
Monographs on Physics, vol. 83, Clarendon Press, 1995.[13] N. Robertson, L. Cronin, Coord. Chem. Rev. 227 (2002) 93.[14] M. Bousseau, L. Valade, J.P. Legros, P. Cassoux, M. Garbauskas, L.V. Interrante, J.
Am. Chem. Soc. 108 (1986) 1908.[15] G.C. Papavassiliou, G.C. Anyfantis, G.A. Mousdis, Crystals 2 (2012) 762.[16] C.T. Vance, R.D. Bereman, J. Bordner, W.E. Hatfield, J.H. Helms, Inorg. Chem. 24
(1985) 2905.[17] U.T. Mueller-Westerhoff, B. Vance, D. Ihl Yoon, Tetrahedron 47 (1991) 909.[18] G. Soras, N. Psaroudakis, G.A. Mousdis, M.J. Manos, A.J. Tasiopoulos, P. Aloukos,
S. Couris, P. Labéguerie, J. Lipinski, A. Avramopoulos, M.G. Papadopoulos,Chem. Phys. 372 (2010) 33.
[19] G. Steimecke, H.J. Sieler, R. Kirmse, E. Hoyer, Phosphorus Sulfur 7 (1979) 49.[20] B.S. Furniss, A.J. Hannaford, P.W.G. Smith, A.R. Tatchell, Vogel’s Textbook of
Practical Organic Chemistry, fifth ed., Longman Scientific & Technical, GreatBritain, 1989.
[21] A. Bou, M.A. Pericàs, F. Serratosa, Tetrahedron 37 (1981) 1441.[22] Oxford Diffraction CrysAlis CCD and CrysAlis RED, (2008).[23] G.M. Sheldrick, SHELXS-97, University of Göttingen, Germany, 1997.[24] L. Farrugia, J. Appl. Crystallogr. 32 (1999) 837.[25] F. Neese, Wiley Interdiscip. Rev: Comput. Mol. Sci. 2 (2012) 73.[26] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato,X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M.Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y.Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery Jr., J.E. Peralta, F. Ogliaro,M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J.
Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M.Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo,J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C.Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth,P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, Ö. Farkas, J.B. Foresman,J.V. Ortiz, J. Cioslowski, D.J. Fox, GAUSSIAN 03, Revision A.1, GAUSSIAN, Inc.,Wallingford CT, 2003.
[27] A.D. Becke, Phys. Rev. A 38 (1988) 3098.[28] C.T. Lee, W.T. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 85.[29] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.[30] J.P. Perdew, Phys. Rev. B 33 (1986) 8822.[31] A.J.H. Wachters, J. Chem. Phys. 52 (1970) 1033.[32] A.D. McLean, G.S. Chandler, J. Chem. Phys. 72 (1980) 5639.[33] M.M. Francl, W.J. Pietro, W.J. Hehre, J.S. Binkley, M.S. Gordon, D.J. DeFrees, J.A.
Pople, J. Chem. Phys. 77 (1982) 3654.[34] A. Schafer, H. Horn, R. Ahlrichs, J. Chem. Phys. 97 (1992) 2571.[35] F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 7 (2005) 3297.[36] S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 132 (2010) 154104.[37] A. Klamt, G. Schuurmann, J. Chem. Soc., Perkin Trans. 2 (1993) 799.[38] S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen, F. Neese, J. Phys. Chem. A
110 (2006) 2235.[39] G.A. Mousdis, N. Psaroudakis, G.C. Anyfantis, Z. Naturforsch. 59b (2004) 839.[40] R. Kato, Chem. Rev. 104 (2004) 5319.[41] P. Deplano, L. Pilia, D. Espa, M.L. Mercuri, A. Serpe, Coord. Chem. Rev. 254
(2010) 1434.[42] G.N. Schrauzer, V.P. Mayweg, J. Am. Chem. Soc. 87 (1965) 1483.[43] P. Deplano, M.L. Mercuri, G. Pintus, E.F. Trogu, Inorg. Chem. 22 (2001) 353.[44] Q. Miao, J. Gao, Z. Wang, H. Yu, Y. Luo, T. Ma, Inorg. Chim. Acta 376 (2011) 619.[45] F. Bigoli, P. Deplano, M.L. Mercuri, M.A. Pellinghelli, L. Pilia, G. Pintus, A. Serpe,
E.F. Trogu, Inorg. Chem. 41 (2002) 5241.[46] Y.S.J. Veldhuizen, N. Veldman, A.L. Spek, P. Cassoux, R. Carlier, M.J.J. Mulder,
J.G. Haasnoot, J. Reedijk, J. Chem. Soc., Dalton Trans. (1998) 2989.[47] E. Watanabe, M. Fujiwara, J.-I. Yamaura, R. Kato, J. Mater. Chem. 11 (2001)
2131.[48] J.E. Jones, Proc. R. London Ser. A 106 (1924) 463.[49] K. Ray, T. Weyhermüller, F. Neese, K. Wieghardt, Inorg. Chem. 44 (2005) 5345.[50] L. Noodleman, J. Chem. Phys. 74 (1981) 5737.[51] G. Jonkers, C.A. de Lange, L. Noodleman, E.J. Baerends, Mol. Phys. 46 (1982)
609.[52] L. Noodleman, J.G. Norman, J.H. Osborne, A. Aizman, D.A. Case, J. Am. Chem.
Soc. 107 (1985) 3418.[53] L. Noodleman, E.R. Davidson, Chem. Phys. 109 (1986) 131.[54] F. Neese, Coord. Chem. Rev. 253 (2009) 526.[55] F. Neese, J. Phys. Chem. Solids 65 (2004) 781.[56] M.L. Kirk, R.L. McNaughton, M.E. Helton, The electronic structure and
spectroscopy of metallo-dithiolene complexes, in: A. Daniel (Ed.), DithioleneChemistry, John Wiley & Sons, Inc., 2004, p. 111.