synthesis and layer-by-layer deposition of spherical poly(3,4-ethylenedioxythiophene) nanoparticles...
TRANSCRIPT

Full Paper
1394
Synthesis and Layer-by-Layer Deposition ofSpherical Poly(3,4-ethylenedioxythiophene)Nanoparticles - Toward Fast Switching Timesbetween Reduced and Oxidized States
Kevin Muller, Mi-Kyoung Park, Markus Klapper, Wolfgang Knoll,Klaus Mullen*
A microemulsion polymerization for the synthesis of poly(3,4-ethylenedioxythiophene)(PEDOT) nanoparticles is described. Spherical and electrically conducting PEDOT particleswith diameters as small as 27� 8 nm were obtained by using decyltrimethylammoniumbromide as a cationic surfactant. PEDOT nanoparticle multi-layers, alternated with poly(styrene sulfonate), were preparedby the layer-by-layer deposition technique. Electrochemicalsurface plasmon resonance experiments revealed a ten-foldincrease in the switching time between redox states as com-pared with Baytron P/poly(ethyleneimine) multilayers. Theenhanced charge transport can be attributed to an improveddiffusion of the charge-balancing counterions into the PEDOTnanoparticles.
Introduction
Due to its high stability, high conductivity in the doped
state, and transparency in thin oxidized films poly(3,4-
ethylenedioxythiophene) (PEDOT) is one of the most
promising examples of conducting polymers.[1,2] Many
applications of PEDOT have been reported ranging from
antistatic and conductive coatings for various devices to
hole injecting layers used in organic light emitting
diodes.[3–5] Today, PEDOT is commercially available as a
water-soluble PEDOT-poly(styrene sulfonate) (PEDOT:PSS,
Baytron P) complex.[6]
K. Muller, M.-K. Park, M. Klapper, W. Knoll, K. MullenMax-Planck Institute for Polymer Research, Ackermannweg 10,D-55128 Mainz, GermanyFax: (þ49) 6131 379 100; E-mail: [email protected]
Macromol. Chem. Phys. 2007, 208, 1394–1401
� 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
An interesting aspect of Baytron P is the opportunity to
affect the oxidation level reversibly by chemical or electro-
chemical doping/dedoping (switching between the oxi-
dized and reduced state). This feature allows Baytron P to
be utilized as an electroactive material in various sensory
devices, such as glucose biosensors or hydrogen chloride
and ammonia vapor sensor devices.[7–10] The switching
time between the oxidation states is determined by elect-
ron hopping (between neighboring oxidized and reduced
sites of the PEDOT segments) and the ionic diffusion of
charge-compensating counterions through the polymer
film to the electroactive PEDOT sites.[11] In this process,
during oxidation, counterions are incorporated into the
PEDOT film in order to compensate the positive charge.
On the other hand during reduction, as the PEDOT
segments are neutralized, the counterions are released.
It was demonstrated in the literature that the charge-
compensating counterions, as well as the analytes,
DOI: 10.1002/macp.200700142

Synthesis and Layer-by-Layer Deposition of Spherical Poly(3,4-ethylenedioxythiophene) Nanoparticles . . .
penetrate only slowly through the Baytron P film.[12,13]
This can be attributed to the low accessibility of the PEDOT
segments due to high amount of PSS in the Baytron P
complex.[14,15] Due to this fact, the switching time of such
devices is rather slow (about 20 s).[12,13] An approach for
increasing the switching time would be to enhance the
accessibility of the electroactive PEDOT sites for the coun-
terions. One way to increase the accessibility is through
the use of dispersible PEDOT latex particles. In this case, no
PSS is needed to keep the rather insoluble PEDOT segments
in solution and the latex particles can be processed directly
out of dispersion.[16] The majority of PEDOT nanoparticles
have been produced using iron(III) salts and different anio-
nic emulsifiers such as sodium dodecylsulfate (SDS) and
sodium dodecylbenzenesulfonate (SDBS) in oxidative
emulsion polymerization processes.[16–18] Disadvantages
of these nanoparticles are their undefined shapes and
relatively large size with broad size distributions. This is
presumably due to the interaction between the anionic
emulsifier and the iron(III) cations which destabilizes the
emulsion system during the polymerization. As such, the
use of a cationic emulsifier would circumvent the inter-
action between the surfactant and the positively charged
oxidant.
The second major factor in the construction of sensory
devices from electroactive polymers is their processability
as well-defined layers. An approach which produces well-
defined films is the layer-by-layer (LbL) ionic self-assembly
method,[19,20] wherein a charged substrate is alternatively
exposed to solutions containing oppositely charged species
to form integrated ultrathin films. It has been used to
create highly tuned, functional thin films with the control
of composition and structure.[21]
Herein, we report the synthesis of positively charged
PEDOT nanoparticles by an oxidative microemulsion
polymerization in the presence of cationic surfactants.
The positively charged surface of PEDOT nanoparticles
permitted the use of the LbL self-assemblymethod offering
a flexible procedure for the preparation of PEDOT devices
with a high surface area. The multilayer growth of
positively charged PEDOT nanoparticles and negatively
charged PSS was monitored by UV-vis spectroscopy. In
addition, for the determination of the switching behavior,
the redox properties of the PEDOT nanoparticle layered
architecture, compared with a Baytron P multilayer, were
evaluated using chronoamperometry and electrochemical
surface plasmon resonance (SPR) measurements.
Experimental Part
Materials
3,4-Ethylenedioxythiophene (EDOT) and Baytron P (PEDOT:PSS)
were provided by H.C. Starck (Leverkusen, Germany). Decyltri-
Macromol. Chem. Phys. 2007, 208, 1394–1401
� 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
methylammonium bromide (DETAB), iron(III) p-toluenesulfo-
nate hexahydrate, poly(diallyldimethylammonium chloride)
(PDADMAC; Mw < 200000), poly(sodium 4-styrenesulfonate)
(PSS; Mw 70 000), poly(ethyleneimine) (PEI; Mw 25 000), 3-
aminopropyltriethoxysilane (APS), and sodium 3-mercapto-1-
propanesulfonate (MPS) were obtained by Sigma-Aldrich. Iron(III)
chloride hexahydrate was purchased by Acros Organics. All
materials were used without further purification. A phosphate
buffer solution was prepared with Na2HPO4 and NaH2PO4 (0.05 M,
pH¼ 7.40). Millipore water (18 MV) was used throughout the
experiments.
Characterization
FT-IR spectra were obtained with a Nicolet 730 FT-IR spectrometer
using a Thermo Electron Endurance ATR single-reflection ATR
crystal. UV-vis spectra were obtained using a Perkin-Elmer
Lambda 900 spectrometer. Dynamic light scattering measure-
ments were performed on a Malvern Zetasizer 3000. For the
determination of electrical conductivity, PEDOT films were
prepared using a blade coater (200mmslit). Conductivitymeasure-
ments were carried out using a Jandel microposition four-point
probe connected to a Keithley 2700DMM digital multimeter.
Transmission electron microscopy (TEM) studies were performed
on carbon meshes using a LEO EM 912 microscope. The average
particle diameters were measured directly from each TEM image.
The diameters of a minimum of 100 particles were measured and
the values averaged by using an image analysis software (Soft
Imaging System). For atomic forcemicroscopy (AFM), a Dimension
3100 scanning probe microscope (Digital Instruments, Santa
Barbara, CA) was used in tapping mode employing Olympus
cantilevers with spring constants ranging between 33.2 and
65.7 N �m�1 and resonant frequencies of 277–346 Hz (as specified
by the manufacturer).
Preparation of PEDOT Nanoparticles
DETAB (2.65 g, 9.5mmol)was stirred inwater (45mL) for 1 h. EDOT
(1.00 g, 7.04 mmol) was added dropwise to the surfactant solution
and stirringwas continued for 2 h. The polymerizationwas started
by the addition of the iron(III) salt (molar ratio of iron(III)/EDOT
being 2:1) dissolved in water (5 mL). The microemulsion was
stirred for 3 h at room temperature. After the addition of excess
methanol to the reaction mixture, separation of the PEDOT
nanoparticles was achieved by centrifugation. The particles were
washed with methanol and redispersed in methanol (15 mL) by
ultrasonification for 20 min.
Preparation of PEDOT Nanoparticle Multilayers
The gold substrates were prepared by thermal evaporation of gold
(450 nm) on top of a chromium (1.5 nm) layer onto LaSFN9 glass
substrates. The substrates were then immersed in an ethanolic
MPS solution (1�10�3M) overnight, rinsed with ethanol, and
dried under a stream of nitrogen in order to obtain a negatively
charged surface. The MPS functionalized gold substrates were
alternatively dipped in PDADMAC [1 mg �mL�1 in 0.2 M NaCl (aq)]
www.mcp-journal.de 1395

K. Muller, M.-K. Park, M. Klapper, W. Knoll, K. Mullen
Figure 1. Preparation of PEDOT nanoparticles in microemulsion.
1396
and PSS [1 mg �mL�1 in 0.2 M NaCl (aq)] for 15 min each. The
substrates were thoroughly rinsed with water between dippings.
Two bilayers of PDADMAC and PSSwere deposited to give a higher
charge density and a smooth surface. For UV-vis spectroscopy
measurements, a quartz slide was used as the substrate. The
quartz slide was functionalized with APS (1� 10�3M in toluene) to
give a positively charged surface. Before the deposition of PEDOT
nanoparticles on the (PSS/PDADMAC)2-gold substrate, methanol
was exchanged for water by centrifugation, removal of the
supernatant, and redispersion of the positively charged PEDOT
nanoparticles in ethanol and then in water. The PEDOT dispersion
was sonificated for 5 min prior to the deposition. The substrates
were dipped in the PEDOT dispersion for 2 h and then rinsed with
water. For the assembly of Baytron P multilayer, PEI (1 mg �mL�1)
was used as a polycation. A (PSS/PDADMAC)2-gold substrate was
alternatively dipped into PEI solution and diluted Baytron P
solution (20 times diluted with MilliQ water from the original
solution).
Electrochemical Measurement and Electrochemical
Surface Plasmon Spectroscopy
Chronoamperometry was performed with a mAutolab type-III
potentiostat (Eco Chemie B. V., Netherlands) in a three-electrode
Teflon cell with the gold substrate as the working electrode
(0.502 cm2), a platinum wire as a counterelectrode, and an Ag/
AgCl reference electrode. The attenuated total reflection (ATR)
setup in the Kretschmann configuration, combined with an
electrochemical cell, was used for the in situ electrochemical SPR
measurements. Details about this setup are published else-
where.[22,23] The Au/glass substrates were clamped against the
Teflon cell with an O-ring providing a liquid-tight seal. Surface
plasmonswere excited at themetal-dielectric interface, upon total
internal reflection of a polarized He-Ne laser (632.8 nm) beam. The
optical/electrochemical processes on the gold were detected by
monitoring the reflectivity as a function of the incident angle or at
a fixed angle as a function of time while a constant voltage was
applied to the multilayers.
Figure 2. FT-IR spectrum of PEDOT nanoparticles (sample 2).
Results and Discussion
PEDOT Nanoparticle Synthesis
For the preparation of the PEDOT nanoparticles, a
microemulsion procedure was chosen. Due to high surfac-
tant concentrations, microemulsions are thermodynami-
cally stable and the monomer droplets are sufficiently
small that they become the locus of polymerization.[24]
Emulsification of the EDOT monomer in an aqueous phase
using different emulsifiers, such as octyltrimethylammo-
nium bromide (OTAB), DETAB, and tetradecyltrimethyl-
ammonium bromide (TETAB) was investigated. OTAB did
not appear to be suitable for the stabilization of the
microemulsion system since it gave undefined PEDOT
nanoparticles with mean diameters as large as 1 mm. The
octyl chain of the OTAB detergent may be too short to
Macromol. Chem. Phys. 2007, 208, 1394–1401
� 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
stabilize the monomer droplets. Analogous findings were
made for the TETAB emulsifier, which formed aqueous
solutions with high viscosities. In contrast, DETAB
was found to be well suited for the stabilization of the
monomer-filled micelles. Polymerization was then per-
formed upon the addition of iron(III) chloride or iron(III)
p-toluenesulfonate to the EDOT microemulsion (Figure 1).
After polymerization, the nanoparticles were separated
from the residual iron salts and redispersed inmethanol or
water, respectively.
The resulting dispersion was stable at room tempera-
ture for several weeks at various solid concentrations
(2–10% by weight). FT-IR spectroscopy of the precipitated
particles showed the characteristic ring vibration of the
thiophene ring at 1 460 cm�1 and of PEDOT at 1 380 cm�1,
which can be attributed to the quinoidal C–C and C––C
structure (Figure 2). Further vibrations at 1 170 and
DOI: 10.1002/macp.200700142

Synthesis and Layer-by-Layer Deposition of Spherical Poly(3,4-ethylenedioxythiophene) Nanoparticles . . .
Figure 3. TEM image of PEDOT nanoparticles (sample 2).
Figure 4. Particle size distribution (by number) of sample 1,obtained from TEM images.
1 000 cm�1 were assigned to the C–O–C bond stretching,
whereby the C–S bond vibrations in the thiophene ring
were found at 974 and 850 cm�1, respectively.[25] Morpho-
logy studies of the PEDOT nanoparticles were performed
by applying TEM. The obtained nanoparticles showed a
spherical shape and an average particle diameter of 27 nm
(�8 nm) (Figure 3 and 4). This was measured directly from
TEM images by counting the diameters of more than 100
particles using an image analysis software.
It was possible to tune the size of the particles by vary-
ing the surfactant concentration. As expected for micro-
emulsion polymerization, the sizes of the PEDOT particles
decreased with increase in emulsifier concentration
(Table 1: samples 3 and 4). Oxidative polymerization
using iron(III) p-toluenesulfonate proceeded faster than the
polyreaction using iron(III) chloride and was accomplished
after 2 h. This is consistent with the higher solubility of
iron(III) p-toluenesulfonate in the EDOT monomer leading
to a better diffusion into the monomer-filled micelles.
To reveal that the obtained PEDOT nanoparticles are
electrically conductive, conductivity measurements were
Table 1. Oxidative Polymerization of EDOT in microemulsion polyme
Sample Oxidant [DETAB] [EDOT]
mol � LS1 mmol � LS1
1 FeCl3 0.40 141
2 FeCl3 0.38 149
3 Fe(OTs)3 0.38 144
4 Fe(OTs)3 0.30 153
a)Optained By TEM.
Macromol. Chem. Phys. 2007, 208, 1394–1401
� 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
performed by the four-point probe on blade coated PEDOT
nanoparticle films. Measurements show the conductivity
to be 0.30 S � cm�1 (�0.08 S � cm�1) at room temperature. In
comparison, the commercially available Baytron P exhi-
bits, depending on the way of processing, a conductivity
ranging from 1� 10�5 S � cm�1 (used for passive matrix
displays) to 1 S � cm�1 (used for antistatic devices).[5] The
similar conductivities of the PEDOT nanoparticles and the
Baytron P demonstrates that conductivities in the metallic
range could be obtained and sufficient percolation path-
ways through the prepared nanoparticle film were
present. This might be attributed to a migration of the
low molecular weight surfactant from the nanoparticle to
the surface of the film, a behavior which has been reported
for various traditional latex particles.[26,27]
It was verified that well defined, spherical, and electri-
cally conductive PEDOT nanoparticles were obtained by
facile, oxidative microemulsion polymerization, using
cationic emulsifiers. As such, the applicability of these
colloids for the construction of PEDOT multilayer devices
by LbL ionic self-assembly was investigated using their
positively charged surface due to residual cationic emulsi-
fiers. For a possible application as sensors, the PEDOT
rization.
Oxidant/EDOT Yield Particle sizea)
% nm
202:100 42 27 (W8)
195:100 37 29 (W7)
200:100 50 34 (W8)
350:100 50 45 (W5)
www.mcp-journal.de 1397

K. Muller, M.-K. Park, M. Klapper, W. Knoll, K. Mullen
1398
nanoparticle multilayers were electrochemically charac-
terized. Thereby, the switching time between the oxidized
and the reduced PEDOT state was investigated as this is a
key parameter for these applications. The results were
subsequently compared to the switching time of Baytron
P multilayers, prepared and characterized under similar
conditions.
Figure 6. UV-vis spectra of the LbL films of (PEDOT/PSS)n (n¼ 1–5)on (PDADMAC/PSS)2-coated glass slide (inset: Absorbance at 1 170nm at a function of the number of bilayers).
LbL Deposition of PEDOT Nanoparticles
Multilayer assemblies were prepared through the LbL
deposition of positively charged PEDOT nanoparticles with
negatively charged PSS. The first layer of PEDOT nano-
particles was formed on an LbL self-assembled layer of
(PSS/PDADMAC)2, where the outermost layer was nega-
tively charged. Precoating of substrates with a (PSS/
PDADMAC)n multilayer was performed as this system is
known to promote the association of a higher charge
density and a uniform surface, improving adsorption of
the nanoparticles on the negatively charged surface.[28]
The straightforward formation of the nanoparticle LbL
films is attributed to the combination of two factors: (i)
electrostatic interactions of the positively charged PEDOT
nanoparticles with the PSS layer and (ii) van der Waals
attractive forces.[29,30] First, the morphology of nanopar-
Figure 5. AFM image of PEDOT nanoparticles (sample 4) on (PSS/PDADMAC)2 on a gold substrate (5� 5 mm2). A cross-section alongthe horizontal line indicated by the arrow is shown beneath theimage.
Macromol. Chem. Phys. 2007, 208, 1394–1401
� 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ticles on a precoated layer was studied by AFM (Figure 5).
The majority of the nanoparticles were observed as iso-
lated objects in a 5� 5 mm2 area. The cross-section of the
AFM image shows that the nanoparticles have a height of
30–45 nm, which is comparable to their lateral size
measured by TEM.
Incomplete surface coverage with nanoparticles was
also observed, presumably due to the electrostatic repul-
sion. The density of surface-charged particles often can be
increased by the addition of salt which screens the electro-
static repulsions.[31] However, the addition of NaCl (0.2 M)
to the nanoparticle dispersion induced coagulation of
the nanoparticles. Nonetheless, a well dispersed layer of
PEDOT nanoparticles was deposited on the (PSS/PDAD-
MAC)2-coated gold substrate. Ultrasonication of the
nanoparticle solution prior to deposition significantly
reduced the aggregation of the nanoparticles on the
substrate.
The LbL deposition of PEDOT nanoparticles and PSS up to
five bilayers on a quartz substrate was monitored by
UV-vis spectroscopy (Figure 6). The broad peaks near 750
and 1 150 nm correspond to the oxidized PEDOT.[32]
Absorbance at 1 170 nm (Figure 6, inset) of the films was
directly proportional to the number of layers. Similar
results were observed by SPR for the linear growth of the
nanoparticle/PSS multilayers on gold electrodes. Thus, LbL
films of PEDOT nanoparticles and PSS were successfully
deposited on both quartz substrates and gold electrodes.
Electrochemical SPR
The changes in optical properties and the swelling/
shrinking process of PEDOT nanoparticles upon applica-
tions of oxidative and reductive potentials were studied by
DOI: 10.1002/macp.200700142
Synthesis and Layer-by-Layer Deposition of Spherical Poly(3,4-ethylenedioxythiophene) Nanoparticles . . .
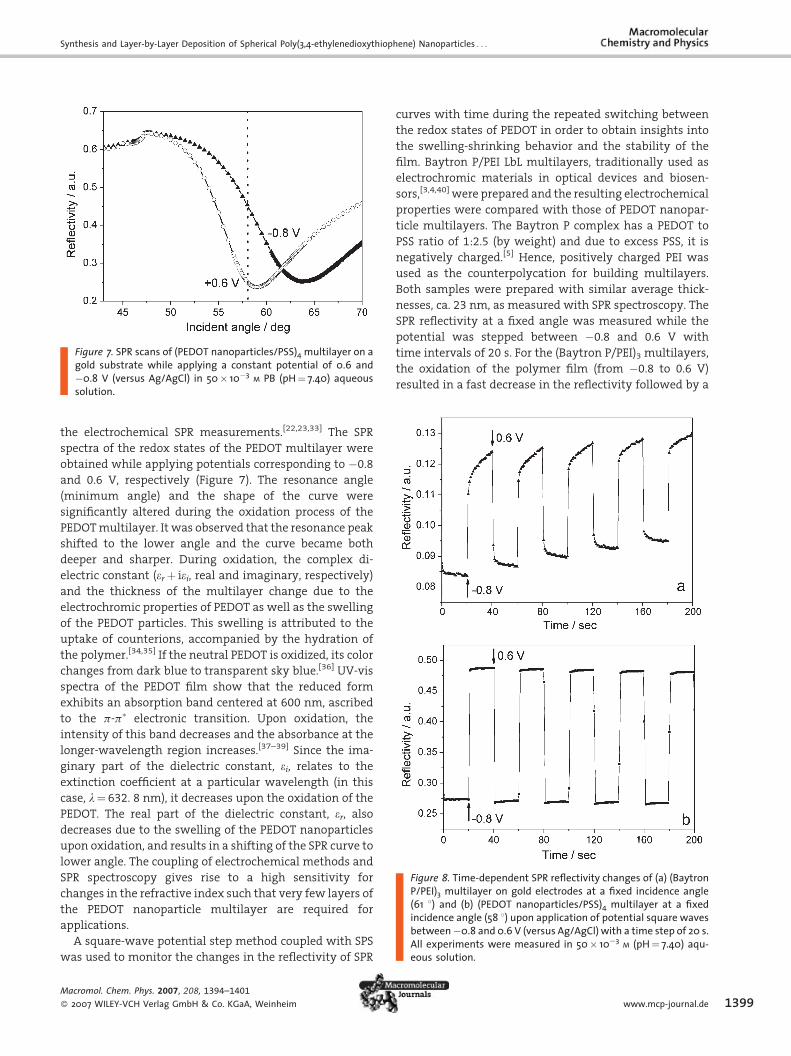
Figure 7. SPR scans of (PEDOT nanoparticles/PSS)4 multilayer on agold substrate while applying a constant potential of 0.6 and�0.8 V (versus Ag/AgCl) in 50� 10�3 M PB (pH¼ 7.40) aqueoussolution.
Figure 8. Time-dependent SPR reflectivity changes of (a) (BaytronP/PEI)3 multilayer on gold electrodes at a fixed incidence angle(61 8) and (b) (PEDOT nanoparticles/PSS)4 multilayer at a fixedincidence angle (58 8) upon application of potential square wavesbetween �0.8 and 0.6 V (versus Ag/AgCl) with a time step of 20 s.All experiments were measured in 50� 10�3 M (pH¼ 7.40) aqu-eous solution.
the electrochemical SPR measurements.[22,23,33] The SPR
spectra of the redox states of the PEDOT multilayer were
obtained while applying potentials corresponding to �0.8
and 0.6 V, respectively (Figure 7). The resonance angle
(minimum angle) and the shape of the curve were
significantly altered during the oxidation process of the
PEDOTmultilayer. It was observed that the resonance peak
shifted to the lower angle and the curve became both
deeper and sharper. During oxidation, the complex di-
electric constant (erþ iei, real and imaginary, respectively)
and the thickness of the multilayer change due to the
electrochromic properties of PEDOT as well as the swelling
of the PEDOT particles. This swelling is attributed to the
uptake of counterions, accompanied by the hydration of
the polymer.[34,35] If the neutral PEDOT is oxidized, its color
changes from dark blue to transparent sky blue.[36] UV-vis
spectra of the PEDOT film show that the reduced form
exhibits an absorption band centered at 600 nm, ascribed
to the p-p� electronic transition. Upon oxidation, the
intensity of this band decreases and the absorbance at the
longer-wavelength region increases.[37–39] Since the ima-
ginary part of the dielectric constant, ei, relates to the
extinction coefficient at a particular wavelength (in this
case, l¼ 632. 8 nm), it decreases upon the oxidation of the
PEDOT. The real part of the dielectric constant, er, alsodecreases due to the swelling of the PEDOT nanoparticles
upon oxidation, and results in a shifting of the SPR curve to
lower angle. The coupling of electrochemical methods and
SPR spectroscopy gives rise to a high sensitivity for
changes in the refractive index such that very few layers of
the PEDOT nanoparticle multilayer are required for
applications.
A square-wave potential step method coupled with SPS
was used to monitor the changes in the reflectivity of SPR
Macromol. Chem. Phys. 2007, 208, 1394–1401
� 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
curves with time during the repeated switching between
the redox states of PEDOT in order to obtain insights into
the swelling-shrinking behavior and the stability of the
film. Baytron P/PEI LbL multilayers, traditionally used as
electrochromic materials in optical devices and biosen-
sors,[3,4,40] were prepared and the resulting electrochemical
properties were compared with those of PEDOT nanopar-
ticle multilayers. The Baytron P complex has a PEDOT to
PSS ratio of 1:2.5 (by weight) and due to excess PSS, it is
negatively charged.[5] Hence, positively charged PEI was
used as the counterpolycation for building multilayers.
Both samples were prepared with similar average thick-
nesses, ca. 23 nm, as measured with SPR spectroscopy. The
SPR reflectivity at a fixed angle was measured while the
potential was stepped between �0.8 and 0.6 V with
time intervals of 20 s. For the (Baytron P/PEI)3 multilayers,
the oxidation of the polymer film (from �0.8 to 0.6 V)
resulted in a fast decrease in the reflectivity followed by a
www.mcp-journal.de 1399

K. Muller, M.-K. Park, M. Klapper, W. Knoll, K. Mullen
Figure 9. Chronoamperometric transients measured upon appli-cations of (a) a reductive potential step from 0.8 to �0.8 V ongold electrodes modified with LbL films of (PEDOT nanoparticle/PSS)4 (solid line) and (Baytron P/PEI)3 (dotted line) multilayers.(Inset: the Cottrell plots (current versus t�1/2) of PEDOT nanopar-ticle (closed square) and Baytron P (open triangle) multilayers.The data were recorded in 50� 10�3 M phosphate buffer, pH¼ 7.4.
1400
slow decreasewhich did not reach a plateau even after 20 s
[Figure 8(a)]. Upon the reduction (from 0.6 to �0.8 V), the
reflectivity showed an initial rapid increase and continued
with a slow decrease over 20 s. [Figure 8(a)]. The immediate
changes in reflectivity upon switching potentials are
attributed to the changes in the complex dielectric cons-
tant of Baytron P as a result of its oxidation and reduction.
The subsequent slow changes result from swelling and
shrinking of the film. The swelling is attributed to the
uptake of counterions in order to compensate the charges
of the oxidized film and solution molecules. On the other
hand, the film releases the counterions upon the reduction
resulting in the shrinking of the film. Similar switching
behavior of redox polymer films has been reported.[12,13]
For the (PEDOT nanoparticle/PSS)4 multilayer film, the
reflectivity switched between its maximum and mini-
mumvalues upon the application of reductive (�0.8 V) and
oxidative (0.6 V) potentials in less than 2 s [Figure 8(b)].
This was more than ten times faster than that of the
Baytron P multilayer and indicates that the shrinking and
swelling of the nanoparticles took place immediately after
the application of the potentials. Furthermore, the mini-
mum and maximum values of the reflectivity remained
constant after five cycles of switching indicating the
stability of the nanoparticle films as compared to the
Baytron P films. Faster switching of the PEDOT nanopar-
ticle film, presented in this study, can be attributed to a
better diffusion of the charge-balancing counterions into
the electroactive PEDOT particle layer. A potential ex-
planation for the enhanced diffusion can be that the
PEDOT particles in the outermost layer of the film are not
covered by PSS, whereas in the case of Baytron P the PSS is
located, due to its high hydrophilicity, at the multilayer/
solution interface. Thus, the diffusion of the counterions to
the PEDOT is hindered.
Chronoamperometry
In order to confirm the fast ion transport through the
nanoparticle multilayer, the electrochemical properties of
the (PEDOT nanoparticle/PSS)4 and (Baytron P/PEI)3 multi-
layers were investigated using chronoamperometric mea-
surements. The current transients of the multilayers on
gold electrodes were recorded as a function of time upon
the application of potential steps. First, current transients
were measured upon the application of a reductive
potential step from 0.6 to �0.8 V (Figure 9).
For both multilayers, the current transients did not fall
to zero even after 120 s. The residual currents are due to the
reduction of the electrolyte on the electrode surface,
resulting in the migration of charged species toward the
surrounding solution, which follows the electrical field.
Counterions and solvent molecules in the film are dragged
Macromol. Chem. Phys. 2007, 208, 1394–1401
� 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
along due to an electro-osmotic effect, promoting a conti-
nuous compacting of the polymeric structure.[41] The
chronoamperometric transients of the multilayers were
analyzed by replotting the current transients as a function
of t�1/2 (Figure 9, inset). The linear portion of each curve
obeys the Cottrell equation,[42]
IðtÞ ¼ nFAD1=2 C
p1=2
� �t�1=2
where I(t) is the current transient that decays in the course
of time t(s), n is the number of electrons per unit of the
redox polymer, F is the Faraday constant, A is the electrode
area, D is the diffusion coefficient for the charge propa-
gation in the polymer film, and C is the concentration of
the redox units in the polymer film which is calculated
from the electrochemically active volume and the total
amount of charge passed following a potential step. The
total charge was obtained by integration of the current I(t)
from t¼ 0 to infinity upon the application of an oxidative
potential, which was found to give similar values for both
multilayers. The current transients versus t�1/2 of the first
5 s upon the application of �0.8 V showed a linear depen-
dence, whereby the slope of the linear decay for the PEDOT
nanoparticle multilayer was steeper than that for Baytron
multilayer, indicating a faster decay of the current
transient. In the reduction of the PEDOT nanoparticle
multilayer, large capacitive charges were delivered in the
early stage (in 5 s) due to the high surface area.[43] Upon the
application of the Cottrell equation to the linear portion of
the current versus t�1/2, the ratio of the charge diffusion
coefficients for PEDOT nanoparticles/PSS to Baytron/PEI
multilayer films was calculated to be ca. 3. This enhanced
DOI: 10.1002/macp.200700142

Synthesis and Layer-by-Layer Deposition of Spherical Poly(3,4-ethylenedioxythiophene) Nanoparticles . . .
rate of charge transport of the PEDOT nanoparticles is
attributed to an increase in the rate of ion transport from
solution to the redox polymer due to high surface area of
the nanoparticles, which resulted in fast switching
behavior of the nanoparticle multilayers.
Conclusion
A versatile microemulsion polymerization for the fabrica-
tion of electrically conducting PEDOT nanoparticles was
presented. By applying cationic surfactants as stabilizers,
i.e. DETAB, electrostatic interactions between the oxidant,
i.e. iron(III), and the emulsifier could be circumvented. Thus,
spherical PEDOT colloids with average diameters as low as
27 nm were obtained.
A PEDOT nanoparticle device was prepared by the LbL
ionic self-assembly method due to the positively charged
nature of the nanoparticles. Electrochemical SPR measure-
ments demonstrated a switching time of less than 2 s for
the PEDOT nanoparticle/PSS multilayers. This is more than
ten times faster than the switching time of traditional
Baytron P/PEI multilayers. The improved switching time
can be attributed to an enhanced diffusion of the
counterions into the PEDOT nanoparticles. Fast switching
between the oxidized and reduced PEDOT states is ofmajor
importance, as this is a prerequisite for sensor applica-
tions. As such, further directions of this work will include
the application of PEDOT particle multilayers for enzy-
matic biosensors.
Acknowledgements: The authors acknowledge Walter Scholdeifor IR-measurements and the German Science Foundation (SFB625) as well as the Bayer AG for financial support. M.-K. Parkacknowledges the Alexander von Humboldt Foundation for aresearch fellowship.
Received: March 13, 2007; Accepted: March 21, 2007; DOI:10.1002/macp.200700142
Keywords: conducting polymers; chronoamperometry; electro-chemical surface plasmon resonance spectroscopy; layer-by-layerdeposition; PEDOT nanoparticles; TEM
[1] F. Jonas, L. Schrader, Synth. Met. 1991, 831, 41.[2] L. Groenedaal, F. Jonas, D. Freitag, H. Pielartzik, J. R. Reynolds,
Adv. Mater. 2000, 12, 481.[3] A. Kros, N. A. J. M. Sommerdijk, R. J. M. Nolte, Sens. Actuators, B
2005, 106, 289.[4] A. Kros, S. W. F. M. van Hoevell, N. A. J. M. Sommerdijk,
R. J. M. Nolte, Adv. Mater. 2001, 13, 1555.[5] S. Kirchmeyer, K. Reuter, J. Mater. Chem. 2005, 15, 2077.
Macromol. Chem. Phys. 2007, 208, 1394–1401
� 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
[6] H. W. Heuer, R. Wehrmann, S. Kirchmeyer, Adv. Funct. Mater.2002, 12, 89.
[7] J. Jang, M. Chang, H. Yoon, Adv. Mater. 2005, 17, 1616.[8] A. Kros, S. W. F. M. van Hoevell, N. A. J. M. Sommerdijk,
R. J. M. Nolte, Adv. Mater. 2001, 13, 1555.[9] A. A. Argun, P. H. Aubert, B. C. Thompson, I. Schwendeman,
C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G.MacDiarmid, J. R. Reynolds, Chem. Mater. 2004, 16, 4401.
[10] R. J. Mortimer, A. L. Dyer, J. R. Reynolds, Displays 2006, 27, 2.[11] S. A. Sapp, G. A. Sotzing, J. R. Reynolds, Chem. Mater. 1998, 10,
2101.[12] S. Tian, J. Liu, T. Zhu, W. Knoll, Chem. Mater. 2004, 16, 4103.[13] S. Tian, A. Baba, J. Liu, Z. Wang, W. Knoll, M.-K. Park,
R. Advincula, Adv. Funct. Mater. 2003, 13, 473.[14] J. Lu, N. J. Pinto, A. G. MacDiarmid, J. Appl. Phys 2002, 92, 6033.[15] A. J. Epstein, F.-C. Hsu, N.-R. Chiou, V. N. Prigodin, Curr. Appl.
Phys. 2002, 2, 339.[16] Y. Kudoh, K. Akami, Y. Matsuya, Synth. Met. 1998, 98, 65.[17] Y. Kudoh, K. Akami, K. Kusayanagi, Y. Matsuya, Synth. Met.
2001, 123, 541.[18] J. W. Choi, M. G. Han, S. K. Oh, S. S. Im, Synth. Met. 2004, 141,
293.[19] G. Decher, J. D. Hong, Ber. Bunsen-ges. 1991, 95, 1430.[20] G. Decher, J. D. Hong, Macromol. Chem., Macromol. Symp.
1991, 46, 321.[21] G. Decher, Science 1997, 277, 1232.[22] A. Baba, R. C. Advincula, W. Knoll, J. Phys. Chem. B 2002, 106,
1581.[23] A. Baba, M.-K. Park, R. C. Advincula, W. Knoll, Langmuir 2002,
18, 4648.[24] P. Y. Chow, L. M. Gan, Adv. Polym. Sci. 2005, 175, 257.[25] C. Kvarnstrom, H. Neugebauer, S. Blomquist, H. J. Ahonen,
J. Kankare, A. Ivaska, Electrochim. Acta 1999, 44, 2739.[26] T. A. Thorstenson, L. K. Tebelius, M. W. Urban, J. Appl. Polym.
Sci. 1993, 50, 1207.[27] K. W. Evanson, T. A. Thorstenson, M. W. Urban, J. Appl. Polym.
Sci. 1991, 42, 2297.[28] R. A. McAloney, M. Sinyor, V. Dudnik, M. C. Goh, Langmuir
2001, 17, 6655.[29] N. A. Kotov, Nanostruct. Mater. 1999, 12, 789.[30] S. T. Dubas, J. B. Schlenoff, Macromolecules 1999, 32, 8153.[31] Y. Lvov, K. Ariga, M. Onda, I. Ichinose, T. Kunitake, Langmuir
1997, 13, 6195.[32] D. M. DeLongchamp, P. T. Hammond, Adv. Mater. 2001, 19,
1455.[33] O. A. Raitman, E. Katz, A. F. Buckmann, I. Willner, J. Am. Chem.
Soc. 2002, 124, 6487.[34] C. Xia, A. Baba, R. C. Advincula, W. Knoll, Langmuir 2002, 18,
3555.[35] A. Baba, J. Lubben, K. Tamada, W. Knoll, Langmuir 2003, 19,
9058.[36] Q. Pei, G. Zuccarello, M. Ahlskog, O. Inganas, Polymer 1994, 35,
1347.[37] B. Sankaran, J. R. Reynolds, Macromolecules 1997, 30, 2582.[38] C. A. Cutler, M. Bouguettaya, T.-S. Kang, J. R. Reynolds,Macro-
molecules 2005, 38, 3068.[39] D. M. DeLongchamp, M. Kastantin, P. T. Hammond, Chem.
Mater. 2003, 15, 1575.[40] Z. Tang, S. T. Donohoe, J. M. Robinson, P. A. Chiarelli,
H.-L. Wang, Polymer 2005, 46, 9043.[41] H. Grande, T. F. Otero, Electrochim. Acta 1999, 44, 1893.[42] F. G. Z. Cottrell, Phys. Chem. 1902, 42, 385.[43] L. S. Van Dyke, C. R. Martin, Langmuir 1990, 6, 1118.
www.mcp-journal.de 1401