synthese von naturstoffen durch olefinkreuzmetathese und ... · generation 3 ist beispielsweise die...
TRANSCRIPT

Synthese von Naturstoffen durch
Olefinkreuzmetathese und Cyclisierungsreaktion
vorgelegt von
Diplom-Chemiker Julian Gebauer
aus Berlin
der Fakultät II
Mathematik und Naturwissenschaften
- Institut für Chemie -
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
- Dr. rer. nat. -
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. rer. nat. M. Lerch
Erster Berichter: Prof. Dr. rer. nat. S. Blechert
Zweiter Berichter: Prof. Dr. rer. nat. K. Rück-Braun
Tag der mündlichen Prüfung: 09.05.2006
Berlin 2006
D 83


Abstract
Das Synthesepotential der Olefinkreuzmetathese (CM) konnte in den letzten Jahren durch die
Entwicklung definierter Ruthenium-Katalysatoren mit einem N-heterocyclischen Carben- und
chelatisierenden o-Isopropoxybenzylidenliganden, die sich durch eine außergewöhnliche
Chemo- und Diastereoselektivität in Kupplungsreaktionen zwischen elektronisch neutralen
und elektronenarmen Alkenen auszeichnen, erheblich gesteigert werden.
Im ersten Teil der vorliegenden Arbeit wurde die effizient durch den Hoveyda-Blechert-
Katalysator katalysierte, hochselektive CM zwischen Alkenylaminen und α,β-ungesättigten
Carbonylverbindungen in Kombination mit einer reduktiven Cyclisierung zum modularen
Aufbau von N-Heterocyclen genutzt. Nach methodischen Arbeiten zur diastereoselektiven
Synthese von substituierten Pipecolinsäure- und Prolinol-Derivaten, konnte das große
Potential dieser Strategie anschließend erfolgreich in der Totalsynthese einiger Naturstoffe
unter Beweis gestellt werden. Neben dem aus neotropischen Fröschen der Gattung
Dendrobatidae isolierten, neurotoxischen Indolizidin 209D, ließen sich auf diese Weise auch
zwei natürlich vorkommende Piperidine synthetisieren. Während (–)-Pinidinol, ein biologisch
aktives Alkaloid aus der nordamerikanischen Fichte Picea engelmannii, effizient in 6 Stufen
ausgehend von käuflichem (S)-4-Penten-2-ol aufgebaut wurde, konnte das aus der gelben
Wasserlilie Nuphar japonica isolierte, sesquiterpenoide Piperidin-Alkaloid (–)-Nupharamin in
einer 9-stufigen Chiral-Pool Synthese aus (–)-Isopinocampheol zugänglich gemacht werden.
Im zweiten Teil der vorliegenden Arbeit wurde eine neuartige chemo- und stereoselektive CM
von langkettigen Hydroxy- und Aminoalkenen mit einem Vinyldioxolenon gefolgt von einer
thermischen Makrocyclisierung zum Aufbau von Makroliden genutzt. Methodische Arbeiten
zeigten, dass monomere γ,δ-ungesättigte-β-Ketolactone ab einer Ringgröße von 16 zugänglich
sind, wohingegen die Synthese azaanaloger Verbindungen nur ausgehend von gesättigten
Cyclisierungsvorläufern gelang. Eine asymmetrische Dihydroxylierung der CM-generierten
Doppelbindung ermöglichte anschließend die effiziente Totalsynthese des aus dem Pilz
Penicillium turbatum isolierten, 16-gliedrigen Makrolid-Antibiotikums (–)-A26771B in 11
Stufen aus (R)-Methyloxiran. In einer auf derselben Strategie basierenden Studie wurde
abschließend ein kurzer und effizienter Zugang zum 18-gliedrigen Flechten-Makrolid
(+)-Aspicilin via Ringerweiterung eines 17-gliedrigen β-Ketolacton-Intermediats aufgezeigt.


Der praktische Teil der vorliegenden Arbeit wurde unter der Leitung von Herrn Prof. Dr.
Siegfried Blechert in der Zeit von Juni 2002 bis Dezember 2005 am Institut für Chemie der
Fakultät II der Technischen Universität Berlin angefertigt.
Herrn Prof. Dr. Siegfried Blechert danke ich für die Aufnahme in seinen Arbeitskreis, die
interessante Themenstellung und die hervorragenden Arbeitsbedingungen. Im Besonderen
möchte ich mich jedoch für die wertvollen fachlichen Hinweise und Diskussionen sowie die
außergewöhnlich große kreative Freiheit bei der Durchführung dieser Arbeit bedanken.
Frau Prof. Dr. Karola Rück-Braun danke ich für die Übernahme der zweiten Berichterstattung
und die gute Zusammenarbeit während meiner Zeit als wissenschaftlicher Mitarbeiter.
Allen gegenwärtigen und ehemaligen Kollegen im Arbeitskreis danke ich für das gute
Arbeitsklima und die ständige Hilfsbereitschaft. Hervorgehoben seien hier besonders meine
Laborkollegen Nicole Holub, Mustafa Biyikal, Martin Lichtenheldt, Purnama Dewi und Peter
Brüchner sowie Stefan Randl und Mirko Zaja. Meinen Arbeitskollegen Simon Michaelis und
David Koch danke ich für die gute Zusammenarbeit.
Unter den Mitarbeitern des Instituts für Chemie möchte ich mich besonders bei Michael
Grenz, Monika Ulrich, Roswitha Hentschel, Marianne Lehmann und Dietmar Spindler für die
Unterstützung in technischen und organisatorischen Angelegenheiten bedanken. Desweiteren
danke ich Herrn Dr. Zeisberg für seine große Hilfsbereitschaft bei NMR-spektroskopischen
Problemen, Herrn Dr. Höhne für die Aufnahme der Massenspektren, Frau Klose für die
Aufnahme der Infrarotspektren, Frau Becker für die Anfertigung der Elementaranalysen und
Herrn Grimm für die schnellen Reparaturen.
Für das Korrekturlesen dieser Arbeit bedanke ich mich bei Nicole Holub.
Mein außerordentlicher Dank gilt meiner Familie und meiner Freundin Zerrin.


INHALTSVERZEICHNIS
I. ALLGEMEINER TEIL ....................................................................................................... 1
1. Einleitung .......................................................................................................................... 1
1.1. Die Olefinmetathese................................................................................................... 1
1.2. Olefinkreuzmetathese mit elektronenarmen Alkenen ................................................ 4
2. Synthese von Azacyclen ................................................................................................... 6
2.1. Motivation und Synthesekonzept ............................................................................... 6
2.2. Diastereoselektive Synthese 6-substituierter Pipecolinsäure-Derivate ...................... 9
2.2.1. Synthese der Metathesevorläufer ....................................................................... 9
2.2.2. Synthese ........................................................................................................... 11
2.3. Synthese 5-substituierter Prolinol-Derivate ............................................................. 14
2.3.1. Synthese ........................................................................................................... 15
2.4. Synthese von (±)-Indolizidin 209D.......................................................................... 17
2.4.1. Retrosynthetische Analyse ............................................................................... 19
2.4.2. Synthese ........................................................................................................... 19
2.5. Synthese des Piperidin-Alkaloids (–)-Pinidinol....................................................... 20
2.5.1. Retrosynthetische Analyse ............................................................................... 21
2.5.2. Synthese ........................................................................................................... 22
2.6. Synthese des Piperidin-Alkaloids (–)-Nupharamin.................................................. 24
2.6.1. Retrosynthetische Analyse ............................................................................... 25
2.6.2. Synthese ........................................................................................................... 26
2.7. Studie zur Synthese 6-substituierter 4,5-Didehydropipecolate ................................ 31
2.8. Zusammenfassung und Ausblick ............................................................................. 33
3. Synthese von Makroliden .............................................................................................. 35
3.1. Motivation und Synthesekonzept ............................................................................. 35
3.2. Synthese von γ,δ-ungesättigten-β-Ketolactonen ...................................................... 38
3.2.1. Synthese der Metathesevorläufer ..................................................................... 38
3.2.2. Synthese ........................................................................................................... 40
3.3. Synthese des Makrolid-Antibiotikums (–)-A26771B .............................................. 42
3.3.1. Retrosynthetische Analyse ............................................................................... 44
3.3.2. Synthese ........................................................................................................... 45

3.4. Studie zur Synthese des Flechten-Makrolids (+)-Aspicilin ..................................... 48
3.4.1. Retrosynthetische Analyse ............................................................................... 49
3.4.2. Synthese ........................................................................................................... 50
3.5. Studie zur Synthese von γ,δ-ungesättigten-β-Ketolactamen .................................... 51
3.6. Zusammenfassung und Ausblick ............................................................................. 54
II. EXPERIMENTELLER TEIL.......................................................................................... 56
1. Allgemeines ..................................................................................................................... 56
2. Versuchsvorschriften und spektroskopische Daten.................................................... 58
2.1. Zu Kapitel 2.2........................................................................................................... 58
2.2. Zu Kapitel 2.3........................................................................................................... 69
2.3. Zu Kapitel 2.4........................................................................................................... 74
2.4. Zu Kapitel 2.5........................................................................................................... 75
2.5. Zu Kapitel 2.6........................................................................................................... 80
2.6. Zu Kapitel 2.7........................................................................................................... 87
2.7. Zu Kapitel 3.2........................................................................................................... 88
2.8. Zu Kapitel 3.3........................................................................................................... 95
2.9. Zu Kapitel 3.4......................................................................................................... 100
2.10. Zu Kapitel 3.5......................................................................................................... 102
III. ANHANG ....................................................................................................................... 107
1. Röntgenstrukturanalyse .............................................................................................. 107
2. Abkürzungen ................................................................................................................ 108
3. Literaturverzeichnis..................................................................................................... 111
4. Lebenslauf ..................................................................................................................... 119
5. Publikationsliste ........................................................................................................... 120

I. Allgemeiner Teil
1
I. Allgemeiner Teil
1. Einleitung
1.1. Die Olefinmetathese
Die C-C-Bindungsknüpfung stellt die wohl größte Herausforderung auf dem Weg zu
komplexen organischen Strukturen dar. Hierzu stehen dem organischen Chemiker heutzutage
eine Vielzahl von zum Teil hochselektiven Transformationen zur Verfügung, die eine
rationale Syntheseplanung ermöglichen. Aus ökonomischen und ökologischen Gründen sind
dabei vornehmlich katalytische Verfahren von Interesse, unter denen insbesondere die
übergansmetallkatalysierten Reaktionen das Methodenarsenal des organischen Chemikers in
den letzten Jahrzehnten in ungeahntem Ausmaß erweitert haben. Eines dieser Verfahren ist
der seit über 50 Jahren bekannte und als „Olefinmetathese“ bezeichnete, wechselseitige
Austausch der Alkylideneinheiten zweier Olefine.1 Die Vermutung, dass während dieser
Kreuzkupplungsreaktion ein Metallacyclobutan als Zwischenstufe durchlaufen wird, konnte
schließlich von Chauvin bestätigt werden, der eine bis heute akzeptierte Abfolge von
[2+2]-Cycloaddition und [2+2]-Cycloreversion zwischen einem Metallalkyliden-Komplex
und einem Olefin formulierte (Schema 1).2
+ [M]
R1 R2
R3[2+2]-CA
[2+2]-CR[M]
R1 R2
R3 [2+2]-CR
[2+2]-CA
[M]
R1 R2
R3
+
Schema 1. Allgemeiner Mechanismus der Olefinmetathese nach Chauvin.
Um eine optimale Ausschöpfung des großen Potentials der Metathese zur C-C-Verknüpfung
in der organischen Synthese3 zu ermöglichen, wurde besonders die Entwicklung definierter,
homogener Molybdän- und Ruthenium-Katalysatoren in den letzten Jahren entscheidend
angetrieben, was schließlich auch durch die Verleihung des Nobelpreises an die Chemiker R.
H. Grubbs, R. R. Schrock und Y. Chauvin gewürdigt werden sollte.4
Vielseitige Verwendung in zahlreichen Metathesereaktionen fanden beispielsweise der von
Schrock et al. beschriebene Molybdän-Komplex 15a sowie der von Grubbs et al. entwickelte
Ruthenium-Komplex 2 (Abbildung 1).5b Letzterer zeigt zwar eine in einigen Fällen geringere
Aktivität, besitzt jedoch eine höhere Toleranz gegenüber funktionellen Gruppen und ist relativ
unempfindlich gegenüber Sauerstoff, Wasser und Lösungsmittelverunreinigungen.

I. Allgemeiner Teil
2
MoN
iPr iPr
Ph(F3C)2MeCO
(F3C)2MeCO
1
PCy3
RuPhCl
Cl
PCy3
2
RuClCl
O
MesN NMes
3 4 5
RuClCl
O
PCy3
RuPhCl
ClMesN NMes
PCy3
Abbildung 1. Ausgewählte Metathese-Katalysatoren.
Da die Stabilität und die Reaktivität der sich im Verlauf des Katalysecyclus durch
Ligandendissoziation bildenden, katalytisch aktiven Ruthenium-Zwischenstufen von den
sterischen und elektronischen Eigenschaften der verbleibenden Neutral-Liganden bestimmt
wird, konnten diesbezüglich verbesserte Katalysatoren durch den gezielten Austausch einer
Phosphineinheit erhalten werden. Für die hohe Reaktivität des sogenannten Grubbs-
Katalysators der 2. Generation 3 ist beispielsweise die Substitution durch einen stark
basischen und sterisch anspruchsvollen N-heterocyclischen Carbenliganden (NHC-Ligand)
entscheidend, der eine gute Stabilisierung der nach Phosphin-Dissoziation gebildeten
14-Elektronen-Spezies bewirkt.5c Eine erhöhte Stabilität des Präkatalysators in Lösung
wurde hingegen nach Einführung eines chelatisierenden o-Isopropoxybenzylidenliganden
beobachtet, die den Monophosphin-Komplex 4 sogar chromatographiestabil und somit
wiederverwendbar macht.5d Als logische Konsequenz wurde in parallelen Arbeiten von
Hoveyda et al.5e und Blechert et al.5f schließlich der mittlerweile käufliche, äußerst stabile
phosphinfreie Chelatkomplex 5 synthetisiert, dessen bemerkenswerte Reaktivität teilweise die
des Grubbs-Katalysators 3 übertrifft und bereits auch erfolgreich in einigen Naturstoff-
synthesen genutzt werden konnte.6
Obwohl alle Teilschritte des Metathesecyclus prinzipiell reversibel sind, kann das thermo-
dynamische Gleichgewicht in der Regel leicht auf die Produktseite verschoben werden
(Schema 2). Die als Ringschlußmetathese (RCM) bezeichnete Cyclisierung eines Diens zu
einem Cycloolefin stellt die mit Abstand am häufigsten eingesetzte Metatheseart dar,7 wobei
sogar mittlere Ringe, deren Bildung infolge ungünstiger transannularer Wechselwirkungen oft
Probleme bereitet, in zum Teil hervorragenden Ausbeuten zugänglich sind.7d Neben dem
Entropiegewinn trägt hier die Bildung eines flüchtigen und folglich aus der Reaktions-
mischung entweichenden Nebenprodukts wie Ethylen oder Propylen entscheidend zur
Triebkraft der Reaktion bei. Unter Ringöffnungsmetathese (ROM) versteht man demnach die
formale Rückreaktion eines möglichst gespannten Cycloolefins, die in Abwesenheit eines
externen Olefins auch zur Alken-Polymerisation genutzt werden kann (ROMP).8

I. Allgemeiner Teil
3
R1 R2
+RCM
ROMR1 R2
RRM
R1R2+
**
CM
− C2H4R1
+R2 R1+R1 R2
R2
C2H4
Schema 2. Grundlegende Metathesearten.
Bei der als Ringumlagerungsmetathese (RRM) bezeichneten Kombination aus einer ROM
und einer RCM, wird ein alkenylsubstituiertes Cycloolefin unter Chiralitätstransfer in ein
ringumgelagertes Strukturisomer überführt, wobei das Gleichgewicht dieses reversiblen
Prozesses ausschließlich durch die Differenz der freien Enthalpien bestimmt wird. In der
Naturstoffsynthese konnte diese atomökonomische Tandemmetathese bislang vor allem zum
stereoselektiven Aufbau von N-Heterocyclen genutzt werden.9
Die Kreuzmetathese (CM) ist die intermolekulare Variante der Olefinmetathese, bei der es zur
Kupplung zweier Alkylideneinheiten unter Abspaltung eines möglichst flüchtigen Alkens
kommt. Da bei der CM zwischen elektronisch und sterisch ähnlichen Substraten oft ein
statistisches Gemisch aller möglichen Produkte resultiert, die zumal als E/Z-Isomere
vorliegen können, spielte die CM in der organischen Synthese lange Zeit eine eher
untergeordnete Rolle. Industriell wird die Reaktion in großtechnischen Prozessen wie der
Disproportionierung von Propen zu 2-Buten und Ethylen (Philips-Triolefin-Prozess)10a sowie
dem Shell-Higher-Olefin-Prozess (SHOP)10b genutzt.

I. Allgemeiner Teil
4
1.2. Olefinkreuzmetathese mit elektronenarmen Alkenen
Die der CM inhärenten Probleme der Chemo- und Stereoselektivität hatten lange Zeit zur
Folge, dass gute Ausbeuten in der Regel nur durch einen großen Überschuß eines CM-
Partners erzielt werden konnten und die CM in der präparativen organischen Synthese somit
vergleichsweise wenig Beachtung fand. Zum entscheidenden Durchbruch auf diesem Gebiet
verhalf erst die Entdeckung, dass Ruthenium-Komplexe vom Typ 3 und 5 äußerst effiziente
Katalysatoren für die hochselektive Metathese zwischen elektronisch neutralen und elektro-
nenarmen Alkenen sind. So wurden beispielsweise in CM mit Acrolein, Vinylketonen,
Acrylaten oder Acrylnitril häufig hohe Kreuzprodukt/Dimer-Verhältnisse und ausgezeichnete
E/Z-Selektivitäten erzielt.11 Die seitdem stark anwachsende Zahl an Publikationen über
selektive CM und deren Anwendung in der organischen Synthese12 hat letztendlich dazu
geführt, dass von Grubbs et al. ein allgemeines Modell zur produktselektiven CM entwickelt
wurde.13 Aufgrund ihrer im Rahmen der vorliegenden Arbeit zentralen Rolle, sollen im
Folgenden kurz einige Beispiele für die Anwendung der selektiven CM mit elektronenarmen
Alkenen in der Naturstoffsynthese vorgestellt werden.
Eines der ersten, von Hoveyda et al. publizierten Beispiele ist die zum Aufbau der Seitenkette
des marinen Diterpens (+)-Erogorgiaen genutzte, regioselektive CM des Diens 6 mit
Methylvinylketon (Schema 3).14a
(+)-Erogorgiaen
10 mol% 5
CH2Cl2, 22 °C
74%6 7
OCOMe
Schema 3. Synthese von (+)-Erogorgiaen nach Hoveyda.
Gute Ausbeuten und E/Z-Selektivitäten (> 95% E) wurden dabei allerdings nur mit dem
phosphinfreien Katalysator 5 erreicht, während die Verwendung des Grubbs-Katalysators der
2. Generation 3 zu Nebenprodukten und deutlich verlängerten Reaktionszeiten führte.
Im Zuge der Totalsynthese des antiprotozoalen Pyrans (–)-Centrolobin konnten Cossy et al.
das Lacton 9 effizient durch eine Eintopf-Reaktion aus CM und anschließender Hydrierung
ausgehend vom chiralen Homoallylalkohol 8 synthetisieren (Schema 4).14b Abschließende
diastereoselektive Einführung des Arylrestes lieferte den Naturstoff in hoher Gesamtausbeute.

I. Allgemeiner Teil
5
2) H2, Pd/C, 96 h
56%
4 mol% 5, CH2Cl2
OH
OBn
O
OH
O O
OH
OMe
(−)-Centrolobin8 9
CO2H1)
Schema 4. Synthese von (–)-Centrolobin nach Cossy.
An dieser Stelle sei erwähnt, dass die auf eine Metathese folgende Hydrierung auch in situ
mittels des Metathese-Katalysators erreicht werden kann.15
Des Weiteren nutzten Blechert und Michaelis kürzlich eine regioselektive CM zwischen dem
Dien 10 und dem aus Milchsäure gewonnenen Hydroxyenon 11 als Schlüsselschritt in der
Synthese von (+)-Phomopsolid C (Schema 5).14c
73%
5 mol% 5, CH2Cl2
(+)-Phomopsolid C10
OTr
HO
12
OTr
HOO
OTBSO
O
OH
O O
O
11
40 °C, 6 h
O
OTBS
Schema 5. Synthese von (+)-Phomopsolid C nach Blechert.
Das neben dem gewünschten Produkt 12 (E/Z > 20:1) isolierte Homodimer von 10 konnte
anschließend wiederverwendet und unter Erhöhung der Ausbeute auf 89% ebenfalls in das
Kreuzprodukt 12 überführt werden. Eine im weiteren Verlauf zum Zuge kommende
regioselektive RCM lieferte den Naturstoff schließlich in insgesamt 9 Stufen mit einer
Gesamtausbeute von 8%.

I. Allgemeiner Teil
6
2. Synthese von Azacyclen
2.1. Motivation und Synthesekonzept
Seit der Isolierung des Morphins im Jahre 1805 werden als Alkaloide inzwischen mehr als
10000 stickstoffhaltige Naturstoffe überwiegend pflanzlicher Herkunft bezeichnet.16 In den
menschlichen oder tierischen Organismus gebracht entfalten sie oft ausgeprägte biologische
Aktivitäten, weshalb heute etwa 25% der kommerziellen Pharmawirkstoffe Alkaloide oder
strukturell modifizierte Derivate sind (Abbildung 2).
(−)-Morphin
N
O
OH
OH
Pentazocin
N
OH
(+)-Epibatidin
HN
NCl
H
Abbildung 2. Morphin, ein synthetisches Äquivalent und eine natürliche Alternative.
Durch den stetig zunehmenden Bedarf an natürlichen Wirkstoffen und Derivaten mit
optimierten Eigenschaften ist die Entwicklung effizienter, stereoselektiver Synthesemethoden
bis heute von unvermindert großem Interesse geblieben.
Das als Piperidin bezeichnete Azacyclohexyl-Gerüst gehört zu den in natürlichen und
synthetischen Wirkstoffen am weitesten verbreiteten stickstoffhaltigen Bausteinen, weshalb
im Laufe der letzten Jahre erhebliche Arbeit in die Isolierung, Strukturaufklärung und
stereoselektive Synthese funktionalisierter Piperidin-Derivate investiert wurde (Schema 6).17
R1 NH
R2
R
[4+2]CA
R1 N R2
PG
RRCM
* *
R1 N R2
PG
* *
R1 NHPG
R
**
R1 NHPG
R
*R2
O
R2
X
SN2Reduktion
Schema 6. Strategien zum Aufbau des Piperidinrings.

I. Allgemeiner Teil
7
Die als reduktive Aminierung bezeichnete intramolekulare Kondensation eines Aminoketons
mit nachfolgender Reduktion des Imins ist aufgrund ihrer zumeist außergewöhnlich hohen
Diastereoselektivität dabei besonders für den cis-selektiven Aufbau von 2,6-substituierten
Piperidinen prädestiniert.18 Ein elegantes Beispiel für die Anwendung als Schlüsselschritt in
der Naturstoffsynthese stellt die Synthese des antifungalen Hydroxypiperidins (+)-Spectalin
nach Ham et al. dar, in der eine abschließende Hydrierung des in 13 Stufen aus dem Serinol
13 erhaltenen geschützten Aminoalkohols 14 im essigsauren Medium den Naturstoff via
Hydrogenolyse des Oxazolins, reduktiver Aminierung und Ketalspaltung diastereomerenrein
in guter Ausbeute ergab (Schema 7).18e
MeOH/AcOH70%
NH
(+)-Spectalin
HO
( )11
( )11
O
O
NPh
70 psi H2, Pd(OH)2HO
BzHN
OTBS
OOO
1413
Schema 7. Synthese von (+)-Spectalin durch reduktive Aminierung nach Ham.
Dass die stereoselektive Reduktion des Imin-Intermediats jedoch nicht notwendigerweise in
situ verlaufen muß, zeigt die Synthese des Quinolizidin-Alkaloids (–)-Lasubin II nach Davis
et al. (Schema 8). Hierbei lieferte ein alternatives zweistufiges Verfahren aus Entschützung
des Sulfinamids 15 gefolgt von Hydridreduktion des resultierenden Imins das cis-Piperidin 16
in einer Ausbeute von 72%, welches anschließend in wenigen Schritten in den Naturstoff
überführt werden konnte.19
2) NH4OH
72%
1) HCl
N
(−)-Lasubin II
OH
MeO
MeOAr N
H
OH
16
3) LAHNH
OH
MeO
MeO
BnO
15
O
SOpTolyl
OBn
Schema 8. Synthese von (–)-Lasubin II durch reduktive Aminierung nach Davis.
Wir waren folglich daran interessiert, die hohe Effizienz der selektiven CM elektronenarmer
Alkene in Kombination mit einer reduktiven Cyclisierung zum modularen Aufbau von
gesättigten N-Heterocyclen zu nutzen. Dazu sollten geeignet geschützte Allyl- und
Homoallylamine mit α,β-ungesättigten Carbonylverbindungen vom allgemeinen Typ 18
gekuppelt und anschließend mittels Wasserstoff und einem heterogenen Palladium-
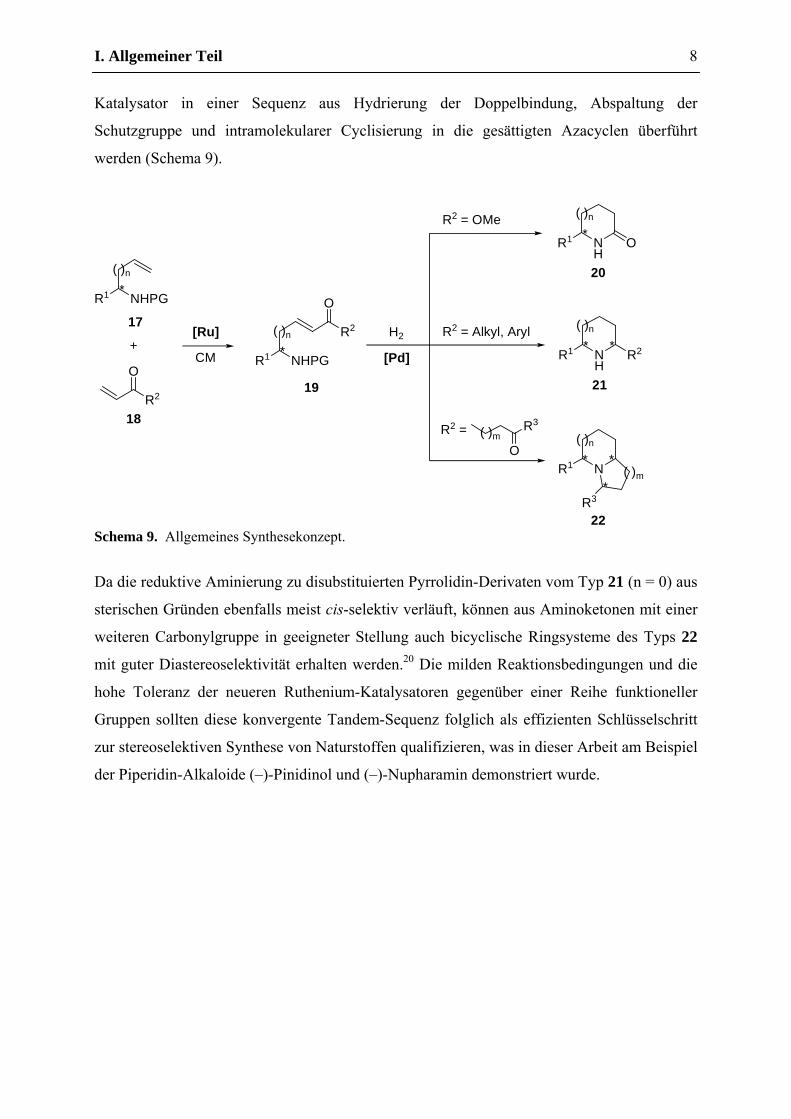
I. Allgemeiner Teil
8
Katalysator in einer Sequenz aus Hydrierung der Doppelbindung, Abspaltung der
Schutzgruppe und intramolekularer Cyclisierung in die gesättigten Azacyclen überführt
werden (Schema 9).
R1 NHPG
R2
OR1 NHPG
O
R2
R1 NH
R2
R1 N
R1 NH
O
+
R3
R2 = OMe
R2 = Alkyl, Aryl
R2 = R3
O
[Pd]
H2[Ru]
CM
17
18
19
20
21
22
( )n
( )n
( )m ( )n
( )n
( )n
( )m
*
* * *
*
* *
*
Schema 9. Allgemeines Synthesekonzept.
Da die reduktive Aminierung zu disubstituierten Pyrrolidin-Derivaten vom Typ 21 (n = 0) aus
sterischen Gründen ebenfalls meist cis-selektiv verläuft, können aus Aminoketonen mit einer
weiteren Carbonylgruppe in geeigneter Stellung auch bicyclische Ringsysteme des Typs 22
mit guter Diastereoselektivität erhalten werden.20 Die milden Reaktionsbedingungen und die
hohe Toleranz der neueren Ruthenium-Katalysatoren gegenüber einer Reihe funktioneller
Gruppen sollten diese konvergente Tandem-Sequenz folglich als effizienten Schlüsselschritt
zur stereoselektiven Synthese von Naturstoffen qualifizieren, was in dieser Arbeit am Beispiel
der Piperidin-Alkaloide (–)-Pinidinol und (–)-Nupharamin demonstriert wurde.

I. Allgemeiner Teil
9
2.2. Diastereoselektive Synthese 6-substituierter Pipecolinsäure-Derivate
Der Pipecolinsäure (Piperidin-2-carbonsäure) kommt als natürlicher aber nichtproteinogener
Aminosäure besondere Bedeutung zu, da sie neben ihrer Verwendung als Prolinanalogon21
auch Bestandteil zahlreicher pharmakologisch bedeutsamer Substanzen wie beispielsweise
den Immunsuppressiva FK506 und Rapamycin, dem HIV-Protease Inhibitor Palinavir oder
dem Antitumor-Antibiotikum Tetrazomin ist.22 Während sich alkylsubstituierte Pipecolate
auch als potente Dihydropicolinsäure-Synthase (DHDPA) Inhibitoren23 sowie als N-Methyl-
D-Aspartat-Rezeptor (NMDA) Agonisten24a und Antagonisten24b erwiesen haben, wurden
funktionalisierte Pipecolinsäuren als Lysinmimetika eingesetzt und in eine Vielzahl bioaktiver
Peptide wie Vasopressin, Oxytocin oder Angiotensin II integriert.25
Neuere Methoden zur stereoselektiven Synthese 6-substituierter Pipecolinsäuren beinhalten
die Kondensation geschützter β-Aminoketone vom Typ 23 mit Alkylglyoxylaten26a (Schema
10, Gleichung 1) sowie die Vinylsilan-Imminiumion Cyclisierung von Morpholinonen des
Typs 26 (Schema 10, Gleichung 2).26b
pTsOH
60−75%> 80% cis
R N
26
1)
2)
R NH2
23
OO
+ O CO2EtR N
H24
OO
CO2Et R NH25a
CO2H
OPh
SiMe3
1) LiHMDS, PhSSPh
> 95% trans
O
2) ZnCl2, 62−83%R N
27
OPh
O H2
Pd(OH)2R N
H25b
CO2H
Schema 10. Neuere Pipecolinsäure-Synthesen.
Mittels der beschriebenen Tandem-Sequenz aus CM und reduktiver Aminierung würden sich
ausgehend von Z-geschützten Allylglycin-Derivaten cis-konfigurierte mono- und bicyclische
Pipecolate effizient in lediglich zwei Schritten aufbauen lassen.
2.2.1. Synthese der Metathesevorläufer
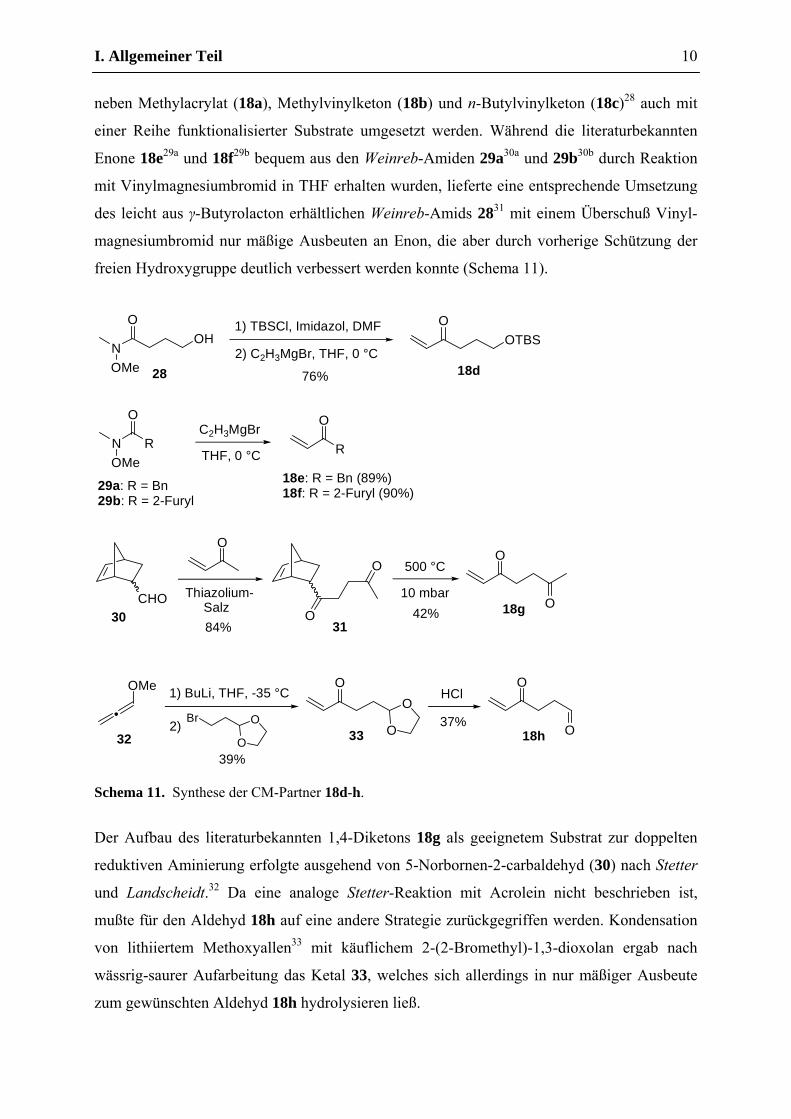
Zur Demonstration der breiten Anwendbarkeit des allgemeinen Synthesekonzepts sollte der
literaturbekannte Z-geschützte Allylglycinmethylester 17a (R1 = CO2Me, n = 1, PG = Z)27
I. Allgemeiner Teil
10
neben Methylacrylat (18a), Methylvinylketon (18b) und n-Butylvinylketon (18c)28 auch mit
einer Reihe funktionalisierter Substrate umgesetzt werden. Während die literaturbekannten
Enone 18e29a und 18f29b bequem aus den Weinreb-Amiden 29a30a und 29b30b durch Reaktion
mit Vinylmagnesiumbromid in THF erhalten wurden, lieferte eine entsprechende Umsetzung
des leicht aus γ-Butyrolacton erhältlichen Weinreb-Amids 2831 mit einem Überschuß Vinyl-
magnesiumbromid nur mäßige Ausbeuten an Enon, die aber durch vorherige Schützung der
freien Hydroxygruppe deutlich verbessert werden konnte (Schema 11).
N
O
OMe
OH1) TBSCl, Imidazol, DMF
2) C2H3MgBr, THF, 0 °C
O
N
O
R
O1) BuLi, THF, -35 °C
O
37%
OMe
•
O
ROMe
29a: R = Bn29b: R = 2-Furyl
THF, 0 °C
2)
39%
HCl
C2H3MgBr
28 18d
32 33 18h
CHO30
Thiazolium- Salz
O
500 °CO
18g84%
42%31
O
O
OTBS
76%
10 mbar
18e: R = Bn (89%)18f: R = 2-Furyl (90%)
O
OO
OBr
O
O
Schema 11. Synthese der CM-Partner 18d-h.
Der Aufbau des literaturbekannten 1,4-Diketons 18g als geeignetem Substrat zur doppelten
reduktiven Aminierung erfolgte ausgehend von 5-Norbornen-2-carbaldehyd (30) nach Stetter
und Landscheidt.32 Da eine analoge Stetter-Reaktion mit Acrolein nicht beschrieben ist,
mußte für den Aldehyd 18h auf eine andere Strategie zurückgegriffen werden. Kondensation
von lithiiertem Methoxyallen33 mit käuflichem 2-(2-Bromethyl)-1,3-dioxolan ergab nach
wässrig-saurer Aufarbeitung das Ketal 33, welches sich allerdings in nur mäßiger Ausbeute
zum gewünschten Aldehyd 18h hydrolysieren ließ.

I. Allgemeiner Teil
11
2.2.2. Synthese
Während die Substrate 18a und 18b in der CM mit Allylglycinmethylester 17a aufgrund ihrer
erhöhten Flüchtigkeit im Überschuß (2 Äquivalente) eingesetzt werden sollten, war ein
stöchiometrischer Einsatz der nichtkäuflichen Enone 18c-h wünschenswert. Mit Ausnahme
von Furylvinylketon 18f, welches zur Unterdrückung der Homodimerisierung von 17a
ebenfalls mit einem Überschuß von 2 Äquivalenten eingesetzt werden mußte, konnten die
Kupplungsprodukte 19a-h auf diese Weise mit maximal 5 mol% des Hoveyda-Blechert-
Katalysators 5 in siedendem CH2Cl2 (0.05 M) stereoselektiv (E/Z > 20:1) in guten bis
akzeptablen Ausbeuten erhalten werden (Tabelle 1).
CH2Cl2, 40 °C
2.5−5 mol% 5
MeO2C NHZ
17a
MeO2C NHZ
O
R2
19a-h
O
R2
18a-h
+
Enon R2 Äquiv. mol% 5 t Ausbeute 19
18a OMe 2 5 3 h 76%
18b Me 2 2.5 3 h 85%
18c n-Bu 1 2.5 3 h 83%
18d (CH2)3OTBS 1 5 16 h 64%
18e CH2Ph 1 5 16 h 78%
18f 2-Furyl 2 5 16 h 73%
18g (CH2)2COMe 1 5 16 h 74%
18h (CH2)2CHO 1 5 16 h 62% Tabelle 1. Ergebnisse der CM von Allylglycinmethylester 17a.
Die nachfolgenden reduktiven Cyclisierungen der Kupplungsprodukte 19a-h wurden mit
5 mol% Pd/C (10%) unter Wasserstoff-Atmosphäre bei Raumtemperatur durchgeführt.
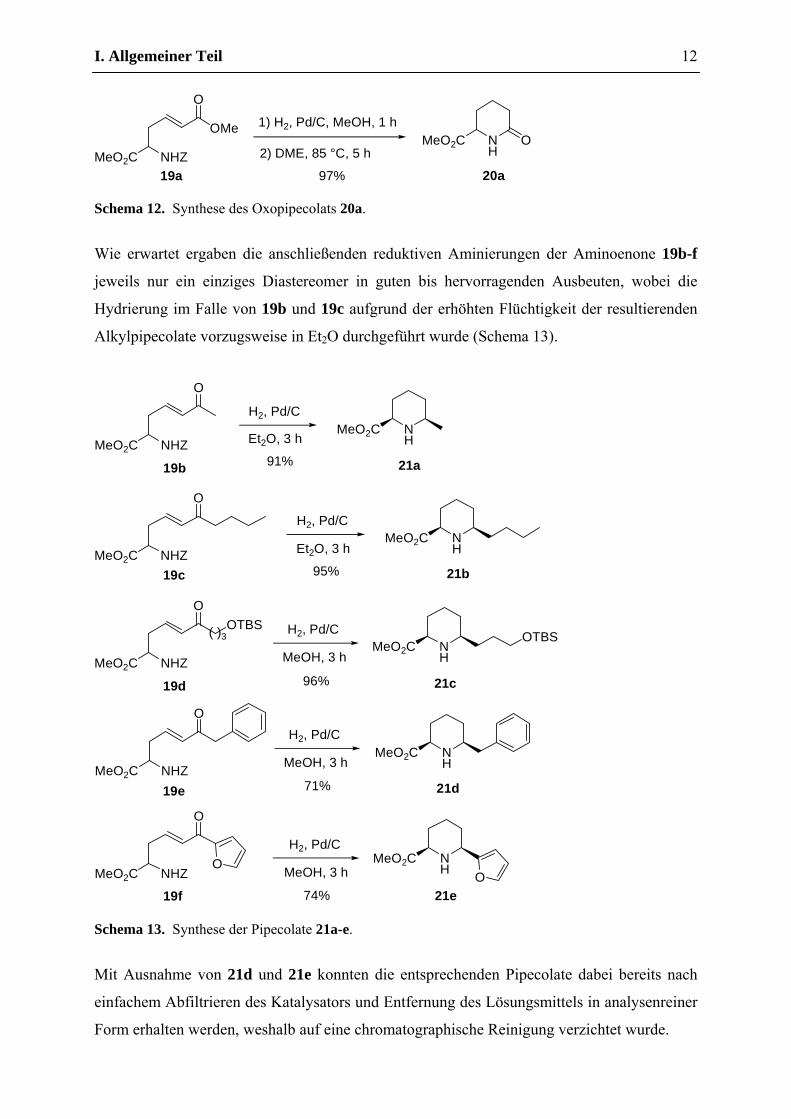
Ausgehend vom Aminoester 19a lieferte eine kurze Hydrierung gefolgt von thermischer
Cyclisierung des Rohprodukts das literaturbekannte Oxopipecolat 20a in hervorragender
Ausbeute, welches sich leicht über das Lactamenolat weiter funktionaliseren lässt und somit
ein interessantes Ausgangsmaterial für die Synthese von höher substituierten Piperidinen
darstellt (Schema 12).34
I. Allgemeiner Teil
12
2) DME, 85 °C, 5 h
1) H2, Pd/C, MeOH, 1 h
MeO2C NHZ
O
OMe
19a 97%
MeO2C NH
20a
O
Schema 12. Synthese des Oxopipecolats 20a.
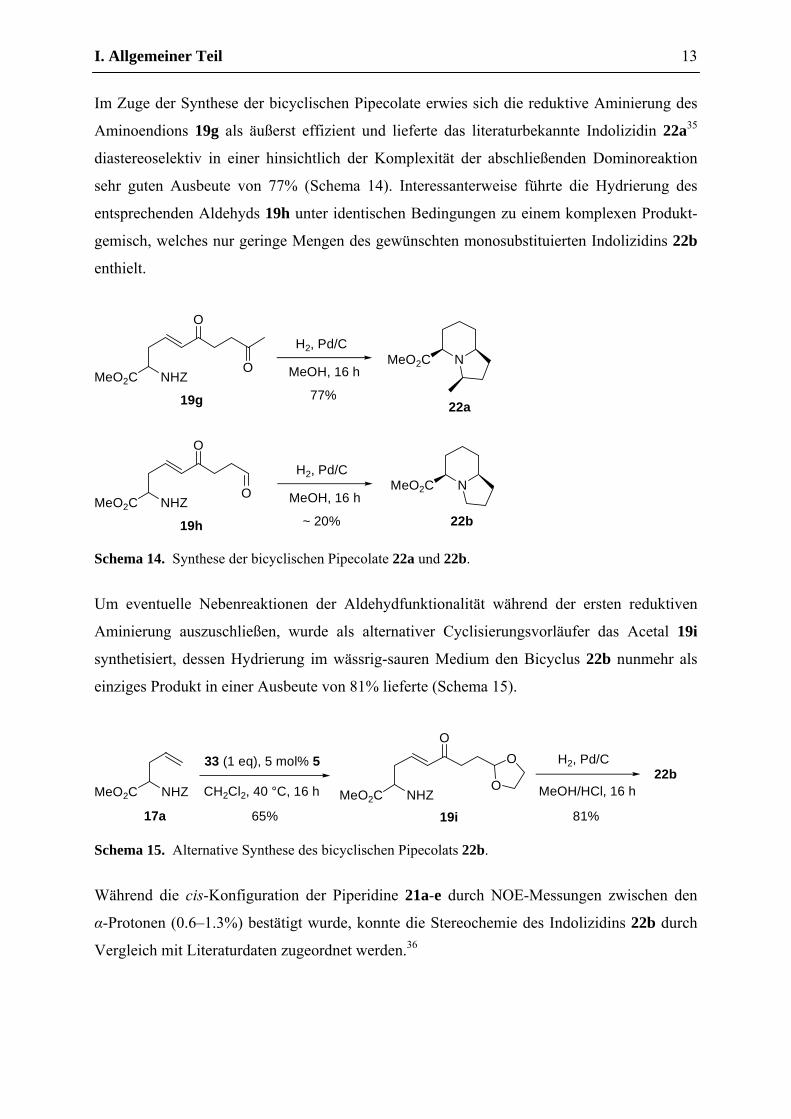
Wie erwartet ergaben die anschließenden reduktiven Aminierungen der Aminoenone 19b-f
jeweils nur ein einziges Diastereomer in guten bis hervorragenden Ausbeuten, wobei die
Hydrierung im Falle von 19b und 19c aufgrund der erhöhten Flüchtigkeit der resultierenden
Alkylpipecolate vorzugsweise in Et2O durchgeführt wurde (Schema 13).
MeO2C NHZ
O
MeO2C NH
19d 21c
OTBS
MeO2C NHZ
O
19e
MeO2C NHZ
O
O
MeO2C NH
21d
MeO2C NH
21eO
H2, Pd/C
MeOH, 3 h
H2, Pd/C
MeOH, 3 h
H2, Pd/C
MeOH, 3 h
( )3
96%
71%
74%19f
OTBS
MeO2C NHZ
O
MeO2C NH
19b 21a
MeO2C NHZ
O
19c
MeO2C NH
21b
H2, Pd/C
Et2O, 3 h
H2, Pd/C
Et2O, 3 h
91%
95%
Schema 13. Synthese der Pipecolate 21a-e.
Mit Ausnahme von 21d und 21e konnten die entsprechenden Pipecolate dabei bereits nach
einfachem Abfiltrieren des Katalysators und Entfernung des Lösungsmittels in analysenreiner
Form erhalten werden, weshalb auf eine chromatographische Reinigung verzichtet wurde.
I. Allgemeiner Teil
13
Im Zuge der Synthese der bicyclischen Pipecolate erwies sich die reduktive Aminierung des
Aminoendions 19g als äußerst effizient und lieferte das literaturbekannte Indolizidin 22a35
diastereoselektiv in einer hinsichtlich der Komplexität der abschließenden Dominoreaktion
sehr guten Ausbeute von 77% (Schema 14). Interessanterweise führte die Hydrierung des
entsprechenden Aldehyds 19h unter identischen Bedingungen zu einem komplexen Produkt-
gemisch, welches nur geringe Mengen des gewünschten monosubstituierten Indolizidins 22b
enthielt.
MeO2C N
22b
MeO2C N
22a
H2, Pd/C
MeOH, 16 h
MeO2C NHZ
O
19h
MeO2C NHZ
O
19g 77%
H2, Pd/C
MeOH, 16 h
~ 20%
O
O
Schema 14. Synthese der bicyclischen Pipecolate 22a und 22b.
Um eventuelle Nebenreaktionen der Aldehydfunktionalität während der ersten reduktiven
Aminierung auszuschließen, wurde als alternativer Cyclisierungsvorläufer das Acetal 19i
synthetisiert, dessen Hydrierung im wässrig-sauren Medium den Bicyclus 22b nunmehr als
einziges Produkt in einer Ausbeute von 81% lieferte (Schema 15).
NHZMeO2C
O
19i
O
O
CH2Cl2, 40 °C, 16 h
33 (1 eq), 5 mol% 5
65%
H2, Pd/C
MeOH/HCl, 16 h
81%17a
22bNHZMeO2C
Schema 15. Alternative Synthese des bicyclischen Pipecolats 22b.
Während die cis-Konfiguration der Piperidine 21a-e durch NOE-Messungen zwischen den
α-Protonen (0.6–1.3%) bestätigt wurde, konnte die Stereochemie des Indolizidins 22b durch
Vergleich mit Literaturdaten zugeordnet werden.36

I. Allgemeiner Teil
14
2.3. Synthese 5-substituierter Prolinol-Derivate
Obwohl im Vergleich zu den entsprechenden Piperidinen in der Natur weit weniger
verbreitet, wurde seit ihrer erstmaligen Isolierung aus Feuerameisen der Gattung Solenopsis
nicht weniger Arbeit in die stereoselektive Synthese von 2,5-disubstituierten Pyrrolidinen
investiert.37 Neben ihren interessanten biologischen Eigenschaften sind Pyrrolidin-Derivate
aber vor allem auch wegen ihrer Verwendung als Katalysatoren, Auxiliare oder Liganden in
der asymmetrischen Synthese38 von großem Interesse (Abbildung 3).
NH
Pyrrolidin 197B
NPh
(+)-Preussin
HO
SAMP
NMeO
NH2
NO B
CBS-Reagenz
PhPh
Abbildung 3. Natürliche Pyrrolidine und Derivate zur asymmetrischen Synthese.
Des Weiteren können enantiomerenreine 5-substituierte Prolinol-Derivate auch als attraktive
Synthesebausteine zur Darstellung biologisch aktiver dialkylsubstituierter Pyrrolizidin- und
Indolizidin-Alkaloide dienen. Beispiele sind die Synthese von (+)-Xenovenin nach Takahata
et al.39a sowie die Synthese des Spurenpheromons (+)-Monomorin nach Bäckvall et al.39b
(Schema 16).
NBnO N
(+)-Monomorin
NHO N
(+)-Xenovenin
ZHN
CO2H
BnO OH
Z
Ts34%
28%
Schema 16. 5-Alkylprolinole als Intermediate in der Naturstoffsynthese.
Ausgehend von Z-geschützten Vinylglycinolen sollte die beschriebene Tandem-Sequenz aus
CM und reduktiver Aminierung daher auch auf die Synthese von 2-Pyrrolidinonen und
cis-konfigurierten 5-Alkylprolinolen angewendet werden, für die in der Literatur bislang nur
wenige Beispiele beschrieben sind.40
I. Allgemeiner Teil
15
2.3.1. Synthese
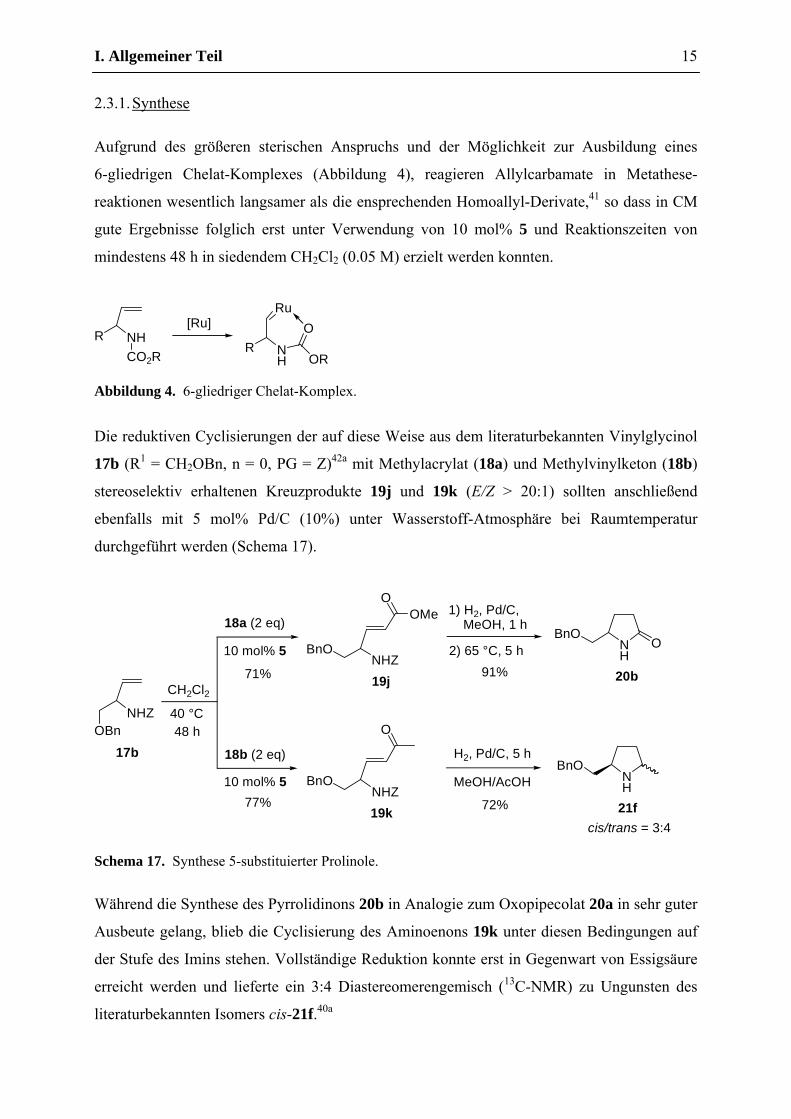
Aufgrund des größeren sterischen Anspruchs und der Möglichkeit zur Ausbildung eines
6-gliedrigen Chelat-Komplexes (Abbildung 4), reagieren Allylcarbamate in Metathese-
reaktionen wesentlich langsamer als die ensprechenden Homoallyl-Derivate,41 so dass in CM
gute Ergebnisse folglich erst unter Verwendung von 10 mol% 5 und Reaktionszeiten von
mindestens 48 h in siedendem CH2Cl2 (0.05 M) erzielt werden konnten.
NHRCO2R
NH
ORu
ROR
[Ru]
Abbildung 4. 6-gliedriger Chelat-Komplex.
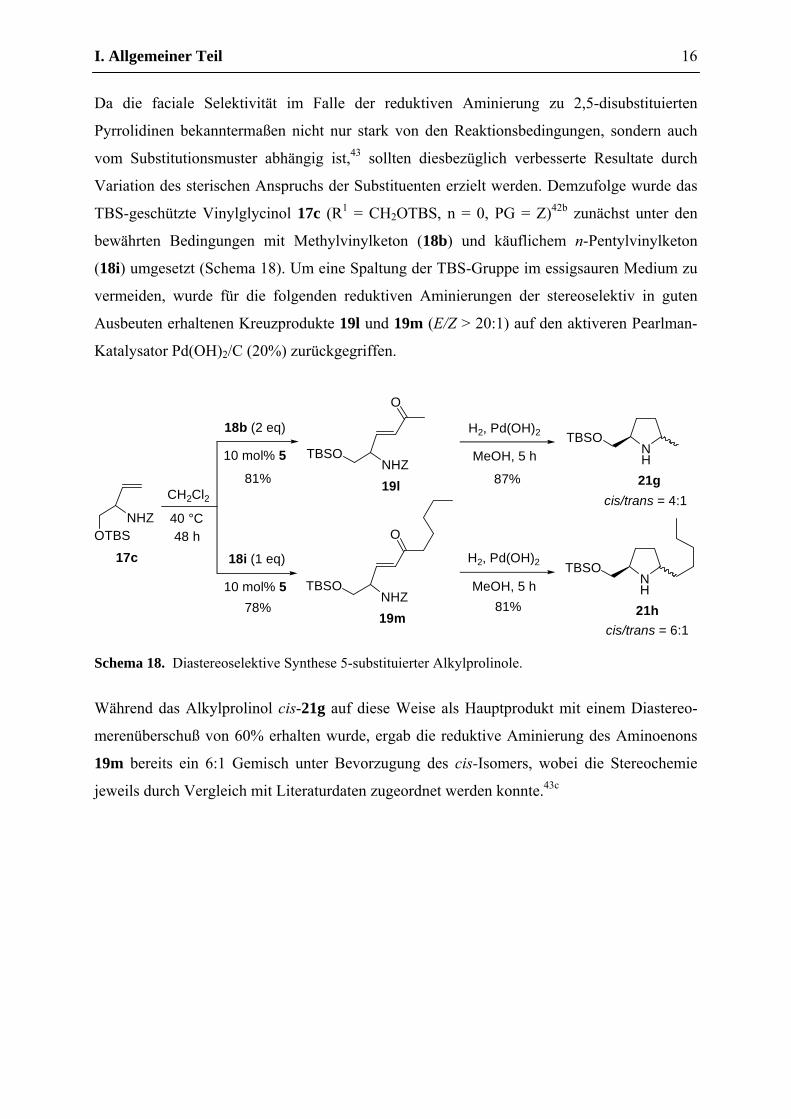
Die reduktiven Cyclisierungen der auf diese Weise aus dem literaturbekannten Vinylglycinol
17b (R1 = CH2OBn, n = 0, PG = Z)42a mit Methylacrylat (18a) und Methylvinylketon (18b)
stereoselektiv erhaltenen Kreuzprodukte 19j und 19k (E/Z > 20:1) sollten anschließend
ebenfalls mit 5 mol% Pd/C (10%) unter Wasserstoff-Atmosphäre bei Raumtemperatur
durchgeführt werden (Schema 17).
17b
18a (2 eq)
CH2Cl240 °C
10 mol% 5
48 h
18b (2 eq)
10 mol% 5
H2, Pd/C, 5 h
MeOH/AcOH
72%
71%
77%
20b
BnONH
O
21f
BnONH
19jNHZ
BnO
OOMe
19kNHZ
BnO
O
cis/trans = 3:4
NHZOBn
1) H2, Pd/C, MeOH, 1 h
2) 65 °C, 5 h91%
Schema 17. Synthese 5-substituierter Prolinole.
Während die Synthese des Pyrrolidinons 20b in Analogie zum Oxopipecolat 20a in sehr guter
Ausbeute gelang, blieb die Cyclisierung des Aminoenons 19k unter diesen Bedingungen auf
der Stufe des Imins stehen. Vollständige Reduktion konnte erst in Gegenwart von Essigsäure
erreicht werden und lieferte ein 3:4 Diastereomerengemisch (13C-NMR) zu Ungunsten des
literaturbekannten Isomers cis-21f.40a
I. Allgemeiner Teil
16
Da die faciale Selektivität im Falle der reduktiven Aminierung zu 2,5-disubstituierten
Pyrrolidinen bekanntermaßen nicht nur stark von den Reaktionsbedingungen, sondern auch
vom Substitutionsmuster abhängig ist,43 sollten diesbezüglich verbesserte Resultate durch
Variation des sterischen Anspruchs der Substituenten erzielt werden. Demzufolge wurde das
TBS-geschützte Vinylglycinol 17c (R1 = CH2OTBS, n = 0, PG = Z)42b zunächst unter den
bewährten Bedingungen mit Methylvinylketon (18b) und käuflichem n-Pentylvinylketon
(18i) umgesetzt (Schema 18). Um eine Spaltung der TBS-Gruppe im essigsauren Medium zu
vermeiden, wurde für die folgenden reduktiven Aminierungen der stereoselektiv in guten
Ausbeuten erhaltenen Kreuzprodukte 19l und 19m (E/Z > 20:1) auf den aktiveren Pearlman-
Katalysator Pd(OH)2/C (20%) zurückgegriffen.
17c
18b (2 eq)
CH2Cl240 °C
10 mol% 5
48 h
18i (1 eq)
10 mol% 5
H2, Pd(OH)2
MeOH, 5 h
H2, Pd(OH)2
MeOH, 5 h81%
87%81%
78%
21g
TBSONH
21h
TBSONH
19lNHZ
TBSO
O
19mNHZ
TBSO
O
cis/trans = 4:1
cis/trans = 6:1
NHZOTBS
Schema 18. Diastereoselektive Synthese 5-substituierter Alkylprolinole.
Während das Alkylprolinol cis-21g auf diese Weise als Hauptprodukt mit einem Diastereo-
merenüberschuß von 60% erhalten wurde, ergab die reduktive Aminierung des Aminoenons
19m bereits ein 6:1 Gemisch unter Bevorzugung des cis-Isomers, wobei die Stereochemie
jeweils durch Vergleich mit Literaturdaten zugeordnet werden konnte.43c

I. Allgemeiner Teil
17
2.4. Synthese von (±)-Indolizidin 209D
Mit mittlerweile mehr als 800 Vertretern aus über 20 strukturellen Klassen haben sich die
Häute von Amphibien als eine scheinbar unerschöpfliche Quelle von mono-, bi- und
tricyclischen Alkaloiden mit interessanten biologischen Eigenschaften erwiesen.44 Das
aufgrund der Vielzahl dieser zumeist mit der Nahrung aufgenommenen Substanzen eigens
eingeführte Nomenklatursystem setzt sich aus der jeweiligen Molekülmasse und
kennzeichnenden Buchstaben zusammen, wobei Konfigurationsisomere durch die Zusätze
cis-, trans-, epi- und iso- unterschieden werden (Abbildung 5).45
NH
HO
NH
trans-219A
NH
OH
cis-241D
H
H
HTX 283A
N
PTX 209F
OH
Abbildung 5. Ausgewählte Amphibien-Alkaloide.
Die in großer Zahl aus neotropischen Fröschen der Gattung Dendrobatidae isolierten,
lipophilen 3,5- und 5,8-disubstituierten Indolizidine stellen eine pharmakologisch interessante
Unterklasse der bicyclischen Alkaloide dar, da sie als atypische Mediatoren neuromuskulärer
Transmission fungieren (Abbildung 6).46
5
N
8
Indolizidin 207A
5
N3
Indolizidin 223AB
N
Indolizidin 209D (34) Abbildung 6. Indolizidine aus Fröschen der Gattung Dendrobatidae.
Als Ursache für diese potente biologische Aktivität konnte die Interaktion des basischen
Indolizidin-Gerüstes mit dem nikotinischen Acetylcholin-Rezeptor (nAChR) verantwortlich
gemacht werden,47 für die sich der Alkylsubstituent an C-3 jedoch als nicht essentiell
erwiesen hat.48a Folglich ist die Synthese des einfacheren monosubstituierten Indolizidins
209D (34) bis heute von besonderem Interesse geblieben,48 da es zumal in nur äußerst
geringen Mengen aus lediglich einer einzigen Dendrobatidae-Spezies isoliert werden
konnte.49

I. Allgemeiner Teil
18
In einer kürzlich publizierten Synthese von Yamamoto et al. wurde das Indolizidin-Gerüst in
wenigen Schritten aus dem käuflichen Aldehyd 35 erhalten, wobei als Schlüsselschritt eine
Pd(0)/PhCO2H-katalysierte hochdiastereoselektive Hydroaminierung des Alkinylamins 36
Verwendung fand (Schema 19).48g
74%
H2, Pd/C
85%
Pd(PPh3)4, PhCO2HN
37
HN
36NEt3, Dioxan, 100 °C MeOH
34
35
NO Boc 32%
Schema 19. Synthese von (–)-Indolizidin 209D nach Yamamoto.
Abschließende katalytische Hydrierung des Cyclisierungsprodukts 37 ergab Indolizidin 209D
in 7 Stufen mit einer Gesamtausbeute von 20%.
In einer der wenigen Arbeiten, die nicht von enantiomerenreinen Aminosäure-Derivaten
ausgehen, konnte erstmals auch das Prinzip der doppelten reduktiven Aminierung erfolgreich
zum Aufbau des Indolizidin-Gerüstes genutzt werden (Schema 20).48e
NHZ
O
NHZ
41
(R)-40
1) 9-BBN-H, THF
57%
H2, Pd/C
MeOH, 10 h
63%
O
2) 2-Iodfuran, Pd(PPh3)4, AsPh3, CsCO3, DMF/H2O
3) NBS, Pyridin, THF/Aceton
OHO
(S)-3938
B(OMe)3, CH2Cl2, 0 °C
70%
1) MsCl, NEt3, CH2Cl2
3) LAH, THF
82%
AllylSnBu3, (R)-BINOL-Ti(IV) 2) NaN3, HMPA, 40 °C
4) ZCl, K2CO3, THF
N
34 Schema 20. Totalsynthese von (–)-Indolizidin 209D nach Kim.
Allerdings lieferte die von Kim et al. durchgeführte Suzuki-Kreuzkupplung des aus Heptanal
enantiomerenrein erhaltenen Carbamats (R)-40 mit 2-Furyliodid gefolgt von oxidativer
Spaltung des Furanringes den als direkten Cyclisierungsvorläufer dienenden Ketoaldehyd 41
in nur mäßiger Ausbeute. Auch die abschließende doppelte reduktive Aminierung mittels
katalytischer Hydrierung erwies sich mit 63% Ausbeute als nur wenig effizient, so dass die
Eleganz dieses Konzepts erheblich geschmälert wurde.

I. Allgemeiner Teil
19
2.4.1. Retrosynthetische Analyse
Mit der bereits erfolgreich auf die Synthese des bicyclischen Pipecolats 22b angewandten
Sequenz aus CM und doppelter reduktiver Aminierung würde sich das Indolizidin-Gerüst
ausgehend vom gleichen Homoallylamin-Intermediat 40 in lediglich zwei Stufen effizient
aufbauen lassen (Schema 21).
NHZ
O
NHZCM
42 40
O
O
(±)-34
33
N
Schema 21. Retrosynthese von (±)-Indolizidin 209D.
Dabei sollte besonders die vergleichsweise einfach auszuführende CM als Schlüsselschritt zur
Darstellung des alternativen Cyclisierungsvorläufers 42 dem aufwendigen zweistufigen
Verfahren von Kim et al. überlegen sein.
2.4.2. Synthese
Wie erwartet, ergab die CM des nach Kim et al. aus racemischem 1-Decen-4-ol (39)50
synthetisierten Carbamats 40 mit einem Äquivalent des Enons 33 unter den bewährten
Bedingungen (5 mol% 5, CH2Cl2, 40 °C, 16 h) den gewünschten Cyclisierungsvorläufers 42
stereoselektiv (E/Z > 20:1) in guter Ausbeute (Schema 22).
NHZ
O
42
O
O(±)-34
CH2Cl2, 40 °C, 16 h
33 (1 eq), 5 mol% 5
78%
H2, Pd/C
MeOH/HCl, 16 h
88%NHZ
40 Schema 22. Synthese von (±)-Indolizidin 209D.
Abschließende reduktive Aminierung des Aminoenons 42 unter den bewährten Bedingungen
lieferte (±)-Indolizidin 209D diastereomerenrein in einer Ausbeute von 88%, was die
beschriebene konvergente Sequenz ausgehend vom enantiomerenreinen Intermediat (R)-40 zu
einem der effizientesten und kürzesten enantioselektiven Zugänge zu (–)-Indolizidin 209D
machen sollte.

I. Allgemeiner Teil
20
2.5. Synthese des Piperidin-Alkaloids (–)-Pinidinol
Nordamerikanische Nadelgewächse der Gattungen Picea und Pinus enthalten neben den
üblichen Terpenen, Flavonoiden, Stilbenen und Tanninen auch eine Reihe 2,6-disubstituierter
Piperidin-Alkaloide.51 Das aufgrund seiner 1,3-Aminoalkohol-Einheit strukturell mit den
Sedum52a und Lobelia52b Alkaloiden verwandte (–)-Pinidinol (43) wurde hauptsächlich aus der
Fichte Picea engelmannii isoliert53 und hat sich als fraßhemmend auf die Larven der in den
USA und Kanada heimischen Motte Choristoneura fumiferana erwiesen (Abbildung 7).54
NH
(−)-Pinidinol (43)
N
(+)-Sedamin (−)-Lobelin
OH
Ph NPh
OOH OH
Ph
Abbildung 7. Piperidin-Alkaloide mit 1,3-Aminoalkohol-Einheit.
Die ersten beiden enantioselektiven Totalsynthesen sind in Schema 22 kurz zusammengefasst.
In der von Takahata et al. publizierten Synthese55a wurden die Stereozentren des chiralen
Cyclisierungsvorläufers 44 durch zweifache asymmetrische Dihydroxylierung ausgehend von
1,6-Heptadien aufgebaut (Schema 23, Gleichung 1). Das durch doppelte Alkylierung von
Benzylamin aus dem entsprechenden Ditosylat erhaltene Piperidin 45 konnte nach Um-
schützung durch eine dritte asymmetrische Dihydroxylierung und anschließende regio-
selektive Desoxygenierung in den Naturstoff überführt werden. Die aufgrund der hohen
Stufenzahl sowie den meist nur moderaten Enantio- und Diastereoselektivitäten äußerst
geringe Gesamtausbeute von 1% konnte in der folgenden Arbeit von Molander et al. bereits
erheblich verbessert werden (Schema 23, Gleichung 2).55b
N
OHOMOM
Bn
431) H3O+
2) TsCl3) BnNH2
OTBDPS 9 mol%ONH2
4445
46
NH47
OTBDPS
Cp*2NdCH(TMS)2
KOH41%
90%
31% 10%
66%
42%
( )31)
( )32)
Schema 23. (–)-Pinidinol-Synthesen nach Takahata und Molander.

I. Allgemeiner Teil
21
Der Schlüsselschritt dieser Synthese ist die von einem sterisch anspruchsvollen Neodym-
Komplex katalysierte, hoch diastereoselektive Hydroaminierung (cis/trans > 100:1) des aus
(R)-Propylenoxid erhaltenen Alkenylamins 46. Abschließende Entfernung der TBDPS-
Schutzgruppe ergab (–)-Pinidinol in 25 % Gesamtausbeute über insgesamt 8 Stufen.
Im Zuge einer Studie zur stereoselektiven Reduktion von β-Enaminonen konnte das Epi-
pinidinol 50 diastereoselektiv in zwei Schritten aus dem homochiralen Lactimether 48
erhalten werden (Schema 24).55c
N
48
NH50
OH
Ni(acac)2,60%
OMe
O O
∆NH
49 OMeOH
H2, Ra-Ni43
Schema 24. Synthese von (–)-Pinidinol nach Lhommet.
Die erforderliche Inversion der Hydroxygruppe lieferte (–)-Pinidinol nach 4 zusätzlichen
Stufen in enantiomerenreiner Form, wobei auf eine Angabe von Ausbeuten verzichtet wurde.
2.5.1. Retrosynthetische Analyse
Entsprechend dem allgemeinen Synthesekonzept wurde die Zielstruktur 43 auf das
Aminoenon 51 und damit auf die beiden CM-Partner 52 und 54 zurückgeführt, zu deren
Darstellung auf die beiden kommerziell erhältlichen Substrate 53 und 55 zurückgegriffen
werden könnte (Schema 25).
NHZ
O
CM
51
52+
O
54
43
OR
OR
MeO
O
55
OH
NHZ53OH
Schema 25. Retrosynthese von (–)-Pinidinol.
Obgleich die Synthese des Enantiomers von 52 via dem entsprechenden 4-Azido-1-penten
bereits literaturbeschrieben ist,56 sollten die mit dessen hoher Flüchtigkeit verbundenen
Probleme durch eine alternative Mitsunobu-Reaktion von 53 mit Phthalimid (PhthNH) und
nachfolgende Eintopf-Umschützung umgangen werden.

I. Allgemeiner Teil
22
2.5.2. Synthese
Die Synthese der CM-Vorläufer 52 und 54 ist in Schema 26 dargestellt. Wie erwartet ergab
die Mitsunobu-Reaktion von (S)-4-Penten-2-ol (53) das Phthalimid 56 in guter Ausbeute.
Die geplante Eintopf-Umschützung zum Benzylcarbamat mittels einer Sequenz aus NaBH4-
Reduktion, Spaltung des resultierenden o-Hydroxymethylbenzamids57 und Umsetzung mit
ZCl gelang problemlos und lieferte das gewünschte Homoallylamin 52 in einer Gesamtaus-
beute von 81%, dessen physikalische Daten {[α]D20 = +13.5 (c = 1.0, CHCl3)} gut mit denen
des Enantiomers übereinstimmten {[α]D20 = –13 (c = 1.09, CHCl3)}.56
N
O
57a: R = H
OR
53PhthNH, DEAD, PPh3
THF, 0 °C, 2 h
1) NaBH4, iPrOH/H2O, 16 h
2) HCl, 80 °C, 2 h
81%
3) ZCl, K2CO3, 16 h
55MeO-NHMe.HCl, iPrMgCl
THF, -30 °C−RT, 80% OMe
57b: R = TBSTBSCl, Imidazol,DMF, 16 h, 92%
THF, 0 °C, 2 h
C2H3MgBr O
54a: R = H (52%)
OR
54b: R = TBS (85%)
82% 56NPhth
52NHZ
Schema 26. Synthese der CM-Partner 52 und 54.
Reaktion des literaturbekannten Weinreb-Amids 57a58 mit Vinylmagnesiumbromid in THF
ergab den Kupplungspartner 54a in einer nur mäßigen Ausbeute von 52%, die jedoch durch
vorherige Einführung einer TBS-Schutzgruppe deutlich verbessert werden konnte.
Zur Ermittlung der optimalen Bedingungen für die CM wurde das Amin 52 zunächst mit
stöchiometrischen Mengen der Enone 54a und 54b unter den bewährten Bedingungen
(5 mol% 5, CH2Cl2, 40 °C, 16 h) umgesetzt (Schema 27). Während mit dem TBS-geschützten
Hydroxyenon 54b auf diese Weise ein kompletter Umsatz zum Aminoenon 51b erfolgte
(E/Z > 20:1), wurde in Gegenwart des freien Alkohols 54a eine verstärkte Dimerisierung der
Homoallyamin-Komponente beobachtet. Durch Einsatz eines leichten Überschusses an Enon
(1.5 Äquivalente) konnte das gewünschte Aminoenon 51a schließlich in einer Ausbeute von
86% isoliert werden (E/Z > 20:1). Die abschließenden reduktiven Aminierungen zu 43
erfolgten problemlos mit jeweils 5 mol% Pd/C (10%) unter Wasserstoff-Atmosphäre bei
Raumtemperatur in Methanol, wobei die Entfernung der TBS-Gruppe im Falle von 51b durch
anschließende saure Hydrolyse des Rohprodukts 58 gelang.

I. Allgemeiner Teil
23
52CH2Cl2, 40 °C
5 mol% 5
16 h
54a (1−1.5 eq)
54b (1 eq)
58
H2, Pd/C
MeOH, 3 h
H2, Pd/C
MeOH, 3 h97%
NHZ
O
51b
OTBS
NHZ
O
51a
OH
65−86%
NH
OTBS
quant.
43
NH
OH
HCl/THF89%
80%
Schema 27. Synthese von (–)-Pinidinol.
In beiden Fällen konnte der Naturstoff diastereoselektiv in hoher Ausbeute und ohne
säulenchromatographische Reinigung als farbloser Feststoff erhalten werden, dessen
spektroskopische und physikalische Daten {[α]D20 = –14 (c = 0.83, CHCl3)} gut mit der
Literatur übereinstimmten {[α]D26 = –15 (c = 0.55, CHCl3)}.55a
Mit einer längsten linearen Sequenz von 6 beziehungsweise 7 Stufen und einer Gesamt-
ausbeute von 55 beziehungsweise 47% ausgehend von käuflichem (S)-4-Penten-2-ol stellt die
beschriebene konvergente Synthese den bisher kürzesten und weitaus effizientesten Zugang
zu (–)-Pinidinol dar.

I. Allgemeiner Teil
24
2.6. Synthese des Piperidin-Alkaloids (–)-Nupharamin
Seerosengewächse (Nymphaeaceae) der Gattung Nuphar haben sich als Quelle einer großen
Klasse seltener Terpen-Alkaloide mit vielfältigen biologischen Eigenschaften erwiesen.59
Gemeinsames Merkmal dieser sogenannten Nuphar Alkaloide sind eine 3-Furyl Gruppe und
fast immer ein bicyclisches Chinolizidin-Grundgerüst (Abbildung 8).
N
O
(−)-Nupharidin
ON
O
(−)-Nupharolutin
OH
N
O
5-(3-Furyl)-8-methyl- octahydroindolizin
N
O
(+)-Nuphacristin
HO
CHO
Abbildung 8. Ausgewählte Nuphar Alkaloide.
Eine interessante Ausnahme stellt ein 5-(3-Furyl)-8-methyloctahydroindolizin dar, welches in
Spuren (0,0002%) im Duftdrüsensekret (Castoreum) des kanadischen Bibers (Castor fiber)
detektiert werden konnte.60
Neben (–)-Nuphamin und (–)-Anhydronupharamin wurde (–)-Nupharamin (59) als eines von
wenigen sesquiterpenoiden Piperidin-Alkaloiden aus dem getrockneten Rhizom der gelben
Wasserlilie Nuphar japonica isoliert,61 welchem in der asiatischen Volksmedizin schmerz-
lindernde Wirkung zugeschrieben wird (Abbildung 9).
NHOH O
(−)-Nupharamin (59)
NH
O
(−)-Nuphamin
NH
O
(−)-Anhydronupharamin
OH
Abbildung 9. Piperidin-Alkaloide aus Nuphar japonica.
Obgleich bislang keine konkrete biologische Aktivität bekannt ist, hat sein außergewöhn-
liches, trisubstituiertes Piperidin-Gerüst einige synthetische Aufmerksamkeit erregt.62 Im
Schlüsselschritt einer früheren Synthese von Kibayashi et al. wurde die aus (R)-Citronellol
erhaltene Hydroxamsäure 60 mittels einer Nitroso-Diels-Alder-Reaktion in ein cis/trans-
Gemisch des Oxazinolactams 61 überführt (Schema 28, Gleichung 1).62c Ausgehend vom
bevorzugt gebildeten trans-Addukt konnte (–)-Nupharamin schließlich in einer Gesamt-
ausbeute von 7% erhalten werden.

I. Allgemeiner Teil
25
HN O 597%
60
OHN O
61
OOH
(R)-Citronellolcis/trans = 1:2
CHCl3, 0 °C
88%
Pr4NIO4
N
ONH
Ar
NOMe
TMS
O
+1) ZnCl2, THF, -78°C
5928%
OTMS
2) NaHCO3
3) SiO2
51%, > 99% ee64
6362
1)
2)
OH
Schema 28. (–)-Nupharamin-Synthesen nach Kibayashi und Barluenga.
In der zuletzt von Barluenga et al. publizierten Synthese,62e lieferte eine asymmetrische Diels-
Alder-Reaktion des Diens 62 mit dem Aldimin 63 das enantiomerenreine Piperidinon 64,
welches sich anschließend in einer 12-stufigen Reaktionssequenz mit einer Gesamtausbeute
von 28% in den Naturstoff überführen ließ (Schema 28, Gleichung 2).
2.6.1. Retrosynthetische Analyse
Gemäß der erfolgreich auf die enantiospezifische Synthese von (–)-Pinidinol angewandten
Synthesestrategie, wurde das trisubstituierte Piperidin-Gerüst von (–)-Nupharamin (59) auf
das Aminoenon 65 zurückgeführt, welches aus dem enantiomerenreinen Homoallylamin 67
durch eine CM mit 3-Furylvinylketon (66) zugänglich wäre (Schema 29).
NHZ
O
CM
65
59O
OH
O
66 O
NHZ
67OH 68
HO
Schema 29. Retrosynthese von (–)-Nupharamin.
Als Quelle für die benötigte stereochemische Information könnte dabei ausschließlich auf
kommerziell erhältliches (–)-Isopinocampheol (68) zurückgegriffen werden, das sich nach
Thermolyse unter regioselektiver [2+2]-Cycloreversion des Cyclobutanringes in wenigen
Schritten in den gewünschten Metathesevorläufer 67 überführen lassen sollte.

I. Allgemeiner Teil
26
2.6.2. Synthese
Obgleich die thermische Fragmentierung des Cyclobutanringes von Pinanderivaten inklusive
(+)-Isopinocampheol beschrieben ist,63 bedurfte es zunächst einiger Modifikationen, um mit
der zur Verfügung stehenden Pyrolyse-Apparatur reproduzierbare und den Literaturangaben
entsprechende Ergebnisse zu erzielen. Schließlich ergab die Flash-Pyrolyse von (–)-Isopino-
campheol (68) bei 480 °C und 15 mbar in der Gasphase neben 50% reisoliertem Edukt auch
ein 20:1 Gemisch (GC-MS) der beiden acyclischen Dienole 69a und 69b, wobei sich das
unerwünschte Konstitutionsisomer 69b vorerst säulenchromatographisch nicht abtrennen ließ
(Schema 30).
OH
69a
OH
69b
+
23%20 : 1
480 °C, 15 mbar
50% Umsatz
68HO
Schema 30. Flash-Pyrolyse von (–)-Isopinocampheol.
Als Entschädigung für die geringe Ausbeute von 23% sei hier die hohe Verfügbarkeit an
enantiomerenreinem Startmaterial, sowie dessen einstufige Transformation in einen äußerst
vorteilhaft substituierten und korrekt konfigurierten Vorläufer angeführt.
Die ursprünglich zur Synthese des Aminoalkohols 67 anvisierten Strategien sind in Schema
31 dargestellt. Eine nach chemoselektiver Epoxidierung des Dienolgemisches 69a/b geplante
regioselektive Öffnung des Oxiranringes gelang erst bei erhöhter Temperatur unter
Verwendung von LAH und lieferte das Diol 70 konstitutionsisomerenrein nach säulen-
chromatographischer Reinigung.
69a/b
1) MsCl, Pyridin, 16 h
1) mCPBA, CH2Cl2, 1 h
88%
N3
72
2) NaN3, DMF, 50 °C, 60 h
OH
70OH
2) LAH, THF, 65 °C, 4 h
90%
1) mCPBA, CH2Cl2
2) LAH, THF, 65 °C
1) MsCl, Pyridin, 16 h
2) NaN3, DMF, 50 °C
NH2
73OH
N3
71OH
Schema 31. Strategien zur Synthese des Aminoalkohols 67.

I. Allgemeiner Teil
27
Die anschließende Transformation zum gewünschten Azidoalkohol 71 durch selektive
Mesylierung der sekundären Hydroxyfunktion und Umsetzung mit NaN3 schlug jedoch fehl,
da bereits bei Raumtemperatur die konkurrierende intramolekulare Substitution durch die
tertiäre Hydroxyfunktion zum entsprechenden Tetrahydrofuran dominierte. Um dieses
Problem zu umgehen, sollte die Reihenfolge umgekehrt und der Stickstoff zuerst eingeführt
werden. Dementsprechend wurde das nach Mesylierung des Dienolgemisches 69a/b und
Substitution mit NaN3 erhaltene Azid 72 in Analogie zur Darstellung des Diols 70 epoxidiert
und mit LAH in refluxierendem THF reduziert. Wider Erwarten enthielt das unter diesen
drastischen Bedingungen erhaltene komplexe Produktgemisch jedoch nicht den gewünschten
Aminoalkohol 73, weshalb auch diese Syntheseroute verworfen werden mußte.
Da die regioselektive Hydratisierung von trisubstituierten Alkenen zu tertiären Alkoholen
auch durch Addition von Trifluoressigsäure (TFA) und anschließende Acetatspaltung
gelingt,64 wurde zunächst eine Lösung des Azids 72 portionsweise mit TFA versetzt.
Leider erfolgte die Zersetzung der Azidgruppe dabei stets schneller als die erwünschte
Funktionalisierung der Doppelbindung (Schema 32).
N3
OCOCF3
73
74
TFA in CH2Cl2
0 °CN3
72 Schema 32. Versuch der Hydratisierung von 72 durch Addition von TFA.
Zur Anwendung dieser Strategie war es daher erforderlich, die Hydroxygruppe des Dienols
69a unter Inversion in eine säurestabilere Aminofunktion zu überführen, welche gleichzeitig
eine intramolekulare N-Alkylierung ausschließen würde. Als ideale einstufige Variante bot
sich hierzu die Einführung einer Phtalimidogruppe (NPhth) unter Mitsunobu-Bedingungen an,
die nach TFA-Addition außerdem eine simultane N,O-Entschützung unter basischen
Bedingungen ermöglichen würde. Leider führte die Mitsunobu-Reaktion des Dienolgemisches
69a/b mit Phthalimid, PPh3 und DEAD in THF unabhängig von den Reaktionsbedingungen
zu einer ausgeprägten Eliminierung (ca. 60%), was auf den hohen sterischen Anspruch der
intermediären Phosphoniumspezies zurückgeführt wurde. Während leicht verbesserte
Resultate mit MePPh2 oder Me2PPh als Phosphin-Komponente erzielt werden konnten, ließ
sich das gewünschte Phthalimid 75 alternativ auch aus dem Azid 72 durch Reduktion zum
Amin, Umsetzung mit Phthalsäureanhydrid (PhthO) und Cyclisierung des resultierenden
Phthalsäuremonoamids mit Carbonyldiimidazol (CDI) synthetisieren (Schema 33).65

I. Allgemeiner Teil
28
69a/bPhthNH, MePPh2, DEAD
50%
NPhth
75
THF, 0 °C, 1 h
1) 30% TFA in CHCl3, 16 h
2) MeNH2, EtOH, 55 °C, 24 h
NHZ
67OH
3) ZCl, NaHCO3, CHCl3, 16 h
79%
72
1) LAH, Et2O, 0 °C, 1 h2) PhthO, THF, 60 °C, 2 h
3) Im2CO, THF, 1 h68%
Schema 33. Synthese des Metathesevorläufers 67.
Säulenchromatographische Reinigung ergab das Phthalimid 75 in beiden Fällen konstitutions-
und diastereoisomerenrein mit einer Ausbeute von 50 beziehungsweise 60% ausgehend von
69a/b. Wie erwartet verlief die geplante Addition von TFA nunmehr problemlos und lieferte
den Metathesevorläufer 67 in einer Gesamtausbeute von 79% nach Aminolyse mit MeNH2
(50 Äquivalente) und Einführung der Z-Schutzgruppe.
Die Darstellung des CM-Partners 66 gelang durch Grignard-Reaktion von 3-Furfural (76) mit
Vinylmagnesiumbromid gefolgt von milder Oxidation des resultierenden Vinylcarbinols 77
mit einem Überschuß (10 Äquivalente) MnO2 in refluxierendem CH2Cl2 (Schema 34).
H
O
Et2O, 0 °C, 1 h
C2H3MgBr
O76 94%
OH
40 °C, 24 h
MnO2, CH2Cl2
O77 65%
O
O66
Schema 34. Synthese des CM-Partners 66.
Da sich das Enon 66 auch bei tiefen Temperaturen und unter Lichtausschluß nicht lange
lagern ließ, wurde es vorzugsweise frisch aus dem stabilen Allylalkohol 77 hergestellt.
Wie erste Testansätze zeigten, erfolgte die CM des Carbamats 67 mit dem Enon 66
unabhängig von den Reaktionsbedingungen (40–70 °C, 1–5 Äquivalente Enon) nur sehr
langsam, was auf den sterischen Einfluß der Methylgruppe in Allylposition zurückgeführt
wurde. Weil gleichzeitig aber auch keine Homodimerisierung der Amin-Komponente zu
beobachten war, konnte ein nahezu vollständiger Umsatz zum Aminoenon 65 (E/Z > 20:1)
schließlich mit 10 mol% 5 nach 72 h Reaktionszeit unter Verwendung eines leichten
Überschusses an Enon (1.2 Äquivalente) in siedendem CH2Cl2 erreicht werden (Schema 35).

I. Allgemeiner Teil
29
NHZ
O
65
67CH2Cl2, 40 °C, 72 h
66 (1.2 eq), 10 mol% 5
73%
H2, 2−5% Pd/C
verschiedeneLösungsmittel
O
OH
NHOH O59
Schema 35. Versuch der Synthese von (–)-Nupharamin mittels reduktiver Aminierung.
Im Zuge der abschließenden reduktiven Aminierung des Aminoenons 65 mußte jedoch
festgestellt werden, dass die Spaltung des Benzylcarbamats unabhängig von den Reaktions-
bedingungen stets langsamer als die Reduktion des Furylvinylketons erfolgte, wodurch eine
Bildung des Piperidinrings verhindert wurde.
Da sich die Hydrierung der nichtaromatischen Doppelbindung dabei als weitaus schnellste
Reaktion erwies, schien es sinnvoll, den Piperidinring alternativ durch eine diastereoselektive
Hydridreduktion des entsprechenden cyclischen Imins aufzubauen. Zu diesem Zwecke wurde
das Phthalimid 75 zunächst zum Boc-geschützten Aminoalkohol 78 umgesetzt, der sich unter
den bewährten Bedingungen in 74% Ausbeute zum ensprechenden Aminoenon 79 kuppeln
ließ (Schema 36).
75
1) 30% TFA in CHCl3, 16 h
2) MeNH2, EtOH, 55 °C, 24 h
NHBoc
78OH3) Boc2O, NaHCO3, CHCl3, 16 h
81%
NHBoc
O
79
CH2Cl2, 40 °C, 72 h
66 (1.2 eq), 10 mol% 5
74%
1) H2, 5% Pd/C, Aceton, 15 min
O
OH
NHOH O59
2) 10% TFA in CH2Cl2, 1 h
3) NaBH4, EtOH, 0 °C, 1 h
75% Schema 36. Synthese von (–)-Nupharamin.
Wie erwartet ergab die kurze Hydrierung von 79 in einem aprotischen Lösungsmittel das
entsprechende gesättigte Aminoketon, welches nach Spaltung der Boc-Gruppe und NaBH4-
Reduktion des resultierenden Imins mit 75% Gesamtausbeute in ein einziges Stereoisomer
überführt werden konnte. Die auffallend hohe Diastereoselektivität läßt sich dabei durch eine
stabile Konformation des Imin-Intermediats erklären, in der der sterisch anspruchsvolle
Hydroxyalkyl-Substituent quasi-äquatorial angeordnet ist und das Hydrid bevorzugt axial
angreift (Abbildung 10).66

I. Allgemeiner Teil
30
NO
OH
HN
OH
H HO
H
NaBH4
Abbildung 10. Hydridreduktion des Imin-Intermediats.
Säulenchromatographische Reinigung über basischem Aluminiumoxid ergab (–)-Nupharamin
ausgehend vom Dienolgemisch 69a/b folglich in 8 beziehungsweise 12 Stufen mit einer
Gesamtausbeute von 22 beziehungsweise 27%, wobei dessen spektroskopische und
physikalische Daten {[α]D20 = –38.7 (c = 0.75, CHCl3)} gut mit der Literatur übereinstimmten
{[α]D22 = –35.4 (CHCl3)}.61,67
Die beschriebene konvergente Sequenz stellt somit einen einfachen und effizienten
enantiospezifischen Zugang zu (–)-Nupharamin sowie auch eine formale Totalsynthese von
(–)-Anhydronupharamin dar, da sich die Dehydratisierung von (–)-Nupharamin bereits als
unproblematisch erwiesen hat (Schema 37).68
NHOH O
(−)-Nupharamin
NH
O
(−)-Anhydronupharamin
SOCl2, CHCl3
95%
Schema 37. Dehydratisierung von (–)-Nupharamin.
Während die einfache Wahl des Startmaterials zudem die Synthese beider Enantiomere
ermöglicht, sollte die Variation des CM-Partners auch die Einführung anderer Arylreste wie
beispielsweise einem 2-Furyl-Substituenten erlauben.69

I. Allgemeiner Teil
31
2.7. Studie zur Synthese 6-substituierter 4,5-Didehydropipecolate
Während in der Natur eher weniger verbreitet, stellt das auch als Piperidein oder
Tetrahydropyridin bezeichnete Didehydropiperidin-Gerüst wegen seiner vielfältigen
Funktionalisierbarkeit ein wichtiges Intermediat auf dem Weg zu höher substituierten
Piperidinen dar.70 Zur Synthese von Tetrahydropyridin-Intermediaten haben sich neben
sigmatropen Umlagerungen und Aza-Diels-Alder-Reaktionen besonders die RCM und die
RRM bewährt,71 wie hier am Beispiel der Synthese der beiden polyhydroxylierten
Glycosidase Inhibitoren (+)-Fagomin72a und (+)-Castanospermin72b gezeigt (Schema 38).
NAc
NAc
NBoc
NBoc
OTBDPSOTBDPSNH
(+)-Fagomin
OH
OHOH5 mol% 2
97%
OTBS
BnO
TBSO
OBn
H
N
(+)-Castanospermin
OHHO OH
HHO
10 mol% 3, C2H4
CH2Cl2, 40 °C
72%
CH2Cl2
Schema 38. Aufbau von Tetrahydropyridin-Intermediaten via Metathese.
Durch Kombination mit einer kationischen 6-endo-trig-Cyclisierung konnten Blechert und
Mix erstmals auch die CM zum Aufbau der Olefineinheit von 4,5-Didehydropipecolinsäure-
Derivaten nutzen. Hierbei wurden N-geschützte Allylglycinmethylester vom Typ 80 mit
3-Buten-2-ol gekuppelt und anschließend in einer BF3·Et2O-induzierten SN1-Reaktion
trans-selektiv cyclisiert (Schema 39).
NHCO2R
80
3 mol% 3
CH2Cl2, 45 °C
74−80%
MeO2C NHCO2R
8180−90%
MeO2C
OH
BF3.Et2O
NCO2R
82
MeO2C
cis/trans = 1:5
CH2Cl2
3-Buten-2-ol
Schema 39. Aufbau von 4,5-Didehydropipecolaten via CM und kationischer Cyclisierung.

I. Allgemeiner Teil
32
Ergänzend zu den Studien von Mix sollte nun erstmals auch eine alternative Palladium-
katalysierte 6-endo-trig-Cyclisierung zum diastereoselektiven Aufbau entsprechender 4,5-Di-
dehydropipecolate untersucht werden. Hierzu wurde das geeignet geschützte Allylglycin 8373
bei erhöhter Temperatur in Gegenwart von 5 mol% 5 mit zwei Äquivalenten (3-Buten-2-yl)-
methylcarbonat74 umgesetzt (Schema 40).
NHpNs
83
5 mol% 5, DCE
65%
MeO2C NHpNs
84 quant.
MeO2C
MeO2CO
1 mol% Pd(PPh3)4NpNs
85
MeO2CCH2Cl2 oder THFRT−60 °C, 30 min
70 °C, 16 h
OCO2Me
cis/trans = 1:1 Schema 40. Aufbau von 4,5-Didehydropipecolaten via CM und Pd-katalysierter Cyclisierung.
Anschließende Behandlung des neben erhöhten Mengen beider Homodimere erhaltenen
Kupplungsprodukts 84 mit 1 mol% Pd(PPh3)4 ergab jedoch unabhängig von den Reaktions-
bedingungen jeweils ein 1:1 Gemisch beider Diastereomere, aus dem sich das cis-Isomer
selektiv umkristallisieren ließ (Abbildung 11).
Abbildung 11. Kristallstruktur des Didehydropipecolats cis-85.
Abschließend sei erwähnt, dass sich auch bei Verwendung einer ortho-Nosyl-Schutzgruppe
keine Diastereoselektivität beobachten ließ, weshalb dieses Synthesekonzept verworfen und
nicht weiter verfolgt wurde.

I. Allgemeiner Teil
33
2.8. Zusammenfassung und Ausblick
Die neuartige Kombination aus CM und reduktiver Cyclisierung konnte zunächst erfolgreich
zum Aufbau mono- und bicyclischer Pipecolinsäure-Derivate sowie 5-substituierter Prolinole
genutzt werden und sollte somit einen allgemeinen, diastereoselektiven Zugang zur Natur-
stoffklasse der 2,6-disubstituierten Piperidine, 2,5-disubstituierten Pyrrolidine und der 3,5-
disubstituierten Indolizidine eröffnen.
Im Anschluß an die methodischen Arbeiten wurde die hocheffiziente Sequenz aus
Kreuzkupplung zwischen elektronisch neutralen und akzeptorsubstituierten Alkenen gefolgt
von reduktiver Aminierung auf die racemischen Synthese des Indolizidins 209D sowie die
enantiospezifische Totalsynthese der Piperidin-Alkaloide (–)-Pinidinol und (–)-Nupharamin
angewandt (Schema 41).
NHZ
40OH
39
NHZ
52
O
54a
OH
O
33 O
O
N
OH
53
(±)-Indolizidin 209D
NH
(−)-Pinidinol
OH
CM − red. Aminierung
4 Stufen
82% CM − red. Aminierung
4 Stufen
66%
69%
83%
NH
78
O
66NH
(−)-Nupharamin
CM − red. Aminierung
4 Stufen
40%
55%OH
O
OOH69a
BocOH
Schema 41. CM – reduktive Aminierung als Schlüsselschritt in der Naturstoffsynthese.
Während die Darstellung von (±)-Indolizidin 209D und (–)-Pinidinol jeweils in 6 Stufen
ausgehend von den Allylalkoholen 39 und 53 mit hervorragenden Gesamtausbeuten von 56%
beziehungsweise 55% gelang, wurde die Effizienz der Synthese von (–)-Nupharamin durch
das Scheitern der direkten reduktiven Aminierung leicht geschmälert. Eine alternative
mehrstufige Transformation lieferte (–)-Nupharamin schließlich in einer Gesamtausbeute von
22 beziehungsweise 27% ausgehend vom Allylalkohol 69a.

I. Allgemeiner Teil
34
Eine attraktive Erweiterung des allgemeinen Synthesekonzepts könnte darin bestehen, die
nach der CM mit hoher E-Selektivität erhaltene, elektronenarme Doppelbindung vor der
Cyclisierung stereoselektiv, beispielsweise durch eine asymmetrische Dihydroxylierung,
Epoxidierung oder Michael-Addition, zu funktionalisieren, um somit auch Zugang zu höher
substituierten N-Heterocyclen zu ermöglichen. Durch den Einsatz geminal disubstituierter
Olefine als CM-Partner würde sich, eine stereoselektive Funktionalisierung des resultierenden
trisubstituierten Alkens vorausgesetzt, die Bandbreite an zugänglichen Strukturen ebenfalls
erweitern lassen.
Im letzten Abschnitt wurde schließlich untersucht, ob die CM von geeignet geschützen
Homoallylaminen mit Allylcarbonaten gefolgt von einer Palladium-katalysierten 6-endo-trig-
Cyclisierung zum diastereoselektiven Aufbau von 4,5-Didehydropipecolinsäure-Derivaten
genutzt werden kann. Die Cyclisierung des Kupplungsprodukts 84 zeigte jedoch, dass in
diesem Fall keine Substratkontrolle vorliegt.

I. Allgemeiner Teil
35
3. Synthese von Makroliden
3.1. Motivation und Synthesekonzept
Als Makrolactone oder Makrolide werden mittlerweile mehr als 2000 natürlich vorkommende
Lactone mit einer Ringgröße zwischen 8 und 62 bezeichnet.75 Unter den Produzenten sind
neben Bakterien (Actinomyceten, Myxobakterien) mit über 900 Substanzen vor allem
Pflanzen, Pilze, Invertebraten, Algen und Insekten hervorzuheben, wobei sich auch
zunehmend marine Organismen als Quelle von Makroliden erweisen (Abbildung 12).
OO
OH
HO
HO
(−)-Macrolactin A
O
OHO
O OH
O
(−)-Octalactin A Abbildung 12. Ausgewählte Marine Makrolide.
Aufgrund ihrer außergewöhnlichen biologischen Eigenschaften und meist bemerkenswerten
strukturellen Komplexität ist die Totalsynthese von Makroliden seit ihren Anfängen mit
Woodward’s Erythromycin-Synthese im Jahre 1973 von unvermindert großem Interesse
geblieben,75 wobei insbesondere die Entwicklung effizienter Methoden zum Aufbau des
Makrocyclus vorangetrieben wurde. Unter den C-C-verknüpfenden Verfahren haben sich
hierzu neben intramolekularen Aldol-, Radikal- oder Wittig-Reaktionen auch die Stille-
Kupplung und vor allem die RCM etabliert,3b,7 wie am Beispiel der Synthese des Mykotoxins
(–)-Zearalenon nach Nicolaou et al.76a bzw. Fürstner et al.76b gezeigt (Schema 42).
(−)-Zearalenon
2) H3O+
1) Stille (54%) 1) RCM (91%)O
O
O
HO OH
O
O
RO OR
O O
O I
O
RO OR
SnR3
O
2) H3O+
Schema 42. Synthese von Zearalenon: Stille-Kupplung versus RCM.

I. Allgemeiner Teil
36
Die bei weitem am häufigsten angewandte Methode zum Aufbau von Makrolactonen ist
jedoch die von Corey bereits in den siebziger Jahren erstmals angewandte intramolekulare
Cyclisierung von Hydroxycarbonsäuren geblieben, von der mittlerweile zahlreiche effektive
Varianten zur Verfügung stehen (Schema 43).77
Corey: (2-PyS)2, PPh3
Mukaiyama: N-Methyl-2-chlorpyridiniumiodid, NEt3
Mitsunobu: DEAD, PPh3
Yamaguchi: 2,4,6-Cl3PhCOCl, NEt3, DMAPO
O
ROHOH
O
R
Schema 43. Klassische Verfahren zur Lactonisierung von Hydroxycarbonsäuren.
Eine interessante Alternative zu diesen klassischen Lactonisierungsmethoden stellt die
intramolekulare Addition eines Nucleophils an in situ generierte Ketene dar. So konnten
Quinkert et al. beispielsweise die photochemische Spaltung von o-Chinolacetaten zu Dien-
ketenen erfolgreich zum Aufbau von Makromonoliden des Typs 87 nutzen (Schema 44).78
OOH
( )n ( )n ( )n
86 87
OAcO
RHO
O •R
OAc
OR
OAc
hν
DABCO
Schema 44. Photolactonisierung nach Quinkert.
Ein weiteres, seit langem bekanntes Verfahren ist die thermisch induzierte [4+2]-Cyclo-
reversion von Dioxolenonen des Typs 88,79 bei der das resultierende Acylketen unter hoher
Verdünnung intramolekular zu einem β-Ketolacton oder -lactam abreagiert (Schema 45).
X
O
O
∆XH OO
O
XH− Aceton
O
O•
X = O, NH( )n ( )n ( )n
R RR
88 89 Schema 45. [4+2]-Cycloreversion von Dioxolenonen.
Obwohl äußerst mild und effizient, sind bis heute nur wenige Anwendungen dieser Methode
in der Naturstoffsynthese bekannt geworden.80 Als ein jüngeres Beispiel sei hier lediglich der
Aufbau der 14-gliedrigen Untereinheit des marinen Antitumor-Makrolids Callipeltosid A
nach Trost et al. genannt (Schema 46).80b

I. Allgemeiner Teil
37
Toluol (0.5 mM)
OOTBS
OTBS
MeO O
O
OH
OTBS
OTBS
MeO
OO
O110 °C, 1 h
Callipeltosid A
82%
Schema 46. Synthese von Callipeltosid A nach Trost.
Aufgrund ihres großen Potentials waren wir daran interessiert, diese generelle Strategie mit
der hohen Effizienz der CM zu kombinieren. Dazu sollten Hydroxyalkene vom Typ 90
zunächst mit dem konjugierten Vinyldioxolenon 91 gekuppelt und anschließend thermisch
zu den entsprechenden γ,δ-ungesättigten-β-Ketolactonen cyclisiert werden (Schema 47).
O
O
OR
CM
OH
R
OO
O
OH
R
90 92 93
− Aceton
OO
O
91
+( )n ( )n ( )n
∆[Ru]
Schema 47. Allgemeines Synthesekonzept.
Zusätzliche Funktionalisierung der CM-generierten Doppelbindung sowie Transformation der
β-Ketoeinheit würde schließlich auch Zugang zu höher substituierten Derivaten ermöglichen,
was in dieser Arbeit am Beispiel der Totalsynthese des Makrolid-Antibiotikums (–)-A26771B
und des Flechten-Makrolids (+)-Aspicilin demonstriert werden konnte.
An dieser Stelle sei erwähnt, dass aufgrund der Probleme bezüglich Chemo- und
Diastereoselektivität bislang nur äußerst wenig Arbeit in die Metathese mit konjugierten
Dienonen investiert wurde. Anhand der selektiven CM eines Sorbinsäureesters konnten
Grubbs et al. jedoch kürzlich zeigen, dass die Reaktivität der einzelnen Doppelbindungen
durch sterische und elektronische Faktoren beeinflußt werden kann (Schema 48).81
Ph 5 mol% 3CO2Et+
Br
CO2Et
Br
PhCH2Cl2, 40 °C
68%
E/Z = 9:1
Schema 48. Beispiel für die selektive CM eines konjugierten Dienons.

I. Allgemeiner Teil
38
3.2. Synthese von γ,δ-ungesättigten-β-Ketolactonen
Der Erfolg einer intramolekularen Addition von Hydroxygruppen an in situ generierte
β-Acylketene unterliegt neben dem sterischen Anspruch des Nucleophils auch konformativen
Effekten und ist somit stark von Größe und Substitutionsmuster des zu bildenden β-Keto-
lactons abhängig. Während die Darstellung von 8-gliedrigen Lactonen aus Hydroxy-
alkyldioxolenonen des Typs 88 in sehr guten Ausbeuten gelang,82a schlug eine entsprechende
Cyclisierung zu gesättigten 10- und 11-Ringen auch unter Hochverdünnung fehl.82b Erst die
Thermolyse des potentiellen 12-Ringvorläufers 88d lieferte neben erheblichen Mengen an
Dimer schließlich auch das erwünschte monomere Lacton 89d (Schema 49).
Toluol, 100 °C
> 90%R
O
O
O
OH OO
O( )n ( )n
88b-d
88a
OH
O
O
O
R
89a
O
1)
2)Toluol, 110 °C
< 10-4 M
O
O
89d (n = 3): 50%89c (n = 2): 0%89b (n = 1): 0%
Schema 49. Einfluß der Ringgröße bei der Lactonisierung von Dioxolenonen.
Unter Berücksichtigung der die konformative Flexibilität zusätzlich einschränkenden, CM-
generierten trans-Doppelbindung schien es daher sinnvoll, das allgemeine Synthesekonzept
zunächst anhand der Synthese einfacher 14- bis 18-gliedriger Makrolactone zu testen.
3.2.1. Synthese der Metathesevorläufer
Als geeignete CM-Substrate wurden neben käuflichem 9-Decen-1-ol (90a) die langkettigen
Alkenole 90b-d favorisiert, da sie leicht aus kommerziell erhältlichen Substanzen zugänglich
sind (Schema 50). Während die Alkenole 90b83 und 90c102g nach Literaturvorschrift
synthetisiert werden konnten, ergab die direkte Reaktion von 11-Brom-1-undecanol mit
Allylgrignard in der Gegenwart von CuI auch erhebliche Mengen an Nebenprodukt. Eine
alternative Umsetzung des entsprechenden THP-Ethers 9684 mit Allylmagnesiumchlorid in
siedendem THF85 ergab schließlich das literaturbekannte Tetradecenol 90d86 in guter
Gesamtausbeute nach Entfernung der Schutzgruppe.

I. Allgemeiner Teil
39
942) Hg(OAc)2, THF/H2O 90b
OHO
95 90c
O
CuCN, THF, -78 °C
BrMg
Br
96
1) C3H5MgCl, THF, 65 °C, 2 h
90d
THPO HO2) pTsOH, MeOH, 1 h
1) MeOCH=PPh3, THF
45 %
88 %
71 %
OH
( )7
( )8
( )9
( )8
( )9
( )7
3) NaBH4, K2CO3, H2O
Schema 50. Synthese der CM-Vorläufer 90b-d.
Bezüglich der Synthese des CM-Partners 91 wurde die ursprünglich geplante Amino-
alkylierung des käuflichen Methyldioxolenons 97 mit nachfolgender Cope-Eliminierung
aufgrund der zu geringen Gesamtausbeute von 20% verworfen (Schema 51, Gleichung 1).
Als eine wesentlich effizientere Strategie, die gleichzeitig auch Zugang zu höher
substituierten Derivaten ermöglichen sollte, erwies sich hingegen die Acetonisierung des aus
3-Chlorpropionsäureethylester durch Kondensation mit tButylacetat erhältlichen β-Ketoesters
99 (Schema 51, Gleichung 2).87
O
Aceton, 16 hO
OtBu O
O
O
99 10076 %
Cl Cl NEt3, CH2Cl2, 3 h
85 %
Ac2O, H2SO4O
O
O
91
O
O
O
97
1) LDA, THF, -78 °C, 1 h
40 %
O
O
O
982) [Me2N=CH2]+Cl-, 16 h
mCPBA1)
2)
Me2N
CH2Cl2, 2 h
50 %
Schema 51. Strategien zur Synthese des Vinyldioxolenons 91.
Eine abschließende basische Chlorid-Eliminierung in Gegenwart von NEt3 ergab schließlich
das gewünschte Vinyldioxolenon 91 in einer akzeptablen Gesamtausbeute von 65%.

I. Allgemeiner Teil
40
3.2.2. Synthese
Die Ergebnisse der sequentiellen CM – Lactonisierung sind in Tabelle 2 zusammengefaßt.
Erfreulicherweise lieferte die Umsetzung der Hydroxyalkene 90a-d mit stöchiometrischen
Mengen des Dienons 91 unter standardisierten Bedingungen (5 mol% 5, CH2Cl2, 40 °C, 24 h)
die gewünschten Kupplungsprodukte 92a-d stereoselektiv (E/Z > 20:1) in guten Ausbeuten
neben geringen Mengen an homodimerisiertem Alkohol.
5, CH2Cl2,40 °C, 24 h
OH
R
OO
O90a-d
92a-d
O
O
OR
93a-d
+
101a-d
( )n ( )nO
O
O
O
O
O
R
R
n+2( )( )n+2
OO
O91 ∆
Alkohol n R eq 91 mol% 5 Ausbeute 92 Ausbeute 93 Ausbeute 101
90a 5 H 1 5 71% – 71%a
90b 7 H 1 5 75% 44%a / 78%b – c
90c 8 Me 1 5 78% 82%a –
90d 9 H 1 5 77% 85%a – a Methode A; b Methode B; c nicht isoliert
Tabelle 2. Ergebnisse der sequentiellen CM – Lactonisierung.
Die nachfolgenden Lactonisierungen der Hydroxyalkenyldioxolenone 92a-d wurden jeweils
im 0.1 mmol Maßstab durchgeführt und bestanden in einfachem Refluxieren einer stark
verdünnten (~ 10-4 M) n-Heptan-Lösung (Methode A). Obwohl die Verwendung von Toluol
aufgrund des höheren Siedepunkts zu deutlich kürzeren Reaktionszeiten führte (3 h), wurde
die säulenchromatographische Reinigung der Rohprodukte jedoch durch erhebliche Mengen
dessen thermischer Zersetzungsprodukte erschwert.
Während die Synthese von 14-gliedrigen Lactonen mittels RCM gewöhnlich einfach ist,88
konnte durch FAB-Massenspektrometrie belegt werden, dass das nach Thermolyse von
Dioxolenon 92a erhaltene Hauptprodukt nicht das erwartete Monomer 93a, sondern das
28-gliedrige Diolid 101a war. Ausgehend von Dioxolenon 92b wurden schließlich zwei
Lactone im Verhältnis von etwa 1:1 (1H-NMR) erhalten, von denen sich das unpolarere
(SiO2; 1% AcMe in CH2Cl2) als das literaturbekannte 16-gliedrige β-Ketolacton 93b erwies.89
Um dessen Bildung zu begünstigen, wurde die Makrocyclisierung mittels tropfenweiser

I. Allgemeiner Teil
41
Zugabe einer verdünnten Toluol-Lösung von 92b zu refluxierendem Toluol während eines
Zeitraums von 4 h durchgeführt (Methode B). Unter diesen Bedingungen (< 10-4 M) wurden
nur noch Spuren des dimeren Lactons 101b gebildet, wohingegen sich das monomere Lacton
93b in einer deutlich verbesserten Ausbeute von 78% isolieren ließ.
Interessanterweise ergab die folgende Thermolyse der Hydroxyalkenyldioxolenone 92c und
92d die entsprechenden Lactone 93c und 93d bereits mittels Methode A in hervorragenden
Ausbeuten, was auf die bei Cycloalkanen beobachtete abrupte Abnahme der Ringspannung
zwischen 16- und 17-Ring zurückzuführen sein könnte.
Abschließend sei gesagt, dass die beschriebene CM – Makrolactonisierungs-Sequenz im Falle
des 16-gliedrigen β-Ketolactons 93b der literaturbekannten Synthese via Cyclisierung der
entsprechenden aktivierten Hydroxycarbonsäure bezüglich ihrer Effizienz deutlich überlegen
ist (Schema 52).
OO
StBu
1032) CuOCOCF3, CH2Cl2
1) HF, MeCN O
O
O
93b
( )9 ( )7
(MeO)2POO
StBu
102
O
NaH, THF
OTBS
84%
O( )9
OTBS
34% Schema 52. Alternative Synthese des β-Ketolactons 93b.
So lieferte die direkte Cyclisierung des mittels Wittig-Reaktion stereoselektiv aus dem
Undecanal 102 gewonnenen Thioesters 103 in Gegenwart von Kupfer(I)-Trifluoracetat das
entsprechende monomere Lacton 93b in lediglich 34% Ausbeute nach Entfernung der
Schutzgruppe.89

I. Allgemeiner Teil
42
3.3. Synthese des Makrolid-Antibiotikums (–)-A26771B
Die trans-γ-Oxo-Crotonsäure-Einheit ist Bestandteil einiger makrocyclischer Lactone mit
interessanten biologischen Eigenschaften wie beispielsweise dem antifungalen 12-gliedrigen
(+)-Patulolid A,90a dem antibiotischen Dilacton (–)-Grahamimycin A90b oder dem antitumor-
aktiven Triolid Macrosphelid B (Abbildung 13).90c
Macrosphelid B
O
OO
O
O
OO
O
OH
O O
O
OO
(+)-Patulolid A (−)-Grahamimycin A
O
HOO
Abbildung 13. Ausgewählte α,β-ungesättigte-γ-Ketolactone.
Das 16-gliedrige, vom Pilz Penicillium turbatum (Westling) nach dem Muster eines
polyketidischen Sekundärmetaboliten synthetisierte91 Makrolid-Antibiotikum (–)-A26771B
(104) weist antimikrobielle Aktivität gegen gram-positive Bakterien, Mycoplasmen sowie
einige Pilze auf,92 und war daher Gegenstand zahlreicher racemischer93 und enantioselektiver
Totalsynthesen (Abbildung 14).94
(−)-A26771B (104)
O
OO
O
O
HO2CStaphylococcus aureus < 1.56Staphylococcus faecalis 25Mycoplasma gallisepticum 3.12Mycoplasma synoviae 12.5Erwinia amylovora < 0.78Pasteurella multocida 6.25Candida tropicalis 100Ceratocystis ulmi 6.25
Organismus MIC (µg/ml)
Abbildung 14. Antibiotikum (–)-A26771B und sein antimikrobielles Profil.
Die Synthese von γ-Oxo-Crotonsäure-Derivaten durch zweistufige Oxidation 2-substituierter
Furane ist weit verbreitet und hat sich in Kombination mit einer Lactonisierung auch zum
Aufbau von entsprechenden Makroliden bewährt.95 So konnte der von Kobayashi et al.94f
durch oxidative Spaltung aus dem Furylcarbinol 106 erhaltene Aldehyd 107 nach Weiter-
oxidation zur Carbonsäure durch Cyclisierung unter Yamaguchi-Bedingungen in guter
Ausbeute in den MOM-Ether 108 überführt werden (Schema 53).

I. Allgemeiner Teil
43
O
OO
OMOM
( )7
OHCOH
OOMOM
OH
OOMOM
1041) TFA, CH2Cl2
2) Succinanhydrid, DMAP, CH2Cl2
1) NBS, NaHCO3, AcMe/H2O, -15 °C
1) NaClO2, Isopren, tBuOH/H2O
Br OH
2) Pyridin, RT70%
21%
2) 2,4,6-Cl3C6H2COCl, NEt3, DMAP, Toluol, 70 °C
66% 63%
105106 107
108 Schema 53. Totalsynthese von (–)-A26771B nach Kobayashi.
Eine abschließende Acetalspaltung und Succinylierung der Hydroxyfunktion in Gegenwart
von DMAP ergab (–)-A26771B in einer Gesamtausbeute von 6% nach 12 Stufen ausgehend
vom käuflichen Bromnonanol 105.
In einer darauf folgenden formalen Totalsynthese von Chang et al.94g wurde die entsprechend
dem Kobayashi-Protokoll aus dem Furylcarbinol 110 in mäßiger Ausbeute erhaltene
Carbonsäure 111 zunächst mit (R)-Octen-7-en-2-ol verestert (Schema 54). Eine anschließende
RCM des resultierenden Triens 112 in Gegenwart von 12 mol% des Grubbs-Katalysators 2
gefolgt von chemoselektiver Hydrierung der RCM-generierten Doppelbindung ergab den
MOM-Ether 108 bereits nach 8 Stufen in einer Gesamtausbeute von 5% ausgehend von
2-Furfural (109).
O CHO
1) 12 mol% 2, CH2Cl2, 76%
2) H2, PtO2, EtOAc, 55%
O
OO
OMOM
OOMOM
OOMOM 1) NBS, NaHCO3, AcMe/H2O,
-15 °C dann Pyridin, RT
2) NaClO2, Isopren, tBuOH/H2O
55%39%
2,4,6-Cl3C6H2COCl, NEt3, DMAP, Toluol
52%
109110 111
112
O
OH
OH
O
OO
OMOM
108 Schema 54. Totalsynthese von (–)-A26771B nach Chang.
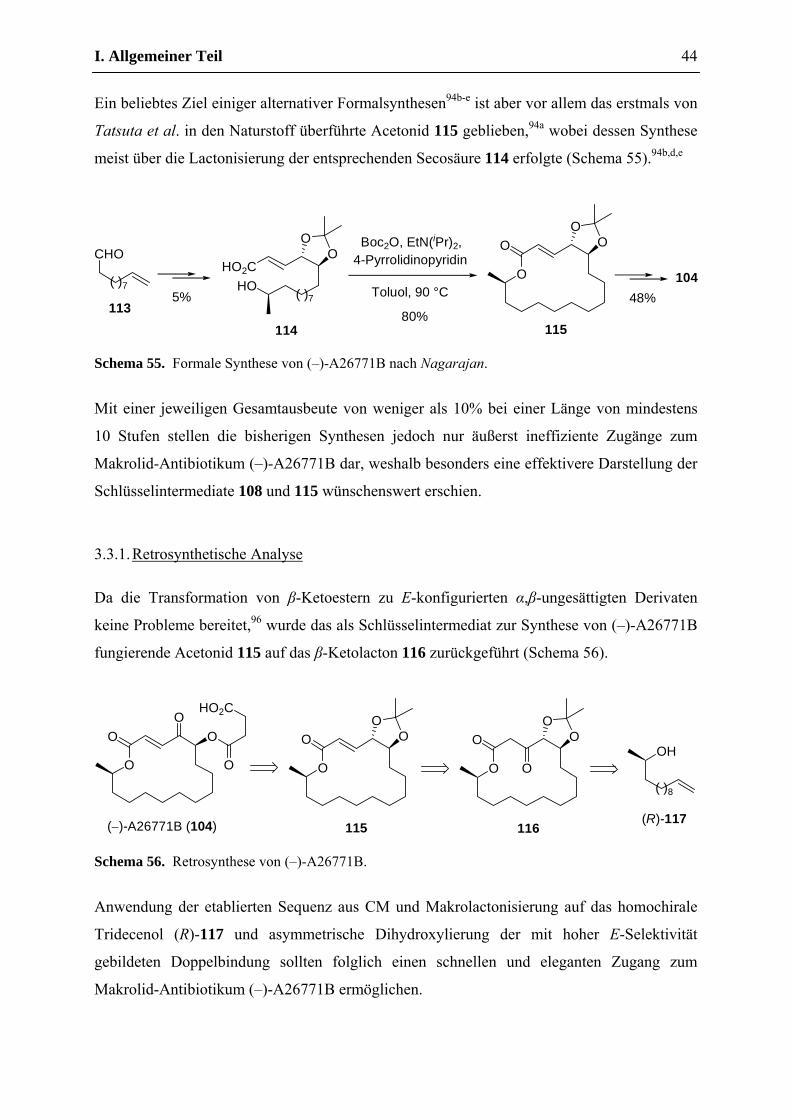
I. Allgemeiner Teil
44
Ein beliebtes Ziel einiger alternativer Formalsynthesen94b-e ist aber vor allem das erstmals von
Tatsuta et al. in den Naturstoff überführte Acetonid 115 geblieben,94a wobei dessen Synthese
meist über die Lactonisierung der entsprechenden Secosäure 114 erfolgte (Schema 55).94b,d,e
Boc2O, EtN(iPr)2, 4-Pyrrolidinopyridin
Toluol, 90 °C
80%5%
113
104O
O
115
OO
CHO
( )7( )7
HO2C
114
OO
HO48%
Schema 55. Formale Synthese von (–)-A26771B nach Nagarajan.
Mit einer jeweiligen Gesamtausbeute von weniger als 10% bei einer Länge von mindestens
10 Stufen stellen die bisherigen Synthesen jedoch nur äußerst ineffiziente Zugänge zum
Makrolid-Antibiotikum (–)-A26771B dar, weshalb besonders eine effektivere Darstellung der
Schlüsselintermediate 108 und 115 wünschenswert erschien.
3.3.1. Retrosynthetische Analyse
Da die Transformation von β-Ketoestern zu E-konfigurierten α,β-ungesättigten Derivaten
keine Probleme bereitet,96 wurde das als Schlüsselintermediat zur Synthese von (–)-A26771B
fungierende Acetonid 115 auf das β-Ketolacton 116 zurückgeführt (Schema 56).
O
O
O
OO
O
O OO
OH
115 116(R)-117(−)-A26771B (104)
O
OO
O
O
HO2C
( )8
Schema 56. Retrosynthese von (–)-A26771B.
Anwendung der etablierten Sequenz aus CM und Makrolactonisierung auf das homochirale
Tridecenol (R)-117 und asymmetrische Dihydroxylierung der mit hoher E-Selektivität
gebildeten Doppelbindung sollten folglich einen schnellen und eleganten Zugang zum
Makrolid-Antibiotikum (–)-A26771B ermöglichen.

I. Allgemeiner Teil
45
3.3.2. Synthese
Wie ein erster Test ausgehend von racemischem 12-Tridecen-2-ol (117)97 zeigte, verlief die
Sharpless-Dihydroxylierung der E-konfigurierten Doppelbindung des konjugierten Dienons
118 zum Triol 119 unter Verwendung eines modifizierten AD-Mix98 in tBuOH/H2O als
Lösungsmittel mit hoher Chemoselektivität und wurde somit einer substratkontrollierten
Dihydroxylierung des entsprechenden β-Ketolactons vorgezogen (Schema 57).
CH2Cl2, 40 °C, 24 h
79%
91 (1 eq), 5 mol% 5
118
OO
O
OH
91% 119
OO
O
OH OH
OHtBuOH/H2O, 16 h
AD-Mix-α, K2OsO2(OH)4, NaHCO3, MeSO2NH2
( )9( )9117
Schema 57. Sequentielle CM – Dihydroxylierung.
Demzufolge begann die Synthese des als Schlüsselintermediat dienenden Acetonids 115 mit
der Darstellung des homochiralen Tridecenols (R)-117 durch Ringöffnung von käuflichem
(R)-Methyloxiran (Schema 58). Die zur Vermeidung von Homodimerisierung mit einem
Überschuß von 1.5 Äquivalenten Dienon 91 durchgeführte CM ergab das Kupplungs-
produkt (R)-118 stereoselektiv (E/Z > 20:1) in einer leicht verbesserten Ausbeute von 88%.
Anschließende asymmetrische Dihydroxylierung unter den bewährten Bedingungen,
Schützung des Diols und direkte Thermolyse des Reaktionsgemisches in Heptan (Methode A)
lieferten das β-Ketolacton 116 diastereomerenrein in 65% Gesamtausbeute als 2:1 Mischung
aus Keto- und Enolform.
CH2Cl2, 40 °C, 24 h
88%
91 (1.5 eq), 5 mol% 5
1) AD-Mix-α, K2OsO2(OH)4, NaHCO3, MeSO2NH2, tBuOH/H2O, 16 h
(R)-95 (R)-117
BrMgO
CuCN, THF, -78 °C85%
2) Aceton, pTsOH, 60 h
65%
O
O OO
O
O OO
116 1153) Heptan, 98 °C, 7 h
2) MsCl, Pyridin, 2 h
80%
1) NaBH4, MeOH, 5 min
3) DBU, CH2Cl2, 1 h
O
OH( )8
( )8
OH
( )8
OO
O
(R)-118
Schema 58. Synthese des Acetonids 115.

I. Allgemeiner Teil
46
Mesylierung der nach NaBH4-Reduktion erhaltenen diastereomeren β-Hydroxylactone und
Eliminierung mittels DBU ergab das gewünschte Acetonid 115 erwartungsgemäß in einer
hervorragenden Gesamtausbeute von 80%, wobei dessen spektroskopische und physikalische
Daten {[α]D20 + 5.4 (c 1.0, CHCl3)} gut mit den literaturbeschriebenen übereinstimmten
{[α]D20 + 4.5 (c 0.58, CHCl3)}.94c Verglichen mit den bisherigen Formalsynthesen94b-e stellt
die beschriebene 8-stufige Sequenz mit einer Gesamtausbeute von 39% folglich einen
einfachen und wesentlich effizienteren Zugang zum Schlüsselintermediat 115 dar.
Während die ursprüngliche Synthese von (–)-A26771B nach Tatsuta et al. mit der
regioselektiven Monosuccinylierung des nach saurer Acetalhydrolyse erhaltenen Diols 120
und anschließender Swern-Oxidation endete (Schema 59),94a sollte die Reihenfolge in einer
darauf folgenden Arbeit von Quinkert et al. umgekehrt werden.
O
O
12051%
O
O
104
2) Ac2O, DMSO
OO
HO2C
OMeOH/H2O
F2HCCO2HOH
OH
94%
1) Succinanhydrid, CCl4, EtN(iPr)2
115
Schema 59. Endphase der Synthese von (–)-A26771B nach Tatsuta.
Interessanterweise resultierte die naheliegende selektive Oxidation der allylischen Hydroxy-
funktion des Diols 120 mit MnO2, PDC oder Fetizons-Reagenz jedoch ausschließlich in
oxidativer Spaltung der Glykoleinheit zum Dialdehyd 121 (Schema 60).94c
O
O
120
MeOH/H2O
F2HCCO2HOH
OH
86%
MnO2, CH2Cl2O
O
121
OO
82%115
Schema 60. Versuch der selektiven Oxidation des Diols 120 nach Quinkert.
Im Zuge einer jüngeren Synthese von Grahamimycin A aus Colletodiol zeigte es sich zudem,
dass außer Braunstein auch die Verwendung von DDQ oder Dess-Martin-Periodinan für eine
entsprechende Oxidation ungeeignet ist. Auf der Suche nach einem geeigneten Reagenz,
konnte eine selektive Transformation schließlich problemlos mittels TEMPO in Gegenwart
von pTsOH erreicht werden (Schema 61).99

I. Allgemeiner Teil
47
O
O
O
Colletodiol
OHOH
TEMPO
76%pTsOH
O
O
O
Grahamimycin A
OHO
O O
Schema 61. Synthese von Grahamimycin A nach O’Doherty.
Auch im Falle des Acetonids 115 ergab die direkte Umsetzung des nach Entschützung
erhaltenenen Diols mit zwei Äquivalenten TEMPO und pTsOH das Hydroxyketon 122 in
akzeptabler Ausbeute, welches sich schließlich unter den bewährten, racemisierungsfreien
Bedingungen in 74% Ausbeute zum Naturstoff {[α]D20 –13.2 (c 0.19, MeOH)} succinylieren
ließ (Schema 62).94f
115 O
O
122
2) TEMPO, pTsOH, CH2Cl2, 5 h
60%
1) TFA, MeCN/H2O, 0 °C, 1 h
OHO
Succinanhydrid
74%
DMAP, CH2Cl2, 24 hO
O
104
OO
HO2C
O
Schema 62. Synthese von (–)-A26771B.
Mit einer Länge von 11 Stufen und einer Gesamtausbeute von 17% ausgehend von
kommerziell erhältlichem (R)-Methyloxiran stellt die beschriebene Synthese den bislang
effizientesten enantioselektiven Zugang zum Makrolid-Antibiotikum (–)-A26771B dar. Über
die einfache Wahl des Startmaterials sowie des AD-Mix sollte überdies auch ein Zugang zu
allen nichtnatürlichen Isomeren wie beispielsweise dem (5R,15R)-Diastereomer ermöglicht
werden, dessen antibakterielle Aktivität sich als etwa doppelt so groß wie die des Naturstoffs
erwiesen hat.94a

I. Allgemeiner Teil
48
3.4. Studie zur Synthese des Flechten-Makrolids (+)-Aspicilin
Das makrocyclische Lacton (+)-Aspicilin (123) wurde bereits vor über 100 Jahren als erstes
Makrolid aus einer Flechte (Aspicilia gibbosa) isoliert,100 wobei die Bestimmung dessen
relativer und absoluter Konfiguration erst wesentlich später erfolgte.101 Obgleich bisher keine
biologischen Eigenschaften bekannt sind, ist die Synthese dieses polyhydroxylierten,
18-gliedrigen Makrocyclus bis heute von großen Interesse geblieben (Abbildung 15).102
Lepranthin
O
OOH
OH
OH
(+)-Aspicilin (123)
O OOAc OAcOH OH
OAc
AcO
O
O
Abbildung 15. Ausgewählte Flechten-Makrolide.
In einer früheren Synthese von Kobayashi et al. lieferte eine diastereoselektive Reduktion
des erneut durch oxidative Spaltung in wenigen Schritten aus dem Furan 124 erhaltenen
γ-Oxo-Crotonsäureesters 125 (ds > 15:1) gefolgt von MOM-Schützung und chemoselektiver
Desilylierung den Cyclisierungsvorläufer 126 in guter Ausbeute (Schema 63).102g
OH
1) LiOH, MeOH/H2O2) 2,4,6-Cl3C6H2COCl, NEt3, DMAP, Toluol
3) HS(CH2)2SH, BF3.Et2O
OHMeO2C
MOMO
MOMO
OMOM
30%
MeO2C
O
MOMO
OMOM
( )9
TBSO
1) Zn(BH4)2, Et2O, -78°C
2) MOMCl, EtN(iPr)2, CH2Cl2
3) NBS, DMSO/H2O
60%
TBSO
( )9
OOH
OH
123
(S)-90c 124125
80% 37%
126 Schema 63. Totalsynthese von (+)-Aspicilin nach Kobayashi.
Abschließende Makrocyclisierung der nach Esterverseifung erhaltenen Hydroxycarbonsäure
unter Yamaguchi-Bedingungen und Acetalspaltung ergaben (+)-Aspicilin in 15 Stufen und
einer Gesamtausbeute von 5% ausgehend vom homochiralen Tetradecenol (S)-90c.

I. Allgemeiner Teil
49
O
128
O
OO
OMe
OMeMOMO
127
O
OOMe
OMe
OH
OH
1) 10 mol% 2, CH2Cl2, 73%
2) H2, Pd/BaSO4, EtOAc, 100%
3) HS(CH2)2SH, BF3.Et2O, 73%
19%
123
Schema 64. Totalsynthese von (+)-Aspicilin nach Ley.
In einer darauf folgenden Arbeit von Ley et al. konnte auch die RCM erfolgreich zum Aufbau
des Makrocyclus genutzt werden (Schema 64).102h Chemoselektive Hydrierung des aus dem
Metathesevorläufer 128 erhaltenen Diens und Entfernung der Schutzgruppen ergab den
Naturstoff ausgehend vom Butantetrol 127 nach 10 Stufen in einer Gesamtausbeute von 10%.
3.4.1. Retrosynthetische Analyse
Die von Zercher et al. entwickelte, einstufige Homologisieurung von β-Ketoestern via
Fragmentierung eines in situ generierten Zink-Cyclopropanolats103a konnte kürzlich auch zur
direkten Synthese von α,β-ungesättigten-γ-Ketolactonen genutzt werden (Schema 65)103b
RO R'
OO
RO R'
O[Zn]OZnEt2
CH2I2
Retro-
Aldol ROR'
O
O[Zn]RO
R'O
O
1) I2
2) DBU
Schema 65. Synthese von α,β-ungesättigten-γ-Ketolactonen nach Zercher.
Das als potentielles Schlüsselintermediat zur Synthese von (+)-Aspicilin anvisierte Keton 129
sollte demnach auf das entsprechende β-Ketolacton 130 zurückgeführt werden, welches
ebenfalls vom Tetradecenol (S)-90c ausgehend via der beschriebenen Sequenz aus CM,
asymmetrischer Dihydroxylierung und Makrolactonisierung zugänglich wäre (Schema 66).
OH
129 130 (S)-90c
( )9
O
OOH
OH
OH
(+)-Aspicilin (123)
O
OO O
O O
O
OO
O
Schema 66. Retrosynthese von (+)-Aspicilin.
Eine auf die Ringerweiterung folgende, diastereoselektive Reduktion der γ-Ketogruppe von
129 und Spaltung des Acetonids würde (+)-Aspicilin schließlich in lediglich 7 Stufen liefern.

I. Allgemeiner Teil
50
3.4.2. Synthese
Entsprechend der Synthese des β-Ketolactons 116 lieferte die CM des Tetradecenols (S)-90c
mit einem Überschuß des Dienons 91 das Kupplungsprodukt (S)-92c stereoselektiv (E/Z >
20:1) in einer hervorragenden Ausbeute von 86% (Schema 67). In einer analogen Sequenz aus
asymmetrischer Dihydroxylierung in Gegenwart von AD-Mix-β, Acetonisierung und direkter
Thermolyse (Methode A) konnte das 17-gliedrige β-Ketolacton 130 anschließend ebenfalls
diastereomerenrein in einer vergleichbaren Gesamtausbeute von 66% als 3:1 Mischung aus
Keto- und Enolform erhalten werden.
86%
5 mol% 5
1) AD-Mix-β, K2OsO2(OH)4, NaHCO3, MeSO2NH2, tBuOH/H2O, 16 h
2) Aceton, pTsOH, 60 h
66%
(S)-90c
3) Heptan, 98 °C, 7 h
OH
( )9
OO
O(S)-92c
91 (1.5 eq)
40 °C, 24 hCH2Cl2
130
O
O
OO
O
Schema 67. Synthese des β-Ketolactons 130.
Obgleich die elektrophile Cyclopropanierung eines β-Ketolactons neben sterischen Faktoren
vor allem von dessen Enolisierbarkeit abhängig ist und daher durch vorheriges Deprotonieren
mit ZnEt2 beschleunigt werden kann,103b wurde die Ringerweiterung des bereits teilweise
enolisiert vorliegenden β-Ketolactons 130 zunächst mittels Zugabe zum vorgebildeten
Cyclopropanierungs-Reagenz (EtZnCH2I)104 durchgeführt (Schema 68).
ZnEt2, CH2I2
CH2Cl2, 0 °C, 3 h
65%130
O
O
OO
O
O
OO O
O
131 Schema 68. Testversuch zur Ringerweiterung des β-Ketolactons 130.
Erfreulicherweise ergab eine einfache wässrige Aufarbeitung103a nach 3 h Reaktionszeit auf
diese Weise das 18-gliedrige gesättigte γ-Ketolacton 131 als Hauptprodukt in diastereomeren-
reiner Form. Auf eine entsprechende Darstellung des α,β-ungesättigten-γ-Ketolactons 129 und
den Abschluß der Synthese von (+)-Aspicilin mußte jedoch aus Zeitgründen leider verzichtet
werden.

I. Allgemeiner Teil
51
3.5. Studie zur Synthese von γ,δ-ungesättigten-β-Ketolactamen
Von cyclischen Peptiden abgesehen, sind Makrolactame in der Natur vergleichsweise wenig
verbreitet. Eine interessantes Beispiel stellen die aus Actinomyceten isolierten 14-gliedrigen
Fluvirucine dar,105 welche aufgrund ihrer vielversprechenden biologischen Eigenschaften
einige Aufmerksamkeit erregt haben.106 So hat sich diese Klasse von Makrolactam-
Antibiotika beispielsweise als wirksam gegen das Influenza-A Virus erwiesen, wobei die
Vertreter der A-Reihe wegen ihrer geringeren Toxizität von besonderer Bedeutung sind
(Abbildung 16).107
Fluvirucin A1
NH
O
O
OOH
NH2
OHFluvirucin B1
HN
O
OOH
NH2
OH
O
Abbildung 16. Ausgewählte Fluvirucine.
Zwecks einer ersten Anwendung der CM – Makrocyclisierungs-Sequenz auf die Darstellung
von entsprechenden γ,δ-ungesättigten-β-Ketolactamen sollten zunächst die Carbamate 132a
und 132b synthetisiert werden, da sie nach CM mit dem Vinyldioxolenon 91 unter relativ
milden, nichthydrolytischen Bedingungen entschützt werden könnten (Schema 69).
N
O
O
CM
NHPGOO
O
133a/b 134
1) Entschützen91( )8
( )8 ( )72) Thermolyse
132a: PG = Fmoc132b: PG = Boc
PGHNH
Schema 69. Konzept zur Synthese von γ,δ-ungesättigten-β-Ketolactamen.
Demzufolge begann die Synthese der Cyclisierungsvorläufer 133a und 133b ausgehend von
kommerziell erhältlichem 11-Brom-1-undecen (135) mit einer Sequenz aus Cyanid-
Substitution, LAH-Reduktion und Einführung der Schutzgruppe (Schema 70). Anschließende
Umsetzung der Carbamate 132a und 132b mit stöchiometrischen Mengen des Dienons 91
unter den bewährten Bedingungen ergab die gewünschten Kreuzprodukte 133a und 133b
stereoselektiv (E/Z > 20:1) in hervorragenden Ausbeuten, wobei in beiden Fällen keine
Homodimerisierung der Amin-Komponente beobachtet wurde.

I. Allgemeiner Teil
52
OO
O133a
( )8
( )8
135
CH2Cl2, 40 °C
5 mol% 5
24 h
3) FmocCl, THF
3) Boc2O, NEt3
90%132a65%
61%
1) KCN, Glykol, 100 °C, 16 h
NHFmoc
( )8
91 (1 eq)
Br
2) LAH, Et2O, 1 h
OO
O
133b
( )8CH2Cl2, 40 °C
5 mol% 5
24 h88%
132b
NHBoc
( )8
91 (1 eq)NHBoc
NHFmoc
CH2Cl2, 2 h
H2O, NaHCO3
Schema 70. Synthese der Cyclisierungsvorläufer 133a und 133b.
Im Zuge der abschließenden Makrolactamisierung erwies sich die geplante Entschützung der
Cyclisierungsvorläufer 133a/b zum freien Amin 136 jedoch als problematisch (Schema 71).
Während die Behandlung des Fluorenyl-Carbamats 133a mit Piperidin oder DBU in einem
komplexen Produktgemisch resultierte, führte die Spaltung des tButylcarbamats 133b mit
10% TFA in CH2Cl2 zu einem teilweisen Verlust der CM-generierten Doppelbindung, was
auf eine intramolekulare Aza-Michael-Reaktion zurückgeführt wurde.
OO
O133a
Piperidin, DMF
( )8
OO
O
136
( )8
NH2NHFmocOO
O
133b
( )8
NHBoc
DBU, CH2Cl2
10% TFA
CH2Cl2
Schema 71. Entschützung der Cyclisierungsvorläufer 133a und 133b.
Dementsprechend konnte dieses Problem durch vorherige chemoselektive Hydrierung der
exocyclischen Doppelbindung von 133b umgangen werden. Anschließende Spaltung des
entsprechenden gesättigten tButylcarbamats gefolgt von Thermolyse (Methode A) lieferte das
β-Ketolactam 137b als einziges Produkt in guter Gesamtausbeute (Schema 72).
NH
O
O
NHBocOO
O
133b137b
1) H2, Pd/C, MeOH, 30 min
( )8
2) 10% TFA in CH2Cl2, 1 h
3) Heptan (10-4 M), 98 °C, 7 h
61%
Schema 72. Synthese von gesättigten β-Ketolactamen.

I. Allgemeiner Teil
53
Um zu prüfen, ob auf diese Weise auch das 14-gliedrige Grundgerüst der Fluvirucin A-Reihe
zugänglich ist (Abbildung 16), wurde das bereits zuvor synthetisierte Hydroxyalkenyl-
dioxolenon 92a zunächst zum entsprechenden Azid umgesetzt (Schema 73).
NH
O
O
137a
3) H2, Pd/C, MeOH, 30 min4) Toluol (< 10-4 M), 110 °C, 3 h
OO
O
92a
( )5
OH
1) MsCl, Pyridin, 1 h
2) NaN3, DMF, 50 °C, 16 h
52% Schema 73. Synthese eines 14-gliedrigen β-Ketolactams.
Thermolyse (Methode B) des nach simultaner Reduktion der Doppelbindung und der Azid-
Gruppe erhaltenen Aminoalkyldioxolenons lieferte das 14-gliedrige β-Ketolactam 137a als
Hauptprodukt in einer Gesamtausbeute von 52%.
Da die Reduktion der Carbonylgruppe von substituierten 14-gliedrigen β-Ketolactonen aus
konformativen Gründen mit hoher Diastereoselektivität verläuft,108 sollte die beschriebene
Sequenz aus CM, Hydrierung oder Funktionalisierung der E-selektiv gebildeten Doppel-
bindung und Makrocyclisierung einen allgemeinen und flexiblen Zugang zu natürlichen oder
synthetischen Fluvirucinen der A-Reihe ermöglichen (Schema 74).
NH
OR''
O
CM NHPGOO
O
Fluvirucine
( )6
NHPG
( )6
R R
R'
R'R
Schema 74. Konzept zur Synthese von Fluvirucinen der A-Reihe.
Die Frage, ob die Synthese von γ,δ-ungesättigten-β-Ketolactamen alternativ durch direkte
Cyclisierung geeignet geschützter Aminoalkyldioxolenone oder die Wahl anderer Schutz-
gruppen ermöglicht werden kann, konnte aus Zeitgründen nicht mehr beantwortet werden.
Eine Verwendung der unter ausgesprochen milden und neutralen Bedingungen spaltbaren
Alloc-Schutzgruppe erschien bezüglich des CM-Schritts aufgrund ihrer terminalen Doppel-
bindung jedoch als eher ungeeignet.

I. Allgemeiner Teil
54
3.6. Zusammenfassung und Ausblick
Die neuartige Sequenz aus CM und anschließender Makrocyclisierung konnte ausgehend von
Hydroxyalkenen und dem Vinyldioxolenon 91 erfolgreich zum Aufbau monomerer 16- bis
18-gliedriger γ,δ-ungesättigter-β-Ketolactone genutzt werden und stellt somit eines der ersten
Beispiele für eine hoch chemo- und stereoselektive CM mit einem konjugierten Dienon dar.
Im Anschluß an die methodischen Arbeiten konnte eine asymmetrische Dihydroxylierung der
während der CM mit hoher E-Selektivität gebildeten Doppelbindung als Schlüsselschritt für
eine effiziente Totalsynthese des Makrolid-Antibiotikums (–)-A26771B genutzt werden,
welche den Naturstoff ausgehend von kommerziell erhältlichem (R)-Methyloxiran nach nur
insgesamt 11 Stufen in einer Gesamtausbeute von 17% lieferte (Schema 75).
(−)-A26771B
O
OO
O
O
O
O
OO
OO
HO2C
5 Stufen
49%
6 Stufen
35%
130
O
O
OO
OO 5 Stufen
50%O
OOH
OH
OH
(+)-Aspicilin
3 Stufen
?
116
(R)-95
(S)-95
Schema 75. CM – Dihydroxylierung als Schlüsselschritt in der Naturstoffsynthese.
Die auf dem selben Konzept basierende Totalsynthese des Flechten-Makrolids (+)-Aspicilin
konnte aus Zeitgründen leider nicht beendet werden. Ein finaler Testversuch zeigte jedoch,
dass die vorgesehene Ringerweiterungsreaktion des β-Ketolacton-Intermediats 130 prinzipiell
funktioniert und somit einen einfachen Zugang zum Naturstoff ermöglichen sollte.
Eine interessante Erweiterung des allgemeinen Synthesekonzepts könnte darin bestehen, die
CM-generierte Doppelbindung nach der Makrocyclisierung für eine intramolekulare Michael-
Addition zu nutzen, um somit Zugang zu bicyclischen Systemen zu eröffnen. Durch den
Einsatz geminal disubstituierter Olefine als CM-Partner würde sich die Bandbreite an

I. Allgemeiner Teil
55
darstellbaren Strukturen ebenfalls erweitern lassen, wobei eine Funktionalisierung der
trisubstituierten Alkeneinheit nach Makrocyclisierung diastereoselektiv verlaufen sollte.
Abschließend wurde untersucht, ob die sequentielle CM – Makrocyclisierung auch zum
Aufbau von entsprechenden Makrolactamen genutzt werden kann. Während die Synthese von
γ,δ-ungesättigten-β-Ketolactamen an der Entschützung der Cyclisierungsvorläufer scheiterte,
konnten die gesättigten Analoga bequem durch vorherige Hydrierung der CM-generierten
Doppelbindung erhalten werden.

II. Experimenteller Teil
56
II. Experimenteller Teil
1. Allgemeines
1H-NMR-Spektren wurden mit den Geräten DRX 500 (500 MHz) und AC 200 (200 MHz)
der Firma Bruker bei Raumtemperatur aufgenommen, und die chemischen Verschiebungen
sind in dimensionslosen δ-Werten (ppm) relativ zum internen Lösungsmittelpeak angegeben.
In Klammern vermerkt sind die Signalmultiplizität, die Kopplungskonstanten J in [Hz] und
die durch elektronische Integration ermittelte Protonenzahl, wobei die Multiplizitäten wie
folgt vermerkt sind: s (Singulett), d (Dublett), t (Triplett), q (Quartett), quin (Quintett), sext
(Sextett), m (Multiplett), br (breites Signal).
13C-NMR-Spektren wurden mit den Geräten DRX 500 (125 MHz) und AC 200 (50 MHz) der
Firma Bruker bei Raumtemperatur aufgenommen. Die chemischen Verschiebungen sind den
protonenbreitbandentkoppelten Spektren entnommen und in dimensionslosen δ-Werten (ppm)
relativ zum internen Lösungsmittelpeak angegeben. Die Anzahl der direkt gebundenen
Protonen wurde durch DEPT ermittelt und ist wie folgt vermerkt: Cq (s), CH (d), CH2 (t),
CH3 (q).
IR-Spektren wurden mit dem Infrarot-Spektrophotometer 881 der Firma Perkin-Elmer als
ATR (Attenuated Total Reflectance) aufgenommen. Die Absorptionsbanden sind in
Wellenzahlen [cm-1] angegeben, und ihre Intensitäten sind wie folgt vermerkt: ss (sehr stark),
s (stark), m (mittel), w (schwach) und br (breit).
LR-MS und HR-MS-Spektren wurden auf einem Finnigan MAT 95 SQ oder Varian MAT
711 aufgenommen, wobei die Ionisierung durch Elektronenstoß (EI) bei einem
Ionisierungspotential von 70 eV beziehungsweise durch Atombeschuß (FAB) aus Glycerol
erfolgte. Die Verdampfungstemperaturen sind vermerkt und die relativen Signalintensitäten
prozentual in Klammern angegeben.
GC-MS-Spektren wurden mit dem Gaschromatographen HP 6890 der Firma Hewlett-
Packard (Säule HP-5MS, crosslinked 5% PH ME Siloxan, Helium als Trägergas, 250 °C
Injektionstemperatur) unter Verwendung eines HP 5973 massenselektiven Detektors mit
Elektronenstoß-Ionisation (EI) durchgeführt.

II. Experimenteller Teil
57
Elementaranalysen wurden mit dem Elementar Vario EI der Firma Analytik Jena
durchgeführt.
Röntgenstrukturanalysen wurden mit einem Siemens SMART CCD Diffraktometer bei RT
durchgeführt.
Schmelzpunkte wurden mittels eines Leica Galen III Heiztischmikroskops der Firma Leica
mit einer Steuereinheit von Wagner-Munz bestimmt und sind nicht korrigiert.
Drehwerte wurden mit dem Polarimeter 341 der Firma Perkin-Elmer bei Raumtemperatur
und einer Wellenlänge von 589 nm (Natrium-D-Linie) gemessen. Das jeweilige Lösungs-
mittel und die Konzentration [g/100 ml] sind in Klammern angegeben.
Dünnschichtchromatogramme wurden mit DC-Folien der Firma Merck (Kieselgel 60 F
254, Schichtdicke 0.2 mm) durchgeführt, wobei zur Detektion UV-Licht der Wellenlänge 254
nm und Kaliumpermanganat dienten.
Säulenchromatographie wurde mit Flash-Kieselgel der Firma Merck (Korngröße 0.03-0.06
mm) oder basischem Aluminiumoxid der Firma Fluka (5016A, Aktivität III) durchgeführt.
Lösungsmittel wurden vor Gebrauch destilliert und gegebenenfalls wie folgt getrocknet:
THF, Diethylether sowie Toluol über Natrium und Dichlormethan über Calciumhydrid oder
Sicapent. Alle anderen Lösungsmittel wurden über Molekularsieb (4 Å) getrocknet.

II. Experimenteller Teil
58
2. Versuchsvorschriften und spektroskopische Daten
2.1. Zu Kapitel 2.2.
6-(tButyldimethylsilyloxy)-hex-1-en-3-on (18d)
Eine Lösung von 500 mg (3.4 mmol) Amid 28 und 463 mg (6.8
mmol) Imidazol in 5 ml DMF wurde bei 0 °C portionsweise mit 560
mg (3.7 mmol) TBSCl versetzt und über 2 h auf RT erwärmt. Nach
Zugabe von 50 ml Et2O wurde mit 1 N HCl (3 x 20 ml) gewaschen und über Na2SO4
getrocknet. Der nach Entfernen des Lösungsmittels erhaltene Rückstand wurde in 15 ml
absolutem THF gelöst und unter Stickstoff-Atmosphäre bei 0 °C tropfenweise mit 5 ml (1 M
in THF, 5.0 mmol) Vinylmagnesiumbromid-Lösung versetzt. Das Reaktionsgemisch wurde 2
h bei RT gerührt und nach Zugabe von 10 ml 1 N HCl mit Et2O (3 x 10 ml) extrahiert.
Trocknen der organischen Phasen über Na2SO4, Entfernen des Lösungsmittels und säulen-
chromatographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:10) ergab 590 mg
(76%) Produkt als gelbliches Öl. 1H-NMR (500 MHz, CDCl3): δ = 6.36 (dd, J = 18, 10.6 Hz, 1H), 6.23 (dd, J = 18, 1 Hz, 1H),
5.82 (dd, J = 10.6, 1 Hz, 1H), 3.64 (t, J = 6 Hz, 2H), 2.67 (t, J = 7.3 Hz, 2H), 1.83 (quin, J =
6.8 Hz, 2H), 0.88 (s, 9H), 0.04 (s, 6H). 13C-NMR (125 MHz, CDCl3): δ = 200.8 (Cq), 136.7 (CH), 128.0 (CH2), 62.1 (CH2), 35.9
(CH2), 27.0 (CH2), 26.0 (CH3), 18.4 (Cq), -5.3 (CH3).
IR (ATR): ν = 2955 (s), 2929 (s), 2857 (s), 1704 (s), 1684 (s), 1472 (m), 1402 (m), 1361 (m),
1256 (s), 1102 (ss), 961 (s), 836 (ss), 776 (ss).
LR-MS (RT): m/z = 213 (2), 171 (82), 115 (4), 97 (10), 75 (100), 55 (12).
HR-MS (C11H21O2Si; M+– CH3): ber. 213.1310; gef. 213.1311.
1-Phenylbut-3-en-2-on (18e)
Eine Lösung von 100 mg (558 µmol) Amid 29a in 3 ml absolutem THF
unter Stickstoff-Atmosphäre wurde bei 0 °C tropfenweise mit 0.7 ml (1
M in THF, 700 µmol) Vinylmagnesiumbromid-Lösung versetzt. Das
Reaktionsgemisch wurde 2 h bei RT gerührt und nach Zugabe von 3 ml 1 N HCl mit Et2O (3
x 3 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4, Entfernen des Lösungs-
mittels und Filtration über Kieselgel (CH2Cl2) ergab 73 mg (89%) Produkt als gelbliches Öl.
OOTBS
O

II. Experimenteller Teil
59
1H-NMR (200 MHz, CDCl3): δ = 7.40-7.20 (m, 5H), 6.49-6.24 (m, 2H), 5.83 (dd, J = 9, 2 Hz,
1H), 3.88 (s, 2H).
1-(Furan-2-yl)-propenon (18f)
Eine Lösung von 100 mg (645 µmol) Amid 29b in 3 ml absolutem THF
unter Stickstoff-Atmosphäre wurde bei 0 °C tropfenweise mit 0.8 ml (1 M
in THF, 800 µmol) Vinylmagnesiumbromid-Lösung versetzt. Das Reak-
tionsgemisch wurde 2 h bei RT gerührt und nach Zugabe von 3 ml 1 N HCl
mit Et2O (3 x 3 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4, Entfernen des
Lösungsmittels und Filtration über Kieselgel (CH2Cl2) ergab 71 mg (90%) Produkt als
gelbliches Öl. 1H-NMR (200 MHz, CDCl3): δ = 7.64 (dd, J = 2.4, 1 Hz, 1H), 7.27 (dd, J = 3.4, 1 Hz, 1H),
7.07 (dd, J = 17, 10 Hz, 1H), 6.59-6.50 (m, 2H), 5.87 (dd, J = 10, 2 Hz, 1H).
5-([1.3]Dioxolan-2-yl)-pent-1-en-3-on (33)
Eine Lösung von 1.4 g (20 mmol) Methoxyallen in 20 ml absolutem
THF unter Stickstoff-Atmosphäre wurde bei -35 °C zunächst mit 12
ml (1.6 M in Hexan, 19 mmol) BuLi und nach 1 h mit einer Lösung
von 1.8 g (10 mmol) 2-(2-Bromethyl)-1,3-dioxolan in 10 absolutem
THF versetzt. Das Reaktionsgemisch wurde über 3 h auf RT erwärmt und nach Zugabe von
25 ml 1 N HCl mit Et2O (3 x 25 ml) extrahiert. Trocknen der organischen Phasen über
Na2SO4, Entfernen des Lösungsmittels und säulenchromatographische Reinigung des
Rückstands (SiO2, Et2O/Pentan 1:1) ergab 610 mg (39%) Produkt als gelbliches Öl. 1H-NMR (500 MHz, CDCl3): δ = 6.36 (dd, J = 18, 10.6 Hz, 1H), 6.23 (dd, J = 18, 1 Hz, 1H),
5.82 (dd, J = 10.6, 1 Hz, 1H), 4.93 (t, J = 4.3 Hz, 1H), 3.80-4.00 (m, 4H), 2.72 (t, J = 7.4 Hz,
2H), 2.02 (m, 2H). 13C-NMR (125 MHz, CDCl3): δ = 199.9 (Cq), 136.6 (CH), 128.1 (CH2), 103.4 (CH), 65.0
(CH2), 33.4 (CH2), 27.7 (CH2).
IR (ATR): ν = 3399 (br), 2956 (m), 2936 (m), 2892 (m), 1713 (ss), 1408 (m), 1364 (m), 1139
(ss), 1032 (ss), 947 (s), 897 (m).
LR-MS (RT): m/z = 155 (2), 129 (3), 111 (1), 99 (4), 86 (10), 73 (100), 55 (22).
HR-MS (C8H11O3; M+– H): ber. 155.0708; gef. 155.0710.
O
O
O
O
O

II. Experimenteller Teil
60
4-Oxohex-5-enal (18h)
312 mg (2 mmol) Acetal 33 wurden bei 0 °C tropfenweise zu 5 ml 1 N
HCl gegeben. Das Reaktionsgemisch wurde 4 h bei RT gerührt und mit
Et2O (3 x 5 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4,
Entfernen des Lösungsmittels und säulenchromatographische Reinigung
des Rückstands (SiO2, Et2O/Pentan 3:1) ergab 83 mg (37%) Produkt als gelbliches Öl. 1H-NMR (500 MHz, CDCl3): δ = 9.83 (s, 1H), 6.38 (dd, J = 17, 10.5 Hz, 1H), 6.27 (dd, J =
17, 1 Hz, 1H), 5.88 (dd, J = 10.5, 1 Hz, 1H), 2.93 (t, J = 6.5 Hz, 2H), 2.81 (t, J = 6.5 Hz, 2H). 13C-NMR (125 MHz, CDCl3): δ = 200.5 (CH), 198.3 (Cq), 136.2 (CH), 128.8 (CH2), 37.4
(CH2), 31.7 (CH2).
IR (ATR): ν = 3444 (br), 2954 (s), 2924 (ss), 2854 (s), 1702 (s), 1680 (s), 1615 (w), 1403
(m), 1363 (m), 1070 (m), 987 (s), 928 (m).
LR-MS (RT): m/z = 112 (< 1), 96 (1), 85 (8), 70 (1), 58 (100), 55 (10).
HR-MS (C6H8O2; M+): ber. 112.0524; gef. 112.0521.
Allgemeine Versuchsvorschrift A für die Kreuzmetathese von Homoallylaminen:
Eine Lösung von Homoallylamin (0.05 M) und α,β-ungesättigter Carbonylverbindung (1–2
eq) in absolutem CH2Cl2 unter Stickstoff-Atmosphäre wurde mit dem Ruthenium-Katalysator
5 (2.5–5 mol%) versetzt und 3–16 h bei 40 °C gerührt. Das Lösungsmittel wurde entfernt und
der Rückstand säulenchromatographisch gereinigt (SiO2, MTB/Hexan).
(E)-5-Benzyloxycarbonylaminohex-2-endicarbonsäuredimethylester (19a)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden aus
200 mg (760 µmol) Homoallylamin 17a, 130 mg (1.52 mmol)
Methylacrylat (18a) und 12 mg (5 mol%) 5 nach 3 h 194 mg
(79%) Produkt als grünliches Harz erhalten. 1H-NMR (200 MHz, CDCl3): δ = 7.35 (m, 5H), 6.80 (dt, J = 15.6, 7.3 Hz, 1H), 5.90 (d, J =
15.6 Hz, 1H), 5.35 (br d, J = 7 Hz, 1H), 5.11 (s, 2H), 4.54 (m, 1H), 3.76 (s, 3H), 3.72 (s, 3H),
2.72 (m, 2H). 13C-NMR (50 MHz, CDCl3): δ = 171.3 (Cq), 166.0 (Cq), 155.5 (Cq), 142.0 (CH), 135.9 (Cq),
128.4 (CH), 128.1 (CH), 127.9 (CH), 124.6 (CH), 67.0 (CH2), 52.8 (CH), 52.5 (CH3), 51.4
(CH3), 34.8 (CH2).
OMe
NHZMeO2C
O
O
O

II. Experimenteller Teil
61
IR (ATR): ν = 3340 (br), 3033 (w), 2953 (w), 1722 (ss), 1660 (m), 1527 (s), 1437 (s), 1321
(m), 1271 (s), 1215 (s), 1048 (s), 983 (m), 740 (w), 699 (m).
LR-MS (125 °C): m/z = 321 (< 1), 222 (2), 178 (3), 154 (2), 111 (6), 108 (8), 91 (100), 79
(6), 59 (6).
HR-MS (C16H19NO6; M+): ber. 321.1212; gef. 321.1214.
CHN-Analyse (C16H19NO6): ber. C 59.81, H 5.96, N 4.36; gef. C 59.86, H 5.99, N 4.74.
(E)-2-Benzyloxycarbonylamino-6-oxohept-4-ensäuremethylester (19b)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden aus 80 mg
(304 µmol) Homoallylamin 17a, 42 mg (608 µmol) Methylvinyl-
keton (18b) und 5 mg (2.5 mol%) 5 nach 3 h 79 mg (85%) Produkt
als grünliches Harz erhalten. 1H-NMR (200 MHz, CDCl3): δ = 7.33 (m, 5H), 6.63 (dt, J = 16, 7.3 Hz, 1H), 6.08 (d, J = 16
Hz, 1H), 5.47 (br d, J = 7 Hz, 1H), 5.10 (s, 2H), 4.54 (m, 1H), 3.74 (s, 3H), 2.86-2.52 (m,
2H), 2.20 (s, 3H). 13C-NMR (50 MHz, CDCl3): δ = 197.9 (Cq), 171.4 (Cq), 155.6 (Cq), 141.0 (CH), 135.9 (Cq),
134.1 (CH), 128.4 (CH), 128.1 (CH), 127.9 (CH), 66.9 (CH2), 52.8 (CH), 52.5 (CH3), 35.4
(CH2), 26.8 (CH3).
IR (ATR): ν = 3317 (br), 3032 (w), 2916 (w), 1717 (ss), 1697 (ss), 1672 (ss), 1521 (s), 1436
(m), 1359 (m), 1251 (ss), 1209 (ss), 1176 (s), 1045 (s), 976 (ss), 737 (m), 696 (s).
LR-MS (160 °C): m/z = 305 (< 1), 246 (4), 222 (8), 199 (62), 178 (10), 156 (12), 107 (8), 91
(100), 69 (12).
HR-MS (C16H19NO5; M+): ber. 305.1263; gef. 305.1267.
CHN-Analyse (C16H19NO5): ber. C 62.94, H 6.27, N 4.59; gef. C 62.84, H 6.38, N 4.53.
(E)-2-Benzyloxycarbonylamino-6-oxodec-4-ensäuremethylester (19c)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden
aus 100 mg (380 µmol) Homoallylamin 17a, 43 mg (380
µmol) n-Butylvinylketon (18c) und 6 mg (2.5 mol%) 5 nach
3 h 109 mg (83%) Produkt als grünliches Harz erhalten. 1H-NMR (200 MHz, CDCl3): δ = 7.34 (m, 5H), 6.65 (dt, J = 16, 7.3 Hz, 1H), 6.11 (d, J = 16
Hz, 1H), 5.41 (br d, J = 7 Hz, 1H), 5.10 (s, 2H), 4.54 (m, 1H), 3.75 (s, 3H), 2.85-2.57 (m,
2H), 2.49 (t, J = 7.8 Hz, 2H), 1.56 (quin, J = 7.3 Hz, 2H), 1.30 (m, 2H), 0.89 (t, J = 7.3 Hz,
3H).
NHZMeO2C
O
NHZMeO2C
O

II. Experimenteller Teil
62
13C-NMR (125 MHz, CDCl3): δ = 200.2 (Cq), 171.6 (Cq), 155.7 (Cq), 139.6 (CH), 136.1 (Cq),
133.6 (CH), 128.6 (CH), 128.4 (CH), 128.2 (CH), 67.2 (CH2), 53.0 (CH), 52.8 (CH3), 40.0
(CH2), 35.6 (CH2), 26.2 (CH2), 22.4 (CH2), 13.9 (CH3).
IR (ATR): ν = 3331 (br), 3034 (w), 2956 (m), 2932 (m), 2873 (m), 1721 (ss), 1700 (ss), 1672
(ss), 1631 (s), 1525 (ss), 1437 (m), 1346 (s), 1258 (ss), 1212 (ss), 1179 (s), 1050 (ss), 980 (s),
739 (m), 697 (s).
LR-MS (140 °C): m/z = 347 (< 1), 242 (1), 212 (3), 199 (15), 178 (2), 126 (6), 91 (100), 69
(2), 65 (5).
HR-MS (C19H25NO5; M+): ber. 347.1733; gef. 347.1731.
CHN-Analyse (C16H19NO5): ber. C 65.69, H 7.25, N 4.03; gef. C 65.63, H 7.40, N 4.00.
(E)-2-Benzyloxycarbonylamino-9-(tbutyldimethylsilyloxy)-6-oxonon-4-ensäuremethyl-
ester (19d)
Entsprechend der allgemeinen Arbeitsvorschrift A
wurden aus 50 mg (190 µmol) Homoallylamin 17a, 44
mg (190 µmol) Enon 18d und 6 mg (5 mol%) 5 nach 16
h 56 mg (64%) Produkt als bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.35 (m, 5H), 6.66 (dt, J = 15.8, 7.3 Hz, 1H), 6.14 (d, J =
15.8 Hz, 1H), 5.34 (br d, J = 7 Hz, 1H), 5.11 (s, 2H), 4.55 (m, 1H), 3.76 (s, 3H), 3.62 (t, J =
6.2 Hz, 2H), 2.80-2.63 (m, 2H), 2.59 (t, J = 7.3 Hz, 2H), 1.80 (quin, J = 6.6 Hz, 2H), 0.88 (s,
9H), 0.03 (s, 6H). 13C-NMR (125 MHz, CDCl3): δ = 199.7 (Cq), 171.5 (Cq), 155.7 (Cq), 139.6 (CH), 136.1 (Cq),
133.6 (CH), 128.6 (CH), 128.3 (CH), 128.2 (CH), 67.2 (CH2), 62.2 (CH2), 53.0 (CH), 52.7
(CH3), 36.6 (CH2), 35.5 (CH2), 27.1 (CH2), 26.0 (CH3), 18.4 (Cq), -5.3 (CH3).
IR (ATR): ν = 3336 (br), 3034 (w), 2954 (s), 2929 (s), 2857 (m), 2104 (m), 1725 (ss), 1703
(ss), 1676 (s), 1634 (m), 1526 (s), 1438 (m), 1347 (m), 1256 (ss), 1214 (ss), 1099 (ss), 1057
(s), 979 (m), 837 (ss), 777 (s), 698 (m).
LR-MS (150 °C): m/z = 406 (4), 362 (2), 297 (3), 255 (100), 181 (4), 149 (2), 110 (6), 91
(90), 75 (10).
HR-MS (C20H28NO6Si; M+– C4H9): ber. 406.1686; gef. 406.1679.
CHN-Analyse (C24H37NO6Si): ber. C 62.17, H 8.04, N 3.02; gef. C 62.18, H 8.03, N 2.97.
NHZMeO2C
OOTBS
II. Experimenteller Teil
63
(E)-2-Benzyloxycarbonylamino-6-oxo-7-phenylhept-4-ensäuremethylester (19e)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden
aus 50 mg (190 µmol) Homoallylamin 17a, 28 mg (190
µmol) Enon 18e und 6 mg (5 mol%) 5 nach 16 h 57 mg
(78%) Produkt als bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.38-7.17 (m, 10H), 6.74 (dt, J = 16, 7.3 Hz, 1H), 6.16 (d, J
= 16 Hz, 1H), 5.35 (br d, J = 7 Hz, 1H), 5.10 (s, 2H), 4.53 (m, 1H), 3.79 (s, 2H), 3.70 (s, 3H),
2.80-2.58 (m, 2H). 13C-NMR (125 MHz, CDCl3): δ = 196.9 (Cq), 171.5 (Cq), 155.7 (Cq), 141.0 (CH), 136.1 (Cq),
134.2 (Cq), 132.5 (CH), 129.5 (CH), 128.8 (CH), 128.7 (CH), 128.6 (CH), 128.4 (CH), 128.2
(CH), 127.1 (CH), 67.2 (CH2), 53.0 (CH), 52.7 (CH3), 47.8 (CH2), 35.5 (CH2).
IR (ATR): ν = 3330 (br), 3031 (w), 2953 (w), 2849 (w), 1717 (ss), 1700 (ss), 1628 (m), 1521
(s), 1497 (s), 1436 (m), 1343 (m), 1246 (s), 1211 (ss), 1048 (s), 978 (s), 736 (s), 698 (ss).
LR-MS (160 °C): m/z = 381 (< 1), 290 (4), 275 (1), 246 (8), 160 (1), 91 (100), 65 (2).
HR-MS (C15H16NO5; M+– CH2Ph): ber. 290.1028; gef. 290.1033.
(E)-2-Benzyloxycarbonylamino-6-(furan-2-yl)-6-oxohex-4-ensäuremethylester (19f)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden aus
50 mg (190 µmol) Homoallylamin 17a, 47 mg (380 µmol) Enon
18f und 6 mg (5 mol%) 5 nach 16 h 50 mg (73%) Produkt als
bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.61 (d, J = 0.8 Hz, 1H), 7.34 (m, 5H), 7.23 (d, J = 3.4 Hz,
1H), 6.97 (dt, J = 15.5, 7.3 Hz, 1H), 6.85 (d, J = 15.5 Hz, 1H), 6.55 (dd, J = 3.4, 1.5 Hz, 1H),
5.41 (br d, J = 7 Hz, 1H), 5.11 (s, 2H), 4.60 (m, 1H), 3.76 (s, 3H), 2.89-2.74 (m, 2H). 13C-NMR (125 MHz, CDCl3): δ = 177.4 (Cq), 171.6 (Cq), 155.7 (Cq), 153.1 (Cq), 146.9 (CH),
141.4 (CH), 136.1 (Cq), 128.6 (CH), 128.4 (CH), 128.3 (CH), 128.2 (CH), 118.2 (CH), 112.6
(CH), 67.2 (CH2), 53.0 (CH), 52.8 (CH3), 35.5 (CH2).
IR (ATR): ν = 3323 (br), 3033 (w), 2953 (w), 2850 (w), 1718 (ss), 1667 (ss), 1623 (s), 1525
(s), 1465 (ss), 1395 (m), 1266 (s), 1214 (ss), 1044 (s), 1027 (s), 981 (m), 883 (w), 757 (m),
698 (m).
LR-MS (150 °C): m/z = 357 (< 1), 298 (3), 251 (26), 222 (4), 181 (4), 156 (4), 136 (70), 107
(10), 95 (16), 91 (100), 65 (8).
HR-MS (C19H19NO6; M+): ber. 357.1212; gef. 357.1218.
NHZMeO2C
O
NHZMeO2C
O
O

II. Experimenteller Teil
64
(E)-2-Benzyloxycarbonylamino-6,9-dioxodec-4-ensäuremethylester (19g)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden
aus 50 mg (190 µmol) Homoallylamin 17a, 24 mg (190
µmol) Enon 18g und 6 mg (5 mol%) 5 nach 16 h 51 mg
(74%) Produkt als bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.33 (m, 5H), 6.70 (dt, J = 16, 8 Hz, 1H), 6.14 (d, J = 16
Hz, 1H), 5.40 (br d, J = 7 Hz, 1H), 5.11 (s, 2H), 4.55 (m, 1H), 3.76 (s, 3H), 2.85-2.60 (m,
6H), 2.19 (s, 3H). 13C-NMR (125 MHz, CDCl3): δ = 207.2 (Cq), 198.0 (Cq), 171.5 (Cq), 155.7 (Cq), 140.3 (CH),
136.1 (Cq), 133.3 (CH), 128.6 (CH), 128.5 (CH), 128.3 (CH), 128.2 (CH), 67.2 (CH2), 53.0
(CH), 52.8 (CH3), 36.9 (CH2), 35.6 (CH2), 33.7 (CH2), 30.0 (CH3).
IR (ATR): ν = 3333 (br), 3033 (w), 2954 (w), 2851 (w), 1715 (ss), 1675 (s), 1633 (m), 1526
(s), 1437 (m), 1359 (s), 1256 (s), 1213 (ss), 1051 (s), 1028 (m), 980 (m), 740 (m), 700 (m).
LR-MS (200 °C): m/z = 362 (1), 302 (1), 254 (4), 237 (8), 212 (2), 156 (3), 140 (26), 107 (2),
99 (8), 91 (100), 65 (4).
HR-MS (C19H24NO6; MH+): ber. 362.1604; gef. 362.1612.
CHN-Analyse (C19H23NO6): ber. C 63.15, H 6.42, N 3.88; gef. C 62.80, H 6.52, N 3.81.
(E)-2-Benzyloxycarbonylamino-6,9-dioxonon-4-ensäuremethylester (19h)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden aus
50 mg (190 µmol) Homoallylamin 17a, 22 mg (190 µmol)
Enon 18h und 6 mg (5 mol%) 5 nach 16 h 41 mg (62%)
Produkt als bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 9.81 (s, 1H), 7.35 (m, 5H), 6.73 (dt, J = 16, 7 Hz, 1H), 6.16
(d, J = 16 Hz, 1H), 5.39 (br d, J = 7 Hz, 1H), 5.11 (s, 2H), 4.55 (m, 1H), 3.77 (s, 3H), 2.90-
2.60 (m, 6H). 13C-NMR (125 MHz, CDCl3): δ = 200.5 (CH), 197.3 (Cq), 171.5 (Cq), 155.7 (Cq), 140.7
(CH), 136.1 (Cq), 133.0 (CH), 128.6 (CH), 128.3 (CH), 128.2 (CH), 67.2 (CH2), 53.0 (CH),
52.8 (CH3), 37.4 (CH2), 35.7 (CH2), 32.2 (CH2).
IR (ATR): ν = 3335 (br), 2924 (w), 2852 (w), 1715 (ss), 1700 (ss), 1673 (s), 1633 (m), 1525
(s), 1437 (m), 1344 (m), 1212 (ss), 1049 (s), 1027 (m), 980 (m), 740 (m), 699 (m).
LR-MS (140 °C): m/z = 348 (< 1), 317 (1), 287 (4), 269 (4), 241 (6), 213 (16), 156 (3), 126
(12), 91 (100), 65 (4).
HR-MS (C18H22NO6; MH+): ber. 348.1447; gef. 348.1455.
NHZMeO2C
O
O
NHZMeO2C
O
O

II. Experimenteller Teil
65
(E)-2-Benzyloxycarbonylamino-8-([1.3]dioxolan-2-yl)-6-oxooct-4-ensäuremethylester
(19i)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden
aus 100 mg (380 µmol) Homoallylamin 17a, 60 mg (380
µmol) Enon 33 und 12 mg (5 mol%) 5 nach 16 h 96 mg
(65%) Produkt als bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.35 (m, 5H), 6.67 (dt, J = 16, 8 Hz, 1H), 6.13 (d, J = 16
Hz, 1H), 5.43 (br d, J = 6 Hz, 1H), 5.10 (s, 2H), 4.90 (t, J = 4.3 Hz, 1H), 4.54 (m, 1H), 3.95-
3.80 (m, 4H), 3.75 (s, 3H), 2.80-2.60 (m, 2H), 2.64 (t, J = 7 Hz, 2H), 1.97 (m, 2H). 13C-NMR (125 MHz, CDCl3): δ = 198.9 (Cq), 171.5 (Cq), 155.7 (Cq), 139.9 (CH), 136.1 (Cq),
133.5 (CH), 128.6 (CH), 128.3 (CH), 128.2 (CH), 103.4 (CH), 67.2 (CH2), 65.0 (CH2), 53.0
(CH), 52.8 (CH3), 35.5 (CH2), 34.0 (CH2), 27.8 (CH2).
IR (ATR): ν = 3328 (br), 3033 (w), 2954 (m), 2889 (m), 2730 (w), 1720 (ss), 1700 (ss), 1674
(s), 1633 (m), 1526 (s), 1437 (m), 1347 (m), 1259 (s), 1213 (ss), 1139 (s), 1028 (s), 980 (s),
904 (m), 741 (m), 699 (m).
LR-MS (110 °C): m/z = 391 (< 1), 332 (2), 300 (4), 284 (2), 223 (2), 199 (6), 170 (20), 129
(3), 91 (100), 73 (46), 65 (4).
HR-MS (C20H25NO7; M+): ber. 391.1631; gef. 391.1650.
CHN-Analyse (C20H25NO7): ber. C 61.37, H 6.44, N 3.58; gef. C 60.98, H 6.47, N 3.54.
6-Oxopiperidin-2-carbonsäuremethylester (20a)
Eine Lösung von 88 mg (274 µmol) Aminoester 19a in 4 ml MeOH
wurde entgast, unter Stickstoff-Atmosphäre mit 15 mg Pd/C (10%)
versetzt und 1 h unter Wasserstoff-Atmosphäre gerührt. Der nach
Filtration und Entfernen des Lösungsmittels erhaltene Rückstand
wurde in 4 ml DME gelöst und 5 h refluxiert. Entfernen des Lösungsmittels ergab 42 mg
(97%) Produkt als gelbliches Öl. 1H-NMR (200 MHz, CDCl3): δ = 6.47 (br s, 1H), 4.08 (m, 1H), 3.75 (s, 3H), 2.35 (m, 2H),
2.18 (m, 1H), 1.95-1.75 (m, 3H). 13C-NMR (125 MHz, CDCl3): δ = 171.7 (Cq), 171.5 (Cq), 54.8 (CH), 52.7 (CH3), 31.1 (CH2),
25.4 (CH2), 19.5 (CH2).
IR (ATR): ν = 3231 (br), 2953 (m), 2877 (w), 1736 (ss), 1660 (ss), 1437 (s), 1400 (s), 1297
(s), 1203 (ss), 1163 (s), 1060 (m), 999 (s), 943 (s), 765 (s), 688 (s).
LR-MS (RT): m/z = 157 (4), 99 (6), 98 (100), 70 (8), 69 (2), 59 (3), 55 (60).
NH
MeO2C O
NHZMeO2C
O
O
O

II. Experimenteller Teil
66
HR-MS (C7H11NO3; M+): ber. 157.0739; gef. 157.0731.
(cis)-6-Methylpiperidin-2-carbonsäuremethylester (21a)
Eine Lösung von 65 mg (203 µmol) Aminoenon 19b in 3 ml Et2O
wurde entgast, unter Stickstoff-Atmosphäre mit 11 mg Pd/C (10%)
versetzt und 3 h unter Wasserstoff-Atmosphäre gerührt. Filtration und
Entfernen des Lösemittels ergab 30 mg (91%) Produkt als gelbes Öl. 1H-NMR (200 MHz, CDCl3): δ = 3.71 (s, 3H), 3.37 (dd, J = 11, 3 Hz, 1H), 2.65 (m, 1H),
2.00-1.80 (m, 3H), 1.65-1.57 (m, 1H), 1.47-1.30 (m, 2H), 1.11 (d, J = 6.3 Hz, 3H), 1.15-0.94
(m, 1H). 13C-NMR (125 MHz, CDCl3): δ = 173.8 (Cq), 59.4 (CH), 52.0 (CH), 51.9 (CH3), 33.8 (CH2),
29.0 (CH2), 24.6 (CH2), 22.9 (CH3).
IR (ATR): ν = 2929 (s), 2856 (m), 2793 (w), 1741 (ss), 1437 (m), 1379 (w), 1296 (w), 1215
(s), 1176 (m), 1131 (w), 1057 (w), 1021 (w), 750 (w).
LR-MS (RT): m/z = 157 (1), 142 (6), 99 (8), 91 (100), 82 (8), 70 (12), 55 (18).
HR-MS (C8H15NO2; M+): ber. 157.1103; gef. 157.1103.
(cis)-6-Butylpiperidin-2-carbonsäuremethylester (21b)
Eine Lösung von 88 mg (253 µmol) Aminoenon 19c in 5 ml
Et2O wurde entgast, unter Stickstoff-Atmosphäre mit 13 mg
Pd/C (10%) versetzt und 3 h unter Wasserstoff-Atmosphäre
gerührt. Filtration und Entfernen des Lösungsmittels ergab 48
mg (95%) Produkt als gelbliches Öl. 1H-NMR (200 MHz, CDCl3): δ = 3.70 (s, 3H), 3.33 (dd, J = 11, 3 Hz, 1H), 2.45 (m, 1H),
2.00-1.80 (m, 3H), 1.70-1.60 (m, 1H), 1.50-1.20 (m, 9H), 1.10-0.95 (m, 1H), 0.88 (t, J = 6 Hz,
3H). 13C-NMR (125 MHz, CDCl3): δ = 173.9 (Cq), 59.4 (CH), 56.5 (CH), 52.0 (CH3), 36.9 (CH2),
32.1 (CH2), 29.4 (CH2), 28.1 (CH2), 24.6 (CH2), 22.9 (CH3), 14.1 (CH3).
IR (ATR): ν = 2930 (ss), 2857 (s), 2789 (w), 1743 (ss), 1436 (s), 1378 (w), 1296 (m), 1206
(s), 1159 (w), 1130 (w), 1057 (w), 1017 (w), 750 (w).
LR-MS (RT): m/z = 199 (< 1), 142 (76), 140 (100), 112 (4), 96 (3), 82 (30), 68 (6), 55 (18).
HR-MS (C11H21NO2; M+): ber. 199.1572; gef. 199.1576.
NH
MeO2C
NH
MeO2C

II. Experimenteller Teil
67
(cis)-6-[3-(tButyldimethylsilyloxy)-propyl]-piperidin-2-carbonsäuremethylester (21c)
Eine Lösung von 67 mg (145 µmol) Aminoenon 19d in 2.5
ml MeOH wurde entgast, unter Stickstoff-Atmosphäre mit
5 mg Pd/C (10%) versetzt und 3 h unter Wasserstoff-
Atmosphäre gerührt. Filtration und Entfernen des Lösungs-
mittels ergab 44 mg (96%) Produkt als gelbliches Öl. 1H-NMR (200 MHz, CDCl3): δ = 3.71 (s, 3H), 3.61 (t, J = 6.3 Hz, 2H), 3.36 (dd, J = 11, 3
Hz, 1H), 2.52 (m, 1H), 2.00 (m, 1H), 1.90 (m, 1H), 1.70-1.35 (m, 8H), 1.10-0.95 (m, 1H),
0.88 (s, 9H), 0.04 (s, 6H). 13C-NMR (125 MHz, CDCl3): δ = 173.6 (Cq), 63.4 (CH2), 59.4 (CH), 56.4 (CH), 52.0 (CH3),
33.5 (CH2), 31.8 (CH2), 29.2 (CH2), 29.1 (CH2), 26.0 (CH3), 24.5 (CH2), 18.4 (Cq),-5.2 (CH3).
IR (ATR): ν = 2930 (ss), 2856 (s), 2793 (w), 1744 (ss), 1436 (m), 1361 (w), 1297 (m), 1252
(s), 1205 (s), 1098 (ss), 960 (w), 837 (ss), 775 (s).
LR-MS (60 °C): m/z = 315 (1), 300 (2), 256 (74), 240 (4), 198 (48), 182 (4), 142 (100), 124
(50), 96 (12), 81 (20), 69 (36), 57 (18).
HR-MS (C16H33NO3Si; M+): ber. 315.2229; gef. 315.2230.
CHN-Analyse (C16H33NO3Si): ber. C 60.91, H 10.54, N 4.44; gef. C 61.00, H 10.33, N 4.07.
(cis)-6-Benzylpiperidin-2-carbonsäuremethylester (21d)
Eine Lösung von 43 mg (113 µmol) Aminoenon 19e in 2 ml
MeOH wurde entgast, unter Stickstoff-Atmosphäre mit 4 mg
Pd/C (10%) versetzt und 3 h unter Wasserstoff-Atmosphäre
gerührt. Filtration, Entfernen des Lösungsmittels und säulen-
chromatographische Reinigung des Rückstands (Al2O3, MTB/Hexan 1:5) ergab 19 mg (72%)
Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 7.31-7.20 (m, 5H), 3.68 (s, 3H), 3.27 (dd, J = 11, 3 Hz,
1H), 2.75-2.68 (m, 3H), 2.00-1.86 (m, 3H), 1.65 (m, 1H), 1.44-1.36 (m, 2H), 1.18 (m, 1H). 13C-NMR (125 MHz, CDCl3): δ = 173.7 (Cq), 138.9 (Cq), 129.3 (CH), 128.6 (CH), 125.4
(CH), 59.4 (CH), 57.9 (CH), 52.0 (CH3), 43.7 (CH2), 31.9 (CH2), 29.1 (CH2), 24.5 (CH2).
IR (ATR): ν = 3026 (w), 2933 (s), 2855 (m), 2793 (w), 1740 (ss), 1603 (w), 1495 (m), 1454
(m), 1435 (s), 1371 (m), 1296 (m), 1205 (ss), 1133 (m), 1051 (m), 1010 (m), 749 (s), 700 (ss).
LR-MS (50 °C): m/z = 234 (2), 174 (40), 142 (100), 131 (6), 115 (6), 91 (36), 82 (86), 65 (6),
55 (24).
HR-MS (C14H20NO2; MH+): ber. 234.1494; gef. 234.1500.
NH
MeO2COTBS
NH
MeO2C

II. Experimenteller Teil
68
(cis)-6-(Furan-2-yl)-piperidin-2-carbonsäuremethylester (21e)
Eine Lösung von 40 mg (112 µmol) Aminoenon 19f in 2 ml MeOH
wurde entgast, unter Stickstoff-Atmosphäre mit 3 mg Pd/C (10%)
versetzt und 3 h unter Wasserstoff-Atmosphäre gerührt. Filtration,
Entfernen des Lösungsmittels und säulenchromatographische Rei-
nigung des Rückstands (Al2O3, MTB/Hexan 1:5) ergab 17 mg (74%) Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 7.33 (s, 1H), 6.30 (m, 1H), 6.18 (d, J = 3 Hz, 1H), 3.80 (dd,
J = 11, 2 Hz, 1H), 3.72 (s, 3H), 3.49 (dd, J = 11, 2 Hz, 1H), 2.15 (br s, 1H), 2.07-1.91 (m,
3H), 1.60-1.45 (m, 3H). 13C-NMR (125 MHz, CDCl3): δ = 173.2 (Cq), 156.8 (Cq), 141.5 (CH), 110.1 (CH), 104.8
(CH), 59.1 (CH), 54.3 (CH), 52.1 (CH3), 30.2 (CH2), 28.8 (CH2), 24.3 (CH2).
IR (ATR): ν = 2941 (m), 2856 (m), 2788 (w), 1738 (ss), 1597 (w), 1507 (m), 1434 (s), 1365
(m), 1295 (s), 1210 (ss), 1153 (s), 1056 (w), 1010 (m), 915 (w), 884 (w), 804 (m), 735 (ss).
LR-MS (50 °C): m/z = 209 (20), 180 (1), 150 (92), 122 (22), 107 (100), 101 (8), 94 (16), 86
(52), 79 (6), 58 (8).
HR-MS (C11H15NO3; M+): ber. 209.1052; gef. 209.1055.
(all-cis)-3-Methyloctahydroindolizin-5-carbonsäuremethylester (22a)
Eine Lösung von 40 mg (111 µmol) Aminoenon 19g in 2 ml MeOH
wurde entgast, unter Stickstoff-Atmosphäre mit 3 mg Pd/C (10%)
versetzt und 16 h unter Wasserstoff-Atmosphäre gerührt. Filtration,
Entfernen des Lösungsmittels und säulenchromatographische Reinigung
des Rückstands (Al2O3, MTB/Hexan 1:5) ergab 17 mg (77%) Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 3.70 (s, 3H), 2.73 (dd, J = 11, 3 Hz, 1H), 2.33 (m, 1H), 1.96
(m, 1H), 1.85-1.65 (m, 6H), 1.49 (m, 1H), 1.40-1.25 (m, 3H), 1.00 (d, J = 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 175.0 (Cq), 67.4 (CH), 66.1 (CH), 61.2 (CH), 51.6 (CH3),
30.9 (CH2), 30.4 (CH2), 30.2 (CH2), 28.1 (CH2), 23.8 (CH2), 19.8 (CH3).
IR (ATR): ν = 2944 (ss), 2862 (s), 2807 (s), 2769 (w), 2731 (w), 1750 (ss), 1734 (ss), 1462
(m), 1435 (m), 1374 (m), 1314 (m), 1277 (s), 1195 (s), 1162 (ss), 1110 (s), 1091 (m), 1031
(m), 822 (w), 774 (w), 736 (w).
LR-MS (60 °C): m/z = 197 (2), 182 (16), 138 (100), 122 (4), 110 (8), 95 (2), 82 (4), 55 (8).
HR-MS (C11H19NO2; M+): ber. 197.1416; gef. 197.1419.
NH
MeO2CO
NMeO2C

II. Experimenteller Teil
69
(cis)-Octahydroindolizin-5-carbonsäuremethylester (22b)
Eine Lösung von 35 mg (90 µmol) Aminoenon 19i in 2 ml MeOH
wurde entgast, unter Stickstoff-Atmosphäre mit 2 mg Pd/C (10%)
versetzt und 4 h unter Wasserstoff-Atmosphäre gerührt. Nach Zugabe
von 0.5 ml 3 N HCl wurde weitere 12 h gerührt, mit 5 ml gesättigter
NaHCO3-Lösung neutralisiert und mit CH2Cl2 (3 x 5 ml) extrahiert. Trocknen der organi-
schen Phasen über Na2SO4, Entfernen des Lösemittels und säulenchromatographische Reini-
gung des Rückstands (Al2O3, MTB/Hexan 1:5) ergab 13 mg (81%) Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 3.72 (s, 3H), 3.19 (dt, J = 8, 2 Hz, 1H), 2.77 (dd, J = 11, 2
Hz, 1H), 1.97 (m, 1H), 1.90-1.75 (m, 6H), 1.70-1.60 (m, 2H), 1.50 (m, 1H), 1.40-1.25 (m,
2H). 13C-NMR (125 MHz, CDCl3): δ = 173.8 (Cq), 67.4 (CH), 64.2 (CH), 52.3 (CH2), 51.9 (CH3),
30.1 (CH2), 29.8 (CH2), 29.7 (CH2), 24.3 (CH2), 20.5 (CH2).
IR (ATR): ν = 2934 (ss), 2858 (m), 2793 (m), 2715 (w), 1746 (ss), 1436 (m), 1379 (m), 1276
(s), 1197 (ss), 1166 (ss), 1153 (ss), 1068 (m), 1024 (w), 813 (w).
LR-MS (RT): m/z = 182 (1), 124 (100), 96 (16), 67 (2), 55 (4).
HR-MS (C10H16NO2; M+– H): ber. 182.1181; gef. 182.1185.
2.2. Zu Kapitel 2.3.
Allgemeine Versuchsvorschrift B für die Kreuzmetathese von Allylaminen:
Eine Lösung von Allylamin (0.05 M) und α,β-ungesättigter Carbonylverbindung (1–2 eq) in
absolutem CH2Cl2 unter Stickstoff-Atmosphäre wurde mit dem Ruthenium-Katalysator 5 (10
mol%) versetzt und 48 h bei 40 °C gerührt. Das Lösungsmittel wurde entfernt und der
Rückstand säulenchromatographisch gereinigt (SiO2, MTB/Hexan).
(E)-4-Benzyloxycarbonylamino-5-(benzyloxy)-pent-2-encarbonsäuremethylester (19j)
Entsprechend der allgemeinen Arbeitsvorschrift B wurden aus 50
mg (160 µmol) Allylamin 17b, 27 mg (320 µmol) Methylacrylat
(18a) und 10 mg (10 mol%) 5 42 mg (71%) Produkt als bräunliches
Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.36-7.26 (m, 10H), 6.94 (dd, J = 16, 5 Hz, 1H), 6.00 (d, J
= 16 Hz, 1H), 5.28 (br s, 1H), 5.11 (m, 2H), 4.55-4.50 (m, 3H), 3.74 (s, 3H), 3.60 (m, 2H).
NHZBnO
OOMe
NMeO2C

II. Experimenteller Teil
70
13C-NMR (125 MHz, CDCl3): δ = 166.5 (Cq), 155.8 (Cq), 145.8 (CH), 137.4 (Cq), 136.3 (Cq),
128.6 (CH), 128.5 (CH), 128.3 (CH), 128.2 (CH), 128.0 (CH), 127.8 (CH), 122.0 (CH), 73.5
(CH2), 71.1 (CH2), 67.1 (CH2), 52.0 (CH), 51.7 (CH3).
IR (ATR): ν = 3327 (br), 3032 (w), 2949 (w), 2862 (w), 1720 (ss), 1662 (w), 1524 (s), 1454
(m), 1274 (s), 1241 (s), 1176 (m), 1100 (m), 1050 (m), 980 (w), 737 (m), 698 (s).
LR-MS (150 °C): m/z = 370 (< 1), 248 (26), 218 (8), 204 (22), 172 (8), 91 (100), 65 (10).
HR-MS (C21H24NO5; MH+): ber. 370.1654; gef. 370.1670.
CHN-Analyse (C21H23NO5): ber. C 68.28, H 6.28, N 3.79; gef. C 68.15, H 6.32, N 3.56.
[(E)-1-(Benzyloxymethyl)-4-oxopent-2-enyl]-carbaminsäurebenzylester (19k)
Entsprechend der allgemeinen Arbeitsvorschrift B wurden aus 50 mg
(160 µmol) Allylamin 17b, 23 mg (320 µmol) Methylvinylketon (18b)
und 10 mg (10 mol%) 5 44 mg (77%) Produkt als bräunliches Harz
erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.36-7.26 (m, 10H), 6.74 (dd, J = 16, 5 Hz, 1H), 6.21 (d, J
= 16 Hz, 1H), 5.29 (br s, 1H), 5.11 (m, 2H), 4.55-4.50 (m, 3H), 3.62 (m, 2H), 2.25 (s, 3H). 13C-NMR (125 MHz, CDCl3): δ = 198.0 (Cq), 155.8 (Cq), 144.3 (CH), 137.4 (Cq), 136.2 (Cq),
131.4 (CH), 128.7 (CH), 128.6 (CH), 128.3 (CH), 128.2 (CH), 128.1 (CH), 127.8 (CH), 73.5
(CH2), 71.0 (CH2), 67.2 (CH2), 52.0 (CH), 27.5 (CH3).
IR (ATR): ν = 3321 (br), 3032 (w), 2925 (w), 2862 (w), 1699 (ss), 1678 (ss), 1631 (w), 1525
(s), 1454 (m), 1361 (m), 1253 (s), 1239 (s), 1096 (s), 1027 (m), 979 (w), 740 (s), 698 (s).
LR-MS (140 °C): m/z = 353 (< 1), 322 (1), 280 (2), 232 (6), 188 (6), 91 (100), 65 (4), 51 (2).
HR-MS (C21H23NO4; M+): ber. 353.1627; gef. 353.1621.
CHN-Analyse (C21H23NO4): ber. C 71.37, H 6.56, N 3.96; gef. C 70.87, H 6.49, N 3.85.
5-(Benzyloxymethyl)-pyrrolidin-2-on (20b)
Eine Lösung von 42 mg (144 µmol) Aminoester 19j in 2.3 ml MeOH
wurde entgast, unter Stickstoff-Atmosphäre mit 6 mg Pd/C (10%)
versetzt und 1 h unter Wasserstoff-Atmosphäre gerührt. Der Kataly-
sator wurde abfiltriert und die Lösung 5 h refluxiert. Entfernen des Lösungsmittels und
säulenchromatographische Reinigung des Rückstands (SiO2; EtOAc) ergab 21 mg (91%)
Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 7.31 (m, 5H), 6.10 (br s, 1H), 4.52 (s, 2H), 3.86 (m, 1H),
3.49 (m, 1H), 3.32 (m, 1H), 2.33 (m, 2H), 2.18 (m, 1H), 1.74 (m, 1H).
NHZBnO
O
BnONH
O

II. Experimenteller Teil
71
13C-NMR (125 MHz, CDCl3): δ = 177.9 (Cq), 137.7 (Cq), 128.6 (CH), 128.0 (CH), 127.8
(CH), 74.0 (CH2), 73.5 (CH2), 53.8 (CH), 29.7 (CH2), 23.2 (CH2).
IR (ATR): ν = 3225 (br), 2923 (w), 2856 (w), 1686 (ss), 1453 (m), 1381 (w), 1317 (w), 1262
(w), 1206 (w), 1104 (s), 1089 (s), 1028 (w), 735 (s), 697 (s).
LR-MS (50 °C): m/z = 205 (< 1), 99 (18), 91 (20), 84 (100), 65 (4), 56 (2).
HR-MS (C12H15NO2; M+): ber. 205.1103; gef. 205.1107.
2-(Benzyloxymethyl)-5-methylpyrrolidin (21f)
Eine Lösung von 90 mg (255 µmol) Aminoenon 19k in 5 ml MeOH und
0.5 ml AcOH wurde entgast, unter Stickstoff-Atmosphäre mit 15 mg
Pd/C (10%) versetzt und 5 h unter Wasserstoff-Atmosphäre gerührt. Der
nach Filtration und Entfernen des Lösungsmittels erhaltene Rückstand wurde in 10 ml CH2Cl2
gelöst und mit 5 ml gesättigter NaHCO3-Lösung gewaschen. Trocknen der organischen
Phase, Entfernen des Lösungsmittels und säulenchromatographische Reinigung des Rück-
stands (Al2O3; MTB/Hexan 1:5) ergab 38 mg (72%) Produkt (cis/trans 3:4) als farbloses Öl. 1H-NMR (500 MHz, CDCl3): cis: δ = 7.33 (m, 5H), 4.52 (m, 2H), 3.51 (m, 1H), 3.41 (m,
1H), 3.29 (m, 1H), 3.10 (m, 1H), 2.05 (s, 1H), 1.82 (m, 2H), 1.50 (m, 1H), 1.26 (m, 1H), 1.15
(d, J = 6 Hz, 3H). trans: δ = 7.33 (m, 5H), 4.52 (m, 2H), 3.51 (m, 1H), 3.37 (m, 2H), 3.22 (m,
1H), 2.05 (s, 1H), 1.93 (m, 2H), 1.49 (m, 1H), 1.26 (m, 1H), 1.12 (d, J = 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): cis: δ = 138.5 (Cq), 128.4 (CH), 127.7 (CH), 127.6 (CH), 74.3
(CH2), 73.3 (CH2), 58.5 (CH), 54.7 (CH), 33.0 (CH2), 28.1 (CH2), 21.3 (CH3). trans: δ =
138.5 (Cq), 128.4 (CH), 127.7 (CH), 127.6 (CH), 74.1 (CH2), 73.2 (CH2), 57.1 (CH), 53.2
(CH), 34.1 (CH2), 28.6 (CH2), 21.6 (CH3).
IR (ATR): ν = 2958 (s), 2926 (m), 2865 (s), 1496 (w), 1453 (s), 1365 (m), 1206 (w), 1100
(ss), 1028 (m), 736 (ss), 697 (ss).
LR-MS (RT): m/z = 205 (< 1), 149 (1), 137 (2), 91 (16), 84 (100), 69 (8), 57 (6).
HR-MS (C13H19NO; M+): ber. 205.1467; gef. 205.1455.
[(E)-1-(tButyldimethylsilyloxymethyl)-4-oxopent-2-enyl]-carbaminsäurebenzylester (19l)
Entsprechend der allgemeinen Arbeitsvorschrift B wurden aus 100 mg
(300 µmol) Allylamin 17c, 42 mg (600 µmol) Methylvinylketon (18b)
und 18 mg (10 mol%) 5 91 mg (81%) Produkt als bräunliches Harz
erhalten. NHZTBSO
O
BnONH

II. Experimenteller Teil
72
1H-NMR (500 MHz, CDCl3): δ = 7.33 (m, 5H), 6.73 (dd, J = 16, 5 Hz, 1H), 6.20 (d, J = 16
Hz, 1H), 5.24 (br s, 1H), 5.12 (m, 2H), 4.44 (m, 1H), 3.76 (m, 2H), 2.25 (s, 3H), 0.87 (s, 9H),
0.04 (s, 6H). 13C-NMR (125 MHz, CDCl3): δ = 197.9 (Cq), 155.8 (Cq), 144.7 (CH), 136.3 (Cq), 131.1
(CH), 128.6 (CH), 128.3 (CH), 67.1 (CH2), 64.7 (CH2), 53.6 (CH), 27.4 (CH3), 25.8 (CH3),
18.3 (Cq), -5.4 (CH3).
IR (ATR): ν = 3326 (br), 2954 (m), 2929 (m), 2857 (m), 1701 (ss), 1680 (ss), 1527 (s), 1464
(w), 1361 (w), 1253 (ss), 1110 (s), 980 (m), 837 (ss), 778 (s), 697 (m).
LR-MS (100 °C): m/z = 377 (< 1), 347 (6), 320 (12), 276 (4), 230 (4), 212 (3), 115 (4), 91
(100), 73 (20).
HR-MS (C20H31NO4Si; M+): ber. 377.2022; gef. 377.2020.
[(E)-1-(tButyldimethylsilyloxymethyl)-4-oxonon-2-enyl]carbaminsäurebenzylester (19m)
Entsprechend der allgemeinen Arbeitsvorschrift B wurden aus
157 mg (467 µmol) Allylamin 17c, 59 mg (467 µmol) n-Pentyl-
vinylketon (18i) und 29 mg (10 mol%) 5 158 mg (78%) Produkt
als bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.33 (m, 5H), 6.75 (dd, J = 16, 5 Hz, 1H), 6.22 (d, J = 16
Hz, 1H), 5.20 (br s, 1H), 5.12 (m, 2H), 4.43 (m, 1H), 3.76 (m, 2H), 2.52 (m, 2H), 1.60 (q, J =
7 Hz, 2H), 1.30 (m, 4H), 0.89 (t, J = 6.8 Hz, 3H), 0.87 (s, 9H), 0.04 (s, 6H). 13C-NMR (125 MHz, CDCl3): δ = 200.2 (Cq), 155.8 (Cq), 143.4 (CH), 136.3 (Cq), 130.2
(CH), 128.6 (CH), 128.3 (CH), 67.1 (CH2), 64.7 (CH2), 53.7 (CH), 40.7 (CH2), 31.5 (CH2),
25.8 (CH3), 23.8 (CH2), 22.5 (CH2), 18.3 (Cq), 14.0 (CH3), -5.4 (CH3).
IR (ATR): ν = 3325 (br), 2955 (s), 2929 (s), 2858 (s), 1700 (ss), 1678 (s), 1527 (s), 1464 (m),
1361 (w), 1254 (ss), 1110 (s), 981 (m), 836 (ss), 778 (s), 697 (m).
LR-MS (120 °C): m/z = 433 (< 1), 403 (4), 376 (14), 332 (6), 268 (2), 225 (3), 115 (4), 91
(100), 73 (16).
HR-MS (C24H39NO4Si; M+): ber. 433.2648; gef. 433.2657.
2-(tButyldimethylsilyloxymethyl)-5-methylpyrrolidin (21g)
Eine Lösung von 50 mg (132 µmol) Aminoenon 19l in 2.5 ml MeOH
wurde entgast, unter Stickstoff-Atmosphäre mit 10 mg Pd(OH)2 (20%)
versetzt und 5 h unter Wasserstoff-Atmosphäre gerührt. Filtration,
NHZTBSO
O
TBSONH

II. Experimenteller Teil
73
Entfernen des Lösungsmittels und säulenchromatographische Reinigung des Rückstands
(Al2O3; MTB/Hexan 1:5) ergab 17 mg (87%) Produkt (cis/trans 4:1) als farbloses Öl. 1H-NMR (500 MHz, CDCl3): cis: δ = 3.63 (dd, J = 10, 4.7 Hz, 1H), 3.55 (dd, J = 10, 4.7 Hz,
1H), 3.16-3.08 (m, 2H), 1.90-1.70 (m, 3H), 1.55 (m, 1H), 1.25 (m, 1H), 1.15 (d, J = 6.3 Hz,
3H), 0.89 (s, 9H), 0.05 (s, 6H). trans: δ = 3.48 (m, 2H), 3.33 (m, 1H), 3.23 (m, 1H), 1.90-1.70
(m, 3H), 1.43 (m, 1H), 1.30 (m, 1H), 1.11 (d, J = 6.3 Hz, 3H), 0.89 (s, 9H), 0.05 (s, 6H). 13C-NMR (125 MHz, CDCl3): cis: δ = 65.9 (CH2), 60.6 (CH), 55.0 (CH), 33.7 (CH2), 27.9
(CH2), 26.0 (CH3), 21.2 (CH3), 18.4 (Cq), -5.3 (CH3). trans: δ = 66.2 (CH2), 59.3 (CH), 53.0
(CH), 34.0 (CH2), 28.0 (CH2), 26.0 (CH3), 21.7 (CH3), 18.4 (Cq), -5.3 (CH3).
IR (ATR): ν = 2956 (ss), 2928 (ss), 2857 (s), 1472 (m), 1463 (m), 1361 (w), 1254 (m), 1097
(s), 1006 (w), 836 (ss), 776 (ss).
LR-MS (RT): m/z = 229 (< 1), 214 (2), 172 (10), 116 (1), 98 (2), 84 (100), 73 (4).
HR-MS (C12H27NOSi; M+): ber. 229.1862; gef. 229.1867.
2-(tButyldimethylsilyloxymethyl)-5-pentylpyrrolidin (21h)
Eine Lösung von 43 mg (100 µmol) Aminoenon 19m in 2 ml MeOH
wurde entgast, unter Stickstoff-Atmosphäre mit 8 mg Pd(OH)2
(20%) versetzt und 5 h unter Wasserstoff-Atmosphäre gerührt.
Filtration, Entfernen des Lösungsmittels und säulenchromatogra-
phische Reinigung des Rückstands (Al2O3; MTB/Hexan 1:5) ergab 23 mg (81%) Produkt
(cis/trans 6:1) als farbloses Öl. 1H-NMR (500 MHz, CDCl3): cis: δ = 3.62 (dd, J = 10, 4.7 Hz, 1H), 3.54 (dd, J = 10, 4.7 Hz,
1H), 3.14 (m, 1H), 2.97 (m, 1H), 1.90-1.70 (m, 3H), 1.50 (m, 1H), 1.40-1.20 (m, 9H), 0.89 (s,
9H), 0.88 (t, J = 7 Hz, 3H), 0.05 (s, 6H). trans: δ = 3.46 (d, J = 6 Hz, 2H), 3.30 (m, 1H), 3.05
(m, 1H), 1.90-1.70 (m, 3H), 1.50 (m, 1H), 1.40-1.20 (m, 9H), 0.89 (s, 9H), 0.88 (t, J = 7 Hz,
3H), 0.05 (s, 6H). 13C-NMR (125 MHz, CDCl3): cis: δ = 66.1 (CH2), 60.3 (CH), 59.8 (CH), 36.6 (CH2), 32.1
(CH2), 31.8 (CH2), 27.5 (CH2), 27.2 (CH2), 26.0 (CH3), 22.7 (CH2), 18.4 (Cq), 14.1 (CH3), -
5.3 (CH3). trans: δ = 66.2 (CH2), 59.1 (CH), 57.6 (CH), 37.0 (CH2), 32.0 (CH2), 31.8 (CH2),
27.7 (CH2), 27.1 (CH2), 26.1 (CH3), 22.8 (CH2), 18.4 (Cq), 14.1 (CH3), -5.3 (CH3).
IR (ATR): ν = 2956 (ss), 2928 (ss), 2857 (s), 1472 (m), 1463 (m), 1361 (w), 1255 (m), 1089
(s), 1006 (w), 837 (ss), 776 (s).
LR-MS (RT): m/z = 270 (4), 228 (12), 214 (5), 198 (1), 156 (1), 140 (100), 116 (2), 73 (2).
HR-MS (C15H32NOSi; M+– CH3): ber. 270.2253; gef. 270.2254.
TBSONH

II. Experimenteller Teil
74
2.3. Zu Kapitel 2.4.
[(E)-7-([1.3]Dioxolan-2-yl)-1-hexyl-5-oxohept-3-enyl]-carbaminsäurebenzylester (42)
Entsprechend der allgemeinen Arbeitsvorschrift A wurden aus
100 mg (345 µmol) Homoallylamin 40, 54 mg (345 µmol)
Enon 33 und 11 mg (5 mol%) 5 nach 16 h 113 mg (78%)
Produkt als bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 7.31 (m, 5H), 6.77 (dt, J = 16, 7 Hz, 1H), 6.11 (d, J = 16
Hz, 1H), 5.07 (s, 2H), 4.91 (t, J = 4.3 Hz, 1H), 4.62 (br d, J = 8 Hz, 1H), 3.95-3.80 (m, 4H),
3.78 (m, 1H), 2.64 (m, 2H), 2.45-2.30 (m, 2H), 1.98 (m, 2H), 1.50-1.20 (m, 10H), 0.87 (t, J =
7 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 199.2 (Cq), 155.9 (Cq), 142.7 (CH), 136.6 (Cq), 132.7
(CH), 128.6 (CH), 128.2 (CH), 128.1 (CH), 103.5 (CH), 66.7 (CH2), 65.0 (CH2), 50.5 (CH),
38.4 (CH2), 34.8 (CH2), 33.9 (CH2), 31.8 (CH2), 29.1 (CH2), 27.9 (CH2), 25.9 (CH2), 22.6
(CH2), 14.1 (CH3).
IR (ATR): ν = 3330 (br), 3033 (w), 2954 (s), 2930 (ss), 2857 (s), 1719 (ss), 1698 (ss), 1675
(s), 1630 (m), 1530 (s), 1455 (w), 1375 (w), 1241 (s), 1140 (s), 1037 (s), 1028 (s), 979 (m),
905 (w), 739 (m), 698 (m).
LR-MS (140 °C): m/z = 417 (< 1), 262 (1), 248 (10), 218 (2), 204 (26), 170 (6), 129 (1), 91
(100), 73 (16), 65 (2).
HR-MS (C24H35NO5; M+): ber. 417.2515; gef. 417.2526.
CHN-Analyse (C24H35NO5): ber. C 69.04, H 8.45, N 3.36; gef. C 68.89, H 8.39, N 3.05.
(±)-Indolizidin 209D (34)
Eine Lösung von 50 mg (120 µmol) Aminoenon 42 in 2.5 ml MeOH wurde
entgast, unter Stickstoff-Atmosphäre mit 6 mg Pd/C (10%) versetzt und 4 h
unter Wasserstoff-Atmosphäre gerührt. Nach Zugabe von 0.5 ml 3 N HCl
wurde weitere 20 h gerührt, mit 5 ml gesättigter NaHCO3-Lösung neutra-
lisiert und mit CH2Cl2 (3 x 5 ml) extrahiert. Trocknen der organischen
Phasen über Na2SO4, Entfernen des Lösungsmittels und säulenchromatographische Reinigung
des Rückstands (Al2O3, MTB/Hexan 1:5) ergab 22 mg (88%) Produkt als farbloses Öl. 1H-NMR (200 MHz, CDCl3): δ = 3.25 (dt, J = 8, 2 Hz, 1H), 2.00-1.50 (m, 10H), 1.50-1.10
(m, 13H), 0.87 (t, J = 7 Hz, 3H).
NHZ
O
O
O
N

II. Experimenteller Teil
75
13C-NMR (125 MHz, CDCl3): δ = 65.2 (CH), 64.0 (CH), 51.6 (CH2), 34.6 (CH2), 31.9 (CH2),
31.0 (CH2), 30.9 (CH2), 30.6 (CH2), 29.8 (CH2), 25.9 (CH2), 24.7 (CH2), 22.7 (CH2), 20.5
(CH2), 14.2 (CH3).
IR (ATR): ν = 3324 (br), 2927 (ss), 2857 (ss), 2780 (s), 2708 (m), 2670 (w), 2595 (w), 1742
(w), 1457 (s), 1380 (m), 1332 (w), 1227 (w), 1173 (w), 1128 (s), 1052 (m), 810 (w), 724 (w).
LR-MS (50 °C): m/z = 208 (82), 184 (50), 170 (68), 154 (36), 142 (28), 124 (100), 96 (14),
82 (8), 69 (8), 55 (12).
HR-MS (C14H26N; M+– H): ber. 208.2065; gef. 208.2069.
2.4. Zu Kapitel 2.5.
2-[(R)-1-Methylbut-3-enyl]-isoindol-1,3-dion (56)
Eine Lösung von 430 mg (5.0 mmol) (S)-4-Penten-2-ol (53), 810 mg (5.5
mmol) Phthalimid und 1.45 g (5.5 mmol) PPh3 in 20 ml absolutem THF unter
Stickstoff-Atmosphäre wurde bei 0 °C tropfenweise mit 960 mg (5.5 mmol)
DEAD versetzt und über 1 h auf RT erwärmt. Das Reaktionsgemisch wurde mit Et2O
verdünnt und das Präzipitat abfiltriert. Entfernung des Lösungsmittels und säulenchromato-
graphische Reinigung des Rückstands (SiO2, MTB/Hexan 1:10) ergab 880 mg (82%) Produkt
als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 7.80 (m, 2H), 7.69 (m, 2H), 5.71 (m, 1H), 5.06-4.95 (m,
2H), 4.44 (m, 1H), 2.80 (m, 1H), 2.51 (m, 1H), 1.50 (d, J = 7 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 168.5 (Cq), 134.8 (CH), 133.8 (CH), 132.0 (Cq), 123.1
(CH), 117.8 (CH2), 47.1 (CH), 38.3 (CH2), 18.4 (CH3).
IR (ATR): ν = 2978 (w), 2938 (w), 1773 (m), 1706 (ss), 1468 (w), 1394 (s), 1365 (s), 1333
(m), 1060 (w), 920 (w), 873 (w), 720 (s).
LR-MS (100 °C): m/z = 215 (< 1), 174 (100), 147 (8), 130 (22), 104 (6), 76 (10), 51 (2).
HR-MS (C13H13NO2; M+): ber. 215.0946; gef. 215.0943.
CHN-Analyse (C13H13NO2): ber. C 72.54, H 6.09, N 6.51, gef. C 72.28, H 5.85, N 6.43.
Drehwert: [α]D20 = –19.5° (c = 1.0, CHCl3).
[(R)-1-Methylbut-3-enyl]-carbaminsäurebenzylester (52)
Eine Lösung von 500 mg (2.32 mmol) Phthalimid 56 in 17 ml iPrOH und 3 ml
H2O wurde mit 440 mg (11.6 mmol) NaBH4 versetzt und 16 h bei RT gerührt.
Das Reaktionsgemisch wurde mit 1.5 ml 32%iger HCl angesäuert (pH 1) und
NPhth
NHZ

II. Experimenteller Teil
76
anschließend 2 h bei 80 °C gerührt. Nach Abkühlen auf RT und Neutralisation mit K2CO3
(pH ~ 8) wurden 475 mg (2.78 mmol) ZCl tropfenweise hinzugegeben und es wurde weitere 2
h bei RT gerührt. Das Reaktionsgemisch wurde mit 30 ml gesättigter NaCl-Lösung verdünnt
und mit Et2O (3 x 30 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4,
Entfernen des Lösungsmittels und säulenchromatographische Reinigung des Rückstands
(SiO2, MTB/Hexan 1:5) ergab 410 mg (81%) Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 7.35 (m, 5H), 5.77 (m, 1H), 5.08 (m, 4H), 4.62 (br s, 1H),
3.80 (m, 1H), 2.22 (m, 2H), 1.16 (d, J = 6.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 155.7 (Cq), 136.7 (Cq), 134.3 (CH), 128.6 (CH), 128.1
(CH), 118.0 (CH2), 66.6 (CH2), 46.6 (CH), 41.2 (CH2), 20.6 (CH3).
IR (ATR): ν = 3327 (br), 3034 (w), 2973 (w), 2933 (w), 1695 (ss), 1533 (ss), 1454 (m), 1335
(m), 1255 (s), 1223 (s), 1061 (s), 916 (m), 783 (m), 697 (m).
LR-MS (50 °C): m/z = 178 (10), 149 (2), 134 (10), 91 (100), 79 (6), 73 (14), 65 (8), 51 (2).
HR-MS (C10H12NO2; M+– C3H5): ber. 178.0868; gef. 178.0861.
CHN-Analyse (C13H17NO2): ber. C 71.26, H 7.82, N 6.39, gef. C 70.96, H 7.54, N 6.50.
Drehwert: [α]D20 = +13.5° (c = 1.0, CHCl3).
(R)-5-Hydroxyhex-1-en-3-on (54a)
Eine Lösung von 100 mg (680 µmol) Amid 57a in 3 ml absolutem Et2O
unter Stickstoff-Atmosphäre wurde bei 0 °C tropfenweise mit 1.7 ml (1 M
in THF, 1.7 mmol) Vinylmagnesiumbromid-Lösung versetzt. Das Reak-
tionsgemisch wurde 2 h bei RT gerührt und nach Zugabe von 5 ml 1 N HCl mit Et2O (3 x 5
ml) extrahiert. Trocknen der organischen Phasen über Na2SO4, Entfernen des Lösungsmittels
und säulenchromatographische Reinigung des Rückstands (SiO2, Et2O/Pentan 1:1) ergab 40
mg (52%) Produkt als gelbliches Öl. 1H-NMR (500 MHz, CDCl3): δ = 6.34 (dd, J = 17.5, 10.5 Hz, 1H), 6.25 (d, J = 17.5 Hz, 1H),
5.90 (d, J = 10.5 Hz, 1H), 4.28 (m, 1H), 3.11 (br s, 1H), 2.79 (dd, J = 17.5, 3 Hz, 1H), 2.68
(dd, J = 17.5, 9 Hz, 1H), 1.22 (d, J = 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 201.4 (Cq), 136.8 (CH), 129.3 (CH2), 63.9 (CH), 47.2
(CH2), 22.4 (CH3).
IR (ATR): ν = 3436 (br), 2971 (s), 2932 (m), 1677 (ss), 1612 (s), 1403 (ss), 1375 (s), 1278
(m), 1191 (m), 1116 (s), 1064 (s), 986 (ss), 941 (s), 825 (w).
LR-MS (RT): m/z = 99 (5), 86 (6), 73 (8), 70 (26), 58 (4), 55 (100), 53 (2).
HR-MS (C5H7O2; M+– CH3): ber. 99.0446; gef. 99.0441.
O OH

II. Experimenteller Teil
77
[(E)-(1R,7R)-7-Hydroxy-1-methyl-5-oxooct-3-enyl]-carbaminsäurebenzylester (51a)
Eine Lösung von 70 mg (320 µmol) Amin 52 und 55 mg (480 µmol)
Enon 54a in 6.4 ml absolutem CH2Cl2 unter Stickstoff-Atmosphäre
wurde mit 10 mg (5 mol%) Ruthenium-Katalysator 5 versetzt und 16
h bei 40 °C gerührt. Entfernen des Lösungsmittels und säulenchro-
matographische Reinigung des Rückstands (SiO2, MTB/Hexan 3:1) ergab 84 mg (86%)
Produkt als bräunliches Harz. 1H-NMR (500 MHz, CDCl3): δ = 7.33 (m, 5H), 6.80 (dt, J = 16, 7.5 Hz, 1H), 6.10 (d, J = 16
Hz, 1H), 5.08 (m, 2H), 4.65 (br s, 1H), 4.23 (m, 1H), 3.92 (m, 1H), 3.19 (br s, 1H), 2.70 (d, J
= 17 Hz, 1H), 2.60 (dd, J = 17, 9 Hz, 1H), 2.41 (m, 2H), 1.21 (d, J = 6.5 Hz, 3H), 1.19 (d, J =
6.8 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 200.7 (Cq), 155.7 (Cq), 144.0 (CH), 136.5 (Cq), 133.0
(CH), 128.6 (CH), 128.3 (CH), 128.2 (CH), 66.8 (CH2), 64.1 (CH), 47.7 (CH2), 46.3 (CH),
40.0 (CH2), 22.5 (CH3), 20.8 (CH3).
IR (ATR): ν = 3324 (br), 3033 (w), 2969 (m), 2931 (w), 1693 (ss), 1659 (s), 1627 (m), 1530
(s), 1454 (m), 1334 (m), 1248 (ss), 1103 (m), 1056 (s), 980 (m), 944 (w), 738 (m), 697 (m).
LR-MS (170 °C): m/z = 306 (< 1), 198 (1), 178 (18), 155 (4), 134 (20), 128 (4), 112 (2), 91
(100), 65 (6).
HR-MS (C17H24NO4; MH+): ber. 306.1705; gef. 306.1716.
CHN-Analyse (C17H23NO4): ber. C 66.90, H 7.60, N 4.59, gef. C 66.82, H 7.34, N 4.53.
Drehwert: [α]D20 = –9.8° (c = 1.4, CHCl3).
(R)-3-(tButyldimethylsilyloxy)-N-methoxy-N-methylbutyramid (57b)
Eine Lösung von 162 mg (1.1 mmol) Amid 57a und 225 mg (3.3 mmol)
Imidazol in 1.5 ml DMF wurde bei 0 °C portionsweise mit 248 mg (1.5
mmol) TBSCl versetzt und 16 h bei RT gerührt. Nach Zugabe von 10 ml
Et2O wurde mit 1 N HCl (2 x 5 ml) gewaschen und über Na2SO4
getrocknet. Entfernen des Lösungsmittels und säulenchromatographische Reinigung des
Rückstands (SiO2, MTB/Hexan 1:2) ergab 265 mg (92%) Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 4.35 (m, 1H), 3.69 (s, 3H), 3.17 (s, 3H), 2.76 (m, 1H), 2.35
(m, 1H), 1.21 (d, J = 6 Hz, 3H), 0.86 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H). 13C-NMR (125 MHz, CDCl3): δ = 66.1 (CH), 61.4 (CH3), 41.8 (CH2), 32.0 (CH3), 25.9
(CH3), 24.2 (CH3), 18.1 (Cq), -4.6 (CH3), -4.8 (CH3).
N
O OTBS
OMe
NHZ
O OH

II. Experimenteller Teil
78
IR (ATR): ν = 2957 (s), 2930 (s), 2857 (m), 1666 (ss), 1473 (m), 1385 (s), 1256 (s), 1136 (s),
1091 (s), 1004 (ss), 939 (w), 835 (ss), 777 (ss).
LR-MS (50 °C): m/z = 246 (5), 214 (4), 204 (100), 159 (12), 155 (10), 129 (6), 115 (10), 89
(26), 73 (34), 56 (8).
HR-MS (C11H24NO3Si; M+– CH3): ber. 246.1525; gef. 246.1520.
(R)-5-(tButyldimethylsilyloxy)-hex-1-en-3-on (54b)
Eine Lösung von 68 mg (260 µmol) Amid 57b in 1.5 ml absolutem THF
unter Stickstoff-Atmosphäre wurde bei 0 °C tropfenweise mit 0.3 ml (1
M in THF, 300 µmol) Vinylmagnesiumbromid-Lösung versetzt. Das
Reaktionsgemisch wurde 2 h bei RT gerührt und nach Zugabe von 3 ml 1 N HCl mit Et2O (3
x 3 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4, Entfernen des Lösungs-
mittels und Filtration über Kieselgel (CH2Cl2) ergab 50 mg (85%) Produkt als gelbliches Öl. 1H-NMR (500 MHz, CDCl3): δ = 6.35 (dd, J = 17.5, 10.5 Hz, 1H), 6.22 (d, J = 17.5 Hz, 1H),
5.84 (d, J = 10.5 Hz, 1H), 4.33 (m, 1H), 2.85 (dd, J = 15, 7 Hz, 1H), 2.54 (dd, J = 15, 5 Hz,
1H), 1.19 (d, J = 6 Hz, 3H) 0.85 (s, 9H), 0.05 (s, 3H), 0.01 (s, 3H). 13C-NMR (125 MHz, CDCl3): δ = 199.8 (Cq), 137.5 (CH), 128.6 (CH2), 65.9 (CH), 49.2
(CH2), 25.9 (CH3), 24.3 (CH3), 18.0 (Cq), -4.5 (CH3), -4.9 (CH3).
IR (ATR): ν = 2958 (m), 2929 (m), 2857 (m), 1684 (m), 1615 (w), 1473 (w), 1375 (m), 1258
(s), 1130 (m), 1080 (s), 1003 (s), 836 (ss), 808 (ss), 776 (ss).
LR-MS (RT): m/z = 227 (< 1), 213 (2), 171 (80), 159 (1), 127 (100), 115 (2), 101 (6), 85
(12), 75 (38), 55 (8).
HR-MS (C12H23O2Si; M+– H): ber. 227.1467; gef. 227.1474.
[(E)-(1R,7R)-7-(tButyldimethylsilyloxy)-1-methyl-5-oxooct-3-enyl]-carbaminsäureben-
zylester (51b)
Eine Lösung von 44 mg (200 µmol) Amin 52 und 46 mg (200 µmol)
Enon 54b in 4 ml absolutem CH2Cl2 unter Stickstoff-Atmosphäre
wurde mit 6 mg (5 mol%) Ruthenium-Katalysator 5 versetzt und 16
h bei 40 °C gerührt. Entfernen des Lösungsmittels und säulenchro-
matographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:3) ergab 67 mg (80%)
Produkt als bräunliches Harz. 1H-NMR (500 MHz, CDCl3): δ = 7.33 (m, 5H), 6.75 (dt, J = 16, 7.5 Hz, 1H), 6.14 (d, J = 16
Hz, 1H), 5.09 (m, 2H), 4.66 (br s, 1H), 4.30 (m, 1H), 3.90 (m, 1H), 2.76 (dd, J = 15, 7 Hz,
O OTBS
NHZ
O OTBS

II. Experimenteller Teil
79
1H), 2.50 (dd, J = 15, 5 Hz, 1H), 2.40 (m, 2H), 1.17 (d, J = 6 Hz, 6H), 0.84 (s, 9H), 0.04 (s,
3H), 0.01 (s, 3H). 13C-NMR (125 MHz, CDCl3): δ = 199.0 (Cq), 155.6 (Cq), 142.7 (CH), 136.5 (Cq), 133.8
(CH), 128.6 (CH), 128.2 (CH), 128.1 (CH), 66.7 (CH2), 66.0 (CH), 49.9 (CH2), 46.3 (CH),
39.7 (CH2), 25.9 (CH3), 24.3 (CH3), 20.7 (CH3), 18.0 (Cq), -4.5 (CH3), -4.8 (CH3).
IR (ATR): ν = 3329 (br), 3034 (w), 2956 (s), 2929 (s), 2856 (m), 1719 (ss), 1697 (ss), 1668
(s), 1628 (m), 1531 (s), 1455 (m), 1375 (m), 1251 (ss), 1130 (m), 1060 (s), 999 (s), 836 (s),
776 (s), 697 (m).
LR-MS (100 °C): m/z = 404 (< 1), 362 (3), 242 (5), 211 (30), 178 (12), 159 (8), 134 (28), 125
(6), 91 (100), 73 (10).
HR-MS (C22H34NO4Si; M+– CH3): ber. 404.2257; gef. 404.2250.
CHN-Analyse (C23H37NO4Si): ber. C 65.89, H 8.89, N 3.34, gef. C 65.69, H 8.76, N 3.18.
Drehwert: [α]D20 = –3.5° (c = 1.0, CHCl3).
(–)-Pinidinol (43)
Aus 51a: Eine Lösung von 28 mg (92 µmol) Aminoenon 51a in 1.8 ml
MeOH wurde entgast, unter Stickstoff-Atmosphäre mit 5 mg Pd/C
(10%) versetzt und 3 h unter Wasserstoff-Atmosphäre gerührt. Filtration
und Entfernen des Lösungsmittels ergab 14 mg (97%) Produkt als
farblosen Feststoff.
Aus 51b: Eine Lösung von 42 mg (100 µmol) Aminoenon 51b in 2 ml MeOH wurde entgast,
unter Stickstoff-Atmosphäre mit 5 mg Pd/C (10%) versetzt und 3 h unter Wasserstoff-
Atmosphäre gerührt. Der nach Filtration und Entfernen des Lösungsmittels erhaltene
Rückstand wurde in 2 ml THF aufgenommen und bei 0 °C tropfenweise mit 0.4 ml 37%iger
HCl versetzt. Nach 15 min wurde mit 5 ml H2O verdünnt und mit CH2Cl2 (2 x 5 ml)
gewaschen bevor mit 5 ml 2 N NaOH basifiziert und mit CH2Cl2 (4 x 5 ml) extrahiert wurde.
Trocknen der organischen Phasen und Entfernen des Lösungsmittels ergab 14 mg (89%)
Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 4.12 (m, 1H), 2.93 (m, 1H), 2.60 (m, 1H), 1.80 (m, 1H),
1.58 (m, 2H), 1.52 (m, 1H), 1.46 (m, 1H), 1.35 (m, 2H), 1.16 (d, J = 6 Hz, 3H), 1.03 (d, J = 6
Hz, 3H), 1.00 (m, 1H). 13C-NMR (125 MHz, CDCl3): δ = 65.1 (CH), 55.1 (CH), 52.6 (CH), 43.8 (CH2), 33.9 (CH2),
30.4 (CH2), 24.8 (CH2), 23.7 (CH3), 23.2 (CH3).
NH
OH

II. Experimenteller Teil
80
IR (ATR): ν = 3272 (br), 2962 (ss), 2927 (ss), 2856 (s), 2722 (w), 1455 (m), 1442 (m), 1374
(m), 1320 (m), 1115 (s), 1058 (m), 1013 (w), 932 (w), 819 (w).
LR-MS (RT): m/z = 157 (2), 142 (10), 124 (3), 112 (4), 98 (100), 82 (6), 70 (12), 55 (10).
HR-MS (C9H19NO; M+): ber. 157.1467; gef. 157.1472.
Drehwert: [α]D20 = –14.0° (c = 0.83, CHCl3).
Schmelzpunkt: 74–75 °C.
2.5. Zu Kapitel 2.6.
(3R,4R)-3,7-Dimethylocta-1,6-dien-4-ol (69a)
10.0 g (65 mmol) verflüssigtes (–)-Isopinocampheol wurden bei einem
Unterdruck von 15 mbar mittels eines Tropftrichters über ca. 1 h in ein
mit Raschigringen gefülltes, auf 480 °C erhitztes Quarzrohr von 80 cm
Länge und 1 cm Breite gegeben. Säulenchromatographische Reinigung
(SiO2, Et2O/Pentan 1:10) des in einer nachgeschaltenen Kühlfalle (Eisbad) kondensierten
Pyrolysats ergab neben 5 g Edukt 2.3 g (23%) Produkt als gelbliche Flüssigkeit, das 5% des
Konstitutionsisomers 69b enthielt. 1H-NMR (500 MHz, CDCl3): δ = 5.81 (m, 1H), 5.17 (t, J = 7 Hz, 1H), 5.10-5.06 (m, 2H) 3.48
(m, 1H), 2.30 (m, 1H), 2.20 (m, 1H), 2.13 (m, 1H), 1.74 (s, 3H), 1.64 (s, 3H), 1.55 (br, 1H),
1.06 (d, J = 7 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 141.2 (CH), 135.2 (Cq), 120.5 (CH), 115.0 (CH2), 74.7
(CH), 43.0 (CH), 33.1 (CH2), 26.0 (CH3), 18.0 (CH3) 14.6 (CH3).
IR (ATR): ν = 3405 (br), 3078 (w), 2968 (ss), 2927 (ss), 2883 (s), 1640 (m), 1452 (s), 1376
(s), 1104 (m), 1045 (s), 996 (ss), 912 (ss), 867 (m).
LR-MS (RT): m/z = 154 (< 1), 139 (2), 136 (8), 98 (12), 81 (60), 70 (100), 55 (46).
HR-MS (C10H18O; M+): ber. 154.1358; gef. 154.1351.
Drehwert: [α]D20 = +36.7° (c = 1.5, CHCl3).
(5R,6R)-2,6-Dimethyloct-7-en-2,5-diol (70)
Eine Lösung von 154 mg (1.0 mmol) Alkohol 69a in 3 ml CHCl3 wurde
mit 260 mg (1.05 mmol) 70%iger mCPBA versetzt und 1 h bei RT
gerührt. Nach Zugabe von 10 ml CH2Cl2 wurde mit 10 ml gesättigter
NaHCO3-Lösung gewaschen und die organische Phase wurde über
Na2SO4 getrocknet. Der nach Entfernen des Lösungsmittels erhaltene Rückstand wurde in 5
OH
OHOH

II. Experimenteller Teil
81
ml absolutem THF gelöst, unter Stickstoff-Atmosphäre mit 114 mg (3.0 mmol) LAH versetzt
und 4 h refluxiert. Das Reaktionsgemisch wurde abgekühlt, mit 5 ml 1 N HCl versetzt und mit
CH2Cl2 (4 x 5 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4, Entfernen des
Lösungsmittels und Filtration über Kieselgel (Et2O) ergab 155 mg (90%) Produkt als
farbloses Harz. 1H-NMR (500 MHz, CDCl3): δ = 5.79 (m, 1H), 5.10-5.06 (m, 2H) 3.49 (m, 1H), 2.29 (m,
1H), 2.08 (br s, 2H), 1.70-1.59 (m, 2H), 1.57-1.45 (m, 2H), 1.23 (s, 6H), 1.04 (d, J = 7 Hz,
3H). 13C-NMR (125 MHz, CDCl3): δ = 141.1 (CH), 115.3 (CH2), 75.3 (CH), 70.8 (Cq), 43.8 (CH),
40.3 (CH2), 29.9 (CH3), 29.3 (CH3), 28.7 (CH2) 14.5 (CH3).
IR (ATR): ν = 3356 (br), 3079 (w), 2970 (ss), 2930 (s), 2877 (m), 1640 (m), 1456 (m), 1378
(s), 1365 (s), 1215 (m), 1151 (m), 1047 (s), 996 (s), 910 (ss).
LR-MS (60 °C): m/z = 139 (6), 121 (4), 99 (52), 81 (100), 68 (10), 59 (32), 55 (38).
HR-MS (C10H21O2; MH+): ber. 173.1542; gef. 173.1557.
(3R,4S)-4-Azido-3,7-dimethylocta-1,6-dien (72)
Eine Lösung von 770 mg (5 mmol) Alkohol 69a in 5 ml Pyridin wurde
mit 685 mg (6 mmol) MsCl versetzt und 16 h bei RT gerührt. Nach
Zugabe von 75 ml Et2O wurde mit 1 N HCl (3 x 25 ml) gewaschen und
die organische Phase wurde über Na2SO4 getrocknet. Der nach Entfernen
des Lösungsmittels erhaltene Rückstand wurde in 5 ml DMF gelöst, unter Stickstoff-
Atmosphäre mit 1.6 g (25 mmol) NaN3 versetzt und 60 h bei 50 °C gerührt. Nach Zugabe von
75 ml Et2O wurde mit H2O (3 x 25 ml) gewaschen und über Na2SO4 getrocknet. Entfernen
des Lösungsmittels und säulenchromatographische Reinigung des Rückstands (SiO2, Pentan)
ergab 790 mg (88%) Produkt als farblose Flüssigkeit. 1H-NMR (500 MHz, CDCl3): δ = 5.76 (m, 1H), 5.15 (t, J = 6 Hz, 1H), 5.10-5.06 (m, 2H) 3.24
(m, 1H), 2.37 (m, 1H), 2.24 (m, 2H), 1.73 (s, 3H), 1.64 (s, 3H), 1.09 (d, J = 7 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 139.3 (CH), 134.9 (Cq), 119.8 (CH), 116.0 (CH2), 67.7
(CH), 41.8 (CH), 30.9 (CH2), 25.9 (CH3), 18.0 (CH3) 17.3 (CH3).
IR (ATR): ν = 3079 (w), 2969 (s), 2929 (s), 2874 (m), 2099 (ss), 1641 (w), 1452 (m), 1377
(m), 1263 (s), 1108 (w), 998 (m), 918 (s), 844 (w).
LR-MS (RT): m/z = 136 (3), 96 (4), 69 (100), 55 (36).
HR-MS (C9H14N; M+– CH3, N2): ber. 136.1126; gef. 136.1130.
N3

II. Experimenteller Teil
82
2-[(S)-4-Methyl-1-((R)-1-methylallyl)-pent-3-enyl]-isoindol-1,3-dion (75)
Aus 69a: Eine Lösung von 154 mg (1.0 mmol) Alkohol 69a, 162 mg
(1.1 mmol) Phthalimid und 220 mg (1.1 mmol) MePPh2 in 5 ml
absolutem THF unter Stickstoff-Atmosphäre wurde bei 0 °C
tropfenweise mit 191 mg (1.1 mmol) DEAD versetzt und über 1 h auf
RT erwärmt. Das Reaktionsgemisch wurde mit Et2O verdünnt und das Präzipitat abfiltriert.
Entfernung des Lösungsmittels und säulenchromatographische Reinigung des Rückstands
(SiO2, MTB/Hexan 1:20) ergab 140 mg (50%) Produkt als farbloses Öl.
Aus 72: Eine Lösung von 358 mg (2 mmol) Azid 72 in 20 ml absolutem Et2O unter
Stickstoff-Atmosphäre wurde bei 0 °C portionsweise mit 300 mg (8 mmol) LAH versetzt und
über 1 h auf RT erwärmt. Nach Zugabe von 50 ml 2 N NaOH wurde mit Et2O extrahiert (2 x
50 ml) und die organischen Phasen wurden über Na2SO4 getrocknet. Der nach vorsichtigem
Abdestillieren des Lösungsmittels erhaltene Rückstand wurde in 10 ml THF gelöst, mit 296
mg (2 mmol) Phthalsäureanhydrid versetzt und 2 h bei 60 °C gerührt. Das Reaktionsgemisch
wurde abgekühlt, mit 325 mg (1 mmol) CDI versetzt und 1 h bei RT gerührt. Entfernen des
Lösungsmittels und säulenchromatographische Reinigung des Rückstands (SiO2, MTB/Hexan
1:20) ergab 385 mg (68%) Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 7.78 (m, 2H), 7.67 (m, 2H), 5.58 (m, 1H), 4.97 (t, J = 7.0
Hz, 1H), 4.91 (dd, J = 17, 1.0 Hz, 1H), 4.76 (dd, J = 10, 1.0 Hz, 1H), 3.99 (dt, J = 11.0, 4 Hz,
1H), 3.00 (m, 1H), 2.85 (m, 1H), 2.45 (m, 1H), 1.53 (s, 3H), 1.52 (s, 3H), 1.14 (d, J = 7 Hz,
3H). 13C-NMR (125 MHz, CDCl3): δ = 168.8 (Cq), 141.2 (CH), 134.6 (Cq), 133.7 (CH), 131.8
(Cq), 123.0 (CH), 120.3 (CH), 115.1 (CH2), 56.8 (CH), 41.1 (CH), 28.4 (CH2), 25.7 (CH3),
18.4 (CH3), 17.8 (CH3).
IR (ATR): ν = 2973 (w), 2928 (w), 1772 (m), 1710 (ss), 1390 (s), 1361 (s), 1088 (m), 919
(m), 874 (w), 721 (s).
LR-MS (100 °C): m/z = 283 (3), 228 (54), 214 (100), 196 (10), 160 (76), 148 (12), 136 (22),
130 (26), 104 (14), 93 (16), 81 (18).
HR-MS (C18H21NO2; M+): ber. 283.1572; gef. 283.1577.
CHN-Analyse (C18H21NO2): ber. C 76.30, H 7.47, N 4.94, gef. C 75.92, H 7.55, N 4.64.
Drehwert: [α]D20 = –18.8° (c = 0.85, CHCl3).
NPhth

II. Experimenteller Teil
83
[(S)-4-Hydroxy-4-methyl-1-((R)-1-methylallyl)-pentyl]-carbaminsäurebenzylester (67)
Eine Lösung von 312 mg (1.1 mmol) Phthalimid 75 in 2.5 ml CHCl3
wurde bei 0 °C tropfenweise mit 0.84 ml (11 mmol) TFA versetzt und
16 h bei RT gerührt. Die flüchtigen Bestandteile wurden entfernt bevor
der Rückstand mit 7 ml einer 8 M MeNH2-Lösung in EtOH (55 mmol)
versetzt und 24 h bei 55 °C gerührt wurde. Der nach Entfernen der flüchtigen Bestandteile
erhaltene feste Rückstand wurde mit Et2O versetzt und die unlöslichen Bestandteile wurden
abfiltriert. Das Filtrat wurde eingeengt, in 5 ml CHCl3 gelöst und tropfenweise zu einer
Lösung von 206 mg (1.2 mmol) ZCl in 5 ml CHCl3 und 1 ml gesättigter NaHCO3-Lösung
gegeben. Nach 16 h bei RT wurde das Reaktionsgemisch mit 30 ml CH2Cl2 verdünnt und die
organische Phase über Na2SO4 getrocknet. Entfernen des Lösungsmittels und säulen-
chromatographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:1) ergab 265 mg (79%)
Produkt als farbloses Harz. 1H-NMR (500 MHz, CDCl3): δ = 7.33 (m, 5H), 5.76 (m, 1H), 5.09 (m, 4H), 4.54 (br d, J = 9
Hz, 1H), 3.64 (m, 1H), 2.37 (m, 1H), 1.65-1.30 (m, 5H), 1.20 (s, 6H), 1.04 (d, J = 7 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 156.7 (Cq), 139.2 (CH), 136.7 (Cq), 128.6 (CH), 128.1
(CH), 128.0 (CH), 116.1 (CH2), 70.7 (Cq), 66.7 (CH2), 55.6 (CH), 41.6 (CH), 39.9 (CH2),
29.4 (CH3), 27.8 (CH2), 16.3 (CH3).
IR (ATR): ν = 3327 (br), 2967 (s), 2932 (m), 1697 (ss), 1536 (s), 1454 (m), 1252 (s), 1065
(w), 912 (m), 737 (m), 697 (m).
LR-MS (130 °C): m/z = 288 (1), 232 (6), 188 (16), 142 (3), 99 (32), 91 (100), 81 (8), 65 (10).
HR-MS (C18H26NO2; M+– OH): ber. 288.1964; gef. 288.1970.
Drehwert: [α]D20 = –6.0° (c = 1.25, CHCl3).
1-(Furan-3-yl)-prop-2-en-1-ol (77)
Eine Lösung von 1.92 g (20 mmol) 3-Furfural 76 in 50 ml absolutem Et2O
unter Stickstoff-Atmosphäre wurde bei 0 °C tropfenweise mit 22 ml (1 M in
THF, 22 mmol) Vinylmagnesiumbromid-Lösung versetzt. Das Reaktions-
gemisch wurde 1 h gerührt und nach Zugabe von 50 ml 1 N HCl mit Et2O
(2 x 50 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4, Entfernen des Lö-
sungsmittels und Filtration über Kieselgel (CH2Cl2) ergab 2.33 g (94%) Produkt als gelbes Öl. 1H-NMR (500 MHz, CDCl3): δ = 7.39 (s, 2H), 6.40 (s, 1H), 6.07 (m, 1H), 5.36 (d, J = 17 Hz,
1H), 5.21 (d, J = 10.5 Hz, 1H), 5.17 (d, J = 5.5 Hz, 1H), 1.87 (br s, 1H).
NHZOH
OH
O

II. Experimenteller Teil
84
13C-NMR (125 MHz, CDCl3): δ = 143.5 (CH), 139.5 (CH), 139.4 (CH), 127.6 (Cq), 115.5
(CH2), 109.0 (CH), 68.2 (CH).
IR (ATR): ν = 3373 (br), 2981 (w), 2873 (w), 1704 (w), 1503 (m), 1422 (w), 1159 (m), 1117
(w), 1021 (s), 992 (m), 928 (m), 874 (ss), 772 (s), 734 (w).
LR-MS (RT): m/z = 124 (36), 106 (8), 95 (100), 81 (26), 77 (26), 69 (30), 55 (30), 51 (30).
HR-MS (C7H8O2; M+): ber. 124.0524; gef. 124.0529.
1-(Furan-3-yl)-propenon (66)
Eine Lösung von 621 mg (5 mmol) Carbinol 77 in 15 ml absolutem CH2Cl2
unter Stickstoff-Atmosphäre wurde portionsweise mit 4.35 g (50 mmol)
MnO2 versetzt und 24 h bei 40 °C gerührt. Filtration über Celite, Entfernen
des Lösungsmittels und säulenchromatographische Reinigung des Rück-
stands (SiO2, CH2Cl2) ergab 400 mg (65%) Produkt als gelbliches Öl. 1H-NMR (500 MHz, CDCl3): δ = 8.06 (s, 1H), 7.47 (t, J = 1.5 Hz, 1H), 6.85-6.80 (m, 2H),
6.44 (dd, J = 17, 1.5 Hz, 1H), 5.84 (dd, J = 10.5, 1.5 Hz, 1H). 13C-NMR (125 MHz, CDCl3): δ = 184.6 (Cq), 147.6 (CH), 144.4 (CH), 133.4 (CH), 128.9
(CH2), 127.8 (Cq), 109.1 (CH).
IR (ATR): ν = 3132 (w), 2954 (w), 1660 (ss), 1606 (m), 1560 (m), 1511 (m), 1408 (m), 1208
(m), 1156 (s), 1015 (m), 915 (m), 873 (s), 767 (w).
LR-MS (RT): m/z = 122 (36), 95 (100), 67 (4), 55 (6).
HR-MS (C7H6O2; M+): ber. 122.0368; gef. 122.0361.
[(E)-(1S,2R)-5-(Furan-3-yl)-1-(3-hydroxy-3-methylbutyl)-2-methyl-5-oxopent-3-enyl]-
carbaminsäurebenzylester (65)
Eine Lösung von 31 mg (100 µmol) Amin 67 und 15 mg (120
µmol) Enon 66 in 2 ml absolutem CH2Cl2 unter Stickstoff-
Atmosphäre wurde mit 6 mg (10 mol%) Ruthenium-
Katalysator 5 versetzt und 72 h bei 40 °C gerührt. Entfernen
des Lösungsmittels und säulenchromatographische Reinigung
des Rückstands (SiO2, MTB/Hexan 3:1) ergab 29 mg (73%) Produkt als bräunliches Harz. 1H-NMR (500 MHz, CDCl3): δ = 8.04 (s, 1H), 7.42 (s, 1H), 7.30 (m, 5H), 6.95 (dd, J = 15.5,
8 Hz, 1H), 6.81 (s, 1H), 6.54 (d, J = 15.5 Hz, 1H), 5.08 (s, 2H), 4.93 (br d, J = 9 Hz, 1H), 3.75
(m, 1H), 2.60 (m, 1H), 1.73 (br s, 1H), 1.61 (m, 1H), 1.55-1.40 (m, 3H), 1.18 (s, 6H), 1.12 (d,
J = 7 Hz, 3H).
O
O
NHZ
O
O
OH

II. Experimenteller Teil
85
13C-NMR (125 MHz, CDCl3): δ = 184.3 (Cq), 156.7 (Cq), 148.5 (CH), 147.6 (CH), 144.3
(CH), 136.6 (Cq), 128.6 (CH), 128.2 (CH), 128.1 (Cq), 128.0 (CH), 127.7 (CH), 109.1 (CH),
70.6 (Cq), 66.8 (CH2), 55.6 (CH), 41.4 (CH), 39.8 (CH2), 29.6 (CH3), 29.5 (CH3), 27.6 (CH2),
16.2 (CH3).
IR (ATR): ν = 3323 (br), 2967 (m), 2933 (w), 1699 (ss), 1666 (s), 1618 (s), 1560 (m), 1535
(s), 1454 (w), 1329 (m), 1251 (s), 1157 (ss), 1062 (m), 983 (w), 873 (m), 739 (m), 697 (m).
LR-MS (180 °C): m/z = 400 (< 1), 322 (1), 268 (1), 250 (6), 232 (1), 188 (8), 150 (10), 121
(6), 99 (18), 91 (100), 65 (3).
HR-MS (C23H30NO5; MH+): ber. 400.2124; gef. 400.2120.
[(S)-4-Hydroxy-4-methyl-1-((R)-1-methylallyl)-pentyl]-carbaminsäure-tbutylester (78)
Eine Lösung von 140 mg (0.5 mmol) Phthalimid 75 in 1.2 ml CHCl3
wurde bei 0 °C tropfenweise mit 0.38 ml (5 mmol) TFA versetzt und
16 h bei RT gerührt. Die flüchtigen Bestandteile wurden entfernt
bevor der Rückstand mit 3 ml einer 8 M MeNH2-Lösung in EtOH (24
mmol) versetzt und 24 h bei 55 °C gerührt wurde. Der nach Entfernen der flüchtigen
Bestandteile erhaltene feste Rückstand wurde mit Et2O versetzt und die unlöslichen
Bestandteile wurden abfiltriert. Das Filtrat wurde eingeengt, in 4 ml CHCl3 gelöst und nach
Zugabe von 0.5 ml gesättigter NaHCO3-Lösung tropfenweise mit einer Lösung von 120 mg
(0.54 mmol) ZCl in 1 ml CHCl3 versetzt. Nach 16 h bei RT wurde das Reaktionsgemisch mit
15 ml CH2Cl2 verdünnt und die organische Phase über Na2SO4 getrocknet. Entfernen des
Lösungsmittels und säulenchromatographische Reinigung des Rückstands (SiO2, MTB/Hexan
2:3) ergab 97 mg (81%) Produkt als farbloses Harz. 1H-NMR (500 MHz, CDCl3): δ = 5.76 (m, 1H), 5.08 (m, 2H), 4.29 (br d, J = 8 Hz, 1H), 3.57
(m, 1H), 2.34 (m, 1H), 1.65-1.30 (m, 5H), 1.44 (s, 9H), 1.21 (s, 6H), 1.03 (d, J = 7 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 156.2 (Cq), 139.5 (CH), 115.9 (CH2), 79.1 (Cq) 70.7 (Cq),
54.8 (CH), 41.6 (CH), 39.9 (CH2), 29.6 (CH3), 29.4 (CH3), 28.5 (CH3), 27.9 (CH2), 16.3
(CH3).
IR (ATR): ν = 3345 (br), 2969 (s), 2932 (m), 1690 (ss), 1504 (s), 1454 (m), 1365 (s), 1250
(m), 1173 (ss), 996 (w), 913 (m), 864 (w).
LR-MS (110 °C): m/z = 256 (10), 160 (46), 145 (60), 117 (16), 98 (40), 81 (36), 57 (100).
HR-MS (C14H26NO3; M+– CH3): ber. 256.1913; gef. 256.1929.
CHN-Analyse (C15H29NO3): ber. C 66.38, H 10.77, N 5.16, gef. C 66.67, H 10.58, N 5.16.
Drehwert: [α]D20 = –7.0° (c = 1.0, CHCl3).
NHBocOH

II. Experimenteller Teil
86
[(E)-(1S,2R)-5-(Furan-3-yl)-1-(3-hydroxy-3-methylbutyl)-2-methyl-5-oxopent-3-enyl]-
carbaminsäure-tbutylester (79)
Eine Lösung von 27 mg (100 µmol) Amin 78 und 15 mg (120
µmol) Enon 66 in 2 ml absolutem CH2Cl2 unter Stickstoff-
Atmosphäre wurde mit 6 mg (10 mol%) Ruthenium-
Katalysator 5 versetzt und 72 h bei 40 °C gerührt. Entfernen
des Lösungsmittels und säulenchromatographische Reinigung
des Rückstands (SiO2, MTB/Hexan 2:1) ergab 27 mg (74%) Produkt als braunen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 8.07 (s, 1H), 7.45 (s, 1H), 6.95 (dd, J = 15.5, 8 Hz, 1H),
6.83 (s, 1H), 6.56 (d, J = 15.5 Hz, 1H), 4.44 (br d, J = 9 Hz, 1H), 3.69 (m, 1H), 2.58 (m, 1H),
1.70-1.35 (m, 5H), 1.42 (s, 9H), 1.20 (s, 6H), 1.13 (d, J = 7 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 184.4 (Cq), 156.1 (Cq), 148.9 (CH), 147.6 (CH), 144.3
(CH), 128.0 (Cq), 127.7 (CH), 109.2 (CH), 79.4 (Cq) 70.6 (Cq), 54.8 (CH), 41.6 (CH), 39.8
(CH2), 29.6 (CH3), 29.4 (CH3), 28.4 (CH3), 27.7 (CH2), 16.3 (CH3).
IR (ATR): ν = 3345 (br), 2970 (m), 2932 (w), 1692 (ss), 1668 (s), 1619 (s), 1561 (m), 1512
(s), 1454 (w), 1365 (m), 1250 (m), 1158 (ss), 1055 (w), 985 (w), 873 (m).
LR-MS (190 °C): m/z = 309 (5), 274 (3), 232 (10), 160 (72), 142 (30), 121 (84), 98 (50), 81
(32), 57 (100).
HR-MS (C16H23NO5; MH+– C4H9): ber. 309.1576; gef. 309.1580.
CHN-Analyse (C20H31NO5): ber. C 65.73, H 8.55, N 3.38, gef. C 65.28, H 8.07, N 3.63.
Drehwert: [α]D20 = –24.4° (c = 0.5, CHCl3).
Schmelzpunkt: 92–94 °C.
(–)-Nupharamin (59)
Eine Lösung von 35 mg (96 µmol) Aminoenon 79 in 2 ml
Aceton wurde entgast, unter Stickstoff-Atmosphäre mit 6 mg
Pd/C (10%) versetzt und 15 min unter Wasserstoff-Atmosphäre
gerührt. Der nach Filtration und Entfernen des Lösungsmittels
erhaltene Rückstand wurde in 1 ml CH2Cl2 gelöst, tropfenweise mit 0.1 ml TFA versetzt und
1 h bei RT gerührt. Das Reaktionsgemisch wurde mit 20 ml CH2Cl2 verdünnt und mit 5 ml
gesättigter NaHCO3-Lösung gewaschen. Trocknen der organischen Phase über Na2SO4 und
Entfernen des Lösungsmittels ergab einen Rückstand, der in 1 ml EtOH gelöst, bei 0 °C mit 4
mg (100 µmol) NaBH4 versetzt und 1 h gerührt wurde. Entfernen des Lösungsmittels und
NHBoc
O
O
OH
NHOH O

II. Experimenteller Teil
87
säulenchromatographische Reinigung des Rückstands (Al2O3, EtOAc/CH2Cl2 1:3) ergab 18
mg (75%) Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 7.35 (m, 2H), 6.42 (s, 1H), 3.62 (dd, J = 11.5, 2 Hz, 1H),
2.39 (m, 1H), 1.85 (m, 2H), 1.75 (m, 2H), 1.57 (m, 2H), 1.45 (m, 2H), 1.23 (m, 1H), 1.21 (s,
3H), 1.19 (s, 3H), 0.90 (d, J = 6.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 143.1 (CH), 138.5 (CH), 129.0 (Cq), 109.3 (CH), 68.9
(Cq), 63.0 (CH), 53.1 (CH), 39.7 (CH2), 34.3 (CH), 34.0 (CH2), 33.7 (CH2), 30.3 (CH3), 29.3
(CH3), 28.5 (CH2), 18.6 (CH3).
IR (ATR): ν = 3385 (br), 2966 (ss), 2926 (ss), 2871 (m), 2850 (m), 1501 (m), 1458 (m), 1377
(m), 1161 (m), 1025 (m), 976 (w), 912 (w), 874 (s), 794 (m), 765 (m).
LR-MS (RT): m/z = 251 (1), 236 (6), 192 (2), 164 (100), 136 (6), 107 (26), 94 (36), 81 (7).
HR-MS (C15H25NO2; M+): ber. 251.1885; gef. 251.1890.
Drehwert: [α]D20 = –38.7° (c = 0.75, CHCl3).
2.6. Zu Kapitel 2.7.
6-Methoxycarbonyloxy-2-(4-nitrobenzylsulfonylamino)-hept-4-ensäuremethylester (84)
Eine Lösung von 31 mg (100 µmol) Amin 83 und 26 mg (200
µmol) (3-Buten-2-yl)-methylcarbonat in 1 ml absolutem DCE
unter Stickstoff-Atmosphäre wurde mit 3 mg (5 mol%)
Ruthenium-Katalysator 5 versetzt und 16 h bei 70 °C gerührt.
Entfernen des Lösemittels und säulenchromatographische Reini-
gung des Rückstands (SiO2, MTB/Hexan 1:1) ergab 27 mg (65%) Produkt als braunes Harz. 1H-NMR (500 MHz, CDCl3): δ = 8.34 (d, J = 9 Hz, 2H), 8.03 (m, 2H), 5.54 (m, 3H), 5.07 (m,
1H), 4.12 (m, 1H), 3.77 (m, 3H), 3.56 (m, 3H), 2.50 (m, 2H), 1.32 (d, J = 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 170.8 (Cq), 155.3 (Cq), 155.1 (Cq), 150.2 (Cq), 150.1 (Cq),
146.1 (Cq), 145.9 (Cq), 135.0 (CH), 134.8 (CH), 128.5 (CH), 125.9 (CH), 125.4 (CH), 124.4
(CH), 124.3 (CH), 74.9 (CH), 74.3 (CH), 55.5 (CH), 55.2 (CH), 54.7 (CH3), 52.8 (CH3), 36.0
(CH2), 35.9 (CH2), 20.3 (CH3), 20.2 (CH3).
IR (ATR): ν = 3276 (br), 2957 (w), 1742 (ss), 1607 (w), 1531 (s), 1442 (m), 1350 (s), 1266
(ss), 1166 (s), 1092 (m), 1038 (w), 940 (w), 854 (m), 792 (w), 737 (m), 686 (w).
LR-MS (130 °C): m/z = 341 (2), 281 (48), 273 (36), 186 (50), 154 (18), 139 (16), 122 (44),
94 (26), 68 (100).
HR-MS (C14H17N2O6S; M+– OCO2CH3): ber. 341.0807; gef. 341.0801.
NHpNs
MeO2C
OCO2Me

II. Experimenteller Teil
88
(cis)-6-Methyl-1-(4-nitrobenzylsulfonyl)-1,2,3,6-tetrahydropyridin-2-carbonsäureme-
thylester (cis-85)
Eine Lösung von 24 mg (57 µmol) Carbonat 84 in 1 ml absolutem THF
unter Stickstoff-Atmosphäre wurde mit 0.7 mg (1 mol%) Pd(PPh3)4
versetzt und 30 min bei 60 °C gerührt. Entfernen des Lösungsmittels
und Umkristallisation des Rohprodukts (MTB) ergab 8 mg (41%) des
cis-Isomers als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 8.34 (d, J = 8.5 Hz, 2H), 8.04 (d, J = 8.5 Hz, 2H), 5.73 (m,
1H), 5.55 (m, 1H), 4.88 (d, J = 7 Hz, 1H), 4.41 (m, 1H), 3.69 (s, 3H), 2.66 (dd, J = 17.5, 6 Hz,
1H), 2.10 (m, 1H), 1.26 (d, J = 7 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 171.1 (Cq), 150.1 (Cq), 146.5 (Cq), 128.4 (CH), 128.2
(CH), 124.4 (CH), 122.4 (CH), 52.6 (CH3), 51.8 (CH), 49.8 (CH), 24.7 (CH2), 21.5 (CH3).
IR (ATR): ν = 2920 (w), 1738 (s), 1606 (w), 1528 (s), 1435 (w), 1348 (ss), 1213 (m), 1165
(ss), 1099 (s), 1021 (m), 980 (m), 847 (m), 739 (s), 684 (w).
LR-MS (150 °C): m/z = 341 (< 1), 325 (10), 281 (100), 265 (4), 186 (10), 154 (4), 122 (16),
94 (24), 80 (36), 69 (22), 57 (12).
HR-MS (C14H17N2O6S; MH+): ber. 341.0807; gef. 341.0809.
Schmelzpunkt: 174–175 °C.
2.7. Zu Kapitel 3.2.
Tetradec-13-en-1-ol (90d)
Eine Lösung von 1.19 g (3.6 mmol) Bromid 96 in 10 ml absolutem
THF unter Stickstoff-Atmosphäre wurde tropfenweise mit 9 ml (2 M
in THF, 18 mmol) Allylmagnesiumchlorid-Lösung versetzt und 2 h refluxiert. Das Reaktions-
gemisch wurde anschließend noch weitere 16 h bei RT gerührt, mit 30 ml gesättigter NH4Cl-
Lösung versetzt und mit MTB (2 x 30 ml) extrahiert. Der nach Trocknen der organischen
Phasen über Na2SO4 und Entfernen des Lösungsmittels erhaltene Rückstand wurde in 10 ml
MeOH gelöst, mit 35 mg (0.2 mmol) pTsOH versetzt und 1 h bei RT gerührt. Entfernen des
Lösungsmittels und säulenchromatographische Reinigung des Rückstands (SiO2, MTB/Hexan
1:3) ergab 535 mg (71%) Produkt als farbloses Wachs. 1H-NMR (500 MHz, CDCl3): δ = 5.81 (m, 1H), 5.00-4.90 (m, 2H), 3.64 (t, J = 6.5 Hz, 2H),
2.04 (m, 2H), 1.56 (quin, J = 7 Hz, 2H), 1.40-1.25 (m, 19H).
HR-MS (C14H28O; M+): ber. 212.2140; gef. 212.2149.
NpNs
MeO2C
HO ( )9

II. Experimenteller Teil
89
6-(2-Dimethylaminoethyl)-2,2-dimethyl-[1.3]dioxin-4-on (98)
Eine Lösung von 1.7 ml (12 mmol) HN(iPr)2 in 20 ml absoluten THF
unter Stickstoff-Atmosphäre wurde bei -78 °C mit 7.5 ml (1.6 M in
Hexan, 12 mmol) BuLi versetzt. Nach 15 min wurden 1.42 g (10
mmol) Dioxolenon 97 und nach weiteren 30 min 1.4 g (15 mmol)
[Me2N=CH2]+Cl- hinzugegeben. Das Reaktionsgemisch wurde 16 h bei RT gerührt und mit
50 ml 1 N HCl extrahiert. Die wässrige Phase wurde mit 50 ml MTB gewaschen, mit 50 ml 2
N NaOH basifiziert und mit CH2Cl2 (3 x 50 ml) extrahiert. Trocknen der organischen Phasen
über Na2SO4 und Entfernen des Lösungsmittels ergab 800 mg (40%) oranges Rohprodukt. 1H-NMR (200 MHz, CDCl3): δ = 5.27 (s, 1H), 2.50-2.38 (m, 4H), 2.23 (s, 6H), 1.71 (s, 6H).
6-(2-Chlorethyl)-2,2-dimethyl-[1.3]dioxin-4-on (100)
Eine Lösung von 1.03 g (5 mmol) Ketoester 99 und 1.5 g (15 mmol)
Ac2O in 0.75 ml (10 mmol) Aceton wurde bei 0 °C tropfenweise mit 0.28
ml (5 mmol) konzentrierter H2SO4 versetzt und 16 h bei RT gerührt. Das
Reaktionsgemisch wurde mit 50 ml H2O verdünnt und mit CH2Cl2 (3 x 50
ml) extrahiert. Trocknen der organischen Phasen über Na2SO4, Entfernen des Lösungsmittels
und säulenchromatographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:2) ergab 730
mg (76%) Produkt als gelbliches Öl. 1H-NMR (500 MHz, CDCl3): δ = 5.36 (s, 1H), 3.69 (t, J = 6.4 Hz, 2H), 2.70 (t, J = 6.4 Hz,
2H), 1.71 (s, 6H). 13C-NMR (125 MHz, CDCl3): δ = 167.1 (Cq), 160.8 (Cq), 107.0 (Cq), 95.8 (CH), 39.4 (CH2),
36.6 (CH2), 25.1 (CH3).
IR (ATR): ν = 2999 (w), 1728 (ss), 1638 (s), 1392 (s), 1376 (s), 1273 (s), 1203 (s), 1016 (m).
LR-MS (RT): m/z = 190 (18), 160 (4), 133 (36), 115 (6), 97 (100), 69 (86), 59 (46), 43 (46).
HR-MS (C8H11ClO3; M+): ber. 190.0397; gef. 190.0388.
2,2-Dimethyl-6-vinyl-[1,3]dioxin-4-on (91)
Aus 98: Eine Lösung von 800 mg (4 mmol) Amin 98 in 10 ml CH2Cl2
wurde portionsweise mit 1.5 g (6 mmol) 70%iger mCPBA versetzt und 2 h
bei RT gerührt. Das Reaktionsgemisch wurde mit 50 ml CH2Cl2 verdünnt
und mit 20 ml gesättigter NaHCO3-Lösung gewaschen. Trocknen der
organischen Phase über Na2SO4, Entfernen des Lösungsmittels und säulenchromatogra-
O
O
OMe2N
O
O
O
O
O
OCl

II. Experimenteller Teil
90
phische Reinigung des Rückstands (SiO2, MTB/Hexan 1:2) ergab 310 mg (50%) Produkt als
gelbliches Öl.
Aus 100: Eine Lösung von 720 mg (3.8 mmol) Chlorid 100 in 10 ml CH2Cl2 wurde
tropfenweise mit 1.05 ml (7.55 mmol) NEt3 versetzt und 3 h bei RT gerührt. Das
Reaktionsgemisch wurde mit 70 ml H2O verdünnt und mit 20 ml 1 N HCl gewaschen.
Trocknen der organischen Phase über Na2SO4, Entfernen des Lösungsmittels und säulen-
chromatographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:2) ergab 500 mg (85%)
Produkt als gelbliches Öl. 1H-NMR (500 MHz, CDCl3): δ = 6.20 (dd, J = 17, 10.5 Hz, 1H), 6.01 (d, J = 17 Hz, 1H),
5.59 (d, J = 10.5 Hz, 1H), 5.34 (s, 1H), 1.71 (s, 6H). 13C-NMR (125 MHz, CDCl3): δ = 162.9 (Cq), 161.8 (Cq), 129.4 (CH), 123.9 (CH2), 106.6
(Cq), 95.3 (CH), 25.1 (CH3).
IR (ATR): ν = 2999 (w), 1728 (ss), 1645 (m), 1581 (m), 1391 (s), 1376 (s), 1273 (s), 1205 (s),
1045 (m), 999 (m), 903 (m), 814 (w).
LR-MS (RT): m/z = 154 (30), 139 (2), 111 (6), 97 (64), 69 (18), 59 (10), 55 (100).
HR-MS (C8H10O3; M+): ber. 154.0630; gef. 154.0631.
Allgemeine Versuchsvorschrift C für die Kreuzmetathese mit Vinyldioxolenon 91:
Eine Lösung von Hydroxyalken (0.1 M) und Vinyldioxolenon 91 (1–1.5 eq) in absolutem
CH2Cl2 unter Stickstoff-Atmosphäre wurde mit dem Ruthenium-Katalysator 5 (5 mol%)
versetzt und 24 h bei 40 °C gerührt. Das Lösungsmittel wurde entfernt und der Rückstand
säulenchromatographisch gereinigt (SiO2, 2–5% AcMe in CH2Cl2).
6-[(E)-10-Hydroxydec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on (92a)
Entsprechend der allgemeinen Arbeitsvorschrift C wurden aus 78 mg
(500 µmol) Decenol 90a, 77 mg (500 µmol) Dienon 91 und 15 mg (5
mol%) 5 in 5 ml CH2Cl2 100 mg (71%) Produkt als bräunliches Harz
erhalten. 1H-NMR (500 MHz, CDCl3): δ = 6.55 (dt, J = 15.5, 7.0 Hz, 1H), 5.90 (d, J = 15.5 Hz, 1H),
5.24 (s, 1H), 3.64 (t, J = 6.5 Hz, 2H), 2.20 (m, 2H), 1.71 (s, 6H), 1.55 (m, 2H), 1.45 (m, 2H),
1.30 (m, 8H), 1.21 (br s, 1H). 13C-NMR (125 MHz, CDCl3): δ = 163.5 (Cq), 162.2 (Cq), 142.7 (CH), 122.5 (CH), 106.3
(Cq), 93.3 (CH), 63.0 (CH2), 32.8 (CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 29.1 (CH2),
28.4 (CH2), 25.8 (CH2), 25.1 (CH3).
OH OO
O( )5

II. Experimenteller Teil
91
IR (ATR): ν = 3434 (br), 2928 (s), 2855 (s), 1724 (ss), 1652 (ss), 1592 (m), 1390 (ss), 1374
(ss), 1274 (ss), 1205 (s), 1019 (s), 970 (w), 903 (w), 801 (w).
LR-MS (70 °C): m/z = 282 (< 1), 224 (8), 181 (6), 155 (4), 123 (40), 110 (100), 97 (20), 81
(30), 69 (20), 55 (28).
HR-MS (C16H26O4; M+): ber. 282.1831; gef. 282.1837.
CHN-Analyse (C16H26O4): ber. C 68.05, H 9.28, gef. C 67.45, H 9.06.
6-[(E)-12-Hydroxydodec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on (92b)
Entsprechend der allgemeinen Arbeitsvorschrift C wurden aus 92 mg
(500 µmol) Dodecenol 90b, 77 mg (500 µmol) Dienon 91 und 15 mg
(5 mol%) 5 in 5 ml CH2Cl2 116 mg (75%) Produkt als bräunliches
Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 6.55 (dt, J = 15.5, 7.0 Hz, 1H), 5.88 (d, J = 15.5 Hz, 1H),
5.23 (s, 1H), 3.63 (t, J = 6.5 Hz, 2H), 2.18 (m, 2H), 1.70 (s, 6H), 1.55 (m, 2H), 1.44 (m, 2H),
1.28 (m, 13H). 13C-NMR (125 MHz, CDCl3): δ = 163.5 (Cq), 162.2 (Cq), 142.8 (CH), 122.5 (CH), 106.3
(Cq), 93.3 (CH), 63.1 (CH2), 32.8 (CH2), 32.8 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4 (CH2),
29.4 (CH2), 29.2 (CH2), 28.4 (CH2), 25.8 (CH2), 25.1 (CH3).
IR (ATR): ν = 3433 (br), 2926 (s), 2854 (s), 1726 (ss), 1653 (ss), 1592 (m), 1390 (s), 1374
(s), 1274 (s), 1206 (s), 1019 (s), 970 (w), 903 (w), 800 (w).
LR-MS (70 °C): m/z = 310 (2), 282 (2), 252 (8), 210 (4), 192 (6), 165 (6), 123 (58), 110
(100), 97 (34), 81 (40), 69 (24), 55 (42).
HR-MS (C18H30O4; M+): ber. 310.2144; gef. 310.2150.
CHN-Analyse (C18H30O4): ber. C 69.64, H 9.74, gef. C 69.10, H 9.35.
6-[(E)-13-Hydroxytetradec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on (92c)
Entsprechend der allgemeinen Arbeitsvorschrift C wurden aus 212
mg (1 mmol) Tetradecenol 90c, 154 mg (1 mmol) Dienon 91 und 30
mg (5 mol%) 5 in 10 ml CH2Cl2 265 mg (78%) Produkt als
bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 6.54 (dt, J = 15.5, 7.0 Hz, 1H), 5.88 (d, J = 15.5 Hz, 1H),
5.22 (s, 1H), 3.78 (m, 1H), 2.19 (m, 2H), 1.70 (s, 6H), 1.40 (m, 6H), 1.26 (m, 13H), 1.17 (d, J
= 6 Hz, 3H).
OH OO
O( )7
OH OO
O( )8

II. Experimenteller Teil
92
13C-NMR (125 MHz, CDCl3): δ = 163.5 (Cq), 162.2 (Cq), 142.8 (CH), 122.5 (CH), 106.3
(Cq), 93.2 (CH), 68.2 (CH), 39.4 (CH2), 32.8 (CH2), 29.7 (CH2), 29.6 (CH2), 29.6 (CH2), 29.5
(CH2), 29.4 (CH2), 29.3 (CH2), 28.4 (CH2), 25.8 (CH2), 25.1 (CH3), 23.6 (CH3).
IR (ATR): ν = 3438 (br), 2925 (ss), 2854 (s), 1726 (ss), 1653 (ss), 1592 (m), 1390 (s), 1374
(s), 1274 (s), 1205 (m), 1019 (m), 970 (w), 902 (w), 801 (w).
LR-MS (150 °C): m/z = 339 (10), 321 (16), 281 (68), 263 (100), 221 (8), 179 (2), 137 (3),
123 (12), 97 (4), 81 (2), 55 (4).
HR-MS (C20H35O4; MH+): ber. 339.2535; gef. 339.2529.
CHN-Analyse (C20H34O4): ber. C 70.97, H 10.12, gef. C 70.63, H 9.97.
6-[(E)-14-Hydroxytetradec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on (92d)
Entsprechend der allgemeinen Arbeitsvorschrift C wurden aus 49 mg
(230 µmol) Tetradecenol 90d, 36 mg (230 µmol) Dienon 91 und 7
mg (5 mol%) 5 in 2.3 ml CH2Cl2 60 mg (77%) Produkt als
bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 6.55 (dt, J = 15.5, 7.0 Hz, 1H), 5.90 (d, J = 15.5 Hz, 1H),
5.23 (s, 1H), 3.63 (t, J = 6.5 Hz, 2H), 2.20 (m, 2H), 1.70 (s, 6H), 1.56 (m, 2H), 1.45 (m, 2H),
1.26 (m, 17H). 13C-NMR (125 MHz, CDCl3): δ = 163.5 (Cq), 162.2 (Cq), 142.8 (CH), 122.5 (CH), 106.3
(Cq), 93.3 (CH), 63.1 (CH2), 32.9 (CH2), 32.8 (CH2), 29.6 (CH2), 29.6 (CH2), 29.5 (CH2),
29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 28.4 (CH2), 25.8 (CH2), 25.1 (CH3).
IR (ATR): ν = 3431 (br), 2925 (ss), 2854 (s), 1727 (ss), 1653 (s), 1592 (m), 1390 (s), 1374
(m), 1274 (m), 1206 (m), 1019 (m), 970 (w), 902 (w), 799 (w).
LR-MS (100 °C): m/z = 338 (2), 280 (8), 220 (4), 165 (6), 123 (56), 110 (100), 97 (38), 81
(44), 69 (32), 55 (60).
HR-MS (C20H34O4; M+): ber. 338.2457; gef. 338.2461.
CHN-Analyse (C20H34O4): ber. C 70.97, H 10.12, gef. C 70.60, H 10.20.
Allgemeine Versuchsvorschrift D für die Lactonisierung der Hydroxyalkenyldioxolenone:
Methode A: Eine Lösung des Hydroxyalkenyldioxolenons (100 µmol) in 800 ml n-Heptan
wurde 9 h unter Stickstoff-Atmosphäre refluxiert und das Lösungsmittel wurde entfernt.
Methode B: Eine Lösung des Hydroxyalkenyldioxolenons (100 µmol) in 50 ml Toluol wurde
unter Stickstoff-Atmosphäre tropfenweise über 4 h zu 800 ml refluxierendem Toluol gegeben.
Die Lösung wurde weitere 3 h refluxiert und das Lösungsmittel wurde entfernt.
OH OO
O( )9

II. Experimenteller Teil
93
(5E,19E)-1,15-Dioxacyclooctacosa-5-19-dien-2,4,16,18-tetraon (101a)
Säulenchromatographische Reinigung (SiO2, 2% AcMe in
CH2Cl2) des entsprechend der allgemeinen Arbeitsvorschrift D
(Methode A) aus 28 mg (100 µmol) Dioxolenon 92a
erhaltenen Rückstands ergab 16 mg (71%) dimeres Produkt als
farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 6.89 (dt, J = 16, 7 Hz, 2H), 6.16 (d, J = 16 Hz, 2H), 4.13 (t,
J = 6 Hz, 4H), 3.56 (s, 4H), 2.23 (m, 4H), 1.60 (m, 4H), 1.45 (m, 4H), 1.28 (m, 16H). 13C-NMR (125 MHz, CDCl3): δ = 192.0 (Cq), 167.4 (Cq), 150.1 (CH), 129.7 (CH), 65.5
(CH2), 47.5 (CH2), 32.6 (CH2), 29.4 (CH2), 29.2 (CH2), 29.1 (CH2), 28.5 (CH2), 28.0 (CH2),
26.0 (CH2).
IR (ATR): ν = 2928 (ss), 2855 (s), 1738 (ss), 1695 (m), 1674 (s), 1627 (m), 1420 (w), 1239
(s), 1149 (w), 978 (w).
LR-MS (140 °C): m/z = 448 (36), 224 (44), 207 (12), 165 (16), 123 (40), 110 (78), 95 (48),
81 (100), 67 (44), 55 (90).
HR-MS (C26H40O6; M+): ber. 448.2825; gef. 448.2829.
CHN-Analyse (C26H40O6): ber. C 69.61, H 8.99, gef. C 69.48, H 8.78.
Schmelzpunkt: 59–60 °C.
(E)-Oxacyclohexadec-5-en-2,4-dion (93b)
Säulenchromatographische Reinigung (SiO2, CH2Cl2) des entsprechend
der allgemeinen Arbeitsvorschrift D (Methode B) aus 31 mg (100 µmol)
Dioxolenon 92b erhaltenen Rückstands ergab 20 mg (78%) monomeres
Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 6.92 (dt, J = 16, 7 Hz, 1H), 6.24 (d, J = 16 Hz, 1H), 4.17 (t,
J = 6 Hz, 2H), 3.52 (s, 2H), 2.30 (m, 2H), 1.65 (m, 2H), 1.56 (m, 2H), 1.35-1.25 (m, 12H). 13C-NMR (125 MHz, CDCl3): δ = 191.5 (Cq), 167.6 (Cq), 150.6 (CH), 129.4 (CH), 65.2
(CH2), 48.3 (CH2), 31.4 (CH2), 28.0 (CH2), 26.8 (CH2), 26.6 (CH2), 26.5 (CH2), 26.2 (CH2),
25.9 (CH2), 25.0 (CH2).
IR (ATR): ν = 2929 (ss), 2857 (s), 1737 (ss), 1694 (m), 1675 (s), 1624 (m), 1461 (w), 1256
(s), 1160 (w), 985 (m).
LR-MS (100 °C): m/z = 225 (32), 234 (4), 210 (20), 192 (16), 123 (16), 110 (26), 95 (56), 81
(100), 68 (56), 55 (86).
HR-MS (C15H24O3; M+): ber. 225.1725; gef. 225.1730.
O
O
O
( )7
O
O
O
O
O
O 7( )( )7

II. Experimenteller Teil
94
(E)-17-Methyloxacycloheptadec-5-en-2,4-dion (93c)
Säulenchromatographische Reinigung (SiO2, MTB/Hexan 1:10) des
entsprechend der allgemeinen Arbeitsvorschrift D (Methode A) aus 34 mg
(100 µmol) Dioxolenon 92c erhaltenen Rückstands ergab 23 mg (82%)
monomeres Produkt als farbloses Harz. 1H-NMR (500 MHz, CDCl3): δ = 6.92 (dt, J = 16, 7.0 Hz, 1H), 6.22 (d, J = 16 Hz, 1H), 4.99
(m, 1H), 3.51 (s, 2H), 2.28 (m, 2H), 1.60-1.40 (m, 4H), 1.35-1.20 (m, 14H), 1.21 (d, J = 6.5
Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 191.7 (Cq), 166.9 (Cq), 150.5 (CH), 129.8 (CH), 72.3
(CH), 48.1 (CH2), 35.8 (CH2), 31.8 (CH2), 27.8 (CH2), 27.3 (CH2), 27.0 (CH2), 26.9 (CH2),
26.6 (CH2), 26.4 (CH2), 26.0 (CH2), 24.4 (CH2), 20.3 (CH3).
IR (ATR): ν = 2929 (ss), 2857 (s), 1734 (ss), 1695 (m), 1675 (s), 1626 (m), 1459 (w), 1258
(m), 1127 (w), 980 (w).
LR-MS (80 °C): m/z = 280 (32), 262 (4), 220 (18), 178 (14), 123 (18), 109 (28), 95 (48), 81
(86), 68 (50), 55 (100).
HR-MS (C17H28O3; M+): ber. 280.2038; gef. 280.2033.
CHN-Analyse (C17H28O3): ber. C 72.82, H 10.06, gef. C 72.39, H 10.13.
(E)-Oxacyclooctadec-5-en-2,4-dion (93d)
Säulenchromatographische Reinigung (SiO2, MTB/Hexan 1:10) des
entsprechend der allgemeinen Arbeitsvorschrift D (Methode A) aus 34 mg
(100 µmol) Dioxolenon 92d erhaltenen Rückstands ergab 24 mg (85%)
monomeres Produkt als farbloses Harz. 1H-NMR (500 MHz, CDCl3): δ = 6.91 (dt, J = 16, 7 Hz, 1H), 6.19 (d, J = 16 Hz, 1H), 4.14 (t,
J = 6 Hz, 2H), 3.53 (s, 2H), 2.26 (m, 2H), 1.60 (m, 2H), 1.51 (m, 2H), 1.29 (m, 16H). 13C-NMR (125 MHz, CDCl3): δ = 191.9 (Cq), 167.5 (Cq), 150.6 (CH), 129.5 (CH), 65.6
(CH2), 47.5 (CH2), 32.0 (CH2), 28.2 (CH2), 27.7 (CH2), 27.4 (CH2), 27.3 (CH2), 27.0 (CH2),
26.9 (CH2), 26.9 (CH2), 26.8 (CH2), 26.7 (CH2), 25.3 (CH2).
IR (ATR): ν = 2928 (ss), 2856 (s), 1740 (ss), 1695 (m), 1676 (s), 1626 (m), 1461 (w), 1261
(m), 1151 (w), 980 (w).
LR-MS (70 °C): m/z = 280 (22), 262 (4), 220 (10), 178 (6), 123 (18), 110 (26), 95 (52), 81
(100), 68 (50), 55 (80).
HR-MS (C17H28O3; M+): ber. 280.2038; gef. 280.2033.
CHN-Analyse (C17H28O3): ber. C 72.82, H 10.06, gef. C 72.12, H 9.86.
O
O
O
( )8
O
O
O
( )9

II. Experimenteller Teil
95
2.8. Zu Kapitel 3.3.
6-[(E)-12-Hydroxytridec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on (118)
Entsprechend der allgemeinen Arbeitsvorschrift C wurden aus 50
mg (250 µmol) Tridecenol 117, 39 mg (250 µmol) Dienon 91 und
8 mg (5 mol%) 5 in 2.5 ml CH2Cl2 64 mg (79%) Produkt als
bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 6.54 (dt, J = 15.5, 7.0 Hz, 1H), 5.87 (d, J = 15.5 Hz, 1H),
5.22 (s, 1H), 3.77 (m, 1H), 2.18 (m, 2H), 1.70 (s, 6H), 1.41 (m, 6H), 1.27 (m, 11H), 1.16 (d, J
= 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 163.5 (Cq), 162.2 (Cq), 142.8 (CH), 122.5 (CH), 106.3
(Cq), 93.2 (CH), 68.2 (CH), 39.4 (CH2), 32.8 (CH2), 29.7 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4
(CH2), 29.3 (CH2), 28.4 (CH2), 25.8 (CH2), 25.1 (CH3), 23.6 (CH3).
IR (ATR): ν = 3434 (br), 2926 (ss), 2854 (s), 1726 (ss), 1653 (ss), 1592 (m), 1390 (s), 1374
(s), 1274 (s), 1206 (m), 1019 (m), 970 (w), 903 (w), 800 (w).
LR-MS (130 °C): m/z = 324 (14), 280 (10), 251 (12), 222 (8), 155 (20), 123 (74), 110 (100),
97 (30), 81 (40), 69 (30), 55 (40).
HR-MS (C19H32O4; M+): ber. 324.2300; gef. 324.2301.
2,2-Dimethyl-6-[(1R,2S)-1,2,12-trihydroxytridecyl]-[1.3]dioxin-4-on (119)
Eine Suspension von 140 mg AD-Mix-α, 0.22 mg K2OsO2(OH)4,
25 mg (300 µmol) NaHCO3 und 9.5 mg (100 µmol) MeSO2NH2 in
0.5 ml tBuOH/H2O (1:1) wurde bei 0 °C tropfenweise mit einer
Lösung von 32 mg (100 µmol) Dienon 118 in 0.5 ml tBuOH/H2O
(1:1) versetzt und 16 h bei RT gerührt. Das Reaktionsgemisch wurde mit 5 ml wässriger
Na2SO3-Lösung verdünnt und mit MTB (4 x 5 ml) extrahiert. Trocknen der organischen
Phasen über Na2SO4, Entfernen des Lösungsmittels und säulenchromatographische Reinigung
des Rückstands (SiO2, MTB) ergab 33 mg (91%) Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 5.62 (s, 1H), 4.00 (m, 1H), 3.81 (m, 2H), 2.60 (br s, 1H),
1.85 (br s, 1H), 1.71 (s, 6H), 1.57 (m, 3H), 1.50-1.25 (m, 16H), 1.19 (d, J = 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 171.4 (Cq), 161.6 (Cq), 107.2 (Cq), 93.5 (CH), 72.8 (CH),
71.5 (CH), 68.3 (CH), 39.3 (CH2), 33.7 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4 (CH2), 29.4
(CH2), 29.3 (CH2), 25.7 (CH2), 25.6 (CH2), 25.5 (CH3), 24.6 (CH3), 23.5 (CH3).
( )9
OH OO
O
( )9
OH OO
O
OH
OH

II. Experimenteller Teil
96
IR (ATR): ν = 3377 (br), 2926 (ss), 2854 (m), 1714 (ss), 1636 (m), 1391 (s), 1378 (s), 1276
(m), 1203 (s), 1012 (m), 908 (w), 818 (w).
FAB-MS: m/z = 359 (MH+, < 1), 327 (2), 281 (10), 221 (14), 147 (44), 136 (6), 73 (100).
(R)-Tridec-12-en-2-ol (117)
Eine aus 510 mg (2.3 mmol) 10-Brom-1-decen und 62 mg Magnesium-
Spänen (2.55 mmol) hergestellte 9-Decenylmagnesiumbromid-Lösung in
THF (3 ml) wurde unter Stickstoff-Atmosphäre bei -78 °C zu einer
Suspension von 90 mg (1.55 mmol) (R)-Methyloxiran und 7 mg (78 µmol) CuCN in 2 ml
THF gegeben. Das Reaktionsgemisch wurde über 2 h auf RT erwärmt, mit 10 ml 1 N HCl
versetzt und mit MTB (3 x 10 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4,
Entfernen des Lösungsmittels und säulenchromatographische Reinigung des Rückstands
(SiO2, MTB/Hexan 1:4) ergab 260 mg (85%) Produkt als farbloses Öl. 1H-NMR (500 MHz, CDCl3): δ = 5.80 (m, 1H), 5.00-4.90 (m, 2H), 3.79 (m, 1H), 2.04 (m,
2H), 1.45-1.25 (m, 17H), 1.18 (d, J = 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 139.3 (CH), 114.2 (CH2), 68.3 (CH), 39.4 (CH2), 33.9
(CH2), 29.7 (CH2), 29.7 (CH2), 29.6 (CH2), 29.5 (CH2), 29.2 (CH2), 29.0 (CH2), 25.8 (CH2),
23.6 (CH3).
IR (ATR): ν = 3349 (br), 2925 (ss), 2854 (s), 1641 (w), 1465 (w), 1373 (w), 909 (m).
LR-MS (RT): m/z = 198 (< 1), 180 (2), 152 (4), 138 (8), 123 (8), 109 (20), 96 (46), 82 (60),
69 (74), 55 (100).
HR-MS (C13H26O; M+): ber. 198.1984; gef. 198.1980.
6-[(E)-(R)-12-Hydroxytridec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on (118)
Entsprechend der allgemeinen Arbeitsvorschrift C wurden aus 100
mg (500 µmol) Tridecenol (R)-117, 116 mg (750 µmol) Dienon 91
und 16 mg (5 mol%) 5 in 5 ml CH2Cl2 142 mg (88%) Produkt als
bräunliches Harz erhalten. 1H-NMR (500 MHz, CDCl3): δ = 6.55 (dt, J = 15.5, 7.0 Hz, 1H), 5.88 (d, J = 15.5 Hz, 1H),
5.23 (s, 1H), 3.78 (m, 1H), 2.19 (m, 2H), 1.70 (s, 6H), 1.45 (m, 6H), 1.28 (m, 11H), 1.18 (d, J
= 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 163.5 (Cq), 162.2 (Cq), 142.8 (CH), 122.5 (CH), 106.3
(Cq), 93.3 (CH), 68.2 (CH), 39.4 (CH2), 32.8 (CH2), 29.7 (CH2), 29.6 (CH2), 29.5 (CH2), 29.4
(CH2), 29.3 (CH2), 28.4 (CH2), 25.8 (CH2), 25.1 (CH3), 23.6 (CH3).
OH
( )8
OH
( )8
OO
O

II. Experimenteller Teil
97
IR (ATR): ν = 3429 (br), 2926 (ss), 2854 (s), 1725 (ss), 1653 (ss), 1592 (m), 1390 (s), 1374
(s), 1274 (s), 1205 (m), 1019 (m), 970 (w), 903 (w), 800 (w).
LR-MS (100 °C): m/z = 324 (4), 280 (2), 251 (12), 222 (6), 206 (16), 182 (80), 155 (14), 123
(94), 110 (100), 95 (44), 81 (62), 69 (48), 55 (42).
HR-MS (C19H32O4; M+): ber. 324.2301; gef. 324.2306.
CHN-Analyse (C19H32O4): ber. C 70.34, H 9.94, gef. C 69.97, H 9.86.
Drehwert: [α]D20 = –4.2° (c = 1.0, CHCl3).
(5R,6S,16R)-5,6-(Isopropylidendioxy)-16-methyloxacyclohexadeca-2,4-dion (116)
Eine Suspension von 280 mg AD-Mix-α, 0.44 mg K2OsO2(OH)4, 50
mg (600 µmol) NaHCO3 und 19 mg (200 µmol) MeSO2NH2 in 1 ml tBuOH/H2O (1:1) wurde bei 0 °C tropfenweise mit einer Lösung von
65 mg (200 µmol) Dienon (R)-118 in 1 ml tBuOH/H2O (1:1) versetzt
und 16 h bei RT gerührt. Das Reaktionsgemisch wurde mit 5 ml
wässriger Na2SO3-Lösung verdünnt und mit MTB (4 x 5 ml)
extrahiert. Der nach Trocknen der organischen Phasen über Na2SO4 und Entfernen des
Lösungsmittels erhaltene Rückstand wurde in 4 ml Aceton gelöst und 60 h in Gegenwart von
4 mg (20 µmol) pTsOH gerührt, bevor die Lösung zu 1600 ml n-Heptan gegeben und 7 h
unter Stickstoff-Atmosphäre refluxiert wurde. Entfernen des Lösungsmittels und säulenchro-
matographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:10) ergab 44 mg (65%)
Produkt (2:1 Gemisch aus Keto- und Enolform) als farbloses Harz. 1H-NMR (500 MHz, CDCl3): δ = 11.9 (s, 1H, Enolform), 5.23 (s, 1H, Enolform), 5.20 (m,
1H, Enolform), 4.94 (m, 1H, Ketoform), 4.24 (m, 1H, Keto- und Enolform), 4.09 (d, J = 7 Hz,
1H, Ketoform), 3.85 (d, J = 8.5 Hz, 1H, Enolform), 3.70 (d, J = 15 Hz, 1H, Ketoform) 3.60
(d, J = 15 Hz, 1H, Ketoform), 1.80-1.20 (m, 27H, Keto- und Enolform). 13C-NMR (125 MHz, CDCl3): δ = 202.0 (Cq), 172.1 (Cq), 171.8 (Cq), 166.4 (Cq), 110.3 (Cq),
110.0 (Cq), 93.4 (CH), 85.4 (CH), 82.5 (CH), 77.7 (CH), 76.7 (CH), 73.0 (CH), 70.2 (CH),
45.9 (CH2), 35.6 (CH2), 35.3 (CH2), 32.1 (CH2), 32.1 (CH2), 28.3 (CH2), 28.1 (CH2), 27.7
(CH2), 27.4 (CH2), 27.3 (CH3), 27.0 (CH3), 26.9 (CH2), 26.8 (CH2), 26.8 (CH2), 26.7 (CH2),
26.4 (CH2), 26.0 (CH3), 25.9 (CH3), 25.8 (CH2), 24.5 (CH2), 23.5 (CH2), 23.3 (CH2), 23.0
(CH2), 20.6 (CH3), 20.1 (CH3).
IR (ATR): ν = 2985 (m), 2932 (ss), 2859 (s), 1747 (s), 1721 (s), 1651 (s), 1460 (m), 1381
(m), 1235 (ss), 1167 (w), 1063 (s), 868 (w), 804 (m).
O
O OO
O

II. Experimenteller Teil
98
LR-MS (110 °C): m/z = 340 (10), 325 (36), 312 (18), 297 (10), 253 (8), 177 (10), 158 (100),
140 (24), 117 (50), 95 (48), 81 (40), 69 (44), 59 (64), 55 (74).
HR-MS (C19H32O5; M+): ber. 340.2250; gef. 340.2252.
CHN-Analyse (C19H32O5): ber. C 67.03, H 9.47, gef. C 66.71, H 9.11.
Drehwert: [α]D20 = +14.5° (c = 1.0, CHCl3).
(E)-(5S,6S,16R)-5,6-(Isopropylidendioxy)-16-methyloxacyclohexadec-3-en-2-on (115)
Eine Lösung von 34 mg (100 µmol) Ketolacton 116 in 1 ml MeOH
wurde mit 4 mg (100 µmol) NaBH4 versetzt und 5 min bei RT
gerührt, bevor mit 5 ml gesättigter NaCl-Lösung verdünnt und mit
Et2O (3 x 5 ml) extrahiert wurde. Der nach Trocknen der organischen
Phasen über Na2SO4 und Entfernen des Lösungsmittels erhaltene
Rückstand wurde in 1 ml Pyridin gelöst, mit 23 mg (200 µmol) MsCl
versetzt und 2 h bei RT gerührt. Das Reaktionsgemisch wurde mit 5 ml 1 N HCl verdünnt und
mit CH2Cl2 (3 x 5 ml) extrahiert. Trocknen der organischen Phasen über Na2SO4 und
Entfernen der flüchtigen Bestandteile ergab einen Rückstand, der in 1 ml CH2Cl2 gelöst, mit
30 mg (200 µmol) DBU versetzt und 1 h bei RT gerührt wurde. Entfernen des Lösungsmittels
und säulenchromatographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:10) ergab 26
mg (80%) Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 6.88 (dd, J = 15.5, 7 Hz, 1H), 6.12 (d, J = 15.5 Hz, 1H),
5.03 (m, 1H), 4.13 (t, J = 7.5 Hz, 1H), 3.75 (m, 1H), 1.80 (m, 1H), 1.63 (m, 2H), 1.50-1.35
(m, 2H) 1.43 (s, 3H), 1.42 (s, 3H), 1.35-1.15 (m, 13H), 1.25 (d, J = 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 165.6 (Cq), 144.4 (CH), 123.7 (CH), 109.3 (Cq), 80.9
(CH), 80.2 (CH), 71.2 (CH), 35.4 (CH2), 31.1 (CH2), 27.9 (CH2), 27.4 (CH2), 27.4 (CH2),
27.3 (CH3), 27.1 (CH2), 26.7 (CH3), 26.6 (CH2), 24.9 (CH2), 23.4 (CH2), 20.6 (CH3).
IR (ATR): ν = 2986 (m), 2929 (ss), 2858 (s), 1709 (ss), 1661 (m), 1458 (m), 1378 (m), 1257
(s), 1182 (s), 1121 (m), 1055 (s), 990 (m), 862 (m), 809 (w).
LR-MS (70 °C): m/z = 324 (12), 309 (98), 295 (4), 267 (4), 249 (8), 203 (4), 183 (6), 165 (8),
142 (100), 109 (10), 97 (34), 84 (86), 69 (14), 59 (58), 55 (20).
HR-MS (C19H32O4; M+): ber. 324.2301; gef. 324.2297.
Drehwert: [α]D20 = +5.4° (c = 1.0, CHCl3).
Schmelzpunkt: 71–72 °C.
O
O OO

II. Experimenteller Teil
99
(E)-(6S,16R)-6-Hydroxy-16-methyloxacyclohexadec-3-en-2,5-dion (122)
Eine Lösung von 25 mg (77 µmol) Acetonid 115 in 0.6 ml MeCN
und 0.3 ml H2O wurde bei 0 °C tropfenweise mit 0.6 ml TFA
versetzt und über 1 h auf RT erwärmt. Das Reaktionsgemisch
wurde mit 60 ml CH2Cl2 verdünnt und mit 30 ml gesättigter
NaHCO3-Lösung gewaschen. Trocknen der organischen Phase über
Na2SO4 und Entfernen des Lösungsmittels ergab einen Rückstand, der in 3 ml absolutem
CH2Cl2 gelöst und unter Stickstoff-Atmosphäre bei 0 °C mit 29 mg (150 µmol) pTsOH und
24 mg (150 µmol) TEMPO versetzt wurde. Nach 5 h bei RT wurde das Lösungsmittels
entfernt und der Rückstand säulenchromatographisch gereinigt (SiO2, MTB/Hexan 1:4).
Umkristallisation aus Hexan ergab 13 mg (60%) Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 7.25 (d, J = 16 Hz, 1H), 6.79 (d, J = 16 Hz, 1H), 5.18 (m,
1H), 4.54 (m, 1H), 3.45 (br s, 1H), 1.85 (m, 2H), 1.73 (m, 1H), 1.52 (m, 3H) 1.45-1.05 (m,
14H), 1.30 (d, J = 6 Hz, 3H), 0.98 (m, 1H). 13C-NMR (125 MHz, CDCl3): δ = 201.5 (Cq), 165.1 (Cq), 135.0 (CH), 132.6 (CH), 76.5
(CH), 72.7 (CH), 34.3 (CH2), 31.2 (CH2), 28.2 (CH2), 28.1 (CH2), 27.4 (CH2), 27.1 (CH2),
26.9 (CH2), 23.5 (CH2), 20.7 (CH2), 19.7 (CH3).
IR (ATR): ν = 3490 (br), 2928 (ss), 2856 (s), 1714 (ss), 1697 (ss), 1642 (w), 1460 (m), 1354
(m), 1287 (ss), 1191 (s), 1059 (m), 983 (m).
LR-MS (160 °C): m/z = 283 (2), 265 (1), 183 (4), 165 (6), 123 (4), 109 (12), 100 (100), 81
(16), 72 (18), 55 (34).
HR-MS (C16H27O4; MH+): ber. 283.1909; gef. 283.1914.
Drehwert: [α]D20 = +22.4° (c = 0.25, CHCl3).
Schmelzpunkt: 84–85 °C.
(–)-A26771B (104)
Eine Lösung von 5 mg (18 µmol) Hydroxyketon 122 in 0.3 ml
absolutem CH2Cl2 wurde unter Stickstoff-Atmosphäre mit 3.5
mg (35 µmol) Succinanhydrid und 2.4 mg (20 µmol) DMAP
versetzt und 24 h bei RT gerührt. Entfernen des Lösungsmittels
und säulenchromatographische Reinigung des Rückstands (SiO2,
5% MeOH in CH2Cl2) ergab 5 mg (74%) Produkt als farblosen Feststoff.
O
O OHO
O
O OO
HO2C
O

II. Experimenteller Teil
100
1H-NMR (500 MHz, CDCl3): δ = 7.22 (d, J = 16, 1H), 6.75 (d, J = 16 Hz, 1H), 5.32 (t, J =
5.5 Hz, 1H), 5.14 (m, 1H), 2.73 (m, 4H), 1.90 (m, 2H), 1.68 (m, 1H), 1.55 (m, 1H) 1.50-1.10
(m, 15H), 1.30 (d, J = 6 Hz, 3H). 13C-NMR (125 MHz, CDCl3): δ = 195.5 (Cq), 176.2 (Cq), 171.5 (Cq), 164.9 (Cq), 135.3 (CH),
132.6 (CH), 78.0 (CH), 72.6 (CH), 34.6 (CH2), 28.9 (CH2), 28.7 (CH2), 28.6 (CH2), 28.0
(CH2), 27.9 (CH2), 27.3 (CH2), 27.1 (CH2), 27.0 (CH2), 23.6 (CH2), 22.3 (CH2), 19.9 (CH3).
IR (ATR): ν = 2928 (s), 2857 (m), 1742 (s), 1703 (ss), 1381 (w), 1300 (s), 1196 (m), 1165 (s).
FAB-MS: m/z = 383 (MH+, 12), 243 (46), 154 (40), 137 (52), 121 (24), 107 (40), 93 (66), 69
(80), 55 (100).
Drehwert: [α]D20 = –13.2° (c = 0.19, MeOH).
Schmelzpunkt: 121–122 °C.
2.9. Zu Kapitel 3.4.
6-[(E)-(S)-13-Hydroxytetradec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on (92c)
Entsprechend der allgemeinen Arbeitsvorschrift C wurden aus 106
mg (500 µmol) Tetradecenol (S)-90c, 116 mg (750 µmol) Dienon
91 und 16 mg (5 mol%) 5 in 5 ml CH2Cl2 146 mg (86%) Produkt
als bräunlicher Feststoff erhalten. 1H-NMR (500 MHz, CDCl3): δ = 6.54 (dt, J = 15.5, 7.0 Hz, 1H), 5.88 (d, J = 15.5 Hz, 1H),
5.22 (s, 1H), 3.78 (m, 1H), 2.19 (m, 2H), 1.70 (s, 6H), 1.40 (m, 6H), 1.26 (m, 13H), 1.17 (d, J
= 6 Hz, 3H).
Drehwert: [α]D20 = +4.4° (c = 1.0, CHCl3).
Schmelzpunkt: 50–51 °C.
(5S,6R,17S)-5,6-(Isopropylidendioxy)-17-methyloxacycloheptadeca-2,4-dion (130)
Eine Suspension von 280 mg AD-Mix-β, 0.44 mg K2OsO2(OH)4, 50
mg (600 µmol) NaHCO3 und 19 mg (200 µmol) MeSO2NH2 in 1 ml tBuOH/H2O (1:1) wurde bei 0 °C tropfenweise mit einer Lösung
von 68 mg (200 µmol) Dienon (S)-92c in 1 ml tBuOH/H2O (1:1)
versetzt und 16 h bei RT gerührt. Das Reaktionsgemisch wurde mit
5 ml wässriger Na2SO3-Lösung verdünnt und mit MTB (4 x 5 ml) extrahiert. Der nach
Trocknen der organischen Phasen über Na2SO4 und Entfernen des Lösungsmittels erhaltene
Rückstand wurde in 4 ml Aceton gelöst und 60 h in Gegenwart von 4 mg (20 µmol) pTsOH
OH
( )9
OO
O
O
O
OO
O

II. Experimenteller Teil
101
gerührt, bevor die Lösung zu 1600 ml n-Heptan gegeben und 7 h unter Stickstoff-Atmosphäre
refluxiert wurde. Entfernen des Lösungsmittels und säulenchromatographische Reinigung des
Rückstands (SiO2, MTB/Hexan 1:10) ergab 47 mg (66%) Produkt (3:1 Gemisch aus Keto-
und Enolform) als farbloses Harz. 1H-NMR (500 MHz, CDCl3): δ = 12.1 (s, 1H, Enolform), 5.21 (s, 1H, Enolform), 5.10 (m,
1H, Enolform), 5.03 (m, 1H, Ketoform), 4.25 (m, 1H, Keto- und Enolform), 4.06 (d, J = 7 Hz,
1H, Ketoform), 3.87 (d, J = 8 Hz, 1H, Enolform), 3.78 (d, J = 15.5 Hz, 1H, Ketoform) 3.57
(d, J = 15.5 Hz, 1H, Ketoform), 1.90-1.20 (m, 29H, Keto- und Enolform). 13C-NMR (125 MHz, CDCl3): δ = 203.0 (Cq), 172.2 (Cq), 171.9 (Cq), 166.7 (Cq), 110.4 (Cq),
110.1 (Cq), 93.0 (CH), 85.4 (CH), 82.5 (CH), 78.0 (CH), 76.8 (CH), 72.2 (CH), 71.2 (CH),
45.6 (CH2), 35.9 (CH2), 34.9 (CH2), 32.9 (CH2), 32.6 (CH2), 29.1 (CH2), 28.4 (CH2), 28.2
(CH2), 28.1 (CH2), 27.9 (CH2), 27.7 (CH2), 27.4 (CH2), 27.3 (CH3), 27.2 (CH3), 27.1 (CH2),
27.0 (CH2), 26.9 (CH2), 26.9 (CH2), 26.8 (CH2), 26.0 (CH3), 24.7 (CH2), 24.6 (CH2), 24.1
(CH2), 23.7 (CH2), 20.6 (CH3), 20.2 (CH3).
IR (ATR): ν = 2985 (m), 2931 (ss), 2858 (s), 1745 (s), 1719 (s), 1651 (s), 1460 (m), 1381
(m), 1237 (ss), 1164 (w), 1063 (s), 868 (w), 807 (w).
LR-MS (110 °C): m/z = 354 (8), 339 (36), 326 (18), 311 (14), 267 (10), 225 (4), 191 (12),
158 (100), 140 (26), 117 (80), 95 (50), 81 (48), 69 (58), 59 (70), 55 (82).
HR-MS (C20H34O5; M+): ber. 354.2406; gef. 354.2411.
(6S,7R,18S)-6,7-(Isopropylidendioxy)-18-methyloxacyclooctadeca-2,5-dion (131)
Eine Lösung von 0.2 ml (1 M in Hexan, 200 µmol) ZnEt2 in 0.5
ml absolutem CH2Cl2 unter Stickstoff-Atmosphäre wurde bei 0
°C tropfenweise mit 54 mg (200 µmol) CH2I2 versetzt. Nach der
Bildung eines weißen Präzipitats (10 min) wurde eine Lösung
von 15 mg (42 µmol) Ketoester 130 in 0.5 ml absolutem CH2Cl2
hinzugegeben und über 3 h auf RT erwärmt. Das Reaktionsgemisch wurde mit 5 ml
gesättigter NH4Cl-Lösung versetzt, mit CH2Cl2 (3 x 5 ml) extrahiert und die organischen
Phasen wurden über Na2SO4 getrocknet. Entfernen des Lösungsmittels und säulenchromato-
graphische Reinigung des Rückstands (SiO2, MTB/Hexan 1:20) ergab 10 mg (65%) Produkt
als farbloses Harz. 1H-NMR (500 MHz, CDCl3): δ = 5.03 (m, 1H), 4.11 (m, 1H), 3.95 (d, J = 8 Hz, 1H), 3.30 (m,
1H), 2.76 (m, 1H), 2.55 (m, 1H), 2.37 (m, 1H), 1.77 (m, 1H), 1.64 (m, 2H), 1.50 (s, 3H), 1.46
(s, 3H), 1.50-1.15 (m, 17H), 1.19 (d, J = 6 Hz, 3H).
O
OO O
O

II. Experimenteller Teil
102
13C-NMR (125 MHz, CDCl3): δ = 209.1 (Cq), 172.6 (Cq), 110.3 (Cq), 85.8 (CH), 77.4 (CH),
70.0 (CH), 35.9 (CH2), 33.2 (CH2), 32.3 (CH2), 28.0 (CH2), 27.9 (CH2), 27.8 (CH2), 27.6
(CH2), 27.4 (CH3), 27.3 (CH2), 27.1 (CH2), 27.0 (CH2), 26.3 (CH3), 24.6 (CH2), 24.2 (CH2),
20.6 (CH3).
IR (ATR): ν = 2983 (m), 2930 (ss), 2858 (s), 1721 (ss), 1461 (w), 1380 (m), 1372 (s), 1209
(ss), 1162 (m), 1072 (s), 865 (w), 805 (w).
LR-MS (120 °C): m/z = 368 (20), 353 (24), 340 (10), 325 (100), 311 (50), 293 (24), 265 (10),
225 (8), 191 (10), 155 (18), 109 (10), 98 (12), 81 (10), 55 (14).
HR-MS (C21H36O5; M+): ber. 368.2563; gef. 368.2566.
2.10. Zu Kapitel 3.5.
Dodec-11-enylcarbaminsäure-(9-fluorenylmethyl)-ester (132a)
Eine Lösung von 1.17 g (5 mmol) 11-Brom-1-undecen in 5 ml
Glycol wurde unter Stickstoff-Atmosphäre mit 650 mg (10 mmol)
KCN versetzt und 16 h bei 100 °C gerührt. Das Reaktionsgemisch wurde mit 100 ml MTB
verdünnt und mit gesättigter NaCl-Lösung gewaschen (2 x 50 ml). Trocknen der organischen
Phase über Na2SO4 und Entfernen des Lösungsmittels ergab einen Rückstand, der in 10 ml
absolutem Et2O gelöst und tropfenweise zu einer Suspension von 190 mg (5 mmol) LAH in
10 ml absolutem Et2O gegeben wurde. Nach 1 h wurde das Reaktionsgemisch langsam mit 20
ml 1 N NaOH versetzt und mit MTB (3 x 20 ml) extrahiert. Der nach Trocknen der
organischen Phasen über Na2SO4 und Entfernen des Lösungsmittels erhaltene Rückstand
wurde in einer Mischung aus 5 ml THF und 5 ml H2O gelöst und nach Zugabe von 425 mg (5
mmol) NaHCO3 portionsweise mit 1.3 g (5 mmol) FmocCl versetzt. Nach 16 h bei RT wurde
mit 100 ml MTB verdünnt und die organische Phase wurde über Na2SO4 getrocknet.
Entfernen des Lösungsmittels und säulenchromatographische Reinigung des Rückstands
(SiO2, MTB/Hexan 1:5) ergab 1.3 g (65%) Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 7.77 (d, J = 7.5 Hz, 2H), 7.60 (d, J = 7.5 Hz, 2H), 7.40 (t, J
= 7.5 Hz, 2H), 7.32 (t, J = 7.5 Hz, 2H), 5.82 (m, 1H), 5.00-4.90 (m, 2H), 4.74 (br s, 1H), 4.40
(m, 2H), 4.22 (m, 1H), 3.19 (m, 2H), 2.04 (m, 2H), 1.50 (m, 2H), 1.40-1.20 (m, 14H). 13C-NMR (125 MHz, CDCl3): δ = 156.5 (Cq), 144.1 (Cq), 141.4 (Cq), 139.3 (CH), 127.7
(CH), 127.1 (CH), 125.1 (CH), 120.0 (CH), 114.2 (CH2), 66.5 (CH2), 47.4 (CH), 41.2 (CH2),
33.9 (CH2), 30.1 (CH2), 29.6 (CH2), 29.5 (CH2), 29.5 (CH2), 29.4 (CH2), 29.2 (CH2), 29.0
(CH2), 26.8 (CH2).
( )8FmocHN

II. Experimenteller Teil
103
IR (ATR): ν = 3337 (s), 2923 (s), 2852 (s), 1689 (ss), 1535 (s), 1451 (m), 1445 (w), 1269 (s),
1143 (w), 990 (w), 913 (w), 757 (m), 740 (s).
LR-MS (180 °C): m/z = 405 (< 1), 322 (< 1), 294 (< 1), 178 (100), 165 (6), 69 (2), 55 (4).
HR-MS (C27H35NO2; M+): ber. 405.2668; gef. 405.2675.
Schmelzpunkt: 90–91 °C.
Dodec-11-enylcarbaminsäure-tbutylester (132b)
Eine Lösung von 1.17 g (5 mmol) 11-Brom-1-undecen in 5 ml Glycol
wurde unter Stickstoff-Atmosphäre mit 650 mg (10 mmol) KCN
versetzt und 16 h bei 100 °C gerührt. Das Reaktionsgemisch wurde mit 100 ml MTB verdünnt
und mit gesättigter NaCl-Lösung gewaschen (2 x 50 ml). Trocknen der organischen Phase
über Na2SO4 und Entfernen des Lösungsmittels ergab einen Rückstand, der in 10 ml
absolutem Et2O gelöst und tropfenweise zu einer Suspension von 190 mg (5 mmol) LAH in
10 ml absolutem Et2O gegeben wurde. Nach 1 h wurde das Reaktionsgemisch langsam mit 20
ml 1 N NaOH versetzt und mit MTB (3 x 20 ml) extrahiert. Der nach Trocknen der
organischen Phasen über Na2SO4 und Entfernen des Lösungsmittels erhaltene Rückstand
wurde in 20 ml CH2Cl2 gelöst, nach Zugabe von 700 mg (5 mmol) K2CO3 portionsweise mit
1.1 g (5 mmol) Boc2O versetzt und 2 h bei RT gerührt. Entfernen des Lösungsmittels und
säulenchromatographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:10) ergab 864 mg
(61%) Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 5.81 (m, 1H), 5.00-4.90 (m, 2H), 4.48 (br s, 1H), 3.10 (m,
2H), 2.04 (m, 2H), 1.44 (m, 11H), 1.37 (m, 2H), 1.26 (m, 12H). 13C-NMR (125 MHz, CDCl3): δ = 156.0 (Cq), 139.3 (CH), 114.2 (CH2), 79.0 (Cq), 40.7
(CH2), 33.9 (CH2), 30.1 (CH2), 29.6 (CH2), 29.6 (CH2), 29.5 (CH2), 29.3 (CH2), 29.2 (CH2),
29.0 (CH2), 28.5 (CH3), 26.9 (CH2).
IR (ATR): ν = 3352 (br), 2977 (m), 2926 (ss), 2854 (s), 1693 (ss), 1641 (w), 1520 (s), 1456
(m), 1391 (w), 1365 (m), 1250 (s), 1174 (ss), 993 (w), 909 (m), 781 (w).
LR-MS (170 °C): m/z = 284 (4), 270 (6), 244 (52), 200 (30), 183 (12), 155 (8), 83 (8), 69
(18), 55 (100).
HR-MS (C17H34NO2; MH+): ber. 284.2590; gef. 284.2593.
Schmelzpunkt: 27–28 °C.
BocHN ( )8

II. Experimenteller Teil
104
6-[(E)-12-(9-Fluorenyloxycarbonyl)aminododec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on
(133a)
Eine Lösung von 81 mg (200 µmol) Amin 132a und 31 mg (200
µmol) Dienon 91 in 2 ml absolutem CH2Cl2 unter Stickstoff-
Atmosphäre wurde mit 6 mg (5 mol%) des Ruthenium-Katalysators 5
versetzt und 24 h bei 40 °C gerührt. Entfernen des Lösungsmittels
und säulenchromatographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:2) ergab 96
mg (90%) Produkt als bräunliches Harz. 1H-NMR (500 MHz, CDCl3): δ = 7.76 (d, J = 7.5 Hz, 2H), 7.59 (d, J = 7.5 Hz, 2H), 7.39 (t, J
= 7.5 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 6.55 (dt, J = 16, 7.0 Hz, 1H), 5.88 (d, J = 16 Hz, 1H),
5.24 (s, 1H), 4.80 (br s, 1H), 4.40 (m, 2H), 4.21 (m, 1H), 3.19 (m, 2H), 2.19 (m, 2H), 1.70 (s,
6H), 1.50-1.40 (m, 4H), 1.28 (m, 12H). 13C-NMR (125 MHz, CDCl3): δ = 163.5 (Cq), 162.2 (Cq), 156.5 (Cq), 144.1 (Cq), 142.8 (CH),
141.4 (Cq), 127.7 (CH), 127.1 (CH), 125.1 (CH), 122.5 (CH), 120.0 (CH), 106.3 (Cq), 93.3
(CH), 66.6 (CH2), 47.4 (CH), 41.2 (CH2), 32.8 (CH2), 30.1 (CH2), 29.6 (CH2), 29.5 (CH2),
29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 28.4 (CH2), 26.8 (CH2), 25.1 (CH3).
IR (ATR): ν = 3340 (br), 2926 (s), 2854 (m), 1723 (ss), 1652 (s), 1592 (w), 1532 (m), 1451
(w), 1390 (s), 1374 (s), 1273 (s), 1249 (s), 1206 (m), 1019 (m), 903 (w), 759 (m), 741 (m).
FAB-MS: 532 (MH+, 6), 474 (10), 355 (4), 252 (16), 179 (100), 147 (30), 73 (76), 55 (24).
6-[(E)-12-tButyloxycarbonylaminododec-1-enyl]-2,2-dimethyl-[1.3]dioxin-4-on (133b)
Eine Lösung von 80 mg (280 µmol) Amin 132b und 43 mg (280
µmol) Dienon 91 in 2.8 ml absolutem CH2Cl2 unter Stickstoff-
Atmosphäre wurde mit 9 mg (5 mol%) des Ruthenium-Katalysators 5
versetzt und 24 h bei 40 °C gerührt. Entfernen des Lösungsmittels
und säulenchromatographische Reinigung des Rückstands (SiO2, MTB/Hexan 1:3) ergab 100
mg (88%) Produkt als bräunliches Harz. 1H-NMR (500 MHz, CDCl3): δ = 6.55 (dt, J = 16, 7.0 Hz, 1H), 5.88 (d, J = 16 Hz, 1H), 5.23
(s, 1H), 4.49 (s, 1H), 3.10 (m, 2H), 2.20 (m, 2H), 1.70 (s, 6H), 1.44 (m, 13H), 1.27 (m, 12H). 13C-NMR (125 MHz, CDCl3): δ = 163.5 (Cq), 162.2 (Cq), 156.1 (Cq), 142.8 (CH), 122.5
(CH), 106.3 (Cq), 93.3 (CH), 79.1 (Cq), 40.6 (CH2), 32.8 (CH2), 30.1 (CH2), 29.6 (CH2), 29.5
(CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 28.5 (CH3), 28.4 (CH2), 26.8 (CH2), 25.1 (CH3).
IR (ATR): ν = 3358 (br), 2927 (s), 2855 (m), 1723 (ss), 1653 (s), 1593 (w), 1517 (m), 1456
(w), 1390 (s), 1374 (m), 1273 (s), 1250 (m), 1174 (m), 1019 (m), 902 (w).
OO
O( )8
NHFmoc
NHBocOO
O( )8

II. Experimenteller Teil
105
LR-MS (180 °C): m/z = 410 (< 1), 336 (14), 308 (18), 295 (34), 278 (24), 250 (60), 142 (26),
123 (22), 110 (40), 97 (10), 81 (12), 69 (8), 57 (100).
HR-MS (C23H40NO5; MH+): ber. 410.2906; gef. 410.2918.
Azacyclohexadecan-2,4-dion (137b)
Eine Lösung von 33 mg (80 µmol) Dienon 133b in 1.6 ml MeOH
wurde entgast, unter Stickstoff-Atmosphäre mit 4 mg Pd/C (10%)
versetzt und 30 min unter Wasserstoff-Atmosphäre gerührt. Der nach
Filtration und Entfernen des Lösungsmittels erhaltene Rückstand
wurde in 1 ml CH2Cl2 gelöst, tropfenweise mit 0.1 ml TFA versetzt
und 1 h bei RT gerührt. Das Reaktionsgemisch wurde mit 20 ml CH2Cl2 verdünnt und mit 5
ml gesättigter NaHCO3-Lösung gewaschen. Trocknen der organischen Phase über Na2SO4
und Entfernen des Lösungsmittels ergab einen Rückstand, der in 800 ml n-Heptan gelöst und
7 h unter Stickstoff-Atmosphäre refluxiert wurde. Entfernen des Lösungsmittels und säulen-
chromatographische Reinigung des Rückstands (SiO2, MTB) ergab 12 mg (61%) Produkt als
farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 6.98 (br s, 1H), 3.39 (s, 2H), 3.33 (m, 2H), 2.57 (t, J = 6.5
Hz, 2H), 1.67 (quin, J = 6.5 Hz, 2H), 1.48 (m, 2H), 1.35-1.15 (m, 16H). 13C-NMR (125 MHz, CDCl3): δ = 208.5 (Cq), 165.0 (Cq), 49.3 (CH2), 43.6 (CH2), 39.1
(CH2), 29.1 (CH2), 27.4 (CH2), 27.3 (CH2), 27.2 (CH2), 26.7 (CH2), 26.6 (CH2), 25.9 (CH2),
25.4 (CH2), 24.7 (CH2), 23.0 (CH2).
IR (ATR): ν = 3296 (br), 2928 (s), 2855 (m), 1717 (s), 1640 (ss), 1552 (m), 1459 (w).
LR-MS (70 °C): m/z = 253 (66), 235 (22), 210 (16), 196 (14), 168 (20), 115 (28), 97 (26), 81
(28), 69 (60), 55 (100).
HR-MS (C15H27NO2; M+): ber. 253.2042; gef. 253.2050.
Schmelzpunkt: 121–122 °C.
Azacyclotetradecan-2,4-dion (137a)
Eine Lösung von 23 mg (80 µmol) Dienon 92a in 0.5 ml Pyridin wurde
mit 10 mg (90 µmol) MsCl versetzt und 2 h bei RT gerührt. Das
Reaktionsgemisch wurde mit 10 ml MTB verdünnt und mit 1 N HCl (3
x 5 ml) gewaschen. Der nach Trocknen der organischen Phase über
Na2SO4 und Entfernen des Lösungsmittels erhaltene Rückstand wurde
in 0.5 ml DMF gelöst, unter Stickstoff-Atmosphäre mit 21 mg (320 µmol) NaN3 versetzt und
NH
O
O
NH
O
O

II. Experimenteller Teil
106
16 h bei 50 °C gerührt. Nach Zugabe von 10 ml MTB wurde mit H2O (3 x 5 ml) gewaschen,
die organische Phase wurde über Na2SO4 getrocknet und das Lösungsmittel wurde entfernt.
Eine Lösung des Rückstands in 1.5 ml MeOH wurde entgast, unter Stickstoff-Atmosphäre mit
4 mg Pd/C (10%) versetzt und 30 min unter Wasserstoff-Atmosphäre gerührt. Der nach
Filtration und Entfernen des Lösungsmittels erhaltene Rückstand wurde in 50 ml Toluol
gelöst und unter Stickstoff-Atmosphäre tropfenweise über 4 h zu 800 ml refluxierendem
Toluol gegeben. Die Lösung wurde weitere 3 h refluxiert und das Lösungsmittel wurde
entfernt. Säulenchromatographische Reinigung des Rückstands (SiO2, MTB) ergab 9 mg
(52%) Produkt als farblosen Feststoff. 1H-NMR (500 MHz, CDCl3): δ = 6.17 (br s, 1H), 3.36 (s, 2H), 3.33 (m, 2H), 2.64 (t, J = 6.5
Hz, 2H), 1.71 (quin, J = 6.5 Hz, 2H), 1.54 (m, 2H), 1.35-1.20 (m, 12H), 1.15 (m, 2H). 13C-NMR (125 MHz, CDCl3): δ = 207.2 (Cq), 165.0 (Cq), 51.9 (CH2), 41.9 (CH2), 38.7
(CH2), 27.7 (CH2), 27.1 (CH2), 26.2 (CH2), 26.1 (CH2), 25.7 (CH2), 25.4 (CH2), 24.3 (CH2),
21.4 (CH2).
IR (ATR): ν = 3288 (s), 2935 (s), 2859 (m), 1717 (s), 1641 (ss), 1560 (m), 1456 (w).
LR-MS (110 °C): m/z = 225 (66), 195 (22), 154 (18), 140 (18), 128 (22), 99 (100), 86 (26),
69 (36), 55 (84).
HR-MS (C13H23NO2; M+): ber. 225.1729; gef. 225.1727.
Schmelzpunkt: 118–119 °C.

III. Anhang
107
III. Anhang
1. Röntgenstrukturanalyse
Kristalldaten und Strukturverfeinerung von cis-85:
Summenformel C14H16N2O6S
Molekulargewicht 340.35
Temperatur 293(2) K
Wellenlänge 0.71073 Å
Kristallsystem triklin
Raumgruppe P-1
Zelldimensionen a = 6.627(5) Å α = 91.38(3)°
b = 10.756(8) Å β = 89.51(3)°
c = 11.051(10) Å γ = 76.88(3)°
Zellvolumen, Z 766.9(10) Å3, 2
Berechnete Dichte 1.474 Mg/m3
Absorptionskoeffizient 0.244 mm-1
F(000) 356
Kristallgröße 0.42 x 0.44 x.0.70 mm
Gemessener θ-Bereich 1.84 bis 25.00°
Indexgrenzen -7 < h < 7
-12 < k < 12
-13 < l < 11
Anzahl der gem. Reflexe 4482
Unabhängige Reflexe 2540 (Rint = 0.3658)
Vollständigkeit bis θ = 25.00° 94.2%
Strukturverfeinerungsmethode Vollmatrix Least-Squares in F2
Daten / Restraints / Parameter 2540 / 0 / 208
Goodness-of-Fit in F2 1.102
Endgültige R-Werte [I>2σ(I)] R1 = 0.2567, wR2 = 0.4972
R-Werte (sämtliche Daten) R1 = 0.5104, wR2 = 0.6334
Größtes Maximum u. Minimum 0.840 und -0.739 eÅ-3

III. Anhang
108
2. Abkürzungen
Ac Acetyl
acac Acetylacetonat
AD Asymmetric Dihydroxylation
Alloc Allyloxycarbonyl
Ar Aryl
ATR Attenuated Total Reflectance
BBN Borabicyclo[3.3.1]nonan
BINOL Binaphtol
Bn Benzyl
Boc t-Butyloxycarbonyl
ber. berechnet
Bu Butyl
BuLi n-Butyllithium
Bz Benzoyl
CA Cycloaddition
CDI Carbonyldiimidazol
CHN Elementaranalyse
CM Cross Metathesis
Cp Cyclopentadienyl
CR Cycloreversion
Cy Cyclohexyl
DABCO 1,4-Diazabicyclo[2.2.2]octan
DBU 1,8-Diazabicyclo[5.4.0]undec-7-en
DC Dünnschichtchromatographie
DCE 1,2-Dichlorethan
DDQ 2,3-Dichlor-5,6-dicyan-1,4-benzochinon
DEAD Diethylazodicarboxylat
DEPT Distortionless Enhancement by Polarisation Transfer
DMAP 4-Dimethylaminopyridin
DME 1,2-Dimethoxyethan
DMF N,N-Dimethylformamid
DMSO Dimethylsulfoxid
ds diastereomeric excess

III. Anhang
109
ee enantiomeric excess
eq Äquivalent(e)
Et Ethyl
FAB Fast Atom Bombardment
Fmoc 9-Fluorenylmethoxycarbonyl
g Gramm
GC Gaschromatographie
gef. gefunden
gem. gemessen
h Stunde(n)
HMPA Hexamethylphosphorsäuretriamid
HR High Resolution
Hz Hertz
i iso
IR Infrarotspektroskopie
Im Imidazol
J Kopplungskonstante
LiHMDS Lithiumhexamethyldisilazid
LAH Lithiumaluminiumhydrid
LDA Lithiumdiisopropylamid
LR Low Resolution
M Molar
mCPBA meta-Chlorperbenzoesäure
Me Methyl
Mes Mesityl
MIC Minimal Inhibitory Concentration
min Minuten
MOM Methoxymethyl
Ms Mesyl
MS Massenspektrometrie
MTB Methyl-t-Butylether
m/z Masse-Ladungs-Verhältnis
N Normal
NBS N-Bromsuccinimid

III. Anhang
110
NHC N-Heterocyclisches Carben
NMR Nuclear Magnetic Resonance
NOE Nuclear Overhauser Effect
Ns Nosyl
o ortho
p para
PDC Pyridiniumdichromat
PG Protecting Group
Ph Phenyl
Phth Phthaloyl
ppm parts per million
Pr Propyl
quant. quantitativ
R Rest
R Rectus (rechts)
RCM Ring Closing Metathesis
ROM Ring Opening Metathesis
RRM Ring Rearrangement Metathesis
RT Raumtemperatur
S Sinister (links)
SN1 Nucleophile Substitution 1. Ordnung
SN2 Nucleophile Substitution 2. Ordnung
t tertiär
TBDPS t-Butyldiphenylsilyl
TBS t-Butyldimethylsilyl
TEMPO 2,2,6,6-Tetramethylpiperidin-N-oxid
TFA Trifluoressigsäure
THF Tetrahydrofuran
THP Tetrahydropyranyl
TMS Trimethylsilyl
Tr Trityl
Ts Tosyl
X Abgangsgruppe
Z Benzyloxycarbonyl

III. Anhang
111
3. Literaturverzeichnis
[1] Grubbs, R. H. Handbook of Metathesis, Wiley-VCH: Weinheim, 2003.
[2] Herisson, J.; Chauvin, Y. Makromol. Chem. 1970, 141, 161-176.
[3] Ausgewählte Übersichten: (a) Fürstner, A. Alkene Metathesis in Organic Chemistry,
Springer: Berlin, 1998. (b) Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem.
2005, 117, 4564-4601.
[4] www.nobelprize.org/chemistry/laureates/2005
[5] (a) Schrock, R. R.; Murdzek, J. S.; Bazan, G. C.; Robbins, J.; DiMare, M.; O’Regan,
M. J. Am. Chem. Soc. 1990, 112, 3875-3886. (b) Schwab, P.; Grubbs, R. H.; Ziller, J.
W. J. Am. Chem. Soc. 1996, 118, 100-110. (c) Scholl, M.; Ding, S.; Lee, C. W.;
Grubbs, R. H. Org. Lett. 1999, 1, 953-956. (d) Kingsbury, J. S.; Harrity, J. P. A.;
Bonitatebus, P. J.; Hoveyda, A. H. J. Am. Chem. Soc. 1999, 121, 791-799. (e) Garber,
S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122,
8168-8179. (f) Gessler, S.; Randl, S.; Blechert, S. Tetrahedron Lett. 2000, 41, 9973-
9976.
[6] Für eine kurze Übersicht siehe: Hoveyda, A. H.; Gillingham, D. G.; Veldhuizen, J. J.;
Kataoka, O.; Garber, S. B.; Kingsbury, J. S.; Harrity, J. P. A. Org. Biomol. Chem.
2004, 2, 8-23.
[7] Ausgewählte Übersichten: (a) Fürstner, A.; Langemann, K. Synthesis 1997, 7, 792-
803. (b) Grubbs, R. H.; Chang, S. Tetrahedron 1998, 54, 4413-4450. (c) Armstrong, S.
K. J. Chem. Soc., Perkin Trans. 1 1998, 2, 371-388. (d) Maier, M. E. Angew. Chemie
2000, 112, 2153-2157. (d) Fürstner, A. Angew. Chem. 2000, 112, 3140-3172. (e)
Deiters, A.; Martin, S. F. Chem. Rev. 2004, 104, 2199-2238. (f) McReynolds, M. D.;
Dougherty, J. M.; Hanson, P. R. Chem. Rev. 2004, 104, 2239-2258.
[8] Ausgewählte Übersicht: Harned, A. M.; Zhang, M.; Vedantham, P.; Mukherjee, S.;
Herpel, R. H.; Flynn, D. L.; Hanson, P. R. Aldrichimica Acta 2005, 38, 3-16.
[9] Für eine kurze Übersicht siehe: Basra, S.; Blechert, S. Strategies and Tactics in
Organic Synthesis 2004, 4, 315-346.
[10] (a) Philips Petroleum Company, Hydrocarbon Processing 1967, 46, 232. (b) Freitas,
E. R.; Gum, C. R. Chem. Eng. Proc. 1979, 75, 73.
[11] Ausgewählte Beispiele: (a) Chatterjee, A. K.; Morgan, J. P.; Scholl, M.; Grubbs, R. H.
J. Am. Chem. Soc. 2000, 122, 3783-3784. (b) Randl, S.; Gessler, S.; Wakamatsu, H.;
Blechert, S. Synlett 2001, 3, 430-432. (c) Cossy, J.; BouzBouz, S.; Hoveyda, A. H. J.
Organomet. Chem. 2001, 624, 327-332.

III. Anhang
112
[12] Für eine kurze Übersicht siehe: Connon, S. J.; Blechert, S. Angew. Chem. 2003, 115,
1944-1968.
[13] Chatterjee, A. K.; Choi, T.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003,
125, 11360-11370.
[14] (a) Cesati, R. R.; Armas, J.; Hoveyda, A. H. J. Am. Chem. Soc. 2004, 126, 96-101. (b)
Boulard, L.; BouzBouz, S.; Cossy, J.; Franck, X.; Figadère, B. Tetrahedron Lett. 2004,
45, 6603-6605. (c) Michaelis, S.; Blechert, S. Org. Lett. 2005, 7, 5513-5516.
[15] Louie, J.; Bielawski, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 11312-
11313.
[16] Hesse, M. Alkaloids, Wiley-VCH: Weinheim, 2001.
[17] Ausgewählte Übersichten: (a) Laschat, S.; Dickner, T. Synthesis 2000, 13, 1781-1813.
(b) Buffat, M. G. P. Tetrahedron 2004, 60, 1701-1729.
[18] Ausgewählte Beispiele: (a) Nagasaka. T; Kato, H.; Hayashi, H.; Shioda, M.; Hikasa,
H.; Hamaguchi, F. Heterocycles 1990, 30, 561-566. (b) Davis, F. A.; Szewczyk, J. M.
Tetrahedron Lett. 1998, 39, 5951-5954. (c) Yamauchi, T.; Fujikura, H.; Higashiyama,
K.; Takahashi, H.; Ohmiya, S. J. Chem. Soc., Perkin Trans. 1 1999, 19, 2791-2794.
(d) Enders. D.; Kirchhoff, J. H. Synthesis 2000, 14, 2099-2105. (d) Boeglin, D.; Heitz,
A.; Martinez, J.; Fehrentz, J. Eur. J. Org. Chem. 2003, 16, 3139-3146. (e) Lee, Y.;
Shin, Y.; Kim, Y.; Lee, K.; Pyun, S.; Park, H.; Jeong, J.; Ham, W. Tetrahedron:
Asymmetry 2003, 14, 87-93.
[19] Davis, F. A.; Chao, B. Org. Lett. 2000, 2, 2623-2625.
[20] Kim, G.; Jung, S.; Lee, E.; Kim, N. J. Org. Chem. 2003, 68, 5395-5398.
[21] Ausgewählte Beispiele: (a) Matsoukas, J. M.; Agelis, G.; Hondrelis, J.; Yamdagni, R.
Wu, Q.; Ganter, R.; Smith, J. R.; Moore, D. J. J. Med. Chem. 1993, 36, 904-911. (b)
Tsuda, Y.; Cygler, M.; Gibbs, B. F.; Pedyczak, A.; Féthière, J.; Yue, S. Y.; Konishi,
Y. Biochemistry 1994, 33, 14443-14451. (c) Hanson, G. J.; Vuletich, J. L.; Bedell, L.
J.; Bono, C. P.; Howard, S. C.; Welpy, J. K.; Woulfe, S. L.; Zacheis, M. L. Bioorg.
Med. Chem. Lett. 1996, 6, 1931-1936.
[22] Für eine kurze Übersicht siehe: Kadouri-Puchot, C.; Comesse, S. Amino Acids 2005,
29, 101-130.
[23] Couper, L.; McKendrick, J. E.; Robins, D. J.; Chrystal, E. J. T. Bioorg. Med. Chem.
Lett. 1994, 4, 2267-2272.
[24] (a) Mannaioni, G.; Alesiani, M.; Carlà, V.; Natalini, B.; Marinozzi, M.; Pelliciari, R.;
Moroni, F. Eur. J. Pharmacol. 1994, 251, 201-207. (b) Ornstein, P. L.; Schoepp, D.

III. Anhang
113
D.; Arnold, M. B.; Jones, N. D.; Deeter, J. B.; Lodge, D.; Leander, J. D. J. Med.
Chem. 1992, 35, 3111-3115.
[25] Murray, P. J.; Starkey, I. D. Tetrahedron Lett. 1996, 37, 1875-1878.
[26] (a) Carbonnel, S.; Fayet, C.; Gelas, J.; Troin, Y. Tetrahedron Lett. 2000, 41, 8293-
8296. (b) Agami, C.; Comesse, S.; Kadouri-Puchot, C. J. Org. Chem. 2002, 67, 2424-
2428.
[27] Caplan, J. F.; Sutherland, A.; Vederas, J. C. J. Chem. Soc., Perkin Trans. 1 2001, 18,
2217-2220.
[28] Vavrecka, M.; Hesse, M. Helv. Chim. Acta 1991, 74, 438-444.
[29] (a) Ahlbrecht, H.; Marcellinus, I. Synthesis 1988, 3, 210-214. (b) Shoji, W.; Tsutomu,
F.; Kyoichi, S.; Norio, S. Yukagaku 1976, 25, 110-112.
[30] (a) Ghosh, U.; Ganessunker, D.; Sattigeri, V. J.; Carlson, K. E.; Mortensen, D. J.;
Katzenellenbogen, B. S.; Katzenellenbogen, J. A. Bioorg. Med. Chem. 2003, 11, 629-
658. (b) Takamasa, K.; Daisuke, I.; Junko, S. J. Heterocycl. Chem. 1996, 33, 1313-
1318.
[31] Huang, P.; Zheng, X.; Deng, X. Tetrahedron Lett. 2001, 42, 9039-9041.
[32] Stetter, H.; Landscheidt, A. Chem. Ber. 1979, 112, 1410-1419.
[33] Für eine kurze Übersicht siehe: Zimmer, R. Synthesis 1993, 2, 165-178.
[34] Ausgewählte Beispiele: (a) Hermitage, S. A.; Moloney, G. Tetrahedron: Asymmetry
1994, 5, 1463-1464. (b) Ezquerra, J.; Pedregal, C.; Escribano, A.; Carreno, M. C.;
Ruano, J. L. G. Tetrahedron Lett. 1995, 36, 3247-3250.
[35] Vavrecka, M.; Hesse, M. Helv. Chim. Acta 1989, 72, 847-855.
[36] Jefford, C. W.; Sienkiewicz, K.; Thornton, S. R. Helv. Chim. Acta 1995, 78, 1511-
1524.
[37] Für eine kurze Übersicht siehe: Pichon, M.; Figadère, B. Tetrahedron: Asymmetry
1996, 7, 927-964.
[38] Ausgewählte Übersicht: Fache, F.; Schulz, E.; Tommasino, M. L.; Lemaire M. Chem.
Rev. 2000, 100, 2159-2231.
[39] (a) Takahata, H.; Bandoh, H.; Momose, T. J. Org. Chem. 1992, 57, 4401-4404. (b)
Riesinger, S. V.; Löfstedt, J.; Pettersson-Fasth, H.; Bäckvall, J. Eur. J. Org. Chem.
1999, 12, 3277-3280.
[40] (a) Pettersson-Fasth, H.; Riesinger, S. V.; Bäckvall, J. J. Org. Chem. 1995, 60, 6091-
6096. (b) Andrés, J. M.; Herráiz, I.; Pedrosa R.; Pérez-Encabo, A. Synlett 2004, 11,
2016-2018.

III. Anhang
114
[41] (a) Choi, T.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem. 2001, 113, 1317-1319.
(b) Fürstner, A.; Thiel, O. R.; Lehmann, C. W. Organometallics 2002, 21, 331-335.
[42] (a) Cooper, T. S.; Larigo, A. S.; Laurent, P.; Moody, C. J.; Takle, A. K. Synlett 2002,
10, 1730-1732. (b) Krebs, A.; Ludwig, V.; Pfizer, J.; Dürner, G.; Göbel, W. Chem.
Eur. J. 2004, 10, 544-553.
[43] Ausgewählte Beispiele: (a) Ho, T. L.; Gopalan, B.; Nestor, J. J. J. Org. Chem. 1986,
51, 2405-2408. (b) Jegham, S.; Das, B. C. Tetrahedron Lett. 1989, 30, 2801-2804. (c)
Mota, A. J.; Chiaroni, A.; Langlois, N. Eur. J. Org. Chem. 2003, 21, 4187-4198.
[44] Daly, J. W.; Spande, T. F.; Garraffo, H. M. J. Nat. Prod. 2005, 68, 1556-1575.
[45] Daly, J. W.; Brown, G. B.; Mensah-Dwumah, M.; Myers, C. W. Toxicon 1978, 16,
163.
[46] Daly, J. W.; Myers, C. W.; Whittaker, N. Toxicon 1987, 25, 1023.
[47] Aronstam, R. S.; Daly, J. W.; Spande, T. F.; Narayanan, T. K.; Albuquerque, E. X.
Neurochem. Res. 1986, 11, 1227-1240.
[48] Ausgewählte Beispiele: (a) Takahata, H.; Kubota, M.; Ihara, K.; Okamoto, N.;
Momose, T.; Azer, N.; Eldefrawi, A. T.; Eldefrawi, M. E. Tetrahedron: Asymmetry
1998, 9, 3289-3301. (b) Yamazaki, N.; Ito, T.; Kibayashi, C. Org. Lett. 2000, 2, 465-
467. (c) Back, T. G.; Nakajima, K. J. Org. Chem. 2000, 65, 4543-4552. (d) Carbonnel,
S.; Troin, Y. Heterocycles 2002, 57, 1807-1830. (e) Kim, G.; Shim, J. H.; Kim, J. H.
Bull. Korean Chem. Soc. 2003, 24, 1832-1834. (f) Reddy, P. G.; Baskaran, S. J. Org.
Chem. 2004, 69, 3093-3101. (g) Patil, N. T.; Pahadi, N. K.; Yamamoto, Y.
Tetrahedron Lett. 2005, 46, 2101-2103. (h) Takahata, H.; Ichinose, M. Heterocycles
2006, 67, 407-411.
[49] Daly, J. W.; Spande, T. F. Alkaloids: Chemical and Biological Perspectives, Pelletier,
S. W., Ed.; Wiley: New York, 1986, 4, 1-274.
[50] Wilson, S. R.; Guazzaroni, M. E. J. Org. Chem. 1989, 54, 3087-3091.
[51] (a) Tawara, J. N.; Blokhin, A.; Foderaro, T. A.; Stermitz, F. R. J. Org. Chem. 1993,
58, 4813-4818. (b) Stermitz, F. R.; Tawara, J. N.; Boeckl, M.; Pomeroy, M.; Foderaro,
T. A.; Todd, F. G. Phytochemistry 1994, 35, 951-953.
[52] (a) Bates, R. W.; Kanicha, S. Tetrahedron 2002, 58, 5957-5978. (b) Felpin, F.;
Lebreton, J. Tetrahedron 2004, 60, 10127-10153.
[53] Schneider, M. J.; Stermitz, F. R. Phytochemistry 1990, 29, 1811-1814.
[54] Schneider, M. J.; Montali, J. A.; Hazen, D.; Stanton, C. E. J. Nat. Prod. 1991, 54, 905-
909.

III. Anhang
115
[55] (a) Takahata, H.; Yotsui, Y.; Momose, T. Tetrahedron 1998, 54, 13505-13516. (b)
Molander, G. A.; Dowdy, E. D.; Pack, S. K. J. Org. Chem. 2001, 66, 4344-4347. (c)
Fréville, S.; Delbecq, P.; Thuy, V. M.; Petit, H.; Célérier, J. P.; Lhommet, G.
Tetrahedron Lett. 2001, 42, 4609-4611.
[56] Randl, S.; Blechert, S. J. Org. Chem. 2003, 68, 8879-8882.
[57] Osby, J. O.; Martin, M. G.; Ganem, B. Tetrahedron Lett. 1984, 25, 2093-2096.
[58] Cohen, F.; Overman, L. E. J. Am. Chem. Soc. 2001, 123, 10782-10783.
[59] Ausgewählte Übersichten: (a) Cybulski, J.; Wróbel, J. T. in: The Alkaloids; Brossi, A.
Ed.; Academic Press: New York, 1989; Vol. 35, pp. 215-257. (b) Miyazawa, M.;
Yoshio, K.; Ishikawa, Y.; Kameoka, H. J. Agric. Food Chem. 1998, 46, 1059-1063.
(c) Michael, J. P. Nat. Prod. Rep. 1999, 16, 675-696. (d) Matsuda, H.; Shimoda, H.;
Yoshikawa, M. Bioorg. Med. Chem. 2001, 9, 1031-1035. (e) Matsuda, H.; Yoshida,
K.; Miyagawa, K.; Nemoto, Y.; Asao, Y.; Yoshikawa, M. Bioorg. Med. Chem. 2006,
16, 1567-1573.
[60] Maurer, B.; Ohloff, G. Helv. Chim. Acta 1976, 59, 1169-1185.
[61] Arata, Y.; Ohashi, T. Yakugaku Zasshi 1957, 77, 792-793.
[62] (a) Shimizu, I.; Yamazaki, H. Chem. Lett. 1990, 5, 777-778. (b) Leniewsky, A.;
Szychowsky, J. Coll. Czech. Chem. Commun. 1991, 56, 1309-1316. (c) Aoyagi, S.;
Shishido, Y.; Kibayashi, C. Tetrahedron Lett. 1991, 32, 4325-4328. (d) Honda, T.;
Ishikawa, F.; Yamane, S. J. Chem. Soc. Chem. Commun. 1994, 4, 499-500. (e)
Barluenga, J.; Aznar, F.; Ribas, C.; Valdés, C. J. Org. Chem. 1999, 64, 3736-3740.
[63] Schulte-Elte, K. H.; Gadola, M.; Ohloff, G. Helv. Chim. Acta 1971, 54, 1813-1822.
[64] Cabbalero, E.; Avendaño, C.; Menéndez, J. C. J. Org. Chem. 2003, 68, 6944-6951.
[65] Miller, D. J.; Hammond, S. M.; Anderluzzi, D.; Bugg, T. D. H. J. Chem. Soc., Perkin
Trans. 1 1998, 1, 131-142.
[66] Honda, T.; Ishikawa, F.; Yamane, S. Heterocycles 2000, 52, 313-323.
[67] Itatani, Y.; Yasuda, S.; Hanaoka, M.; Arata, Y. Chem. Pharm. Bull. 1976, 24, 2521-
2524.
[68] Arata, Y.; Ohashi, T. Chem. Pharm. Bull. 1965, 13, 1365-1368.
[69] Szychowski, J.; Wróbel, J. T.; Leniewski, A. Can. J. Chem. 1977, 55, 3105-3110.
[70] Ausgewählte Übersichten: (a) Angle, S. R.; Breitenbucher, J. G. Studies in Natural
Products Chemistry: Stereoselective Synthesis; Atta-ur Rahman (Ed.) Elsevier: New
York, 1995; Vol. 16, Part J, S. 453-502. (b) Leclercq, S.; Daloze, D.; Braekman, J.
Org. Prep. Proced. Int. 1996, 28, 501.

III. Anhang
116
[71] Felpin, F.; Lebreton, J. Curr. Org. Synth. 2004, 1, 83-109.
[72] (a) Takahata, H.; Banba, Y.; Ouchi, H.; Nemoto, H.; Kato, A.; Adachi, I. J. Org.
Chem. 2003, 68, 3603-3607. (b) Zhao, Z.; Song, L.; Mariano, P. S. Tetrahedron 2005,
61, 8888-8894.
[73] Witulski, B.; Gößmann, M. Chem. Commun. 1999, 18, 1879-1880.
[74] Xu, Y.; Zhou, B. J. Org. Chem. 1987, 52, 974-977.
[75] Omura, S. Macrolide Antibiotics: Chemistry, Biology and Practice (2nd Edition),
Academic Press: San Diego, 2002, 181-284.
[76] (a) Nicolaou, K. C.; Winssinger, N.; Pastor, J.; Murphy, F. Angew. Chem. Int. Ed.
1998, 37, 2534-2537. (b) Fürstner, A.; Thiel, O. R.; Kindler, N.; Bartkowska, B. J.
Org. Chem. 2000, 65, 7990-7995.
[77] Parenty, A.; Moreau, X.; Campagne, J. Chem. Rev. 2006, 106, 911-939.
[78] Quinkert, G.; Billhardt, U. M.; Jakob, H.; Fischer, G.; Glenneberg, J.; Nagler, P.;
Autze, V.; Heim, N.; Wacker, M.; Schwalbe, T.; Kurth, Y.; Bats, J. W.; Dürner, G.;
Zimmermann, G.; Kessler, H. Helv. Chim. Acta 1987, 70, 771-861.
[79] Hyatt, J. A.; Feldman, P. L.; Clemens, R. J. J. Org. Chem. 1984, 49, 5105-5108.
[80] Ausgewählte Beispiele: (a) Boeckman, R. K.; Weidner, C. H.; Perni, R. B.; Napier, J.
J. J. Am. Chem. Soc. 1989, 111, 8036-8037. (b) Trost, B. M.; Gunzner, J. L.; Dirat, O.;
Rhee, Y. H. J. Am. Chem. Soc. 2002, 124, 10396-10415. (c) Hart, A. C.; Phillips, A. J.
J. Am. Chem. Soc. 2006, 128, 1094-1095.
[81] Funk, T. W.; Efskind, J.; Grubbs, R. H. Org. Lett. 2005, 7, 187-190.
[82] (a) Petasis, N. A.; Patane, M. A. J. Chem. Soc., Chem. Commun. 1990, 11, 836-837.
(b) Eisenberg, S. W. E.; Chen, C.; Wu, J.; Lebrilla, C.; Kurth, M. J. Tetrahedron Lett.
1996, 37, 7683-7686.
[83] Crouch, D. R.; Mehlmann, J. F.; Herb, B. R.; Mitten, J. V.; Dai, G. H. Synthesis 1999,
4, 559-561.
[84] Reetz, M. T.; Ruggeberg, C. J.; Droge, M. J.; Quax, W. J. Tetrahedron 2002, 58,
8465-8473.
[85] Taber, D. F.; Green, J. H.; Geremia, J. M. J. Org. Chem. 1997, 62, 9342-9344.
[86] Yamanaka, T.; Imai, T. Bull. Chem. Soc. Jpn. 1981, 54, 1585-1586.
[87] Hoffman, R. V.; Kim, H.; Wilson, A. L. J. Org. Chem. 1990, 55, 2820-2822.
[88] Ausgewählte Beispiele: (a) Goldring, W. P. D.; Hodder, S. A.; Weiler, L. Tetrahedron
Lett. 1998, 39, 4955-4958. (b) Lee, C. W.; Grubbs, R. H. Org. Lett. 2000, 2, 2145-
2147.

III. Anhang
117
[89] Booth, P. M.; Broughton, H. B.; Ford, M. J.; Fox, C. M. J.; Ley, S. V.; Slawin, A. M.
Z.; Williams, D. J.; Woodward, P. R. Tetrahedron 1989, 45, 7565-7580.
[90] (a) Sekiguchi, J.; Kuroda, H.; Yamada, Y.; Okada, H. Tetrahedron Lett. 1985, 26,
2341-2342. (b) Gurusiddaiah, S.; Ronald, R. C. Antimicrob. Agents Chemother. 1981,
19, 153-165. (c) Hayashi, M.; Kim, Y. P.; Hiraoka, H.; Natori, M.; Takamatsu, S.;
Kawakubo, T.; Masuma, R.; Komiyama, K.; Omura, S. J. Antibiot. 1995, 48, 1435-
1493.
[91] Arai, K.; Rawlings, B. J.; Yoshizawa, Y.; Vederas, J. C. J. Am. Chem. Soc. 1989, 111,
3391-3399.
[92] Michel, K. H.; Demarco, P. V.; Nagarajan, R. J. Antibiot. 1977, 30, 571-575.
[93] Übersicht: Synform 1984, 2, 125-144.
[94] (a) Tatsuta, K.; Amemiya, Y.; Kanemura, Y.; Kinoshita, M. Bull. Chem. Soc. Jpn.
1982, 55, 3248-3253. (b) Ichimoto, I.; Sato, M.; Tsuji, H.; Kirahata, M.; Ueda, H.
Chem. Express 1988, 3, 499-502. (c) Quinkert, G.; Kueber, F.; Knauf, W.; Wacker,
M.; Koch, U.; Becker, H.; Nestler, H. P.; Dürner, G.; Zimmermann, G.; Bats, J. W.;
Egert, E. Helv. Chim. Acta 1991, 74, 1853-1923. (d) Sinha, S. C.; Sinha-Bagchi, A.;
Keinan, E. J. Org. Chem. 1993, 58, 7789-7796. (e) Nagarajan, M. Tetrahedron Lett.
1999, 40, 1207-1210. (f) Kobayashi, Y.; Okui, H. J. Org. Chem. 2000, 65, 612-615.
(g) Lee, W.; Shin, H. J.; Chang, S. Tetrahedron: Asymmetry 2001, 12, 29-31.
[95] Kobayashi,Y. Recent Res. Devel. in Org. Chem. 2000, 4, 169-182.
[96] Ausgewählte Beispiele: (a) Taber, D. F.; Neubert, T. D. J. Org. Chem. 2001, 66, 143-
147. (b) Kimura, Y.; Atarashi, S.; Takahashi, M.; Hayakawa, I. Chem. Pharm. Bull.
1994, 42, 1442-1454.
[97] Paterne, M.; Brown, E. J. Chem. Res. Miniprint 1985, 9, 2924-2947.
[98] Sharpless, B. K.; Walsh, P. J. Synlett 1993, 8, 605-606.
[99] Hunter, T. J.; O’Doherty, G. A. Org. Lett. 2002, 4, 4447-4450.
[100] (a) Hesse, O. J. Prakt. Chem. 1900, 62, 430-480. (b) Hesse, O. J. Prakt. Chem. 1904,
70, 449-502.
[101] (a) Huneck, S.; Schreiber, K.; Steglich, W. Tetrahedron 1973, 29, 3687-3693. (b)
Quinkert, G.; Heim, N.; Bats, J. W.; Oschkinat, H.; Kessler, H. Angew. Chem. 1985,
97, 985-986.
[102] (a) Quinkert, G.; Heim, N.; Glenneberg, J.; Döller, U.; Eichhorn, M.; Billhardt, U. M.;
Schwarz, C.; Zimmermann, G.; Bats, J. W.; Dürner, G. Helv. Chim. Acta 1988, 71,
1719-1794. (b) Solladié, G.; Fernandez, I.; Maestro, C. Tetrahedron: Asymmetry 1991,

III. Anhang
118
2, 801-819. (c) Oppolzer, W.; Radinow, R. N.; Brabander, J. D. Tetrahedron Lett.
1995, 36, 2607-2610. (d) Enders, D.; Prokopenko, O. F. Liebigs Ann. 1995, 7, 1185-
1191. (e) Sinha, S. C.; Keinan, E. J. Org. Chem. 1997, 62, 377-386. (f) Nishioka, T.;
Iwabuchi, Y.; Irie, H.; Hatakeyama, S. Tetrahedron Lett. 1998, 39, 5597-5600. (g)
Kobayashi, Y.; Nakano, M.; Kumar, G. B.; Kishihara, K. J. Org. Chem. 1998, 63,
7505-7515. (h) Dixon, D. J.; Foster, A. C.; Ley, S. V. Org. Lett. 2000, 2, 123-125. (i)
Maezaki, N.; Li, Y.; Ohkubo, K.; Goda, S.; Iwata, C.; Tanaka, T. Tetrahedron 2000,
56, 4405-4413. (j) Banwell, M. G.; McRae, K. J. Org. Lett. 2000, 2, 3583-3586.
[103] (a) Brogan, J. B.; Zercher, C. K. J. Org. Chem. 1997, 62, 6444-6446. (b) Ronsheim,
M. D.; Zercher, C. K. J. Org. Chem. 2003, 68, 1878-1885.
[104] Furukawa, J.; Kawabata, N.; Nishimura, J. Tetrahedron Lett. 1966, 7, 3353-3354.
[105] Naruse, N.; Tenmyo, O.; Kawano, K.; Tomita, K.; Ohkusa, N.; Miyaki, T.; Konishi,
M.; Oki, T. J. Antibiot. 1991, 44, 733-740.
[106] Ausgewählte Beispiele: (a) Trost, B. M.; Ceschi, M. A.; König, B. Angew. Chem. Int.
Ed. Engl. 1997, 36, 1486-1489. (b) Suh, Y.; Kim, S.; Jung, J.; Shin, D.; Min, K.; Koo,
B.; Kim, H. Angew. Chem. 1999, 111, 3753-3755. (c) Baltrusch, A. W.; Bracher, F.
Synlett 2002, 10, 1724-1726.
[107] Tomita, K.; Oda, N.; Hoshino, Y.; Ohkusa, N.; Chikazawa, H. J. Antibiot. 1991, 44,
940-948.
[108] Neeland, E. G.; Ounsworth, J. P.; Russel, J. S.; Weiler, L. J. Org. Chem. 1994, 59,
7383-7394.

III. Anhang
119
4. Lebenslauf
Persönliche Daten
Name: Julian Gebauer
Geboren am: 15.09.1976 in Berlin
Nationalität: deutsch
Familienstand: ledig
Schulausbildung
1984–1989 Wald-Grundschule Berlin
1990–1996 Herder-Oberschule Berlin
29.05.1996 Abitur
Studium und Promotion
10/1996–04/1999 Grundstudium der Chemie an der TU Berlin
26.04.1999 Vordiplom
04/1999–07/2001 Hauptstudium der Chemie an der TU Berlin
09/2001–04/2002 Diplomarbeit unter der Leitung von Prof. Dr. S. Blechert
02.05.2002 Diplom
06/2002–05/2006 Promotion in der Arbeitsgruppe von Prof. Dr. S. Blechert
Berufliche Tätigkeiten
06/2002–06/2005 Wissenschaftlicher Mitarbeiter am Institut für Chemie der TU Berlin

III. Anhang
120
5. Publikationsliste
Julian Gebauer, Purnama Dewi und Siegfried Blechert „Stereoselective synthesis of
substituted N-heterocycles via sequential cross metathesis – reductive cyclization”,
Tetrahedron Lett. 2005, 46, 43-46.
Julian Gebauer und Siegfried Blechert „A Short and Enantiospecific Synthesis of
(–)-Nupharamine”, Synlett 2005, 18, 2826-2828.
Purnama Dewi-Wülfing, Julian Gebauer und Siegfried Blechert „Concise Enantiospecific
Synthesis of (+)-Calvine”, Synlett 2006, 3, 487-489.
Julian Gebauer und Siegfried Blechert „Synthesis of γ,δ-unsaturated-β-keto lactones via
sequential cross metathesis – lactonization: A facile entry to the Macrolide Antibiotic
(–)-A26771B”, J. Org. Chem. 2006, 71, 2021-2025.