sx mielodisplasico (2)
DESCRIPTION
Sx Mielodisplasico (2)medicinahematologiasindrome mielodisplasicoDiagnosticoTTOmedicinaTRANSCRIPT

SÍNDROME MIELODISPLASICO
Integrantes:Darlay MachacónGénesis Martínez
Jaulanerth MedinaPaola Mejías
Lisandra ParraPaola TabordaNertis Trujillo
Dr. Asdrúbal SarasMaracaibo, Marzo 2015
Citopenias Riesgo de Transformación en Leucemia Aguda
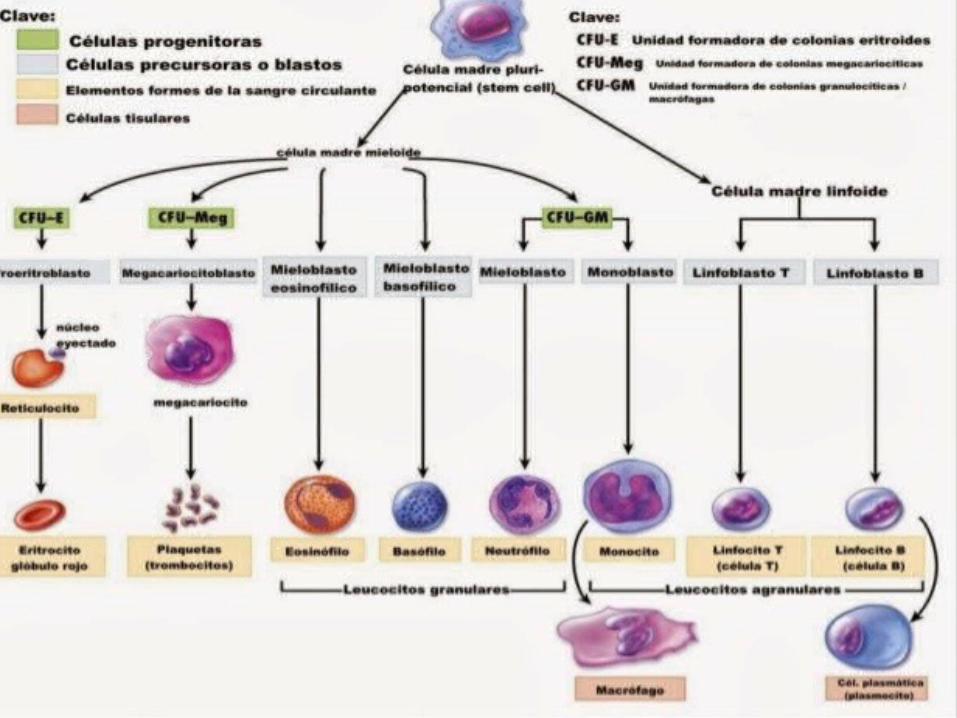
Síndrome Mielodisplásico
Dishemopoyesis
DiseritropoyesisDisgranulopoyesisDismegacariocitopoy
esis
Epidemiología- Edad Avanzada >50 años.- Ligero
predominio en Hombres.
- No se ha encontrado
relación con la raza.
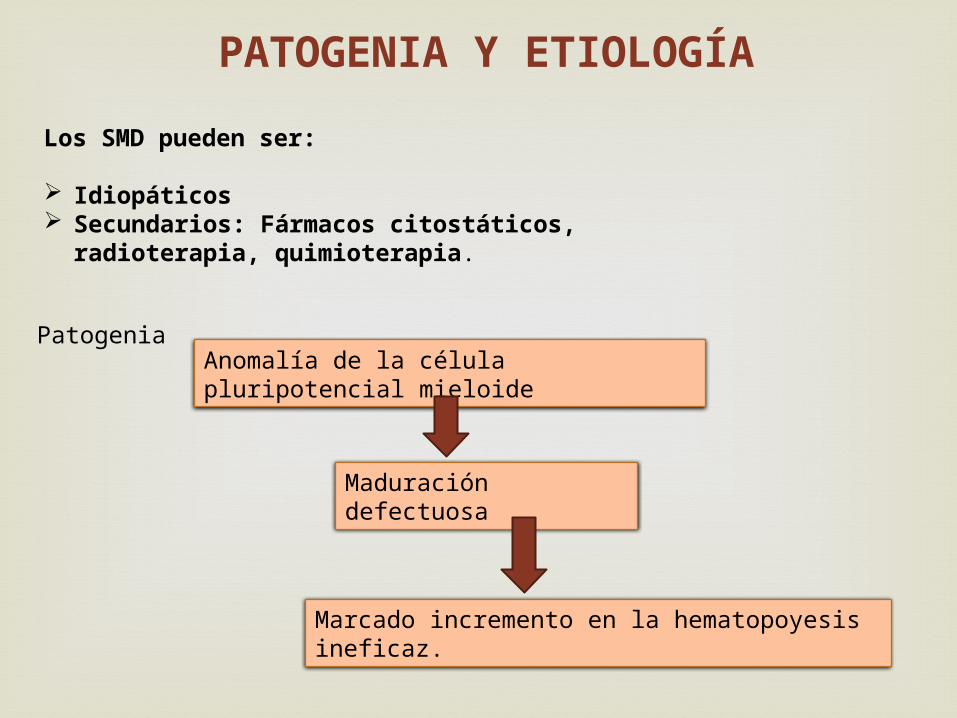
Los SMD pueden ser: Idiopáticos Secundarios: Fármacos citostáticos, radioterapia,
quimioterapia.
PATOGENIA Y ETIOLOGÍA
Anomalía de la célula pluripotencial mieloide
Maduración defectuosa
Marcado incremento en la hematopoyesis ineficaz.
Patogenia
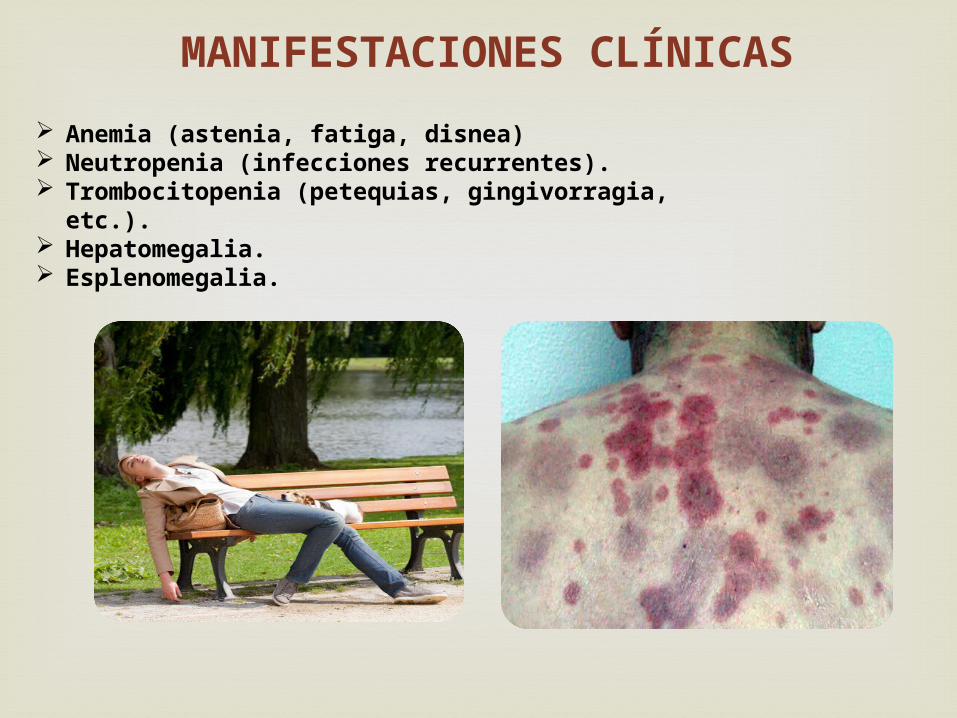
Anemia (astenia, fatiga, disnea) Neutropenia (infecciones recurrentes). Trombocitopenia (petequias, gingivorragia, etc.). Hepatomegalia. Esplenomegalia.
MANIFESTACIONES CLÍNICAS

DIAGNÓSTICOS Y ESTUDIOS DE LABORATORIO
Alteraciones de la serie roja(diseritropoyesis)
Sangre Periférica: *Macrocitosis(redondos)
*Anisocitosis *Poikiolocitosis
*Punteada basofilo *Presencia de células nucleadas
Médula ósea:*Cambios Megaloblastoides
*Gigantismo*Asincronia de la maduracion nucleo-
citoplasma*Mutinucleosidad
*Fragmentación nuclear*Puentes internucleares

ALTERACIONES DE LA SERIE GRANULOCITICA (DISGRANULOPOYESIS)
Sangre Periférica:
*Neutrofilos hipogranulares o agranulares
*Hiposegmentacion del nucleo
*Hipersegmentacion nuclear
*Núcleos en anillo
Médula ósea.
*Hipogranulacion
*Tinción anormal de los gránulos
*Neutrofilos de aspecto monocitoide

ALTERACIONES DE LA SERIE MEGACARIOCITICA(DISMEGACARIOPOYESI
S)
Sangre Periférica:
*Plaquetas gigantes
*Hipogranulacion
*Hipergranulacion central
Medula osea:
*Micromegacariocitos(megacariocitos enanos)
*Megacariocitos con nucleo uni o bilobulado
*Megacariocitos con múltiples núcleos pequeños

BIOPSIA DE MEDULA ÓSEA Brinda información útil respecto
En el 50 % de los 9 pacientes la fibrosis es de tipo reticulínica ligera o moderada
Es de suma importancia advertir la disposición de estas células blasticas inmaduras, que tienden a agruparse en cinco o más elementos constituyendo los ALIPS (anormal localization immature precursors), que son agrupaciones de mieloblastos y/o promielocitos ubicados en un área central de la médula ósea. La presencia de tres o más de estos focos se considera ALIP-positiva, siempre que cumplan con el requisito de ser mieloperoxidasa positivo, ya que existen agrupaciones de elementos eritroides que no tienen significado patológico.(seudo ALIPS).
Además, en el estudio histológico se deben evaluar los fibroblastos, adipocitos, células endoteliales y estructuras vasculares para mejor conocimiento del proceso.
Celularidad global, a la disposición alterada de las distintas series en comparación con el patrón normal : Granulopoyesis central en vez de estar paratrabecular o perivascular
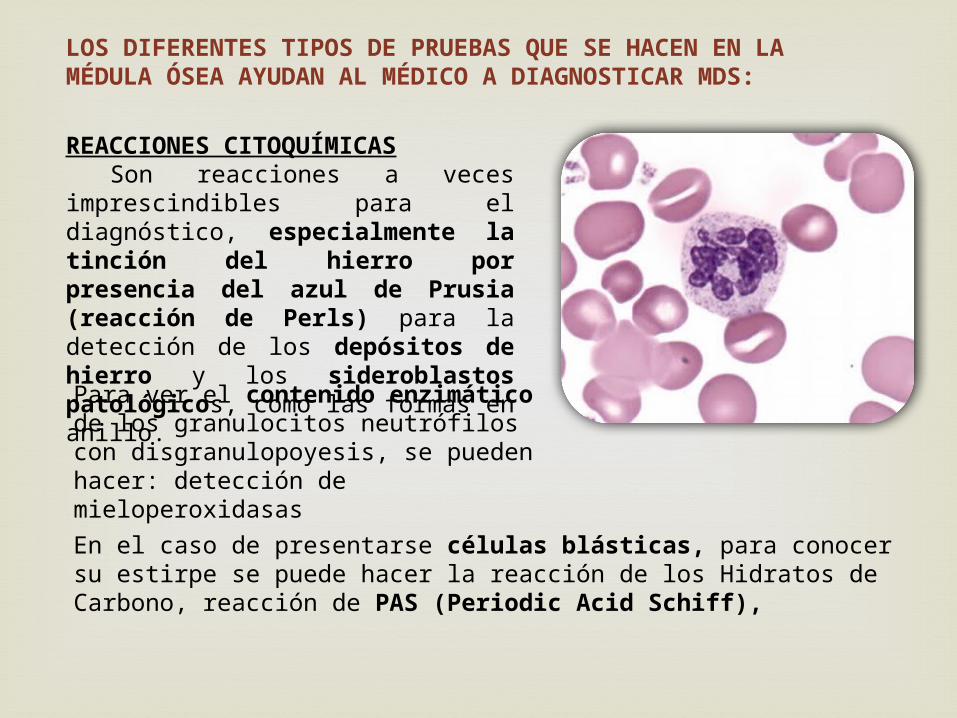
REACCIONES CITOQUÍMICAS Son reacciones a veces imprescindibles para el diagnóstico, especialmente la tinción del hierro por presencia del azul de Prusia (reacción de Perls) para la detección de los depósitos de hierro y los sideroblastos patológicos, como las formas en anillo.Para ver el contenido enzimático de los granulocitos neutrófilos con disgranulopoyesis, se pueden hacer: detección de mieloperoxidasas
En el caso de presentarse células blásticas, para conocer su estirpe se puede hacer la reacción de los Hidratos de Carbono, reacción de PAS (Periodic Acid Schiff),
LOS DIFERENTES TIPOS DE PRUEBAS QUE SE HACEN EN LA MÉDULA ÓSEA AYUDAN AL MÉDICO A DIAGNOSTICAR MDS:

Las alteraciones del cariotipo, consideradas marcadores clonales de malignidad, están presentes en el 30-50 % de los SMD primarios y en el 80 % de los secundarios, sin que ninguna de éstas sea específica de esta entidad
Se ha observado una mayor incidencia de la pérdida total o parcial de un cromosoma, seguida de la existencia de trisomías. Las traslocaciones son menos frecuentes y afectan fundamentalmente los cromosomas 1,3,5,7 y 17 ocurren en el 15 % de los SMD primarios y en el 50 % de los secundarios; es más frecuente la afectación de los cromosomas 5 y 7.
CITOGENÉTICA

Son frecuentes deleciones parciales o totales de los cromosomas 5, 7, 20 y anomalías del 3. La deleción 5q- también se puede hallar en el 10-15% de lo SMD secundarios. Como única anomalía, da un pronóstico favorable. Este síndrome se acompaña generalmente con anemia que requiere soporte transfusional. Los puntos de rotura varían en cada caso pero se coincide que la región crítica de la deleción está situada entre 5q31 y 5q33. La adquisición de anomalías cariotípicas nuevas son de mal pronóstico.
En los SMD secundarios generalmente se reúnen varias alteraciones sobre todo en los cromosomas 5, 7, 8 y 12 y la evolución leucémica cursa con cariotipos complejos que incluyen monosomía o deleción de los cromosomas 5 y 7. La monosomía 7 (-7) ocurre en el 15% de los casos y la trisomía del 8 (+8) en el 19%.
CITOGENÉTICA

El significado pronóstico del cariotipo en los SMD está bien establecido considerándose tres categorías de riesgo:
favorable: cariotipo normal, del 5q- y del 20q; desfavorable: -7, del 7q y cariotipos complejos.
Las otras anomalías son consideradas de riesgo intermedio en cuanto a supervivencia y evolución a leucemia aguda.
CITOGENÉTICA

Detectar antigeno CD61 que es un marcador de la serie megacariocitica. Y también CD34
BIOLOGÍA MOLECULAR Su valor es leucemógeno ya que a veces las alteraciones ocurren en áreas donde existen oncogenes y genessupresores tumorales Estos pacientes muestran mutaciones génicas detectables en alrededor del 60% de los casos. El más frecuente mutado es el RAS, después le siguen el FMS y el p53.
INMUNOHISTOQUIMICA:

CITOMETRIA DE FLUJOLa búsqueda de aberraciones ayuda a concretar el diagnostico y el pronostico en aquellos casos donde el citogenetico es normal.
La cantidad de metastasis es insuficiente, el numero de blastos bajos o las caracteristicas morfológicas son confusas.
Expresión anormal de la maduración de la granulocitica: CD10, CD33, CD56
El análisis inmunofenotípico de la ferritina mitocondrial es de gran ayuda en la anemia sideroblástica, y la ferritina citosólica indica sobrecarga de hierro.

EPIGENÉTICA
el Fluorescence In Situ Hybridization (FISH), que permiten localizar un gen o grupos de genes (llamados target) dentro del ADN celular. Combina la citogenética clásica con la biología molecular. Estos estudios se están realizando en los SMD tanto para diagnóstico como para seguimiento y evaluación de la respuesta a la terapéutica
Clasificación FAB
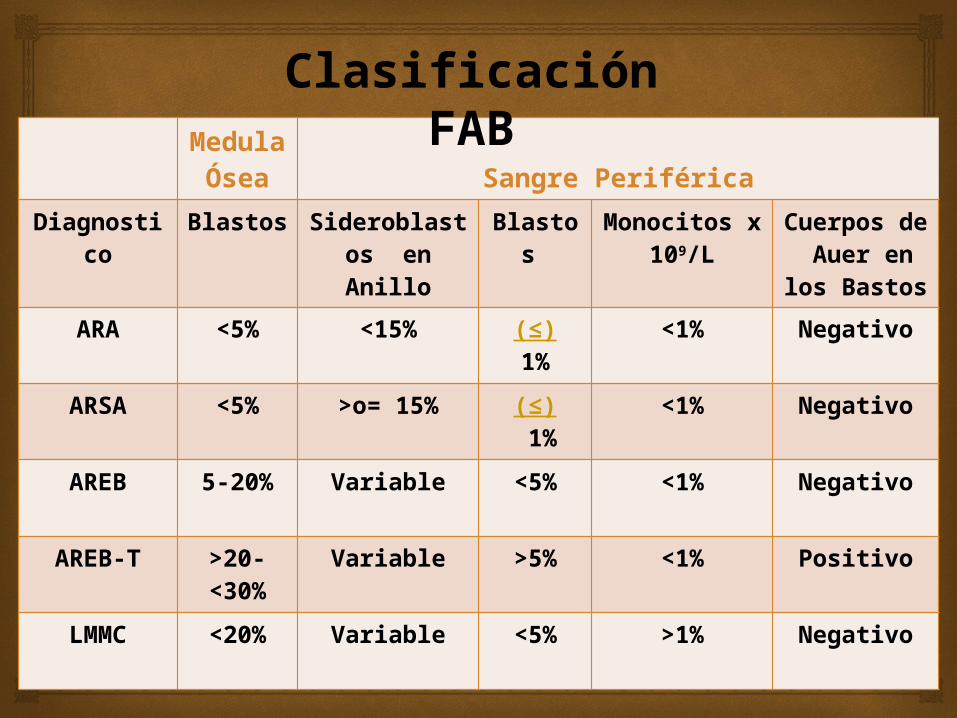
Medula
ÓseaSangre Periférica
Diagnostico
Blastos Sideroblastos en Anillo
Blastos
Monocitos x 109/L
Cuerpos de Auer en los Bastos
ARA <5% <15% (≤)1%
<1% Negativo
ARSA <5% >o= 15% (≤) 1%
<1% Negativo
AREB 5-20% Variable <5% <1% Negativo
AREB-T >20-<30%
Variable >5% <1% Positivo
LMMC <20% Variable <5% >1% Negativo
Clasificación FAB

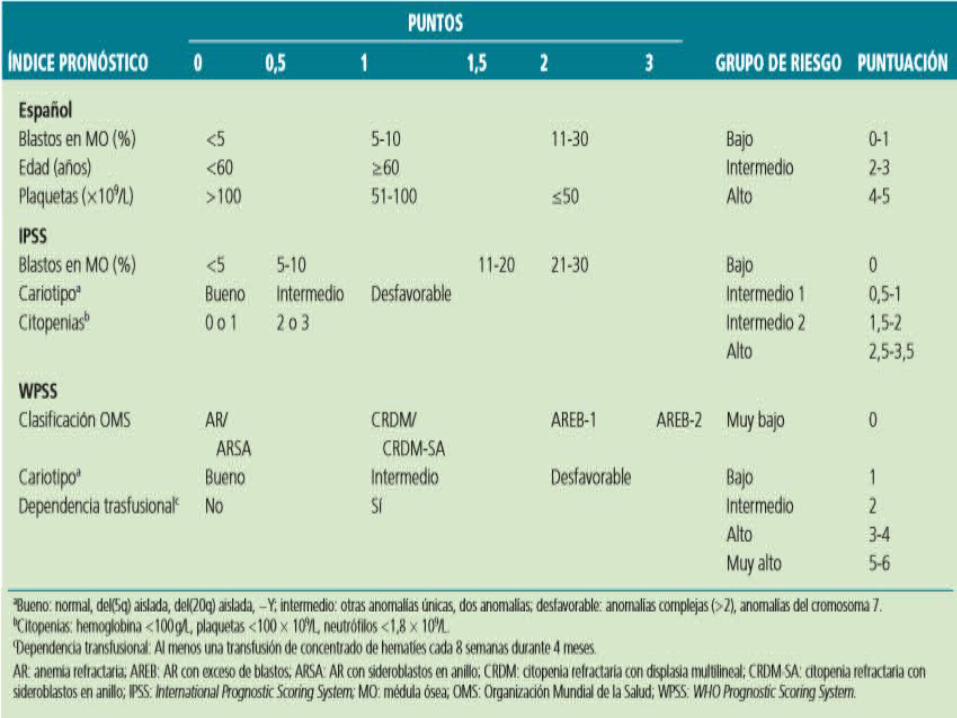
Mediana de supervivencia es de 18-24 meses. Riesgo de evolucion a leucemia aguda
mielodisplasica 30 % - 40% en 5 años. Principales causas de muertes son las
infecciones y las hemorragias, secundaria al fallo medular o la evolucion a leucemia aguda.
PRONÓSTICO

La proporción medular de blastos.
La citogenéticas y el numero.
Grado de citopenias.
PRONÓSTICO

Edad Avanzada.
Comorbilidad.
Displasia multilineal.
Dependencia transfusional.
LDH aumentada.
Mielofibrosis.
PRONÓSTICO

Un riesgo bajo o intermedio 1 según el IPSS. Una concentracion de Hb > 10gr/dl y un
conteo de plaquetas >50.000 microlitros o 100.000 por microlitros sin necesidad de transfusión.
Estos pacientes puedes permanecer sin tratamiento y ser evaluado por un hematólogo oncólogo.
OBSERVACIÓN DE LOS CONTEOS SANGUÍNEOS.
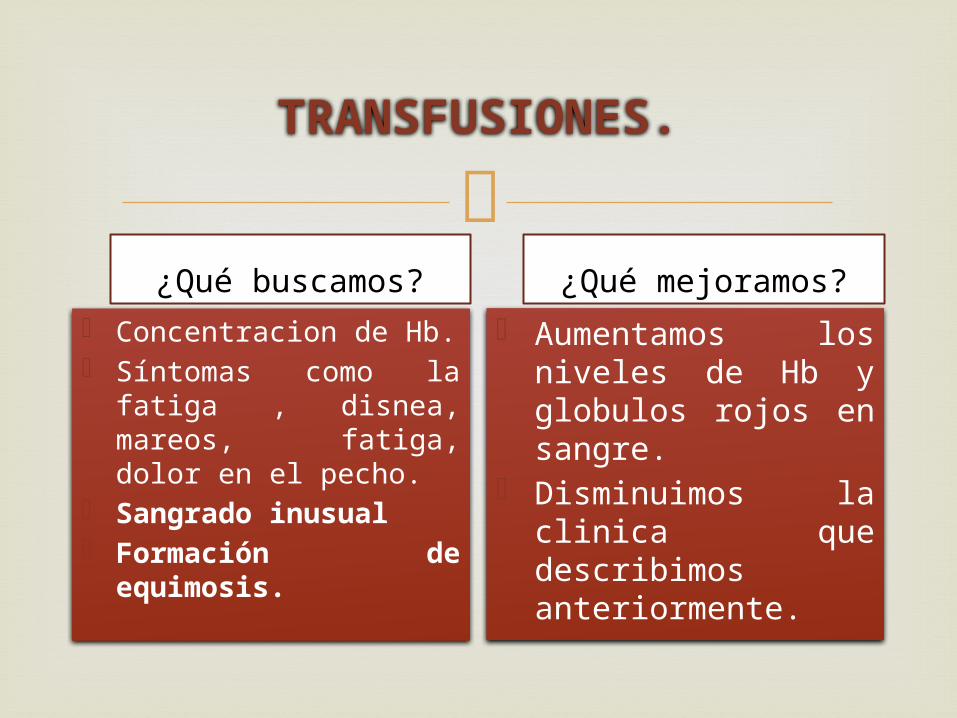
TRANSFUSIONES.
¿Qué buscamos?
Concentracion de Hb. Síntomas como la
fatiga , disnea, mareos, fatiga, dolor en el pecho.
Sangrado inusual Formación de
equimosis.
¿Qué mejoramos?
Aumentamos los niveles de Hb y globulos rojos en sangre.
Disminuimos la clinica que describimos anteriormente.

Deferasirox (Exjade) OD Deferiprona (Ferriprox) OD Mesilato de Deferoxamina (DFO; Desferal) se
administra como infusion lenta en forma SC, IM o EV.
TERAPIA DE QUELACION DE HIERRO.

FÁRMACOS ESTIMULANTES DE LA ERITROPOYESIS.
La epoyetina alfa (Procrit) SC
La darbepoyetina alfa (Araneps) SC (acción mas prolongada)
Tratamiento de pacientes con deficit de eritropoyetina.
Pacientes de bajo riesgo o intermedio I según el IPPS (evitamos las constantes transfusiones)

El factor estimulante de colonias de
granulocitos (G-CSF) combinado con un fármaco estimulador de la eritropoyesis pueden aumentar los niveles de Hb.
Un factor estimulante de colonias de granulocitos o un factor estimulante de colonias de granulocitos y macrofagos (GM-CSF) sirven en el tratamiento de pacientes que padecen infecciones por bajos conteos de neutrofilos.
FACTOR DE CRECIMIENTO DE GLOBULOS BLANCOS.

El Romiplostim (Nplate). El trombopag (Promacta).Indicado en pacientes con trombocitopenia, purpura trombocitopenica inmune crónica, que han tenido una respuesta insuficiente a corticoesteroides, inmunoglobulinas o esplenectomía.
FACTOR DE CRECIMIENTO DE PLAQUETAS.

FÁRMACOS DISPONIBLES PARA EL TRATAMIENTO SÍNDROME
MIELODISPLASICO
Los enfoques de tratamiento con farmacos individuales que han sido aprobados por la FDA para los pacientes con sindromes mielodisplasicos incluyen azacitidina (VidazaR), decitabina (DacogenR), lenalidomida (RevlimidR) y mesilato de imatinib (GleevecR).

Es un fármaco “hipometilante” o “demetilante”. Esta aprobado para el tratamiento de pacientes de
bajo y alto riesgo. Mejora el funcionamiento de la medula ósea.
Se administra EV o SC El tratamiento dura siete días consecutivos y se repite
cada cuatro semanas por lo menos cuatro ciclos. Mejor calidad de vida, menor necesidad de
transfusiones. EA: Nauseas, vómitos, diarrea
Valores bajos en los conteos de células sanguíneas
AZACITIDINA (VIDAZA)

Es un fármaco hipometilante/demetilante. Esta aprobado para el tratamiento de pacientes de bajo y alto
riesgo. Se administra por vía intravenosa de dos maneras diferentes: Durante un periodo de 3 horas cada 8 horas durante 3 días
(el ciclo debe repetirse cada 6 semanas) Durante un periodo de 1 hora una vez al día durante 5 días
(el ciclo debe repetirse cada 4 semanas)El tratamiento produce:
Menor necesidad de transfusiones. Mejores conteos de células sanguíneas en 30 al 40 por ciento de
los pacientes.
DECITABINA (DACOGEN®)

Es un fármaco inmunomodulador (IMiD, por sus siglas en ingles).Forma parte de la misma clase de fármacos que la talidomida.
Es la terapia de preferencia para pacientes con anemia dependiente detransfusiones debido a un caso de síndrome mielodisplasico de riesgo bajo
o intermedio asociado a la deleción del brazo largo del cromosoma 5.
LENALIDOMIDA (REVLIMID®)
No es neurotóxica. No es sedante.
Reduce la necesidad de transfusiones de glóbulos rojos en pacientes de bajo
riesgo.

El propósito de la quimioterapia es eliminar las células madre anormales y
permitir el crecimiento de nuevas células normales.
QUIMIOTERAPIA
Es posible que los pacientes con síndromes mielodisplasicos en las categorias de riesgo intermedio 2 y alto requieran tratamiento con el mismo tipo de quimioterapia que se usa para tratar la leucemia mieloide aguda.
Citarabina Darrubicina
Daunorrubicina Mitoxantrona

Debido a que el MDS puede convertirse en leucemia mieloide aguda (AML), es posible que los pacientes con MDS reciban el mismo tratamiento que los pacientes con AML.
El medicamento de quimioterapia citarabina (ara-C) es el que se administra con más frecuencia para MDS. Se puede administrar por sí solo en una baja dosis, la cual puede ayudar a controlar la enfermedad, aunque a menudo no provoca que entre en remisión.
Este tratamiento también se usa en pacientes de edad más avanzada que padecen AML.
Otra opción consiste en administrar la misma quimioterapia que se emplea en pacientes más jóvenes que padecen AML. Esto significa administrar citarabina a una mayor dosis junto con otros medicamentos de quimioterapia. Esto se emplea con más frecuencia en MDS avanzados.
Para el tratamiento de MDS, los medicamentos de quimioterapia que más frecuentemente se combinan con citarabina son: Idarubicina, Topotecán, Fludarabina.
QUIMIOTERAPIA CONVENCIONAL

Entre los efectos secundarios comunes a quimioterapia se encuentran:
· Pérdida del cabello
· Úlceras en la boca
· Pérdida de apetito
· Náuseas y vómitos
· Bajos recuentos sanguíneos.

El único tratamiento que puede curar el síndrome mielodisplásico (MDS) es el trasplante de células madre. En este tratamiento, el paciente recibe altas dosis de quimioterapia y/o irradiación corporal total para destruir las células en la médula ósea (incluyendo las células anormales de la médula ósea). Luego el paciente recibe células madre nuevas y sanas productoras de sangre.
TRASPLANTE DE CÉLULAS MADRE PARA EL SÍNDROME MIELODISPLÁSICO
Alotransplante
Autotransplante
En el autotrasplante de células madre, después de destruir la médula ósea, el paciente recibe nuevamente sus propias células madre. Este tipo de trasplante no es un tratamiento convencional para pacientes con MDS, ya que sus médulas óseas contienen células madre anormales.
*Alotransplante de células madre no mieloablativo
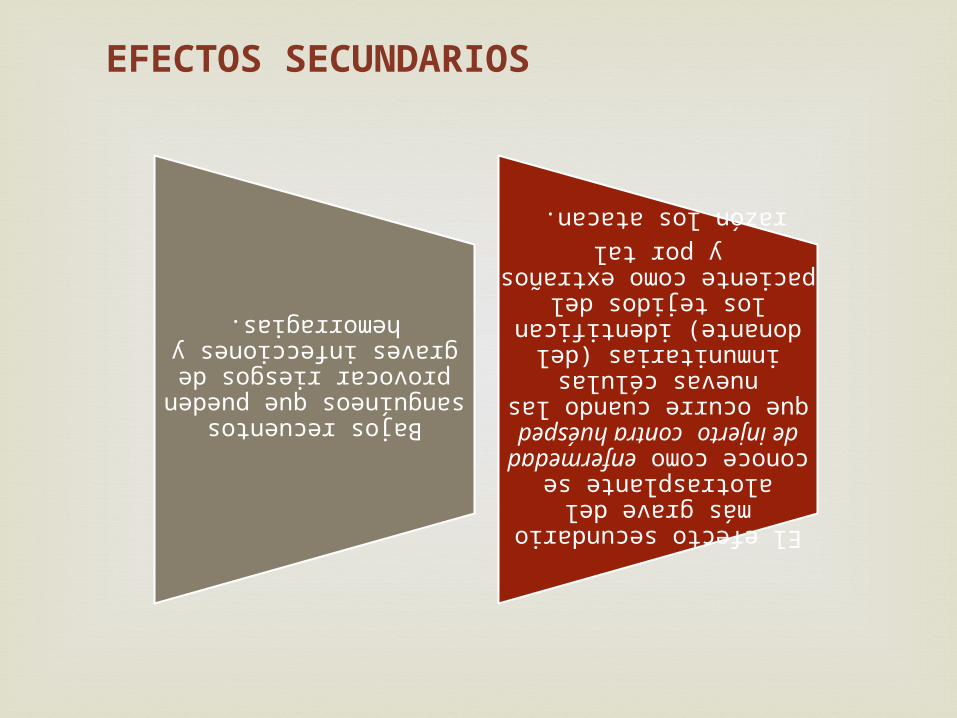
EFECTOS SECUNDARIOS
Bajos recuentos sanguíneos que pueden provocar riesgos de graves
infecciones y hemorragias.
El efecto secundario más grave del alotrasplante se conoce como
enfermedad de injerto contra huésped que ocurre cuando las nuevas células inmunitarias (del donante) identifican
los tejidos del paciente como extraños y por tal
razón los atacan.

GRACIAS POR SU ATENCIÓN