swog cancer research network title s1933 agents
TRANSCRIPT

S1933 Page 1
Version Date 6/1/2021
PRIVILEGED COMMUNICATION FOR INVESTIGATIONAL USE ONLY
SWOG CANCER RESEARCH NETWORK TITLE
S1933, A Pilot Study of Hypofractionated Radiotherapy Followed by Atezolizumab Consolidation in Stage II or III NSCLC Patients with Borderline Performance Status
This trial is part of the National Clinical Trials Network (NCTN) program, which is sponsored by the National Cancer Institute (NCI). The trial will be led by SWOG with the participation of the network of NCTN organizations: Alliance for Clinical Trials in Oncology; ECOG-ACRIN Cancer
Research Group; and NRG Oncology.
NCT# 04310020
This study is being conducted under DCTD IND #138328
STUDY CHAIRS: Raid Aljumaily, M.D. (Primary Chair, Medical Oncology) University of Oklahoma Health Sciences Center Stephenson Cancer Center 800 NE 10th Street, SCC 6019 Oklahoma City, OK 73104 Phone: 405/271-4022 FAX: 405/271-4221 E-mail: [email protected] Timur Mitin, M.D., Ph.D. (Co-Chair, Radiation Oncology) Knight Cancer Institute Department of Radiation Medicine Oregon Health and Science University 3181 SW Sam Jackson Park Road, KPV4 Portland, OR 97239 Phone: 503/681-4200 FAX: 503/681-4210 E-mail: [email protected] Antoinette Wozniak, M.D. (Co-Chair, Medical Oncology) Hillman Cancer Center University of Pittsburgh Medical Center Cancer Pavilion Room 568 5150 Centre Avenue Pittsburgh, PA 15232 Phone: 412/623-2696 E-mail: [email protected]
AGENTS: NCI Supplied Investigational Agent: Atezolizumab (NSC-783608, IND #138328) BIOSTATISTICIANS: Mary Redman, Ph.D. (Biostatistics) Jieling Miao, M.S. (Biostatistics) SWOG Statistics and Data Management Center Fred Hutchinson Cancer Research Center 1100 Fairview Avenue North, M3-C102 P.O. Box 19024 Seattle, WA 98109-1024 Phone: 206/667-4623 FAX: 206/667-4408 E-mail: [email protected] E-mail: [email protected] STUDY CHAIRS continued: Roy Decker, M.D., Ph.D. (Co-Chair, Radiation Oncology) Department of Therapeutic Radiology Hunter Building 15 York Street, Ste HRT 134 New Haven, CT 06510 Phone: 203/737-2758 E-mail: [email protected] COMMUNITY MEDICAL ONCOLOGIST: Suzanne Cole, M.D. UT Southwestern Medical Center Simmons Cancer Center 5323 Harry Hines Blvd NB02.102 Dallas TX 75390-8852 Phone: 214/648-4180 FAX: 972/669-7190 E-mail: [email protected]

S1933 Page 2
Version Date 6/1/2021
PARTICIPANTS
ALLIANCE/Alliance for Clinical Trials in Oncology
ECOG-ACRIN/ECOG-ACRIN Cancer Research Group NRG/NRG Oncology
SWOG/SWOG Cancer Research Network

S1933 Page 3
Version Date 6/1/2021
TABLE OF CONTENTS
TITLE ......................................................................................................................................................... 1 PARTICIPANTS ............................................................................................................................................ 2 TABLE OF CONTENTS ............................................................................................................................... 3 CANCER TRIALS SUPPORT UNIT (CTSU) ADDRESS AND CONTACT INFORMATION ....................... 6 SCHEMA ....................................................................................................................................................... 7 1.0 OBJECTIVES .................................................................................................................................. 8 1.1 Primary Objective ............................................................................................................................. 8 1.2 Secondary Objective(s) .................................................................................................................... 8 1.3 Additional Objective ......................................................................................................................... 8 2.0 BACKGROUND ............................................................................................................................... 8 2.1 Lack of evidence-based data to guide treatment recommendations for patients with Stage III
NSCLC with performance status 2 and patients with Stage II who are not surgical candidates ..... 8 2.2 The role for immune checkpoint inhibitors in lung cancer ................................................................ 9 2.3 Immune checkpoint inhibitors in patients with NSCLC and performance status (PS) 2 ................ 10 2.4 Radiation therapy regimens in locally advanced NSCLC and PS 2 patients ................................. 11 2.5 Radiation therapy and immune checkpoint inhibitors .................................................................... 12 2.6 Inclusion of Women and Minorities and Planned Enrollment Report ............................................ 14 3.0 DRUG INFORMATION .................................................................................................................. 14 3.1 Atezolizumab (NSC-783608, IND # 138328) ................................................................................. 15 4.0 STAGING CRITERIA ..................................................................................................................... 23 5.0 ELIGIBILITY CRITERIA ................................................................................................................ 24 5.1 Registration Step 1 ......................................................................................................................... 25 5.2 Registration Step 2 ......................................................................................................................... 27 6.0 STRATIFICATION FACTORS ....................................................................................................... 28 7.0 TREATMENT PLAN ...................................................................................................................... 28 7.1 Treatment Overview ....................................................................................................................... 28 7.2 Radiation Therapy (Registration Step 1) ........................................................................................ 28 7.3 Atezolizumab (Registration Step 2) ............................................................................................... 33 7.4 Prohibited and cautionary medications .......................................................................................... 35 7.5 Disease Assessment ...................................................................................................................... 35 7.6 Complete DMU Reporting Requirement ........................................................................................ 36 7.7 Criteria for Removal from Protocol Treatment ............................................................................... 36 7.8 Discontinuation of Treatment ......................................................................................................... 36 7.9 Follow-Up Period ............................................................................................................................ 36 8.0 TOXICITIES TO BE MONITORED AND DOSE MODIFICATIONS .............................................. 37 8.1 NCI Common Terminology Criteria for Adverse Events ................................................................ 37 8.2 Radiation-Related Toxicities .......................................................................................................... 37 8.3 Dose Interruptions for Atezolizumab .............................................................................................. 39 8.4 Dose modification and treatment decision contacts ...................................................................... 57 8.5 Adverse Event Reporting Requirements ........................................................................................ 57 8.6 Serious Adverse Event Reporting Requirements .......................................................................... 58 9.0 STUDY CALENDAR ...................................................................................................................... 65 10.0 CRITERIA FOR EVALUATION AND ENDPOINT ANALYSIS ..................................................... 67 10.1 Measurability of lesions .................................................................................................................. 67 10.2 Objective status at each disease evaluation .................................................................................. 68 10.3 Best Response ............................................................................................................................... 70 10.4 Performance Status ....................................................................................................................... 71 10.5 Progression-free survival ............................................................................................................... 71 10.6 Overall Survival .............................................................................................................................. 71 11.0 STATISTICAL CONSIDERATIONS .............................................................................................. 71 11.1 Sample size and power justifications ............................................................................................. 71 11.2 Analysis Populations ...................................................................................................................... 72 11.3 Analysis Plans ................................................................................................................................ 73 11.4 Accrual ........................................................................................................................................... 73 11.5 Data and safety monitoring ............................................................................................................ 73

S1933 Page 4
Version Date 6/1/2021
12.0 DISCIPLINE REVIEW .................................................................................................................... 74 12.1 Radiation Therapy Review ............................................................................................................. 74 13.0 REGISTRATION GUIDELINES ..................................................................................................... 76 13.1 Registration Timing ........................................................................................................................ 76 13.2 CTEP Registration Procedures ...................................................................................................... 76 13.3 CTSU Registration Procedures ...................................................................................................... 77 13.4 Oncology Patient Enrollment Network (OPEN) Registration Requirements .................................. 79 13.5 Exceptions to SWOG registration policies will not be permitted. ................................................... 81 14.0 DATA SUBMISSION SCHEDULE ................................................................................................ 81 14.1 Data Submission Requirement ...................................................................................................... 81 14.2 Master Forms ................................................................................................................................. 82 14.3 Data Submission Procedures ........................................................................................................ 82 14.4 Data Submission Overview and Timepoints .................................................................................. 83 15.0 SPECIAL INSTRUCTIONS............................................................................................................ 86 15.1 Specimen Submission for Banking (REQUIRED IF PARTICIPANT CONSENTS) ....................... 86 16.0 ETHICAL AND REGULATORY CONSIDERATIONS ................................................................... 89 17.0 BIBLIOGRAPHY ............................................................................................................................ 92 18.0 APPENDIX ..................................................................................................................................... 95 18.1 Instructions for SWOG Biospecimen Bank .................................................................................... 96

S1933 Page 5
Version Date 6/1/2021
PROTOCOL CONTACT INFORMATION
Patient Advocate:
Judy Johnson, M.B.A. E-mail: [email protected] Phone: 314/477-6139
Eligibility, RAVE, Data Submission: SWOG Statistics and Data Management Center E-mail: [email protected] or Phone: 206/652-2267
Regulatory, Protocol, Informed Consent: SWOG Operations Office E-mail: [email protected] or Phone: 210/614-8808
Medical Queries (treatment or toxicity related questions):
E-mail or call the study chairs listed on the title page
Investigational Drug questions: Requests for Investigator’s Brochures: Access issues for the PMB Online Agent Ordering Processing (OAOP) application:
See Protocol Section 3.0 or [email protected] See Protocol Section 3.0 or http://ctep.cancer.gov/branches/pmb/agent_order_processing.htm [email protected]
Specimen Tracking System (STS) Amendments, Errors, Connectivity Issues and Technical issues with the SWOG CRA Workbench:
Cancer Therapy and Evaluation Program - Identity and Access Management (CTEP-IAM)
To review CTEP-IAM account (new requests, reset passwords): https://ctepcore.nci.nih.gov/iam/index.jsp
Access to iMedidata Rave See Protocol Section 14.0 or contact CTSU Help Desk: Phone: 1-888-823-5923 or Email: [email protected]
Oncology Patient Enrollment Network (OPEN)
See Protocol Section 13.3 or contact CTSU Help Desk: Phone: 1-888-823-5923 or Email: [email protected]
TRIAD installations: https://triadinstall.acr.org/triadclient/ Questions: [email protected]
Serious Adverse Event Reporting questions: See Protocol Section 8.5 Email: [email protected]
S1933 Page 6
Version Date 6/1/2021
CANCER TRIALS SUPPORT UNIT (CTSU) ADDRESS AND CONTACT INFORMATION CONTACT INFORMATION
For regulatory requirements: For patient enrollments: For study data submission: Regulatory documentation must be submitted to the CTSU via the Regulatory Submission Portal. (Sign in at www.ctsu.org, and select the Regulatory > Regulatory Submission.) Institutions with patients waiting that are unable to use the Portal should alert the CTSU Regulatory Office immediately at 1-866-651-2878 to receive further instruction and support. Contact the CTSU Regulatory Help Desk at 1-866-651-2878 for regulatory assistance.
Refer to the patient enrollment section of the protocol for instructions on using the Oncology Patient Enrollment Network (OPEN). OPEN can be accessed at https://www.ctsu.org/OPEN_SYSTEM/ or https://OPEN.ctsu.org. Contact the CTSU Help Desk with any OPEN-related questions at [email protected].
Data collection for this study will be done exclusively through Medidata Rave. Refer to the data submission section of the protocol for further instructions. Other Tools and Reports: Institutions participating through the CTSU continue to have access to other tools and reports available on the SWOG CRA Workbench via the SWOG website (www.swog.org).
The most current version of the study protocol and all supporting documents must be downloaded from the protocol-specific Web page of the CTSU Member Web site located at https://www.ctsu.org. Access to the CTSU members’ website is managed through the Cancer Therapy and Evaluation Program - Identity and Access Management (CTEP-IAM) registration system and requires user log on with CTEP-IAM username and password. For patient eligibility or data submission questions contact the SWOG Statistics and Data Management Center (SDMC) by phone or email: 206/652-2267 [email protected] For treatment or toxicity related questions contact one of the Study Chairs listed on the title page by phone or email. For non-clinical questions (i.e. unrelated to patient eligibility, treatment, or clinical data submission) contact the CTSU Help Desk by phone or e-mail: CTSU General Information Line – 1-888-823-5923, or [email protected]. All calls and correspondence will be triaged to the appropriate CTSU representative.
The CTSU Website is located at https://www.ctsu.org.
S1933 Page 7
Version Date 6/1/2021
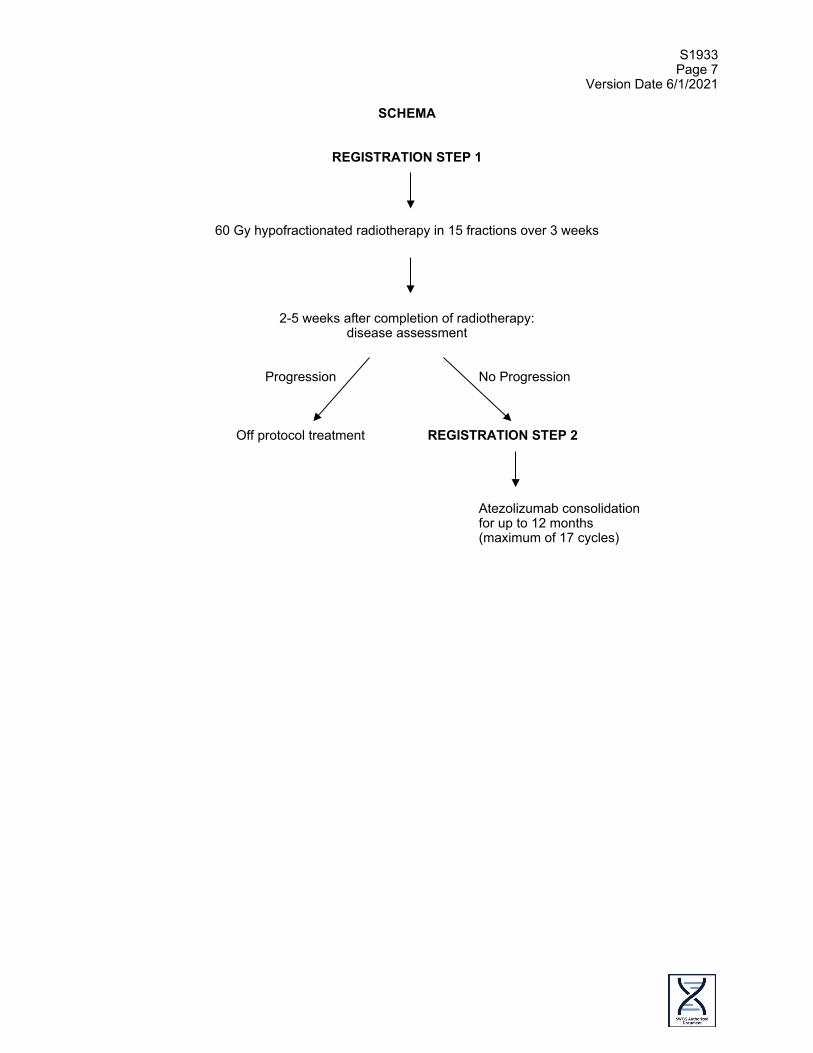
SCHEMA
REGISTRATION STEP 1
60 Gy hypofractionated radiotherapy in 15 fractions over 3 weeks
2-5 weeks after completion of radiotherapy: disease assessment
Progression No Progression
Off protocol treatment REGISTRATION STEP 2 Atezolizumab consolidation
for up to 12 months (maximum of 17 cycles)

S1933 Page 8
Version Date 6/1/2021
1.0 OBJECTIVES
1.1 Primary Objective To evaluate the rate of Grade 3-5 Treatment-Related Adverse Events (TRAEs) in patients who are not candidates for surgery or concurrent chemoradiation and who have either performance status 0-2 and Stage II or performance status 2 and Stage III non-small cell lung cancer (NSCLC), treated with hypofractionated thoracic radiotherapy followed by atezolizumab.
1.2 Secondary Objective(s)
a. To evaluate response rate (confirmed and unconfirmed, complete and partial by RECIST 1.1) from Registration Step 2 in the subset of patients with measurable disease.
b. To evaluate response rate (confirmed and unconfirmed, complete and partial by
RECIST 1.1) during radiation therapy in the subset of patients with measurable disease.
c. To evaluate progression free survival (PFS) from Registration Step 2 by RECIST 1.1.
d. To evaluate overall survival (OS) from Registration Step 2.
e. To evaluate the frequency and severity of toxicities.
1.3 Additional Objective
a. To bank blood and archival tissue for future research.
2.0 BACKGROUND
2.1 Lack of evidence-based data to guide treatment recommendations for patients with Stage III NSCLC with performance status 2 and patients with Stage II who are not surgical candidates
Lung cancer is the most common cause of cancer related mortality in the United States accounting for more than 150,000 deaths a year. More than 200,000 new cases of lung cancer are diagnosed each year in the United States and about 85% of these are NSCLC. (1) Approximately 30% of patients with NSCLC seen in the out-patient setting present with performance status 2 (PS 2). (2) Most large randomized studies in patients with Stage III NSCLC using concurrent chemoradiotherapy excluded patients with performance status equivalent to 2 or more. (3) S9712, a Phase II study evaluating the role of consolidation paclitaxel after chemoradiation in poor risk Stage III NSCLC patients included 43% PS 2 patients. (4) That regimen was associated with increased toxicity and no survival advantage was demonstrated. Eight toxic deaths (9%) occurred, four during concurrent chemoradiation and four during consolidation chemotherapy. A retrospective review of newly diagnosed Stage III NSCLC patients showed that more than half of these patients did not receive curative intent treatment. In about one third of those patients who did not receive curative intent therapy, poor performance status was cited as a reason and most of these patients received palliative radiotherapy only. (5) The improved local control and survival achieved by adding chemotherapy to radiation therapy comes at the expense of increased toxicity. RTOG 9410 showed that concurrent chemoradiotherapy in patients with Stage III NSCLC was associated with more frequent Grade 3 or higher toxicities compared to sequential therapy. (6) This leads to approximately 11% of patients with node positive non-metastatic NSCLC referred for definitive chemoradiotherapy eventually receiving

S1933 Page 9
Version Date 6/1/2021
radiation only. (7) In summary, there is a large population of patients with NSCLC who cannot receive curative therapy primarily due to the toxicities from concurrent chemoradiotherapy. The paucity of data and the lack of modern clinical trials leave physicians struggling on how to guide treatment decisions for these patients as no standard regimen exists. Immune checkpoint inhibitors are ideal agents to study with radiation because of the strong preclinical evidence supporting increased tumor destruction, their success in treating metastatic disease and their mild toxicity profile. Potentially curative treatment regimens must be available to all patients. Our study is very timely in that it aligns with the newly released NCI guideline for expanding trial eligibility to ensure studies reflect a real-world population. A positive finding from this study would be the first step in achieving our goal of offering curative intent therapy to patients with borderline performance status. We are optimistic that a confirmatory Phase III randomized trial comparing thoracic radiotherapy followed by atezolizumab consolidation to sequential thoracic radiation and chemotherapy would be positive for our approach in terms of efficacy and safety. Hence a new potentially curative treatment would become available to a large patient population with NSCLC.
2.2 The role for immune checkpoint inhibitors in lung cancer
Lung cancer has long been considered poorly immunogenic, with resultant lack of activity in clinical trials evaluating immunotherapies including interleukin 2, interferon and vaccines. One reason for that lack of activity of immunotherapeutic agents was that some tumors express Programed cell Death Ligand-1 (PD-L1) that interacts with PD-1 on cytotoxic T lymphocytes and renders them not able to recognize tumor cells. Agents that target the PD-1/PD-L1 pathway were developed to reinvigorate anticancer immunity. Four agents (nivolumab, pembrolizumab, atezolizumab and durvalumab) are FDA approved for the treatment of NSCLC in various settings. One of these agents, atezolizumab, is a humanized anti-PD-L1 monoclonal antibody that inhibits PD-L1 interaction with PD-1 and B7-1. A Phase III trial comparing atezolizumab to docetaxel in patients with previously treated Stage IV NSCLC showed statistically significant improvement in median overall survival in favor of atezolizumab (median overall survival 13.8 months versus 9.6 months, P=0.0003). (8) An updated analysis from that study with a follow up duration of about 2 years showed continued overall survival benefit in favor of atezolizumab. (9) In the first line setting, in patients with Stage IV NSCLC and SCLC atezolizumab plus chemotherapy provided superior outcomes compared to chemotherapy alone. (10, 11) Evaluation of atezolizumab in early stage and locally advanced stage NSCLC is ongoing. Durvalumab is the first immune checkpoint inhibitor to show efficacy in the curative setting of locally advanced Stage III disease. An interim analysis of the Phase III PACIFIC trial showed that the use of durvalumab, another anti PD-L1 antibody, after concurrent chemoradiotherapy in patients with Stage III NSCLC was associated with more than 11 months improvement in progression free survival. (12) Durvalumab consolidation after chemoradiotherapy was well tolerated with Grade 3 or 4 adverse events of 29.9% and 26.1% for durvalumab and placebo respectively of which 3.4% and 2.6% were pneumonitis. Based on the results of this study, the FDA approved durvalumab consolidation for patients with unresectable Stage III NSCLC who have no disease progression after chemoradiotherapy on February 16, 2018. An update from the PACIFIC trial reported statistically significant improvement in overall survival with the use of durvalumab consolidation compared to placebo in patients with Stage III NSCLC who received chemoradiotherapy with a 24-month overall survival rate of 66.3% and 55.6% in favor of durvalumab. (13) With regard to atezolizumab in this setting, early safety data from the Phase II DETERRED study using atezolizumab consolidation after chemoradiotherapy demonstrated this approach to be feasible and well tolerated with manageable toxicity. (14) Data from 10 evaluable patients noted three patients with potential immune related AEs: Grade 3 arthralgia, Grade 3 dyspnea due to COPD
S1933 Page 10
Version Date 6/1/2021
exacerbation and Grade 2 radiation-induced pneumonitis that resolved with steroids. In addition to their efficacy, immune checkpoint inhibitors have a mild toxicity profile. Grade 3-4 adverse events in the studies using immune checkpoint inhibitors were less than half of what was observed in the chemotherapy arms. (15)This safety profile heightens our enthusiasm for these agents because of the potential to treat less robust patients who have been excluded from trials because of toxicity concerns. Thus, it is critical that trials are urgently conducted in poor risk patients to ascertain their risks and benefits.
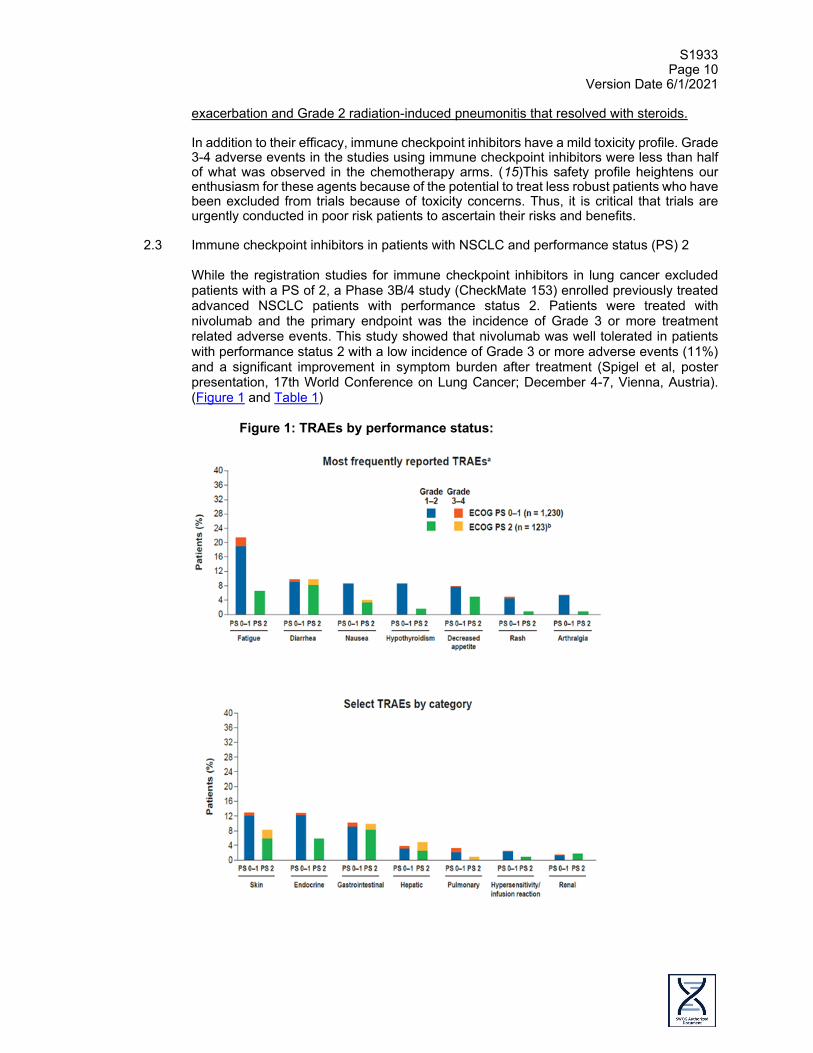
2.3 Immune checkpoint inhibitors in patients with NSCLC and performance status (PS) 2
While the registration studies for immune checkpoint inhibitors in lung cancer excluded patients with a PS of 2, a Phase 3B/4 study (CheckMate 153) enrolled previously treated advanced NSCLC patients with performance status 2. Patients were treated with nivolumab and the primary endpoint was the incidence of Grade 3 or more treatment related adverse events. This study showed that nivolumab was well tolerated in patients with performance status 2 with a low incidence of Grade 3 or more adverse events (11%) and a significant improvement in symptom burden after treatment (Spigel et al, poster presentation, 17th World Conference on Lung Cancer; December 4-7, Vienna, Austria). (Figure 1 and Table 1)
Figure 1: TRAEs by performance status:

S1933 Page 11
Version Date 6/1/2021
A retrospective review from Japan showed that nivolumab was effective in the treatment of patients with performance status 2 with a response rate of 20%, disease control rate of 50% and median survival was not reached after 10 months of follow up (Watanabe et al, poster presentation in the 17th World Conference on Lung Cancer; December 2016, Vienna, Austria) (Table 2 and Figure 2).
In summary, these data suggest that the administration of immune checkpoint inhibitors have similar efficacy and safety regardless of performance status
2.4 Radiation therapy regimens in locally advanced NSCLC and PS 2 patients Conventionally fractionated radiation therapy alone as definitive treatment for locally advanced NSCLC has poor survival rates, with a median survival of about 11 months. (16) Iyengar et al reported an interim analysis that included the first 60 patients from a Phase III randomized trial of conventionally fractionated (60 Gy in 30 fractions) versus

S1933 Page 12
Version Date 6/1/2021
hypofractionated thoracic radiotherapy (60 Gy in 15 fractions) in patients with Stage II and III NSCLC with poor performance status during the ASTRO meeting in 2016. (17) Of note, this is an ongoing study using modern techniques in radiation therapy with the goal of enrolling 226 patients. After a median follow up of 24 months for the 48 evaluable patients, there were no statistical differences between the two arms in terms of overall and progression free survival which were about 14 months and 11 months respectively. Hypofractionated radiotherapy was better tolerated with fewer Grade 3 toxicities compared to conventional radiotherapy (19% vs. 36% respectively). The most recent data using hypofractionated thoracic radiotherapy in the same patient population targeted in this study come from the above-mentioned UTSW study by Iyengar et al. This study showed that hypofractionated radiotherapy was well-tolerated with reported Grade 3 or higher treatment related adverse events (TRAEs) of 22% (95% CI 9%-40%). Data with the use of PD-1/PD-L1 inhibitors alone in this patient population showed Grade 3 or higher TRAEs of 11% (95% CI 6%-17%). (18) Data from the PACIFIC study in Stage III patients with PS 0-1 showed Grade 3 or higher TRAEs of 12% (95% CI 9%-15%). (19,20) Studies that included the use of concurrent chemoradiotherapy in the treatment of patients with NSCLC with borderline performance status showed poor outcomes in general with significant toxicity. SWOG S9712 was a Phase II study to treat patients with poor risk Stage III NSCLC that enrolled 43% of patients with PS2 and investigated the role of consolidation paclitaxel after concurrent chemoradiation. (21) This study showed that concurrent chemoradiotherapy was not well tolerated in this patient population with 8 toxic deaths and a high percentage of Grade 3 or higher adverse events (52% neutropenia and 26% thrombocytopenia). A review article of studies that used concurrent chemoradiotherapy in this patient population showed poor outcomes in general with significant toxicity. (22) A retrospective analysis of studies using concurrent chemoradiotherapy in locally advanced NSCLC identified poor performance status as an important predictive factor for death within 180 days after therapy with an odds ratio of 2.84. (23) Although weekly carboplatin and paclitaxel concurrent with radiation has generally a lesser percentage of hematologic adverse events, this regimen is usually associated with a higher percentage of pneumonitis compared to platinum and etoposide (66% vs. 38%) and this adverse event could be a significant problem in patients with borderline performance status and comorbidities. (24) Based on the above-mentioned points, we do not think that concurrent chemotherapy and thoracic radiation is a good option for this patient population and the use of thoracic radiotherapy followed by atezolizumab consolidation could be an effective, well-tolerated alternative.
2.5 Radiation therapy and immune checkpoint inhibitors Radiation therapy has been reported to modulate the immune system resulting in both local and systemic effects. (25) Regarding local effects of radiation on the immune system, radiotherapy upregulates MHC class I molecules that are essential for antigen presentation to T cells. Radiation also causes the release of death associated molecular patterns like HMGB1 and calreticulin that in turn activate dendritic cells that play a role in the priming of the adaptive (T cell mediated) immune system. Other reported local effects of radiation therapy are to increase the density of tumor-infiltrating lymphocytes and increase expression of PD-L1. These 2 factors were cited as potential causes for the reported synergistic effects of radiotherapy and anti-PD-1 agents in preclinical studies. (26) Regarding the systemic effects of radiotherapy, irradiation has been reported to be able to induce tumor regression at non-irradiated, distant tumor sites. This phenomenon is called the “abscopal effect” and potentially has an important role in eradicating micrometastases. A review article of case reports of the abscopal effect documented this phenomenon in different types of cancers including lung cancer with progression free survival after response of up to 39 months. (27) This paper also cited cases of the abscopal effect after the use of immunotherapy and radiotherapy. Preclinical studies showed that combining

S1933 Page 13
Version Date 6/1/2021
radiation with PD-1/PD-L1 inhibitors improve outcomes. (28) A secondary analysis of patients with advanced NSCLC treated with pembrolizumab on the KEYNOTE-001 Phase I trial showed that patients who received radiation therapy prior to pembrolizumab had significantly better progression free survival (median progression free survival 4.4 months vs. 2.1 months) and overall survival (median overall survival 10.7 months vs. 5.3 months). (29) The outcome in terms of progression free and overall survival was even better in patients who had extra-cranial radiotherapy, (6.3 months vs. 2 months) and (11.6 months vs. 5.3 months) respectively. Although there was an increase in treatment related pulmonary toxicity in patients who received prior thoracic radiation, the incidence of Grade 3 or more pulmonary toxicity was low in both groups (less than 5%). The authors of that analysis concluded that further studies are needed to investigate the combination of radiotherapy and PD-1 inhibitors in treatment of patients with NSCLC. Many preclinical and early clinical studies have shown that higher doses of radiation therapy are more likely to illicit immune response and hypofractionated radiation therapy regimens are believed to work better with immune check-point inhibitors. (30) Radiation pneumonitis is a possible adverse event of thoracic radiotherapy. Cho et al reported their experience treating patients with early stage NSCLC who are not candidates for surgical resection with single modality radiotherapy. Results were reported for groups of patients where Group 1 and 2 had 60 Gy in 20 fractions and Group 3 had 60 Gy in 15 fractions. Grade 3 or more radiation pneumonitis occurred in 3-4% of these patients. (31) Concurrent chemoradiotherapy for localized NSCLC usually results in a greater incidence of Grade 3 or higher radiation pneumonitis of about 10% as reported by Warner and colleagues. (32) A more recent larger review by Steuer et al. reported a similar incidence of radiation pneumonitis with the use of concurrent chemoradiotherapy in this patient population. (33) Grade 3 or more radiation pneumonitis was reported in about 2% of patients with NSCLC treated with an immune checkpoint inhibitor as a single agent. (34) Esophagitis is another possible adverse event of using thoracic radiotherapy. The incidence of Grade 1 or 2 esophagitis was 37.1% with no reported Grade 3 or higher of this adverse event in patients who received 60 Gy single modality thoracic radiation. (35) Patients who received concurrent chemoradiotherapy had a higher incidence of esophagitis (50% Grade 1 or 2 and 30% Grade 3 or 4). (36) A retrospective review noted the safety and efficacy of hypofractionated radiation therapy concurrent or within 8 weeks of the use of immune checkpoint inhibitors in patients with solid tumors including NSCLC. (37) In this review that included patients with PS 2, the incidence of Grade 3 or higher TRAEs was 20% of which 5% were pneumonitis. The efficacy and safety of durvalumab after concurrent chemotherapy and thoracic radiation in the PACIFIC trial as described above are encouraging. (38, 39) Having said that, possible adverse events of the use of atezolizumab consolidation after thoracic radiotherapy will be monitored carefully during this study. After patients complete radiation therapy, atezolizumab consolidation will be used for up to 12 months. That time frame was chosen based on data from the PACIFIC clinical trial that showed statistically significant improvement in progression free and overall survival with the use of durvalumab after chemoradiotherapy in Stage III NSCLC patients. Moreover, durvalumab was well tolerated with good compliance. (40, 41) In summary, compared to other solid tumors, a higher percentage of lung cancer patients present with borderline performance status. These patients were excluded from most randomized Phase III studies that set the standard of care in the treatment of Stage II and III NSCLC. As such, there is no evidence based curative option in the treatment of this patient population. Atezolizumab, a PD-L1 inhibitor showed activity in the treatment of patients with Stage IV NSCLC and is now FDA approved in the first-line setting with chemotherapy and bevacizumab and the second-line setting as a single agent in the management of this patient population. Moreover, this agent was well tolerated in general with a significantly lower incidence of adverse events compared to chemotherapy. Radiation therapy was shown in preclinical studies to induce the immune system and

S1933 Page 14
Version Date 6/1/2021
increase the expression of molecules involved in cytotoxic T cell activity. This, together with data from the PACIFIC trial using another PD-L1 inhibitor durvalumab, makes the use of atezolizumab consolidation after thoracic radiotherapy a promising approach in our patient population. We are optimistic that hypofractionated thoracic radiotherapy followed by atezolizumab consolidation would be well tolerated with good efficacy. A positive result from this study would lead to a definitive Phase III study that would hopefully provide a curative well tolerated option for a large population of real world less robust patents with Stage II and III NSCLC. The success of atezolizumab in the metastatic setting warrant its evaluation in earlier, potentially curable stages of lung cancer. The main reasons why patients are not cured are due to micrometastatic disease and/or the inability to eradicate the primary tumor with current modalities that require improved systemic therapies. The PACIFIC trial showed that the use of immune checkpoint inhibitors after concurrent chemoradiotherapy improves survival and is a, well tolerated approach. Considering that a large percentage of NSCLC patients are not candidates for concurrent chemotherapy and thoracic radiation, our approach of thoracic radiotherapy followed by atezolizumab consolidation would hopefully provide an alternative, effective, better tolerated option for this patient population.
2.6 Inclusion of Women and Minorities and Planned Enrollment Report
This study was designed to include women and minorities, but was not designed to measure differences of intervention effects. The anticipated accrual in the ethnicity/race and sex categories is shown in the table below.
DOMESTIC PLANNED ENROLLMENT REPORT
Racial Categories
Ethnic Categories Total Not Hispanic or Latino Hispanic or Latino
Female Male Female Male American Indian/ Alaska Native
0 0 0 0 0
Asian 0 1 0 0 1 Native Hawaiian or Other Pacific Islander
0 0 0 0 0
Black or African American
2 4 0 0 6
White 15 24 0 1 40 More Than One Race
0 0 0 0 0
Total 17 29 0 1 47
3.0 DRUG INFORMATION
Investigator Brochures For information regarding Investigator Brochures, please refer to SWOG Policy 15. For this study, atezolizumab is investigational and is being provided under an IND held by the National Cancer Institute. The current version of the Investigator Brochure (IB) will be accessible to site investigators and research staff through the PMB Online Agent Order Processing (OAOP) application: (http://ctep.cancer.gov/branches/pmb/agent_order_processing.htm). Access to OAOP requires the establishment of a CTEP Identity and Access Management (IAM) account and the maintenance of an “active” account status and a “current” password. Questions about IB access

S1933 Page 15
Version Date 6/1/2021
may be directed via email to [email protected] or by phone (240) 276-6575 Monday through Friday between 8:30 am and 4:30 pm (ET).
3.1 Atezolizumab (NSC-783608, IND # 138328)
a. PHARMACOLOGY
Mechanism of Action: Atezolizumab is a humanized immunoglobulin (IgG1) monoclonal antibody that is produced in Chinese hamster ovary (CHO) cells. Atezolizumab targets programmed death-ligand 1 (PD-L1) on immune cells or tumor cells and prevents interaction with either programmed death-1 (PD-1) receptor or B7.1 (CD80), both of which function as inhibitory receptors expressed on T cells. Interference of the PD-L1: PD-1 and PD-L1: B7.1 interactions may enhance the magnitude and quality of the tumor-specific T-cell response through increased T-cell priming, expansion, and/or effector function. Atezolizumab was engineered to eliminate Fc-effector function via a single amino acid substitution at position 298 on the heavy chain, which results in a non-glycosylated antibody that has minimal binding to Fc receptors and, consequently, eliminates detectable Fc-effector function. By eliminating Fc-effector function and antibody dependent cell-mediated cytotoxicity (ADCC), antibody-mediated clearance of activated effector T cells is also eliminated. Atezolizumab shows anti-tumor activity in both nonclinical models and cancer patients, and is being investigated as a potential therapy in a wide variety of malignancies.
b. PHARMACOKINETICS
On the basis of available preliminary PK data (0.03-20 mg/kg), atezolizumab appeared to show linear pharmacokinetics at doses ≥ 1 mg/kg. For the 1 mg/kg and 20 mg/kg dose groups, the mean apparent CL and the mean Vss had a range of 3.20 to 4.44 mL/day/kg and 48.1 to 65.7 mL/kg, respectively, which is consistent with the expected profile of an IgG1 antibody in humans. Serum atezolizumab concentrations exhibited a biphasic disposition with an initial rapid distribution phase followed by a slow elimination phase. Atezolizumab exhibited nonlinear pharmacokinetics at doses <1 mg/kg (i.e., 0.03-0.3 mg/kg), likely due to target-mediated CL at lower concentrations. Atezolizumab exhibited linear pharmacokinetics at doses ≥ 1 mg/kg. At doses ≥ 1 mg/kg, the mean Cmax increased in a dose-proportional manner and was 26.0 mcg/mL for the 1-mg/kg dose group and 472 mcg/mL for the 20 mg/kg dose group. Similarly, at doses ≥ 1 mg/kg, the group mean AUC0-∞ had a range of 340-6050 Day x mcg/mL for 1 mg/kg and 20 mg/kg dose group and was approximately dose proportional, by similar CL across the dose range. The observed CL and Vss for atezolizumab at doses ≥ 1 mg/kg are consistent with these of a typical IgG1 antibody in humans.
Currently available PK and ATA data from Study PCD4989g suggest that the 15-mg/kg atezolizumab q3w regimen (or fixed-dose equivalent) for Phase II and Phase III studies would be sufficient to both maintain trough concentration (Ctrough) ³ 6 mg/mL and further safeguard against both interpatient variability and the potential effect of ATAs that could lead to subtherapeutic levels of atezolizumab relative to the 10-mg/kg atezolizumab q3w regimen (or fixed-dose equivalent). From inspection of available observed Ctrough data, moving further to the 20-mg/kg atezolizumab q3w regimen does not appear to be warranted to maintain targeted Ctrough levels relative to the proposed 15-mg/kg atezolizumab q3w level.
Refer to the atezolizumab Investigator’s Brochure for details regarding nonclinical and clinical pharmacology of atezolizumab.
S1933 Page 16
Version Date 6/1/2021
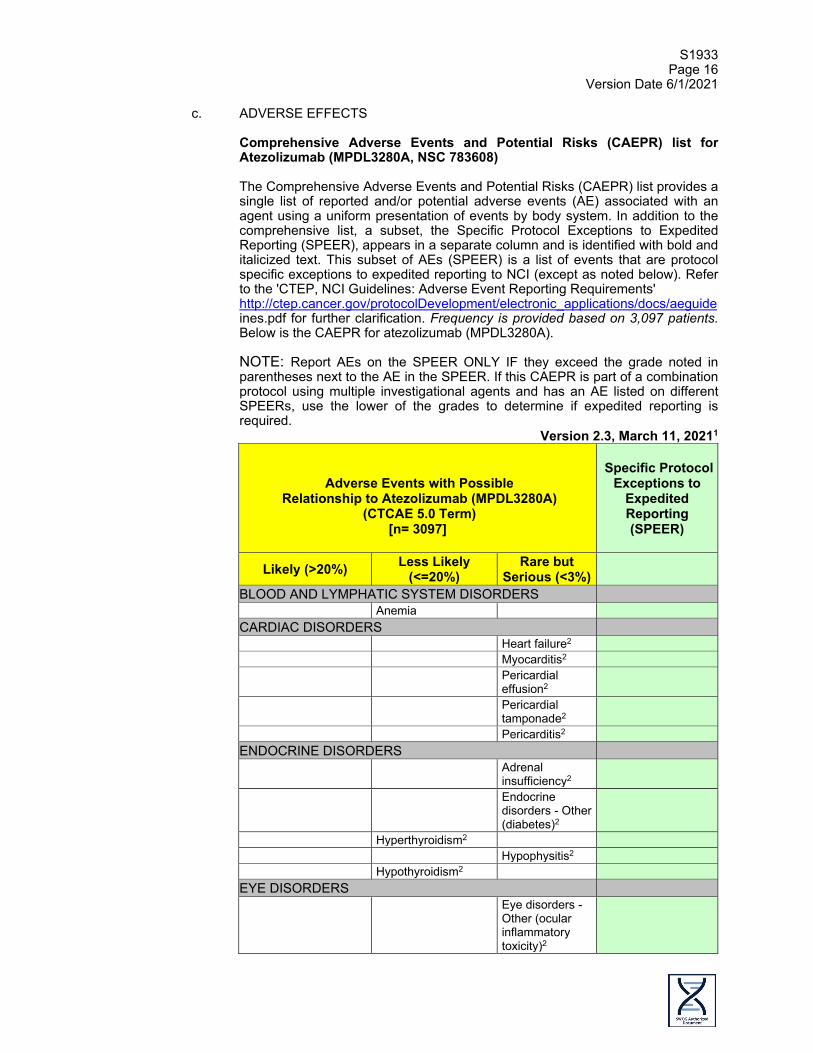
c. ADVERSE EFFECTS
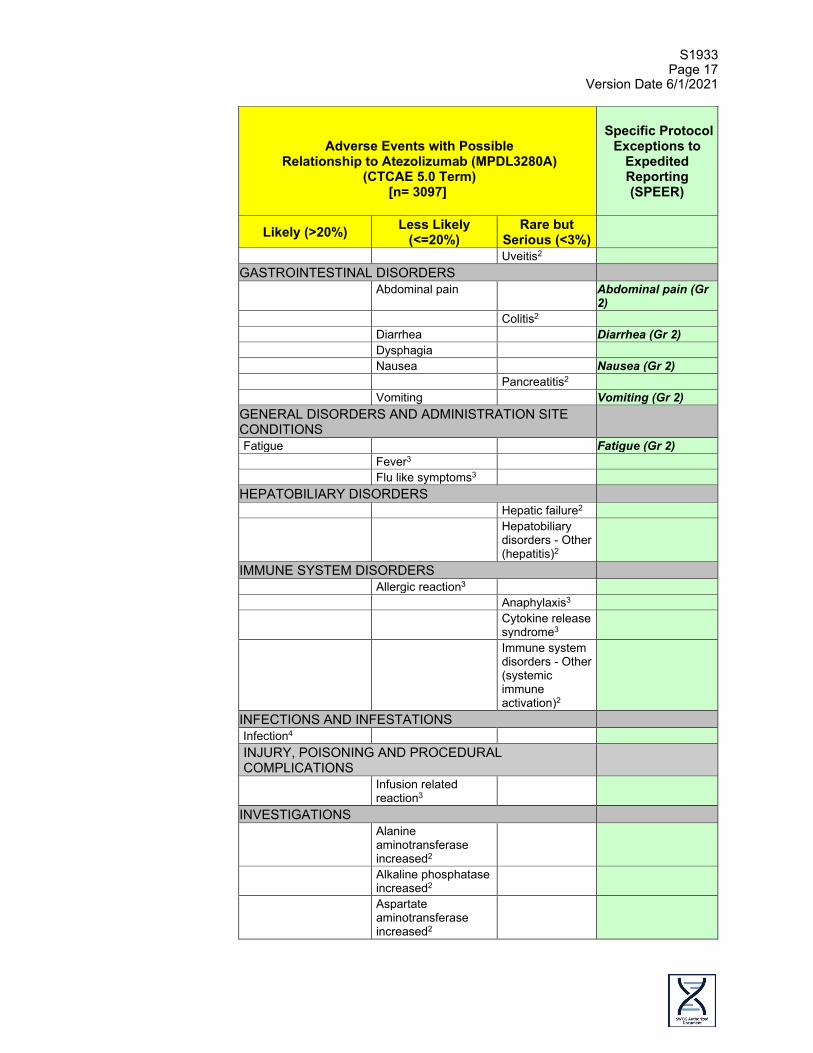
Comprehensive Adverse Events and Potential Risks (CAEPR) list for Atezolizumab (MPDL3280A, NSC 783608) The Comprehensive Adverse Events and Potential Risks (CAEPR) list provides a single list of reported and/or potential adverse events (AE) associated with an agent using a uniform presentation of events by body system. In addition to the comprehensive list, a subset, the Specific Protocol Exceptions to Expedited Reporting (SPEER), appears in a separate column and is identified with bold and italicized text. This subset of AEs (SPEER) is a list of events that are protocol specific exceptions to expedited reporting to NCI (except as noted below). Refer to the 'CTEP, NCI Guidelines: Adverse Event Reporting Requirements' http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/aeguide ines.pdf for further clarification. Frequency is provided based on 3,097 patients. Below is the CAEPR for atezolizumab (MPDL3280A). NOTE: Report AEs on the SPEER ONLY IF they exceed the grade noted in parentheses next to the AE in the SPEER. If this CAEPR is part of a combination protocol using multiple investigational agents and has an AE listed on different SPEERs, use the lower of the grades to determine if expedited reporting is required.
Version 2.3, March 11, 20211
Adverse Events with Possible
Relationship to Atezolizumab (MPDL3280A) (CTCAE 5.0 Term)
[n= 3097]
Specific Protocol
Exceptions to Expedited Reporting (SPEER)
Likely (>20%) Less Likely (<=20%)
Rare but Serious (<3%)
BLOOD AND LYMPHATIC SYSTEM DISORDERS Anemia CARDIAC DISORDERS Heart failure2 Myocarditis2 Pericardial
effusion2
Pericardial tamponade2
Pericarditis2 ENDOCRINE DISORDERS Adrenal
insufficiency2
Endocrine disorders - Other (diabetes)2
Hyperthyroidism2 Hypophysitis2 Hypothyroidism2 EYE DISORDERS Eye disorders -
Other (ocular inflammatory toxicity)2
S1933 Page 17
Version Date 6/1/2021
Adverse Events with Possible
Relationship to Atezolizumab (MPDL3280A) (CTCAE 5.0 Term)
[n= 3097]
Specific Protocol
Exceptions to Expedited Reporting (SPEER)
Likely (>20%) Less Likely (<=20%)
Rare but Serious (<3%)
Uveitis2 GASTROINTESTINAL DISORDERS Abdominal pain Abdominal pain (Gr
2) Colitis2 Diarrhea Diarrhea (Gr 2) Dysphagia Nausea Nausea (Gr 2) Pancreatitis2 Vomiting Vomiting (Gr 2) GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS
Fatigue Fatigue (Gr 2) Fever3 Flu like symptoms3 HEPATOBILIARY DISORDERS Hepatic failure2 Hepatobiliary
disorders - Other (hepatitis)2
IMMUNE SYSTEM DISORDERS Allergic reaction3 Anaphylaxis3 Cytokine release
syndrome3
Immune system disorders - Other (systemic immune activation)2
INFECTIONS AND INFESTATIONS Infection4 INJURY, POISONING AND PROCEDURAL COMPLICATIONS
Infusion related reaction3
INVESTIGATIONS Alanine
aminotransferase increased2
Alkaline phosphatase increased2
Aspartate aminotransferase increased2

S1933 Page 18
Version Date 6/1/2021
Adverse Events with Possible
Relationship to Atezolizumab (MPDL3280A) (CTCAE 5.0 Term)
[n= 3097]
Specific Protocol
Exceptions to Expedited Reporting (SPEER)
Likely (>20%) Less Likely (<=20%)
Rare but Serious (<3%)
Blood bilirubin increased2
Creatinine increased
GGT increased2 Lipase increased* Platelet count
decreased
Serum amylase increased*
METABOLISM AND NUTRITION DISORDERS Anorexia Anorexia (Gr 2) Hyperglycemia2 Hypokalemia Hyponatremia MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS
Arthralgia2 Back pain Generalized
muscle weakness
Myalgia Myositis2 NERVOUS SYSTEM DISORDERS Ataxia2 Encephalopathy2 Nervous system
disorders - Other (encephalitis non-infective)2
Guillain-Barre syndrome2
Nervous system disorders - Other (meningitis non-infective)2
Myasthenia gravis2
Paresthesia2 Peripheral motor
neuropathy2
Peripheral sensory neuropathy2

S1933 Page 19
Version Date 6/1/2021
Adverse Events with Possible
Relationship to Atezolizumab (MPDL3280A) (CTCAE 5.0 Term)
[n= 3097]
Specific Protocol
Exceptions to Expedited Reporting (SPEER)
Likely (>20%) Less Likely (<=20%)
Rare but Serious (<3%)
RENAL AND URINARY DISORDERS Acute kidney
injury
Renal and urinary disorders - Other (nephritis)2
RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS
Cough Cough (Gr 2) Dyspnea Hypoxia Nasal congestion Nasal congestion
(Gr 2) Pleural effusion2 Pneumonitis2 SKIN AND SUBCUTANEOUS TISSUE DISORDERS Bullous
dermatitis2
Erythema multiforme2
Pruritus Rash acneiform Rash maculo-papular Skin and
subcutaneous tissue disorders - Other (drug reaction with eosinophilia and systemic symptoms [DRESS])2
Skin and subcutaneous tissue disorders - Other (lichen planus)2
Skin and subcutaneous tissue disorders - Other (exanthematous pustulosis)2
Stevens-Johnson syndrome2
Toxic epidermal necrolysis2

S1933 Page 20
Version Date 6/1/2021
Adverse Events with Possible
Relationship to Atezolizumab (MPDL3280A) (CTCAE 5.0 Term)
[n= 3097]
Specific Protocol
Exceptions to Expedited Reporting (SPEER)
Likely (>20%) Less Likely (<=20%)
Rare but Serious (<3%)
* Denotes adverse events that are <3%.
1 This table will be updated as the toxicity profile of the agent is revised. Updates will be distributed to all Principal Investigators at the time of revision. The current version can be obtained by contacting [email protected]. Your name, the name of the investigator, the protocol and the agent should be included in the e-mail.
2 Atezolizumab, being a member of a class of agents involved in the inhibition of “immune checkpoints,” may result in severe and possibly fatal immune-mediated adverse events probably due to T-cell activation and proliferation. Immune-mediated adverse reactions have been reported in patients receiving atezolizumab. Adverse events potentially related to atezolizumab may be manifestations of immune-mediated adverse events. In clinical trials, most immune-mediated adverse reactions were reversible and managed with interruptions of atezolizumab, administration of corticosteroids and supportive care.
3 Infusion reactions, including high-grade hypersensitivity reactions, anaphylaxis, and cytokine release syndrome, which have been observed following administration of atezolizumab, may manifest as fever, chills, shakes, itching, rash, hypertension or hypotension, or difficulty breathing during and immediately after administration of atezolizumab.
4 Infection includes all 75 sites of infection under the INFECTIONS AND INFESTATIONS SOC.
Adverse events reported on atezolizumab (MPDL3280A) trials, but for which there is insufficient evidence to suggest that there was a reasonable possibility that atezolizumab (MPDL3280A) caused the adverse event: BLOOD AND LYMPHATIC SYSTEM DISORDERS - Blood and lymphatic system disorders - Other (pancytopenia); Febrile neutropenia CARDIAC DISORDERS - Cardiac arrest; Ventricular tachycardia GASTROINTESTINAL DISORDERS - Constipation; Dry mouth; Ileus GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS - Chills; Edema limbs; Malaise; Multi-organ failure HEPATOBILIARY DISORDERS - Portal vein thrombosis INVESTIGATIONS - Lymphocyte count decreased; Neutrophil count decreased; Weight loss; White blood cell decreased METABOLISM AND NUTRITION DISORDERS - Hypophosphatemia; Tumor lysis syndrome MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS - Bone pain; Muscle cramp: Pain in extremity NERVOUS SYSTEM DISORDERS - Headache PSYCHIATRIC DISORDERS - Confusion; Insomnia; Suicide attempt REPRODUCTIVE SYSTEM AND BREAST DISORDERS - Breast pain RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS - Bronchopulmonary hemorrhage; Pulmonary hypertension; Respiratory failure

S1933 Page 21
Version Date 6/1/2021
SKIN AND SUBCUTANEOUS TISSUE DISORDERS - Alopecia; Dry skin2; Hyperhidrosis VASCULAR DISORDERS - Hypertension; Hypotension; Thromboembolic event
Note: Atezolizumab (MPDL3280A) in combination with other agents could cause an exacerbation of any adverse event currently known to be caused by the other agent, or the combination may result in events never previously associated with either agent. 1. Pregnancy and Lactation: No developmental or reproductive toxicity
studies have been conducted with atezolizumab as several nonclinical studies have already demonstrated that the PD-L1/PD-1 signaling pathway is essential in establishing maternal/fetal tolerance, which is necessary for embryo-fetal survival during gestation. Based on the critical role that PD-L1/PD1 pathway plays in the maintenance of maternal-fetal tolerance, atezolizumab should not be administered to pregnant women. The effects of atezolizumab on human reproduction or on the fetus or the developing infant are unknown but expected to have an adverse effect.
It is not known whether atezolizumab is excreted in human milk. However, antibodies are known to cross the placenta and are excreted in breast milk during lactation. Atezolizumab should not be administered to nursing mothers. Male patients and female patients of childbearing potential should utilize contraception and take active measures to avoid pregnancy while undergoing atezolizumab treatment and for at least 5 months (150 days) after the last dose of atezolizumab.
2. Drug Interactions: Cytochrome P450 enzymes as well as
conjugation/glucuronidation reactions are not involved in the metabolism of atezolizumab. No drug interaction studies for atezolizumab have been conducted or are planned. There are no known interactions with other medicinal products or other form of interactions.
d. DOSING & ADMINISTRATION
See Section 7.0 Treatment Plan.
e. HOW SUPPLIED
Atezolizumab is provided by Genentech/F. Hoffmann-La Roche LTD and distributed by the Pharmaceutical Management Branch, CTEP, NCI. The agent is supplied in a single-use, 20-mL glass vial as a colorless-to-slightly-yellow, sterile, preservative-free clear liquid solution intended for IV administration. Atezolizumab is formulated as 60 mg/mL atezolizumab in 20 mM histidine acetate, 120 mM sucrose, 0.04% polysorbate 20, at a pH of 5.8. The vial is designed to deliver 20 mL (1200 mg) of atezolizumab solution but may contain more than the stated volume to enable delivery of the entire 20 mL volume.
f. STORAGE, PREPARATION & STABILITY
Atezolizumab must be refrigerated at 2°C-8°C (36°F- 46°F) upon receipt until use. No preservative is used in atezolizumab and therefore, the vial is intended for single use only. Discard any unused portion of drug remaining in a vial. Vial contents should not be frozen or shaken and should be protected from direct sunlight.

S1933 Page 22
Version Date 6/1/2021
Atezolizumab (1200 mg per vial) will be administered in 250 mL 0.9% NaCl IV infusion bags and infusion lines equipped with 0.2 micron in-line filters. The IV bag may be constructed of polyvinyl chloride (PVC) or polyolefin (PO); the IV infusion line may be constructed of polyvinyl chloride (PVC) or polyethylene (PE); and the 0.2 micron in-line filter may be constructed of polyehersulfone (PES). Atezolizumab must be prepared/diluted under appropriate aseptic conditions as it does not contain antimicrobial preservatives. The solution for infusion should be used immediately to limit microbial growth in case of potential accidental contamination. If not used immediately, in-use storage time and conditions prior to use are the responsibility of the user. The dose solution prepared for IV bag delivery may be stored at 2°C to 8°C (36°F to 46°F) and/or at room temperature for up to a total in-use storage time of 8 hours.
g. DRUG ORDERING & ACCOUNTABILITY
NOTE: Atezolizumab cannot be ordered until the patient is registered to Step 2. To ensure drug supplies are received in time to treat within the 7-day window described in Section 7.3, sites should order atezolizumab immediately after registration to Step 2 and coordinate carefully with the Pharmaceutical Management Branch.
1. Drug ordering: Atezolizumab is an investigational agent supplied to
Investigators by the Division of Cancer Treatment and Diagnosis (DCTD), NCI. NCI-supplied agents may be requested by the Principal Investigator (or their authorized designees) at each participating institution. Pharmaceutical Management Branch (PMB) policy requires that the agent be shipped directly to the institution where the patient is to be treated. PMB does not permit the transfer of agents between institutions (unless prior approval from PMB is obtained). The CTEP assigned protocol number must be used for ordering all CTEP supplied investigational agents. The responsible investigator at each participating institution must be registered with CTEP, DCTD through an annual submission of FDA form 1572 and a CV. If there are several participating investigators at one institution, CTEP supplied investigational agents for the study should be ordered under the name of one lead investigator at that institution. Active CTEP-registered investigators and investigator-designated shipping designees and ordering designees can submit agent requests through the PMB Online Agent Order Processing (OAOP) application https://eapps- ctep.nci.nih.gov/OAOP/. Access to OAOP requires the establishment of a CTEP Identity and Access Management (IAM) account https://eapps-ctep.nci.nih.gov/iam/ and the maintenance of an “active” account status and a “current” password. For questions about drug orders, transfers returns or accountability, call 240/276-6575 Monday through Friday between 8:30 am and 4:30 pm (ET) or e-mail [email protected] any time.
2. Drug Handling and Accountability
Drug Accountability: The investigator, or a responsible party designated by the investigator, must maintain a careful record of the receipt, disposition and return or disposal of all drugs received from the PMB using the NCI Drug Accountability Record Form (DARF) available on the NCI home page (http://ctep.cancer.gov).
Electronic logs are allowed as long as a print version of the log process is the exact same appearance as the current NCI DARF.

S1933 Page 23
Version Date 6/1/2021
3. Drug Return and/or Disposition Instruction
Drug Returns: All unused drug supplies must be returned to the PMB. When it is necessary to return study drug (e.g., sealed vials remaining when expired vials are recalled by the PMB), investigators should return the study drug to the PMB using the NCI Return Agent Form available on the NCI home page (http://ctep.cancer.gov).
Drug expiration: If packaging has expiration date, indicate drug expiration date on the DARF under Manufacturer and Lot # and use the drug lots with shorter expiration date first.
4. Contact Information
Useful Links and Contacts
• CTEP Forms, Templates, Documents: http://ctep.cancer.gov/forms/
• RCR Help Desk: [email protected] • PMB policies and guidelines:
http://ctep.cancer.gov/branches/pmb/agent_management.htm • PMB Online Agent Order Processing (OAOP) application:
https://eapps-ctep.nci.nih.gov/OAOP/pages/login.jspx • CTEP Identity and Access Management (IAM) account:
https://ctepcore.nci.nih.gov/iam/index.js p • CTEP Associate Registration and IAM account help:
[email protected] • PMB IB Coordinator: [email protected] • PMB e-mail: [email protected]
PMB phone and hours of service: 240/276-6675 Monday through Friday between 8:30 am and 4:30 pm (ET)
4.0 STAGING CRITERIA
Patients must have Stage II or III disease as outlined below (AJCC Cancer Staging Manual, 8th Edition, 2017): Stage IIA: T2b N0 M0 Stage IIB T1a-c N1 M0 T2a N1 M0 T2b N1 M0 T3 N0 M0 Stage IIIA T1a-c N2 M0 T2a-b N2 M0 T3 N1 M0 T4 N0 M0 T4 N1 M0 Stage IIIB T1a-c N3 M0 T2a-b N3 M0 T3 N2 M0 T4 N2 M0 Stage IIIC T3 N3 M0 T4 N3 M0

S1933 Page 24
Version Date 6/1/2021
T: Primary tumor
T1 Tumor ≤3 cm in greatest dimension surrounded by lung or visceral pleura without
bronchoscopic evidence of invasion more proximal than the lobar bronchus (i.e., not in the main bronchus)*
T1a(mi) Minimally invasive adenocarcinoma T1a Tumor ≤1 cm in greatest dimension* T1b Tumor >1 cm but ≤2 cm in greatest dimension* T1c Tumor >2 cm but ≤3 cm in greatest dimension* T2 Tumor >3 cm but ≤5 cm or tumor with any of the following features: Involves main
bronchus regardless of distance from the carina but without involvement of the carina, invades visceral pleura, associated with atelectasis or obstructive pneumonitis that extends to the hilar region, involving part or all of the lung.
T2a Tumor >3 cm but ≤4 cm in greatest dimension T2b Tumor >4 cm but ≤5 cm in greatest dimension T3 Tumor >5 cm but ≤7 cm in greatest dimension or associated with separate tumor nodule(s)
in the same lobe as the primary tumor or directly invades any of the following structures: chest wall (including the parietal pleura and superior sulcus tumors), phrenic nerve, parietal pericardium
T4 Tumor >7 cm in greatest dimension or associated with separate tumor nodule(s) in a different ipsilateral lobe than that of the primary tumor or invades any of the following structures: diaphragm, mediastinum, heart, great vessels, trachea, recurrent laryngeal nerve, esophagus, vertebral body, and carina
N: Regional lymph node involvement N0 No regional lymph node metastasis N1 Metastasis in ipsilateral peribronchial and/or ipsilateral hilar lymph nodes and
intrapulmonary nodes, including involvement by direct extension N2 Metastasis in ipsilateral mediastinal and/or subcarinal lymph node(s) N3 Metastasis in contralateral mediastinal, contralateral hilar, ipsilateral or contralateral
scalene, or supraclavicular lymph node(s) M: Distant metastasis M0 No distant metastasis
5.0 ELIGIBILITY CRITERIA
Each of the criteria in the following section must be met in order for a patient to be considered eligible for registration in OPEN. Section 5 may be printed and used by the site, but is not to be uploaded in RAVE (unless specially stated). For each criterion requiring test results and dates, please record this information on the Onstudy Form and submit via Medidata Rave® (see Section 14.0). Any potential eligibility issues should be addressed to the SWOG SDMC in Seattle at 206/652-2267 or [email protected] prior to registration. NCI policy does not allow for waiver of any eligibility criterion (http://ctep.cancer.gov/protocolDevelopment/policies_deviations.htm).
In calculating days of tests and measurements, the day a test or measurement is done is considered Day 0. Therefore, if a test is done on a Monday, the Monday 4 weeks later would be considered Day 28. This allows for efficient patient scheduling without exceeding the guidelines. If Day 14, 28, 42, or 90 falls on a weekend or holiday, the limit may be extended to the next working day.

S1933 Page 25
Version Date 6/1/2021
5.1 Registration Step 1
Disease Related Criteria
a. Participants must have pathologic (cytological or histological) proof of non-small
cell lung cancer (NSCLC).
b. Participants must have Stage III NSCLC with Zubrod Performance Status of 2 or Stage II NSCLC with Zubrod Performance Status of 0-2 (see Sections 4.0 and 10.4).
c. Participants must not be candidates for surgical resection in the opinion of the
treating investigator. Participants whose disease was previously resected must have experienced local or regional recurrence at least 12 months after resection.
d. Participants must not be candidates for concurrent chemoradiation in the opinion
of the treating investigator.
e. Participants must have measurable or non-measurable disease (see Section 10.1) documented by CT or MRI. Measurable disease must be assessed within 28 days prior to Registration Step 1. Non-measurable disease must be assessed within 42 days prior to Step 1 registration. The CT from a combined PET/CT may be used only if it is of diagnostic quality as defined in Section 10.1a. All known sites of disease must be assessed and documented on the Baseline Tumor Assessment Form (RECIST 1.1).
f. Participants must have an MRI or CT scan of the brain with contrast within 28 days
prior to Registration Step 1.
g. Participants’ disease must fit within the radiation constraints detailed in Section 7.2 in the opinion of a local radiation oncologist.
Prior/Concurrent Therapy Criteria
h. Participants may have received prior treatment for their lung cancer, including
surgery, chemotherapy, targeted agents, and/or radiation treatment. At least 12 months must have elapsed since last treatment.
i. Participants may have had prior radiation therapy as long as the irradiated area
does not overlap with the radiation field targeted for this study.
Clinical/Laboratory Criteria
j. Participants must have recovered from any adverse effects of prior major surgery to the satisfaction of the treating physician. Biopsies and central IV access placement are not considered major surgery.
k. Participants must have adequate bone marrow function as evidenced by all of the
following: ANC ≥ 1500/mcl; platelet count ≥ 100,000/mcl; and hemoglobin ≥ 9 grams/dL. These results must be obtained within 28 days prior to Registration Step 1.
l. Participants must have adequate liver function as evidenced by the following: total
bilirubin ≤ 1.5 x institutional upper limit of normal (IULN), and aspartate transaminase (AST) and alanine transaminase (ALT) ≤ 2.5 x IULN. These results must be obtained within 28 days prior to Registration Step 1.

S1933 Page 26
Version Date 6/1/2021
m. Participants must have adequate renal function as evidenced by ONE of the following: serum creatinine ≤ 1.5 x IULN OR measured or calculated creatinine clearance ≥ 40 mL/min. This result must have been obtained within 28 days prior to Registration Step 1.
Calculated Creatinine Clearance = (140 - age) X (weight in kg) † 72 x serum creatinine * Multiply this number by 0.85 if the patient is a female. † The kilogram weight is the patient weight with an upper limit of 140% of the IBW. * Actual lab serum creatinine value with a minimum of 0.8 mg/dL.
n. Participants must have percent predicted diffusing capacity of the lungs for carbon monoxide (DLCO) of at least 50% documented within 90 days prior to Registration Step 1.
o. Patient must not have had a prior history of interstitial lung disease or > Grade 2
(CTCAE Version 5) pneumonitis.
p. Participants must not have active autoimmune disease requiring therapy within the past 6 months.
q. Participants must not have an active infection requiring therapy.
r. Participants must be ≥ 18 years old. s. Participants must not be pregnant or nursing because atezolizumab has not been
studied in pregnant or nursing women and the mechanism of action is expected to cause fetal harm. Women/men of reproductive potential must have agreed to use an effective contraceptive method while on protocol treatment and for five months after last dose of atezolizumab. A woman is considered to be of "reproductive potential" if she has had menses at any time in the preceding 12 consecutive months. In addition to routine contraceptive methods, "effective contraception” also includes heterosexual celibacy and surgery intended to prevent pregnancy (or with a side-effect of pregnancy prevention) defined as a hysterectomy, bilateral oophorectomy or bilateral tubal ligation. However, if at any point a previously celibate patient chooses to become heterosexually active during the time period for use of contraceptive measures outlined in the protocol, he/she is responsible for beginning contraceptive measures.
t. Participants with known human immunodeficiency virus (HIV) infection must be on
effective anti-retroviral therapy and must have undetectable viral load at their most recent viral load test and within 6 months prior to Registration Step 1.
u. Patient must be tested for hepatitis B within 28 days prior to Registration Step 1.
Patient must not have active (chronic or acute) hepatitis B virus (HBV) infection. Patients may have past or resolved HBV infection.
Active HBV is defined as having a positive hepatitis B surface antigen (HBsAg) test. Past or resolved HBV is defined as having a negative HBsAG test and a positive total hepatitis B core antibody (HBcAb) test.
v. Patients must not have active hepatitis C virus (HCV) infection. Active HCV is
defined as having a positive HCV antibody test followed by a positive HCV RNA test. Patient must have an HCV antibody test within 28 days prior to Registration

S1933 Page 27
Version Date 6/1/2021
Step 1. If the HCV antibody test is positive, the patient must also have an HCV quantitative RNA test within 28 days prior to Registration Step 1.
w. No other prior malignancy is allowed except for the following: adequately treated
basal cell or squamous cell skin cancer, in situ cervical cancer, adequately treated Stage I or II cancer from which the patient is currently in complete remission, or any other cancer from which the patient has been disease free for three years. Participants with localized prostate cancer who are being followed by an active surveillance program are also eligible.
Specimen Submission Criteria
x. Participants must be offered optional participation in banking of specimens for
future research as described in Section 15.1.
Regulatory Criteria
y. Participants must be informed of the investigational nature of this study and must sign and give written informed consent in accordance with institutional and federal guidelines.
z. As a part of the OPEN registration process (see Section 13.3 for OPEN access
instructions) the treating institution's identity is provided in order to ensure that the current (within 365 days) date of institutional review board approval for this study has been entered in the system.
5.2 Registration Step 2
a. Participants must be registered to Step 2 within 42 days after completion of radiation
treatment. Participants must have received at least 44 Gy of radiation treatment.
b. Participants must have no evidence of progression per RECIST 1.1 on CT scan of the chest, abdomen, and pelvis performed between 2 and 5 weeks after completion of radiation therapy.
c. Any toxicities from radiation therapy must have resolved to < Grade 2.
d. Participants must have adequate bone marrow function as evidenced by all of the
following: ANC ≥ 1500/mcl; platelet count ≥ 100,000/mcl; and hemoglobin ≥ 9 grams/dL. These results must be obtained within 28 days prior to Registration Step 2.
e. Participants must have adequate liver function as evidenced by the following: total bilirubin ≤ 1.5 x institutional upper limit of normal (IULN), and AST and ALT ≤ 2.5 x IULN. These results must be obtained within 28 days prior to Registration Step 2.
f. Participants must have adequate renal function as evidenced by ONE of the following: serum creatinine ≤ 1.5 x IULN OR measured or calculated creatinine clearance ≥ 40 mL/min. This result must have been obtained within 28 days prior to Registration Step 2. Calculated Creatinine Clearance = (140 - age) X (weight in kg) † 72 x serum creatinine * Multiply this number by 0.85 if the patient is a female. † The kilogram weight is the patient weight with an upper limit of 140% of the IBW. * Actual lab serum creatinine value with a minimum of 0.8 mg/dL.

S1933 Page 28
Version Date 6/1/2021
g. Participants must not have received steroids in doses of more than prednisone 10
mg daily or equivalent within 14 days prior to Registration Step 2.
h. Participants must not have received a live vaccine within 28 days prior to Registration Step 2.
6.0 STRATIFICATION FACTORS
This study does not include any stratification factors.
7.0 TREATMENT PLAN
For radiation treatment questions, please contact Dr. Timur Mitin or Dr. Roy Decker (contact information on title page). For atezolizumab treatment or dose modification questions, please contact Dr. Raid Aljumaily or Dr. Antoinette Wozniak (contact information on title page). For dosing principles or questions, please consult the SWOG Policy #38 "Dosing Principles for Participants on Clinical Trials" at https://www.swog.org/sites/default/files/docs/2017-11/Policy38.pdf. 7.1 Treatment Overview
Following registration to Step 1, participants will receive radiation therapy as described in Section 7.2. Between two and five weeks following completion of radiation therapy, disease assessment will be performed using the same modality as baseline. Participants with no disease progression who meet the remaining eligibility criteria in Section 5.2 will be registered to Step 2 and receive atezolizumab as described in Section 7.3.
7.2 Radiation Therapy (Registration Step 1)
a. Timing of Radiation Therapy
Radiation treatment must begin within 28 calendar days after Step 1 Registration. Simulation can take place before registration, but treatment must begin within 3 weeks after simulation.
b. Technical Factors
Photon beams produced by linear accelerators with photon energies 6-10 MV will be allowed. Only IMRT/VMAT modes of treatment are allowed. Proton beams, Cobalt-60 and other charged particle beams are not allowed. Multi-leaf collimation (MLC) or individually-shaped custom blocks should be used to protect normal tissues outside of the target volume.
c. Localization, Simulation and Immobilization
Participants will be positioned supine in a stable but comfortable position. Any immobilization system may be used. A volumetric four-dimensional (4-D) treatment planning CT study is required to define gross tumor volume (GTV), clinical target volume (CTV), internal target volume (ITV) and planning target volume (PTV). Contiguous CT slices of 3 mm or less thickness should be obtained starting from the level of cricoid cartilage and extending inferiorly through the entire lung volume. IV contrast during planning CT is encouraged but optional, if a diagnostic chest CT was done with contrast to delineate the major blood vessels.

S1933 Page 29
Version Date 6/1/2021
A treatment planning PET/CT with the Participant in the treatment position is encouraged for treatment planning.
d. Image-guidance, adaptive radiation therapy, and motion control
The use of four-dimensional (4D) radiation treatment planning is required. Daily cone-beam computed tomography (CBCT) will be used for image-guided radiation therapy. With daily imaging, the change in tumor size during the course of treatment can be easily monitored. At physician discretion, the treatment plan can be modified if enough response is attained to meaningfully reduce dose to normal tissue. One of the motion control techniques (abdominal compression, accelerator beam gating with the respiratory cycle, tumor tracking, and active breath-holding) is mandatory if the tumor motion is determined to be > 1 cm as determined by the 4D simulation planning scan. Of note, NCTN studies, including RTOG 0915, have used similar techniques and guidelines.
e. Dose Fractionation
Participants will receive 4 Gy per fraction for 15 fractions of radiation. All fields must be treated daily. In the event all fields cannot be treated on the same day (equipment malfunction, or other reasons), the remaining non-delivered radiation will be given as a separate fraction at the end of the treatment.
f. Normalization and Prescribing Dose
The prescription dose will cover at least 95% of the PTV60. 90% of the prescription dose should cover at least 99% of the PTV60. All radiation doses will be calculated with tissue density (heterogeneity) corrections that take into account the density differences within the irradiated volume (air in the lung and bone). A list of acceptable heterogeneity dose calculation algorithms can be found at http://rpc.mdanderson.org/RPC/Services/Anthropomorphic_%20Phantoms/TPS%20-%20algorithm%20list%20updated.pdf. Pencil beam and Clarkson’s method are not allowed as heterogeneity correction algorithms.
g. Target Volumes
The primary tumor and clinically and/or pathologically involved lymph nodes as determined by diagnostic and/or planning CT (> 1 cm short axis) or pretreatment PET scan judged involved or suspicious by diagnostic radiologist), or a pathologically involved lymph node station based on EBUS or mediastinoscopy will make up the Gross Tumor Volume (GTV). This volume may be disjointed. The GTV will be expanded into a clinical target volume (CTV) by adding between 5 and 10 mm in any direction with trimming expansions into normal structures and bone. Uninvolved lymph nodes will not be targeted electively. The motion quantified from the 4-dimensional (4-D) scan will be added to the CTV to constitute the ITV. Maximum Intensity Projection Image (MIP) from simulation with regular comfortable breathing will be used to determine the ITV. If the motion is greater than 1 cm in any direction, special maneuvers such as abdominal compression, gating, chasing or regimented breath hold will be required to reduce the final motion below 1 cm. ITV will be expanded by 5 mm in all directions to create the PTV. These techniques and guidelines have been used in previous NCTN studies including RTOG 0915. PTV60 is PTV as created above minus select Organs at Risk with expansion margins as following:

S1933 Page 30
Version Date 6/1/2021
OARs=spinal cord + 10 mm; esophagus + 5 mm, trachea + 3 mm, heart + 3 mm, brachial plexus + 5 mm, great vessels + 3 mm, rib + 3 mm, skin + 3 mm PTV45 is PTV without subtraction of Organs at Risk (OARs)
h. Critical Structures and Constraints
Structures will be named according to the recommendations of AAPM Task Group 263. The following table indicates the standard names to be used for organs at risk as well as the dose constraints to be used for planning.
Standard Name Description DVH Metric Constraint Endpoint (≥Grade 3)
Serial Tissues Spinal Cord Spinal Cord D5cc[Gy] <39 Gy myelitis
D0.03cc[Gy]* <42.3 Gy Esophagus Esophagus D5cc[Gy] <51.3 Gy stenosis/
fistula D0.03cc[Gy]* <55.3 Gy Brachial Plexus Brachial Plexus D3cc[Gy] <44.5 Gy neuropathy
D0.03cc[Gy]* <50.6 Gy Heart Heart/Pericardium D15cc[Gy] <39.5 Gy pericarditis
D0.03cc[Gy]* <60 Gy GreatVes Great Vessels D10cc[Gy] <48.9 Gy aneurysm
D0.03cc[Gy]* <60 Gy Trachea Trachea and Large
Bronchus D5cc[Gy] <39.5 Gy stenosis/
fistula D0.03cc[Gy]* <60 Gy Rib Rib D5cc[Gy] <48.9 Gy pain or fracture
D0.03cc[Gy]* <60 Gy Skin Skin D10cc[Gy] <49 Gy ulceration
D0.03cc[Gy]* <55.4 Gy
Parallel Tissues
Lungs Lungs (Right and Left minus GTV)
D1500cc[Gy] <15.5 Gy
Basic Lung Function Pneumonitis D1000cc[Gy] <16.3 Gy
Mean Dose <18 Gy V18Gy[%] <37%
*A maximum point dose is defined as the highest dose to 0.03 cc of tissue within the critical structure.
i. Normal Tissue Structures Contouring
Spinal Cord: Spinal cord is contoured based on the bony limits of the spinal canal, starting at least 10 cm above and superior extent of the PTV and continuing on every CT slice to at least 10 below the inferior extent of the PTV. Lungs minus GTV: Contour right and left lung as one structure including all parenchymal lung tissue but excluding the GTV and major airways (trachea, main and lobar bronchi).

S1933 Page 31
Version Date 6/1/2021
Esophagus: Contour on mediastinal windowing starting at least 10 cm above the superior extent of PTV and continuing on every CT slice to at least 10 cm below the inferior extent of the PTV. Brachial Plexus: Contour brachial plexus starting proximally at the neuroforamina on the involved side from C5 down to T2, following along the route of the subclavian vein ending after it crosses the second rib. Heart: Contour heart, along with the pericardial sac, from its base to apex, beginning superiorly at the level of the inferior aspect of the aortic arch. Great Vessels: Contour the wall and lumen of the named vessel at least 10 cm superior and inferior to the PTV. Trachea and Large Bronchus: Contour the trachea and cartilage rings starting 10 cm superior to the PTV extending inferiorly to the bronchi ending at the first bifurcation of the named lobar bronchus. Rib: contour each rib separately within 5 cm of the PTV in any direction Skin: Skin is defined as the outer 0.5 cm of the body surface.
j. Planning Radiation Therapy
Priorities: Participant cannot be treated on the protocol if the dosimetric goals for the spinal cord are not met. Meeting protocol metrics for the Organs at Risk should be prioritized over meeting PTV60 and PTV45 protocol metrics. Deviations: Per Protocol: ≥ 95% of the PTVs receives the prescribed dose, ≥ 99% of the PTVs receives ≥ 90% of the prescribed dose, and a contiguous volume of no more than 2 cc anywhere within the Participant receives no more than 66 Gy. Spinal cord constraints are met. Constraints for Organs at risk listed in Table below are met. Variation Acceptable: > 95% of the PTVs receives ≥ 97% of the prescribed dose, ≥ 99% of the PTVs receives ≥ 87% of the prescribed dose, or a contiguous volume of no more than 2 cc anywhere in the body receives >66 Gy but <69 Gy. Spinal cord constraints are met. Constraints for Organs at risk listed in Table below are exceeded by no more than 10%.

S1933 Page 32
Version Date 6/1/2021
The compliance criteria are summarized in the following table.
Compliance Criteria Target/OAR Metric Per Protocol Variation
Acceptable Deviation Unacceptable
PTV60
D95%[%] >100% of protocol dose
>97% of protocol dose
<97% of protocol dose
D99%[%] >90% of protocol dose
>87% of protocol dose
<87% of protocol dose
D2cc[%] <110% of protocol dose
>110% of protocol dose
>115% of protocol dose
PTV45 D95%[%] >100% of
protocol dose >97% of protocol dose
<97% of protocol dose
D99%[%] >90% of protocol dose
>87% of protocol dose
<87% of protocol dose
Spinal Cord
D5cc[Gy] <39 Gy >39 Gy D0.03cc[Gy]* <42.3 Gy >42.3 Gy
Brachial Plexus D3cc[Gy] <44.5 Gy <49.0 Gy >49.0 Gy D0.03cc[Gy]* <50.6 Gy <55.7 Gy >55.7 Gy
Lungs (Right and Left minus GTV)
D1500cc[Gy] <15.5 Gy <17.1 Gy >17.1 Gy D1000cc[Gy] <16.3 Gy <17.9 Gy >17.9 Gy Mean Dose <18 Gy <19.8 Gy >19.8 Gy V18 Gy <37% <40.7% >40.7%
Heart D15cc[Gy] <39.5 Gy <43.5 Gy >43.5 Gy D0.03cc[Gy]* <60.0 Gy <66.0 Gy >66.0 Gy
Esophagus D5cc[Gy] <51.3 Gy <56.4 Gy >56.4 Gy D0.03cc[Gy]* <55.3 Gy <60.8 Gy >60.8 Gy
Great Vessels D10cc[Gy] <48.9 Gy <53.8 Gy >53.8 Gy D0.03cc[Gy]* <60.0 Gy <66.0 Gy >66.0 Gy
Trachea D5cc[Gy] <39.5 Gy <43.5 Gy >43.5 Gy D0.03cc[Gy]* <60.0 Gy <66.0 Gy >66.0 Gy
Rib D5cc[Gy] <48.9 Gy <53.8 Gy >53.8 Gy D0.03cc[Gy]* <60.0 Gy <66.0 Gy >66.0 Gy
Skin D10cc[Gy] <49 Gy <53.9 Gy >53.9 Gy D0.03cc[Gy]* <55.4 Gy <60.9 Gy >60.9 Gy
k. Documentation Requirements
Treatment interruptions should be avoided by preventative medical measures and nutritional, psychological and emotional support. Treatment breaks, including indications, length, must be clearly documented on the treatment record. Any re-simulation and re-planning (both due to anatomy changes as well as for technical reasons, such as switching participants to a different Linac, replacing a ruptured body bag, etc.) should be clearly documented on the treatment record with explanation.
l. Submission of treatment plans
Digital submission of treatment plans is required, including 4DCT, planning CT, RT plan, RT dose and structure set.

S1933 Page 33
Version Date 6/1/2021
m. Radiation Quality Assurance Review
IROC will perform a rapid review of each treatment plan. Institutions should allow 5 business days for each case to be received, processed, and reviewed. If the plan must be resubmitted it will be given a rapid review (within 3 business days). All treatment plans (CT images, contours and dose distribution), will be submitted to IROC Rhode Island electronically, using an encrypted transmission. See Section 12 for submission instructions and for the list of items to be submitted.
7.3 Atezolizumab (Registration Step 2)
After registration to Step 2, participants will receive atezolizumab consolidation. Treatment should begin within 7 calendar days after Registration Step 2. NOTE: Atezolizumab cannot be ordered until the patient is registered to Step 2. To ensure drug supplies are received in time to treat within the 7-day window, sites should order atezolizumab immediately after registration to Step 2 and coordinate carefully with the Pharmaceutical Management Branch.
a. Atezolizumab will be given as follows:
Agent Dose Route Schedule*
Atezolizumab
1200 mg
IV over 60 minutes
Day 1 of each cycle for up to 12 months regardless of dose delays (maximum of 17 cycles)
*One cycle = 21 days
Refer to the preparation instructions in Section 3.1f.
Atezolizumab is administered as an intravenous infusion over 60 minutes. If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes. Do not administer atezolizumab as an intravenous push or bolus. No premedication is indicated for administration of Cycle 1 of atezolizumab. Patients who experience an infusion related reaction with Cycle 1 of atezolizumab may receive premedication with antihistamines or antipyretics/analgesics (e.g. acetaminophen) for subsequent infusions. Atezolizumab treatment should be administered after all procedures and assessments have been completed. Atezolizumab treatment may be administered up to 3 days before or after the protocol-specified Q 3 week interval.
Atezolizumab treatment will be administered on an outpatient basis. No premedication is indicated for the administration of Cycle 1 of atezolizumab. However, participants who experience an infusion-related reaction (IRR) with Cycle 1 of atezolizumab may receive premedication with antihistamines or antipyretics/analgesics (e.g., acetaminophen) for subsequent infusions. Metamizole (dipyrone) is prohibited in treating atezolizumab-associated IRRs because of its potential for causing agranulocytosis.

S1933 Page 34
Version Date 6/1/2021
b. Infusion Related Reactions
Administration of atezolizumab must be performed in a setting with emergency medical facilities and staff who are trained to monitor for and respond to medical emergencies. For the first infusion, the participant’s vital signs (heart rate, respiratory rate, blood pressure, and temperature) should be determined within 60 minutes before, during (every 15 [±5] minutes), and 30 (±10) minutes after the infusion. For subsequent infusions, vital signs will be collected within 60 minutes before and within 30 minutes after the infusion. Vital signs should be collected during the infusion only if clinically indicated. Participants will be informed about the possibility of delayed post-infusion symptoms and instructed to contact their study physician if they develop such symptoms. The management of Infusion Related Reactions will be according to severity as follows:
• In the event that a participant experiences a Grade 1 Infusion Related Reaction during Cycle 1, the infusion rate should be reduced to half the rate being given at the time of event onset. Once the event has resolved, the investigator should wait for 30 minutes while delivering the infusion at the reduced rate. If tolerated, the infusion rate may then be increased to the original rate.
• In the event that a participant experiences a Grade 2 Infusion Related
Reaction, or flushing, fever, or throat pain, the infusion should be immediately interrupted, and the participant should receive aggressive symptomatic treatment. The infusion should be restarted only after the symptoms have adequately resolved to baseline grade. The infusion rate at restart should be half of the infusion rate that was in progress at the time of the onset of the Infusion Related Reaction. For subsequent infusions, administer oral premedication with antihistamine and anti-pyretic and monitor closely for Infusion Related Reactions.
• For Grade 3 or 4 Infusion Related Reactions, the infusion should be
stopped immediately, and aggressive resuscitation and supportive measures should be initiated (e.g., oral or IV antihistamine, anti-pyretic, glucocorticoids, epinephrine, bronchodilators, oxygen). Atezolizumab should be permanently discontinued. Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-related event; retreatment requires consultation with, and consent of, the Study Chair.
For anaphylaxis precautions, use the following procedure: Equipment Needed
• Tourniquet • Oxygen • Epinephrine for subcutaneous, intravenous, and/or endotracheal use in
accordance with standard practice • Antihistamines • Corticosteroids • Intravenous infusion solutions, tubing, catheters, and tape

S1933 Page 35
Version Date 6/1/2021
Procedures In the event of a suspected anaphylactic reaction during atezolizumab infusion, the following procedures should be performed:
1. Stop the study drug infusion. 2. Apply a tourniquet proximal to the injection site to slow systemic
absorption of study drug. Do not obstruct arterial flow in the limb. 3. Maintain an adequate airway. 4. Administer antihistamines, epinephrine, or other medications as required
by Participant status and directed by the physician in charge. 5. Continue to observe the participant and document observation.
7.4 Prohibited and cautionary medications
Participants are prohibited from receiving the following therapies while on protocol treatment:
• Anti-cancer systemic chemotherapy or biological therapy. • Immunotherapy not specified in this protocol. • Any non-study anti-cancer agent (investigational or non-investigational). • Investigational agents other than atezolizumab. • Radiation therapy other than protocol-specified radiation therapy • Live vaccines: Examples of live vaccines include, but are not limited to, the
following: measles, mumps, rubella, chicken pox, shingles, yellow fever, rabies, BCG, and typhoid (oral) vaccine. Seasonal influenza vaccines for injection are generally killed virus vaccines and are allowed; however, intranasal influenza vaccines (e.g. Flu-Mist®) are live attenuated vaccines, and are not allowed.
• Glucocorticoids long term use (more than 21 days) in doses higher than prednisone 10 mg daily or equivalent for any purpose other than to modulate symptoms from an event of suspected immunologic etiology. The use of physiologic doses of corticosteroids (defined as 10 mg prednisone or equivalent) are acceptable, however site investigators must consult with the Study Chair for any dose higher than 10 mg prednisone or equivalent.
Metamizole (dipyrone) is prohibited in treating atezolizumab-associated IRRs because of its potential for causing agranulocytosis. Participants who, in the assessment by the investigator, require the use of any of the aforementioned treatments for clinical management must be removed from protocol treatment. Participants may receive other medications that the investigator deems to be medically necessary.
7.5 Disease Assessment
CT or MRI (the same method used at pre-study to meet the eligibility criteria in Section 5.1) must be done 2-5 weeks after completion of radiotherapy. The same imaging studies are to be done 9 weeks (± 7 days) after Registration Step 2, then every 9 weeks (± 7 days) while on atezolizumab. After discontinuation of atezolizumab and prior to progression, the same imaging studies are to be done every 12 weeks (± 14 days) until the end of the second year after Registration Step 2, then every 6 months until the end of the third year. Participants who do not go on to Registration Step 2 must have imaging studies performed every 9 weeks (± 7 days) until progression.

S1933 Page 36
Version Date 6/1/2021
7.6 Complete DMU Reporting Requirement Because this study contains an investigational drug for which CTEP holds the IND, it falls under CTEP requirements for complete reporting. This involves required submission of cycle- specific toxicity and dose information (see Section 14, the S1933 Treatment Form, and the S1933 Adverse Event Form). A cycle is defined as 21 days.
7.7 Criteria for Removal from Protocol Treatment
a. Criteria for Removal from Protocol Treatment before Registration Step 2
1. Progression of disease or symptomatic deterioration (as defined in Section 10.2)
2. Radiation treatment delay > 28 days due to side effects related to
radiotherapy
3. The participant may withdraw from the study at any time for any reason.
4. The investigator may discontinue treatment if they determine that the participant’s continued treatment on the study is detrimental to their long-term health, or due to poor compliance with the study’s required visits and treatments.
b. Criteria for Removal from Protocol Treatment after Registration Step 2
1. Progression of disease or symptomatic deterioration (as defined in Section 10.2). However, the participant may continue protocol treatment as long as the participant is continuing to clinically benefit from treatment in the opinion of the treating investigator. Participants should still be removed from protocol treatment for criteria below.
2. Unacceptable toxicity 3. Atezolizumab treatment delay > 84 days. If atezolizumab is being held due
to an adverse event and the AE resolves within 84 days and the participant is receiving corticosteroid therapy for the event, atezolizumab may be held for up to 28 additional days in order to allow tapering of the steroid dose to < 10 mg oral prednisone or equivalent.
4. The participant may withdraw from the study at any time for any reason. 5. The investigator may discontinue treatment if they determine that the
participant’s continued treatment on the study is detrimental to their long-term health, or due to poor compliance with the study’s required visits and treatment.
6. Completion of protocol treatment.
7.8 Discontinuation of Treatment
All reasons for discontinuation of treatment must be documented in the Off Protocol Treatment Notice.
7.9 Follow-Up Period
All participants will be followed until death or 3 years after Registration Step 1.

S1933 Page 37
Version Date 6/1/2021
8.0 TOXICITIES TO BE MONITORED AND DOSE MODIFICATIONS
8.1 NCI Common Terminology Criteria for Adverse Events
This study will utilize the CTCAE (NCI Common Terminology Criteria for Adverse Events) Version 5.0 for toxicity and Serious Adverse Event reporting. A copy of the CTCAE Version 5.0 can be downloaded from the CTEP home page (http://ctep.cancer.gov). All appropriate treatment areas should have access to a copy of the CTCAE Version 5.0.
8.2 Radiation-Related Toxicities
Radiotherapy should be interrupted for Grade 4 in-field toxicity and resumed when that toxicity has decreased to Grade ≤ 2 as detailed below. If treatment is interrupted for > 28 days, the participant should be removed from study treatment. Reversible or permanent alopecia, bone marrow toxicity, skin pigmentation, and esophagitis are expected side effects of radiation therapy. Radiation induced myocarditis or transverse myelitis rarely occur at doses lower than 50 Gy. Radiographic evidence of radiation change and subsequent fibrosis of the lung will occur within lung volume receiving ≥ 20 Gy, usually within the first six months after initiation of treatment. It is essential to spare as much normal lung as possible in order to avoid symptomatic lung injury.
a. Esophagitis
Esophageal complaints are common with thoracic radiation therapy. Esophagitis does not constitute a reason to interrupt or delay radiotherapy provided oral intake is sufficient to maintain hydration. Participants should be advised to avoid alcoholic, acidic, or spicy foods or beverages. Viscous Xylocaine, Carafate, or other medications should be used for symptomatic relief. Occasionally, narcotics may be required. It is not necessary to biopsy acute esophagitis in the first 2 weeks of therapy since it is rarely due to underlying viral or fungal disease. Acute esophagitis may persist for 4-6 weeks. If Grade 3 or 4 esophagitis occurs, and a treatment interruption is being considered, every effort should be made to limit it to 3 treatment days or less. Participants requiring hospitalization because of esophagitis may have their treatment interrupted. In this event, immediately notify the protocol PI. Treatment should be interrupted for Grade 4 (CTCAE Version 5.0) dysphagia or odynophagia. Acute esophageal toxicity, which typically can occur within two weeks of the initiation of treatment and manifests as dysphagia, odynophagia, reflux symptoms, etc. should be pharmacologically managed with the following approach and should be initiated at the first signs or symptoms of esophageal toxicity. Suggested treatments include the following:
• Ketoconazole 200 mg PO q day OR Fluconazole 100 mg PO q day until the completion of radiation.
• Mixture of 2% viscous lidocaine: 60 cc, Mylanta: 30 cc, sucralfate (1 gm/cc): 10 cc; Take 15-30 cc PO q3-4 hrs prn. (Contraindications: pts on Dilantin, Cipro, Digoxin)
• Ranitidine 150 mg PO BID (or other H2 blocker or a proton pump inhibitor such as omeprazole) until the completion of radiation
• Grade 4 esophagitis: hold XRT until Grade 2 or less. We expect a significant portion of participants will experience Grade 3 esophagitis.

S1933 Page 38
Version Date 6/1/2021
b. Pneumonitis
Radiation pneumonitis is a subacute (weeks to months from treatment) inflammation of the end bronchioles and alveoli. The infiltrate on chest x-ray should include the area treated to high dose, but may extend outside of these regions. The infiltrates may be characteristically “geometric” corresponding to the radiation portal, but may also be ill defined. Participants reporting symptoms as above will be promptly evaluated and treated. Mild radiation pneumonitis may be treated with nonsteroidal anti-inflammatory agents or steroid inhalers. For mild pneumonitis, consider non-steroidal treatment (e.g. 600-800 mg/day Ibuprofen in divided doses) with or without an inhaled steroid (e.g. Vanceril or Azmacort 2 puffs qid). Use H-2 blocker, H+ pump blocker, or Sucralfate for gastric prophylaxis. Consider Bactrim (if not allergic) in severely immunocompromised participants to avoid opportunistic infection. Consider other appropriate antibiotics if cough is productive of greenish sputum or if clinical suspicion indicates bacterial supra-infection within the areas of pneumonitis.
More significant pneumonitis will be treated with systemic steroids, bronchodilators, and pulmonary toilet. Supra- and concurrent infections should be treated with antibiotics. Consideration of prophylaxis of opportunistic infections should be considered in immunocompromised participants.
A suggested course of prednisone is shown below.
Prednisone dose (mg), several times per day Days
20-20-20 2 20-20-15 2 20-15-15 2 15-15-15 2 15-15-10 2 15-10-10 2 10-10-10 2 10-10-5 2 10-5-5 2 5-5-5 2 5-5-2.5 3 5-2.5-2.5 3 2.5-2.5-2.5 3 2.5-2.5 3 2.5 10 STOP Total: 42 days
If participant has return of pneumonitis symptoms during weaning of steroids, go back to dose level where participant was last comfortable, and resume wean at that level. It may be necessary to lengthen interval at each dose (e.g. double from 3 to 6 days at a particular level) in order to successfully wean the participant. Be participant with steroid taper as participants may experience rebound pneumonitis if weaning proceeds too quickly. Periodically check blood sugars, especially in obese participants or those with overt or borderline diabetes.
Properly performed incentive spirometry appears to help re-expand collapsed alveoli. Excessive coughing should be treated with antitussives (e.g. Tessalon Perles 100 tid PRN).

S1933 Page 39
Version Date 6/1/2021
Consider inhaled bronchodilator in participants who have signs of bronchospasm or reactive airways (e.g. Albuterol inhaler 2 puffs qid). It is unlikely that symptomatic pneumonitis will occur during the three weeks of radiation therapy. However, if a participant experiences pneumonitis before completing therapy, therapy will be put on hold until symptoms resolve and PI should be notified immediately for decision on further treatment.
8.3 Dose Interruptions for Atezolizumab
There will be no dose reduction for atezolizumab in this study. Participants may temporarily suspend study treatment for up to 84 days (12 weeks) beyond the scheduled date of delayed infusion if study drug-related toxicity requiring dose suspension is experienced. If atezolizumab is held because of AEs for > 84 days beyond the scheduled date of infusion, the participant will be discontinued from atezolizumab and will be followed for safety and efficacy as specified in this protocol. If the AE resolves within 84 days and the participant is receiving corticosteroid therapy for the event, atezolizumab may be held for an additional 28 days in order to allow tapering of the steroid dose to ≤ 10 mg oral prednisone or equivalent. Therefore, the maximum treatment delay allowed is 112 days. Atezolizumab must be permanently discontinued if the participant experiences any of the following events, regardless of benefit:
• Grade 4 pneumonitis • AST or ALT >5×ULN or total bilirubin >3×ULN • Grade 4 diarrhea or colitis • Grade 4 hypophysitis • Any grade myasthenic syndrome/myasthenia gravis, Guillain-Barré or
meningoencephalitis • Grade 4 ocular inflammatory toxicity • Grade 4 pancreatitis or any grade of recurrent pancreatitis • Grade 4 rash • Any grade myocarditis • Grade 3 or 4 immune-mediated neuropathy • Grade 3 or 4 renal event • Grade 4 immune-mediated myositis
Treatment may, under limited and compelling circumstances, be resumed in participants who have recovered from the following events, but only after consultation with the Study Chair:
• Grade 3 pneumonitis • Grade 3 ocular inflammatory toxicity • Grade 3 or 4 infusion-related reactions
Any toxicity associated or possibly associated with atezolizumab treatment should be managed according to standard medical practice. Additional tests, such as autoimmune serology or biopsies, may be used to determine a possible immunogenic etiology. Although most immune-related adverse events (irAEs) observed with immunomodulatory agents have been mild and self-limiting, such events should be recognized early and treated promptly to avoid potential major complications (Di Giacomo et al., 2010). Discontinuation of atezolizumab may not have an immediate therapeutic effect, and there is no available antidote for atezolizumab. In severe cases, immune-related toxicities may be acutely managed with topical corticosteroids, systemic corticosteroids, or other immunosuppressive agents. The investigator should consider the benefit-risk balance prior to further administration of atezolizumab.

S1933 Page 40
Version Date 6/1/2021
For detailed information regarding management of adverse events associated with atezolizumab, please refer to the most current version of the Atezolizumab Investigator’s Brochure and the FDA product label. The primary approach to Grade 1 to 2 irAEs is supportive and symptomatic care with continued treatment with atezolizumab; for higher-grade irAEs, atezolizumab should be withheld and oral and/or parenteral steroids administered. Recurrent Grade 2 irAEs may also mandate withholding atezolizumab or the use of steroids. Assessment of the benefit-risk balance should be made by the investigator, with consideration of the totality of information as it pertains to the nature of the toxicity and the degree of clinical benefit a given participant may be experiencing prior to further administration of atezolizumab. Atezolizumab should be permanently discontinued in participants with life-threatening irAEs. Participants should be assessed clinically (including review of laboratory values) for toxicity prior to, during, and after each infusion. If unmanageable toxicity due to atezolizumab occurs at any time during the study, treatment with atezolizumab should be discontinued. a. Systemic Immune Activation
Systemic immune activation is a rare condition characterized by an excessive immune response. Given the mechanism of action of atezolizumab, systemic immune activation is considered a potential risk when given in combination with other immunomodulation agents. Systemic immune activation should be included in the differential diagnosis for participants who, in the absence of an alternative etiology, develop a sepsis-like syndrome after administration of atezolizumab, and the initial evaluation should include the following:
• CBC with peripheral smear • PT, PTT, fibrinogen, and D-dimer • Ferritin • Triglycerides • AST, ALT, and total bilirubin • LDH • Complete neurologic and abdominal examination (assess for
hepatosplenomegaly)
If systemic immune activation is still suspected after the initial evaluation, contact the Study Chair for additional recommendations.
b. Management of Specific Adverse Events (AEs)
Toxicities associated or possibly associated with atezolizumab treatment should be managed according to standard medical practice. Additional tests, such as autoimmune serology or biopsies, should be used to evaluate for a possible immunogenic etiology. Although most immune-mediated adverse events observed with immunomodulatory agents have been mild and self-limiting, such events should be recognized early and treated promptly to avoid potential major complications. Discontinuation of atezolizumab may not have an immediate therapeutic effect, and in severe cases, immune-mediated toxicities may require acute management with topical corticosteroids, systemic corticosteroids, or other immunosuppressive agents. The treating investigator should consider the benefit-risk balance a given participant may be experiencing prior to further administration of atezolizumab. In participants

S1933 Page 41
Version Date 6/1/2021
who have met the criteria for permanent discontinuation, resumption of atezolizumab may be considered if the participant is deriving benefit and has fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after approval has been discussed with the Study Chair. Pulmonary Events Dyspnea, cough, fatigue, hypoxia, pneumonitis, and pulmonary infiltrates have been associated with the administration of atezolizumab. Participants will be assessed for pulmonary signs and symptoms throughout the study and will have computed tomography (CT) scans of the chest performed at every tumor assessment. All pulmonary events should be thoroughly evaluated for other commonly reported etiologies such as pneumonia or other infection, lymphangitic carcinomatosis, pulmonary embolism, heart failure, chronic obstructive pulmonary disease, or pulmonary hypertension. Management guidelines for pulmonary events are provided in the following table. Management Guidelines for Pulmonary Events, Including Pneumonitis
Event Management
Pulmonary event, Grade 1
• Continue atezolizumab and monitor closely. • Re-evaluate on serial imaging. • Consider participant referral to pulmonary
specialist. Pulmonary event, Grade 2
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Refer participant to pulmonary and infectious disease specialists and consider bronchoscopy or BAL.
• Initiate treatment with corticosteroids equivalent to 1-2 mg/kg/day oral prednisone.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
• For recurrent events, treat as a Grade 3 or 4 event.
Pulmonary event, Grade 3 or 4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Bronchoscopy or BAL is recommended. • Initiate treatment with corticosteroids equivalent
to 1−2 mg/kg/day oral prednisone. • If event does not improve within 48 hours after
initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month.
BAL= bronchoscopic alveolar lavage. a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after
event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.

S1933 Page 42
Version Date 6/1/2021
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
Hepatic Events Immune-mediated hepatitis has been associated with the administration of atezolizumab. Eligible participants must have adequate liver function, as manifested by measurements of total bilirubin and hepatic transaminases, and liver function will be monitored throughout study treatment. Management guidelines for hepatic events are provided in the following table. Participants with right upper-quadrant abdominal pain and/or unexplained nausea or vomiting should have liver function tests (LFTs) performed immediately and reviewed before administration of the next dose of study drug. For participants with elevated LFTs, concurrent medication, viral hepatitis, and toxic or neoplastic etiologies should be considered and addressed, as appropriate. Management Guidelines for Hepatic Events
Event Management
Hepatic event, Grade 1
• Continue atezolizumab. • Monitor LFTs until values resolve to within normal limits
or to baseline values. Hepatic event, Grade 2
All events: • Monitor LFTs more frequently until return to baseline
values. Events of > 5 days’ duration: • Withhold atezolizumab for up to 12 weeks after event
onset. a • Initiate treatment with corticosteroids equivalent to 1−2
mg/kg/day oral prednisone. • If event resolves to Grade 1 or better, resume
atezolizumab. b • If event does not resolve to Grade 1 or better while
withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
Hepatic event, Grade 3 or 4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Consider participant referral to gastrointestinal specialist for evaluation and liver biopsy to establish etiology of hepatic injury.
• Initiate treatment with corticosteroids equivalent to 1−2 mg/kg/day oral prednisone.
• If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month.
LFT = liver function tests. a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after
event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the

S1933 Page 43
Version Date 6/1/2021
extended period of time must be discussed with the Study Chair. b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the
equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed. c Resumption of atezolizumab may be considered in participants who are deriving
benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
Gastrointestinal Events Immune-mediated colitis has been associated with the administration of atezolizumab. Management guidelines for diarrhea or colitis are provided in the following table. All events of diarrhea or colitis should be thoroughly evaluated for other more common etiologies. For events of significant duration or magnitude or associated with signs of systemic inflammation or acute-phase reactants (e.g., increased C-reactive protein, platelet count, or bandemia): Perform sigmoidoscopy (or colonoscopy, if appropriate) with colonic biopsy, with three to five specimens for standard paraffin block to check for inflammation and lymphocytic infiltrates to confirm colitis diagnosis. Management Guidelines for Gastrointestinal Events (Diarrhea or Colitis)
Event Management Diarrhea or colitis, Grade 1
• Continue atezolizumab. • Initiate symptomatic treatment. • Endoscopy is recommended if symptoms persist for
> 7 days. • Monitor closely.
Diarrhea or colitis, Grade 2
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Initiate symptomatic treatment. • Participant referral to GI specialist is recommended. • For recurrent events or events that persist > 5 days,
initiate treatment with corticosteroids equivalent to 1−2 mg/kg/day oral prednisone.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
Diarrhea or colitis, Grade 3
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Refer participant to GI specialist for evaluation and confirmatory biopsy.
• Initiate treatment with corticosteroids equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c

S1933 Page 44
Version Date 6/1/2021
Event Management Diarrhea or colitis, Grade 4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Refer participant to GI specialist for evaluation and confirmation biopsy.
• Initiate treatment with corticosteroids equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month.
GI = gastrointestinal. a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after
event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
Endocrine Events Thyroid disorders, adrenal insufficiency, diabetes mellitus, and pituitary disorders have been associated with the administration of atezolizumab. Management guidelines for endocrine events are provided in the following table. Participants with unexplained symptoms such as headache, fatigue, myalgias, impotence, constipation, or mental status changes should be investigated for the presence of thyroid, pituitary, or adrenal endocrinopathies. The participant should be referred to an endocrinologist if an endocrinopathy is suspected. Thyroid-stimulating hormone (TSH) and free triiodothyronine and thyroxine levels should be measured to determine whether thyroid abnormalities are present. Pituitary hormone levels and function tests (e.g., TSH, growth hormone, luteinizing hormone, follicle-stimulating hormone, testosterone, prolactin, adrenocorticotropic hormone [ACTH] levels, and ACTH stimulation test) and magnetic resonance imaging (MRI) of the brain (with detailed pituitary sections) may help to differentiate primary pituitary insufficiency from primary adrenal insufficiency.
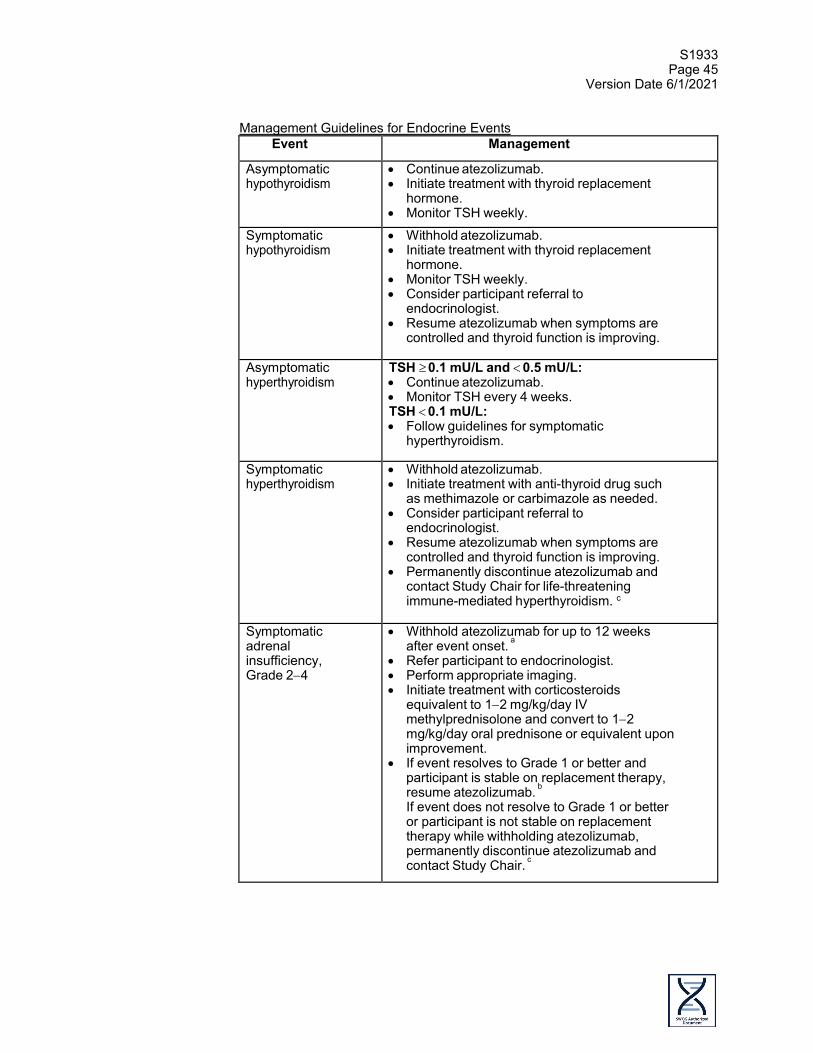
S1933 Page 45
Version Date 6/1/2021
Management Guidelines for Endocrine Events
Event Management
Asymptomatic hypothyroidism
• Continue atezolizumab. • Initiate treatment with thyroid replacement
hormone. • Monitor TSH weekly.
Symptomatic hypothyroidism
• Withhold atezolizumab. • Initiate treatment with thyroid replacement
hormone. • Monitor TSH weekly. • Consider participant referral to
endocrinologist. • Resume atezolizumab when symptoms are
controlled and thyroid function is improving.
Asymptomatic hyperthyroidism
TSH ≥ 0.1 mU/L and < 0.5 mU/L: • Continue atezolizumab. • Monitor TSH every 4 weeks. TSH < 0.1 mU/L: • Follow guidelines for symptomatic
hyperthyroidism.
Symptomatic hyperthyroidism
• Withhold atezolizumab. • Initiate treatment with anti-thyroid drug such
as methimazole or carbimazole as needed. • Consider participant referral to
endocrinologist. • Resume atezolizumab when symptoms are
controlled and thyroid function is improving. • Permanently discontinue atezolizumab and
contact Study Chair for life-threatening immune-mediated hyperthyroidism. c
Symptomatic adrenal insufficiency, Grade 2−4
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Refer participant to endocrinologist. • Perform appropriate imaging. • Initiate treatment with corticosteroids
equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event resolves to Grade 1 or better and participant is stable on replacement therapy, resume atezolizumab. b If event does not resolve to Grade 1 or better or participant is not stable on replacement therapy while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c

S1933 Page 46
Version Date 6/1/2021
Event Management
Hyperglycemia, Grade 1 or 2
• Continue atezolizumab. • Investigate for diabetes. If participant has
Type 1 diabetes, treat as a Grade 3 event. If participant does not have Type 1 diabetes, treat as per institutional guidelines. Monitor for glucose control.
Hyperglycemia, Grade 3 or 4
• Withhold atezolizumab. • Initiate treatment with insulin. • Monitor for glucose control.
Resume atezolizumab when symptoms resolve, and glucose levels are stable.
Hypophysitis (pan-hypopituitarism), Grade 2 or 3
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Refer participant to endocrinologist. • Perform brain MRI (pituitary protocol). • Initiate treatment with corticosteroids
equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• Initiate hormone replacement if clinically indicated.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c For recurrent hypophysitis, treat as a Grade 4 event.
Hypophysitis (pan-hypopituitarism), Grade 4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Refer participant to endocrinologist. • Perform brain MRI (pituitary protocol). • Initiate treatment with corticosteroids
equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement. Initiate hormone replacement if clinically indicated.
IV = intravenous; MRI = magnetic resonance imaging; TSH = thyroid-stimulating hormone.
a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only discussion with the Study Chair.
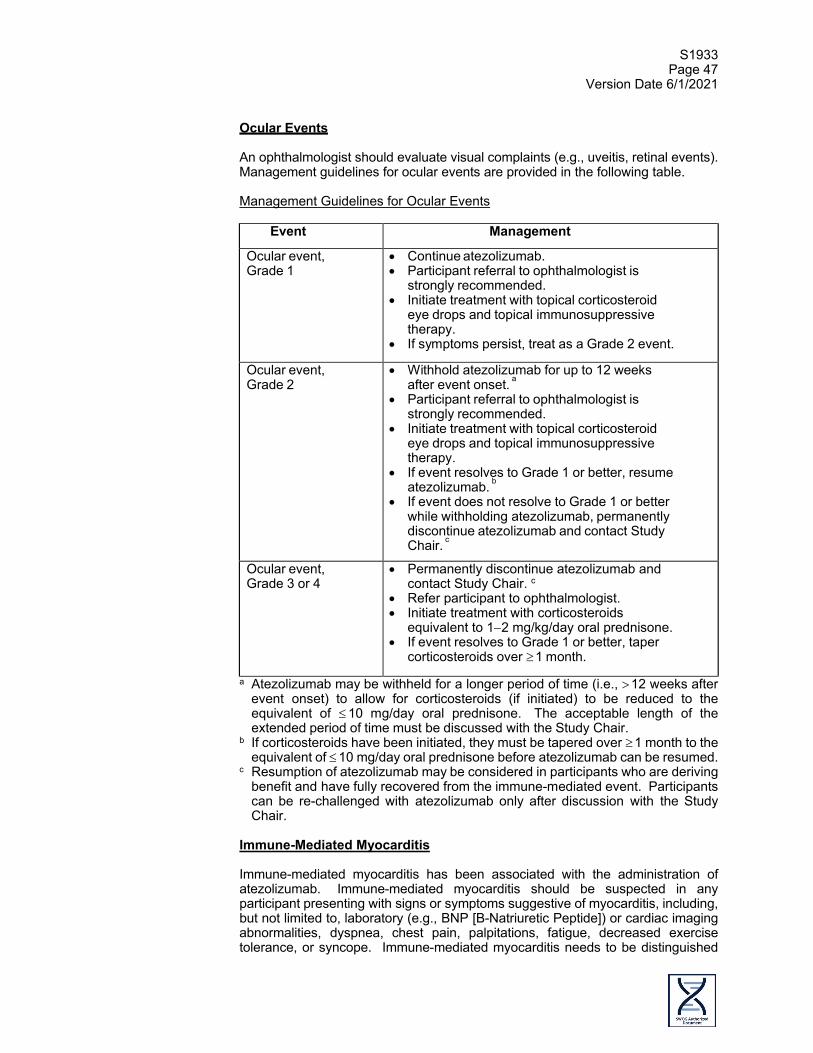
S1933 Page 47
Version Date 6/1/2021
Ocular Events An ophthalmologist should evaluate visual complaints (e.g., uveitis, retinal events). Management guidelines for ocular events are provided in the following table. Management Guidelines for Ocular Events
Event Management
Ocular event, Grade 1
• Continue atezolizumab. • Participant referral to ophthalmologist is
strongly recommended. • Initiate treatment with topical corticosteroid
eye drops and topical immunosuppressive therapy.
• If symptoms persist, treat as a Grade 2 event.
Ocular event, Grade 2
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Participant referral to ophthalmologist is strongly recommended.
• Initiate treatment with topical corticosteroid eye drops and topical immunosuppressive therapy.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
Ocular event, Grade 3 or 4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Refer participant to ophthalmologist. • Initiate treatment with corticosteroids
equivalent to 1−2 mg/kg/day oral prednisone. • If event resolves to Grade 1 or better, taper
corticosteroids over ≥ 1 month.
a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
Immune-Mediated Myocarditis Immune-mediated myocarditis has been associated with the administration of atezolizumab. Immune-mediated myocarditis should be suspected in any participant presenting with signs or symptoms suggestive of myocarditis, including, but not limited to, laboratory (e.g., BNP [B-Natriuretic Peptide]) or cardiac imaging abnormalities, dyspnea, chest pain, palpitations, fatigue, decreased exercise tolerance, or syncope. Immune-mediated myocarditis needs to be distinguished

S1933 Page 48
Version Date 6/1/2021
from myocarditis resulting from infection (commonly viral, e.g., in a participant who reports a recent history of gastrointestinal illness), ischemic events, underlying arrhythmias, exacerbation of preexisting cardiac conditions, or progression of malignancy. All participants with possible myocarditis should be urgently evaluated by performing cardiac enzyme assessment, an ECG, a chest X-ray, an echocardiogram, and a cardiac MRI as appropriate per institutional guidelines. A cardiologist should be consulted. An endomyocardial biopsy may be considered to enable a definitive diagnosis and appropriate treatment, if clinically indicated. Participants with signs and symptoms of myocarditis, in the absence of an identified alternate etiology, should be treated according to the guidelines in the following table. Management Guidelines for Immune-Mediated Myocarditis
Event Management
Immune-mediated myocarditis, Grade 2
• Withhold atezolizumab for up to 12 weeks after event onset a and contact Study Chair.
• Refer participant to cardiologist. • Initiate treatment as per institutional
guidelines and consider antiarrhythmic drugs, temporary pacemaker, ECMO, or VAD as appropriate.
• Consider treatment with corticosteroids equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
Immune-mediated myocarditis, Grade 3-4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Refer participant to cardiologist. • Initiate treatment as per institutional
guidelines and consider antiarrhythmic drugs, temporary pacemaker, ECMO, or VAD as appropriate.
• Initiate treatment with corticosteroids equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 12 mg/kg/day oral prednisone or equivalent upon improvement.
• If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month.
ECMO = extracorporeal membrane oxygenation; VAD = ventricular assist device. a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after
event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the

S1933 Page 49
Version Date 6/1/2021
extended period of time must be discussed with the Study Chair. b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the
equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed. c Resumption of atezolizumab may be considered in participants who are deriving
benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
Infusion-Related Reactions No premedication is indicated for the administration of Cycle 1 of atezolizumab. However, participants who experience an infusion-related reaction (IRR) with Cycle 1 of atezolizumab may receive premedication with antihistamines or antipyretics/analgesics (e.g., acetaminophen) for subsequent infusions. Metamizole (dipyrone) is prohibited in treating atezolizumab-associated IRRs because of its potential for causing agranulocytosis. Guidelines for medical management of IRRs during Cycle 1 are provided in the following table. For subsequent cycles, IRRs should be managed according to institutional guidelines. Management Guidelines for Infusion-Related Reactions
Event Management
IRR, Grade 1 • Reduce infusion rate to half the rate being given at the time of event onset.
• After the event has resolved, the investigator should wait for 30 minutes while delivering the infusion at the reduced rate.
• If the infusion is tolerated at the reduced rate for 30 minutes after symptoms have resolved, the infusion rate may be increased to the original rate.
IRR, Grade 2 • Interrupt atezolizumab infusion. • Administer aggressive symptomatic treatment (e.g.,
oral or IV antihistamine, anti-pyretic medication, glucocorticoids, epinephrine, bronchodilators, oxygen, IV fluids).
• After symptoms have resolved to baseline, resume infusion at half the rate being given at the time of event onset.
• For subsequent infusions, consider administration of oral premedication with antihistamines, anti-pyretics, and/or analgesics and monitor closely for IRRs.
IRR, Grade 3 or 4
• Stop infusion. • Administer aggressive symptomatic treatment (e.g.,
oral or IV antihistamine, anti-pyretic medication, glucocorticoids, epinephrine, bronchodilators, oxygen, IV fluids).
• Permanently discontinue atezolizumab and contact Study Chair. a
IRR = infusion-related reaction. a Resumption of atezolizumab may be considered in participants who are deriving
benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.

S1933 Page 50
Version Date 6/1/2021
Pancreatic Events Symptoms of abdominal pain associated with elevations of amylase and lipase, suggestive of pancreatitis, have been associated with the administration of atezolizumab. The differential diagnosis of acute abdominal pain should include pancreatitis. Appropriate workup should include an evaluation for ductal obstruction, as well as serum amylase and lipase tests. Management guidelines for pancreatic events, including pancreatitis, are provided in the following table.
Management Guidelines for Pancreatic Events, Including Pancreatitis
Event Management
Amylase and/or lipase elevation, Grade 2
Amylase and/or lipase > 1.5−2.0 × ULN: • Continue atezolizumab. • Monitor amylase and lipase weekly. • For prolonged elevation (e.g., > 3
weeks), consider treatment with corticosteroids equivalent to 10 mg/day oral prednisone.
Asymptomatic with amylase and/or lipase > 2.0−5.0 × ULN: • Treat as a Grade 3 event.
Amylase and/or lipase elevation, Grade 3 or 4
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Refer participant to GI specialist. • Monitor amylase and lipase every other
day. • If no improvement, consider treatment
with corticosteroids equivalent to 1−2 mg/kg/day oral prednisone.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
• For recurrent events, permanently discontinue atezolizumab and contact Study Chair. c
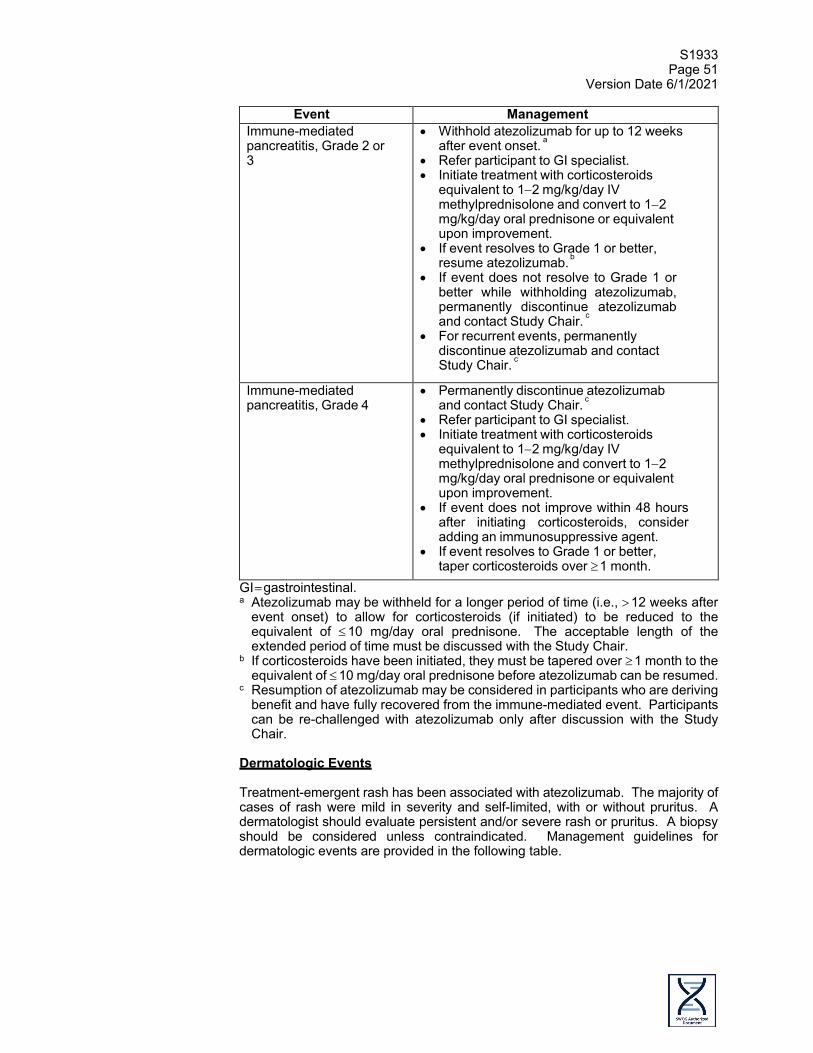
S1933 Page 51
Version Date 6/1/2021
Event Management Immune-mediated pancreatitis, Grade 2 or 3
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Refer participant to GI specialist. • Initiate treatment with corticosteroids
equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
• For recurrent events, permanently discontinue atezolizumab and contact Study Chair. c
Immune-mediated pancreatitis, Grade 4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Refer participant to GI specialist. • Initiate treatment with corticosteroids
equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month.
GI = gastrointestinal. a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after
event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
Dermatologic Events Treatment-emergent rash has been associated with atezolizumab. The majority of cases of rash were mild in severity and self-limited, with or without pruritus. A dermatologist should evaluate persistent and/or severe rash or pruritus. A biopsy should be considered unless contraindicated. Management guidelines for dermatologic events are provided in the following table.

S1933 Page 52
Version Date 6/1/2021
Management Guidelines for Dermatologic Events
Event Management Dermatologic event, Grade 1
• Continue atezolizumab. • Consider treatment with topical
corticosteroids and/or other symptomatic therapy (e.g., antihistamines).
Dermatologic event, Grade 2
• Continue atezolizumab. • Consider participant referral to dermatologist. • Initiate treatment with topical corticosteroids. • Consider treatment with higher-potency
topical corticosteroids if event does not improve.
Dermatologic event, Grade 3
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Refer participant to dermatologist. • Initiate treatment with corticosteroids
equivalent to 10 mg/day oral prednisone, increasing dose to 1−2 mg/kg/day if event does not improve within 48−72 hours.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
Dermatologic event, Grade 4
• Permanently discontinue atezolizumab and contact Study Chair. c
a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re- challenged with atezolizumab only after discussion with the Study Chair.
Neurologic Disorders Myasthenia gravis and Guillain-Barré syndrome have been observed with single-agent atezolizumab. Participants may present with signs and symptoms of sensory and/or motor neuropathy. Diagnostic workup is essential for an accurate characterization to differentiate between alternative etiologies. Management guidelines for neurologic disorders are provided in the following table.

S1933 Page 53
Version Date 6/1/2021
Management Guidelines for Neurologic Disorders
Event Management
Immune- mediated neuropathy, Grade 1
• Continue atezolizumab. • Investigate etiology.
Immune- mediated neuropathy, Grade 2
• Withhold atezolizumab for up to 12 weeks after event onset. a
• Investigate etiology. • Initiate treatment as per institutional
guidelines. • If event resolves to Grade 1 or better,
resume atezolizumab. b • If event does not resolve to Grade 1 or
better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
Immune- mediated neuropathy, Grade 3 or 4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Initiate treatment as per institutional guidelines.
Myasthenia gravis and Guillain-Barré syndrome (any grade)
• Permanently discontinue atezolizumab and contact Study Chair. c
• Refer participant to neurologist. • Initiate treatment as per institutional
guidelines. • Consider initiation of corticosteroids
equivalent to 1−2 mg/kg/day oral or IV prednisone.
a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re- challenged with atezolizumab only after discussion with the Study Chair.
Immune-Mediated Meningoencephalitis Immune-mediated meningoencephalitis is an identified risk associated with the administration of atezolizumab. Immune-mediated meningoencephalitis should be suspected in any participant presenting with signs or symptoms suggestive of meningitis or encephalitis, including, but not limited to, headache, neck pain, confusion, seizure, motor or sensory dysfunction, and altered or depressed level of consciousness. Encephalopathy from metabolic or electrolyte imbalances needs to be distinguished from potential meningoencephalitis resulting from infection (bacterial, viral, or fungal) or progression of malignancy, or secondary to a paraneoplastic process.

S1933 Page 54
Version Date 6/1/2021
All participants being considered for meningoencephalitis should be urgently evaluated with a CT scan and/or MRI scan of the brain to evaluate for metastasis, inflammation, or edema. If deemed safe by the treating physician, a lumbar puncture should be performed, and a neurologist should be consulted. Participants with signs and symptoms of meningoencephalitis, in the absence of an identified alternate etiology, should be treated according to the guidelines in the following table.
Management Guidelines for Immune-Mediated Meningoencephalitis
Event Management
Immune-mediated meningoencephalitis, all grades
• Permanently discontinue atezolizumab and contact Study Chair. a
• Refer participant to neurologist. • Initiate treatment with corticosteroids equivalent to 1−2
mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month.
a Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
Renal Events Immune-mediated nephritis has been associated with the administration of atezolizumab. Eligible participants must have adequate renal function. Renal function, including serum creatinine, should be monitored throughout study treatment. Participants with abnormal renal function should be evaluated and treated for other more common etiologies (including prerenal and postrenal causes, and concomitant medications such as non-steroidal anti-inflammatory drugs). Refer the participant to a renal specialist if clinically indicated. A renal biopsy may be required to enable a definitive diagnosis and appropriate treatment. Participants with signs and symptoms of nephritis, in the absence of an identified alternate etiology, should be treated according to the guidelines in the following table.

S1933 Page 55
Version Date 6/1/2021
Management Guidelines for Renal Events
Event Management
Renal event, Grade 1
• Continue atezolizumab. • Monitor kidney function, including creatinine, closely until
values resolve to within normal limits or to baseline values.
Renal event, Grade 2
• Withhold atezolizumab for up to 12 weeks after event onset. a • Refer participant to renal specialist. • Initiate treatment with corticosteroids equivalent to 1−2
mg/kg/day oral prednisone. • If event resolves to Grade 1 or better, resume atezolizumab. b • If event does not resolve to Grade 1 or better while
withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
Renal event, Grade 3 or 4
• Permanently discontinue atezolizumab and contact Study Chair.
• Refer participant to renal specialist and consider renal biopsy.
• Initiate treatment with corticosteroids equivalent to 1−2 mg/kg/day oral prednisone.
• If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month.
a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
Immune-Mediated Myositis Immune-mediated myositis has been associated with the administration of atezolizumab. Myositis or inflammatory myopathies are a group of disorders sharing the common feature of inflammatory muscle injury; dermatomyositis and polymyositis are among the most common disorders. Initial diagnosis is based on clinical (muscle weakness, muscle pain, skin rash in dermatomyositis), biochemical (serum creatine kinase increase), and imaging (electromyography/MRI) features, and is confirmed with a muscle biopsy. Participants with signs and symptoms of myositis, in the absence of an identified alternate etiology, should be treated according to the guidelines in the following table.

S1933 Page 56
Version Date 6/1/2021
Management Guidelines for Immune-Mediated Myositis
Event Management
Immune- mediated myositis, Grade 1
• Continue atezolizumab. • Refer participant to rheumatologist or neurologist. • Initiate treatment as per institutional guidelines.
Immune- mediated myositis, Grade 2
• Withhold atezolizumab for up to 12 weeks after event onset a and contact Study Chair.
• Refer participant to rheumatologist or neurologist. • Initiate treatment as per institutional guidelines. • Consider treatment with corticosteroids
equivalent to 1−2 mg/kg/day IV methylprednisolone and convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If corticosteroids are initiated and event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
Immune- mediated myositis, Grade 3
• Withhold atezolizumab for up to 12 weeks after event onset a and contact Study Chair.
• Refer participant to rheumatologist or neurologist.
• Initiate treatment as per institutional guidelines.
• Respiratory support may be required in more severe cases.
• Initiate treatment with corticosteroids equivalent to 1−2 mg/kg/day IV methylprednisolone, or higher-dose bolus if participant is severely compromised (e.g., cardiac or respiratory symptoms, dysphagia, or weakness that severely limits mobility); convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, resume atezolizumab. b
• If event does not resolve to Grade 1 or better while withholding atezolizumab, permanently discontinue atezolizumab and contact Study Chair. c
• For recurrent events, treat as a Grade 4 event.

S1933 Page 57
Version Date 6/1/2021
Event Management
Immune- mediated myositis, Grade 4
• Permanently discontinue atezolizumab and contact Study Chair. c
• Refer participant to rheumatologist or neurologist.
• Initiate treatment as per institutional guidelines.
• Respiratory support may be required in more severe cases.
• Initiate treatment with corticosteroids equivalent to 1−2 mg/kg/day IV methylprednisolone, or higher-dose bolus if participant is severely compromised (e.g., cardiac or respiratory symptoms, dysphagia, or weakness that severely limits mobility); convert to 1−2 mg/kg/day oral prednisone or equivalent upon improvement.
• If event does not improve within 48 hours after initiating corticosteroids, consider adding an immunosuppressive agent.
• If event resolves to Grade 1 or better, taper corticosteroids over ≥ 1 month.
a Atezolizumab may be withheld for a longer period of time (i.e., > 12 weeks after event onset) to allow for corticosteroids (if initiated) to be reduced to the equivalent of ≤ 10 mg/day oral prednisone. The acceptable length of the extended period of time must be discussed with the Study Chair.
b If corticosteroids have been initiated, they must be tapered over ≥ 1 month to the equivalent of ≤ 10 mg/day oral prednisone before atezolizumab can be resumed.
c Resumption of atezolizumab may be considered in participants who are deriving benefit and have fully recovered from the immune-mediated event. Participants can be re-challenged with atezolizumab only after discussion with the Study Chair.
8.4 Dose modification and treatment decision contacts
For treatment or dose modification questions, please contact Dr. Aljumaily at 405/271-4022 or [email protected] or Dr. Mitin at 503/681-4200 or [email protected]. For dosing principles or questions, please consult the SWOG Policy #38 "Dosing Principles for Participants on Clinical Trials" at http://swog.org (then click on "Policies and Manuals" under the "Visitors" menu and choose Policy 38)
8.5 Adverse Event Reporting Requirements
This protocol utilizes Rave®/Cancer Therapy Evaluation Program Adverse Event Reporting System (CTEP-AERS) integration for expedited reporting of serious adverse events. The CTEP-AERS integration enables evaluation of post-baseline Adverse Events (AE) entered in Rave to determine whether they require expedited reporting. All AEs that occur after baseline are collected in Medidata Rave using the Adverse Event form, which is available for entry at each treatment or reporting period, and used to collect AEs that start during the period or persist from the previous reporting period. The Clinical Research Associate (CRA) will enter AEs that occur prior to the start of treatment on a baseline form that is not included in the Rave-CTEP-AERS integration. AEs that occur prior to enrollment must begin and end on the baseline Adverse Events form and should not be included on the standard Adverse Events form that is available at treatment unless there has been an increase in grade.

S1933 Page 58
Version Date 6/1/2021
Prior to sending AEs through the rules evaluation process, site staff should verify the following on the Adverse Event form in Rave:
• The reporting period (course/cycle) is correct; and • AEs are recorded and complete (no missing fields) and the form is query free
(fields added to the form during study build do not need to be query free for the integration call with CTEP-AERS to be a success).
The CRA reports AEs in Rave at the time the Investigator learns of the event. If the CRA modifies an AE, it must be re-submitted for rules evaluation. Upon completion of AE entry in Medidata Rave, the CRA submits the AE for rules evaluation by completing the Expedited Reporting Evaluation form. Both NCI and protocol-specific reporting rules evaluate the AEs submitted for expedited reporting. A report is initiated in CTEP-AERS using information entered in Medidata Rave for AEs that meet reporting requirements. The CRA completes the report by accessing CTEP-AERS via a direct link on the Medidata Rave Expedited Reporting Evaluation form. In the rare occurrence that Internet connectivity is lost, a 24-hour notification is to be made to CTEP by telephone at 301-897-7497. Once Internet connectivity is restored, the 24-hour notification that was phoned in must be entered immediately into CTEP-AERS using the deep link from Medidata Rave. Additional information about the CTEP-AERS integration is available on the CTSU website:
• Study specific documents: Protocols > Documents> Education and Promotion; and • Expedited Safety Reporting Rules Evaluation user guide: Resources > CTSU
Operations Information> User Guides.
NCI requirements for SAE reporting are available on the CTEP website: • NCI Guidelines for Investigators: Adverse Event Reporting Requirements is
available at https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/aeguidelines.pdf.
If you have questions about this process, please contact the SAE Program Manager 210-614-8808 or email [email protected]. The CTEP-AERS electronic reporting system “Help” feature has detailed instructions in the section "Submitting Reports for RAVE Users".
8.6 Serious Adverse Event Reporting Requirements a. Definition and Purpose
Definition: Adverse event means any untoward medical occurrence associated with the use of a drug in humans, whether or not considered drug related. (FDA, 21 CFR 312.32). See Table 8.6a for definition of a Serious Adverse Event (SAE) and reporting requirements. Adverse event data collection and reporting, which are required as part of every clinical trial, are done to ensure the safety of patients enrolled in the studies as well as those who will enroll in future studies using similar agents. Adverse events are reported in a routine manner at scheduled times during a trial. (Directions for routine reporting are provided in Section 14.0.) Additionally, certain adverse events must be reported in an expedited manner to allow for more timely monitoring of patient safety and care. The following guidelines prescribe expedited adverse event reporting for this protocol.

S1933 Page 59
Version Date 6/1/2021
b. Reporting method
This study requires that expedited adverse events be reported to SWOG Operations Office using the Cancer Therapy Evaluation Program Adverse Event Reporting System (CTEP-AERS). CTEP's guidelines for CTEP-AERS can be found at http://ctep.cancer.gov. NOTE: For this study, all adverse events requiring expedited reporting must initially be reported on the Adverse Event Form in the appropriate Treatment Cycle folder in Medidata Rave. The CTEP-AERS report must then be initiated directly from the Adverse Event Form in Medidata Rave. Do not initiate the CTEP-AERS report via the CTEP-AERS website.
c. When to report an event in an expedited manner
Some adverse events require 24-hour notification (refer to Table 8.6a) via CTEP-AERS.
In the rare event when internet connectivity is disrupted a 24-hour notification is made to NCI by telephone at 301-897-7497. An electronic report MUST be submitted immediately upon re-establishment of internet connection. When the adverse event requires expedited reporting, submit the report via CTEP-ARS within the number of calendar days of learning of the event specified in Table 8.6a or Table 8.6b. Any supporting documentation requested by CTEP should be submitted in accordance with instructions provided by the CTEP-AERS system.
d. Other recipients of adverse event reports
CTEP will forward reports and documentation to the appropriate regulatory agencies and drug companies as required. Adverse events determined to be reportable to the Institutional Review Board responsible for oversight of the patient must be reported according to local policy and procedures.
e. Expedited reporting for investigational agents
Expedited reporting is required if the patient has received at least one dose of the investigational agent(s) as part of the trial. Reporting requirements are provided in Table 8.6a. The investigational agent(s) used in this study is atezolizumab. If there is any question about the reportability of an adverse event or if Internet connectivity is disrupted please telephone or email the SAE Program Manager at the Operations Office, 210/614-8808 or [email protected], before preparing the report. NOTE: For this study, all adverse events requiring expedited reporting must initially be reported on the Adverse Event Form in the appropriated Treatment Cycle folder in Medidata Rave. Once the adverse event is entered into RAVE, the Rules Engine will confirm whether or not the adverse event requires expedited reporting. The CTEP-AERS report must then be initiated directly from the Adverse Event Form in Medidata Rave. Do not initiate the CTEP-AERS report via the CTEP-AERS website. Sites are encouraged to confirm the Expedited Reporting Evaluation Recommendation with the reporting criteria outlined in Table 8.6a.
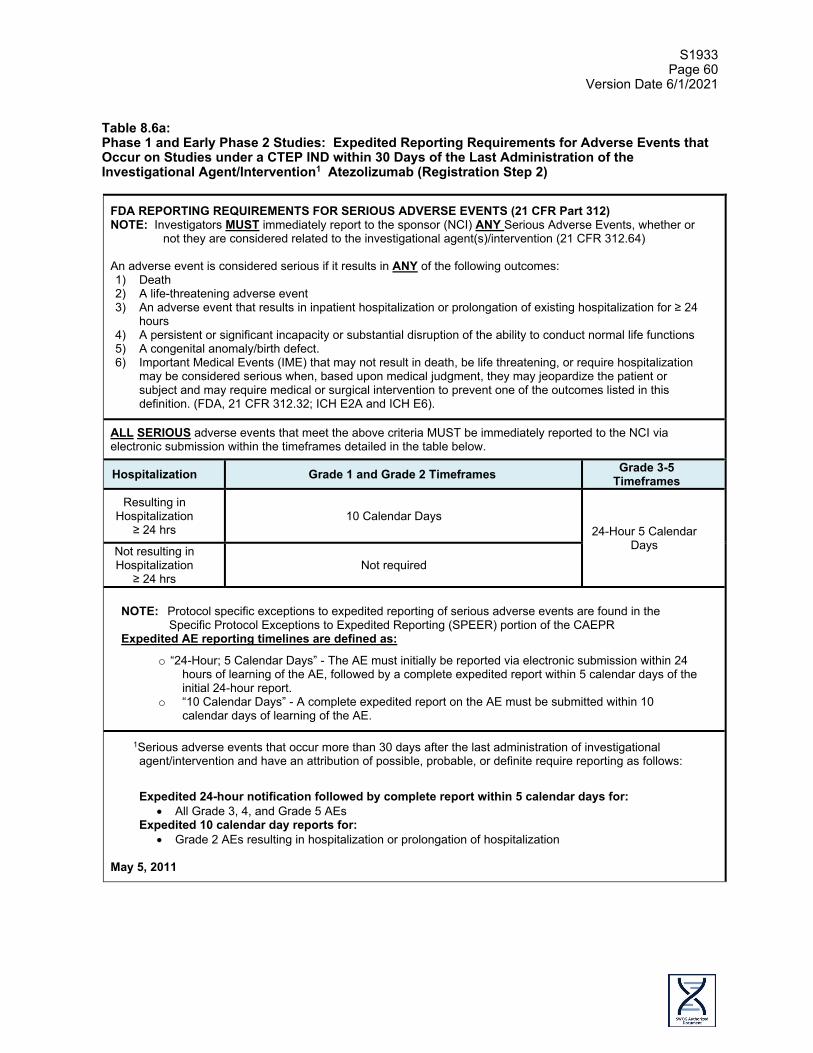
S1933 Page 60
Version Date 6/1/2021
Table 8.6a: Phase 1 and Early Phase 2 Studies: Expedited Reporting Requirements for Adverse Events that Occur on Studies under a CTEP IND within 30 Days of the Last Administration of the Investigational Agent/Intervention1 Atezolizumab (Registration Step 2)
FDA REPORTING REQUIREMENTS FOR SERIOUS ADVERSE EVENTS (21 CFR Part 312) NOTE: Investigators MUST immediately report to the sponsor (NCI) ANY Serious Adverse Events, whether or
not they are considered related to the investigational agent(s)/intervention (21 CFR 312.64) An adverse event is considered serious if it results in ANY of the following outcomes: 1) Death 2) A life-threatening adverse event 3) An adverse event that results in inpatient hospitalization or prolongation of existing hospitalization for ≥ 24
hours 4) A persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions 5) A congenital anomaly/birth defect. 6) Important Medical Events (IME) that may not result in death, be life threatening, or require hospitalization
may be considered serious when, based upon medical judgment, they may jeopardize the patient or subject and may require medical or surgical intervention to prevent one of the outcomes listed in this definition. (FDA, 21 CFR 312.32; ICH E2A and ICH E6).
ALL SERIOUS adverse events that meet the above criteria MUST be immediately reported to the NCI via electronic submission within the timeframes detailed in the table below.
Hospitalization Grade 1 and Grade 2 Timeframes Grade 3-5 Timeframes
Resulting in Hospitalization
≥ 24 hrs 10 Calendar Days
24-Hour 5 Calendar Days Not resulting in
Hospitalization ≥ 24 hrs
Not required
NOTE: Protocol specific exceptions to expedited reporting of serious adverse events are found in the Specific Protocol Exceptions to Expedited Reporting (SPEER) portion of the CAEPR
Expedited AE reporting timelines are defined as:
o “24-Hour; 5 Calendar Days” - The AE must initially be reported via electronic submission within 24 hours of learning of the AE, followed by a complete expedited report within 5 calendar days of the initial 24-hour report.
o “10 Calendar Days” - A complete expedited report on the AE must be submitted within 10 calendar days of learning of the AE.
1Serious adverse events that occur more than 30 days after the last administration of investigational agent/intervention and have an attribution of possible, probable, or definite require reporting as follows:
Expedited 24-hour notification followed by complete report within 5 calendar days for:
• All Grade 3, 4, and Grade 5 AEs Expedited 10 calendar day reports for:
• Grade 2 AEs resulting in hospitalization or prolongation of hospitalization May 5, 2011

S1933 Page 61
Version Date 6/1/2021
f. Additional Instructions or Exceptions to CTEP-AERS Expedited Reporting
Requirements for Phase 1 and Early Phase 2 Studies Utilizing an Agent under a CTEP-IND: 1. Group-specific instructions
Submission of the on-line CTEP-AERS report plus any necessary amendments generally completes the reporting requirements. In addition, you may be asked to submit supporting clinical data to the SWOG Operations Offices in order to complete the evaluation of the event. If requested by the SAE Program Manager, the supporting data should be sent within 5 calendar days by fax to 210-614-0006.
2. Adverse events of special interest (AESI)
The following adverse events are considered adverse events of special interest (AESI) for this protocol and require expedited reporting via CTEP-AERS. Unless otherwise specified in the list below, events are to be reported regardless of grade or attribution: • Cases of potential drug-induced liver injury that include an elevated
ALT or AST in combination with either an elevated bilirubin or clinical jaundice, as defined by Hy's Law and based on the following observations:
o Treatment-emergent ALT or AST > 3 x ULN (or > 3 x baseline value in disease states where LFTs may be elevated at baseline) in combination with total bilirubin > 2 x ULN (of which ≥ 35% is direct bilirubin)
o Treatment-emergent ALT or AST > 3 x ULN (or > 3 x baseline value in disease states where LFTs may be elevated at baseline) in combination with clinical jaundice
• Suspected transmission of an infectious agent by the study treatment, as defined below:
o Any organism, virus, or infectious particle (e.g., prion protein transmitting transmissible spongiform encephalopathy), pathogenic or non-pathogenic, is considered an infectious agent. A transmission of an infectious agent may be suspected from clinical symptoms or laboratory findings that indicate an infection in a patient exposed to a medicinal product. This term applies only when a contamination of study treatment is suspected.
• Systemic lupus erythematosus • Events suggestive of hypersensitivity, infusion-related reactions,
cytokine release syndrome, macrophage activating syndrome and hemophagocytic lympohistiocytosis
• Nephritis • Ocular toxicities (e.g., uveitis, retinitis, optic neuritis) • Grade ≥ 2 cardiac disorders (e.g., atrial fibrillation, myocarditis,
pericarditis) • Vasculitis • Autoimmune hemolytic anemia • Severe cutaneous reactions (e.g., Stevens-Johnson syndrome,
dermatitis bullous, toxic epidermal necrolysis)

S1933 Page 62
Version Date 6/1/2021
g. Expedited reporting for Radiation Therapy (Registration Step 1)
Reporting requirements are provided in Table 8.6b for radiation therapy administered in Registration Step 1. If there is any question about the reportability of an adverse event or if Internet connectivity is disrupted please telephone or email the SAE Program Manager at the Operations Office, 210-614-8808 or [email protected], before preparing the report. NOTE: For this study, all adverse events requiring expedited reporting must initially be reported on the Adverse Event Form in the appropriate Treatment Cycle folder in Medidata Rave. Once the adverse event is entered into RAVE, the Rules Engine will confirm whether or not the adverse event requires expedited reporting. The CTEP-AERS report must then be initiated directly from the Adverse Event Form in Medidata Rave. Do not initiate the CTEP-AERS report via the CTEP-AERS website. Sites are encouraged to confirm the Expedited Reporting Evaluation Recommendation with the reporting criteria outlined in Table 8.6b. Table 8.6b: Expedited reporting requirements for adverse events experienced by patients who have received radiation therapy while on Registration Step 1 within 30 days after the last radiation treatment
ATTRIBUTION
Grade 4
Grade 5a Unexpected Expected Unexpected Expected
Unrelated or Unlikely CTEP-AERS CTEP-AERS
Possible, Probable, Definite
CTEP-AERS CTEP-AERS CTEP-AERS
CTEP-AERS: Indicates an expedited report is to be submitted via CTEP-AERS within 10 calendar days of learning of the eventb.
a This includes all deaths within 30 days of the last dose of radiotherapy regardless of attribution. Any death that occurs more than 30 days after the last dose of radiotherapy and is attributed (possibly, probably, or definitely) to the agent(s) and is not due to cancer recurrence must be reported according to the instructions above.
b Submission of the on-line CTEP-AERS report plus any necessary amendments
generally completes the reporting requirements. You may, however, be asked to submit supporting clinical data to the Operations Office in order to complete the evaluation of the event. If requested, the specified data should be sent within 5 calendar days by fax to 210-614-0006.
h. Reporting Secondary Malignancies, including AML/ALL/MDS 1. A secondary malignancy is a cancer caused by treatment for a previous
malignancy (e.g., treatment with investigational agent/intervention, radiation or chemotherapy). A secondary malignancy is not considered a metastasis of the initial neoplasm.
CTEP requires all secondary malignancies that occur following treatment with an agent under an NCI IND to be reported via CTEP-AERS. Three options are available to describe the event.

S1933 Page 63
Version Date 6/1/2021
• Leukemia secondary to oncology chemotherapy (e.g., Acute Myelocytic Leukemia [AML])
• Myelodysplastic syndrome (MDS) • Treatment-related secondary malignancy
Any malignancy possibly related to cancer treatment (including AML/MDS) should also be reported via the routine reporting mechanisms outlined in each protocol.
Second Malignancy: A second malignancy is one unrelated to the treatment of a prior malignancy (and is NOT a metastasis from the initial malignancy). Second malignancies require ONLY routine reporting.
For more information see: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/aeguidelines.pdf.
2. Supporting documentation should be submitted to CTEP by fax at 301-897-7404 in accordance with instructions provided by the CTEP-AERS system. A copy of supporting documentation must also be submitted to SWOG Operations Office by fax to 210-614-0006. NOTE: If a patient has been enrolled in more than one NCI-sponsored study, the report must be submitted for the most recent trial.
i. Reporting Pregnancy, Pregnancy Loss, and Death Neonatal
1. Pregnancy: Study participants who become pregnant while on study; that
pregnancy should be reported in an expedited manner via CTEP-AERS as Grade 3 “Pregnancy, puerperium and perinatal conditions – Other (pregnancy)” under the Pregnancy, puerperium and perinatal conditions SOC.
Additionally, the pregnancy outcome for patients on study should be reported via CTEP-AERS at the time the outcome becomes known, accompanied by the same Pregnancy Report Form used for the initial report.
2. Pregnancy Loss: Pregnancy loss is defined in CTCAE as “Death in utero.”
Pregnancy loss should be reported expeditiously as Grade 4 “Pregnancy loss” under the Pregnancy, puerperium and perinatal conditions SOC. A Pregnancy loss should NOT be reported as a Grade 5 event under the Pregnancy, puerperium and perinatal conditions SOC, as currently CTEP-AERS recognizes this event as a patient death.
3. DEATH NEONATAL: Death neonatal is defined in CTCAE as “Newborn
death occurring during the first 28 days after birth. A neonatal death should be reported expeditiously as Grade 4 “Death neonatal” under the General disorders and administration SOC. Neonatal death should NOT be reported as a Grade 5 event under the General disorders and administration SOC as currently CTEP-AERS recognizes this event as a patient death.

S1933 Page 64
Version Date 6/1/2021
NOTE: When submitting CTEP-AERS reports for “Pregnancy, “Pregnancy loss”, or “Neonatal loss”, the Pregnancy Information Form should also be completed and faxed with any additional medical information to 301-897-7404. The potential risk of exposure of the fetus to the investigational agent(s) or chemotherapy agent(s) should be documented in the “Description of Event” section of the CTEP-AERS report. The Pregnancy Information Form is available at: http://ctep.cancer.gov/protocolDevelopment/adverse_effects.htm.
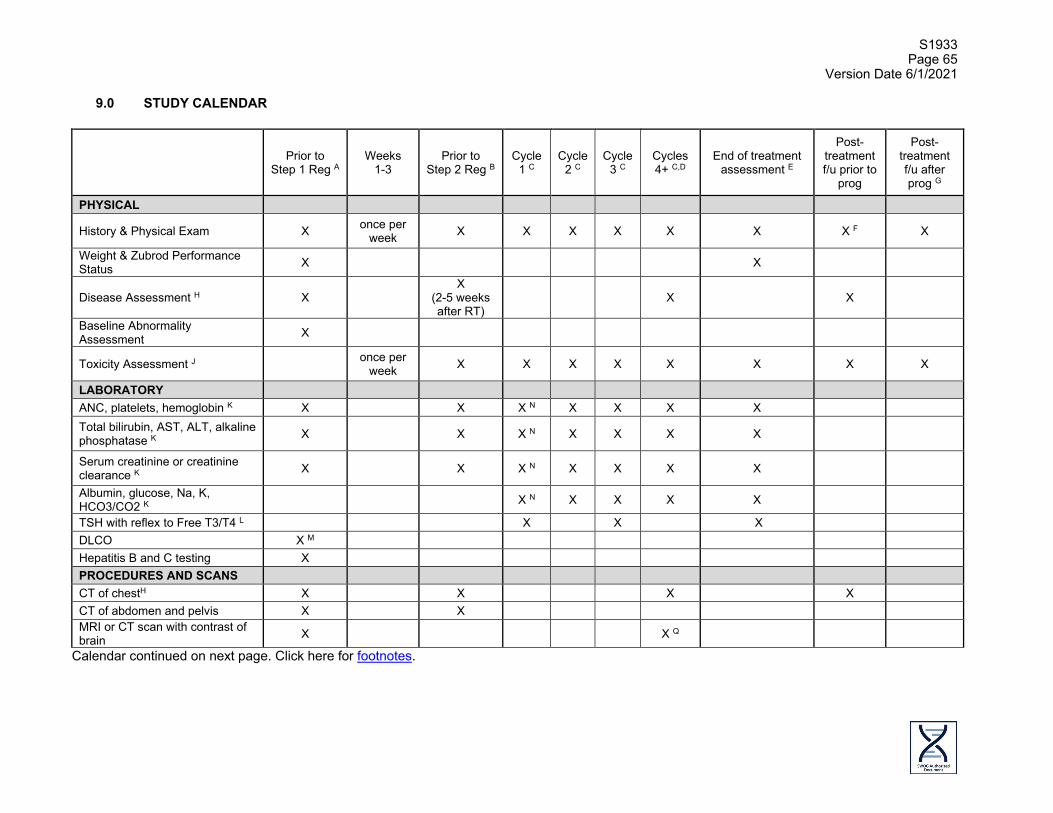
S1933 Page 65
Version Date 6/1/2021
9.0 STUDY CALENDAR
Prior to Step 1 Reg A
Weeks 1-3
Prior to Step 2 Reg B
Cycle 1 C
Cycle 2 C
Cycle 3 C
Cycles 4+ C,D
End of treatment assessment E
Post-treatment f/u prior to
prog
Post-treatment f/u after prog G
PHYSICAL
History & Physical Exam X once per week X X X X X X X F X
Weight & Zubrod Performance Status X X
Disease Assessment H X X
(2-5 weeks after RT)
X X
Baseline Abnormality Assessment X
Toxicity Assessment J once per week X X X X X X X X
LABORATORY ANC, platelets, hemoglobin K X X X N X X X X Total bilirubin, AST, ALT, alkaline phosphatase K X X X N X X X X
Serum creatinine or creatinine clearance K X X X N X X X X
Albumin, glucose, Na, K, HCO3/CO2 K X N X X X X
TSH with reflex to Free T3/T4 L X X X DLCO X M Hepatitis B and C testing X PROCEDURES AND SCANS CT of chestH X X X X CT of abdomen and pelvis X X MRI or CT scan with contrast of brain X X Q
Calendar continued on next page. Click here for footnotes.
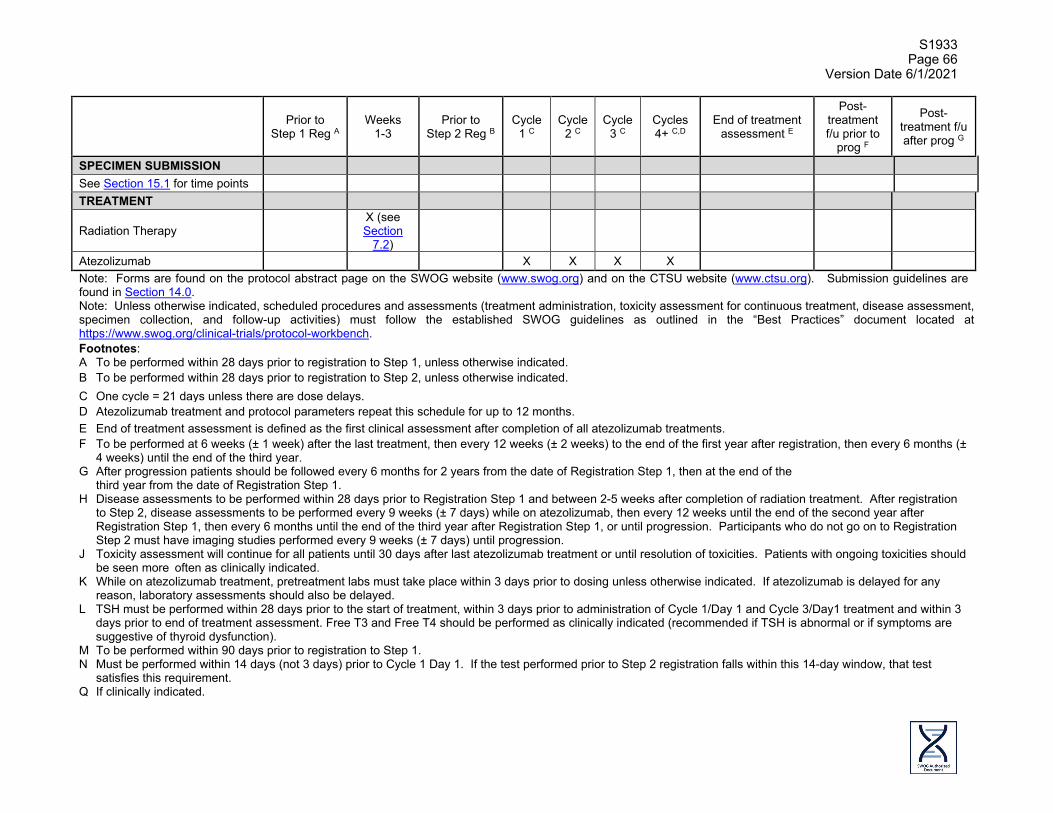
S1933 Page 66
Version Date 6/1/2021
Prior to Step 1 Reg A
Weeks 1-3
Prior to Step 2 Reg B
Cycle 1 C
Cycle 2 C
Cycle 3 C
Cycles 4+ C,D
End of treatment assessment E
Post-treatment f/u prior to
prog F
Post-treatment f/u after prog G
SPECIMEN SUBMISSION See Section 15.1 for time points TREATMENT
Radiation Therapy X (see Section
7.2)
Atezolizumab X X X X Note: Forms are found on the protocol abstract page on the SWOG website (www.swog.org) and on the CTSU website (www.ctsu.org). Submission guidelines are found in Section 14.0. Note: Unless otherwise indicated, scheduled procedures and assessments (treatment administration, toxicity assessment for continuous treatment, disease assessment, specimen collection, and follow-up activities) must follow the established SWOG guidelines as outlined in the “Best Practices” document located at https://www.swog.org/clinical-trials/protocol-workbench. Footnotes: A To be performed within 28 days prior to registration to Step 1, unless otherwise indicated. B To be performed within 28 days prior to registration to Step 2, unless otherwise indicated. C One cycle = 21 days unless there are dose delays. D Atezolizumab treatment and protocol parameters repeat this schedule for up to 12 months. E End of treatment assessment is defined as the first clinical assessment after completion of all atezolizumab treatments. F To be performed at 6 weeks (± 1 week) after the last treatment, then every 12 weeks (± 2 weeks) to the end of the first year after registration, then every 6 months (±
4 weeks) until the end of the third year. G After progression patients should be followed every 6 months for 2 years from the date of Registration Step 1, then at the end of the
third year from the date of Registration Step 1. H Disease assessments to be performed within 28 days prior to Registration Step 1 and between 2-5 weeks after completion of radiation treatment. After registration
to Step 2, disease assessments to be performed every 9 weeks (± 7 days) while on atezolizumab, then every 12 weeks until the end of the second year after Registration Step 1, then every 6 months until the end of the third year after Registration Step 1, or until progression. Participants who do not go on to Registration Step 2 must have imaging studies performed every 9 weeks (± 7 days) until progression.
J Toxicity assessment will continue for all patients until 30 days after last atezolizumab treatment or until resolution of toxicities. Patients with ongoing toxicities should be seen more often as clinically indicated.
K While on atezolizumab treatment, pretreatment labs must take place within 3 days prior to dosing unless otherwise indicated. If atezolizumab is delayed for any reason, laboratory assessments should also be delayed.
L TSH must be performed within 28 days prior to the start of treatment, within 3 days prior to administration of Cycle 1/Day 1 and Cycle 3/Day1 treatment and within 3 days prior to end of treatment assessment. Free T3 and Free T4 should be performed as clinically indicated (recommended if TSH is abnormal or if symptoms are suggestive of thyroid dysfunction).
M To be performed within 90 days prior to registration to Step 1. N Must be performed within 14 days (not 3 days) prior to Cycle 1 Day 1. If the test performed prior to Step 2 registration falls within this 14-day window, that test
satisfies this requirement. Q If clinically indicated.

S1933 Page 67
Version Date 6/1/21
10.0 CRITERIA FOR EVALUATION AND ENDPOINT ANALYSIS
10.1 Measurability of lesions
a. Measurable disease
Measurable disease is defined differently for lymph nodes compared with other disease and will be addressed in a separate section below. 1. Lesions that can be accurately measured in at least one dimension
(longest diameter to be recorded) as ≥ 2.0 cm by chest x-ray, by ≥ 1.0 cm with CT or MRI scans, or ≥ 1.0 cm with calipers by clinical exam. All tumor measurements must be recorded in decimal fractions of centimeters (or millimeters).
2. The defined measurability of lesions on CT scan is based on the
assumption that CT slice thickness is 0.5 cm or less. It is strongly recommended that CT slice of 0.5 cm be used. If CT scans have slice thickness greater than 0.5 cm, the minimum size for a measurable lesion should be twice the slice thickness.
3. Malignant lymph nodes are to be considered pathologically enlarged and
measurable if it measures ≥ 1.5 cm in SHORT AXIS (greatest diameter perpendicular to the long axis of the lymph node) when assessed by scan (CT scan slice recommended being no greater than 0.5 cm).
b. Non-measurable disease
All other lesions (or sites of disease), including small lesions (longest diameter <1.0 cm or pathologic lymph nodes with ≥ 1.0 cm to <1.5 cm short axis), are considered non-measurable disease. Bone lesions, leptomeningeal disease, ascites, pleural/pericardial effusions, lymphangitis cutis/pulmonitis, inflammatory breast disease, and abdominal masses (not followed by CT or MRI), are considered as non-measurable as are previously radiated lesions that have not progressed.
c. Notes on measurability
1. For CT and MRIs, the same type of scanner should be used and the image
acquisition protocol should be followed as closely as possible to prior scans. It is no longer necessary to distinguish between spiral and conventional CT.
2. Body scans should be performed with breath-hold scanning techniques, if
possible.
3. PET-CT: At present, the low dose or attenuation correction CT portion of a PET-CT is not always of optimal diagnostic CT quality for use with RECIST measurements. However, if the site can document that the CT performed as part of a PET-CT is of identical diagnostic quality to a diagnostic CT, then the CT portion of the PET-CT can be used for RECIST measurements and can be used interchangeably with stand-alone CT. The slice thickness of 0.5 cm or less is highly recommended. If CT scans have slice thickness > 0.5 cm, the minimum size for a measurable lesion should be twice the slice thickness.

S1933 Page 68
Version Date 6/1/2021
4. Ultrasound: Ultrasound is not useful in assessment of lesion size and should not be used as a method of measurement.
5. Cystic lesions that meet the criteria for radiographically defined simple
cysts should not be considered as malignant lesions (neither measurable nor non-measurable) since they are, by definition simple cysts.
6. If a target lesion becomes very small some radiologists indicate that it is
too small to measure. If the lesion is actually still present, a default measurement of 0.5 cm should be applied. If the radiologist believes the lesion has gone, a default measurement of 0.0 cm should be recorded.
10.2 Objective status at each disease evaluation
Objective Status is to be recorded at each evaluation. All measurable lesions up to a maximum of 2 lesions per organ, 5 lesions in total, representative of all involved organs, should be identified as target lesions at baseline. All other lesions (or sites of disease) including any measurable lesions over and above the 5 target lesions should be identified as non-target lesions. Measurements must be provided for target measurable lesions, while presence or absence must be noted for non-target measurable and non-measurable disease. For studies that use disease progression as an endpoint, all potential sites of metastases should be evaluated at each time point rather than following only sites of disease identified at baseline. It is acceptable to image only the areas of the body most likely to be involved with metastatic disease for the tumor type (chest, abdomen, pelvis, and/or bone scan are typical), with the addition of any areas with suspected involvement based upon clinical symptoms. For study-specific imaging requirements, see the Study Calendar in Section 9.0. a. Complete Response (CR): Complete disappearance of all target and non-target
lesions (with the exception of lymph nodes mentioned below). No new lesions. No disease related symptoms. Any lymph nodes (whether target or non-target) must have reduction in short axis to < 1.0 cm. All disease must be assessed using the same technique as baseline.
b. Partial Response (PR): Applies only to patients with at least one measurable lesion. Greater than or equal to 30% decrease under baseline of the sum of appropriate diameters of all target measurable lesions. No unequivocal progression of non-measurable disease. No new lesions. All target measurable lesions must be assessed using the same techniques as baseline.
c. Stable: Does not qualify for CR, PR, Progression or Symptomatic Deterioration.
All target measurable lesions must be assessed using the same techniques as baseline.
d. Progression: One or more of the following must occur: 20% increase in the sum of appropriate diameters of target measurable lesions over smallest sum observed (over baseline if no decrease during therapy) using the same techniques as baseline, as well as an absolute increase of at least 0.5 cm. Unequivocal progression of non-measurable disease in the opinion of the treating physician (an explanation must be provided). Appearance of any new lesion/site. Death due to disease without prior documentation of progression and without symptomatic deterioration (see 10.2e).
NOTE: For patients who have received at least one dose of atezolizumab, an initial report of progression must be confirmed by a second determination of progression at least 4 weeks after the initial report of progression. If the second assessment

S1933 Page 69
Version Date 6/1/2021
documents an absence of progression, the patient’s disease is assumed not to have progressed and the initial report of progression is coded as pseudo-progression. If there are no additional disease assessments at least 4 weeks after the initial assessment documenting absence of progression (either due to inadequate or missing assessment) then the initial assessment will be coded as a progression. Any indication of progression (per the definition above) reported after the determination of pseudo-progression will be coded a progression and will not require a second assessment to confirm progression.
Notes on progression and new lesions: • For equivocal findings of progression (e.g. very small and uncertain new
lesions; cystic changes or necrosis in existing lesions), treatment may continue until the next scheduled assessment. If at the next scheduled assessment, progression is confirmed, the date of progression should be the earlier date when progression was suspected.
• FDG-PET imaging can complement regular scans in identifying new lesions according to the following algorithm.
• Negative FDG-PET at baseline, with a positive FDG-PET at follow-up is a sign of progression based on a new lesion.
• No FDG-PET at baseline and a positive FDG-PET at follow-up corresponding to a potential new site of disease must have a confirmation by anatomical assessment (e.g. CT, MRI, x-ray) as new site of disease to be considered progressive disease. In such a case, the date of progressive disease will be the date of the initial abnormal FDG-PET.
• A previous abnormal target lymph node that became normal and subsequently enlarged in size meeting the criteria for a pathologic and measurable lymph node (a short axis of ≥ 1.5 cm) should be added to the sum of diameters to determine if criteria for progression are met based on target lesions.
• A previously abnormal non-target lymph node that became normal and subsequently recurred must meet the criteria for progression based on non-target lesions to be considered progression.
• A normal lymph node at baseline (<1.0 cm) that subsequently becomes pathologic is considered a new lesion and should be considered progression.
• If a single pathologic lymph node is driving the progression event, continuation of treatment/follow-up and confirmation by a subsequent exam should be contemplated. If it becomes clear that the new lymph node has not resolved, or has increased in size, the date of progression would be the date the new lymph node was first documented.
e. Symptomatic deterioration: Global deterioration of health status requiring discontinuation of treatment without objective evidence of progression. Efforts should be made to obtain objective evidence of progression after discontinuation.
f. Assessment inadequate, objective status unknown. Progression or symptomatic deterioration has not been documented, and one or more target measurable lesions have not been assessed or inconsistent assessment methods were used.
Objective status notes: 1. Non-measurable and non-target measurable disease do not affect
Objective Status in determination of CR (must be absent--a patient who otherwise has a CR, but who has non-measurable or non-target measurable disease present or not assessed, will be classified as having a PR). However, non-measurable and non-target lesions are included in

S1933 Page 70
Version Date 6/1/2021
determination of progression (if new sites of disease develop or if unequivocal progression occurs in the opinion of the treating physician).
2. An objective status of PR or stable cannot follow one of CR. Stable can
follow PR only in the rare case that tumor increases too little to qualify as progression, but enough that a previously documented 30% decrease no longer holds.
3. In cases for which initial flare reaction is possible (hypercalcemia,
increased bone pain, erythema of skin lesions), objective status is not progression unless either symptoms persist beyond 4 weeks or there is additional evidence of progression.
4. Lesions that appear to increase in size due to presence of necrotic tissue
will not be considered to have progressed. 5. For bone disease documented on bone scan only, increased uptake does
not constitute unequivocal progression. However, increase in the soft tissue component of a lesion as measured by CT or MRI would constitute progression.
6. Appearance of new pleural effusions does not constitute unequivocal
progression unless cytologically proven of neoplastic origin, since some effusions are a toxicity related to therapy or other medical conditions. Increase in the size of an existing effusion does not constitute unequivocal progression, since the fluid status of the patient could alter the size of the effusion.
7. If CR determination depends on a lesion for which the status is unclear by
the required tests, it is recommended the residual lesion be investigated with biopsy or fine needle aspirate.
8. Lymph nodes are considered one organ. Only two lymph nodes should
be selected as target lesions. Other involved lymph nodes should be assessed and followed as non-target lesions.
9. “Paired” organs, i.e. lungs, kidneys and ovaries, are considered one organ.
Pleural-based lung lesions are considered part of the lung in determining target lesions (a maximum of two lung lesions should be selected), whereas pleural effusions/thickening can be reported as a separate site.
10.3 Best Response
Best response should be documented for as long as the patient remains on protocol treatment. a. CR: Two or more consecutive objective statuses of CR a minimum of four weeks
apart.
b. PR: Two or more consecutive statuses of PR or better a minimum of four weeks apart, but not qualifying as CR.
c. Unconfirmed CR: One objective status of CR documented but not qualifying as CR
or PR.
d. Unconfirmed PR: One objective status of PR documented but not qualifying as CR, PR or unconfirmed CR.

S1933 Page 71
Version Date 6/1/2021
e. Stable/no response: At least one objective status of stable/no response
documented at least 6 weeks after registration, but not qualifying as anything else above.
f. Increasing disease: Objective status of progression within 12 weeks of registration,
not qualifying as anything else above.
g. Symptomatic deterioration: Objective status of symptomatic deterioration within 12 weeks of registration, not qualifying as anything else above.
h. Inadequate assessment, response unknown: Progression or symptomatic
deterioration greater than 12 weeks after registration and no other response category applies.
10.4 Performance Status Patients will be graded according to the Zubrod performance status scale.
POINT DESCRIPTION 0 Fully active, able to carry on all pre-disease performance without
restriction. 1 Restricted in physically strenuous activity but ambulatory and able to
carry out work of a light or sedentary nature, e.g., light housework, office work.
2 Ambulatory and capable of self-care but unable to carry out any work
activities; up and about more than 50% of waking hours. 3 Capable of limited self-care, confined to bed or chair more than 50% of
waking hours. 4 Completely disabled; cannot carry on any self-care; totally confined to
bed or chair.
10.5 Progression-free survival
From date of Step 2 registration to date of first documentation of progression (see note in Section 10.2) or symptomatic deterioration (as defined in above), or death due to any cause. Progression-free survival for patients last known to be alive and progression free are censored at date of last contact.
10.6 Overall Survival
From date of Step 2 registration to date of death due to any cause. Overall survival for patients last known to be alive are censored at date of last contact.
11.0 STATISTICAL CONSIDERATIONS
11.1 Sample size and power justifications The primary study objective is to evaluate the rate of Grade 3-5 adverse events possibly, probably, or likely related to treatment among patients with medically inoperable Stage II or III NSCLC which is not a candidate for concurrent chemoradiation treated with hypofractionated thoracic radiotherapy followed by atezolizumab therapy. It is assumed
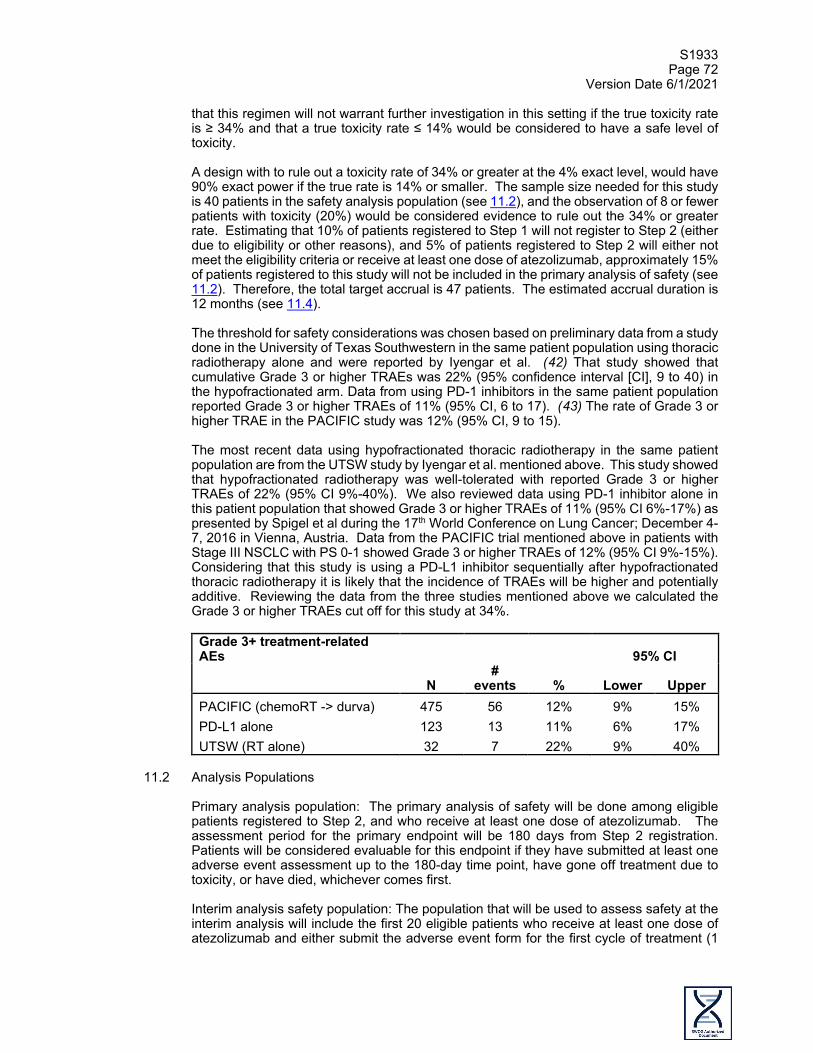
S1933 Page 72
Version Date 6/1/2021
that this regimen will not warrant further investigation in this setting if the true toxicity rate is ≥ 34% and that a true toxicity rate ≤ 14% would be considered to have a safe level of toxicity. A design with to rule out a toxicity rate of 34% or greater at the 4% exact level, would have 90% exact power if the true rate is 14% or smaller. The sample size needed for this study is 40 patients in the safety analysis population (see 11.2), and the observation of 8 or fewer patients with toxicity (20%) would be considered evidence to rule out the 34% or greater rate. Estimating that 10% of patients registered to Step 1 will not register to Step 2 (either due to eligibility or other reasons), and 5% of patients registered to Step 2 will either not meet the eligibility criteria or receive at least one dose of atezolizumab, approximately 15% of patients registered to this study will not be included in the primary analysis of safety (see 11.2). Therefore, the total target accrual is 47 patients. The estimated accrual duration is 12 months (see 11.4). The threshold for safety considerations was chosen based on preliminary data from a study done in the University of Texas Southwestern in the same patient population using thoracic radiotherapy alone and were reported by Iyengar et al. (42) That study showed that cumulative Grade 3 or higher TRAEs was 22% (95% confidence interval [CI], 9 to 40) in the hypofractionated arm. Data from using PD-1 inhibitors in the same patient population reported Grade 3 or higher TRAEs of 11% (95% CI, 6 to 17). (43) The rate of Grade 3 or higher TRAE in the PACIFIC study was 12% (95% CI, 9 to 15). The most recent data using hypofractionated thoracic radiotherapy in the same patient population are from the UTSW study by Iyengar et al. mentioned above. This study showed that hypofractionated radiotherapy was well-tolerated with reported Grade 3 or higher TRAEs of 22% (95% CI 9%-40%). We also reviewed data using PD-1 inhibitor alone in this patient population that showed Grade 3 or higher TRAEs of 11% (95% CI 6%-17%) as presented by Spigel et al during the 17th World Conference on Lung Cancer; December 4-7, 2016 in Vienna, Austria. Data from the PACIFIC trial mentioned above in patients with Stage III NSCLC with PS 0-1 showed Grade 3 or higher TRAEs of 12% (95% CI 9%-15%). Considering that this study is using a PD-L1 inhibitor sequentially after hypofractionated thoracic radiotherapy it is likely that the incidence of TRAEs will be higher and potentially additive. Reviewing the data from the three studies mentioned above we calculated the Grade 3 or higher TRAEs cut off for this study at 34%.
Grade 3+ treatment-related AEs 95% CI
N #
events % Lower Upper PACIFIC (chemoRT -> durva) 475 56 12% 9% 15% PD-L1 alone 123 13 11% 6% 17% UTSW (RT alone) 32 7 22% 9% 40%
11.2 Analysis Populations
Primary analysis population: The primary analysis of safety will be done among eligible patients registered to Step 2, and who receive at least one dose of atezolizumab. The assessment period for the primary endpoint will be 180 days from Step 2 registration. Patients will be considered evaluable for this endpoint if they have submitted at least one adverse event assessment up to the 180-day time point, have gone off treatment due to toxicity, or have died, whichever comes first. Interim analysis safety population: The population that will be used to assess safety at the interim analysis will include the first 20 eligible patients who receive at least one dose of atezolizumab and either submit the adverse event form for the first cycle of treatment (1

S1933 Page 73
Version Date 6/1/2021
cycle = 21 days and dose is given on Day 1) and/or discontinue treatment due to toxicity. The interim analysis population will include patients assessable for the primary endpoint (as defined above). Secondary analyses of toxicity, response, PFS, and OS will be performed on all eligible patients registered to Step 1 who receive at least one fraction of radiation, and among eligible patients registered to Step 2.
11.3 Analysis Plans
An interim analysis will take place when 20 patients are evaluable for toxicity in the safety analysis population. Accrual to the study may be placed on a temporary hold if the data to support the interim decision needs additional follow-up and reporting. The SWOG SDMC will generate a report summarizing the adverse event data (all reports and grades) to be used to determine the interim decision (continue accrual or close the study to further accrual). The study team along with the SWOG Lung Committee chair and representatives from CTEP will review the data to make a joint decision. However, independent of this review, if 7 or more patients (35%) experience a Grade 3 or worse adverse event attributable to treatment, then the regimen may be used as the threshold to interpret the regimen as too toxic, in which case both Steps 1 and 2 would be closed to further accrual (see Section 11.2 for population definitions). Binary endpoints will be summarized as proportions with 95% Clopper-Pearson confidence intervals. Individual toxicities with possible, probable or likely attribution to treatment along with the overall rate of Grade 3, 4 or 5 toxicities attributable to treatment (as previously defined) will be summarized. With 40 eligible patients, binary proportions can be estimated to within 15% with 95% confidence interval. Any toxicity with at least 5% prevalence is likely to be observed (with 87% probability). OS and PFS will be estimated using the method of Kaplan-Meier. 95% confidence for the medians will be constructed using the method of Brookmeyer-Crowley. With 40 patients, OS (or PFS) at a particular time point (e.g. 1 year) can be estimated to within ± 15% with 95% confidence interval.
11.4 Accrual
The estimated monthly average accrual rate for this study is 4 patients/ month. The accrual duration is estimated to be 12 months. This overall estimate is based on estimates within the separate Stage II and III populations as follows. For Stage III patients, based on the actual accrual rate from S9712 in which 43% of patients enrolled were PS 2, the estimated accrual rate for PS 2 Stage III patients is 2 patients per month. For Stage II patients, assuming similar percentage of lung cancers diagnosed as Stage II or Stage III NSCLC (30% Stage II, 25% Stage III) and assuming that approximately 50% of patients will not be surgical candidates, the estimated accrual rate is 2 patients per month.
11.5 Data and safety monitoring
There is no formal data and safety monitoring committee for this study. Toxicity and accrual are primarily monitored by the Study Chair, Study Statistician and the Disease Committee Chair. Weekly batch report of treatment related AE will be generated by Study Statisticians and reviewed by the Study Statisticians, Study Chairs, Study Coordinators, the Adverse Event Coordinators at the Operation Office and the Disease Committee Chair. Endpoint monitoring is done by the Study Statistician and Study Coordinator. The Adverse Event Coordinator at the Operations Office and the SAE Physician Reviewer are notified immediately whenever a serious adverse event is reported using the NCI CTEP-AERS system (see Section 16.0 for details). The Adverse Event Coordinator then provides the

S1933 Page 74
Version Date 6/1/2021
Study Chair and Study Statisticians a cumulative report of all SAEs within 24 hours with the most recent event highlighted. Regular monitoring of all adverse events is performed monthly by the Study Chair and Study Statistician(s). The Study Chair and Study Statistician(s) receive monthly reports summarizing all adverse events and serious adverse events. In addition, the Study Chair and Study Statistician(s) receive a monthly summary of treatment status for patients on the study. Accrual reports are generated weekly and formal toxicity reports are available to all SWOG group members are generated every 6 months.
12.0 DISCIPLINE REVIEW
12.1 Radiation Therapy Review
a. Submission of Digital Radiation Therapy Data
Submission of treatment plans in digital format as DICOM RT is required. Digital data must include CT scans, structures, plan, and dose files. This study uses TRIAD for RT data submission. Use of TRIAD requires several preliminary steps (see Section 12.1b). Additional information is available at: http://triadhelp.acr.org/ClinicalTrials/NCISponsoredTrials.aspx Use of SFTP will also be accepted as an alternate method of data submission on this study. See the instructions for submission of data via SFTP at https://www.qarc.org under Digital Data. Any items on the list below that are not part of the digital submission may be included with the transmission of the digital RT data. At least 5 days prior to the start of radiotherapy, the following data must be submitted for all patients: • RT treatment plans including treatment planning CT, structures, dose and
plan files. These items are included in the digital plan. • The full 4DCT study used to define the target volume.
• Treatment planning system summary report that includes the monitor unit
calculations, beam parameters, calculation algorithm, and volume of interest dose statistics.
• Copies (in DICOM format) and reports of all imaging studies used to define the target volume.
• Prescription sheet for entire treatment.
• RT-1 Dosimetry Summary Form • Motion Management Reporting Form Within one week following the completion of radiotherapy, the following data must be submitted for all patients:
• The RT-2 Radiotherapy Total Dose Record Form
• A copy of the patient's radiotherapy record including the prescription, and the
daily and cumulative doses to all required areas.

S1933 Page 75
Version Date 6/1/2021
Supportive data and forms may be included with the transmission of the digital RT data or submitted separately via e-mail to [email protected]. Questions regarding the dose calculations or documentation should be directed to: Protocol Dosimetrist IROC Rhode Island QA Center Phone: (401) 753-7600 Email: [email protected]
b. Digital RT Data Submission Using TRIAD
TRIAD is the American College of Radiology’s (ACR) image exchange application. TRIAD provides sites participating in clinical trials a secure method to transmit DICOM RT and other objects. TRIAD anonymizes and validates the images as they are transferred. TRIAD Access Requirements: Site staff who will submit images through TRIAD will need to be registered with the Cancer Therapy Evaluation Program (CTEP) and have a valid and active CTEP Identity and Access Management (IAM) account. Please refer to CTEP Registration Procedures of the protocol for instructions on how to request a CTEP-IAM account. To submit digital RT data and images (DICOM and DICOM RT), the site user must be on the site’s roster and be assigned the 'TRIAD site user' role. Users should contact the site’s roster Administrator to request assignment of the TRIAD site user role. TRIAD Installations: When a user applies for a CTEP-IAM account with the proper user role, he/she will need to have the TRIAD application installed on his/her workstation to be able to submit images. TRIAD installation documentation can be found by following this link https://triadinstall.acr.org/triadclient/ This process can be done in parallel to obtaining your CTEP-IAM account username and password. If you have any questions regarding this information, please send an e-mail to the TRIAD Support mailbox at [email protected].
c. Credentialing
RT Credentialing Requirements
Web Link for Procedures and Instructions: http//irochouston.mdanderson.org
Key Information
Facility Questionnaire
The IROC Houston electronic facility questionnaire (FQ) should be completed or updated with the most recent information about your institution. To access this FQ go to http://irochouston.mdanderson.org/questionnaires.
Credentialing Status Inquiry Form
To determine whether your institution needs to complete any further credentialing requirements, please complete the “Credentialing Status Inquiry Form” found under

S1933 Page 76
Version Date 6/1/2021
RT Credentialing Requirements
Web Link for Procedures and Instructions: http//irochouston.mdanderson.org
Key Information
credentialing on the IROC Houston QA Center website (http://irochouston.mdanderson.org)
Phantom Irradiation
The lung phantom available from IROC Houston must be successfully irradiated (if the institution has not previously met this credentialing requirement). Tomotherapy and Cyberknife treatment delivery modalities must be credentialed individually. Instructions for requesting and irradiating the phantom are available on the IROC Houston website under credentialing (http//irochouston.mdanderson.org). The phantom must be irradiated with the motion management technique (e.g., tracking, gating) to be used for protocol treatments.
Credentialing Notification
IROC Houston QA Center will notify the a) institution and b) IROC Rhode Island once credentialing requirements have been met.
13.0 REGISTRATION GUIDELINES
13.1 Registration Timing Patients must be registered to Step 1 prior to initiation of radiation treatment (no more than 28 calendar days prior to planned start of radiation treatment).
Patients must be registered to Step 2 prior to initiation of atezolizumab treatment (no more than 7 calendar days prior to planned start of atezolizumab).
13.2 CTEP Registration Procedures
Food and Drug Administration (FDA) regulations and National Cancer Institute (NCI) policy require all individuals contributing to NCI-sponsored trials to register and to renew their registration annually. To register, all individuals must obtain a Cancer Therapy Evaluation Program (CTEP) Identity and Access Management (IAM) account at https://ctepcore.nci.nih.gov/iam. In addition, persons with a registration type of Investigator (IVR), Non-Physician Investigator (NPIVR), or Associate Plus (AP) (i.e., clinical site staff requiring write access to OPEN, Rave, or acting as a primary site contact) must complete their annual registration using CTEP’s web-based Registration and Credential Repository (RCR) at https://ctepcore.nci.nih.gov/rcr. RCR utilizes five-person registration types. • IVR — MD, DO, or international equivalent; • NPIVR — advanced practice providers (e.g., NP or PA) or graduate level researchers
(e.g., Ph.D.; • AP — clinical site staff (e.g., RN or CRA) with data entry access to CTSU applications
(e.g., Roster Update Management System (RUMS), OPEN, Rave,); • Associate (A) — other clinical site staff involved in the conduct of NCI-sponsored trials;
and • Associate Basic (AB) — individuals (e.g., pharmaceutical company employees) with
limited access to NCI-supported systems.
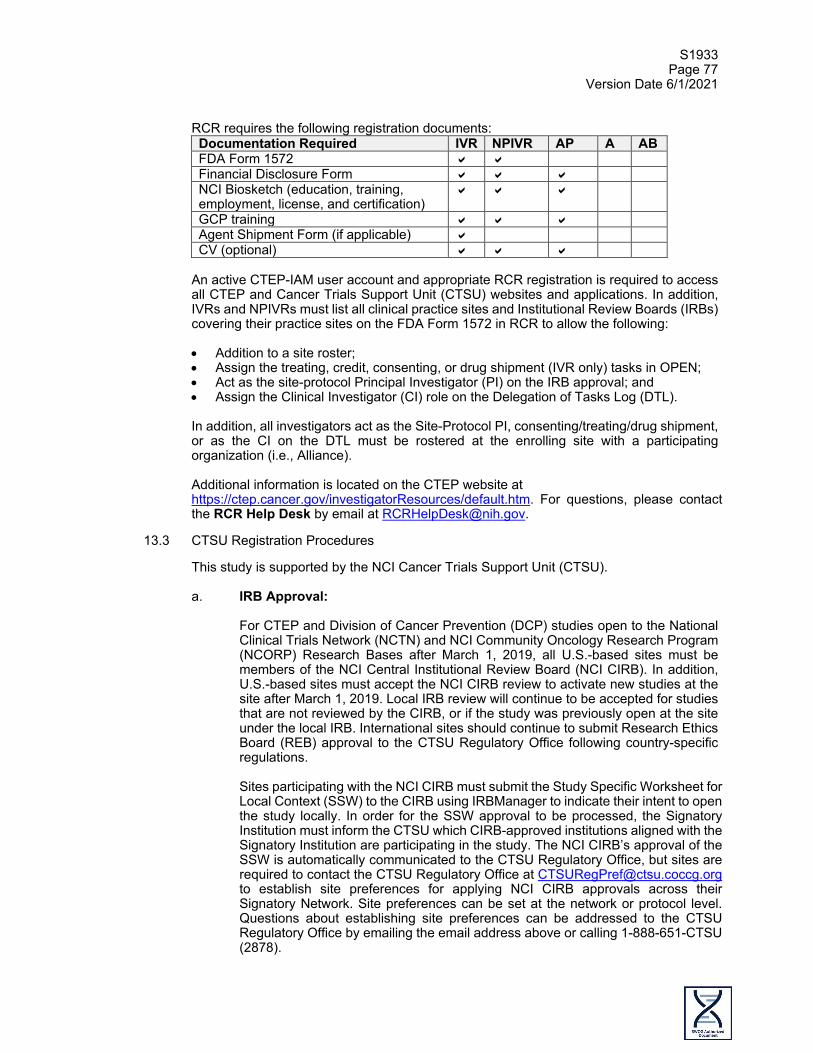
S1933 Page 77
Version Date 6/1/2021
RCR requires the following registration documents: Documentation Required IVR NPIVR AP A AB FDA Form 1572 Financial Disclosure Form NCI Biosketch (education, training, employment, license, and certification)
GCP training Agent Shipment Form (if applicable) CV (optional)
An active CTEP-IAM user account and appropriate RCR registration is required to access all CTEP and Cancer Trials Support Unit (CTSU) websites and applications. In addition, IVRs and NPIVRs must list all clinical practice sites and Institutional Review Boards (IRBs) covering their practice sites on the FDA Form 1572 in RCR to allow the following: • Addition to a site roster; • Assign the treating, credit, consenting, or drug shipment (IVR only) tasks in OPEN; • Act as the site-protocol Principal Investigator (PI) on the IRB approval; and • Assign the Clinical Investigator (CI) role on the Delegation of Tasks Log (DTL). In addition, all investigators act as the Site-Protocol PI, consenting/treating/drug shipment, or as the CI on the DTL must be rostered at the enrolling site with a participating organization (i.e., Alliance). Additional information is located on the CTEP website at https://ctep.cancer.gov/investigatorResources/default.htm. For questions, please contact the RCR Help Desk by email at [email protected].
13.3 CTSU Registration Procedures
This study is supported by the NCI Cancer Trials Support Unit (CTSU). a. IRB Approval:
For CTEP and Division of Cancer Prevention (DCP) studies open to the National Clinical Trials Network (NCTN) and NCI Community Oncology Research Program (NCORP) Research Bases after March 1, 2019, all U.S.-based sites must be members of the NCI Central Institutional Review Board (NCI CIRB). In addition, U.S.-based sites must accept the NCI CIRB review to activate new studies at the site after March 1, 2019. Local IRB review will continue to be accepted for studies that are not reviewed by the CIRB, or if the study was previously open at the site under the local IRB. International sites should continue to submit Research Ethics Board (REB) approval to the CTSU Regulatory Office following country-specific regulations. Sites participating with the NCI CIRB must submit the Study Specific Worksheet for Local Context (SSW) to the CIRB using IRBManager to indicate their intent to open the study locally. In order for the SSW approval to be processed, the Signatory Institution must inform the CTSU which CIRB-approved institutions aligned with the Signatory Institution are participating in the study. The NCI CIRB’s approval of the SSW is automatically communicated to the CTSU Regulatory Office, but sites are required to contact the CTSU Regulatory Office at [email protected] to establish site preferences for applying NCI CIRB approvals across their Signatory Network. Site preferences can be set at the network or protocol level. Questions about establishing site preferences can be addressed to the CTSU Regulatory Office by emailing the email address above or calling 1-888-651-CTSU (2878).

S1933 Page 78
Version Date 6/1/2021
In addition, the Site-Protocol Principal Investigator (PI) (i.e. the investigator on the IRB/REB approval) must meet the following criteria to complete processing of the IRB/REB approval record:
• Holds an Active CTEP status; • Rostered at the site on the IRB/REB approval (applies to US and Canadian
sites only) and on at least one participating roster; • If using NCI CIRB, rostered on the NCI CIRB Signatory record; • Includes the IRB number of the IRB providing approval in the Form FDA
1572 in the RCR profile; and • Holds the appropriate CTEP registration type for the protocol.
Additional Requirements Assignment of site registration status in the CTSU Regulatory Support System (RSS) uses extensive data to make a determination of whether a site has fulfilled all regulatory criteria including but not limited to: the following:
• An active Federal Wide Assurance (FWA) number; • An active roster affiliation with the Lead Protocol Organization (LPO) or a
Participating Organization (PO); and • Compliance with all protocol-specific requirements (PSRs).
b. Protocol Specific Requirements (PSR) for S1933 Site Registration
1. PSR for Radiation and/or Imaging (RTI) Component
This is a study with a radiation and/or imaging (RTI) component and the enrolling site must be aligned to an RTI provider. To manage provider associations or to add or remove associated providers, access the Provider Association page from the Regulatory section on the CTSU members’ website at https://www.ctsu.org/RSS/RTFProviderAssociation. Sites must be linked to at least one Imaging and Radiation Oncology Core (IROC) provider to participate on trials with an RTI component. Enrolling sites are responsible for ensuring that the appropriate agreements and IRB approvals are in place with their RTI provider. A primary role on any roster is required to update provider associations, though all individuals at a site may view provider associations. To find who holds primary roles at your site, please view the Person Roster Browser under the RUMS link on the CTSU website.
2. PSR for RT Modality Credentialing (IROC-Houston)
IROC Credentialing Status Inquiry (CSI) Form – this form is submitted to IROC Houston to verify credentialing status or to begin a new modality credentialing process.
3. PSR for Radiation or Imaging (RT/I) Provider Credentialed (IROC Integration suite) To complete protocol-specific credentialing the RTI provider or enrolling site should follow instructions in the protocol to submit documentation or other materials to the designated IROC Quality Assurance (QA) center. Upon the IROC QA center approving the RTI provider for the study modality, IROC will automatically send the approval to the Regulatory Support System (RSS) to comply the protocol specific requirement. IROC will continue to copy the provider and/or enrolling site on modality approvals.

S1933 Page 79
Version Date 6/1/2021
Upon site registration approval in RSS, the enrolling site may access OPEN to complete enrollments. The enrolling site will select their credentialed provider treating the subject in the OPEN credentialing screen and may need to answer additional questions related to treatment in the eligibility checklist.
c. Downloading Site Registration Documents:
Download the site registration forms from the protocol-specific page located on the CTSU members’ website. Permission to view and download this protocol and its supporting documents is restricted and is based on person and site roster assignment. To participate, the institution and its associated investigators and staff must be associated with the LPO or a PO on the protocol.
• Log on to the CTSU members’ website (https://www.ctsu.org) using your CTEP-IAM username and password;
• Click on Protocols in the upper left of your screen o Enter the protocol number in the search field at the top of the
protocol tree, or o Click on the By Lead Organization folder to expand, then select
[Corresponding Organization], and protocol number [NCI Protocol #];
• Click on Documents, select Site Registration, and download and complete the forms provided. (Note: For sites under the CIRB initiative, IRB data will load automatically to the CTSU as described above.)
d. Submitting Regulatory Documents:
Submit required forms and documents to the CTSU Regulatory Office via the Regulatory Submission Portal on the CTSU website. To access the Regulatory Submission Portal log on to the CTSU members’ website Regulatory Regulatory Submission. Institutions with patients waiting that are unable to use the Regulatory Submission Portal should alert the CTSU Regulatory Office immediately at 1-866-651-2878 in order to receive further instruction and support.
e. Checking Your Site’s Registration Status:
You can verify your site’s registration status on the members’ side of the CTSU website.
• Log on to the CTSU members’ website; • Click on Regulatory at the top of your screen; • Click on Site Registration; • Enter your 5-character CTEP Institution Code and click on Go.
Note: The status shown only reflects institutional compliance with site registration requirements as outlined above. It does not reflect compliance with protocol requirements for individuals participating on the protocol or the enrolling investigator’s status with the NCI or their affiliated networks.
13.4 Oncology Patient Enrollment Network (OPEN) Registration Requirements
The individual registering the patient must have completed the appropriate SWOG Registration Worksheet. The completed form must be referred to during the registration but should not be submitted as part of the patient data.

S1933 Page 80
Version Date 6/1/2021
The Oncology Patient Enrollment Network (OPEN) is a web-based registration system available on a 24/7 basis. OPEN is integrated with CTSU regulatory and roster data and with the Lead Protocol Organization (LPOs) registration/randomization systems or Theradex Interactive Web Response System (IWRS) for retrieval of patient registration/randomization assignment. OPEN will populate the patient enrollment data in NCI’s clinical data management system, Medidata Rave. Requirements for OPEN access:
• A valid CTEP-IAM account; • To perform enrollments or request slot reservations: Be on a LPO roster, ETCTN
Corresponding roster, or PO roster with the role of Registrar. Registrars must hold a minimum of an AP registration type;
• If a Delegation of Tasks Log (DTL) is required for the study, the registrar(s) must hold the OPEN Registrar task on the DTL for the site; and
• Have an approved site registration for a protocol prior to patient enrollment.
To assign an Investigator (IVR) or Non-Physician Investigator (NPIVR) as the treating, crediting, consenting, drug shipment (IVR only), or receiving investigator for a patient transfer in OPEN, the IVR or NPIVR must list the IRB number used on the site’s IRB approval on their Form FDA 1572 in RCR. If a DTL is required for the study, the IVR or NPIVR must be assigned the appropriate OPEN-related tasks on the DTL. Prior to accessing OPEN, site staff should verify the following:
• Patient has met all eligibility criteria within the protocol stated timeframes and the affirmation of eligibility on the Registration Worksheet has been signed by the registering investigator or another investigator designate. Site staff should refer to Section 5.0 to verify eligibility.
• All patients have signed an appropriate consent form and HIPAA authorization form (if applicable).
OPEN will also ask additional questions that are not present on the SWOG Registration Worksheet. The individual registering the patient must be prepared to provide answers to the following questions: a. Institution CTEP ID b. Protocol Number c. Registration Step d. Treating Investigator e. Credit Investigator f. Patient Initials
g. Patient’s Date of Birth
h. Patient SSN (SSN is desired, but optional. Do not enter invalid numbers.)
i. Country of Residence
j. ZIP Code
k. Gender (select one):
• Female Gender • Male Gender

S1933 Page 81
Version Date 6/1/2021
l. Ethnicity (select one):
• Hispanic or Latino • Not Hispanic or Latino • Unknown
m. Method of Payment (select one): • Private Insurance • Medicare • Medicare and Private Insurance • Medicaid • Medicaid and Medicare • Military or Veterans Sponsored NOS • Military Sponsored (Including Champus & Tricare) • Veterans Sponsored • Self Pay (No Insurance) • No Means of Payment (No Insurance) • Other • Unknown
n. Race (select all that apply): • American Indian or Alaska Native • Asian • Black or African American • Native Hawaiian or other Pacific Islander • White • Unknown
Note: The OPEN system will provide the site with a printable confirmation of registration and treatment information. Please print this confirmation for your records. Access OPEN at https://open.ctsu.org or from the OPEN link on the CTSU members’ website. Further instructional information is in the OPEN section of the CTSU website at https://www.ctsu.org, https://open.ctsu.org, or from the OPEN Patient Registration link on the SWOG CRA Workbench. For any additional questions, contact the CTSU Help Desk at 1-888-823-5923 or [email protected].
13.5 Exceptions to SWOG registration policies will not be permitted.
a. Patients must meet all eligibility requirements.
b. Institutions must be identified as approved for registration.
c. Registrations may not be cancelled.
d. Late registrations (after initiation of treatment) will not be accepted.
14.0 DATA SUBMISSION SCHEDULE
14.1 Data Submission Requirement
Data must be submitted according to the protocol requirements for ALL patients registered, whether or not assigned treatment is administered, including patients deemed to be ineligible. Patients for whom documentation is inadequate to determine eligibility will generally be deemed ineligible.

S1933 Page 82
Version Date 6/1/2021
14.2 Master Forms
Master forms can be found on the protocol abstract page on the SWOG website (www.swog.org) and the CTSU website (www.ctsu.org) and (with the exception of the sample consent form and the Registration Worksheet) must be submitted on-line via the Web; see below for details.
14.3 Data Submission Procedures
a. Data collection for this study will be done exclusively through the Medidata Rave®
clinical data management system. Medidata Rave is a clinical data management system being used for data collection for this trial/study. Access to the trial in Rave is controlled through the CTEP-IAM system and role assignments. To access Rave via iMedidata: • Site staff will need to be registered with CTEP and have a valid and active
CTEP-IAM account; and • Assigned one of the following Rave roles on the relevant Lead Protocol
Organization (LPO) or Participating Organization roster at the enrolling site: Rave CRA, Rave Read Only, Rave CRA (LabAdmin), Rave SLA, or Rave Investigator. Refer to https://ctep.cancer.gov/investigatorResources/default.htm for registration types and documentation required.
o To hold Rave CRA or Rave CRA (Lab Admin) role, site staff must hold a minimum of an AP registration type;
o To hold Rave Investigator role, the individual must be registered as an NPIVR or IVR; and
o To hold Rave Read Only role, site staff must hold an Associates (A) registration type.
Upon initial site registration approval for the study in Regulatory Support System (RSS), all persons with Rave roles assigned on the appropriate roster will be sent a study invitation e-mail from iMedidata. To accept the invitation, site staff must log into the Select Login (https://login.imedidata.com/selectlogin) using their CTEP-IAM user name and password, and click on the “accept” link in the upper right-corner of the iMedidata page. Site staff will not be able to access the study in Rave until all required Medidata and study specific trainings are completed. Trainings will be in the form of electronic learnings (eLearnings), and can be accessed by clicking on the link in the upper right pane of the iMedidata screen. If an eLearning is required and has not yet been taken, the link to the eLearning will appear under the study name in iMedidata instead of the Rave EDC link; once the successful completion of the eLearning has been recorded, access to the study in Rave will be granted, and a Rave EDC link will display under the study name. Site staff that have not previously activated their iMedidata/Rave account at the time of initial registration approval for the study in RSS will also receive a separate invitation from iMedidata to activate their account. Account activation instructions are located on the CTSU website, Rave tab under the Rave resource materials (Medidata Account Activation and Study Invitation Acceptance). Additional information on iMedidata/Rave is available on the CTSU members’ website in the Data Management > Rave section at www.ctsu.org/RAVE/ or by contacting the CTSU Help Desk at 1-888-823-5923 or by e-mail at [email protected].
b. You may also access Rave® via the SWOG CRA Workbench via the SWOG
website (www.swog.org).
For difficulties with the CRA Workbench, please email [email protected].

S1933 Page 83
Version Date 6/1/2021
c. Institutions participating through the Cancer Trials Support Unit (CTSU), please
refer to the CTSU Participation Table. 14.4 Data Submission Overview and Timepoints
a. STEP 1 REGISTRATION: RADIATION
1. WITHIN 15 DAYS OF STEP 1 REGISTRATION, SUBMIT:
Vital Status Form S1933 Eligibility Criteria Step 1
S1933 Onstudy Form
Baseline Abnormalities Form
Baseline Tumor Assessment Form (RECIST 1.1)
Radiology reports from all scans performed to assess disease at baseline**
Pathology report documenting cytological or histologic confirmation of Stage II-III NSCLC**
**NOTE: Upload reports via the Source Documentation: Baseline form in Rave®.
2. AT TIME POINTS SPECIFIED IN SECTION 15, IF PATIENT CONSENTS:
Submit specimens 3. WITHIN 5 DAYS PRIOR TO PLANNED START OF RADIATION
THERAPY:
Submit materials for radiation treatment plan review as described in Section 12.1a.
4. WITHIN 7 DAYS OF COMPLETION OF RADIATION THERAPY:
Submit materials for radiation treatment plan review as describe in Section 12.1a.
5. WITHIN 15 DAYS AFTER COMPLETION OF RADIATION THERAPY, SUBMIT:
Vital Status Form S1933 Radiation Therapy Summary Form S1933 Adverse Event Form

S1933 Page 84
Version Date 6/1/2021
6. WITHIN 15 DAYS AFTER DISEASE ASSESSMENT 2-5 WEEKS AFTER COMPLETION OF RADIATION, SUBMIT:
Vital Status Form Follow-Up Tumor Assessment Form S1933 Post Radiation Evaluation Form Radiology reports from all scans performed to assess disease*.
*NOTE: Upload reports via the Source Documentation: Follow-Up form in Rave®
If patient meets the eligibility for step 2 (Section 5.2), complete the Step2 registration in OPEN.
b. STEP 2 REGISTRATION: ATEZOLIZUMAB
1. WITHIN 15 DAYS OF STEP 2 REGISTRATION SUBMIT:
Vital Status Form S1933 Eligibility Criteria Step 2 S1933 Onstudy Step 2 Form
2. WITHIN 15 DAYS AFTER EACH CYCLE OF ATEZOLIZUMAB TREATMENT (1 CYCLE = 21 DAYS) SUBMIT:
Vital Status Form S1933 Atezolizumab Treatment Form
S1933 Adverse Event Form
3. WITHIN 15 DAYS OF DISCONTINUATION OF PROTOCOL
TREATMENT, SUBMIT: Vital Status Form Off Treatment Notice documenting reasons for off protocol treatment S1933 Atezolizumab Treatment Form S1933 Adverse Event Form
4. WITHIN 14 DAYS AFTER EVERY DISEASE ASSESSMENT (INCLUDE BOTH ON TREATMENT AND OFF TREATMENT PRIOR TO DISEASE PROGRESSION), SUBMIT: Vital Status Form Follow-Up Tumor Assessment Form Radiology reports from all scans performed to assess disease*. *NOTE: Upload reports via the Source Documentation: Follow-Up form in Rave®

S1933 Page 85
Version Date 6/1/2021
5. WITHIN 30 DAYS OF PROGRESSION OR RELAPSE, SUBMIT:
Vital Status Form Site(s) of Progression or Relapse Form Follow-Up Tumor Assessment Form Radiology reports from all scans performed to assess disease*. If patient was on protocol treatment at the time of progression, also submit the materials specified in Section 14.4b.3. *NOTE: Upload reports via the Source Documentation: Follow-Up form in Rave®.
6. ONCE OFF ALL PROTOCOL TREATMENT SUBMIT EVERY 6 MONTHS
FOR THE FIRST 2 YEARS, THEN YEARLY FOR A TOTAL OF 3 YEARS FROM INITIAL REGISTRATION:
Vital Status Form Follow-Up Form Late Adverse Events (if prior to treatment for progression or relapse or a second primary, and prior to non-protocol treatment, the patient experiences any severe [Grade ≥ 3] long term toxicity that has not been previously reported).
7. WITHIN 30 DAYS OF KNOWLEDGE OF DEATH: Notice of Death and all of the items listed in Section 14.4b.3 (if the patient was still on protocol treatment) or Follow-Up Form (if the patient was off protocol treatment) documenting death information
c. FOR PATIENTS WHO DO NOT GO TO STEP2:
1. WITHIN 15 DAYS OF DISCONTINUATION OF PROTOCOL
TREATMENT AND DECIDE NOT GO TO STEP 2, SUBMIT:
Vital Status Form Off Treatment Notice documenting reasons for off protocol treatment S1933 Radiation Therapy Form S1933 Adverse Event Form
2. WITHIN 15 DAYS AFTER EVERY DISEASE ASSESSMENT, SUBMIT:
Vital Status Form Follow-Up Tumor Assessment Form (RECIST 1.1) Radiology reports from all scans performed to assess disease*.

S1933 Page 86
Version Date 6/1/2021
*NOTE: Upload reports via the Source Documentation: Follow-Up form in Rave®
3. WITHIN 30 DAYS OF PROGRESSION OR RELAPSE, SUBMIT:
Vital Status Form Site(s) of Progression or Relapse Form Follow-Up Tumor Assessment Form Radiology reports from all scans performed to assess disease*. If patient was on protocol treatment at the time of progression, also submit the materials specified in Section 14.4c.1.
*NOTE: Upload reports via the Source Documentation: Follow-Up form in Rave®.
4. ONCE OFF ALL PROTOCOL TREATMENT SUBMIT EVERY 6 MONTHS
FOR THE FIRST 2 YEARS, THEN YEARLY FOR A TOTAL OF 3 YEARS FROM INITIAL REGISTRATION:
Vital Status Form Follow-Up Form Late Adverse Events (if prior to treatment for progression or relapse or a second primary, and prior to non-protocol treatment, the patient experiences any severe [Grade ≥ 3] long term toxicity that has not been previously reported).
5. WITHIN 30 DAYS OF KNOWLEDGE OF DEATH:
Notice of Death and all of the items listed in Section 14.4 c1 (if the patient was still on protocol treatment) or Follow-Up Form (if the patient was off protocol treatment) documenting death information
15.0 SPECIAL INSTRUCTIONS
15.1 Specimen Submission for Banking (REQUIRED IF PARTICIPANT CONSENTS)
Sites are required to seek additional participant consent for the submission of specimens for banking. With participant’s consent, specimens must be submitted as described in this section. No translational medicine analyses will be performed until a translational medicine plan is approved by SWOG and CTEP. a. Archived tumor tissue
Within 28 days after Registration Step 1, submit one or two representative formalin-fixed paraffin-embedded (FFPE) tissue block(s) containing tumor from time of diagnosis.

S1933 Page 87
Version Date 6/1/2021
If blocks are unavailable for submission, slides are to be provided according to the following requirements: • One (1) H&E slide • Minimum of 20 consecutive, recently obtained (< 2 weeks), unstained 10-
micron thick FFPE sections placed in air-dried uncharged slides. Tumor content in each specimen >30%.
Specimen size requirement is as follows: • Surface area > 25mm2 is optimal. Minimum is 5mm2. • Volume: -1mm3 optimal. The success of the research assessments decreases
at suboptimal tissue volume. Block and slides must be labeled according to instructions below.
b. Blood samples Blood is to be collected at the following time points: • Prior to beginning study radiation treatment • After completion of study radiation treatment, but prior to beginning
atezolizumab • At Cycle 3 Day 1, prior to atezolizumab dosing • At progression or at end of study treatment, whichever comes first At each of these time points, collect the following: • Approximately 20 ml of blood in two 10-ml Streck tubes.
o Label blood collection tube according to instructions below. o Heparin should be avoided in pre-collection flush procedures. If
therapeutic heparin dosing contamination is a possibility, then venipuncture is recommended as a first choice collection method. If a Streck tube immediately follows a heparin tube in the draw order, then collecting an EDTA tube as a waste tube prior to collection in the Streck tube is recommended.
o Fill the tube completely (10 mL) o After collection, immediately mix by gentle inversion 8-10 times.
Inadequate or delayed mixing may result in inaccurate test results. o After collection, blood Streck tubes must never be refrigerated, as cold
temperatures damage the tube reagent. Keep at room temperature. o Do not process. Ship overnight on the same day as collection at
room temperature.
Collect approximately 10 ml of blood in a green top (sodium heparin) tube. • Label blood collection tube according to instructions below. • Gently invert 8-10 times to mix. • Do not process. Ship overnight on the same day as collection at room
temperature. Blood samples can only be drawn Monday through Friday in order to complete processing within 24-48 hours. When shipping blood on Friday, please ship for Saturday Delivery.
c. Specimen collection kits will be provided for the blood collection/submission. Kits
may be ordered by using the SWOG Biospecimen Bank Kit Management Application at http://ricapps.nationwidechildrens.org/KitManagement. Sites will use institutional supplies for the tissue submission.

S1933 Page 88
Version Date 6/1/2021
d. SWOG Specimen Tracking System (STS)
All specimen submissions for this study must be entered and tracked using the SWOG online Specimen Tracking system. SWOG members may log on the online system via the CRA Workbench. To access the CRA Workbench, go to the SWOG Web site (http://swog.org) Non- SWOG users may log into SpecTrack using their CTSU UserID and password on the SpecTrack login page located at https://spectrack.crab.org (select the option “SWOG – SWOG – CTSU”). SpecTrack start-up instructions (both written and demo) are available after signing in to SpecTrack. A copy of the Shipment Packing List produced by the online Specimen Tracking system should be printed and placed in the pocket of the specimen bag if it has one, or in a separate resealable bag. ALL SPECIMENS MUST BE LOGGED VIA THIS SYSTEM; THERE ARE NO EXCEPTIONS. NOTE: If a specimen had an incomplete submission, this must be documented in the Specimen Tracking System under “Special Instructions” at time of specimen submission. If no specimen was available, this must be documented in the Specimen Tracking System by choosing “Notify that Specimen Cannot be Submitted”. To report technical problems with Specimen Tracking, such as database errors or connectivity issues, please send an email to [email protected]. For procedural help with logging and shipping specimens, there is an introduction to the system on the Specimen Tracking main page (https://spectrack.crab.org/Instructions); or contact the SWOG Statistics and Data Management Center at 206/652-2267 to be routed to the Data Coordinator for further assistance. In the online specimen tracking system, the appropriate SWOG laboratory for this study is:
Lab #201: SWOG Biospecimen Bank – Solid Tissue, Myeloma & Lymphoma
Division Phone: 614/722-2865 Email: [email protected]
e. Specimen Labeling
Liquid specimens must be labeled with the following: • SWOG patient number • Patient initials • Collection date (date the specimen was collected from the patient) • Specimen type (e.g. blood, serum, etc.)
Solid tissue specimens must be labeled with the following: • SWOG patient number • Patient initials • Collection date or procedure date • Specify whether tissue is from primary (P) or metastatic (M) • Surgical Pathology ID # (Accession#) and block number (e.g. A2, 3E, etc.)
must be on both the specimen label and the pathology report for the Bank to adequately match the specimen with any findings in the pathology report. If

S1933 Page 89
Version Date 6/1/2021
slides are submitted instead of blocks, they should be labeled with the section number.
f. Specimen Shipping Instructions
Refer to the SWOG Specimen Submission webpage for instructions regarding specimen packaging and shipment: (https://www.swog.org/clinical-trials/biospecimen-resources/biospecimen-processing-and-submission-procedures)
16.0 ETHICAL AND REGULATORY CONSIDERATIONS
The following must be observed to comply with Food and Drug Administration regulations for the conduct and monitoring of clinical investigations; they also represent sound research practice: Informed Consent The principles of informed consent are described by Federal Regulatory Guidelines (Federal Register Vol. 46, No. 17, January 27, 1981, part 50) and the Office for Protection from Research Risks Reports: Protection of Human Patients (Code of Federal Regulations 45 CFR 46). They must be followed to comply with FDA regulations for the conduct and monitoring of clinical investigations. Institutional Review This study must be approved by an appropriate institutional review committee as defined by Federal Regulatory Guidelines (Ref. Federal Register Vol. 46, No. 17, January 27, 1981, part 56) and the Office for Protection from Research Risks Reports: Protection of Human Patients (Code of Federal Regulations 45 CFR 46). Drug Accountability An investigator is required to maintain adequate records of the disposition of investigational drugs according to procedures and requirements governing the use of investigational new drugs as described in the Code of Federal Regulations 21 CFR 312. Publication and Industry Contact The agent(s) supplied by CTEP, DCTD, NCI used in this protocol is/are provided to the NCI under a Collaborative Agreement (CRADA, CTA, CSA) between Merck (hereinafter referred to as "Collaborator(s)") and the NCI Division of Cancer Treatment and Diagnosis. Therefore, the following obligations/guidelines in addition to the provisions in the "Intellectual Property Option to Collaborator" (http://ctep.cancer.gov/industryCollaborations2/intellectual_property.htm) contained within the terms of award apply to the use of the Agent in this study:
1. Agent(s) may not be used for any purpose outside the scope of this protocol, nor can
Agent(s) be transferred or licensed to any party not participating in the clinical study. Collaborator(s) data for Agent(s) are confidential and proprietary to Collaborator(s) and shall be maintained as such by the investigators. The protocol documents for studies utilizing Agents contain confidential information and should not be shared or distributed without the permission of the NCI. If a copy of this protocol is requested by a patient or patient’s family member participating on the study, the individual should sign a confidentiality agreement. A suitable model agreement can be downloaded from: http://ctep.cancer.gov.

S1933 Page 90
Version Date 6/1/2021
2. For a clinical protocol where there is an investigational Agent used in combination with
(an)other investigational Agent(s), each the patient of different Collaborative Agreements, the access to and use of data by each Collaborator shall be as follows (data pertaining to such combination use shall hereinafter be referred to as "Multi-Party Data"):
a. NCI will provide all Collaborators with prior written notice regarding the existence
and nature of any agreements governing their collaboration with NCI, the design of the proposed combination protocol, and the existence of any obligations which would tend to restrict NCI's participation in the proposed combination protocol.
b. Each Collaborator shall agree to permit use of the Multi-Party Data from the clinical trial by any other Collaborator solely to the extent necessary to allow said other Collaborator to develop, obtain regulatory approval or commercialize its own Agent.
c. Any Collaborator having the right to use the Multi-Party Data from these trials must agree in writing prior to the commencement of the trials that it will use the Multi- Party Data solely for development, regulatory approval, and commercialization of its own Agent.
3. Clinical Trial Data and Results and Raw Data developed under a Collaborative Agreement
will be made available exclusively to Collaborator(s), the NCI, and the FDA, as appropriate and unless additional disclosure is required by law or court order as described in the IP Option to Collaborator (http://ctep.cancer.gov/industryCollaborations2/intellectual_property.htm). Additionally, all Clinical Data and Results and Raw Data will be collected, used and disclosed consistent with all applicable federal statutes and regulations for the protection of human patients, including, if applicable, the Standards for Privacy of Individually Identifiable Health Information set forth in 45 C.F.R. Part 164.
4. When a Collaborator wishes to initiate a data request, the request should first be sent to
the NCI, who will then notify the appropriate investigators (Group Chair for Cooperative Group studies, or PI for other studies) of Collaborator's wish to contact them.
5. Any data provided to the Collaborator(s) for Phase III studies must be in accordance with
the guidelines and policies of the responsible Data Monitoring Committee (DMC), if there is a DMC for this clinical trial.
6. Any manuscripts reporting the results of this clinical trial must be provided to CTEP by the
Group office for Cooperative Group studies or by the principal investigator for non- Cooperative Group studies for immediate delivery to Collaborator(s) for advisory review and comment prior to submission for publication. Collaborator(s) will have 30 days from the date of receipt for review. Collaborator shall have the right to request that publication be delayed for up to an additional 30 days in order to ensure that Collaborator's confidential and proprietary data, in addition to the Collaborator(s)'s intellectual property rights, are protected. Copies of abstracts must be provided to CTEP for forwarding to Collaborator(s) for courtesy review as soon as possible and preferably at least three (3) days prior to submission, but in any case, prior to presentation at the meeting or publication in the proceedings. Press releases and other media presentations must also be forwarded to CTEP prior to release. Copies of any manuscript, abstract and/or press release/media presentation should be sent to:
E-mail: [email protected]
The Regulatory Affairs Branch will then distribute them to the Collaborator(s). No publication, manuscript or other form of public disclosure shall contain any of the Collaborator's confidential/proprietary information.

S1933 Page 91
Version Date 6/1/2021
Monitoring Data for this study will be submitted via the Data Mapping Utility (DMU). Cumulative protocol- and patient-specific data will be submitted weekly to CTEP electronically via the DMU. DMU Complete reporting consists of Patient Demographics, Baseline Abnormalities, On/Off Treatment/Study Status, Treatment/Course/Dosing information, Adverse Events, Late Adverse Events, and Response data as applicable. Instructions for setting up and submitting data via DMU are available on the CTEP Website: (https://ctep.cancer.gov/protocolDevelopment/dmu.html). Note: All adverse events (both routine and serious) that meet the protocol mandatory reporting requirements must be reported via DMU in addition to expedited reporting of serious adverse events via CTEP-AERS. Confidentiality Please note that the information contained in this protocol is considered confidential and should not be used or shared beyond the purposes of completing protocol requirements until or unless additional permission is obtained.

S1933 Page 92
Version Date 6/1/2021
17.0 BIBLIOGRAPHY 1 Siegel RL, Miller KD, Jemal A, et al. Cancer Statistics 2016. CA Cancer J Clin 2016; 66:7. 2 Blagden SP, Charman SC, Sharples LD, et al. Performance Status Score: Do Patients and Their
Oncologists Agree? Br J Cancer 89:1022. 3 Albain KS, Swann RS, Rusch VW, et al. Radiotherapy plus Chemotherapy with or without Surgical
Resection for Stage III Non-Small-Cell Lung Cancer: A Phase III Randomized Trial. Lancet 2009; 374:379.
4 Davies AM, Chansky K, Derick HM, et al. Phase II Study of Consolidation Paclitaxel After
Concurrent Chemoradiotherapy in Poor-Risk Stage III Non-Small-Cell Lung Cancer: SWOG S9712. J Clin Oncol 2006; 24:5242.
5 Robinson AG, Young K, Balchin K, et al. Reasons for Palliative Treatment in Stage III Non-Small-
Cell Lung Cancer: What Contribution is Made by Time-Dependent Changes in Tumor or Patient Status? Curr Oncol 2015; 22:399.
6 Curran WJ, Paulus R, Langer CJ, et al. Sequential vs Concurrent Chemoradiation for Stage III Non-
Small Cell Lung Cancer: Randomized Phase III Trial RTOG 9410. J Natl Cancer Inst. 2011; 103:1452.
7 Kim E, Daly ME, Westover KD, et al. Modern Non-Operative Treatment Strategies for Patients with
Stage II-III Non-Small Cell Lung Cancer in the United States. Int J Radiat Oncol Biol Phys; 99(25): Suppl 2017 E469.
8 Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus Docetaxel in Patients with
Previously Treated Non-Small-Cell Lung Cancer (OAK): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet 2017; 389(10066):255.
9 Fehrenbacher L, Von Pawel J, Park K, et al. Updated Efficacy Analysis Including Secondary
Population Results for OAK: A Randomized Phase III Study of Atezolizumab versus Docetaxel in Patients with Previously Treated Advanced Non-Small Cell Lung Cancer. J Thorac Oncol 2018; 13(8):1156.
10 Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for First-Line Treament of Metastatic
Nonsquamous NSCLC. N Engl J Med 2018; 378(24):2288 11 Horn L, Mansfield AS, Szczesna A, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-
Stage Small-Cell Lung Cancer. N Engl J Med 2018; 379(23):2220. 12 Antonia SJ, Villegas A, Daniel D, et al. Durvalumab After Chemoradiotherapy in Stage III Non-
Small-Cell Lung Cancer. N Engl J Med 2017. 13 Antonia SJ, Villegas A, Daniel D, et al. Overall Survival with Durvalumab after Chemoradiotherapy
in Stage III NSCLC. N Engl J Med September 25th, 2018. 14 Lin SH, Lin Y, Price J, et al. DETERRED: PD-L1 Blockade to Evaluate the Safety of Lung Cancer
Therapy Using Carboplatin, Paclitaxel, and Radiation Combined with MPDL3280A (atezolizumab). J Clin Oncol 35,2017 (suppl; abstr 3064).
15 Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus Docetaxel in Patients with
Previously Treated Non-Small-Cell Lung Cancer (OAK): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet 2017; 389(10066):255.

S1933 Page 93
Version Date 6/1/2021
16 Sause WT, Scott Ch, Taylor S, et al. Radiation Therapy Oncology Group (RTOG) 88-08 and
Eastern Cooperative Oncology Group (ECOG) 4588: Preliminary Results of a Phase III Trial in Regionally Advanced, Unresectable Non-Small Cell Lung Cancer. J Natl Cancer Inst 1995; 87:198.
17 Iyengar KD, Westover LE, Court MK, et al. A Phase III Randomized Study of Image Guided
Conventional (60 Gy/30fx) Versus Accelerated Hypofractionated (60Gy/15fx) Radiation for Poor Performance Status Stage II and III NSCLC Patients-An Interim Analysis. Int J Radiat Oncol Biol Phys; 96(2): E451.
18 Spigel et al. Poster presentation, 17th World Conference on Lung Cancer; December 4-7, Vienna,
Austria). 19 Antonia SJ, Villegas A, Daniel D, et al. Durvalumab After Chemoradiotherapy in Stage III Non-
Small-Cell Lung Cancer. N Engl J Med 2017. 20 Antonia SJ, Villegas A, Daniel D, et al. Overall Survival with Durvalumab after Chemoradiotherapy
in Stage III NSCLC. N Engl J Med September 25th, 2018. 21 Davies AM, Chansky K, Derick HM, et al. Phase II Study of Consolidation Paclitaxel After
Concurrent Chemoradiotherapy in Poor-Risk Stage III Non-Small-Cell Lung Cancer: SWOG S9712. J Clin Oncol 2006; 24:5242.
22 Cardenal F, Nadal E, Jove M, et al. Concurrent Systemic Therapy with Radiotherapy for the
Treatment of Poor-Risk Patients with Unresectable stage III Non-Small-Cell Lung Cancer: A Review of the Literature. Annals Oncol 2015; 26:278.
23 Warner A, Dahele M, Hu B, et al. Factors Associated with Early Mortality in Patients With
Concurrent Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. Int J Radiation Oncol Biol Phys 2016; 94:612.
24 Liew MS, Sia J, Starmans MH, et al. Comparison of Toxicity and Outcomes of Concurrent
Radiotherapy with Carboplatin/Paclitaxel or Cisplatin/Etoposide in Stage III Non-Small-Cell Lung Cancer. Cancer Medicine 2013; 2:916.
25 Sharabi AB, Lim M, DeWeese T, et al. Radiation and Checkpoint Blockade Immunotherapy:
Radiosensitisation and Potential Mechanisms of Synergy. Lancet Oncol 2015; 16:e498. 26 Sharabi AB, Lim M, DeWeese T, et al. Radiation and Checkpoint Blockade Immunotherapy:
Radiosensitisation and Potential Mechanisms of Synergy. Lancet Oncol 2015; 16:e498. 27 Reynders K, Illidge T, Siva S, et al. The abscopal effect of local radiotherapy: using immunotherapy
to make a rare event clinically relevant. Cancer Treatment Rev 2015; 41:503. 28 Sharabi AB, Lim M, DeWeese T, et al. Radiation and Checkpoint Blockade Immunotherapy:
Radiosensitisation and Potential Mechanisms of Synergy. Lancet Oncol 2015; 16:e498. 29 Shaverdian N, Lisberg A, Bamazyan K, et al. Previous Radiotherapy and the Clinical Activity and
Toxicity of Pembrolizumab in the Treatment of Non-Small-Cell Lung Cancer: A Secondary Analysis of the KEYNOTE-001 Phase 1 Trial. Lancet Oncol 2017; 18:895.
30 Kang J, Demaria S, Formenti S. Current Clinical Trials Testing the Combination of Immunotherapy
with Radiotherapy. J Immunother Cancer. 2016 Sep 20; 4:51. 31 Cho WK, Noh JM, Ahn YC, et al. Radiation Therapy Alone in cT1-3N0 Non-Small Cell Lung Cancer
Patients Who Are Unfit for Surgical Resection or Stereotactic Radiation Therapy: Comparison of Risk-Adaptive Dose Schedule. Cancer Res Treat. 2016; 48:1187.

S1933 Page 94
Version Date 6/1/2021
32 Warner A, Dahele M, Hu B, et al. Factors Associated With Early Mortality in Patients With
Concurrent Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. Int J Radiation Oncol Biol Phys 2016; 94:612.
33 Steuer CE, Behera M, Ernani V, et al. Comparison of Concurrent Use of Thoracic Radiation With
Either Carboplatin-Paclitaxel or Cisplatin-Etoposide for Patients With Stage III Non-Small-Cell Lung Cancer: A Systematic Review. JAMA Oncol Dec 15, 2016.
34 Fehrenbacher L, Von Pawel J, Park K, et al. Updated Efficacy Analysis Including Secondary
Population Results for OAK: A Randomized Phase III Study of Atezolizumab versus Docetaxel in Patients with Previously Treated Advanced Non-Small Cell Lung Cancer. J Thorac Oncol 2018; 13(8):1156.
35 Kang J, Demaria S, Formenti S. Current Clinical Trials Testing the Combination of Immunotherapy
with Radiotherapy. J Immunother Cancer. 2016 Sep 20; 4:51. 36 Atagi S, Kawahara A, Okamoto H, et al. Thoracic Radiotherapy with or without Daily Low-Dose
Carboplatin in Elderly Patients with Non-Small-Cell Lung Cancer: A Randomised, Controlled, Phase 3 Trial by the Japan Clinical Oncology Group (JCOG0301). Lancet Oncol 2012; 13:671
37 Mohamad O, De Leon AD, Schroeder S, et al. Safety and Efficacy of Concurrent Immune
Checkpoint Inhibitors and Hypofractionated Body Radiotherapy. Oncoimmunology 2018; Vol 7, No 7, e1440168
38 Antonia SJ, Villegas A, Daniel D, et al. Durvalumab After Chemoradiotherapy in Stage III Non-
Small-Cell Lung Cancer. N Engl J Med 2017. 39 Antonia SJ, Villegas A, Daniel D, et al. Overall Survival with Durvalumab after Chemoradiotherapy
in Stage III NSCLC. N Engl J Med September 25th, 2018. 40 Antonia SJ, Villegas A, Daniel D, et al. Durvalumab After Chemoradiotherapy in Stage III Non-
Small-Cell Lung Cancer. N Engl J Med 2017. 41 Antonia SJ, Villegas A, Daniel D, et al. Overall Survival with Durvalumab after Chemoradiotherapy
in Stage III NSCLC. N Engl J Med September 25th, 2018. 42 Iyengar KD, Westover LE, Court MK, et al. A Phase III Randomized Study of Image Guided
Conventional (60 Gy/30fx) Versus Accelerated Hypofractionated (60Gy/15fx) Radiation for Poor Performance Status Stage II and III NSCLC Patients-An Interim Analysis. Int J Radiat Oncol Biol Phys; 96(2): E451.
43 Spigel et al, poster presentation in the 17th World Conference on Lung Cancer; December 4-7,
Vienna, Austria.

S1933 Page 95
Version Date 6/1/2021
18.0 APPENDIX
18.1 Instructions for SWOG Biospecimen Bank

S1933 Page 96
Version Date 6/1/2021
18.1 Instructions for SWOG Biospecimen Bank
No translational medicine analyses will be performed until a translational medicine plan is approved by SWOG and CTEP. Upon receipt of FFPE tissue, the SWOG Biospecimen Bank will co-extract RNA and DNA from the most representative FFPE tissue block (if more than one is received). Following QC, RNA will be stored as 5 aliquots, and DNA will be stored in one stock vial. Nucleic acids will be stored in a -80°C freezer. Remaining FFPE tissue (blocks or slides) will be stored at room temperature. Upon receipt of blood in green top (sodium heparin) tubes, the SWOG Bank will process for peripheral blood mononuclear cells (PBMCs), and slow freeze cell vials in freezing media before long-term storage in a liquid nitrogen vapor phase freezer. Upon receipt of cfDNA Streck tubes, the Bank will process the tubes for plasma and buffy coat. Plasma will be divided into 1-mL aliquots). Both plasma and buffy coat will be stored in a -80°C freezer.