submicroscopic deletion in 14q32.3 through a de novo tandem translocation between 14q and 21p
TRANSCRIPT

Submicroscopic Deletion in 14q32.3 Through a DeNovo Tandem Translocation Between 14q and 21p
Dieter Meschede, Rita Exeler, Barbel Wittwer, and Jurgen Horst*Institute of Human Genetics of the University, Munster, Germany
We describe a male child with craniofacialanomalies, postnatal onset growth retarda-tion, microcephaly, multiple minor anoma-lies, hearing loss, and moderate delay ofmental and statomotor development. Hecarries a previously undescribed tandemtranslocation between the long arm of chro-mosome 14 and the short arm of chromo-some 21 that arose de novo. As proven byfluorescence in situ hybridization a microde-letion not detectable with high-resolutionG-banding occured in 14q32.3, the terminalband on the long arm of chromosome 14. Theresulting phenotype includes most abnor-malities encountered in patients with termi-nal 14q32.3 deletions but in addition in-cludes some characteristics of the ring chro-mosome 14 syndrome. Am. J. Med. Genet.80:443–447, 1998. © 1998 Wiley-Liss, Inc.
KEY WORDS: chromosome 14; chromosome21; tandem translocation; ter-minal deletion
INTRODUCTION
Translocations between acrocentric chromosomesare usually of the Robertsonian type with breakpointslocalized either on both short arms or on one short andone proximal long arm [Gravholt et al., 1992]. In con-trast, tandem type rearrangements juxtaposing theterminal long arm of one acrocentric with the shortarm of another are exceedingly rare. We report the firstcase of a tandem translocation (14q;21p), demonstratea minute deletion of terminal 14q at the translocationbreakpoint, and discuss the resulting phenotype.
CLINICAL REPORT
The patient was born as the second child of healthynonconsanguineous parents. At a gestational age of 35
weeks the infant was delivered by cesarean section forsigns of fetal distress. Apgar scores at 1, 5, and 10minutes were 8, 9, and 10, respectively. Birth length,weight, and head circumference (OFC) corrected forgestational age were at the 50, 10, and 10th centile,respectively.
When first examined by us at age 9 months the boypresented with developmental delay, profound muscu-lar hypotonia, high forehead with mild frontal bossing,apparently low-set and abnormal ears, broad and flatnasal bridge, bilateral epicanthal folds, marked eyelidptosis, long philtrum, down-turned corners of themouth, and micrognathia (Figure 1A and B). The headwas micro- and dolichocephalic. Other findings in-cluded postnatal growth retardation, hypospadias withchordee, wide internipple distance, bilateral transversepalmar creases, and marbled skin. A hemodynamicallyrelevant patent ductus arteriosus was demonstrated byechocardiography.
A follow-up examination at the age of 3 years and 4months showed a pleasant, moderately retarded boy(Figure 1C and D). Motor and social skills were betterthan language abilities (developmental age between 13months for active speech and 22 months for gross mo-tor skills). The patient had learned to walk at the age of21 months, and muscular hypotonia was markedly im-proved. While height was at the 25th centile (95 cm),weight (10.5 kg) and OFC (46.5 cm) were below thethird centile. Marked hearing loss of the mixed typehad necessitated the prescription of hearing aids. Thecharacteristic facial phenotype remained essentiallyunchanged. The parents reported that a single seizurehad occured, triggered by a hypoglycemic episode.There was no indication of epilepsy.
Chromosome analysis performed on G-banded lym-phocyte metaphases showed a tandem translocationbetween the terminal long arm of a chromosome 14 andthe short arm of a chromosome 21 (Fig. 2). Even inhigh-resolution prometaphase spreads a deletion of14q32.3, the terminal band of 14q, could not be dem-onstrated unequivocally. C-banding showed two C-positive bands on the translocation chromosome indi-cating the presence of two centromeres. Fluorescencein situ hybridization (FISH) with a commercially avail-able probe (Oncor, Gaithersburg, MD) specific for thesubtelomeric locus D14S308 yielded a strong fluores-cence signal at the long arm’s end of the normal chro-
*Correspondence to: Prof. Dr. J. Horst, Institute of Human Ge-netics, Vesaliusweg 12-14, D-48149 Munster, Germany. E-mail:[email protected]
Received 12 November 1997; Accepted 26 August 1998
American Journal of Medical Genetics 80:443–447 (1998)
© 1998 Wiley-Liss, Inc.

Fig. 1 (A and B) Patient at age 9 months. (C and D) Patient at age 3 years and 4 months.
444 Meschede et al.
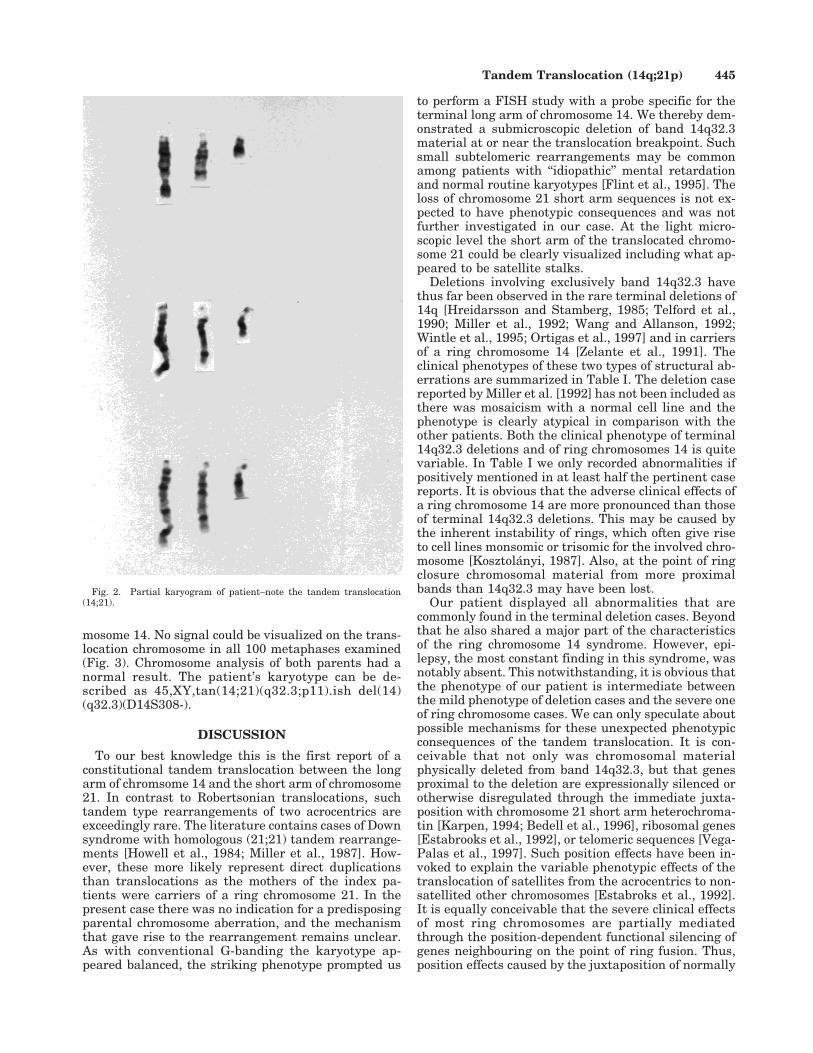
mosome 14. No signal could be visualized on the trans-location chromosome in all 100 metaphases examined(Fig. 3). Chromosome analysis of both parents had anormal result. The patient’s karyotype can be de-scribed as 45,XY,tan(14;21)(q32.3;p11).ish del(14)(q32.3)(D14S308-).
DISCUSSION
To our best knowledge this is the first report of aconstitutional tandem translocation between the longarm of chromsome 14 and the short arm of chromosome21. In contrast to Robertsonian translocations, suchtandem type rearrangements of two acrocentrics areexceedingly rare. The literature contains cases of Downsyndrome with homologous (21;21) tandem rearrange-ments [Howell et al., 1984; Miller et al., 1987]. How-ever, these more likely represent direct duplicationsthan translocations as the mothers of the index pa-tients were carriers of a ring chromosome 21. In thepresent case there was no indication for a predisposingparental chromosome aberration, and the mechanismthat gave rise to the rearrangement remains unclear.As with conventional G-banding the karyotype ap-peared balanced, the striking phenotype prompted us
to perform a FISH study with a probe specific for theterminal long arm of chromosome 14. We thereby dem-onstrated a submicroscopic deletion of band 14q32.3material at or near the translocation breakpoint. Suchsmall subtelomeric rearrangements may be commonamong patients with ‘‘idiopathic’’ mental retardationand normal routine karyotypes [Flint et al., 1995]. Theloss of chromosome 21 short arm sequences is not ex-pected to have phenotypic consequences and was notfurther investigated in our case. At the light micro-scopic level the short arm of the translocated chromo-some 21 could be clearly visualized including what ap-peared to be satellite stalks.
Deletions involving exclusively band 14q32.3 havethus far been observed in the rare terminal deletions of14q [Hreidarsson and Stamberg, 1985; Telford et al.,1990; Miller et al., 1992; Wang and Allanson, 1992;Wintle et al., 1995; Ortigas et al., 1997] and in carriersof a ring chromosome 14 [Zelante et al., 1991]. Theclinical phenotypes of these two types of structural ab-errations are summarized in Table I. The deletion casereported by Miller et al. [1992] has not been included asthere was mosaicism with a normal cell line and thephenotype is clearly atypical in comparison with theother patients. Both the clinical phenotype of terminal14q32.3 deletions and of ring chromosomes 14 is quitevariable. In Table I we only recorded abnormalities ifpositively mentioned in at least half the pertinent casereports. It is obvious that the adverse clinical effects ofa ring chromosome 14 are more pronounced than thoseof terminal 14q32.3 deletions. This may be caused bythe inherent instability of rings, which often give riseto cell lines monsomic or trisomic for the involved chro-mosome [Kosztolanyi, 1987]. Also, at the point of ringclosure chromosomal material from more proximalbands than 14q32.3 may have been lost.
Our patient displayed all abnormalities that arecommonly found in the terminal deletion cases. Beyondthat he also shared a major part of the characteristicsof the ring chromosome 14 syndrome. However, epi-lepsy, the most constant finding in this syndrome, wasnotably absent. This notwithstanding, it is obvious thatthe phenotype of our patient is intermediate betweenthe mild phenotype of deletion cases and the severe oneof ring chromosome cases. We can only speculate aboutpossible mechanisms for these unexpected phenotypicconsequences of the tandem translocation. It is con-ceivable that not only was chromosomal materialphysically deleted from band 14q32.3, but that genesproximal to the deletion are expressionally silenced orotherwise disregulated through the immediate juxta-position with chromosome 21 short arm heterochroma-tin [Karpen, 1994; Bedell et al., 1996], ribosomal genes[Estabrooks et al., 1992], or telomeric sequences [Vega-Palas et al., 1997]. Such position effects have been in-voked to explain the variable phenotypic effects of thetranslocation of satellites from the acrocentrics to non-satellited other chromosomes [Estabroks et al., 1992].It is equally conceivable that the severe clinical effectsof most ring chromosomes are partially mediatedthrough the position-dependent functional silencing ofgenes neighbouring on the point of ring fusion. Thus,position effects caused by the juxtaposition of normally
Fig. 2. Partial karyogram of patient–note the tandem translocation(14;21).
Tandem Translocation (14q;21p) 445

Fig.3. Fluorescence in situ hybridization with a probe specific for the subtelomeric locus D14S308. Arrows indicate the translocation chromosome andthe normal chromosomes 14 and 21, respectively. Note absence of fluorescent signal from translocation chromosome.
TABLE I. Core Characteristics of Clinical Phenotypes of Ring Chromosomes 14 and Terminal Deletions ofBand 14q32.3
Type of abnormality Ring chromosome 14a Terminal deletion 14q32.3b Present case
Intrauterine growth retardation + −Postnatal growth retardation + +Microcephaly + +Dolichocephaly + +Flat occiput + −Retinal abnormalities + −Oval face + +High forehead/frontal bossing + + +Narrow palpebral fissures + −Eyelid ptosis +Epicanthus + + +Broad nasal bridge + + +Long philtrum +High arched palate + + +Down-turned corners of mouth +Micrognathia +Low-set and/or dysplastic ears +Short neck + −Widely spaced nipples +Congenital heart disease +Genital anomaly +Transverse palmar crease +Marbled skin +Proneness to infection +Developmental delay + + +Muscular hypotonia + + +Epilepsy + −Ataxia + −Hearing loss +
aModified from Zelante et al. [1991].bModified from Hreidarsson and Stamberg [1985]; Telford et al. [1990]; Wang and Allanson [1992]; Wintle et al. [1995]; Ortigas et al. [1997].+, denotes findings mentioned in at least half of the case reports; −, denotes the absence of the respective anomaly in our patient.

separated chromosomal domains could be a shared fea-ture of both ring chromosomes and tandem type rear-rangements as observed in our patient.
REFERENCESBedell MA, Jenkins NA, Copeland NG (1996): Good genes in bad neigh-
bourhoods. Nat Genet 12:229–232.
Estabrooks LL, Lamb AN, Kirkman HN, Callanan NP, Rao KW (1992): Amolecular deletion of distal chromosome 4p in two families with a sat-ellited chromosome 4 lacking the Wolf-Hirschhorn syndrome pheno-type. Am J Hum Genet 51:971–978.
Flint J, Wilkie AOM, Buckle V, Winter RM, Holland AJ, McDermid HE(1995): The detection of subtelomeric chromosomal rearrangements inidiopathic mental retardation. Nat Genet 9:132–140.
Gravholt CH, Friedrich U, Caprani M, Lund Jorgensen A (1992): Break-point in Robertsonian translocations are localized to satellite III DNAby fluorescence in situ hybridization. Genomics 14:924–930.
Howell RT, McDermott A, Gardner A, Dickinson V (1984): Down’s syn-drome with a recombinant tandem duplication of chromosome 21 de-rived from a maternal ring. J Med Genet 21:310–314.
Hreidarsson SJ, Stamberg J (1985): Distal monosomy 14 not associatedwith ring formation. J Med Genet 20:147–149.
Karpen GH (1994): Position-effect variegation and the new biology of het-erochromatin. Curr Opin Genet Dev 4:281–291.
Kosztolanyi G (1987): Does ‘‘ring syndrome’’ exist? An analysis of 207 casereports on patients with a ring autosome. Hum Genet 75:174–179.
Miller BA, Jayakar P, Capo H (1992): Child with multiple congenitalanomalies and mosaicism 46,XX/46,XX,del(14)(q32.2). Am J Med Genet44:635–637.
Miller K, Reimer A, Schulze B (1987): Tandem duplication chromosome 21in the offspring of a ring chromosome 21 carrier. Ann Genet 30:180–182.
Ortigas AP, Stein CK, Thomson LL, Hoo JJ (1997): Delineation of 14q32.3deletion syndrome. J Med Genet 34:515–517.
Telford N, Thomson DAG, Griffiths MJ, Ilett S, Watt JL (1990): Terminaldeletion (14)(q32.3): A new case. J Med Genet 27:261–263.
Vega-Palas MA, Venditti S, Di Mauro E (1997): Telomeric transcriptionalsilencing in a natural context. Nat Genet 15:232–233.
Wang HS, Allanson JE (1992): A further case of terminal (14)(q32.2) in achild with mild dysmorphic features. Ann Genet 35:171–173.
Wintle RF, Costa T, Haslam RHA, Teshima IE, Cox DW (1995): Molecularanalysis redefines three human chromosome 14 deletions. Hum Genet95:495–500.
Zelante L, Torricelli F, Calvano S, Mignarellir, Dallapiccola B (1991): Ringchromosome 14 syndrome: Report of two cases, including extendedevaluation of a previously reported patient and review. Ann Genet34:93–97.
Tandem Translocation (14q;21p) 447