study of the lewis acid catalyzed povarov reaction for the ... · 1 abstract during last years,...
TRANSCRIPT

Study of the Lewis Acid catalyzed Povarov reaction
for the synthesis of polycyclic tetrahydroquinoline derivatives
Riccardo Vallesi
Thesis to obtain the Master of Science
Degree in
Chemistry
Supervisors: Dr. Cristina Cimarelli
Dr. Alexandra Antunes
Examination Committee
Chairman: Profª. Isabel M. Marrucho
Supervisor: Dra. Alexandra Antunes
Member of committee: Prof. João Paulo Cabral Telo
December 2017


1
Abstract
During last years, tetrahydroquinolines have improved their synthetic importance due to their
biological activity from a pharmaceutical and medicinal point of view. A great number of substituted
tetrahydroquinolines has been discovered in nature, especially in plants. Their biological activity and
industrial applications stimulate researchers all over the world and, as a result, a huge number of
synthetic approaches have been developed. One of the most used and well-known strategies involves
pericyclic reactions and, among all this type of reactions, aza-Diels-Alder approach is a valid
alternative; Povarov reactions can be included in this area. Lewis catalyzed Povarov processes have
awakened attention especially when Lanthanide salts are used. Among them, Cerium(III) has attracted
researcher’s interest as its salt CeCl3∙7H2O because it is an ecofriendly and cheap reagent, which can
be easily employed also in air and non-anhydrous conditions. The aims of this work are: (i) study of
some aspects of the mechanism of a Povarov reaction of 3,4-dihydro-2H-pyran (DHP), aniline and
benzaldehyde; (ii) study of catalyst and temperature effect of the Povarov reaction of indolines and N-
Vinyl-2-pyrrolidone; (iii) synthetic applications of Povarov reaction to these substrates. This last point
afforded to the synthesis of unprecedented products.
Keywords
Povarov reaction, Lewis acids, Tetrahyroquinolines derivatives, Metal iodides, Cerium
trichloride heptahydrated.

2
Resumo
Nos últimos anos, as tetrahidroquinolinas melhoraram a sua importância sintética devido à
sua atividade biológica do ponto de vista farmacêutico e medicinal. Um grande número de
tetrahidroquinolinas substituídas foi descoberto na natureza, especialmente em plantas. Sua atividade
biológica e aplicações industriais estimulam pesquisadores em todo o mundo e, como resultado, um
grande número de abordagens sintéticas foram desenvolvidas. Uma das estratégias mais utilizadas e
bem conhecidas envolve reações pericíclicas e, entre todo esse tipo de reações, a abordagem aza-
Diels-Alder é uma alternativa válida; As reações de Povarov podem ser incluídas nesta área. Os
processos de Povarov catalisados por Lewis despertaram atenção especialmente quando os sais de
Lantanídeos são usados. Entre eles, o Cerium (III) atraiu o interesse do pesquisador como seu sal
CeCl3∙7H2O porque é um reagente ecofriendly e barato, que pode ser facilmente empregado também
em condições de ar e não anidras. Os objetivos deste trabalho são: (i) estudar alguns aspectos do
mecanismo de uma reação de Povarov de 3,4-dihidro-2H-pirano (DHP), anilina e benzaldeído; (ii)
estudo do efeito de catalisador e temperatura da reação de Povarov de indolinas e N-vinil-2-
pirrolidona; (iii) aplicações sintéticas da reação de Povarov a esses substratos. Este último ponto
conferido à síntese de produtos sem precedents.
Palavras-Chave
Reação de Povarov, ácidos de Lewis, Derivados de Tetrahroquinolinas, iodetos de metais, Tricloreto
de cerio heptaidratado.

3
Table of contents
Abstract 1
Keywords 1
Resumo 2
Palavras-Chave 2
Index of Figures 5
Index of Schemes 6
Index of Tables 9
Index of Equations 10
Abbreviation List 11
1. Pericyclic reactions 13
1.1 Sigmatropic reactions 14
[3,3]-Rearrangement 14
Claisen rearrangement 15
Cope rearrangement 15
[2,3]-Rearrangements 15
1.2 Electrocyclic reactions 17
1.3 Cycloadditions 17
Diels-Alder reaction 17
Imino Diels-Alder reactions (Povarov reactions) 17
1.3.1 Applications of Povarov reactions 19
Brønsted acids and other catalysts 19
Lewis acids as catalysts 21
2. CeCl3, CeCl3∙7H2O and CeCl3∙7H2O/NaI 25
2.1 Organocerium compounds 25
2.2 Application of CeCl3∙7H2O in reductions 28
2.3 CeCl3∙7H2O /NaI 30

4
Applications of CeCl3∙7H2O /NaI 31
3. Tetrahydroquinolines: applications and synthesis 34
3.1 Application of tetrahydroquinolines 35
Chemotherapeutic targets 35
Pharmacodynamic targets 36
Application in Chemistry 38
3.2 Synthesis of tetrahydroquinolines 39
Intramolecular oxidative-cyclization/Lactamization (Formation of N-C2 bond) 39
Conjugate addition-cyclization sequence (Formation of C2-C3 bond) 40
Intramolecular cyclization of ene-C=N functionality (formation of C3-C4 bond) 42
Intramolecular Friedel-Crafts related reactions (formation of C4-C4a bond) 42
Photochemical reactions (formation of C8a-N bond) 44
Diels-Alder approach (Formation of N-C2 and C3-C4 bonds) 44
Brønsted acid-catalyzed reactions (Formation of three or more bonds) 45
4. Experimental section 46
Aim of the work 46
Results and discussion 47
Materials and methods for the synthesis of compounds 52
Characterization of compounds 55
5. Conclusions 63
6. References 64
7. Annexes 71

5
Index of Figures
Figure 1. Intermediate of the Brønsted acid catalyzed Povarov reaction between N-phenyl-C-
methoxycarbonyl imine and methylenecycloprpopane.
Figure 2. Benzastatins C 140 and D 141.
Figure 3. Penigequinolone A 142(S) and B 142(R) and Peniprequinolone 143.
Figure 4. (+)-Sceletium A-4 144, (+)-Tortuosamine 145 and (+)-N-Formyltortuosamine 146.
Figure 5. Molecules showing efficacy against HIV virus.
Figure 6. Interesting antibacterial agents.
Figure 7. Molecules used in the treatment of Malaria (Plasmonium falciparum’s farnesyltransferase).
Figure 8. Molecules having interesting activity on ion channels.
Figure 9. Molecules with important role on membranes and neurotransmitter receptors.
Figure 10. Selective Estrogen Receptors Modulator (SERM).
Figure 11. Selective Androgen Receptor Modulator (SARM).
Figure 12. Tetrahydroquinolines used as electron donors in dye-sensitized solar cells (DSSC).
Figure 13. Chiral ligand for Rhodium-catalyzed hydrogenation of amino acrylates.

6
Index of Schemes
Scheme 1. Formation of lactones.
Scheme 2. Chlorination of methane.
Scheme 3. Claisen rearrangement.
Scheme 4. Cope rearrangement.
Scheme 5. Oxy-Cope reaarangement.
Scheme 6. [2,3]-Rearrangement.
Scheme 7. Synthesis of allylic sulfoxides by [2,3]-rearrangement.
Scheme 8. Stereospecific conversion of 3,4-dimethylcyclobutene to two different isomeric dienes.
Scheme 9. Reverse process that affords to syn-3,4-dimethylcyclobutene.
Scheme 10. Synthesis of Periplanone B.
Scheme 11. Classification of Diels-Alder reactions.
Scheme 12. General Povarov reaction.
Scheme 13. Original Povarov reaction.
Scheme 14. The two possible Povarov reaction mechanism.
Scheme 15. Brønsted acid catalyzed Povarov reaction between N-phenyl-C-methoxycarbonyl imine
and methylenecycloprpopane.
Scheme 16. Three component Povarov reaction catalyzed by a chiral BINOL type phpsphoric acid
derivative.
Scheme 17. Povarov reaction catalyzed by an achiral Brønsted acid and a chiral urea derivative.
Scheme 18. Povarov reaction catalyzed by Titanium(IV) oxide and UV light staring from ethanol and
3-nitrotoluene.
Scheme 19. Synthesis of 4-aryl-3-methyltetrahydroquinoline from N-benzylanilines and formaldehyde.
Scheme 20. Synthesis of 2-sypiro-tetrahydroquinoline from isoeugenol and 3-aryliminosatins.
Scheme 21. Synthesis of 2-aryl-1,2,3,4-tetrahydroquinolines using N-vinyl-2-pirrolindone as
dienophile.
Scheme 22. Sc(OTf)3 catalyzed Povarov reaction.
Scheme 23.Synthesis of the first G protein-coupled estrogen receptor using Povarov chemistry.

7
Scheme 24. Povarov reaction using catalytic amount of ytterbium complex.
Scheme 25. Synthesis of trans-(2S,5S)-(1,1-diphenylmethyl)pyrrolidine.
Scheme 26. Synthesis of VEGF-R2 kinase inhibitor.
Scheme 27. Examples of the activity of Cerium(III) trichloride on C=C bonds.
Scheme 28. Luche’s mechanism with NaBH4 and MeOH.
Scheme 29. Synthesis of 1,9-Deoxypreaxinellamine.
Scheme 30. Synthesis of WRC-0571.
Scheme 31. Synthesis of biologically active 3-mercapto-2(1H)-pyridinones.
Scheme 32. Use of CeCl3∙7H2O/NaI in the Knoevagel condensation of ethyl-tert-butylmalonate with
aromatic or heteroaromatic aldehydes.
Scheme 33. Synthesis of furan derivatives by cyclization reaction of homobenzylic alcohols.
Scheme 34. Synthesis of S-(-)-Pulegone.
Scheme 35. Synthesis of the N-protected nine component of Griseoviridin.
Scheme 36. Different strategies for tetrahydroquinolines synthesis.
Scheme 37. Oxidative cyclization of aromatic aminoalcohols to tetrahydroquinoline derivatives.
Scheme 38. Plausible mechanism of oxidative cyclization of aromatic aminoalcohols to
tetrahydroquinoline derivatives.
Scheme 39. Conjugate addition-cyclization sequence with electron-deficient alkenes catalyzed by
Rhodium.
Scheme 40. Tandem conjugate addition-cyclization sequence of lithium (R)-N-benzyl-N-(α-
methylbenzyl)amide to aromatic imines having an electron-deficient alkene in ortho position.
Scheme 41. Typical Radical Addition Cyclization Elimination(RACE) reaction.
Scheme 42. Synthesis of 1,2,3,4-tetrahydroquinolines from ω-vinylimines.
Scheme 43. Synthesis of biquinoline derivatives from homoallylamines from primary amines and
quinolonecarboxyaldehyde.
Scheme 44. Acid-catalyzed reaction of 1-allyl-1-N-arylaminocyclohexanones.
Scheme 45. Synthesis of tetrahydroquinolines starting from primary amines sulfonamides having an
aryl group in ϒ-position under photochemical conditions.
Scheme 46. Corey’s Diels-Alder approach to tetrahydroquinolines synthesis.

8
Scheme 47. Brønsted acid catalyzed Povarov reaction with a range of different imines and
methylenecyclopropanes.
Scheme 48. Alternative one pot synthesis of isoindolo[2,1-a]quinolone derivative based on a Povarov
cyclocondensation domino approach.
Scheme 49. Povarov reaction between3-aminoacetophenone, benzaldehyde, cyclopentadiene and
TFA:
Scheme 50. Synthesis of spiro-tetrahydroquinolines from arylamines and keto sugars.
Scheme 51. Povarov reaction between aniline, benzaldehyde and 3,4-dihydro-2H-pyran.
Scheme 52. Povarov reaction between indolines, benzaldehyde and N-Vinyl-2-pirrolidinone.
Scheme 53. Povarov reaction with different indolines.
Scheme 54. Results of the trial for the stability of the diastereoisomers.
Scheme 55. Synthesis of indoline.
Scheme0 56. Povarov reaction with different temperatures and metal iodide sources.
Scheme 57. Synthesis of substituted indolines.
Scheme 58. Povarov reaction starting with different indolines.

9
Index of Tables
Table 1. Syn : anti ratio of Povarov reaction at -10°C in solvent/solventless conditions.
Table 2. Syn : anti ratio and yield of Povarov reaction at different temperatures and catalyst.
Table 3. Synthesis of the substituted indolines.
Table 4. Result of the Povarov reaction with substituted indolines.

10
Index of Equations
Equation 1. Preparation of organocerium compounds by reaction of Grignard with organolithium
reagents.
Equation 2. Exchange equation of CeCl3 and NaI.
Equation 3. Exchange equation of the 1:1 combination of CeCl3 and NaI.

11
Abbreviation List
DHP 3,4-dihydro-2H-pyran
HAD Hetero Diels Alder
DFT Density Functional Theory
MCP Methylenecyclopropane
BINOL 1,1’-Bi-2-naphthol
dr Diastereomeric ratio
ee Enantiomeric ratio
UV Ultra Violet light
DBU 1,5-Diazabiciclo(5.4.0)undec-5-ene
DTBP 2,6-di-tert-butylpyrindine
Ce Cerium
NaBH4 Sodium BoroHydride
XPS X-Ray Photoelectron Spectroscopy
MCR Multi Component Reaction
ETBM Ethyl-Tert-ButylMalonate
HIV Human Immunodeficiency Virus
RT Reverse Transcriptase
DNA Deoxyribonucleic Acid
tRNA Transfer RiboNucleic Acid
SERM Selective Estrogen Receptor Modulator
SARM Selective Androgen Receptor Modulator
DSSC Dye-Sensitizied Solar Cells
RACE Radical Addition Cyclization Elimination
GC-MS Gas Chromatography-Mass Spectroscopy
TLC Thin Layer Chromatography
MeCN Acetonitrile

12
r.t. Room Temperature
NaBH3CN Sodium Cyano BoroHydride
N2 Molecular Nitrogen
CH3COOH Acetic Acid
XI Metal Iodide
LiI Lithium Iodide
KI Potassium Iodide
CuI Copper Iodide
NaI Sodium Iodide
NMR Nuclear Magnetic Resonance
CH2Cl2 DichloroMethane
Hex Hexane
EtOAc Etil Acetate
MgSO4 Magnesium Sulphate
HCl Chloride Acid
NaHCO3 Sodium Bicarbonate
Na2SO4 Sodium Sulphate
NaOH Sodium Hydroxide
Et2O DiEthyl Ether
Mp Melting point

13
1. Pericyclic Reactions
Organic reactions may take place in different ways. Breaking and forming of bonds may involve
polar (ionic) intermediates (heterolytic pathway) or radicals (homolytic pathway). In the ionic processes,
electrons “flow” from an electron-rich atom to an electron-poor one: formation of lactones is a typical
example in which, the formation of the desired product involves five steps and four cationic
intermediates: it’s acid-catalyzed and, because it is an ionic reaction, electrons move towards the
positive charge, as reported in Scheme 1.
Scheme 1
Radical reactions have a different pathway: radicals are formed from the homolytic scission of a
covalent bond generally caused by heating or irradiation and they are very reactive. Typically, a
radical reaction has three steps:
1. Initiation, where there is formation of the radicals;
2. Propagation, where the reaction happens;
3. Termination, where the reaction stops and the free radicals are not present anymore.
Classical example of a radical reaction is the chlorination of methane (Scheme 2):
Scheme 2

14
There is another kind of organic processes that differ from the first two: pericyclic reactions in which
electrons do not flow linearly but they move roundly without anionic or cationic intermediates and for this
reason, these reactions are called “pericyclic”.1 There are three main classes of pericyclic reactions:
Sigmatropic rearrangements;
Electrocyclic reactions;
Cycloadditions.
1.1 Sigmatropic Reactions
A sigmatropic reaction is a pericyclic reaction wherein the net result is that a σ-bond is changed
to another σ-bond in an intramolecular process: as a consequence, a substituent moves from one part
of a π-bonded system to another part with simultaneous rearrangement of the π system itself.
[3,3]-Rearrangement
-Claisen rearrangement: This sigmatropic process is called [3,3] because the new-formed σ-bond
has a [3,3] relationship with the previous one. The most famous is the Claisen rearrangement of allyl
vinyl ethers, under heating conditions, which give a γ,δ-unsaturated carbonyl compound, that is more
difficult to obtain respect to the α,β ones. The process occurs under a six-member cyclic chair-like
transition state, which allows predicting the stereochemistry of the final alkene (Scheme 3).2
Scheme 3
-Cope rearrangement: The Cope Rearrangement is the thermal isomerization of a 1,5-diene to a
thermodynamically more stable isomer (Scheme 4):
Scheme 4
An interesting variant is the Oxy-Cope rearrangement (Scheme 5), in which a hydroxy group in a
suitable position affords to an enolether that then tautomerizes to the corresponding carbonyl
compound. The tautomerization is the driving force for this process. The use of a strong base promotes
and speeds up the reaction.3

15
Scheme 5
[2,3]-Rearrangements
This type of rearrangement has a five-membered transition state. The substrate is a benzyl allyl ether
that isomerizes to a final product with a new C-C σ bond (Scheme 6):
Scheme 6
Heteroatoms like sulfur (S) or selenium (Se) can change their oxidation state by two, providing two bond-
forming electrons. This makes the reaction particularly useful for the synthesis of allylic sulfoxides. The
starting alcohol reacts with PhSCl and gives a not stable sulfenate ester; pyridine promotes the [2,3]
rearrangement involving O and S. The product is an allylic sulfoxide (Scheme 7):4
Scheme 7
1.2 Electrocyclic reactions
The main feature of these pericyclic reactions is the formation or breaking of a ring. In this
process, only one σ-bond is broken (or formed) across the end of conjugated π-system.5 One of the
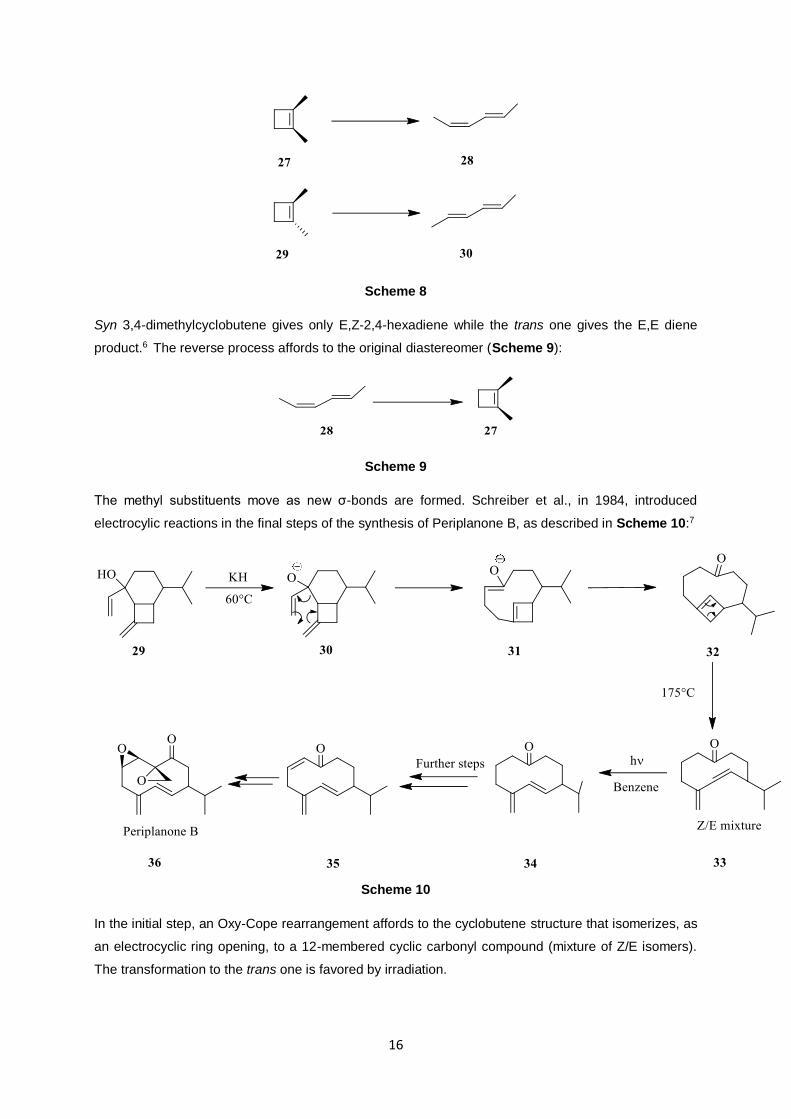
most known example is the stereospecific conversion of 3,4-dimethylcyclobutene to two different
isomeric dienes (Scheme 8):
16
Scheme 8
Syn 3,4-dimethylcyclobutene gives only E,Z-2,4-hexadiene while the trans one gives the E,E diene
product.6 The reverse process affords to the original diastereomer (Scheme 9):
Scheme 9
The methyl substituents move as new σ-bonds are formed. Schreiber et al., in 1984, introduced
electrocylic reactions in the final steps of the synthesis of Periplanone B, as described in Scheme 10:7
Scheme 10
In the initial step, an Oxy-Cope rearrangement affords to the cyclobutene structure that isomerizes, as
an electrocyclic ring opening, to a 12-membered cyclic carbonyl compound (mixture of Z/E isomers).
The transformation to the trans one is favored by irradiation.

17
1.3 Cycloadditions
According to the IUPAC Gold Book,8 in a cycloaddition reaction two or more molecules reacts
together and a cyclic product with a reduction of bond multiplicity is formed. The most famous
cycloaddition reaction is the “Diels-Alder” reaction, discovered in 1928 by Otto Diels and Kurt Alder, that
were awarded the Chemistry Nobel prize in 1950.9
Diels-Alder reaction
During the years, Diels-Alder’s reaction has been used in a wide range of synthetic applications
and it has become an important part in processes of synthesis of polycarbo- and polyheterocyclic
derivatives. This reaction is an excellent alternative for the construction of six-membered rings with a
great control of enantio-and diastereoselectivity. This family of reactions can be distinguished in Carbo-
Diels Alder and Hetero-Diels Alder. The most important of the latter one is imino(aza)-Diels Alder
reactions (Scheme 11):
Scheme 11
Imino Diels-Alder reactions (Povarov reactions)
An inverse electron-demand Diels–Alder reaction is a particular type of Diels–Alder
cycloaddition between an electron-rich dienophile and an electron-poor diene. In the early 1960s, the
Russian chemist Povarov developed an alternative acid-catalyzed inverse electron demand [4+2]

18
cycloaddition between electron-rich dienophiles and N-arylimines, in order to synthetize 1,2,3,4-
tetrahydroquinolines (Scheme 12).10
Scheme 12
This reaction is generally classified as aza- or imino-Diels Alder and allows very versatile applications
of a wide range of catalysts and a huge number of substrates. Furthermore, it can be also performed in
a three-component way, using a dienophile and N-arylimines generated in situ, that are often unstable
and difficult to isolate. In his first pioneering work, Povarov made the reaction of ethyl vinyl ether or
sulfide and N-arylaldimine under BF3-OEt2 to obtain 2,4 substituted tetrahydroquinolines, lately
converted into the corresponding quinolone (Scheme 13):11,12
Scheme 13
Based on this work, the same author and his research group discussed and proposed two possible
mechanisms that are still debated until now (Scheme 14):13

19
Scheme 14
The first is stepwise and goes through the ionic intermediate A with a final intramolecular electrophilic
substitution of a carbenium ion; the second one passes through the asynchronous transition state B.
Researchers believe that the first one is more probable.14
1.3.1 Applications of Povarov reactions
Brønsted acids and other catalysts
Brønsted acid are widely applied in Povarov chemistry. Recently, Rios-Gutierrez and coworkers
made a DFT study of the Brønsted acid Povarov reaction molecular mechanism between N-phenyl-C-
methoxycarbonyl imine and methylenecyclopropane (MCP) (Scheme 15):15
Scheme 15
The study demonstrates that the electrophilic character of the starting imine seems not to be sufficiently
electrophilic for the nucleophilic attack at MCP and that Brønsted acid catalyzed Povarov reaction are
domino processes. The electrophilicity of the imine is enhanced by protonation of the nitrogen and the
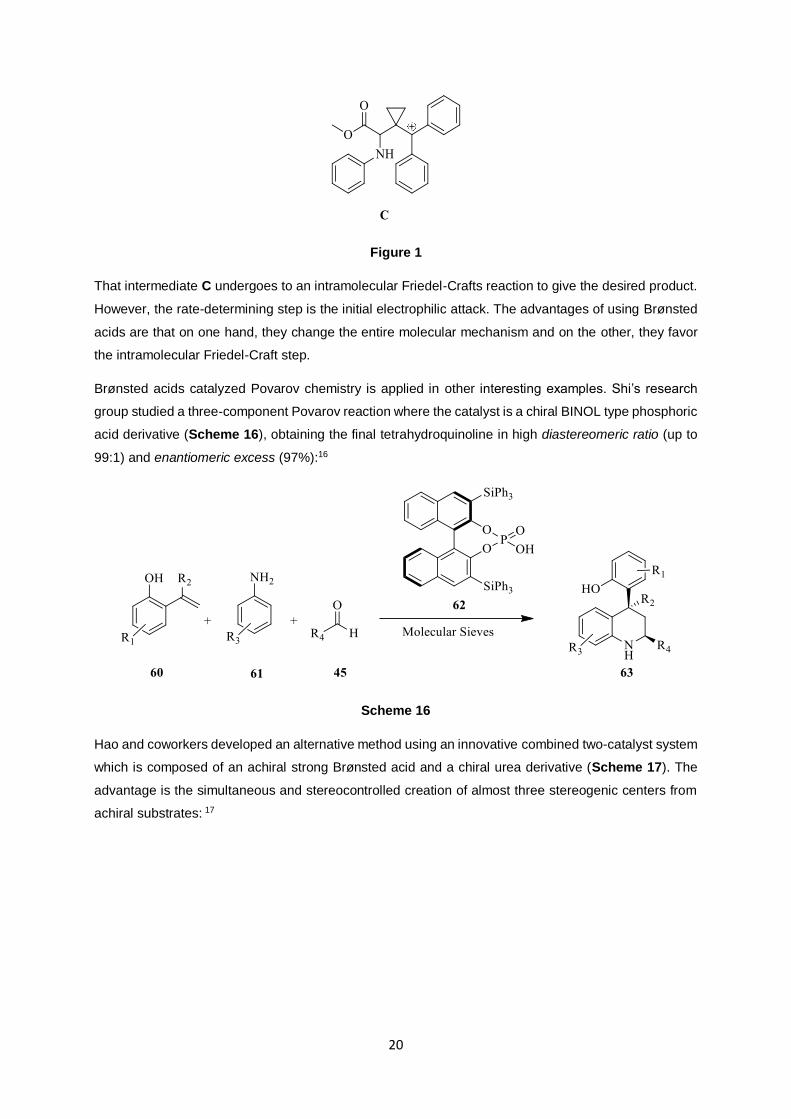
reaction starts with a nucleophilic attack on MCP that affords to a cationic intermediate C (Figure 1):
20
Figure 1
That intermediate C undergoes to an intramolecular Friedel-Crafts reaction to give the desired product.
However, the rate-determining step is the initial electrophilic attack. The advantages of using Brønsted
acids are that on one hand, they change the entire molecular mechanism and on the other, they favor
the intramolecular Friedel-Craft step.
Brønsted acids catalyzed Povarov chemistry is applied in other interesting examples. Shi’s research
group studied a three-component Povarov reaction where the catalyst is a chiral BINOL type phosphoric
acid derivative (Scheme 16), obtaining the final tetrahydroquinoline in high diastereomeric ratio (up to
99:1) and enantiomeric excess (97%):16
Scheme 16
Hao and coworkers developed an alternative method using an innovative combined two-catalyst system
which is composed of an achiral strong Brønsted acid and a chiral urea derivative (Scheme 17). The
advantage is the simultaneous and stereocontrolled creation of almost three stereogenic centers from
achiral substrates: 17

21
Scheme 17.
Lewis acids as catalysts
Lewis acids have been applied in Povarov chemistry since first Povarov’s publication. In last
years, CAN-catalyzed reactions have gained attention as an alternative and efficient method to
synthetize substituted tetrahydroquinolines. A wide range of anilines and vinyl ethers has been tested:
the method can be applied due to its convience.18 Similarly, a heterogeneous solution of 3-nitrotoluene,
ethanol and titanium(IV) oxide upon UV irradiation formed 4-ethoxytetrahydroquinoline (Scheme 18):19
Scheme 18
Interestingly, the reaction mechanism includes the formation of ethyl vinyl ether, aniline and N-
ethyldene-p-methylaniline.
Vladimir Kouznetzov, one of the most important researchers in the field of heterocycles synthesis,
studied a process for the synthesis of 4-aryl-3-methyltetrahydroquinoline starting from substituted N-
benzylanilines and formaldehyde in the presence of BF3∙OEt2. In this process iminium ions are used as
dienophiles (Scheme 19):20

22
Scheme 19
2-spiro-tetrahydroquinoline can be synthetized in moderate yield from the BF3∙OEt2 catalyzed reaction
between isoeugenol and 3-aryliminoisatins (Scheme 20):21
Scheme 20
N-Vinyl-2-pyrrolidone is an important and interesting dienophile in Povarov chemistry. A recent
application on the synthesis of 2-aryl-1,2,3,4-tetrahydroquinolines was reported by Astudillo (Scheme
21): 22
Scheme 21
Also Sc(OTf)3 was used as Lewis acid catalyst in reactions for the synthesis of tetrahydroquinolines
using as dienophiles unsaturated lactams with endo- and exocyclic C-C bonds (Scheme 22):

23
Scheme 22
The overall diastereoselectivity was very low (from dr:50:50 to dr:85:15) but in the case of 2-furyl
substituent a single diastomer was obtained.23 A similar Sc(OTf)3 approach was used for the synthesis
of the first G protein-coupled estrogen receptor and its analogues (Scheme 23):
Scheme 23
A single crystal X-Ray analysis confirmed the structure of the tetrahydroquinoline derivative and
additional work on this type of compounds using Povarov chemistry is present in literature.24-26
The enantioselective Povarov reaction with chiral Lewis acids is an alternative approach, although up
to date it is still an unexplored area of research, because most chiral Lewis acids cannot work in a basic
environment being the catalytic cycle blocked.27,28 Kobayashi and Ishitani developed an efficient method
using a catalytic amount of a chiral ytterbium complex (Scheme 24):

24
Scheme 24
The catalyst was synthetized starting from DBU, (R)-(+)-BINOL, Yb(OTf)3 and 2,6-di-tert-butylpyrindine
(DTBP). The products have high diastereoselectivity (from dr: 94:6 to dr: 98:2).29

25
2. CeCl3, CeCl3∙7H2O and CeCl3∙7H2O/NaI
More ecofriendly chemical processes are one of the most desired aspirations for organic
chemists. Over the last years, a great number of attempts has been done in order to study and stimulate
the utilization of Lewis acid promoters in common organic reactions, because of their high resistance to
air and water, low cost and toxicity, stability towards oxygen and easy availability, that makes them really
environment friendly catalysts.30,31 In particular, Lanthanides possesses all of this properties and the
corresponding salts can be used as ecofriendly Lewis acid promoters. Recently, their importance has
been improved in a huge part of organic transformations. Among all the “rare earths”, Cerium is the most
abundant: even Tin, Zinc and Cobalt are present on Earth in lower amount respect to it.
Cerium has two stable oxidation states: Ce4+ is used as one-electron oxidant for a wide range of
processes.32 The electronic configuration of Ce atom is [Xe] 4f15d16s2, where the 4f shell remains
inactive and it is shielded by the 5s2 and 5p6 orbitals. No s-donor-p-acceptor bonding mode is possible
because Cerium(III) is a tripositively charged ion.33 According to Pearson’s principle,34 Ce3+ is a hard
cation and, as result, has a deep affinity to hard bases. The oxophilicity of Cerium(III) cation is due to
the high Lewis acidity combined with ionic bond contributions,35 Lewis acidity decreases as ionic radius
increases, so CeCl3 has to be seen as a mild Lewis acidic promoter.36 In organic reactions, several
Cerium salts have been used but the most common are the ones with halides, nitrate and triflate. The
cost and the use of triflates promoters is not practicable at industrial scale: in addition, there are some
concerns about its application in experimental procedures. These considerations shift the attention to
CeCl3, which has large applications both in hydrated and anhydrous forms.
2.1 Organocerium compounds
Firstly employed by Inamoto,37 organocerium derivatives are used to be added to electrophilic
compounds, which have acidic hydrogens or a high reduction potential.38 They must be prepared freshly
and display interesting properties such as high nucleophilicity and low basicity. They can be prepared
by reaction of Grignard with organolithium reagents (Equation 1):
RLi + CeCl3 ⟶ RCeCl2 + LiCl
RMgX + CeCl3 ⟶ RMgX ∙ CeCl3
Equation 1
This strategy can be applied in different situations: organocerium addition is a valid alternative in a series
of carbon-carbon bond forming reaction.39 Using this type of promoter, alcohols can be obtained from
carbonyl compounds even from starting materials capable of enolization.40 The superiority of
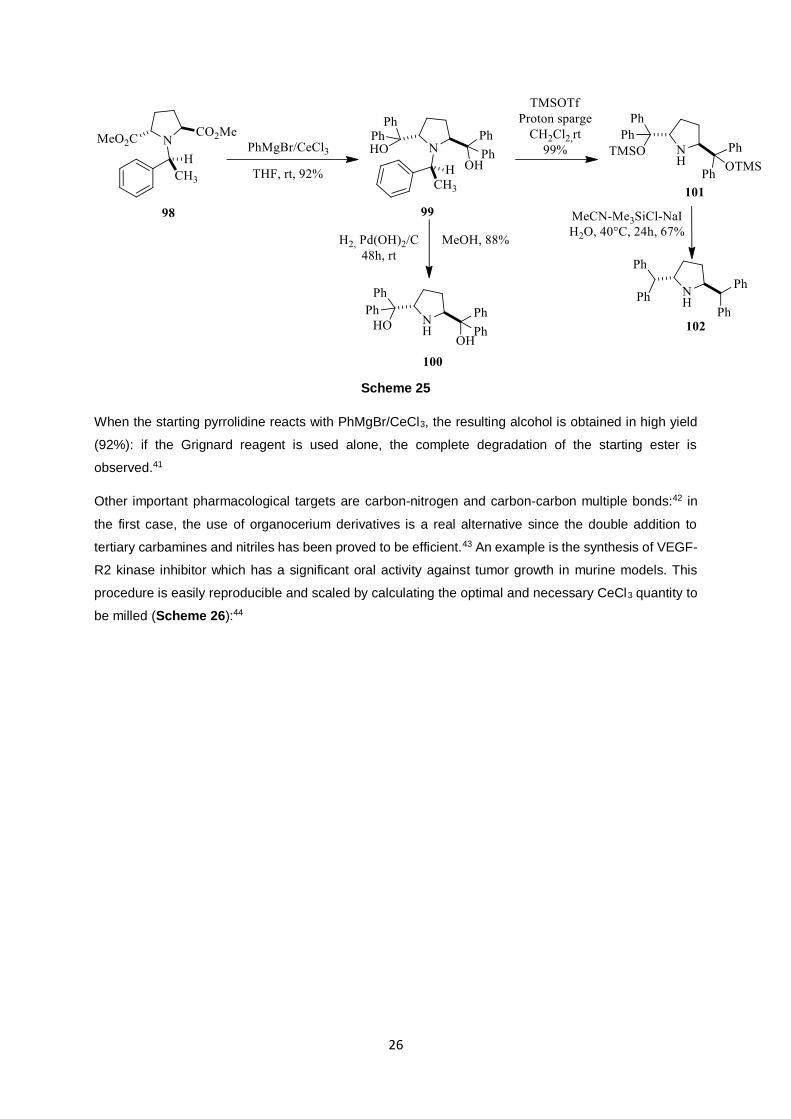
organocerium can be seen in the synthesis of trans-(2S,5S)-(1,1-diphenylmethyl)pyrrolidine as shown
in Scheme 25:
26
Scheme 25
When the starting pyrrolidine reacts with PhMgBr/CeCl3, the resulting alcohol is obtained in high yield
(92%): if the Grignard reagent is used alone, the complete degradation of the starting ester is
observed.41
Other important pharmacological targets are carbon-nitrogen and carbon-carbon multiple bonds:42 in
the first case, the use of organocerium derivatives is a real alternative since the double addition to
tertiary carbamines and nitriles has been proved to be efficient.43 An example is the synthesis of VEGF-
R2 kinase inhibitor which has a significant oral activity against tumor growth in murine models. This
procedure is easily reproducible and scaled by calculating the optimal and necessary CeCl3 quantity to
be milled (Scheme 26):44

27
Scheme 26
Some methods have been studied in order to overcome the inertness of C=C bonds towards nucleophilic
attack and they are still studied because of the very harsh reaction conditions.45 However, some
example have been published and they are reported in Scheme 27. The procedure is efficient and the
products are obtained without chromatographic purification in a very good yield (respectively 97%, 86%
and 48%).46
Scheme 27

28
2.2 Application of CeCl3∙7H2O in reductions
Reduction in organic synthesis is the removal of oxygen from and/or the addition of hydrogen to
a certain functional group of a molecule. The ideal goal is the selectivity: the complete reduction of
insaturations in a molecule is achieved without any particular trouble, while reducing one particular part
respect to another is a useful but a hard challenge. As a rule of thumb, the more active is the catalyst,
the less selective is his action: a very low active catalyst, mild conditions and a good operation rate are
the ideal guidelines to obtain high selectivity in this kind of reactions. In order to achieve it, additives as
Brønsted or Lewis acids, play an important role,47 in particular, Lewis acids shows a significant
improvement on selectivity.48 CeCl3∙7H2O is one of the most effective among all the lanthanide salts and
it has been employed in the reduction of carbonyl groups.
The utility of CeCl3 combined with NaBH4 was firstly demonstrated by Luche (Scheme 28):49
Scheme 28
CeCl3∙7H2O is crucial in the conversion of NaBH4 to [BH4-n(OMe)n]-. The carbonyl group is activated by
hydrogen bonding with methanol and the borohydride attacks the carbonyl carbon to give the
corresponding alcohol. This strategy maintains the stereochemistry of the starting material unaltered.50
Luche’s work on the combination of CeCl3 with metal hydride is a valid alternative to classical reductions
and it has been used for the synthesis of complex biologically active molecules. Baran’s research group
applied it in an easy pathway for the synthesis of 1,9-deoxypreaxinellamine, a marine-derived natural
product, in which Cerium(III) chloride allows the successful reduction of the carbonyl group because of
the high covalent character of cerium-oxygen bond that makes the reduction irreversible and kinetically
favored (Scheme 29): 51

29
Scheme 29
In the pharmaceutical industry, adenosine is a very important nucleoside: during the years, a great
number of adenosine receptors ligands have been made.52 Among all of them, WRC-0571, an
antagonist of adenosine’s A1 receptor, is probably the one with the best selectivity and potency.53 Jin’s
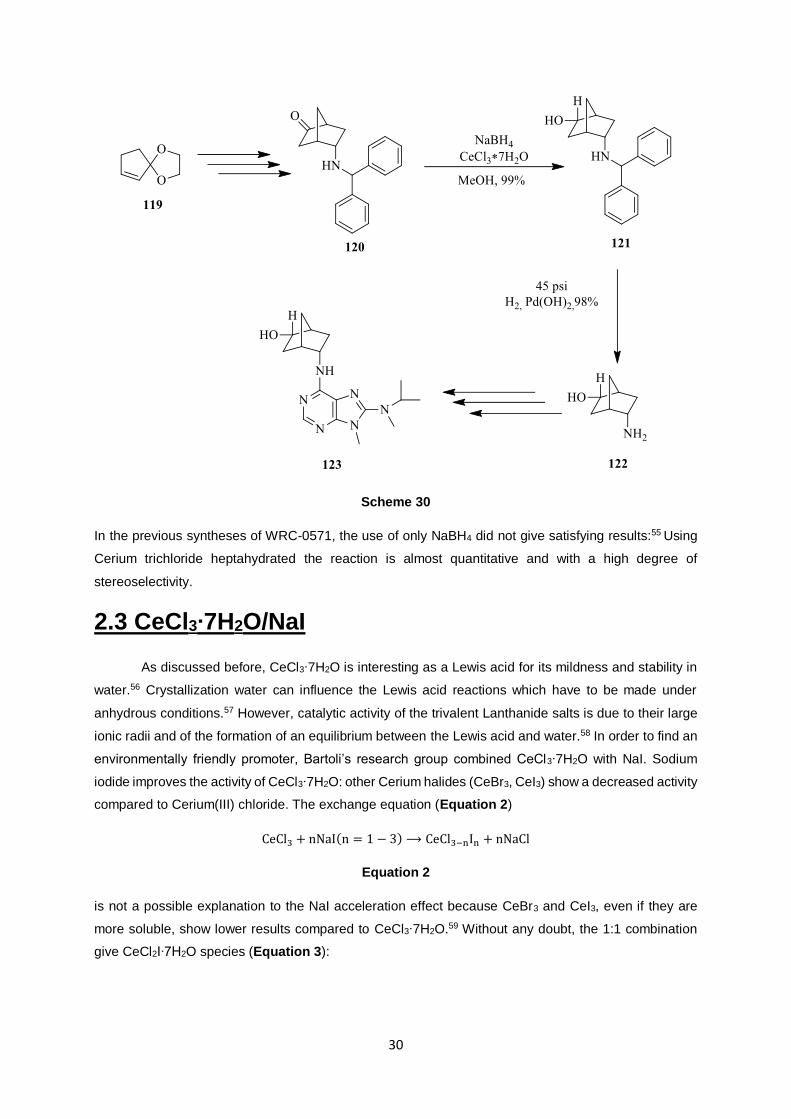
research group studied a new alternative synthesis of WRC-0571, using CeCl3∙7H2O (Scheme 30):54
30
Scheme 30
In the previous syntheses of WRC-0571, the use of only NaBH4 did not give satisfying results:55 Using
Cerium trichloride heptahydrated the reaction is almost quantitative and with a high degree of
stereoselectivity.
2.3 CeCl3∙7H2O/NaI
As discussed before, CeCl3∙7H2O is interesting as a Lewis acid for its mildness and stability in
water.56 Crystallization water can influence the Lewis acid reactions which have to be made under
anhydrous conditions.57 However, catalytic activity of the trivalent Lanthanide salts is due to their large
ionic radii and of the formation of an equilibrium between the Lewis acid and water.58 In order to find an
environmentally friendly promoter, Bartoli’s research group combined CeCl3∙7H2O with NaI. Sodium
iodide improves the activity of CeCl3∙7H2O: other Cerium halides (CeBr3, CeI3) show a decreased activity
compared to Cerium(III) chloride. The exchange equation (Equation 2)
CeCl3 + nNaI(n = 1 − 3) ⟶ CeCl3−nIn + nNaCl
Equation 2
is not a possible explanation to the NaI acceleration effect because CeBr3 and CeI3, even if they are
more soluble, show lower results compared to CeCl3∙7H2O.59 Without any doubt, the 1:1 combination
give CeCl2I∙7H2O species (Equation 3):

31
CeCl3 ∙ 7H2O + NaI ⟶ CeCl2I ∙ 7H2O + NaCl
Equation 3
Even if this complex hasn’t been characterized, it could be a better Lewis acid than its only hydrated
precursor.60 The formation of the complex can be explained using Pearson’s principle: the hard CeCl3
can probably interacts with the iodide ion, forms this species and, as a result, the nucleophilicity of the
donor improves the electrophilic property of the Lanthanide metal.61,62
An X-ray photoelectron spectroscopy (XPS) analysis has been made in order to understand how NaI
interacts with CeCl3∙7H2O. Bartoli and co-workers found that the reaction takes place in heterogeneous
phase on the Cerium salt surface and not in the solution phase.63 A chlorinated bridge oligomeric
structure of Cerium(III) trichloride is formed and broken by an I- species. The result of this process is a
more powerful Lewis acid promoter.64 An other important aspect is crystallization water: CeCl3∙7H2O
/NaI promoted procedures work better in presence of water and the overall activity is even amplified.
This is demonstrated by using anhydrous CeCl3 instead of CeCl3∙7H2O: catalytic activity of the CeCl3-
NaI is absent. However, adding one or more equivalent of water to the dry mixture generates more
active Lewis acid species and the reaction goes with good results.65 During the last years, this catalytic
system has been applied in numerous organic reactions and processes in order to demonstrate its
efficiency and economy.
Applications of CeCl3∙7H2O/NaI
During last years, industrial companies have been demanding to organic chemists the
improvement or the development of new synthetic pathways for compounds. In this area or research,
MCR (Multi Component Reactions) have increase their importance as an efficient procedure in terms of
economical and ecological impact.66,67 Moreover, MCR catalyzed by Lewis acids have increased their
synthetic interest.
Recently, biologically active 3-mercapto-2(1H)-pyridinones with anti-bacterial and anti-fungal activity
have been synthetized by a CeCl3∙7H2O/NaI three-component diastereoselective reaction (Scheme
31):68-70

32
Scheme 31
CeCl3∙7H2O/NaI catalytic combination can also be useful in Michael additions: this reaction has
interesting applications in organic and bioorganic chemistry.71,72 However, side reactions such as
condensation and polymerization are the big drawback of processes.73 Bartoli and coworkers tried
CeCl3∙7H2O/NaI in the Knoevagel condensation of ethyl-tert-butylmalonate (ETBM) with aromatic or
heteroaromatic aldehydes (Scheme 32):74
Scheme 32
This kind of strategy overcomes two of the major obstacles in Knoevagel reaction: firstly, the inability to
stop the coupling of the aldehydes at the mono addition stage; secondly, the spontaneous
decarboxylation that happens during the reaction of malonic acid monoester.75,76 However, this
procedure does not work for aliphatic aldehydes because of the retro-aldol reaction that brings back to
the starting aldehyde.77 Another interesting application is the cyclization reaction of homobenzylic
alcohols to furan derivatives that was studied by Yeh’s research group (Scheme 33):78
Scheme 33

33
This example demonstrates that CeCl3∙7H2O/NaI is an alternative reagent because it is safer and it can
be used in mild conditions although yields are not very high.79
One of the advantage of CeCl3∙7H2O/NaI is its role of promoting selectivity in processes: Bartoli’s
research group applied this protocol to the synthesis of S-(-)-Pulegone (Scheme 34)
Scheme 34
and of a N-protected nine-membered component of Griseoviridin (Scheme 35):79,80
Scheme 35
34
3. Tetrahydroquinolines: applications and synthesis
In the agrochemical and pharmaceutical industry, one of the most important and interesting
family of compounds is constituted by nitrogen containing heterocycles that constitutes about the 60%
of all drugs. The tetrahydroquinoline moiety (a benzene ring fused to a piperidine one) is found
commonly in biologically active substances and it is involved in important therapeutic strategies. A very
broad range of 1,2,3,4-tetrahydroquinoline-based natural products has been discovered during the last
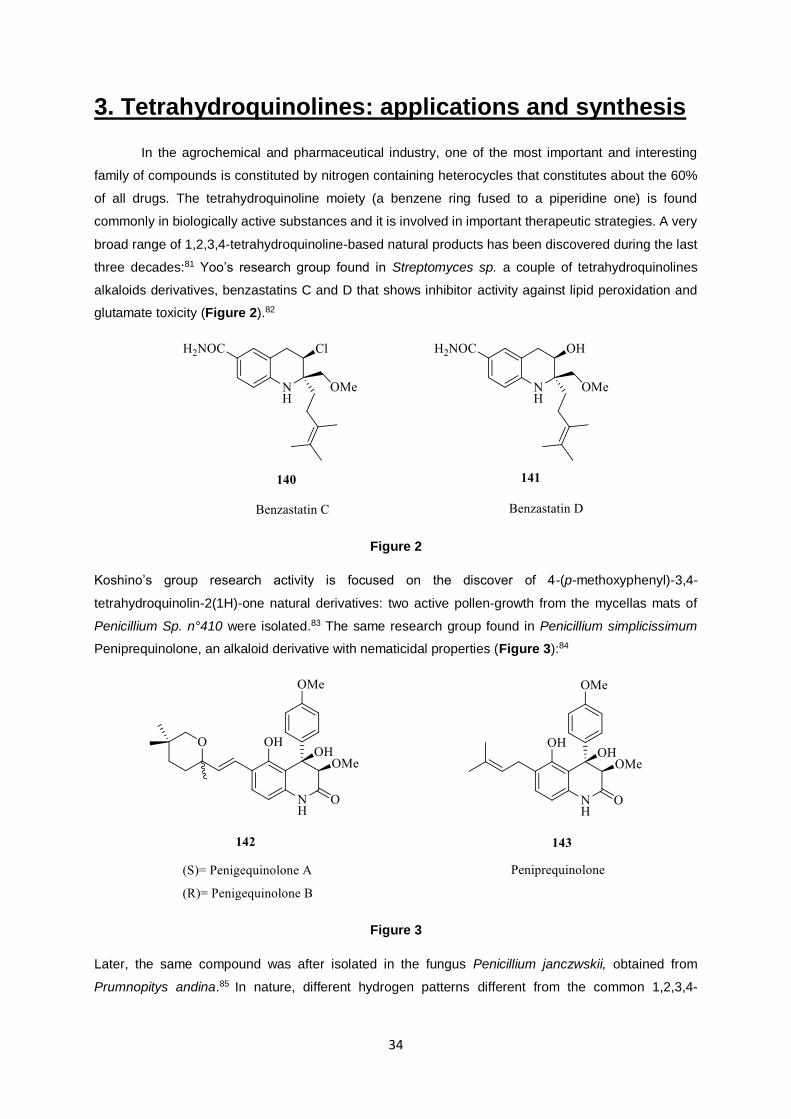
three decades:81 Yoo’s research group found in Streptomyces sp. a couple of tetrahydroquinolines
alkaloids derivatives, benzastatins C and D that shows inhibitor activity against lipid peroxidation and
glutamate toxicity (Figure 2).82
Figure 2
Koshino’s group research activity is focused on the discover of 4-(p-methoxyphenyl)-3,4-
tetrahydroquinolin-2(1H)-one natural derivatives: two active pollen-growth from the mycellas mats of
Penicillium Sp. n°410 were isolated.83 The same research group found in Penicillium simplicissimum
Peniprequinolone, an alkaloid derivative with nematicidal properties (Figure 3):84
Figure 3
Later, the same compound was after isolated in the fungus Penicillium janczwskii, obtained from
Prumnopitys andina.85 In nature, different hydrogen patterns different from the common 1,2,3,4-
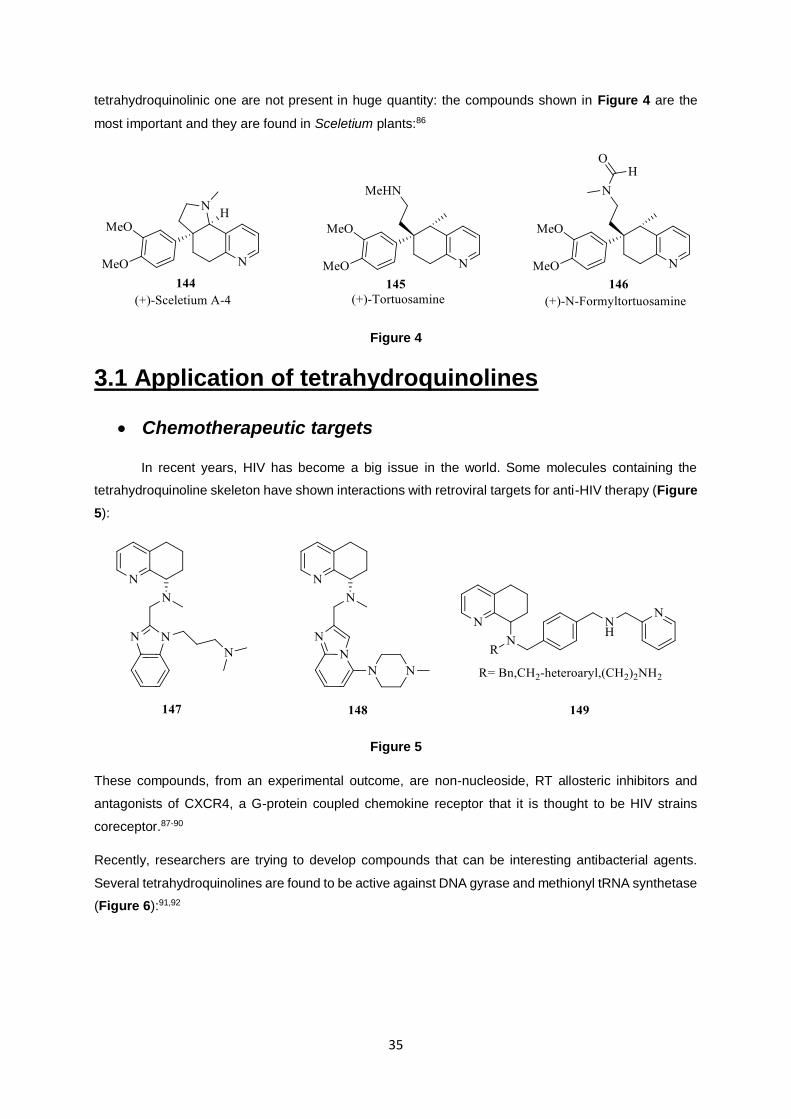
35
tetrahydroquinolinic one are not present in huge quantity: the compounds shown in Figure 4 are the
most important and they are found in Sceletium plants:86
Figure 4
3.1 Application of tetrahydroquinolines
Chemotherapeutic targets
In recent years, HIV has become a big issue in the world. Some molecules containing the
tetrahydroquinoline skeleton have shown interactions with retroviral targets for anti-HIV therapy (Figure
5):
Figure 5
These compounds, from an experimental outcome, are non-nucleoside, RT allosteric inhibitors and
antagonists of CXCR4, a G-protein coupled chemokine receptor that it is thought to be HIV strains
coreceptor.87-90
Recently, researchers are trying to develop compounds that can be interesting antibacterial agents.
Several tetrahydroquinolines are found to be active against DNA gyrase and methionyl tRNA synthetase
(Figure 6):91,92

36
Figure 6
The center compound can be an alternative in infections treatment because of the so-called “Gram
positive bacterial resitance”.93
Imidazole ring is an important tool in pharmaceutical chemistry and its derivatives showed potential
antimalarial activity: they display a potent cytotoxic effect against Plasmonium falciparum’s
farnesyltransferase (Figure 7):94-98
Figure 7
Pharmacodynamic targets
Tetrahydroquinolines having a guanidine functionality in N-1 position show interesting activity
on ion channels (Figure 8):

37
Figure 8
The first group of compounds possesses neuronal Na+ channel antagonist activity: the second one is
Na+/H+ exchanger inhibitor, important in the treatment of ischemia-reperfusion after a myocardial
infarction, and the third one derives from Povarov chemistry and it is an agonist of BKCa (large-
conductance calcium-activated potassium channel).99-101 It was also demonstrated the important role of
tetrahydroquinolines on membranes and neurotransmitter receptors (Figure 9):
Figure 9
In this sense, the first tetrahydroquinoline derivative 158 is a positive allosteric modulator of the α7
nicotinic acetylcholine receptor; Sumanirole is dopaminergic D2 receptors agonist and 160 is an
antagonist of the serotonin 5-HT3 receptor.102-104 This class of compounds is also acting at steroid
hormone receptors: they can be selective estrogen receptor modulator (SERMs), where the active part
is 6-hydroxy-2-phenyl-1,2,3,4-tetrahydroquinoline system (Figure 10):105

38
Figure 10
Another type of tetrahydroquinoline is found to have a potent anabolic activity and to be a SARM
(Selective Androgen Receptor Modulator) (Figure 11):106
Figure 11
Applications in Chemistry
In recent years, tetrahydroquinolines derivatives with a cyano-acrylic moiety as electron acceptor have
been used as electron donors in dye-sensitized solar cells (DSSC) (Figure 12): 107
Figure 12
Asymmetric synthesis, nowadays, is one of the most interesting and studied topic in organic chemistry.
In this view, also tetrahydroquinoline derivatives have showed to be suitable chiral ligands for Rh-

39
catalyzed hydrogenation acrylates of amino acrylates having a product with a 99% of enantiomeric
excess and for asymmetric Friedel-Crafts indoles reaction are shown in Figure 13:108,109
Figure 13
3.2 Synthesis of tetrahydroquinolines
During the years, researchers have developed different strategies for tetrahydroquinolines
synthesis as shown in Scheme 36:
Scheme 36
In the next sub-sections, some strategies will be discussed.
Intramolecular oxidative-cyclization/Lactamization (Formation
of N-C2 bond)
Fujika’s research group worked on the oxidative cyclization of aromatic aminoalcohols to
tetrahydroquinoline derivatives catalyzed by iridium catalyst (Scheme 37):
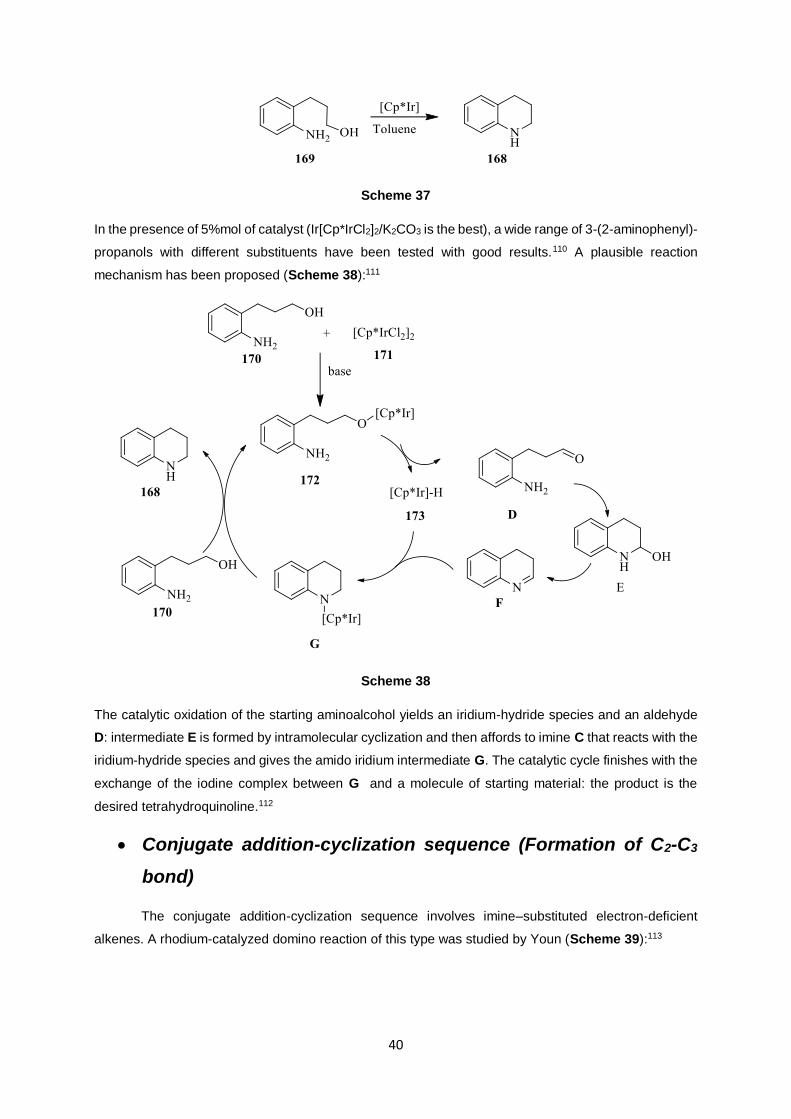
40
Scheme 37
In the presence of 5%mol of catalyst (Ir[Cp*IrCl2]2/K2CO3 is the best), a wide range of 3-(2-aminophenyl)-
propanols with different substituents have been tested with good results.110 A plausible reaction
mechanism has been proposed (Scheme 38):111
Scheme 38
The catalytic oxidation of the starting aminoalcohol yields an iridium-hydride species and an aldehyde
D: intermediate E is formed by intramolecular cyclization and then affords to imine C that reacts with the
iridium-hydride species and gives the amido iridium intermediate G. The catalytic cycle finishes with the
exchange of the iodine complex between G and a molecule of starting material: the product is the
desired tetrahydroquinoline.112
Conjugate addition-cyclization sequence (Formation of C2-C3
bond)
The conjugate addition-cyclization sequence involves imine–substituted electron-deficient
alkenes. A rhodium-catalyzed domino reaction of this type was studied by Youn (Scheme 39):113
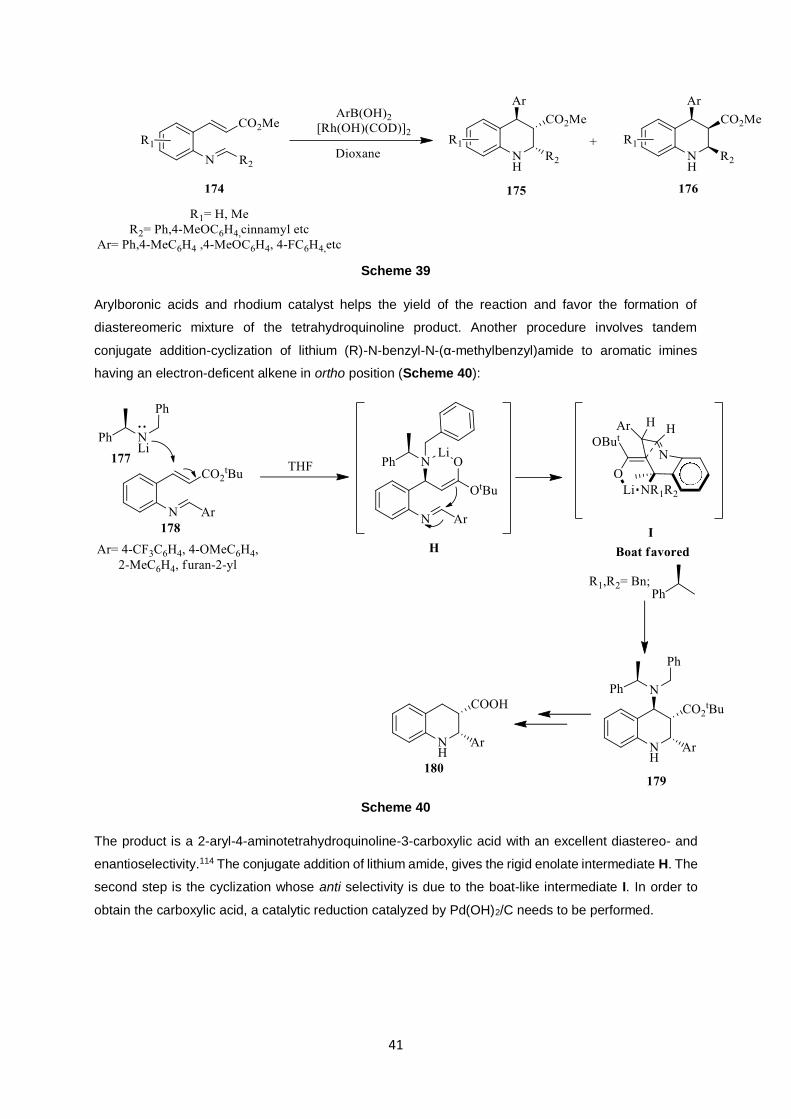
41
Scheme 39
Arylboronic acids and rhodium catalyst helps the yield of the reaction and favor the formation of
diastereomeric mixture of the tetrahydroquinoline product. Another procedure involves tandem
conjugate addition-cyclization of lithium (R)-N-benzyl-N-(α-methylbenzyl)amide to aromatic imines
having an electron-deficent alkene in ortho position (Scheme 40):
Scheme 40
The product is a 2-aryl-4-aminotetrahydroquinoline-3-carboxylic acid with an excellent diastereo- and
enantioselectivity.114 The conjugate addition of lithium amide, gives the rigid enolate intermediate H. The
second step is the cyclization whose anti selectivity is due to the boat-like intermediate I. In order to
obtain the carboxylic acid, a catalytic reduction catalyzed by Pd(OH)2/C needs to be performed.
42
Intramolecular cyclization of ene-C=N functionality (formation
of C3-C4 bond)
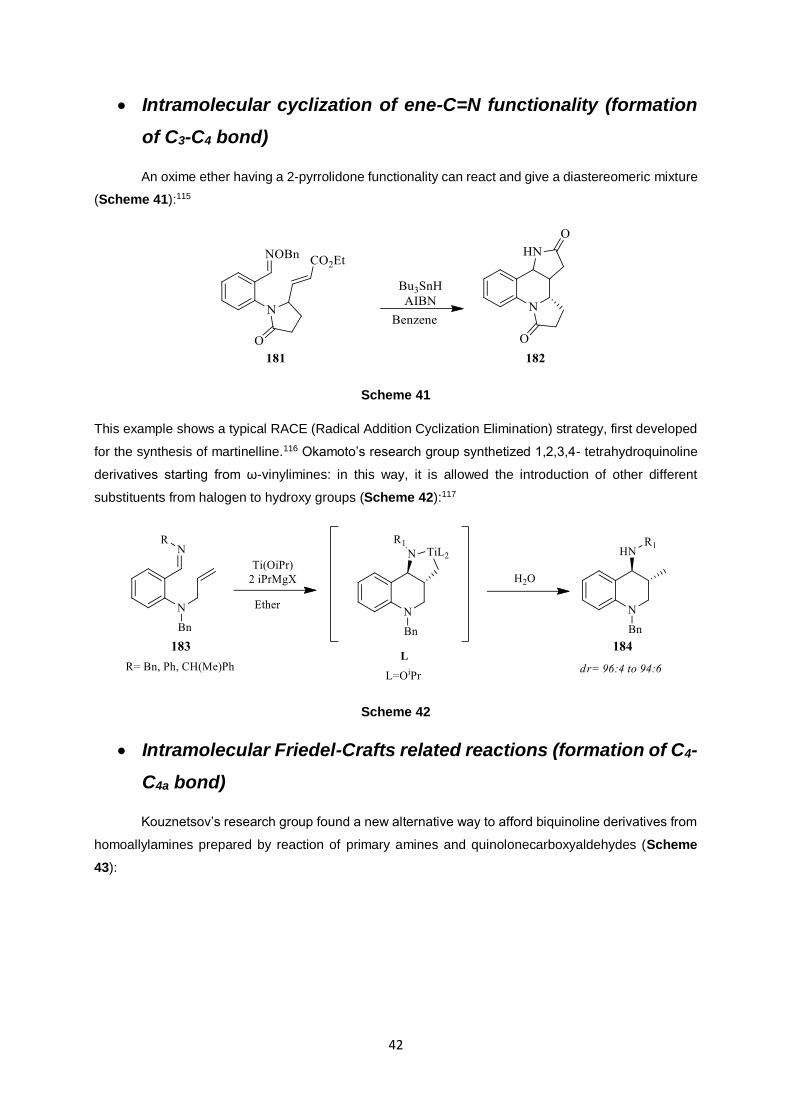
An oxime ether having a 2-pyrrolidone functionality can react and give a diastereomeric mixture
(Scheme 41):115
Scheme 41
This example shows a typical RACE (Radical Addition Cyclization Elimination) strategy, first developed
for the synthesis of martinelline.116 Okamoto’s research group synthetized 1,2,3,4- tetrahydroquinoline
derivatives starting from ω-vinylimines: in this way, it is allowed the introduction of other different
substituents from halogen to hydroxy groups (Scheme 42):117
Scheme 42
Intramolecular Friedel-Crafts related reactions (formation of C4-
C4a bond)
Kouznetsov’s research group found a new alternative way to afford biquinoline derivatives from
homoallylamines prepared by reaction of primary amines and quinolonecarboxyaldehydes (Scheme
43):
43
Scheme 43
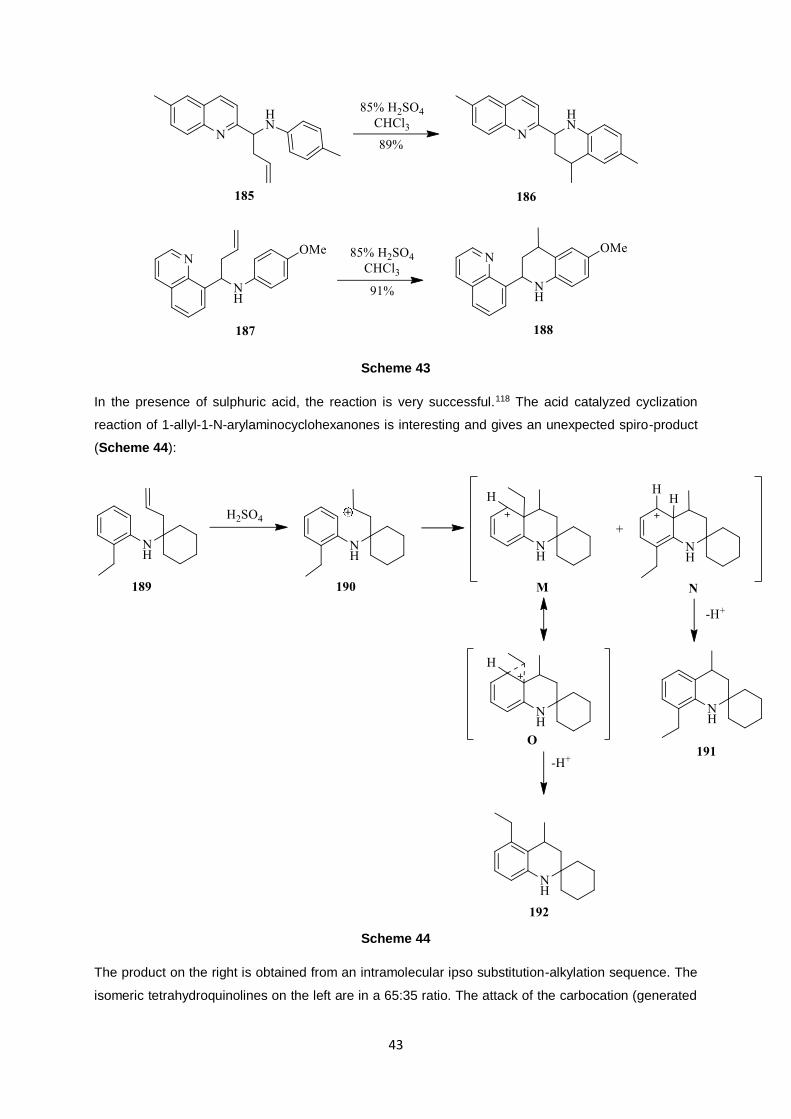
In the presence of sulphuric acid, the reaction is very successful.118 The acid catalyzed cyclization
reaction of 1-allyl-1-N-arylaminocyclohexanones is interesting and gives an unexpected spiro-product
(Scheme 44):
Scheme 44
The product on the right is obtained from an intramolecular ipso substitution-alkylation sequence. The
isomeric tetrahydroquinolines on the left are in a 65:35 ratio. The attack of the carbocation (generated
44
in acid conditions) followed by 1,2-shift of the ethyl substituent could be the reason for the presence of
the other product.119
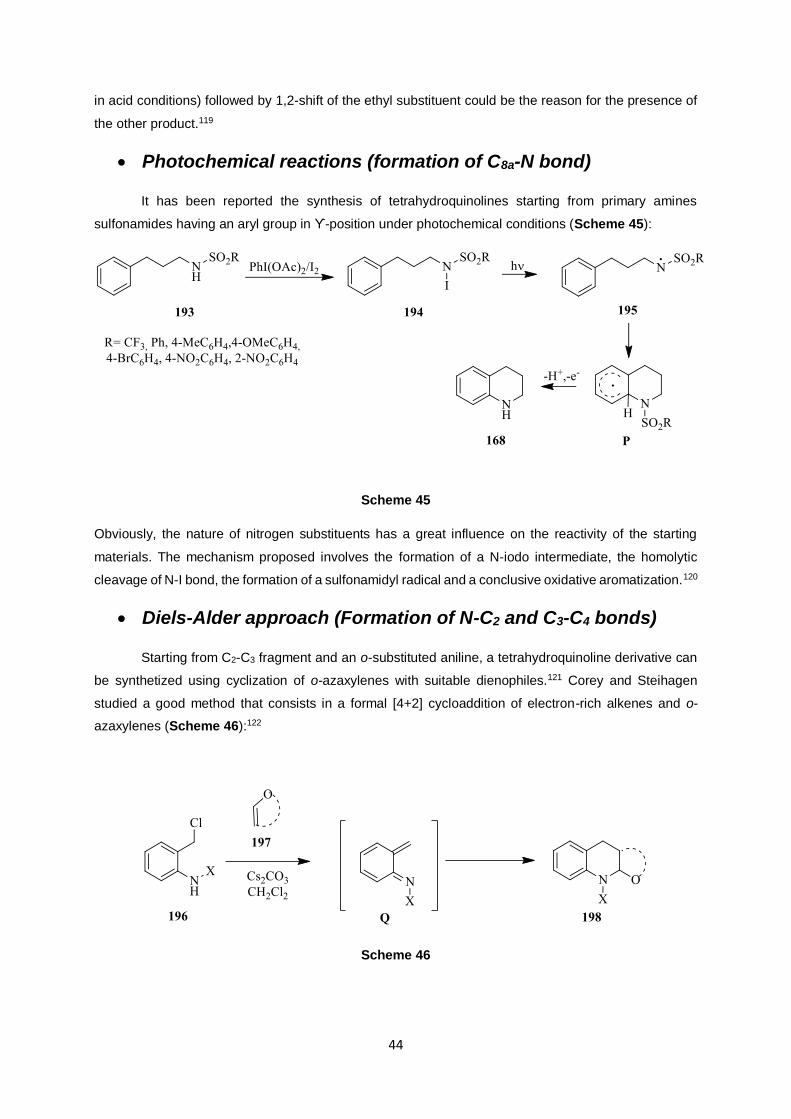
Photochemical reactions (formation of C8a-N bond)
It has been reported the synthesis of tetrahydroquinolines starting from primary amines
sulfonamides having an aryl group in ϒ-position under photochemical conditions (Scheme 45):
Scheme 45
Obviously, the nature of nitrogen substituents has a great influence on the reactivity of the starting
materials. The mechanism proposed involves the formation of a N-iodo intermediate, the homolytic
cleavage of N-I bond, the formation of a sulfonamidyl radical and a conclusive oxidative aromatization.120
Diels-Alder approach (Formation of N-C2 and C3-C4 bonds)
Starting from C2-C3 fragment and an o-substituted aniline, a tetrahydroquinoline derivative can
be synthetized using cyclization of o-azaxylenes with suitable dienophiles.121 Corey and Steihagen
studied a good method that consists in a formal [4+2] cycloaddition of electron-rich alkenes and o-
azaxylenes (Scheme 46):122
Scheme 46

45
Brønsted acid-catalyzed reactions (Formation of three or more
bonds)
Brønsted acids are capable of catalyzing Povarov reaction for the synthesis of
tetrahydroquinoline derivatives. The treatment of different imines and a wide range of
methyleneciclopropanes in the presence of montmorillonite K-10 or triflic acid gives very satisfactory
yields of the desired product (Scheme 47):123
Scheme 47
Khadem and co-workers studied an alternative one-pot synthesis of isoindolo[2,1-a]quinoline derivative
based on a Povarov-cyclocondensation domino approach (Scheme 48):124
Scheme 48
The advantage of this reaction is the fact that only one diastereomer is formed. It can be seen also in
the reaction between 3-aminoacetophenone, benzaldehyde, and cyclopentadiene with TFA (1
equivalent) (Scheme 49):
Scheme 49
The adjacent carbonyl group is responsible for the stabilization of the carbocationic intermediate
generated during the reaction and this fact explains the diasteroselectivity of this reaction. Important
spiro-tetrahydroquinolines can be synthetized starting from arylamines and keto sugars in presence of
p-toluensulfonic acid (Scheme 50):

46
Scheme 50
The reaction starts with the formation of the imine S which is in equilibrium with the enamine form T.
The next Povarov-like [4+2] cycloaddition in the presence of acid catalyst gives the tetrahydroquinoline
product in good yield: this compound showed immunobiological and cytotoxic activity.125
4. Experimental section
Aim of the work
In the first part of my work, I studied some mechanistic aspects of Povarov reaction of aniline,
benzaldehyde and 3,4-dihydro-2H-pyran (Scheme 51):
Scheme 51

47
The aim of this study was to understand the effect of the amount of solvent (acetonitrile) on the
stereoselectivity of the products.
In the second part of my thesis work, I focused my attention on the optimization of the Povarov reaction
between indolines, benzaldehyde and N-vinyl-pyrrolidone (Scheme 52):
Scheme 52
When the optimization study was finished, I attempted to synthetize tetrahydroquinoline derivatives from
substituted indolines (Scheme 53):
Scheme 53
The obtained tetrahydroquinolines can be applied in useful applications.
Result and discussion
The pilot reaction was carried out in solvent/solventless conditions using aniline, benzaldehyde and 3,4-
dihydro-2H-pyran (DHP), exploring CeCl3·7H2O/NaI as promoting system. The reaction was monitored
by GC-MS and TLC until the starting reagents were consumed or a constant composition of the mixture
was reached, and the final tetrahydroquinolines were obtained as racemates in syn/anti diastereomer
mixture. Based on the previous work of the research group, the reaction in CeCl3∙7H2O/NaI in dry
acetonitrile at 50°C for one hour led to an excellent syn:anti ratio of 15:85 and 82% yield while in
solventless conditions and lowering the temperature at -10°C it afforded to a 73% yield with an
interesting inverted diastereoselectivity (syn:anti 77:23). Generally the reactions performed in solvent
show a strong preference for the anti diastereomer together with slightly lower yields with respect to the
corresponding ones in solventless conditions. The aim of this part of my thesis is to understand if the
different selectivity is due to a different selectivity of the reaction in solvent or solventless conditions and
48
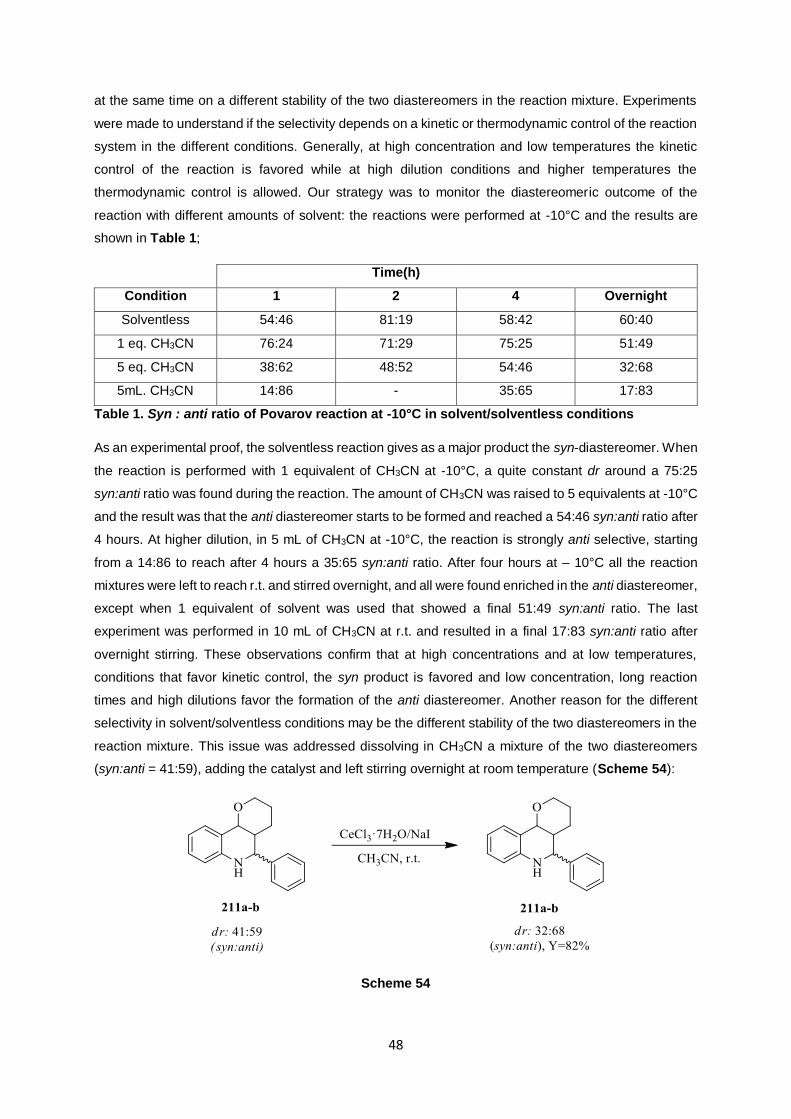
at the same time on a different stability of the two diastereomers in the reaction mixture. Experiments
were made to understand if the selectivity depends on a kinetic or thermodynamic control of the reaction
system in the different conditions. Generally, at high concentration and low temperatures the kinetic
control of the reaction is favored while at high dilution conditions and higher temperatures the
thermodynamic control is allowed. Our strategy was to monitor the diastereomeric outcome of the
reaction with different amounts of solvent: the reactions were performed at -10°C and the results are
shown in Table 1;
Time(h)
Condition 1 2 4 Overnight
Solventless 54:46 81:19 58:42 60:40
1 eq. CH3CN 76:24 71:29 75:25 51:49
5 eq. CH3CN 38:62 48:52 54:46 32:68
5mL. CH3CN 14:86 - 35:65 17:83
Table 1. Syn : anti ratio of Povarov reaction at -10°C in solvent/solventless conditions
As an experimental proof, the solventless reaction gives as a major product the syn-diastereomer. When
the reaction is performed with 1 equivalent of CH3CN at -10°C, a quite constant dr around a 75:25
syn:anti ratio was found during the reaction. The amount of CH3CN was raised to 5 equivalents at -10°C
and the result was that the anti diastereomer starts to be formed and reached a 54:46 syn:anti ratio after
4 hours. At higher dilution, in 5 mL of CH3CN at -10°C, the reaction is strongly anti selective, starting
from a 14:86 to reach after 4 hours a 35:65 syn:anti ratio. After four hours at – 10°C all the reaction
mixtures were left to reach r.t. and stirred overnight, and all were found enriched in the anti diastereomer,
except when 1 equivalent of solvent was used that showed a final 51:49 syn:anti ratio. The last
experiment was performed in 10 mL of CH3CN at r.t. and resulted in a final 17:83 syn:anti ratio after
overnight stirring. These observations confirm that at high concentrations and at low temperatures,
conditions that favor kinetic control, the syn product is favored and low concentration, long reaction
times and high dilutions favor the formation of the anti diastereomer. Another reason for the different
selectivity in solvent/solventless conditions may be the different stability of the two diastereomers in the
reaction mixture. This issue was addressed dissolving in CH3CN a mixture of the two diastereomers
(syn:anti = 41:59), adding the catalyst and left stirring overnight at room temperature (Scheme 54):
Scheme 54

49
The diastereomers were isolated in mixture and the yield and the dr were measured, showing that the
initial dr ratio passed to 32:68 demonstrating that the mixture was enriched in the anti diastereomer.
Significative is the fact that only the 82% of the initial amount of the mixture was recovered, that in more
detail revealed a 5% loss of the anti product and a 37% loss of the syn. These results confirm that the
selectivity of this reaction depends both on kinetic or thermodynamic control in the different reaction
conditions and on a different stability of the two diastereomers in the reaction mixture.
In the second part of my thesis work, I studied the effect of different additives of the reaction in Scheme
52 at different temperatures (0°C-r. t.-40°C). The first step is the synthesis of the starting indoline by
reduction of indole with sodium cyanoborohydride in acetic acid at room temperature (Scheme 55):
Scheme 55
The procedure is efficient and the yields are high. The indoline was synthetized, purified by filtration on
basic alumina (Al2O3) then used in the successive Povarov reaction (Scheme 56):
Scheme 56
These reactions are performed at three different temperatures and with the promoting system
CeCl3∙7H2O/different iodide sources (LiI, KI, CuI and NaI) to evaluate their effect on this Povarov
reaction. The results are shown in Table 2:

50
T NaI CuI KI LiI
dr(syn:anti) Yield dr(syn:anti) Yield dr(syn:anti) Yield dr(syn:anti) Yield
0°C 92.8 60 % 94:6 51 % 91:9 27 % 92:8 18 %
rt 91:9 48 % 95:5 42 % 93:7 48 % 93:7 45 %
40°C 90:10 51 % 95:5 51 % 91:9 45 % 93:7 42 %
Table 2. Syn : anti ratio and yeld of Povarov reaction at different temperatures and catalyst
When the reaction was judged to be complete, the solution was diluted with dichloromethane, filtered
with celite and purified with flash chromatography; the dr vas evaluated by NMR analysis on the
chromatographic fraction of the diasteroisomers isolated in mixture. As shown in the Table 2, except for
a few cases, yields are quite similar and the syn adduct is favored over the anti one. The reason for this
behavior is the electron-donating dienophile, N-vinyl-2-pyrrolidone. According to literature,126,127 we
thought that the syn adduct in high quantity respect to the anti one: this hypothesis was confirmed during
these trials. The optimal reaction conditions derived from these trials are the CeCl3∙7H2O/NaI as catalyst
and 0°C as operating conditions that we apply to the synthesis of a series of substituted indolines that
were prepared by reduction of the corresponding indole with NaCNBH3 in acetic acid at room
temperature under inert atmosphere (Scheme 57), according to literature.128 The results are shown in
Table 3.
Scheme 57
Indoline Yield (%)
214b 92
214c 76
214d 81
214e 89
214f 84
Table 3. Synthesis of the substituted indolines.
Scheme 58 shows the successive Povarov reaction and the results are shown on Table 4:

51
Scheme 58
Indoline Yield (%) dr (syn:anti)
213b 72 80:20
213c 53 90:10
213d 64 95:5
213e 50 92:8
213f 65 95:5
Table 4. Results of the Povarov reaction with substituted indolines.
The reaction mixture was diluted with CH2Cl2, filtered with celite and the product were purified by flash
chromatography (Hex : EtOAc). Unexpectedly, yields are quite high compared to the ones with
unsubstituted indoline and as expected, dr are also quite good in these reactions.

52
Materials and methods for the synthesis of compounds
All reagents and solvents were purchased from commercial suppliers and used without further
purification, unless mentioned otherwise. All reactions were monitored by thin-layer chromatography
using EMD/Merck silica gel 60 pre-coated plates (0.25 mm), and the compounds were visualized either
by using UV light (254 nm), and vanillin stains as developing agent. Purification of the reaction products
was carried out by column flash-chromatography using silica gel (0.040–0.063 mesh). 1H and 13C NMR
spectra were recorded on a Varian Mercury 400 (400 MHz, 100 MHz or 377 MHz respectively). Chemical
shifts are given in ppm with reference to residual H in deuterated solvents as the internal standard.
Coupling constants J are reported in Hz. Diastereomeric ratio was determined by integration of 1H-NMR
signals (H4 and H6) of the mixture of diasteromers. The compounds are described as ”M” for the major
isomer and ”m” for the minor one. Mass spectra were obtained using a gas chromatograph Agilent 6850
equipped with a HP5MS column (0.25 mm diameter) and a mass-selective detector Agilent 5973N, or
using a LC-MS Hewlett Packard series 1100 MSD.

53
Method for the synthesis of 5-phenyl-3,4,4a,5,6,10b-hexahydro-2H-
pyrano[3,2-c]quinoline (211a-b):
In a round bottom flask, aniline (1 mmol) and benzaldehyde (1 mmol) were stirred together in different
amounts of CH3CN according to Table 1 in the presence of anhydrous MgSO4 (200 mg). The mixture
was stirred until imine was completely formed. Then, CeCl3·7H2O (112 mg, 0.3 mmol) and NaI (45 mg,
0.3 mmol) were added, followed by 2,3 dihydropyran (1 mmol) and the mixture was stirred at -10°C until
consumption of starting materials (TLC, hexane/EtOAc, vanillin stain). The mixture was diluted with
CH2Cl2 and washed with HCl 0.5 M. The aqueous layer was extracted with CH2Cl2, then treated with
satured solution of NaHCO3 until basic pH was reached and extracted again with CH2Cl2. The organic
layers were unified, washed with brine and dried over anhydrous Na2SO4. The crude was purified by
flash chromatography over silica (eluent hexane/EtOAc) to afford the pure tetrahydroquinoline products.
The reaction was also performed in solventless conditions.
Method for the synthesis of indolines (212a-f):
In inert atmosphere, to a stirred solution of the starting indole (2-5 mmol) in acetic acid (4-10 mL) was
added NaBH3CN (3-7.5 mmol, 1.5 eqiv.). The reaction was allowed to stir at room temperature and
reaction progress was monitored by TLC. After the reaction was judged to be complete, the reaction
mixture was diluted with H2O, brought to pH~9 with a 2M NaOH solution and extracted with Et2O or
EtOAc. The combined organic layers were washed with H2O, brine, dried over Na2SO3, filtered and
concentrated unfer reduced pressure. The crude product was filtered on basic alumina (Al2O3) or purified
by flash column cromatography (Hex:EtOAc) to yield the corresponding indoline.

54
Method for the synthesis of 1-(3-phenyl-1,2,3,5,6,7-
hexahydropyrido[3,2,1-ij]quinolin-1-yl)pyrrolidin-2-one and its
derivatives (213a-f):
In a round bottom flask, indoline (substituted or not) (1 mmol) and benzaldehyde (1 mmol) were stirred
together in 5 ml of CH3CN in the presence of anhydrous MgSO4 (200 mg). The mixture was stirred until
imine was completely formed. Then, CeCl3·7H2O (112 mg, 0.3 mmol) and MI (X=Na, Li, Cu, K, 0.3
mmol) were added, followed by N-vinyl-2-pyrrolidinone (1 mmol) and the mixture was stirred at 0, 40°C
or room temperature until consumption of starting materials (TLC, hexane/EtOAc, vanillin stain). The
mixture was diluted with CH2Cl2, filtered on a gooch and Celite and evaporate to dryness. The crude
was purified by flash chromatography over silica (eluent hexane/EtOAc) to afford the pure
tetrahydroquinoline derivative products.

55
Characterization of compounds
5-phenyl-3,4,4a,5,6,10b-hexahydro-2H-pyrano[3,2-c]quinoline (211a)
White solid. Mp: 130 °C. 1H NMR (400 MHz, CDCl3) δH (ppm) 7.44-7.28 (m, 6H),
7.10 (t, J= 7.4 Hz, 1H), 6.80 (t, J = 7.4 Hz, 1H), 6.61 (d ,J= 7.9 Hz, 1H), 5.33 (d,
J= 5.4 Hz, 1H), 4.69 (d, J= 2.1 Hz, 1H), 3.88 (m, 1H), 3.58 (dd, J1 = 11.3 Hz, = 3.8
Hz, 1H), 3.43 (td, J= 11.0, 2.3 Hz, 1H), 2.17-2.16 (m, 1H), 1.58-1.42(m, 3H), 1.31-
1.25 (m, 1H); 13C NMR (400 MHz, CDCl3) δC (ppm) 145.1, 141.1, 128.3, 128.1, 127.6, 127.5, 126.8,
119.8, 118.2, 114.4, 72.7, 60.6, 59.3, 38.9, 25.4, 18.0. MW: 265.350 g/mol. MS m/z: 265 (M)+; 234; 220;
194; 129; 117; 91; 77. Data are in accordance with literature.129
5-phenyl-3,4,4a,5,6,10b-hexahydro-2H-pyrano[3,2-c]quinoline (211b)
Viscous oil. 1H NMR (400 MHz, CDCl3) δH (ppm) 7.44-7.29 (m, 5H), 7.24-7.21 (m,
1H), 7.10 (dt, J= 8.0, 1.4 Hz, 1H), 6.70 (t, J= 7.4 Hz, 1H), 6.51 (d, J= 8.0 Hz, 1H),
4.71 (d, J= 10.8 Hz, 1H), 4.38 (d, J= 2.6 Hz, 1H), 4.13-4.08 (m, 2H), 3.71 (td, J=
11.5, 2.5 Hz, 1H), 2.12-2.05 (m, 1H), 1.93-1.77 (m, 1H), 1.71-1.59 (m, 1H), 1.49-
1.44 (m, 1H), 1.36-1.26(m, 1H); 13C NMR (400 MHz, CDCl3) δC (ppm) 144.8, 142.4,
130.9, 129.4, 128.7, 127.9, 127.9, 120.6, 117.4, 114.2, 74.5, 68.6, 54.8, 38.9, 24.2, 22.1. MW: 265.15
g/mol. MS m/z: 265 (M)+, 234; 220; 194; 129; 117; 91; 77. Data are in accordance with literature.129
Indoline 212a
Pale yellow oil. Yields from 74 to 90%. 1H NMR (400 MHz, CDCl3) δH 7.03 (d, J= 7.2 Hz,
1H), 6.93 (t, J=7.6 Hz, 1H), 6.62 (t, J=7.4 Hz, 1H), 6.55 (d, J=7.7 Hz, 1H), 3.44 (t, J= 8.3
Hz, 3H), 2.93 (t, J=8.3 Hz, 3H). 13C NMR (400 MHz, CDCl3) δC 151.64, 129.37, 127.25,
124.68, 118.71, 109.51, 47.37, 29.89; MW: 119.166 g/mol. Data are in accordance with literature.130

56
(±)-3-Methylindoline 212b
Pale red/brown oil. Yield: 92 %. 1H NMR (400 MHz, CDCl3) δH 7.14-6.98 (m, 2H), 6.75 (t,
J=7.3 Hz, 1H), 6.66 (d, J= 7.8 Hz, 1H), 3.71 (t, J= 8.7 Hz, 1H), 3.38 (q, J= 7.7 Hz, 1H), 3.12
(t, J=8.6 Hz, 2H), 1.33 (d, J=6.7 Hz, 3H); 13C NMR (400 MHz, CDCl3) δC 151.2, 134.5,
127.4, 123.5, 118.9, 109.7, 55.5, 36.7, 18.7; MW: 133,193 g/mol. Data are in accordance with
literature.131
(±)-2-Phenylindoline 212c
Yellow oil. Yield: 76 %. 1H NMR (400 MHz, CDCl3) δH 7.51-7.41 (m, 2H), 7.40-7.33
(m, 2H), 7.32-7.26 (m, 1H), 7.16-7.03 (m, 2H), 6,76 (td, J=7.4,1.0 Hz, 1H), 6.69 (d,
J= 7.7 Hz, 1H), 4.97 (t, J=9.0 Hz, 1H), 4.03 (s, 1H), 3.46 (dd, J=15.6, 9.2 Hz, 1H),
3.01 (ddt, J= 15.6, 8.7, 1.1 Hz, 1H); ). 13C NMR (400 MHz, CDCl3) δC 150.5, 144.3, 129.1, 128.7, 128.4,
127.7, 127.6, 126.5, 125.3, 124.7, 119.3, 109.4, 63.6, 39.6; MW: 195,264 g/mol. Data in accordance
with literature.132
5-Bromoindoline 212d
Yellow oil. Yield: 81%. 1H NMR (400 MHz, CDCl3) δH 7.19 (dt, J=2.2, 1.2 Hz, 1H), 7.12-
7.07 (m, 1H), 6.49 (d, J=8.3 Hz, 1 H), 3.73-3.61 (m, 1H), 3.56 (t, 8.4 Hz, 2H), 3.01 (t,
J=8.3,1.0, 2H); 13C NMR (400 MHz, CDCl3) δC 150.7, 131.9, 129.9, 127.7, 110.6, 110.2, 47.7, 29.8;
MW: 198,062 g/mol. Data in accordance with literature.132
5-Methoxyindoline 212e
Pale yellow oil. Yield: 89%. 1H NMR (400 MHz, CDCl3) H 6.72 (dd, J= 2.4, 1.2 Hz,
1H), 6.59 (ddt, J= 8.5, 2.6, 0.9 Hz, 1H), 6.54 (d, J= 8.4 Hz, 1H), 3.97 (tq, J= 8.1, 6.2
Hz, 1H), 3.74 (s, 3H), 3.38 (s, 1H), 3.11 (m, 1H), 2.62 (ddt, J= 15.5, 7.9, 1.0 Hz, 1H), 1.29 (d, J= 6.2 Hz,
3H);13C NMR (400 MHz, CDCl3) C 153.4, 145.2, 131.11, 112.0, 111.4, 110.1, 55.8, 47.7, 30.4; MW:
149,192 g/mol. Data are in accordance with literature.133

57
(±)-5-Methoxy-2-methylindoline 212f
. Yellow oil. Yield: 84%. 1H NMR (400 MHz, CDCl3) δH 6.71–6.70 (m, 1H), 6.59–
6.51 (m, 2H), 4.00–3.90 (m, 1H), 3.73 (s, 1H), 3.40 (m, 1H), 3.10 (dd, J = 15.6,
8.4 Hz, 1H), 2.60 (dd, J = 15.4, 7.6Hz, 1H), 1.27 (d, J = 6.0 Hz, 3H); 13C NMR (400 MHz, CDCl3) δC
153.4, 144.7, 130.6, 112.0,111.6, 109.8, 55.9, 55.6, 38.2, 22.1. MW: 163,219 g/mol. Data are in
accordance in literature.134
1-(4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213a (M)
White foam. Yield from 18 to 60%. FT-IR (neat, cm-1) 2959, 2928, 2860,
1717,1267; 1H NMR (400 MHz, CDCl3) δH 7.47–7.40 (m, 2H), 7.37 ( t, J = 7.5
Hz, 2H), 7.34–7.27 (m, 1H), 7.03 (d, J= 7.0 Hz, 1H), 6.83–6.66 (m, 2H), 5.69
(dd, J= 11.3, 6.5 Hz, 1H), 4.02 (dd, J=11.0, 2.5 Hz, 1H), 3.42–3.21 (m, 3H),
3.04–2.92 (m, 1H), 2.90–2.81 (m, 1H), 2.79–2.71 (m, 1H), 2.56–2.39 (m, 2H), 2.27-2.11 (m, 2H), 2.08–
1.94 (m, 2H); 13C NMR (400 MHz, CDCl3) δC 175.8, 148.8, 141.6, 129.9, 128.7, 127.7, 126.9, 124.3,
123.6, 119.3 , 117.3, 116.2, 63.0, 56.7, 53.4, 47.8, 42.4, 36.8, 31.4, 28.8, 18.2; Anal Calcd for
C21H22N2O; MS m/z: 318.2 (M+), 232 (100), 204, 185, 156, 135, 115, 91, 65, 41; MW: 318.418 g/mol; C
79.21% H 6.96% N 8.80% Found C 79.25% H 6.94% N 8.78%. Data are in accordance with literature.135
1-(4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213a (m)
White foam. Yield from 18 to 60%. FT-IR (neat, cm-1) 2959, 2928, 2860,
1717,1267; 1H NMR (400 MHz, CDCl3) δH 7.47–7.40 (m, 2H), 7.37 ( t, J = 7.5
Hz, 2H), 7.34–7.27 (m, 1H), 7.03 (d, J= 7.0 Hz, 1H), 6.83–6.66 (m, 2H), 5.42
(dd, J= 6.4, 2.3 Hz, 1H), 3.82 (dd, J=11.1, 3.1 Hz, 1H), 3.42–3.21 (m, 3H), 3.04–
2.92 (m, 1H), 2.90–2.81 (m, 1H), 2.79–2.71 (m, 1H), 2.56–2.39 (m, 2H), 2.27-2.11 (m, 2H), 2.08–1.94
(m, 2H); 13C NMR (400 MHz, CDCl3) δC 175.8, 148.8, 141.6, 129.9, 128.7, 127.7, 126.9, 124.3, 123.6,
119.3 , 117.3, 116.2, 63.0, 56.7, 53.4, 47.8, 42.4, 36.8, 31.4, 28.8, 18.2; Anal Calcd for C21H22N2O; MS
m/z: 318.2 (M+), 232 (100), 204, 185, 156, 135, 115, 91, 65, 41; MW: 318.418 g/mol; C 79.21% H 6.96%
N 8.80% Found C 79.25% H 6.94% N 8.78%. Data are in accordance with literature.134

58
1-(3-methyl-4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213b
(M)
Light red foam. Yield: 72%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267; 1H
NMR (400 MHz, CDCl3) δH 7.47 (d, J=7.26 Hz, 2H), 7.40 (t, J= 7.5,7.5 Hz, 2H),
7.3-7.27 (m, 1H), 7.02 (d, J=7.1 Hz, 1H), 6.73 (t, J=8.9 Hz, 1H), 6.67 (t, J=7.4 Hz,
1H), 5.70 (dd, J=11.5, 5.9 Hz, 1H), 5.31 (s, 1H), 4.47 (dd, J=11.0,2.6 Hz, 1H),
3.84-3.74 (m, 1H), 3.35-3.19 (m, 2H), 2.60-2.42 (m, 3H), 2.25-2.09 (m, 2H), 2.09-
1,95 (m, 2H), 0.90 (dd, J=6.5, 3.8 Hz, 3H); 13C NMR (400 MHz, CDCl3) δC 175.3, 148.8, 128.7, 128.5,
128.24, 128.1, 127.0, 124,1, 123.9, 118.1, 116.8, 77.3, 77.1, 76.9, 57.3, 56.8, 53.45, 36.5, 43.7, 42.4,
37.2, 31.5. 18.3, 15.9; Anal Calcd for C22H24N2O; MS m/z: 332.2 (M+), 246 (100), 213, 196, 170, 142,
115, 91, 69, 41; MW: 332.170 g/mol; C 79.48% H 7.28% N 8.43% Found: C 79.51% H 7.31% N 9.37%.
1-(3-methyl-4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213b
(m)
Light red foam.. Yield: 72%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267; 1H
NMR (400 MHz, CDCl3) δH 7.47 (d, J=7.26 Hz, 2H), 7.40 (t, J= 7.5,7.5 Hz, 2H),
7.3-7.27 (m, 1H), 7.02 (d, J=7.1 Hz, 1H), 6.73 (t, J=8.9 Hz, 1H), 6.67 (t, J=7.4 Hz,
1H), 5.42 (dd, J= 6.4, 2.3 Hz, 1H), 5.31 (s, 1H), 3.80 (dd, J=11.1, 3.1 Hz, 1H),
3.84-3.74 (m, 1H), 3.35-3.19 (m, 2H), 2.60-2.42 (m, 3H), 2.25-2.09 (m, 2H), 2.09-
1,95 (m, 2H), 0.90 (dd, J=6.5, 3.8 Hz, 3H); 13C NMR (400 MHz, CDCl3) δC 175.3, 148.8, 128.7, 128.5,
128.24, 128.1, 127.0, 124,1, 123.9, 118.1, 116.8, 77.3, 77.1, 76.9, 57.3, 56.8, 53.45, 36.5, 43.7, 42.4,
37.2, 31.5. 18.3, 15.9; Anal Calcd for C22H24N2O; MS m/z: 332.2 (M+), 246 (100), 213, 196, 170, 142,
115, 91, 69, 41; MW: 332.170 g/mol; C 79.48% H 7.28% N 8.43% Found: C 79.51% H 7.31% N 9.37%.

59
1-(2,4-diphenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213c (M)
Light green oil. Yield: 53%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267; 1H
NMR (400 MHz, CDCl3) δH 7.37-6.88 (m, 10 H), 6.79-6.65 (m, 3H), 5.66 (dd, J=
11.8, 5.4 Hz, 1H), 4.54 (t, J= 10.1 Hz, 1H), 4.15-4.08 (m, 1H), 3.69-3.58 (m,
1H), 3.43-3.18 (m, 2H), 3.01 (t, J=11.8 Hz, 1H), 2.60-2.40 (m, 2H), 2.31-2.22
(m, 1H), 2.12-1.92 (m, 2H), 1.28 (t, J=7.1 Hz, 1H); 13C NMR (400 MHz, CDCl3)
δC 175.9, 149.7,145.6, 141.0, 140.7, 128.5, 128.3, 127.8, 127.6, 127.3, 126.68, 124.1, 123.7, 118.3,
116.0, 65.4, 57.7, 47.5, 42.5, 37.6, 36.8, 31.4, 18.3; Anal Calcd for C27H26N2O; MS m/z: 394.2 (M+), 309
(100), 232, 205, 178, 154, 115, 91, 65, 41; MW: 394.205 g/mol; C 88.20% H 6.64% N 7.10%; Found C
82.23 % H 6.66% N 7.05%.
1-(2,4-diphenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213c (m)
Light green oil. Yield: 53%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267; 1H
NMR (400 MHz, CDCl3) δH 7.37-6.88 (m, 10 H), 6.79-6.65 (m, 3H), 5.42 (dd, J=
6.4, 2.3 Hz, 1H), 4.54 (t, J= 10.1 Hz, 1H), 3.79 (dd, J=11.1, 3.1 Hz, 1H), 3.69-
3.58 (m, 1H), 3.43-3.18 (m, 2H), 3.01 (t, J=11.8 Hz, 1H), 2.60-2.40 (m, 2H),
2.31-2.22 (m, 1H), 2.12-1.92 (m, 2H), 1.28 (t, J=7.1 Hz, 1H); 13C NMR (400
MHz, CDCl3) δC 175.9, 149.7,145.6, 141.0, 140.7, 128.5, 128.3, 127.8, 127.6, 127.3, 126.68, 124.1,
123.7, 118.3, 116.0, 65.4, 57.7, 47.5, 42.5, 37.6, 36.8, 31.4, 18.3; Anal Calcd for C27H26N2O; MS m/z:
394.2 (M+), 309 (100), 232, 205, 178, 154, 115, 91, 65, 41; MW: 394.205 g/mol; C 88.20% H 6.64% N
7.10%; Found C 82.23 % H 6.66% N 7.05%.

60
1-(8-bromo-4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213d
(M)
White foam. Yield: 50%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267; 1H
NMR (400 MHz, CDCl3) δH 7.47-7.24 (m, 5H), 7.17-7.04 (m, 1H), 6.87 (s, 1H),
5.67 (dd, J= 11.4, 6.4 Hz, 1H), 4.01 (dd, J= 11.1, 2.5 Hz, 1H), 3.31 (dt, J= 14.0,
7.7 Hz, 4H), 3.08-2.69 (m, 2H), 2.61-2.37 (m, 2H), 2.26-1.91 (m, 4H); 13C NMR
(400 MHz, CDCl3) δC 175.9, 150.4, 141.2, 132.2, 129.9, 128.8, 127.9, 126.9,
126.8, 126.7, 118.9, 111.0, 77.4, 77.1, 76.8, 62.8, 53.0, 47.5, 42.3, 36.5, 31.3, 28.6, 18.2; Anal cald for
C21H21BrN2O; MS m/z: 396.1 (M+), 312 (100), 234, 204, 178, 154, 115, 91, 63, 41; MW: 397,122 g/mol;
C 63.48% H 5.33% Br 20.11% N 7.05%; Found C 63.50% H 5.31% Br 20.13% N 7.03%.
1-(8-bromo-4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213d
(m)
White foam. Yield: 50%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267; 1H
NMR (400 MHz, CDCl3) δH 7.47-7.24 (m, 5H), 7.17-7.04 (m, 1H), 6.87 (s, 1H),
5.42 (dd, J= 6.4, 2.3 Hz, 1H), 3.78 (dd, J=11.1, 3.1 Hz, 1H), 3.31 (dt, J= 14.0, 7.7
Hz, 4H), 3.08-2.69 (m, 2H), 2.61-2.37 (m, 2H), 2.26-1.91 (m, 4H); 13C NMR (400
MHz, CDCl3) δC 175.9, 150.4, 141.2, 132.2, 129.9, 128.8, 127.9, 126.9, 126.8,
126.7, 118.9, 111.0, 77.4, 77.1, 76.8, 62.8, 53.0, 47.5, 42.3, 36.5, 31.3, 28.6, 18.2; Anal cald for
C21H21BrN2O; MS m/z: 396.1 (M+), 312 (100), 234, 204, 178, 154, 115, 91, 63, 41; MW: 397,122 g/mol;
C 63.48% H 5.33% Br 20.11% N 7.05%; Found C 63.50% H 5.31% Br 20.13% N 7.03%.
1-(8-methoxy-4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213e
(M)
Yellow oil. Yield: 64%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267; 1H NMR
(400 MHz, CDCl3) δH 7.59-7.19 (m, 5H), 6.72 (s, 1H), 6.36 (s, 1H), 5.69 (dd, J=
10.4, 7.4, 1H), 3.91 (t, J= 10.4 Hz, 1H), 3.75 (s, 3H), 3.34 (dt, J= 13.5, 7.6 Hz,
3H), 2.97 (d, J= 4.5 Hz, 1H), 2.87-2.77 (m, 1H), 2.71 (s, 1H), 2.57-2.39 (m, 2H),
2.26-2.12 (m, 2H), 2.08-1.97 (m, 2H); 13C NMR (400 MHz, CDCl3) δC 175.8, 150.4, 148.8, 141.2, 132.2,
129.9, 128.7, 127.7, 127.2, 127.0, 111.3, 109.1, 56.2, 54.1, 47.9, 42.9, 36.7, 31.4, 29.1, 18.2; Anal Calcd
for C22H24N2O2; MS m/z: 348.2 (M+), 262 (100), 232, 204, 186, 160, 143, 115, 91, 69, 41; MW: 348,123
g/mol; C 75.83% H 6.94% N 8.04%; Found C 75.84% H 6.93% N 8.02%.

61
1-(8-methoxy-4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-one 213e
(m)
Yellow oil. Yield: 64%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267; 1H NMR
(400 MHz, CDCl3) δH 7.59-7.19 (m, 5H), 6.72 (s, 1H), 6.36 (s, 1H), 5.42 (dd, J=
6.4, 2.3 Hz, 1H), 3.85 (dd, J=11.1, 3.1 Hz, 1H), 3.75 (s, 3H), 3.34 (dt, J= 13.5,
7.6 Hz, 3H), 2.97 (d, J= 4.5 Hz, 1H), 2.87-2.77 (m, 1H), 2.71 (s, 1H), 2.57-2.39
(m, 2H), 2.26-2.12 (m, 2H), 2.08-1.97 (m, 2H); 13C NMR (400 MHz, CDCl3) δC 175.8, 150.4, 148.8,
141.2, 132.2, 129.9, 128.7, 127.7, 127.2, 127.0, 111.3, 109.1, 56.2, 54.1, 47.9, 42.9, 36.7, 31.4, 29.1,
18.2; Anal Calcd for C22H24N2O2; MS m/z: 348.2 (M+), 262 (100), 232, 204, 186, 160, 143, 115, 91, 69,
41; MW: 348,123 g/mol; C 75.83% H 6.94% N 8.04%; Found C 75.84% H 6.93% N 8.02%.
1-(8-methoxy-2-methyl-4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-
one 213f (M)
Brown/red foam. Yield: 65%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267;
1H NMR (400 MHz, CDCl3) δH 7.57-7.26 (m, 5H), 6.70 (s, 1H), 6.32 (s, 1H), 5.68
(dd, J=10.4, 6.8 Hz, 1H), 5.32 (s, 1H), 4.37 (d, J=12.2 Hz, 1H), 3.78 (d, J= 30.1
Hz, 3H), 3.35-3.11 (m,2H), 2.61-2.37 (m, 2H), 2.18 (d, J= 7.0 Hz, 2H), 2.14 (dd,
J= 37.3, 16.6 Hz, 2H), 2.08-1.95 (m, 2H), 1.09-0.67 (m,3H); 13C NMR (400 MHz,
CDCl3) δC 175.9, 129.9, 128.7, 128.5, 128.3, 127.7, 127.0, 111.8, 108.9, 57.8, 57.1, 56.2, 53.4, 47.9,
42.5, 37.3, 36.7, 31.4, 18.4, 18.3, 15.4; Anal. calcd for C23H26N2O2; MS m/z: 362.2 (100, M+), 276, 249,
218, 200, 172, 147, 115, 91, 69, 41; MW: 362,154 g/mol; C 76.21% H 7.23% N 7.73%; Found C 76.22%
H 7.25% N 7.70%.

62
1-(8-methoxy-2-methyl-4-phenyl-2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-6-yl)pyrrolidin-2-
one 213f (m)
Brown/red foam. Yield: 65%. FT-IR (neat, cm-1) 2959, 2928, 2860, 1717,1267;
1H NMR (400 MHz, CDCl3) δH 7.57-7.26 (m, 5H), 6.70 (s, 1H), 6.32 (s, 1H), 5.42
(dd, J= 6.4, 2.3 Hz, 1H), 5.32 (s, 1H), 4.03 (dd, J=11.1, 3.1 Hz, 1H), 3.78 (d, J=
30.1 Hz, 3H), 3.35-3.11 (m,2H), 2.61-2.37 (m, 2H), 2.18 (d, J= 7.0 Hz, 2H), 2.14
(dd, J= 37.3, 16.6 Hz, 2H), 2.08-1.95 (m, 2H), 1.09-0.67 (m,3H); 13C NMR (400
MHz, CDCl3) δC 175.9, 129.9, 128.7, 128.5, 128.3, 127.7, 127.0, 111.8, 108.9, 57.8, 57.1, 56.2, 53.4,
47.9, 42.5, 37.3, 36.7, 31.4, 18.4, 18.3, 15.4; Anal. calcd for C23H26N2O2; MS m/z: 362.2 (100, M+), 276,
249, 218, 200, 172, 147, 115, 91, 69, 41; MW: 362,154 g/mol; C 76.21% H 7.23% N 7.73%; Found C
76.22% H 7.25% N 7.70%.

63
5. Conclusions
The main purpose of this experimental thesis was to study some Lewis acid catalyzed Povarov
reactions for the synthesis of important and biologically active polycyclic tetrahydroquinolines
derivative. The results are quite interesting: on one hand, the choice of dienophile (DHP or N-vinyl-
2.pyrrolidone) had a great influence on the reaction outcome, on the other reaction conditions, such as
temperature and the presence/absence of solvent, played an important role. In the first part of the
work, a confirmation of the previous research group’s work has been obtained. In the second one,
among all the metal iodide tested, NaI was the one with better results in combination with CeCl3∙7H2O:
this is another experimental proof of the optimal activity of this catalytic couple. In the third and last
part of the work, the synthesis of new polycyclic tetrahydroquinoline derivatives starting from
substituted indolines gave good results. Each compound has been analyzed and characterized. For
the future research work, parallel to the synthesis of new compounds of this type, mew methods can
be studied in order to obtain the desired diastereomer.

64
6. References
Pericylic Reactions
1 Tietze, L. F.; Brasche, G.; Gericke, K. Domino Reactions in Organic Synthesis, 2006, Wiley-VCH;
2 Claisen, L. Chem. Ber., 1912, 45, 3157;
3 Reid, J. P.; McAdam, C. A.; Johnston, A. J. S.; Grayson, M. N.; Goodman, J. M.; Cook, M. J. J. Org.
Chem., 2015, 80, 1472-1498;
4 Nishibayashi, Y.; Uemura, S. Top. Curr. Chem., 2000, 208, 201-235;
5 Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry, Second Edition, 2012, Oxford University
Press;
6 Ernst, R,; Winter, K. Tetrahedron Letters, 1965, 6, 1207-1212;
7 Schreiber, S. L.; Santini, C., J. Am. Chem. Soc., 1984, 106, 4038;
8 IUPAC, Compendium of Chemical Terminology, Gold Book, 2014;
9 Diels, O.; Alder, K. Justus Liebigs Ann. Chem., 1928, 460, 98–122;
10 Povarov, L. S., Russ. Chem. Rev., 1967, 36, 656;
11 Povarov, L. S.; Mikhailov, B.M.; Izv. Akad. Nauk, S.S.R. Ser. Khim., 1963, 953–956;
12 Povarov, L. S.; Grigos, V. I.; Mikhailov, B. M.; Izv. Akad. Nauk, S.S.R. Ser. Khim. 1963, 2039–2041;
13 Buonora, P.; Olsen, J. C.; Oh, T. Tetrahedron, 2001, 57, 6099–6138;
14 (a) Beifuss, U.; Ledderhose, S.; Ondrus, V. Arkivoc, 2005, 147–173; (b) Alves, M. J.; Azoia, N. G.;
Fortes, A. G. Tetrahedron, 2007, 63, 727–734; (c) Mayr, H.; Ofial, A. R.; Sauer, J.; Schmied, B. Eur. J.
Org. Chem., 2000, 2013–2020;
15 Ríos-Gutierrez, M.; Layeb, H.; Domingo, L. R. Tetrahedron, 2015, 71, 9339-9345;
16 Shi, F.; Xing, G.J.; Tao, Z.L.; Luo, S.W.; Tu, S.J.; Gong, L.Z. J. Org. Chem., 2012, 77, 6970-6979;
17 Hao, X.; Zhang, H.; Jacobsen, E. N. Nat Protoc., 2014, 9, 1860–1866;
18 Sridharan, V.; Avendano, C.; Menendez, J. C. Tetrahedron, 2007, 63, 673–681;
19 Park, K. H.; Joo, H. S.; Ahn, K. I.; Jun, K. Tetrahedron Lett., 1995, 36, 5943–5946;
20 Bohorquez, A. R. R.; Kouznetsov, V. V. Synlett, 2010, 6, 970;
21 Kouznetsov, V. V.; Forero, J. S. B.; Torres, D. F. A. Tetrahedron Lett., 2008, 49, 5855;
22 Astudillo, L. S.; Gutierrez, M.; Gaete, H.; Kouznetsov, V. V.; Melendez, C. M.; Palenzuela, J. A.;
Vallejos, G. Lett. Org. Chem., 2009, 6, 208;

65
23 Vicente-García, E.; Catti, F.; Ramon, R.; Lavilla, R. Org. Lett., 2010, 4, 860.
24 Dennis, M. K.; Burai, R.; Ramesh, C.; Petrie, W. K.; Alcon, S. N.; Nayak, T. K.; Bologa, C. G.; Leitao,
A.; Brailoiu, E.; Deliu, E.; Dun, N. J.; Sklar, L. A.; Hathaway, H. J.; Arterburn, J. B.; Oprea, T. I.; Prossnitz,
E. R.; Nat. Chem. Biol., 2009, 5, 421;
25 Burai, R.; Ramesh, C.; Shorty, M.; Curpan, R.; Bologa, C.; Sklar, L. A.; Oprea, T.; Prossnitz, E. R.;
Arterburn, J. B. Org. Biomol. Chem., 2010, 8, 2252;
26 Ramesh, C.; Nayak, T. K.; Burai, R.; Dennis, M. K.; Hathaway, H. J.; Sklar, L. A.; Prossnitz, E. R.;
Arterburn, J. B. J. Med. Chem., 2010, 53, 1004;
27 Kobayashi, S.; Jørgensen, K. A. Cycloaddition Reactions in Organic Synthesis, 2002, Wiley-VCH:
Weinheim;
28 Povarov, L. S.; Mikhailov, B. M. Ser. Khim. 1963, 953–956;
29 Ishitani, H.; Kobayashi, S. Tetrahedron Lett., 1996, 37, 7357–7360;
CeCl3, CeCl3∙7H2O and CeCl3∙7H2O/NaI
30 Yamamoto, H. Lewis acid reagents: A Pratical Approach, 1999, Oxford Press: New York;
31 Santelli, M.; Pons, J-M. Lewis Acids and Selectivity in Organic Synthesis, 1995, CRC Press: Boca
Ranton;
32 (a) Nair, V.; Deepthi, A. Chem. Rev., 2007, 107, 1862. (b) Mehdi, H.; Bodor, A.; Lantos, D.; Horva´th,
I. T.; De Vos, D. E.; Binnemans, K. J. Org. Chem., 2007, 72, 517;
33 Anwander, R.; Herrmann, W. A. Top. Curr. Chem., 1996, 179, 1;
34 Pearson, R. G. J. Chem. Educ., 1987, 64, 561;
35 Murad, E.; Hildenbrand, D. L. J. Chem. Phys., 1980, 73, 4005;
36 Tsuruta, H.; Yamaguchi, K.; Imamoto, T. Chem. Commun., 1999, 1703;
37 Imamoto, T.; Kusumoto, T.; Yokoyama, N. J. Chem. Soc. Chem. Commun., 1982, 1042;
38 Molander, G. A. In Chemistry of the Metal-Carbon Bond, Hartley, 1989, John Wiley: New York,;
39 Bartoli, G.; Marcantoni, E.; Sambri, L. Synlett, 2003, 5, 2101;
40 Imamoto, T.; Kusumoto, T.; Tawarayama, Y.; Sugiura, Y.; Mita, T.; Hatamaka, Y.; Yokoyama M. J.
Org. Chem., 1984, 49, 3904;
41 Shi, M.; Satoh, Y.; Masaki, Y. J. Chem. Soc., Perkin Trans, 1998, 1, 2547;
42 Nakanishi, K.; Goto, T.; Ito, S.; Natori, S.; Nozoe, S. Natural Product Chemistry; 1983, University
Press: Oxford;

66
43 (a) Dahlenburg, L.; Treffert, H.; Dannhauser, J.; Heinemann, F. W. Inorg. Chem. Acta, 2007, 360,
1474. (b) Ciganek, E. J. Org. Chem,. 1992, 57, 4521;
44 Reuman, M.; Beish, S.; Davis, J.; Batchelor, M. J.; Hutchings, M. C.; Moffat, D. F. C.; Connolly, P. J.;
Russell, R. K. J. Org. Chem., 2008, 73, 1121;
45 Moret, E.; Schlosser, M. Tetrahedron Lett., 1985, 26, 4423;
46 (a) Bartoli, G.; Dalpozzo, R.; De Nino, A.; Procopio, A.; Sambri, L.; Tagarelli, A. Tetrahedron Lett.
2001, 42, 8833. (b) Bartoli, G.; Bellucci, M. C.; Bosco, M.; Dalpozzo, R.; De Nino, A.; Sambri, L.;
Tagarelli, A. Eur. J. Org. Chem. 2000, 99. (c) Bartoli, G.; Bellucci, M. C.; Bosco, M.; Dalpozzo, R.; De
Nino, A.; Sambri, L.; Tagarelli, A. J. Org. Chem. 1998, 63, 9559;
47 Wang, C.; Xi, Z. Chem. Soc. Rev,. 2007, 36, 1395;
48 Johnstone, R. A. W.; Wilby, A. H.; Entwistle, I. D. Chem. Rev., 1985, 85, 129;
49 Luche, J.-L. J. Am. Chem. Soc., 1978, 100, 2226;
50 Iacazio, G. Chem. Phys. Lipids, 2003, 125, 115;
51 Yamaguchi, J.; Seiple, I. B.; Young, I. S.; O’Malley, D. P.; Mane, M.; Baran, P. S. Angew. Chem. Int.
Ed., 2008, 47, 3578;
52 Williams, M. Adenosine and Adenosine Receptors, 1990, Humana Press: Clifton;
53 Martin, P. L.; Wysocki, R. J.; Barrett, R. J. Jr.; May, J. M.; Linden, J. J, Pharmacol. Exp. Ther., 1996,
276, 490;
54 Jin, C.; Burgess, J. P.; Rehder, K. S.; Brine, G. A. Synthesis, 2007, 219;
55 Peck, J. V.; Wysock, R. J.; Uwaydah, I. M.; Cusak, N. J. U.S. Patent No. 5670501, 1997; Chem. Abstr.
1996, 125, 58207;
56 Khodaei, M. M.; Khosropour, A. R.; Kookhazadeh, M. Russ. J. Org. Chem., 2005, 41, 1445;
57 Schinzer, D. Selective in Leiws Acid Promoted Reactions; 1989, Kluwer Academic Publisher:
Dordrect;
58 Iimura, S.; Manabe, K.; Kobayashi, S. Tetrahedron, 2004, 60, 7673;
59 (a) Fukuzawa, S.; Tsurute, T.; Fujinami, T.; Sakai, S.; J. Chem. Soc., Perkin Trans., 1987, 1473. (b)
Fukuzawa, S.; Fujinami, T.; Sakai, S. J. Chem. Soc., Chem. Commun., 1985, 777;
60 Myers, E.; Butts, C. P.; Aggarwal, V. K. Chem. Commun. 2006, 4434;
61 Cappa, A.; Marcantoni, E.; Torregiani, E.; Bartoli, G.; Bellucci, M. C.; Bosco, M.; Sambri, L. J. Org.
Chem., 1999, 64, 5696.
62 a) Denmark, S. E.; Wynn, T.; Bentner, G. L. J. Am. Chem. Soc. 2002, 124, 13405-13407. (b) Denmark,
S. E.; Wynn, T. J. Am. Chem. Soc. 2001, 123, 6199-6200;

67
63 Bartoli, G.; Fernández-Bolaños, J. G.; Di Antonio, G.; Foglia, G.; Giuli, S.; Gunnella, R.; Mancinelli,
M.; Marcantoni, E.; Paoletti, M. J. Org. Chem., 2007, 72, 6029–6036;
64 Evans, W. J.; Feldman, J. D.; Ziller, J. W. J. Am. Chem. Soc., 1996, 118, 4581;
65 Narayau, S.; Muldoon, J.; Finn, M. G.; Fokin, V. V.; Kolb, H. C.; Sharpless, K. B. Angew. Chem., Int.
Ed., 2005, 44, 3275;
66 (a) Schreiber, S. L. Science, 2000, 287, 1964. (b) Posner, G. H. Chem. Rev. 1986, 86, 831;
67 (a) Dax, S. L.; Nally, J.-J. M. C.; Youngman, M. A. Curr. Med.Chem., 1999, 6, 255 (b) Gallop, M. A.;
Banett, R. W.; Dower, W. J.; Fodor, S. P. A. J. Med. Chem,. 1994, 37, 1385;
68 Yadav, L. D. S.; Kapoor, R. Tetrahedron Lett., 2008, 49, 4840;
69 Teshima, Y.; Shiya, K.; Shimazu, A.; Furihata, K.; Chul, H. S.; Hayakawa, Y.; Nagi, K.; Seto, H.J.
Antibiot., 1991, 44, 685;
70 Cox, R. J.; O’Hagan, D. J. Chem. Soc., Perkin Trans. 1, 1991, 2537;
71 Duval, D.; Geribaldi, S. In The Chemistry of Enones Part 1, 1989, Interscience: New York;
72 Talalay, P.; De long, M. J.; Prochaska, H. J. Proc. Natl. Acad. Sci. U.S.A., 1988, 85, 8261;
73 Christoffers, J. J. Chem. Soc., Perkin Trans. 1, 1997, 3141;
74 Bartoli, G.; Beleggia, R.; Giuli, S.; Giuliani, A.; Marcantoni, E.; Massaccesi, M.; Paoletti, M.
Tetrahedron Lett,. 2006, 47, 6501;
75 Tietze, L. F.; Beifuss, U. Comprehensive Organic Synthesis, 1991, Pergamon Press: Oxford;
76 Tanaka, M.; Oota, O.; Miramatsu, H.; Fujiwara, K. Bull. Chem. Soc. Jpn., 1988, 61, 2473;
77 Bartoli, G.; Bosco, M.; Carlone, A.; Dalpozzo, R.; Galzerano, P.; Melchiorre, P.; Sambri, L.
Tetrahedron Lett., 2008, 49, 2555;
78 Yeh, M.-C. P.; Yeh, W.-J.; Tu, L.-H. Wu J.-R., Tetrahedron, 2006, 62, 7466;
79 Marcantoni, E.; Massaccesi, M.; Petrini, M.; Bartoli, G.; Bellucci, M. C.; Bosco, M.; Sambri, L. J. Org.
Chem., 2000, 65, 4553;
80 Bartoli, G.; Bosco, M.; Dalpozzo, R.; Giuliani, A.; Marcantoni, E.; Mecozzi, T.; Sambri, L.; Torregiani,
E. J. Org. Chem., 2002, 67, 9111;
Tetrahydroquinolines: applications and synthesis
81 Katritzky, A. R.; Rachwal, S.; Rachwal, B. Tetrahedron, 1996, 52, 15031;
82 (a) Kim, W.-G.; Kim, J.-P.; Kim, C.-J.; Lee, K.-H.; Yoo, I.-D.; J. Antibiot., 1996, 49, 20. (b) Kim, W.-G.;
Kim, J.-P.; Yoo, I.-D. J. Antibiot., 1996, 49, 26;

68
83 Kimura, Y.; Kusano, M.; Koshino, H.; Uzawa, J.; Fujioka, S.; Tani, K. Tetrahedron Lett., 1996, 37,
4961.
84 Kusano, M.; Koshino, H.; Uzawa, J.; Fujioka, S.; Kawano, T.; Kimura, Y. Biosci., Biotechnol.,
Biochem., 2000, 64, 2559;
85 Schmeda-Hirschmann, G.; Hormazabal, E.; Astudillo, L.; Rodriguez, J.; Theoduloz, C. World J.
Microbiol. Biotechnol., 2005, 21, 27;
86 Yamada, O.; Ogasawara, K. Tetrahedron Lett., 1998, 39, 7747;
87 Gudmundsson, K. S.; Sebahar, P. R.; Richardson, L. D.; Miller, J. F.; Turner, E. M.; Catalano, J. G.;
Spaltenstein, A.; Lawrence, W.; Thomson, M.; Jenkinson, S. Bioorg. Med. Chem. Lett., 2009, 19, 5048;
88 Catalano, J. G.; Gudmundsson, K. S.; Svolto, A.; Boggs, S. D.; Miller, J. F.; Spaltenstein, A.; Thomson,
M.; Wheelan, P.; Minick, D. J.; Phelps, D. P.; Jenkinson, S. Bioorg. Med. Chem. Lett., 2010, 20, 2186;
89 Miller, J. F.; Turner, E. M.; Gudmundsson, K. S.; Jenkinson, S.; Spaltenstein, A.; Thomson, M.;
Wheelan, P. Bioorg. Med. Chem. Lett., 2010, 20, 2125;
90 Skerlj, R. T.; Bridger, G. J.; Kaller, A.; McEachern, E. J.; Crawford, J. B.; Zhou, Y.; Atsma, B.; Langille,
J.; Nan, S.; Veale, D.; Wilson, T.; Hartwig, C.; Hatse, S.; Princen, K.; De Clercq, E.; Schols, D. J. Med.
Chem., 2010, 53, 3376;
91 Ramesh, E.; Manian, R. D. R. S.; Raghunathan, R.; Sainath, S.; Raghunathan, M.; Bioorg. Med.
Chem., 2009, 17, 660;
92 Jarvest, R. L.; Berge, J. M.; Berry, V.; Boyd, H. F.; Brown, M. J.; Elder, J. S.; Forrest, A. K.; Fosberry,
A. P.; Gentry, D. R.; Hibbs, M. J.; Jaworski, D. D.; O’Hanlon, P. J.; Pope, A. J.; Rittenhouse, S.;
Sheppard, R. J.; Slater-Radosti, C.; Worby, A. J. Med. Chem., 2002, 45, 1959;.
93 Jarvest, R. L.; Armstrong, S. A.; Berge, J. M.; Brown, P.; Elder, J. S.; Brown, M. J.; Copley, R. C. B.;
Forrest, A. K.; Hamprecht, D. W.; O’Hanlon, P. J.; Rittenhouse, S.; Witty, D. R. Bioorg. Med. Chem.
Lett., 2004, 14, 3937;
94 Bendale, P.; Olepu, S.; Suryadevara, P. K.; Bulbule, V.; Rivas, K.; Nallan, L.; Smart, B.; Yokoyama,
K.; Ankala, S.; Pendyala, P. R.; Floyd, D.; Lombardo, L. J.; Williams, D. K.; Buckner, F. S.; Chakrabarthi,
D.; Verlinde, C. L. M. J.; Voorhis, W. C. V.; Gelb, M. H. J. Med. Chem., 2007, 50, 4585;
95 Eastman, R. T.; White, J.; Hucke, O.; Yokoyama, K.; Verlinde, C. L. M. J.; Hast, M. A.; Beese, L. S.;
Gelb, M. H.; Rathod, P. K; Voorhis, W. C. V. Mol. Biochem. Parasitol., 2007, 152, 66;
96 Gupta, M. K.; Prabhakar Y. S. Eur. J. Med. Chem., 2008, 43, 2751;.
97 Kaur, K.; Jain, M.; Reddy, R. P.; Jain, R. Eur. J. Med. Chem., 2010, 45, 3245;
98 Pagliero, R. J.; Lusvarghi, S.; Pierini, A. B.; Brun, R.; Mazzieri, M. R. Bioorg. Med. Chem., 2010, 18,
142;

69
99 Maillard, M. C.; Perlman, M. E.; Amitay, O.; Baxter, D.; Berlove, D.; Connaughton, S.; Fischer, J. B.;
Guo, J. Q.; Hu, L.-Y.; McBurney, R. N.; Nagy, P. I.; Subbarao, K.; Yost, E. A.; Zhang, L.; Durant, G. J.
J. Med. Chem., 1998, 41, 3048;
100 Fukumoto, S.; Imamiya, E.; Kusumoto, K.; Fujiwara, S.; Watanabe, T.; Shiraishi, M.; J. Med. Chem.,
2002, 45, 3009;.
101 Gore, V. K.; Ma, V. V.; Yin, R.; Ligutti, J.; Immke, D.; Doherty, E. M.; Norman, M. H. Bioorg. Med.
Chem. Lett., 2010, 20, 3573;.
102 Faghih, R.; Gopalakrishnan, M.; Briggs, C. A. J. Med. Chem., 2008, 51, 701;.
103 Romero,,A. G.; Darlington, W. H.; McMillan, M. W. J. Org. Chem., 1997, 62, 6582;
104 Ohta, M.; Suzuki, T.; Ohmori, J.; Koide, T.; Matsuhisa, A.; Furuya, T.; Miyata, K.; Yanagisawa, I.
Chem. Pharm. Bull., 1996, 44, 1000;
105 Wallace, O. B.; Lauwers, K. S.; Jones, S. A.; Dodge, J. A. Bioorg. Med. Chem. Lett., 2003, 13, 1907
106 Thevis, M.; Kohler, M.; Schanzer, W. Drug Discovery Today, 2008, 13, 59;
107 Chen, R.; Yang, X.; Tian, H.; Sun, L. J. Photochem. Photobiol. A, 2007, 189, 295;.
108 Pullmann, T.; Engendahl, B.; Zhang, Z.; Holscher, M.; Zanotti-Gerosa, A.; Dyke, A.; Francio, G.;
Leitner, W. Chem.—Eur. J., 2010, 16, 7517;.
109 Liu, W.-B.; He, H.; Dai, L.-X.; You, S.-L. Synthesis, 2009, 12, 2076;
110 Fujita, K.; Yamamoto, K.; Yamaguchi, R. Org. Lett., 2002, 4, 2691;
111 Fujita, K.; Takahashi, Y.; Owaki, M.; Yamamoto, K.; Yamaguchi, R. Org. Lett., 2004, 6, 2785;.
112 Fujita, K.; Furukawa, S.; Yamaguchi, R. J. Organomet. Chem., 2002, 649, 289;
113 Youn, S. W.; Song, J.-H.; Jung, D.-I. J. Org. Chem., 2008, 73, 5658;
114 Davies, S. G.; Mujtaba, N., Roberts, P. M.; Smith, A. D.; Thomson, J. E. Org. Lett., 2009, 9, 1959;
115 Sridharan, V.; Suryavanshi, P. A; Menendez, J. C. Chem. Rev., 2011, 111, 7157–7259;
116 (a) Takeda, Y.; Nakabayashi, T.; Shirai, A.; Fukumoto, D.; Kiguchi, T.; Naito, T. Tetrahedron Lett.,
2004, 45, 3481; (b) Miyata, O.; Shirai, A.; Yoshino, S.; Nakabayashi, T.; Takeda, Y.; Kiguchi, T.;
Fukumoto, D.; Ueda, M.; Naito, T. Tetrahedron, 2007, 63, 10092;
117 Uchikawa, W.; Matsuno, C.; Okamoto, S. Tetrahedron Lett., 2004, 45, 9037;
118 Saavedra, L. A.; Vallejos, G.; Kouznetsov, V. V.; Gutierrez, M.; Gomez, C. M. M.; Mendez, L. Y. V.;
Jaimes, J. H. B. Synthesis, 2010, 4, 593;
119 (a) Kouznetsov, V. V.; Zubkov, F. I.; Nikitina, E. V.; Duarte, L. D. A. Tetrahedron Lett., 2004, 45, 1981;
(b) Zubkov, F. I.; Nikitina, E. V.; Kouznetsov, V. V.; Duarte, L. D. A. Eur. J. Org. Chem., 2004, 24, 5064;

70
120 Togo, H.; Hoshina, Y.; Muraki, T.; Nakayama, H.; Yokoyama, M. J. Org. Chem., 1998, 63, 5193;
121 Wojciechowski, K. Eur. J. Org. Chem., 2001, 19, 3587;
122 Steinhagen, H.; Corey, E. J. Angew. Chem., Int. Ed., 1999, 38, 1928;
123 Zhu, Z.-B.; Shao, L.-X.; Shi, M. Eur. J. Org. Chem., 2009, 15, 2576;
124 Khadem, S.; Udachin, K. A.; Enright, G. D.; Prakesch, M.; Arya, P. Tetrahedron Lett., 2009, 50, 6661;
125 Liu, H.-M.; Liu, F.-W.; Zou, D.-P.; Dai, G.-F. Bioorg. Med. Chem. Lett., 2005, 15, 1821;
Result and discussion
126 Jha, A.; -Yi Chou, T.; Al Jaroudi, Z.; Ellis, B. D.; Cameron, T. S. Beilstein J. Org. Chem. 2014, 10,
848–857;
127 Tarantin, A.V.; Glushkov, V.A.; Mayorova, O. A.; Shcherbininab, I. A.; Tolstikova, A. G. Mendeleev
Commun., 2008, 18, 188–190;
128 Jiang, X.; Wang, C.; Wei, Y.; Xue, D.; Liu, Z.; Xiao, J. Chemistry - A European Journal, 2014 , 20,
58–63;
Characterization of compounds
129 Abu, T. K.;.Musawwer K. Tetrahedron Lett., 2011, 52, 4539–4542;
130 Talwar, D.; Li, H.; Durham,E.; Xiao, J. Chemistry- A European Journal, 2015, 21, 5370-5379;
131 Compain, G.; Bonneau, C.; Martin.Mingot, A.; Thibaudeau, S. J. Org. Chem., 2013, 78, 4463-4472;
132 Ding, F.; Zhang, Y.; Zhao, R.; Jiang, Y.; Bao, R. L.-Y.; Lin, K.; Shi, L. Chem. Comm., 2017, 53, 9262-
9234;
132 Maier, A. F. G.; Tussing, S.; Schneider, T.; Florke, U.; Qu, Z.-W.; Grimme, S.; Paradies, J. Angew.
Chem,- Int. Ed., 2016, 55, 12219-12223; Maier, A. F. G.; Tussing, S.; Schneider, T.; Florke, U.; Qu, Z.-
W.; Grimme, S.; Paradies, J. Angew. Chem., 2016, 128, 12407-12411,5;
134 Gong, T.-J.; Cheng, W.-M.; Su, W.; Xiao, B.; Fu, Y. Tethaedron Lett., 2014, 55, 1859-1862;
135 Min, C.; Mittal, N.; Sun, D. X.; Seidel, D. Angew. Chem.- Int. Ed., 2013, 52, 14084-14088; Min, C.;
Mittal, N.; Sun, D. X.; Seidel, D Angew. Chem., 2013, 125, 14334-14338,5.

71
Annexes
In the Annexes section, 1H and 13C spetra of un precendented products (213b-f) are reported. The
values are reported in Characterization of Compounds section. Other experimental data are
reported in Characterization of Compounds section.
213b
1H
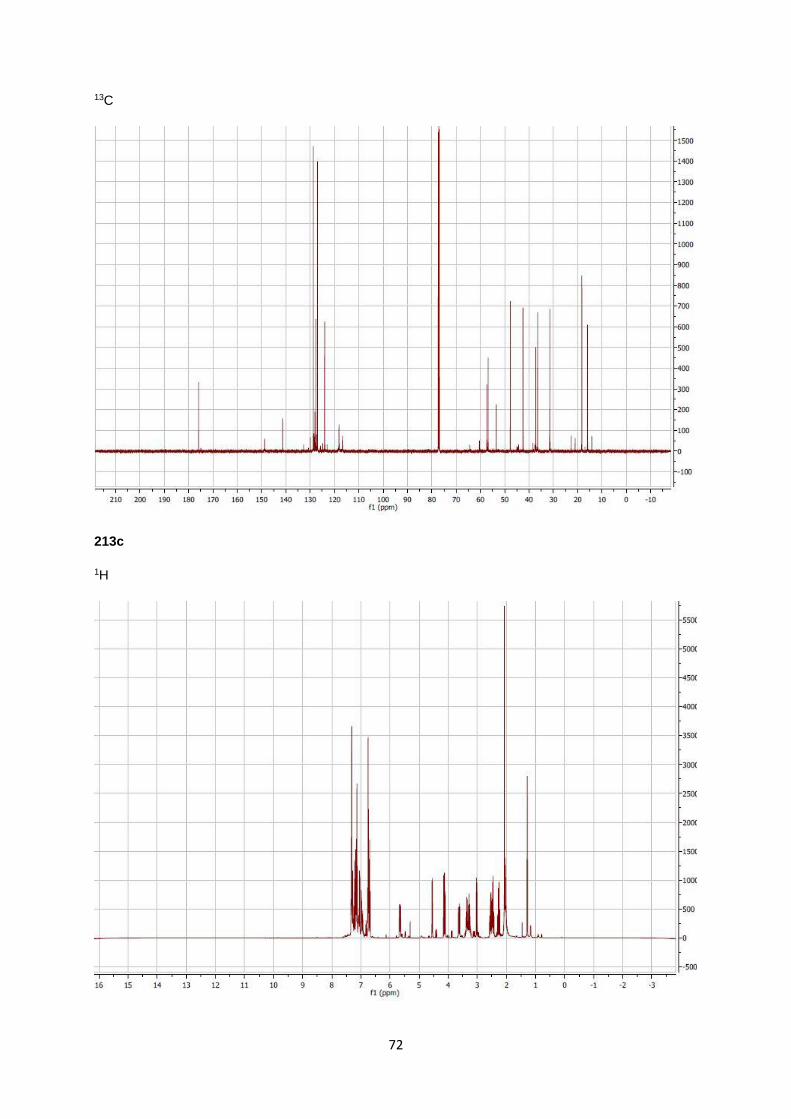
72
13C
213c
1H

73
13C
213d
1H

74
13C
213e
1H

75
13C
213f
1H

76
13C