study of resistance to artemisinins in plasmodium...
TRANSCRIPT

i
STUDY OF RESISTANCE TO ARTEMISININS IN PLASMODIUM FALCIPARUM
ISOLATES FROM KILIFI COUNTY
NDUNGU DUNCAN NDEGWA
A Research Thesis Submitted to the Graduate School in Partial Fulfillment for the
Requirements of the Master of Science Degree in Biochemistry of Egerton University
EGERTON UNIVERSITY
JANUARY, 2011

ii
DECLARATION AND RECOMMENDATION
DECLARATION
I declare that this thesis is my original work and has not been submitted wholly or in part
in this form or any form for a degree in this or any other university
Ndungu Duncan Ndegwa
Signature ……………….. Date ……………………
RECOMMENDATION
We wish to confirm that this research thesis has been prepared under our supervision and
have our approval to be presented for examination as per the Egerton University
Dr. A. Orina Isaac
Signature ……………………………… Date……………………………..
Egerton University, Kenya.
Dr. Steffen Borrmann
Signature ……………………………… Date……………………………..
Heidelberg University, Germany.
Dr. Maina Joseph
Signature ……………………………… Date……………………………..
Moi University, Kenya.

iii
COPYRIGHT
All rights reserved. No part of this thesis may be reproduced, stored in a retrieval system or transmitted, in any form or by any means, electronic, mechanical, photocopying or otherwise, without permission from the author or Egerton University.
© Ndungu Duncan Ndegwa 2012

iv
ACKNOWLEDGEMENT
I owe my unwavering gratitude and appreciation to the following for their great support
without which I could not have made it this far:
1. My immediate and extended family members for their financial and moral support.
2. The German Academic Exchange Services - DAAD for the in-country scholarship and
the short study fellowship to Heidelberg University, Germany.
3. Dr. Steffen Borrmann, for hosting and allowing me to work in his laboratory in
Heidelberg University, Germany.
4. My supervisors Dr. A. Orina Isaac, Dr. Steffen Borrmann and the late Dr. Maina
Joseph for their endless support.
5. The entire AG. Borrmann, for their moral and technical support: Anja Rippert for her
technical support in carrying out the project; Elise Schieck for her support in making the
statistical analysis; Not forgetting Yvonne Maier, Judith Straimer, Mamadou Tekete and
Patricia Jebet.
Above all, the almighty God and may He bless them all.

v
ABSTRACT
Malaria is a health problem and drug resistance to antimalarial poses a great challenge to
its treatment and control. Having a rapid activity against malaria parasites, artemisinins are the
front line antimalarials. However, already there are reports on reduced response to artemisinins.
Thus it’s important to monitor resistance in malaria risky areas. Merozoite surface protein (MSP)
1 and 2 genotyping has shown difference between isolates from primary and recurrent infections
but how this relates to their in vitro sensitivity is not known. The study aimed at determining
resistance of P. falciparum to artemisinins in Kilifi County. Specifically to determine: 1. in vitro
sensitivity of P. falciparum to artemisinins; 2. the difference between their in vitro activity with
that of other antimalarial drugs; 3. the existence of in vitro cross-resistance; and 4. whether there
is difference in the in vitro response to artemisinins in isolates from primary and recurrent
infection. Parasites isolates were cultured in vitro and drug response assay carried out by
subculturing them in various drug concentrations. In vitro activity was determined as the drug
concentration that inhibited 50% of parasite growth (IC50) using SYBR green I microtest.
Kruskal Wallis and Dunn’s post test were used to compare the in vitro activity of artemisinins
and that of other antimalarial drugs. Mann Whitney test was used to determine the difference in
the in vitro response of the isolates from initial and recurrent infection. Spearman correlation
analysis was used to determine existence of in vitro cross-resistance patterns between
artemisinins and other antimalarials. The mean IC50s (in nM) of Dihydroartemisinin (DHA),
Piperaquine (PQ), Lumefantrine (LMF), Chloroquine (CQ), Quinine (Q), Pyrimethamine (PYR),
Desethylamodiaquine (DE-A) and Mefloquine (MFQ) were 1.861, 30.34, 46.5, 24.29, 46.7,
18074, 37.8 and 41.7 nM respectively. There was significant difference in the in vitro activities
of the antimalarial drugs, DHA being the most active drug. There was no significant difference
in the in vitro response of the isolates from initial and recurrent malarial infections towards the
antimalarials. There was no significant correlation in the IC50s of the eight antimalarial drugs.
Therefore, artemisinins are still active in Kilifi County; more active than other standard
antimalarials and no cross-resistance was observed with other antimalarials. Consequently, as
ACTs they should be continued to be used as first line antimalarials.

vi
TABLE OF CONTENTS DECLARATION AND RECOMMENDATION .................................................................... ii
ACKNOWLEDGEMENT ...................................................................................................... iv
ABSTRACT...............................................................................................................................v
LIST OF FIGURES .............................................................................................................. viii
LIST OF TABLES .................................................................................................................. ix
LIST OF ABBREVIATIONS ...................................................................................................x
CHAPTER ONE .......................................................................................................................1
INTRODUCTION .....................................................................................................................1
1.1 Background Information ....................................................................................................1
1.1.2 Malaria Control Strategies ...........................................................................................3
1.2 Statement of the Problem ...................................................................................................5
1.3 Objectives ..........................................................................................................................6
1.3.1 General objective.........................................................................................................6
1.3.1 Specific objectives .......................................................................................................6
1.4 Hypotheses ........................................................................................................................6
1.5 Justification .......................................................................................................................7
1.6 Expected Outputs……………………………………………………………………………7
CHAPTER TWO ......................................................................................................................8
LITERATURE REVIEW .........................................................................................................8
2.1 Artemisinins ......................................................................................................................8
2.2 Mechanisms of Artemisinins’ Action .................................................................................8
2.3 Resistance to Artemisinins .................................................................................................9
2.4 In Vitro Drug Assays .........................................................................................................9
CHAPTER THREE ................................................................................................................ 12
MATERIALS AND METHODOLOGY ................................................................................ 12
3.1 Study Area. ...................................................................................................................... 12
3.2. Plasmodium falciparum Parasites Samples...................................................................... 12
3.3 Methods ........................................................................................................................... 13
3.3.1 In Vitro Culturing of P. falciparum ............................................................................ 13
3.3.1.1 Thawing of Parasites isolates..…………………………………………………...13

vii
3.3.1.2 Adaptation of Parasites into Culture.…………………………………………….14
3.3.1.3 Routine Parasite Culture.………………………………………………………...14
3.3.1.4 Splitting of Culture.……………………………………………………………...15
3.3.1.5 Giemsa Staining....……………………………………………………………….15
3.3.1.6 Parasite Freezedown.…………………………………………………………….15
3.3.1.7 Synchronization of Cultures..……………………………………………………16
3.3.2 In Vitro Drug Sensitivity Assay ................................................................................. 16
3.3.2.1 Drug Sensitivity Assay..…………………………………………………………16
3.3.2.2 Preparation of Stock Drug solution...…………………………………………….16
3.3.2.3 Preparation of the Mother Plate...………………………………………………..17
3.3.2.4 Dilution of Culture...……………………………………………………………..18
3.3.2.5 Preparation of the Working Plate...………………………………………………18
3.3.3 SYBR Green I Assay ................................................................................................. 18
3.3.3.2 Growth Inhibition Assay...……………………………………………………….19
RESULTS AND DISCUSSION .............................................................................................. 21
4.1 RESULTS ....................................................................................................................... 21
4.1.1 The In Vitro response of P. falciparum isolates to Artemisinins and other Antimalarials. .................................................................................................................... 21
4.1.2 The Difference between the In Vitro Activities of Artemisinins with that of other Antimalarial Drugs. ............................................................................................................ 26
4.1.3 The Difference in the In Vitro response to Artemisinins in P. falciparum Isolates from Primary and Recurrent Malaria infection in Kilifi County. ................................................. 29
4.1.4 The Correlation Patterns between Artemisinins and other Antimalarials .................... 34
4.2 DISCUSSION.................................................................................................................. 37
CHAPTER FIVE..................................................................................................................... 46
CONCLUSIONS ..................................................................................................................... 46
RECOMMENDATIONS ........................................................................................................ 46
REFFERENCES ..................................................................................................................... 47

viii
LIST OF FIGURES
Figure 1: Plasmodium falciparum endemicity map ......................................................................1
Figure 2: Lifecycle of P. falciparum in the host and vector ..........................................................2
Figure 3: Artemisinins .................................................................................................................8
Figure 4: The proposed mode of action of artemisinins................................................................9
Figure 5: Study site. .................................................................................................................. 12
Figure 6: Rate of in vitro resistance or susceptibility of Plasmodium falciparum to
the eight antimalarial drugs tested. .............................................................................. 25
Figure 7: Difference between the in vitro activities of artemisinins with that of other
standard antimalarial drug. .......................................................................................... 27
Figure 8: A comparison of the in vitro response of the parasite isolates from initial
infection (d0) and recurrent infection (dRC)…………………………………………..30
Figure 9: A comparison of the in vitro response of the parasite isolates from initial
infection (d0) and recurrent infection (dRC).. ............................................................. 31
Figure 10: A comparison of the in vitro response of the parasite isolates from initial
infection (d0) and recrudescent infection (dRD).. ........................................................ 32
Figure 11: A comparison of the in vitro response of the parasite isolates from initial
infection (d0) and re-infection (dRI) ........................................................................... 33
Figure 12: Correlation between the IC50s values of the eight antimalarial drugs tested.............. 35

ix
LIST OF TABLES
Table 1: Plasmodium falciparum parasites samples ................................................................... 13
Table 2: Preparation of stock drug solution ............................................................................... 17
Table 3: The In vitro response of P. falciparum isolates to antimalarials ................................... 22
Table 4: Mean IC50s of the eight antimalarial drugs tested against field isolates of
P. falciparum in Kilifi County .................................................................................... 23
Table 5: Number of resistant or susceptible parasites ................................................................. 24
Table 6: Difference between the in vitro activities of the antimalarial drugs tested .................... 28
Table 7: Correlation between the IC50s values of the eight antimalarial drugs tested ................. 36

x
LIST OF ABBREVIATIONS
ACTs: Artemisinin combination therapies
CO2 : Carbon dioxide
CQ: Chloroquine
DDT: Dichlorodiphenyltrichloroethane
DELI: Double-site enzyme-linked LDH immunodetection assay
DEA: Desethylamodiaquine
DHA: Dihydroartemisinin
DMSO: Dimethyl sulfoxide
DV: Digestive vacuole
EDTA: Ehtylenediaminetetraacetic acid
ELISA: Enzyme-linked immunosorbent assays
HCl: Hydrochloric acid
HRP- 2: Histidine-rich protein-2
KEMRI-WTRP: Kenya Medical Research institute – Wellcome Trust Research Programme
LMF: Lumefantrine
O2 : Oxygen
MHC – I: Major Histocompatibility class I
MFQ: Mefloquine
MSF: Malaria SYBR green I Fluorimetric Assay
MSP: Merozoite surface protein
NaCl: Sodium Chloride
N2 : Nitrogen
PfEMP1: P. falciparum erythrocyte membrane protein-1
pLDH: P. falciparum lactate dehydrogenase

xi
PQ: Piperaquine
PYR: Pyrimethamine
Rpm: Revolutions per minute
Q: Quinine
SP or PYR-SD: Sulphadoxine – Pyrimethamine
WHO: World health organisation

1
CHAPTER ONE
INTRODUCTION
1.1 Background Information
Malaria is a health problem in the world (Figure1) out of which 3.3 billion people are at
risk, over 300 million every year get infected and killing between 2 - 3 millions (Snow et al.,
2005). Which is a big number caused by only a single species of Plasmodium (Van Dooren et al.,
2002). Mostly it affects children less than 5 years (Breman et al., 2004; Tärning 2007) and
pregnant women. As a result of this huge number of casualty, malaria restricts development in
poor countries (Tripathi et al., 2005) due to mobilization of resources to fight it in place of other
priority needs.
In humans, malaria is caused by four species of Plasmodium; P. falciparum, P. vivax, P.
malariae and P. ovale (Day and Marsh, 1991); and P. falciparum kills more than one million
people annually (Roos et al., 2002). The tropical areas of the world provide good environment
Figure 1: Plasmodium falciparum endemicity map (Hay et al., 2009; Straimer 2009)

2
for the vectors and thus transmission of malaria, as a result the continent of Africa is the most
affected, with 60% of all malaria infections and 80% deaths (WHO, 2005). South East Asia and
South America are also highly affected (Tärning 2007).
1.1.1 Malaria Lifecycle
Figure 2: Lifecycle of P. falciparum in the host (a) and vector (b) (Menard 2005, Straimer 2009).
Malaria infection begins by an infected mosquito biting a human host. At the same time,
sporozoites from the salivary gland of the mosquito enter the bloodstream. They migrate and
within 45 (Tärning 2007) to 60 minutes (Tripathi et al., 2005) invade the liver cells, where they
multiply and develop into exo-erythrocytic schizonts (Wernsdorfer and Mc Gregor, 1988). These
schizonts burst releasing merozoites which invade erythrocytes, replicate and develop into ring
stages. Ring stages mature and develop into trophozoites. These inturn develop into erythrocytic
schizonts which bursts releasing merozoites that initiates another string of infections by invading
other erythrocytes. In the process, some merozoites develop into male or female gametocytes.
These are then taken up by a mosquito during a blood meal, and they contribute to the sexual
reproduction phase. Whereby, inside the mosquito gut the gametes fuse as a form of fertilization
resulting into a zygote which develops into oocysts. These burst and produce sporozoites that
migrate through the haemolyph to the salivary glands (Griffith et al., 2007; Tärning 2007). The
ability to cause the malaria disease is due to the high rate of multiplication which results in

3
rupturing of the erythrocytes and release of toxins. The parasite is able to modify the erythrocyte
which leads to complication of the disease whereby, by forming knobs on the erythrocytes the
parasite is able to sequester on the blood vessels avoiding clearance by the spleen (Tripathi et al.,
2005).
1.1.2 Malaria Control Strategies
The control and elimination of malaria rely on the control of the vector and management
of malaria infections. By controlling the vector it helps in reducing transmission of the disease.
The control of the vector involves the use of insecticide-treated nets, in-door and out-door
spraying. Both approaches have an impact, whereby treated nets can be maintained for long
periods and regular spraying can provide a long lasting protection (WHO, 2005). To achieve
complete vector control, is however expensive if not impractical (Miller et al., 2007; Tärning
2007).
Management of infections relies on prevention of transmission and treatment. A malaria
vaccine would prevent infection with malaria. Hence, there is need for an effective vaccine. But
this is still far from being achieved as there is currently no any functional and acceptable
vaccine. Of the many vaccines that have been presented only a few have passed the early clinical
trials (Aide et al., 2007; Snounou and Renia, 2007) and many are yet to be presented due to the
availability of P. falciparum genome sequence (Gardner et al., 2002).
The prevention of infection with malaria is further weakened by the immune evasion
challenges the parasite poses to the immune system preventing parasite clearance consequently
resulting in continual malarial infections:
The sporozites avoid immune detection and destruction as once they are in the
bloodstream they move quickly to the liver where they change into merozoites, thus they don’t
become exposed to antibodies and T cells respectively (Taubes, 2000).
The erythrocytes lack antigen presenting receptors i.e. MHC – I receptors as such, the
parasites become hidden from T cells (Janeway et al., 2005).
The parasite modifies erythrocytes by making them rigid, interfering with transport
mechanisms and PfEMP1 encoded by var genes is deposited into the membrane forming Knobs.

4
Through the knobs infected erythrocytes attach to walls of the blood vessels preventing their
destruction by the spleen. These knobs also result in infected erythrocytes binding to uninfected
ones, forming a complex that leads to blockage of blood vessels. These modifications result in
complication of malaria. PfEMP1 is involved in antigenic variation whereby as a result of the
immune pressure the parasite changes the sequence of var genes expressed resulting in immune
evasion (Hastings et al., 2004). Additionally, PfEMP1 can attach to dendritic cells, interfering
with the immune response process (Craig and Scherf, 2001). To avoid destruction gametocytes
remain inactive while in the human host (Taubes, 2000).
With the above complications in the control and management of malarial, an alternative
to avoiding the malaria burden is the use of effective, safe and cheap antimalarials (Tärning
2007). Treatment of malaria has always relied on antimalarials such as chloroquine,
sulphadoxine-pyrimethamine etc.
After chloroquine was discovered, its use together with the insecticide DDT boosted the
fight and elimination of malaria in the western world. However, in developing countries political
instabilities, shortage of funds among other factors hindered this fight, resulting in resistance to
CQ and SP (Wellems and Plowe, 2001; Gregson and Plowe, 2005). Antimalarial drug resistance
later spread to MFQ or Q (Wongsrichanalai et. al., 2002) worsening the malaria burden.
The wide geographic distribution of P. falciparum (Tärning 2007) influences the
dynamics of antimalarial drug resistance (Hastings and Watkins, 2005) greatly inhibiting the
fight against malaria. There are low and high transmission areas e.g. South east of Asia and
Africa respectively. People in these areas thus experience different infectious bites per person per
year e.g. 2 and up to 1500 respectively (Hay et al., 2000). Consequently, people in low
transmission areas develop malaria symptoms as opposed to those in the high. This is a result of
the latter developing partial immunity to malaria but which wanes out when they migrate to low
transmission areas. Thus, in low transmission areas there is frequent treatment with antimalarials
than from the high (Breman et al., 2004). As a result there is a lot of drug pressure in the low
transmission areas which selects resistant parasites that inturn spread to the high transmission
areas e.g. CQ and SP first emerged in south east asia and spread to africa. These drugs were
affordable and easily available (Touré et al., 2008; Lim and McFadden, 2010), worsening the

5
malaria fight (Greenwood et al., 2005) thus necessitating new and different antimalarials (White,
2004).
ACTs are effective against parasites resistant to other common antimalarials and they are
recommended by the WHO as the front-line drugs. Indeed in combination with other control
strategies they are proving useful (Feachem and Sabot, 2008; Eastman and Fidock, 2009).
Hence, maintaining the optimism in malaria elimination (Kaddouri et al., 2006).
The basis of ACTs is, since resistance results from mutations in the target or transport
proteins, it would be hard for a parasite to become resistance at the same time to two drugs that
act differently in comparison to a single drug usage (White, 1999; Nosten and White, 2007; Cui
and Su, 2009). Examples of ACTs include; Artemether–lumefantrine and dihydroartemisinin–
Piperaquine.
However, there are accounts of reduced response to these antimalarials in the South East
of Asia (Cui and Su, 2009; Dondorp et al., 2009; Leah et al., 2009). Hence generating fears
towards spread of this resistance and erosion of their utility (Wootton et al., 2002; Roper et al.,
2004). Assessment of P. falciparum drug susceptibility in Kilifi is thus necessary to sustain
health recommendations for malaria treatment and prophylaxis in Kenya and other countries
where malaria is endemic (Kaddouri et al., 2006).
During the time course of malaria tropica infection different clones of P. falciparum
parasites have been isolated and differentiated by genotyping of the MSP 1 and 2. These result in
primary and recurrent infections. Primary referring to the initial infection and recurrent to
subsequent infections, which is associated with two different parasite clones; recrudescent,
which are similar genotypically to those of the primary infection and new different parasites.
Thus, does this difference translate into differences in drug response or polymorphisms in
Pfatpase6 and Pfmdr1 genes associated in artemisinin resistance?
1.2 Statement of the Problem
Antimalarial drug resistance is a great limitation in the fight against malaria, since there
is a high rate of resistance to antimalarials developing and spreading. The lack of a new and
different antimalarial worsens the situation. As a result the possibility of parasites developing
resistance to artemisinins threatens the usefulness of ACTs. Making it mandatory to monitor

6
artemisinins susceptibility and hence researching more on this, hence, the need to determine
artemisinins resistance in endemic areas such as Kilifi.
Additionally, through MSP 1 and 2 genotyping there is difference between parasite
clones from initial and recurrent malaria infection but how this relates to their in vitro sensitivity
toward artemisinins needs to be determined.
1.3 Objectives
1.3.1 General objective
To determine resistance of P. falciparum to artemisinins in Kilifi County
1.3.1 Specific objectives
1. To determine in vitro response of P. falciparum isolates to artemisinins and to establish
the sensitivity level of artemisinins in Kilifi County.
2. To determine difference between the in vitro activity of artemisinins with that of other
antimalarial drugs.
3. To determine whether there is a difference in the in vitro response to artemisinins in P.
falciparum isolates from primary and recurrent malaria infection in Kilifi County.
4. To determine existence of in vitro cross-resistance between artemisinins and other
antimalarials.
1.4 Hypotheses
1. P. falciparum isolates from Kilifi County have reduced in vitro response to
artemisinins?
2. There is difference between the in vitro activity of artemisinins and other antimalarials
towards P. falciparum isolates from Kilifi County?
3. There is difference in the in vitro sensitivity towards artemisinins in P. falciparum
isolates from primary and recurrent malaria infection in Kilifi County?
4. There is in vitro cross resistance between artemisinins and other antimalarials towards
P. falciparum isolates from Kilifi County?

7
1.5 Justification
Guiding principles for malaria treatment rely on in vitro drug tests to provide information
on the susceptibility profiles of parasites. In this way the study will demonstrate the status on P.
falciparum response to artemisinins in Kilifi County, generating background information which
can be used to asses and improve treatment guidelines.
1.6 Expected Outputs
1. The sensitivity level of artemisinins in Kilifi County will be established to inform
policy on treatment programs.
2. Master of Science Degree in Biochemistry.
3. Publication in peer reviewed journals.

8
CHAPTER TWO
LITERATURE REVIEW
2.1 Artemisinins
The artemisinins is a product of research by Chinese scientists (Klayman, 1985; Cui and
Su, 2009). Besides malaria, they are able to treat schistosomiasis and cancer (Krishna et al.
2008). Their activity against malaria is due to an endoperoxide bridge they posses, and can kill
parasites in several minutes (Woodrow et al., 2005; White, 2008; Cui and Su, 2009). They are
extracted from Artemisia annua and then modified to other derivatives due to their instability and
insolubility (Touré et al., 2008). Alternatively, they can also be obtained through genetic
engineering using the yeast cell (Ro et al., 2006; Eastman and Fidock 2009).
Figure 3: Artemisinins (Cui and Su, 2009). Artemisinins are broken down to DHA, their active component (Wongsrichanalai et al., 1999; Schlitzer 2008; Mwai et al. ., 2009).
2.2 Mechanisms of Artemisinins’ Action
The mode of action of artemisinins is not yet well understood and there are two possible
ways how this happens (Figure 4): either, the endoperoxide bridge is reduced by haem iron in the
digestive vacuole (Hartwig et al., 2009) generating toxic radicals (Krungkrai and Yuthavong,
1987; Asawamahasakda et al., 1994; Kannan et al., 2005) or the bridge is reduced by iron-
sulphur redox centres (Wu, 2002).

9
Figure 4: The proposed mode of action of artemisinins: The Endoperoxide Bridge is the active
component of artemisinins which undergoes reduction to produce radicals whose target is not yet
clear (Schlitzer 2008).
2.3 Resistance to Artemisinins
Artemisinin resistance has been demonstrated in P. falciparum lab strain, F32-ART
(Witkowski et al., 2009). And despite the lack of demonstrable resistance in isolates from
patients, there are reports of reduced response to these antimalarials in different parts of the
world, which include: western Thailand (Luxemburger et al., 1998), Cambodia (Denis et al.,
2006; Vijaykadga et al., 2006; Cui and Su, 2009), India (Gogtay et al., 2000), Sierra Leone
(Sahr et al., 2001), Nigeria and Madagascar (Oduola et al., 1992; Randrianarivelojosia et al.,
2001), and Yunnan province of western China (Yang et al., 2003), French Guiana and Senegal
(Jambou et al., 2005).
As we await more confirmation on the same issue, these are proof of resistance to
artemisinins in P. falciparum. This is a threat to the fight against malaria (O'Neill et al.. 2010),
consequently, the need to determine artemisinins resistance in endemic areas such as Kilifi.
2.4 In Vitro Drug Assays
Techniques for assessing artemisinins resistance include in vivo and in vitro assays. In
vitro enable determination of inherent antimalarial drug resistance, they are advantageous in
being cheap and from free from host factors (Bacon et al., 2007), While the in vivo, are
applicable in determining clinical resistance (Noedl et al., 2004). In vitro assays include:

10
1. The WHO microtest, in which results are, assessed microscopically, a time-consuming
process.
2. Isotopic microtest; which requires high parasitemia (Druile et al., 2001), its expensive
and dangerous involving radioactive materials.
3. Molecular marker determination which sometimes doesn’t connect to in vitro drug
resistance (Bacon et al., 2007).
4. Colorimetric assays based on biochemical reactions resulting in colour development.
This is available in various types:
a) Fluorescent-based techniques that use fluorescent dyes e.g. SYBR green I that
intercalate with DNA (Johnson et al., 2007). It is specific to the erythrocytic stage since
erythrocytes lack Nucleic acids (Bennett et al., 2004, Johnson et al., 2007).
b) Sensitive quantification of major parasite proteins by sandwich ELISA based on
labeled primary and secondary antibodies directed to distinct epitopes of the same antigen
giving greater sensitivity (Druilhe et al., 2001). The antibodies are for antigen capture
and identification respectively.
Antigens targeted are either only found in a specific Plasmodium species or are found in
all of them. Those specific to P. falciparum are HRP-2 and pLDH used in HRP2 ELISA (Noedl
et al., 2004, 2005) and DELI (Druilhe et al., 2001) respectively. On the contrary pLDH and
aldolase are found in all species. Aldolase is used in sandwich aldolase ELISA (Murray and
Bennett 2009).
The discrimination on the type of assay to use involves the time taken to culture parasite.
Hypoxanthine, pLDH and SYBR green I assay needs 48 or 72 hour. HRPII assay needs 72-hour
inhibiting its application in assays requiring shorter times (Bacon et al., 2007). Additionally,
mutations in the targeted antigens can lead to wrong results (Murray and Bennett, 2009).
Aldolase based tests are applicable in a parasitemia of atleast 0.03% (Tritten et al., 2009).
While in the DELI, a parsitemia of atleast 0.005% (Druilhe et al., 2001).

11
Even though ELISA-based techniques are precise and consistent, they are however,
costly and tiresome. On the other hand, fluorescence-based techniques provide a less costly and
fast approach.
The SYRB green I assay for example: Is performed on one plate consuming fewer
reagents in comparison to the standard isotopic microtest. It’s carried out in a single process. The
SYBR green dye itself is stable. It doesn’t use any antibodies. It is less costly (Smilkstein et al.,
2004; Bacon et al., 2007; Johnson et al., 2007). Thus MSF assay will be used.

12
CHAPTER THREE
MATERIALS AND METHODOLOGY
3.1 Study Area.
The study site is Pingilikani (Figure 5) which is under a partnership involving Heidelberg
University, Germany and the KEMRI-WTRP Kilifi. It is about 20 km away southwards of
Kilifi. It experiences two rainy seasons, April - June and November – December, during which
malaria transmission maximizes (Straimer 2009). The inoculation rate is about 22 to 53 infective
bites per person per year (Mbogo et al., 2003). Artemether-lumefantrine is the current front-line
drug replacing amodiaquine which has been in use since 2003 (Sasi et al., 2009).
Figure 5: Study site: The figure on the left is a map of Kilifi County, Kenya. And the right one is
Pingilikani dispensary (Straimer 2009).
3.2. Plasmodium falciparum Parasites Samples
The samples for the study were randomly selected from a huge collection of samples
collected from Pingilikani between 2006 – 2008 in a study that was determining safety and
efficacy of Artekin in African children, among which Kilifi was one of the study site.
During the study, the parasites were isolated from the patients during the initial day of the
study (This was the day zero- d0) and subsequently during the follow up days of the study in

13
patients presenting with recurrent parasitemia (This were named according to the day of parasite
isolation).
To differentiate the P. falciparum isolates whether they are the same or different ones,
isolates from patients with recurrent parasitemia were MSP 1 / 2 genotyped; those with similar
sequences as isolates from day zero were referred to as recrudescent and those that were
different; re-infection. From this pool of isolates from day zero and day of recurrence the
following 8 isolates for this study were randomly selected.
Table 1: Plasmodium falciparum parasites samples
Day Zero Recurrence Remarks
27d0 27d21 Recrudescent
171d0 171d21 Recrudescent
115d0 115d49 Re-infection
191d0 191d54 Re-infection
The laboratory strain 3D7 was used as the control.
3.3 Methods
3.3.1 In Vitro Culturing of P. falciparum
In vitro culturing of P. falciparum was done according to Trager and Jensen (Jensen
2002) with few modifications.
3.3.1.1 Thawing of Parasites Isolates
To prepare frozen parasites for culture, cryovials stored in liquid nitrogen were
transferred into - 800C and left to thaw overnight. Then after 12-24 hours they were removed
and thawing was done by rapidly rolling the cryovials between two gloved hands. Then 200µl of
12% NaCl was added slowly to the vial and mixed by shaking gently. The mixture was
transferred to a 50 ml falcon tube. Nine millimeters of 1.6% NaCl was added drop by drop for
the first 3 ml and mixed. This was then centrifuged at 1800 rpm for 3 minutes. The supernatant
was then carefully removed by drawing it out with Pasteur pipettes connected to the vacuum
pump and this was replaced by dropwise addition of 7 ml of 0.9% NaCl and 0.2% glucose with

14
gentle agitation. This was then centrifuged at 1800 rpm for 3 minutes and the supernatant
removed as above. Then the parasite pellet was introduced directly into culture as described
below.
3.3.1.2 Adaptation of Parasites into Culture
P. falciparum culture medium was prepared by mixing 500 ml of RPMI 1640
(supplemented with L glutamine and HEPES), 250µl gentamicin, 10ml human AB serum, 5ml
hypoxanthine and 50ml Albumax II. The solution was then filtered through 0.22µ Millipore filter
connected to a vacuum pump. This was then stored at 40C.
Under sterile conditions and working under a culture hood, 10 ml of culture medium was
pipetted into the above parasite isolate then mixed by gently pipetting the mixture up and down.
This was then transferred into a T25 culture flask and 1 µl of fresh uninfected erythrocytes
pipetted into the solution and mixed again. The flask was placed in an incubator and the parasites
were incubated at 370C, in constant flowing gas mixture of 90% N2, 5% CO2 and 5% O2. The
culture media was changed after every two days and the parasitemia was checked daily by
giemsa staining as described below. When at least a single parasite was spotted the culture was
split into two portions and one portion maintained in culture and the other frozen down as
described below (Section 3.3.1.4 and 3.3.1.6 respectively);
Also after splitting of the culture to encourage faster growth the remaining portion was
transferred to T75 culture flask and 1.5 ml of fresh erythrocytes and 30 ml of fresh culture
medium was added; in all maintaining a hematocrit of 5% and a parasitemia of less than 1%. The
parasites were then maintained in culture with regular media change after two days and daily
monitoring of the parasitemia.
3.3.1.3 Routine Parasite Culture
Routine parasite culture refers to the continued culture of parasites after they had been
adapted to culture. It was done by maintaining the parasites in culture for a longer period 1 to 3
weeks through regular media change after two days and daily monitoring of the parasitemia. This
was continued until the parasites attained a two, three or four folds multiplication of the
parasitemia; which is regarded as the normal parasite cycle. During this whole period (2-3
weeks), the parasitemia was not allowed to exceed 5%, otherwise it was splitted as described

15
below (some portion being used for parasite freeze down, DNA extraction or stored as a parasite
pellet for other studies). In cases where there was a mixture of trophozoites and ring stages the
culture was made synchronous by sorbitol synchronization as described below (Section 3.3.1.7).
3.3.1.4 Splitting of Culture
Splitting of the parasites was done by mixing the culture by gentle swirling and then
using a pipette and accu-jet: In T25 culture flasks 1, 2 and utmost 3 ml of the solution was
removed and the rest transferred to 15 ml falcon tube (for freeze down). The removed solution
was returned to the same culture flask or a new one, and then 500µl of fresh erythrocytes and
10ml of fresh culture medium was added.
In T75 culture flasks 5 or 10 ml of the solution was removed and the rest transferred to
50ml falcon tube (for freeze down). The removed solution was returned to the same culture flask
or a new one, and then 1.5ml of fresh erythrocytes and 30 ml of fresh culture medium was added.
The solution was mixed gently then returned to culture.
3.3.1.5 Giemsa Staining
From the culture solution and using a 1ul pipette a drop of blood was taken and smeared
on a clean glass slide and air dried. This was then fixed by immersion in methanol for a minute
and then air-dried; then immersed for 10–30 minutes in Giemsa solution, washed under running
water and then dried. This was then examined on a microscope at 100 objectives with immersion
oil by quickly scanning through the slide and choosing an area where the red blood cells were
uniformly distributed (with no overlapping cells). One thousand erythrocytes were counted
randomly in different fields while partially filled fields were avoided. Also infected erythrocytes
were counted and they were expressed per a thousand of the uninfected cells to give the
parasitemia.
3.3.1.6 Parasite Freezedown
During parasite adaptation when at least a single parasite was spotted and the culture was
split into two portions, one portion was maintained in culture and the other frozen down by
centrifuging the culture solution at 2100 rpm for 3 minutes at 200C to obtain an erythrocyte
pellet. Then the supernatant was carefully sucked out using the vacuum pump. The volume of the
packed cells was estimated and depending on the volume, 500µl or less was transferred to a

16
cryovial, labeled with the parasite’s code and date. Then equal volume of the freezing solution (3
% sorbitol; 0.65 % NaCl and 28 % glycerol) was added followed with a gentle mixing and then
the cryovial was frozen down immediately at – 800C at least for one day and then transferred to
liquid nitrogen for long-term storage.
3.3.1.7 Synchronization of Cultures
Asynchronous culture was split as described above (Section 3.3.1.4), one portion returned
to culture and the larger portion was centrifuged at 2100 rpm for 3 minutes at 200C to precipitate
the erythrocytes. The supernatant was removed and then 10ml of sterilized 5% D-sorbitol added
to the precipitate. This was incubated in a waterbath for 10 minutes, and then centrifuged again
as later. Parasitemia was determined and the pellets returned to culture. The procedure was
repeated after one cycle (48 hours) and once a week for 2 weeks.
3.3.2 In Vitro Drug Sensitivity Assay
In vitro drug sensitivity assay was done as described in Druilhe et al. (2001)
3.3.2.1 Drug sensitivity Assay
The sensitivity of the isolates was determined for Artemisinins as DHA (this was the test
compound and it is the active compound in Artemisinins), LMF, MFQ, PQ (Because these are
used with artemisinins as ACTs), CQ, PYR, Q and DEA. The non artemisinin drugs were for
comparison and cross resistance determination.
3.3.2.2 Preparation of Stock Drug Solution
PQ, LMF, Q and DEA were dissolved in 90% methanol plus 10% HCl; DHA in 70%
ethanol; PYR in DMSO; MFQ in 99% methanol and 1% acetic acid and then CQ in distilled
water, as shown in the following table. These solutions were then diluted by culture media to
obtain a working solution. The solutions were covered with aluminum paper foil since the drugs
are light sensitive. PYR and DHA were prepared immediately when they were being used due to
their high light sensitivity and unstable nature.

17
Table 2: Preparation of stock drug solution
Compound Solvent Stock
Solution
(mg/ml)
Dilution
Factor
Working
Solution
(mg/ml)
1st
concentratio
n (ng/ml)
PQ 90% Meth
10% HCL
5 x 100 0.05 1851.85
LMF 90% Meth
10% HCL
1 x 10 0.10 3703.70
DHA 70% ethanol 1 x 200 0.005 185.18
CQ Distilled
water
5 x 100 0.05
1851.85
Q 90% Meth
10% HCL
2 x 10 0.20 7407.41
PYR DMSO 5 x 10 0.50
18518.52
DEA 90% Meth
10% HCL
5 x 100 0.05 925.925
MFQ 99% Meth
1% HCL
Acetic acid
5 x 100 0.05 1851.85
NB: Drugs were dissolved into respective solvents to obtain stock solutions. These were then
diluted with culture medium to obtain working solution. This working solution when added in a
threefold dilution to the mother plate (Section 3.3.2.3) resulted in X27 dilution of the drug from
the second well to the last well, giving the indicated first drug concentration on the second well.
3.3.2.3 Preparation of the Mother Plate
The microtiter plate that was used consisted of 96 flat-bottom wells, arranged in a matrix
of eight rows (A through H) and 12 columns (1 through 12). Two mother plates were prepared
each containing four drugs in duplicate rows, successively following the order shown in the
above table. Two hundred microliters of culture medium was then added to each well. From the

18
working drug solutions 100µl of last stock drug solution was added to each well in the second
column. These were then mixed by gentle up and down pipetting. Then 100µl of the mixture was
transferred to each adjacent well in the third column. This was also transferred to the wells in the
second column and so on until the last column, from which 100 µl of the mixture was removed.
3.3.2.4 Dilution of Culture
Dilution of culture was performed by first determining the parasitemia as described in
Section 3.3.1.5 then diluting it to 1% by first spinning down the culture solution to obtain an
erythrocyte pellet as described below. The supernatant was removed and the infected
erythrocytes mixed with uninfected ones in a ratio according to the determined parasitemia to
obtain a total count of 600µl erythrocytes. Then 30ml of media was added to the cells, in total
getting a parasitemia of 1% and a hematocrit of 2%.
Note: For IC50, lab strain parasitemia was maintained at 0.5 to 1%, mostly less than 1% this is
because they grew very fast i.e. they are culture adapted. Thus the dilution was made in 1:4 (1+3
parts of infected and uninfected erythrocytes, respectively). For field isolates, the parasitemia
was maintained at 1%, because they grow very slowly.
3.3.2.5 Preparation of the Working Plate
One hundred microliters of the culture solution (diluted to 1% parasitemia and 2%
hematocrit as described in section 3.3.2.4) was added to each well except the last three wells in
the first column. These three wells were loaded with 100µl of uninfected erythrocytes acting as
negative controls. Then 12.5 µl of the drug solution from the mother plate was added to the
working plate, transferring the contents from wells in one column to the similar wells in the other
column, respectively taking care that each well was correctly matched. The first five wells in the
first column contained no drug solution and these acted as the positive controls. The plates were
then incubated for 72 hours.
3.3.3 SYBR Green I Assay
SYBR Green I Assay was done according to Smilkstein et al. (2004). This was done to
determine growth inhibition, whereby SYBR green dye intercalates with double stranded DNA
and fluoresces (Johnson et al., 2007). As such, positive control wells will give 100%

19
fluorescence due to complete growth, while test wells will yield reduced fluorescence due to
reduced growth of the parasites.
3.3.3.1 Growth Inhibition Assay
MSF lysis buffer was prepared by mixing 1 L cell culture water with 2.423 g of Tris base.
This was then dissolved completely using a magnetic stirrer. Then the pH was adjusted to 7.5
using concentrated HCl. To this solution 10 ml 0.5 M EDTA was then added. Then 80 mg of
saponin was added, followed with 0.8 ml Triton X-100. Then the solution was mixed thoroughly,
avoiding the production of bubbles. This was then Vacuum filtered to remove particulate matter
and stored at Room Temperature.
After 72 hours of growth, 100µl of SYBR Green I in lysis buffer (2.2 µl of SYBR Green
I/11ml of lysis buffer (Tris HCl, EDTA, Triton X-100 and Saponin)) was added to each well and
the contents were mixed until no visible erythrocyte sediment remained. After 1 h of incubation
in the dark at room temperature, fluorescence was measured with Fluostar optima fluorescence
multiwell plate reader with excitation and emission wavelength bands centered at 485 and 530
nm, respectively, and a gain setting equal to 46.
3.3.4 Data Analysis
In vitro activity was assessed as the drug concentration that inhibited 50% of parasite
growth (IC50). Data was collected as fluorescence units and matching drug concentrations in
nano Moles (nM).
a) To investigate in vitro response of P. falciparum isolates from Kilifi County to
artemisinins: IC50 (ng/ml) obtained after incubation times of 48 hours was determined
using excel sheets (Microsoft corporation, USA) based on non linear regression model.
These were then converted to nM. Parasite isolates were identified as resistant or
susceptible based on IC50 cutoff of reference IC50s: DHA >10.5 nM (Bruno et al.,
2010), Q > 800 nM (Ndong et al., 2003) (Bruno P. et al., 2010), MFQ >30 nM (Ndong et
al., 2003)(Bruno P. et al., 2010), LMF >150 nM, DEA >80 nM, 100 nM (Kaddouri et al.,
2006), PYR >2,000 nM (Touré et al., 2008; Cui and Su, 2009) and PQ > 100nM
(Leonardo and Pascal, 2003; Mwai et al., 2009).

20
b) To determine difference between the in vitro activity of artemisinins with that of
other standard antimalarial drugs. The above calculated IC50s for each drug for the
samples were compared by Kruskal Wallis test and Dunn’s post test. The level of
significance was set at a P value of <0.05.
c) To determine whether there is a difference in the in vitro response to artemisinins
in P. falciparum isolates from primary and recurrent malaria infection in Kilifi
County. Mean IC50s for each drug for each group of the samples were compared by
Mann Whitney test. The level of significance was set at a P value of <0.05.
d) To determine existence of in vitro cross-resistance patterns between artemisinins
and other standard antimalarials: Correlation analysis of the above calculated IC50s
for all drugs for the samples was performed using Spearman corelation analysis. The
statistical analysis was carried out using Prism 4.0 for windows (Graphpad software Inc.
CA).

21
CHAPTER FOUR
RESULTS AND DISCUSSION
4.1 RESULTS
4.1.1 The In Vitro response of P. falciparum isolates to Artemisinins and other
Antimalarials.
Eight field isolates of P. falciparum were adapted for long-term culture and their in
vitro drug chemosensitivity profiles analyzed (Table 3). Laboratory strain 3D7 was used as the
control. The mean IC50s of PQ, LMF, DHA, CQ, Q, PYR, DE-A and MFQ were 30.34 nM
(95% CI 26.96-33.72nM), 46.5nM (95% CI 31.40-61.60nM), 1.861 nM (95% CI 1.489-2.233
nM), 24.29 nM (95% CI 13.2-35.46 nM), 46.7 nM (95% CI 36.78-56.62 nM), 18074 nM (95%
CI 9483-26665), 37.8 nM (95% CI 19.46-56.14nM) and 41.7 nM (95% CI 23.64-59.75 nM)
respectively (Table 4).
No isolate was resistant to PQ, LMF and DHA. Also considering the internationally used
IC50 cut off of > 100 nM all (8) isolates were sensitive to CQ, but considering the IC50 cutoff of
> 25 nM recommended for the study site (Mwai et al., 2009) one (1/8) isolate was resistant to
CQ. No isolate was resistant to Q considering both the high IC50 cutoff value of > 800 nM and
lower > 500 nM (Okombo et al., 2010). All (8) isolates were resistant to PYR. One (1/8) isolate
was resistant to DE-A. Five (5/8) isolates were resistant to MFQ (Table 5 and Figure 6).

22
Table 3: The In vitro response of P. falciparum isolates to antimalarials
Drugs tested
PQ LMF DHA CQ Q PYR DE-A MFQ
Control
3D7 33.03 70.31 2.04 9.31 49.43 20.58 83.07 70.95
Isolates
27d0 24.05 36.01 1.58 57.09 68.70 5347.00 38.06 53.06
27d21 34.04 53.02 2.62 22.39 51.28 29295.00 50.61 66.30
171d0 30.81 68.35 2.06 18.54 38.05 23301.00 85.83 54.41
171d21 33.58 58.58 2.38 19.15 31.85 33452.00 27.44 65.28
115d0 32.25 68.85 1.54 18.17 47.43 18168.00 19.30 41.70
115d49 24.44 35.48 1.76 21.56 56.03 17468.00 30.46 24.44
191d0 29.81 21.73 1.38 20.10 37.70 11463.00 18.81 15.33
191d54 33.77 29.99 1.57 17.35 42.58 6097.00 31.90 13.04
The in vitro response of field isolates of P. falciparum from Kilifi County to eight antimalarials expressed as IC50 in nM. The isolates are named using the number of patient it was isolated from and the day of isolation.

23
Table 4: Mean IC50s of the eight antimalarial drugs tested against field isolates of P. falciparum in Kilifi County
Drugs No. of isolates Mean IC50 95% Confidence Range (nM)
tested nM interval (nM) Minimum Maximum
PQ 8 30.34 26.96-33.72 24.05 34.04
LMF 8 46.50 31.40-61.60 21.73 68.85
DHA 8 1.86 1.49-2.23 1.38 2.62
CQ 8 24.29 13.20-35.46 17.35 57.09
Q 8 46.70 36.78-56.62 31.85 68.70
PYR 8 18074.00 9483.00-26665.0 5347.00 33452.00
DE-A 8 37.80 19.46-56.14 18.81 85.83
MFQ 8 41.7 0 23.64-59.75 13.04 66.30

24
Table 5: Number of resistant or susceptible parasites
PQ LMF DHA CQ CQ Q Q PYR DE-A MFQ
CUTOFF >100 >150 >10.5 >100 >25 >800 >500 >2,000 >80 >30
Total 8 8 8 8 8 8 8 8 8 8
Susceptible 8 8 8 8 7 8 8 0 7 3
Resistant 0 0 0 0 1 0 0 8 1 5
Parasites are determined to be sensitive or resistant based on their IC50 value to a particular drug being lower or upper than the cutoff
value respectively. The IC50 cutoff values are in nM. No isolate was resistant to PQ, LMF, DHA, CQ (>100 nM) and Q. One isolates
was resistant to CQ considering the >25nM recommended for the study site, five to MFQ and all were resistant to PYR.

25
Figure 6: Rate of in vitro resistance or susceptibility of Plasmodium falciparum to the eight antimalarial drugs tested.

26
4.1.2 The Difference between the In Vitro Activities of Artemisinins with that of other
Antimalarial Drugs.
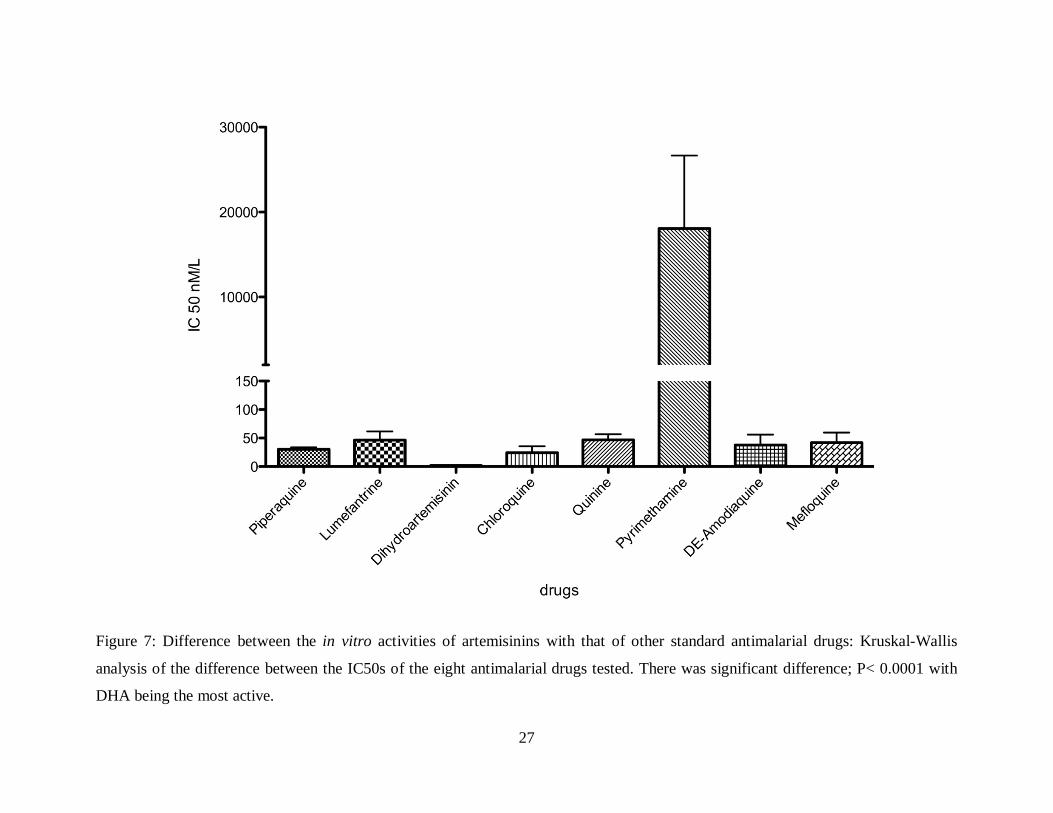
There was significant difference (P< 0.0001) in the in vitro activities of the eight
antimalarial drugs towards the isolates (Kruskal-Wallis analysis, P<0.05) (Figure 7). As
expected, DHA was the most active drug, followed by CQ, PQ, DE-A, MFQ, LMF, Q and lastly
PYR (Table 5). Specifically there was significant difference between the in vitro activity of DHA
compared to LMF, CQ, Q, PYR, and MFQ. But there was no significant difference when
compared to PQ and DEA (Table 6).
27
Figure 7: Difference between the in vitro activities of artemisinins with that of other standard antimalarial drugs: Kruskal-Wallis
analysis of the difference between the IC50s of the eight antimalarial drugs tested. There was significant difference; P< 0.0001 with
DHA being the most active.

28
Table 6: Difference between the in vitro activities of the antimalarial drugs tested; Dunn’s post test, P<0.05
Drug pair Summary Drug pair Summary
PQ-LMF ns DHA-Q **
PQ-DHA ns DHA-PYR ***
PQ-CQ ns DHA-DEA ns
PQ-Q ns DHA-MFQ *
PQ-PYR * CQ-Q ns
PQ-DEA ns CQ-PYR ***
PQ-MFQ ns CQ-DEA ns
LMF-DHA ** CQ-MFQ ns
LMF-CQ ns Q-PYR ns
LMF-Q ns Q-DEA ns
LMF-PYR ns Q-MFQ ns
LMF-DEA ns PYR-DEA *
LMF-MFQ ns PYR-MFQ ns
DHA-CQ ns DEA-MFQ ns
Dunn's Multiple Comparison Test; post test of the Kruskal-Wallis analysis of the difference between the IC50s of the eight antimalarial drugs tested. ns; non-significant = P > 0.05.*; significant = P < 0.05. **; very significant = P < 0.01, ***; highly significant = P < 0.001.

29
4.1.3 The Difference in the In Vitro response to Artemisinins in P. falciparum Isolates from
Primary and Recurrent Malaria infection in Kilifi County.
There was no significant difference (P = 0.9591) in the in vitro response of the isolates
from initial and recurrent malarial (both recrudescent and re-infections) infections towards
dihydroartemisinin and the other antimalarial drugs tested (Mann Whitney test, P <0.05) (Figure
8, 9, 10 and 11).

30
Figure 8: A comparison of the in vitro response of the parasite isolates from initial infection (d0) and recurrent infection (dRC).

31
Figure 9: A comparison of the in vitro response of the parasite isolates from initial infection (d0) and recurrent infection (dRC). There
was non-significance difference (P = 0.9591). Mann Whitney test P <0.05.

32
Figure 10: A comparison of the in vitro response of the parasite isolates from initial infection (d0) and recrudescent infection (dRD).
There was non-significance difference (P = 0.9591). Mann Whitney test P <0.05.
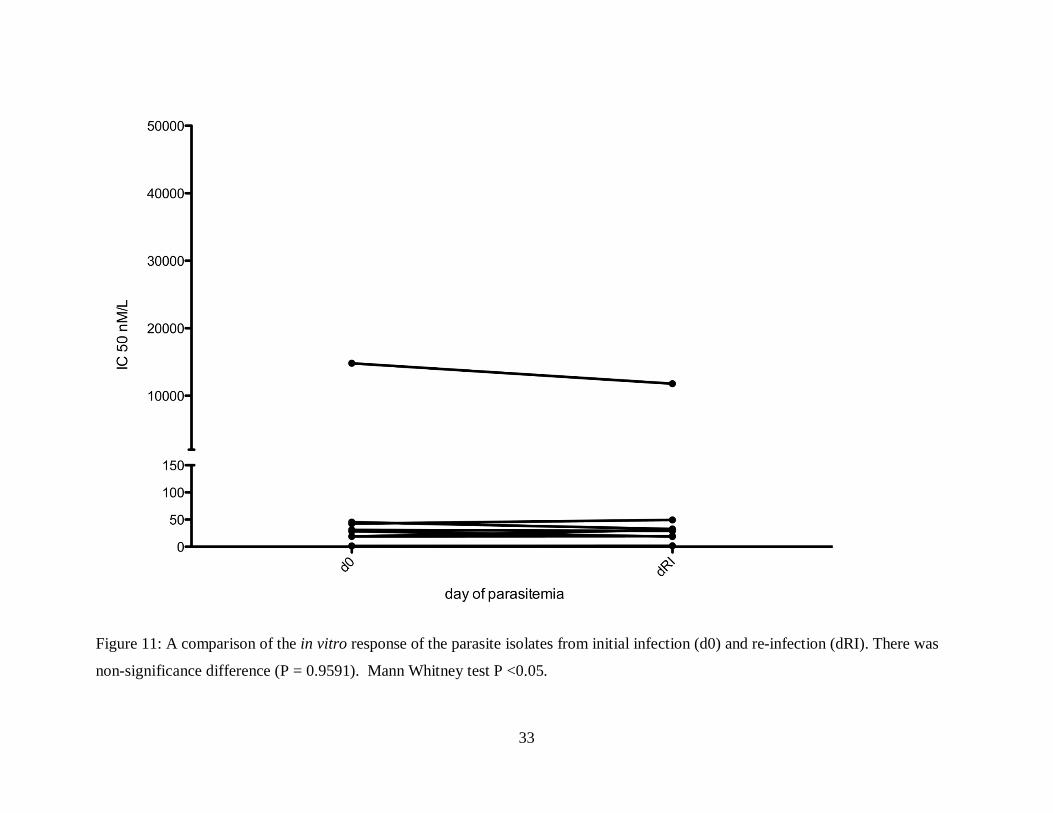
33
Figure 11: A comparison of the in vitro response of the parasite isolates from initial infection (d0) and re-infection (dRI). There was
non-significance difference (P = 0.9591). Mann Whitney test P <0.05.

34
4.1.4 The Correlation Patterns between Artemisinins and other Antimalarials
There was no significant correlation in the IC50s of the eight antimalarial drugs towards
the isolates (Spearman correlation analysis, P<0.05) (Figure 12 and Table 7).

35
Figure 12: Correlation between the IC50s values of the eight antimalarial drugs tested.

36
Table 7: Correlation between the IC50s values of the eight antimalarial drugs tested; Spearman correlation, P<0.05
Except between DHA vs MFQ and PYR vs MFQ there was no significant correlation in the IC50s of the eight antimalarial drugs towards the isolates. But the DHA vs MFQ and PYR vs MFQ correlation is highly unlikely (Spearman correlation analysis, P<0.05). ns; non-significant = P > 0.05.*; significant = P < 0.05.
Drug pair r2 P Drug pair r2 P
DHA-MFQ 0.83 * PQ-MFQ 0.29 ns
PYR-MFQ 0.74 * LMF-DEA 0.24 ns
DHA-PYR 0.69 ns PQ-LMF 0.21 ns
DHA-DEA 0.64 ns CQ-DEA 0.21 ns
LMF-MFQ 0.64 ns PQ-DEA 0.14 ns
LMF-PYR 0.62 ns PYR-DEA 0.10 ns
CQ-Q 0.55 ns DHA-Q 0.05 ns
PQ-PYR 0.55 ns Q-MFQ 0.00
DEA-MFQ 0.48 ns LMF-Q -0.05 ns
PQ-DHA 0.38 ns CQ-PYR -0.10 ns
LMF-DHA 0.38 ns LMF-CQ -0.21 ns
CQ-MFQ 0.40 ns PQ-Q -0.36 ns
Q-DEA 0.36 ns PQ-CQ -0.43 ns
DHA-CQ 0.33 ns Q-PYR -0.45 ns

37
4.2 DISCUSSION
As a result of P. falciparum developing resistance to common antimalarials such as CQ
(Le Bras et al., 2006) there is need of a novel antimalarial to boost the fight against malaria.
ACTs are currently recommended as the front line dugs (Nosten and White 2007; Eastman and
Fidock 2009). The partner drugs used in the combination protect each other in terms of resistance
(White 2001). However, the parasite as stubborn as it is has started developing resistance (Noedl
et al., 2008; Dondorp et al., 2009; Rogers et al., 2009). This generates fear that ACTs will
become inefficient (Pradines et al., 2010).
Artemisinins are normally metabolized into DHA which is their active compound
(Wongsrichanalai et al., 1999; Mwai et al., 2009). It is through this compound that artemisinins
exert their effect to malarial parasites. Furthermore, in aqueous media artemisinins are broken
down to dihydroartemisinin. Thus artemisinins have similar IC50 values (Wongsrichanalai et al.,
1999).
DHA with a mean IC50 of 1.861 nM (95% CI 1.489-2.233 nM) (Table 3), was the most
active drug (Table 4 and Figure 7); all parasites being susceptible to the lowest IC50 value
among all the drugs that were tested (Table 5 and Figure 6) in line with Mwai et al., (2009). It
gave significant statistical difference in the in vitro activities with all the other drugs except PQ
and CQ (Table 6).
Artemisinins have a short half-life and target all the stages of P. falciparum having much
more effect on the ring stages as compared to other drugs (Sompob et al., 2011). They are able to
reduce the parasite number up to as much as 10000 in one cycle (Woodrow et al., 2005). This
makes them the most active (Schlitzer 2008) accounting for the rapid activity of DHA compared
with all other antimalarial drugs.
The ability of P. falciparum to develop resistance to antimalarials is normally due to gene
polymorphism in the target or transport molecules (White, 1999; Tärning 2007). But the
mechanism of resistance to artemisinins is uncertain and polymorphisms in several candidate
genes have been proposed (Eastman and Fidock, 2009; Gama et al. 2010), which include:

38
(i.) Single nucleotide polymorphism (SNPs) in pfatpase6, a gene which codes for a
SERCA-type calcium-translocating ATPase (Jambou et al., 2005; Eastman and Fidock,
2009; Gama et al., 2010).
(ii.) SNPs or amplification in codons 86, 184, 1034, 1042 and 1246 of pfmdr1, which
codes for P. falciparum multidrug resistance (PfMDR1) (Cecilia et al., 2004; Price et al.,
2004; Sidhu et al., 2006; Eastman and Fidock, 2009).
(iii.) SNPs in codons 74, 75, 76, 220, 271, 326 and 371 of pfcrt which codes for P.
falciparum chloroquine resistance transporter (PfCRT) (Cecilia et al., 2004)
(iv.) Polymorphisms in the gene coding for P. falciparum multidrug resistance –
associated protein (PfMRP) (Raj et al., 2009).
PfMDR1 and PfCRT are transport proteins located on the membrane of the DV of the
parasite, and since this is one of the proposed active site of artemisinins, it thus controls drug
accretion (Eastman and Fidock, 2009) as any mutation in these genes results in a modified
protein which impairs drug accumulation and consequently their potencies (Valderramos and
Fidock, 2006).
Kilifi isolate have been characterised to have one mutation and copy number of pfmdr1
(Mwai et al., 2009) further explaining the high activity of DHA. Pfmdr1 amplification is not
commonly found in Africa (Holmgren et al., 2006; Ursing, et al., 2006; Sisowath et al., 2007;
Mwai et al., 2009) due to low usage of MFQ (Mwai et al., 2009).
Additionally, as drug pressure contributes greatly in promoting the development and
spread of drug resistance due to their short elimination life (Schlitzer 2008), artemisinins exert
little drug pressure (Wongsrichanalai et al., 1999) consequently reducing the chances of
resistance, hence the observed high activity towards all the isolates.
Despite the lack of demonstrate able artemisinins resistance there are reports of delayed
clearance times (Noedl et al., 2008; Dondorp et al., 2009; Witkowski et al., 2010) whereby
parasites survive in presence of artemisinins longer times as would be expected but later get
killed. As such, as opposed to the classical resistant phenotype this points out a different
resistance mechanism (Witkowski et al., 2010). Thus reduced artemisinins response in the ring-
stage could provide an answer to the reports of resistance in Cambodia (Benoit-Vical et al.,
2007; Witkowski et al., 2010) whereby a proportion of rings remain dormant in presence of the
drug and resume growth after withdrawal of the drug (Sompob et al. 2011). In other words, P.

39
falciparum uses a quiescence mechanism that allows it to survive ART treatment (Witkowski et
al., 2010). Indeed, even though artemisinins are still effective resistance will eventually emerge
(Yavo et al., 2010).
When the IC50s of two antimalarials are positively correlated, this indicates cross
resistance (Yavo et al., 2010). However, the positive correlation in the in vitro activities of DHA
and other antimalarials, except for MFQ, was insignificant reassuring the use of ACTs as first
line antimalarial drugs. The significant correlation with MFQ can be attributed to technical
failures that resulted in reduced MFQ response. Despite this observation, polymorphisms and
amplification of pfmdr1 indeed does reduce DHA and MFQ sensitivity (Pickard et al., 2003;
Reed et al., 2004; Sidhu et al., 2005; Gama et al., 2010). As the causation of this polymorphism
is the usage of MFQ, which is rare in Africa and Kenya indeed, in addition to the observed low
pfmdr1 copy number and polymorphism in Kilifi isolates (Mwai et al., 2009), this correlation is
highly unlikely. Based on this observation of unlikely MFQ reduced response, it is also unlikely
for the observed correlation between MFQ and PYR. Indeed, they have different modes of
action, PYR, an antifolate, inhibits dihydrofolate reductase in the folate biosynthetic pathway
while MFQ, an arylaminoalcohol, inhibits heme digestion (Schlitzer et al., 2008).
Unexpectedly CQ demonstrated a higher IC50 value second to DHA (Table 4 and
figure 13). With a mean IC50 of 24.29 nM (95% CI 13.2-35.46 nM) all isolates were sensitive to
CQ (Table 3, 5 and figure 12) in line with Mwai et al., (2009). But based on the lower IC50
cutoff value, one isolate was resistant to CQ. There was no significant statistical difference
between its in vitro activities as compared to the other antimalarials except PYR (Table 6). This
demonstrates that CQ is regaining its activities in this part of Kenya after its withdrawal.
Just as in the case of artemisinins, PfMDR1 and PfCRT control accumulation of CQ and
consequently the pH in the DV. Hence they are involved in chloroquine resistance too, whereby
polymorphisms in pfmdr1 and pfcrt have been found in CQ resistant parasites (Cecilia et al.,
2004; Gama et al., 2010). Infact, the response to CQ is much reduced when polymorphisms in
both genes are present (Cecilia et al., 2004). In a study on Kilifi isolates only one Pfmdr1 and the
pfcrt mutation in codon 76 were present in Kilifi isolates (Mwai et al., 2009). This explains the
low levels of CQ resistance observed in the isolates in addition to the reduced CQ drug pressure
since its ban more than a decade ago.

40
PQ was also very active; with a mean IC50 value of 30.34 nM (95% CI 26.96-33.72 nM)
all isolates were sensitive to it in line with another study of isolates from Kilifi (Mwai et al.,
2009). Similar results were obtained for isolates from other malaria endemic areas (Deloron et
al., 1985; Barends et al., 2007; Basco and Ringwald 2003). Like CQ there was no significant
statistical difference between its in vitro activities as compared to the other antimalarials except
PYR.
PQ is a CQ based antimalarial consisting of two CQ molecules and is used in
combination with DHA in the ACT, Artekin, which is a possible substitute of Coartem
(Karunajeewa et al., 2008; Yeka et al., 2008; Thanh et al., 2009, Mwai et al., 2009).
Polymorphisms in pfcrt- 76 and pfmdr1- 86 don’t contribute to piperaquine resistance despite the
contribution of pfcrt to reduced response to piperaquine (Muangnoicharoen et al., 2009; Mwai et
al., 2009).
As piperaquine is effective against CQ resistant parasites (Basco and Ringwald 2003;
Mwai et al 2009) and with the current observed regaining of CQ sensitivity, this predicts
development of resistance to it (Karunajeewa et al., 2008; Muangnoicharoen et al., 2009; Mwai
et al., 2009). As a result of this, the effectiveness of PQ in areas of high CQ resistance e.g. the
South east of Asia will be short lived as compared to Africa (Karunajeewa et al., 2008; Yeka et
al., 2008; Thanh et al., 2009; Mwai et al., 2009).
Amodiaquine is only suggested for treatment of falciparum malaria (WHO 2000; Sasi et
al., 2009) due to its toxicity (Hatton et al., 1986; Neftel et al., 1986; Sasi et al., 2009). As it
demonstrated effectiveness against CQ resistant parasites, which was prevalent, amodiaquine use
was more around the 1990s (Fadat et al., 1991; Sasi et al., 2009). And due to this, it has been
adopted for CQ replacement in most African countries or better still as second line.
Consequently, it is a commonly used drug in Africa (WHO 2007; Sasi et al., 2009). On the same
note, it has been used in Kenya as a second line drug between 1998 and 2008 (Shretta et al.,
2000; Sasi et al., 2009) and as a front line between 2004 and 2006 (Zurovac 2008; Sasi et al.,
2009).
The use of ACTs which combines amodiaquine results in resistance to
monodesethylamodiaquine which predicts resistance to amodiaquine (Nawaz et al., 2009).

41
Further emphasizing the need for a new and different antimalarial (Bruno et al., 2010 a).
However, despite some of the isolates from Kilifi being resistant to DE-A and MFQ, 12.5% and
62.5% respectively they were still more active than LMF and Q (These had 100% sensitivity).
The mean IC50 of DE-A was 37.8 nM (19.46-56.14 nM), more active following PQ. Like CQ
and PQ there was no significant statistical difference between its in vitro activities as compared
to the other antimalarials except PYR.
DE-A is the active compound of AQ and having a similar structure as CQ, it also
inhibits haem detoxification (Legrand et al., 2008; Eastman and Fidock 2009) and it has efficacy
towards CQ-resistant parasites (Olliaro and Mussano 2003; Eastman and Fidock, 2009).
Polymorphisms in pfcrt and pfmdr1 are associated with its resistance (Bray1996; Ochong 2003;
Fitch 2004; Dokomajilar et al., 2006; Mwai et al., 2009)
Furthermore, AQ has been the second line of treatment in Kenya since SP was introduced in
1999 and has remained so until now. This drug has partially been used in several sites in Kenya
(Sidhu et al., 2002; Mwai et al 2009), including Kilifi, even before the withdrawal of CQ (Sidhu
et al., 2005; Mwai et al 2009).
The observed high DE-A sensitivity results from high CQ sensitivity (Kokwaro et al.,
2007; Kobbe et al., 2008) and the similar chemical structure between amodiaquine and
chloroquine. Furthermore, there has been less amodiaquine drug pressure in Kenya as it has been
always used as a backup drug (Sidhu et al., 2002; Sidhu et al., 2005; Mwai et al 2009).
MFQ had a mean IC50 of 41.7 nM (95% CI 23.64-59.75 nM). Except DHA it had no
significant statistical difference between its in vitro activities as compared to the other
antimalarials.
Polymorphisms in pfcrt-76 and pfnhe (a sodium hydrogen exchanger gene) are not
associated with MFQ resistance (Price et al., 2004; Okombo et al., 2010). While polymorphisms
and amplification of pfmdr1 reduce MFQ sensitivity (Pickard et al., 2003; Reed et al., 2004;
Sidhu et al., 2005; Gama et al., 2010). Indeed pfmdr1 amplification mainly determines MFQ
susceptibility (Pukrittayakamee et al., 2000; Price et al., 2004; Okombo et al., 2010).
Despite the uncommon use of MFQ compared to other antimalarial drugs in Africa
(Okombo et al., 2010) and Kilifi County for that matter, 62.5% MFQ-resistant isolates were

42
observed in this study. This observation is in contrast to another study on isolates from Kilifi
County whereby the isolates were found to be sensitive to MFQ (Okombo et al., 2010).
Furthermore, the isolates were found to have 1 copy of the pfmdr1 gene (Mwai et al., 2009),
explaining the susceptibility of parasites to MFQ.
A more plausible explanation for this difference can be attributed to a greater disparity
in the threshold IC50 for MFQ resistance. Some literatures cites > 30 nM (Ndong et al., 2003;
Bruno et al., 2010), others > 40 nM (Hatabu et al., 2010) and > 50 nM (Randrianarivelojosia et
al., 2004). But still based on the previous two IC50 cutoff values, a high MFQ resistance was
observed, with 62.5% (5 out of 8 isolates) and 50% (4 out of 8 isolates) resistant, respectively.
Thus the observation of a high failure of MFQ sensitivity to isolates from Kilifi is
highly unlikely and may be attributed to technical failures. Or perhaps, the presence of isolates
that are inherently resistant to MFQ could help to explain this observation (Gari-Toussaint et al.,
2002; Henry et al, 2006; Yavo et al., 2010). However, there is a possibility of MFQ resistance as
it can be in rodent malaria (Sidhu et al., 2005, 2006). And if this were to happen in P.
falciparum, it would complicate the fight against malaria (Gama et al. 2010).
In Kenya, currently Coartem is the front line drug (Kokwaro et al., 2007; Mwai et al
2009). But LMF usage results in resistance to Coartem (Dokomajilar et al., 2006; Humphreys et
al., 2007; Happi et al., 2009; Mwai et al 2009). And bearing in mind the reported artemisinins
resistance in South East Asia, it’s predictable that this drug will lose its efficacy (Dondorp et al .,
2009; Mwai et al., 2009).
LMF was more active than Q and PYR following MFQ. With a mean IC50 of 46.5
nM (95% CI 31.40-61.60 nM), all (100%) isolates were sensitive to it in line with another study
on Kilifi field isolates (Mwai et al., 2009) and studies in other endemic areas (Anderson et al.,
2005; Basco and Ringwald 2007; Kaddouri et al., 2008; Mayxay et al., 2007, Parola et al., 2007;
Pradines et al., 2006; Mwai et al., 2009). Like MFQ it had no significant statistical difference
between its in vitro activities as compared to the other antimalarials except DHA. It had positive
correlation with all the drugs except CQ and Q. This inverse relationship between the activities
of LM and CQ is in line with Mwai et al. studies suggesting that selection of LM resistance
would be associated with an increase in CQ activity (2009).

43
Parasites with wild type pfmdr1 and pfcrt are less sensitive to LM and on the contrary,
mutants of these genes are resistant to CQ (Dokomajilar et al., 2006; Humphreys et al., 2007;
Happi et al., 2009; Sisowath et al., 2005, 2007, 2009; Mwai et al., 2009). This helps to explain
the inverse relationship between CQ and LM activity (Pradines et al., 1999, Price et al., 2006;
Mwai et al., 2009). However, at the moment Coartem is still effective (Falade et al., 2008 a,b;
Kobbe et al., 2008; Yeka, et al., 2008; Mwai et al., 2009).
For the treatment of complicated malaria and CQ resistance, Q is the recommended drug
(Ndong et al., 2003).
When compared to the other antimalarial drugs Q was among the least active drugs
despite 100% sensitivity among the isolates. But it was more active than PYR. This total
sensitivity is in line with other studies (Je ˆ rome et al., 2003; Djaman et al., 2004; Toure et al.,
2008; Yavo et al., 2010). Other studies carried out in the coastal region of Kenya including Kilifi
showed a similar Q activity (Watkins et al., 1987; Pasvol et al., 1991: Haruki et al., 1998;
Okombo et al., 2010). And so as from other parts of Kenya and other African countries (Ndong
et al., 2003; Odhiambo and Odulaja 2005; Agnamey et al., 2006; Pradines et al., 2006; Tinto et
al., 2006; Quashie et al., 2007; Okombo et al., 2010). With a mean IC50 value of 46.7 nM (95%
CI 36.78-56.62 nM) and like MFQ and LMF it had no significant statistical difference between
its in vitro activities as compared to the other antimalarials except DHA.
Polymorphisms in pfmdr1 and pfcrt as well as pfmdr1 amplification are associated with
Q resistance (Pickard et al., 2003; Reed et al., 2003; Lakshmanan et al., 2005; Sidhu et al., 2005
Nsobya et al., 2010; Chaijaroenkul et al., 2010).
Polymorphism in pfnhe modify the in vitro response to Q whereby an increase in the
number of DNNND repeats (1–5) in ms4760 microsatellite is associated with less sensitivity to
Q (Ferdig et al., 2004; Henry et al., 2009; Okombo et al., 2010) and discrepancy in its copy
number occur (Vinayak et al., 2007; Okombo et al., 2010).
In a study of 29 isolates from Kilifi characterised them to have 1- 3 DNNND (Aspartic
acid-asparagine- asparagine-asparagine-aspartic acid) repeats. 17 and 6 isolates had 2 and 1
repeats, respectively, and the remaining 6 isolates had 3 repeats. The increase from 1 to 2 repeats
was associated with decreasing susceptibility to Q, and the change from 2 to 3 repeats rendered

44
parasites more susceptible (Okombo et al., 2010). However, another study shows no decrease in
Q activity in parasites with 3 DNNND repeats (Henry et al., 2009; Okombo et al., 2010).
Pfmdr1 is an important contributing factor in Q resistance and the isolates have been
characterised to have pfmdr1-86 mutation and 1 pfmdr1 copy number (Mwai et al., 2009;
Okombo et al., 2010). The potency of Q can be altered by the degree of accumulation inside the
DV, which is the site of haem detoxification (Valderramos and Fidock, 2006). The low number
of DNNND repeats and the low rate of Pfmdr1 polymorphism could explain the high activity of
QN, in addition to less drug pressure due to correct use (Bustos et al., 1994; Okombo et al.,
2010).
There is lack of acquiescence in the correct use of Q (Fungladda et al., 1998; Okombo
et al., 2010) resulting in less drug pressure and hence resistance not developing towards it, and
further explaining the observed high Q activity. Thus QN is still effective (Okombo et al., 2010).
Unfortunately there are accounts of Q resistance (Brasseur et al., 1988; Kilimali et al.,
1990; Mutanda 1999; Pettinelli et al., 2004; Okombo et al., 2010) and reduced response
(Pukrittayakamee et al., 1994, 2000; Parola et al., 2001; Roche, et al., 2003; Molinier et al.,
2004; Adam et al., 2004; Achan et al., 2009; (Okombo et al., 2010). Which even if are however,
based on effectiveness studies and that it is possible it could be out of the lack of acquiescence in
correct Q use (Okombo et al., 2010), but still it would be better to undertake resistance
surveillance of the same (Tinto et al., 2001; Okombo et al., 2010). To emphasize on this, drug
pressure resulting from the usage of Q selects resistant parasites (Randrianarivelojosia et al.,
2004; Henry et al., 2006; Pradines et al., 2006; Tinto et al., 2006; Parola et al., 2007; Yavo et al.,
2010).
PYR was the least active drug, with a mean IC50 value of 18074 nM (95% CI 9483-
26665 nM) all isolates were resistant to it. Surprisingly, except PQ, DHA, CQ and DE-A it had
no significant statistical difference between its in vitro activities as compared to the other
antimalarials.
PYR resistance is associated with mutations in the dihydrofolate reductase (DHFR) gene
dhfr, whereby the mutations at codon 108 (Serine-108-Asparagine) leads to PYR resistance, with

45
level increasing when additional mutations, Asparagine-51-Isoleucine and Cystine-59-Arginine,
occurs. This becomes even higher with the Isoleucine-164-Leucine mutation (Kiara et al., 2009).
Infact, mutations in dhfr are the major determinant of resistance towards Pyrimethamine-
Sulfadoxine (PYR-SD), which occurs as a triple mutant involving the mutations; (Serine-108-
Asparagine, Asparagine-51-Isoleucine, and Cystine-59-Arginine) (Nzila 2006; Kiara et al.,
2009). And the addition of the mutation Isoleucine-164-Leucine (quadruple mutant parasites)
leads to higher resistance of the same (Wilairatana et al., 1997; Watkins et al., 1997; Kiara et al.,
2009). This mutation is indeed found in Africa (Hame et al., 2008; Kiara et al., 2009), no wonder
the high PYR-SD resistance.
Polymorphism in Guanine triphosphates (GTP)-cyclohydrolase also leads to PYR-SD
resistance, which is also an enzyme in the folate metabolism pathway (Nair et al., 2008; Kiara et
al., 2009).
The above dhfr gene mutations have been documented in isolates from Kilifi (Kiara et
al., 2009) explaining the resistance towards PYR.
Besides, there was resistance towards PYR-SD in Kilifi County as early as the mid-1990s
(Shretta et al., 2000; Kiara et al., 2009) which proved futile in early 2000s and its use was
eventually banned in 2004 (Amin et al., 2007; Kiara et al., 209).
Despite its ban, there is still usage of PYR-SD (Clarke et al., 2008; Temperley et al.,
2008; Kiara et al., 2009) and based on its affordability its abused leading to drug pressure
(Wichmann et al., 2003; Ochong et al., 2003 b; Parola et al., 2007; Kiara et al., 2009) and
consequently the observed high PYR resistance (Kiara et al., 2009). Therefore, the use of PYR-
SD should be highly regulated (Spalding et al., 2010).

46
CHAPTER FIVE
CONCLUSIONS
Artemisinins are still active in Kilifi County. They are more active than other standard
antimalarials. No cross-resistance was observed with other antimalarials. Isolates from initial and
recurrent malaria have similar in vitro response to artemisinins and other antimalarials.
RECOMMENDATIONS
One; continued public awareness should be done to encourage vector and parasite control
to help fight malaria. Two; to avoid drug pressure, effective and correct use of antimalarials
should be reinforced to avoid resistance emergence. Three; in addition to the need of more
studies on polymorphisms of the antimalarials candidate genes in isolates from Kilifi County
more studies should be carried out in other parts of Kenya and other endemic countries to
provide unremitting surveillance of response to artemisinins. This will be useful in providing
information that will advice the making of treatment policy. Four; understanding how
artemisinins act and the resistance mechanisms as well as development of new and different
antimalarials to avoid relying only on artemisinins will help protect artemisinins against
resistance. Five; hopes in the fight against malaria rely on an effective and acceptable vaccine.
Thus the search towards such development should be accelerated.

47
REFFERENCES
Achan J., Tibenderana J., K., Kyabayinze D., Wabwire Mangen F., Kamya M. R., Dorsey G.,
D’Alessandro U., Rosenthal P. J., and A. O. Talisuna. (2009). Effectiveness of quinine
versus artemether-lumefantrine for treating uncomplicated falciparum malaria in
Ugandan children: randomized trial. British Medical Journal 339: 2763.
Adam I., Ali D. M., Noureldien W., and Elbashir M. I. (2005). Quinine for the treatment of
chloroquine-resistant Plasmodium falciparum malaria in pregnant and non-pregnant
Sudanese women. Annals of Tropical Medicine and Parasitology 99: 427–429.
Agnamey P., Brasseur P., de Pecoulas P. E., Vaillant M., and Olliaro P. (2006). Plasmodium
falciparum in vitro susceptibility to antimalarial drugs in Casamance (southwestern
Senegal) during the first 5 years of routine use of artesunate-amodiaquine. Antimicrobial
Agents and Chemotherapy 50: 1531–1534.
Aide P., Bassat Q. and Alonso P.L. (2007). Towards an effective malaria vaccine. Archives of
Disease in Childhood 92: 476-479.
Amin A. A., Zurovac D., Kangwana B. B., Greenfield J., Otieno D. N., Akhwale W. S., and
Snow R. W. (2007). The challenges of changing national malaria drug policy to a
rtemisinin-based combinations in Kenya. Malaria Journal 6: 72.
Anderson T.J., Nair S., Qin H., Singlam S., Brockman A., Paiphun L., Nosten F. (2005). Are
transporter genes other than the chloroquine resistance locus (pfcrt) and multidrug
resistance gene (pfmdr) associated with antimalarial drug resistance? Antimicrobial
Agents and Chemotherapy 49: 2180-2188.
Asawamahasakda W., Ittarat I., Pu Y.M., Ziffer H., Meshnick S.R. (1994). Reaction of
antimalarialendoperoxides with specific parasite proteins. Antimicrobial Agents and
Chemotherapy 38: 1854–1858.
Auerbach T. (2002). Antibiotics targeting ribosomes: crystallographic studies. Current Drug
Targets. Infectious Disorders 2: 169–186.

48
Bacon D.J, Latour C., Lucas C., Colina O., Ringwald P. and Picot S. (2007). Comparison of a
SYBR Green I-Based Assay with a Histidine-Rich Protein II Enzyme-Linked
Immunosorbent Assay for In VitroAntimalarial Drug Efficacy Testing and Application to
Clinical Isolates. Antimicrobial Agents and Chemotherapy 51: 1172–1178.
Barends M., Jaidee A., Khaohirun N., Singhasivanon P. and Nosten F. (2007). In vitro activity of
ferroquine (SSR 97193) against Plasmodium falciparum isolates from the Thai-Burmese
border. Malaria Journal 6: 81.
Basco L.K. and Ringwald P. (2003). In vitro activities of piperaquine andother 4-
aminoquinolines against clinical isolates of Plasmodium falciparum in Cameroon.
Antimicrobial Agents and Chemotherapy 47: 1391–1394.
Basco L.K. and Ringwald P. (2007). Molecular epidemiology of malaria in Cameroon. XXIV.
Trends of in vitro antimalarial drug responses in Yaounde, Cameroon. The American
Journal of Tropical Medicine and Hygiene 76: 20–26.
Bennett T. N., Paguio M., Gligorijevic B., Seudieu C., Kosar A. D., Davidson E. and Roepe P.
D. (2004). Novel, rapid, and inexpensive cell-based quantification of antimalarial drug
efficacy. Antimicrobial Agents and Chemotherapy 48: 1807–1810.
Benoit-Vical F., Lelievre J., Berry A., Deymier C., Dechy-Cabaret O., Cazelles J., Loup C.,
Robert A., Magnaval J. and Meunier B. (2007). Trioxaquines:new antimalarial agents
active on all erythrocytic forms including gametocytes. Antimicrobial Agents and
Chemotherapy 51: 1463–1472.
Brasseur P., Kouamouo J., Brandicourt O., Moyou-Somo R. and Druilhe P. (1988). Patterns of in
vitro resistance to chloroquine, quinine, and mefloquine of Plasmodium falciparum in
Cameroon, 1985–1986. The American Journal of Tropical Medicine and Hygiene 39:
166–172.
Bray P.G., Hawley S.R., Mungthin M., Ward S.A. (1996). Physicochemical properties correlated
with drug resistance and the reversal of drug resistance in Plasmodium falciparum.
Molecular Pharmacology 50: 1559–1566.

49
Breman J.G., Alilio M.S. and Mills A. (2004). Conquering the intolerable burden of malaria:
what’s new, what’s needed: a summary. The American Journal of Tropical Medicine and
Hygiene 71: 1–15.
Bruno P., Sébastien B., Maud H., Claude O., Eric B., Rémy A.,Eric D. and Christophe R. (2010).
Absence of association between pyronaridine in vitro responses and polymorphisms in
genes involved in quinoline resistance in Plasmodium falciparum. Malaria Journal 9: 1-
7. (a)
Bruno P., Thierry P., Khaled E., Sébastien B., Lionel B., Marie-Catherine R., Daniel P., Pascal
M., Christophe R.,and Denis M. (2010). Quinine-Resistant Malaria in Traveler Returning
from Senegal, 2007. Emerging Infectious Disease 16: 546-548. (b)
Bustos M.D.G., Gay P.F. and B. Diquet. (1994). In-vitro tests on Philippine isolates of
Plasmodium falciparum against four standard antimalarials and four qinghaosu
derivatives. Bulletin of the World Health Organization 72: 729-735
Chaijaroenkul W., Wisedpanichkij R. and Na-Bangchang K. (2010). Monitoring of in vitro
susceptibilities and molecular markers of resistance of Plasmodium falciparum isolates
from Thai-Myanmar border to chloroquine, quinine, mefloquine and artesunate. Acta
Tropica 113: 190–194.
Clarke S.E., Jukes M.C., Njagi J. K., Khasakhala L., Cundill B., Otido J., Crudder C., Estambale
B. B., and Brooker S. (2008). Effect of intermittent preventive treatment of malaria on
health and education in schoolchildren: a cluster-randomised, double-blind, placebo-
controlled trial. Lancet 372: 127–138.
Craig A. and Scherf A. (2001). Molecules on the surface of the Plasmodium falciparum infected
erythrocyte and their role in malaria pathogenesis and immune evasion. Molecular &
Biochemical Parasitology 115: 129–143.
Cui L and Su X. (2009). Discovery, mechanisms of action and combination therapy of
artemisinin. Expert Review of Anti-Infective Therapy 7: 999–1013.

50
Day K.P. and Marsh K. (1991). Naturally acquired immunity to Plasmodium falciparum.
Immunology Today 12: 68-71.
Deloron P., Le Bras J., Ramanamirija J. A. and Coulanges P. (1985). Plasmodium falciparum in
Madagascar: in vivo and in vitro sensitivity to seven drugs. Annals of Tropical Medicine
and Parasitology 79, 357–365.
Denis M.B., Tsuyuoka R., Lim P., Lindegardh N., Yi P., Top S.N., Socheat D., Fandeur T.,
Annerberg A., Christophel E.M. and Ringwald P. (2006). Efficacy of artemether-
lumefantrine for the treatment of uncomplicated falciparum malaria in northwest
Cambodia. Tropical Medicine and International Health 11: 1800-1807.
Djaman J., Abouanou S., Basco L. and Kone M. (2004). Limits of the efficacy of chloroquine
and sulfadoxine-pyrimethamine in Northern Abidjan (Cote d'Ivoire): Combined in vivo
and in vitro studies. Sante 14: 205–209.
Dokomajilar C., Nsobya S.L., Greenhouse B., Rosenthal P.J. and Dorsey G. (2006). Selection of
Plasmodium falciparum pfmdr1 alleles following therapy with artemether-lumefantrine in
an area of Uganda where malaria is highly endemic. Antimicrobial Agents and
Chemotherapy 50: 1893–1895.
Dondorp A.M., Nosten F., Yi P., Das D., Phyo A.P., Tarning J., Lwin K.M., Ariey F.,
Hanpithakpong W., Lee S.J., Ringwald P., Silamut K., Imwong M., Chotivanich K., Lim
P., Herdman T., An S.S., Yeung S., Singhasivanon P., Day N.P., Lindegardh N., Socheat
D., White N.J. (2009). Artemisinin resistance in Plasmodium falciparum malaria. The
New England Journal of Medicine 361: 455-467.
Druile P., Moreno A., Blanc C., Brasseur P.H. and Jacquier P. (2001). A colometric in vitro drug
sensitivity assay for Plasmodium based on a highly sensitive double site lactate
dehydrogenase antigen-capture Elisa. The American Journal of Tropical Medicine and
Hygiene 64: 233–241.
Eastman R.T. and Fidock D.A. (2009). Artemisinin-based combination therapies: a vital tool in
efforts to eliminate malaria. Nature Reviews. Microbiology 7: 864–874.

51
Fadat G., Le Bras J., Hengy C., Louis J.P., Gimou M.M. and Verdier F. (1991). Efficacy of
amodiaquine against chloroquine-resistant malaria in Cameroon. Lancet 338: 1092.
Falade C.O., Ogundele A.O., Yusuf B.O., Ademowo O.G. and Ladipo S.M. (2008). High
efficacy of two artemisinin-based combinations (artemether- lumefantrine and artesunate
plus amodiaquine) for acute uncomplicated malaria in Ibadan, Nigeria. Tropical Medicine
and International Health 13: 635–643. (a)
Falade C.O., Ogunkunle O.O., Dada-Adegbola H.O., Falade A.G., Ibarra de Palacios P., Hunt P.,
Virtanen M., Oduola A. M. and Salako L. A. (2008). Evaluation of the efficacy and
safety of artemether-lumefantrine in the treatment of acute uncomplicated Plasmodium
falciparum malaria in Nigerian infants and children. Malaria Journal 7: 246. (b)
Feachem R. and Sabot O. (2008). A new global malaria eradication strategy. Lancet 371: 1633–
1635.
Ferdig M.T., Cooper R.A., Mu J., Deng B., Joy D.A., Su X.Z. and Wellems T.E. (2004).
Dissecting the loci of low-level quinine resistance in malaria parasites. Molecular
Microbiology 52: 985–997.
Fitch C.D. (2004). Ferriprotoporphyrin IX, phospholipids and the antimalarial actions of
quinoline drugs. Life Sciences 74: 1957–1972.
Fungladda, W., Honrado E. R., Thimasarn K., Kitayaporn D., Karbwang J., Kamolratanakul P.
and Masngammueng R. (1998). Compliance with artesunate and quinine + tetracycline
treatment of uncomplicated falciparum malaria in Thailand. Bulleting of the World
Health Organisation 76: Supplementary 1:59-66.
Gama B.E., Almeida de Oliveira N.K., de Souza J.M, Santos F., de Carvalho L.M.J., Melo
Y.F.C., Rosenthal F.P., Daniel-Ribeiro C.T. and Ferreira-da-Cruz M.F. (2010). Brazilian
Plasmodium falciparum isolates:investigation of candidate polymorphisms for
artemisinin resistance before introduction ofartemisinin-based combination therapy.
Malaria Journal 9: 1-5.

52
Gardner M.J., Shallom S.J., Carlton J.M., Salzberg S.L., Nene V., Shoaibi A., Ciecko A., Lynn
J., Rizzo M., Weaver B., Jarrahi B., Brenner M., Parvizi B., Tallon L., Moazzez A.,
Granger D., Fujii C.,Hansen C., Pederson J., Feldblyum T., Peterson J., Suh B. and
Angiuoli S. (2002). Sequence of Plasmodium falciparum chromosomes 2, 10, 11 and 14.
Nature 419: 531-534.
Gari-Toussaint M., Pradines B., Mondain V., Keundjian A., Dellamonica P., Le Fichoux Y.
(2002). Senegal and malaria. True prophylactic failure of mefloquine. Presse Medicale
31: 1136
Gogtay N.J., Kadam V.S, Karnad D.R., Kanbur A., Kamtekar K.D. and Kshirsagar N.A. (2000).
Probable resistance to parenteral artemether in Plasmodium falciparum: case reports from
Mumbai (Bombay), India. Annals of Tropical Medicine and Parasitoogy 94: 519–520.
Greenwood B.M., Bojang K., Whitty C.J. and Targett G.A. (2005). Malaria. Lancet 365: 487–
498.
Gregson A. and Plowe C.V. (2005). Mechanisms of resistance of malaria parasites to antifolates.
Pharmacological Reviews 57: 117–145.
Griffith K.S., Lewis L.S., Mali S. and Parise M.E. (2007). Treatment of malaria in the United
States: a systematic review. JAMA: the journal of american medical association 297:
2264-2277.
Happi C.T., Gbotosho G.O., Folarin O.A., Sowunmi A., Hudson T., O’Neil M., Milhous W.,
Wirth D.F. and Oduola A.M.. (2009). Selection of Plasmodium falciparum multidrug
resistance gene 1 alleles in asexual stages and gametocytes by artemether-lumefantrine in
Nigerian children with uncomplicated falciparum malaria. Antimicrobial Agents and
Chemotherapy 53: 888– 895.
Hatton C.S., Peto T.E. and Bunch C. (1986). Frequency of severe neutropenia associated with
amodiaquine prophylaxis against malaria. Lancet 1: 411– 414.
Hartwig C.L., Rosenthal A.S., D'Angelo J., Griffin C.E., Posner G.H., Cooper R.A. (2009).
Accumulation of artemisinin trioxane derivatives within neutral lipids of Plasmodium

53
falciparum malaria parasites is endoperoxide-dependent. Biochemical Pharmacology 77:
322-336.
Haruki K., Winstanley P. A., Watkins W. M. and Marsh K. (1998). Quinine sensitivity of
isolates of Plasmodium falciparum from the coast of Kenya. Transactions of the Royal
Society of Tropical Medicine and Hygiene 92:195–196.
Hastings I. M., Paget-McNicol S. and Saul A. (2004). Can mutation and selection explain
virulence in human P.falciparum infections? Malaria Journal 3: 2.
Hastings I.M. and Watkins W.M. (2005). Intensity of malaria transmission and the evolution of
drug resistance. Acta Tropica 94: 218–229.
Hay S.I., Rogers D.J., Toomer J.F. and Snow R.W. (2000). Annual Plasmodium falciparum
entomological inoculation rates (EIR) across Africa: literature survey, internet access and
review. Transactions of the Royal Society of Tropical Medicine and Hygiene 94: 113–
127.
Hatabu T., Kawazu S., Kojima S., Singhasivanon P., Krudsood S., Looareesuwan S. and Kano S. (2004). A pilot field trialof an in vitro drug suscpetibility test using the anaeropack malaria culture system on the Thai-Myanmar border. Tropical medicine and Hygiene 32: 335-337.
Hay S.I., Guerra C.A., Gething P.W., Patil A.P., Tatem A.J., Noor A.M., Kabaria C.W., Manh
B.H., Elyazar I.R., Brooker S., Smith D.L., Moyeed R.A. and Snow R.W. (2009). A
world malaria map: Plasmodium falciparum endemicity in 2007. PLoS Medicine 6:
e1000048.
Henry M., Diallo I. and Bordes J., Ka S., Pradines B., Diatta B., M'Baye P.S., Sane M., Thiam
M., Gueye P.M., Wade B., Touze J.E., Debonne J.M., Rogier C. and Fusai T. (2006).
Urban malaria in Dakar, Senegal: chemosusceptibility and genetic diversity of
Plasmodium falciparum isolates. The American Journal of Tropical Medicine and
Hygiene 75: 146–151.
Henry M., Briolant S., Zettor A., Pelleau S., Baragatti M., Baret E., Mosnier J., Amalvict R.,
Fusai T., Rogier C. and Pradines B. (2009). Plasmodium falciparum Na/Hexchanger 1

54
transporter is involved in reduced susceptibility to quinine. Antimicrobial Agents and
Chemotherapy 53: 1926– 1930.
Holmgren G., Bjorkman A., and Gil J. P. (2006). Amodiaquine resistance is not related to rare
findings of pfmdr1 gene amplifications in Kenya. Tropical Medicine and Inernational
Health 11: 1808–1812.
Humphreys G. S., Merinopoulos I., Ahmed J., Whitty C. J., Mutabingwa T. K., Sutherland C. J.
and Hallett R. L. (2007). Amodiaquine and artemether- lumefantrine select distinct alleles
of the Plasmodium falciparum mdr1 gene in Tanzanian children treated for
uncomplicated malaria. Antimicrobial Agents and Chemotherapy 51: 991–997.
Jambou R., Legrand E., Niang M., Khim N., Lim P., Volney B., Ekala M.T., Bouchier C.,
Esterre P., Fandeur T. and Mercereau-Puijalon O. (2005). Resistance of Plasmodium
falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type
PfATPase6. Lancet 366: 1960-1963.
Janeway C., Travers P., Walport M. and Schlomnik M. J. (2005). Immuno Biology: The Immune
System in Health and Disease. 6th Ed. New York: Garland Science Publishing.
Jensen J. B. (2002). In Vitro Culture of Plasmodium Parasites. In D. L. Doolan, Malaria Methods
and Protocols (pp. 477-488). Totowa, NJ: Humana Press, Inc.,.
Johnson J.D., Dennull R.A., Gerena L., Lopez-Sanchez M., Roncal N. E. and Waters N.C.
(2007). Assessment and Continued Validation of the Malaria SYBR Green I-Based
Fluorescence Assay for Use in Malaria Drug Screening. Antimicrobial Agents and
Chemotherapy 51: 1926–1933.
Kaddouri H., Nakache S., Houzé S., Mentré F. and Le Bras J. (2006). Assessment of the Drug
Susceptibility of Plasmodium falciparum Clinical Isolates from Africa by Using a
Plasmodium Lactate DehydrogenaseImmunodetection Assay and an Inhibitory Maximum
Effect Model for Precise Measurement of the 50-Percent Inhibitory Conc. Antimicrobial
Agents and Chemotherapy 33: 3343–3349.

55
Kaddouri H., Djimde A., Dama S., Kodio A., Tekete M., Hubert V., Kone A., Maiga H., Yattara
O., Fofana B., Sidibe B., Sangare C.P., Doumbo O. and Le Bras J. (2008). Baseline in
vitro efficacy of ACT component drugs on Plasmodium falciparum clinical isolates from
Mali. International Journal of Parasitology 38: 791–798.
Kannan R., Kumar K., Sahal D., Kukreti S. and Chauhan V.S. (2005). Reaction of artemisinin
with haemoglobin:implications for antimalarial activity. The Biochemical Journal 385:
409–418.
Karunajeewa H.A., Mueller I., Senn M., Lin E., Law I., Gomorrai P.S., Oa O., Griffin S., Kotab
K., Suano P., Tarongka N., Ura A., Lautu D., Page-Sharp M., Wong R., Salman S., Siba
P., Ilett K. F. and Davis T.M. (2008). A trial of combination antimalarial therapies in
children from Papua New Guinea. The New England Journal of Medicine 359: 2545–
2557.
Kiara S.M., Okombo J., Masseno V., Mwai L., Ochola I., Borrmann S. and Nzila A. (2009). In
Vitro Activity of Antifolate and Polymorphism in Dihydrofolate Reductase of
Plasmodium falciparum Isolates from the Kenyan Coast: Emergence of Parasites with
Ile-164-Leu Mutation. Antimicrobial Agents and Chemotherapy 53: 3793–3798.
Kilimali V. A. (1990). The in vitro response of Plasmodium falciparum to amodiaquine, quinine
and quinidine in Tanga region, Tanzania. East Africa Medical Journal 67: 336–340.
Klayman D.L. (1985). Qinghaosu (artemisinin): an antimalarial drug from China. Science 228:
1049–1055.
Kobbe R., Klein P., Adjei S., Amemasor S., Thompson W. N., Heidemann H., Nielsen M.V.,
Vohwinkel J., Hogan B., Kreuels B., Buhrlen M., Loag W., Ansong D. and May J.
(2008). A randomized trial on effectiveness of artemether-lumefantrine versus artesunate
plus amodiaquine for unsupervised treatment of uncomplicated Plasmodium falciparum
malaria in Ghanaian children. Malaria Journal 7: 261.
Kokwaro G., Mwai L. and Nzila A. (2007). Artemether/lumefantrine in the treatment of
uncomplicated falciparum malaria. Expert Opinion on Pharmacotherapy 8: 75–94.

56
Krishna S., Bustamante L., Haynes R.K. and Staines H. M. (2008). Artemisinins: their growing
importance in medicine. Trends in Pharmacological Sciences 29: 520–527.
Krungkrai S.R. and Yuthavong Y. (1987). The antimalarial action on Plasmodium falciparum of
qinghaosu and artesunate in combination with agents which modulate oxidant stress.
Transactions of the Royal Society of Tropical Medicine and Hygiene 81: 710–714.
Lakshmanan V., Bray P.G., Verdier-Pinard D., Johnson D.J., Horrocks P., Muhle R.A., Alakpa
G.E., Hughes R.H., Ward S.A., Krogstad D.J., Sidhu A.B. and Fidock D.A. (2005). A
critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible
chloroquine resistance. The EMBO Journal 24: 2294–2305.
Le Bras J., Musset L. and Clain J. (2006). Antimalarial drug resistance. Medecine Maladies
Infectieuses 36: 401-405.
Legrand E., Volney B., Meynard J.B., Mercereau-Puijalon O., Esterre P. (2008). In vitro
monitoring of Plasmodium falciparum drug resistance in French Guiana: a synopsis of
continuous assessment from 1994 to 2005. Antimicrobial Agents and Chemotherapy 52:
288–298.
Leonardo K.B. and Pascal R. (2003). In Vitro Activities of Piperaquine and Other 4-
Aminoquinolines against Clinical Isolates of Plasmodium falciparum in Cameroon.
Antimicrobial Agents and Chemotherapy 47: 1391–1394.
Lim I. and Macfadden G.I. (2010). The evolution, metabolism and functions of the apicoplast.
Philosophical Transactions of the Royal Society of london 365: 749-763.
Luxemburger C., Brockman A., Silamut K., Nosten F., van Vugt M., Gimenez F.,
Chongsuphajaisiddhi T. and White N.J. (1998). Two patients with falciparum malaria and
poor in vivo responses to artesunate. Transactions of the Royal Society Tropical Medicine
and Hygiene 92: 668-669.
Mayxay M., Barends M., Brockman A., Jaidee A., Nair S., Sudimack D., Pongvongsa T.,
Phompida S., Phetsouvanh R., Anderson T., White N. J. and Newton P.N. (2007). In vitro
antimalarial drug susceptibility and pfcrt mutation among fresh Plasmodium falciparum

57
isolates from the Lao PDR (Laos). The American Journal of Tropical Medicine and
Hygiene 76: 245–250.
Mbogo C.M., Mwangangi J.M., Nzovu J., Gu W., Yan G., Gunter J.T., Swalm C., Keating
J.,Regens J.L., Shililu J.I., Githure J.I. and Beier J.C. (2003). Spatial and temporal
heterogeneity of Anopheles mosquitoes and Plasmodium falciparum transmission along
the Kenyan coast. The American journal of tropical medicine and hygiene 68: 734-742.
Menard R. (2005). Medicine: knockout malaria vaccine? Nature 433: 113-114.
Miller J.M., Korenromp E.L., Nahlen B.L. and R W.S. (2007). Estimating the number of
insecticidetreatednets required by African households to reach continent-wide malaria
coverage targets. JAMA 297: 2241-2250.
Molinier S., Imbert P., Verrot D., Morillon M., Parzy D. and Touze J.E. (1994). Plasmodium
falciparum malaria: type R1 quinine resistance in East Africa. Presse Medicale 23: 1494.
Murray C. K. and Bennett J.W. (2009). Rapid Diagnosis of Malaria. Interdisciplinary
Perspectives on Infectious Disease 2009: 1- 7.
Muangnoicharoen S., Johnson D. J., Looareesuwan S., Krudsood S. and Ward S. A. (2009). Role
of known molecular markers of resistance in the antimalarial potency of piperaquine and
dihydroartemisinin in vitro. Antimicrobial Agents and Chemotherapy 53: 1362–1366.
Mutanda L. N. (1999). Assessment of drug resistance to the malaria parasite in residents of
Kampala, Uganda. East African Medicine Journal 76: 421–424.
Mwai L., Steven M. K., Abdi A., Lewa P., Anja R., Abdi D., Pete B., Kevin M., Steffen B. and
Alexis N. (2009). In Vitro Activities of Piperaquine, Lumefantrine, and
Dihydroartemisinin in Kenyan Plasmodium falciparum Isolates and Polymorphisms in
pfcrt and pfmdr1. Antimicrobial Agents and Chemotherapy 53: 5069–5073.
Nair, S., Miller B., Barends M., Jaidee A., Patel J., Mayxay M., Newton P., Nosten F., Ferdig M.
T. and Anderson T.J. (2008). Adaptive copy number evolution in malaria parasites. PLoS
Genetics 4:e1000243.

58
Nawaz F., Nsobya S.L., Kiggundu M., Joloba M., Rosenthal P.J. (2009). Selection of parasites
with diminished drug susceptibility by amodiaquine-containing antimalarial regimens in
Uganda. The Journal of Infectious Disease 200: 1650-1657.
Ndong J. M., Atteke C., Aubouy A., Bakary M., Lebibi J. and Deloron P. (2003). In vitro
activity of chloroquine, quinine, mefloquine and halofantrine against Gabonese isolates
of Plasmodium falciparum. Tropical Medicine and International Health 8: 25–29.
Neftel K.A., Woodtly W., Schmid M., Frick P.G. and Fehr J. (1986). Amodiaquine induced
agranulocytosis and liver damage. British Medical Journal 292: 721–723.
Noedl H., Attlmayr B., Wernsdorffer W. H, Kollaritsch H., and Miller S. (2004). A Histidine-
Rich Protein 2−Based Malaria Dug Sensitivity Assay For Field Use. The American
Journalof Tropical Medicine and Hygiene 71: 711–714.
Noedl H., Bronnert J., Yingyuen K., Attlmayr B., Kollaritsch H. and Fukuda M. (2005). Simple
Histidine-Rich Protein 2 Double-Site Sandwich Enzyme-Linked. Antimicrobial Agents
and Chemotherapy 35: 3575–3577.
Noedl H., Se Y., Schaecher K., Smith B.L., Socheat D. and Fukuda M.M. (2008). Evidence of
artemisinin-resistant malaria in western Cambodia. The New England Journal of
Medicine 359: 2619-2620.
Nosten F. and White N.J. (2007). Artemisinin-based combination treatment of falciparum
malaria. The American Journal of Tropical Medicine and Hygiene 77: 181–192.
Nsobya S.L., Kiggundu M., Nanyunja S., Joloba M., Greenhouse B. and Rosenthal P.J. (2010).
In vitro sensitivities of Plasmodium falciparum to different antimalarial drugs in Uganda.
Antimicrobial Agents and Chemotherapy 54: 1200–1206.
Nzila, A. (2006). The past, present and future of antifolates in the treatment of Plasmodium
falciparum infection. The Journal of Antimicrobial Chemotherapy 57: 1043–1054.
Ochong E.O., Broek v.d., Keus K. and Nzila A. (2003). Short report: association between
chloroquine and amodiaquine resistance and allelic variation in the Plasmodium
falciparum multiple drug resistance 1 gene and the chloroquine resistance transporter

59
gene in isolates from the upper Nile in southern Sudan. The American Journal of
Tropical Medicine and Hygiene 69: 184–187.
Ochong E., Nzila A., Kimani S., Kokwaro G., Mutabingwa T., Watkins W., and Marsh K.
(2003). Molecular monitoring of the Leu-164 mutation of dihydrofolate reductase in a
highly sulfadoxine/pyrimethamine-resistant area in Africa. Malaria Journal 2: 46.
Odhiambo R.A. and Odulaja A. (2005). Parasite lactate dehydrogenase assay for the
determination of antimalarial drug susceptibility of Kenyan field isolates. East African
Medical Journal 82: 118–122.
Oduola A.M., Sowunmi A., Milhous W.K., Kyle D.E., Martin R.K., Walker O. and Salako L.A.
(1992). Innate resistance to new antimalarial drugs in Plasmodium falciparum from
Nigeria. Transactions of the Royal Society and Tropical Medicine Hygiene 86: 123-126.
Okombo J., Kiara S. M., Rono J., Mwai L., Pole L., Ohuma E., Borrmann S., Ochola L.I. and
Nzila A. (2010). In Vitro Activities of Quinine and Other Antimalarials and pfnhe
Polymorphisms in Plasmodium Isolates from Kenya. Antimicrobial Agents and
Chemotherapy 54: 3302–3307
Olliaro P. and Mussano P. (2003). Amodiaquine for treating malaria. Cochrane Database System
Review CD000016.
O'Neill P.M., Barton V.E. and Ward S.A. (2010). The molecular mechanism of action of
artemisinin--the debate continues. Molecules 15: 1705-1721.
Parola P., Ranque S., Badiaga S., Niang M., Blin O., Charbit J. J., Delmont J. and Brouqui P.
(2001). Controlled trial of 3-day quinine-clindamycin treatment versus 7-day quinine
treatment for adult travelers with uncomplicated falciparum malaria imported from the
tropics. Antimicrobial Agents and Chemotherapy 45: 932–935.
Parola, P., Pradines B., Simon F., Carlotti M.P., Minodier P., Ranjeva M.P., Badiaga S., Bertaux
L., Delmont J., Morillon M., Silai R., Brouqui P. and Parzy D. (2007). Antimalarial drug
susceptibility and point mutations associated with drug resistance in 248 Plasmodium
falciparum isolates imported from Comoros to Marseille, France in 2004–2006. The
American Journal of Tropical Medicine and Hygiene 77: 431–437.

60
Pasvol G., Newton C. R.,Winstanley P. A., Watkins W. M., Peshu N. M., Were J. B., Marsh K.
and Warrell D. A. (1991). Quinine treatment of severe falciparum malaria in African
children: a randomized comparison of three regimens. The American Journal of Tropical
Medicine and Hygiene 45: 702–713.
Pettinelli F., M.E. Pettinelli M.E., Eldin de P.P., Millet J., Michel D., Brasseur P. and Druilhe P.
(2004). Short report: high prevalence of multidrug resistant Plasmodium falciparum
malaria in the French territory of Mayotte. The American Journal of Tropical Medicine
and Hygiene 70: 635–637.
Pickard A.L., Wongsrichanalai C., Purfield A., Kamwendo D., Emery K., Zalewski C.,
Kawamoto F., Miller R.S., Meshnick S.R. (2003). Resistance to antimalarials in
Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrobial Agents and
Chemotherapy 47: 2418-2423.
Pradines B., Tall A., Fusai T., Spiegel A., Hienne R., Rogier C., Trape J. F., Le Bras J. and Parzy
D. (1999). In vitro activities of benflumetol against 158 Senegalese isolates of
Plasmodium falciparum in comparison with those of standard antimalarial drugs.
Antimicrobial Agents and Chemotherapy 43: 418–420.
Pradines B., Hovette P., Fusai T., Atanda H.L., Baret E., Cheval P., Mosnier J., Callec A., Cren
J., Amalvict R., Gardair J.P. and Rogier C. (2006). Prevalence of in vitro resistance to
eleven standard or new antimalarial drugs among Plasmodium falciparum isolates from
Pointe-Noire, Republic of the Congo. Journal of Clinical Microbiology 44: 2404–2408.
Pradines B., Briolant S., Henry M., Oeuvray C., Baret E., Amalvict R.,Didillon E., and Rogier C.
(2010). Absence of association between pyronaridine in vitro responses and
polymorphisms in genes involved in quinoline resistance in Plasmodium falciparum.
Malaria Journal 9: 339.
Price R.N., Uhlemann A.C., Brockman A., McGready R., Ashley E., Phaipun L., Patel R., Laing
K., Looareesuwan S., White N.J., Nosten F. and Krishna S. (2004). Mefloquine resistance
in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364: 438–
447.

61
Price, R.N., Uhlemann A.C., van Vugt M., Brockman A., Hutagalung R., Nair S., Nash D.,
Singhasivanon P., Anderson T.J., Krishna S., White N.J. and Nosten F. (2006). Molecular
and pharmacological determinants of the therapeutic response to artemether-lumefantrine
in multidrug-resistant Plasmodium falciparum malaria. Clinical Infectious Disease 42:
1570–1577.
Pukrittayakamee S., Supanaranond W., Looareesuwan S., Vanijanonta S. and White N.J. (1994).
Quinine in severe falciparum malaria: evidence of declining efficacy in Thailand.
Transactions of the Royal Society Tropical Medicine and Hygiene 88: 324–327.
Pukrittayakamee S., Viravan C., Charoenlarp P.,Yeamput C., Wilson R.J. and White N.J.
(1994). Antimalarial effects of rifampin in Plasmodium vivax malaria. Antimicrobial
Agentsand Chemotherapy 38: 511-514.
Pukrittayakamee S., Chantra A., Vanijanonta S., Clemens R., Looareesuwan S. and White N.J.
(2000). Therapeutic responses to quinine and clindamycin in multidrug-resistant
falciparum malaria. Antimicrobial Agents and Chemotherapy 44: 2395–2398.
Quashie N.B., Duah N.O., Abuaku B. and Koram K.A. (2007). The in-vitro susceptibilities of
Ghanaian Plasmodium falciparum to antimalarial drugs. Annals of Tropical Medicine and
Parasitology 101: 391–398.
Randrianarivelojosia M., Randrianasolo L., Randriamanantena A., Ratsimbasoa A., Rakotoson
J.D. (2004). Susceptibility of Plasmodium falciparum to the drugs used to treat severe
malaria (quinine) and to prevent malaria (mefloquine, cycloguanil) in Comoros Union
and Madagascar. South African Medical Journal 94: 47–51.
Raj D.K., Mu J., Jiang H., Kabat J., Singh S., Sullivan M., Fay M.P., McCutchan T.F. and Su
X.Z. (2009). Disruption of a Plasmodium falciparum multidrug resistance-associated
protein (PfMRP) alters its fitness and transport of antimalarial drugs and glutathione.
Journal of Biological Chemistry 284: 7687-7696.
Randrianarivelojosia M., Raharimalala L.A., Randrianasolo L., Ratsimbasoa A., Rason M.A.,
Ariey F., Jambou R. (2001). Madagascan isolates of Plasmodium falciparum showing

62
low sensitivity to artemether in vitro. Annals of Tropical Medicine and Parasitology 95:
237-43.
Randrianarivelojosia M., Randrianasolo L., Randremanana R. V., Randriamanantena A., Ratsimbasoa A., Rakotoson J.D. (2004). Susceptibility of Plasmodium falciparum to the drugs used to treat severe malaria (quinine) and to prevent malaria (mefloquine, cycloguanil) in Comoros Union and Madagascar. South Africa Medical Journal 94: 47-51.
Reed M.B., Saliba K.J, Caruana S.R., Kirk K., Cowman A.F. (2000). Pgh1 modulates sensitivity
and resistance to multiple antimalarials in Plasmodium falciparum. Nature 403: 906–909.
Roche J., Guerra-Neira A., Raso J. and Benito A. (2003). Surveillance of in vivo resistance of
Plasmodium falciparum to antimalarial drugs from 1992 to 1999 in Malabo (Equatorial
Guinea). The American Journal of Tropical Medicine and Hygiene 68: 598–601.
Ro D.K., Paradise E.M., Ouellet M., Fisher K.J., Newman K.L., Ndungu J.M., Ho K.A., Eachus
R.A., Ham T.S., Kirby J., Chang M.C., Withers S.T., Shiba Y., Sarpong R. and Keasling
J.D. (2006). Production of the antimalarial drug precursor artemisinic acid in engineered
yeast. Nature 440: 940-943.
Rogers W.O., Sem R., Tero T., Chim P., Lim P., Muth S., Socheat D., Ariey F. and
Wongsrichanalai C. (2009). Failure of artesunate-mefloquine combination therapy for
uncomplicated Plasmodium falciparum malaria in southern Cambodia. Malaria Journal
8: 10.
Roos D.S., Crawford M. J., Donald R. G. K, Fraunholz M., Harb O. S., He C. Y., Kissinger J. C.,
Shaw M. K. and Striepen B. (2002). Mining the Plasmodium genome database to define
organellar function: what does the apicoplast do? Phillosophical Transactions of the
Royal Society Biological Science 357: 35 - 46.
Roper C., Pearce R., Nair S., Sharp B., Nosten F. and Anderson T. (2004). Intercontinental
spread of pyrimethamine-resistant malaria. Science 305: 1124.

63
Sahr F., Willoughby V.R., Gbakima A.A. and Bockarie M.J. (2001). Apparent drug failure
following artesunate treatment of Plasmodium falciparum malaria in Freetown, Sierra
Leone: four case reports. Annals of Tropical Medicine and Parasitology 95: 445–449.
Sasi P., Abdulrahaman A, Mwai L., Muriithi S., Straimer J., Schieck E., Rippert A., Bashraheil
M., Salim A., Peshu J., Awuondo K., Lowe B., Pirmohamed M., Winstanley P., Ward S.,
Nzila A. and Borrmann S. (2009). In Vivo and In Vitro Efficacy of Amodiaquine against
Plasmodium falciparum in an Area of Continued Use of 4-Aminoquinolines in East
Africa. The Journal of Infectious Diease 199: 1575 – 1582.
Schlitzer M. (2008). Antimalarial Drugs – What is in Use and What is in the Pipeline. Archives
de pharmacy 341: 149 – 163.
Shretta R., Omumbo J., Rapuoda B. and Snow R.W. (2000). Using evidence to change
antimalarial drug policy in Kenya. Tropical Medicine and International Health 5: 755–
64.
Sidhu A.B., Verdier-Pinard D. and Fidock D.A. (2002). Chloroquine resistance in Plasmodium
falciparum malaria parasites conferred by pfcrt mutations. Science 298: 210–213.
Sidhu A.B., Valderramos S.G. and D.A. Fidock. (2005). pfmdr1 mutations contribute to quinine
resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum.
Molecular Microbiology 57: 913–926.
Sidhu A.B., Uhlemann A.C., Valderramos S.G., Valderramos J.C., Krishna S. and Fidock D.A.
(2006). Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens
susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. Journal
of Infectious Disease 194: 528-535.
Sisowath C., Strömberg J., Mårtensson A., Msellem M., Obondo C., Björkman A., Gil J.P.
(2005). In vivo selection of Plasmodium falciparum pfmdr1 86N coding alleles by
artemether-lumefantrine (Coartem). Journal of Infectious Disease 191: 1014-1017.
Sisowath C., Ferreira P. E., Bustamante L. Y., Dahlstrom S., Martensson A., Bjorkman A.,
Krishna S., and Gil J. P. (2007). The role of pfmdr1 in Plasmodium falciparum tolerance

64
to artemether-lumefantrine in Africa. Tropical Medicine and International Health 12:
736–742.
Smilkstein M., Sriwilaijaroen N, Kelly J.X., Wilairat P. and Riscoe M. (2004). Simple and
Inexpensive Fluorescence-Based Technique for High-Throughput Antimalarial Drug
Screening. Antimicrobial Agents and Chemotherapy 48: 1803–1806.
Snounou G. and Renia L. (2007). The vaccine is dead--long live the vaccine. Trends in
Parasitology 23: 129-132.
Snow R.W, Guerra C.A, Noor A.M, Myint H.Y and Hay S.I. (2005). The global distribution of
clinical episodes of Plasmodium falciparum malaria. Nature 434: 214–217.
Sompob S., Wirichada P., Richard J. M, Sue J. L., Joel T., Niklas L., Kesinee C., François N.,
Nicholas P. J. D., Duong S., Nicholas J. W., Arjen M. D., and Lisa J. W. (2011). Intrahost
modeling of artemisinin resistance in Plasmodium falciparum. PNAS. 108: 397–402.
Spalding M.D., Eyase F.L., Akala H.M., Bedno S.A., Prigge S.T, Coldren R.L, Moss W.J. and
Waters N.C. (2010). Increased prevalence of the pfdhfr/phdhps quintuple mutant and
rapid emergence of pfdhps resistance mutations at codons 581 and 613 in Kisumu,
Kenya. Malaria Journal 9: 338
Straimer J. (2009). Studies on evolution of drug resistance in P. falciparum malaria in vivo and
in vitro. Doctor of natural sciences thesis, Heidelberg University, Germany 5-6.
Tärning J. (2007). Piperaquine; Bioanalysis, drug metabolism and pharmacokinetics. Göteborg,
Sweden: ISBN. 11-13
Taubes G. (2000). Searching for a parasite’s weak spot. Science 290: 434-437.
Thanh N.X., Trung T.N., Phong N.C., Thien N.X., Dai B., Shanks G.D., Chavchich M. and
Edstein M.D. (2009). Open label randomized comparison of dihydroartemisinin-
piperaquine and artesunate-amodiaquine for the treatment of uncomplicated Plasmodium
falciparum malaria in central Vietnam. Tropical Medicine and International Health 14:
504–511.

65
Temperley M., Mueller D.H., Njagi J.K., Akhwale W., Clarke S.E., Jukes M.C., Estambale B.B.,
and Brooker S. (2008). Costs and cost-effectiveness of delivering intermittent preventive
treatment through schools in western Kenya. Malaria Journal 7:196.
Tinto H., Ouedraogo J.B., Traore B., Coulibaly S.O., Guiguemde T.R. (2001). In vitro
susceptibility of 232 isolates of Plasmodium falciparum to antimalarials in Burkina Faso
(West Africa). Bulleting de la Societe de Pathologie Exotique. 94: 188–191
Tinto, H., C. Rwagacondo, C. Karema, D. Mupfasoni, W. Vandoren, E. Rusanganwa, A. Erhart,
C. Van Overmeir, E. Van Marck, and U. D’Alessandro. (2006). In-vitro susceptibility of
Plasmodium falciparum to monodesethylamodiaquine, dihydroartemisinin and quinine in
an area of high chloroquine resistance in Rwanda. Transactions of the Royal Society
Tropical Medicine and Hygiene 100: 509 –514.
Touré A.O., Koné L.P., Jambou R., Konan T.D., Demba S., Beugre G.E. and Koné M. (2008). In
vitro susceptibility of P. falciparum isolates from Abidjan (Côte d'Ivoire) to quinine,
artesunate and chloroquine. Sante. 18: 43-47.
Tripathi R.P., Mishra R.C., Dwivedi N., Tewari N. and Verma S.S. (2005). Current Status of
Malaria Control. Current Medicinal Chemistry 12: 2643-2659.
Tritten T., Matile T., Brun R. and Wittlin S. (2009). A new double-antibody sandwich ELISA
targeting Plasmodium. Malaria Journal 8: 226.
Ursing, J., Kofoed P. E., Rombo L. and Gil J. P. (2006). No pfmdr1 amplifications in samples
from Guinea-Bissau and Liberia collected between 1981 and 2004. Journal of Infectious
Disease 194: 716–718.
Valderramos S.G. and Fidock D. (2006). Transporters involved in resistance to antimalarial
drugs. Trends in Pharmacological Scence 27: 594–601.
Van Dooren G.G., Su V., D'Ombrain M.C., McFadden G.I. (2002). Processing of an Apicoplast
Leader Sequence in Plasmodium falciparum and the Identification of a Putative Leader
Cleavage Enzyme. Journal of Biological Chemistry 277: 23612-23619.

66
Vijaykadga S., Rojanawatsirivej C., Cholpol S., Phoungmanee D., Nakavej A. and
Wongsrichanalai C. (2006). In vivo sensitivity monitoring of mefloquine monotherapy
and artesunate-mefloquine combinations for the treatment of uncomplicated falciparum
malaria in Thailand in 2003. Tropical Medicine and International Health 11: 211-219.
Vinayak, S., M. T. Alam, M. Upadhyay, M. K. Das, V. Dev, N. Singh, A. P. Dash, and Y. D.
Sharma. (2007). Extensive genetic diversity in the Plasmodium falciparum Na/H
exchanger 1 transporter protein implicated in quinine resistance. Antimicrobial Agents
and Chemotherapy 51: 4508–4511.
Watkins W.M., Mberu E.K., Winstanley P.A. and Plowe C.V. (1997). The efficacy of antifolate
antimalarial combinations in Africa: a predictive model based on pharmacodynamic and
pharmacokinetic analyses. Parasitology Today 13: 459 – 464.
Wellems T.E. and Plowe C.V. (2001). Chloroquine-resistant malaria. Journal of Infectious
Disease 184: 770–776.
Wernsdorfer W.H. and Mc Gregor I. (1988). Malaria: Principles and Practice of Malariology .
London: Churchill Livingstone.
Watkins W. M., Howells R. E., Brandling-Bennett A. D., and Koech D. K. (1987). In vitro
susceptibility of Plasmodium falciparum isolates from Jilore, Kenya, to antimalarial
drugs. The American Journal of Tropical Medicine and Hygiene 37: 445–451.
White N. (1999). Antimalarial drug resistance and combination chemotherapy. Philosophical
Transactions of the Royal Societyof London 354: 739–749.
White N.J. (2001). Preventing antimalarial drug resistance through combinations. Drug
Resistance Updates 1: 3-9.
White N.J., Pongtavornpinyo W. (2003). The de novo selection of drug-resistant malaria
parasites. Proceedings of the Biological Sciences 270: 545–554.
White N.J. (2008). Qinghaosu (artemisinin): the price of success. Science 320: 330–334.
WHO. (2005). World malaria report 2005. Roll Back Malaria, World Health Organization.

67
Wichmann, O., T. Jelinek, G. Peyerl-Hoffmann, N. Muhlberger, M. P. Grobusch, J. Gascon, A.
Matteelli, C. Hatz, H. Laferl, M. Schulze, G. Burchard, S. da Cunha, J. Beran, P.
McWhinney, H. Kollaritsch, P. Kern, J. Cuadros, M. Alifrangis, and I. Gjorup. (2003).
Molecular surveillance of the antifolateresistant mutation I164L in imported African
isolates of Plasmodium falciparum in Europe: sentinel data from TropNetEurop. Malaria
Journal 2: 17.
Wilairatana, P., D. E. Kyle, S. Looareesuwan, K. Chinwongprom, S. Amradee, N. J. White, and
W. M. Watkins. (1997). Poor efficacy of antimalarial biguanide-dapsone combinations in
the treatment of acute, uncomplicated, falciparum malaria in Thailand. Annals of Tropical
Medicine and Parasitology 91: 125–132.
Witkowski B., Lelie`vre J., Barraga´n M.J.L., Laurent V., Su X., Berry A.and Benoit-Vical F.
(2010). Increased Tolerance to Artemisinin in Plasmodium falciparum Is Mediated by a
Quiescence Mechanism. Antimicrobial Agents and Chemotherapy 54: 1872–1877.
Wongsrichanalai C., Wimonwattrawatee T. , Sookto P., Laoboonchai A., Heppner D.G., Kyle
D.E. and Wernsdorfer W.H. (1999). In vitro sensitivity of Plasmodium falciparum to
artesunate in Thailand. Bulleting of the World Health Organisation 77: 392-398.
Wongsrichanalai C., Pickard A.L., Wernsdorfer W.H. and Meshnick S.R. (2002). Epidemiology
of drug-resistant malaria. Lancet 2: 209–218.
Woodrow C.J., Haynes R.K. and Krishna S. (2005). Artemisinins. Postgraduate Medical Journal
81: 71–78.
Wootton J.C., Feng X., Ferdig M.T., Cooper R.A., Mu J., Baruch D.I., Magill A.J. and Su X.Z.
(2002). Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum.
Nature 418: 320-323.
World Health Organization (WHO). (2000). WHO Expert Committee on Malaria, twentieth
report. 30. Geneva: WHO.
World Health Organization (WHO). (2007). Antimalarial treatment policies. Vol 2008. Geneva:
WHO.

68
Wu Y. (2002). How might qinghaosu (artemisinin) and related compounds kill the
intraerythrocytic malaria parasite? A chemist’s view. Accounts of Chemical Research 35:
255–259.
Yang H., Liu D., Yang Y., Fan B., Yang P., Li X., Li C., Dong Y. and Yang C. (2003). Changes
in susceptibility of Plasmodium falciparum to artesunate in vitro in Yunnan Province,
China. Transactions of the Royal Society Tropical Medicine and Hygiene 97: 226-228.
Yavo W., K.B. Bla, A.J. Djaman, S.B. Assi, L.K. Basco, A. Mazabraud, and M. Koné. (2010). In
vitro susceptibility of Plasmodium falciparum to monodesethylamodiaquine, quinine,
mefloquine and halofantrine in Abidjan (Côte d'Ivoire). African Health Sciences 10: 111–
116.
Yeka A., Dorsey G., Kamya M.R., Talisuna A., Lugemwa M., Rwakimari J.B., Staedke S.G.,
Rosenthal P. J., Wabwire-Mangen F. and Bukirwa H. (2008). Artemether-lumefantrine
versus dihydroartemisinin-piperaquine for treating uncomplicated malaria: a randomized
trial to guide policy in Uganda. PLoS One 3: e2390.
Zurovac D., Njogu J., Akhwale W., Hamer D.H., Snow R.W. (2008). Translation of artemether-
lumefantrine treatment policy into paediatric clinical practice: an early experience from
Kenya. Tropical Medicine and International Health 13: 99–107.