studies on a novel powder formulation for nasal...
TRANSCRIPT

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2008
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Pharmacy 81
Studies on a Novel PowderFormulation for Nasal DrugDelivery
NELLY FRANSÉN
ISSN 1651-6192ISBN 978-91-554-7288-7urn:nbn:se:uu:diva-9292

���������� �������� �� ������ �������� � �� �������� ������� � ���� ������������� �� ������� ������� ����� !�� !""# �� "$%�& '� �(� ������ ' ���� ')(����(� *������� ' )(������+, -(� �������� .��� �� ������� � /����(,
��������
����0� 1, !""#, 2������ � 1��� ).��� �������� '� 1���� ���� ��������, 3��� ����������� ���������, ������� ��� � ���� ����� � � ������� ���� ������� ���� ������� � ������� #�, 4" ��, ������, 52�1 $4#6$�6&&�64!##64,
1���� ����������� (�� ������� '� �(� �������� ' �������� ��7����� � '��� ��� '�''��� � '� ����� .��( �. ��� ��������������, 8�7��� ���� ������ ��� ���������� ������� �� �� ��������� .��( ��������� ������� '�� �(� ���� ������9 �(�� �(���� �(.� �(������ ������� �� �� ����'������ �(���� .��( � ��� �.��� '�������,5� .�� �(. �(�� ���������� ��������� ������� ' '�� ���� ��������� ��(���� � �(�
���'��� ' �����(����� ������� ���������� ���� �� ������� � � �������� ��:� �������� '� ���������������, 2���� �����( �������� *22;+� � ��� ������ ��������� .�� ���� ��������� ��������, �� ���� �������� ' �(� '������� �������� �(�� �(� �����(��� ' �(�������� .�� ���<��� � �� �''����� �� �(� ������ ' � ����, -(� �.��� '������� ��� ������� �(� �� ���� ����'�� ' ��(����������� ����� ����� ���� ����� �������.��( � ��7��� '�������9 (.����� �(� ������� .��� ��������� .��( ���(��������(�������, -(� ����� ' ���� ��������� � 22; �� �(��� �(�� ��������� .�����������, 5 ���(��� ��������� .��� '� ������� �������� ���.�� 22; �� ������������ ��� �(��� ��������� .��� ��� �������� �� �. ���� ��������� �� �(������<��� �''��� �� ��� �������������� �� �(��������� ���� ���������, 3������ ' �(�������� ���� ���������� '�� �(� 22; ���� '�������� '�� � ��� ��������� ������'������� �� '�� � ��������� ���� ��7��� ����� .�� ��������� � � ������� �����, =(��� ��������� ��� �(� ��7��� ����� .�� ��� .��( �(� ��������� ������� ��������������� �'��� �(� ���� �.��� '������� .��� �(��� ����� (��(�� �(� �(�� �'��� �(���7��� �����, 3�� '�������� .��� .��� �������� �� �(� ��������, -(� ��� ' ����������������� �����(����� ������� ��������� � ���������� �������� ''��� ������� '� � �.���(� ' ������� ���� �.��� '�������� �(�� �(��� ��� �� ���������� � ����� ������������,
� ������ ���� ���� ��������� ���� �.��� ������ ���������� �������� �����(��������� �����( ��������� ���������������� ��������������� ������� �����
� ��� ����� �! � ���� �� � �������! "# $%&! ������� ���� �����! �'()$*+, �������!�� � �
> 1���� ����0 !""#
5221 �?&�6?�$!52�1 $4#6$�6&&�64!##64��%�%��%��%����6$!$! *(���%@@��,<�,��@������A��B��%�%��%��%����6$!$!+

”Det vill gärna bli bra det jag gör.”
Morfar Tage Ryman


Papers discussed
This thesis is based on the following papers, which will be referred to by the Roman numerals assigned below:
I Fransén, N., Björk, E. and Nyström, C., Development and characterisation of interactive mixtures with a fine-particulate mucoadhesive carrier for nasal drug delivery. European Journal of Pharmaceutics and Biopharmaceutics, 2007, 67 (2): 370-376. Reproduced with permission ©2007 Elsevier
II Fransén, N., Espefält Westin, U., Nyström, C. and Björk, E., The in vitro transport of dihydroergotamine across por-cine nasal respiratory and olfactory mucosa and the effect of a novel powder formulation. Journal of Drug Delivery Science and Technology, 2007, 17(4): 267-271. Issue devoted to
Uppsala University, Department of Pharmacy. Reproduced with permission
©2007 Editions de Santé
III Fransén, N., Björk, E. and Edsman, K., Changes in the mu-coadhesion of powder formulations after drug application investigated with a simplified method. Journal of Pharma-ceutical Sciences, 2008, 97 (9): 3855-3864. Reprinted with per-
mission ©2008 Wiley-Liss, Inc. a subsidiary of John Wiley & Sons, Inc.
IV Fransén, N., Morin, M., Björk, E. and Edsman, K., Physico-chemical interactions between drugs and superdisinte-grants. Accepted for publication in the Journal of Pharmacy and Pharmacology.
V Fransén, N., Bredenberg, S. and Björk E., Clinical study shows improved absorption of desmopressin with novel formulation. Submitted.
Further publications co-authored by Nelly Fransén can be found on the fol-lowing page.

Additional papers:
• Jansson, B., Hägerström, H., Fransén, N., Edsman, K. and Björk, E., The influence of gellan gum on the transfer of fluo-rescein dextran across rat nasal epithelium in vivo. European Journal of Pharmaceutics and Biopharmaceutics, 2005, 54 (3): 557-564.
• Mihranyan, A., Frenning, G., Fransén, N., Welch, K. and Strømme, M., Order and disorder in powder mixtures: Spatial distribution functions as tools to assess powder homogeneity. Particle & Particle Systems Characterization, In press.
• Ljungberg, C., Fransén, N., Björnsson, M. and Kettis Lind-blad, Å., The importance of the dosage form – a qualitative study of migraine patients. In manuscript.
Patent
• Nyström C., Fransén N. and Björk, E., Pharmaceutical com-positions useful in the transmucosal administration of drugs. European Patent WO2006GB00481

Contents
1. Introduction to nasal powder sprays ..............................................11 1.1. Physiological aspects ..............................................................11 1.2. Mucoadhesion ..........................................................................13 1.3. Interactive mixtures .................................................................14
1.3.1. Definitions.............................................................................14 1.3.2. Formation .............................................................................15 1.3.3. Characterisation ..................................................................15 1.3.4. Applications..........................................................................16
1.4. Superdisintegrants...................................................................17 1.5. Nasal drug delivery..................................................................19
1.5.1. Drug candidates ..................................................................19 1.5.2. Olfactory transfer.................................................................21 1.5.3. Comparisons with sublingual administration...................21 1.5.4. Powders or liquids?.............................................................22 1.5.5. Important characteristics for nasal powders ...................23
1.6. Assessing the efficacy of a powder formulation..................25 1.6.1. Mucoadhesion in vitro.........................................................25 1.6.2. Uptake in vitro ......................................................................26
2. Aims of the thesis..............................................................................27
3. Description of essential methods....................................................28 3.1. Powder materials .....................................................................28
3.1.1. Obtaining the required particle size..................................28 3.1.2. Determining the particle size .............................................28 3.1.3. Determining the ability of the particles to swell ..............29
3.2. Interactive mixtures .................................................................29 3.2.1. Theoretical models..............................................................29 3.2.2. Practical methods................................................................30
3.3. In vitro uptake ...........................................................................31 3.3.1. Porcine nasal respiratory and olfactory mucosa ............31 3.3.2. Horizontal Ussing chamber measurements ....................32
3.4. Mucoadhesion ..........................................................................33 3.4.1. Test materials ......................................................................33 3.4.2. Tensile strength measurements .......................................33 3.4.3. Development of a mucosal substitute ..............................34

3.5. Physicochemical interactions.................................................35 3.5.1. Test materials ......................................................................35 3.5.2. Binding study .......................................................................35 3.5.3. Release study ......................................................................36
3.6. Clinical study ............................................................................37 3.6.1. Study basics.........................................................................37 3.6.2. Administration of the study drugs .....................................37 3.6.3. Evaluation of the volunteers’ views ..................................38
3.7. Statistical analysis ...................................................................38
4. Key findings .......................................................................................40 4.1. Interactive mixtures with small carrier particles ..................40
4.1.1. Characterisation of mixtures in Papers I-III .....................40 4.2. In vitro absorption of dihydroergotamine..............................42
4.2.1. The porcine olfactory mucosa ...........................................42 4.2.2. The effect of the powder formulation ...............................43 4.2.3. Differences in mucosal transfer ........................................44
4.3. Mucoadhesion measurements...............................................45 4.3.1. The simplified method ........................................................45 4.3.2. The effect of surface coverage .........................................47
4.4. Investigation of physicochemical interactions .....................48 4.4.1. Binding and release at different salt concentrations......49 4.4.2. In vivo correlations and effect in vitro...............................51
4.5. Clinical trial................................................................................52 4.5.1. In vivo absorption of desmopressin..................................53 4.5.2. Volunteers’ opinions ...........................................................55
5. Concluding remarks..........................................................................56
6. Future outlook....................................................................................57
7. Populärvetenskaplig sammanfattning ............................................59
8. Acknowledgements...........................................................................60
9. References.........................................................................................62

Abbreviations
ANOVA Analysis of variance AUC Area under the plasma concentration-time curve BBB Blood-brain barrier cac Critical aggregation concentration CCS Croscarmellose sodium clogP Calculated octanol-water coefficient Cmax Maximum plasma concentration cmc Critical micelle concentration CPC Cetylpyridinium chloride CPP Crospovidone CV Coefficient of variation Dapp Apparent diffusion coefficient DHE Dihydroergotamine FPF Fine particle fraction GLM General linear model HPLC High performance liquid chromatography IM Interactive mixture I.N. Intranasal Isc Short circuit current KRB Krebs Ringer Bicarbonate buffer MW Molecular weight Papp Apparent permeability coefficient PBS Phosphate buffered saline PD Mucosal resting potential difference pKa Acid dissociation constant PPS Partly pregelatinised maize starch RH Relative humidity RM Random mixture Rs Surface area ratio (degree of surface area coverage) S.D. Standard deviation SEM Scanning electron microscopy S.L. Sublingual SPC Summary of product characteristics SSG Sodium Starch Glycolate TB Tris buffer tmax Time to reach maximum plasma concentration


11
1. Introduction to nasal powder sprays
Why nasal administration and why, if you are not a drug addict, would you want to have powder up your nose? This may seem a good question and in the end it is of course a matter of taste –perhaps literally, as will be discussed later. This thesis concerns the nasal administration of systemically active drugs and the development of a nasal powder formulation with the objective of improving drug delivery. Nasal powders can also be used for local treat-ment of nasal congestion or allergic rhinitis; the only nasal powder formula-tion currently sold in Swedish pharmacies, Rhinocort® Turbuhaler®, is in fact a corticosteroid for the treatment of rhinitis. The nasal formulations intended for systemic effect are all liquid sprays; we have, for example, the antimi-graine drugs Imigran® and Zomig® and peptide hormone analogues such as oxytocin and desmopressin. The reasons for administering these drugs via the nasal route will be examined later, as will the possibilities of improving administration by using a powder spray. However, to facilitate these discus-sions, some fundamental concepts are introduced first.
1.1. Physiological aspects The main functions of the nose are olfaction and the conditioning of inspired air. The physiology of the nose is well suited for these purposes; the nasal cavity contains turbinates comprising a surface area of 150-180 cm2 but al-lowing only a narrow pathway for the inspired air [1] (Figure 1). Inhaled particles larger than 10 μm are thus efficiently kept in the nose [2] at the same time as the air is heated and moistened.
Three types of mucosa cover the surface of the nasal cavity. The stratified squamous epithelium is found in close proximity to the nostrils and gradu-ally transforms into a pseudostratified columnar epithelium, which covers most of the nasal cavity. The olfactory epithelium is situated in the upper posterior part of the nasal cavity (Figure 1) and covers approximately 10 cm2 of the human nasal cavity; in comparison, the olfactory mucosa in rodents constitutes about 50% of the nasal cavity [3].
The pseudostratified columnar epithelium of the respiratory nasal mucosa consists of a single layer of four main cell types: ciliated and nonciliated columnar cells, basal cells and goblet cells (Figure 1). The cells are covered by microvilli. Many, especially those in the posterior half of the nasal cavity,

Introduction to nasal powder sprays
12
also have approximately 200 5 μm long cilia [1, 4], whose synchronised movements enable mucociliary clearance of unwanted particles from the nose. The epithelial cells are covered by a layer of mucus, which is thought to consist of two distinct layers, each approximately 5 μm in depth [5, 6]. The cilia move in the low viscosity layer and as they project into the upper gel layer they push the mucus back to the nasopharynx at a speed of 3-25 mm/min [1]. The mucus, produced by the submucosal glands and goblet cells, is composed of >90% water, 0.5-5% mucins (mucous glycoproteins), 1-2% salts and 0.5-1% free proteins [2, 7]. The mucosa is slightly acidic (pH 5.5-6.5), which is thought to be important for its antibacterial properties [2].
The epithelial cells are secured in the basement membrane, a layer of col-lagen fibrils. The submucosa, which is highly vascularised and thus plays an important role in the systemic absorption of drugs, is situated under the basement membrane. The passive absorption of large, hydrophilic molecules is likely achieved paracellularly, as opposed to more lipophilic molecules which may diffuse through the cells. Paracellular absorption is limited by the tight junctions connecting the epithelial cells on the apical side (Figure 1). The nasal mucosa is relatively permeable to drug molecules; the extent of absorption is lower than that from the lung [8] but in the same order as that from the small intestine [8, 9]. Absorption has been shown to decrease sig-nificantly with size for hydrophilic molecules weighing more than 300 Da [9] and is in general restricted for molecules weighing more than 1 000 Da [9, 10]. Absorption across the nasal mucosa can be further restricted by en-zymatic degradation. The metabolic capacity is considered to be rather high [11], especially in the olfactory mucosa [11, 12]. It may be of consequence for the delivery of peptides and proteins, but fast absorption and high local concentration of the compound will decrease the risk of metabolism [11].
Figure 1. A sagittal view of the human nasal cavity and a schematic drawing of the nasal respiratory epithelium with mucus. The specific elements shown are: The approximate place of the olfactory epithelium (Olf), the superior turbinate (ST), middle turbinate (MT) and inferior turbinate (IT) and to the right a ciliated columnar cell (CC), basal cell (BC), goblet cell (GC), nonciliated columnar cell (NC), tight junctions (TJ) and mucus layer (ML). A comparison can be made with the frontal section of the porcine snout shown in Figure 13.

Introduction to nasal powder sprays
13
1.2. Mucoadhesion When administering drugs to mucosal tissues, such as in the nasal cavity, it is helpful if the formulation stays in the right place long enough for the drug to be absorbed across the mucosa. It may, therefore, be beneficial to use a mucoadhesive agent in the formulation to achieve sufficient residence time.
Mucoadhesion, as the word suggests, refers to adhesion of matter to a mucus layer for an extended period of time [13]. A mucoadhesive agent is thus a substance that adheres to mucus. The term bioadhesion is less specific and can be used to denote adhesion to any biological surface [13-15].
Mucoadhesive agents are usually polymers containing hydrogen bonding groups that can be used in wet formulations or, as in this thesis, in dry pow-ders. The mechanisms behind mucoadhesion have not yet been fully eluci-dated, but a theory that has stuck is that close contact must first be estab-lished between the mucoadhesive agent and the mucus, followed by inter-penetration of the mucoadhesive polymer and the mucin and finishing with the formation of entanglements and chemical bonds between the macro-molecules [14]. In the case of a dry polymer powder, the initial adhesion is most likely achieved by water movement from the mucosa into the formula-tion [15], which has also been shown to lead to dehydration and strengthen-ing of the mucus layer [16]. The subsequent formation of van der Waals, hydrogen and, in the case of a positively charged polymer, electrostatic bonds between the mucins and the hydrated polymer promotes prolonged adhesion.
A predicament with trying to increase the contact time by adhesion to mucus is that the residence time of the mucus itself is limited by mucociliary clearance. The normal transit time of a particle deposited on top of the nasal mucus layer is approximately 12-15 min [17]. However, dehydration of the mucus layer on contact with the mucoadhesive powder will increase the mucus viscosity and subsequently decelerate its clearance [18, 19]. Dry powder formulations should hence be especially well suited for nasal ad-ministration as increased mucus viscosity would lower the normal require-ments for the formation of secondary chemical bonds in order to prolong residence time.

Introduction to nasal powder sprays
14
1.3. Interactive mixtures The concept of an “interactive mixture” is central to this thesis. Before con-tinuing with further discussions about nasal administration, we will therefore linger a while in the literally dry world of powder technology.
1.3.1. Definitions A powder is an assembly of particles dispersed in air. The diameter of the powder particles is generally below 1 000 μm, if larger conglomerates are not formed by granulation. A powder with good flowability contains free-flowing primary particles whereas the particles in a cohesive powder are more likely to stick together to form loose agglomerates or harder aggre-gates. Cohesion is caused by a combination of mechanical, electrostatic, molecular and surface tension forces and is consequently not only dependent on the particle size. Powder particles below 100 μm in diameter have been shown empirically to be more cohesive than larger particles, although parti-cles half this size or less can also be noncohesive [20, 21].
A random mixture can be obtained if two free-flowing powders of ap-proximately the same particle size, density and shape are mixed (Figure 2a). Travers and White [22] were the first to report that another type of mixture resulted from mixing a free-flowing powder with a micronised cohesive powder where the latter appeared to adhere to the surface of the larger parti-cles. Hersey [23] later introduced the term “ordered mixture” to denote a completely homogeneous mixture where the two components adhere to each other to form ordered units. The concept of ordered mixing became a hot topic during the late 1970s and 1980s with numerous articles published on the subject. The disorder in the nomenclature of the mixtures was noted by Egermann [24]; the terms interactive mixture [25] or adhesive mixture [26] were later suggested to represent a mixture where a certain degree of disor-der is allowed because a different number of fine particles can be attached to each carrier particle, as shown in Figure 2b. Furthermore, the particles in the two powders do not need to be monodispersed. This terminology describes a more realistic situation than the complete homogeneity of the ordered mix-ture. Nonetheless, even though an interactive mixture allows some degree of disorder, it still represents an ideal state. Further disorder in the mixture may be caused by segregation of the interactive units [27]. Staniforth [28] sug-gested that the term “total mixing” could be used to describe the process resulting in a randomised mixture of carrier particles, agglomerates of fine particles and interactive units.

Introduction to nasal powder sprays
15
Figure 2. The schematic formation of a random mixture (A) and an interactive mix-ture (B) by dry mixing two powder materials.
1.3.2. Formation The formation of interactive mixtures cannot automatically be assumed, especially if smaller carrier particles [29] or a greater proportion of fine par-ticles [30, 31] are used. If an interactive mixture is to be formed, it is neces-sary that enough force is exerted by the carrier particles during dry mixing to break up the aggregates formed by the fine particles. Adhesion can then be achieved if the adhesive forces exceed the gravitational forces that otherwise lead to separation of the constituents. The movement of the carrier particles during mixing has been compared with that of balls in a ball mill [32]. Large (generally >200 μm in diameter), dense particles with good flowability are consequently well suited as carrier particles. The deagglomeration of the fine particles will be the rate limiting step in the formation of an interactive mix-ture; prolonged mixing times are thus likely needed when using smaller or lighter carrier particles [29, 33], or smaller more cohesive adherent particles [21, 34]. Mixing times of 1 000 min may well be required [35, 36], but can be reduced with larger batch sizes for large scale production runs [33].
The main initial driving forces for adhesion of fine particles onto the car-riers are electrostatic forces [37]. Once close contact has been established, stronger adhesion of the fine particles can be achieved by the formation of van der Waals bonds [38, 39]. Capillary forces can take over as the predomi-nant adhesive force at high humidity [39], with resultant stronger adhesion possible even after the mixture has been restored to low RH [40, 41]. The fine particles are thought to adhere especially well to so-called “active” sites on the carrier particles. The number of active sites has been associated with surface rugosity and the more extensive binding to large carrier particles than to smaller particles despite their lower total surface area [42, 43].
1.3.3. Characterisation Several methods have been employed to establish whether an interactive mixture has been achieved. These include qualitative determination by visual inspection using scanning electron microscopy (SEM) or X-ray microanaly-

Introduction to nasal powder sprays
16
sis [30, 44] and quantitative evaluation using sieve tests [35, 36] or meas-urement of the mixture homogeneity [32, 45, 46]. The adhesive strengths have been determined by ultracentrifugation [30, 47] and atomic force mi-croscopy [48].
The homogeneity of a powder mixture can be described as the relative standard deviation or coefficient of variation (CV) of the amount of drug in a collection of powder samples [49]. The homogeneity of an ordered mixture is independent of the size of the sample extracted from the powder mixture as long as it exceeds the size of an ordered unit, i.e., CV = 0 [32]. This is in contrast to the homogeneity of random or interactive mixtures, for which the measured homogeneity will increase with increasing sample size since more particles are extracted from the mixture. The theoretical homogeneity of random or interactive mixtures can be calculated using mathematical equa-tions. Comparisons between these theoretical values and the measured CV from different sample sizes can then be used to distinguish between mixture types. Equations for the theoretical homogeneities of random or interactive mixtures and the use thereof are further described in Section 3.2.1.
1.3.4. Applications The concept of interactive mixtures has often been applied for direct com-pression vehicles [45, 50], the development of low dose tablet formulations [46] and enhancing the dissolution of poorly soluble drugs [51, 52]. Interac-tive mixtures have also been used to make inert carrier particles mucoadhe-sive [53]. One of the main applications is in the field of inhalation therapy, where the concept has been used in the development of alternatives to pres-surised metered dose inhalers.
The objective of using interactive mixtures in inhalation therapy is to in-crease the fine particle fraction (FPF), i.e., the fraction of the fine drug parti-cles that reaches the lower respiratory tract. Particles between 1-5 μm have the potential of reaching the lung, but are also very cohesive because of the fine particle size. An interactive mixture could increase the FPF by prevent-ing aggregation of the drug particles and adhesion to the surfaces of the inha-lation device. However, this requires that the adhesive forces between the drug and carrier particles are not too strong. A test of commercial inhalation devices containing interactive mixtures revealed that only 20-30% of the dose would reach the lower respiratory tract [54]. Several studies, most of which used lactose as the carrier particle, have thus been conducted in order to understand and optimise the interactive forces. Attempts have, for exam-ple, been made to increase the FPF by using smaller carrier particles [55-57].
The success rate for mixtures used for inhalation therapy is usually meas-ured as their ability to deliver the FPF. The characterisation of the mixture itself has, as a consequence, been less than thorough and the mixing times have been kept short to decrease the risk of over-strong adhesion to active

Introduction to nasal powder sprays
17
sites on the carrier particle [58]. The practices of taking large powder sam-ples, of taking too few samples and of making mixtures with a high drug content can result in low CV values even if an interactive mixture has not been formed [31]. This may merely be of terminological importance in inha-lation therapy, but it may be difficult to draw conclusions regarding the ac-tual effect of an interactive mixture when the characterisation of mixtures for other purposes, such as dissolution enhancement, is also insufficient.
1.4. Superdisintegrants Superdisintegrants are commonly used in tablets and capsules to decrease their disintegration time in the gastro-intestinal tract. The Swedish term “su-persprängmedel”, i.e., super-explosive, is vividly descriptive. The excipient should cause rapid disintegration of the solid formulation by quickly draw-ing water into it. The disintegration may be helped further by swelling of the disintegrant.
Three superdisintegrants are discussed in this thesis: sodium starch glyco-late (SSG), croscarmellose sodium (CCS) and crospovidone (CPP) (Figure 3). In their dry powder forms, SSG particles are smooth and egg-shaped, CCS particles are elongated and fibrous and CPP particles are raisin-shaped; CPP particles flow relatively poorly compared with the others. SSG and CCS both form anionic hydrogels on contact with water; the charged groups on the polymer chains (Figure 3) repel each other and this, along with the effects of osmosis, results in swelling of the gel. The semisolid gel particles that are formed are mainly composed of water, stabilised by the polymer network. SSG has an extensive capacity for absorbing water and can swell up to 300 times its original volume [59] (Figure 3). However, the swelling effect will not be quite as marked in a salt solution because the repelling charges are shielded. CPP differs somewhat from the other two superdisinte-grants in that it is nonionic and does not swell so markedly upon hydration.
Figure 3. The molecular structure of SSG (A), CPP (B), CCS (C) and PPS (D). (Derived from Rowe el al. [59].) The swelling of SSG in water is shown to the right as a micrograph of the same particle before and after the addition of a drop of deion-ised water. The resulting gel particle has been made darker to increase its visibility as it mainly consists of water. The scale bar represents 100 μm.

Introduction to nasal powder sprays
18
SSG is prepared either by chemical crosslinking and substitution by car-boxymethylation of starch through a reaction with sodium chloroacetate (Williamson ether synthesis) (Type A and B: e.g. Primojel® and Explotab®) or crosslinking by physical dehydration after the substitution (Type C: e.g. Vivastar P) [59]. The unsubstituted starch form shown in Figure 3d can also be used as a disintegrant but is not a superdisintegrant because of its less extensive swelling capacity. The different manufacturing methods result in differences in the amounts of by-products such as sodium chloride and so-dium glycolate. Most NaCl has been shown to be present on the surface of the particles of the SSG brand Primojel®[60] and the amount of salt may therefore also depend on the size fraction of the powder used. Information on the exact composition of or batch-to-batch variations in pharmaceutical ex-cipients is generally lacking. A salt content of up to 7% is approved for SSG [59], but it should be limited to 4% [61]. The degree of crosslinking of Pri-mojel® has been reported to be 100% and the degree of substitution 27% [61].
The capacity of SSG to absorb water makes it a plausible mucoadhesive agent for dry formulations and its natural particle size of around 40 μm is suitable for nasal administration. SSG forms a slightly acidic gel (pH 6) [59], which matches the pH of the nasal mucosa [2]. The excipient has also previ-ously been used by one research group to increase the dissolution of poorly soluble drugs [62, 63], which indicates that it has a good deagglomeration capacity.

Introduction to nasal powder sprays
19
1.5. Nasal drug delivery Let us return to the question we started with: why nasal administration? Be-cause of the physiological features described under Section 1.1, nasal ad-ministration offers a way of achieving faster, more extensive absorption of some drugs than, for example, oral administration. A molecule can quickly be transferred across the thin epithelial cell layer to the systemic blood circu-lation from the well vascularised submucosa without first-pass hepatic and intestinal metabolism. The effect is often reached within 5 min for smaller drug molecules. Complete bioavailability, i.e., the same as after the intrave-nous infusion, was reported for the low molecular weight drug propranolol in one of the first studies on systemic absorption after nasal administration to humans [64], while the bioavailability after oral administration was only 25%. However, in this case, the nasal absorption was probably improved by the use of a 2% methylcellulose vehicle. Favourable absorption after nasal administration has also been seen for opioid drugs, with maximum plasma concentrations achieved between 5 and 50 min and bioavailabilities of 46-71% [65].
There are, of course, also drawbacks associated with nasal administration. The volume that can be sprayed into the nasal cavity is limited to approxi-mately 150 μL, which subsequently limits the dose of the compound to be administered. Further, diseases such as the common cold or allergic rhinitis can alter the absorption characteristics of the area, although there are many reports that show the opposite [66-68]. Nasal administration has been associ-ated with a high variability in the mount of drug absorbed. Upper airway infections may increase the variability as may the extent of sensory irritation of the nasal mucosa, differences in the amount of liquid spray that is swal-lowed and not kept in the nasal cavity and differences in the spray actuation process [69]. Nonetheless, variability after nasal administration should be comparable to that after oral administration [70, 71].
1.5.1. Drug candidates Potential drug candidates for nasal administration include anaesthetics, an-tiemetics and sedatives that all benefit from a fast onset of effect. Nasal ad-ministration is particularly suitable for antimigraine drugs, because a fast effect is desired and oral administration can be prohibited by nausea; many of these drugs are already available as nasal sprays. Peptide drugs are also available as nasal sprays, in this case to avoid drug degradation after oral administration. Drugs for nasal administration should also preferably be potent enough to allow administration of a small volume and for administra-tion when needed rather than chronic treatments, to avoid long term effects on the nasal mucosa. See the recent review by Cosantino et al. [72] for fur-ther aspects of this topic.

Introduction to nasal powder sprays
20
Two compounds, dihydroergotamine (DHE) and desmopressin, were used in this work as model drugs in the nasal formulations. DHE is an ergot alkaloid used for migraine (Figure 4a). It is currently available as a tablet formulation in Sweden, but is marketed as a liquid nasal spray in other countries. The absolute bioavailability of nasal DHE, relative to the intramuscular formula-tion, is 38.4% [66], whereas the corresponding bioavailability of the oral tablet has been reported to be below 1% [73]. The substance does, however, have active metabolites that improve the effect somewhat despite the exten-sive first-pass metabolism after oral delivery. Although absorption from the liquid spray is quite high, the substance is chemically unstable in solution and the administration is inconvenient because of the large volume required, which necessitates repeated dosing [66].
The second compound, desmopressin or 1-desamino-8-D-arginine vaso-pressin (Figure 4b), is a synthetic peptide analogue of the posterior pituitary hormone vasopressin and is used as an antidiuretic or, at higher doses, in the control of bleeding disorders. Desmopressin can also be administered by either nasal or oral routes. The tablet formulation of desmopressin, which has very low bioavailability, is unusual; other peptide analogues are avail-able only as injections or nasal sprays. The bioavailability of the commercial tablet is 0.1% while that of the nasal spray is 3-5% according to the SPC (summary of product characteristics) [74]. Desmopressin has been shown to be degraded by intracellular metabolism in rabbit nasal mucosa homogenates [75]. No metabolite was detected after nasal administration to humans in the same study, which could be explained by paracellular absorption of the compound. The absorption of desmopressin is highly variable, with coeffi-cients of variation reported in the order of 60% after nasal [76] and oral [77] administration. Hence, not only increased absorption but also reduced vari-ability would be beneficial for future formulations of desmopressin.
Figure 4. The molecular structures of DHE (A) and desmopressin (B).

Introduction to nasal powder sprays
21
1.5.2. Olfactory transfer The nasal administration route is associated with the unique possibility of achieving olfactory transfer of drugs, which may be considered as either an exclusive advantage or a potential risk. The objective of this thesis is not, however, to review the many interesting aspects of the olfactory uptake of drugs as it is mainly concerned with systemic absorption; nonetheless, a short introduction will be provided and the interested reader can find more detailed discussions in the theses by Jansson [78] and Espefält Westin [79].
The nerve cells of the olfactory epithelium project into the olfactory bulb of the brain, thus providing a direct connection between the brain and the external environment. The transfer of drugs to the brain is normally hindered by the blood-brain barrier (BBB), which is virtually impermeable to passive diffusion of all but small, lipophilic substances. However, if drug substances can be transferred along the olfactory nerve cells, they can bypass the BBB and enter the brain directly. The olfactory transfer of drugs into the brain is thought to occur by either slow transport inside the olfactory nerve cells to the olfactory bulb or by faster transfer along the perineural space surround-ing the olfactory nerve cells into the cerebrospinal fluid surrounding the olfactory bulbs and the brain [80, 81].
As implied above, olfactory transfer could be a great opportunity for drugs that have a required effect in the central nervous system such as those for Parkinson’s or Alzheimer’s diseases. It could also be seen as a risk if these or other substances were to accumulate in the brain; however, it is questionable whether sufficient quantities to cause concern could be trans-ferred by this route. Studies have been presented that show that direct trans-fer of drugs is achievable [81, 82], but the possibility of olfactory delivery of therapeutically relevant doses to humans remains to be demonstrated.
1.5.3. Comparisons with sublingual administration It is also possible to achieve fast, extensive absorption with sublingual ad-ministration. The drug is transferred directly to the systemic circulation without first-pass hepatic and intestinal metabolism and the nonkeratinised sublingual mucosa is more permeable than other areas in the oral cavity and absorption of larger molecules such as peptide drugs is possible [83]; yet, the surface area under the tongue is smaller than in the nasal cavity and the mu-cosa has a multiple epithelial cell layer. In comparison, the buccal epithelium is 500-800 μm thick [84], the sublingual 100-200 μm [84] and the nasal 25 μm [1]. Absorption will therefore not be as fast or as extensive as after nasal administration [83]. Nevertheless, the convenience of manufacturing and administrating a tablet is greater than that for a nasal spray, which may outweigh these shortcomings. For example, desmopressin is available as a sublingual tablet in addition to the oral tablet and nasal spray mentioned

Introduction to nasal powder sprays
22
above. The SPC states a bioavailability for the sublingual formulation of 0.25%, which is about two times higher than after oral administration but around twenty times lower than after nasal administration [74]. A mucoad-hesive sublingual tablet based on the formation of interactive mixtures has been developed for the administration of fentanyl [85], resulting in exten-sive, rapid absorption from the oral cavity. The concentrated deposition and reduced risk of swallowing may also be beneficial for sublingual absorption of other drugs.
1.5.4. Powders or liquids? Drugs in liquid nasal sprays can, as mentioned, be very well absorbed and liquid sprays are relatively simple, cost-effective and generally well accepted by the patient. It should also be possible to produce sprays that are preserva-tive-free with the help of modern devices. However, a liquid spray has a tendency to run down the oesophagus, which may result in a bad taste. Most of us have probably tasted the slightly bitter flavour of the common decon-gestant sprays but other drugs, such as the antimigraine drug sumatriptan (Imigran®), can taste quite foul [86]. A perhaps more severe effect of the swallowing of nasally administered fluid is decreased and more variable absorption. We can again take the example of Imigran® where most of the nasally administered dose is actually absorbed from the stomach [87]. The bioavailabilities from the commercial products are consequently almost equal (16% and 14% from the nasal spray and oral tablet, respectively [87, 88]), but the effect is achieved faster after nasal administration (15 compared with 30 min [88]). Moreover, absorption of polar and/or large drug mole-cules such as peptides from a liquid formulation may be suboptimal because of a short residence time in the nasal cavity or low permeability of the mu-cosa to the active component. Permeation enhancers such as cyclodextrins, bile salts and chelating agents can be added, but there is an associated risk of harmful effects on the epithelium and/or mucociliary clearance [89].
The use of dry powder formulations presents an alternative to achieve bet-ter deposition and residence time in the nasal cavity, increased stability and possible absorption enhancement. The administration of nasal powder for-mulations has been associated with greater sensory irritation than liquid sprays and the amount of powder should be kept as low as possible, prefera-bly below 20 mg. The sensation will also depend on the type of powder and may even be less irritating than a liquid in some cases [90]. Mucoadhesive particle formulations have generally proven to be safe delivery systems with few harmful effects even after repeated dosing [91-93]. Dry powder formula-tions are also associated with better stability than liquid ones, both microbio-logically and chemically [90, 94].
The effect of water-insoluble, mucoadhesive powder mixtures on the ab-sorption of insulin was first investigated by Nagai et al. [95], who concluded
Introduction to nasal powder sprays
23
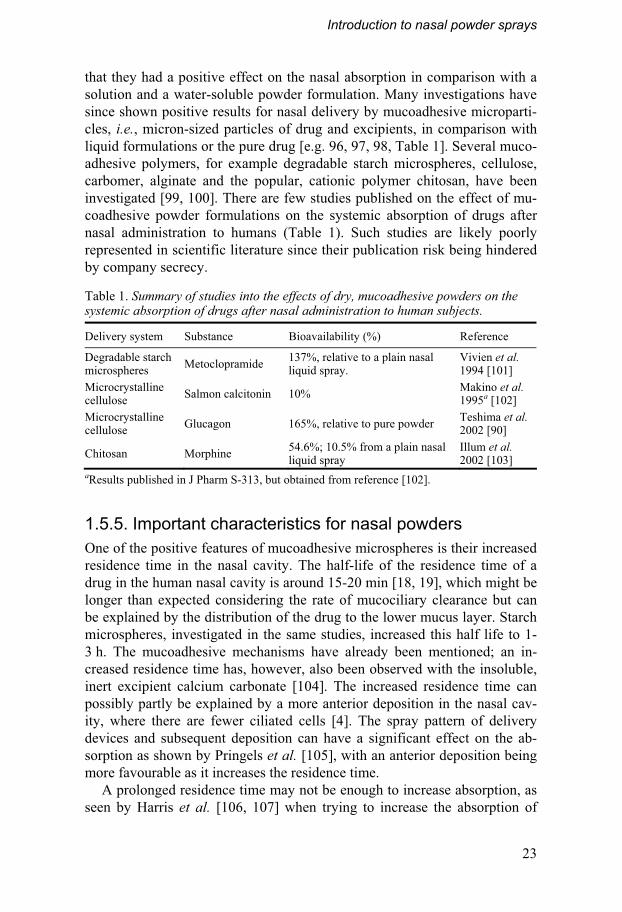
that they had a positive effect on the nasal absorption in comparison with a solution and a water-soluble powder formulation. Many investigations have since shown positive results for nasal delivery by mucoadhesive microparti-cles, i.e., micron-sized particles of drug and excipients, in comparison with liquid formulations or the pure drug [e.g. 96, 97, 98, Table 1]. Several muco-adhesive polymers, for example degradable starch microspheres, cellulose, carbomer, alginate and the popular, cationic polymer chitosan, have been investigated [99, 100]. There are few studies published on the effect of mu-coadhesive powder formulations on the systemic absorption of drugs after nasal administration to humans (Table 1). Such studies are likely poorly represented in scientific literature since their publication risk being hindered by company secrecy.
Table 1. Summary of studies into the effects of dry, mucoadhesive powders on the systemic absorption of drugs after nasal administration to human subjects.
Delivery system Substance Bioavailability (%) Reference
Degradable starch microspheres
Metoclopramide 137%, relative to a plain nasal liquid spray.
Vivien et al. 1994 [101]
Microcrystalline cellulose
Salmon calcitonin 10% Makino et al. 1995a [102]
Microcrystalline cellulose
Glucagon 165%, relative to pure powder Teshima et al. 2002 [90]
Chitosan Morphine 54.6%; 10.5% from a plain nasal liquid spray
Illum et al. 2002 [103]
aResults published in J Pharm S-313, but obtained from reference [102].
1.5.5. Important characteristics for nasal powders One of the positive features of mucoadhesive microspheres is their increased residence time in the nasal cavity. The half-life of the residence time of a drug in the human nasal cavity is around 15-20 min [18, 19], which might be longer than expected considering the rate of mucociliary clearance but can be explained by the distribution of the drug to the lower mucus layer. Starch microspheres, investigated in the same studies, increased this half life to 1-3 h. The mucoadhesive mechanisms have already been mentioned; an in-creased residence time has, however, also been observed with the insoluble, inert excipient calcium carbonate [104]. The increased residence time can possibly partly be explained by a more anterior deposition in the nasal cav-ity, where there are fewer ciliated cells [4]. The spray pattern of delivery devices and subsequent deposition can have a significant effect on the ab-sorption as shown by Pringels et al. [105], with an anterior deposition being more favourable as it increases the residence time.
A prolonged residence time may not be enough to increase absorption, as seen by Harris et al. [106, 107] when trying to increase the absorption of

Introduction to nasal powder sprays
24
desmopressin by using a viscous solution. Increased absorption from powder formulations may also be explained by a higher concentration gradient of the active component from a coherent particle than from a drop or a quickly dissolving particle, exemplified by the positive effect obtained with insolu-ble, inert excipients [104, 108]. Mucoadhesive microspheres can also lead to increased paracellular absorption. Dry starch microspheres, for example, induced temporary opening of the tight junctions between the epithelial cells as a result of dehydration of the mucosa during hydration of the powder [109]. The effect was corroborated in vivo, where the dry spheres improved the absorption of insulin [110], but pre-hydrated spheres did not have the same effect [111] which was also reported by Wang et al. [112]. The cal-cium binding capacity of the polymers may also influence absorption, as the integrity of the tight junctions is dependent on the extracellular calcium lev-els [113]. This effect has been strongly associated with the absorption en-hancing effect of carbomer [114]. However, Oechslein et al. [115] predicted a delayed onset of this effect when dry particles are used as it requires full hydration of the polymers and should therefore not affect instant absorption. Because a protracted hydration phase could also slow the release of the ac-tive component, surface deposition of the drug is favoured as shown by Pereswetoff-Morath and Edman [116].
Considering the positive results achieved with microspheres, it may be hard to understand their poor representation on the pharmaceutical markets. There is one nasal powder spray containing cellulose available for sale in Sweden and other countries as a remedy for allergic rhinitis. The spray is, however, drug free and is only intended to protect the mucosa from allergens by gel formation [117]. Some spray devices that can deliver a powder dose into the nasal cavity in the same manner as conventional liquid sprays, i.e., without manual inhalation of the powder, are also available. A possible ex-planation for the relative lack of nasal powder formulations may lie in the difficulties associated with achieving a cost effective and reproducible prod-uct for large scale manufacturing. The most common methods of producing microspheres involve dissolution of the drug to incorporate it into the muco-adhesive polymer, usually followed by spray drying or lyophilisation. This can result in unwanted residues in the formulation and difficulties in control-ling the particle size. Particles smaller than 10 μm in diameter risk not being approved by medical product agencies because of the increased likelihood of pulmonary deposition [2]. In contrast, particles that are too big (generally above 50 μm) can be deposited too far in front of the nasal cavity [2, 118], where absorption is poor.
An effective and reproducible way of producing mucoadhesive particles for nasal drug delivery in the size range of 10-50 μm is thus called for. If fast absorption of the drug is desired, it would also be beneficial if a method was devised where the drug is deposited on the surface of the microparticles.

Introduction to nasal powder sprays
25
1.6. Assessing the efficacy of a powder formulation How can we measure the effect of a novel nasal powder formulation on the systemic absorption of a drug? Studies in human volunteers are time con-suming, expensive and, as a consequence, scarce (Table 1). Studies on depo-sition characteristics and residence time in the nasal cavity (not presented in Table 1) have been performed using gamma-scintigraphy [18, 19, 119]. Animal studies, which are more common, can also provide information on the effects of a formulation on plasma concentrations and/or residence time. A direct correlation to results in humans is, however, not possible because of species differences [3, 120]. Even comparisons between powders and liquids can be troublesome in some cases; for example, if the animal is sedated and in the supine position, the residence time of a liquid formulation can be in-creased. Although in vitro methods cannot replace in vivo studies, they can reduce the requirement for these, since they can be used for mechanistic studies and comparisons.
1.6.1. Mucoadhesion in vitro In vitro mucoadhesion measurements of dry particle formulations are based on the extent of adhesion, the molecular interactions between polymers and mucin or the residence time. The latter measurements require the most ad-vanced experimental setup since functional mucociliary clearance is re-quired. Frog palate studies, such as those described by Pritchard et al. [121], have often been used to compare the mucociliary clearance of various parti-cles. The air interface and limited fluid access should reflect the in vivo situation, although the clearance rate will not be directly comparable to that in the human nasal cavity.
Measurement of molecular interactions may seem superfluous for dry par-ticle systems if the primary cause of mucoadhesion is water movement into the particle. Rheological methods can be employed for this purpose and have also been used to measure the mucoadhesion of dry powder formulations [122, 123]. Measuring mucoadhesion by rheology has been questioned pre-viously [124] and did in the investigations by Callens et al. [123] not con-firm the results of the powder formulations in vivo [97], which was ex-plained by overhydration of the particles in the in vitro method [123].
Perhaps the most common way of measuring the adhesion of dry particles is to estimate the force needed for detachment, which can occur through failure of the mucus layer, the formulation or the area in between. Tensile strength measurements, which have been thoroughly discussed and evaluated by Hägerström [125], are popular. It has even been suggested that the type of mucosa used is less crucial in such measurements on dry powder formula-tions owing to the primary importance of the hydration of the formulation [126, 127].

Introduction to nasal powder sprays
26
1.6.2. Uptake in vitro The simplest way of roughly estimating the in vivo performance of a powder formulation is probably to measure its effect on the release of the active component. Still, the release of the drug is unlikely to be the same in vivo as it is in vitro, because of factors such as the degree of hydration of the parti-cles and surface-volume relationships. Furthermore, release of the drug from the formulation may not be the rate-limiting step of absorption. In vitro cell models provide a more sophisticated method of estimating nasal absorption [128] using primary cultures of cells obtained from healthy human nasal mucosa [129] or nasal cell lines such as RPMI 2650, which is of human ori-gin [128]. Due to its poor differentiation and lack of polarisation, this cell line is best used for metabolism studies; however, a polarised monolayer containing tight junctions and potentially suitable for drug transfer studies has recently been achieved by culturing RPMI 2650 cells at an air-liquid interface [130].
Freshly excised nasal mucosa has often been used to evaluate the effect of powder delivery systems [e.g. 104, 131]. These studies usually include ani-mal mucosa (rabbit, sheep, cow or pig); human mucosa is understandably more rarely used [132]. The use of mucosa from slaughter animals provides a cheap and humane alternative to the sacrifice of research animals. A fur-ther advantage with this experimental setup is the utilisation of intact mucosa comprising different cell types, tight junctions and mucus. Regional differ-ences in uptake can also be evaluated by excising mucosa from different parts of the nasal cavity such as the olfactory region [133, 134], an advan-tage not achievable in vivo. The integrity of the excised mucosa should al-ways be ascertained; this can be achieved, for example, by electrophysio-logical measurements [132, 135]. A horizontal Ussing chamber setup using excised porcine mucosa has been developed [136], where the viability of the mucosa can be meticulously monitored. However, species differences, and possibly the lack of blood circulation, prohibit extrapolation of the results to absorption in humans. The substance will have to cross both the epithelial cells and the submucosa, if they are not separated, in the Ussing chamber measurements; in contrast, blood is flowing directly beneath the epithelial cells in vivo. Correlations with passively diffusing drugs should be more reliable in this context as shown by Wadell et al. [135].
The air interface in the nasal cavity, which is important for evaluation of the functionality of a dry formulation, can be mimicked using in vitro meth-ods. However, the effect of mucociliary clearance or deposition characteris-tics cannot be reproduced in vitro, and these methods will thus only provide information on absorption enhancing effects.

27
2. Aims of the thesis
The main objective of this doctoral thesis was to develop and evaluate a novel powder formulation for improved nasal systemic drug delivery. The specific aims of Papers I-V were as follows:
• To investigate whether interactive mixtures can be created using a small mucoadhesive carrier suitable for nasal administration and to determine how such mixtures could best be characterised.
• To evaluate the effect of the formulation on the uptake of a relevant drug in vitro using the horizontal Ussing chamber system and also to compare transport of the drug across nasal respiratory and olfactory mucosa.
• To develop a simplified method for measuring mucoadhesion and to use it to establish whether the surface coverage of the carrier particles in inter-active mixtures has an effect on their mucoadhesion.
• To determine whether the results of in vitro and in vivo studies can be affected by chemical interactions between superdisintegrants and certain drug substances and, if so, which molecular characteristics are of impor-tance for these interactions.
• To compare the nasal absorption of desmopressin from a formulation containing SSG as the carrier particle with absorption from a mucoadhe-sive sublingual tablet and from the commercial nasal spray in a clinical trial and also to record the volunteers’ opinions on the different formula-tions.

28
3. Description of essential methods
The results included in this thesis are based on a number of methods. The methods that have been central for the thesis and/or that require more than a few words to explain are included in this section to facilitate critical evalua-tion of the results. Other methods are mentioned in the Key findings section and further details can be found in Papers I-V.
For all studies, the carrier material SSG was obtained from DMV Interna-tional, where it is marketed under the proprietary name Primojel®.
3.1. Powder materials
3.1.1. Obtaining the required particle size The required sizes of the carrier particles were obtained by dry sieving or air classification. The latter method was especially useful for retrieving the smaller particle size fractions, i.e., below 32 μm, when efficient dry sieving was impeded by the flowability of the powder.
Milling was employed to create powders with sufficiently small particle sizes of the fine particulate component for the interactive mixtures. A mortar grinder was used for milling sodium salicylate in Paper I, a centrifugal ball mill was used for DHE in Paper II and oxazepam was micronised in a pin disc mill in Paper III. The amounts of micronised sodium salicylate and oxazepam were sufficient to allow final air classification, to ensure that only the finest particles were used in the mixtures.
3.1.2. Determining the particle size The size of the carrier particles was evaluated by laser diffraction. Because SSG swells extensively in contact with water, dry laser diffraction was used in Paper I (Sympatec Helos H0321, Sympatec GmbH, Germany). In Papers II and III, SSG and PPS were dispersed in 99.5% ethanol and measured us-ing wet laser diffraction (LS 230, Coulter, USA) as this gave exactly the same results as dry laser diffraction (unpublished data). The external surface area of the carrier particles was measured with steady-state permeametry [137, 138] and calculations were made according to Eriksson et al. [139].

Description of essential methods
29
Dry laser diffraction was also used for measuring the particle size of the fine particulate component sodium salicylate in Paper I. However, laser diffrac-tion is associated with problems in obtaining correct estimates of particle sizes below 10 μm if the refractive index of the substance is not known. Sur-face measurements alone were thus used for characterisation of the fine par-ticles in the other papers. The external surface areas were measured with Blaine permeametry [140] and were corrected for slip flow [141].
3.1.3. Determining the ability of the particles to swell The capacity of the powders to absorb liquid was evaluated by placing a known weight of the dry powder on top of a pre-weighed moist membrane and letting the powder absorb the liquid from underneath the membrane. The swellability was expressed as the increase in weight per mg powder.
3.2. Interactive mixtures
3.2.1. Theoretical models Homogeneity evaluation. The homogeneity of a powder can be expressed as the CV of the amount of drug substance in a number of powder samples extracted from the mixture. Larger powder samples will lead to a lower CV as each sample contains more particles.
According to the theoretical models, described in more detail in Paper I, an interactive mixture should be more homogeneous than a random mixture and the measured homogeneity should also not increase as much with in-creasing size of the powder samples. This can be seen when comparing the equation for the homogeneity of a random mixture [142] (Eq. 1) with the equation that describes the homogeneity of an interactive mixture [143-146] (Eq. 2). The homogeneity is here expressed as the CV:
Mwqwp
pqCV pq +⋅⋅=100RM (Eq. 1)
Mw
pCV p⋅⋅= 1
100IM (Eq. 2)
where p and q are the proportions by weight of fine and coarse particles, respectively. The weight of the mixture sample is represented by M and the mean particle weights of the two components by pw and qw .
Description of essential methods
30
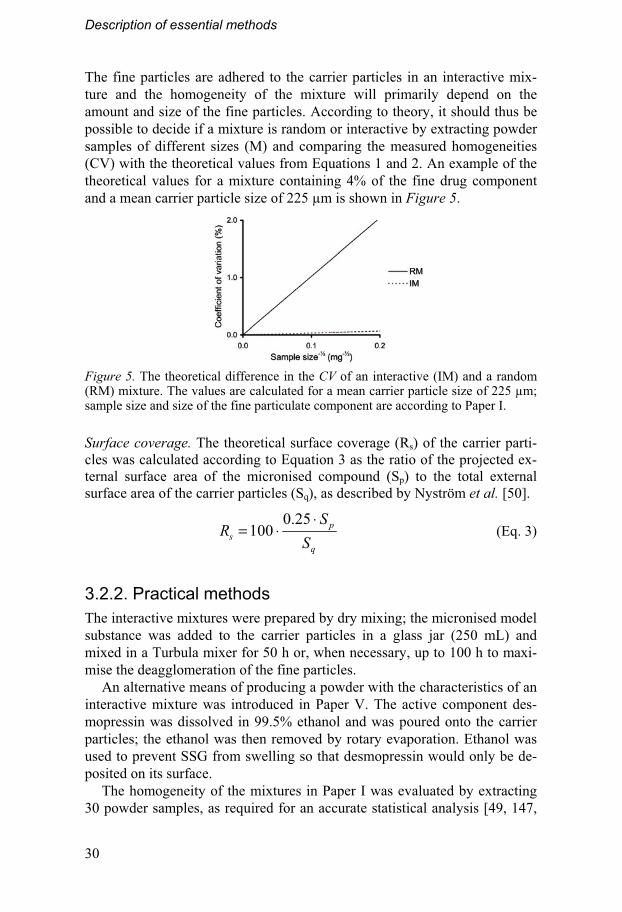
The fine particles are adhered to the carrier particles in an interactive mix-ture and the homogeneity of the mixture will primarily depend on the amount and size of the fine particles. According to theory, it should thus be possible to decide if a mixture is random or interactive by extracting powder samples of different sizes (M) and comparing the measured homogeneities (CV) with the theoretical values from Equations 1 and 2. An example of the theoretical values for a mixture containing 4% of the fine drug component and a mean carrier particle size of 225 μm is shown in Figure 5.
Figure 5. The theoretical difference in the CV of an interactive (IM) and a random (RM) mixture. The values are calculated for a mean carrier particle size of 225 μm; sample size and size of the fine particulate component are according to Paper I.
Surface coverage. The theoretical surface coverage (Rs) of the carrier parti-cles was calculated according to Equation 3 as the ratio of the projected ex-ternal surface area of the micronised compound (Sp) to the total external surface area of the carrier particles (Sq), as described by Nyström et al. [50].
q
ps S
SR
⋅⋅=
25.0100 (Eq. 3)
3.2.2. Practical methods The interactive mixtures were prepared by dry mixing; the micronised model substance was added to the carrier particles in a glass jar (250 mL) and mixed in a Turbula mixer for 50 h or, when necessary, up to 100 h to maxi-mise the deagglomeration of the fine particles.
An alternative means of producing a powder with the characteristics of an interactive mixture was introduced in Paper V. The active component des-mopressin was dissolved in 99.5% ethanol and was poured onto the carrier particles; the ethanol was then removed by rotary evaporation. Ethanol was used to prevent SSG from swelling so that desmopressin would only be de-posited on its surface.
The homogeneity of the mixtures in Paper I was evaluated by extracting 30 powder samples, as required for an accurate statistical analysis [49, 147,

Description of essential methods
31
148], in three different sizes, using powder thieves (Figure 6). The amount of fine particulate component in each sample was measured with UV-absorption after dissolving the samples in water and the results were com-pared with the theoretical calculations. The 95% confidence limits were cal-culated for the coefficients of variation, which were assumed to follow a χ2 distribution. The homogeneity of the mixtures in Papers II and III was evalu-ated by collecting 30 samples with the smallest powder thief (15 mg) and the UV-absorption was measured in 99.5% ethanol.
Figure 6. Drawing of a powder thief in closed (top) and open (bottom) position.
3.3. In vitro uptake For a more detailed description of the horizontal Ussing chamber measure-ments, please see Paper II and the theses by Östh [149], Jansson [78] and Espefält Westin [79].
3.3.1. Porcine nasal respiratory and olfactory mucosa Both nasal respiratory and olfactory mucosa were used in Paper II. The mu-cosae were isolated at the local slaughterhouse (Swedish Meats, Uppsala) from 6-month-old domestic pigs. The olfactory mucosa was obtained from the upper part of the septum, the roof of the nasal cavity and the superior turbinate. Respiratory mucosa was obtained from the ventral nasal concha at the anterior part of the nasal cavity. The excised mucosae were placed in pre-oxygenated, ice cold Krebs Ringer bicarbonate buffer (KRB), supple-mented with 15 mM NaHCO3, 1.2 mM CaCl2 and 138 mM NaCl, within 10 min after the animal had been killed and were transported to the labora-tory. Two pieces of either nasal respiratory or nasal olfactory mucosa were obtained from each snout.
Mucosae from the Ussing chamber experiments and further fresh samples were embedded in plastic (Technovit 7100, Leica Microsystems, Germany) for histological evaluation. Thin sections were stained with toluidine blue and examined in a light microscope. The mucosae were studied to detect possible damage to the epithelial cell layer. The presence of elements spe-cific to olfactory mucosa, i.e., Bowman’s glands, dendritic knobs with bun-dle formations of long cilia and nonciliated epithelial cells, was also investi-gated to determine whether the mucosa had been successfully isolated.

Description of essential methods
32
3.3.2. Horizontal Ussing chamber measurements The horizontal Ussing chamber measurements were performed according to a method described by Östh et al. [136]. The stability of DHE and its poten-tial to bind to the Ussing chamber were studied prior to the transport experi-ments and adjusted so as not to affect the study results. All solutions that were to contain DHE were supplemented with Na-EDTA (1 mg/mL) and Na-Metabisulfite (0.05 mg/mL) to improve its chemical stability. The muco-sae were mounted in the Ussing chambers, exposing a surface area of 0.55 cm2 per sample (Figure 7). The viability measurements commenced within 100 min of slaughter of the animal. The tissues were oxygenated and kept on a circular shaker at a temperature of 37º C during the experiments. The viability of the mucosae was measured after equilibration with KRB on both the mucosal and serosal side. Only viable mucosa, that fulfilled prede-termined criteria regarding the electrophysiological parameters resistance (R), potential difference (PD) and short circuit current (Isc), were included in the study.
Figure 7. Schematic drawing of a horizontal Ussing chamber with the excised mu-cosal sample, including both epithelial cells and submucosa, mounted with an air interface on the mucosal side (1) and buffer solution on the serosal side (2).
The KRB was removed from the mucosal side before the transport experi-ments and the buffer on the receiver side was exchanged for a KRB solution containing the stabilising agents. A volume of 50 μL of the reference solu-tion (1 mg/mL DHE in 0.05 mg/mL stabilised mannitol solution) or 1 mg of the powder formulation (SSG mixture with 5% DHE) was applied to the donor side. Samples were taken from the receiver side and stored at -18° C pending analysis by UV-HPLC by Mikrokemi AB, Uppsala. The second viability measurement was performed after adding 1.2 mL of the stabilised KRB buffer on the donor side, when the transport study was finalised.
The apparent permeability coefficients [Papp (cm/s)] were calculated in accordance with the model for non-sink conditions suggested by Palm et al. [150] and the results were used for comparison of the transfer of DHE across the two types of mucosa. The efficacy of the delivery system was evaluated

Description of essential methods
33
as the amount of DHE transferred to the receiver compartment per surface area of the mucosa (ng/cm2). Papp was not used for this purpose as it can be underestimated after application of the powder formulation, if the available concentration of DHE is decreased as a result of delayed dissolution.
3.4. Mucoadhesion
3.4.1. Test materials Only respiratory mucosa was used for the mucoadhesion measurements. Fresh snouts were obtained from 6-month-old domestic pigs at Swedish Meats (Uppsala). The snouts were divided by an incision along the septum and were transported on ice to the laboratory where the mucosa was care-fully removed from the nasal turbinates within 2 h of slaughter, similar to the procedure described by Wadell et al. [151]. Each sample of mucosa was only used once and the rest were kept in pre-oxygenated, ice cold KRB for a maximum of 6 h awaiting the experiments.
Partly pregelatinised maize starch (PPS, Starch 1500® from Colorcon) was included in the study as a comparator with a less extensive swelling capacity than SSG. As the aim of the study was to investigate the effect of surface coverage on the mucoadhesion of the carrier particles, SSG and PPS with different degrees of surface coverage were used in the measurements. Maximum hydrophobic surface coverage was achieved by mixing SSG and PPS with 2% magnesium stearate for 100 h. The surface coverage of SSG was further evaluated by creating interactive mixtures with varying amounts of the hydrophobic oxazepam or the hydrophilic sodium salicylate. Glass beads in the same particle size range were used as negative standards.
3.4.2. Tensile strength measurements A texture analyzer, TA.HDi (Stable Micro Systems), was used for the tensile strength measurements. A type of absorbent paper with a plastic coating (Whatman Benchkote) was evaluated as a substitute for the nasal mucosa, as suggested by Caramella et al. [152].
A sample of fresh mucosa or a 2x2 cm piece of the paper substitute was fastened on the lower platform of the texture analyser (Figure 8). The pow-der was attached to the upper movable probe (10 mm ø) with double-sided adhesive tape. The amount of powder attached to the probe was evaluated to ensure that a reproducible monoparticulate layer was obtained. The meas-urement commenced as the upper probe triggered at a force of 2 mN upon contact with the lower surface. The probe was kept in contact at a constant force of 10 mN for a predetermined time, after which it was raised at the

Description of essential methods
34
speed of 0.1 mm/s and the force needed for detachment was registered. A lid was placed on the measuring system to prevent water evaporation during contact times above 120 s. The mucoadhesive characteristics were described by the fracture strength (mN/cm2), determined as the peak detachment force per square centimetre, and the tensile work (μJ), i.e., the area under the force-distance curve (Figure 8).
Figure 8. Schematic drawing of the powder on the upper movable probe (1), which is lowered to the mucosa or substitute paper (2). The detachment force and distance is recorded as the movable probe is raised.
3.4.3. Development of a mucosal substitute The measurements on the nasal mucosa after contact times of 1, 120 and 600 s were used as reference results for the measurements on the substitute. The absorbent paper was wetted by either pure KRB or mucin solution (Por-cine gastric mucin type II in KRB) of different concentrations and volumes. The measurements were performed on pure SSG and on SSG covered with magnesium stearate.
The effect of the addition of mucin was also evaluated using freshly pre-pared pre-swelled SSG and PPS particles of different concentrations. In these measurements, the upper probe was covered with absorbent paper which was wetted with KRB and lowered to the gel on the stationary plat-form. After triggering, the probe was lowered 0.3 mm into the gel and kept there for 10 s after which it was raised and the detachment force was regis-tered. These measurements were performed at a controlled depth instead of a constant force to prevent the probe from penetrating too deep into the softer gels.

Description of essential methods
35
3.5. Physicochemical interactions
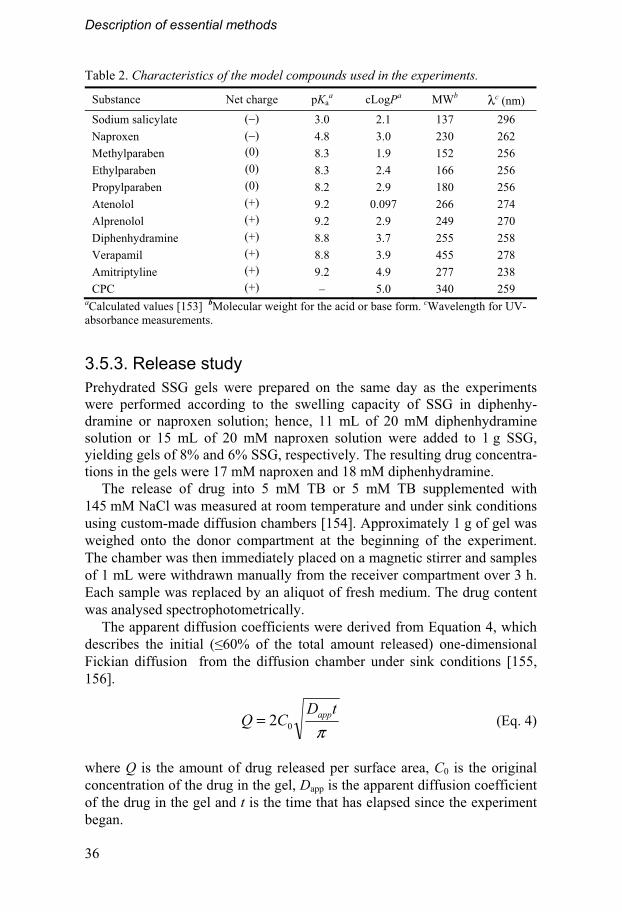
3.5.1. Test materials The characteristics of the disintegrants evaluated in the study are described in Section 1.4. The model substances included in the study are shown in Figure 9 and further details are given in Table 2.
Figure 9. The molecular structures of the model compounds included in Paper IV. R in the paraben structure represents CH3, CH2CH3 and CH2CH2CH3 for methyl-, ethyl- and propylparaben, respectively. CPC stands for cetylpyridinium chloride.
3.5.2. Binding study The compounds were dissolved in 5 mM tris buffer (TB) to give a final con-centration of 0.2 or 2 mM and a pH of 7.4 ± 0.1. The effect of salt was stud-ied by adding NaCl to the 5 mM TB. All use of plastic utensils was avoided to prevent sorption of the amphiphilic substances.
The extent of binding was studied by adding 15 mL of drug solution to test tubes containing 50 mg of dry disintegrant powder. The equilibrium concentration in the supernatant was measured spectrophotometrically and was compared to the drug concentration in the original solution. Any ab-sorbance from the disintegrant was compensated for by subtracting the ab-sorbance from a drug-free experiment.
Description of essential methods
36
Table 2. Characteristics of the model compounds used in the experiments.
Substance Net charge pKaa cLogPa MWb λc (nm)
Sodium salicylate (−) 3.0 2.1 137 296
Naproxen (−) 4.8 3.0 230 262
Methylparaben (0) 8.3 1.9 152 256
Ethylparaben (0) 8.3 2.4 166 256
Propylparaben (0) 8.2 2.9 180 256
Atenolol (+) 9.2 0.097 266 274
Alprenolol (+) 9.2 2.9 249 270
Diphenhydramine (+) 8.8 3.7 255 258
Verapamil (+) 8.8 3.9 455 278
Amitriptyline (+) 9.2 4.9 277 238
CPC (+) – 5.0 340 259 aCalculated values [153] bMolecular weight for the acid or base form. cWavelength for UV-absorbance measurements.
3.5.3. Release study Prehydrated SSG gels were prepared on the same day as the experiments were performed according to the swelling capacity of SSG in diphenhy-dramine or naproxen solution; hence, 11 mL of 20 mM diphenhydramine solution or 15 mL of 20 mM naproxen solution were added to 1 g SSG, yielding gels of 8% and 6% SSG, respectively. The resulting drug concentra-tions in the gels were 17 mM naproxen and 18 mM diphenhydramine.
The release of drug into 5 mM TB or 5 mM TB supplemented with 145 mM NaCl was measured at room temperature and under sink conditions using custom-made diffusion chambers [154]. Approximately 1 g of gel was weighed onto the donor compartment at the beginning of the experiment. The chamber was then immediately placed on a magnetic stirrer and samples of 1 mL were withdrawn manually from the receiver compartment over 3 h. Each sample was replaced by an aliquot of fresh medium. The drug content was analysed spectrophotometrically.
The apparent diffusion coefficients were derived from Equation 4, which describes the initial (�60% of the total amount released) one-dimensional Fickian diffusion from the diffusion chamber under sink conditions [155, 156].
πtD
CQ app02= (Eq. 4)
where Q is the amount of drug released per surface area, C0 is the original concentration of the drug in the gel, Dapp is the apparent diffusion coefficient of the drug in the gel and t is the time that has elapsed since the experiment began.

Description of essential methods
37
3.6. Clinical study
3.6.1. Study basics The absorption of desmopressin from the nasal powder formulation was compared with the absorption from a sublingual tablet and from a commer-cial nasal liquid spray (Desmopressin Alpharma). The nasal powder formu-lation was prepared as described in Section 3.2.2; SSG, which was air classi-fied to obtain particles between 10 and 50 μm, was used as carrier material. The sublingual tablet was prepared by direct compression after dry mixing desmopressin and mannitol. Carbomer was added for mucoadhesion and absorption enhancement in this formulation [157].
The clinical study was performed at the Berzelius Clinical Research Cen-ter, Linköping, Sweden, as an open-label, randomised, three-period cross-over pharmacokinetic study in healthy female and male volunteers. The study was approved by the Swedish medical agency and the regional inde-pendent ethics committee in Linköping (EudraCT number 2006-006774-25). A total of 13 volunteers, 6 male and 7 female, were included in the study after undergoing a medical examination and giving their written, informed consent. The three administrations were separated by a wash-out period of at least 3 days. The plasma concentrations of desmopressin from the venous blood samples were evaluated with a validated radioimmunoassay by MDS Pharma Services Switzerland AG. The lower limit of quantification was 2.5 pg/mL. The pharmacokinetic parameters were calculated with non-compartmental analysis using WinNonlin® 4.0. The area under the concen-tration-time curve (AUC) was calculated with the linear/logarithmic trape-zoidal method.
3.6.2. Administration of the study drugs The drug formulations were administered by the trained study personnel, who were provided with an instruction sheet to ensure that the nasal spray devices for powder and liquid administration were held at a constant angle to give an accurate dose straight into the nasal cavity, as shown in Figure 10. The powder formulation was sprayed into one nostril, whereas the liquid formulation was sprayed into both nostrils with a minimal delay between doses. The volunteers were instructed to blow their noses before administra-tion and were asked not to inhale or exhale during the actuation of the spray device. They were also asked to try to avoid sneezing or blowing the nose, and to remain seated in an upright position, for at least 15 min after admini-stration.

Description of essential methods
38
The sublingual tablet was placed under the tongue in the deepest part of the oral cavity and the patients were asked to keep their mouths shut without moving their tongues for 15 min after dosage; the tablet was to dissolve un-der the tongue without chewing or sucking. There was no water intake per-mitted from 1 h before until 1 h after administration of the tablet.
Figure 10. The powder spray device (UniDose DP® from Bespak) and diagrams given to the study personnel to ensure consistent positioning of the nasal powder and liquid spray during administration.
3.6.3. Evaluation of the volunteers’ views The volunteers were asked to fill out an evaluation form after each admini-stration to record their opinions on the different formulations. The question-naire was divided into four parts, one for each dosage form (nasal powder, sublingual tablet and commercial nasal liquid spray) and one for final com-parisons. The final comparative part was completed after all doses had been received. The volunteers were asked if they had previously used a nasal spray or sublingual tablet and were asked to grade and comment on the taste and smell of each dosage form, and on the sensations associated with ad-ministration of the dosage forms. In the final part, they were asked which one of the three formulations they would prefer and why.
3.7. Statistical analysis All statistical analyses were performed with the statistical software Minitab®. A p-value of less than 0.05 was regarded as significant. Statistical signifi-cances were evaluated with General Linear Models (equivalent to ANOVA) and comparisons were performed with Bonferroni’s multiple comparisons test. In the clinical study (Paper V), treatment, dosing sequence and period were included as fixed parameters and subject number within sequence as a random effect in the GLM for statistical evaluation of the pharmacokinetic parameters AUC, maximum concentration (Cmax) and half-life (t½). A loga-rithmic transformation of the AUC and Cmax values was performed to ensure normal distribution. Differences in time to Cmax (tmax) were evaluated with the nonparametric Kruskal-Wallis test.

Description of essential methods
39
The logarithmic values of the pharmacokinetic parameters AUC and Cmax were evaluated in Paper V with Bartlett’s test for equal variances. The stan-dard deviations of logarithmically transformed data can be used instead of the CV to evaluate the homoscedasticity of data in different size ranges [158], which should be especially suitable when the data is normally distrib-uted after transformation.

40
4. Key findings
A summary of the most important results in Papers I-V is given in this chap-ter. Some condensed background information with reference to the relevant articles is given with each new section.
4.1. Interactive mixtures with small carrier particles As discussed in the introduction, powder formulations may have many ad-vantages as nasal drug delivery systems. However, for a powder formulation to reach the pharmaceutical market, it must also have a cost-effective, repro-ducible means of production. Paper I discusses the creation of mucoadhesive interactive mixtures suitable for nasal administration.
4.1.1. Characterisation of mixtures in Papers I-III The creation of interactive mixtures with mucoadhesive carrier particles down to a particle size of approximately 30 μm was shown to be possible in Paper I. However, comparisons between experimental homogeneity meas-urements and the theoretical homogeneity models described in Section 3.2.1 could not be used to distinguish between random and interactive mixtures in this size range. An example is given in Figure 11, which provides the ex-perimental and theoretical CV values for a mixture of SSG and 4% sodium salicylate. Although the experimental CV values are very low, they are still well above the theoretical values for both an interactive and a random mix-ture; it was also not possible to make a correlation between the slopes of the lines. The experimental results would have been affected by both the intrin-sic disorder of the samples, i.e., the distribution of the particles in the mix-ture, and the analytical errors from sampling and concentration measure-ments. The difficulties in discerning between analytical and intrinsic vari-ability have been addressed by Crooks and Ho [159], who proposed that an arbitrary maximum CV value of 5%, from powder samples in a size corre-sponding to the intended dose, could be used to assure good mixture quality. However, the results obtained here show that this limit is much too high for interactive mixtures with small carrier particles. Of the mixtures created during the course of this thesis, only the mixture containing 16 μm SSG particles resulted in a CV above 5% (Table 3).

Key findings
41
Figure 11. Homogeneity of a mixture of SSG with a mean particle size of 45 μm and 4% sodium salicylate (mixture 45:4 in Table 3). The theoretical CV values for a random mixture (RM) and an interactive mixture (IM) are shown together with the experimental results.
The homogeneity measurements were complemented by visual inspection using SEM, which showed that, as expected, aggregates remained in the mixture containing 16 μm SSG, but also in the mixtures containing SSG plus 6% sodium salicylate and SSG plus 5% DHE in Paper II (Figure 12). The surface coverage (Rs, Eq.3) of these carriers was also not as high as pre-dicted. It may be worth mentioning that, in these cases, random mixtures were not formed either, as aggregates of the fine particles were still present.
Table 3. The characteristics of the mixtures in Papers I-III Substance Sodium salicylate DHE Oxazepam
Mixturea 59:1 45:1 30:1 16:1 45:2 45:4 45:6 36:5 45:2 45:4 CV (%)b 1.77 1.32 1.29 8.15 0.917 1.18 3.04 4.0 2.13 1.88 Rs (%) 5.9 4.9 3.5 1.9 9.8 19 29 26 18 35 aDescribes the median carrier size (μm): amount of fine particulate component (w/w %). bResults from the small sample size of approximately 15 mg.
Figure 12. SEM pictures of SSG (A), SSG plus 4% sodium salicylate (B), SSG plus 6% sodium salicylate (C), SSG plus 5% DHE (D) and SSG plus 4% oxazepam (E). Scale bars represent 20 μm.

Key findings
42
The mixtures that had CV values in the order of 2%, which is within the error range for analytical procedures of this type [33, 46], were all interac-tive after visual inspection (Figure 12, Table 3). In Paper I, it was therefore suggested that homogeneity measurements can be used as an indication of the kind of mixture, but that they should be used in conjunction with visual inspection, at least for mixtures of new compounds. In this context, a limit for the CV within the order of analytical error, e.g. 2%, would be more ap-propriate, provided that the sampling has been satisfactorily carried out.
The use of sieve tests to characterise the interactive mixtures, as per-formed by Malmqvist et al. [36], was not applicable to the present mixtures since the size of the carrier particles was too small for successful sieving. Spatial statistical models and especially radial distribution functions were later suggested as a means of determining the distribution of the fine parti-cles and thus the degree of order in the micrographs [160]. These methods could be used to obtain an objective evaluation of the degree of order in the mixtures with small carrier particles.
4.2. In vitro absorption of dihydroergotamine The horizontal Ussing chamber setup has previously been used by our re-search group to evaluate wet delivery systems [161, 162]. The air interface on the mucosal side in the chamber also provides an opportunity for measur-ing the absorption of drugs from dry powders. The effect of prolonged resi-dence time cannot be evaluated since it is a closed system and a liquid for-mulation will consequently also remain in the chamber throughout the ab-sorption phase. The method should, however, be useful for evaluating ab-sorption enhancing effects. In Paper II, the horizontal Ussing chamber system was used to investigate the absorption of DHE from the novel pow-der formulation described in Paper I; differences in the absorption of DHE across nasal respiratory and olfactory mucosa were also evaluated.
4.2.1. The porcine olfactory mucosa It had been shown in a previous study that nasal respiratory mucosa can be excised from the ventral nasal concha of the pig [149]. The olfactory mucosa can be differentiated because it is visibly thicker than the respiratory mucosa and has a yellowish colour. The histological evaluation also demonstrated that it had been correctly excised as it contained Bowman’s glands (Figure 13b), the typical ciliated dendritic knobs of the olfactory neurons (Figure 13c) and nonciliated cells, which are rare in the porcine respiratory mucosa [163]. The dendritic knobs differ from the ciliated epithelial cells in the res-piratory epithelium shown in Figure 13a.

Key findings
43
Figure 13. The drawing shows a frontal section of the porcine snout, which has been divided along septum, and the points of excision for nasal respiratory mucosa (A) and olfactory mucosa (B and C). The nasal respiratory mucosa has ciliated epithelial cells (A) and the olfactory mucosa contains Bowman’s glands (B) as well as the dendritic knobs of olfactory neurons (C) and unciliated epithelial cells.
4.2.2. The effect of the powder formulation There was no statistically significant difference in the transfer of DHE to the receiver compartment between the powder formulation and the reference solution (Figure 14). It has been suggested that swelling particle systems can facilitate initial paracellular absorption by widening of the tight junctions [109]. This should be favourable for the absorption of DHE since it is most likely absorbed paracellularly because of its size and polarity (Figure 4). However, the addition of EDTA in the reference solution, which was neces-sary to keep the drug stable, may have had a similar effect on the absorption since EDTA can also be used as an absorption enhancer [164]. During the viability measurement performed after the transport study, it was noted that the resistance of the mucosae was significantly lower after contact with the liquid than after the powder. The resistance can be used as a measure of the integrity of the tight junctions and a decrease in resistance could thus indi-cate that facilitated paracellular transfer occurred from the reference solu-tion, as has been suggested in previous investigations [78].
A drawback of the horizontal Ussing chamber system for a powder such as SSG, is that only a small amount of liquid is available. In the in vivo situa-tion, the powder can be distributed over a larger surface area and there will be a continuous production of mucus. The powder appeared to be partly dry after application onto the mucosa in the Ussing system, and this could have impeded the absorption of DHE. The insufficient deaggregation, noticed in the SEM micrographs, would also have been disadvantageous for the disso-lution. It is also possible that DHE interacted with the negatively charged hydrogel formed by SSG to prevent absorption. However, the risk of this happening appeared insignificant when the possibility was further addressed in Paper IV. The horizontal Ussing chamber technique may be better suited for powders with less extensive swelling capacity.

Key findings
44
Figure 14. The cumulative amount of DHE per square centimetre transferred to the receiver compartment from the liquid (�) and the powder (�) formulations (n = 8 and 10, respectively).
4.2.3. Differences in mucosal transfer Because the antimigraine drug DHE is unlikely to be absorbed across the BBB, olfactory transfer to the brain could be beneficial for its effect. In fact, the Papp for DHE was significantly higher across porcine olfactory mucosa than across nasal respiratory mucosa (5.04·10-7 ± 4.59·10-7 and 1.48·10-7 ± 1.56·10-7 cm/s, respectively, p = 0.043). The olfactory mucosa has greater enzymatic activity than the nasal respiratory mucosa [12, 165] and DHE is metabolised by Cyp 3A4, which has been detected in the porcine olfactory mucosa [165]. Hence, low olfactory uptake would have been expected. It is possible that enzymatic degradation was decreased in the artificial setup of the horizontal Ussing chambers [79]; however, this should at best result in comparable absorption to that across respiratory mucosa. The epithelial cells are not separated from the submucosa when the mucosa is mounted in the Ussing chamber (Figure 7). The substance must therefore cross both layers to reach the receiver chamber. It is thus possible that the increased absorp-tion across the olfactory mucosa was caused by facilitated submucosal trans-port along the perineural space surrounding the olfactory nerve cells. This effect may not be as pronounced in vivo, since DHE can then be transported to the systemic blood circulation via the blood vessels underneath the epithe-lial cells. Corroborating results were, however, obtained in an in vivo study by Wang et al. [166] which indicated that DHE is transferred to the brain from the cerebrospinal fluid, which one would anticipate if perineural trans-port occurs across the olfactory mucosa.

Key findings
45
4.3. Mucoadhesion measurements It has been shown that interactive mixtures can be used to make inert carrier particles mucoadhesive by covering their surface with mucoadhesive fines [53]. The powder formulation for nasal drug delivery, on the contrary, con-sists of mucoadhesive carrier particles that are covered by inert particles, which may instead have a negative effect on mucoadhesion. The correlation between surface coverage and mucoadhesion was therefore investigated in Paper III. It has been suggested that substitutes for mucosa can be used to measure the mucoadhesion of swelling particle systems [126, 127] and this concept was evaluated in Paper III to create a simple, reproducible method. Mucoadhesion was evaluated for two kinds of carrier particles with different capacities for absorbing liquids: highly swellable SSG and partly prege-latinised maize starch (PPS).
4.3.1. The simplified method Measurements of mucoadhesion onto nasal mucosa showed that the magne-sium stearate-covered SSG particles had significantly lower tensile work values and fracture strength than the pure SSG after 1 s of contact, but that the differences were insignificant after 120 s (Figure 15). Remains of gel were found at both the upper movable probe and the lower surface after the measurements, indicating that detachment occurred through cohesive failure between the swollen gel particles, even though weaker fracture strengths have previously been recorded for the mucus layer itself [167]. It was there-fore assumed that the dry powder strengthened the mucus layer by dehydra-tion, as has previously been reported by Mortazavi and Smart for other dry dosage forms [16]. Because the failure occurred between the gel particles, the tensile work could be measured using a mucosal substitute comprising a 2x2 cm piece of absorbent paper to which 60 μL of 4% mucin solution was added directly before the measurement (Figure 15). Evaluation of the vol-ume and concentration of the solution showed that these settings gave the most consistent results with the least number of failures. The water retention of the mucosal substitute and, thus, the rate and degree of hydration of the particles were too low when less mucin was added and too high when the concentration of mucin was increased.
The momentary contact time with the substitute was prolonged to 3 s (Figure 15) as this gave comparable results to those obtained with the mu-cosa after 1 s of contact. The mucosa is softer and more uneven than the flat absorbent paper, which prolonged the time from the first contact until the contact force of 10 mN was reached and the measurement began.
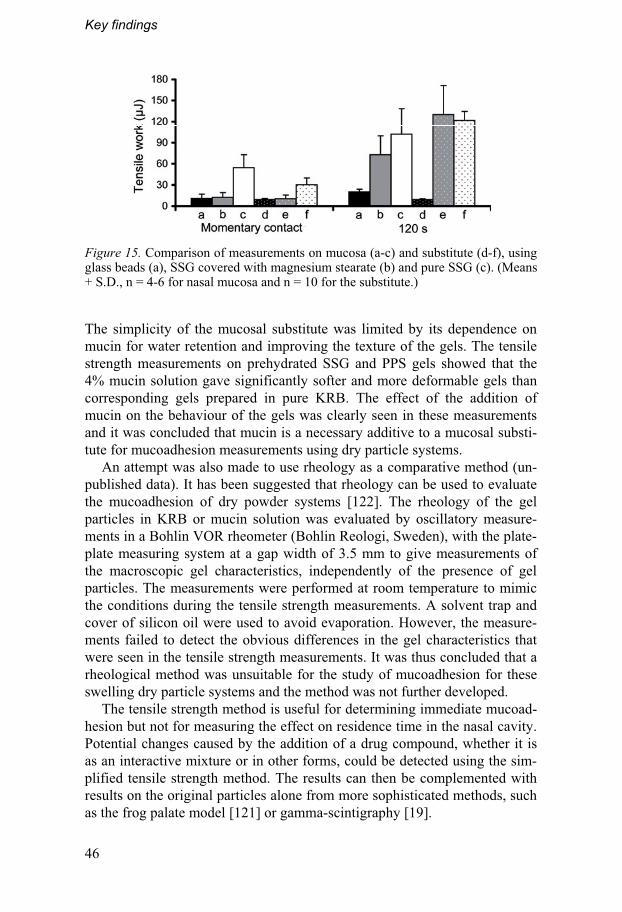
Key findings
46
Figure 15. Comparison of measurements on mucosa (a-c) and substitute (d-f), using glass beads (a), SSG covered with magnesium stearate (b) and pure SSG (c). (Means + S.D., n = 4-6 for nasal mucosa and n = 10 for the substitute.)
The simplicity of the mucosal substitute was limited by its dependence on mucin for water retention and improving the texture of the gels. The tensile strength measurements on prehydrated SSG and PPS gels showed that the 4% mucin solution gave significantly softer and more deformable gels than corresponding gels prepared in pure KRB. The effect of the addition of mucin on the behaviour of the gels was clearly seen in these measurements and it was concluded that mucin is a necessary additive to a mucosal substi-tute for mucoadhesion measurements using dry particle systems.
An attempt was also made to use rheology as a comparative method (un-published data). It has been suggested that rheology can be used to evaluate the mucoadhesion of dry powder systems [122]. The rheology of the gel particles in KRB or mucin solution was evaluated by oscillatory measure-ments in a Bohlin VOR rheometer (Bohlin Reologi, Sweden), with the plate-plate measuring system at a gap width of 3.5 mm to give measurements of the macroscopic gel characteristics, independently of the presence of gel particles. The measurements were performed at room temperature to mimic the conditions during the tensile strength measurements. A solvent trap and cover of silicon oil were used to avoid evaporation. However, the measure-ments failed to detect the obvious differences in the gel characteristics that were seen in the tensile strength measurements. It was thus concluded that a rheological method was unsuitable for the study of mucoadhesion for these swelling dry particle systems and the method was not further developed.
The tensile strength method is useful for determining immediate mucoad-hesion but not for measuring the effect on residence time in the nasal cavity. Potential changes caused by the addition of a drug compound, whether it is as an interactive mixture or in other forms, could be detected using the sim-plified tensile strength method. The results can then be complemented with results on the original particles alone from more sophisticated methods, such as the frog palate model [121] or gamma-scintigraphy [19].
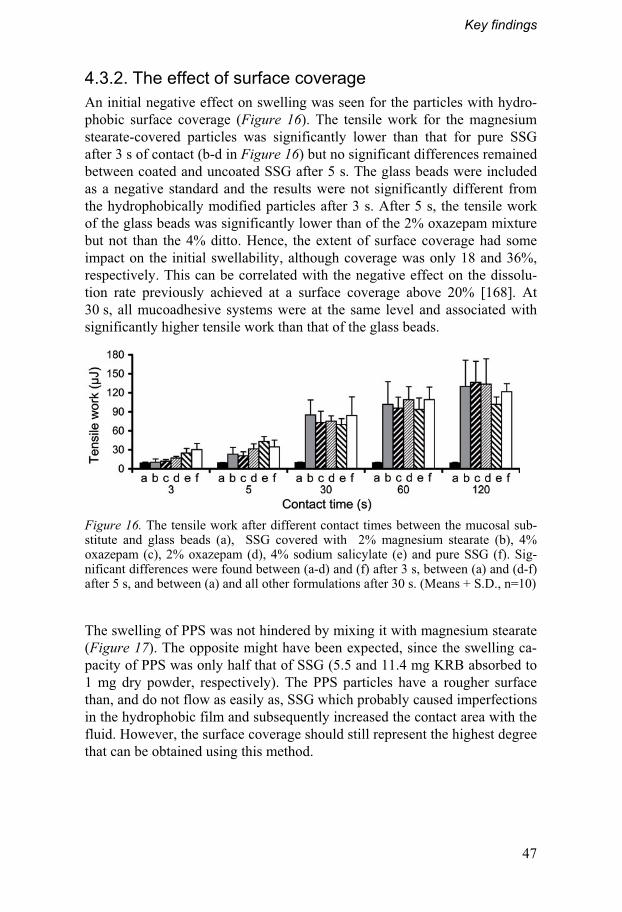
Key findings
47
4.3.2. The effect of surface coverage An initial negative effect on swelling was seen for the particles with hydro-phobic surface coverage (Figure 16). The tensile work for the magnesium stearate-covered particles was significantly lower than that for pure SSG after 3 s of contact (b-d in Figure 16) but no significant differences remained between coated and uncoated SSG after 5 s. The glass beads were included as a negative standard and the results were not significantly different from the hydrophobically modified particles after 3 s. After 5 s, the tensile work of the glass beads was significantly lower than of the 2% oxazepam mixture but not than the 4% ditto. Hence, the extent of surface coverage had some impact on the initial swellability, although coverage was only 18 and 36%, respectively. This can be correlated with the negative effect on the dissolu-tion rate previously achieved at a surface coverage above 20% [168]. At 30 s, all mucoadhesive systems were at the same level and associated with significantly higher tensile work than that of the glass beads.
Figure 16. The tensile work after different contact times between the mucosal sub-stitute and glass beads (a), SSG covered with 2% magnesium stearate (b), 4% oxazepam (c), 2% oxazepam (d), 4% sodium salicylate (e) and pure SSG (f). Sig-nificant differences were found between (a-d) and (f) after 3 s, between (a) and (d-f) after 5 s, and between (a) and all other formulations after 30 s. (Means + S.D., n=10)
The swelling of PPS was not hindered by mixing it with magnesium stearate (Figure 17). The opposite might have been expected, since the swelling ca-pacity of PPS was only half that of SSG (5.5 and 11.4 mg KRB absorbed to 1 mg dry powder, respectively). The PPS particles have a rougher surface than, and do not flow as easily as, SSG which probably caused imperfections in the hydrophobic film and subsequently increased the contact area with the fluid. However, the surface coverage should still represent the highest degree that can be obtained using this method.

Key findings
48
Figure 17. The tensile work obtained after different contact times with pure SSG (�), pure PPS (� ), and magnesium stearate-covered SSG (�) and PPS (�). The error bars represent the standard deviations of 10 measurements.
The results shown in Figure 17 cannot be used to predict whether SSG or PPS is the better mucoadhesive agent. While SSG has a good swelling ca-pacity and creates a rigid gel that is likely to increase the residence time in the nasal cavity, its swelling is very rapid and the slower water absorption by PPS could possibly result in prolonged mucoadhesion [169], even though the gel is softer than SSG. The results do, however, show that the effect of hy-drophobic surface coverage is very short and should be negligible for muco-adhesion in vivo. New uncovered surface areas will appear as the particles begin to swell and the fluid absorption can continue rapidly. If the residence time is measured for the original powder material, it should therefore be safe to assume that it will not change after the creation of an interactive mixture.
4.4. Investigation of physicochemical interactions A suspicion was raised during the in vitro study (Paper II) that the cationic compound DHE may have interacted with the anionic gel formed by SSG, preventing it from being absorbed. Interactions between anionic gels and surfactants are well known; see for example the review by Hansson [170].
Interactions between disintegrants and drug substances can occur through ion exchange, lipophilic interactions or a combination of these if the sub-stance for example is surface active. Strong surfactants are known to aggre-gate within the gel structure above the critical aggregation concentration (cac), which generally occurs below their critical micelle concentration (cmc). The binding of drugs with different physicochemical characteristics, i.e., charge and lipophilicity, was therefore evaluated in Paper IV using SSG, PPS, croscarmellose sodium (CCS) and crospovidone (CPP). The objective was to investigate which characteristics were of importance and whether the interactions could affect in vivo absorption.
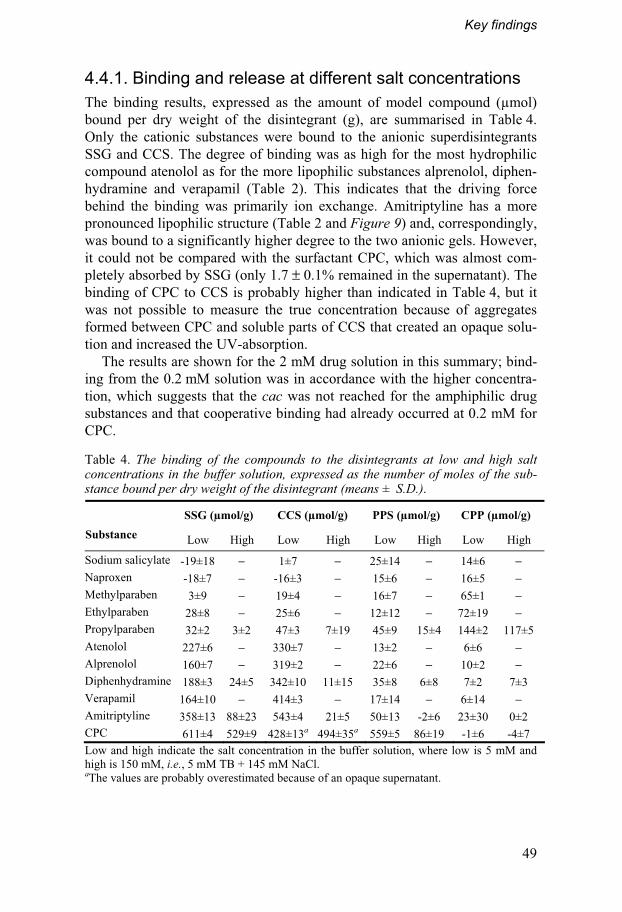
Key findings
49
4.4.1. Binding and release at different salt concentrations The binding results, expressed as the amount of model compound (μmol) bound per dry weight of the disintegrant (g), are summarised in Table 4. Only the cationic substances were bound to the anionic superdisintegrants SSG and CCS. The degree of binding was as high for the most hydrophilic compound atenolol as for the more lipophilic substances alprenolol, diphen-hydramine and verapamil (Table 2). This indicates that the driving force behind the binding was primarily ion exchange. Amitriptyline has a more pronounced lipophilic structure (Table 2 and Figure 9) and, correspondingly, was bound to a significantly higher degree to the two anionic gels. However, it could not be compared with the surfactant CPC, which was almost com-pletely absorbed by SSG (only 1.7 ± 0.1% remained in the supernatant). The binding of CPC to CCS is probably higher than indicated in Table 4, but it was not possible to measure the true concentration because of aggregates formed between CPC and soluble parts of CCS that created an opaque solu-tion and increased the UV-absorption.
The results are shown for the 2 mM drug solution in this summary; bind-ing from the 0.2 mM solution was in accordance with the higher concentra-tion, which suggests that the cac was not reached for the amphiphilic drug substances and that cooperative binding had already occurred at 0.2 mM for CPC.
Table 4. The binding of the compounds to the disintegrants at low and high salt concentrations in the buffer solution, expressed as the number of moles of the sub-stance bound per dry weight of the disintegrant (means ± S.D.).
SSG (μmol/g) CCS (μmol/g) PPS (μmol/g) CPP (μmol/g) Substance Low High Low High Low High Low High
Sodium salicylate -19±18 − 1±7 − 25±14 − 14±6 −
Naproxen -18±7 − -16±3 − 15±6 − 16±5 −
Methylparaben 3±9 − 19±4 − 16±7 − 65±1 −
Ethylparaben 28±8 − 25±6 − 12±12 − 72±19 −
Propylparaben 32±2 3±2 47±3 7±19 45±9 15±4 144±2 117±5
Atenolol 227±6 − 330±7 − 13±2 − 6±6 −
Alprenolol 160±7 − 319±2 − 22±6 − 10±2 −
Diphenhydramine 188±3 24±5 342±10 11±15 35±8 6±8 7±2 7±3
Verapamil 164±10 − 414±3 − 17±14 − 6±14 −
Amitriptyline 358±13 88±23 543±4 21±5 50±13 -2±6 23±30 0±2
CPC 611±4 529±9 428±13a 494±35a 559±5 86±19 -1±6 -4±7
Low and high indicate the salt concentration in the buffer solution, where low is 5 mM and high is 150 mM, i.e., 5 mM TB + 145 mM NaCl. aThe values are probably overestimated because of an opaque supernatant.
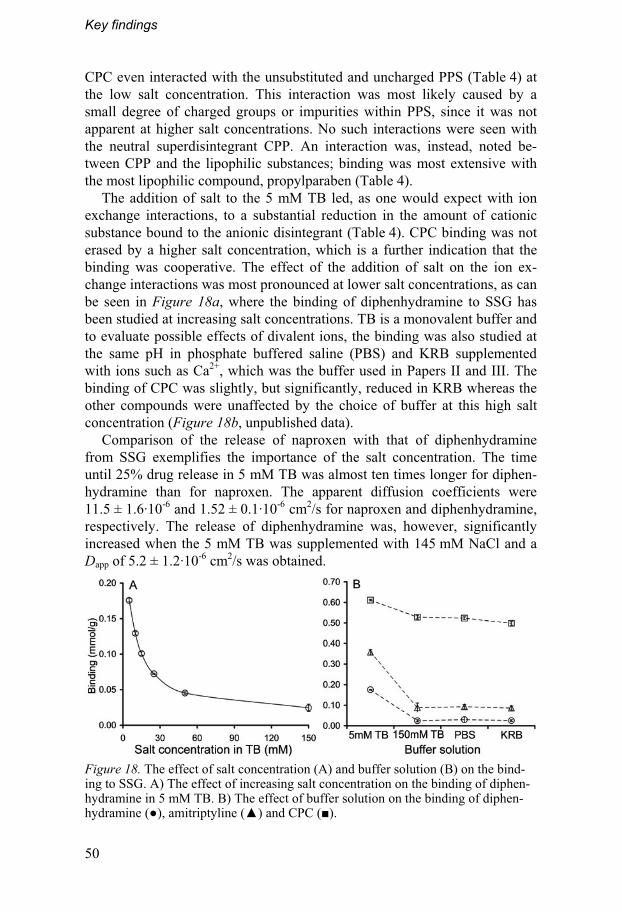
Key findings
50
CPC even interacted with the unsubstituted and uncharged PPS (Table 4) at the low salt concentration. This interaction was most likely caused by a small degree of charged groups or impurities within PPS, since it was not apparent at higher salt concentrations. No such interactions were seen with the neutral superdisintegrant CPP. An interaction was, instead, noted be-tween CPP and the lipophilic substances; binding was most extensive with the most lipophilic compound, propylparaben (Table 4).
The addition of salt to the 5 mM TB led, as one would expect with ion exchange interactions, to a substantial reduction in the amount of cationic substance bound to the anionic disintegrant (Table 4). CPC binding was not erased by a higher salt concentration, which is a further indication that the binding was cooperative. The effect of the addition of salt on the ion ex-change interactions was most pronounced at lower salt concentrations, as can be seen in Figure 18a, where the binding of diphenhydramine to SSG has been studied at increasing salt concentrations. TB is a monovalent buffer and to evaluate possible effects of divalent ions, the binding was also studied at the same pH in phosphate buffered saline (PBS) and KRB supplemented with ions such as Ca2+, which was the buffer used in Papers II and III. The binding of CPC was slightly, but significantly, reduced in KRB whereas the other compounds were unaffected by the choice of buffer at this high salt concentration (Figure 18b, unpublished data).
Comparison of the release of naproxen with that of diphenhydramine from SSG exemplifies the importance of the salt concentration. The time until 25% drug release in 5 mM TB was almost ten times longer for diphen-hydramine than for naproxen. The apparent diffusion coefficients were 11.5 ± 1.6·10-6 and 1.52 ± 0.1·10-6 cm2/s for naproxen and diphenhydramine, respectively. The release of diphenhydramine was, however, significantly increased when the 5 mM TB was supplemented with 145 mM NaCl and a Dapp of 5.2 ± 1.2·10-6 cm2/s was obtained.
Figure 18. The effect of salt concentration (A) and buffer solution (B) on the bind-ing to SSG. A) The effect of increasing salt concentration on the binding of diphen-hydramine in 5 mM TB. B) The effect of buffer solution on the binding of diphen-hydramine (�), amitriptyline (�) and CPC (�).

Key findings
51
4.4.2. In vivo correlations and effect in vitro Binding to CPC was strongly cooperative and was not eradicated by in-creased salt concentrations. The amphiphilic drug substances were, however, not surface active enough to reach the cac and achieve such strong interac-tions. The most lipophilic of the compounds, amitriptyline (Table 2), has a cmc of 25 mM at 150 mM NaCl [171], compared with the cmc of 0.79 mM for CPC already at 100 mM NaCl [172]. Although the lipophilicity of the compounds should be comparable (Table 2), the rigidity of the drug sub-stance is likely to reduce its ability to aggregate.
A binding of 100 μmol/g, which is the approximate maximum amount ob-tained for the lipophilic or ion exchange interactions at physiological salt concentrations, would correspond to �1μg of a small molecular compound (MW�500 g/mol) being bound to 20 mg disintegrant. The dose must thus either be very low or the amount of disintegrant very high for this to affect in vivo absorption. The release of the compound may be somewhat delayed by remaining ionic interactions. However, the gel will be spread over a larger surface area in vivo and the release will therefore be quicker than in the in vitro experiments. The carrier particles in interactive mixtures will further-more not need to be fully hydrated before the drug compound can be re-leased as the drug is only deposited on the surface. The results of the in vitro experiments can therefore be seen as a worst-case scenario for the in vivo release profile.
The results of in vitro experiments can, nevertheless, be greatly affected if pure water is used, as only a small amount of salt is necessary to give a sub-stantial decrease in the binding of ionic substances. SSG may contain up to 7% NaCl [59] and the amount of salt can vary from batch to batch and al-most certainly between the different brands [60]. In vitro experiments should therefore preferably be performed at physiological salt concentrations to avoid unnecessary variations.
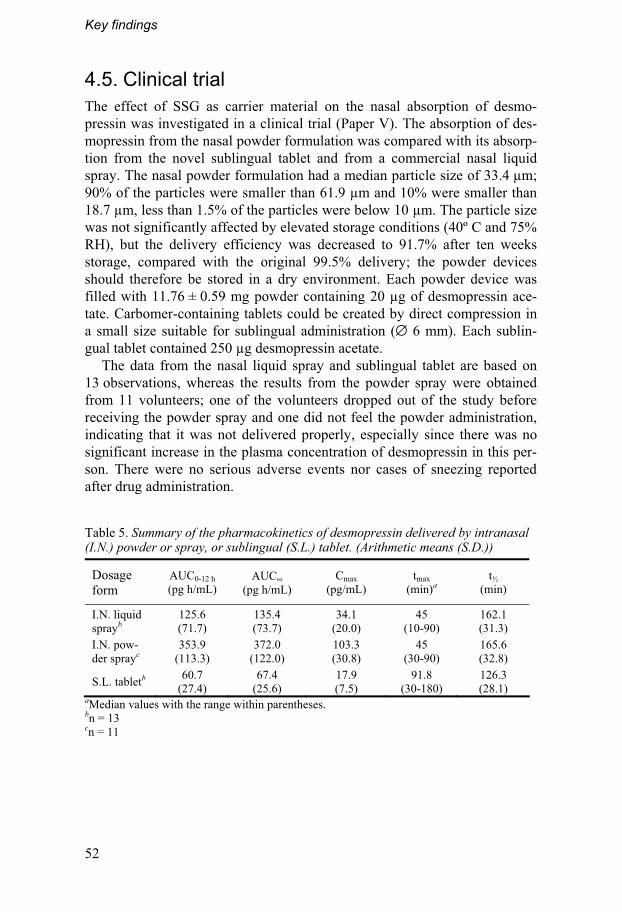
Key findings
52
4.5. Clinical trial The effect of SSG as carrier material on the nasal absorption of desmo-pressin was investigated in a clinical trial (Paper V). The absorption of des-mopressin from the nasal powder formulation was compared with its absorp-tion from the novel sublingual tablet and from a commercial nasal liquid spray. The nasal powder formulation had a median particle size of 33.4 μm; 90% of the particles were smaller than 61.9 μm and 10% were smaller than 18.7 μm, less than 1.5% of the particles were below 10 μm. The particle size was not significantly affected by elevated storage conditions (40º C and 75% RH), but the delivery efficiency was decreased to 91.7% after ten weeks storage, compared with the original 99.5% delivery; the powder devices should therefore be stored in a dry environment. Each powder device was filled with 11.76 ± 0.59 mg powder containing 20 μg of desmopressin ace-tate. Carbomer-containing tablets could be created by direct compression in a small size suitable for sublingual administration (∅ 6 mm). Each sublin-gual tablet contained 250 μg desmopressin acetate.
The data from the nasal liquid spray and sublingual tablet are based on 13 observations, whereas the results from the powder spray were obtained from 11 volunteers; one of the volunteers dropped out of the study before receiving the powder spray and one did not feel the powder administration, indicating that it was not delivered properly, especially since there was no significant increase in the plasma concentration of desmopressin in this per-son. There were no serious adverse events nor cases of sneezing reported after drug administration.
Table 5. Summary of the pharmacokinetics of desmopressin delivered by intranasal (I.N.) powder or spray, or sublingual (S.L.) tablet. (Arithmetic means (S.D.))
Dosage form
AUC0-12 h (pg h/mL)
AUC∞ (pg h/mL)
Cmax (pg/mL)
tmax
(min)a
t½ (min)
I.N. liquid sprayb
125.6 (71.7)
135.4 (73.7)
34.1 (20.0)
45 (10-90)
162.1 (31.3)
I.N. pow-der sprayc
353.9 (113.3)
372.0 (122.0)
103.3 (30.8)
45 (30-90)
165.6 (32.8)
S.L. tabletb 60.7 (27.4)
67.4 (25.6)
17.9 (7.5)
91.8 (30-180)
126.3 (28.1)
aMedian values with the range within parentheses. bn = 13 cn = 11

Key findings
53
4.5.1. In vivo absorption of desmopressin The plasma desmopressin concentration-time profiles are presented in Figure 19 and other pharmacokinetic parameters are given in Table 5. Plasma desmopressin concentrations (AUC0-12 h, AUC� and Cmax) after the sublingual tablet were significantly lower than those after the nasal liquid spray (p <0.001) The median tmax was significantly longer after sublingual administration (p <0.001), which is to be expected considering the differ-ences in mucosal constitution. Given the higher dose, the bioavailability would be approximately 25 times lower from the sublingual tablet than from the nasal liquid spray. Plasma desmopressin concentrations of approximately the same magnitude as those measured here have been obtained after sublin-gual administration previously [173, 174], but absorption was not enhanced with the novel formulation. The sublingual formulation could be further improved by decreasing its disintegration time, since this was noticed as a disadvantage with the formulation. Carbomer has been associated with both facilitated paracellular absorption and reduced enzymatic degradation [157], but it is not possible to draw any conclusions regarding its effect on the ab-sorption from the sublingual tablet as it was most likely counteracted by a poor dissolution. The advantage of the tablet was rather that it could be pro-duced by direct compression after dry mixing of desmopressin and the car-rier mannitol instead of by more costly methods such as lyophilisation.
The absorption of desmopressin (AUC0-12 h, AUC� and Cmax) from the na-sal powder formulation was significantly more extensive than from the liq-uid formulation (p <0.0001). The median tmax was comparatively long for nasal delivery (45 min), probably because of the size of the molecule, as the same tmax was obtained for both the powder and the liquid formulation. The geometric means showed that absorption from the nasal powder spray was
Figure 19. Plasma pharmacokinetics after administration of desmopressin in the commercial nasal liquid spray (�, n = 13), the nasal powder spray (�, n = 11) or the sublingual tablet (�, n = 13). Mean values +S.D..

Key findings
54
three times higher than from the nasal liquid spray. The bioavailability of the liquid spray is documented as 3-5% [74], which indicates that the bioavail-ability of the powder formulation lies between 9% and 15%. Higher bioavailabilities from nasal liquid sprays have been reported in the scientific literature; one of the most commonly cited is 11.3% [175], which would correspond to 30% bioavailability from the powder formulation.
The results of previous studies have shown that an increased residence time alone is not enough to increase the absorption of desmopressin [106, 107]. The increased absorption from the nasal powder formulation was thus most likely achieved by a combination of three factors: (1) improved deposi-tion in the nasal cavity compared with a liquid, which could run straight down the oesophagus; (2) prolonged residence time in the nasal cavity; and (3) facilitated paracellular absorption, probably as a result of a temporary opening of the tight junctions caused by dehydration of the epithelial cells [109].
The in vivo results support the assumptions made in Papers II and IV. The evaluation of the powder formulation in Paper II was likely negatively af-fected by methodological shortcomings and the supposition that ion ex-change interactions should be negligible at physiological salt concentrations in vivo, is strengthened by the extensive absorption of the positively charged, low dose peptide in the clinical trial.
The variability in the absorption of desmopressin is a further area in need of improvement, as discussed in the introduction (Section 1.5.1). The coeffi-cient of variation is usually used to express variability despite the log normal distribution of the pharmacokinetic parameters. In this study, the CV tended to be lower after administration of the nasal powder spray (32.0 and 29.8% for AUC0-12 h and Cmax, respectively) than after either the nasal liquid spray (57.1 and 58.5%) or the sublingual tablet (45.1 and 42.0%). However, the differences were not statistically significant, as was shown by comparing the standard deviations of the logarithmic values [158].
There did not seem to be an individual inclination for enhanced uptake of desmopressin, as high absorption from one formulation did not automatically mean high absorption from another (Figure 20). As can also be seen in Figure 20, there seemed to be more extensive absorption from the powder formulation in female volunteers. Indications of differences in the absorption of desmopressin correlated with formulation and gender, but not with gender alone, have been reported previously [77]. However, it was not within the scope of this study to evaluate gender-related differences and, as both the number of observations and the age (the female volunteers were in general younger than the male volunteers) differed between the groups, it is not pos-sible to draw any conclusions regarding these effects.
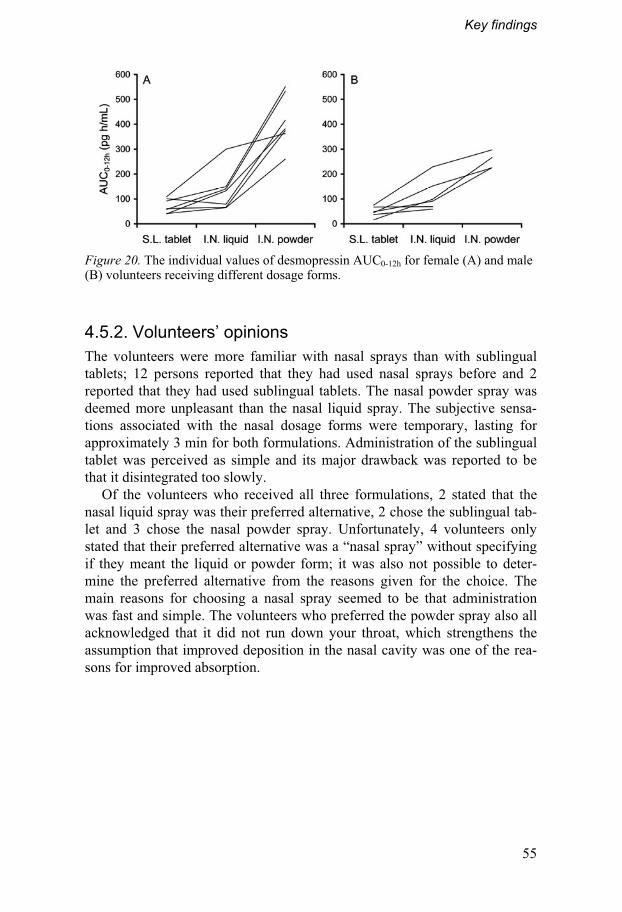
Key findings
55
Figure 20. The individual values of desmopressin AUC0-12h for female (A) and male (B) volunteers receiving different dosage forms.
4.5.2. Volunteers’ opinions The volunteers were more familiar with nasal sprays than with sublingual tablets; 12 persons reported that they had used nasal sprays before and 2 reported that they had used sublingual tablets. The nasal powder spray was deemed more unpleasant than the nasal liquid spray. The subjective sensa-tions associated with the nasal dosage forms were temporary, lasting for approximately 3 min for both formulations. Administration of the sublingual tablet was perceived as simple and its major drawback was reported to be that it disintegrated too slowly.
Of the volunteers who received all three formulations, 2 stated that the nasal liquid spray was their preferred alternative, 2 chose the sublingual tab-let and 3 chose the nasal powder spray. Unfortunately, 4 volunteers only stated that their preferred alternative was a “nasal spray” without specifying if they meant the liquid or powder form; it was also not possible to deter-mine the preferred alternative from the reasons given for the choice. The main reasons for choosing a nasal spray seemed to be that administration was fast and simple. The volunteers who preferred the powder spray also all acknowledged that it did not run down your throat, which strengthens the assumption that improved deposition in the nasal cavity was one of the rea-sons for improved absorption.

56
5. Concluding remarks
The results presented in this thesis show that currently available mucoadhe-sive carrier particles can be used to create dry powder formulations for nasal drug delivery and that the bioavailability of drugs can be significantly im-proved with the powder formulation in comparison with a liquid spray.
More specifically, it was shown that interactive mixtures can be created with mucoadhesive carrier particles of approximately 30 μm in diameter. A simplified method for evaluating the mucoadhesion of dry particle systems was successfully developed; it showed that the surface coverage of the mu-coadhesive carrier led to a very short delay in the swelling of the particles that should not affect mucoadhesion in vivo. Furthermore, ion exchange in-teractions between cationic drugs and the anionic gel formed by SSG were low at physiological salt concentrations and appeared to be insignificant for the absorption in vivo. However, in vitro studies may be greatly affected by the interactions if a solution with low salt concentration is used.
While the in vitro study in horizontal Ussing chambers found no differ-ences in the uptake of DHE between the nasal powder formulation and a liquid, the clinical trial demonstrated significantly improved systemic uptake of the peptide drug desmopressin in the nasal powder spray formulation ver-sus a nasal liquid spray and a sublingual tablet. The in vitro method seems to be unsuitable for a powder with extensive swelling capacity; a more interest-ing result was that the in vitro transfer of DHE was significantly higher across olfactory mucosa than across nasal respiratory mucosa.
The significantly improved absorption of desmopressin from the nasal powder spray compared with the nasal liquid spray was not associated with a delayed absorption. The dosage forms were well accepted by the volunteers in the clinical trial. This is one of few studies that actually show the effect of dry powder formulations administered to human subjects. The improved bioavailability obtained in the in vivo study was probably caused by a com-bination of improved deposition, prolonged residence time and enhanced paracellular uptake.

57
6. Future outlook
The delivery system investigated here provides a new method of producing powder formulations for nasal drug delivery that should be applicable for large-scale production. The particle size of the formulation is determined by the size of the mucoadhesive carrier; the risk of pulmonary deposition can thus be reduced by removing carrier particles below 10 μm in advance by air classification or other means. If the carrier particles are too large, they may need further classification or size reduction to achieve beneficial distribution in the nasal cavity.
The small amount of drug that can be administered by nasal administra-tion limits the number of drugs that can be delivered by this route. With the formulation studied in this work, the drug amount in the powder spray would be restricted to approximately 2 mg, depending on the surface coverage achieved and the total amount of powder in each spray dose. On the other hand, the powder formulation is intended to improve the absorption, which will subsequently reduce the drug amount needed. The 12 mg powder dose in Paper V was perceived as more irritating than the liquid equivalent; yet, the sensation was only felt for a short time and was not irritating enough to induce sneezing in any of the volunteers. A powder of lower swelling capac-ity would probably be less irritating. The irritation of the powder must also be compared with the possible drawbacks of a liquid formulation. The pow-der formulation has the advantage over a bad tasting liquid spray of not run-ning down the throat.
A generally positive attitude towards trying new dosage forms such as na-sal powder sprays and sublingual tablets was revealed in a qualitative survey of the opinions of 20 migraine patients on different dosage forms [176]. An administration technique that is simple and discreet, and assurance that the same effect will always be obtained, were mentioned as important factors by the patients. A nasal powder spray may well fulfil these requirements, as it can be taken at any time, independent of nausea or water access and, as also seen in Paper V, could decrease variability in the extent of absorption as well as the possible bad taste caused by the liquid running down the oesophagus. An interesting continuation would be to investigate prescribing physicians’ views on different dosage forms and their willingness to inform their patients of the available alternatives.
There may be many reasons for choosing nasal administration. In my opinion, it is best to make use of at least one of the advantages with the ad-
Future outlook
58
ministration route, that is attempt to achieve a faster effect or a higher bioavailability than obtainable with oral administration or a more convenient administration than, for example, injections. The presented powder formula-tion could facilitate faster absorption as the active component is deposited on the surface of the carrier. The release of the drug is therefore not dependent on the full hydration of the carrier particles and subsequent diffusion to the mucosa.
Potential therapeutic areas for application of this formulation would, for example, include pain, nausea or insomnia. The nasal powder spray formula-tion would also be suitable for drugs that are unstable in solution and/or poorly absorbed, such as peptide drugs. It would be interesting to compare the positive effects achieved with the peptide formulation in this work with the effects of an interactive mixture containing SSG and a micronised pow-der of, for example, an antimigraine drug. The possibility of using other mucoadhesive carrier materials, such as unsubstituted starch or chitosan, would also be worth investigating both concerning the subjective sensations associated with the spray dose and their effect on drug absorption.

59
7. Populärvetenskaplig sammanfattning
Många av oss har använt nässprejer för lokal effekt mot nästäppa. Ytterligare några har kanske använt nässprejer för effekt ute i kroppen som ett alternativ till tabletter, till exempel mot migrän eller under en hormonbehandling. I den här avhandlingen beskrivs en ny pulversprej från inledande undersökningar till en studie i människa som visade att pulvret ökade upptaget av ett läke-medel mot inkontinens i jämförelse med den vanliga flytande nässprejen.
Mediciner som sväljs måste först stå emot nedbrytning i mage och tarm och därefter i levern, som är kroppens främsta organ för nedbrytning av sub-stanser. Den del av substansen som inte bryts ned kan fortsätta ut i kroppen och utöva sin effekt. Nässprejer ger generellt både ett snabbt och högt upptag av den aktiva substansen, eftersom den tas upp över den tunna nässlemhin-nan och åker direkt ut i kroppen utan att först passera levern. Fördelaktiga användningsområden för nässprejer är därför där en snabb effekt är fördelak-tig (till exempel i behandling mot smärta, illamående eller sömnsvårigheter) eller för aktiva substanser som har dåligt upptag från mage och tarm, som exempelvis peptidläkemedel (hormoner).
Det finns en del nackdelar med flytande nässprejer. En stor del av voly-men i en flytande sprejdos riskerar till exempel att försvinna direkt ned i svaljet, vilket både kan ge en bitter smak och göra att upptaget av den aktiva substansen försämras. En pulversprej skulle kunna förbättra läkemedelsupp-taget eftersom chansen är större att hela dosen blir kvar i näshålan. Används dessutom ett pulver, som kan suga åt sig vätska, kan det fastna på den fukti-ga slemhinnan och leda till längre uppehållstid i näshålan och bättre upptag.
Den här avhandlingen visar att en pulversprej kan tillverkas genom att blanda ett pulver med partiklar i storleksordningen 30 μm, som har förmågan att ta upp vätska, med ungefär tio gånger mindre partiklar av den aktiva sub-stansen. Substansen kan då fastna på ytan av de större bärarpartiklarna och bilda en så kallad interaktiv blandning. Pulverblandningen utvärderades på olika sätt för att undersöka dess funktionalitet. I det sista steget sattes ett peptidläkemedel mot inkontinens (desmopressin, Minirin®) på ytan av bärar-partiklarna. Pulversprejen jämfördes med en flytande nässprej i en klinisk studie med friska frivilliga försökspersoner. Upptaget visade sig vara lika snabbt och tre gånger högre från pulvret än från den flytande sprejen. Till-verkningen av pulversprejen är relativt enkel, eftersom färdiga bärarpar-tiklar, som normalt används i tabletter, används. Pulversprejen skulle kunna bli ett nytt sätt att förbättra upptaget av flera olika aktiva substanser.

60
8. Acknowledgements
Funding from Orexo AB is gratefully acknowledged, as is the company’s devoted support of and belief in research. The travel grants from the CD Carlsson Foundation and IF Foundation for Pharmaceutical Research are thankfully recognised. I also wish to express my sincere appreciation to all the people who have shown an interest in my work and helped in the compiling of this thesis, es-pecially: My great supervisors: Dr Erik Björk, my main supervisor, for a comforting, free and fun PhD education. I’ve learned to always be critical of research results and uncritical of opportunities (such as free gifts at conferences). Assoc Prof. Katarina Edsman, my co-supervisor, for adding the extra sci-entific knowledge, niceness and inspiration. Professor Christer Nyström, my initial supervisor, for giving me an enthusiastic beginning to my PhD studies and also for providing the possibility of working with a very exciting project.
Professor Göran Alderborn and Professor Martin Malmsten for providing research facilities at the Department of Pharmacy, Uppsala University.
Dr Helene Hägerström and Dr Björn Jansson for a terrific undergraduate project that became an important base for this thesis.
Co-authors Dr Ulrika Espefält Westin and Dr Susanne Bredenberg for great cooperation, and co-pioneer Christina Ljungberg for introducing me to a more qualitative world.
The fantastic people at Orexo AB for letting me feel like a part of your com-pany and sharing both your knowledge and fun events. Special thanks to Dr Susanne Bredenberg (and of course the whole DDI group), Thomas Lundqvist and Dr Nils-Otto Ahnfelt for all your support, and last but not least to all the skilled, optimistic and stubborn people who worked so hard with the OX19 project!
My exam workers Marcus Söderberg, Anna Willander Ventorp, Malin Morin and Maria Björnsson for your excellent, dedicated, patient work with powders, texture analysis, gels, interviews and more.

Acknowledgements
61
Elisabet Börjesson and the late Leif Dahlberg for helping me with new methods and old machines in the laboratory. I’m so lucky to have started the PhD project while you two were still there!
Christin Magnusson for doing the tough job of administering the teaching assignments and letting me keep lab C. Dr Göran Ocklind, Johan Gråsjö, Assoc Prof Göran Frenning, Dr Albert Mihranyan and Assoc Prof Per Hansson for contributing knowledge about computers, statistics and/or sci-ence.
Eva Nises Ahlgren, Ulla Wästberg Galik, Harriet Östlund, Eva Lide, Birgitta Rylén and Lotta Wahlberg for your kind help with small and big things and especially for giving the Department such a friendly foundation.
All the kind professionals who have contributed to the thesis, especially Be-spak for great cooperation with the nasal devices, Swedish Meats through Erik Lindberg for all snouts and Antona Wagstaff for linguistic revision of the manuscript.
The PhD students at the Department of Pharmacy for all the fun happenings and the great group spirit. Special thanks to past and present colleagues in the “Nose Group” and its neighbour the “Gel Group”: Dr Karin Östh, Dr Björn Jansson, Dr Ulrika Espefält Westin, Dr Helene Hägerström, Dr Tobias Bramer and Noel Dew. To Dr Frauke Fichtner, my enduring workroom mate, for not letting me forget my German.
The board of the Association of Pharmaceutics and Biopharmaceutics within the Swedish Academy of Pharmaceutical Sciences, the Alumni association at the Department of Pharmacy and the WC-club for important, inspiring and fun experiences.
All of you who have contributed with your wonderful noses for this thesis! They (= you) are something that I will remember when the written content is long forgotten.
All my friends – old, new, near-by and further away. No names are men-tioned but no one is ever forgotten. You make this thesis very unimportant and for that I am extremely grateful!
My family. Mamma Gunilla and pappa Carl Gustav, I’ve kept this for the last and most certain conclusion of the thesis: You are the best!

62
9. References
1. Proctor, D.F. and Andersen, I. The nose. Upper airway physiology and the atmospheric environment, Elsevier Biomedical Press, Amsterdam, 1982.
2. Chien, Y.W., Su, K.S.E., and Chang, S.-F. Nasal systemic drug delivery, Marcel Dek-ker, Inc., New York, 1989.
3. Gizurarson, S. The relevance of nasal physiology to the design of drug absorption studies. Adv Drug Deliv Rev, 1993, 11:329-347.
4. Marttin, E., Schipper, N.G., Verhoef, J.C., and Merkus, F.W. Nasal mucociliary clear-ance as a factor in nasal drug delivery. Adv Drug Deliv Rev, 1998, 29:13-38.
5. Mygind, N. and Dahl, R. Anatomy, physiology and function of the nasal cavities in health and disease. Adv Drug Deliv Rev, 1998, 29:3-12.
6. Sanderson, M.J. and Sleigh, M.A. Ciliary activity of cultured rabbit tracheal epithe-lium: beat pattern and metachrony. J Cell Sci, 1981, 47:331-347.
7. Carlstedt, I., Sheehan, J.K., Corfield, A.P., and Gallagher, J.T. Mucous glycoproteins: A gel of a problem. Essays Biochem, 1985, 20:40-76.
8. Yamamoto, A., Iseki, T., Ochi-Sugiyama, M., Okada, N., Fujita, T., and Muranishi, S. Absorption of water-soluble compounds with different molecular weights and [Asu1.7]-eel calcitonin from various mucosal administration sites. J Control Release, 2001, 76:363-374.
9. Donovan, M.D., Flynn, G.L., and Amidon, G.L. Absorption of polyethylene glycols 600 through 2000: The molecular weight dependence of gastrointestinal and nasal absorp-tion. Pharm Res, 1990, 7:863-868.
10. McMartin, C., Hutchinson, L.E.F., Hyde, R., and Peters, G.E. Analysis of structural requirements for the absorption of drugs and macromolecules from the nasal cavity. J Pharm Sci, 1987, 76:535-540.
11. Sarkar, M.A. Drug metabolism in the nasal mucosa. Pharm Res, 1992, 9:1-9. 12. Thornton-Manning, J.R. and Dahl, A.R. Metabolic capacity of nasal tissue interspecies
comparisons of xenobiotic-metabolizing enzymes. Mutat Res, 1997, 380:43-59. 13. Gu, J.M., Robinson, J.R., and Leung, S.H. Binding of acrylic polymers to
mucin/epithelial surfaces: structure-property relationships. Crit Rev Ther Drug Carrier Syst, 1988, 5:21-67.
14. Mathiowitz, E. and Chickering, D.E. Definitions, mechanisms, and theories of bioadhe-sion. In Mathiowitz, E., D.E. Chickering, and C.M. Lehr (eds.), Bioadhesive drug de-livery systems: Fundamentals, novel approaches, and development, Marcel Dekker, New York, 1999, pp. 1-10.
15. Smart, J.D. The basics and underlying mechanisms of mucoadhesion. Adv Drug Deliv Rev, 2005, 57:1556-1568.
16. Mortazavi, S.A. and Smart, J.D. An investigation into the role of water movement and mucus gel dehydration in mucoadhesion. J Control Release, 1993, 25:197-203.
17. Schipper, N.G., Verhoef, J.C., and Merkus, F.W. The nasal mucociliary clearance: relevance to nasal drug delivery. Pharm Res, 1991, 8:807-814.
18. Illum, L., Jorgensen, H., Bisgaard, H., Krogsgaard, O., and Rossing, N. Bioadhesive microspheres as a potential nasal drug delivery system. Int J Pharm, 1987, 39:189-199.

References
63
19. Soane, R.J., Frier, M., Perkins, A.C., Jones, N.S., Davis, S.S., and Illum, L. Evaluation of the clearance characteristics of bioadhesive systems in humans. Int J Pharm, 1999, 178:55-65.
20. Bridgewater, J. Fundamental powder mixing mechanisms. Powder Technol, 1976, 15:215-236.
21. Orr, N.A. and Shotton, E. The mixing of cohesive powders. Chemical engineer, 1973, 269:12-18.
22. Travers, D.N. and White, R.C. The mixing of micronized sodium bicarbonate with sucrose crystals. J Pharm Pharmacol, 1971, 23:S260-S261.
23. Hersey, J.A. Ordered mixing: a new concept in powder mixing practice. Powder Tech-nol, 1975, 11:41-44.
24. Egermann, H. Suggestions on the nomenclature of powder mixtures. Powder Technol, 1980, 26:235-237.
25. Egermann, H. Ordered mixtures- interactive mixtures. Powder Technol, 1983, 36:117-118.
26. Staniforth, J.N. Order out of chaos. J Pharm Pharmacol, 1987, 39:329-334. 27. Yip, C.W. and Hersey, J.A. Segregation in ordered powder mixtures. Powder Technol,
1977, 16:149-150. 28. Staniforth, J.N. Total mixing. Int J Pharm Tech & Prod Mfr, 1981, 2:7-12. 29. Yeung, C.C. and Hersey, J.A. Ordered powder mixing of coarse and fine particulate
systems. Powder Technol, 1979, 22:127-131. 30. Kulvanich, P. and Stewart, P.J. The effect of particle size and concentration on the
adhesive characteristics of a model drug-carrier interactive system. J Pharm Pharma-col, 1987, 39:673-678.
31. de Boer, A.H., Le Brun, P.P., van der Woude, H.G., Hagedoorn, P., Heijerman, H.G., and Frijlink, H.W. Dry powder inhalation of antibiotics in cystic fibrosis therapy, part 1: development of a powder formulation with colistin sulfate for a special test inhaler with an air classifier as de-agglomeration principle. Eur J Pharm Biopharm, 2002, 54:17-24.
32. Hersey, J.A. Preparation and properties of ordered mixtures. Aust J Pharm Sci, 1977, 6:29-31.
33. Malmqvist, K. and Nyström, C. Studies on direct compression of tablets IX. The effect of scaling-up on the preparation of ordered mixtures in double-cone mixers. Acta Pharm Suec, 1984, 21:21-30.
34. Nyström, C. and Malmqvist, K. Studies on direct compression of tablets I. The effect of particle size in mixing finely divided powders with granules. Acta Pharm Suec, 1980, 17:282-287.
35. Malmqvist, K. and Nyström, C. Studies on direct compression of tablets VIII. A sieve classification method for the determination of agglomerates and the distribution of fine particles in ordered mixing. Acta Pharm Suec, 1984, 21:9-21.
36. Malmqvist, K. and Nyström, C. Studies on direct compression of tablets VII. Sieve methods for the characterization of the adhesion tendency in ordered mixing. Acta Pharm Suec, 1982, 19:437-446.
37. Bailey, A.G. Electrostatic phenomena during powder handling. Powder Technol, 1984, 37:71-85.
38. Staniforth, J.N. Ordered mixing or spontaneous granulation? Powder Technol, 1985, 45:73-77.
39. Rumpf, H. Die Wissenschaft des agglomerierens. Chem Ing Techn, 1974, 46:1-11. 40. Padmadisastra, Y., Kennedy, R.A., and Stewart, P.J. Influence of carrier moisture
adsorption on the degree of adhesion of interactive mixtures. Int J Pharm, 1994, 104:R1-R4.

References
64
41. Podczeck, F., Newton, J.M., and James, M.B. Variations in the adhesion force between a drug and carrier particles as a result of changes in the relative humidity of the air. Int J Pharm, 1997, 149:151-160.
42. Travers, D.N. Some observations on the ordered mixing of micronized sodium bicar-bonate with sucrose crystals. Powder Technol, 1975, 12:189-190.
43. de Boer, A.H., Hagedoorn, P., Gjaltema, D., Goede, J., Kussendrager, K.D., and Fri-jlink, H.W. Air classifier technology (ACT) in dry powder inhalation. Part 2. The effect of lactose carrier surface properties on the drug-to-carrier interaction in adhesive mix-tures for inhalation. Int J Pharm, 2003, 260:201-216.
44. Rees, J.E. and Staniforth, J.N. Characterisation of ordered mixes using X-ray micro-analysis. Powder Technol, 1979, 23:135-138.
45. Thanomkiat, P. and Stewart, P.J. Influence of carrier particle size on prednisone-direct compression vehicle ordered mixes. Powder Technol, 1979, 24:97-98.
46. Sundell-Bredenberg, S. and Nyström, C. The possibility of achieving an interactive mixture with high dose homogeneity containing an extremely low proportion of a mi-cronised drug. Eur J Pharm Sci, 2001, 12:285-295.
47. Podczeck, F. and Newton, J.M. Development of an ultracentrifuge technique to deter-mine the adhesion and friction properties between particles and surfaces. J Pharm Sci, 1995, 84:1067-1071.
48. Young, P.M., Cocconi, D., Colombo, P., Bettini, R., Price, R., Steele, D.F., and Tobyn, M.J. Characterization of a surface modified dry powder inhalation carrier prepared by "particle smoothing". J Pharm Pharmacol, 2002, 54:1339-1344.
49. Williams, J.C. The mixing of dry powders. Powder Technol, 1968, 2:13-20. 50. Nyström, C., Mazur, J., and Sjögren, J. Studies on direct compression of tablets II. The
influence of the particle size of a dry binder on the mechanical strength of tablets. Int J Pharm, 1982, 10:209-218.
51. Lee, C.C., Ong, C.L., Heng, P.W., Chan, L.W., and Wong, T.W. Interactive mixture as a rapid drug delivery system. Drug Dev Ind Pharm, 2008, 34:206-214.
52. Nystrom, C. and Westerberg, M. The use of ordered mixtures for improving the dissolu-tion rate of low solubility compounds. J Pharm Pharmacol, 1986, 38:161-165.
53. Bredenberg, S. and Nyström, C. In-vitro evaluation of bioadhesion in particulate sys-tems and possible improvement using interactive mixtures. J Pharm Pharmacol, 2003, 55:169-177.
54. Steckel, H. and Müller, B. In vitro evaluation of dry powder inhalers I: drug deposition of commonly used devices. Int J Pharm, 1997, 154:19-29.
55. Steckel, H. and Müller, B.W. In vitro evaluation of dry powder inhalers II: influence of carrier particle size and concentration on in vitro deposition. Int J Pharm, 1997, 154:31-37.
56. Larhrib, H., Zeng, X.M., Martin, G.P., Marriott, C., and Pritchard, J. The use of differ-ent grades of lactose as a carrier for aerosolised salbutamol sulphate. Int J Pharm, 1999, 191:1-14.
57. Zeng, X.M., Martin, G.P., Marriott, C., and Pritchard, J. The effects of carrier size and morphology on the dispersion of salbutamol sulphate after aerosolization at different flow rates. J Pharm Pharmacol, 2000, 52:1211-1221.
58. Dickhoff, B.H., de Boer, A.H., Lambregts, D., and Frijlink, H.W. The effect of carrier surface and bulk properties on drug particle detachment from crystalline lactose car-rier particles during inhalation, as function of carrier payload and mixing time. Eur J Pharm Biopharm, 2003, 56:291-302.
59. Rowe, R.C., Sheskey, P.J., and Owen, S.C. (eds.). Handbook of Pharmaceutical excipi-ents, fifth edition, Pharmaceutical Press, Somerset, UK, 2006.
60. Edge, S., Belu, A.M., Potter, U.J., Steele, D.F., Young, P.M., Price, R., and Staniforth, J.N. Chemical characterisation of sodium starch glycolate particles. Int J Pharm, 2002, 240:67-78.

References
65
61. Bolhuis, G.K., van Kamp, H.V., and Lerk, C.F. Effect on variation of degree of substi-tution, crosslinking and purity on the disintegration efficiency of sodium starch glyco-late. Acta Pharm Technol, 1984, 30:24-32.
62. Bolhuis, G.K., Zuurman, K., and te Wierik, G.H.P. Improvement of dissolution of poorly soluble drugs by solid deposition on a super disintegrant. II. The choice of super disintegrants and effect of granulation. Eur J Pharm Sci, 1997, 5:63-69.
63. te Wierik, G.H.P., Bolhuis, G.K., Zuurman, K., and Lerk, C.F. Improvement of dissolu-tion of poorly soluble drugs by solid deposition on a super disintegrant I. Physical mix-tures. Acta Pharm Nord, 1992, 4:239-244.
64. Hussain, A., Foster, T., Hirai, S., Kashihara, T., Batenhorst, R., and Jones, M. Nasal absorption of propranolol in humans. J Pharm Sci, 1980, 69:1240.
65. Dale, O., Hjortkjaer, R., and Kharasch, E.D. Nasal administration of opioids for pain management in adults. Acta Anaesthesiol Scand, 2002, 46:759-770.
66. Humbert, H., Cabiac, M.D., Dubray, C., and Lavene, D. Human pharmacokinetics of dihydroergotamine administered by nasal spray. Clin Pharmacol Ther, 1996, 60:265-275.
67. Shyu, W.C., Pittman, K.A., Robinson, D.S., and Barbhaiya, R.H. The absolute bioavailability of transnasal butorphanol in patients experiencing rhinitis. Eur J Clin Pharmacol, 1993, 45:559-562.
68. Greiff, L., Andersson, M., Svensson, J., Wollmer, P., Lundin, S., and Persson, C.G. Absorption across the nasal airway mucosa in house dust mite perennial allergic rhini-tis. Clin Physiol Funct Imaging, 2002, 22:55-57.
69. Kublik, H. and Vidgren, M.T. Nasal delivery systems and their effect on deposition and absorption. Adv Drug Deliv Rev, 1998, 29:157-177.
70. Coda, B.A., Rudy, A.C., Archer, S.M., and Wermeling, D.P. Pharmacokinetics and bioavailability of single-dose intranasal hydromorphone hydrochloride in healthy vol-unteers. Anesth Analg, 2003, 97:117-123.
71. Studd, J., Pornel, B., Marton, I., Bringer, J., Varin, C., Tsouderos, Y., and Christiansen, C. Efficacy and acceptability of intranasal 17 beta-oestradiol for menopausal symp-toms: randomised dose-response study. Aerodiol Study Group. Lancet, 1999, 353:1574-1578.
72. Costantino, H.R., Illum, L., Brandt, G., Johnson, P.H., and Quay, S.C. Intranasal deliv-ery: physicochemical and therapeutic aspects. Int J Pharm, 2007, 337:1-24.
73. Little, P.J., Jennings, G.L., Skews, H., and Bobik, A. Bioavailability of dihydroergo-tamine in man. Br J Clin Pharmacol, 1982, 13:785-790.
74. Ferring. SPC: Minirin nasal spray, Minirin Freeze-dried tablet and Minirin tablet, 2005.
75. Jönsson, K., Alfredsson, K., Söderberg-Ahlm, C., Critchley, H., Broeders, A., and Ohlin, M. Evaluation of the degradation of desamino1,D-arginine8-vasopressin by na-sal mucosa. Acta Endocrinol (Copenh), 1992, 127:27-32.
76. Joukhadar, C., Schenk, B., Kaehler, S.T., Kollenz, C.J., Bauer, P., Muller, M., and Eichler, H.G. A replicate study design for testing bioequivalence: a case study on two desmopressin nasal spray preparations. Eur J Clin Pharmacol, 2003, 59:631-636.
77. Kaehler, S.T., Steiner, I.M., Sauermann, R., Scheidl, H., Mueller, M., and Joukhadar, C. A bioequivalence study of two oral desmopressin tablet formulations. Pharmacology, 2006, 77:46-52.
78. Jansson, B. Models for the transfer of drugs from the nasal cavity to the central nervous system, Department of Pharmacy, Uppsala University, Uppsala, 2004*.
79. Westin, U.E. Olfactory transfer of analgesic drugs after nasal administration, Depart-ment of Pharmacy, Uppsala University, Uppsala, 2007*.
80. Mathison, S., Nagilla, R., and Kompella, U.B. Nasal route for direct delivery of solutes to the central nervous system: Fact or fiction? J Drug Target, 1998, 5:415-441.

References
66
81. Illum, L. Is nose-to-brain transport of drugs in man a reality? J Pharm Pharmacol, 2004, 56:3-17.
82. Westin, U.E., Boström, E., Gråsjö, J., Hammarlund-Udenaes, M., and Björk, E. Direct nose-to-brain transfer of morphine after nasal administration to rats. Pharm Res, 2006, 23:565-572.
83. Gedulin, B.R., Smith, P.A., Jodka, C.M., Chen, K., Bhavsar, S., Nielsen, L.L., Parkes, D.G., and Young, A.A. Pharmacokinetics and pharmacodynamics of exenatide follow-ing alternate routes of administration. Int J Pharm, 2008, 356:231-238.
84. Harris, D. and Robinson, J.R. Drug delivery via the mucous membranes of the oral cavity. J Pharm Sci, 1992, 81:1-10.
85. Bredenberg, S., Duberg, M., Lennernäs, B., Lennernäs, H., Pettersson, A., Westerberg, M., and Nyström, C. In vitro and in vivo evaluation of a new sublingual tablet system for rapid oromucosal absorption using fentanyl citrate as the active substance. Eur J Pharm Sci, 2003, 20:327-334.
86. Ryan, R., Elkind, A., Baker, C.C., Mullican, W., DeBussey, S., and Asgharnejad, M. Sumatriptan nasal spray for the acute treatment of migraine. Results of two clinical studies. Neurology, 1997, 49:1225-1230.
87. Duquesnoy, C., Mamet, J.P., Sumner, D., and Fuseau, E. Comparative clinical pharma-cokinetics of single doses of sumatriptan following subcutaneous, oral, rectal and in-tranasal administration. Eur J Pharm Sci, 1998, 6:99-104.
88. GalaxoSmithKline. SPC: Imigran nasal spray and Imigran Novum filmcoated tablet, 2007.
89. Davis, S.S. and Illum, L. Absorption enhancers for nasal drug delivery. Clin Pharma-cokinet, 2003, 42:1107-1128.
90. Teshima, D., Yamauchi, A., Makino, K., Kataoka, Y., Arita, Y., Nawata, H., and Oishi, R. Nasal glucagon delivery using microcrystalline cellulose in healthy volunteers. Int J Pharm, 2002, 233:61-66.
91. Holmberg, K., Björk, E., Bake, B., and Edman, P. Influence of degradable starch mi-crospheres on the human nasal mucosa. Rhinology, 1994, 32:74-77.
92. Pereswetoff-Morath, L., Bjurström, S., Khan, R., Dahlin, M., and Edman, P. Toxico-logical aspects of the use of dextran microspheres and thermogelling ethyl(hydroxyethyl) cellulose (EHEC) as nasal drug delivery systems. Int J Pharm, 1996, 128:9-21.
93. Callens, C., Adriaens, E., Dierckens, K., and Remon, J.P. Toxicological evaluation of a bioadhesive nasal powder containing a starch and Carbopol 974 P on rabbit nasal mu-cosa and slug mucosa. J Control Release, 2001, 76:81-91.
94. Marttin, E., Romeijn, S.G., Verhoef, J.C., and Merkus, F.W. Nasal absorption of dihy-droergotamine from liquid and powder formulations in rabbits. J Pharm Sci, 1997, 86:802-807.
95. Nagai, T., Nishimoto, Y., Nambu, N., Suzuki, Y., and Sekine, K. Powder dosage form of insulin for nasal administration. J Control Release, 1984, 1:15-22.
96. Critchley, H., Davis, S.S., Farraj, N.F., and Illum, L. Nasal absorption of desmopressin in rats and sheep. Effect of a bioadhesive microsphere delivery system. J Pharm Phar-macol, 1994, 46:651-656.
97. Callens, C. and Remon, J.P. Evaluation of starch-maltodextrin-Carbopol 974 P mix-tures for the nasal delivery of insulin in rabbits. J Control Release, 2000, 66:215-220.
98. Gavini, E., Hegge, A.B., Rassu, G., Sanna, V., Testa, C., Pirisino, G., Karlsen, J., and Giunchedi, P. Nasal administration of Carbamazepine using chitosan microspheres: in vitro/in vivo studies. Int J Pharm, 2006, 307:9-15.
99. Pereswetoff-Morath, L. Microspheres as nasal drug delivery systems. Adv Drug Deliv Rev, 1998, 29:185-194.
100. Vasir, J.K., Tambwekar, K., and Garg, S. Bioadhesive microspheres as a controlled drug delivery system. Int J Pharm, 2003, 255:13-32.

References
67
101. Vivien, N., Buri, P., Balant, L., and Lacroix, S. Nasal absorption of metoclopramide administered to man. Eur J Pharm Biopharm, 1994, 40:228-231.
102. Suzuki, Y. and Makino, Y. Mucosal drug delivery using cellulose derivatives as a functional polymer. J Control Release, 1999, 62:101-107.
103. Illum, L., Watts, P., Fisher, A.N., Hinchcliffe, M., Norbury, H., Jabbal-Gill, I., Nanker-vis, R., and Davis, S.S. Intranasal delivery of morphine. J Pharmacol Exp Ther, 2002, 301:391-400.
104. Ishikawa, F., Katsura, M., Tamai, I., and Tsuji, A. Improved nasal bioavailability of elcatonin by insoluble powder formulation. Int J Pharm, 2001, 224:105-114.
105. Pringels, E., Callens, C., Vervaet, C., Dumont, F., Slegers, G., Foreman, P., and Re-mon, J.P. Influence of deposition and spray pattern of nasal powders on insulin bioavailability. Int J Pharm, 2006, 310:1-7.
106. Harris, A.S., Ohlin, M., Svensson, E., Lethagen, S., and Nilsson, I.M. Effect of viscosity on the pharmacokinetics and biological response to intranasal desmopressin. J Pharm Sci, 1989, 78:470-471.
107. Harris, A.S., Svensson, E., Wagner, Z.G., Lethagen, S., and Nilsson, I.M. Effect of viscosity on particle size, deposition, and clearance of nasal delivery systems contain-ing desmopressin. J Pharm Sci, 1988, 77:405-408.
108. Ishikawa, F., Murano, M., Hiraishi, M., Yamaguchi, T., Tamai, I., and Tsuji, A. Insolu-ble powder formulation as an effective nasal drug delivery system. Pharm Res, 2002, 19:1097-1104.
109. Björk, E., Isaksson, U., Edman, P., and Artursson, P. Starch microspheres induce pul-satile delivery of drugs and peptides across the epithelial barrier by reversible separa-tion of the tight junctions. J Drug Target, 1995, 2:501-507.
110. Björk, E. and Edman, P. Degradable starch microspheres as a nasal delivery system for insulin. Int J Pharm, 1988, 47:233-238.
111. Björk, E. Starch microspheres as a nasal delivery system for drugs, Department of pharmaceutics, Uppsala University, Uppsala, 1993.
112. Wang, J., Tabata, Y., and Morimoto, K. Aminated gelatin microspheres as a nasal delivery system for peptide drugs: evaluation of in vitro release and in vivo insulin ab-sorption in rats. J Control Release, 2006, 113:31-37.
113. Bhat, M., Toledo-Velasquez, D., Wang, L., Malanga, C.J., Ma, J.K., and Rojanasakul, Y. Regulation of tight junction permeability by calcium mediators and cell cytoskeleton in rabbit tracheal epithelium. Pharm Res, 1993, 10:991-997.
114. Kriwet, B. and Kissel, T. Interactions between bioadhesive poly(acrylic acid) and calcium ions. Int J Pharm, 1996, 127:135-145.
115. Oechslein, C.R., Fricker, G., and Kissel, T. Nasal delivery of octreotide: Absorption enhancement by particulate carrier systems. Int J Pharm, 1996, 139:25-32.
116. Pereswetoff-Morath, L. and Edman, P. Dextran microspheres as a potential nasal drug delivery system for insulin - in vitro and in vivo properties. Int J Pharm, 1995, 124:37-44.
117. Emberlin, J.C. and Lewis, R.A. A double blind, placebo controlled trial of inert cellu-lose powder for the relief of symptoms of hay fever in adults. Curr Med Res Opin, 2006, 22:275-285.
118. Matsuyama, T., Morita, T., Horikiri, Y., Yamahara, H., and Yoshino, H. Influence of fillers in powder formulations containing N-acetyl-l-cysteine on nasal peptide absorp-tion. J Control Release, 2007, 120:88-94.
119. Tafaghodi, M., Abolghasem Sajadi Tabassi, S., Jaafari, M.R., Zakavi, S.R., and Mo-men-Nejad, M. Evaluation of the clearance characteristics of various microspheres in the human nose by gamma-scintigraphy. Int J Pharm, 2004, 280:125-135.
120. Lindhardt, K., Olafsson, D.R., Gizurarson, S., and Bechgaard, E. Intranasal bioavail-ability of diazepam in sheep correlated to rabbit and man. Int J Pharm, 2002, 231:67-72.

References
68
121. Pritchard, K., Lansley, A.B., Martin, G., Helliwell, M., Marriott, C., and Benedetti, L. Evaluation of the bioadhesive properties of hyaluronan derivatives: detachment weight and mucociliary transport rate studies. . Int J Pharm, 1996, 129:137-145.
122. Ceulemans, J. and Ludwig, A. Development of a rheometric technique to measure the mucoadhesive capacity of a dry powder formulation. Pharmazie, 2002, 57:181-185.
123. Callens, C., Ceulemans, J., Ludwig, A., Foreman, P., and Remon, J.P. Rheological study on mucoadhesivity of some nasal powder formulations. Eur J Pharm Biopharm, 2003, 55:323-328.
124. Hägerström, H. and Edsman, K. Limitations of the rheological mucoadhesion method: the effect of the choice of conditions and the rheological synergism parameter. Eur J Pharm Sci, 2003, 18:349-357.
125. Hägerström, H. Polymer gels as pharmaceutical dosage forms, Department of phar-macy, Uppsala University, Uppsala, 2003*.
126. Jacques, Y. and Buri, P. An investigation of the physical behaviour of moisture-activated mucoadhesive hydrogels upon contact with biological and non-biological substrates. Pharm Acta Helv, 1997, 72:225-232.
127. Mortazavi, S.A. and Smart, J.D. An investigation of some factors influencing the in vitro assessment of mucoadhesion. Int J Pharm, 1995, 116:223-230.
128. Dimova, S., Brewster, M.E., Noppe, M., Jorissen, M., and Augustijns, P. The use of human nasal in vitro cell systems during drug discovery and development. Toxicol In Vitro, 2005, 19:107-122.
129. Schmidt, M.C., Peter, H., Lang, S.R., Ditzinger, G., and Merkle, H.P. In vitro cell models to study nasal mucosal permeability and metabolism. Adv Drug Deliv Rev, 1998, 29:51-79.
130. Bai, S., Yang, T., Abbruscato, T.J., and Ahsan, F. Evaluation of human nasal RPMI 2650 cells grown at an air-liquid interface as a model for nasal drug transport studies. J Pharm Sci, 2008, 97:1165-1178.
131. Russo, P., Sacchetti, C., Pasquali, I., Bettini, R., Massimo, G., Colombo, P., and Rossi, A. Primary microparticles and agglomerates of morphine for nasal insufflation. J Pharm Sci, 2006, 95:2553-2561.
132. Merkle, H.P., Ditzinger, G., Lang, S.R., Peter, H., and Schmidt, M.C. In vitro cell models to study nasal mucosal permeability and metabolism. Adv Drug Deliv Rev, 1998, 29:51-79.
133. Kandimalla, K.K. and Donovan, M.D. Transport of hydroxyzine and triprolidine across bovine olfactory mucosa: role of passive diffusion in the direct nose-to-brain uptake of small molecules. Int J Pharm, 2005, 302:133-144.
134. Du, G., Gao, Y., Nie, S., and Pan, W. The permeation of nalmefene hydrochloride across different regions of ovine nasal mucosa. Chem Pharm Bull (Tokyo), 2006, 54:1722-1724.
135. Wadell, C., Björk, E., and Camber, O. Permeability of porcine nasal mucosa correlated with human nasal absorption. Eur J Pharm Sci, 2003, 18:47-53.
136. Östh, K., Gråsjö, J., and Björk, E. A new method for drug transport studies on pig nasal mucosa using a horizontal Ussing chamber. J Pharm Sci, 2002, 91:1259-1273.
137. Johansson, B., Nicklasson, F., and Alderborn, G. Effect of pellet size on degree of de-formation and densification during compression and on compactability of microcrystal-line cellulose pellets. Int J Pharm, 1998, 163:35-48.
138. Tunon, A. and Alderborn, G. Granule deformation and densification during compres-sion of binary mixtures of granules. Int J Pharm, 2001, 222:65-76.
139. Eriksson, M., Nyström, C., and Alderborn, G. The use of air permeametry for the as-sessment of external surface area and sphericity of pelletized granules. Int J Pharm, 1993, 99:197-207.
140. Kaye, B.H. Permeability techniques for characterizing fine powders. Powder Technol, 1967, 1:11-22.

References
69
141. Alderborn, G., Duberg, M., and Nyström, C. Studies on direct compression of tablets X. Measurement of tablet surface area by permeametry. Powder Technol, 1985, 41:49-56.
142. Poole, K.R., Taylor, R.F., and Wall, G.P. Mixing powders to fine-scale homogeneity: Studies of batch mixing. Trans Instn Chem Engrs, 1964, 42:T305-T315.
143. Johnson, M.C.R. Particle size distribution of the active ingredient for solid dosage form of low dosage. Pharm Acta Helv, 1972, 47:546-559.
144. Egermann, H. Effects of adhesion on mixing homogeneity part I: Ordered adhesion- random adhesion. Powder Technol, 1980, 27:203-206.
145. Egermann, H. Extension of Johnson's equation of homogeneity of random mixtures. J Pharm Pharmacol, 1985, 37:491-492.
146. Egermann, H. Effects of adhesion on mixing homogeneity II: Highest attainable degree of mixing of a polydisperse ingredient and a monodisperse diluent. J Pharm Sci, 1985, 74:999-1000.
147. Valentin, F.H.H. The mixing of powders and pastes: Some basic concepts. Chem Eng, 1967, 5:CE99-CE106.
148. Johnson, M.C.R. Powder mixing in direct compression formulation by ordered and random processes. J Pharm Pharmacol, 1979, 31:273-276.
149. Östh, K. The horizontal Ussing chamber method in studies of nasal drug delivery, Department of Pharmacy, Uppsala University, Uppsala, 2002*.
150. Palm, K., Luthman, K., Ros, J., Grasjo, J., and Artursson, P. Effect of molecular charge on intestinal epithelial drug transport: pH-dependent transport of cationic drugs. J Pharmacol Exp Ther, 1999, 291:435-443.
151. Wadell, C., Björk, E., and Camber, O. Nasal drug delivery--evaluation of an in vitro model using porcine nasal mucosa. Eur J Pharm Sci, 1999, 7:197-206.
152. Caramella, C., Bonferoni, M.C., Rossi, S., and Ferrari, F. Rheological and tensile tests for the assessment of polymer-mucin interactions. Eur J Pharm Biopharm, 1994, 40:213-217.
153. SciFinder. Property values were calculated using advanced chemistry development (ACD/Labs) software V8.14 for solaris ((c) 1994-2008 ACD/Labs) and delivered through SciFinder Scholar; reproduced with permission of CAS.
154. Björk, E. and Edman, P. Characterization of degradable starch microspheres as a nasal delivery system for drugs. Int J Pharm, 1990, 62:187-192.
155. Ritger, P.L. and Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release, 1987, 5:23-36.
156. Higuchi, W.I. Analysis of data on the medicament release from ointments. J Pharm Sci, 1962, 51:802-804.
157. Singla, A.K., Chawla, M., and Singh, A. Potential applications of carbomer in oral mucoadhesive controlled drug delivery system: a review. Drug Dev Ind Pharm, 2000, 26:913-924.
158. Lewontin, R.C. On the measurement of relative variability. Systematic Zoology, 1966, 15:141-142.
159. Crooks, M.J. and Ho, R. Ordered mixing in direct compression of tablets. Powder Technol, 1976, 14:161-167.
160. Mihranyan, A., Frenning, G., Fransén, N., Welch, K., and Strömme, M. Order and disorder in powder mixtures: Spatial distribution functions as tools to assess powder homogeneity. Part Part Syst Charact, 2008, In Press.
161. Östh, K., Paulsson, M., Björk, E., and Edsman, K. Evaluation of drug release from gels on pig nasal mucosa in a horizontal Ussing chamber. J Control Release, 2002, 83:377-388.
162. Östh, K., Strindelius, L., Larhed, A., Ahlander, A., Roomans, G.M., Sjöholm, I., and Björk, E. Uptake of ovalbumin-conjugated starch microparticles by pig respiratory na-sal mucosa in vitro. J Drug Target, 2003, 11:75-82.

References
70
163. Martineau-Doize, B. and Caya, I. Ultrastructural characterization of the nasal respira-tory epithelium in the piglet. Anat Rec, 1996, 246:169-175.
164. Cassidy, M.M. and Tidball, C.S. Cellular mechanism of intestinal permeability altera-tions produced by chelation depletion. J Cell Biol, 1967, 32:685-698.
165. Marini, S., Longo, V., Mazzaccaro, A., and Gervasi, P.G. Xenobiotic-metabolizing enzymes in pig nasal and hepatic tissues. Xenobiotica, 1998, 28:923-935.
166. Wang, Y., Aun, R., and Tse, F.L. Brain uptake of dihydroergotamine after intravenous and nasal administration in the rat. Biopharm Drug Dispos, 1998, 19:571-575.
167. Hägerström, H. and Edsman, K. Interpretation of mucoadhesive properties of polymer gel preparations using a tensile strength method. J Pharm Pharmacol, 2001, 53:1589-1599.
168. Westerberg, M. and Nyström, C. Physicochemical aspects of drug release XVII. The effect of drug surface area coverage to carrier materials on drug dissolution from or-dered mixtures. Int J Pharm, 1993, 90:1-17.
169. Mortazavi, S.A. and Smart, J.D. An in-vitro method for assessing the duration of muco-adhesion. J Control Release, 1994, 31:207-212.
170. Hansson, P. Interaction between polyelectrolyte gels and surfactants of opposite charge. Curr Oppin Colloid Interface Sci, 2006, 11:351-362.
171. Bramer, T., Paulsson, M., Edwards, K., and Edsman, K. Cationic Drug-Surfactant Mixtures: Phase behavior and sustained release from gels. Pharm Res, 2003, 20:1661-1667.
172. Varade, D., Joshi, T., Aswal, V.K., Goyal, P.S., Hassan, P.A., and Bahadur, P. Effect of salt on the micelles of cetyl pyridinium chloride. Colloids and surfaces, 2005, 259:95-101.
173. Steiner, I.M., Kaehler, S.T., Sauermann, R., Rinosl, H., Muller, M., and Joukhadar, C. Plasma pharmacokinetics of desmopressin following sublingual administration: an ex-ploratory dose-escalation study in healthy male volunteers. Int J Clin Pharmacol Ther, 2006, 44:172-179.
174. Osterberg, O., Savic, R.M., Karlsson, M.O., Simonsson, U.S., Norgaard, J.P., Walle, J.V., and Agerso, H. Pharmacokinetics of desmopressin administrated as an oral ly-ophilisate dosage form in children with primary nocturnal enuresis and healthy adults. J Clin Pharmacol, 2006, 46:1204-1211.
175. Vilhardt, H. and Lundin, S. Biological effect and plasma concentrations of DDAVP after intranasal and peroral administration to humans. Gen Pharmacol, 1986, 17:481-483.
176. Ljungberg, C., Fransén, N., Björnsson, M., and Kettis Lindblad, Å. The importance of the dosage form - a qualitative study of migraine patients, In manuscript.
* The doctoral theses can be obtained from: http://publications.uu.se/theses/


Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Pharmacy 81
Editor: The Dean of the Faculty of Pharmacy
A doctoral dissertation from the Faculty of Pharmacy, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofPharmacy. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Pharmacy”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-9292
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2008