studies of the cag repeat in the machado-joseph disease gene in taiwan
TRANSCRIPT

Abstract Machado-Joseph disease (MJD) is an autoso-mal dominant spinocerebellar degeneration characterizedby cerebellar ataxia and pyramidal signs associated invarying degrees with a dystonic-rigid extrapyramidal syn-drome or peripheral amyotrophy. Unstable CAG trinu-cleotide repeat expansion in the MJD gene on the longarm of chromosome 14 has been identified as the patho-logical mutation for MJD. While investigating the distrib-ution of CAG repeat lengths of the MJD gene in Taiwan’spopulation, we have identified 18 MJD-affected patientsand 12 at-risk individuals in seven families. In addition,we have analyzed the range of CAG repeat lengths in 96control individuals. The CAG repeat number ranged from13 to 44 in the controls and 72–85 in the affected and at-risk individuals. Our results indicated that the CAG repeatnumber was inversely correlated with the age of onset.The differences in CAG repeat length between parent andchild and between siblings are greater with paternal trans-mission than maternal transmission. Our data show a ten-dency towards the phenomenon of anticipation in theMJD families but do not support unidirectional expansionof CAG repeats during transmission. We also demon-strated that PCR amplification of the CAG repeats in theMJD gene from villous DNA was possible and might
prove useful as a diagnostic tool for affected families inthe future.
Introduction
Machado-Joseph disease (MJD) is an autosomal dominantspinocerebellar degeneration characterized by a widerange of clinical manifestations, including ataxia, progres-sive external ophthalmoplegia, pyramidal and extrapyra-midal signs, dystonia with rigidity, and distal muscular at-rophies (Coutinho et al. 1977; Lima and Coutinho 1980).The disease locus has been mapped to chromosome14q32.1 in Japanese families (Takiyama et al. 1993). Re-cently, the gene has been identified (Kawaguchi et al. 1994)and shown to contain a CAG repeat motif in the 5′ regionof the coding sequence, which is selectively expanded inMJD patients. Therefore, MJD is one of the at least tendiseases that result from abnormally large expansions oftrinucleotide repeats (Sutherland and Richards 1995). Asubgroup of these diseases results from CAG repeat ex-pansions in coding sequences that are translated intopolyglutamine tracts. These diseases include Huntington’sdisease (HD; Huntington’s Disease Collaborative ResearchGroup 1993), spinocerebellar ataxia type 1 (SCA 1; Orr etal. 1993), spinal and muscular atrophy (SBMA or Kennedydisease; La Spada et al. 1991), Machado-Joseph disease(MJD; Kawaguchi et al. 1994), dentatorubral-pallido-luysian atrophy (DRPLA; Koide et al. 1994; Nagafuchi etal. 1994) and spinocerebellar ataxia type 2 (SCA 2; Pulstet al. 1996; Sanpei et al. 1996; Imbert et al. 1996). As yetthere is little understanding of how the polyglutaminesfunction either normally or when expanded.
MJD has been reported to be characterized by a widevariety of clinical features (Coutinho and Andrade 1978).Detailed clinical examination of the variability in MJDpatients has led to its classification into three subpheno-types (Maciel et al. 1995). The type I patient is character-ized by marked pyramidal and extrapyramidal signs, inaddition to the common features of cerebellar ataxia andophthalmoplegia, as well as a rapid and more severe clin-
Mingli Hsieh · Hui-Fang Tsai · Tsong-Ming Lu ·Chien-Ying Yang · Hung-Ming Wu · Shuan-Yow Li
Studies of the CAG repeat in the Machado-Joseph disease gene in Taiwan
Hum Genet (1997) 100 :155–162 © Springer-Verlag 1997
Received: 4 December 1996 / Accepted: 5 March 1997
ORIGINAL INVESTIGATION
M. Hsieh · H.-F. Tsai · C.-Y. YangInstitute of Medicine, Chung Shan Medical and Dental College, Taichung, Taiwan, Republic of China
T.-M. LuDepartment of Neurology, Chung Shan Medical and Dental College Hospital, Taichung, Taiwan, Republic of China
H.M. WuDepartment of Neurology, Kuang-Tien General Hospital, Taichung, Taiwan, Republic of China
S.-Y. Li (Y)Cytogenetics Laboratory, Chung Shan Medical and Dental College, Taichung, Taiwan, Republic of ChinaTel.: +886-4-3896190, ext. 50610 or 50617; Fax: +886-4-3892412

ical course, and a mean age at onset of 24.3 years. Thetype II patient has cerebellar and pyramidal signs, mainlycerebellar ataxia and ophthalmoplegia, with an intermedi-ate mean age at onset of 40.5 years. The type III patientshows cerebellar and distal amyotrophic signs, markedperipheral signs, weakness, and amyotrophy, and a latermean age at onset of 46.8 years.
An inverse correlation between the expanded repeatlength and the age at onset of the trinucleotide disease hasbeen reported in HD (Andrew et al. 1993; Duyao et al.1993; Snell et al. 1993), DRPLA (Koide et al. 1994), SCA1 (Jodice et al. 1994; Ranum et al. 1994), SBMA (LaSpada et al. 1991), MJD (Maruyama et al. 1995; Maciel etal. 1995) and SCA 2 (Pulst et al. 1996; Sanpei et al. 1996;Imbert et al. 1996). In addition, the trinucleotide repeatmutation in most of these diseases is associated with thephenomenon of anticipation, where the disease tends topresent at an earlier age in successive generations. Thiscan be explained by the inverse correlation between re-peat size of the disease gene and age at onset of symp-toms; also the abnormally expanded repeats are unstableand tend to increase in size when passed from one gener-ation to the next (Sutherland and Richards 1995). Yet, thecorrelation of repeat length with clinical anticipation isnot perfect in the case of MJD. Maruyama et al. (1995) re-ported clinical anticipation, but Maciel et al. (1995) in astudy of 100 patients showed the anticipation to be an ar-tifact. Therefore, the association between variation in repeatnumber and anticipation in MJD remains to be clarified.
Although MJD was originally described in a Por-tuguese Azorean family (Coutinho and Andrade 1978), ithas been reported to have a wide distribution in differentethnic populations (Sequeiros et al. 1994; Twist et al.1995; Maruyama et al. 1995; Cancel et al. 1995; Maciel etal. 1995). Nevertheless, the distribution of the MJD genein the Chinese population has not been well documented.We report here on the distribution of CAG repeats in nor-mal population and in MJD patients in Taiwan. We haveexamined the CAG repeat length using genomic PCRanalysis to compare the size range of the CAG repeat inthe MJD and control populations. Furthermore, we haveinvestigated the relationship between the size of the ex-panded allele and the clinical presentation. Our data haveindicated that the repeat number is inversely correlated
with the age at onset. In addition, we have shown a mildinstability in both an increase and a decrease in repeat sizein the transmission from parent to progeny. There was asuggestion of anticipation in the five-generation familyincluded in this study. Finally, we demonstrated that PCRamplification of the CAG repeats of the MJD gene fromthe villous DNA was successful.
Materials and methods
Subjects
This study was performed using DNA samples from 62 membersof seven MJD-affected families. The diagnosis of MJD was deter-mined after clinical examination by experienced neurologists usingestablished diagnostic criteria. Ages at onset were based on infor-mation provided by the patient or a close relative. For determina-tion of the frequency of the normal alleles, a total of 192 normal al-leles were used, including a group of unrelated unaffected spousesfrom MJD families and a group of controls from the general popu-lation. Since both groups had similar allele sizes, the data werepooled and the controls were considered as single group.
Methods
Genomic DNA was isolated from peripheral lymphocytes andchorionic villi by standard methods(Ko et al. 1992). The CAG-containing fragment of the MJD gene was amplified by PCR usingprimers MJD52 and MJD25a (a slight modification of MJD25 withsequence ATCCATGTGCAAAGGCCAGCC; Kawaguchi et al.1994). PCR was performed in a final volume of 20 µl, containing40 ng of genomic DNA; 10 mM Tris-HCl (pH 8.8); 1.5 mMMgCl2; 50 mM KCl; 0.1% Triton X-100; 10% dimethylsulfoxide;250 µM each dCTP, dGTP, and dTTP; 25 µM dATP; 1.5 µCi 35S-alpha-dATP; 125 ng each primer; and 3 units of Taq DNA poly-merase (Perkin Elmer). The DNA was denatured at 95°C for 8min; then 30 cycles at 94°C for 1 min, 61°C for 1 min, and 72°Cfor 1 min were performed, followed by a final extension at 72°Cfor 10 min.
For determination of allele size, PCR products were analyzedon denaturing 6% polyacrylamide gels in parallel with a pGEM se-quencing ladder (–40 primer) and were visualized by autoradiog-raphy. Allele size (S) was determined by comparison with the se-quence ladder and was converted to number of CAG units (N), us-ing the formula N = (S–163)/3, assuming that the variation in thesize of the PCR product occurred within the repetitive CAGstretch.
Linear regression techniques were used to determine the asso-ciation between repeat number and age at onset.
156
Table 1 Clinical features of10 MJD patients in families Aand B (CA cerebellar ataxia;Amy amyotrophy; Py pyrami-dal signs; Ex extrapyramidalsigns; PEO progressive exter-nal ophthalmoplegia; SD sen-sory disturbance; and Nys nys-tagmus, N/D not determined)
Family Patient Sex CAG Age Age of Major Minorrepeats onset symptoms symptoms
Family A II-9 M 15, 76 79 52 CA, Py Nys, PEOII-15 M 31, 75 65 42 CA, Py Nys, SDII-19 F 30, 78 60 47 CA, Py Nys
III-27 M 16, 83 45 31 CA, Py Nys, SD, PEOIII-38 M 29, 76 45 32 CA, Py Nys, SD, MyokymiaIII-45 F 31, 82 41 N/D Ca, Py, Amy, Ex Nys, SD, MyokymiaIII-48 M 27, 82 38 29 CA, Py, Ex Nys, SD, MyokymiaIV-3 M 16, 84 29 25 CA, Py, Amy, Ex Nys, Myokymia
Family B II-3 F 21, 74 44 41 CA, Py NysII-5 M 16, 74 40 30 CA, Py, Amy, Ex Nys

Results
Clinical features of MJD patients
Table 1 summarizes the clinical features of ten MJD pa-tients from two families. All patients from the seven fam-ilies are Chinese and have family histories consistent withautosomal dominant inheritance (Fig.1). Since the previ-
ously reported patients were clinically indistinguishablefrom others with ADCA type I, each genomic sample wasanalyzed by the method described in Maciel et al. (1995),with a slight modification of the PCR conditions (for de-tails see Materials and Methods). As expected, all ge-nomic samples from the 18 patients showed two PCRproducts, demonstrating expanded PCR fragments as wellas normal lengths (for examples, see Fig.2A). We nextperformed the analysis on the patients’ offspring who did
157
Fig. 1A–G Pedigrees for seven Machado-Joseph disease (MJD)families. Solid symbols indicate affected individuals; *, sampledindividuals; open symbols indicate normal individuals; half-filledsymbols indicate asymptomatic carriers
C
B
A
D
E
F
G
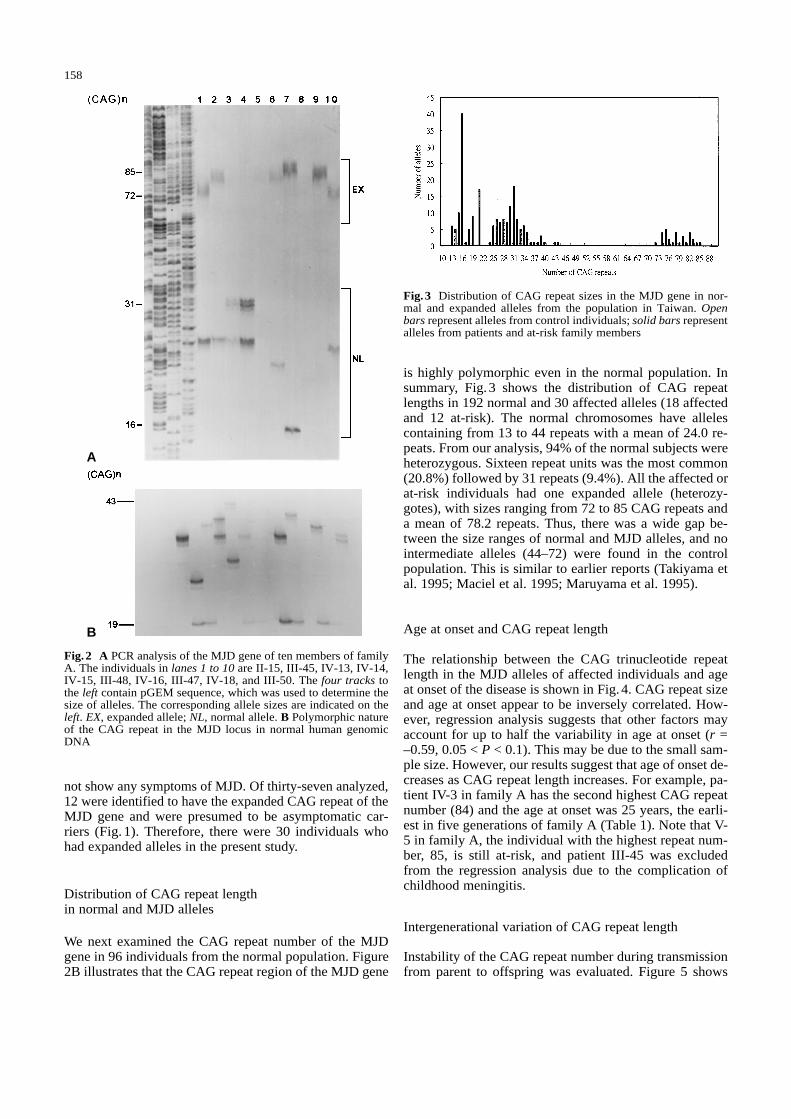
not show any symptoms of MJD. Of thirty-seven analyzed,12 were identified to have the expanded CAG repeat of theMJD gene and were presumed to be asymptomatic car-riers (Fig.1). Therefore, there were 30 individuals whohad expanded alleles in the present study.
Distribution of CAG repeat length in normal and MJD alleles
We next examined the CAG repeat number of the MJDgene in 96 individuals from the normal population. Figure2B illustrates that the CAG repeat region of the MJD gene
is highly polymorphic even in the normal population. Insummary, Fig. 3 shows the distribution of CAG repeatlengths in 192 normal and 30 affected alleles (18 affectedand 12 at-risk). The normal chromosomes have allelescontaining from 13 to 44 repeats with a mean of 24.0 re-peats. From our analysis, 94% of the normal subjects wereheterozygous. Sixteen repeat units was the most common(20.8%) followed by 31 repeats (9.4%). All the affected orat-risk individuals had one expanded allele (heterozy-gotes), with sizes ranging from 72 to 85 CAG repeats anda mean of 78.2 repeats. Thus, there was a wide gap be-tween the size ranges of normal and MJD alleles, and nointermediate alleles (44–72) were found in the controlpopulation. This is similar to earlier reports (Takiyama etal. 1995; Maciel et al. 1995; Maruyama et al. 1995).
Age at onset and CAG repeat length
The relationship between the CAG trinucleotide repeatlength in the MJD alleles of affected individuals and ageat onset of the disease is shown in Fig.4. CAG repeat sizeand age at onset appear to be inversely correlated. How-ever, regression analysis suggests that other factors mayaccount for up to half the variability in age at onset (r =–0.59, 0.05 < P < 0.1). This may be due to the small sam-ple size. However, our results suggest that age of onset de-creases as CAG repeat length increases. For example, pa-tient IV-3 in family A has the second highest CAG repeatnumber (84) and the age at onset was 25 years, the earli-est in five generations of family A (Table 1). Note that V-5 in family A, the individual with the highest repeat num-ber, 85, is still at-risk, and patient III-45 was excludedfrom the regression analysis due to the complication ofchildhood meningitis.
Intergenerational variation of CAG repeat length
Instability of the CAG repeat number during transmissionfrom parent to offspring was evaluated. Figure 5 shows
158
Fig.2 A PCR analysis of the MJD gene of ten members of familyA. The individuals in lanes 1 to 10 are II-15, III-45, IV-13, IV-14,IV-15, III-48, IV-16, III-47, IV-18, and III-50. The four tracks tothe left contain pGEM sequence, which was used to determine thesize of alleles. The corresponding allele sizes are indicated on theleft. EX, expanded allele; NL, normal allele. B Polymorphic natureof the CAG repeat in the MJD locus in normal human genomicDNA
Fig.3 Distribution of CAG repeat sizes in the MJD gene in nor-mal and expanded alleles from the population in Taiwan. Openbars represent alleles from control individuals; solid bars representalleles from patients and at-risk family members
A
B

the distribution of the differences in CAG repeat lengthbetween the 16 parent-child pairs. For 13 pairs (81%) therepeat number was unchanged or increased after transmis-sion, whereas for 3 pairs (19%) the repeat number de-creased. The difference varied from –3 to +7, with a meanof 1.8 repeats more per transmission.
Instability of CAG repeat length as a function of the sex of the transmitting parent
There were nine father-offspring pairs and seven mother-offspring pairs (Fig.5). Seven of the nine paternal trans-missions resulted in an increase or no change in the lengthof CAG repeats, and two resulted in a decrease. The rangewas from –3 to +7, and the average was an increase of 3.0repeats. Of the seven maternal transmissions, six resultedin an increase or no change, and one resulted in a 3 repeat
decrease. The average was a 0.2 repeat increase and therange was from –3 to 2. The slight increase or decrease inCAG repeats after transmission is consistent with the re-port by Maciel et al. (1995). In addition, we also com-pared CAG repeat number between siblings (Fig.6). Eventhough there is not enough data for a statistic analysis,there is at most a 3 repeat difference in sibslings from anaffected mother, but a 10 repeat difference in sibslingsfrom an affected father. These results support the sugges-tion that paternal transmissions are less stable than mater-nal ones.
Genomic amplification of CAG repeats in the MJD gene from villous DNA
In an effort to provide prenatal screening for affected fam-ilies in the future, we here demonstrated PCR amplifica-tion of the CAG repeats from four different normal villousDNA samples (Fig.7). The results showed clear signalswith the repeat number ranging from 16 to 29 on the poly-acrylamide gel. However, whether the expansion of theMJD gene occurs in the chorionic villi and whether thematerial is sufficiently free of mosaicism to be diagnosti-cally useful is still not known.
Discussion
In this study, we reported a first systematic study of MJD-affected families in Taiwan. The CAG repeat numberranged from 13 to 44 in the controls and 72 to 85 in theexpanded individuals. In general, the distribution of theCAG repeat number in a normal population is similar tothe 13–40 repeats reported by Ranum et al. (1995), andthe distribution of the expanded alleles is also within thereported 61–84 repeat range (Takiyama et al. 1995; Ma-ciel et al. 1995). The distribution of repeat lengths of the
159
Fig. 4 Correlation between age of onset and CAG repeat numbers.A correlation coefficient of r = –0.59 was calculated based on thedata from 17 MJD patients. Patient III-45 was excluded from thisanalysis. Regression line is Y = 84.4–0.18X (Y CAG repeat length,X age at onset)
Fig. 5 Intergenerational varia-tion of repeat number in mater-nal and paternal transmission.Repeat variation is shown as adecrease (–) or an increase (+)of repeat units in the next gen-eration. Maternal transmissionis represented by shaded bars;and paternal transmissions, byopen bars
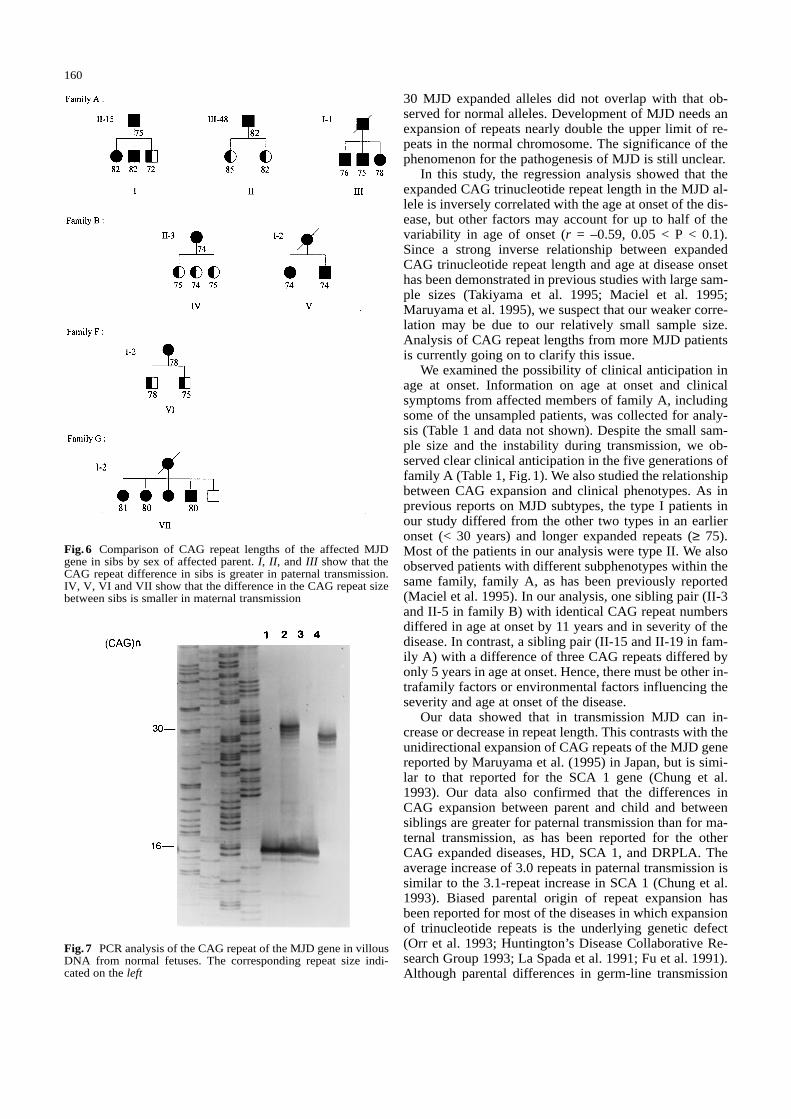
30 MJD expanded alleles did not overlap with that ob-served for normal alleles. Development of MJD needs anexpansion of repeats nearly double the upper limit of re-peats in the normal chromosome. The significance of thephenomenon for the pathogenesis of MJD is still unclear.
In this study, the regression analysis showed that theexpanded CAG trinucleotide repeat length in the MJD al-lele is inversely correlated with the age at onset of the dis-ease, but other factors may account for up to half of thevariability in age of onset (r = –0.59, 0.05 < P < 0.1).Since a strong inverse relationship between expandedCAG trinucleotide repeat length and age at disease onsethas been demonstrated in previous studies with large sam-ple sizes (Takiyama et al. 1995; Maciel et al. 1995;Maruyama et al. 1995), we suspect that our weaker corre-lation may be due to our relatively small sample size.Analysis of CAG repeat lengths from more MJD patientsis currently going on to clarify this issue.
We examined the possibility of clinical anticipation inage at onset. Information on age at onset and clinicalsymptoms from affected members of family A, includingsome of the unsampled patients, was collected for analy-sis (Table 1 and data not shown). Despite the small sam-ple size and the instability during transmission, we ob-served clear clinical anticipation in the five generations offamily A (Table 1, Fig.1). We also studied the relationshipbetween CAG expansion and clinical phenotypes. As inprevious reports on MJD subtypes, the type I patients inour study differed from the other two types in an earlieronset (< 30 years) and longer expanded repeats (≥ 75).Most of the patients in our analysis were type II. We alsoobserved patients with different subphenotypes within thesame family, family A, as has been previously reported(Maciel et al. 1995). In our analysis, one sibling pair (II-3and II-5 in family B) with identical CAG repeat numbersdiffered in age at onset by 11 years and in severity of thedisease. In contrast, a sibling pair (II-15 and II-19 in fam-ily A) with a difference of three CAG repeats differed byonly 5 years in age at onset. Hence, there must be other in-trafamily factors or environmental factors influencing theseverity and age at onset of the disease.
Our data showed that in transmission MJD can in-crease or decrease in repeat length. This contrasts with theunidirectional expansion of CAG repeats of the MJD genereported by Maruyama et al. (1995) in Japan, but is simi-lar to that reported for the SCA 1 gene (Chung et al.1993). Our data also confirmed that the differences inCAG expansion between parent and child and betweensiblings are greater for paternal transmission than for ma-ternal transmission, as has been reported for the otherCAG expanded diseases, HD, SCA 1, and DRPLA. Theaverage increase of 3.0 repeats in paternal transmission issimilar to the 3.1-repeat increase in SCA 1 (Chung et al.1993). Biased parental origin of repeat expansion hasbeen reported for most of the diseases in which expansionof trinucleotide repeats is the underlying genetic defect(Orr et al. 1993; Huntington’s Disease Collaborative Re-search Group 1993; La Spada et al. 1991; Fu et al. 1991).Although parental differences in germ-line transmission
160
Fig.6 Comparison of CAG repeat lengths of the affected MJDgene in sibs by sex of affected parent. I, II, and III show that theCAG repeat difference in sibs is greater in paternal transmission.IV, V, VI and VII show that the difference in the CAG repeat sizebetween sibs is smaller in maternal transmission
Fig.7 PCR analysis of the CAG repeat of the MJD gene in villousDNA from normal fetuses. The corresponding repeat size indi-cated on the left

of the expanded MJD alleles has been explained by themitotic potential model (Chong et al. 1995), the basis forsuch biased transmission is still under investigation. Al-though the limited number of samples in the present studydid not enable us to conduct a thorough statistical analy-sis, our data suggested that parent-child analysis and sib-sib analysis exhibited a similar distribution of the mild re-peat length instability.
All identified CAG expansions in inherited neurode-generative diseases are located in the translated regions ofgenes suggesting that the CAG expansions affect the pro-tein function through abnormally long stretches of gluta-mines. It has been suggested that such disorders as SCA 1,SBMA, and HD involve gain-of-function mutations re-lated to the expansion of the repeats because (1) patientswith complete deletions of the androgen receptor geneshow no manifestation of SBMA (Quigle et al. 1992; Tri-firo et al. 1991) and (2) two HD homozygous individualsexist with undistinguished phenotypes compared to het-erozygous ones (Kremer et al. 1994). On the other hand, ithas been reported that in MJD the homozygote is more se-verely affected than the heterozygote (Takiyama et al.1995; Lerer et al. 1996), indicating that the expanded al-lele may work by a dominant negative effect. As yet whatfunction is altered in each disorder is still unknown. Re-cently, it has been demonstrated that the expanded polyg-lutamine in the Machado-Joseph disease protein inducescell death in vitro and in vivo (Ikeda et al. 1996). Ataxiatransgenic mice were created in the study by expressingonly the expanded polyglutamine stretch. Their results in-dicated the potential involvement of the expanded polyg-lutamine as the common etiological agent for inheritedneurodegenerative disorders with CAG expansions. How-ever, the precise role of the CAG trinucleotide repeat ex-pansion in the pathogenesis of these diseases remains tobe determined.
Finally, although there is no cure to neurodegenerativediseases like MJD, the accurate molecular analysis of theMJD gene using trinucleotide repeat PCR amplificationdemonstrates that predictive testing and prenatal diagno-sis based on this technique is possible.
Acknowledgements We thank the MJD families for their cooper-ation; Drs. Akira Kakizuka, Yoshiya Kawaguchi, Guy A. Rouleauand Chin-San Lui for their MJD genomic DNA to initiate ourwork; Dr. Claudia Gaspar for the valuable comments; and Dr.Chuan Li for critical reading of the manuscript. This work wassupported by grants from the National Science Council of the Re-public of China, NSC 85–2331-B-040–010; the National Health ofResearch Institute of the Republic of China, DOH85-HR-405; andthe Chung Shan Medical and Dental College Research Fund,CSMC 84-OM-A-016.
References
Andrew SE, Goldberg YP, Kremer B, Telenius H, Theimann J,Adam S, Starr E, Squitieri F, Lin B, Kalchman MA, GrahamRK, Hayden MR (1993) The relationship between trinucleotide(CAG) repeat length and clinical features of Huntington’s dis-ease. Nat Genet 4 :398–403
Cancel G, Abbas N, Stevanin G, Durr A, Chneiweiss H, Neri C,Duyckaerts C, Penet C, Cann HM, Agid Y, Brice A (1995)Marked phenotypic heterogeneity associated with expansion ofa CAG repeat sequence at the spinocerebellar ataxia 3/Machado-Joseph disease locus. Am J Hum Genet 57 :809–816
Coutinho P, Andrade C (1978) Autosomal dominant system de-generation in Portuguese families of the Azores Islands: a newgenetic disorder involving cerebellar, pyramidal, extrapyrami-dal and spinal cord motor functions. Neurology 28 :703–709
Coutinho P, Calheiros JM, Andrade C (1977) On a new degenera-tive disorder of the central nervous system, inherited in an au-tosomal dominant mode and affecting people of Azorean ex-traction. O Medico 82 :446–448
Chong SS, McCall AE, Cota J, Subramony SH, Orr HT, HughesMR, Zoghbi HY (1995) Gametic and somatic tissue-specificheterogeneity of the expanded SCA1 CAG repeat in spinocere-bellar ataxia type 1. Nat Genet 10 :344–350
Chung M, Ranum LPW, Duvic LA, Servadio A, Zoghbi HY, OrrHT (1993) Evidence for a mechanism predisposing to intergen-erational CAG repeat instability in spinocerebellar ataxia typeI. Nat Genet 5 :254–258
Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F,Fontali M, Folstein S, et al (1993) Trinucleotide repeat lengthinstability and age of onset in Huntington’s disease. Nat Genet4 :387–892
Fu Y-H, Kuhl DPA, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S,Verkerk AJMH, Holden JJA, Fenwick RG, Warren ST, OostraBA, Nelson DL, Caskey CT (1991) Variation of the CGG re-peat at the fragile X site results in genetic instability: resolutionof the Sherman paradox. Cell 67 :1047–1058
Huntington’s Disease Collaborative Research Group (1993) Anovel gene containing a trinucleotide repeat that is expandedand unstable on Huntington’s disease chromosome. Cell 72 :971–983
Ikeda H, Yamuguchi M, Sugai S, Aze Y, Narumiya S, Kakizuka A(1996) Expanded polyglutamine in the Machado-Joseph dis-ease protein induces cell death in vitro and in vivo. Nat Genet13 :196–202
Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, et al (1996)Cloning of the gene for spinocerebellar ataxia 2 reveals a locuswith high sensitivity to expanded CAG/glutamine repeats. NatGenet 14 :284–290
Jodice C, Malaspina P, Persichetti F, Novelletto A, Spadaro M,Giunti P, Morocutti C, et al (1994) Effect of trinucleotide re-peat length and parental sex on phenotypic variation in spin-ocerebellar ataxia 1. Am J Hum Genet 54 :959–965
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M,Katayama S, Kawakami H, et al (1994) CAG expansions in anovel gene from Machado-Joseph disease at chromosome14q32.1. Nat Genet 8 :221–227
Ko TM, Tseng LH, Hsieh FJ, Hsu PM, Lee TY (1992) Carrier de-tection and prenatal diagnosis of alpha-thalassemia of South-east Asian deletion by polymerase chain reaction. Hum Genet88 :245–248
Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K,Takahashi H, Kondo R, Ishiawa A, Hayashi T, Saito M, To-moda A, Miike T, Naito H, Ikuta F, Tsuji S (1994) Unstableexpansion of CAG repeat in hereditary dentatorubral-palli-doluysian atrophy (DRPLA). Nat Genet 6 :9–13
Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H,Zeisler J, Squitieri F, Lin B, Bassett A, Almqvist E, Bird TD,Hayden MR (1994) A worldwide study of the Huntington’sdisease mutation: the sensitivity and specificity of measuringCAG repeats. N Engl J Med 330 :1401–1406
La Spada AR, Wilson EM, Lubahn DB, Harding AE, FischbeckKH (1991) Androgen receptor gene mutations in X-linkedspinal and bulbar muscular atrophy. Nature 352 :77–79
Lerer I, Merims D, Abeliovich D, Zlotogora J, Gadoth N (1996)Machado-Joseph disease: correlation between the clinical fea-tures, the CAG repeat length and homozygosity for the muta-tion. Eur J Hum Genet 4 :3–7
161

Lima L, Coutinho P(1980) Clinical criteria for diagnosis of Machado-Joseph disease: report of a non-Azorean Portuguese family.Neurology 30 :319–322
Maciel P, Gaspar C, DeStefano AL, Silveira L, Coutinho P, Rad-vany J, Dawson DM, Sundarsky L, Guimaraes J, Loureiro JEL,Nezarati MM, Corwin LL, Lopes-Cendes I, Rooke K, Rosen-berg R, MacLeod P, Farrer LA, Sequeiros J, Rouleau GA(1995) Correlation between CAG repeat length and clinicalfeatures in Machado-Joseph disease. Am J Hum Genet 57 :54–61
Maruyama H, Nakamura S, Matsuyama Z, Sakai T, Doyu M,Sobue G, Seto M, Tsujihata M, Oh-i T, Nishino T, Sunohara N,Takahashi R, Hayashi M, Nishino I, Ohtake T, Oda T,Nishimura M, Saida T, Matsumoto H, Baba M, Kawaguchi Y,Kakizuka A, Kawakami H (1995) Molecular features of theCAG repeats and clinical manifestation of Machado-Josephdisease. Hum Mol Genet 4 :807–812
Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E,Bunda M, Takeda T, Tadokoro L, Murayama N, Tanaka Y,Kikushima H, Umino K, Kurosawa H, Furukawa T, Nihei K,Inoue T, Sano A, Komure O, Takahashi M, Yoshizawa T,Kanazawa I, Yamada M (1994) Dentatorubral and pallido-luysian atrophy expansion of an unstable CAG trinucleotide onchromosome 12p. Nat Genet 6 :14–18
Orr HT, Chung M, Banfi S, Kwiatkowski TJ, Servadio A, BeaudetAL, McCall AE, Duvick LA, Ranum LPW, Zoghbi HY (1993)Expansion of an unstable trinucleotide CAG repeat in spin-ocerbellar ataxia type 1. Nat Genet 4 :221–226
Pulst S-M, Nechiporuk A, Nechiporuk T, Gispert S, Chen X-N,Lopes-Cendes I, Pearlman S, Starkman S, et al (1996) Moder-ate expansion of a normally biallelic trinucleotide repeat inspinocerebellar ataxia type 2. Nat Genet 14 :269–276
Quigle CA, et al (1992) Complete deletion of the androgen recep-tor gene: definition of the null phenotype of the androgen in-sensitivity syndrome and determination of carrier status. MolCell Endocrinol 74 :927–933
Ranum LPW, Chung M, Banfi S, Bryer A, Schut LJ, Rameser R,Duvick LA, et al (1994) Molecular and clinical correlations inspinocerebellar ataxia type 1: evidence for familial effects onthe age at onset. Am J Hum Genet 55 :244–252
Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wak-isaka A, et al (1996) Identification of the spinocerebellar ataxiatype 2 gene using a direct identification of repeat expansionand cloning technique, DIRECT. Nat Genet 14 :277–284
Sequeiros J, Silveira I, Maciel P, Coutinho P, Manaia A, Gaspar C,Burlet P, et al (1994) Genetic linkage studies of Machado-Joseph disease with chromosome 14q STRPs in 16 Portuguese-Azorean kindreds. Genomics 21 :645–648
Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP,Davies P, MacDonald ME, Gusella JF, Harper PS, Shaw DJ(1993) Relationship between trinucleotide repeat expansionand phenotypic variation in Huntington’s disease. Nat Genet 4 :393–397
Sutherland GR, Richards RI (1995) Simple tandem repeats and hu-man genetic disease. Proc Natl Acad Sci USA 92 :3636–3641
Takiyama Y, Nishizawa M, Tanaka H, Kawashima S, SakamotoH, Karube Y, Shimazaki H, et al (1993) The gene for Machado-Joseph disease is mapped to chromosome 14q. Nat Genet 4 :300–304
Takiyama Y, Igarashi S, Rogaeva EA, Endo K, Rogaev EI, TanakaH, Sherrington R, et al (1995) Evidence for intergenerationalinstability in the CAG repeat in the MJD1 gene and for con-served haplotypes at flanking markers amongst Japanese andCaucasian subjects with Machado-Joseph disease. Hum MolGenet 4 :1137–1146
Trifiro M, Gottlieb B, Pinsky L, Kaufman M, Prior L, BelshamDD, Wrogemann K, RoBrown CJ, Willard HF, Trapman J,Brinkmann AO, Chang C, Liao S, Sergovich F, Jung J (1991)The 56/58 kDa androgen-binding in male genital skin fibro-blasts with a deleted androgen receptor gene. Mol Cell En-docrinol 75 :37–47
Twist EC, Causaubon LK, Ruttledge M, Rao VS, MacLeod PM,Radvany J, Zhao Z, et al (1995) Machado-Joseph disease mapsto the same region of chromosome 14 as the spinocerebellarataxia type 3 locus. J Med Genet 31 :823–829
162