student seminar
DESCRIPTION
Student Seminar. Tutor: Professor V. CN Wong Group B(1) Chiang Chi Lin, Chilene Lai Yi Lam Lee Yin Yin, Candice Mui Wing Ho. Case. Chan Yu Kai M/ 7 ½ years old First consultation - 1yr 8mths Presentation – Poor feeding since birth. History of Presenting Illness. Artificial formula - PowerPoint PPT PresentationTRANSCRIPT

Student Seminar
Tutor: Professor V. CN WongGroup B(1) Chiang Chi Lin, Chilene Lai Yi Lam Lee Yin Yin, Candice Mui Wing Ho

Case
Chan Yu KaiM/ 7 ½ years old
First consultation - 1yr 8mths
Presentation – Poor feeding since birth

History of Presenting Illness
Artificial formulaAfter birth: stayed in nursery for 20 days due to poor feeding:
Feeding 1 ½ oz X3 X 8 30 min to finish No choking or cyanosis
2 mths: “Does not want to eat” Obstruction by tongue Some vomiting after feeding Consulted QE Paediatrics
Feed by syringe ? GE reflux
8 mths: Head trauma with right epidural haemorrhage
Required tube feeding1yr 8mths: referred to feeding team of DK

1 yr 8 mthsObs Hx – G1P1, 37wks, LSCS, 2kgAntenatal –IUGR since 4 mths. 46 XYImmunization – up to dateFamily History
No parental consangunity. No MR, psychiatric illness, hearing problem, motor problem, speech problem
Father: 49, air conditioner techinician Mother: 33, housewife Only child
Past medical History Feeding problem since birth 8 mth – head trauma with right epidural haemorrhage.
Fall from low chair while sitting without support Operated in QEH. Required enteral feed for 1 year after operation. Left sided clumsiness Physiotherapy and occupational therapy

1 yr 8 mths (20 mths)Developmental History
Gross motor Sit without support 9-10 mths (6mths) Stand without support 14 mths (10mths) Walk without support 17 mths (15mths)
Fine motor Right hand dominance > 8 mths
Language First word 12 mths (9mths) At 20mths – about 10 single words (uses 2 or more words to
make simple phrases) Social
Feeding – requires tube feeding Drank from cup with aid with water only. Refused milk or sol
id food. (18mths – holds spoon and gets food safely to mouth)
Gross hearing and vision – normal

1 yr 8 mths (20 mths)Griffiths Mental Developmental Scale General IQ = 82
Mental Age = 17 ¼ mth
Mental age (mths) SubquotientA) Locomotor 16 75
B) Personal 19 ¾ 94
C) Hearing & Speech
18 ¼ 88
D) Eye & Hand 20 95
E) Performance 15 56

1 yr 8 mthsNeurological Examination
Toddler’s gait Clumsy left side Upper limb – increased pronater tone on left side Lower limb – phasic spasticity of left hip adductor ? Cranial nerves Vision – normal Hearing – normal
Diagnosis Left mild hemiparesis Global delay Feeding difficulty Failure to thrive ? GE reflux
Etiology Post epidural haemorrhage Poor oromotor function


Causes of poor feedingHealthy children Parental factors Environmental factors Psychological factors
In children with GI tract disorders Oral dysphagia
structural anomalies of the mouth and pharynx central nervous system defects or injury
Pharyngeal dysphagia enlarged tonsils, adenoids, and retropharyngeal abscess
Esophageal dysphagia stricture, stenosis, atresia, stridor, and esophagitis
Gastro-oesophageal reflux

Special needs children Chronic ill health
seizures, frequent upper respiratory infection (decrease motivation to eat, increased energy expenditure)
Cerebral palsy Syndromes
E.g. Down's, Riley-Day, Angelman's, Silver-Russell, Pierre Robin syndromes;
Physical and structural abnormalities pertinent to feeding poor lip closure muscle rigidity or weakness malocclusion high palate poor tongue movement Aspiration Reflux difficulty with oromotor control postural tone abnormalities abnormal oromotor pattern and oral tactile sensitivity

2-7 yrs oldFU at DK for Feeding difficulty Global developmental delay Oromotor dyspraxia
Reduced vertical and horizontal (esp to the left) movement of tongue
No fasciculations or atrophy No other neurological signs

Growth parameters Weight
2-6 yrs old: < 3rd percentile 6-7 yrs old: 25 percentile
Height 2-5 yrs old: ~ 3rd percentile 5-6 yrs old: 10th percentile 6-7 yrs old: ~ 25th percentile
Head circumference 2-4yrs old: 3-10th percentile 4-6 yr old: 10-25th percentile

Attended local kindergarten 2-3 yrs old: Feeding problem at school. Requires teacher
’s assistance. 5-7 yrs old: Poor attention at class. Need to sit in the first r
ow. Average performance. Mother chose to retake K3. Will enter local primary school this year.
Previous IQ scores according to the mother: 70 ( 5 yrs old), 80 (7 yrs old)
Lives with mother and father. Previous caretaker was grandmother until 8 mths old (after head trauma). Chief caretaker now is his mother.Mother describes child has few friends. Poor attention at school but good attention when doing things that interests him e.g. reading picture books and watching cartoons.

7 ½ yrs oldGeneral Examination
Face: Triangular shaped face. Thin lipped. No other dysmorphic features e.g. single looped ears, hyperteleorism
Limbs: No obvious limb asymmetry. Incurved 5th finger of the right hand.
Skin: no neurocutaneous features e.g. café au lait patches.Growth parameters
Weight: 20kg (25th percentile) Height: 118cm (~ 45th percentile) Head circumference: 51.5 cm (~ 40th percentile)
Chest – normalAbdomen – normal. No undescended testes

Neurological Observation: very active. Running around. Could a
scend and descend stairs unaided. Intelligence: normal. Able to tell examiner his nam
e, age, full address and full name of school attended.
Social: blunt sentences. Does not listen to instructions of examiner
Speech: difficulty in pronouncing the “k” sound Vision: Hypermetropia (5D) Astigmatism (2D) Cranial nerves: limited vertical movement of the t
ongue. No fasciculations. No tongue atrophy. Limbs: normal





SummaryChan Yu Kai M/ 7 ½ yrs old
Presented with poor feeding since birth 2 mth: ? GE Reflux.
8 mth: head trauma with right epidural haemorrhage1 yr 8 mth: assessment results show global developmental delay2-7 yrs: weight and height > & ~ 3 percentile, later pick up and now ~ 25 percentile7 yrs
normal IQ but ?hyperactive ? Attention deficit Positive P/E findings: triangular shaped face, thin lips, clin
odactyly

DdxGI disorder GE reflux
Special needs children Developmental delay poor oromotor control
E.g. Cerebral palsy, Silver Russell syndrome, Pierre Robin sequence
Dysphagia after head trauma ( e.g. cranial nerve palsy)
Normal child Psychiatric disorder e.g. ADHD, Aspergers’







Silver-Russell syndromeHeterogeneous group of conditions of varying severity1953, 2 cases described by Silver Low birth weight Asymmetry Growth retardation
1954, 5 Similar cases by Russell

Clinical FeaturesSGALow birth weightReduced postnatal growthClassical facial featuresAsymmetryFifth finger clinodactylyCafé-au-lait spots

Classical facial features, by Russell
High forehead taperingSmall jawProminent nasal bridgeWell-demarcated philtrumMouth angles down-turning

Other problemsSkeletal system Delayed bone age in early childhood Syndactyly of 2nd and 3rd toes
Neurological: Oromotor dyspraxia Feeding difficulty, articulation problem
Urogenital system Renal anomaly Hypospadias
USUALLY normal intelligenceExcessive sweating (esp head and upper trunk)

Natural history
Early childhood slim and underweight for heightGross motor developmental delayGrowth improvement in childhood or adolescent, still short statureLess obvious facial features with ageIQ correlated with OFC(~30 % with learning disability)

Etiology
Mostly sporadic Uniparental disomy of chromosome 7 Isosomy of chromosome 7
Others reported: AR X-linked deletions of distal 15q Ring chromosome 15 Trisomy 18 mosacism Others….

New Diagnostic criteria
Presence of 3 of the following Low birth weight Short stature Classical facial features Asymmetry Clinodactyly

What can be done about?
Specialized schoolingSx for urogenital abnomaliesGH supplement

Mental retardation
AAMR Definition Substantial limitations in present functioning. Significantly subaverage intellectual functionin
g, with related limitations in two or more of the following applicable adaptive skill areas:
Communication, Self-care, Home living, Social skills, Community use, Self-direction, Health and safety, Functional academics, Leisure, and Work.
Mental retardation manifests before age 18. (Luckasson et al., 1992, [25] p.5)"

Mental retardation
MR is subdivided into: mild (IQ between 50 and 70), moderate (IQ between 35 and 50), severe (IQ between 20 and 35), profound (IQ below 20).
Adaptive functioning sometimes relatively good, may mask the extent of the general intellectual functioning.

3 steps of diagnosing MR
Step 1 diagnosis, determines eligibility based on three cirteria:
IQ, adaptive skill level, and age of onset. IQ determination is made on the basis of perfor
mance below 97% of same-age peers with comparable cultural backgrounds if appropriate standardized measure unavailable (Luckasson, 1992) [25].

3 steps of diagnosing MR Adaptive skill level determined with
standardized instruments such as the revised Vineland Adaptive Behavior Scales (Sparrow, Balla, & Cicchetti, 1984) [38]
the Comprehensive Test of Adaptive Behavior (Adams, 1984) [1] in addition to narrative descriptions by the diagnostic team.
Age of onset before 18 reflects U.S. cultural norms about when an individual is expected to assume adult roles; this age may be different for different cultures (Schalock et al., 1994) [36].

3 steps of diagnosing MR
Step 2: Description, identifies a person's strengths and
weaknesses. Strengths and weaknesses of intellectual and a
daptive skills must be described Source identified by the multidisciplinary team f
or 10 skill areas: communication, self-care, home living, social skills, co
mmunity use, self-direction, health and safety, functional academics, leisure, and work (Luckasson et al., 1992) [25].

3 steps of diagnosing MR
Step 3: profile and intensities of needed supports, describes the support function, activity, and
level of intensity for each functional area requiring support identified in Step 2.
For example, if an individual had been identified in Step 2 as having a physical-health diagnosis, such as cerebral palsy, extensive supports identified in Step 3 may include a wheelchair and physical therapy.

Evaluation of the child with global developmental delay (GDD)1. Obtain a detailed history and examination
• Able to identify the etiology in 20-30% of GDD• Accurate prenatal/birth history and family history,
together with a three-generation pedigree• Presence of dysmorphic features, minor anomali
es, skin changes• A thorough neurological assessment• Complemented by photographs and videotaping
(especially valuable in documenting posture, gait, any movement disorders, and behaviour characteristics)

Evaluation of the child with global developmental delay (GDD)
2. Refer for auditory and ophthalmological screening• Children with GDD are at risk to have primary sen
sory impairments of vision and hearing• Speech and language delay is often a feature of G
DD and may be the result of a hearing loss

Evaluation of the child with global developmental delay (GDD)
3. Consider thyroid function test and metabolic screening if universal newborn screening not done
• Thyroid function test (TSH +/- T4)• Metabolic screening (capillary blood
gas, serum lactate and ammonia levels, serum amino acids and urine organic acids)

Evaluation of the child with global developmental delay (GDD)
4. Consider EEG if history of suspected seizures or epilepsy syndrome
5. Consider screening for autism or a language disorder
6. If there is a close family member with GDD• Specific tests for a known metabolic, genetic o
r structural nervous system disorder• Cytogentic screen and subtelomeric rearrange
ments

7. If no family history of GDD but with features suggestive of a specific diagnosis, direct testing may be utilized
Dysmorphic features in Down’s syndrome (karyotype), Fragile X (FMR1), Rett syndrome (MECP2), Praeder-Willi/Angelman (FISH), or hypothyroidism
History of intrapartum asphyxia or physical signs such as microcephaly, cerebral palsy, focal neurological signs or focal seizures may suggest acquired CNS injury or an underlying cerebral malformations (neuroimaging study preferably MRI)
Risk factors for lead poisoning/findings suggestive of lead intoxication (lead screening)
Loss/regression of developmental milestones, history of parental consanguinity, prior unexplained loss of a child or multiple miscarriages (metabolic screening, neuroimaging, EEG, cytogenetic studies, genetic and ophthalmologic consultations)

Evaluation of the child with global developmental delay (GDD)
8. If there is no feature suggestive of any specific diagnosis
• Less likely to be associated with a definable disease and thus a stepwise approach is more appropriate
• Includes initial neuroimaging (MRI), cytogenetic and fragile X screening
• If negative, proceed to metabolic evaluation, testing for subtelomeric rearrangements, and genetic consultation, and test for Rett syndrome (Shevell et al, 2003)

Cytogenetics/molecular cytogenetics in the evaluation of MR
Genetic defects account for 28% of MR (Stevenson et al, 2003)Cytogenetic studies at a 400-550 band resolution is the standard investigation for suspected chromosomal rearrangements and can detect microscopically visible rearrangements (>3MB)4p- (Wolf-Hirschhorn) and 5p- (cri du chat) syndromes are examples of microscopically visible deletions that mostly include the subtelomeric region and cause MR associated with a specific phenotype

Cytogenetics/molecular cytogenetics in the evaluation of MR
Submicroscopic rearrangements (<2-3MB) however cannot be detected by cytogenetics analysis alone, and may account for a significant proportion of MR cases with unknown etiologyThe end of chromosomes (telomeres) are involved in the majority of translocationsThe regions adjacent to telomeres are gene rich so that rearrangements involving telemere-adjacent DNA (subtelomeric) are more likely to have phenotypic consequences

Cytogenetics/molecular cytogenetics in the evaluation of MR
Once recognizable syndromes have been excluded, submicroscopic subtelomeric rearrangements are the commonest cause of undiagnosed moderate to severe MR (Knight et al, 1999)Molecular cytogenetics enable the use of chromosome-specific probes for the detection of submicroscopic subtelomeric rearrangementsTechniques include fluorescent in-situ hybridization (FISH) of subtelomeric probes, and microsatellite markers (which can also detect uniparental disomy)

Cytogenetics/molecular cytogenetics in the evaluation of MR
Molecular cytogenetic techniques are complex, costly and not widely availablePreselection of patients with a 5-item checklist (cut-off score of ≥3) can increase the diagnostic yield (De Vries et al, 2001)


Submicroscopic subtelomeric rearrangements in MR
(De Vries et al, 2003)

Subtelomeric rearrangements in MR
Some of the subtelomeric deletions result in a specific phenotype (4p, 5p, 9p more consistent)

Management
Treat any treatable cause (T4 for hypothyroidism)Explanation of prognosis and management plan to parentsMultidisciplinary approach (paediatricians, PT, OT, speech therapist, MSW etc)

Etiology of MR
2 overlapping populations of retarded children: environmental influences mild MR biologic causes severe MR

Identification of Cause in Children with Mild Mental Retardation
offspring of women who have not completed high school than in women who have graduatedboth genetic (i.e., children may inherit a cognitive impairment) and socioeconomic (i.e., poverty, undernutrition) factorsThe specific causes of mild MR, however, are currently identifiable in less than half of affected individuals.

The most common biologic causes of mild mental retardation include:
Genetic syndromes with multiple minor congenital anomalies Fetal deprivationPerinatal insults Intrauterine exposure to drugs of abuseSex chromosomal abnormalities

Identification of Cause in Children with Severe Mental Retardation
Chromosomal disorder 22%Genetic syndrome 21%Developmental brain abnormality 9%Inborn errors of metabolism/neurodegenerative disorder 8%Congenital infections 4%
Familial retardation 6%Perinatal causes 4%Postnatal causes 5%Unknown 21%

Down’s syndrometrisomy 21- non dysjunction, translocation, mosaicism
Other features Hypotonia (floppy baby) Relatively small stature Hyperreflexible joints Developmental delay Short fingers 5th finger clinodactyly Simian palmar crease Wide gap between 1st and 2nd toe
s CHD: AVSD, VSD, PDA, ASD, valve
prolapse > 20 years Lose neck folds in infant Dry skin folliculitis in aldolescent
s Soft fine pubic hair Small penis and teticular volume Infertility common
Craniofacial features Brachycephaly Mild microcephaly Upslanting palpebral fissures Epicanthic folds Myopia Acquired cataracts Brushfield spots Small ears Mixed hearing loss Glue ear Small nose Protuding tounge Dental hypoplasia Short neck

Which one has Down’s syndrome?

Fragile X syndromeXq27.3 fragile site triplet repeat, single gene inheritance
Craniofacial features Large head with promin
ent forehead Long thin face Thickening of nasal brid
ge Prominent jaw Pale blue irides Large ears; maybe poste
rior rotated
Other features Large testes Hypotonia Joint laxity Cluttering speech Hyperkinetic behaviour Mental retardation

Fragile-X syndrome

Tuberous sclerosis80% new mutation. AD inheritance
Craniofacial and cutaneous features
Amelantotic naevi (ash leaf spots) ~ may need wood light to see
Adenoma sebaceum ~ erythematous papular acneiform rash over nose and cheeks
Shagreen patch ~ raised irregular rough area usually over the lumbar region
Subungual fibroma ~ fleshy outgrowth of nail bed of fingers or toes (rare in childhood)
Gingival fibromas Café-au-lait
Other features Learning difficulties and d
evelopmental delay Palpable kidneys due to r
enal angiomyolipomata or polycystic kidneys
Retinal phakomata ~ raised mushroom-like lesion near optic disc
Parents may have stigmata of disease

Adenoma sebaceum of TS

Angelman syndrome15q11-13 uniparental disomy (paternal)
Craniofacial features Happy appearance +/- i
nappropriate laughter Characteristic arm post
ure (resembling a puppet)
Prominent jaw Maxillary hypoplasia Tongue protrusion Wide spaced teeth
Other features Microbrachycephaly Deep set eyes Jerky movement Hypotonia Growth retardation Ataxic gait Mental retardation Optic atrophy Seizure
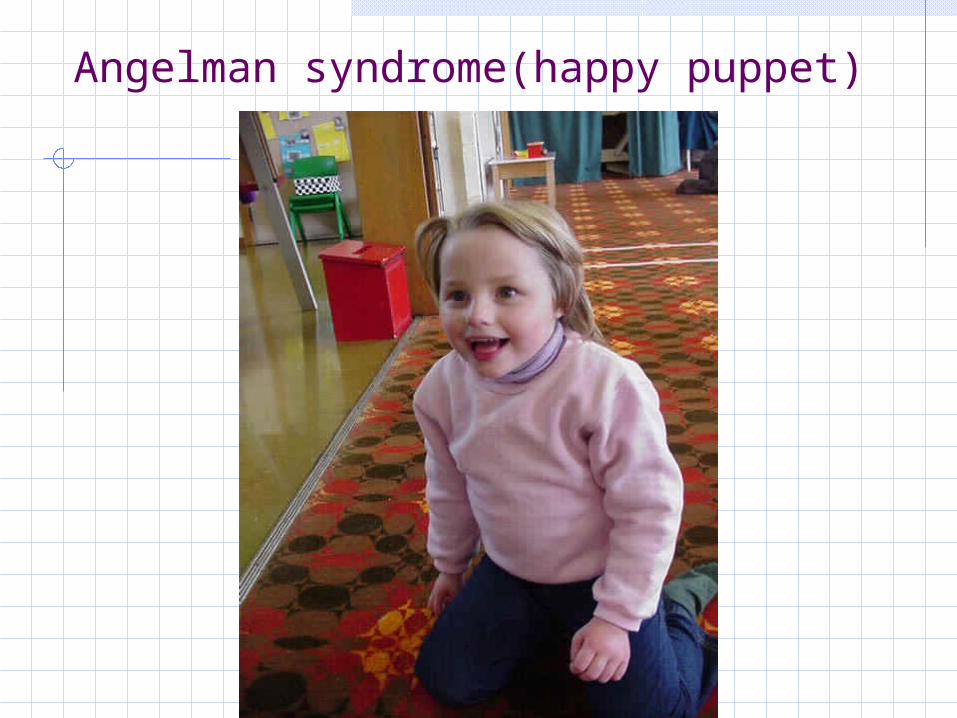
Angelman syndrome(happy puppet)

Williams syndrome7q11microdeletion
Craniofacial features Medial eyebrow flare Depressed nasal bridge Epicanthic folds Blue eyes Stellate pattern iris Prominent lips (fish-sha
ped)
Other features Mental retardation Friendly manner Cocktail party speech Hypersensitivity to sound Short stature Hypoplastic nails Scoliosis Hyphosis Joint limitations Cardiac problem: supravalvula
r aortic stenosis, peripheral pulmonary artery stenosis, VSD, ASD, renal artery stenosis
Nephrocalcinosis Pelvic kidney Urethral stenosis

Williams syndrome

Prader-Willi Syndrome 15q11-13 uniparental disomy (maternal)
Craniofacial features Obesity Almond shaped eyes Prominent forehead wit
h narrow bifrontal diameter
Triangular shaped upper lip
Hypogonadism / cryptorchidism
Short stature
Other features Small hands and feet In neonatal and infants,
major sign is hypotonia Mental retardation Hyperphagia Diabetes mellitus Behavioural difficulties

Prader-Willi syndrome

ReferencesKnight SJL et al. Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet 1999; 354:1676-81De Vries et al. Clinical studies on submicroscopic subtelomeric rearrangements: a checklist. J Med Genet 2001; 38: 145-150Stevenson et al. Genetic syndromes among individuals with MR. AJMG 2003 123A:29-32Shevell et al. Practice parameter: Evaluation of the child with GDD. Neurology 2003; 60:367-80De Vries et al. Telomeres: a diagnosis at the end of chromosomes. J Med Genet 2003; 40:385-98S M Prince et al. The spectrum of Silver-Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J Med Genet 1999; 36:837-842Lai KYC et al. Cognitive abilities associated with Silver-Russell syndrome. Arch Dis Child 1994; 71:490-6