structure-based view of epidermal growth factor receptor regulation
TRANSCRIPT

ANRV343-BB37-17 ARI 24 April 2008 15:30
Structure-Based Viewof Epidermal GrowthFactor Receptor RegulationKathryn M. FergusonDepartment of Physiology, University of Pennsylvania School of Medicine,Philadelphia, Pennsylvania 19104; email: [email protected]
Annu. Rev. Biophys. 2008. 37:353–73
First published online as a Review in Advance onFebruary 7, 2008
The Annual Review of Biophysics is online atbiophys.annualreviews.org
This article’s doi:10.1146/annurev.biophys.37.032807.125829
Copyright c© 2008 by Annual Reviews.All rights reserved
1936-122X/08/0609-0353$20.00
Key Words
EGFR, ligand-induced receptor dimerization, mechanisms ofactivation and inhibition, receptor tyrosine kinase
AbstractHigh-resolution X-ray crystal structures determined in the past sixyears dramatically influence our view of ligand-induced activation ofthe epidermal growth factor receptor (EGFR) family of receptor ty-rosine kinases. Ligand binding to the extracellular region of EGFRpromotes a major domain reorganization, plus local conformationalchanges, that are required to generate an entirely receptor-mediateddimer. In this activated complex the intracellular kinase domainsassociate to form an asymmetric dimer that supports the allostericactivation of one kinase. These models are discussed with emphasison recent studies that add details or bolster the generality of thisview of activation of this family of receptors. The EGFR family isimplicated in several disease states, perhaps most notably in cancers.Activating tumor mutations have been identified in the intracellularand extracellular regions of EGFR. The impact of these tumor muta-tions on the understanding of EGFR activation and of its inhibitionis discussed.
353
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
EGFR: epidermalgrowth factorreceptor
RTK: receptortyrosine kinase
Contents
INTRODUCTION. . . . . . . . . . . . . . . . . 354EGFR FAMILY OF RECEPTOR
TYROSINE KINASES . . . . . . . . . . 355THE UNEXPECTED
RECEPTOR-MEDIATEDDIMER OF THEEXTRACELLULARREGION OF EGFR . . . . . . . . . . . . . 357
A STRUCTURE-BASED MODELFOR LIGAND-INDUCEDEGFR DIMERIZATION . . . . . . . . 359
CONSTITUTIVELY EXTENDEDErbB2 AND RECEPTORHETERODIMERIZATION . . . . . 361
sEGFR AUTOINHIBITION ANDTHE TETHERED STATE . . . . . . 362
EXTRACELLULAR EGFRMUTATIONS IN CANCER . . . . . 363
ACTIVATION OF THEINTRACELLULAR TYROSINEKINASE DOMAIN OF EGFR . . . 363
INTRACELLULAR EGFRMUTATIONS IN CANCER . . . . . 365
MECHANISM OF EGFRACTIVATION AT THECELL MEMBRANE. . . . . . . . . . . . . 365
EGFR FAMILY AS TARGETS FORANTICANCER THERAPY . . . . . 367
OUTSTANDING QUESTIONS . . . 368
INTRODUCTION
The epidermal growth factor receptor(EGFR) is historically the prototypical recep-tor tyrosine kinase (RTK). It was the firstof this large family of transmembrane recep-tors to be cloned; the first for which the im-portance of ligand-mediated oligomerizationin the activation of the enzyme was appreci-ated; and the first for which a clear connec-tion between aberrant receptor function andcancer could be drawn (58, 67, and referencestherein).
The first step in RTK activation involvesligand-induced receptor dimerization or al-teration of pre-existing dimers (57). Thisleads to stimulation of the intracellular ki-nase domain and tyrosine autophosphoryla-tion in trans. Phosphorylated tyrosines act asrecruitment sites for downstream signalingmolecules containing SH2 and/or PTB do-mains. The cascade of events that are initiatedfollowing activation of EGFR represent someof the most extensively studied sets of signaltransduction pathways. Many of the princi-pals governing the regulation of such path-ways were elucidated from studies of EGFRsignaling (15, 69).
In normal physiological settings EGFR,and the other three homologous membersof the EGFR family, regulate key events incoordination of cell growth, differentiation,and migration (15). EGFR itself is criticalin epithelial development, and other mem-bers of the family are essential for cardiacdevelopment and/or have well-studied rolesin mammary glands and the nervous system(7). Aberrant signaling from all four receptors,through misregulation of the receptors or oftheir ligands, has been implicated in diseasesincluding nervous system disorders and manycancers (7, 34, 49). For example, EGFR ac-tivation in epithelial tumors has been linkedwith more aggressive disease and poorer out-comes. Drugs that inactivate EGFR throughinteraction with either the extracellular or in-tracellular regions of EGFR are intensivelystudied in the clinic (49).
This review considers the impact of high-resolution structural studies upon our cur-rent understanding of EGFR regulation. Thestarting point is the model of ErbB receptoractivation that was proposed in 2003 basedon structural data available at that time (8,40). Since 2003, significant new informationhas been published that increases the sophis-tication of this model and/or adds new facetsthat were not previously considered. It is tothese studies that I devote the majority of thisreview.
354 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
EGFR FAMILY OF RECEPTORTYROSINE KINASES
The EGFR family of RTKs comprises fourmembers (collectively referred to as theErbB or HER family): EGFR itself, ErbB2(HER2/Neu), ErbB3 (HER3), and ErbB4(HER4). Like all RTKs, each ErbB receptorcomprises a large extracellular region, a sin-gle spanning transmembrane (TM) domain,
TM:transmembrane
JM: juxtamembrane
EGF: epidermalgrowth factor
an intracellular juxtamembrane (JM) region,a tyrosine kinase domain, and a C-terminalregulatory region (Figure 1a). The ligandsthat regulate ErbB receptors can be separatedinto two main groups (Figure 2): the EGFagonists that activate EGFR, and the neureg-ulins (NRG) that bind ErbB3 and ErbB4(69). There are at least seven different EGFagonists: EGF, transforming growth factor
Figure 1The domains of EGFR. (a) The extracellular region comprises four domains: I–IV, sometimes referred toas L1, CR1, L2, and CR2 or L1, S1, L2, and S2. Domains I (red ) and III ( gray with red outline) shareabout 37% sequence identity, while domains II ( green) and IV ( gray with green outline) are cystine rich.The N-lobe of the kinase domain is in lavender and the C-lobe is in blue. This color scheme is used in allfigures unless otherwise noted. Amino acid numbers are noted for each domain boundary. Theconventional numbering system is used in which amino acid one of EGFR is the assumed first amino acidof the mature protein. In some recent papers, including those defining EGFR cancer mutations,alternative numbering is used where the signal peptide of EGFR is included. To convert to this alternativescheme, add 24 to the numbers used here. (b) Representative ribbon diagrams of the domains of EGFR.Domains I and III adopt a β-helix fold; here domain I from PDB ID 1YY9 is shown. Domains II and IVadopt extended structures comprising a series of disulfide-bonded modules. Domain IV from PDB ID1YY9 is shown with the disulfides in stick representation and the disulfide-bonded modules numbered.There are two types of disulfide-bonded module. One has a single disulfide bond and the interveningloops adopt a bow-like arrangement (modules 2, 3, 5, and 6). The second type has two disulfide bondswith consecutive cysteines linked in the pattern Cys1-Cys3 and Cys2-Cys4 (modules 1, 4, and 7). Theinactive kinase is shown (PDB ID 2GS7) with the ATP analogue (AMP-PNP) in stick representation.
www.annualreviews.org • Structure and Regulation of EGFR 355
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
Figure 2The extracellular regions of ErbB receptors and their activating ligands. Two orthogonal ribbondiagrams are shown for each unliganded ErbB receptor (PDB IDs 1NQL, 1N8Z, 1M6B, and 2AHX).The coordinates of domain III only were used to align the structures. ErbB2 is an outlier adopting anextended rather than tethered arrangement of domains. This extended arrangement of the domains ofErbB2 is similar to the domain arrangement observed in the ligand-induced dimer of sEGFR (Figure 3).Ligands are listed, grouped according to the receptors they activate. Ribbon diagrams of TGFα (left;PDB ID 1MOX) and NRG1α (right; PDB ID 1HRE) are shown in cyan as representative structures ofthe EGF-like domain of ErbB ligands. The scale for the ligands is twice that used for the receptorextracellular regions.
ErbB receptors:members of theEGFR family ofRTKs; EGFR/ErbB1/HER1,ErbB2/HER2/neu,ErbB2/HER3,ErbB4/HER4
α (TGFα), amphiregulin (AR), betacellulin(BTC), epigen (EPN), epiregulin (EPR), andheparin binding EGF-like growth factor (HB-EGF) (30). Of these, a subset can also acti-vate ErbB4 and are known as the bispecificligands (BTC, EPR, and HB-EGF). ErbB3and ErbB4 are regulated by multiple differ-ently spliced variants of the four different
NRG gene products (20). Each ErbB ligandcontains an EGF-like core domain of about60 amino acids (Figure 2) that is sufficientfor its biological activity (30). ErbB2 has noknown soluble ligand and has been proposedto play a role in ErbB receptor activationby forming heterodimers with other ligandedErbB family members (14, 69). ErbB2 is also
356 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
distinguished from other members of this re-ceptor family in that overexpression of ErbB2causes ligand-independent cell transforma-tion (18). As shown in Figure 2, and discussedin detail below, ErbB2 also turns out to be anoutlier structurally.
The extracellular regions of EGFR fam-ily members contain two homologous lig-and binding domains (domains I and III) andtwo cystine-rich domains (domains II and IV;Figure 1). The only other RTKs with asimilar extracellular domain arrangement aremembers of the insulin receptor (IR) family,which share the same domain I/II/III organi-zation, but the membrane proximal cystine-rich domain IV of ErbB receptors is replacedby fibronectin type III domains in the IR fam-ily. By contrast with the EGFR and IR familiesmost other RTKs have extracellular regionscomprised of immunoglobulin or fibronectintype III domains (32). Just as they are distinctin their domain composition, so do the IR andErbB families differ from other RTKs in theirmechanisms of ligand activation (66).
Although high-resolution structural stud-ies of intact RTKs pose technical challengesthat have not yet been overcome, there is awealth of structural data on both the extra-and intracellular regions of the EGFR fam-ily. X-ray crystal structures have been de-termined for the extracellular regions of allfour ErbB receptors (sErbBs) in their unli-ganded state (6, 11, 12, 22, 25) (Figure 2).The structure of the EGFR soluble extra-cellular region (sEGFR) has also been deter-mined in a dimeric—presumably activated—state induced by binding of EGF or TGFα
(26, 54) (Figure 3). Additional insight into themechanisms of extracellular control has alsobeen provided by three different structuresof sErbB proteins in complex with the Fabfragments of inhibitory therapeutic antibod-ies (12, 24, 44). The structure of the intracel-lular kinase domain of EGFR has also been ex-tensively studied in different activation states(62, 68, 71, 74). Structural details of the in-dividual domains of EGFR and their homo-logues have been extensively reviewed else-
TGFα:transforming growthfactor alpha
sEGFR/sErbBs:the solubleextracellular regionof EGFR or of theErbB receptors
where (1, 32, 40) and are summarized brieflyin the legend to Figure 1.
Despite this wealth of structural informa-tion, there are important regions of EGFRfor which relatively little data are available.For example, little is known about the struc-ture of the first ∼30 amino acids of the intra-cellular JM region (Figure 1a), which mayplay an important regulatory role (31, 48).Moreover, the most C-terminal ∼190-amino-acid region of EGFR that contains multi-ple tyrosine phosphorylation sites is poorlycharacterized but is implicated in regulationof receptor activation (45).
THE UNEXPECTEDRECEPTOR-MEDIATED DIMEROF THE EXTRACELLULARREGION OF EGFR
In 2002 two papers published back-to-backin Cell radically changed the mechanistic viewof ligand-induced EGFR dimerization. Theydescribed a dimer in which all contacts be-tween the two molecules were receptor me-diated (26, 54). This contrasts starkly withthe direct contribution of the bound ligandto the dimer interface of other cytokine re-ceptor and RTK dimers that have been stud-ied (5, 8, 32, 57, and references therein). Formany RTKs, such as those of the plateletderived growth factor (PDGF)/Kit receptorfamily, the ligands themselves are dimeric andbivalent. In Kit, each protomer in the lig-and contacts a different receptor molecule, sothat the dimeric ligand effectively cross-linksthe receptor into a dimeric complex (46, 72).In other cases, such as the fibroblast growthfactor (FGF) receptor, accessory molecules(heparan sulfate proteoglycans) link the twoligands to yield a bivalent complex that effec-tively cross-links two receptor molecules (59).
In the dimeric complexes formed whenEGF or TGFα bind to the first three do-mains of sEGFR, the growth factor bind-ing sites are distant from the dimer inter-face and do not contribute directly to dimercontacts (Figure 3b). All contacts across the
www.annualreviews.org • Structure and Regulation of EGFR 357
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
Figure 3Ligand-induced dimerization of the extracellular region of EGFR. (a) Ribbon diagram of the tetheredsEGFR (PDB ID 1YY9) oriented as in the upper panel of Figure 2. (b) Ribbon diagram of theTGFα-induced dimer of sEGFR501 (PDB ID 1MOX). The orientation of domain III is as in the lowerpanels of sErbB ribbon diagrams in Figure 2. The colors of the left-hand molecule have been lightenedfor contrast. (c) A molecular surface representation of tethered sEGFR in the same orientation as in panela, with domains I and III in red, II in green, and IV in gray. An ≈130◦ rotation about the indicated axis(black dot) plus 20 A translation into the plane of the page is required to bring domain I from it position inthe tethered structure (left) to its location in the dimer (right) (22). In this model of the sEGFR dimer,domain IV is included so as to maintain the same domain III/IV relationship as in the tethered structure.Domains I, II, and III in the dimer are from PDB ID 1IVO and are shown in the same orientation as inpanel b.
Tethered sEGFR:The configuration ofthe four domains ofthe extracellularregion of EGFR thatis observed in crystalstructures ofunliganded/inactivesEGFR, sErbB3, andsErbB4
dimer interface are mediated by domain II ofthe receptor—making these dimers receptormediated rather than ligand mediated (58).A beta hairpin, referred to as the dimer-ization arm, in domain II makes extensivecontacts with the domain II of its bindingpartner, reaching at its tip to interact with theopposite domain I (Figure 3b,c). EGF/TGFα
is bivalent; each ligand binds simultaneouslyto domains I and III of the same receptormolecule.
Structures of unliganded sErbB recep-tors indicated that exposure of the dimer-ization arm is a key event in regulation of
receptor dimerization. Indeed, in the inac-tive/unliganded form of sEGFR and sErbB3,the dimerization arm is buried by intramolec-ular interactions with domain IV (11, 22, 44).The intramolecular interactions in this so-called tethered conformation of the receptorand the intermolecular interactions across thedimer interface are mutually exclusive. Thisled to the proposal that the tethered sEGFRand sErbB3 structures represent an autoin-hibited conformation (8, 22), since also seenfor sErbB4 (6).
The tethered configuration of unligandedsEGFR, sErbB3, and sErbB4 also places the
358 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
two ligand-binding sites on domains I and IIIrelatively distant from one another. For a sin-gle EGF molecule to contact these two bind-ing sites simultaneously, a large domain rear-rangement is required in sEGFR (Figure 3c).This domain rearrangement allows domains Iand III to dock onto the same EGF molecule,while simultaneously exposing the dimeriza-tion arm in an extended sEGFR conformationthat closely resembles the structure observedin the dimeric complexes with bound EGF orTGFα (Figure 3c) (22).
A STRUCTURE-BASED MODELFOR LIGAND-INDUCEDEGFR DIMERIZATION
Exposure of the dimerization arm is the mostobvious change induced upon binding ofgrowth factor ligands to the EGFR extracel-lular region (Figure 3), but it remains un-clear exactly how this is achieved. The ligand-binding domains I and III are identical instructure whether bound to ligand or not(21, 44), arguing against an allosteric mech-anism for triggering exposure of the dimer-ization arm. EGF (or TGFα) binds to a trun-cated sEGFR (sEGFR501) that cannot formthe intramolecular domain II/IV tether (withdomain IV removed) only ∼20–30-fold morestrongly than it binds to intact sEGFR (16,19, 22). This observation suggests that thetether provides only a modest energy barrierto the close apposition of domains I and III—of the order of 2 kcal mol−1. If the tetheris this weak, it seems reasonable to suggestthat the EGFR extracellular region could existin a dynamic equilibrium, sampling multipleconformations including tethered and variousuntethered states. Binding of growth factorto domains I and III could then trap unteth-ered receptors in a dimerization-competentextended configuration, driving the system to-ward the active dimeric form (8). There hasbeen no direct analysis of the structural dy-namics of sEGFR invoked by this model, andthis remains an important knowledge gap inthe field.
sEGFR501: atruncated sEGFRthat terminates atamino acid 501, afterthe firstdisulfide-bondedmodule of domain IV
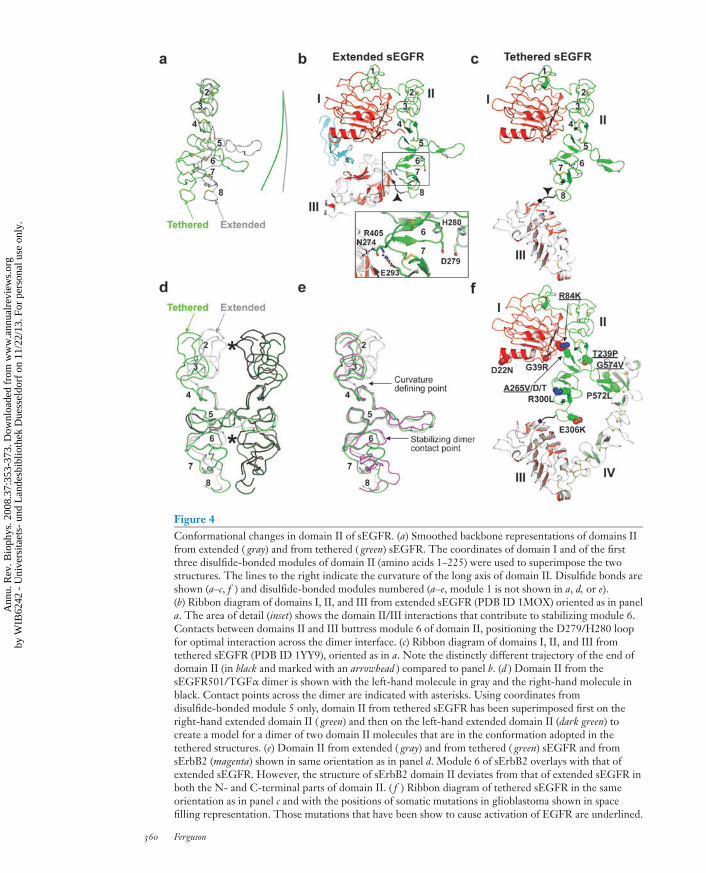
Although there is no doubt that exposingthe domain II dimerization arm is necessaryfor EGFR dimerization, it is not sufficient.Indeed, sEGFR501 remains monomeric un-less EGF/TGFα is added (19). Recent studiesargue that much smaller adjustments in theconformation of domain II at the dimer inter-face are also critical (16). As can be appreciatedin Figure 4, significant local conformationalchanges in domain II accompany the dramaticdomain rearrangement. These changes resultin a different trajectory of the dimerizationarm in the extended compared to the teth-ered receptor, and significantly alter the over-all curvature of the long axis of domain II(Figure 4a). Because domain II forms thedimer interface, this change in curvature uponligand binding could have substantial implica-tions for dimerization strength.
The domain II conformation remainssimilar in the tethered and extended receptorsfor the first three disulfide-bonded modules,which are stabilized by interaction withdomain I. However, the structures beginto deviate significantly at disulfide-bondedmodule 4 (Figure 4a), so that domain II has adifferent overall curvature between disulfide-bonded modules 5 and 8 in the tetheredand extended forms. The consequences ofthe different domain II curvatures can bebetter appreciated if the two conformationsare superimposed using the central disulfide-bonded module 5 as a reference point(Figure 4d ). In dimers constructed fromthese domain II conformations, disulfide-bonded modules 2 and 6 project furtherinto the dimerization interface in the ex-tended or activated structure than in thetethered or inactive structure. The relativeprojection of these two modules into thedimer interface allows them to form di-rect contacts across the dimer interface, asdirectly observed in the crystal structuresof ligand-bound sEGFR (26, 54). Muta-tional analysis demonstrates that interfacialinteractions from module 6 (involvingD279 and H280) contribute substantially todimer stability, while those from module 2
www.annualreviews.org • Structure and Regulation of EGFR 359
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.
ANRV343-BB37-17 ARI 24 April 2008 15:30
Figure 4Conformational changes in domain II of sEGFR. (a) Smoothed backbone representations of domains IIfrom extended ( gray) and from tethered ( green) sEGFR. The coordinates of domain I and of the firstthree disulfide-bonded modules of domain II (amino acids 1–225) were used to superimpose the twostructures. The lines to the right indicate the curvature of the long axis of domain II. Disulfide bonds areshown (a–c, f ) and disulfide-bonded modules numbered (a–e, module 1 is not shown in a, d, or e).(b) Ribbon diagram of domains I, II, and III from extended sEGFR (PDB ID 1MOX) oriented as in panela. The area of detail (inset) shows the domain II/III interactions that contribute to stabilizing module 6.Contacts between domains II and III buttress module 6 of domain II, positioning the D279/H280 loopfor optimal interaction across the dimer interface. (c) Ribbon diagram of domains I, II, and III fromtethered sEGFR (PDB ID 1YY9), oriented as in a. Note the distinctly different trajectory of the end ofdomain II (in black and marked with an arrowhead ) compared to panel b. (d ) Domain II from thesEGFR501/TGFα dimer is shown with the left-hand molecule in gray and the right-hand molecule inblack. Contact points across the dimer are indicated with asterisks. Using coordinates fromdisulfide-bonded module 5 only, domain II from tethered sEGFR has been superimposed first on theright-hand extended domain II ( green) and then on the left-hand extended domain II (dark green) tocreate a model for a dimer of two domain II molecules that are in the conformation adopted in thetethered structures. (e) Domain II from extended ( gray) and from tethered ( green) sEGFR and fromsErbB2 (magenta) shown in same orientation as in panel d. Module 6 of sErbB2 overlays with that ofextended sEGFR. However, the structure of sErbB2 domain II deviates from that of extended sEGFR inboth the N- and C-terminal parts of domain II. ( f ) Ribbon diagram of tethered sEGFR in the sameorientation as in panel c and with the positions of somatic mutations in glioblastoma shown in spacefilling representation. Those mutations that have been show to cause activation of EGFR are underlined.
360 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
(involving Q194) contribute to a smallerextent (16). The conformation of domain II inthe region of disulfide-bonded module 6 is sta-bilized in part by direct interactions with do-main III (Figure 4b) (16, 54). Binding of EGFor TGFα to sEGFR drives a dramatic reori-entation of domain III (compare Figure 3b
with 3c) and promotes domain II/III interac-tions that stabilize the precise conformationof domain II in this region (around module6) that is required for dimerization.
It has also been suggested that domainIV contributes directly to stabilization ofthe ligand-induced sEGFR dimer (8, 22), al-though it was not present in the publisheddimer structures. If the relationship betweendomains III and IV remains fixed in tethered(monomeric) and extended (dimeric) sEGFR,the two copies of domain IV are predictedto make contact across the dimer interface(Figure 3c). However, in studies of solublesEGFR variants, deletion of a putative do-main IV interaction loop or deletion of al-most all of domain IV had only minimal effectson dimerization strength (16). It is possiblethat rather weak domain IV interactions aidin orientating the membrane proximal partsof the EGFR extracellular region in an intactEGFR dimer at the cell surface (4). Indeed,such weak association between the membraneproximal domains of another RTK, Kit, hasrecently been crystallographically visualizedin a ligand-induced dimer (72), and this couldbe an important theme for interactions in theextracellular region of many RTKs.
CONSTITUTIVELY EXTENDEDErbB2 AND RECEPTORHETERODIMERIZATION
Crystal structures of the orphan ErbB2 ex-tracellular region revealed that it adopts anextended configuration in which the arrange-ment of the four domains resembles that seenin each molecule of a sEGFR dimer (Figure 2)(12, 25). Thus, sErbB2 resembles a constitu-tive, or ligand-independent, activated confor-mation. This is consistent with the facts that
Extended sEGFR:The configuration ofthe four domains ofthe extracellularregion of EGFR inthe ligand-boundform
ErbB2 (but not EGFR) overexpression trans-forms cells (14, 18), and that ErbB2 overex-pression is associated with a significant classof human breast cancers (51). Rather thanhaving a growth factor molecule bound be-tween domains I and III—as seen in activatedsEGFR—domains I and III of sErbB2 in-teract directly with one another (Figure 2),and domain I/III interactions appear to sta-bilize the extended configuration of sErbB2.This close proximity of sErbB2 domains Iand III may explain the failure of countlessefforts to identify a high-affinity soluble lig-and for ErbB2. There is no room for a lig-and to bind between domains I and III, so ithas been suggested that no such ligand exists(8, 25).
Despite adopting a constitutively extendedconformation (Figure 2), sErbB2 does nothomodimerize detectably in solution (23) oreven at the very high concentrations presentin the crystals used to determine its structure(25, 40). When a symmetric sErbB2 homod-imer is modeled by overlaying its dimerizationarm with that seen in sEGFR dimers, clashesin the C-terminal portion of domain II (mod-ule 8) preclude satisfactory docking. As shownin Figure 4e, module 6 of domain II of sErbB2adopts the same conformation with respectto the dimerization arm as it does in acti-vated/extended sEGFR. Moreover, the samedomain II/III interactions that stabilize thisregion in the sEGFR dimer are also observedin the sErbB2 structures (25). By contrast theN-terminal part of domain II in sErbB2 doesnot overlay with the equivalent region in ex-tended sEGFR (Figure 4e). Thus, althoughmodule 6 and the dimerization arm appear toadopt conformations in sErbB2 that are rem-iniscent of those required for sEGFR homod-imerization, disulfide-bonded module 2 is notpositioned for optimal interaction across ahomodimeric interface, and module 8 willactually disrupt such a symmetric interface.These observations have been interpreted tosuggest that the extended sErbB2 structuremight be optimal for domain II-mediatedheterodimerization of ErbB2 with another
www.annualreviews.org • Structure and Regulation of EGFR 361
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
ligand-bound (extended) ErbB receptor, inpreference to homodimerization (24). Forexample, X-ray scattering shows that theextracellular region of ErbB3 adopts an ex-tended conformation upon binding to theEGF-like domain of its ligand, neuregulin1β1 (NRG1-β1), without forming homod-imers (17). ErbB2 and ErbB3 are well-knownheterodimerization partners (14), and ex-tended ErbB2 may associate with NRG-bound monomeric ErbB3 in a complex remi-niscent of an asymmetric version of the dimerdepicted in Figure 3. Some evidence hasbeen presented for formation of an NRG-dependent sErbB2/sErbB3 heterodimer (23),and a high-resolution structural view of sucha heterodimer remains an important frontierin this field.
sEGFR AUTOINHIBITION ANDTHE TETHERED STATE
The models described above for EGF-induced dimerization of the EGFR extra-cellular region assume that the crystallo-graphically observed domain II/IV tetherrepresents a set of autoinhibitory interactions.If this is the case, mutations that disrupt thetether should lead to an increased sensitivityof EGFR to activation by ligand. In the ex-treme case, disrupting such an autoinhibitorytether might lead to constitutive activationof the receptor, and we might anticipate thattether mutations will be found in patients withEGFR-dependent cancers. Contrary to theseexpectations, mutations designed to disruptall of the domain II/IV intramolecular inter-actions in intact EGFR did not substantiallyalter cell surface activation of the receptor orthe sensitivity of its EGF dependence (16, 47,64). These results were interpreted to suggestthat the tether has a limited autoinhibitory ef-fect (or even none) on EGFR activity at thecell surface.
Recent studies employing small-angleX-ray scattering (SAXS) suggest that a reeval-uation of the role played by the tethered
configuration of EGFR is required (17). So-lution SAXS studies could readily distinguishbetween the tethered and extended confor-mations of sEGFR, sErbB3, and sErbB2 atlow resolution. However, mutated forms ofsEGFR lacking all of the crystallographicallyobserved domain II/IV interactions thoughtto stabilize the tethered conformation gaveSAXS-derived molecular envelopes that werebarely distinguishable from those seen forwild-type tethered sEGFR. Thus, disruptingthe domain II/IV tether interactions viamutations is insufficient to drive sEGFRinto a significantly extended conformation.The failure of equivalent mutations in full-length EGFR to affect receptor activationis therefore likely to reflect their failure toalter the receptor’s conformation, rather thandiscerning the importance of the tetheredstate at the cell surface.
If direct domain II/IV interactions do nothold sEGFR in a tethered or compact confor-mation, what does? As pointed out by Dawsonet al. (17) the relative orientation of domainsII and III are similar in tethered sEGFRand in domain I/II/III fragments from theIR family (1). This comparison suggests thatthe region of polypeptide linking disulfide-bonded module 8 of domain II with domain III(Figure 4c) may be unexpectedly rigid. Rigid-ity of this linkage could play a significant partin maintaining the EGFR extracellular regionin a tethered-like conformation. Consistentwith this notion, mutation of a cysteine inthe domain II module 8 disulfide bond causesa partial gain of function in the Caenorhab-ditis elegans EGFR ortholog Let-23 (37).Conserved prolines in this region may alsocontribute to local main chain stability, con-sistent with the relatively low crystallographictemperature factors (B-factors) observed inthis region in each tethered sErbB receptor.By contrast the B-factors in disulfide-bondedmodule 8 in the ligand bound dimers arehigher than average, suggesting that inthe extended configuration this region isunder strain. Additional stabilization of the
362 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
tethered configuration could come fromoligosaccharides, not fully visualized in theX-ray crystal structures.
EXTRACELLULAR EGFRMUTATIONS IN CANCER
Somatic mutations were recently identifiedin glioblastomas that map to the extracellularregion of EGFR, and a subset of these en-hanced receptor activation (Figure 4f ) (41).Several of these mutations fall in the vicinityof the intramolecular tether (e.g., P572L andG574V) and could destabilize domain II/IVinteractions—although these mutations arenot likely sufficient for EGFR activationgiven the discussion presented above. Othermutations cluster in different parts of theextracellular region. One group falls closeto disulfide-bonded module 8 of domain IIand could affect the main chain rigidity inthis region—although the effects of thesemutations on EGFR activation was notreported. Another cluster of mutations atthe domain I/II interface is interesting. Inthe tethered receptor, A265 from disulfide-bonded module 5 of domain II packs againstthe aliphatic portion of R84 from domainI (Figure 4f ). Mutations at either of thesepositions lead to EGFR activation (41), whichcould possibly result from alterations in theconformation of domain II.
Further studies of the effects of thesemutations (and their combinations) on theconformational properties of the EGFRextracellular region should provide importantinsight into the structural restraints that keepsEGFR in a tethered-like conformation, andinto the energetic barriers to its extensionand dimerization.
ACTIVATION OF THEINTRACELLULAR TYROSINEKINASE DOMAIN OF EGFR
The unique mechanism of ligand-induceddimerization is not the only feature that
sets EGFR apart from other RTKs. Recentstructural studies of the intracellular EGFRtyrosine kinase domain (EGFR-TK) alsosuggest that it is regulated through an un-expected set of interactions (74); the forma-tion of an asymmetric kinase domain dimer iscritical.
The first reported crystal structures ofEGFR-TK revealed a conformation withcharacteristics of an activated kinase (62),based on the structural features of its acti-vation loop and the orientation of the C-helix (in the N-terminal lobe). Although theapparently constitutive adoption of such anactive conformation was surprising, it wasconsistent with the fact that EGFR is un-usual in not requiring activation-loop phos-phorylation to promote its activity (27). Thesestructures suggested a notable absence of au-toinhibitory interactions in the EGFR kinasedomain, by contrast with the well-defined in-teractions that maintain the kinase domainsfrom the insulin receptor, FGF receptor, andother RTKs in their inactive states (33). Fromamong the possible interpretations of thisstructural view (reviewed in Reference 8), ele-gant studies from the Kuriyan laboratory (74)argue that crystal packing mimics interactionsfound in an active receptor dimer, which leadto activation of EGFR-TK through an al-losteric mechanism.
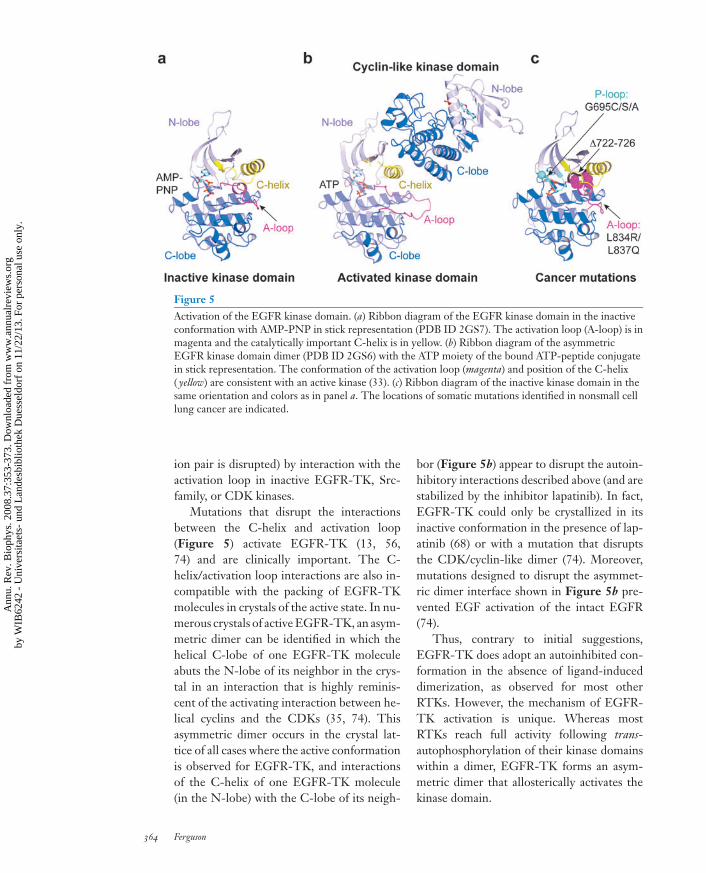
Structures of EGFR-TK bound to thetherapeutic inhibitor lapatinib (68) and of anEGFR kinase mutant bound to AMP-PNP(74) revealed that EGFR-TK can also adopta characteristic inactive structure (Figure 5a)with clear intramolecular autoinhibitory in-teractions. The two inactive EGFR-TK struc-tures are virtually identical to one another andresemble inactive forms of cyclin-dependentkinases (CDKs) and Src-family kinases (33).In each case, a short helical region in the ac-tivation loop is packed against the catalyti-cally critical C-helix, which contains a con-served glutamate that must form an ion pairwith a lysine that coordinates ATP’s α- and β-phosphates. The C-helix is displaced (and this
www.annualreviews.org • Structure and Regulation of EGFR 363
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.
ANRV343-BB37-17 ARI 24 April 2008 15:30
Figure 5Activation of the EGFR kinase domain. (a) Ribbon diagram of the EGFR kinase domain in the inactiveconformation with AMP-PNP in stick representation (PDB ID 2GS7). The activation loop (A-loop) is inmagenta and the catalytically important C-helix is in yellow. (b) Ribbon diagram of the asymmetricEGFR kinase domain dimer (PDB ID 2GS6) with the ATP moiety of the bound ATP-peptide conjugatein stick representation. The conformation of the activation loop (magenta) and position of the C-helix( yellow) are consistent with an active kinase (33). (c) Ribbon diagram of the inactive kinase domain in thesame orientation and colors as in panel a. The locations of somatic mutations identified in nonsmall celllung cancer are indicated.
ion pair is disrupted) by interaction with theactivation loop in inactive EGFR-TK, Src-family, or CDK kinases.
Mutations that disrupt the interactionsbetween the C-helix and activation loop(Figure 5) activate EGFR-TK (13, 56,74) and are clinically important. The C-helix/activation loop interactions are also in-compatible with the packing of EGFR-TKmolecules in crystals of the active state. In nu-merous crystals of active EGFR-TK, an asym-metric dimer can be identified in which thehelical C-lobe of one EGFR-TK moleculeabuts the N-lobe of its neighbor in the crys-tal in an interaction that is highly reminis-cent of the activating interaction between he-lical cyclins and the CDKs (35, 74). Thisasymmetric dimer occurs in the crystal lat-tice of all cases where the active conformationis observed for EGFR-TK, and interactionsof the C-helix of one EGFR-TK molecule(in the N-lobe) with the C-lobe of its neigh-
bor (Figure 5b) appear to disrupt the autoin-hibitory interactions described above (and arestabilized by the inhibitor lapatinib). In fact,EGFR-TK could only be crystallized in itsinactive conformation in the presence of lap-atinib (68) or with a mutation that disruptsthe CDK/cyclin-like dimer (74). Moreover,mutations designed to disrupt the asymmet-ric dimer interface shown in Figure 5b pre-vented EGF activation of the intact EGFR(74).
Thus, contrary to initial suggestions,EGFR-TK does adopt an autoinhibited con-formation in the absence of ligand-induceddimerization, as observed for most otherRTKs. However, the mechanism of EGFR-TK activation is unique. Whereas mostRTKs reach full activity following trans-autophosphorylation of their kinase domainswithin a dimer, EGFR-TK forms an asym-metric dimer that allosterically activates thekinase domain.
364 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
INTRACELLULAR EGFRMUTATIONS IN CANCER
The importance of EGFR-TK autoinhibi-tion is underscored by the growing num-bers of somatic EGFR mutations reported incertain cancers, particularly in nonsmall celllung cancer (NSCLC). In clinical NSCLC tri-als with EGFR-targeted tyrosine kinase in-hibitors (TKIs), a small subset of patientsshowed dramatic initial responses, and thisresponse correlated with the occurrence ofsomatic mutations in exons 18 to 21 in theEGFR kinase domain (56, 61). Point muta-tions in the nucleotide binding loop (the P-loop; exon 18) or in the activation loop (exon21) and deletions immediately preceding thecatalytically important C-helix all lead to en-hanced sensitivity to TKIs. In vitro cellu-lar and biochemical studies have shown thatthese alterations activate the EGFR kinase do-main, leading to ligand-independent signalingthat is effectively inhibited by the TKIs (56).The initial patient response to TKIs there-fore appears to reflect inhibition of constitu-tive, oncogenic signaling by EGFR in theirtumors.
Each class of EGFR-TK mutations foundin NSCLC (Figure 5c) is likely to destabilizethe inactive conformation of the EGFR ki-nase domain (71, 74). For example the L834Rsubstitution (L858R in kinase mutation litera-ture) disrupts interactions between the helicalturn in the activation loop and the C-helix inthe inactive conformation (Figure 5a,c). L834is relatively surface exposed in the active state.Similarly, deletions in the region precedingthe C-helix also remove interactions likely tostabilize the inactive conformation of the acti-vation loop (74). The occurrence and proper-ties of these cancer mutations thus stronglyargue that in inactive EGFR, as for mostother RTKs (32), the kinase domain adoptsan autoinhibited state. Normal activation re-quires ligand-induced dimerization that pro-motes allosteric activation of EGFR-TK. Thecancer mutations circumvent the need for lig-and activation by disrupting interactions that
TKI: tyrosinekinase inhibitor
maintain the kinase in its autoinhibited inac-tive state.
MECHANISM OF EGFRACTIVATION AT THECELL MEMBRANE
In Figure 6 an overall model is presented thatcombines structural information for EGFRon the outside and on the inside of the mem-brane. In the resting state, EGFR is shownwith its extracellular region in the tetheredconfiguration and its kinase domain in the in-active form. Ligand binding to the extracellu-lar region induces receptor-mediated dimer-ization that brings the intracellular domainsinto close proximity, and promotes the as-sociation of the kinase domains in an asym-metric dimer. In the asymmetric EGFR-TKdimer, one molecule is activated through in-teraction of its N-lobe with the C-lobe ofthe cyclin-like activator (shown in the in-active conformation). It is thought that theactivated kinase phosphorylates the C-terminal tail of the activator (cyclin-like) re-ceptor. In a subsequent step (not shown) it isproposed that the roles of the two receptorsswitch, such that both intracellular domainscan become trans-autophosphorylated.
There is structural information for all butthe most C-terminal half dozen amino acids ofthe extracellular region, which link to the pre-sumably helical TM domain. The TM helicesof ErbB receptors self- and hetero-associatein membranes (50). Although TM interac-tions of this sort may aid in stabilizing thedimer, or in orienting its components, muta-tions that disrupt TM domain association donot influence receptor signaling (10, 36; J.M.Mendrola & M.A. Lemmon, unpublisheddata).
On the intracellular side of the mem-brane, several key pieces of information re-main missing—as implied in Figure 6. Thereis no reliable structural information for thefirst ∼30 amino acids of the intracellular JMregion. By analogy with other RTKs, this
www.annualreviews.org • Structure and Regulation of EGFR 365
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
Figure 6Mechanism of EGFR activation. The crystal structures from EGFR are placed so as to provide aframework to consider the mechanism of activation of EGFR at the cell membrane. The same scale isused for the ribbon diagram representations (plus transparent molecular surface) of the extracellularregion and kinase domain. The TM domain is shown as an α-helix (gray), also to this same scale. Regionsthat have not been crystallographically defined are shown with a dashed or solid lines. The missingstretches of the inactive kinase are shown in brown, while those of the active kinase are in black.
region may play a regulatory role—possiblycontributing to autoinhibition (31). A basicstretch at the beginning of this JM regionhas been reported to associate with acidiclipids in the inner leaflet of the membrane andthis could promote association of the kinasedomain itself with the membrane throughelectrostatic interactions (48). Membranetethering of this type is proposed to have anautoinhibitory influence on EGFR, which isreversed when calcium/calmodulin (Ca/CaM)binds to the JM region and dissociates itfrom the membrane. Once released from themembrane the intracellular domains are pro-posed to come together to form the asymmet-ric dimer outlined above. If the asymmetricdimer depicted in Figure 6 forms with its
TM domains in contact, the JM region willhave to adopt an extended structure in order tolink the N termini of the two kinase domains,which are separated in this model by ∼50 A.
The C-terminal ∼190 amino acids ofEGFR have not yet been resolved in any crys-tal structure. Circular dichroism studies indi-cate a significant amount of secondary struc-ture in this region (42). Hydrodynamic studiesshow that the relationship between the largeC-terminal regulatory region and the TK do-main is altered upon autophosphorylation (9).FRET studies indicate that the C-terminaland TK domains become separated—to givea more extended molecule—following acti-vation and phosphorylation (43), possibly re-flecting loss of intramolecular interactions. It
366 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
is intriguing that the side chains of Y974 andY992 in the C-terminal domain, two of theEGFR autophosphorylation sites (60), makewell-defined interactions with the TK domain(68, 74). Disruption of interactions like thiscould be responsible for the changes in overallintracellular region structure upon EGFR au-tophosphorylation and could in turn influenceenzymatic activity of the TK domain. The C-terminal region of EGFR may thus play anautoinhibitory role (65), and autophosphory-lation could reverse this through the typesof conformational changes seen in hydrody-namic and FRET studies. A structural view ofthis region remains one of the key challengesin this field.
EGFR FAMILY AS TARGETS FORANTICANCER THERAPY
Therapeutic agents that target both the ex-tracellular and intracellular regions of ErbBreceptors are in clinical use and/or de-velopment. There are many excellent re-views covering this area (see, for example,References 2, 49, 53, 73). Here, comments arerestricted to the impact of structural studieson understanding the mechanisms of recep-tor inhibition by these drugs. Several mon-oclonal antibodies with clinical efficacy thatbind the EGFR or ErbB2 extracellular regionshave been studied. An EGFR-targeted anti-body (cetuximab/ErbituxTM) binds directly tothe ligand-binding site on domain III (44)and will also sterically block the transitionfrom the tethered to extended states depictedin Figure 3. An ErbB2-targeted antibody(pertuzumab, OmnitargTM) impedes receptordimerization by binding directly to the do-main II (presumed) heterodimerization sitein ErbB2 (24). Perhaps the most well-knownof the ErbB-targeted antibody therapeutics(trastuzumab/HerceptinTM) binds to domainIV of the ErbB2 extracellular domain (12).This appears to block ErbB2 ectodomainshedding that occurs when ErbB2 is expressedat very high levels and that is linked to onco-genesis (52). Although these direct effects on
ErbB receptor signaling contribute to the in-hibitory effects of the antibodies, antibody-dependent cellular cytotoxicity also plays asignificant role in each case. Based on thestructural models for ligand-induced ErbB re-ceptor activation described above, it is highlylikely that antibodies with other modes ofbinding to ErbB receptor extracellular regionswill prove to be valuable. One obvious exam-ple would be antibodies that stabilize the teth-ered conformation.
The small molecule TKIs all bind tothe ATP-binding site of the kinase do-main, and structural details of a number ofthese inhibitors bound to EGFR-TK havebeen reported (62, 68, 71, 74). Some ofthe TKIs, notably erlotinib/TarcevaTM andgefitinib/IressaTM, bind to the active config-uration of EGFR-TK (62, 71). By contrast,lapatinib/TykerbTM binds to (and appears tostabilize) the inactive, autoinhibited form ofEGFR-TK (68). As described above, numer-ous EGFR mutations in NSCLC and head-and-neck cancer have been reported thatappear to destabilize the autoinhibited confor-mation of EGFR-TK (56, 61), and screeningpatients for these mutations can aid in treat-ment decisions. It should be noted that thesemutations destabilize the EGFR-TK confor-mation preferred by lapatinib (68), but sta-bilize the conformation bound by erlotinib(62), gefitinib, and other TKIs (71). The pres-ence of these mutations may therefore indi-cate the use of erlotinib/gefitinib-type drugsrather than the more bulky lapatinib. Thisexample illustrates how a structural under-standing of the effects of both mutations andtherapeutic inhibitors on autoinhibitoryinteractions can have important clinicalimplications.
Finally, a sobering aspect of the use ofTKIs in EGFR-dependent NSCLC is the oc-currence of resistance mutations, in partic-ular the T790M mutation (61). This muta-tion is predicted to clash with inhibitors suchas erlotinib or gefitinib (71). Irreversible in-hibitors of similar chemical structure (anili-noquinazolines) have been reported to inhibit
www.annualreviews.org • Structure and Regulation of EGFR 367
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
T790M EGFR-TK (39) and may representviable treatment alternatives. Further struc-tural studies are needed to define the optimalinhibitors.
OUTSTANDING QUESTIONS
A major outstanding issue is precisely how tocorrelate the structural and biophysical stud-ies that have been the focus of this reviewwith EGFR activation at the cell surface. Sinceearly studies of EGFR it has been known thatthere are two affinity classes for EGF bind-ing to its cell surface receptor (28, 29). Theseclasses are evident in curvilinear Scatchardplots for EGF binding that have been inter-preted as indicating a high-affinity (2%–5%)
class and a low-affinity (the remainder) class ofsites (3). Several studies attempt to correlatethese states with the structure-based modelfor dimerization of the extracellular regionof EGFR with conflicting conclusions (38,47, 64). It is likely that the low-affinity statescorrespond to the binding of EGF to forma symmetric dimer akin to that in the crys-tal structures (55). The nature of the high-affinity states is less clear, yet of critical im-portance as this class of receptors is likelythe mediator of normal EGFR activation (63).One suggestion is that these high-affinityreceptors may be preformed oligomers, forwhich there is some evidence (70). Furtherstudies are needed to resolve this importantissue.
SUMMARY POINTS
1. Ligand binding to the extracellular region of EGFR induces an entirely receptor-mediated symmetric dimer.
2. The arrangement of the domains of the extracellular region of EGFR is dramaticallydifferent in the unliganded and ligand-bound states.
3. Ligand binding induces local conformational changes in domain II of sEGFR thatare critical for dimerization.
4. The EGFR-TK is activated allosterically in an asymmetric homodimer.
5. EGFR is autoinhibited by interactions in the extracellular region, TK domain andpossibly also from the JM and C-terminal regions.
6. Cancer mutations in both the extracellular region and kinase domain can activateEGFR most likely by destabilizing the inactive, autoinhibited state.
FUTURE ISSUES
1. A structure of an intact ErbB receptor, or of the entire intracellular region, would behighly informative.
2. Additional high-resolution structures of ligand-induced homodimers and het-erodimers of sErbB receptors will (or will not) confirm current models proposedbased on available data.
3. Determination of the features that distinguish the population of cell surface EGFRthat binds ligand with high affinity is key to gaining a complete understanding ofnormal receptor activation.
368 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
4. A molecular understanding of differences in cellular responses to stimulation by dif-ferent ErbB ligands is needed.
5. The role of ligand-independent EGFR dimerization/clustering needs to be clarified.
6. Full characterization of the effects of cancer mutations on EGFR (and other ErbB)activity is needed, and this information should be applied to the development ofoptimal therapeutic strategies for patients bearing such mutations.
7. Greater appreciation of similarities and differences in the mechanisms of activationof the EGF and IR receptor family is needed.
DISCLOSURE STATEMENT
The author is not aware of any biases that might be perceived as affecting the objectivity ofthis review.
ACKNOWLEDGMENTS
I thank Mark Lemmon and members of the Ferguson laboratory for valuable discussions andcritical comments on the manuscript. K.F. is the recipient of a Career Award in the BiomedicalSciences from the Burroughs Wellcome Fund and is the Dennis and Marsha DammermanScholar supported by the Damon Runyon Cancer Research Foundation (DRS-52-06).
LITERATURE CITED
1. Adams TE, Epa VC, Garrett TP, Ward CW. 2000. Structure and function of the type 1insulin-like growth factor receptor. Cell. Mol. Life Sci. 57:1050–93
2. Baselga J, Arteaga CL. 2005. Critical update and emerging trends in epidermal growthfactor receptor targeting in cancer. J. Clin. Oncol. 23:2445–59
3. Bellot F, Moolenaar W, Kris R, Mirakhur B, Verlaan I, et al. 1990. High-affinity epider-mal growth factor binding is specifically reduced by a monoclonal antibody, and appearsnecessary for early responses. J. Cell Biol. 110:491–502
4. Berezov A, Chen J, Liu Q, Zhang HT, Greene MI, Murali R. 2002. Disabling receptorensembles with rationally designed interface peptidomimetics. J. Biol. Chem. 277:28330–39
5. Boulanger MJ, Garcia KC. 2004. Shared cytokine signaling receptors: structural insightsfrom the Gp130 system. Adv. Protein Chem. 68:107–46
6. UnligandedsErbB4 is also inthe tetheredconfiguration,supporting ageneral model forall ErbB receptordimerization.6. Bouyain S, Longo PA, Li S, Ferguson KM, Leahy DJ. 2005. The extracellular region
of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad.
Sci. USA 102:15024–297. Bublil EM, Yarden Y. 2007. The EGF receptor family: spearheading a merger of signaling
and therapeutics. Curr. Opin. Cell Biol. 19:124–34
8. Summary ofEGFR dimerizationand activationmechanism fromstructures ofextracellularregions of ErbBreceptors and theEGFR TK domain.
8. Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, et al. 2003. Anopen-and-shut case? Recent insights into the activation of EGF/ErbB receptors.Mol. Cell 12:541–52
9. Cadena DL, Chan CL, Gill GN. 1994. The intracellular tyrosine kinase domain of theepidermal growth factor receptor undergoes a conformational change upon autophospho-rylation. J. Biol. Chem. 269:260–65
www.annualreviews.org • Structure and Regulation of EGFR 369
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
10. Carpenter CD, Ingraham HA, Cochet C, Walton GM, Lazar CS, et al. 1991. Structuralanalysis of the transmembrane domain of the epidermal growth factor receptor. J. Biol.Chem. 266:5750–55
11. Cho HS, Leahy DJ. 2002. Structure of the extracellular region of HER3 reveals an inter-domain tether. Science 297:1330–33
12. Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, et al. 2003. Structure of the extra-cellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421:756–60
13. Choi SH, Mendrola JM, Lemmon MA. 2007. EGF-independent activation of cell-surfaceEGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene26:1567–76
14. Citri A, Skaria KB, Yarden Y. 2003. The deaf and the dumb: the biology of ErbB-2 andErbB-3. Exp. Cell Res. 284:54–65
15. Citri A, Yarden Y. 2006. EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol.Cell Biol. 7:505–16
16. Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. 2005.Epidermal growth factor receptor dimerization and activation require ligand-inducedconformational changes in the dimer interface. Mol. Cell. Biol. 25:7734–42
17. First directevidence for theligand-inducedtransition from thetethered to theextended states forsEGFR andsErbB3.
17. Dawson JP, Bu Z, Lemmon MA. 2007. Ligand-induced structural transitions inErbB receptor extracellular domains. Structure 15:942–54
18. Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. 1987. erbB-2 isa potent oncogene when overexpressed in NIH/3T3 cells. Science 237:178–82
19. Elleman TC, Domagala T, McKern NM, Nerrie M, Lonnqvist B, et al. 2001. Identifica-tion of a determinant of epidermal growth factor receptor ligand-binding specificity usinga truncated, high-affinity form of the ectodomain. Biochemistry 40:8930–39
20. Falls DL. 2003. Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res.284:14–30
21. Ferguson KM. 2004. Active and inactive conformations of the epidermal growth factorreceptor. Biochem. Soc. Trans. 32:742–45
22. Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. 2003. EGFactivates its receptor by removing interactions that autoinhibit ectodomain dimerization.Mol. Cell 11:507–17
23. Ferguson KM, Darling PJ, Mohan MJ, Macatee TL, Lemmon MA. 2000. Extracellulardomains drive homo- but not hetero-dimerization of erbB receptors. EMBO J. 19:4632–43
24. Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. 2004. Insightsinto ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell5:317–28
25. Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, et al. 2003. The crystalstructure of a truncated ErbB2 ectodomain reveals an active conformation, poised tointeract with other ErbB receptors. Mol. Cell 11:495–505
26. Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, et al. 2002. Crystal structureof a truncated epidermal growth factor receptor extracellular domain bound to transform-ing growth factor alpha. Cell 110:763–73
27. Gotoh N, Tojo A, Hino M, Yazaki Y, Shibuya M. 1992. A highly conserved tyrosine residueat codon 845 within the kinase domain is not required for the transforming activity ofhuman epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 186:768–74
28. Greenebaum E, Nicolaides M, Eisinger M, Vogel RH, Weinstein IB. 1983. Bindingof phorbol dibutyrate and epidermal growth factor to cultured human epidermal cells.J. Natl. Cancer Inst. 70:435–41
370 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
29. Gullick WJ, Downward DJ, Marsden JJ, Waterfield MD. 1984. A radioimmunoassay forhuman epidermal growth factor receptor. Anal. Biochem. 141:253–61
30. Harris RC, Chung E, Coffey RJ. 2003. EGF receptor ligands. Exp. Cell Res. 284:2–1331. Hubbard SR. 2004. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev.
Mol. Cell Biol. 5:464–7132. Hubbard SR, Till JH. 2000. Protein tyrosine kinase structure and function. Annu. Rev.
Biochem. 69:373–9833. Huse M, Kuriyan J. 2002. The conformational plasticity of protein kinases. Cell 109:275–
8234. Hynes NE, Lane HA. 2005. ERBB receptors and cancer: the complexity of targeted
inhibitors. Nat. Rev. Cancer 5:341–5435. Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, et al. 1995. Mechanism of CDK
activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376:313–2036. Kashles O, Szapary D, Bellot F, Ullrich A, Schlessinger J, Schmidt A. 1988. Ligand-
induced stimulation of epidermal growth factor receptor mutants with altered transmem-brane regions. Proc. Natl. Acad. Sci. USA 85:9567–71
37. Katz WS, Lesa GM, Yannoukakos D, Clandinin TR, Schlessinger J, Sternberg PW. 1996.A point mutation in the extracellular domain activates LET-23, the Caenorhabditis elegansepidermal growth factor receptor homolog. Mol. Cell Biol. 16:529–37
38. Klein P, Mattoon D, Lemmon MA, Schlessinger J. 2004. A structure-based model forligand binding and dimerization of EGF receptors. Proc. Natl. Acad. Sci. USA 101:929–34
39. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, et al. 2005. Irreversibleinhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl.Acad. Sci. USA 102:7665–70
40. Leahy DJ. 2004. Structure and function of the epidermal growth factor (EGF/ErbB)family of receptors. Adv. Protein Chem. 68:1–27
41. The first reportof activatingmutations in thefull-lengthextracellular regionof EGFR fromtumor samples.
41. Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, et al. 2006. Epidermalgrowth factor receptor activation in glioblastoma through novel missense muta-tions in the extracellular domain. PLoS Med. 3:e485
42. Lee NY, Hazlett TL, Koland JG. 2006. Structure and dynamics of the epidermal growthfactor receptor C-terminal phosphorylation domain. Protein Sci. 15:1142–52
43. Lee NY, Koland JG. 2005. Conformational changes accompany phosphorylation of theepidermal growth factor receptor C-terminal domain. Protein Sci. 14:2793–803
44. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. 2005. Structural basisfor inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 7:301–11
45. Linggia B, Carpenter G. 2006. ErbB receptors: new insights on mechanisms and biology.Trends Cell Biol. 16:649–56
46. Liu H, Chen X, Focia PJ, He X. 2007. Structural basis for stem cell factor-KIT signalingand activation of class III receptor tyrosine kinases. EMBO J. 26:891–901
47. Mattoon D, Klein P, Lemmon MA, Lax I, Schlessinger J. 2004. The tethered configurationof the EGF receptor extracellular domain exerts only a limited control of receptor function.Proc. Natl. Acad. Sci. USA 101:923–28
48. McLaughlin S, Smith SO, Hayman MJ, Murray D. 2005. An electrostatic engine modelfor autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB)family. J. Gen. Physiol. 126:41–53
49. Mendelsohn J, Baselga J. 2006. Epidermal growth factor receptor targeting in cancer.Semin. Oncol. 33:369–85
www.annualreviews.org • Structure and Regulation of EGFR 371
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
50. Mendrola JM, Berger MB, King MC, Lemmon MA. 2002. The single transmembranedomains of ErbB receptors self-associate in cell membranes. J. Biol. Chem. 277:4704–12
51. Meric-Bernstam F, Hung MC. 2006. Advances in targeting human epidermal growthfactor receptor-2 signaling for cancer therapy. Clin. Cancer Res. 12:6326–30
52. Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. 2001. Trastuzumab(herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal andactivated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 61:4744–49
53. Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, et al. 2006. Epidermal growthfactor receptor (EGFR) signaling in cancer. Gene 366:2–16
54. Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, et al. 2002. Crystal structure ofthe complex of human epidermal growth factor and receptor extracellular domains. Cell110:775–87
55. Ozcan F, Klein P, Lemmon MA, Lax I, Schlessinger J. 2006. On the nature of low- andhigh-affinity EGF receptors on living cells. Proc. Natl. Acad. Sci. USA 103:5735–40
56. An excellentsurvey of mutationsin ErbB receptorsand theirinterpretation incontext of modelsfor ErbB receptoractivation.
56. Riese DJ, Gallo RM, Settleman J. 2007. Mutational activation of ErbB family re-ceptor tyrosine kinases: insights into mechanisms of signal transduction and tu-morigenesis. Bioessays 29:558–65
57. Schlessinger J. 2000. Cell signaling by receptor tyrosine kinases. Cell 103:211–2558. Schlessinger J. 2002. Ligand-induced, receptor-mediated dimerization and activation of
EGF receptor. Cell 110:669–7259. Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, et al. 2000. Crystal
structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin inFGFR binding and dimerization. Mol. Cell 6:743–50
60. Schulze WX, Deng L, Mann M. 2005. Phosphotyrosine interactome of the ErbB-receptorkinase family. Mol. Syst. Biol. 1:5–13
61. Sharma SV, Bell DW, Settleman J, Haber DA. 2007. Epidermal growth factor receptormutations in lung cancer. Nat. Rev. Cancer 7:169–81
62. Stamos J, Sliwkowski MX, Eigenbrot C. 2002. Structure of the epidermal growth fac-tor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor.J. Biol. Chem. 277:46265–72
63. Ullrich A, Schlessinger J. 1990. Signal transduction by receptors with tyrosine kinaseactivity. Cell 61:203–12
64. Walker F, Orchard SG, Jorissen RN, Hall NE, Zhang HH, et al. 2004. CR1/CR2 in-teractions modulate the functions of the cell surface epidermal growth factor receptor.J. Biol. Chem. 279:22387–98
65. Walton GM, Chen WS, Rosenfeld MG, Gill GN. 1990. Analysis of deletions of thecarboxyl terminus of the epidermal growth factor receptor reveals self-phosphorylationat tyrosine 992 and enhanced in vivo tyrosine phosphorylation of cell substrates. J. Biol.Chem. 265:1750–54
66. The similaritiesand differencesbetween the EGFand insulinreceptor structuresare wellsummarized.
66. Ward CW, Lawrence MC, Streltsov VA, Adams TE, McKern NM. 2007. The insulinand EGF receptor structures: new insights into ligand-induced receptor activation.Trends Biochem. Sci. 32:129–37
67. Waterfield M. 2007. Cracking the mild, difficult and fiendish codes within and downstreamof the EGFR to link diagnostics and therapeutics. Biochem. Soc. Trans. 35:1–6
68. Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, et al. 2004. A unique structurefor epidermal growth factor receptor bound to GW572016 (Lapatinib): relationshipsamong protein conformation, inhibitor off-rate, and receptor activity in tumor cells. CancerRes. 64:6652–59
372 Ferguson
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

ANRV343-BB37-17 ARI 24 April 2008 15:30
69. Yarden Y, Sliwkowski MX. 2001. Untangling the ErbB signalling network. Nat. Rev. Mol.Cell Biol. 2:127–37
70. Yu X, Sharma KD, Takahashi T, Iwamoto R, Mekada E. 2002. Ligand-independent dimerformation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 13:2547–57
71. Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, et al. 2007. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insightsinto differential inhibitor sensitivity. Cancer Cell 11:217–27
72. Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. 2007. Structuralbasis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 130:323–34
73. Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, et al. 2007. ErbB receptors: fromoncogenes to targeted cancer therapies. J. Clin. Invest. 117:2051–58
74. Confirms thatthe EGFR-TK isautoinhibited andproposes anelegant activationmechanism thatinvolvesasymmetric kinasedomaindimerization.
74. Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. 2006. An allosteric mecha-nism for activation of the kinase domain of epidermal growth factor receptor. Cell
125:1137–49
www.annualreviews.org • Structure and Regulation of EGFR 373
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

AR343-FM ARI 10 April 2008 7:2
Annual Review ofBiophysics
Volume 37, 2008
Contents
FrontispieceRobert L. Baldwin � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �xiv
The Search for Folding Intermediates and the Mechanismof Protein FoldingRobert L. Baldwin � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �1
How Translocons Select Transmembrane HelicesStephen H. White and Gunnar von Heijne � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 23
Unique Rotary ATP Synthase and Its Biological DiversityChristoph von Ballmoos, Gregory M. Cook, and Peter Dimroth � � � � � � � � � � � � � � � � � � � � � � � � 43
Mediation, Modulation, and Consequencesof Membrane-Cytoskeleton InteractionsGary J. Doherty and Harvey T. McMahon � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 65
Metal Binding Affinity and Selectivity in Metalloproteins:Insights from Computational StudiesTodor Dudev and Carmay Lim � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � 97
Riboswitches: Emerging Themes in RNA Structure and FunctionRebecca K. Montange and Robert T. Batey � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �117
Calorimetry and Thermodynamics in Drug DesignJonathan B. Chaires � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �135
Protein Design by Directed EvolutionChristian Jäckel, Peter Kast, and Donald Hilvert � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �153
PIP2 Is A Necessary Cofactor for Ion Channel Function:How and Why?Byung-Chang Suh and Bertil Hille � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �175
RNA Folding: Conformational Statistics, Folding Kinetics,and Ion ElectrostaticsShi-Jie Chen � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �197
Intrinsically Disordered Proteins in Human Diseases: Introducingthe D2 ConceptVladimir N. Uversky, Christopher J. Oldfield, and A. Keith Dunker � � � � � � � � � � � � � � � �215
Crowding Effects on Diffusion in Solutions and CellsJames A. Dix and A.S. Verkman � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �247
vii
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.

AR343-FM ARI 10 April 2008 7:2
Nanobiotechnology and Cell Biology: Micro- and NanofabricatedSurfaces to Investigate Receptor-Mediated SignalingAlexis J. Torres, Min Wu, David Holowka, and Barbara Baird � � � � � � � � � � � � � � � � � � � � � �265
The Protein Folding ProblemKen A. Dill, S. Banu Ozkan, M. Scott Shell, and Thomas R. Weikl � � � � � � � � � � � � � � � � � �289
Translocation and Unwinding Mechanisms of RNAand DNA HelicasesAnna Marie Pyle � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �317
Structure of Eukaryotic RNA PolymerasesP. Cramer, K.-J. Armache, S. Baumli, S. Benkert, F. Brueckner, C. Buchen,G.E. Damsma, S. Dengl, S.R. Geiger, A.J. Jasiak, A. Jawhari, S. Jennebach,T. Kamenski, H. Kettenberger, C.-D. Kuhn, E. Lehmann, K. Leike, J.F. Sydow,and A. Vannini � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �337
Structure-Based View of Epidermal Growth Factor ReceptorRegulationKathryn M. Ferguson � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �353
Macromolecular Crowding and Confinement: Biochemical,Biophysical, and Potential Physiological ConsequencesHuan-Xiang Zhou, Germán Rivas, and Allen P. Minton � � � � � � � � � � � � � � � � � � � � � � � � � � � � �375
Biophysics of Catch BondsWendy E. Thomas, Viola Vogel, and Evgeni Sokurenko � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �399
Single-Molecule Approach to Molecular Biology in Living BacterialCellsX. Sunney Xie, Paul J. Choi, Gene-Wei Li, Nam Ki Lee, and Giuseppe Lia � � � � � � � � �417
Structural Principles from Large RNAsStephen R. Holbrook � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �445
Bimolecular Fluorescence Complementation (BiFC) Analysisas a Probe of Protein Interactions in Living CellsTom K. Kerppola � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �465
Multiple Routes and Structural Heterogeneity in Protein FoldingJayant B. Udgaonkar � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �489
Index
Cumulative Index of Contributing Authors, Volumes 33–37 � � � � � � � � � � � � � � � � � � � � � � � �511
Errata
An online log of corrections to Annual Review of Biophysics articles may be found athttp://biophys.annualreviews.org/errata.shtml
viii Contents
Ann
u. R
ev. B
ioph
ys. 2
008.
37:3
53-3
73. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by W
IB62
42 -
Uni
vers
itaet
s- u
nd L
ande
sbib
lioth
ek D
uess
eldo
rf o
n 11
/22/
13. F
or p
erso
nal u
se o
nly.