structural integrity is essential for the replication of the virusoid
TRANSCRIPT

Structural Integrity is Essential for the Replication of the Virusoid RNA of Lucerne Transient Streak Sobemovirus
by
Kayvan Mirhadi
A thesis submitted in conformity with the requirements for the degree of Master of Science
Cell and Systems Biology University of Toronto
© Copyright by Kayvan Mirhadi 2010

ii
Structural Integrity is Essential for the Replication of the Virusoid
RNA of Lucerne Transient Streak Sobemovirus
Kayvan Mirhadi
Master of Science
Cell and Systems Biology University of Toronto
2010
Abstract
Lucerne transient streak sobemovirus (LTSV) supports the replication of a small, 322-nucleotide,
untranslated virusoid (vLTSV) that has an extensively base-paired, viroid-like structure. Since
vLTSV does not code for its own proteins or share sequence homology with its helper virus
(LTSV), it is presumed that it uses structural motifs to signal the helper virus (and host)
machinery for its replication. In order to elucidate these structural domains, insertion-deletion
mutations were introduced to disrupt the secondary structure. Infectivity assays of these mutants
showed that they were all lethal, except a 9-nucleotide, palindromic insertion, which preserved
the overall rod-like structure of the virusoid. Sequence analysis of cDNA clones prepared from
progeny virusoid RNA revealed that the palindromic sequence was replicated up to twelve days
of infection but discarded afterwards. Results indicate that vLTSV has an optimum size and
secondary structure for replication and packaging within the LTSV helper virus.

iii
Acknowledgements
I would like to thank Professor Mounir G. AbouHaidar, who was my supervisor and
mentor, for making everything possible. He has given me much support and encouragement and
most importantly an opportunity to experience research first hand.
Many thanks goes to my supervisory committee members, Professor Maurice Ringuette
and Professor Keiko Yoshioka for their continuous help and support. I would not have reached
this level without their guidance and helpful remarks.
Thanks goes to my current and former colleagues: Taqueer, Vidya, Amanda, and Huda. A
special thanks goes to Taqueer for helping my out with this project as well as the Begomovirus
side project. You are truly an example of hard work and determination.
I especially want to thank my parents and bigger brother for their love, support, and
friendship. They gave me confidence to know that I can accomplish anything and they were
always supportive of my goals in life.
And, of course, a especial thanks to my beautiful Niloofar, for her love, patience,
understanding, and helping me out through thick and thin.

iv
Table of Contents
Abstract……………………………………………………………………………………………ii
Acknowledgements........................................................................................................................ iii
Table of Contents ........................................................................................................................... iv
List of Figures ............................................................................................................................... vii
List of Tables ............................................................................................................................... viii
List of Abbreviations ..................................................................................................................... ix
Chapter 1 Literature Review........................................................................................................... 1
1 Viroids and Viroid-like Satellite RNAs (Virusoids)................................................................. 1
1.1 General features and organization...................................................................................... 1
1.2 Viroid and virusoid replication .......................................................................................... 4
2 Structure-Function Relationship ............................................................................................... 9
2.1 Functional domains of viroids............................................................................................ 9
2.2 Functional domains of satellite RNAs ............................................................................. 11
Chapter 2 Introduction .................................................................................................................. 12
3 Lucerne Transient Streak virus ............................................................................................... 12
4 The LTSV Virusoid ................................................................................................................ 13
4.1 Satellite-like nature and symptom induction.................................................................... 13
4.2 Helper and host specificity............................................................................................... 13
4.3 Sequence and structure..................................................................................................... 14
4.4 Replication and ribozyme activity.................................................................................... 15
5 Full-length infectious cDNA clones ....................................................................................... 16
6 Research proposal ................................................................................................................... 18
6.1 Overview of previous work.............................................................................................. 18
6.2 Current objectives ............................................................................................................ 18
6.3 Hypothesis ........................................................................................................................ 19
Chapter 3 Materials and Methods ................................................................................................. 20

v
7 General Molecular Techniques ............................................................................................... 20
7.1 Plasmid DNA isolation from E. coli (miniprep) .............................................................. 20
7.2 Heat shock transformation of E. coli................................................................................ 21
7.3 Glycerol stock preparation ............................................................................................... 21
7.4 Phenol-chloroform extraction of DNA/RNA................................................................... 22
8 Sub-cloning of 322I8 to generate multimers .......................................................................... 23
8.1 Restriction digest and agarose gel electrophoresis........................................................... 23
8.2 Ligation ............................................................................................................................ 23
8.3 Screening of colonies ....................................................................................................... 23
9 In vitro runoff transcription of cDNA clones ......................................................................... 25
10 Purification of viruses ........................................................................................................... 26
10.1 Lucerne transient streak virus......................................................................................... 26
10.2 Turnip rosette virus ........................................................................................................ 26
10.3 Extraction of viral RNA ................................................................................................. 27
11 Infectivity assays of cDNA clones........................................................................................ 28
11.1 Coinoculation of TRosV with (+) and (-) RNA transcripts............................................ 28
11.2 Coinoculation of TRosV with dsDNA ........................................................................... 28
11.3 Coinoculation of TRosV with mutants........................................................................... 29
11.4 Total RNA extraction from B. rapa leaves .................................................................... 29
12 Reverse transcription, PCR, and cloning .............................................................................. 30
12.1 Reverse transcription (RT) ............................................................................................. 30
12.2 Polymerase chain reaction (PCR)................................................................................... 30
12.3 Cloning ........................................................................................................................... 31
Chapter 4 Results .......................................................................................................................... 33
13 Infectivity Assays of vLTSV as RNA or dsDNA................................................................. 33
13.1 RNA transcripts .............................................................................................................. 33
13.2 Double-stranded DNA.................................................................................................... 35
14 Structure-function analysis ................................................................................................... 37
14.1 Infectivity of large sized insertion-deletion mutants ...................................................... 37
14.2 Infectivity of smaller sized insertion-deletion mutants .................................................. 41

vi
14.3 Rod-preserving mutant (322I8) ...................................................................................... 43
15 Stability of foreign sequence in vLTSV ............................................................................... 45
15.1 Progeny RNA from 5 days post inoculation .................................................................. 45
15.2 Progeny RNA from 12 days post inoculation ................................................................ 47
15.3 Progeny RNA from 21 days post inoculation ................................................................ 47
Chapter 5 Discussion .................................................................................................................... 49
16 Infectivity of full-length cDNA clones ................................................................................. 49
17 Structural integrity of vLTSV............................................................................................... 51
18 Summary and Conclusions ................................................................................................... 54
19 Future Directions .................................................................................................................. 55
References..................................................................................................................................... 56

vii
List of Figures
Figure 1: The predicted primary and secondary structures of viroids and virusoids……………...3
Figure 2: Rolling circle model for the replication of circular pathogenic RNAs…………………5
Figure 3: Secondary structure of the plus (left) and minus (right) hammerhead domains for
vLTSV……………………………………………………………………………..………7
Figure 4: A reversible self-cleavage reaction mediated by the hammerhead structure………...…8
Figure 5: Model of viroid domains for the potato spindle tuber viroid (PSTV) group of viroids.10
Figure 6: Schematic diagram of full-length clones of vLTSV…………………………………...17
Figure 7: 2% agarose RNA gel and RT-PCR products of total RNA extracted from purified
virions in RNA transcript infectivity assays……………………………………………..34
Figure 8: 2% agarose RNA gel and RT-PCR products of total RNA extracted from purified
virions in RNA transcript infectivity assays......................................................................36
Figure 9: Predicted primary and secondary structure of deletion mutants show large deviation
from the native rod-like structure of vLTSV..………………………………………..….38
Figure 10: Predicted primary and secondary structure of three large insertion mutants of
vLTSV……………………………………………………………………………………40
Figure 11: Schematic diagram showing different in vitro generated insertion/deletion mutants of
vLTSV…………………………………………………………………………...……….42
Figure 12: Predicted folding at the end of native vLTSV (left) and 322I8 mutant (Right)……...44

viii
List of Tables
Table 1. Sequences of forward and reverse primers used for RT-PCR analysis………………...32
Table 2. Positions of mutations in cloned progeny RNAs from 5 dpi…………………………...46
Table 3. Positions of mutations in cloned progeny RNAs from 12 dpi………………………….48

ix
List of Abbreviations
Amp ampicillin
ASBV avocado sunblotch viroid
bp base pair
CaCl2 calcium chloride
CCR central conserved region
cDNA complementary DNA
CEV citrus exocortis viroid
CfMV cocksfoot mottle sobemovirus
CMV cucumber mosaic cucumovirus
dpi days post-inoculate
DEPC diethylpyrocarbonate
ddH2O double-distilled water
DNA deoxyribonucleic acid
ds double-stranded
EDTA ethylenediaminetetra-acetate
EtBr ethidium bromide
EtOH ethanol
kDa kilodalton
LTSV lucerne transient streak virus

x
mA milliampere
min minute
MgCl2 magnesium chloride
ml milliliter
mM millimolar
NaCl sodium chloride
NaOAc sodium acetate
NaOH sodium hydroxide
ng nanogram
nt nucleotide
ORF open reading frame
pH -log[H+]
PLMV peach latent mosaic viroid
RNA ribonucleic acid
RYMV rice yellow mottle virus
sat satellite
SBMV southern bean mosaic sobemovirus
SCMoV subterranean clover mottle sobemovirus
SDS sodium dodecyl sulfate
SNMV solanum nodiflorum mottle sobemovirus

xi
SoMV sowbane mosaic sobemovirus
TBE 1X: 0.1 M Tris, 0.1 M boric acid and 7 mM EDTA
TBRV tomato black ring nepovirus
TCV turnip crinkle carmovirus
TRSV tobacco ringspot nepovirus
Tris tris(hydroxymethyl)aminomethane
TRosV turnip rosette virus
VTMoV velvet tobacco sobemovirus
YT yeast tryptone

1
Chapter 1 Literature Review
1 Viroids and Viroid-like Satellite RNAs (Virusoids)
1.1 General features and organization
i) Viroids
Viroids are small (~246 – 401 nucleotides), circular, non-encapsidated, autonomous RNA
pathogens. On average, they are up to ten times smaller than the smallest RNA viruses, and are
highly self-complementary, allowing a compact folding structure (Fig. 1). Viroids are believed to
predate viruses because they are not known to code for any functional proteins (Flores et al.,
2004). While viruses hijack the translation apparatus of their hosts, viroids are parasites of the
transcriptional machinery. Since they do not code for their own proteins, viroids are completely
reliant on host factors during their infectious cycle, though some have a self-splicing ribozyme
activity. The ribozyme activity is thought to be involved in autocatalytic processing during a
rolling circle mode of replication (Flores et al., 2005).
Viroids are classified into two families, the Avsunviroidae and the Pospiviroidae, whose
members replicate and accumulate in the nucleus and chloroplast, respectively. The
Avsunviroidae family, whose type species is Avocado sunblotch viroid (ASBV), has a branched
quasi rod-like secondary structure, and can self-splice using the ribozyme activity (Reviewed in
Flores et al., 2005). The Pospiviroidae family, to which most of the viroid species belong, type
species Potato spindle tuber viroid (PSTV), assumes a rod-like secondary structure as well
(Gora-Sochacka, 2004). Most viroids are transmitted mechanically, some through seed or pollen,
and only one is known to be aphid-transmissible. However, the most efficient route of
transmission is vegetative propagation of infected tissue (Flores et al., 2005).

2
ii) Virusoids
Virusoids are a class of satellite RNAs that resemble viroids. Phylogenetic analysis of viroids
and satellite RNAs are consistent with the concept that these RNAs have a common origin and
that ASBV is a connecting link between virusoids and viroids (Diener, 1991). Virusoids are
found in viruses belonging to the Sobemoviruses, Nepoviruses, Poleroviruses and more. They are
small, circular, satellite RNA (scRNA), ranging from 220 – 450 nucleotides (nt) in length, with
high self-complementarity, but depend on helper RNA viruses for replication. They get
encapsidated within helper virions, and are disseminated among hosts along with the genomic
RNA. In general, the genomic RNA of the helper virus is referred to as RNA-1, while the
accompanying scRNA is called RNA-2. Much like viroids, virusoids are not known to encode
functional open reading frames (ORFs), but do possess a self-splicing ribozyme activity.
High internal base pairing present in both viroids and virusoids results in a rod-like secondary
structure (Fig. 1). It is believed that structural motifs and/or important sequences in the RNA are
responsible for replication, host specificity, and packaging. Therefore, machinery provided by
the helper virus and the infected host must recognize such motifs and provide tras-acting factors
for the RNA. It is intriguing that even though virusoids have no sequence homology with their
helper virus, they can successfully make use of the viral replication machinery, and accumulate
to high levels in the presence of the helper virus. Therefore, structural determinants may be more
critical in the lifecycle of virusoids than sequence homology with the helper virus. Together,
virusoids and viroids are the smallest known replicating nucleic acids, and are almost exclusively
plant pathogens, with human hepatitis delta virus satellite RNA being the only known exception
(Flores et al., 2001; Taylor, 2003).

3
Viroids
Virusoids
Figure 1. The predicted primary and secondary structures of viroids and virusoids. Proposed structure of viroids: ASBV, PSTV and virusoids: vVTMoV, vSNMV, two structural isolates of SCMoV: (388) and (332), and vLTSV. Adapted from Franckie, 1987.

4
1.2 Viroid and virusoid replication
i) Replication
Viroids and virusoids replicate through RNA intermediates and do not produce DNA. Because
neither of these infectious RNA pathogens act as mRNA, they must be replicated by preexisting
host or helper virus enzymes. Based on numerous studies investigating viroid/virusoid
replication intermediates and their processing, their replication has been proposed to occur
through a rolling circle mechanism (Branch & Robertson, 1984; Bruening et al., 1991). There are
two types of rolling-circle modes of replication, symmetric and asymmetric variants (Fig. 2). In
the asymmetric replication the monomeric circular (+) RNA (polarity assigned arbitrarily to the
most abundant strand in vivo), is copied by an RNA polymerase into a multimeric (-) strand. The
(-) RNA multimer serves as a template for the RNA polymerase transcription of head-to-tail (+)
RNA multimer, which are cleaved into monomers and then ligated into the circular RNA. DNA-
dependent RNA polymerase (RNA polymerase II) of the host is believed to be responsible for
replicating viroids inside the nucleus or the chloroplast. On the other hand, the RNA-dependent
RNA polymerase of the helper virus, joined with host factors to form a replicase complex, is
believed to replicate virusoids in the cytoplasm. Viroids and virusoids that are presumed to adopt
an asymmetrical rolling circle model are PSTV and other members of the family Pospiviroidae,
velvet mottle virus virusoid (vVTMoV), solanum nodiflorum mottle virus virusoid (vSNMV),
and subterranean clover mottle virus virusoid (vSCMoV) (Branch et al., 1988; Chue et al., 1983;
Hutchins et al., 1985; Davies et al., 1990).
Symmetric replication involves the synthesis of (-) RNA multimers just the same as the first
mechanism, but these are then cleaved into monomeric (-) RNA, which circularize and serve as
templates for (+) RNA multimers. Consequent cleavage and ligation events generate monomeric,
circular (+) RNA progeny. Viroids and virusoids that are believed to replicate through the
symmetrical model include ASBV and other members of the family Avsunviroidae, rice yellow
mottle virus virusoid (vRYMV), lucerne transient streak virus virusoid (vLTSV), and tobacco
ringspot nepovirus (sTRSV) (Daros et al., 1994; Collins et al., 1998; Gellatly, 1994).

5
Figure 2. Rolling circle model for the replication of circular pathogenic RNAs. (A) Model where both the (+) and (-) multimeric RNAs are processed to monomers as indicated by arrows. Steps 3 and 6 involved the circularization of the linear monomers. (B) Model where only the linear multimeric (+) RNA is processed. The unprocessed linear (-) strand is copied to give a linear (+) strand. Adapted from Forster & Symons, 1987.

6
i) Ribozyme activity
Rolling circle replication requires a precise mechanism by which viroids/virusoids monomers are
excised from multimeric replication intermediates and ligated to form circular, monomeric
progeny RNAs. This process is quite analogous to the processing reactions by which introns are
spliced out of precursor RNAs and exons joined to form functional RNAs. While some viroids
require trans factors present in the cell for splicing, other viroids and virusoids are self-cleaving
(Tsagris et al., 1987). These self-cleaving pathogenic RNAs contain a highly conserved series of
short nucleotide sequences that can assume, by base pairing, a characteristic secondary structure
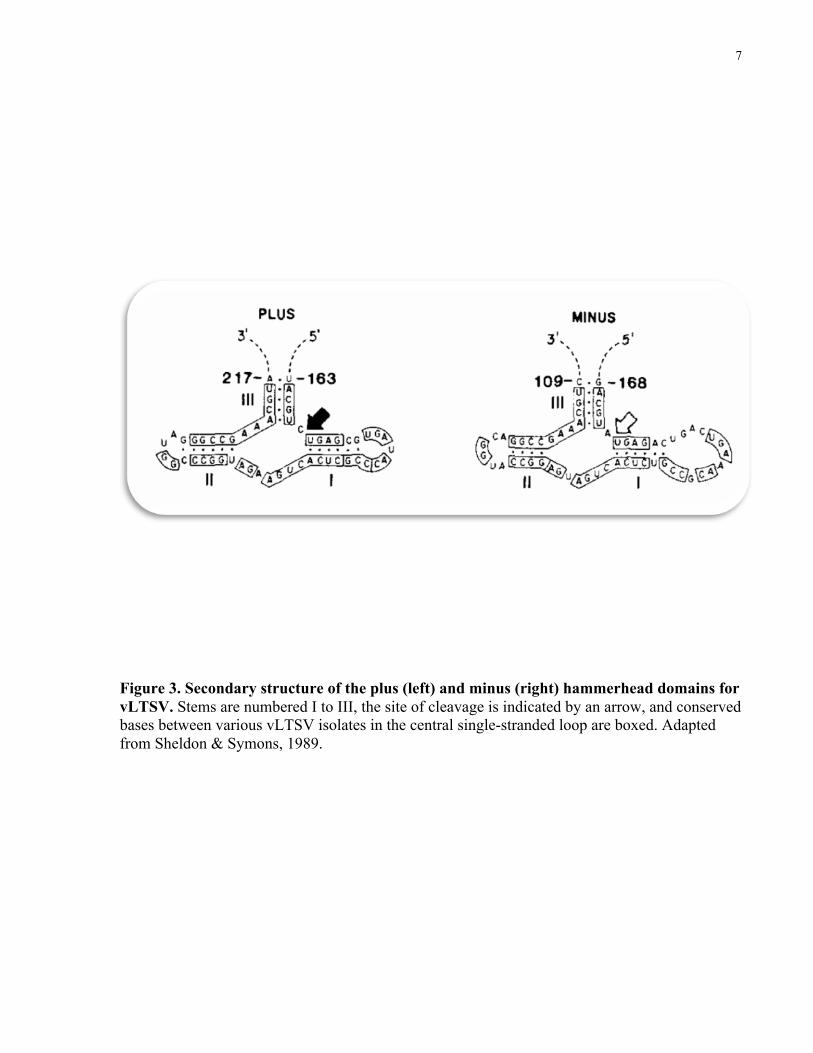
called a “hammerhead” (Fig. 3).
Hammerhead structure forms in both (+) and (-) sense of vLTSV, ASBV, and peach latent
mosaic viroid (PLMV) (Hernandez & Flores, 1992), whereas sTRSV, vVTMoV, and vSNMV
(Symons, 1997) can only form hammerheads in the (+) sense. Therefore, it is safe to assume that
viroids/virusoids, which lack hammerhead formation of the (-) sense will replicate via an
asymmetrical rolling circle pathway. Total RNA extracted from plants infected with these
viroids/virusoids has validated this assumption (Symons, 1997). The formation of the
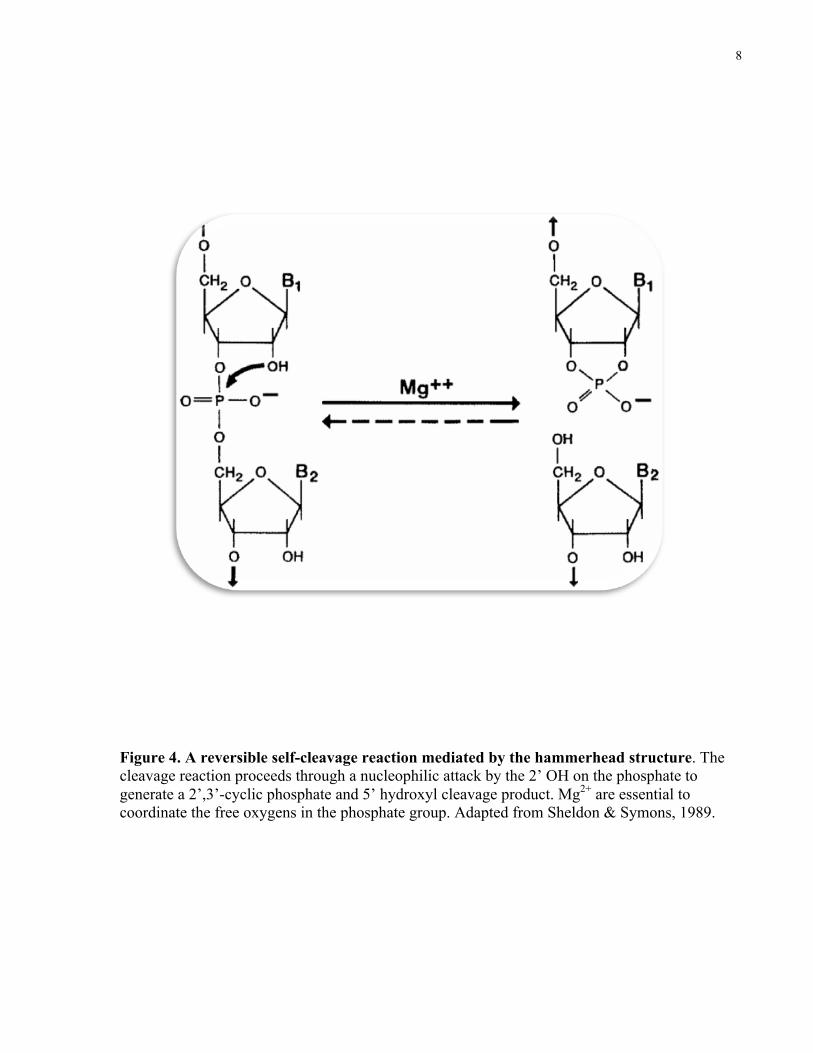
hammerhead structure is necessary for the self-cleavage reaction to occur. This reaction occurs
through a nucleophilic attack by the 2’-hydroxyl at the cleavage site on the inter-nucleotide
phosphate to give a 5’ hydroxyl and 2’, 3’ cyclic phosphodiester termini (Symons, 1997).
Magnesium ions are required for the formation of the active structure by neutralizing and
bridging the negative charged oxygens of the phosphodiester group (Forster & Symons, 1987).
Furthermore, this reaction is reversible, meaning viroids/virusoids have the capability of self-
splicing as well as ligation without the need for plant enzymes.
7
Figure 3. Secondary structure of the plus (left) and minus (right) hammerhead domains for vLTSV. Stems are numbered I to III, the site of cleavage is indicated by an arrow, and conserved bases between various vLTSV isolates in the central single-stranded loop are boxed. Adapted from Sheldon & Symons, 1989.
8
Figure 4. A reversible self-cleavage reaction mediated by the hammerhead structure. The cleavage reaction proceeds through a nucleophilic attack by the 2’ OH on the phosphate to generate a 2’,3’-cyclic phosphate and 5’ hydroxyl cleavage product. Mg2+ are essential to coordinate the free oxygens in the phosphate group. Adapted from Sheldon & Symons, 1989.

9
2 Structure-Function Relationship
2.1 Functional domains of viroids
Since viroids and virusoids do not code for their own proteins, they are completely dependent on
the machinery of the host or the helper virus. To make use of this, viroids and virusoids have
cryptic signals in their sequence and/or their structure. So far, a lot of work has been done on
elucidating structural domains in viroids. For example, from sequence comparison, the rod-like
structure of viroids has been divided into five structural and functional domains: central (C),
pathogenic (P), variable (V), terminal right (TR), and left (TL) (Fig. 5) (Keese & Symons, 1985).
These regions have been further characterized into central conserved region (CCR) located
within the C domain, terminal conserved region (TCR), and a terminal conserved hairpin (TCH)
(Tabler & Tsagris, 2004). Many of these structural domains have been related to specific
functions. For example, the C domain, particularly the upper strand of the CCR, has been
involved in the cleavage and ligation of the multimeric PSTV RNA intermediates in the
replication cycle (Baumstark et al., 1997). Similarly, the P domain has been associated with
pathogenicity of PSTV and other closely related viroids. Almost every nucleotide in these
identified domains appear to be functional and under selection. For example, it was shown that a
few nucleotide exchanges affect not only the pathogenicity of PSTV but also its host range
(Wassenegger, 1996).
Structural determinants of viroids have also been extensively studied. For example, the so-called
loop E, located within the CCR, has been proposed to play a role in the final ligation step of the
PSTV replication cycle (Baumstark et al., 1997). Another example is the pseudoknot element of
the kissing loop class, which have been identified in PLMV by in vitro chemical and enzymatic
probing (Bussiere et al., 2000) where they may contribute to stabilizing the branched
conformation of this viroid. More recently, a genomic map of PSTV was constructed through a
genome-wide mutational analysis (Zhong et al., 2008). In this study, Zhong et al. (2008) were
able to identify multiple loops/bulges essential for single-cell replication and systemic trafficking
throughout plants. Studies on structure-function relationships of viroids are numerous and
valuable for understanding the lifecycle of these RNA molecules.

10
Figure 5. Model of viroid domains for the potato spindle tuber viroid (PSTV) group of viroids. The five domains, T1, P, C, V, and T2 were determined from sequence homologies between the viroids. Adapted from Symons, 1991.

11
2.2 Functional domains of satellite RNAs
It is evident that extensive research on structure-function relationship of circular virusoids is
lacking. A few studies on linear satellites, in particular the linear satellite of cucumber mosaic
virus (sCMV), have been significant. Masuta and Takanami (1989) constructed insertion and
deletion mutant clones of sCMV and analyzed their survival in infected plants. It was found that
although sCMV is tolerant to small insertions (as much as 4 bases), any deletion mutations were
lethal to the satellite RNA (Masuta & Takanami, 1989). More importantly, they were able to
narrow down the specific sequence and secondary structure of a sCMV domain responsible for
the yellowing symptoms observed on tomato leaves (Masuta & Takanami, 1989). Following this
study, Sleat and Palukaitis (1990) found the sequence responsible for the induction of necrosis in
tomatoes by constructing cDNA clones of sCMV mutated at three specific sites.
In the field of circular virusoids, however, structure-function investigation is only found in one
paper. Sheldon and Symons (1993) constructed three cDNA clones of vLTSV with mutations
within the (-) hammerhead. Upon infection of these mutants with a helper virus, they were able
to show that the (-) sense self-cleavage capability of the virusoid was eliminated (Sheldon &
Symons, 1993). As a result, they concluded that the hammerhead structural motif is indeed
involved in the self-splicing ribozyme activity of this virusoid.

12
Chapter 2 Introduction
3 Lucerne Transient Streak virus
Lucerne transient streak virus (LTSV) is a plant virus, which belongs to the Sobemovirus group,
causing chlorotic streaking and distortions on the lateral veins of Medicago sativa leaves (Forster
& Jones, 1979). Isolates of this virus have been found in Canada (LTSV-C; Paliwal, 1983), New
Zealand (LTSV-N; Blackstock, 1978), and Australia (LTSV-A; Forster & Jones, 1979). LTSV
has a wide host range and is mechanically transmitted to most of these hosts.
The LTSV capsid is not enveloped and has a diameter of 27-28 nm, consisting of 180 subunits of
a single 32 kDa coat protein (Forster & Jones, 1979; Paliwal, 1984a). The genome of LTSV
consists of a 4.5 kilobase, single-stranded messenger RNA (RNA-1), covalently bound to a 12
kDa protein (VPg) at its 5’ terminus. The VPg protein has been shown to be essential for its
infectivity (Mang et al., 1982). In addition to RNA-1, LTSV also packages 322-nucleotide (nt),
scRNA molecules, which represent the virusoid of LTSV (vLTSV).

13
4 The LTSV Virusoid
4.1 Satellite-like nature and symptom induction
The LTSV virusoid depends on RNA-1 for its replication and encapsidation. It has been shown
that vLTSV cannot cause a systemic infection in the absence of the LTSV genome (RNA-1)
(Jones et al., 1983). This relationship, however, is not reciprocal as RNA-1 is capable of
replication and encapsidation in the absence of its satellite. The presence of vLTSV in the LTSV
virions does have an influence on the symptoms. In an experiment where Chenopodium quinoa
and Chenopodium amaranticolor plants were infected with either RNA-1 only or unfractionated
LTSV, chlorotic or necrotic lesions were seen on the leaves of plants, respectively (Jones et al.,
1983). More interestingly, once plants infected with RNA-1 were back inoculated with purified
vLTSV, chlorotic lesions converted into necrotic lesions in proportion to the concentration of
vLTSV added (Jones et al., 1983). Likewise, other virusoids from beet black scorch virus
(BBSV; Guo et al., 2005), turnip crinkle virus (TCV; Carpenter et al., 1991), CMV (Kuroda et
al., 1990), and SCMoV (Davies et al., 1990) have been shown to complicate the symptoms of
their respective helper viruses. Further support for vLTSV’s satellite nature came from Paliwal
(1984b) who infected Trigonella foenum-graecum and Trifolium incarnatum with purified
vLTSV and recovered no progeny RNA.
4.2 Helper and host specificity
LTSV is not the only virus that supports the replication and encapsidation of vLTSV. SBMV
(Paliwal, 1984b), cocksfoot mottle virus (CfMV; Sehgal et al., 1993), turnip rosette virus
(TRosV; Jones & Mayo 1984; Sehgal et al., 1993), and sowbane mosaic virus (SoMV; Franckie
et al., 1983a) all supported the replication and encapsidation of vLTSV. These viruses are all
sobemoviruses that are normally devoid of virusoids. It must be noted that LTSV, which has a
virusoid, can also support the replication of vSCMoV (Keese et al., 1984) and vSNMV (Jones &
Mayo, 1983). Therefore, possessing a virusoid does not prevent the RNA-1 of a sobemovirus
from associating with other closely related virusoids.

14
The relationship between vLTSV and its helper virus is also host-dependent. TRosV was
reported to effectively support the replication of vLTSV in Brassica rapa, Raphanus
raphanistrum, and Sinapsis arvensis, but not in Thlaspi arvense or Nicotiana bigelovii (Sehgal et
al., 1993). Furthermore, Paliwal (1984b) reported that vLTSV replicated efficiently in T.
foenum-graecum, but poorly in phaseolus vulgaris. Specific host factors that bind with the RNA-
dependent RNA polymerase of the helper virus to form a replicase unit may be a reason for host
specificity (Sehgal et al., 1993).
4.3 Sequence and structure
The main sequence has been determined for the three known isolates of vLTSV (AbouHaidar &
Paliwal, 1988; Keese et al., 1983). Two of these isolates, from New Zealand and Australia, are
324 nt in length and share 98% sequence identity (Keese et al., 1983). The Canadian isolate is
322 nt in length and shares only 80% similarity with the others (AbouHaidar & Paliwal, 1988).
The sequence of vLTSV encodes seven potential polypeptides (Keese et al., 1983). However,
despite the high level of sequence identity between the three isolates, only two of these
polypeptides are common (Keese et al., 1983). Furthermore, no proteins were recovered from
translating vLTSV RNA in vitro using rabbit reticulocyte lysate and wheat germ extract (Morris-
Krsinich & Forster, 1983). Therefore, vLTSV lacks mRNA activity and depends on its helper
virus and the host for its replication. RNA models of all three isolates of vLTSV show that up to
70% of residues in the molecule can internally base pair. As a result, vLTSV has a rod-like
secondary structure in its native conformation (Fig. 1). This structure is similar to viroids (Gross
and Riesner, 1980) and other virusoids from the sobemovirus group (Haseloff and Symons,
1982). This secondary structure is presumed to aid the virusoid in carrying out its biological
functions.

15
4.4 Replication and ribozyme activity
The virusoid of LTSV replicates through a symmetrical rolling circle model for replication (Fig.
2). Support for this comes from identification of RNA species extracted form plants infected
with vLTSV and a helper virus. When total RNA was extracted from infected plants, (+) or (-)
sense monomers, with a few species of (+) multimers, were found (Hutchins et al., 1985). No (-)
sense multimers were present in these extracts. In vitro and in vivo studies also prove that
vLTSV can self-cleave in both polarities, thus supporting a symmetrical rolling circle mode of
replication for this virusoid (Sheldon & Symons, 1993).

16
5 Full-length infectious cDNA clones
Head-to-tail monomeric, dimeric, and trimeric cDNA copies of vLTSV have been previously
cloned within the NcoI site of a pUC Bluescript phagemid (pBS+) in the AbouHaidar laboratories
(Fig. 6). The infectivities of these clones were analyzed using a virusoid-free helper virus
(TRosV) and Brassica rapa as the host plant. Preliminary data suggest that in vitro RNA
transcripts generated from multimeric clones were infectious with the presence of TRosV
(Gellatly, 1994). However, monomeric RNA transcripts were incapable of replicating inside B.
rapa. Furthermore, recent data suggest that monomeric and multimeric clones are infectious as
intact DNA plasmids or digested inserts with flanking vector bases (Gellatly, 1994). Similarly,
Sheldon and Symons (1993) showed that excised monomeric clones of vLTSV were infectious
as dsDNA with the help of a virusoid-deficient LTSV. Therefore, cloned DNA copies of vLTSV
may be used in infectivity assays instead of RNA. However, the validity of this claim remains to
be tested.

17
Figure 6. Schematic diagram of full-length clones of vLTSV. Monomer (1M5), dimer (2D2), and trimer (29T2) clones are shown with sizes of excised inserts after digestion with either NcoI (for monomer) or EcoRI + HindIII (for multimers). Arrows indicate the direction of (+) strand within the pBS+ vector. Adapted from Gellatly, 1994.

18
6 Research proposal
6.1 Overview of previous work
Viroids and virusoids are both small, covalently closed, circular RNA with a high degree of
base-pairing. Viroids are known to replicate using the cellular RNA polymerase II and virusoids
replicate and package their RNA with the help of a helper virus. There has been a lot of work
done on elucidating important sequences and structural features of viroids and linear satellite
RNAs. Such information is lacking on the circular viroid-like satellites, such as that of LTSV.
This seems to be a particularly interesting group as they are phylogenetically intermediate or
transitional between viroids and linear satellites. Various aspects of the vLTSV replication
(transmission, helper virus, host specificity) have already been investigated. However, aside
from the ribozyme activity, little is known about the specific sequences and/or structures, which
are essential in the lifecycle of this virusoid.
6.2 Current objectives
This study aimed to investigate various aspects of the replication of the virusoid of LTSV,
particularly with respect to:
1. Determining specific sequences and/or structures essential to replication. Since this small
virusoid is replicated by the helper virus (and host) replicase, it is very interesting to determine
functional domains and/or structures involved in this process. A series of induced mutations
(insertions and deletions) were introduced into the circular RNA to disrupt the secondary
structure. Infectivity of these mutants was carried out with a satellite-free helper virus (TRosV).
2. Investigating the ability of vLTSV to support the replication of foreign sequences. Since this
virusoid has an optimal size (322 nucleotides) for packaging and replication, it is interesting to
determine whether foreign introduced nucleotides would be viable in the virusoid. Mutants
having insertions will be used to determine whether they are replicated. Furthermore, the fidelity
of the sequence as well as the stability of the overall rod-like virusoid will be analyzed.

19
6.3 Hypothesis
The virusoid of LTSV is very small in size and has an extensive rod-like secondary structure. It
has no sequence homology with its helper virus yet it is able to make sufficient use of its
replication machinery. It is presumed that vLTSV may have structural signals, which attract the
replicase unit for its replication. We hypothesize that insertion and/or deletion mutants which
cause structural perturbations to the virusoid are lethal to its infectivity. As a result, foreign
sequences introduced into vLTSV will only be tolerated if they maintain the overall rod-like
motif of the molecule. These “rod-preserving” mutants will be recognized by the helper virus
(and host) replicase and be replicated. Progeny RNA originating from such mutant will maintain
foreign bases, as the replicase cannot distinguish them from native vLTSV sequence. Finally,
local structural instability caused by the insertions will be tolerated as long as the overall
integrity of the rod-like structure is preserved.

20
Chapter 3 Materials and Methods
7 General Molecular Techniques
7.1 Plasmid DNA isolation from E. coli (miniprep)
The appropriate E. coli colonies were used to inoculate 3 ml of LB media (1% tryptone, 0.5%
yeast extract, 1% NaCl, pH 7.5) containing 60 µg/ml ampicillin (Amp) and cultured overnight in
a 37oC shaker. Plasmid DNA was then extracted from the overnight cultures using a modified
version of the “mini-prep” alkaline lysis method described in Maniatis et al. (1982). 1.5 ml
aliquots of the E. coli cultures were pelleted at 16,000 g for 2 min, and the supernatant was
discarded. Each pellet was then resuspended in 100 µl of ice-cold Solution I (50 mM glucose, 10
mM EDTA, 25 mM Tris, pH 8.0, containing 5 mg/ml lysozyme added fresh) by vortexing and
incubated for 10 min at room temperature. Next, 200 µl of freshly prepared Solution II (0.2N
NaOH, 1% SDS) was added to the mixture, gently inverted 6-8 times, and stored on ice for 20
min. Finally, 150 µl of ice-cold Solution III (3M Sodium Acetate, pH 4.8) was added to the
solution, inverted 6-8 times, and kept on ice for an additional 45 min. The tubes were centrifuged
at 16,000 g at 4oC for 10 min. The supernatant was transferred into a clean tube and plasmid
DNA was precipitated by the addition of 0.6 volumes of isopropanol and incubating at room
temperature for 1hr. The precipitated plasmid DNA was centrifuged at 16,000 g for 10 min and
the supernatant was discarded. The pellet was washed twice with 70% ethanol (EtOH) and once
with 95% EtOH, and dried by vacuum desiccation. Consequently, the pellet was dissolved in 0.1
M TE buffer (1 mM Tris pH 8.0, 0.1 mM EDTA). The extracted DNA was analyzed by agarose
gel electrophoresis, using 1X TBE buffer (0.1 M Tris, 0.1 M Boric acid, 2 mM EDTA, pH 8) for
the gel and the running buffer. 1 µl of each sample was mixed with 4 µl of TE-1 buffer and 1 µl
of 6X loading dye [0.25% xylene cyanol (XC), 0.25% bromophenol blue (BPB), 40% sucrose],
and electrophoresed at 45 mA constant current through a horizontal 1.2% agarose (molecular
biology-grade, BioRad) gel in a Mini-Sub Cell (BioRad) until the BPB migrated half-way
through the gel. After each run, the gels were stained with ethidium bromide (EtBr) and the DNA
bands visualized and photographed under ultraviolet light (300 nm) using a GelDoc-It® imaging
system

21
7.2 Heat shock transformation of E. coli
Competent E. coli DH5α cells were prepared according to Maniatis et al. (1982). Three ml of LB
media were inoculated from a single colony of DH5α cells and incubated overnight in a 37oC
shaker incubator. A fresh stock of LB media was inoculated from the overnight culture
(culture:broth, 1:100, v/v) and incubated at 37oC with vigorous shaking until an optical density
of 0.4-0.6 units at 600 nm was obtained. Two red-capped tubes were filled with 10 ml of the
cells, chilled on ice for 30 min, and then centrifuged at 1,000 g for 10 min at 4oC. The
supernatant was discarded, the cells were resuspended in 5ml of cold 50 mM calcium chloride
(CaCl2), incubated for 20 min, and the suspension centrifuged as before. The pelleted cells were
resuspended in 670 µl of chilled 100 mM CaCl2 and stored on ice for up to 3 hrs prior to
transformation. 200 µl aliquots of the competent cells were prepared and 50 ng of the appropriate
DNA was added to each tube. The mixture was incubated on ice for 30 min. The cells were then
heat shocked at 42oC for 90 sec and incubated on ice for 2 min. 800 µl of LB media was added to
each tube, vortexed, and incubated for 45 min at 37oC. The cells were centrifuged at 16,000 g for
2 minutes, 700 µl of supernatant was removed, and the pellet was re-dissolved in the remaining
media. The cells were spread onto agar plates [LB media + 15g/L agar (LBA)], containing 60
µg/ml Amp. The plates were incubated overnight at 37oC, in an inverted position.
7.3 Glycerol stock preparation
3 ml of LB were inoculated by a single colony of the desired E. coli clones and incubated
overnight at 37oC with vigorous shaking. A 500 µl aliquot of the overnight culture was mixed by
vortexing with an equal volume of sterile glycerol and stored at -80oC.

22
7.4 Phenol-chloroform extraction of DNA/RNA
An equal volume of Tris-HCl saturated phenol was added to the dissolved DNA or RNA,
vortexed, and centrifuged at 16,000 g for 10 min. The upper phase was transferred into a clean
tube. About 1/3 of the original volume was added with TE-1, vortexed, centrifuged at 16,000 g
for 10 min, and the recovered upper phase was transferred into the new tube. About 2.5X volume
of the obtained DNA or RNA was added with chloroform, vortexed, and centrifuged at 16,000 g
for 5 min. The lower phase was removed and the previous step was repeated once more. The
upper phase was transferred into a clean tube and the DNA or RNA was precipitated by adding
sodium acetate to a final concentration of 0.1 M, followed by mixing with 2.5 volumes of 95%
EtOH, and stored at -80oC for 2hrs or -20oC overnight. The DNA or RNA was pelleted by
centrifugation at 16,000 g for 10 minutes, and the resulting pellet was washed twice with 70%
EtOH and once with 95% EtOH. The pellet was dried with vacuum desiccation and dissolved in
an appropriate volume of TE-1 depending on the pellet size.

23
8 Sub-cloning of 322I8 to generate multimers
8.1 Restriction digest and agarose gel electrophoresis
To generate monomeric dsDNA copies of 322I8 one µg of plasmid DNA from each clone was
digested to completion at 37oC with 5 units of restriction endonucleases NcoI (Fermentas) or pstI
(Fermentas), respectively, in a 50 µl reaction volume using 1X Tango Buffer®. The restriction
reaction was left overnight and terminated the next day by the addition of 1 µl of 20 mM EDTA.
2 µl from the reaction was loaded on a 2% agarose gel and electrophoresed at 45 mA constant
current, followed by EtBr staining and visualization with UV light. Finally, the plasmid digest
was deproteinated using a phenol chloroform extraction procedure as outlined above; however,
the pellet was dissolved in 17.5 µl of double distilled water (ddH2O).
8.2 Ligation
Two µl of 10X Ligation Buffer from Fermentas (400 mM Tris-HCl, 100 mM MgCl2, 100 mM
DTT, 5 mM ATP) and 0.5 µl of T4 DNA Ligase from Fermentas (5 u/µl) were added to digested
and phenol-chloroform extracted 322I8 and AB3 clones. The solution was mixed by vortexing
and incubated at room temperature overnight. 2 µl from the reaction was used to transform E.
coli DH5α (as before) and the cells were incubated at 37oC overnight on agar plates containing
60 µg/ml Amp, in an inverted manner.
8.3 Screening of colonies
Ten colonies were selected from each plate were grown overnight, in 3 ml of LB, at 37oC.
Plasmid DNA was extracted (as before) and 1-2 µg was double-digested at 37oC with 10 units of
each of KpnI (Fermentas) and XbaI (Invitrogen) in a 25 µl reaction containing 10 mM Tris-HCl
(pH 7.5), 10 mM MgCl2, 0.02% Triton X-100, 0.1 mg/ml BSA, pH 7.5. Digested DNA was

24
electrophoresed in a 2% agarose gel alongside a GeneRulerTM 100 bp DNA ladder (Fermentas).
Samples containing dimer or trimer clones were selected. Plasmid DNA from these samples was
digested with SmaI (NEB) at 25oC (in 50 mM potassium acetate, 20 mM Tris-HCl, 1 mM DTT,
and 10 mM magnesium acetate, pH 7.9) followed by an EtOH precipitation step, and was
subsequently digested with PvuII (NEB) at 37oC in 10 mM Tris-HCl, 50 mM NaCl, 10 mM
MgCl2, and 1 mM DTT, pH 7.9. Based on the fragment sizes observed following electrophoresis
in agarose gels, the orientation of the inserts could be determined (i.e. 322I8 and AB3 clones
have an internal SmaI site; pBS+ has two PvuII sites flanking the multiple cloning site). Two
clones denoted 332I8trimer were selected and sent for sequencing at the Centre for Applied
Genomics in SickKids hospital.

25
9 In vitro runoff transcription of cDNA clones
Two µg of plasmid DNA of clones Mono (1M5), Di (2D2), Tri (29T2), and 322I8trimer were
linearized by restriction digest at 37oC with either EcoRI or HindIII. Following digestion, the
reaction mixtures were phenol-chloroform extracted, EtOH precipitated, and the linearized
plasmid DNA was pelleted by centrifugation. Runoff transcription reactions were performed
according to Beck et al. (1990), with several modifications. One µg of linearized template DNA
(resuspended in DEPC treated ddH2O) was added to a 50 µl reaction containing the following:
for HindIII digested DNA, 1X T7 buffer (40 mM Tris-HCl, pH 7.5, 10 mM NaCl, 6 mM MgCl2,
and 2 mM Spermidine) or, for EcoRI-digested DNA, 1X T3 buffer (40 mM Tris-HCl, pH 8.0, 25
mM NaCl, 8 mM MgCl2, and 2 mM Spermidine); 10 mM DTT; 25 units of RiboLockTM RNase
Inhibitor; 5 µg acetylated BSA (NEB); 0.5 mM of each of ATP, CTP, GTP, and UTP; and 50
units of either T7 RNA polymerase (NEB) or T3 RNA Polymerase (NEB). The reaction mixtures
were incubated at 37oC for 1-4 hours. Template DNA was removed by the addition of 1.5 units
of RNase-free DNase I (NEB) with an additional 37oC incubation for 45 min. All reactions were
terminated by the addition of EDTA. 2 µl of the reaction mixtures and 1 µl High Range RNA
ladder (Fermentas) were mixed with 1X loading dye (prepared with DEPC treated ddH2O) and
loaded on a 2% agarose gel prepared with 1X TBE buffer (made with DEPC treated ddH2O) for
the gel and running buffer. The RNA agarose gel was stained with EtBr and subsequently
visualized under UV light.

26
10 Purification of viruses
10.1 Lucerne transient streak virus
LTSV-C was propagated in greenhouse-grown Trigonella foenum-graecum. Virions were
purified according to a modified method of Foster and Jones (1980). Leaves showing signs of
systemic infection, 12-14 days post-inoculation (dpi), were harvested and homogenized in a
Waring blender with 50 mM sodium phosphate buffer, pH 7.0, containing 0.2% 2-
mercaptoethanol (v/v), using a ratio of 5 ml buffer/g leaves. The homogenate was strained
through 4 layers of cheesecloth and the pulp was returned into the blender for further
homogenization. The pooled extract was stirred on ice for 30 min and centrifuged at 7,800 g for
10 min. Virions in the aqueous phase were then sedimented by ultracentrifugation at 90,000 g for
3 hr. Viral pellets were dissolved overnight in 50 mM sodium phosphate buffer, pH 7.0. The
following day, the viral solution was collected in an Eppendorf tube and centrifuged at 16,000 g,
at cold, for 10 min to further clarify the extract. Optical density measurements were taken at 260
and 280 nm to determine virus purity and concentration. 0.04% sodium azide (w/v) was added to
the viral solution and stored at 4oC for up to 3 weeks.
10.2 Turnip rosette virus
TRosV was propagated in greenhouse-grown Brassica rapa (var. Purple Top Globe) and purified
according to a method described by Hollings (1973) with several modifications. Turnip leaves
showing symptoms of systemic infections 12-20 dpi were harvested and homogenized in 50 mM
sodium acetate buffer, pH 5.0 containing 0.1% 2-mercaptoethanol (v/v), using 1.5 ml buffer/g
leaves. The homogenate was expressed through 4 layers of cheesecloth and the pulp was
homogenized and passed through the cheesecloth again. The extract was stirred on ice for 30 min
and subsequently centrifuged at 7,800 g for 30 min. To sediment the virus, the aqueous phase
was ultracentrifuged at 105,000 g for 3 hr. Viral pellets were dissolved in sodium acetate buffer,
pH 5.0 overnight. The following day, the viral solution was collected in an Eppendorf tube and
centrifuged at 16,000 g, at cold, for 10 min to further clarify the extract. Optical density

27
measurements were taken at 260 and 280 nm to determine virus purity and concentration. 0.04%
sodium azide (w/v) was added to the viral solution and stored at 4oC for up to 3 weeks.
10.3 Extraction of viral RNA
Total RNA was extracted from purified virions using a modified procedure of Sehgal (1990).
Approximately 75-100 µg of purified virus (in 50 mM sodium acetate, pH 5.0 or 50 mM sodium
phosphate, pH 7.0) were mixed with 1% sodium dodecyl sulphate (SDS) and 0.2 M sodium
chloride (NaCl), and the solution was incubated at 560C for 15 min. To further dissociate virion
particles, one volume of Tris-HCl saturated phenol was added and the solution was again
incubated at 56oC with occasional vortexing. The solution was centrifuged at 9,100 g for 10 min.
The upper phase was transferred into a new Eppendorf tube and viral RNA was extracted twice
with 2.5X volume of chloroform. RNA in the aqueous phase was transferred into a fresh tube
and precipitated by the addition of 2.5 volumes of 95% EtOH, with incubation for 2hr at -80oC
or overnight at -20oC. Precipitated RNA was pelleted by centrifugation at 9,100 g for 10 min in
cold and the pellets were rinsed with 70% EtOH twice and 95% EtOH once and left to air dry in
the fume hood. The RNA was re-dissolved in diethylpyrocarbonate (DEPC) treated double
distilled water, with the purity and concentration determined RNA agarose gel electrophoresis.

28
11 Infectivity assays of cDNA clones
11.1 Coinoculation of TRosV with (+) and (-) RNA transcripts
Brassica rapa (var. Purple Top Glove), grown under standard conditions in a greenhouse, were
used in all of the coinoculation assays. An identical inoculation procedure was used in all tests.
Leaves of 12-21 day-old healthy seedlings were lightly dusted with Carborundum and the
petioles were wrapped with a thin strip of masking tape. For each test, groups of five plants were
mechanically inoculated, two leaves per plant, by gently rubbing a solution of purified TRosV
virions (100 µg/leaf) plus 1-2 µg of (+) or (-) RNA from one of each of the in vitro runoff
transcription reactions of monomer, dimer, and trimer clones. Inoculated leaves were then
returned to the greenhouse and after the initial appearance of systemic infection (about 10-21
days post-inoculation), unmarked symptomatic leaves were ground in 50 mM sodium acetate
buffer, pH 5.0, and the sap was lightly dusted with Carborundum; this homogenate was used to
further inoculate 25-30 B. rapa seedlings. Virions were subsequently purified from systemically
infected plants from this second passage and assayed for the presence of vLTSV. Controls for
this experiment included: plants inoculated with 100 µg/leaf of TRosV alone; 100 µg TRosV +
100 µg LTSV per leaf; 100 µg LTSV alone; inoculation with 2 µg of (+) or (-) RNA transcripts
from each clone (i.e. no helper virus); or, inoculation with buffer alone (i.e. no helper virus or
RNA transcript).
11.2 Coinoculation of TRosV with dsDNA
To determine whether each of the monomer, dimer, and trimer clones were infectious as dsDNA
in pBS+ plasmid, B. rapa seedlings were coinoculated with a mixture of purified TRosV (100
µg/leaf) and 1-2 µg/leaf of monomer, dimer, and trimer clone. Controls for this test included:
inoculation with 100 µg/leaf TRosV alone; TRosV + LTSV; 100 µg/leaf LTSV alone; 2 µg/leaf
of intact plasmid from each of the clones; or, inoculation with buffer alone.

29
11.3 Coinoculation of TRosV with mutants
Infectivity assays were performed similar to the infectivity assay of monomeric and multimeric
RNA transcripts. Plants were inoculated with 100 µg/leaf of purified TRosV and 2 µg/leaf of
mutant DNA or RNA. Controls included TRosV alone, TRosV + LTSV, and buffer alone.
Inoculated leaves were marked with masking tape and total RNA was extracted from non-
marked leaves 5 days, 12 days, and 20 days post-inoculation.
11.4 Total RNA extraction from B. rapa leaves
One leaf of B. rapa showing signs of systemic infection was harvested and washed with 70%
EtOH. The leaf was cut into smaller pieces using a sterile scissor. The leaf pieces were snap
frozen in liquid nitrogen, and then crushed using a N2 Liq chilled mortar and pestle and sterile
sand. While the powdered leaves was still frozen, 150 µl of phenol:chloroform:isoamyl alcohol
(25:24:1) was added to it. Once the mix thawed, an equal volume of TES buffer (10 mM Tris, 1
mM EDTA, and 0.1 M sodium chloride) was added. The sample was then transferred into a 1.5
ml microfuge tube and stored on ice. The mortar was then rinsed with another 150 µl of the
phenol mix and 150 µl of TES buffer. The wash was pooled with the initially collected sample,
and spun at 16,000 g for 1 min at room temperature. The aqueous top phase was then transferred
to a new tube without disturbing the interface. RNA was then precipitated by adding 2X volume
of 95% EtOH and incubated at -80oC for 2 hrs or -20oC overnight. To pellet the precipitated
nucleic acid, the tube was spun at 16,000 g at 4oC for 15 min. The recovered RNA was then
washed twice with ice-cold 70% EtOH and once with 95% EtOH, followed by dissolution in 15
µl of 10 mM Tris buffer pH 7.2, prepared with DEPC-treated ddH2O. 2-5 µl of total extracted
RNA was subsequently used in a reverse transcription polymerase chain reaction.

30
12 Reverse transcription, PCR, and cloning
12.1 Reverse transcription (RT)
First strand synthesis was carried using RNA extracted from purified virions or plant leaves. 1
µg of extracted RNA was mixed with 5 pmol of the desired antisense primer and sterile DEPC
treated ddH2O was added to a final volume of 30 µl. The mixture was boiled for 5 minutes and
then cooled until the water bath reached 65 oC. The following reagents provided by Invitrogen
were then added to each sample: 2.5 µl of 2.5 mM dNTPs, 10 µl of 5X First Strand buffer, 5 µl
of 0.1 M DTT, 40 units of recombinant RNase inhibitor, and 200 units of Superscript II Reverse
Transcriptase. The samples were then incubated at 40 oC for two hours. The resulting cDNA (1
µl) would serve as a template for PCR amplification.
12.2 Polymerase chain reaction (PCR)
Polymerase Chain Reaction (PCR) was used for the synthesis and amplification of cDNA
products. An aliquot of DNA template (cDNA) was mixed with 2.5 µl 10 X PCR buffer, 2.5 mM
dNTPs, 2.5 mM magnesium chloride, 10 pmol each of sense and antisense primers (please see
Table 1 for primer sequences), 2.5 units of Taq DNA polymerase, and filled to 25 µl with sterile
ddH2O. A layer of mineral oil was placed on top of the reactions to prevent evaporation and the
samples were placed in a thermal cycler for 30 cycles for amplification.

31
12.3 Cloning
PCR products were digested overnight with NcoI as previously described. Digestion reactions
were phenol-chlorofom extracted and ligated into NcoI-cut pBS plasmid using 3 units of T4
DNA ligase with incubation overnight at 160C. The ligation mix was used to transform E. coli
DH5α using the heat shock method, and plated on Amp (60 ug/ml) LBA plates. The resulting
colonies were cultured and their plasmid DNA extracted via miniprep. Plasmids were digested
with NcoI and further sequenced at a nearby sequencing facility.

32
Table 1. Sequences of forward and reverse primers used for RT-PCR analysis.
Primer Sequence (5’ 3’)
TRosVFor TGTGGGCTAAGCTTGGAGTT
TRosVRev TCTCTATCCGCAGCCTCATC
vLTSVFor GATTCCATGGCAAGCTGCGCAGGGGGCTGA
vLTSVRev AGCGCCATGGAAGCTGCCGGTAGGATGATG

33
Chapter 4 Results
13 Infectivity Assays of vLTSV as RNA or dsDNA
13.1 RNA transcripts
Full-length monomeric and multimeric cDNA clones of vLTSV (Fig. 6) had been previously
constructed in our laboratory (kindly provided by M.G. AbouHaidar). RNA transcripts of both
polarities were generated in vitro using T3 or T7 polymerase. Each of the three transcripts was
coinoculated with TRosV to B. rapa (var. Purple Top Globe). Transcripts generated by T7 RNA
polymerase contained 21 nucleotides 5’ to the vLTSV sequence and 37 nucleotides on the 3’ end
that were derived from the vector (i.e. from the +1 site of the T7 promoter and to the HindIII site
in the pBS+). Similarly, transcripts made using T3 RNA polymerase included 45 and 12
nucleotides, upstream and downstream, respectively, derived from pBS+ (i.e. from the T3
promoter +1 site and to the EcoRI site in pBS+).
Agarose gel electrophoresis and RT-PCR of RNA extracted from purified virions recovered in
these tests (Fig. 7, lanes B-F) showed the presence of vLTSV progeny in plants infected with
TRosV + LTSV (Fig. 7, lane C), TRosV + dimer transcript (Fig. 7, lane E), and TRosV + trimer
transcript (Fig. 7, lane F). No vLTSV RNA progeny is observed in plants infected with TRosV
alone (Fig. 7, lane B), so there was no cross-contamination between plants. Finally, no virions
could be recovered from B. rapa infected with LTSV alone, RNA transcript alone, or buffer
alone (i.e. LTSV or RNA transcripts cannot replicate in B. rapa without the presence of TRosV).
Therefore, only multimeric RNA transcripts were infectious once inoculated with TRosV helper
virus. In addition, random cloning and sequencing of vLTSV RT-PCR products showed that the
cDNA progeny were 322 nt in length and identical to the native vLTSV. This meant that foreign
bases originating from the pBS+ plasmid were discarded in vivo during the replication process.

34
A B C D E F
Figure 7. 2% agarose RNA gel and RT-PCR products of total RNA extracted from purified virions in RNA transcript infectivity assays. B: TRosV alone; C: TRosV + LTSV; D: TRosV + monomer RNA; E: TRosV + dimer RNA; F: TRosV + trimer RNA. Forward and reverse primers were specific to TRosV or vLTSV RNA resulting in 349 bp or 322 bp bands, respectively. A: represents a High Range RNA Ladder (Fermentas), top, and a 100 bp DNA Ladder (Fermentas) in the bottom two gels.

35
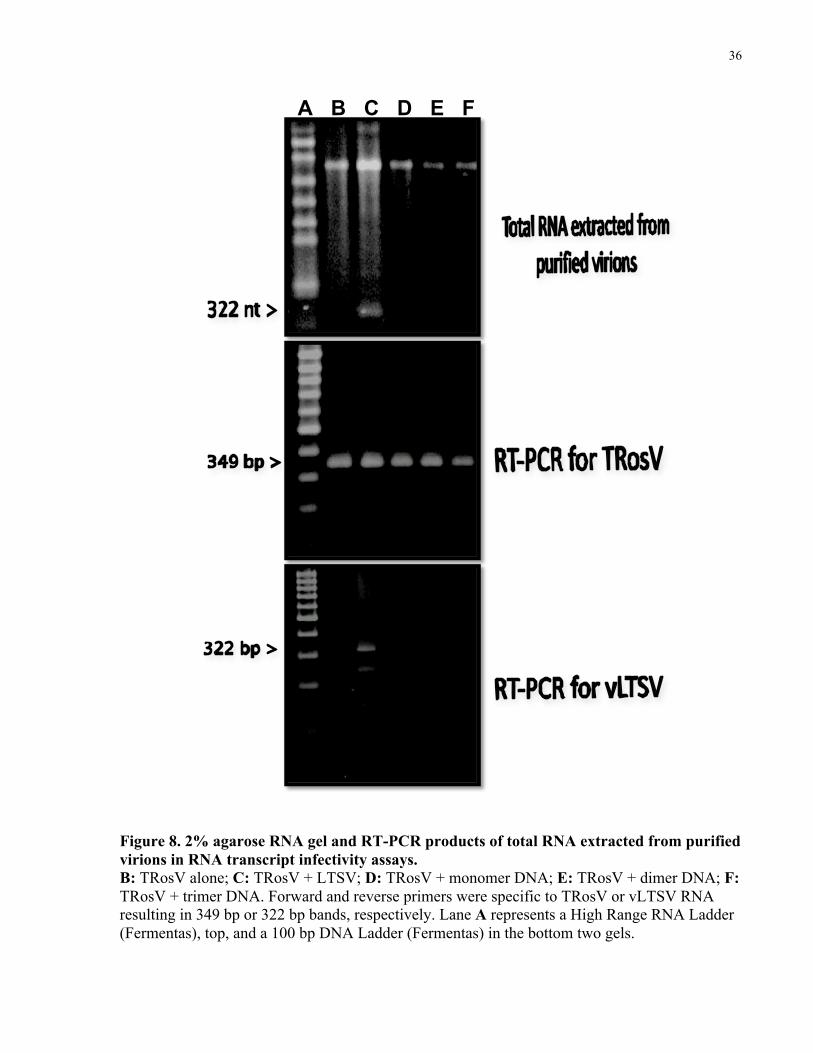
13.2 Double-stranded DNA
Each of the three cDNA clones of vLTSV was coinoculated as plasmid DNA with TRosV to B.
rapa. Virions were purified from systemically infected B. rapa (for each of the coinoculated
tests) after a second passage of the virus. Agarose gel electrophoresis and RT-PCR of RNA
extracted from purified virions in each test showed that vLTSV progeny was only present in
plants infected with TRosV + LTSV (Fig. 8, Lane C). No progeny vLTSV was detected from
TRosV alone (Fig. 8, Lane B) (i.e. no cross-contamination between tests) or plants infected with
TRosV + DNA clones (Fig. 8, Lanes D-F).
No virions were recovered from plants infected with LTSV alone (i.e. LTSV could not replicated
in B. rapa), DNA alone (i.e. none of the DNA clones were capable of replicating on their own),
or buffer alone. Therefore, DNA copies of vLTSV in monomeric and multimeric forms were not
infectious. Our results were in contrast to preliminary data presented by Gellatly (1994) who
found DNA copies of vLTSV to be infectious. Our study shows that only multimeric forms of
vLTSV RNA transcripts are infectious. Obtained results are in agreement with the symmetrical
rolling circle model for replication.
36
A B C D E F
Figure 8. 2% agarose RNA gel and RT-PCR products of total RNA extracted from purified virions in RNA transcript infectivity assays. B: TRosV alone; C: TRosV + LTSV; D: TRosV + monomer DNA; E: TRosV + dimer DNA; F: TRosV + trimer DNA. Forward and reverse primers were specific to TRosV or vLTSV RNA resulting in 349 bp or 322 bp bands, respectively. Lane A represents a High Range RNA Ladder (Fermentas), top, and a 100 bp DNA Ladder (Fermentas) in the bottom two gels.

37
14 Structure-function analysis
14.1 Infectivity of large sized insertion-deletion mutants
In order to determine specific sequences and/or structures in vLTSV, which have a role in its
biological function, a library of mutants has been previously constructed in our laboratory
(courtesy of Duncan L. Gellatly). These mutants have large or small insertion or deletions of the
native vLTSV sequence at various positions of the virusoid. Three deletion mutants, dM1 (-4 nt),
dE1 (-50 nt), and dE4 (-110 nt), with deletions originating at the BglII site of vLTSV were tested
for infectivity using TRosV helper virus. It was observed that none of these mutants could
replicate in B. rapa plants to produced progeny RNA. A computer program that estimates RNA
secondary structure based on the optimal free energy of folding (Zucker & Steigler, 1981),
showed that mutant clones had large deviations from the native rod-like conformation of vLTSV
(Fig. 9).

38
Figure 9. Predicted primary and secondary structure of deletion mutants show large deviation from the native rod-like structure of vLTSV. Mutant dM1 has a -4 nt deletion, dE1 has a -50 nt deletion, and dE4 has a -110 nt deletion originating at the BglII site. Underlined areas are regions that deviate most from the native conformation of vLTSV. All mutants were lethal.

39
Following these results, the infectivity of two insertion mutants was tested. These mutants had a
SnaBI linker (ATACGTAT) inserted into the BglII site of the vLTSV sequence. One mutant had
a 12 bp insertion (178I12 mutant) while the other had only a 4 bp insertion (178I4 mutant) at the
respective position. Infection of B. rapa plants with TRosV and either of these mutants produced
no vLTSV progeny. Both of these insertions were lethal to the replication of the virusoid. In each
case, the secondary structure of the mutant virusoid was significantly disrupted in up to one third
of the molecule (Fig. 10).
Regions of single stranded RNA may be more susceptible to degradation by endoribonucleases
inside the plant. Based on this presumption, another mutant with flanking bases that are
complementary with one another (mutant AB3) was tested. It was believed that the flanking
bases should be able to base-pair and mimic the native vLTSV structure which has a high level
of internal base-pairing. This clone was presumed to be less susceptible to degradation inside the
plant. The mutant was previously constructed by adding pstI and NotI palindromic sequences at
the NcoI ends of the monomeric clone. The resultant mutant would have a hairpin structure
extending from the NcoI origin (Fig. 10). Regardless of complete base-pairing, infectivity assays
for this mutant also proved it to be lethal.

40
Native vLTSV-C
vLTSV mutant 178I4 (+4 bp)
vLTSV mutant 178I12 (+12 bp)
vLTSV mutant AB3 (+28 bp)
Figure 10. Predicted primary and secondary structure of three large insertion mutants of vLTSV. The native form of vLTSV is shown on top for comparison. Mutants 178I4 and 178I12 have a 4 bp and 12 bp insertion at the BglII site, respectively. The AB3 mutant has a hairpin extending from the NcoI site in the right hand corner. All insertion mutants depicted proved to be lethal.

41
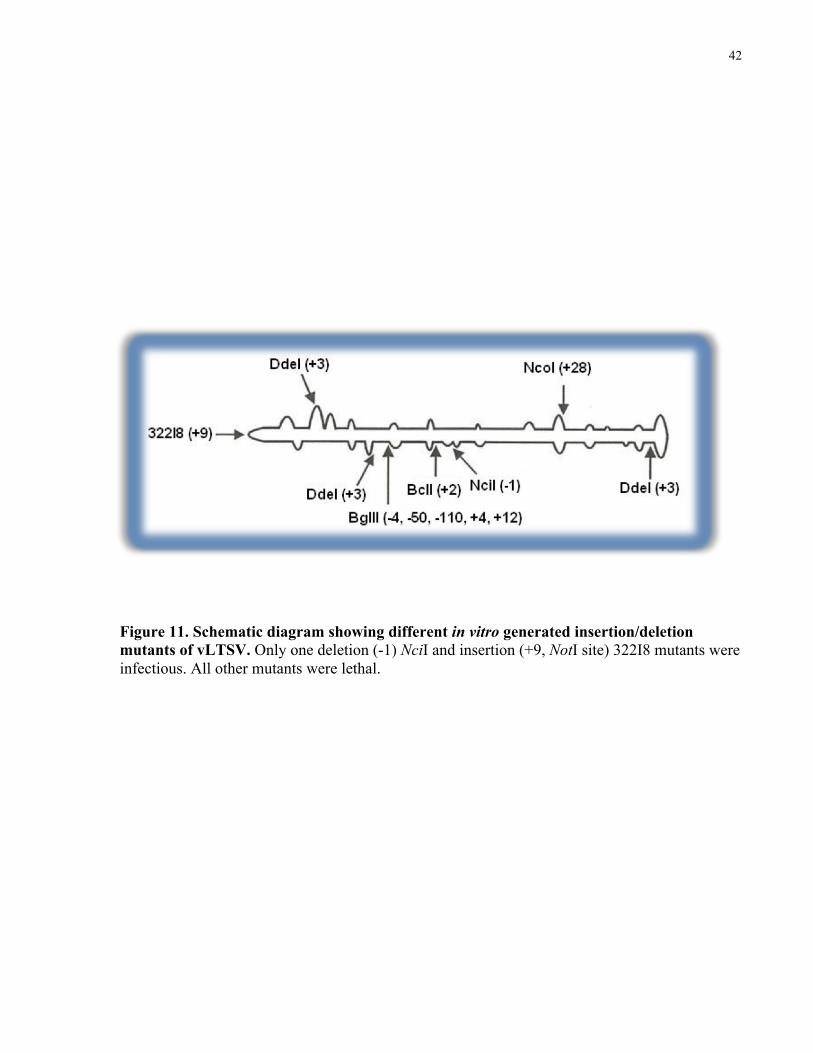
14.2 Infectivity of smaller sized insertion-deletion mutants
Following the above infectivity tests on “medium-large” sized mutations; a series of
progressively “smaller” insertion and deletion mutants were tested for viability. These mutants
were previously constructed at various positions throughout the vLTSV sequence. Three small
insertion mutants, with a 3 bp insertion at three different DdeI sites of vLTSV, were tested for
infectivity. One was located near the end of the rod opposite the hammerhead domain. This
insertion was close to a GAUUUU sequence that is conserved in all the circular virusoids
associated with sobemoviruses. Of the remaining two DdeI sites, one was located about 10 bp
away from the initially mutagenized BglII site while the other was positioned directly at the
hammerhead (+) sense splice site. Infectivity assays showed that all three of the mutants were
deleterious.
To further investigate whether the deleterious effect of these 3 bp insertions was possibly due to
a disruption of the overall rod-like structure, several smaller mutations were tested. One of these
was a -1 bp deletion mutation at the NciI site, which was located approximately in the middle of
the rod and in the region that shows the greatest sequence variability between the Canadian
isolate and the Australian/New Zealand isolates. Infectivity assay suggested that NciI (-1 bp) was
capable of replication. A summary of the above mutations is provided in Figure 11.
42
Figure 11. Schematic diagram showing different in vitro generated insertion/deletion mutants of vLTSV. Only one deletion (-1) NciI and insertion (+9, NotI site) 322I8 mutants were infectious. All other mutants were lethal.

43
14.3 Rod-preserving mutant (322I8)
It is clear that the virusoid of LTSV is not tolerant of any modifications to its overall rod-like
structure. Large deletions were deleterious to the virusoid as well as medium to small sized
insertions at various positions. The only mutant that was viable turned out to be a -1 nt deletion
at the NciI site which did not disturb the overall structure of the virusoid. An additional mutant
was tested for, having a 9-nt palindromic sequence of NotI restriction site (GGCGGCCGC)
inserted at the predicted “end” of the rod opposite the hammerhead domain. The NotI
palindromic sequence is presumed to form an internal hairpin extending the loop created at the
left end of the vLTSV rod (Fig. 12). This inserted sequence can hypothetically maintain the
overall virusoid structure by extending, rather than disrupting the rod.

44
Figure 12. Predicted folding at the end of native vLTSV (left) and 322I8 mutant (Right). The NotI sequence is displayed in a grey color whereas the native vLTSV sequence is in black. The predicted primary and secondary structure of the 322I8 shows almost no deviation from the native conformation.

45
15 Stability of foreign sequence in vLTSV
15.1 Progeny RNA from 5 days post inoculation
The 322I8 mutant was sub-cloned as a trimer, in a head-to-tail fashion, in pBS+ phagemid.
Infectivity assays were carried out with purified TRosV + RNA transcripts generated in vitro
from the trimeric clone. Plants were also inoculated with TRosV + LTSV RNA as a positive
control. Total RNA was extracted periodically (5 dpi, 12 dpi, and 21 dpi) from B. rapa leaves
which were not previously inoculated but showed signs of systemic infection (i.e. to avoid
contamination from the original inoculum). RT-PCR was performed on plant RNA using
vLTSVFor and vLTSVRev. Primers used in this procedure were located at the end of the rod
opposite the NotI insertion, and their back-to-back orientation allowed for full-length virusoid
cDNAs to be synthesized. RT-PCR products were then digested over night with NcoI and ligated
into the pBS+ plasmid. Ligation reactions were used in the transformation of E. coli and
respective colonies were selected and sent for sequencing.
Sequencing revealed that all of the 10 colonies selected from 5 dpi maintained the NotI
restriction site (Table 2, A-D). All of these clones were identical in sequence to the original
322I8 mutant except two (I8T-7 and I8T-8), which possessed additional point mutations close to
the NotI restriction site (Table 2, C-D). Cloning and sequencing cDNA from TRosV + LTSV
infected plants did not show any such mutations. The fidelity of the vLTSV sequence was
maintained in other places of the virusoid.

46
Table 2. Positions of mutations in cloned progeny RNAs from 5 dpi
Clone NotI? Mutations in Progeny RNAs from 5 dpi
A
I8T
1-6
Yes
B
I8T
9
Yes
C
I8T-7
Yes
D
I8T-8
Yes

47
15.2 Progeny RNA from 12 days post inoculation
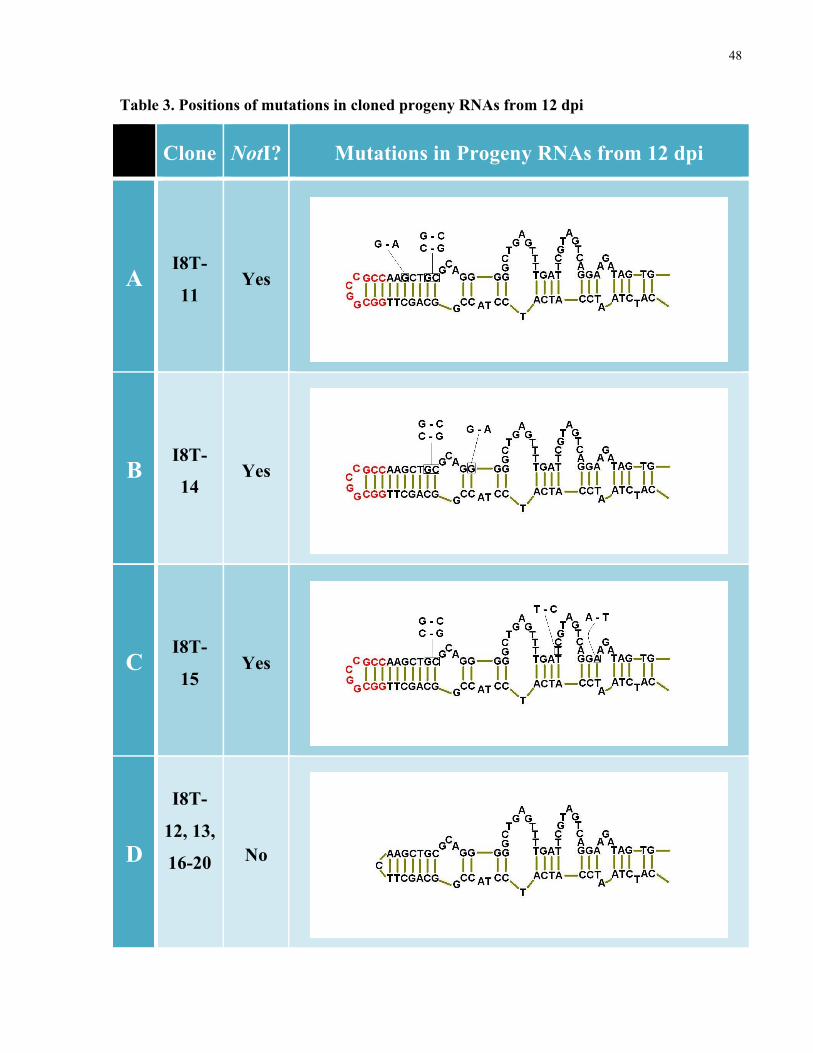
Sequencing of the 10 colonies selected from 12 dpi, showed a different scenario. Progeny from
12 dpi represented a heterogeneous mixture of molecules, some having a cleavable NotI
restriction site and others not. Estimates of the proportion of NotI-cleavable to non-cleavable RT-
PCR products, based on agarose electrophoresis, suggested the original insertion had been
altered (ie., rendered non-cleavable) in a vast majority of the RNAs. Corroborating with this
observation, cloning of a random sample of the RT-PCR products from 12 dpi indicated that only
3 out of 10 clones contained a cleavable NotI site (Table 3, A-C). Sequencing revealed that in
each clone, which maintained the NotI insertion, 2-3 point mutations were present in close
proximity to the end of the rod (Table 3, A-C) while the fidelity of the sequence was maintained
further away from the insertion. Based on sequence fidelity of cDNA progeny from TRosV +
LTSV infection, it was considered unlikely the amplification and cloning process alone could
account for the observed mutations in the NotI maintaining progeny RNAs.
15.3 Progeny RNA from 21 days post inoculation
In contrast to results obtained from 5 dpi and 12 dpi, progeny cDNA obtained from 21 dpi did
not contain the NotI site in any of the clones. There was a complete reversion of the sequence
after 21 days of infection. Furthermore, no mutations were observed. Aside from the absence of
NotI insertion, the sequences of these reversion RT-PCR clones were completely identical to
clone 322I8.
48
Table 3. Positions of mutations in cloned progeny RNAs from 12 dpi
Clone NotI? Mutations in Progeny RNAs from 12 dpi
A
I8T-
11
Yes
B
I8T-
14
Yes
C
I8T-
15
Yes
D
I8T-
12, 13,
16-20
No

49
Chapter 5 Discussion
16 Infectivity of full-length cDNA clones
In this study, the infectivity of full-length clones of vLTSV were tested as RNA or dsDNA.
Head-to-tail, multimeric cDNA clones of vLTSV were shown to be infectious as (+) and (-)
sense transcripts (Fig. 7, lanes E-F). However, the efficiency of infectivity of each RNA species
was not measured quantitatively. All multimeric RNA transcripts contained vector derived bases
(58 nts if generated from T7 promoter or 57 nts if generated from T3 promoter). However,
vLTSV progeny recovered from RNA infectivity assays were identical in size to the natural
vLTSV (Fig. 7, lanes E-F). Infectivity studies with monomeric transcripts of the linear sat-RNAs
of CMV and TBRV (Kurath & Palukaitis, 1987; Matsuta et al., 1988; Greif et al., 1990) showed
that additional vector-derived bases reduced the infectivity of these RNAs, and in cases where
progeny RNAs were recovered; the extra bases were not maintained. It should be noted,
however, that neither sCMV nor sTBRV is thought to replicate via a rolling circle pathway
(reviewed by Matthews, 1991; Roossinck et al., 1992). Our vLTSV sequence was liberated from
surrounding vector sequences to produce unit length progeny RNAs. This can be attributed to the
hammerhead splice sites. One full-length circular vLTSV can be generated from dimeric
transcripts and two from trimeric transcripts (Fig. 6). The remainder of vector or virusoid
sequences may have been discarded after self-cleavage has occurred.
In contrast to results obtained from multimeric transcripts, we observed that neither (-) or (+)
sense in vitro RNA transcripts from a monomeric clone of vLTSV were infectious (Fig. 7, lane
D). Similarly, PSTV was shown to have a low level of infectivity as a monomeric RNA
transcript (Cress et al., 1983), and it lost its infectivity once it was flanked by vector sequence
(Tabler & Sanger, 1985). On the contrary, monomeric RNA transcripts from sCMV and sTBRV
were infectious both as (+) and (-) transcripts (Kurath & Palukaitis, 1987; Greif et al., 1990). Our
monomeric clone had only one hammerhead (and hence only one splice site), and the location of
this structure was at an internal position in the insert (i.e. not at the NcoI cloning site). Therefore,
monomeric transcripts may have been spliced in the middle.

50
Infectivity of monomeric or multimeric vLTSV as dsDNA was not observed. None of the DNA
clones inoculated with TRosV produced vLTSV progeny RNA (Fig. 8, lanes D-F). This was in
contrast to preliminary data reported by Gellatly (1994). Infectivity of full-length DNA copies of
virusoids and viroids has been observed in previous studies. Cress et al. (1983) shows that
dimeric (head-to-tail) cDNA clones of PSTV inserted within the HindIII site of pBR322 are
infectious as intact plasmid DNA. However, a monomeric cDNA clone of PSTV is not infectious
as an intact plasmid (Cress et al., 1983). Later, Tabler and Sanger (1984) show that dimeric and
oligomeric PSTV clones were infectious as dsDNA excised from vectors. Monomeric dsDNA of
PSTV was also infectious, both excised from vector and left present in the BamHI site of
pBR322 plasmid (Tabler & Sanger, 1984).
Similar to viroids, infectivity assays of satellite RNAs and virusoids has been performed as DNA
copies. Gerlach et al. (1986) constructed trimeric clones of sTRSV that were infectious when
inoculated with a helper virus as dsDNA excised from the vector. More recently, Sheldon and
Symons (1993) showed that excised monomeric clones of vLTSV were infectious as dsDNA
with the help of satellite free LTSV. The vLTSV DNA inoculums were double-stranded
monomeric vLTSV cDNA containing 5’ overhangs of four nucleotides derived by excision of
the pGem2 clones with SalI (Sheldon & Symons, 1993). In our present study, vLTSV clones
were not infectious as dsDNA in monomeric or multimeric forms. While these studies were
performed in the early 90s, it is possible that our TRosV helper virus has lost the ability to
recognize vLTSV in a DNA form. It would be worthwhile to find a virusoid-free LTSV and test
its ability to support our DNA clones.

51
17 Structural integrity of vLTSV
Studies on structural domains of viroids are numerous. For example, several mutational analyses
of cloned PSTV, CEV, and tomato apical stunt viroid (TASV) have confirmed the necessity of
the central conserved region in viroid replication (Visvader & Symons, 1986; Hammond &
Owens, 1987; Hammond et al., 1989; Candresse et al., 1990). The aim of this study was to
elucidate structural features or domains in the vLTSV molecule. Three deletion mutants with -4
nt, -50 nt, or -110 nt were shown to be lethal to the infectivity of vLTSV. Other insertion mutants
(+3, +4, +12, hairpin) were also shown to be lethal to the infectivity of vLTSV (Fig. 11). The
deleterious nature of these mutants can be attributed to their location within the sequence as well
as their ability to disrupt the overall secondary structure of the molecule (Fig. 10).
Mutation studies on other sat-RNA molecules have demonstrated varying results. Masuta and
Takanami (1989) constructed insertion or deletion mutant clones of sCMV and analyzed their
survival in infected plants. It was found that although sCMV is tolerant to small insertions (as
much as 4 bases); any deletion mutations were detrimental to the viability of the satellite RNA
(Masuta & Takanami, 1989). On the other hand, Dalmay and Rubino (1995) report that
transcripts from mutant clones of cymbidium ringspot tombusvirus satellite (sCyRSV) which
lacked up to eight nucleotides at the 3’ end were biologically active and yielded RNA progeny
that had the 3’ end restored as in wild-type RNA. However, deletions and substitutions at the 5’
end produced nonviable molecules (Dalmay & Rubino, 1995). Later on, sCyRSV mutants were
constructed with deletions in the internal regions of the molecule. Dalmay & Rubino (1995)
showed that replication of the satellite RNA was affected based on the location of these
deletions. Furthermore, it was observed that viable mutants were much less encapsidated than
wild-type RNA which indicates a size limitation requirement for the encapsidation and survival
of sCyRSV (Dalmay & Rubino, 1995).
It is clear that our deletion mutants were missing essential parts of their sequence. These missing
bases may be signals for the helper virus (and host) replicase to recognize the virusoid RNA. As
well, they may contribute to the packaging and/or transport of vLTSV within the cell. Since
vLTSV has a very small genome (322 nt), it is presumed that every nucleotide has a functional
basis. On the other hand, insertion mutants which possessed the entire vLTSV sequence were

52
still lethal (Fig. 9). It is possible that these insertion mutants were being replicated but their
size/structure was not optimal for packaging within the helper virus. However, this presumption
is false since detection of RNA progeny was directly from plant RNA. Another possibility is that
their rod-like secondary structure was destabilized due to the insertion of foreign nucleotides. As
a result, important structural clues that are important in the replication process were absent or
malformed. It is already known that structural motifs are critical in the replication process of
viroids. A similar reasoning could be attributed to the deleterious nature of insertion mutants.
The assumption that every nucleotide is functional in the replication of vLTSV may not be true.
A -1 nt deletion mutant at the NciI cloning site was not lethal to the vLTSV replication (Fig. 11).
Similarly, Sheldon and Symons (1993) showed single insertion or deletion mutants in the (-)
hammerhead region of the molecule, which were still infectious. It is possible, that in both cases,
the missing nucleotides were not an essential part of the virusoid genome. Except that, the
mutants in Sheldon and Symons (1993) destroyed the (-) sense hammerhead and its self-splicing
capability. Our -1 nt deletion was also located in the hammerhead domain; however, no analysis
was performed on its loss of function. A single base change can have profound implications on
the biology of the virusoid. Nevertheless, our mutational analysis shows that insertion or deletion
mutants can be tolerated as long as the overall structural integrity of the molecule is preserved. It
must be noted that no analysis was done on the level of support for these mutants. It is possible
that although these small mutations are being tolerated, the mutants are not replicated and
encapsidated as much as the native vLTSV. Therefore, there exists a strong selection for the
natural form of this virusoid as seen from the 322I8 infectivity.
Selection towards the native form was observed in the infectivity of the 322I8 insertion mutant.
The NotI insertion was tolerated up to 12 dpi, where it started to be selected against. Progeny
cDNA from 5 dpi (Table 2, A-D) and a few from 12 dpi (Table 3, A-C) still maintained the NotI
insertion. Sequence analysis showed 2-3 point mutations in close proximity to the end of the rod,
while the fidelity of the sequence was maintained further away from the insertion. Presumably,
these mutations have a role in stabilizing the RNA from its local structural stresses created by the
NotI insertion. It must be noted that all of the stabilizing point mutations occur at nucleotides that
are conserved between the various strains of vLTSV. Intriguingly, in a subset of 12 dpi clones
examined by sequence analysis, the NotI insertion was missing entirely, suggesting a complete

53
reversion to wild-type at the insertion point (Table 3, D). The sequences of reversion RT-PCR
clones were identical to clone 322I8 (minus the NotI insertions). One of these clones had a single
point mutation (not shown), although several possibilities can account for this (i.e. it may
represent sequence heterogeneity in RNA populations or an artifact of PCR). We also expected
to find among the non-revertant clones a subset of mutants with partial NotI insertions. It is quite
possible the absence of such mutants is due to our sample size (10 clones were examined for
each stage). Moreover, RNA extracted at an intermediary stage between 5dpi and 12 dpi may
show progeny RNA with partial NotI sequence. However, if the loss of the NotI sequence was a
periodic phenomena, it would be expected that progeny cDNA from 12 dpi would have partial
NotI sites as well. It appears that the NotI site is eliminated abruptly rather than transitionally.
The apparent majority of complete reversion mutants coupled with the heterogeneity of
stabilizing point mutations in NotI maintaining progeny (with the exception of two G-C and C-G
substitutions; Table 3), would appear to indicate that selection for wild-type like virusoid
sequences is considerable. This was evident in cDNA progeny from 21 dpi where all clones were
reverted back to the wild-type sequence. Reversion of mutants to their wild-type sequence has
been observed in previous studies on virusoids and satellite RNAs. By sequencing the progeny
RNA, Sheldon and Symons (1993) report 8-20% reversions and pseudo-reversions of the
introduced mutations in the (-) sense hammerhead region. In other words, the (-) sense
hammerhead was restored in these reversion mutants. Pseudo-reversion mutants were those who
possessed additional mutations in other locations of vLTSV to “compensate” for the introduced
mutation. Interestingly, these pseudo-revertants did not have the (-) sense hammerhead
functionality restored (Sheldon & Symons, 1993). No explanation was given for why these
“compensatory” mutations were unable to restore the (-) sense hammerhead function. Robagli et
al. (1993) also found new variants in insertion-deletion mutants of the cDNA of sTRSV. Three
possible modifications in the progeny RNA were observed: (i) a deletion of the mutated region
followed by sequence duplication, (ii) sequence duplication and deletion outside of the mutated
region, and (iii) limited rearrangement at the site of the mutation (Robaglia et al., 1993). The
original mutation was never recovered, except in one case, which shows a rapid selection of a
well-adapted RNA molecule (Robaglia et al., 1993).

54
Because mutant 322I8 was capable of replication, and the insertion was wholly maintained in
progeny RNA, the notion that overall structural integrity is important to vLTSV replication
appears to be supported. Although the 9-nt insertion was not lethal to the replication of the
virusoid it might not have been optimal for encapsidation within the helper virus. As a result, any
revertants of this mutant, which were closer to the wild-type form, were highly selected for and
preferred by the replicase unit. It was shown that some nucleotides are dispensable in the
replication of vLTSV; however, there exist no tolerance for structural perturbations. It is quite
possible that larger insertions would be viable in the virusoid of LTSV; however, over time these
insertions will get eliminated. This virusoid has an optimal size and sequence. It was shown that
deletions or insertions would eventually revert back to their wild-type form. Therefore, although
mutants can be tolerated as long as the overall rod-like structure is preserved, the virusoid will
return back to its original sequence either with the help of the replicase or by a natural
phenomenon.
18 Summary and Conclusions
The virusoid of LTSV is untranslatable and depends on a helper virus for its replication and
encapsidation. Large deletions of the virusoid were deleterious to its infectivity as important
sequences and/or structural signals were lost. Small deletions (-1 nt), on the other hand, were
tolerated as long as the overall structure of the virusoid was maintained. Small or large insertions
in different parts of the virusoid were deleterious as they disrupt the rod-like motif of the
molecule. The only insertions that were tolerated by the virusoid were “rod-preserving”. Inserted
sequence was replicated along with native sequence by the helper virus (and host) replicase and
incorporated into the progeny RNA. However, after several days of replication, the inserted
sequence was eliminated during the replication cycle and the virusoid reverted back to its wild-
type form.

55
19 Future Directions
Further investigation into structure-function relationship of vLTSV should focus on one
fundamental question: was the infectivity of vLTSV influenced by the sequence of the sites
checked or did the perturbation of the overall rod-like structure abolish infectivity? Or was it a
combination of the two? For future work, mutagenic strategy should be considered at specific
regions such as using mutagenic primers to make mutants that further investigate the tolerance of
small base changes in the sequences that are highly variable between different vLTSV isolates. It
is interesting to find whether these variable domains are similar to ones found in PSTV viroids.
Another possibility is to generate random changes throughout the virusoid sequence, by using a
technique such as mutagenic PCR. The latter strategy may be best approached by screening
populations of mutagenized virusoids in planta thereby possibly identifying sequences that are
tolerant (or in tolerant) to modifications.
In a second approach, an additional “rod-preserving” mutant should be constructed, preferably in
the central region of the virusoid. Infectivity of this clone could shed light on the importance of
the domain where NotI sequence was inserted. Other constructs should be made where the
position of various nucleotides are “flipped” to resolve whether the order of nucleotides are as
important as the overall structure. Alternatively, if these mutants are not infectious the question
arises: why not? Perhaps predicated models of the overall structure were incorrect.

56
References
AbouHaidar, M.G. & Paliwal, Y.C. (1988). Comparison of the nucleotide sequences of the
viroid-like satellite RNA of the Canadian and Australasian strains of Lucerne transient
streak virus. Journal of General Virology, 69: 2369-2372.
Blackstock, J.M. (1978). Lucerne transient streak and Lucerne latent, two new viruses of lucerne.
Australian Journal of Agricultural Research 29, 291-304.
Branch, A. D. & Roberston, H. D. (1984). A replication cycle for viroids and other small
infectious RNAs. Science, 223: 450-455.
Bruening, G., Passmore, B.K., van Tol, H., Buzayan, J.M. & Feldstein, P.A. (1991). Replication
of a plant virus satellite RNA: evidence favors transcription of circular templates of both
polarities. Molecular Plant-Microbe Interactions, 4: 219-225.
Candresse, T, Diener, T.O. & Owens, R.A. (1990). The role of the viroid central conserved
region in cDNA infectivity. Virology, 175: 232-237.
Collins R. F., Gellatly D. L., Sehgal O. P., and AbouHaidar M. G. (1998). Self-cleaving circular
RNA associated with rice yellow mottle virus is the smallest viroid-like RNA. Virology,
241: 269-275.
Cress, D.E., Kiefer, M.C. & Owens, R.A. (1983). Construction of infectious potato spindle tuber
viroid cDNA clones. Nucleic Acids Research, 19: 6821-6835.
Daros J.A., Marcos J.F., Hernandez C., and Flores R. (1994). Replication of avocado sunblotch
viroid: evidence for a symmetric pathway with two rolling circles and hammerhead
ribozyme processings. Proceedings of the National Academy of Sciences, 91: 12813-
12817.
Davies, C., Haseloff, J. & Symons, R.H. (1990). Structure, self-cleavage, and replication of two
viroid-like satellite RNAs (virusoids) of subterranean clover mottle virus. Virology, 177:
216-224.

57
Diener, T.O. (1986). Viroid processing: a model involving the central conserved region and
hairpin I. Proceedings of the National Academy of Science, U.S.A., 83: 58-62.
Diener, T.O. (1991). The frontier of life: the viroids and viroid-like satellite RNAs. In: Viroids
and Satellites: Molecular Parasites at the Frontiers of Life, pp. 1-20. Edited by K.
Maramorosch. Boca Raton, Fl. CRC Press.
Flores R., Hernandez C., Martinez de Alba A.E., Daros J.A, and Di Serio F. (2005). Viroids and
viroid-host interations. Annual Review of Phytopathology, 43: 117-139.
Forster, R.L.S. & Jones, A. T. (1979). Properties of lucerne transient streak virus and evidence of
its affinity to southern bean mosaic virus. Annals of Applied Biology, 93: 181-189.
Forster, R.L.S. & Jones, A. T. (1980). Lucerne transient streak virus. CMI/AAB Descriptions of
Plant Viruses. No. 244
Forster, A.C. & Symons, R.H. (1987). Self-cleavage of plus and minus RNAs of a virusoid and a
structural model for the active sites. Cell 49, 211-220.
Forster, A.C., Davies, C., Sheldon, C.C., Jefferies, A.C. & Symons, R.H. (1988). Self-cleaving
viroid and newt RNAs may only be active as dimmers. Nature, 334: 265-267.
Francki, R.I.B., Randles, J.W., Hatta, T., Davies, C. & Chu, P.W.G. (1983). Subterranean clover
mottle virus: another virus from Australia with encapsidated viroids-like RNA. Plant
Pathology 32, 47-59.
Francki, R.I.B., Grivell, C.J. & Gibb, K.S. (1986). Isolation of velvet mottle virus capable of
replication with and without a viroid-like RNA. Virology, 148: 381-384.
Gellatly, D.L. (1994). Infectivity of full-length clones of the viroids-like satellite RNA of lucerne
transient streak virus. Master’s thesis. University of Toronto, Dept of Cell and Systems
Biology.
Gellatly, D.L. (1995). Structure-function analysis of the circular satellite RNA of lucerne
transient streak virus. Unpublished doctoral dissertation. University of Toronto, Dept of
Cell and Systems Biology.

58
Gerlach, W.L., Buzayan, J.M., Schneider, I.R. & Bruening, G. (1986). Satellite tobacco ringspot
virus RNA: biological activity of DNA clones and their in vitro transcripts. Virology,
151: 172-185.
Gora-Sochacka A. (2004). Viroids: unusual small pathogenic RNAs. Acta Biochimica Polonica,
51: 587-607.
Greif, C., Hemmer, O., Demangeat, G. & Fritsch, C. (1990). In vitro synthesis of biologically
active transcripts of tomato black ring virus satellite RNA. Journal of General Virology,
71: 907-915.
Guo L., Cao Y., Li D., Niu S., Cai Z., Han C., Zhai Y., Yu J. (2004). Analysis of nucleotide
sequences and multimeric forms of a novel satellite RNA associated with Beet Black
Scorch Virus. Journal of Virology, 79: 3664 – 3674.
Hammond, R.W. & Owens, R.A. (1987). Mutational analysis of potato spindle tube viroid
reveals complex relationships between structure and infectivity. Proceedings of the
National Academy of Science, U.S.A. 84: 3967-3971.
Hammond, R.W., Diener, T.O. & Owens, R.A. (1989). Infectivity of chimeric viroid transcripts
reveals the presence of alternative processing sites in potato spindle tube viroid. Virology,
170: 486-495.
Hammond, R.W. (1992). Analysis of the virulence modulating region of potato spindle tube
viroid (PSTVd) by site-directed mutagenesis. Virology, 187: 654-662.
Haseloff, J. & Symons, R.H. (1982). Comparative sequence and structure of viroid-like RNAs of
two plant viruses. Nucleic Acids Research, 10: 3681-3691.
Hernandez, C. & Flores, R. (1992). Plus and minus RNAs of peach latent mosaic viroid self-
cleave in vitro via hammerhead structures. Proceedings of the National Academy of
Science, U.S.A. 89: 3711-3715.
Hollings, M. (1973). Turnip rosette virus. CMI/AAB Descriptions of Plant Viruses No. 125.
Hull, R. (1988). The sobemovirus group. In: The Plant Viruses, Vol. 3. Polyhedral virions with

59
monopartite RNA genomes. pp. 113-146. Edited by R. Koeing. New York. Plenum Press.
Hutchins, C.J., Keese, P., Visvader, J.E., Rathjen, P.D., McInnes, J.L. & Symons, R.H (1985).
Comparison of multimeric plus and minus forms of viroids and virusoids. Plant
Molecular Biology, 4: 293-304.
Hutchins, C.J., Rathjen, P.D., Forster, A.C. & Symons, R.H (1986). Self-cleavage of plus and
minus RNA transcripts of avocado sunblotch viroid. Nucleic Acids Research, 14: 3627-
3640.
Jones, A.T., Mayo, M.A. & Duncan, G.H. (1983). Satellite-like properties of small circular RNA
molecules in particles of Lucerne transient streak virus. Journal of General Virology 64,
1167-1173.
Jones, A.T. & Mayo, M.A. (1984). Satellite nature of the viroids-like RNA-2 of solanum
nodiflorum mottle virus and the ability of other plant viruses to support the replication of
viroids-like RNA molecules. Journal of General Virology 65, 1713-1721.
Keese, P., Bruening, G. & Symons, R.H. (1983). Comparative sequence and structure of circular
RNAs from two isolates of lucerne transient streak virus. FEBS Letters, 159: 185-190.
Keese, P. & Symons, R.H. (1987). The structure of viroids and virusoids. In: Viroids and Viroid-
like Pathogens, pp. 2-47. Edited by J.S.Semancik. Boca Raton, Fl. CRC Press.
Kurath, G. & Palukaitis, P. (1989). Satellite RNAs of cucumber mosaic virus: recombinants
constructed in vitro reveal independent functional domains for chlorosis and necrosis in
tomato. Molecular Plant-Microbe Interactions, 2: 91-96.
Mang, K.Q., Ghosh, A. & Kaesberg, P. (1982). A comparative study of the cowpea and bean
strains of southern bean mosaic virus. Virology 116, 264-269.
Maniatis, T., Fritsch, E.F. & Sambrook, J. (1982). Molecular cloning a laboratory manual. Cold
Spring Harbour, N.Y. Cold Spring Harbour Laboratory Press.
Masuta, C., Kuwata, S. & Takanami, Y. (1988). Effects of extra 5’ non-viral bases on the
infectivity of transcripts form a cDNA clones of satellite RNA (strain Y) of cucumber

60
mosaic virus. Journal of Biochemistry, 104: 841-846.
Masuta, C. & Takanami, Y. (1989). Determination of sequence and structural requirements for
pathogenicity of a cucumber mosaic virus satellite RNA (Y-satRNA). The Plant Cell, 1:
1165-1173.
Matthews, R.E.F. (1991). Viroids, satellite viruses, and satellite RNAs. In: Plant Virology, pp.
306-338. New York. Academic Press.
Morris-Krsinich, B.A.M. & Forster, R.L.S. (1983). Lucerne transient streak virus RNA and its
translation in rabbit reticulocyte lysate and wheat germ extract. Virology 128, 176-185.
Paliwal, Y.C. (1983). Identification and distribution in eastern Canada of Lucerne transient
streak, a virus newly discovered in North America. Canadian Journal of Plant Pathology
5, 75-80.
Paliwal, Y.C. (1984a). Properties of a Canadian isolate of Lucerne transient streak virus and
further evidence of similarity of the virus to sobemoviruses. Canadian Journal of Plant
Pathology 6, 1-92.
Paliwal, Y.C. (1984b). Interaction of the viroids-like RNA-2 of lucerne transient streak virus
with southern bean mosaic virus. Canadian Journal of Plant Pathology 6, 93-184.
Riesner, D. (1991). Viroids: from thermodynamics to cellular structure and function. Molecular
Plant-Microbe Interactions, 4: 122-131.
Rigden, J.E. & Rezaian, M.A. (1992). In vitro synthesis of an infectious viroid: analysis of the
infectivity of monomeric linear CEV. Virology, 186: 201-206.
Robaglia C., Bruening G., Haseloff J., Gerlach W. (1993). Evolution and replication of tobacco
ringspot virus satellite RNA mutants. The EMBO Journal, 12: 2969-2976.
Roossinck, M.J., Sleat, D. & Palukaitis, P. (1992). Satellite RNAs of plant viruses: structures and
biological effects. Microbiological Reviews, 56: 265-279.
Rubino L., & Dalmay T. (1994). Replication of cymvidium ringspot virus satellite RNA mutants.
Virology, 206: 1092-1098.

61
Sehgal, O.P. (1990). Structural transitions in southern bean mosaic virus and their correlation
with infectivity and ribonunclease sensitivity. J. Phytopathology, 129: 69-82.
Sehgal, O.P, Sinha, R.C., Gellatly, D.L., Ivanov, I. & AbouHaidar, M.G. (1993). Replication and
encapsidation of the viroids-like satellite RNA of lucerne transient streak virus are
supported in divergent hosts by cocksfoot mottle virus and turnip rosette virus. Journal of
General Virology 74, 785-788.
Sehgal, O.P, AbouHaidar, M.G, Gellatly, D.L, Ivanov, I & Thottappilly, G. (1993). An
associated small RNA in rice yellow mottle sobemovirus is homologous to the satellite
RNA of lucerne transient streak sobemovirus. (1993). Phytopathology, 83, in press.
Sheldon, C.C. & Symons, R.H. (1993). Is hammerhead self-cleavage involved in the replication
of a virusoid in vivo? Virology 194, 463-474.
Sleat, D.E. & Palukaitis, P. (1992). A single nucleotide change within a plant virus satellite RNA
alters the host specificity of disease induction. The Plant Journal, 2: 43-49.
Sleat D., & Paukaitis P. (1990). Site-directed mutagenesis of a plant virual satellite RNA changes
its phenotype from ameliorative to necrogenic. Proceedings of the National Academy of
Science, 87: 2946-2950.
Symons, R.H. (1992). Small catalytic RNAs. Annual Review of Biochemistry, 61: 641-671.
Symons, R.H. (1997). Plant pathogenic RNAs and RNA catalysis. Nucleic Acids Research 25,
2683-2689.
Tabler, M. & Sanger, H.L. (1984). Cloned single- and double-stranded DNA copies of potato
spindle tuber viroid (PSTV) RNA and co-inoculated subgenomic DNA fragments are
infectious. EMBO Journal, 3: 3055-3062.
Tabler M., & Tsagris M. (2004). Viroids: Petite RNA pathogens with distinguished talents.
Trends in Plant Sciences, 9: 339-328.
Taylor J.M. (2003) Replication of Human Hepatitis Delta Virus: Recent developments. Trends in
Microbiology, 11: 185-190.

62
Tien-Po, Davies, C., Hatta, T. & Francki, R.I.B. (1981). Viroid-like RNA encapsidated in
lucerne transient streak virus. FEBS Letters 132, 353-356.
Wessenegger M., Spieker R. L., Thalmeir S., Gast F. U., Riedel L., Sanger H. L. (1996). A single
nucleotide substitution converts potato spindle tuber viroid (PSTVd) from a noninfectious
to an infectious RNA for nicotiana tabacum. Virology, 15: 191-197.
Zhong X., Archial A. J., Amin A. A., Ding B. (2008). A genomic map of viroid RNA motifs
critical for replication and systemic trafficking. The Plant Cell, 20: 35-47.
Zucker, M. & Stiegler, P. (1981). Optimal computer folding of large RNA sequences using
thermodynamic and auxiliary information. Nucleic Acids Research, 9: 133-148.