structural basis for inhibition of the epidermal growth factor receptor by cetuximab
DESCRIPTION
Structural basis for inhibition of the epidermal growth factor receptor by cetuximabTRANSCRIPT

epidermal growth factor receptor
by: Bundit Boonyar i t
Dept.Chemistry, Fac.Sc ience Pr ince of Songkla Univers i ty
566-544 Drug target
tructural basis for inhibition of theS
by cetuximabLi, S., Schmitz, K. R., Jeffrey, P. D., Wiltzius, J. J. W., Kussie, P., & Ferguson, K. M. (2005). Cancer Cell, 7(4), 301-311.

by: Bundit Boonyar i t566-544 Drug target
Cancer
Lead ing cause o f dea th in economically developed countries and second leading cause of death in development countries
Disease cause of abnormalities in cell division that refers to over 100 distinct clinical pathologies
Jemal et al. (2011); Kufe et al. (2003)
2

Yap et al. (2010)
3
The challenge of anticancer drug development
by: Bundit Boonyar i t566-544 Drug target

Yap et al. (2010)
4
The challenge of anticancer drug development
Pharmacokinetics: what the body does to the drug (ADME)
Pharmacodynamic: what the drug does to the body (biological effects)
Maximum tolerated dose: the highest dose of a drug or treatment that does not cause unacceptable side effects.
by: Bundit Boonyar i t566-544 Drug target

“Selective anticancer drug”
“Targeted cancer therapy”
“Personalized medicine”
5by: Bundit Boonyar i t566-544 Drug target

Cancer pathways
http://www.nature.com/nrc/poster/subpathways

CETUXIMAB
7by: Bundit Boonyar i t566-544 Drug target
monoclonal antibody
metastatic coloractal cancer
Phase II trial
Epidermal Growth Factor Receptor (EGFR)

8by: Bundit Boonyar i t566-544 Drug target
140 Chapter 5: Growth Factors, Receptors, and Cancer
Since these initial analyses of the EGF receptor structure, a large number of other, sim-ilarly structured receptors have been characterized (Figure 5.9). As discussed later, each of these receptors has its own growth factor ligand or set of ligands (Table 5.1). Depending on the particular growth factor–receptor pair, the binding of a ligand to its
Figure 5.9 Structure of tyrosine kinase receptors The EGF receptor (Figure 5.8A) is only one of many similarly structured receptors that are encoded by the human genome. These tyrosine kinase receptors (RTKs) can be placed into distinct families, depending on the details of their structure. Representatives of most of these families are shown here. All have in common quite similar cytoplasmic tyrosine kinase domains (red), although in some cases (e.g., the PDGF receptor) these domains are interrupted by small “insert” regions. The RTK ectodomains (which protrude into the extracellular space, green, gray) have highly variable structures, reflecting the fact that they recognize and bind a wide variety of extracellular ligands. (From B. Alberts et al., Molecular Biology of the Cell, 5th ed. New York: Garland Science, 2008.)
EGFreceptor
SSSS
SS
insulinreceptor,
IGF-1receptor
NGFreceptor
PDGFreceptor,
M-CSFreceptor
FGFreceptor
plasma membraneCYTOPLASM
EXTRACELLULARSPACE
tyrosinekinasedomain
cysteine-richdomain
immunoglobulin-like domain
TBoC2 b5.10/5.09
VEGFreceptor
Ephreceptor
kinase insert region
fibronectin type III-like domain
Table 5.1 Growth factors (GFs) and tyrosine kinase receptors that are often involved in tumor pathogenesis
Name of GF Name of receptor Cells responding to GF
PDGFa PDGF-R endothelial, VSMCs, fibroblasts, other mesenchymal cells, glial cells
EGFb EGF-Rc many types of epithelial cells, some mesenchymal cells
NGF Trk neurons
FGFd FGF-Re endothelial, fibroblasts, other mesenchymal cells, VSMCs, neuroectodermal cells
HGF/SF Met various epithelial cells
VEGFf VEGF-Rg endothelial cells in capillaries, lymph ducts
IGFh IGF-R1 wide variety of cell types
GDNF Ret neuroectodermal cells
SCF Kit hematopoietic, mesenchymal cells
aPDGF is represented by four distinct polypeptides, PDGF-A, -B, -C, and -D. The PDGF-Rs consist of at least two distinct species, α and β, that can homodimerize or heterodimerize and associate with these ligands in different ways. bThe EGF family of ligands, all of which bind to the EGF-R (ErbB1) and/or heterodimers of erbB1 and one of its related receptors (footnote c), includes—in addition to EGF—TGF-α, HB-EGF, amphiregulin, betacellulin, and epiregulin. In addition, other related ligands bind to heterodimers of ErbB2 and ErbB3 or ErbB4; these include epigen and a variety of proteins generated by alternatively spliced neuregulin (NRG) mRNAs, including heregulin (HRG), glial growth factor (GGF), and less well-studied factors such as sensory and motor neuron–derived factor (SMDF). cThe EGF-R family of receptors consists of four distinct proteins, ErbB1 (EGF-R), ErbB2 (HER2, Neu), ErbB3 (HER3), and ErbB4 (HER4). They often bind ligands as heterodimeric receptors, for example, ErbB1 + ErbB3, ErbB1 + ErbB2, or ErbB2 + ErbB4; ErbB3 is devoid of kinase activity and is phosphorylated by ErbB2 when the two form heterodimers. ErbB2 has no ligand of its own but does have strong tyrosine kinase activity. ErbB3 and ErbB4 bind neuregulins, a family of more than 15 ligands that are generated by alternative splicing. dFGFs constitute a large family of GFs. The prototypes are acidic FGF (aFGF) and basic FGF (bFGF); in addition there are other known members of this family. eThere are four well-characterized FGF-Rs.fThere are four known VEGFs. VEGF-A and -B are involved in angiogenesis, while VEGF-C and -D are involved predominantly in lymphangiogenesis.gThere are three known VEGF-Rs: VEGF-R1 (also known as Flt-1) and VEGF-R2 (also known as Flk-1/KDR), involved in angiogenesis; and VEGF-R3, involved in lymphangiogenesis. hThe two known IGFs, IGF-1 and IGF-2, both related in structure to insulin, stimulate cell growth (that is, increase in size) and survival; they also appear to be weakly mitogenic.
Abbreviation: VSMC, vascular smooth muscle cell. Adapted in part from B. Alberts et al., Molecular Biology of the Cell, 5th ed. New York: Garland Science, 2008.
Receptor Tyrosine Kinase (RTK)
The Biology of Cancer, 2ed

9by: Bundit Boonyar i t566-544 Drug target
The Human EGF Receptor (HER)
The Biology of Cancer, 2ed
132 Chapter 5: Growth Factors, Receptors, and Cancer
These signaling processes are part of the larger problem of cell-to-cell communica-tion. Indeed, the evolution of the first multicellular animals (metazoa) 600 to 700 mil-lion years ago depended on the development of biochemical mechanisms that allow cells to receive and process signals arising from their neighbors within tissues. With-out effective intercellular communication, the behavior of individual cells could not be coordinated, and the formation of architecturally complex tissues and organisms was inconceivable. Obviously, such communication depended on the ability of some cells to emit signals and of others to receive them and respond in specific ways.
In very large part, the signals passed between cells are carried by proteins. Hence, signal emission requires an ability by some cells to release proteins into the extracel-lular space. Such release—the process of protein secretion—is also complicated by the imperviousness of the plasma membrane. After these signaling proteins are released into the extracellular space, the designated recipient cells must be able to sense the presence of these proteins in their surroundings. Much of this chapter is focused on
LPAthrombin
ET, etc.TGF-α epiregulin amphi-
regulinNRG-1 cytokinesEGF β–cellulin HB-EGF NRG-2 NRG-3 NRG-4
SRC CBL PLC
PKC BAD S6K
AKT
PI3K SHP2 GRB7GRB2GAP
SHC
SOS
MAPK
RAF
RAC
MEK PAK ABL
JNK
NCK VAV CRK
STATERG1MYCSP1 FOS
JUN
ELK
JAKRAS-GDP
RAS-GTP
JNKK
growthfactorligands
cellsurfacereceptors
adaptorsandenzymes
signalingcascades
transcriptionfactors
INPU
T LAY
ER
APOPTOSIS MIGRATION GROWTH ADHESION DIFFERENTIATION
SIGN
AL PRO
CESSING
OU
TPUT
LAY
ER
= HER1 = HER2 = HER3 = HER4
TBoC2 b5.01/5.01
plasmamembrane
cytoplasm
nucleus
Figure 5.1 The human EGF receptor (HER) signaling network: how cells communicate with their surroundings A variety of protein messengers (growth factor ligands, light green rectangles, top) interact with a complex array of cell surface receptors, which transduce signals across the plasma membrane (gray) into the cytoplasm. There, a complex network of signal-transducing proteins processes these signals, funnels signals into the nucleus (bottom), and ultimately evokes a variety of biological responses (“output layer,” yellow rectangles, bottom). Many of the components of this circuitry, both at the cell surface and in
the cell interior, are involved in cancer pathogenesis. This cartoon focuses on a small subset of the receptors—the EGF receptor and its cousins—that are displayed on the surfaces of mammalian cells. Receptors like these are the main topic of this chapter; the adaptors and signaling cascades will be covered in the next chapter. The X’s associated with the cytoplasmic domain of HER3 indicate the absence of detectable tyrosine kinase activity in contrast to the readily detectable kinase activity of the other three members of this family of receptors. (From Y. Yarden and M.X. Sliwkowski, Nature Rev. Mol. Cell Biol. 2:127–137, 2001.)

10by: Bundit Boonyar i t566-544 Drug target
The Human EGF Receptor (HER)
The Biology of Cancer, 2ed
144 Chapter 5: Growth Factors, Receptors, and Cancer
5.5 A growth factor gene can become an oncogene: the case of sis
The notion that oncoproteins can activate mitogenic signaling pathways received another big boost when the platelet-derived growth factor (PDGF) protein was iso-lated and its amino acid sequence determined. In 1983, the B chain of PDGF was found to be closely related in sequence to the oncoprotein encoded by the v-sis onco-gene of simian sarcoma virus. Once again, study of the oncogenes carried by rapidly transforming retroviruses paid off handsomely.
The PDGF protein was discovered to be unrelated in structure to EGF and to stimu-late proliferation of a different set of cells. PDGF stimulates largely mesenchymal cells, such as fibroblasts, adipocytes, and smooth muscle cells; in the brain, it can also stim-ulate the growth and survival of certain types of glial cells. By contrast, the mitogenic activities of EGF are focused largely (but not entirely) on epithelial cells (for example, see Figure 5.11B). This specificity of action could be understood once the PDGF recep-tor was isolated: the PDGF-R was found to be expressed on the surfaces of mesenchy-mal cells and is not usually displayed by epithelial cells, while the EGF-R largely shows the opposite pattern of expression. (Like the EGF receptor, the PDGF-R uses a tyrosine kinase in its cytoplasmic domain to broadcast signals into the cell; see Figure 5.9.)
The connection between PDGF and the sis-encoded oncoprotein suggested another important mechanism by which oncoproteins could transform cells: when simian sar-coma virus infects a cell, its sis oncogene causes the infected cell to release copious amounts of a PDGF-like Sis protein into the surrounding extracellular space. There, the PDGF-like molecules attach to the PDGF-R displayed by the same cell that just synthesized and released them. The result is strong activation of this cell’s PDGF receptors and, in turn, a flooding of the cell with an unrelenting flux of the growth-stimulating signals released by the ligand-activated PDGF-R.
These discoveries also resolved a long-standing puzzle. Most types of acutely trans-forming retroviruses are able to transform a variety of infected cell types. Simian sar-coma virus, however, was known to be able to transform fibroblasts, but it failed to transform epithelial cells. The cell type–specific display of the PDGF-R explained the differing susceptibilities to transformation by simian sarcoma virus.
Table 5.2 Tyrosine kinase GF receptors altered in human tumorsa
Name of receptor Main ligand Type of alteration Types of tumor
EGF-R/ErbB1 EGF, TGF-α overexpression non-small cell lung cancer; breast, head and neck, stomach, colorectal, esophageal, prostate, bladder, renal, pancreatic, and ovarian carcinomas; glioblastoma
EGF-R/ErbB1 truncation of ectodomain glioblastoma; lung and breast carcinomas
ErbB2/HER2/Neu NRG, EGF overexpression 30% of breast adenocarcinomas
ErbB3, 4 various overexpression oral squamous cell carcinoma
Flt-3 FL tandem duplication acute myelogenous leukemia
Kit SCF amino acid substitutions gastrointestinal stromal tumor
Ret GFL fusion with other proteins; point mutations
papillary thyroid carcinomas; multiple endocrine neoplasias 2A and 2B
FGF-R2 FGF amino acid substitutions breast, gastric, endometrial carcinomas
FGF-R3 FGF overexpression; amino acid substitutions; translocations
multiple myeloma; bladder and cervical carcinomas; acute myelogenous leukemia
PDGF-Rβ PDGF translocations chronic myelomonocytic leukemia
aSee also Figure 5.16.
Tyrosine kinase GF receptors altered in human tumors

11by: Bundit Boonyar i t566-544 Drug target
The Human EGF Receptor (HER)
Flynn et al. (2009)
Epidermal growth factor
4 Journal of Oncology
Trancemembrane
Juxtamembrane
Tyrosine kinase
Regulatory region
Domain I (L1)
Domain II (CR1)
Domain III (L2)
Domain IV (CR2)Extracellular
Extr
acel
lula
r do
mai
ns:
Intr
acel
lula
r do
mai
ns:
Out
In
Intracellular matrix:cell cytosol
Cytoplasmic domain(includes autophosphorylated
tyrosine residues)
Cysteine-richEC domain
(ligand binding site)
N-lobe
L1
L2
NDFG
Leu955
His964
Glycine rich loop
Activation loop
Catalytic loop
CR1
C-lobeY
1
165
310
480
620
643685
953
1186
(a) (c)
(b)
YY
YY
Figure 2: (a) Basic Structure of EGFR demonstrating relevant domains. (I) The extracellular domains: (1) domain I: L1; (2) domain II: CR1;domain III: L2; domain IV: CR2. (II) Transmembrane domains. (III) The intracellular domains (1) juxtamembrane domain; (2) tyrosinekinase domain; (3) regulatory region domain. The phosphorylation of several substrates by the tyrosine kinase domain of the EGFR receptoris responsible for activating the various signaling cascades seen in Figure 1. (b) Structure of domains I–IV of EGFR (no ligand bound). Notethe “protruding loop” in domain II (CR1) directed away from the C-shaped region of the ligand-binding zone formed by domains I, II, andIII. (c) The tyrosine kinase domain of EGFR showing the N-lobe and C-lobe flanking the activation loop and active site cleft [2, 3].
been exploited in the development of small molecule tyrosinekinase inhibitors (smTKIs) for targeting EGFR in variouscancer types including breast cancer.
(4) The Activation Loop, Active Site, and C-Terminal TailRegions of EGFR. The activation loop of the tyrosine kinaseof EGFR is quite distinct from other receptors harboringtyrosine kinases. Namely, whereas most receptor TKDsrequire phosphorylation for tyrosine kinase activation, thisdoes not appear to be the case in EGFR. For example,the phosphorylation of Tyr845 has little affect on theEGFR kinase activity [3], possibly due to a conformationalarrangement that directs the activation loop away fromthe active site rendering it refractory to the state of phos-phorylation of the receptor tyrosine kinase. Activation ofthe EGFR tyrosine kinase phosphorylates numerous targets,including itself (autophosphorylation), a different EGFR(homodimerization), HER-2/neu of the ErbB gene family
(heterodimerization), and nonreceptor substrates such asGrb2/SOS, STATs, PLC, and/or PI3K, which in turn initiatethe signaling cascades of MAPK/ERK, STAT, PIP2, and AKT,respectively. Not surprisingly, therefore, mutations in thisregion can cause a substantial decrease in kinase activity,an outcome considered desirable in cancer therapy and mayunderlie the therapeutic efficacy of smTKIs. By binding tothe TKD of EGFR, smTKIs may act by sterically interferingwith the binding of both the substrate and ATP necessary forphosphorylation, resulting in an overall decreased signalingactivity of the EGFR.
Lastly, it is important to mention that the tyrosinekinase activity of EGFR is tightly regulated via its owninternal regulatory region located at the C-terminal tail ofthe structure, which involves the tyrosine residue cluster withthe potential of being transphosphorylated during EGFR-dimerization. It is noteworthy that EGFR dimerizationinduces phosphorylation of several tyrosine residues includ-ing Tyr1069 Tyr1092 Tyr1110 Tyr1116 Tyr1172 Tyr1197, creating
www.rcsb.org/pdb

12by: Bundit Boonyar i t566-544 Drug target
Cetuximab as anticancer
Bardelli & Jänne (2012)
N E W S A N D V I E W S
200 VOLUME 18 | NUMBER 2 | FEBRUARY 2012 NATURE MEDICINE
discover additional mechanisms12–14. Once these analyses have been completed, the infor-mation could be used to develop a rational framework to guide the treatment of individu-als who relapse while on EGFR-targeting anti-body therapies. There are likely to be multiple mechanisms of drug resistance, each requiring a specific, and often a nonoverlapping, thera-peutic approach. For example, when relapse is linked to the EGFR S492R mutation, the subsequent therapeutic strategy will be likely to involve alternative ways of inhibiting EGFR, including using panitumumab. In cases in which cetuximab resistance is mediated by ERBB2, combination therapies that include an EGFR inhibitor need to be employed, as more
effective EGFR inhibitors, including panitu-mumab, are unlikely to be effective as single therapeutic agents.
In summary, understanding the mecha-nisms of drug resistance through both in vitro models and human tumor tissues can clearly lead to the development of more effective tar-geted therapies, new therapeutic combinations or both. It will be important to personalize such approaches on the basis of the mecha-nisms of resistance of each individual tumor.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
1. Montagut, C. et al. Nat. Med. 18, 221–223 (2012).2. Jonker, D.J. et al. N. Engl. J. Med. 357, 2040–2048
(2007).
3. Vermorken, J.B. et al. N. Engl. J. Med. 359, 1116–1127 (2008).
4. Karapetis, C.S. et al. N. Engl. J. Med. 359, 1757–1765 (2008).
5. De Roock, W. et al. Lancet Oncol. 11, 753–762 (2010).
6. Amado, R.G. et al. J. Clin. Oncol. 26, 1626–1634 (2008).
7. Bardelli, A. & Siena, S. J. Clin. Oncol. 28, 1254–1261 (2010).
8. Wadlow, R.C. et al. Oncologist published online, doi: 10.1634/theoncologist.2011-0452 (30 December 2011).
9. Jänne, P.A., Gray, N. & Settleman, J. Nat. Rev. Drug Discov. 8, 709–723 (2009).
10. Yonesaka, K. et al. Sci. Transl. Med. 3, 99ra86 (2011).11. Bertotti, A. et al. Cancer Discov. 1, 509–523 (2011).12. Benavente, S. et al. Clin. Cancer Res. 15, 1585–1592
(2009).13. Li, C., Iida, M., Dunn, E.F., Ghia, A.J. & Wheeler, D.L.
Oncogene 28, 3801–3813 (2009).14. Lu, Y. et al. Cancer Res. 67, 8240–8247 (2007).
PI3K
AKT
RAS
ERK1/2
Survival Proliferation
EGFR
EGFR ligand
PI3K
AKT
RAS
ERK1/2
Survival Proliferation
a bCetuximab
PI3K
AKT
RAS
ERK1/2
Survival Proliferation
S492Rmutation
PI3K
AKT
RAS
ERK1/2
Survival Proliferation
ERBB2
Heregulin
ERBB3
c d
Figure 1 Cetuximab sensitivity and resistance. (a) EGFR ligands bind the extracellular domain of EGFR, induce receptor dimerization and activate downstream signaling pathways that are crucial for cell survival and proliferation. (b) Cetuximab prevents ligand binding to EGFR, thus blocking EGFR signaling. (c) Montagut et al.1 now show that the EGFR S492R mutation inhibits cetuximab but not EGFR ligand binding to EGFR. (d) Cetuximab resistance can be mediated by activation of alternative signaling pathways. In this scenario, although cetuximab can still block EGFR ligand binding, it does not inhibit downstream signaling. PI3K, phosphoinositide 3-kinase; ERK, extracellular signal-regulated kinase.
epidemics in communities immune to earlier strains1. This is why seasonal influenza vac-cines require frequent updating.
The specificity of the antibody response also underlies the emergence of influenza pandemics. Pandemics occur when gene shuf-fling between animal and human influenza A viruses produces a human-transmissible virus
When it comes to protection from influenza infection, antibodies are best. Neutralizing anti-bodies to the surface glycoproteins of influenza
type A and B viruses, hemagglutinin and neuraminidase, block binding to receptors on the respiratory epithelium, preventing virus uptake and subsequent release of new virus particles (Fig. 1). But influenza viruses move quickly: the selective pressure of the antibody response on seasonal influenza viruses drives the emergence of escape mutants that can cause
Anne Kelso is at the World Health Organization Collaborating Centre for Reference and Research on Influenza, North Melbourne, Victoria, Australia. e-mail: [email protected]
CD4+ T cells limit the damage in influenzaAnne Kelso
Why do some influenza infections cause fatal disease and others barely a sniffle? Although viral virulence can vary, the immunological history of the host is also important. A new study in humans suggests that CD4+ T lymphocytes activated during previous infections can limit disease severity in the absence of specific antibodies (pages 274–280).

13by: Bundit Boonyar i t566-544 Drug target
Cetuximab as anticancer
Graham et. al. (2014)
© 2004 Nature Publishing Group
Cetuximab
CetuximabJoanne Graham, Mohamed Muhsin and Peter Kirkpatrick
FRESH FROM THE PIPELINE
N E W S & A N A L Y S I S
NATURE REVIEWS | DRUG DISCOVERY VOLUME 3 | JULY 2004 | 549
Cetuximab (Erbitux; ImClone Systems/Bristol-Myers Squibb) is a monoclonalantibody that binds to the epidermal growthfactor receptor, which is important in thegrowth of many cancers. In February 2004,it was granted accelerated approval by theFDA for the treatment of metastaticcolorectal cancer on the basis of tumourresponse rates in Phase II trials.
Colorectal cancer is one of the most com-monly diagnosed cancers, and has been esti-mated to be the fourth largest cause of cancerdeaths worldwide1. Recently, there have beenimportant advances in the therapy of this dis-ease, such as the introduction of the cytotoxicdrugs irinotecan (Camptosar; Pfizer) andoxaliplatin (Eloxatin; Sanofi-Synthelabo),and the antibody bevacizumab (Avastin;Genentech), which targets tumour angiogen-esis. Nevertheless, a major need remains fornovel agents for treating colorectal cancer,especially for those patients who fail torespond to current treatments or who developresistance to them.
Basis of discoveryAdvances in the understanding of the aberrantsignalling pathways involved in cancer haveled to considerable efforts to develop thera-pies that target specific elements of thesepathways, in the hope that such agents willhave greater activity and higher specificity fortumour cells than traditional cytotoxic drugs.Receptor tyrosine kinases ⎯ which have keyroles in signalling pathways that regulateprocesses such as cell proliferation, apoptosisand angiogenesis ⎯ have attracted muchattention, as several of them have been stronglyimplicated in tumour growth and progres-sion, including the epidermal growth factorreceptor (EGFR)2,3 (FIG. 1). The EGFR isexpressed in a wide range of solid tumours,including colorectal cancers4, which hasprompted the development of a number ofagents that inhibit its activity2–4.
Drug propertiesCetuximab is a recombinant, human/mousechimeric monoclonal antibody that bindsspecifically to the extracellular domain of thehuman EGFR2,5,6. Bound antibody competi-tively inhibits the binding of epidermal growth
factor (EGF) and other ligands to EGFR,blocking receptor phosphorylation and activa-tion of receptor-associated kinases, therebyinhibiting downstream signal transduction2,5,6
(FIG. 1). In preclinical studies, cetuximabshowed promising anticancer activity bothalone and in combination with traditionalcytotoxic drugs2,5,7, prompting its clinicalevaluation in a range of cancers in whichEGFR is thought to have an important role,including colorectal cancer.
Clinical dataCetuximab was studied in trials involvingpatients with EGFR-expressing metastaticcolorectal cancer, whose disease had pro-gressed after receiving an irinotecan-containingregimen6. In a randomized controlled trialconducted in 329 such patients, patientsreceived either cetuximab (400 mg per m2 initialdose, followed by 250 mg per m2 weekly untildisease progression or unacceptable toxicity)
and irinotecan, or cetuximab alone6. Patientsreceiving cetuximab and irinotecan had anobjective response rate of 22.9%, and thosereceiving cetuximab alone had objectiveresponse rate of 10.8%. The median durationof response was 5.7 months in the combina-tion arm and 4.2 months in the monotherapyarm, and the median time to disease progres-sion was 4.1 months in the combination armand 1.5 months in the monotherapy arm6.
IndicationsCetuximab, used in combination with irino-tecan, is indicated for the treatment of EGFR-expressing, metastatic colorectal carcinoma inpatients who are refractory to irinotecan-basedchemotherapy6.
Cetuximab administered as a single agentis indicated for the treatment of EGFR-expressing, metastatic colorectal carcinoma inpatients who are intolerant to irinotecan-basedchemotherapy6.
TK TK TK TK
Autophosphorylation
Gefitinib
Activation of signal-transductioncascades (for example, MAPK)
Apoptosis Invasion andmetastasis AngiogenesisCell
proliferation
P P
EGFR
Ligand (EGF, TGFα)
Ligand binding
Cetuximab
Figure 1 | EGFR and the mode of action of cetuximab. The epidermal growth factor receptor (EGFR) isone of four members of the erbB family of receptor tyrosine kinases, which consist of an extracellular domainthat can bind ligands, a transmembrane domain and an intracellular tyrosine kinase domain8. A simplifiedillustration of the EGFR signal transduction pathway is shown. Binding of a ligand to EGFR causes receptordimerization (either with another EGFR monomer or with another member of the erbB family), leading totyrosine kinase activation8. The resultant receptor autophosphorylation initiates signal-transduction cascadesinvolved in cell proliferation and survival8. Cetuximab blocks binding of ligands to EGFR, thereby inhibitingreceptor phosphorylation and downstream events. Agents that inhibit the tyrosine kinase activity of EGFRhave also been developed and, of these, one has been approved by the FDA so far: gefitinib (Iressa;AstraZeneca), for the treatment of advanced non-small-cell lung cancer. MAPK, mitogen-activated proteinkinase; TGF-α, transforming growth factor-α; TK, tyrosine kinase domain.

14by: Bundit Boonyar i t566-544 Drug target
Li et al. (2005)
cetuximab
Cetuximab as anticancer
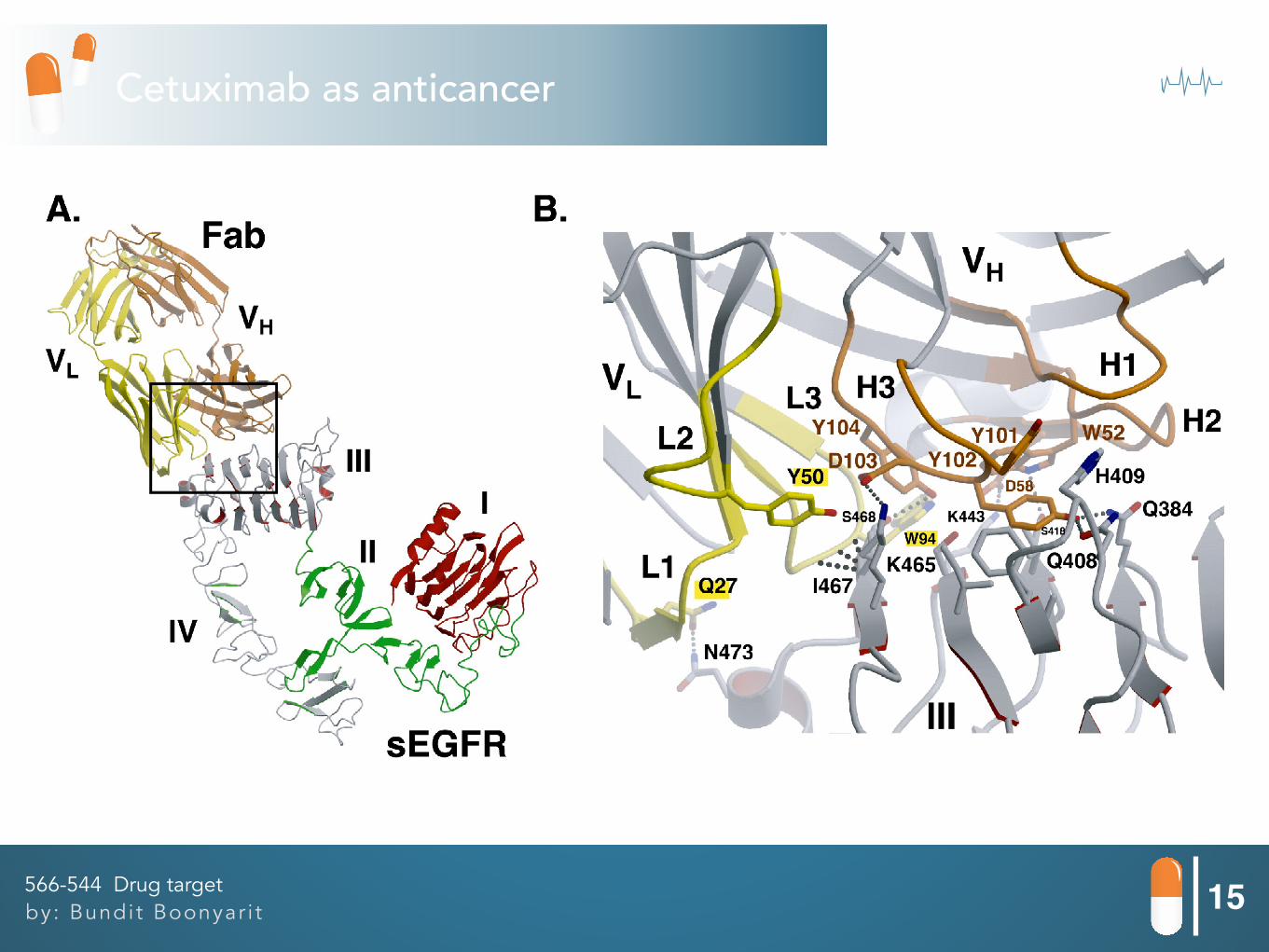
15by: Bundit Boonyar i t566-544 Drug target
Li et al. (2005)
Cetuximab as anticancer
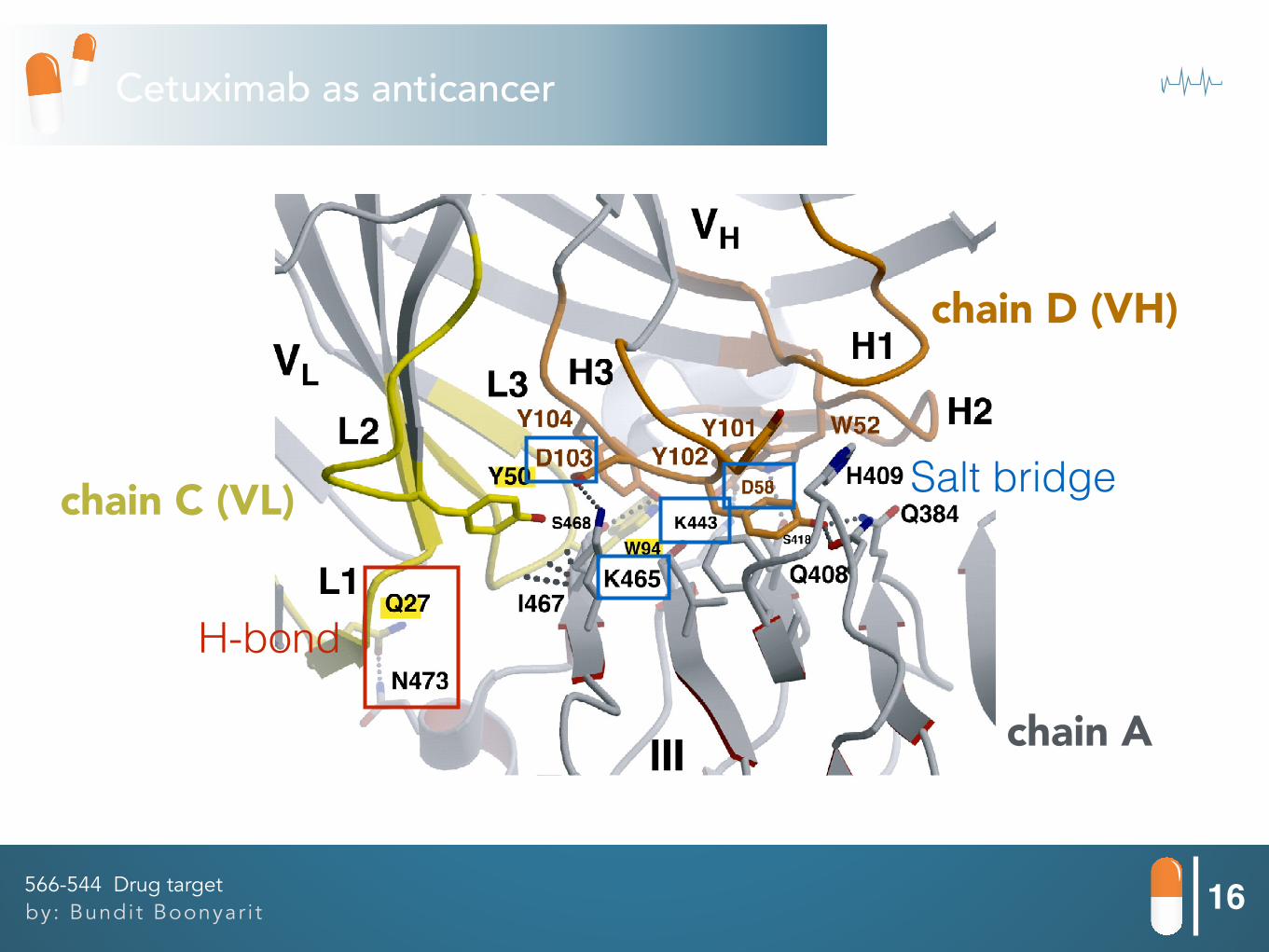
16by: Bundit Boonyar i t566-544 Drug target
Li et al. (2005)
Cetuximab as anticancer
H-bond
Salt bridge
chain A
chain C (VL)
chain D (VH)
PDB: 1YY9 (Let’s go in VMD)

17by: Bundit Boonyar i t566-544 Drug target
Li et al. (2005)
Cetuximab as anticancer

18by: Bundit Boonyar i t566-544 Drug target
Li et al. (2005)
H-bond

19by: Bundit Boonyar i t566-544 Drug target
Li et al. (2005)
Cetuximab as anticancer

20by: Bundit Boonyar i t566-544 Drug target
Li et al. (2005)
Cetuximab as anticancer

References
21
[3] Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., & Forman, D. (2011). Global cancer statistics.
CA: A Cancer Journal for Clinicians, 61(2), 69-90.
[5] Kufe, D. W., Frei, E., Holland, J. F., Society, A. C., & ., N. C. f. B. I. (2003). Holland-Frei Cancer Medicine (6 ed.): B.C. Decker.
[8] Yap, T. A., Sandhu, S. K., Workman, P., & de Bono, J. S. (2010). Envisioning the future of early anticancer drug development. Nat Rev Cancer, 10(7), 514-523.
[7] Weinberg, R. A. (2007). The Biology of Cancer: Garland Science.
[6] Li, S., Schmitz, K. R., Jeffrey, P. D., Wiltzius, J. J. W., Kussie, P., & Ferguson, K. M. (2005). Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell, 7(4), 301-311.
[4] Kirkpatrick, P., Graham, J., & Muhsin, M. (2004). Cetuximab. Nat Rev Drug Discov, 3(7), 549-550.
[2] Flynn, J. F., Wong, C., & Wu, J. M. (2009). Anti-EGFR Therapy: Mechanism and Advances in Clinical Efficacy in Breast Cancer. Journal of Oncology, 2009, 16.
[1] Bardelli, A., & Janne, P. A. (2012). The road to resistance: EGFR mutation and cetuximab. Nat Med, 18(2), 199-200.
by: Bundit Boonyar i t566-544 Drug target

“Thank you for your attention”