staurosporine induces apoptosis of melanoma by both caspase
TRANSCRIPT

Staurosporine induces apoptosis of melanoma by bothcaspase-dependent and -independentapoptotic pathways
Xu Dong Zhang, Susan K. Gillespie, andPeter Hersey
Oncology and Immunology Unit, Newcastle Mater Hospital,Newcastle, New South Wales, Australia
AbstractStaurosporine has long been used in vitro as an initiator ofapoptosis in many different cell types, but the mechanisminvolved remains poorly understood. In the present study,we have examined the apoptosis-inducing potential ofstaurosporine in cultured melanoma cell lines and dissectedthe staurosporine-induced apoptotic signaling pathway.We report that although staurosporine activated Bax andthe mitochondrial caspase-dependent apoptotic pathway,it also induced apoptosis of melanoma by caspase-independent pathways. The caspase-dependent apoptoticpathway was activated relatively soon after exposure tostaurosporine and was associated with release of cyto-chrome c and Smac/DIABLO from mitochondria andcleavage of poly(ADP-ribose) polymerase and inhibitor ofcaspase-activated DNase. This pathway was inhibitable bybroad caspase inhibitors. A second apoptotic pathway thatappeared to be involved in late apoptotic events wascaspase independent in that inhibitors of caspases did notprevent the late onset of apoptosis. Overexpression ofBcl-2 inhibited the early onset of apoptosis but not thelater, caspase-independent pathway. Apoptosis-inducingfactor may be responsible for the late apoptotic executionin that its translocation from mitochondria into the nucleuscoincided with the late onset of apoptosis and could not beinhibited by either a pan-caspase inhibitor or overexpres-sion of Bcl-2. Our results indicate that staurosporine is ableto bypass resistance of melanoma cells to mitochondrialcaspase-dependent apoptotic pathways; hence, deriva-tives of staurosporine may warrant further evaluationeither alone or with other apoptosis-inducing agents. [MolCancer Ther. 2004;3(2):187–197]
IntroductionRecent studies have suggested that many therapeuticagents used against cancer, such as immunotherapy,
chemotherapy, and irradiation, mediate their effects byinduction of apoptosis of the cancer cells (1–3). Threemajor apoptotic pathways originating from three separatesubcellular compartments have been identified as the deathreceptor-mediated pathway, the mitochondrial apoptoticpathway, and the endoplasmic reticulum pathway (4–6).Although each pathway is initially mediated by differentmechanisms, they share a common final phase of apopto-sis, consisting of the activation of the executioner caspasesand dismantling of substrates critical for cell survival (7, 8).
The mechanisms involved in induction of apoptosis bychemotherapeutic agents such as alkylating agents, top-oisomerase inhibitors, and antimitotic agents are believed tobe largely mediated by the mitochondrial apoptoticpathway (1). This involves release of mitochondrialapoptotic proteins such as cytochrome c (9), apoptosis-inducing factor (AIF; 10), second mitochondrial-derivedactivator of caspase/direct inhibitor of apoptosis (IAP)protein binding protein with low pI (Smac/DIABLO;11–12), endonuclease G (13) and Omi1/HtrA2 (14, 15).On release, cytochrome c interacts with apoptotic protein-ase-activating factor-1 and pro-caspase-9 to form apopto-somes. The latter activates caspase-9 and downstreameffector caspases such as caspase-3 that are responsible forapoptotic destruction of the cells (9, 16). In contrast, AIF andendonuclease G translocate directly to the nucleus wherethey induce chromatin condensation and/or DNA frag-mentation (10, 13). Cytosolic Smac/DIABLO and Omi1/HtrA2 mediate apoptosis by binding to IAP protein familymembers that inhibit activation of caspase-9 and inhibit theactivity of activated caspase-3 (11, 12, 14). Although themechanism(s) that underlies the release of mitochondrialapoptotic proteins remains uncertain, the Bcl-2 familymembers play a central role in regulating changes in mito-chondrial outer membrane permeability (17–20). Studieshave shown that the antiapoptotic Bcl-2 family memberssuch as Bcl-2, Bcl-XL, and Mcl-1 appear to preserve the in-egrity of the outer mitochondrial membrane by binding tomitochondrial porin channels (17, 18). Apoptosis proceedswhen proapoptotic BH3-only proteins such as Bid, Bim, andNoxa bind to the antiapoptotic Bcl-2 family members andpromote binding of the multidomain proapoptotic proteinsBax and Bak to the mitochondrial outer membrane, wherethey initiate changes in mitochondrial outer membranepermeability. Overexpression of Bcl-2 inhibits the mito-chondria-dependent pathway to apoptosis (19, 20).
Staurosporine, a protein kinase inhibitor, has beencharacterized as a strong inducer of apoptosis in manydifferent cell types. The mechanism(s) by which staurospor-ine induces apoptosis, however, remains controversial.Although it is generally believed that the mitochondrialapoptotic pathway plays a critical role in staurosporine-induced apoptosis (21, 22), some studies found that Bcl-2
Received 7/31/03; revised 10/13/03; accepted 10/28/03.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
Requests for Reprints: Peter Hersey, Oncology and Immunology Unit,Newcastle Mater Hospital, David Maddison Clinical Sciences Building,Room 443, Corner King and Watt Streets, Newcastle, New South Wales2300, Australia. Phone: 61-2-49-236828; Fax: 61-2-49236184.E-mail: [email protected]
Molecular Cancer Therapeutics 187
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

overexpression was ineffective in protecting cells fromkilling by staurosporine (23). Similarly, while most reportsshowed a requirement for caspase activation in staurospor-ine-induced apoptosis, caspase-independent mechanism(s)was also suggested (24, 25). Multiple mechanisms maytherefore be involved in staurosporine-induced apoptosisand these may vary between different cell types.
Melanoma continues to increase in incidence in manyparts of the world and remains among the top six cancersas a cause of death and morbidity. Treatment of melanomaonce it has spread beyond the skin remains unsatisfactory.This is largely due to its unresponsiveness to availablechemotherapeutic and biologic reagents, which has been at-tributed to development of resistance to apoptosis (26, 27).Understanding and overcoming resistance mechanism(s)of melanoma to apoptosis would therefore facilitate iden-tification of new therapeutic targets and development ofnew treatments.
In the present study, we examined the apoptosis-inducing potential of staurosporine in cultured melanomacell lines and dissected the staurosporine-induced apopto-tic signaling pathway. We report that staurosporineinduced relatively high levels of apoptosis in the majorityof melanoma cell lines through both caspase-dependentand -independent pathways. While caspases play adetermining role in early apoptotic responses, AIF maybe involved in apoptotic execution at late stages afterstaurosporine treatment. Although the mitochondrial apo-ptotic pathway was activated by staurosporine, over-expression of Bcl-2 could only delay and partially inhibitstaurosporine-induced apoptosis. This suggests that staur-osporine induces apoptosis of melanoma by nonconven-tional mitochondrial apoptotic pathways and the latter maybe useful to exploit in treatment of melanoma resistant tothe conventional apoptotic pathway.
Materials andMethodsCell LinesHuman melanoma cell lines Me4405, Me1007, IgR3,
Mel-FH, Mel-RMu, Mel-RM, Mel-CV, and MM200 havebeen described previously (28, 29). The cell lines werecultured in DMEM containing 5% FCS (CommonwealthSerum Laboratories, Melbourne, Victoria, Australia). Mel-anocytes were kindly provided by Dr. P. Parson (Queens-land Institute of Medical Research, Brisbane, Queensland,Australia) and cultured in medium supplied by Clonetics(Edward Keller Australia, Hallam, Victoria, Australia).
Antibodies, Recombinant Proteins, and OtherReagents
Staurosporine was purchased from Sigma Chemical Co.(Castle Hill, New South Wales, Australia). It was dissolvedin DMSO and made up in a stock solution of 1 mM. Thecell-permeable pan-caspase inhibitor Z-Val-Ala-Asp(OMe)-CH2F (z-VAD-fmk), the caspase-3 specificinhibitor Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-CH2F(z-DEVD-fmk), the caspase-9 specific inhibitor Z-Leu-Glu(OMe)-His-Asp(OMe)-CH2F (z-LEHD-fmk), the cas-
pase-8 specific inhibitor Z-Ile-Glu(OMe)-Thr-Asp(OMe)-CH2F (z-IETD-fmk), and the caspase-2 specific inhibitorZ-Val-Ala-Asp(OMe)-Val-Ala-Asp(OMe)-CH2F (z-VDVAD-fmk) were purchased from Calbiochem (La Jolla, CA). Theantioxidant g-glutamylcysteinylglycine (GSH) was pur-chased from Sigma Chemical Co. (St. Louis, MO). Therabbit polyclonal antibodies (Abs) against caspase-3,caspase-8, and Bid, the mouse monoclonal Abs (mAbs)against cytochrome c and poly(ADP-ribose) polymerase(PARP), and the rabbit mAb against the active form ofcaspase-3 were purchased form PharMingen (Bioclone,Marrickville, New South Wales, Australia). The rabbitpolyclonal Abs against inhibitor of caspase-activatedDNase (ICAD) and the mouse mAbs against Bcl-2 werepurchased from Santa Cruz Biotechnology (Santa Cruz,CA). The rabbit polyclonal Ab against Smac/DIABLO wasfrom Calbiochem. The rabbit polyclonal Ab againstcleaved caspase-9 was purchased from New EnglandBiolabs (Beverly, MA). The rabbit polyclonal Ab againstcaspase-2 was from R&D Systems, Inc. (Minneapolis, MN).The rabbit polyclonal anti-Bax against amino acids 1–20was purchased from Upstate Biotechnology, Inc. (LakePlacid, NY). Isotype control Abs used were the ID4.5mouse IgG2a mAb against Salmonella typhi supplied byDr. L. Ashman (Institute for Medical and VeterinaryScience, Adelaide, South Australia, Australia), the 107.3mouse IgG1 mAb purchased from PharMingen (San Diego,CA), and the rabbit IgG from Sigma Chemical (Castle Hill,New South Wales, Australia).
PlasmidVector andTransfectionStable Mel-RM transfectants of Bcl-2 were established by
electroporation of the PEF-puro vector carrying humanBcl-2 provided by Dr. David Vaux (Walter and Eliza HallInstitute, Melbourne, Victoria, Australia) and describedelsewhere (29).
Flow CytometryImmunostaining on intact and permeabilized cells was
carried out as described previously (28). Analysis wascarried out using a Becton Dickinson (Mountain View, CA)FACScan flow cytometer. The percentage of antigen-positive cells was calculated as the difference in positivearea between positive and negative control histograms. Thepositive area was that to the right of the intersection of thetwo curves.
ApoptosisApoptotic cells were determined by the propidium
iodide method as described elsewhere (28).
Mitochondrial Membrane PotentialTumor cells were cultured in 24-well plates and allowed to
reach exponential growth for 24 h before treatment.MitoTracker Red CMXRos (Molecular Probes, Eugene, OR)was added at 100 nM during the last 30 min of treatment. Themedium was removed into a 75-mm Falcon polystyrene tube(Becton Dickinson, Sunnyvale, CA), and the adherent cellswere trypsinized and collected into the same tube. Afterwashing with PBS, the cells were analyzed using a FACScanflow cytometer (Becton Dickinson, Sunnyvale, CA) forMitoTracker uptake. Untreated cells were used as controls.
Apoptosis of Melanoma Induced by Staurosporine188
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from
Western Blot AnalysisMethods used were as described previously (28), with
minor modification. Briefly, the protein content of cellextracts was determined by the Bradford assay (Bio-Rad,Sydney, New South Wales, Australia). A total of 20–30 Ag ofprotein was electrophoresed on 10–15% SDS-PAGE gels andtransferred to nitrocellulose membranes. Membranes wereblocked, incubated with primary Abs at the appropriateconcentration, and subsequently incubated with horserad-ish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (1:3000 dilution; Bio-Rad). Labeled bands weredetected by Renaissance Western Blot ChemiluminescenceReagent (New England Nuclear Life Science Products,Boston, MA) and exposed on Hyper MP autoradiographyfilm (Amersham, Castle Hill, New South Wales, Australia).
ImmunofluorescenceMicroscopyMelanoma cells were seeded onto sterile glass coverslips
in 24-well plates (Falcon 3047; Becton Dickinson, LaneCove, New South Wales, Australia) 16 – 24 h beforetreatment. In some experiments, MitoTracker Red CMXRos(50 nM; Molecular Probes) was added to the culturemedium for 30 min before washing cells with PBS followedby fixation with 2% paraformaldehyde for 10 min. Cellswere then permeabilized in 0.05% saponin diluted in PBScontaining 10% human AB serum. After incubating with amouse mAb against AIF (Santa Cruz Biotechnology) at5 Ag/ml at 4jC for 45 min, the cells were washed with PBScontaining 0.05% saponin followed by incubating withAlexa 488 anti-mouse secondary Ab (1:400; MolecularProbes). In some experiments, the cells were incubatedwith 4V,6-diamidino-2-phenylindole (DAPI; MolecularProbes) at 300 nM for 5 min. Coverslips were mounted inGel-Mount (Biomeda, Foster City, CA) and examined usinga Zeiss Axiophot microscope (Oberkochem, Germany).
PreparationofMitochondrial and Cytosolic FractionsMethods used for subcellular fraction were similar to the
methods described previously (29).
Measurement of Reactive Oxygen SpeciesGeneration
Generation of reactive oxygen species (ROS) wasmonitored by measurement of hydrogen peroxide genera-tion. Cells that were seeded in 24-well plates overnightwith or without treatment with staurosporine wereincubated with the fluorescent probe 2V,7V-dichlorofluores-cein diacetate (DCF-DA; Sigma Chemical, St. Louis, MO)for 30 min. The medium was removed to a 75-mm Falconpolystyrene tube and the adherent cells were trypsinizedand collected into the same tube. After washing twice withPBS, the intensity of DCF-DA fluorescence was determinedby using a FACScan flow cytometer (Becton Dickinson,Sunnyvale, CA), with an excitation wavelength of 480 nmand an emission wavelength of 530 nm.
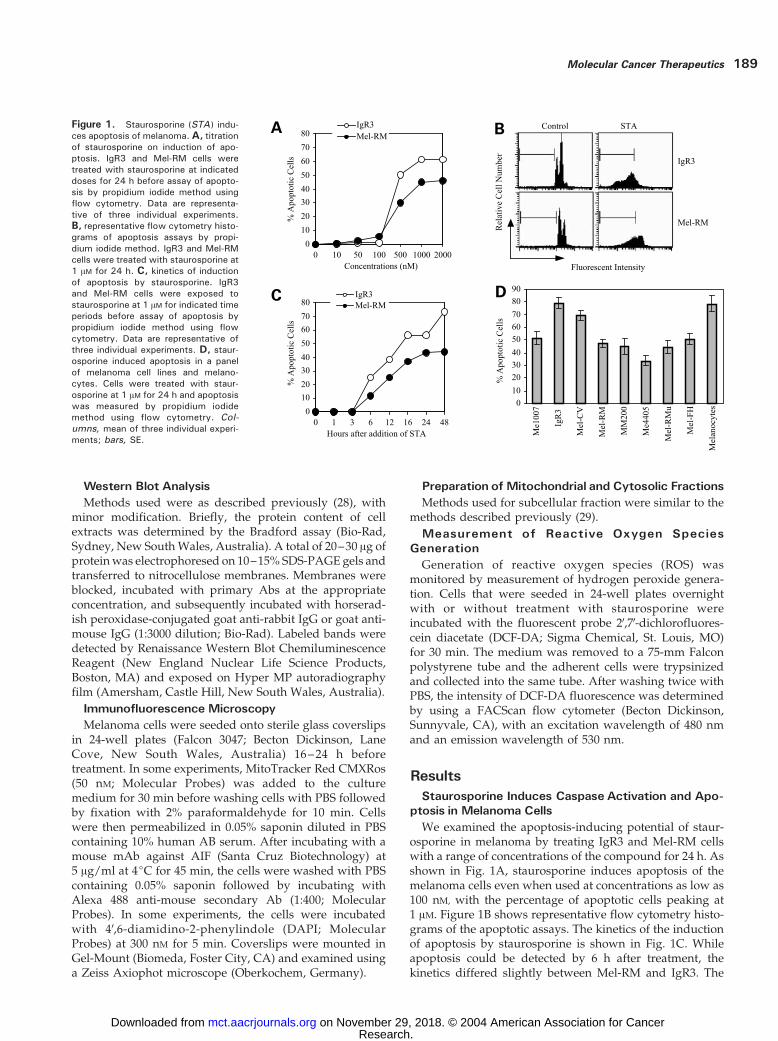
ResultsStaurosporine Induces Caspase Activation and Apo-
ptosis inMelanoma CellsWe examined the apoptosis-inducing potential of staur-
osporine in melanoma by treating IgR3 and Mel-RM cellswith a range of concentrations of the compound for 24 h. Asshown in Fig. 1A, staurosporine induces apoptosis of themelanoma cells even when used at concentrations as low as100 nM, with the percentage of apoptotic cells peaking at1 AM. Figure 1B shows representative flow cytometry histo-grams of the apoptotic assays. The kinetics of the inductionof apoptosis by staurosporine is shown in Fig. 1C. Whileapoptosis could be detected by 6 h after treatment, thekinetics differed slightly between Mel-RM and IgR3. The
Figure 1. Staurosporine (STA ) indu-ces apoptosis of melanoma. A, titrationof staurosporine on induction of apo-ptosis. IgR3 and Mel-RM cells weretreated with staurosporine at indicateddoses for 24 h before assay of apopto-sis by propidium iodide method usingflow cytometry. Data are representa-tive of three individual experiments.B, representative flow cytometry histo-grams of apoptosis assays by propi-dium iodide method. IgR3 and Mel-RMcells were treated with staurosporine at1 AM for 24 h. C, kinetics of inductionof apoptosis by staurosporine. IgR3and Mel-RM cells were exposed tostaurosporine at 1 AM for indicated timeperiods before assay of apoptosis bypropidium iodide method using flowcytometry. Data are representative ofthree individual experiments. D, staur-osporine induced apoptosis in a panelof melanoma cell lines and melano-cytes. Cells were treated with staur-osporine at 1 AM for 24 h and apoptosiswas measured by propidium iodidemethod using flow cytometry. Col-umns, mean of three individual experi-ments; bars, SE.
Molecular Cancer Therapeutics 189
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

percentage of apoptotic cells in the former peaked at 24h after treatment, whereas peak apoptosis of the latter wasobserved at 48 h. A summary of studies on a panel ofmelanoma cell lines and melanocytes treated with staur-osporine at 1 AM for 24 h is shown in Fig. 1D. Staurosporineinduced apoptosis in all the melanoma cell lines, with thepercentage of apoptotic cells ranging from 33% in Me4405to 79% in IgR3. Staurosporine induced apoptosis in about78% of melanocytes.
To study if caspase activation was involved in stauro-sporine-induced apoptosis, we firstly examined caspase-3activation in IgR3 and Mel-RM cells by flow cytometry usinga mAb that specifically recognizes the active form ofcaspase-3. Figure 2A shows representative flow cytometryhistograms of activated caspase-3 and Fig. 2B shows thekinetics of caspase-3 activation. Caspase-3 activation couldbe detected by 3 h with peak activation at 16 h after exposureto staurosporine. Activation of caspase-3 by staurosporinewas confirmed by Western blot analysis as shown in Fig. 2C.
The functional activity of the activated caspase-3 wasexamined against two caspase-3 substrates, ICAD andPARP. As shown in Fig. 2C, ICAD expression was reducedby 6 h and was barely detectable by 16 h in both Mel-RM andIgR3 cells after staurosporine treatment. Similarly, PARPcleavage was also detected by 6 h with the appearance of thecleaved 85-kDa fragment after exposure to staurosporine. At16 h, only the cleaved form of PARP was detectable. Figure2C also shows the effect of treatment with staurosporine onX-linked IAP (XIAP) expression, which was shown to becleaved by activated caspase-3 during apoptosis (33). XIAPexpression was decreased at 6 h and was no longerdetectable by 16 h after exposure to staurosporine.
We examined possible involvement of caspase-8, -9, and-2 in staurosporine-induced apoptosis by Western blotanalysis before and after exposure to the compound. As canbe seen from Fig. 2C, pro-caspase-8 expression remainedunaltered until 6 h after treatment with staurosporine. By
16 h, pro-caspase-8 expression was no longer detectable,but there was no apparent appearance of the active form ofcaspase-8 (data not shown). The activation status ofcaspase-9 was examined by using an Ab that specificallyrecognizes the cleaved p38 fragment of cleaved caspase-9.Treatment with staurosporine for 6 h resulted in expressionof the cleaved caspase-9, and there was a marked increasein the expression levels by 16 h after the addition ofstaurosporine. Figure 2C also shows that pro-caspase-2 wasalmost completely cleaved as early as 6 h after treatmentwith staurosporine.
Staurosporine Induces Apoptosis of Melanomathrough Caspase-Dependent and -IndependentPathways
To confirm the role of caspase activation in staurospor-ine-induced apoptosis of melanoma, we treated IgR3 andMel-RM cells with the pan-caspase inhibitor, z-VAD-fmk, 1h before adding staurosporine at 1 AM for a further 24 h.Figure 3A shows that while z-VAD-fmk completelyinhibited apoptosis of melanoma induced by tumornecrosis factor (TNF)-related apoptosis-inducing ligand(TRAIL), a member of the TNF family that is known toinduce apoptosis in the melanoma cell lines (28, 29), it onlypartially blocked staurosporine-induced apoptosis. Thissuggests that both caspase-dependent and -independentpathways were induced by staurosporine in melanomacells. To further confirm this, we treated Mel-RM cells withz-VAD-fmk 1 h before adding staurosporine for differingtime periods as indicated in Fig. 3B. Staurosporine-inducedapoptosis was markedly delayed by z-VAD-fmk, withnegligible apoptosis being detected at 6 h. The levels ofapoptosis at 12, 16, 24, and 48 h after treatment withstaurosporine in the presence of z-VAD-fmk were mark-edly decreased.
The role of caspase-3, -8, -9, and -2 in staurosporine-induced apoptosis was further studied by using specificinhibitors. Mel-RM and IgR3 cells, were treated with the
Figure 2. Staurosporine (STA) indu-ces caspase activation. A, representa-tive flow cytometry histograms ofassays of caspase-3 activation inducedby staurosporine. Mel-RM and IgR3 cellswere treated with staurosporine (1 AM)for 6 h before activated form of caspase-3 was measured in permeabilized cellsusing flow cytometry. B, kinetics ofstaurosporine-induced caspase-3 activa-tion. Mel-RM and IgR3 cells were treatedwith staurosporine (1 AM) for indicatedtime periods. Caspase-3 activationwasmeasured as described in A. Data arerepresentative of three individual experi-ments. C, staurosporine induced cleav-age of pro-caspase-3, -8, -9, and -2 andICAD, PARP, and XIAP. Mel-RM andIgR3 cells were treated with staurospor-ine (1 AM) for indicated time periods.Whole cell lysates were subjected toWestern blot analysis. Western blotanalysis of h-Actin levels was includedto show that equivalent amounts ofprotein were loaded in each lane.
Apoptosis of Melanoma Induced by Staurosporine190
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

caspase-3 specific inhibitor z-DEVD-fmk, the caspase-9 spe-cific inhibitor z-LEHD-fmk, the caspase-8 specific inhibitorz-IETD-fmk, or the caspase-2 specific inhibitor z-VDVAD-fmk 1 h before adding staurosporine for a further 24 h.As shown in Fig. 3C, pretreatment with z-DEVD-fmk,z-LEHD-fmk, or z-VDVAD-fmk resulted in partial re-duction in staurosporine-induced apoptosis, whereas pre-treatment with z-IETD-fmk, the caspase-8 inhibitor, hadonly a minor effect on the percentage of apoptotic cells.There was no significant difference in the degrees ofreduction of staurosporine-induced apoptosis producedby z-DEVD-fmk, z-LEHD-fmk, or z-VDVAD-fmk (P > 0.05,Fisher’s exact test).
Staurosporine Activates the Conventional Mito-chondrial Apoptotic Pathway
To study the potential effects of staurosporine onthe mitochondrial apoptotic pathway, we measured the
Figure 3. Effects of inhibition of caspases on staurosporine-inducedapoptosis of melanoma. A, inhibition of staurosporine- and TRAIL-inducedapoptosis by a pan-caspase inhibitor, z-VAD-fmk. Mel-RM and IgR3 cellswere treated with z-VAD-fmk (20 AM) 1 h before adding TRAIL (200 ng/ml)or staurosporine (1 AM) for another 24 h. Apoptosis was measured bypropidium iodide method using flow cytometry. Columns, mean of threeindividual experiments; bars, SE. B, effects of z-VAD-fmk on apoptosisinduced by staurosporine at different staurosporineges. Mel-RM cellswere treatedwith z-VAD-fmk (20 AM) 1 h before adding staurosporine (1 AM)for indicated time periods. Apoptosis was measured by propidium iodidemethod using flow cytometry. Data are representative of three individualexperiments. C, effects of inhibition of caspase-3, -9, -2, and -8 onstaurosporine-induced apoptosis. IgR3 and Mel-RM cells were treated withthe caspase-3 specific inhibitor z-DEVD-fmk (30 AM), the caspase-9 specificinhibitor z-LEHD-fmk (30 AM), the capsase-2 specific inhibitor z-VDVAD-fmk (50 AM), and the caspase-8 specific inhibitor z-IETD-fmk (30 AM) 1 hbefore adding staurosporine (1 AM) for another 24 h. Apoptosis wasmeasured by propidium iodide method using flow cytometry. Columns,mean of three individual experiments; bars, SE.
Figure 4. Staurosporine induces changes in MMP. A, induction ofchanges in the DCm by staurosporine in Mel-RM and IgR3 lines. Cells weretreated with staurosporine (1 AM) for indicated time periods. The DCm wasmeasured by uptake of theMitoTracker Red CMXRos using flow cytometry.Data are representative of three individual experiments. B, staurosporine-induced changes in DCm are independent of caspases. Mel-RM (left ) andIgR3 (right) cells were treated with z-VAD-fmk (20 AM) 1 h before addingstaurosporine (1 AM) for indicated time periods. The DCm was measured asdescribed in A. Columns, mean of three individual experiments; bars, SE.C, staurosporine-induced release of cytochrome c and Smac/DIABLO frommitochondria to the cytosol. Mel-RM and IgR3 cells were treated withstaurosporine (1 AM) for 6 h before harvest. Mitochondrial and cytosolicfractions were subjected to Western blot analysis. Data are representativeof two individual experiments. Western blot analysis of COX IV levels wasincluded to show relative purity of mitochondrial fractions.
Molecular Cancer Therapeutics 191
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

mitochondrial membrane potential (DCm) in melanomacells treated with staurosporine. Figure 4A shows thattreatment of melanoma cells with staurosporine inducedchanges in the DCm in melanoma cells, which could bedetected at 3 h and peaked at 6 h after exposure tostaurosporine. The reduction in DCm was markedlydecreased by 12 h and was barely detectable at 16 h aftertreatment. The changes in DCm induced by staurosporineappeared caspase independent in that pretreatment of cellswith z-VAD-fmk before adding staurosporine had anegligible effect as shown in Fig. 4B.
The induction of changes in mitochondrial membranepermeability (MMP) by staurosporine was further con-firmed by studying the release of cytochrome c and Smac/DIABLO from mitochondria into the cytosol in Mel-RMand IgR3 cells as shown in Fig. 4C. Cytochrome c andSmac/DIABLO were localized exclusively in mitochondrial
fractions before treatment, but after exposure to stauro-sporine for 6 h, both cytochrome c and Smac/DIABLOwere observed in the cytosolic fractions with acorresponding decrease in the mitochondrial fractions.
We next studied the possible role of AIF in staurospor-ine-induced apoptosis by immunofluorescence microscopy.As shown in Fig. 5, before treatment, punctuate cytoplas-mic staining of AIF was predominantly colocalized withmitochondria that were labeled by CMXRos. At 6 h afterexposure to staurosporine, there was a marked decrease incellular volume that was associated with perinuclearcondensation of mitochondria in most of the cells. AIFstaining at this stage was still primarily colocalized withmitochondria. In contrast, AIF staining at 16 h after expo-sure to staurosporine appeared predominantly associatedwith the nucleus, as indicated by colocalization with nucleiidentified by DAPI labeling. Figure 5 also shows that
Figure 5. Staurosporine indu-ces translocation of AIF from mi-tochondria to nucleus. A, Mel-RMcells growing on coverslips weretreated with staurosporine (1 AM)for indicated time periods. Forlabeling of mitochondria, the Mito-Tracker Red CMXRos was addedfor the last 30 min of treatment.Cells were then incubated with anAb against AIF followed by label-ing with the Alexa 488 secondaryAb. For nuclear labeling, cells werethen incubated with DAPI. Cover-slips were mounted and examinedusing a fluorescence microscope.B, translocation of AIF induced bystaurosporine is independent ofcaspases. Mel-RM cells were trea-ted with z-VAD-fmk (20 AM) 1 hbefore adding staurosporine (1 AM)for another 16 h. Cells were thenstained as described in A.
Apoptosis of Melanoma Induced by Staurosporine192
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

pretreatment of cells with z-VAD-fmk could not inhibitstaurosporine-induced relocation of AIF from mitochondriainto nuclei. This suggests that translocation of AIF inducedby staurosporine is a caspase-independent process.
Staurosporine Induces Conformational Changesof Bax and Its Relocation from the Cytosol toMitochondria
Bax translocation from the cytosol to mitochondria isbelieved to play a key role in mitochondrion-mediatedapoptosis induced by a variety of apoptotic stimuli (22, 31).Bax translocation involves a conformation change thatexposes the NH2 terminus and the hydrophobic COOHterminus that targets mitochondria (31, 32). The NH2-terminal region is occluded in intact cells and hence is notavailable for binding by Bax-NH2-terminal epitope-specificAbs (31, 32). We studied the conformational status of Bax inmelanoma cells with or without exposure to staurosporineby using an Ab directed against the NH2-terminal region ofBax in flow cytometry. As shown in Fig. 6A, a population ofthe cells was positive for this Ab with weak stainingintensity before treatment. After exposure to staurosporinefor 6 h, there was a marked increase in the levels ofconformationally changed Bax.
We next studied the role of Bax in staurosporine-inducedapoptosis of melanoma by examining the expression of Baxin different subcellular fractions of IgR3 and Mel-RM cellswith or without exposure to staurosporine. As shown inFig. 6B, the Bax protein was predominantly in the cytosol
with only a negligible amount being detected in themitochondrial fractions both before and at 3 h aftertreatment. In contrast, a considerable amount of Bax wasobserved in the mitochondrial fractions with acorresponding decrease in the levels of expression in thecytosol after treatment with staurosporine for 6 h. This wasmost evident in the staurosporine-sensitive IgR3 cells.
Overexpression of Bcl-2 Delays staurosporine-Induced Apoptosis
To further study the role of mitochondria in staurospor-ine-induced apoptosis, we transfected cDNA encoding Bcl-2 into Mel-RM cells. Bcl-2 expression was measured byWestern Blot analyses as shown in Fig. 7A. There was amarked increase in the levels of Bcl-2 in the Bcl-2-trans-fected cells, but the levels in the cells transfected with thevector alone were similar to those in the parental cells. Asshown in Fig. 7B, apoptosis of melanoma induced byTRAIL, which is known to induce apoptosis of melanomapredominantly through the mitochondrial apoptotic path-way (29), was nearly completely inhibited in the Bcl-2transfectants. In contrast, the percentage of apoptotic cellsafter treatment with staurosporine for 24 h was onlypartially decreased in Bcl-2 transfectants compared withcells transfected with the vector alone. Figure 7C shows thatstaurosporine-induced changes in the DCm were reversedin Bcl-2-transfected cells. Similarly, staurosporine-inducedcaspase-3 activation was also inhibited by Bcl-2 over-expression (Fig. 7D).
Given that apoptosis induced by staurosporine atdifferent stages may be associated with different apoptoticsignaling pathways, we studied the effects of overexpres-sion of Bcl-2 on apoptosis at different time points afterexposure to staurosporine. Mel-RM cells transfected withBcl-2 were treated with staurosporine for indicated timeperiods as shown in Fig. 7E. staurosporine-inducedapoptosis was markedly delayed in Bcl-2 transfectantswith significant apoptosis only being detected at 16h compared with 6 h in cells transfected with the vectoralone. The levels of staurosporine-induced apoptosis weremarkedly decreased in the Bcl-2 transfectants with thepercentages of apoptotic cells reaching about 50–70% ofthose in the control cells at 16, 24, and 48 h after treatmentwith staurosporine.
We next studied the effects of overexpression of Bcl-2 onstaurosporine-induced translocation of AIF from mitochon-dria into the nucleus. As shown in Fig. 7F, at 16 h afterexposure to staurosporine, AIF staining in Bcl-2 trans-fectants displayed a diffuse perinuclear staining pattern.Weak diffuse staining was also detected within the nucleus.In contrast, by 24 h after exposure to staurosporine, nuclearstaining pattern of AIF was predominant.
To further understand the mechanism(s) by whichstaurosporine induces changes in mitochondria, we treatedMel-RM and IgR3 cells with staurosporine or TRAIL in thepresence of cyclosporine A (Cyc.A), which is believed to beable to inhibit mitochondrial permeability transition in-duced by various stimuli and thus blocking apoptoticsignaling (33, 34). As shown in Fig. 7G, while Cyc.A
Figure 6. Staurosporine induces a conformational change in Bax andinduces its relocation from the cytosol to mitochondria. A, representativeflow cytometry histograms of assays of induction of a Bax conformationchange by suberic bishydroxamic acid. Mel-RM and IgR3 cells with orwithout treatment with staurosporine (1 AM) for 6 h were subjected to flowcytometry analyses using a Bax-NH2-terminal epitope-specific Ab inpermeabilized cells. B, relocation of Bax from the cytosol to mitochondria.Mel-RM and IgR3 cells were treated with staurosporine (1 AM) for indicatedtime periods before harvest. The mitochondrial and cytosolic fractionswere subjected to Western blot analyses. Data are representative of twoindividual experiments.
Molecular Cancer Therapeutics 193
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

Figure 7. Effects of overexpression of Bcl-2 on staurosporine-induced apoptotic events.A, Bcl-2 was overexpressed in Mel-RM cells transfected with thecDNA encoding Bcl-2 but not in the cells transfected with the vector alone. Whole cell lysates were subjected to Western blot analysis. Data arerepresentative of two individual experiments.B, overexpression of Bcl-2 partially inhibited apoptosis induced by staurosporine.Mel-RM cells transfectedwithcDNA for Bcl-2 or vector alone were treated with staurosporine (1 AM) for 24 h before assay of apoptosis by propidium iodide method using flow cytometry.Columns, mean of three individual experiments; bars, SE. C, overexpression of Bcl-2 inhibited changes in the DCm induced by staurosporine. Mel-RM cellstransfected with cDNA for Bcl-2 or vector alone were treated with staurosporine (1 AM) for 6 h before the DCm was measured by uptake of the MitoTrackerRed CMXRos using flow cytometry. Columns, mean of three individual experiments; bars, SE. D, overexpression of Bcl-2 inhibited caspase-3 activationinduced by staurosporine. Mel-RM cells transfected with cDNA for Bcl-2 or vector alone were treated with staurosporine (1 AM) for 6 h before activated formof caspase-3 was measured in permeabilized cells using flow cytometry. Columns, mean of three individual experiments; bars, SE. E, overexpression ofBcl-2 delayed staurosporine-induced apoptosis. Mel-RM cells transfected with cDNA for Bcl-2 or vector alone were treated with staurosporine (1 AM) forindicated time periods before assay of apoptosis by propidium iodide method using flow cytometry. Data are representative of three individual experiments.F, overexpression of Bcl-2 delayed staurosporine-induced translocation of AIF. Mel-RM cells transfected with cDNA for Bcl-2 that were grown on coverslipswere treated with staurosporine (1 AM) for indicated time periods. Cells were then incubated with an Ab against AIF followed by labeling with the Alexa 488secondary Ab. For nuclear labeling, cells were then incubated with DAPI. Coverslips were mounted and examined using a fluorescence microscope (�400).G, staurosporine-induced apoptosis of melanoma could not be inhibited by Cyc.A. Mel-RM and IgR3 cells with or without pretreatment with Cyc.A (5 AM) for1 h were treated with staurosporine (1 AM) or TRAIL (200 ng/ml) for another 24 h. Apoptosis was measured by the propidium iodide method using flowcytometry. Data are representative of three individual experiments.
Apoptosis of Melanoma Induced by Staurosporine194
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

partially inhibited TRAIL-induced apoptosis, it had noeffect on staurosporine-induced apoptosis at 24 h aftertreatment. Similar results were obtained when cells weretreated with staurosporine for 6 h in the presence of Cyc.A(data not shown). This indicates that staurosporine-induced changes in mitochondria of melanoma cells maybe Cyc.A insensitive (33).
Staurosporine-Induced Apoptosis ofMelanoma Is In-dependent of the Generation of ROS
We studied if the treatment of melanoma cells withstaurosporine may increase the generation of ROS, as itdoes in several other cell types, by using DCF-DA in flowcytometry analysis. As shown in Fig. 8A, the levels of DCF-DA fluorescence in Mel-RM and IgR3 cells treated withstaurosporine were markedly increased, which could beclearly detected at 3 h and maintained until 16 h afterexposure to staurosporine. To study if increased produc-tion of ROS may play a role in staurosporine-inducedapoptosis of melanoma, we treated Mel-RM and IgR3 cells
with the antioxidant, GSH, 2 h before adding staurosporinefor a further 24 h. Figure 8B shows that pretreatment withGSH had negligible effects on staurosporine-inducedapoptosis, while it markedly decreased staurosporine-induced production of ROS (Fig. 8C). This suggests thatthe staurosporine-induced generation of ROS does not playa significant part in staurosporine-induced apoptosis ofmelanoma.
DiscussionStaurosporine has long been used in vitro as an initiator ofapoptosis in many different cell types, but the mechanisminvolved remains poorly understood. A growing body ofevidence suggests that staurosporine differs from otherapoptotic agents such as chemotherapeutic drugs anddeath-inducing ligands in that it induces apoptotic celldeath in tumor cells normally resistant to these agents (24,25, 35). We show in the present study that staurosporineinduces relatively high levels of apoptosis in all themelanoma cell lines including those resistant to TRAIL, amember of the TNF family that induces apoptosis in abouttwo-thirds of melanoma cell lines (28, 29). At least twoapoptotic pathways appeared to be responsible forinduction of apoptosis by staurosporine.
Firstly, staurosporine induced changes in MMP, whichwas evidenced by changes in DCm and release ofcytochrome c and Smac/DIABLO into the cytosol, andcaspase-3 activation, which were maximal at about 6 h afterexposure to staurosporine. The mechanism(s) by whichstaurosporine induced changes in the MMP is not entirelyclear. Caspase-8 was degraded at a relatively late stage(after 6 h), and an inhibitor of caspase-8 did not inhibitapoptosis. Cleavage of caspase-2, -3, and -9 was evident by6 h, consistent with the kinetics of changes in MMP (i.e. ,they were downstream of or parallel with changes inMMP). Changes in the DCm were inhibited by over-expression of Bcl-2, which resulted in a marked delay inthe kinetics of apoptosis. Inhibition of changes in the DCm
by overexpression of Bcl-2 would be consistent withinduction of a BH3-only proapoptotic protein(s) bystaurosporine and induction of changes in Bax. Indeed,the conformational changes of Bax and its translocationfrom the cytosol to mitochondria were detected by 6 h afterexposure to staurosporine. The BH3-only protein(s) in-volved, however, is not clear (see below).
Overexpression of Bcl-2 and inhibition of caspases onlypartially inhibited staurosporine-induced apoptosis ofmelanoma cells, clearly indicating that a second pathwaywas involved. The kinetics of the latter appeared slowerthan the caspase-dependent pathway in that apoptosis wasmuch slower in cells with overexpression of Bcl-2. Thesefindings bear similarity with studies on L1210 cells, whichdemonstrated that the broad-spectrum caspase inhibitorblocked early (<3 h) but not delayed (>12 h) apoptotic celldeath induced by staurosporine (25). Studies on MCF-7cells also demonstrated caspase-dependent and -indepen-dent events (24). In Jurkat cells, a dominant-negative
Figure 8. Staurosporine-induced apoptosis of melanoma is independentof the generation of ROS. A, representative flow cytometry histograms ofassays of ROS production. Mel-RM and IgR3 cells with or withouttreatment with staurosporine (1 AM) for indicated time periods. DCF-DA(10 AM) was added for the last 30 min of incubation. The data shown arethe mean FSE of three individual experiments. B, the antioxidant GSH didnot inhibit staurosporine-induced apoptosis. Mel-RM and IgR3 cells weretreated with GSH for 2 h before adding staurosporine (1 AM) for another24 h. Apoptosis was measured by the propidium iodide method using flowcytometry. Columns, mean of three individual experiments; bars, SE.C, the antioxidant GSH inhibited the generation of ROS induced bystaurosporine. Mel-RM and IgR3 cells were treated with GSH (10 AM) for2 h before adding staurosporine (1 AM) for another 16 h. ROS productionwas measured as described in A. Columns, mean of three individualexperiments; bars, SE.
Molecular Cancer Therapeutics 195
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

caspase-9 mutant inhibited apoptosis induced by FasL or achemotherapeutic drug, etoposide, but had only marginaleffects on apoptosis induced by staurosporine (35). It wasreported that release of AIF is a caspase-independent event(36), and in view of this, we examined whether transloca-tion of AIF from mitochondria into the nucleus may beresponsible for late apoptotic execution of melanoma cellsby staurosporine.
Evidence in support of this possibility was firstly thekinetics of AIF release. Microscopy showed that stauro-sporine-induced release of AIF from mitochondria did notoccur until after 6 h and that its translocation into thenucleus was evident by 16 h. This release was much laterthan that of cytochrome c and Smac/DIABLO. Secondly,translocation of AIF into the nucleus was not inhibited bythe pan-caspase inhibitor (z-VAD-fmk) or by overexpres-sion of Bcl-2. The latter did cause some delay in AIFtranslocation to the nucleus, consistent with the observeddelay in staurosporine-induced apoptosis in melanomacells with overexpressed Bcl-2. These results suggest thatstaurosporine induces release of AIF by mechanisms thatare independent of changes in the DCm. This is also sup-ported by the kinetics of staurosporine-induced changes inthe DCm in that the changes in the DCm had peaked andwere reversing prior to AIF entry into the nucleus. Furtherinvestigation of the mechanism concerned and its relationto the conformational changes of Bax is needed.
An alternative explanation that we considered forcaspase-independent induction of apoptosis by staurospor-ine was generation of ROS. Some apoptotic agents such ascertain histone deacetylase inhibitors can induce apoptosisby the production of ROS independent of caspase activa-tion (37, 38). The present studies, however, showed thatalthough ROS were produced in melanoma cells bytreatment with staurosporine, they did not appear to beinvolved in induction of apoptosis as the antioxidant GSHinhibited generation of ROS but was unable to preventmelanoma from apoptosis induced by staurosporine.
Staurosporine is known to inhibit several protein kinasesin cells. Apparently, inhibition of protein kinases may havebeen responsible for inducing conformational changes inBax and its translocation from the cytosol to mitochondria(39 –44). staurosporine has been shown to inhibit theserine/threonine kinase Akt/protein kinase B leading todecreased phosphorylation of Bad (39, 40). Bad is capable offorming heterodimers with the antiapoptotic proteins Bcl-XL and Bcl-2 and antagonizing their antiapoptotic activity(39). Phosphorylated Bad cannot bind to either Bcl-XL orBcl-2, so that inhibition of Akt-mediated phosphorylation ofBad would increase sensitivity of cells to apoptosis (44).Overexpression of Akt was shown to inhibit staurosporine-induced movement of Bax and apoptosis in HeLa cells (21).Inhibition of Erk1/2 activation by staurosporine may alsoplay a part in staurosporine-induced changes in Bax (43).We have previously shown that inhibition of Erk1/2facilitated apoptosis induction in melanoma by promotingconformational changes of Bax and its relocation tomitochondria (44).
Despite the overall relatively high levels of apoptosisinduced by staurosporine, there was some variation insensitivity to staurosporine among the melanoma celllines, as shown for studies on the IgR3 (sensitive) andMel-RM (partially resistant) cell lines. The mechanism(s)underlying this is currently not clear but was reflected inincreased changes in MMP and caspase-3 activation in theIgR3 compared with the Mel-RM line. Several differentkinases are involved in resistance to apoptosis, such as themitogen-activated protein kinase Erk1/2 and Akt/proteinkinase C pathways. It is therefore possible that thedifferent sensitivities of the melanoma cells to staurospor-ine may reflect the degree to which these pathways wereinvolved in protection of cells against apoptosis.
In summary, these studies show that staurosporine canactivate an apoptotic pathway that is dependent onchanges in MMP and activation of caspases as well as apathway that is independent of caspase activation and notinhibitable by overexpression of Bcl-2. The latter may bedue to AIF release in that the latter was also independent ofcaspase activation and overexpression of Bcl-2. staurospor-ine is therefore able to bypass resistance of melanoma cellsto mitochondrial caspase-dependent pathways, and (non-toxic) derivatives of staurosporine may prove to bevaluable agents against melanoma alone or in combinationwith other agents such as TRAIL.
References
1. Debatin KM, Poncet D, Kroemer G. Chemotherapy: targeting themitochondrial cell death pathway. Oncogene, 2002;21:8786–803.
2. Lewanski CR, Gullick WJ. Radiotherapy and cellular signaling. LancetOncol, 2001;2:366–70.
3. Wesselborg S, Engels IH, Rossmann E, Los M, Schulze-Osthoff K.Anticancer drugs induce caspase-8/FLICE activation and apoptosis in theabsence of CD965 receptor/ligand interaction. Blood, 1999;93:3053–63.
4. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation.Science, 1998;281:1305–8.
5. Green DR, Reed JC. Mitochondria and apoptosis. Science, 1998;281:1309–12.
6. Scorrano L, Oakes SA, Opferman JT, et al. BAX and BAK regulation ofendoplasmic reticulum Ca2+: a control point for apoptosis. Science, 2003;300:135–9.
7. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science, 1998;281:1312–6.
8. Cryns V, Yuan J. Proteases to die for. Genes Dev, 1998;12:1551–70.
9. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptoticprogram in cell-free extracts: requirement for dATP and cytochrome c.Cell, 1996;86:145–57.
10. Susin SA, Lorenzo HK, Zamzami N, et al. Mitochondrial release ofcaspase-2and -9during theapoptoticprocess. JExpMed,1999;189:381–93.
11. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein thatpromotes cytochrome c -dependent caspase activation by eliminating IAPinhibition. Cell, 2000;102:33–42.
12. Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO,a mammalian protein that promotes apoptosis by binding to andantagonizing IAP proteins. Cell, 2000;102:43–53.
13. Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase whenreleased from mitochondria. Nature, 2001;412:95–9.
14. Hegde R, Srinivasula SM, Zhang Z, et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disruptsinhibitor of apoptosis protein-caspase interaction. J Biol Chem, 2002;277:432–8.
15. Verhagen AM, Silke J, Ekert PG, et al. HtrA2 promotes cell death
Apoptosis of Melanoma Induced by Staurosporine196
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

through its serine protease activity and its ability to antagonize inhibitorof apoptosis proteins. J Biol Chem, 2002;277:445–54.
16. Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptoticprotease cascade. Cell, 1997;91:479–89.
17. Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival.Science, 1998;281:1322–6.
18. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate therelease of apoptogenic cytochrome c by the mitochondrial channel VDAC.Nature, 1999;399:483–7.
19. Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3domain-only molecules preventing BAX- and BAK-mediated mitochondrialapoptosis. Mol Cell, 2001;8:705–11.
20. Bouillet P, Strasser A. BH3-only proteins—evolutionarily conservedproapoptotic Bcl-2 family members essential for initiating programmed celldeath. J Cell Sci, 2002;115:1567–74.
21. Tafani M, Minchenko DA, Serroni A, Farber JL. Induction of themitochondrial permeability transition mediates the killing of HeLa cellsby staurosporine. Cancer Res, 2001;61:2459–66.
22. Tafani M, Cohn JA, Karpinich NO, et al. Regulation of intracellularpH mediates Bax activation in HeLa cells treated with staurosporine ortumor necrosis factor-a. J Biol Chem, 2002;277:49569–76.
23. Yuste VJ, Sanchez-Lopez I, Sole C, et al. The prevention of thestaurosporine-induced apoptosis by Bcl-X(L), but not by Bcl-2 or caspaseinhibitors, allows the extensive differentiation of human neuroblastomacells. J Neurochem, 2002;80:126–39.
24. Xue LY, Chiu SM, Oleinick NL. Staurosporine-induced death of MCF-7human breast cancer cells: distinction between caspase-3-dependentsteps of apoptosis and the critical lethal lesions. Exp Cell Res, 2003;283:135–45.
25. Belmokhtar CA, Hillion J, Segal-Bendirdjian E. Staurosporine inducesapoptosis through both caspase-dependent and caspase-independentmechanisms. Oncogene, 2001;20:3354–62.
26. Hersey P. Advances in the non-surgical treatment of melanoma. ExpOpin Investig Drugs, 2002;11:75–85.
27. Hersey P, Zhang XD. Overcoming resistance of cancer cells toapoptosis. J Cell Physiol, 2003;196:9–18.
28. Zhang XD, Franco A, Myers K, Gray C, Nguyen T, Hersey P. Relationof TNF-related apoptosis-inducing ligand (TRAIL) receptor and FLICE-inhibitory protein expression to TRAIL-induced apoptosis of melanoma.Cancer Res, 1999;59:2747–53.
29. Zhang XD, Zhang XY, Gray CP, Nguyen T, Hersey P. Tumor necrosisfactor-related apoptosis-inducing ligand-induced apoptosis of humanmelanoma is regulated by Smac/DIABLO release from mitochondria.Cancer Res, 2001;61:7339–48.
30. Johnson DE, Gastman BR, Wieckowski E, et al. Inhibitor of apoptosisprotein hILP undergoes caspase-mediated cleavage during T lymphocyteapoptosis. Cancer Res, 2000;60:1818–23.
31. Desagher S, Osen-Sand A, Nichols A, et al. Bid-induced conforma-tional change of Bax is responsible for mitochondrial cytochrome c releaseduring apoptosis. J Cell Biol, 1999;144:891–901.
32. Bellosillo B, Villamor N, Lopez-Guillermo A, et al. Spontaneous anddrug-induced apoptosis is mediated by conformational changes of Baxand Bak in B-cell chronic lymphocytic leukemia. Blood, 2002;100:1810–6.
33. Chinopoulos C, Starkov A, Fiskum G. Cyyclosporin A-insensitivepermeability transition in brain mitochondria: inhibition by 2-aminoethox-ydiphenyl borate. J Biol Chem, 2003;278:273–89.
34. Poot M, Hosier S, Swisshelm K. Distinct patterns of mitochondrialchanges precede induction of apoptosis by all-trans -retinoic acid andN -(4-hydroxypheny) retinamide in MCF-7 breast cancer cells. Exp CellRes, 2002;279:128–40.
35. Stepczynska A, Lauber K, Engels IH, et al. Staurosporine andconventional anticancer drugs induce overlapping, yet distinct path-ways of apoptosis and caspase activation. Oncogene, 2001;20:1193–202.
36. Daugas E, Susin SA, Zamzami N, et al. Mitochondrio-nucleartranslocation of AIF in apoptosis and necrosis. FASEB J, 2000;14:729–39.
37. Ruefli AA, Ausserlechner MJ, Bernhard D, et al. The histonedeacetylase inhibitor and chemotherapeutic agent suberoylanilidehydroxamic acid (SAHA) induces a cell-death pathway characterized bycleavage of Bid and production of reactive oxygen species. Proc Natl AcadSci, 2001;98:10833–8.
38. Rosato RR, Almenara JA, Grant S. The histone deacetylaseinhibitor MS-275 promotes differentiation or apoptosis in humanleukemia cells through a process regulated by generation of reactive oxy-gen species and induction of p21CIP1/WAF1 1. Cancer Res, 2003;63:3637–45.
39. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serinephosphorylation of death agonist BAD in response to survival factorresults in binding to 14-3-3 not Bcl-X. Cell, 1996;87:619–28.
40. Franke TF, Cangley LC. Apoptosis. A bad kinase makes good. Nature,1997;390:116–7.
41. Songyang Z, Baltimore D, Cantley LC, Kaplan DR, Franke TF.Interleukin 3-dependent survival by the Akt protein kinase. Proc Natl AcadSci USA, 1997;94:11345–50.
42. Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad,a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotescell death. Cell, 1995;80:285–91.
43. Yamaki K, Hong J, Hiraizumi K, Ahn JW, Zee O, Ohuchi K.Participation of various kinases in staurosporine induced apoptosis RAW264.7 cells. J Pharm Pharmacol, 2002;54:1535–44.
44. Zhang XD, Borrow JM, Zhang XY, Nguyen T, Hersey P. Activation ofERK1/2 protects melanoma cells from TRAIL-induced apoptosis byinhibiting Smac/DIABLO release from mitochondria. Oncogene, 2003;22:2869–81.
Molecular Cancer Therapeutics 197
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from

2004;3:187-197. Mol Cancer Ther Xu Dong Zhang, Susan K. Gillespie and Peter Hersey caspase-dependent and -independent apoptotic pathwaysStaurosporine induces apoptosis of melanoma by both
Updated version
http://mct.aacrjournals.org/content/3/2/187
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/3/2/187.full#ref-list-1
This article cites 42 articles, 21 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/3/2/187.full#related-urls
This article has been cited by 18 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://mct.aacrjournals.org/content/3/2/187To request permission to re-use all or part of this article, use this link
Research. on November 29, 2018. © 2004 American Association for Cancermct.aacrjournals.org Downloaded from