st föreläsning barnneurologi 160210 kopia 3 - svenwiklund.sesvenwiklund.se/onewebmedia/st...
TRANSCRIPT

Barn neurologiSven Wiklund Barnneurolog
SUS Lund 160210
Epilepsi Migrän
HjärntumörCerebral ParesMissbildningar
Utvecklingsstörning
Guillian Barré Neuroborelios
Charcot Marie ToothB12 brist
Spina Bifida
Framhornscells sjukdomar
Muskeldystrofi Mitokondrie sjukdomar
Synaps sjukdomar Dermatomyosit
Muskelsvaghet

Dermatom
Funktionella bortfalls symtom Nerv inklämning

Neurometabola sjukdomar
GaucherAdrenoleukodystrofi
Metakromatisk leukodystrofiCeroid Lipofucinos
och många, många fler

Pattern of recognition
System kunskap

Mål med dagens föreläsningar
Visa på hur man kan nå fram till diagnos genom att tänka på hur nervsystemet är organiserat och fungerar beroende på olika lokaler och åldrar.
Gå igenom några typiska ” pattern of recognition ” inom barnneurologin.Epilepsi - migrän - kardiogen syncopeMuskel dystrofiDyskinesierHereditära neuropatierKongenitala myastenierMyotonier
Framtiden är här inom vissa behandlingar.
Jag kommer inte ta upp: neonatal neurologi, floppy infant, cerebral pares, utvecklings störning, neuroonkologi( jo - när misstänka hjärntumör ), neuropsykiatri m.m.

Viktiga frågor längs vägen
Vilken/a anatomisk/a lokal/er är engagerad/e?
Vilken patologisk mekanism kan förklara symtomen?
Är det en ålders specifik sjukdom?
Hur startade det?
Hur har det utvecklats?
Hereditet?

Epilepsi
ILAE working definition
Patofysiologi - behandling - skillnaden mellan epilepsi och migrän.
Hur skilja epileptiskt utlösta anfall gentemot migrän och kardiogen syncope?
Benign barnepilepsi
Absence epilepsi
Infantil spasm
Partiellt status epileptikus
Är epilepsi farligt?

Olika slags anfall
Film 1
Film2
Film 3
Film 4
Film 5

Hur skilja på syncope och ep utlösta anfall?
At times do you wake with a cut tongue after your spells? 2
At times do you have a sense of deja vu or jamais vu before your spells? 1
At times is emotional stress associated with losing consciousness? 1
Has anyone ever noted your head turning during a spell? 1
Has anyone ever noted that you are unresponsive, have unusual posturing or have jerking limbs during your spells or have no memory of your spells afterwards? (Score as yes for any positive response)
1
Has anyone ever noted that you are confused after a spell? 1
Have you ever had lightheaded spells? −2
At times do you sweat before your spells? −2
Is prolonged sitting or standing associated with your spells? −2
The patient has seizures if the point score is ≥1, and syncope if the point score is <1.


ILAE senaste definition
An epileptic seizure is a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain.
Epilepsy is a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures, and by the neurobiologic, cognitive, psychological, and social consequences of this condition. The definition of epilepsy requires the occurrence of at least one epileptic seizure.

Epilepsy is a disease of the brain defined by any of the following conditions
1. At least two unprovoked (or reflex) seizures occurring >24 h apart
2. One unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years
3. Diagnosis of an epilepsy syndrome
Epilepsy is considered to be resolved for individuals who had an age-dependent epilepsy syndrome but are now past the applicable age or those who have remained seizure-free for the last 10 years, with no seizure medicines for the last 5 years.

Fallgropar vid epilepsi diagnostik
2-4 % av alla friska barn har epileptiforma förändringar på EEG.
EEGs sensitivitet 25 - 50 - 80%

Epilepsi - migränlikheter och skillnader
Comorbiditet; Individer med epilepsi har migrän dubbelt så ofta som de utan epilepsi och individer med migrän har epilepsi tre gånger så ofta som de med spänningshuvudvärk.
Occipital epilepsi och migrän kan vara mycket svåra att skilja åt - migralepsi.
Migränliknande huvudvärk efter anfall ( fr.a. efter tonsik kliniska samt temporallobs utösta )
Trigger faktorer
Familjär hemiplegisk epilepsi. CACNA1A - Ca kanal ATP1A2 - Na - K ATPas SCN1A - Na kanal
Jonkanals sjuka? Arytmier, episodisk paralys, dyskinesier.
AED kan hjälpa vid båda tillstånd. Men de som verkar via spännings känsliga Na kanaler och via GABA systemet gör det inte.Dessutom är upp till 30% av både migrän och epilepsi patienter läkemedels resistenta - andra mekanismer?
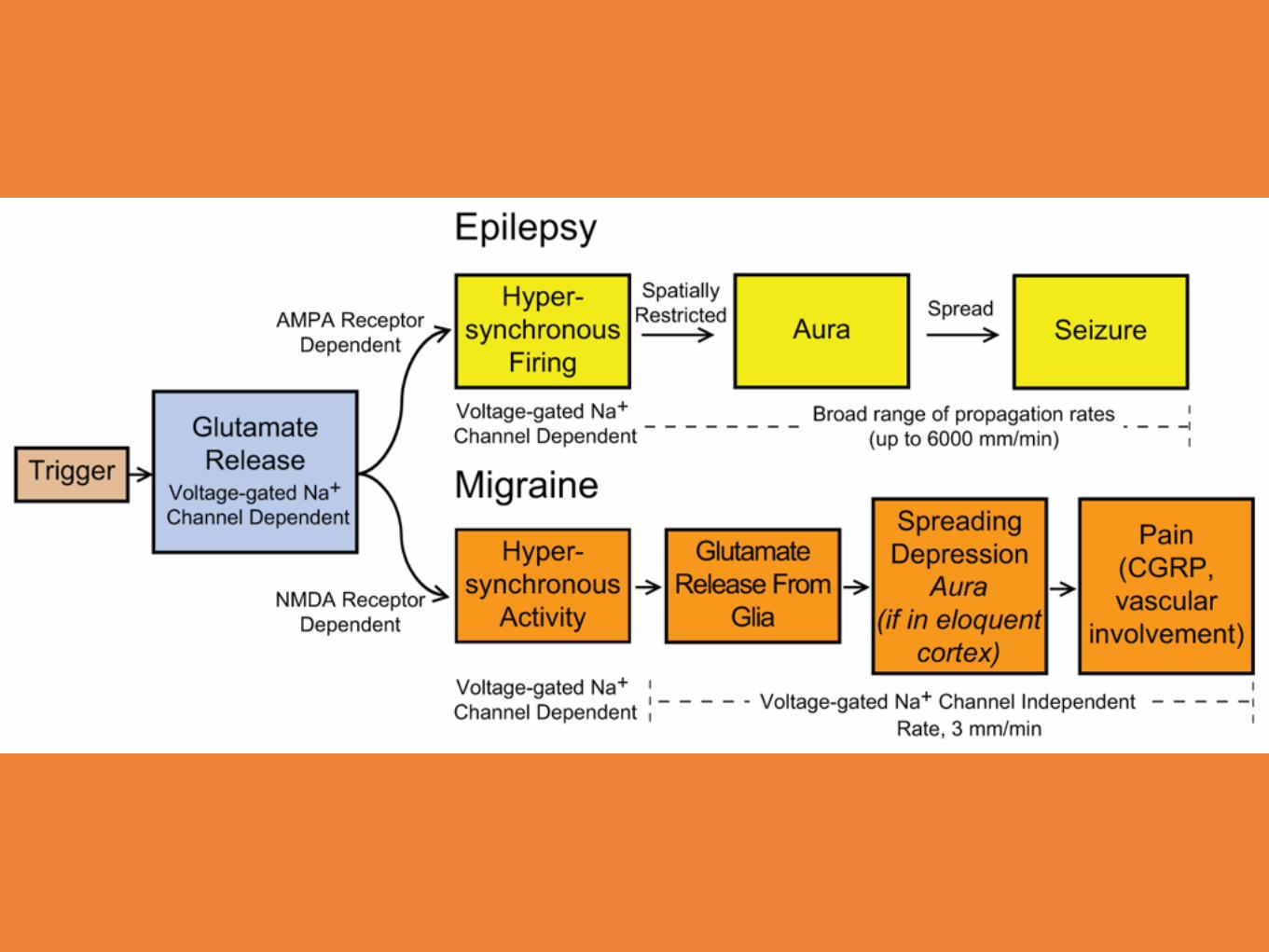
Epilepsi synkrona urladdningar - spreading depression vid migrän.

Migrän
Cortical spreading depression Aristide Leao 1944.
Initial neuronal exitation under ett par sekunder som följs av en 5 - 10 minuter lång period med negativ laddning över cortex området, som sprider sig med en hastighet av 3 - 5 mm.
CSD tros orsaka migrän auran och huvudvärken
Gemensamt för både migrän och epilepsi tros den glutamat utlösta hypersynkroniseringen vara.

Benign barnepilepsi med centrotemporala spikarBRE ( BenignRolandicEpilepsy )
BECTS ( BenignEpilepsywithCentraTemporalSpikes )
Film
Drabbar barn mellan ( 1 ) 3- 13 ( 14 ) år ( peak 7 - 10 )
Normal utvecklade, ingen avvikande neurologi, ev lättare språkliga och inlärnings problem som sannolikt försvinner.
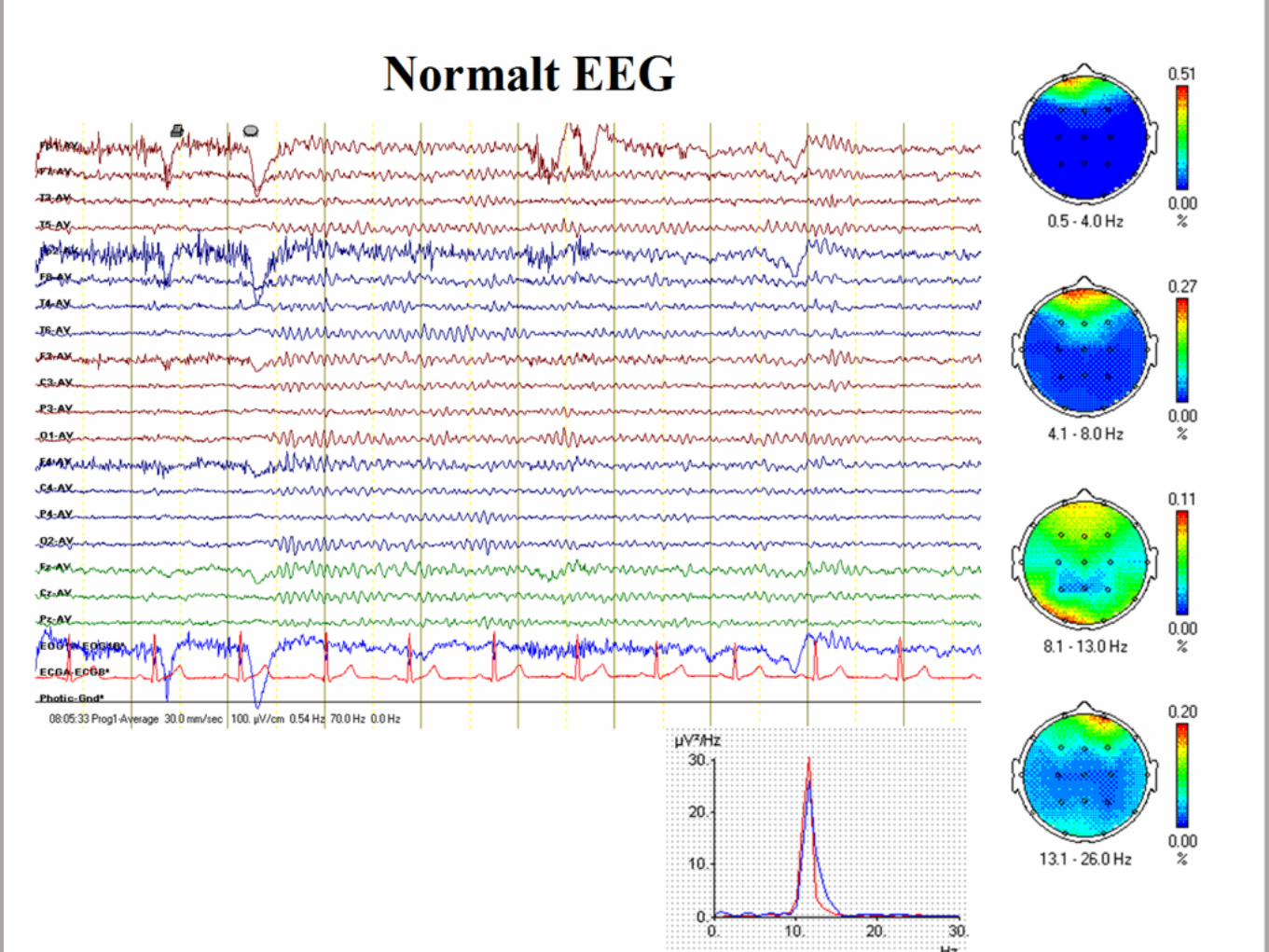
EEG visar fokal ep - aktivitet, klassiskt centro - temporalt, sidoväxlande med markant ökad mängd i sömn. Bakgrundsaktiviteten är normal.
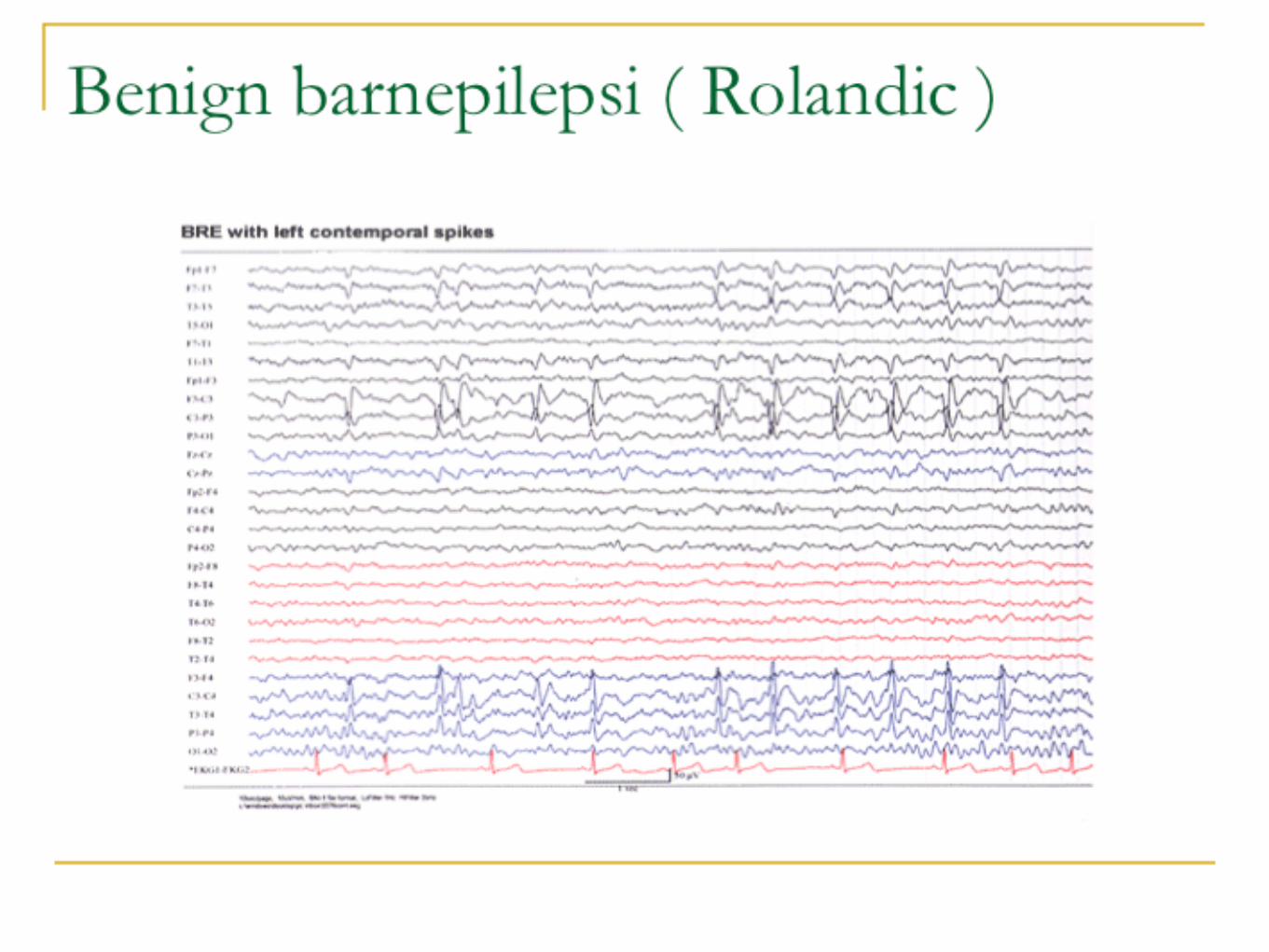

BRE, fortsättning
De flesta anfall är under sömn / uppvaknadet
Ofta partiella anfall men över 50% får GTCS.
De flesta är friska efter 3 år.
Över 90 % fortsätter att vara anfalls fria i 10 år eller längre.
Aktiv expektans alternativt behandling med oxkarbamazepin eller valproat.

BRE - del av fenotypiskt spektrum
BRE är ” idiopatisk” = genetisk
Gener associerade med BRE finns beskrivna
Ett fåtal med BRE utvecklar svårare epilepsier:
Atypisk benign fokal epilepsi
Pseudo Lennox
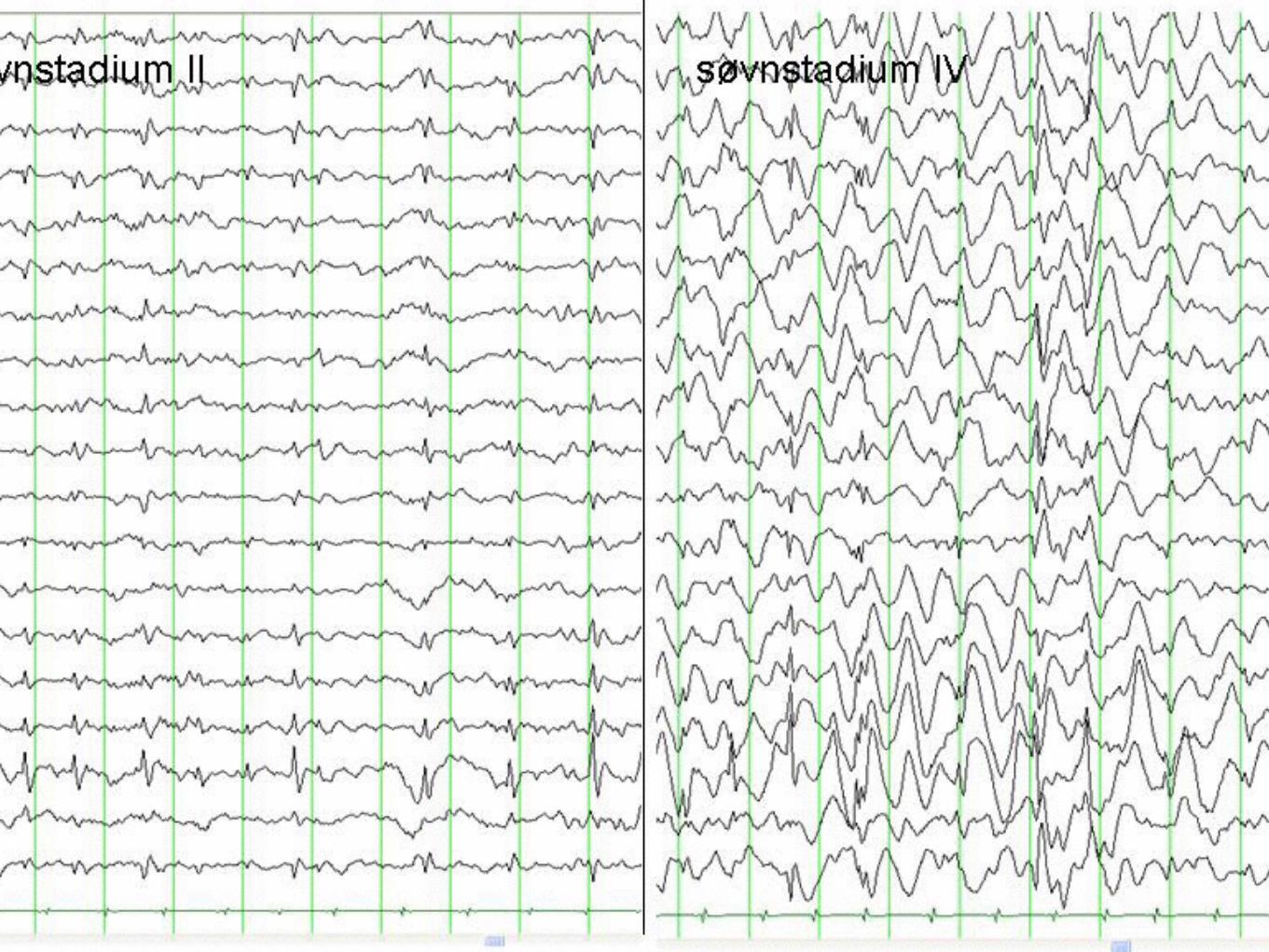
Landau - Kleffner 3- 7 år. Plötslig afasi, verbal agnosi, BECT likände EEG med ESES ( Electrical Status epilepticus during Slow wave Sleep )
CSWS , tiltagande kognitiv och motorisk försämring, sällan anfall, EEG i vakenhet ofta normal mesans ESES bild ( se nästa slide ) under sömn.
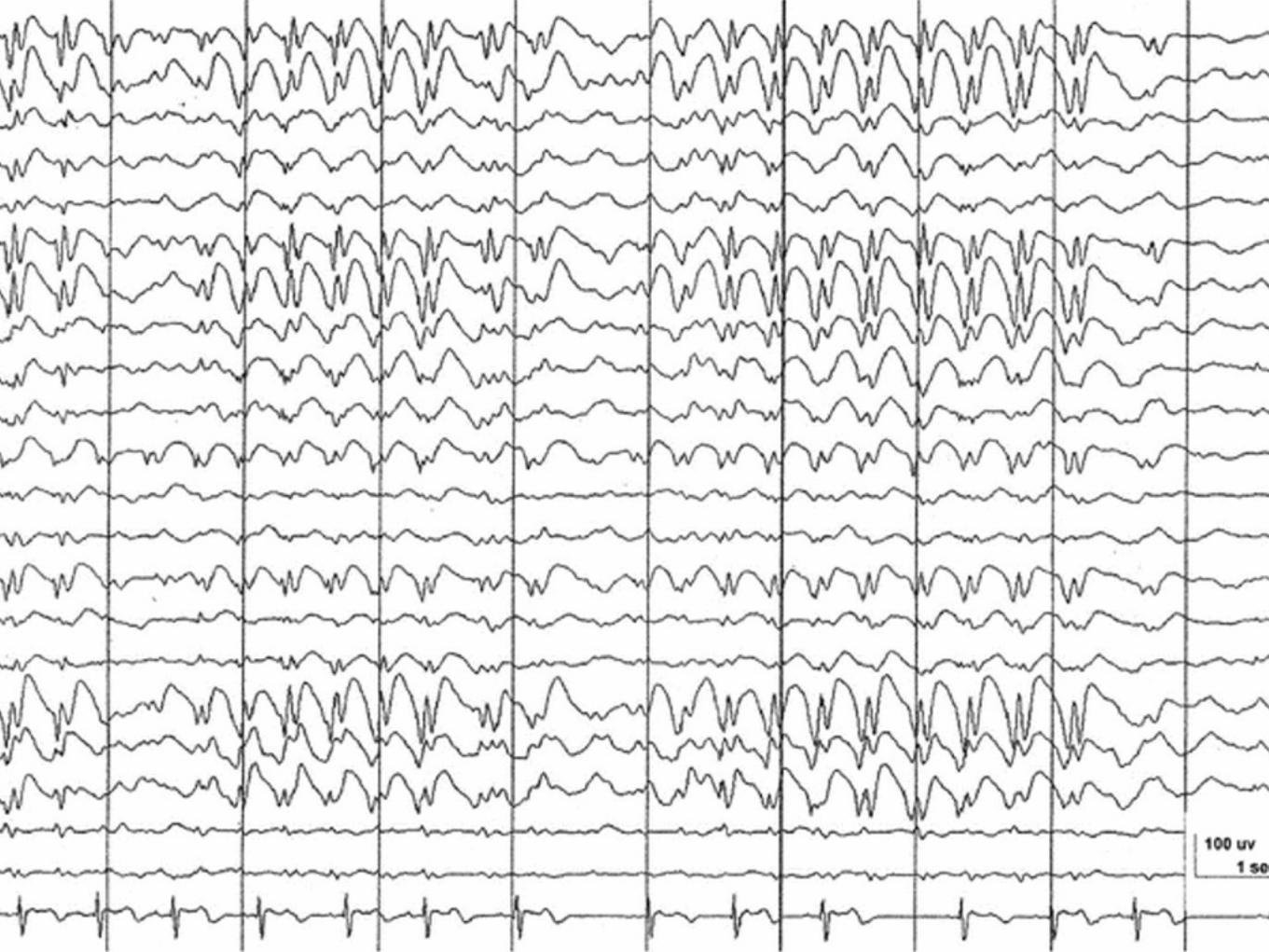

Absence epilepsiChildhood Absence Epilepsy ( CAE )
Film 1
Film 2

Absence epilepsi fortsättning
Debuterar 4 - 10 års åldern peak 5 - 7 ( senare debut betyder sannolikt juvenil absence epilepsi med risk för GTCS och tillägg med myoklonier för juvenil myoklon epilepsi där man, . GTCS, där man mera sällan ser remission ).
klassisk 3 Hz / sekund ( 2-4 )
Anfallen typiskt 4 - 20 sekunder långa, myoklonier, atonier, tonaska komponenter, automatismer
CAE förutsätter intakta kortikothalamiska banor.
Ethosuxemide bäst ( skyddar inte mot GTCS ) , valproat, lamotrigin( Ox ) karbamazepin, gabapentin, vigabatrin och tigabin är kontraindicerade.


Infantil spasm
Barn under 2 år, peak 3-5 månaders ålder
Cerebral pares, tubers skleros , migrations störningar och Downs syndromepredisponerar.
100tals anfall dagligen, ” Salaam” anfall, ofta följt av skrik och ledsenhet.
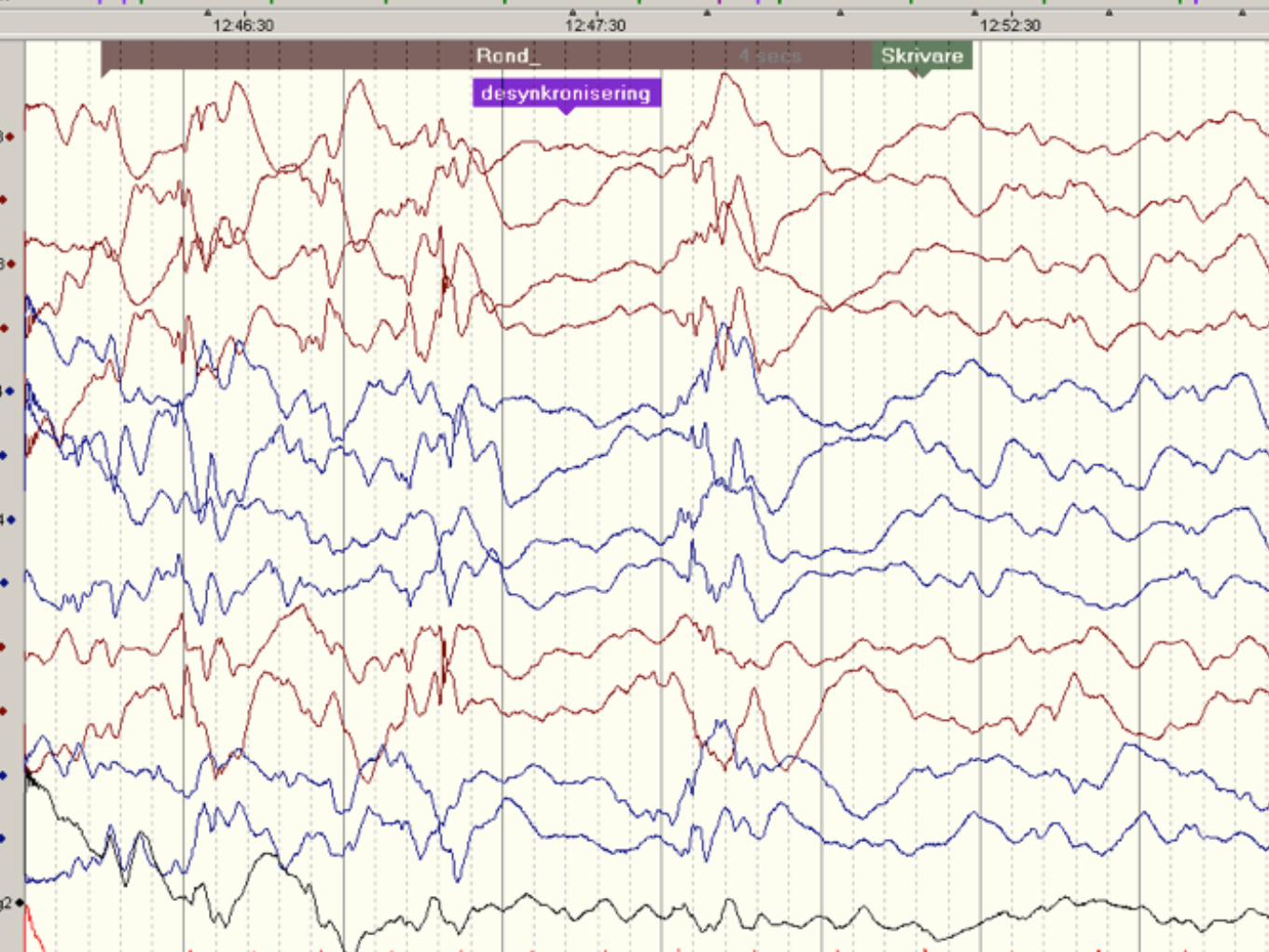

Infantil spasm fortsättning
Behandlas på SUS med ACTH i.m. ( ett par veckor )+ Vigabatrin ( 6 månader )Risk för sepsis och hypertension ( ACTH ) samt synfälts defekt ( Vigabatrin )
Stor risk för negativ kognitiv påverkan på sikt - 80%

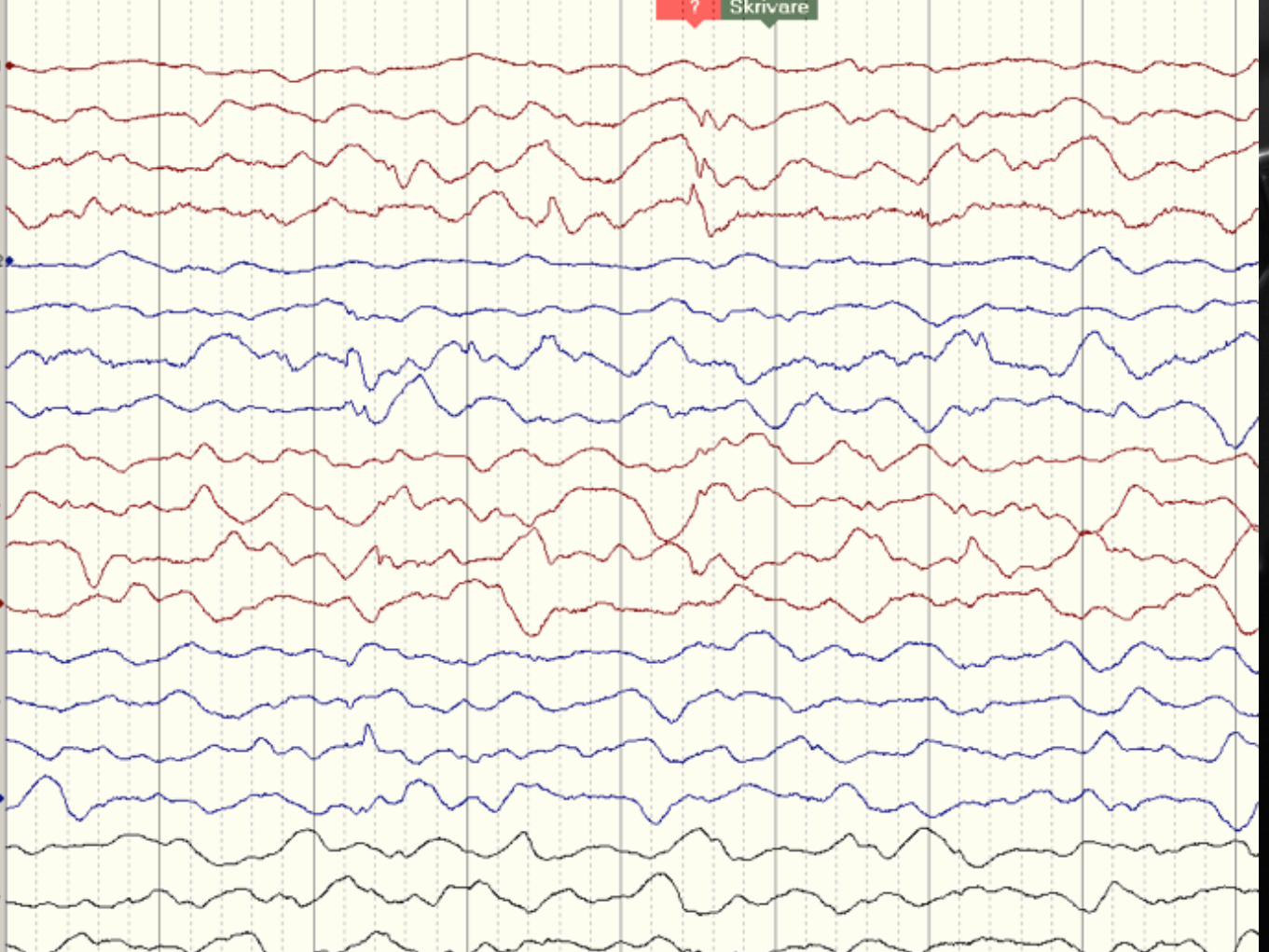
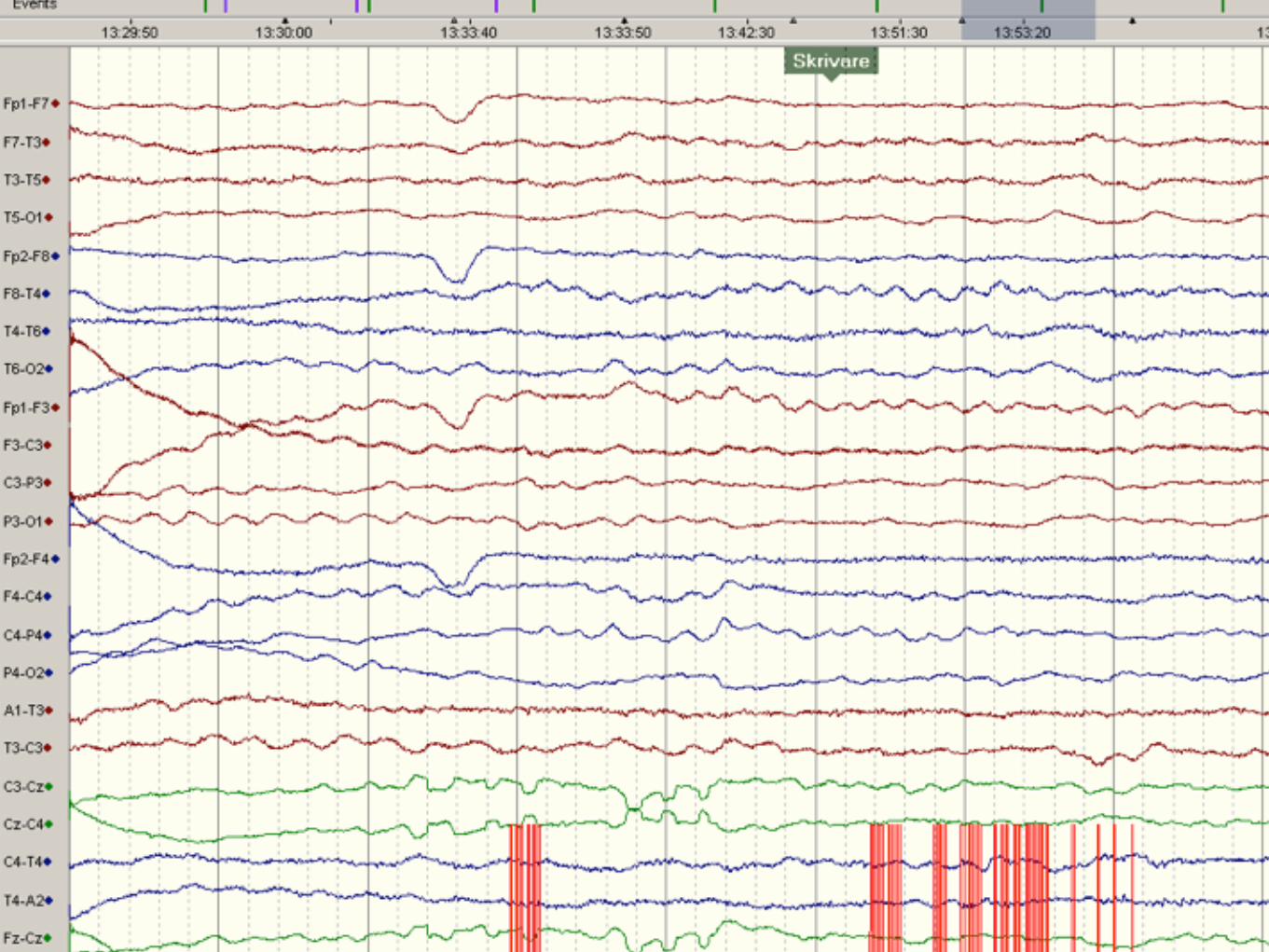

Non konvulsivt status epileptikus
Partiellt status epileptikus


Vad tror ni att föräldrarna undrar?

Är Epilepsi farligt?
Non konvulsion anfall verkar inte ge skador.
Feberkramper hos barn ger ingen negativ kognitiv påverkan på sikt.
Meldrum visade på 1970-talet att konvulsiva status > 45 minuter hos babianer gav hjärn skador.
Mortaliteten hos barn med konvulsivt status är 2,7 - 5,4 %( 5 - 8 % hos de som kräver IVA vård ).
Mortaliteten hos epilepsi patienter är 0,44 / 1000 / år.

Är epilepsi farligt ? Fortsättning
Den relativa risken att skada sig är 1.4
2 % hade fått hjärnskakning
0,2 % av 44000 dödsfall i trafikolyckor i USA bedömdes vara orsakade av epilepsianfall.
14 % uppgav sig ha haft ett anfall under bad.

Long term mortality in childhood onsetepilepsy
Sillanpää NEJM 2010
Inga fall av SUDEP < 14 års ålder.
Barn dog fr.a. p.g.a av andra tillstånd relaterade till grundsjukdomen än epilepsi.
Under 40 års uppföljningen dog 24% vilket är tre gånger mer än normal befolkningen.

Hur länge kan man expektera?
90 % av anfallen upphör inom 2 minuter.
När ett anfall varat 5 minuter och definitivt efter 10 minuter, bryts det sällan spontant.

Risken för fler anfall än ett?
90 % av alla recidiv är inom ett år.
Dutch study group on childhood epilepsy:25 % är friska efter 1 år.50% var anfalls fria år 2; 5 och 15.25 % har epilepsi längre än 12 år.9 % hade terapi resistent epilepsiAv de med till synes glatt förlopp = anfalls fria under de 5 första åren eller enbart recidiv under de första 6 månader efter första anfallet:Av dessa recidiverade 17 % efter long remission och hälften utveckladeterapi resistent epilepsi.
Risk faktorer var : Låg ålder, patologiskt eeg, avvikande neurolog status och eventuellt anfall under sömn.
Idiopatisk epilepsi ger bäst prognos.

Kan man låta barnet sova själv?
Risken för SUDEP är mycket liten.
De flesta anfall slutar inom 2 minuter.
Monitorer har en dålig sensitivitet och specificitet.
Risken att skada sig när man ligger till sängs är lägre.

Däremot…
Inte bada utan särskild tillsyn - även i skolan.
Vid diagnos epilepsi gäller ett års körförbud för moped, traktor och bil.
Klokt med cykelhjälm.

Tillslut
Hjälper febernedsättande att förebygga feberkramper?
Nej; bl.a. Rosenbloom et al 2013


Progressiva sjukdomar
Leukodystrofi
Regression
Myopati
Rhabdomyolys
Psykiatriska symtom

Progressiva sjukdomar fortsättning
Akut encephalopati
Dystoni
Utvecklings försening / störning
Ataxi
Hypotoni

Progressiva sjukdomar fortsättning
Stroke
Epilepsi
Perifer neuropati
Progressivt förlopp
Ålder
Ofta flera organ
Typiska förlopp
Specifik provtagning

Viktiga frågor längs vägen
Vilka anatomiska lokaler är engagerade?
Vilken/a patologisk mekanism/er kan förklara symtomen?
Är det en ålders specifik sjukdom?
Hur startade det?
Hur har det utvecklats?
Hereditet?

Anatomiska lokaler
OpthalmologyCherry red spot –GM1 and 11, NPC A and BRetinitis pigmentosa-peroxisomal disorders, mitochondrial disease, FAOD, MTHFR deficiency, vitamin E deficiencEctopia lentis—homocystinuriaCataract-galactosaemia, peroxisomal disorders, Wilson disease, CTX, Fabry disease
DeafnessZellweger spectrum
HaematologyMacrocytic anaemia—cobalamin disordersPancytopenia—organic acidurias, Gaucher type 1, GSD 1b, cobalamin and folate metabolism disorders, lysinuric protein intolerance

Anatomiska lokaler fortsättning
Abnormalities of skin or hair
Brittle hair—Menkes disease, argininosuccinic aciduria, biotinidase deficiencyAlopecia, seborrhoic dermatitis—biotinidase deficiency
Ichthyosis neutral lipid storage disease, multiple sulfatase deficiency, Sjögren-Larsson syndrome
Angiokeratoma Fabry disease, fucosidosis, sialidosis

Anatomiska lokaler fortsättning
Macrocephaly Alexander disease Canavan disease GM2 L2 hydroxyglutaric acuduria Glutaric aciduria type 1
Microcephaly Congenital—molybdenum cofactor deficiency, SLO Acquired-GLUT 1 deficiency, cerebral folate deficiency,
Dysmorphic features Peroxisomal disorders Lysosomal storage disorders SLO CDG

Cardiac
Conduction defects—FAOD, mitochondrial disease
Cardiomyopathy—Barth syndrome, CDG, FAOD, Pompe disease, GSD 3 and 4, mitochondrial disease
Anatomiska lokaler fortsättning

Anatomiska lokaler fortsättning
Hepatomegaly and/or splenomegaly
Niemann Picks diseaseGaucher diseaseUrea cycle defectsGlycogen storage diseases

Patologisk mekanism
Disorders of intermediary metabolism that give rise to acute or chronic intoxication:Includes organic acidurias, urea cycle disorders, other aminoacidopathies, sugar intolerances, metal disorders and porphyriasMost are treatable, and require the emergency removal of the toxin by special diets, extracorporeal procedures, cleansing drugs or vitamins
Disorders of intermediary metabolism that affect the cytoplasmic and mitochondrial energetic processesIncludes disorders of glycolysis, glycogenosis, gluconeogenesis, creatine and pentose phosphate pathways, neurotransmitter disorders, respiratory chain disorders and pyruvate oxidation defects Some are treatable
Disorders of cellular organellesIncludes peroxisomal disorders, lysosomal storage disorders and disorders of glycosylationSome are treatable

Ålder
is characterized by progressive motor losses and dysfunc-tion. Motor symptoms characteristically occur early andare more prominent than seizures and mental deteriora-tion. A frequent early problem is a gait disorder with un-steadiness. Within months, this leads to the loss of theability to walk and stand. Decrease in deep tendon reflexesoccurs in the early stage, as the peripheral neuropathyworsens and is later replaced by hyperreflexia. Bilateralextensor toe responses occur early and persist. Speech de-teriorates as a result of dysarthria and aphasia. Ataxia andtruncal instability become obvious. Intermittent pain is amanifestation of peripheral neuropathy. Nystagmus ispresent. Optic atrophy and a grayish discoloration of themacula are occasionally observed. Hypotonia is progres-sively replaced by rigidity and spasticity. Megalencephalyis frequently noted. Most children are bedridden by age 3years. All meaningful contact with the surroundings is pro-gressively lost. An opisthotonic posture with flexion of thearms, equinovarus posture, and scissoring of the legs ispresent in later stages of the illness and may last for a fewmonths to several years.
The juvenile form has its onset between age 4 and10 years. The majority of cases develop during the firstyears of school with bradykinesia and poor school per-formance. Daydreaming, confusion, or emotional labilitymay be seen early. Unsteadiness of gait, usually owing topyramidal system involvement, may occur. Extrapyrami-dal dysfunction, as suggested by postural abnormalities,rigidity, and tremor may also develop. Seizures occur in
more than half of the patients. Deep tendon reflexes areusually increased. The rate of deterioration is usuallyslower and more variable than in the late-infantile form.Patients may not be bedridden until 5–50 years aftersymptoms begin.
The adult form has its onset any time after puberty.Initial symptoms consist of personality and mentalchanges. Such symptoms are often misdiagnosed as atten-tion deficit disorder, schizophrenia, or manic-depressiveillness. Seizures are rarely the presenting symptom. Move-ment disorders, ataxia, and paresis appear later. There areusually no clinical signs of peripheral neuropathy. In thefinal stage, the patient is mute, blind, bedridden, andunresponsive.
A rare variant of MLD combines features of the mu-copolysaccharidosis with X-linked ichthyosis, X-linkedchondrodysplasia punctata, and those of MLD. It is nowcalled multiple sulfatase deficiency. The mucopolysac-charidosis-like features include a mild gargoylism (facialchanges with depressed bridge of the nose), hydro-cephalus, growth retardation, limitation in extension ofthe elbows, radiologic changes of chondrodysplasia punc-tata, hepatosplenomegaly, heart defects, and deafness.Corneal opacities do not occur. Ichthyosis, when present,develops at an early age. In general, presymptomatic de-velopment is less advanced than that of late-infantilemetachromatic leukodystrophy. Most children neverachieve normal gait or speech. Neurologic regression fol-lows the pattern of late-infantile MLD. By the age of 5,
Chapter 15 ▼ Inborn Errors of Metabolism I: Neurologic Degenerative Diseases 279
Table 15.12 Age of onset of progressive leukoencephalopathies and leukodystrophies
Early infancy (before 12 months)
! Infantile Krabbe disease
! Canavan disease
! Alexander disease
! Pelizaeus-Merzbacher disease
! Vanishing white matter disease
Early childhood (1–5 years)
! Metachromatic leukodystrophy
! Late infantile forms of Krabbe disease
! Vanishing white matter disease
Late childhood and adolescence (5–15 years)
! X-linked adrenoleukodystrophy
! Juvenile metachromatic leukodystrophy
! Juvenile Krabbe disease
! Vanishing white matter disease
Adulthood
! Metachromatic leukodystrophy (ataxia, dementia)
! Krabbe disease
! Adrenomyeloneuropathy
! Vanishing white matter disease
Pearls and Perils
! Detection of large amounts of urinary sulfatides is essentialfor diagnosis. Urine should be kept at 4°C for collection.
! Low arylsulfatase-A in asymptomatic persons may beseen in two situations. Presymptomatic cases ofmetachromatic leukodystrophy excrete excessiveamounts of urinary sulfatide. Normal amounts of sul-fatide are found in pseudoarylsulfatase-A deficiency.
! Arylsulfatase-A is not always low in metachromaticleukodystrophy. Assay of arylsulfatase-A in intact cellsand detection of large amounts of sulfatide in the urineallow diagnosis of saposin B deficiency.
! Arylsulfatase-A deficiency is deficient in multiple sulfatase deficiency, a rare autosomal recessivemetachromatic leukodystrophy associated with combined features of mucopolysaccharidoses, X-linkedichthyosis, and X-linked chondrodysplasia punctata.
! CT scan and MRI of the head typically demonstrate mildenlargement of the ventricles and demyelination bilaterally, with the maximum at the anterior and posterior poles of the ventricles.
▼Copyright © 2009. Demos Medical. All rights reserved. May not be reproduced in any form without permission from the publisher, except fair uses permitted under U.S. or applicable
copyright law.
EBSCO Publishing : eBook Clinical Collection (EBSCOhost) - printed on 2/4/2016 3:21 PM via SKANEAN: 276618 ; David, Ronald B..; Clinical Pediatric NeurologyAccount: ns071520

402 Section 2 ▼ General Pediatric Neurologic Diseases and Disorders
Annotated bibliographyBrooke MH. A clinician’s view of neuromuscular diseases. Bal-
timore: Williams & Wilkins, 1986.At more than 20 years, this edition is dated, particularly withrespect to molecular genetics. This single-author text, how-ever, remains among the most readable and lucid mono-graphs describing the clinical features of neuromusculardisorders.
Jones HR, DeVivo DC, Darras BT. Neuromuscular disorders ofinfancy, childhood, and adolescence: A clinician’s approach.Philadelphia: Butterworth Heinemann, 2003.A substantial text devoted to pediatric neuromuscular dis-ease. Currently, this is the definitive work in the field.
Engel AG. Myology, 3rd ed. New York: McGraw-Hill, 2004.The two-volume text is a comprehensive reference source re-garding muscle and disorders of that tissue.
Dyck PJ, Thomas PK. Peripheral neuropathy, 4th ed. Philadel-phia: Saunders, 2005.Definitive text regarding the neurobiology of peripheralnerve and peripheral neuropathies.
Table 18.12 Differential diagnosis of neuromusculardisorders presenting in the preadolescent
Anterior horn cell disorders
! Infection (polio, enteroviruses, rabies)
! Genetic (spinal muscular atrophies)
Peripheral neuropathies
! Genetically determined neuropathies
! Hereditary motor and sensory neuropathy (HMSN types Ia, Ib, Ix, II)
! Guillain-Barré syndrome
! Chronic inflammatory demyelinating neuropathy
Disorders affecting neuromuscular transmission
! Myasthenia gravis
! Congenital myasthenic syndromes
! Botulism (food-borne or wound)
Disorders affecting muscle
! Duchenne/Becker muscular dystrophy
! Severe childhood autosomal recessive muscular dystrophy and
other limb-girdle muscular dystrophies
! Myotonic dystrophy
! Congenital myopathy
! Inflammatory myopathy
! Metabolic disorders
! Hypokalemia
! Periodic paralysis
! Metabolic myopathies
Key Clinical Questions
! Is the infant’s diminished spontaneous movement dueto a neuromuscular disorder or a systemic disease?
! Is the hypotonia due to a central or peripheral process?! Does hypotonia improve with stimulation?! Are reflexes normal or diminished?! Is motor development improving, static, or worsening?! Is the pattern of weakness predominantly proximal or
distal?! Is there evidence of associated sensory loss?! Does weakness and sensory loss conform to a length
dependent pattern of severity?! Is diminished sensation predominantly related to
dysfunction of myelinated or unmyelinated axons?! Is motor dysfunction due to weakness or pain?! Is internal or external ophthalmoplegia present?! Do symptoms fluctuate in the course of the day, during
the week, over several months, or at all?! Is ataxia sensory or cerebellar?
▼
Table 18.11 Differential diagnosis of neuromusculardiseases presenting in the infant/toddler
Anterior horn cell disorders! Infection (polio, enteroviruses, rabies)! Genetic (spinal muscular atrophies)Peripheral neuropathies! Genetically determined neuropathy! Hereditary sensory-motor neuropathies (HMSN types Ia, Ib, Ix, II)! Guillain-Barré syndrome! Chronic inflammatory demyelinating neuropathyDisorders affecting the neuromuscular junction! Myasthenia gravis! Congenital myasthenic syndromes! Infant botulismDisorders affecting muscle! Congenital myopathy! (Not myotonic dystrophy in infants)! Congenital muscular dystrophy! Inflammatory myopathy! Metabolic disorders! Hypokalemia! Periodic paralysis! Metabolic myopathies! Pompe disease! Phosfructokinase deficiency! Debrancher deficiency! Phosphoglycerokinase deficiency! Methylglutaconic aciduria! Acyl-CoA-dehydrogenase deficiency (multiple variants)! Mitochondrial disorders
Copyright © 2009. Demos Medical. All rights reserved. May not be reproduced in any form without permission from the publisher, except fair uses permitted under U.S. or applicable
copyright law.
EBSCO Publishing : eBook Clinical Collection (EBSCOhost) - printed on 2/4/2016 3:31 PM via SKANEAN: 276618 ; David, Ronald B..; Clinical Pediatric NeurologyAccount: ns071520

Table 15.24 Differential diagnosis of childhood slowly progressive spinocerebellopathies
Type C* Age of onset Ataxia Sensory Dtr Babinski Muscle Mental Eye Heart Other
IOSCA 10 Infancy Severe Moderate Absent + Hypotonia Late Ptosis, optic No Hypogonadism
and late atrophy, athetosis, seizures,
ophthalmoplegia hearing loss
RL 17 Infancy Moderate Mild Absent + Atrophy (legs) Normal Normal No Pes cavus
SLO 7 Infancy Severe Moderate ↓ + Hypotonia Abnormal Ptosis, cataract Congenital Failure to thrive,
defects malformations
FA 9 1st decade Severe Moderate ↓ in LE + Atrophy (legs) Normal Normal Cardiomyopathy Pes cavus
BKS 4 1st decade Severe Moderate ↓ + late Atrophy (legs) Normal Degenerative Rare Pes cavus
retinopathy cardiomyopathy
Refsum 10 1st decade Severe Severe ↓ + Atrophy Normal Degenerative Hypertrophic Pes cavus, deafness,
retinopathy cardiomegaly ichthyosis
AVED 8 1st or 2nd Severe Moderate ↓ + Late atrophy Normal Cataract, Normal Pes cavus, scoliosis
decade retinopathy
NARP M* 1st decade Mild Mild ↑-early + Proximal Deterioration Degenerative No Seizures
or later ↓-late weakness retinopathy
SCD 1st or 2nd Moderate Paresthesia ↓ + Weakness and Frequent Macular changes No Microcephaly,
decade atrophy (legs) deterioration seizures
CDG 16 1st decade Moderate Mild ↑-early + Weakness and Deterioration Retinopathy, Pericarditis, Skeletal deformity
or later ↓-late atrophy (legs) strabismus hypertrophic
cardiomegaly
AT 11 1st decade Severe Mild ↓-late – Atrophy (late) Late Slow eye No Telangiectasia,
deterioration movements immune deficiency
AOA1 9 1st decade Severe Mild ↓-late – Atrophy (late) Mild Slow eye Dystonia
movements
MS 5 1st decade Moderate Mild ↑-early + in 50% Atrophy (legs) Deterioration Cataracts No Skeletal deformity,
truncal ↓-late (congenital) short stature
CX 2 2nd decade Mild-early Mild-early ↑ + Later Deterioration, Cataracts, Myocardial Tendon xanthomas,
early at times retinopathy infarction fractures, diarrhea
IOSCA, infantile onset spinocerebellar ataxia; C*, abnormal chromosome; RL, Roussy-Levy syndrome; M*, mitochondrial DNA; SLO, Smith-Lemli-Opitz; FA, Friedreich ataxia; BKS, Bassen-Kornzweig syndrome; AVED, ataxia
with isolated vitamin E deficiency; NARP, neuropathy, ataxia, retinitis pigmentosa syndrome; SCD, subacute combined degeneration; CDG, carbohydrate deficiency glycoprotein; AT, ataxia telangiectasia; AOA1, early-onset
ataxia with oculomotor apraxia; MS, Marinesco-Sjögren; CX, cerebrotendinous xanthomatosis.
296
Copyright © 2009. Demos Medical. All rights reserved. May not be reproduced in any form without permission from the publisher, except fair uses permitted under U.S. or applicable
copyright law.
EBSCO Publishing : eBook Clinical Collection (EBSCOhost) - printed on 2/4/2016 3:19 PM via SKANE
AN: 276618 ; David, Ronald B..; Clinical Pediatric Neurology
Account: ns071520
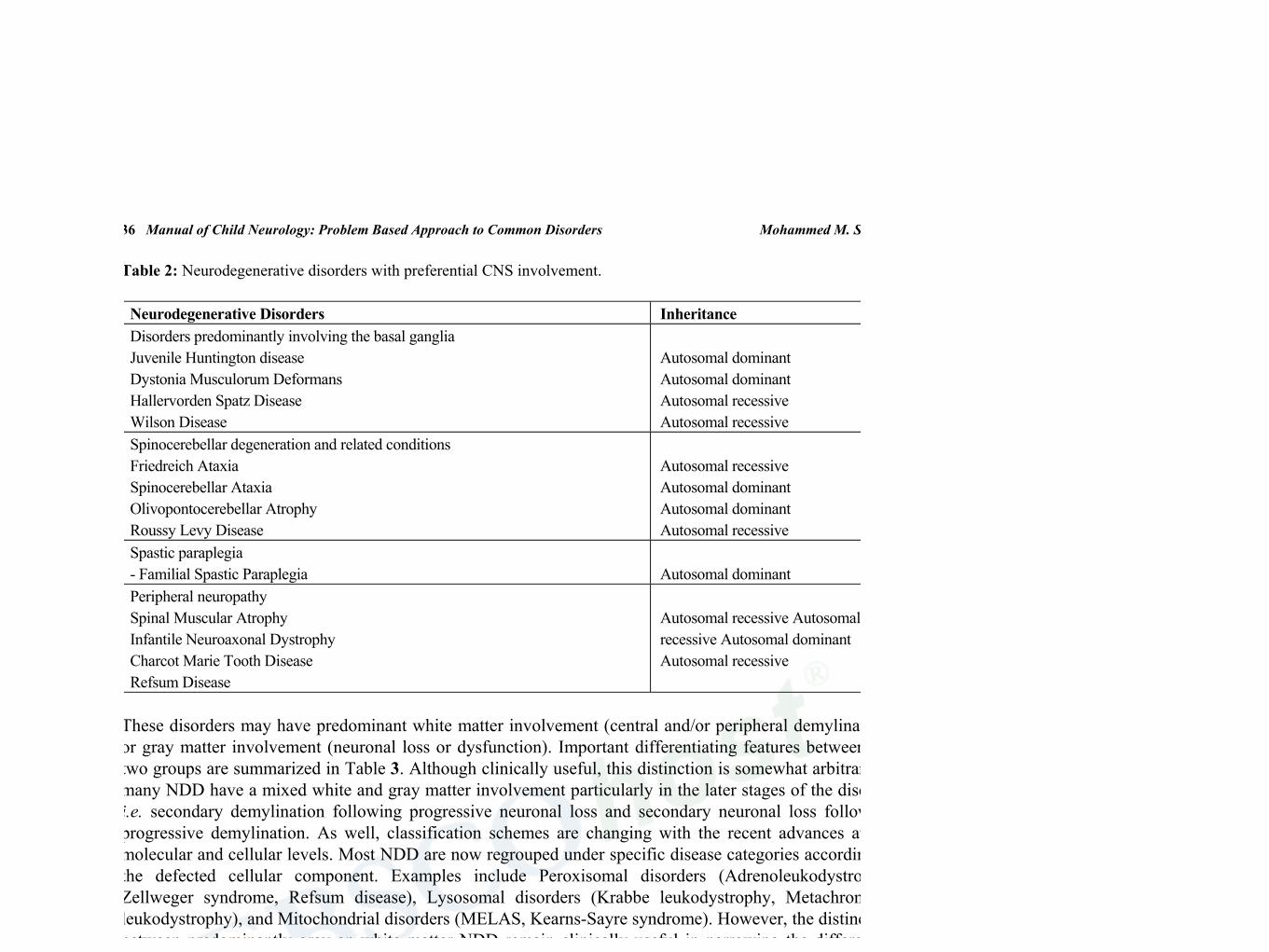
36 Manual of Child Neurology: Problem Based Approach to Common Disorders Mohammed M. S. Jan
Table 2: Neurodegenerative disorders with preferential CNS involvement.
Neurodegenerative Disorders Inheritance Disorders predominantly involving the basal ganglia Juvenile Huntington disease Dystonia Musculorum Deformans Hallervorden Spatz Disease Wilson Disease
Autosomal dominant Autosomal dominant Autosomal recessive Autosomal recessive
Spinocerebellar degeneration and related conditions Friedreich Ataxia Spinocerebellar Ataxia Olivopontocerebellar Atrophy Roussy Levy Disease
Autosomal recessive Autosomal dominant Autosomal dominant Autosomal recessive
Spastic paraplegia - Familial Spastic Paraplegia
Autosomal dominant
Peripheral neuropathy Spinal Muscular Atrophy Infantile Neuroaxonal Dystrophy Charcot Marie Tooth Disease Refsum Disease
Autosomal recessive Autosomal recessive Autosomal dominant Autosomal recessive
These disorders may have predominant white matter involvement (central and/or peripheral demylination) or gray matter involvement (neuronal loss or dysfunction). Important differentiating features between the two groups are summarized in Table 3. Although clinically useful, this distinction is somewhat arbitrary as many NDD have a mixed white and gray matter involvement particularly in the later stages of the disease, i.e. secondary demylination following progressive neuronal loss and secondary neuronal loss following progressive demylination. As well, classification schemes are changing with the recent advances at the molecular and cellular levels. Most NDD are now regrouped under specific disease categories according to the defected cellular component. Examples include Peroxisomal disorders (Adrenoleukodystrophy, Zellweger syndrome, Refsum disease), Lysosomal disorders (Krabbe leukodystrophy, Metachromatic leukodystrophy), and Mitochondrial disorders (MELAS, Kearns-Sayre syndrome). However, the distinction between predominantly gray an white matter NDD remain clinically useful in narrowing the differential diagnosis list, examining for associated and complicating features, and guiding the initial laboratory investigations (Table 3).
Table 3: Differentiation between White and Gray matter NDD.
Features White matter disorders Gray matter disorders
Age of onset Usually late (childhood) Usually early (infancy)
Head size May have megalencephaly Usually microcephaly
Seizures Late, rare Early, severe
Cognitive functions Initially normal Progressive dementia
Peripheral Neuropathy Early, demylination Late, axonal loss
Spasticity Early, severe Later, progressive
Reflexes Absent (neuropathy) Exaggerated (long tracts) Normal or exaggerated
Cerebellar signs Early, prominent Late
Fundal examination May show optic atrophy Retinal degeneration
EEG Diffuse delta slowing Epileptiform spikes
Electromyography (EMG) Slowed nerve conduction velocity Usually normal
Evoked potentials (VEP, ABR)
Prolonged or absent Usually normal
Electroretinograms (ERG) Normal Abnormal
Copy
righ
t © 2
012.
Ben
tham
Sci
ence
Pub
lish
ers.
All
rig
hts
rese
rved
. Ma
y no
t be
rep
rodu
ced
in a
ny f
orm
with
out
perm
issi
on f
rom
the
publ
ishe
r, e
xcep
t fa
ir u
ses
perm
itte
d un
der
U.S.
or
appl
icab
le c
opyr
ight
law
.
EBSCO Publishing : eBook Clinical Collection (EBSCOhost) - printed on 2/4/2016 3:38 PM via SKANEAN: 500550 ; Jan, Mohammed M.S.; Manual of Child Neurology : Problem Based Approach to Common DisordersAccount: ns071520

Vilka skall vi ta upp och varför?
Adrenoleukodystrofi
Duchennes muskel dystrofi
Charcot Marie Tooth


TACK!

Adrenoleukodystrofi
Debuterar med kognitiva/ neuropsykiatriska problem. Kan likna ADHD men progredierar. Alla pojkar har binjurebarks påverkan vid diagnos.
X- bunden. 20% av de kvinnliga bärarna utvecklar neurologiska symtom.
Finns adulta former bl.a. ” ren ” adrenal insufficient variant.
Diagnos: Ökad mängd VLCFA ( 20 % av kvinnliga bärare har normala halter. Genetik: ABCD1 genen.
Radiologi

ALD Behandling
Lorenzos olja: 4 delar Oelic acid + 1 del erucic acid. båda är monounsaturated omega 9 fatty acids.Ingen effekt på pojkar med symtom eller MR fynd.
Steroider + ev mineralkortikoid viktigt.
Benmärgs transplantation. Endast för pojkar med minimal kognitiv påverkan ( IQ > 80 ) och normalt neurologiskt status.

ALD - Framtiden är här.
Pågående studier
Artikel

Duchennes Muskeldystrofi
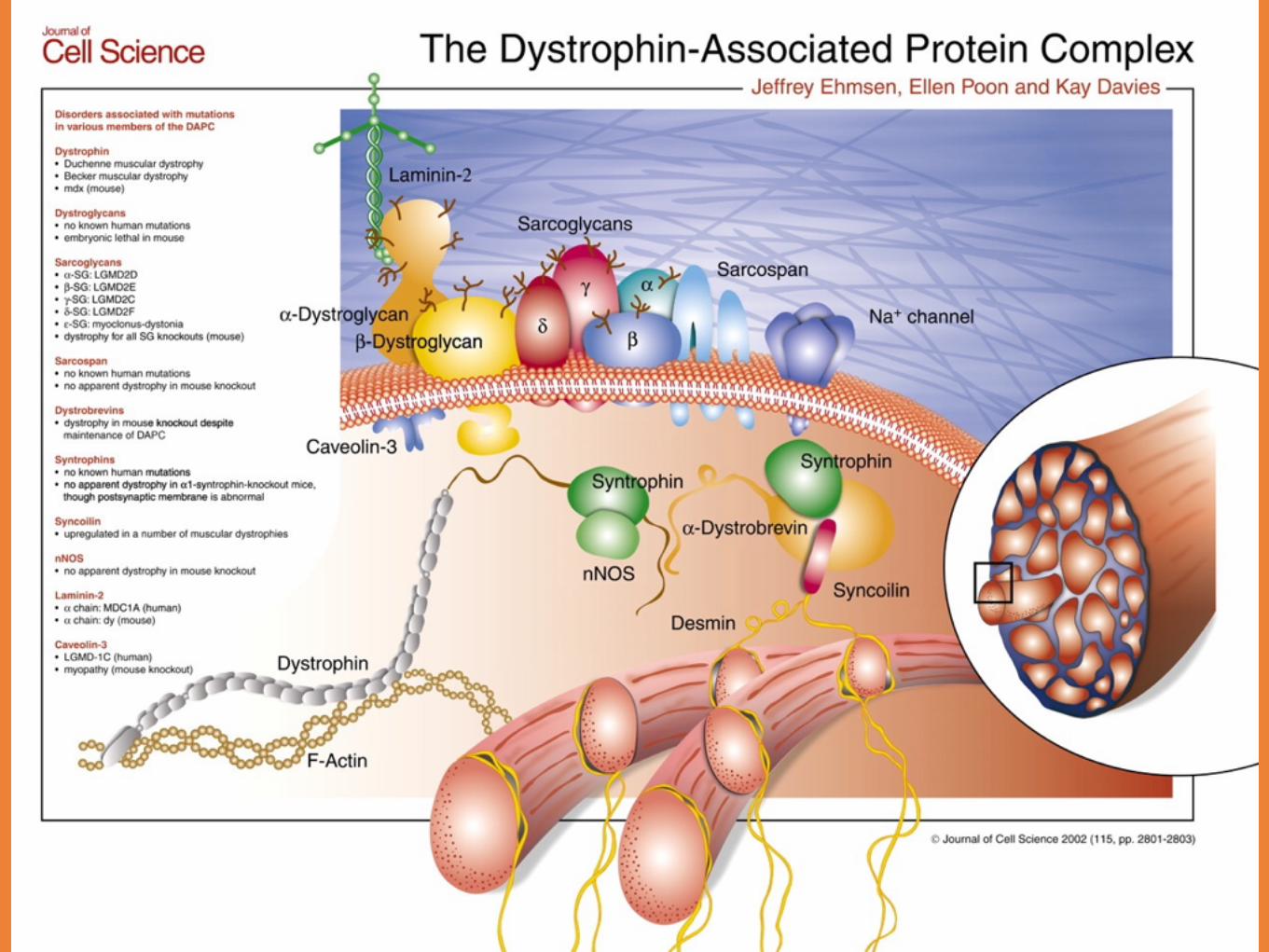
DMD fortsättning

DMD fortsättning
Mutation i Duchenne genen
Beroende på hur lite protein som uttrycks:Duchennes MDIntermediär formBecker
Diagnos vid 3 - 5 års ålder
Utan behandling död vid 19 år

DMD fortsättning
Behandling:SteroiderSjukgymnastikAndnings understöd

Charcot Marie Tooth
Kan vara såväl demyelinerande som axonal skada
Dominant; recessivt och X - bundna former
Vissa rent motoriska, sensoriska och vissa med kognitiv påverkan.
En mängd viktiga differential diagnoser.

Tillsist
Film 1
Film 2
Film 3
Film 4
Film 5

