spurenanalytik von elementspezies: … · resolution mass spectra of the ams. therefore, not msa...
TRANSCRIPT

1
Arbeits- und Ergebnisbericht
des Graduiertenkollegs GRK 826/3
Berichtszeitraum
Juli 2009 – April 2010 (Zeitpunkt der Erstellung des Berichts)
Spurenanalytik von Elementspezies: Methodenentwicklungen und Anwendungen
Trace analysis of elemental species:
Development of methods and applications
FB Chemie, Pharmazie und Geowissenschaften
Prof. Dr. T. Hoffmann Anorganische und Analytische Chemie
Prof. Dr. J.V. Kratz Prof. Dr. T. Reich
Kernchemie Prof. Dr. M. Kersten Geowissenschaften Prof. Dr. W. Wilcke
Geographisches Institut
Max-Planck-Institut für Chemie
Prof. Dr. S. Borrmann Abteilung
Partikelchemie
FB Biologie
Prof. Dr. H. König Mikrobiologie und Weinforschung
FB Medizin
Prof. Dr. H. Duschner Angewandte Struktur-
und Mikroanalytik
FB Physik, Mathematik
und Informatik Prof. Dr. S. Borrmann
Physik der Atmosphäre PD. Dr. Klaus Wendt
Physik
Beteiligte Fachbereiche und Arbeitsgruppen
2
Inhaltsverzeichnis
Jovana-Maria Diesch Formation and Transformation of Marine Aerosol
Species under Continental Influences 3
Daniel Fröhlich Speziation von Neptunium bei der Migration in Tongestein
8
Enrico Gromm Bestimmung von Komplexbildungskonstanten für die Komplexierung von Actiniden mit Huminstoffen sowie die Speziation von Plutonium im Ultraspurenbereich mit CE-(DAD)-ICP-MS und CE-RIMS
15
Elisabeth Kaschak Biochemie der Biomethylierung von Quecksilber durch Organismen aus dem Intestinaltrakt von Invertebraten
22
Michael Kundel Echtzeit-Bestimmung von gasförmigen Iodspezies mittels Flugzeit-Aersolmassenspektrometrie
26
Ka Hei Lui Speciation of Copper enriched in Agricultural Limespeciation and mobility of arsenic in agricultural lime
32
Ute Thorenz, geb. Müller Entwicklung einer TD-GC-MS Methode zur Messung von Halogenkohlenwasserstoffen aus biotischen und abiotischen Quellen
37
Niklas Schaper Entwicklung eines Picolitertropfengenerators für die emissions- und massenspektrometrische Elementspuren- und Speziesanalytik
42
Yvonne Scheller Detektion von Metallspezies in einzelnen lebenden Zellen mit hoher lateraler Auflösung
50
Kathrin Schilling Speziesabhängige Fraktionierung der stabilen Isotope von Selen während der biogenen Alkylierung in Böden
56
Nils Stöbener Elementspeziation von Neptunium im Ultraspurenbereich
63

3
Topic and supervising tutor of the PhD-project Scholarship holder: Jovana-Maria Diesch Formation and Transformation of Marine Aerosol Species under Continental Influences
Supervising tutor: Prof. Dr. Stephan Borrmann, Dr. Frank Drewnick Reporting period: July 2009 – April 2010 Presentation of the PhD-project and results obtained during the reporting period Motivation
The impact of atmospheric aerosol on the environment has an important influence on three different scales - local, regional and global. Air pollution can be caused on a local scale by industrial combustion processes, vehicular emissions and wood burning fires. The origin of the particles can also be natural, for example sea spray particles, photochemically formed particles and particles from resuspended soil. Due to the fact that the aerosol particles, created by such processes, are responsible for many adverse health effects, special attention is given to limit particle emissions. Regionally, urban aerosol originated from the previously mentioned anthropogenic sources is transported from cities to relatively clean rural areas. On a global scale the particles significantly influence our entire planet and the Earth´s climate by scattering and absorption of sunlight and by their influence on the formation of cloud droplets and ice crystals based on their role as cloud and ice condensation nuclei as well as their influence on heterogeneous chemistry in the troposphere and stratosphere. The importance of the impact of atmospheric aerosol in respect to environmental and health issues is a broadly accepted fact, nevertheless, for a better understanding of the formation, transformation and composition of aerosol particles as well as their chemical and physical properties further investigations in these subjects are of crucial importance.
One focus of this work will be the characterization of halogenated species within the
marine aerosol. Iodine is a trace element that is needed in minor amounts by many life-forms on earth including human. Organic and inorganic iodine compounds are also suspected to play an important role in the formation of new particles in the coastal marine boundary layer but it is still not fully understood which iodine species are responsible for nucleation and growth of aerosol particles. Another focus will be the speciation of sulfur components in atmospheric aerosol particles that also participate in particle formation. The ocean is a large source for sulfur species, so it will be interesting to understand the pathways of the formation of sulfur compounds after being emitted from the ocean to the atmospheric marine boundary layer.
I use the recently developed mobile laboratory ´MoLa´. MoLa is equipped with many
state-of-the-art aerosol instruments to study the chemical and physical particle properties and instruments to measure trace gases to get a better understanding of the composition of aerosol particles and trace gases and to study their production, transformation and their impact on the environment. On the one hand the mobile laboratory can be used for stationary measurements at flexible measurement locations on the other hand it is possible to measure during driving. Additionally, there is a meteorological station, a GPS and a webcam installed on the platform.

4
Approach and Results For a better comprehension of a selection of these processes a field campaign, DOMINO (Diel Oxidant Mechanisms In relation to Nitrogen Oxides) took place from 20th of November until 9th of December 2008 at the atmospheric research station "El Arenosillo" in southern Spain. The focus of this campaign was the study of atmospheric oxidation chemistry during the entire diurnal cycle. The station was located in a natural preserve near the city Huelva next to the Atlantic Ocean so that air masses from different directions can be separated and compared to characterize industrial, continental and marine influenced air masses separately. Therefore, the whole measured dataset was divided according to air mass trajectories. In figure 1 there is a map presented including trajectories corresponding to the sectors. So, in the first sector, calculated backtrajectories cross Sevilla before arriving at the station. In the second sector, wind directions include air arriving from the continent. In the third sector, the so-called Portugal+Huelva sector urban pollution was measured caused by Huelva, a city with extensive industrial activities. While these trajectories mainly spread over Portugal before passing Huelva, trajectories corresponding to the Marine+Huelva sector firstly pass the Atlantic Ocean. Finally, the fifth and sixth sectors include marine influenced air masses with the difference that Portugal+Marine trajectories also pass over Portugal. For each sector aerosol as well as gas concentrations were calculated individually.
Figure 1 Map marked with calculated trajectories around the measurement site; the air mass
trajectories are colored according to the associated air mass category Firstly, the diverse measured parameters were averaged individually for each sector, so that averaged values over the whole campaign at the inferface Continental, Ocean and City can be compared (Figure 2). The green bars represent average organic mass concentrations, the red bars average sulfate concentrations. Blue bars represent nitrate concentrations, orange ones the ammonium and the black ones show average black carbon concentrations. In the first sector not only the sulfate but also average black carbon concentrations are enhanced due to industrial emissions originating likely from Sevilla. In sector 2 the calculated organic aerosol concentration mean value is increased probably due to natural and anthropogenic emissions from the continent that lead to secondary aerosol.

5
Figure 2 Average particle composition values from the AMS organics, sulfate, nitrate, ammonium as well as black carbon values for the six sectors In the so-called city sector, the particle loading and composition is mainly determined by particle emissions of traffic and industrial emissions. This leads to high black carbon values as well as enhanced aerosol mass concentration values like the presented organic and nitrate mean values. In the last sector where emissions from the Marine sector are obtained, low aerosol concentrations are observed as expected. However, high sulfate concentrations were measured as the ocean is a large natural source for atmospheric sulfur.
Figure 3 Average particle concentration values (number concentration and mass concentration) for the different sectors In figure 3 average particle concentration values for the sectors were calculated individually. The grey bars represent average number concentrations, the brown bars average mass concentrations. While in the Continental, P+Huelva, M+Huelva sector large particle number concentrations with dependent on that less mass concentrations were measured, over the sea mainly large particles of small number concentrations were determined. The smaller the particles, the less aged they are because they grow during the aging process. In the Sevilla sector particles from the emissions travel a long distance until they reach the station and also in the marine sector particles are aged while in the Continental, P+Huelva, M+Huelva sector many freshly produced small particles occur from the industries, from vehicles etc. The same information is given in the particle size distributions (figure 4) with additional details. The number size distributions for the Sevilla, Continental, Marine+Huelva and Portugal+ Marine sector show a bimodal size distribution. The dominant mode for the Sevilla sector has its maximum at 50 nm while the other sectors have their dominant maxima at 12 nm. Possible reasons are a mixing of aged continental or in the Portugal+Marine sector aged marine aerosol together with freshly created particles originating from urban pollution in the cities Sevilla and Huelva, biomass burning in the continental sector or emissions from fishing boats in the marine sector. The extreme urban pollution in Huelva causes the highest particle number concentrations as well as the major volume concentration of the particles including

6
small particles in a wide size range. Over the sea mainly large aged particles with less number concentration were measured.
Figure 4 Averaged particle size distributions in a size range of 7 until 500 nm for the different sectors In figure 5 correlations between ammonium, sulfate and nitrate for the sectors are shown. In the continental sector most of the time pure ammonium sulfate occurs while over the sea the ammonium concentrations are low, therefore the species sulfuric acid exists because sulfate is not completely neutralized. In the Huelva sector the concentration of ammonium is increased. In figure 5 b the nitrate to sulfate ratio of the sectors is shown. It can be noticed that there is very often a significant additional occurence of ammonium nitrate in the Huelva sector in the opposite to the marine sector where more sulfate than nitrate is observed. Two sulfur species were identified, but another species, MSA was expected.
Figure 5 Correlations of ammonium (a) and nitrate (b) versus sulfate within the sectors MSA is an oxidation product of dimethyl sulfide (DMS) that is the most abundant biological sulfur species emitted to the atmosphere. Emissions occur by phytoplankton, so MSA is a good tracer for sulfur emissions of marine origin. For calculating the MSA concentrations an extraction procedure developed in the framework of the GRK (Zorn et al., Atmos.Chem.Phys.2008) was used. In figure 6 averaged MSA concentrations for the sectors were calculated, however only for the marine sector it is possible to identify MSA. In the Huelva sector a huge organic signal of the same m/z ratio like MSA was identified in the high resolution mass spectra of the AMS. Therefore, not MSA but the nitro methane peak was fitted and calculated.

7
Figure 6 Averaged MSA concentrations for the sectors
In table 1 the sulfate signal was splitted into all sulfate components that have been observed. It can be noticed that there is a significant additional occurence of sulfuric acid in all sectors with the highest abundance in the Marine sector.
Table 1 Different sulfate species – ammonium sulfate, sulfuric acid and methyl sulfuric acid in the individual sectors
Duration of the PhD-project Since July 2009 List of oral presentations [1] J.-M. Diesch, Characterization of the ambient aerosol at the continental, urban and marine interface, GRK-seminar, Bad Münster am Stein, October, 20th, 2009. [2] J.-M. Diesch, S. R. Zorn, S.-L. von der Weiden, F. Drewnick, Investigation of the ambient aerosol at the continental, urban and marine interface, Madrid, November 4th, 2009. [3] J.-M. Diesch, Characterization of the ambient aerosol at the continental, urban, marine interface, Mainz, April, 15th, 2010. Participation in the course program of the GRK “Ringvorlesung I” winter semester 2009/2010 Tutors Supervising tutor: Dr. Frank Drewnick Stay abroad AMS user meeting, Manchester 2008 Domino campaign workshop, Madrid, November 2-4, 2010 Twice times a one-week stay during two campaigns in Paris (Megapoli I + II), which is part of the European Union FP7, July 7-31, 2009 and January 15 until February 15, 2010

8
Thema und Betreuer des Dissertationsprojekts Stipendiat: Daniel Fröhlich Speziation von Neptunium bei der Migration in Tongestein Betreuer: Prof. Dr. T. Reich Berichtszeitraum: März 2009 – Februar 2010 Darstellung des Dissertationsprojekts und der Forschungsergebnisse im Berichtszeitraum Einführung
Die Problematik um die Endlagerung hochradioaktiver Abfälle ist bisher noch nicht geklärt. Eine Möglichkeit stellt die Endlagerung in tiefen geologischen Gesteinsformationen dar. In verschiedenen Ländern Europas (u.a. Deutschland, Frankreich und Schweiz) wird Tongestein als eine mögliche Wirtsgesteinsformation in Betracht gezogen. [1-3] In einem solchen Endlager wird die Aktivität der Abfälle nach einer Lagerzeit von mehr als 100.000 Jahren vor allem durch Plutonium und die minoren Aktiniden Americium, Curium und Neptunium dominiert werden. Im Falle das Neptunium muss auf Grund seiner langen Halbwertszeit (2,14 × 106 a) vor allem das Nuklid Np-237 betrachtet werden. Vor der Errichtung eines Endlagers für hochradioaktive Abfälle müssen genaue Untersuchungen hinsichtlich des Migrationsverhaltens der Radionuklide im umgebenden Wirtsgestein für den Fall eines Austritts aus den Aufbewahrungsbehältern durchgeführt werden.
Zur Untersuchung des Sorptions- und Diffusionsverhaltens von Neptunium wird Opalinuston aus Mont Terri (Schweiz) als natürliches Referenzmaterial verwendet. Dieses natürliche Tongestein besteht zum Großteil (> 60%) aus Tonmineralen (Kaolinit, Smectit, Illit, Chlorit). Dazu kommen Quarz und Calcit mit Anteilen über 10% sowie ~ 4% Eisen(II)-Minerale und Spuren von Albit, Feldspäten und Tonorganik.
Neptunium kann in wässriger Lösung in den Oxidationsstufen III bis VII existieren. Unter natürlichen Bedingungen sind jedoch nur die Oxidationsstufen IV und V von Relevanz.
Aufbauend auf früheren Arbeiten [4] soll das Sorptionsverhalten von Neptunium in Abhängigkeit verschiedener äußerer Parameter weiter untersucht werden. Mittels Batch-Experimenten werden die zugehörigen Verteilungskoeffizienten bestimmt. Diese Migrationsstudien werden durch spektroskopische Speziationsmethoden ergänzt. Die an Tonoberflächen gebundene Neptuniumspezies wird dabei mittels Röntgenabsorptionsspektroskopie (XAS) untersucht. Zum einen steht dabei die Affinität von Neptunium für bestimmte Mineralphasen im Vordergrund, zum anderen werden die Abstände der nächsten Nachbarn des Neptuniums mittels EXAFS-Spektroskopie bestimmt. Die Messung erfolgt sowohl an feuchten Pasten als auch an Ton-Dünnschliffen. Diese Messungen werden an der ROBL Beamline in Grenoble (Frankreich) und an der µ-XAS Beamline der Swiss Light Source (Schweiz) durchgeführt.

9
Ergebnisse
Batch-Experimente
Bei der Endlagerung hochradioaktiver Stoffe wird es durch die Radioaktivität für einen längeren Zeitraum zu einer deutlichen Erhöhung der Temperatur im Umfeld der Abfälle kommen. Im Falle von Tongestein soll die Aufheizung am Kontakt zur Oberfläche der Abfallbehälter nicht mehr als 100 °C betragen. Daher wurde die Sorption von Np(V) im Bereich zwischen 20 und 80 °C untersucht. Die Np-Konzentration betrug in allen Fällen ~ 8 µM und als Hintergrundelektrolyt wurde synthetisches Opalinuston Porenwasser (pH = 7,6, I = 0,4 M) verwendet. Die Experimente wurden im Gleichgewicht mit atmosphärischem CO2 durchgeführt. Ton und Lösung wurden über einen Zeitraum von drei Tagen vorkonditioniert. Die Kontaktzeit zwischen Ton und Radionuklid betrug ebenfalls drei Tage. Während dieses Zeitraums wurde der pH-Wert durchgehend kontrolliert und gegebenenfalls nachgestellt. Die Phasentrennung erfolgt mittels Ultrazentrifugation (30.000 U/min, 100.000 g), und die überstehende Lösung jeder Probe wurde mittel -Spektroskopie und Flüssigszintillationszählung analysiert. Das Ergebnis ist in Abbildung 1 dargestellt.
Abbildung 1: Temperaturabhängigkeit des Verteilungskoeffizienten für die Sorption von Np(V) an Opalinuston in synthetischem Porenwasser, pH = 7,6, Gleichgewicht mit Luft, [Np(V)] = 8 µM
Es ist deutlich erkennbar, dass eine Erhöhung der Temperatur auf 40 °C unter diesen experimentellen Bedingungen keinen Einfluss auf die Sorption von Np(V) an Opalinuston hat, ebenso wenig das Festottverhältnis. Wird die Temperatur jedoch weiter bis auf 80 °C erhöht, so steigt die Sorption von Neptunium und damit der Verteilungskoeffizient (Kd) um mehr als eine Größenordnung. Die aus Abbildung 1 erhaltenen Mittelwerte des Kd sind mit der zugehörigen Standardabweichung (1 ) in Tabelle 1 dargestellt.
Tabelle 1: Mittlere Kd-Werte für die Sorption von Np(V) an Opalinuston in Abhängigkeit der Temperatur
0 2 4 6 8 10 12 14 16 18 20 220,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
RT[5] 40 °C 60 °C 70 °C 80 °C
log
(Kd /
L/kg
)
Feststoffverhältnis / g/L
Temperatur / °C Kd-Mittelwert / L/kg 21 25 ± 5 40 23 ± 4 60 57 ± 8 70 217 ± 67 80 674 ± 245

10
Ein mögliche Erklärung für das Ansteigen der Sorption könnte eine Erhöhung der Oberflächenladung der Tonoberfläche in Folge der Temperaturerhöhung sein [6]. Der endotherme Charakter der Sorption ist des Weiteren im Einklang mit Literaturergebnissen [7,8]. Trägt man den log(Kd) Wert gegen die inverse Temperatur auf, so lässt sich außerdem eine Sorptionsenthalpie, sowie die –entropie nach der Van’t Hoff’schen Gleichung ermitteln.
TRH
RS
K rrd
13,23,2
)log(
Die entsprechende Auftragung für die in Tabelle 1 gezeigten Werte ist in Abbildung 2 dargestellt. Der Bereich zwischen 80 und 40 °C lässt sich gut anpassen. Die Gleichung der zugehörigen Fit-Geraden ist ebenfalls in Abbildung 2 gezeigt. Aus den Fit-Paramtern ergibt sich eine Sorptionsenthalpie rH = 77,3 ±15,8 kJ/mol. Dieser Wert liegt in der gleichen Größenordnung wie für die Sorption von Ni2+ und Eu3+ an Montmorillonit [7].
Abbildung 2: Van't Hoff Plot zur Bestimmung der Sorptionsenthalpie und -entropie für die Sorption von Np(V) an Opalinuston
Neben der Temperatur spielen andere Faktoren eine wichtige Rolle für die Neptunium Sorption an Tongestein, darunter auch die Elektrolytzusammensetzung. Das Opalinuston Porenwasser ist ein komplexer Elektrolyt mit NaCl, CaCl2 und MgCl2 als Hauptbestandteile, einem pH = 7,6 und einer hohen Ionenstärke von I = 0,4 M [9]. Vergangene Studien haben gezeigt, dass die Sorption verschiedener Actiniden (Th, Np) in Porenwasser deutlich geringer ist als in NaClO4 Lösung, einem typischen Hintergrundelektrolyt für Sorptionsstudien. Um diesen Sachverhalt genauer zu untersuchen, wurden verschiedene Sorptionsisothermen unter Variation des Feststoffverhältnisses aufgenommen. Dabei wurden einerseits die Ionenstärke zwischen 0,1 und 0,4 M und zum anderen der Hintergrundelektrolyt selbst variiert (NaCl, NaClO4, CaCl2 und MgCl2). Alle Experimente erfolgten im Gleichgewicht mit dem CO2 der Luft und bei einem pH 7,6, um die Ergebnisse mit denen für Porenwasser vergleichen zu können. Das Ergebnis ist in Abbildung 3 dargestellt.
0,0028 0,0029 0,0030 0,0031 0,0032 0,0033 0,0034 0,0035
1,2
1,4
1,6
1,8
2,0
2,2
2,4
2,6
2,8
3,0
3,2
log(
Kd /
L/kg
)
Temperatur-1 / K-1
log(Kd) = (-4040 ± 826 K) × 1/T + (14 ± 3)
R2 = 0,92
11
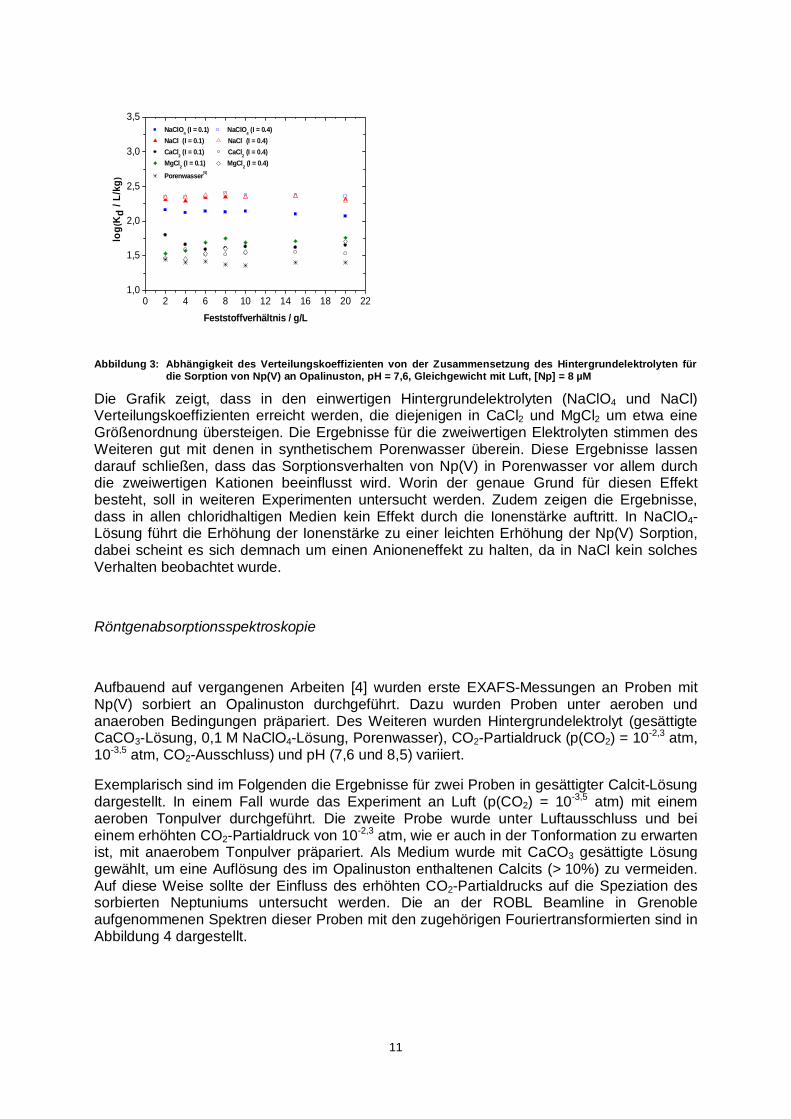
Abbildung 3: Abhängigkeit des Verteilungskoeffizienten von der Zusammensetzung des Hintergrundelektrolyten für die Sorption von Np(V) an Opalinuston, pH = 7,6, Gleichgewicht mit Luft, [Np] = 8 µM
Die Grafik zeigt, dass in den einwertigen Hintergrundelektrolyten (NaClO4 und NaCl) Verteilungskoeffizienten erreicht werden, die diejenigen in CaCl2 und MgCl2 um etwa eine Größenordnung übersteigen. Die Ergebnisse für die zweiwertigen Elektrolyten stimmen des Weiteren gut mit denen in synthetischem Porenwasser überein. Diese Ergebnisse lassen darauf schließen, dass das Sorptionsverhalten von Np(V) in Porenwasser vor allem durch die zweiwertigen Kationen beeinflusst wird. Worin der genaue Grund für diesen Effekt besteht, soll in weiteren Experimenten untersucht werden. Zudem zeigen die Ergebnisse, dass in allen chloridhaltigen Medien kein Effekt durch die Ionenstärke auftritt. In NaClO4-Lösung führt die Erhöhung der Ionenstärke zu einer leichten Erhöhung der Np(V) Sorption, dabei scheint es sich demnach um einen Anioneneffekt zu halten, da in NaCl kein solches Verhalten beobachtet wurde.
Röntgenabsorptionsspektroskopie
Aufbauend auf vergangenen Arbeiten [4] wurden erste EXAFS-Messungen an Proben mit Np(V) sorbiert an Opalinuston durchgeführt. Dazu wurden Proben unter aeroben und anaeroben Bedingungen präpariert. Des Weiteren wurden Hintergrundelektrolyt (gesättigte CaCO3-Lösung, 0,1 M NaClO4-Lösung, Porenwasser), CO2-Partialdruck (p(CO2) = 10-2,3 atm, 10-3,5 atm, CO2-Ausschluss) und pH (7,6 und 8,5) variiert.
Exemplarisch sind im Folgenden die Ergebnisse für zwei Proben in gesättigter Calcit-Lösung dargestellt. In einem Fall wurde das Experiment an Luft (p(CO2) = 10-3,5 atm) mit einem aeroben Tonpulver durchgeführt. Die zweite Probe wurde unter Luftausschluss und bei einem erhöhten CO2-Partialdruck von 10-2,3 atm, wie er auch in der Tonformation zu erwarten ist, mit anaerobem Tonpulver präpariert. Als Medium wurde mit CaCO3 gesättigte Lösung gewählt, um eine Auflösung des im Opalinuston enthaltenen Calcits (> 10%) zu vermeiden. Auf diese Weise sollte der Einfluss des erhöhten CO2-Partialdrucks auf die Speziation des sorbierten Neptuniums untersucht werden. Die an der ROBL Beamline in Grenoble aufgenommenen Spektren dieser Proben mit den zugehörigen Fouriertransformierten sind in Abbildung 4 dargestellt.
0 2 4 6 8 10 12 14 16 18 20 221,0
1,5
2,0
2,5
3,0
3,5 lo
g(K
d / L
/kg)
Feststoffverhältnis / g/L
NaClO4 (I = 0.1) NaClO4 (I = 0.4) NaCl1 (I = 0.1) NaCl2 (I = 0.4) CaCl2 (I = 0.1) CaCl2 (I = 0.4) MgCl2 (I = 0.1) MgCl2 (I = 0.4)
Porenwasser[5]
12
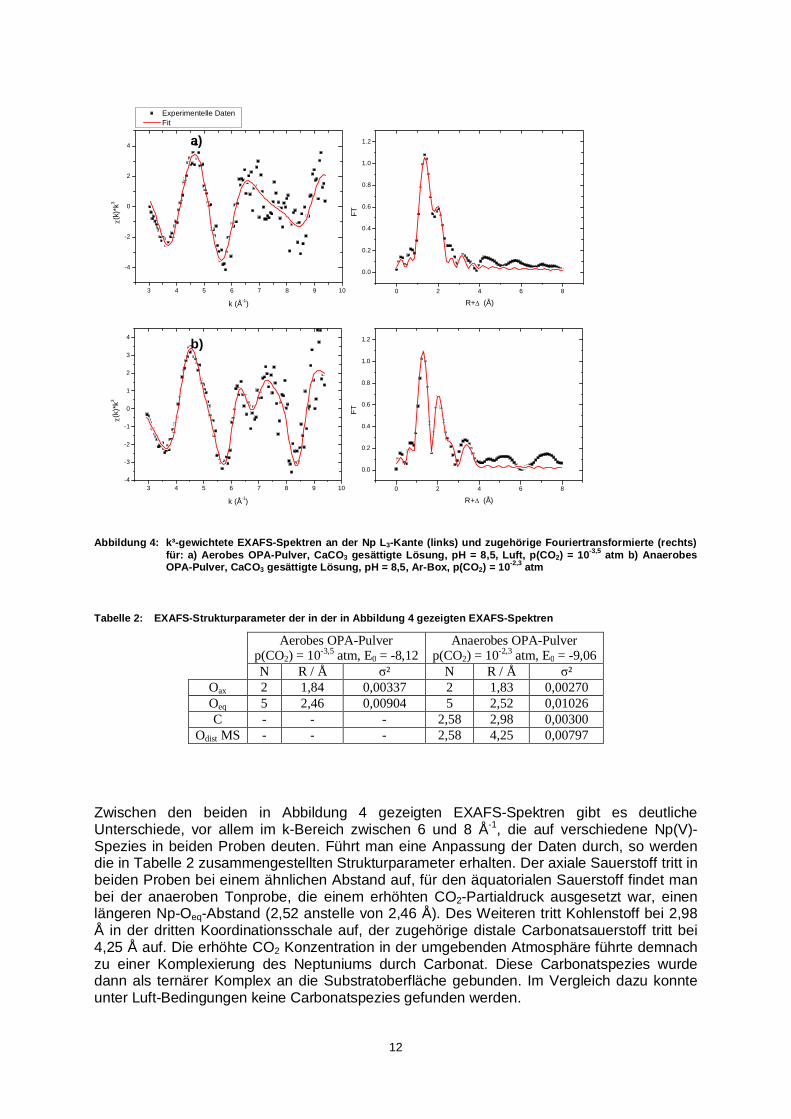
Abbildung 4: k³-gewichtete EXAFS-Spektren an der Np L3-Kante (links) und zugehörige Fouriertransformierte (rechts) für: a) Aerobes OPA-Pulver, CaCO3 gesättigte Lösung, pH = 8,5, Luft, p(CO2) = 10-3,5 atm b) Anaerobes OPA-Pulver, CaCO3 gesättigte Lösung, pH = 8,5, Ar-Box, p(CO2) = 10-2,3 atm
Tabelle 2: EXAFS-Strukturparameter der in der in Abbildung 4 gezeigten EXAFS-Spektren
Aerobes OPA-Pulver p(CO2) = 10-3,5 atm, E0 = -8,12
Anaerobes OPA-Pulver p(CO2) = 10-2,3 atm, E0 = -9,06
N R / Å ² N R / Å ² Oax 2 1,84 0,00337 2 1,83 0,00270 Oeq 5 2,46 0,00904 5 2,52 0,01026 C - - - 2,58 2,98 0,00300
Odist MS - - - 2,58 4,25 0,00797
Zwischen den beiden in Abbildung 4 gezeigten EXAFS-Spektren gibt es deutliche Unterschiede, vor allem im k-Bereich zwischen 6 und 8 Å-1, die auf verschiedene Np(V)-Spezies in beiden Proben deuten. Führt man eine Anpassung der Daten durch, so werden die in Tabelle 2 zusammengestellten Strukturparameter erhalten. Der axiale Sauerstoff tritt in beiden Proben bei einem ähnlichen Abstand auf, für den äquatorialen Sauerstoff findet man bei der anaeroben Tonprobe, die einem erhöhten CO2-Partialdruck ausgesetzt war, einen längeren Np-Oeq-Abstand (2,52 anstelle von 2,46 Å). Des Weiteren tritt Kohlenstoff bei 2,98 Å in der dritten Koordinationsschale auf, der zugehörige distale Carbonatsauerstoff tritt bei 4,25 Å auf. Die erhöhte CO2 Konzentration in der umgebenden Atmosphäre führte demnach zu einer Komplexierung des Neptuniums durch Carbonat. Diese Carbonatspezies wurde dann als ternärer Komplex an die Substratoberfläche gebunden. Im Vergleich dazu konnte unter Luft-Bedingungen keine Carbonatspezies gefunden werden.
3 4 5 6 7 8 9 10
-4
-2
0
2
4
(k
)*k3
k (Å-1)
Experimentelle Daten Fit
0 2 4 6 8
0.0
0.2
0.4
0.6
0.8
1.0
1.2
FT
R+ (Å)
0 2 4 6 8
0.0
0.2
0.4
0.6
0.8
1.0
1.2
FT
R+ (Å)
3 4 5 6 7 8 9 10-4
-3
-2
-1
0
1
2
3
4
(k)*
k3
k (Å-1)
a)
b)

13
[1] Hoth, P., Wirth, H., Reinhold, K., Bräuer, V., Krull, P., Feldrappe, H.: Endlagerung radioaktiver Abfälle in tiefen geologischen Formationen Deutschlands – Untersuchung und Bewertung von Tongesteinsformationen. Bundesanstalt für Geowissenschaften und Rohstoffe BGR. Hannover/Germany 2007.
[2] NAGRA: Projekt Opalinuston – Synthese der geowissenschaftlichen Untersuchungsergebnisse, Entsorgungsnachweis für abgebrannte Brennelemente, verglaste hochaktive sowie langlebige mittelaktive Abfälle. Technical Report NTB 02-03, NAGRA Nationale Genossenschaft für die Lagerung radioaktiver Abfälle, Wettingen/Switzerland 2002.
[3] OECD: Safety of geological disposal of high-level and long-lived radioactive waste in France - An international peer review of the “Dossier 2005 Argile” concerning disposal in the Callovo-Oxfordian formation. NEA No. 6178, OECD 2006.
[4] Fröhlich, D.R.: Sorption von Np(V) an Opalinuston, Diplomarbeit, Universität Mainz, 2008.
[5] Wu, T., Amayri, S., Drebert, J., Van Loon, L.R., Reich, T., Neptunium(V) sorption and diffusion in Opalinus Clay. Environ. Sci. Technol., 43, 6567, (2009).
[6] Ward D. B. and Brady P. V. Effect of Al and organic acids on the surface chemistry of kaolinite. Clays Clay Miner., 46, 453, (1998).
[7] Tertre, E., Berger, G., Castet, S., Loubet, M., Giffaut, E., Experimental sorption of Ni2+, Cs+ and Ln3+ onto montmorillonite up to 150 °C.. Geochim. Cosmochim. Acta, 69, 4937, (2005).
[8] Tertre, E., Berger, G., Simoni, E., Castet, S., Giffaut, E., Loubet, M., Catalette, H., Europium retention onto clay minerals from 25 to 150 °C: Experimental measurements, spectroscopic features and sorption modelling., Geochim. Cosmochim. Acta, 70, 4563, (2006).
[9] Van Loon, L.R., Soler, J.M., Bradbury, M.H., Diffusion of HTO, 36Cl- and 125I- in Opalinus Clay samples from Mont Terri: Effect of confining pressure. J. Contam. Hydrol., 61, 73 (2003).
Dauer der Promotion Beginn Januar 2009, voraussichtlicher Abschluss Januar 2012. Angaben zu entstandenen Publikationen, Kongressbeiträgen etc. (seit Beginn der Promotion)
T. Wunderlich, S. Amayri, R. Buda, D. Fröhlich, E. Gromm, J. V. Kratz, T. Reich, N. Trautmann, T. Wu: Sorption von Neptunium und Plutonium an Opalinuston unter aeroben und anaeroben Bedingungen. Vortrag beim 6. Verbundprojektworkshop “Wechselwirkung und Transport von Actiniden im natürlichen Tongestein unter Berücksichtigung von Huminstoffen und Tonorganika“, Leipzig, 07.-08.04.2009.
T. Reich, S. Amayri, J. Drebert, D. Fröhlich, L.R. Van Loon, T. Wu: Sorption and diffusion of Np(V) in Opalinus clay. Poster, Goldschmidt Conference, Davos, Schweiz, 21.-26.06.2009.

14
S. Amayri, D.R. Fröhlich, T. Wu., J. Drebert, L.R. Van Loon, T. Reich: Untersuchung der Sorption und Diffusion von Neptunium(V) in Opalinuston. Vortrag bei der Jahrestagung der GDCh, Frankfurt, 30.08.-02.09.2009.
D.R. Fröhlich, S. Amayri, J. Drebert, T. Reich: Sorption and speciation ob neptunium(V) on Opalinus clay. Poster; Tracespec Conference, Mainz, 15.-18.09.2009.
D.R. Fröhlich, S. Amayri, J. Drebert, T. Reich: Study of neptunium(V) sorption on Opalinus clay under aerobic/anaerobic conditions. Poster; Migration Conference, Kennwick, Washington, USA, 20.-25.09.2009.
S. Amayri, D.R. Fröhlich, T. Wu., J. Drebert, L.R. Van Loon, T. Reich: Sorption and diffusion behavior of neptunium(V) in Opalinus clay. Poster; Migration Conference, Kennwick, Washington, USA, 20.-25.09.2009.
S. Amayri, R. Buda, J. Drebert, D.R. Fröhlich, E. Gromm, J. V. Kratz, T. Reich, N. Trautmann, L.R. Van Loon, T. Wu, T. Wunderlich: Interaction of neptunium and plutonium with Opalinus clay. Vortrag beim 7. Verbundprojektworkshop “Wechselwirkung und Transport von Actiniden im natürlichen Tongestein unter Berücksichtigung von Huminstoffen und Tonorganika“, Mainz, 06.-07.10.2009.
D.R. Fröhlich, S. Amayri, J. Drebert, L.R. Van Loon, T. Reich: Sorptions- und Diffusions-untersuchungen mit Neptunium. Vortrag beim 8. Verbundprojektworkshop “Wechselwirkung und Transport von Actiniden im natürlichen Tongestein unter Berücksichtigung von Huminstoffen und Tonorganika“, Dresden, 13.-14.04.2010.
Zusammenarbeit mit bzw. Bezug zu anderen Projekten (innerhalb und außerhalb des GRK) Es besteht eine große Unterstützung unserer Forschungsarbeiten durch das Max-Planck-Institut für allgemeine Chemie, Mainz, im speziellen durch die Herren Joachim Huth und Maik Biegler. Im Rahmen dieser Zusammenarbeit werden regelmäßig Ton-Dünnschliffe für röntgenabsorptions-spektroskopische Untersuchungen präpariert. Des Weiteren wurde ein Neptuniumoxid-Standard für die Elektronenmikroskopie hergestellt und man ermöglicht uns die Nutzung der Elektronenmikroskopie zur Untersuchung des Sorptionsverhaltens von Neptunium auf Tonoberflächen.
Teilnahme am Studienprogramm (seit Beginn der Promotion) Ringvorlesung Wintersemester 2009/2010 Chemometriekurs „Statistik, Chemometrie und Qualitätssicherung“, Prof. J. Einax,
Friedrich-Schiller-Universität Jena, Institut für Anorganische und Analytische Chemie, Lehrbereich Umweltanalytik
Betreuung im Graduiertenkolleg Betreuer: Prof. Dr. T. Reich Auslandsaufenthalte (seit Beginn der Promotion) ESRF, Grenoble, Frankreich; Messzeit; 03.04.-06.04.2009 Paul Scherrer Institut (PSI), Villigen, Schweiz; Testexperimente; 11.08.–12.08.2009 Kennwick, Washington, USA; Teilnahme an der Migration Konferenz; 20.09.-25.09.2009 ESRF, Grenoble, Frankreich; Messzeit; 04.11.-06.11.2009

15
Thema und Betreuer des Dissertationsprojekts Stipendiat: Enrico Gromm Thema: Bestimmung von Komplexbildungskonstanten für die Komplexierung von Actiniden mit Huminstoffen sowie die Speziation von Plutonium im Ultraspurenbereich mit CE-(DAD)-ICP-MS und CE-RIMS Betreuer: Prof. Dr. Jens Volker Kratz Berichtszeitraum: März 2009 – Februar 2010 Darstellung des Dissertationsprojekts und der Forschungsergebnisse im Berichtszeitraum Huminsäurekomplexierung:
Bezüglich der Endlagerung von hochradioaktiven Abfällen in tiefen geologischen Formationen ist die Bestimmung von Komplexbildungskonstanten für die Huminstoff-komplexierung von Actiniden und, als Modellsubstanzen, Lanthaniden von großem Interesse. In den meisten aquatischen Systemen sowie in Tonformationen sind natürliche organische Materialien wie Huminstoffe vorhanden. Diese können als Liganden für Metallionen fungieren und somit einen Einfluss auf die Speziation und Migration von Radionukliden haben. Abbildung 1 zeigt hierzu schematisch ein Huminsäuremolekül mit dessen Bindungs- und Komplexierungsmöglichkeiten.
Abb. 1: Strukturschema eines Huminstoffmoleküls und seiner Bindung an die Oberfläche eines Tonminerals (M = Metallkationen).[1]
In dieser Arbeit wird das binäre System Huminstoff/Metallion untersucht. Zur Bestimmung von Komplexbildungskonstanten werden mittels Kapillarelektrophorese (CE) die freien von
16
den komplexierten Metallionen getrennt und die einzelnen Spezies mittels Dioden-Array-Detektor (DAD) und Massenspektrometrie mit induktiv gekoppeltem Plasma (ICP-MS) detektiert. Durch das Anlegen von Druck auf die CE-Kapillare während der Trennung ist es möglich sowohl die kationischen als auch die anionischen Spezies am Kapillarausgang zu detektieren. Während die freien Metallionen und die metallhaltigen Huminstoffspezies durch die ICP-MS nachgewiesen werden können, werden die Huminstoffe sowie eventuelle EOF-Marker (EOF: elektroosmotischer Fluss) aufgrund ihrer Lichtabsorption durch den DAD erfasst. Ausgehend von den Anteilen an freiem und gebundenem Metall können die Komplexbildungskonstanten nach dem Ladungsneutralisationsmodell[2] berechnet werden.
Frühere Arbeiten haben gezeigt, dass die CE-ICP-MS- sowie die CE-DAD-ICP-MS-Kopplung bei der Untersuchung des Komplexierungsverhaltens von Metallionen mit Huminstoffen anwendbar ist.[3,4] Aufgrund der teilweisen Dissoziation der Metall-Humat-Komplexe im elektrischen Feld ist die Interpretation der erhaltenen Elektropherogramme nicht einfach. Neben den kationischen freien Metallionen und dem anionischen Metall-Humat-Komplex ist eine weitere Spezies ersichtlich, welche aus ursprünglich gebundenem Metall besteht. Es wird angenommen, dass diese Metallionen aufgrund des starken elektrischen Feldes während der CE-Trennung von „schwachen Bindungsstellen“ der Huminstoffe dissoziiert werden.
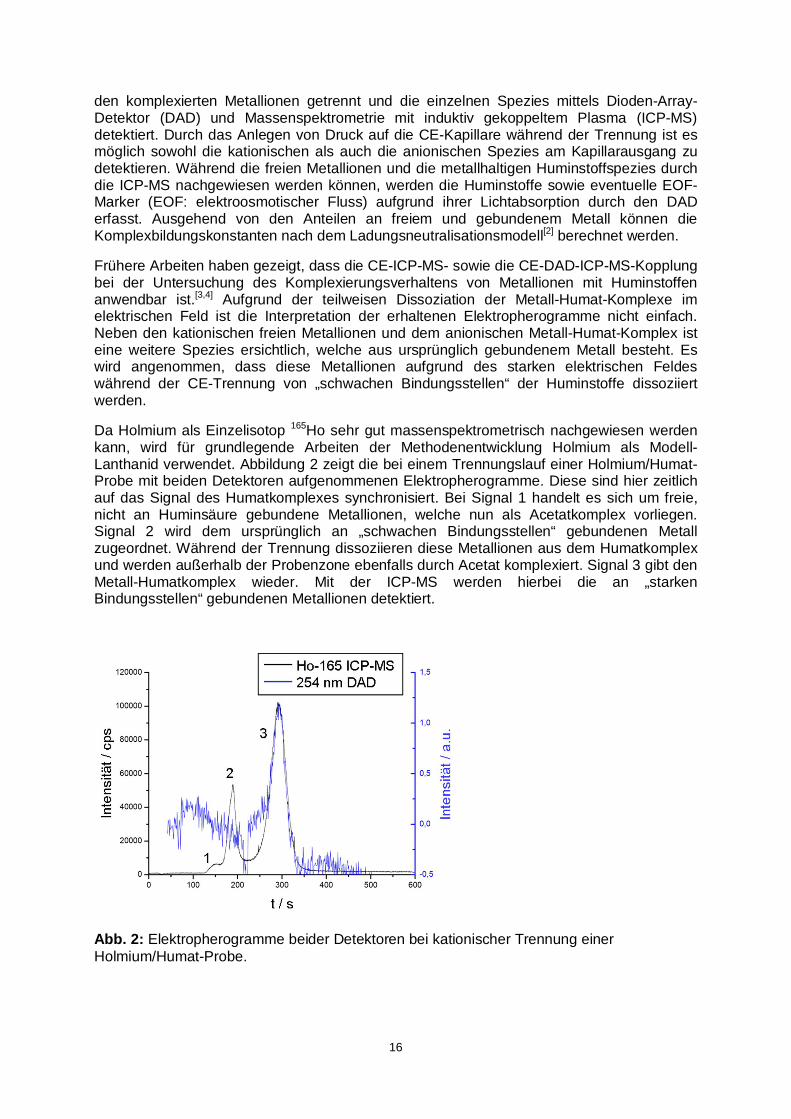
Da Holmium als Einzelisotop 165Ho sehr gut massenspektrometrisch nachgewiesen werden kann, wird für grundlegende Arbeiten der Methodenentwicklung Holmium als Modell-Lanthanid verwendet. Abbildung 2 zeigt die bei einem Trennungslauf einer Holmium/Humat-Probe mit beiden Detektoren aufgenommenen Elektropherogramme. Diese sind hier zeitlich auf das Signal des Humatkomplexes synchronisiert. Bei Signal 1 handelt es sich um freie, nicht an Huminsäure gebundene Metallionen, welche nun als Acetatkomplex vorliegen. Signal 2 wird dem ursprünglich an „schwachen Bindungsstellen“ gebundenen Metall zugeordnet. Während der Trennung dissoziieren diese Metallionen aus dem Humatkomplex und werden außerhalb der Probenzone ebenfalls durch Acetat komplexiert. Signal 3 gibt den Metall-Humatkomplex wieder. Mit der ICP-MS werden hierbei die an „starken Bindungsstellen“ gebundenen Metallionen detektiert.
Abb. 2: Elektropherogramme beider Detektoren bei kationischer Trennung einer Holmium/Humat-Probe.

17
Es ist erkennbar, dass die beiden ersten Holmiumspezies (Signale 1 und 2) bei kationischer Trennung schneller migrieren als der EOF (negatives DAD-Signal bei ca. 218 s). Sie müssen daher eine positive Ladung tragen, dies stimmt mit der Erwartung von nicht durch Huminsäure komplexierten Holmiumkationen überein.
Zur Bestimmung von Komplexbildungskonstanten muss ein Korrekturfaktor, die „loading capacity“ LC, für die vorgegebenen Reaktionsbedingungen (pH, Ionenstärke, Art des Huminstoffs) bekannt sein oder experimentell ermittelt werden. Zur Bestimmung der LC werden unter sonst gleichen Bedingungen bei konstanter Huminsäurekonzentration unterschiedliche Mengen an Metallionen zugesetzt und diese Metall/Humat-Proben mit CE-ICP-MS vermessen.
Für Aldrich Huminsäure wird bisher zur Bestimmung von log -Werten eine Kombination von Ultrafiltration (zur Bestimmung der „loading capacity“ als Funktion des pH-Wertes) mit der CE-ICP-MS verwendet. Für Fulvinsäuren kann die Ultrafiltration aufgrund des im Vergleich zu Huminsäuren niedrigeren Molekulargewichtes nicht angewandt werden. Ziel dieser Arbeit ist die Bestimmung sowohl der LC als auch der log -Werte durch die CE-ICP-MS.
Bei Messungen mit dem im Institut für Kernchemie vorhandenen CE-System ergaben sich zwischenzeitlich größere Probleme. Bei Messungen von Metall/Humat-Proben wies das Signal des Humatkomplexes (Signal 3, Abb. 2) ein sehr starkes Tailing auf. Bei anschließenden Spülschritten fiel auf, dass ein erheblicher Teil des Analyten auf der Kapillare und/oder im Kopplungssystem verblieb. Bedingt durch dieses Tailing ergaben sich erhebliche Ungenauigkeiten bei der Integrierung der Signalflächen. Eine Bestimmung von LC- bzw. log -Werten war mit den vorhandenen Daten nicht möglich. Trennungen an einfachen Elementstandards konnten mit dem vorhandenen CE-System ebenfalls nicht mehr sauber durchgeführt werden.
Es wurden vielfältige Anstrengungen unternommen, die Ursache hierfür ausfindig zu machen. Es wurde leihweise ein anderes CE-System beschafft und in Betrieb gesetzt. Aufgrund größtenteils kompatibler Komponenten konnten parallele Testläufe mit beiden CE-Systemen unter Konstanthaltung verschiedener Parameter (Lösungen, Kapillare mit Zerstäuber bzw. Kopplung mit ICP-MS, Hochspannungsversorgung, Gasversorgung für Probenaufgabe) durchgeführt werden. Ein chemisches Problem bezüglich der verwendeten Kapillaren, Proben bzw. Lösungen konnte letztlich durch verschiedene Tests ausgeschlossen werden. Ebenso zeigte sich, dass bei den verwendeten Systemen für die Kopplung mit der ICP-MS nicht die Ursache für die apparativen Probleme zu finden ist. Derzeit wird das Probenaufgabesystem mit Hilfe der elektronischen und mechanischen Werkstätten des Instituts für Kernchemie neu gestaltet.
Nach Inbetriebnahme des leihweise beschafften Gerätes war schließlich ein funktionierendes Trennsystem vorhanden, mit diesem konnten schließlich nach längerer Zeit Messungen an Metall/Humat-Proben vorgenommen werden.
Um näher an natürlichen Konzentrationsverhältnissen zu sein, wurde eine Messreihe mit einer Huminsäurekonzentration von 20 ppm (frühere Messungen: 100 ppm) angesetzt. Dabei wurde die Konzentration an Metall in einem weiten Konzentrationsbereich (5ppb bis 800 ppb) unter ansonsten gleichen Bedingungen (Ionenstärke 0,1 M, NaClO4; pH 4,2) variiert. Zur Trennung mit CE wurde wie bei früheren Messungen ein Acetatpuffer (0,1 M Essigsäure; 0,01 M Natriumacetat; pH 3,7) als Elektrolyt verwendet.

18
Es zeigte sich, dass für eine akzeptable Trennleistung die Parameter zur Probenaufgabe (Druck, Zeit) aufgrund der niedrigeren Huminsäurekonzentration angepasst werden mussten. Es wurde festgestellt, dass bei niedrigerer Metallionenkonzentration der Anteil an dissoziierendem Metall (2. Signal, Abb. 3) nicht wie erwartet vernachlässigbar gering ist. Eine weitere Auswertung der Daten bezüglich der Berechnung von LC und log steht zurzeit noch aus. Ebenfalls müssen zur Verbesserung der Trennleistung noch weitere Parameter optimiert werden.
Abb. 3: Elektropherogramme bei kationischer Trennung von Holmium/Humat-Proben mit verschiedenen Konzentrationen an Metall.
Eine weitere Messreihe (analog zu o.a. Messungen) mit konstantem Verhältnis der Konzentrationen an Metall und Huminsäure wurde angesetzt und vermessen.
Dabei zeigte sich, dass sich die Anteile an dissoziiertem und komplexiertem Metall (2. und 3. Signal, Abb. 4) in Abhängigkeit von der „Gesamt“-konzentration erheblich ändern. Ebenfalls ist hier ersichtlich, dass bezüglich der Trennung zwischen freiem und dissoziiertem Metall die Probenaufgabeparameter für verschiedene Huminsäurekonzentrationen jeweils angepasst werden müssen (vergleiche Abb. 2 und Abb. 4, rechts; gleiche Konzentrationen und Trennbedingungen, verschiedene Probenaufgabeparameter).
Abb. 4: Elektropherogramme bei kationischer Trennung von Holmium/Humat-Proben mit gleichen Konzentrationensverhältnissen Metall/Huminsäure.

19
CE-RIMS:
Zur Speziation von Plutonium im umweltrelevanten Ultraspurenbereich konnte die RIMS (Resonanzionisations-Massenspektrometrie) erfolgreich offline an die CE gekoppelt und erstmals auf reale Proben angewandt werden.
Bei Untersuchungen zur Wechselwirkung von Pu(III) und Pu(IV) mit Opalinuston und synthetischem Porenwasser unter anaeroben Bedingungen lagen die Konzentrationen des Plutoniums in den überstehenden Lösungen unter den Nachweisgrenzen der sonst üblichen anwendbaren Speziationsmethoden. Daher wurde hier die Speziation mit CE-RIMS durchgeführt. Hierzu wurden die enthaltenen Plutoniumspezies mit der CE getrennt und die einzelnen Fraktionen anschließend jeweils zu Filamenten für die RIMS aufbereitet.
Es wurde festgestellt, dass sich unabhängig davon, ob Pu(III) oder Pu(IV) als Ausgangspezies zu den Suspensionen zugegeben wurde, eine Verteilung über alle Oxidationsstufen des Plutoniums einstellte. Jedoch war die Beobachtungszeit für den Nachweis einer Gleichgewichtseinstellung in der Speziesverteilung zu gering. Die gemessenen Atomzahlen liegen gut im abgeschätzten Größenbereich von 108 der theoretisch zu erwartenden Atomzahlen. Da die untersuchten Konzentrationen im Ultraspurenbereich liegen, ist der Fehler in der Bestimmung der Speziesverteilung etwas größer, als bei Untersuchungen mit der CE-ICP-MS-Kopplung. In synthetischem Porenwasser in Abwesenheit von Opalinuston war Pu(V) die dominante Spezies unabhängig davon, ob Pu(III) oder Pu(IV) als Ausgangsspezies eingesetzt wurden.[5]
Literatur
[1] F. Scheffer, P. Schachtschabel, Lehrbuch der Bodenkunde, 13., durchgesehene Aufl.,
Ferdinand Enke Verlag, Stuttgart, 1992.
[2] J. I. Kim, K. R. Czerwinski, Radiochim. Acta 1996, 73, 5-10.
[3] R. Kautenburger, K. Nowotka, H. P. Beck, Anal. Bioanal. Chem. 2006, 384, 1416.
[4] E. Gromm, Diplomarbeit, Johannes Gutenberg-Universität, Mainz 2008.
[5] Th. Wunderlich, Dissertation, Johannes Gutenberg-Universität, Mainz 2009.
Dauer der Promotion Beginn der Promotion: Juli 2008 Angaben zu entstandenen Publikationen, Kongressbeiträgen etc. (seit Beginn der Promotion)

20
E. Gromm, “Speciation Analysis of Plutonium by CE-ICP-MS and CE-RIMS”, Vortrag, GRK Jahresseminar 2008, Bad Münster am Stein-Ebernburg, Nov. 2008
E. Gromm, „Zur Huminsäurekomplexierung von Lanthanoiden“, Vortrag, Workshop des Instituts für Kernchemie, Bad Münster am Stein-Ebernburg, Nov. 2008
E. Gromm, “Complexation of Lanthanides with Humic Acid”, Vortrag, ACTINET-Workshop, Graz, Dez. 2008
E. Gromm, R.A. Buda, J.V. Kratz, „CE-DAD-ICP-MS zur Bestimmung von Komplexbildungskonstanten für die Komplexierung von Lanthaniden mit Huminstoffen“, Poster, GDCh-Wissenschaftsforum Chemie 2009, Frankfurt a. M., Aug./Sept. 2009
E. Gromm, R.A. Buda, J.V. Kratz, “CE-DAD-ICP-MS for determination of complex formation constants for the complexation of lanthanides with humic substances”, Poster, 12th Workshop on Progress in Analytical Methodologies for Trace Metal Speciation (TraceSpec 2009), Mainz, Sept. 2009
E. Gromm, R.A. Buda, J.V. Kratz, “CE-DAD-ICP-MS for determination of complex formation constants for the complexation of lanthanides with humic substances”, Poster, 12th International Conference on the Chemistry and Migration Behaviour of Actinides and Fission Products in the Geosphere (Migration ’09), Kennewick, USA, Sept. 2009
E. Gromm, “CE-DAD-ICP-MS for determination of complex formation constants for the complexation of lanthanides with humic substances”, Vortrag, GRK Jahresseminar 2009, Bad Münster am Stein-Ebernburg, Okt. 2009
Zusammenarbeit mit bzw. Bezug zu anderen Projekten (innerhalb und außerhalb des GRK)
Bezüglich des RIMS-Systems besteht Kooperation mit S. Raeder (GRK-Stipendiat) und weiteren Mitgliedern des AK Wendt sowie mit N. Stöbener (GRK-Stipendiat) aus dem AK Reich
Bezüglich der CE-ICP-MS ergibt sich eine Kooperation auf europäischer Ebene im Rahmen eines durch das „European Network for Actinide Sciences ACTINET“ eingerichteten „joint research project“ (JRP 05-17 bzw. JRP 06-07): “Development of analytical tools for speciation of plutonium and uranium ions: focus on capillary electrophoresis coupled to ICP-MS”
Desweiteren besteht auf nationaler Ebene eine Kooperation mit verschiedenen Institutionen im BMWi-Verbundprojekt „Wechselwirkung und Transport von Actiniden im natürlichen Tongestein unter Berücksichtigung von Huminstoffen und Tonorganika“
Teilnahme am Studienprogramm (seit Beginn der Promotion)
01.09 - 05.09.2008 Kurs ”Analytische Qualitätskontrolle” Prof. Dr. Wegscheider,
Montanuniversität Leoben Ringvorlesung I, Wintersemester 2009/2010 02.03. – 04.03.2010 Kurs „Chemometrik“ Prof. Dr. Einax, Friedrich-Schiller-
Universität Jena Betreuung im Graduiertenkolleg Betreuer: Prof. Dr. Jens Volker Kratz ggf. Auslandsaufenthalte (seit Beginn der Promotion)

21
12th International Conference on the Chemistry and Migration Behaviour of Actinides
and Fission Products in the Geosphere” (Migration ’09) in Kennewick, USA, 20. - 25. September 2009.

22
Stipendiat: Elisabeth Kaschak Thema: „Biochemie der Biomethylierung von Quecksilber durch Organismen aus dem
Intestinaltrakt von Invertebraten.“ Betreuer: Prof. Dr. Helmut König Berichtszeitraum: Juni 2009 – April 2010 Der Eintrag von Quecksilber und seinen Spezies in die Umwelt wird durch die Herstellung von Chlor, Gewinnung von Gold, aber auch bei der Verbrennung fossiler Rohstoffe und Haushaltsmüll gefördert. Die EU ist derzeit der weltweit größte Exporteur von Quecksilber und liefert einen Anteil von 1.000 Tonnen (der insgesamt 3.600 Tonnen) pro Jahr. Auf der 24. Sitzung des UNEP-Governing-Council im Februar 2007 hat die EU eine Stellungnahme zum Thema Quecksilber abgegeben, wonach der Gebrauch von Quecksilber um 70 Prozent bis 2017 weltweit reduziert werden soll. Die WHO gibt als vorläufige duldbare wöchentliche Aufnahmemenge (provisional tolerable weekly intake, PTWI) für Methylquecksilber einen Wert von 1,6 µg/kg Körpergewicht an (Verordnung der EG Nr. 1881/2006 der Kommission zur Festsetzung der Höchstgehalte für bestimmte Kontaminanten in Lebensmitteln). Methylquecksilber ist hochtoxisch für höhere Organismen, da es die Blut-Hirn und Plazenta-schranke durchdringt und neuronale Schädigungen hervorruft, die sich in Tremor und motorischen Koordinations- und Sprachstörungen äußern, aber auch zum Koma oder Tod des Patienten führen können. Die Spezies-Umwandlung von Quecksilber (Hg0) zu Methylquecksilber führt zu einer Akkumulation in der Nahrungskette von Fischen und Meerestieren, die letztlich den Menschen als Endkonsumenten erreicht [1,2]. Im Vordergrund dieser Arbeit steht die mikrobielle Biomethylierung von Quecksilber aus dem Intestinaltrakt des Invertebraten Eisenia foetida. Bisher wurden aerobe und fakultativ anaerobe Mikroorganismen aus dem Darm des Anneliden isoliert, identifiziert und auf die Produktion von Methylquecksilber untersucht. Die Nährlösungen und –böden für die Kultivierung von Mikroorganismen wurden nach den Vorschriften der Deutschen Sammlung für Mikroorganismen und Zellkulturen (DSMZ, Göttingen) hergestellt. Aus der Beimpfung des Mediums mit Darmmaterial gingen zunächst Mischkulturen hervor, von denen einige mit Vereinzelungstechniken in Reinkultur gebracht werden konnten. Zur Identifizierung wurde nach DNA Extraktion die 16S rDNA mithilfe spezifischer Primer für konservierte Regionen partiell sequenziert. Die Tab.1 zeigt die erfolgreiche Vereinzelung aerober oder fakultativ anaerober Mikroorganismen mit zugehörigem Alignment der NCBI Datenbank (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Die Reinkulturen wurden im jeweiligen Medium 7 Tage mit Quecksilberchlorid inkubiert. Die Analyse der Quecksilberspezies erfolgte mittels der GC-CV-AFS (Gas Chromatography Cold Vapor Atomic Fluorescence Spectrometry). Für die Probenvorbereitung wurde eine alkalische Extraktion angewendet und CuSO4 mit Na-Oxalat als Maskierungsreagenz zu der Probe gegeben, da diese Substanz den Nachweis von Methylquecksilber in sulfidhaltigem Medium erheblich verbessern [3]. Die Vorsäulenderivatisierung wurde mit Natriumtetrapropylborat durchgeführt. Die Nachweisgrenze der CV-AFS liegt bei optimalen Bedingungen bei 1 ppt (ng/kg). Die Reinkulturen, die in Tab.1 aufgelistet sind, wurden in flüssigem Medium auf die Methylierung von Hg2+ zu Methylquecksilber untersucht. Die Analysen zeigten, dass keine der Spezies in Reinkultur diese Fähigkeit aufwies.

23
Tab.1: Reinkulturen, die bisher aus dem Intestinaltrakt von Eisenia foetida isoliert wurden. Das Alignment gibt die Sequenzübereinstimmung in % (nach NCBI) wieder. * 1 Nutrient Agar, 63 Desulfovibrio Medium, 80 Glycerol-Soil Medium, YEPG Pepton, Hefeextrakt, Glucose mit pH = 7, 464 Plate Count Agar, 98 Rhizobium Medium Es konnten jedoch zwei neue Mischkulturen aus dem Intestinaltrakt von Eisenia foetida isoliert werden, die in der Lage waren, Quecksilber zu methylieren. Nach 7tägiger Inkubationszeit und Zugabe von 1 µg/l HgCl2 in das DSMZ Medium (Nr. 63) wurde von Kultur 1 10,57 ng/ml Methylquecksilber produziert (Abb.1).
Medium DSMZ
16S rDNA Sequenz Alignment nach NCBI
Alignment Übereinstimmung in %
1 Bacillus licheniformes 448/450 (99%) 1 Bacillus subtilis 448/449 (99%) 1* Brevibacillus brevis 431/433 (99%) 1 Lysinibacillus fusiformis 481/483 (99%) 1 Lysinibacillus sp. 473/473 (100%) 1 Lysinibacillus sphaericus 451/452 (99%) 1 Lysobacter sp. 453/455 (99%) 1 Pseudomonas alcaligenes 479/481 (99%) 1 Pseudomonas sp. 483/485 (99%) 63 Enterobacter asburiae 814/815 (99%) 63 Enterobacter cancerogenus 486/488 (99%) 63* Enterobacter sp. 471/472 (99%) 63 Pantoea agglomerans 918/921 (99%) 63 Pseudomonas jinjuensis 496/501 (98%) 63 Pseudomonas sp. II 446/456 (98%) 63 Pseudomonas sp. VI 491/491 (100%) 63 Shewanella putrefaciens 495/499 (99%) 63 Shewanella sp. I 475/479 (99%) 80 Pseudomonas sp. IV 499/503 (99%) 80* Shewanella sp. II 500/505 (99%) 80 Sinorhizobium sp. 478/478 (100%)
YEPG Bacillus cereus 418/420 (99%) YEPG Bacillus pumilus 480/481 (99%) YEPG* Microbacterium resistens 479/484 (99%) YEPG Paracoccus sp. 502/505 (99%) YEPG Pseudomonas putida 483/484 (99%) 464 Bacillus thuringiensis 481/484 (99%) 464 Flavobacterium denitrificans 465/466 (99%) 464 Microbacterium thalassium 434/435 (99%) 464* Pseudomonas sp. 472/472 (100%) 98* Pseudomonas fluorescens 455/455 (100%)
Hg2+
CH3Hg+

24
Abb.1: GC-CV-AFS Chromatogramm: Transformation von Hg2+ (1 µg/ ml) zu HgCH3+ (10,6 ng/ ml)
durch eine Mischkultur von Mikroorganismen aus dem Intestinaltrakt von Eisenia foetida. Die Zellzahl in dieser Mischkultur betrug nach Auszählung mit der Neubauer Zählkammer (Tiefenprofil von 0,02 mm) 9,86 109 Zellen/ml. Zur näheren Identifizierung dieser Mikroorganismen wurden molekularbiologische Methoden angewendet, da die Gewinnung von Reinkulturen mittels der Ausstrich-Technik nicht das Gesamtbild beteiligter Spezies in einer Mischkultur wiedergeben kann. Zunächst wurde eine DNA-Extraktion mit dem Kit „Blood & Tissue“ von QIAGEN (Hilden, Germany) durchgeführt. Zur systematischen Identifizierung der konservierten 16S rDNA Region unbekannter Organismen aus einer Kultur wurde nach einer PCR mit den Primern Eub519f und 1070r die Dichtegradientengelelektrophorese (DGGE) durchgeführt. Bereits der Austausch von 1 Base in der Nukleotidsequenz führt zu einem veränderten Laufverhalten im Gradientengel. Einzelne Banden sind daher ein Indiz für verschiedene Spezies. Nach Anfärbung des Gels mittels einer 0,01%igen Ethidiumbromidlösung wurden die Banden unter UV-Licht sichtbar und aus dem Gel ausgeschnitten. Zur Bestätigung der Ergebnisse wurde eine unabhängie Methode der RFLP (Restriktionsfragmentlängenpolymorphismus) angewendet. Nach Amplifizierung der eluierten 16S rDNA wurden die Produkte nach Aufreinigung von der Firma MWG Operon sequenziert. Die partiellen 16S rDNA Sequenzen wurden in respräsentativen Datenbanken wie z.B. NCBI und Ribosomal Database project (http://rdp.cme.msu.edu/) einem Alignment unterzogen. In den Mischkulturen wurden bisher Übereinstimmungen mit folgenden Organismen gefunden: Kultur 1: Enterobacter sp. (643bp/ 645bp, 99%) und Pantoea agglomerans (906bp/ 908bp, 99%). Kultur 2: Pseudomonas azaleica (467bp/ 468bp, 99%), Pseudomonas sp. (683bp /685bp, 99%), möglicherweise Sporomusa ovata und Sporomusa sphaeroides 467bp/ 488bp, 95%) und ein bisher unkultivierbarer Organismus (667bp/ 687bp, 97%). In Mischkultur sind diese Organismen den Analysen zufolge in der Lage in einem HgCl2-haltigem Medium Methlyquecksilber zu produzieren. Die Biochemie der biotischen Quecksilbermethylierung ist bisher in nur wenigen Mikroorganismen untersucht und dargestellt worden [4,5,6]. Das Ziel der Arbeit ist es daher, Isolate aus dem Intestinaltrakt von Eisenia foetida zu identifizieren und auf die Fähigkeit zur Quecksilbermethylierung zu untersuchen. In Zukunft werden anhand dieser Reinkulturen Untersuchungen zur Biochemie der Methylierung von Quecksilber vorgenommen.

25
Literatur [1] J.E.S. Uria, A. Sanz-Medel, Inorganic and methylmercury speciation in environmental samples, Talanta 47 (1998) 509-524. [2] Limper U., Knopf B., Knopf H., Poduction of methyl mercury in the gut of the Australian termite Mastotermes darwiniensis. J. Appl. Entomol. (2008) 132: 168 – 176. [3] D.Y. Yang, H.Y.T. Truong, Y.W. Chen, N. Bezile, Improvements of reliability for methlymercury determination in environmental samples. Anal. Chim. Acta 633 (2009) 157-164. [4] S.C. Choi, T. Chase, Jr., R. Bartha, Enzymatic catalysis of mercury methylation by Desulfovibrio desulfuricans LS, Appl Environ Microbiol 60 (1994) 1342-1346. [5] E.B. Ekstrom, F.M. Morel, Cobalt limitation of growth and mercury methylation in sulfate-reducing bacteria, Environ Sci Technol 42 (2008) 93-99. [6] Knopf B., König H., Biomethylation of heavy metals in soil and terrestrial invertebrates. In: Soil Heavy Metals. I. Sherameti and A. Varma (eds). Springer Heidelberg (2009) pp. 315 – 328. Beginn der Promotion: Juni 2009 Angaben zu entstandenen Publikationen, Kongressbeiträgen etc. (seit Beginn der Promotion) Elisabeth Kaschak, Burkhard Knopf & Helmut König, Poster, 3. Gemeinsame Tagung von
VAAM und DGHM, Hannover. Teilnahme am Studienprogramm (seit Beginn der Promotion) WS 2009/ 2010 Vortrag im Rahmen des Graduiertenseminar „Biochemistry of mercury
methylation“ Ringvorlesung WS 2009/ 2010 08.02. – 12.02.2010 Organisation und Betreuung des Praktikums “Course in Microbiology – Microbiological Practices“ im Institut für Mikrobiologie und
Weinforschung 02.03. – 04.03.2010 „Chemometrik Kurs“ durchgeführt von Prof. Dr. Einax
Betreuung im Graduiertenkolleg Betreuer: Prof. Dr. Helmut König

26
Thema und Betreuer des Dissertationsprojekts Stipendiat: Michael Kundel Echtzeit-Bestimmung von gasförmigen Iodspezies mittels Flugzeit-Aersolmassenspektrometrie Betreuer: Prof. Dr. Thorsten Hoffmann Berichtszeitraum: April 2009 – März 2010 Darstellung des Dissertationsprojekts und der Forschungsergebnisse im Berichtszeitraum 1. Einleitung In der maritimen Atmosphärenchemie besitzen iodhaltige Verbindungen eine große Bedeutung. Vor allem bei der troposphärischen Ozonzerstörung und der Bildung neuer Partikel spielen iodhaltige Verbindungen eine wichtige Rolle. Dabei zeigen aktuelle Untersuchungen, dass molekulares Iod und iodorganische Verbindungen (z.B. Iodmethan und Diiodmethan), die aus Algen und Phytoplankton freigesetzt werden (O´Dowd et al., 2002, McFiggans et al., 2004, Küpper et al., 2008), die wichtigsten Vorläufersubstanzen für sekundäre iodhaltige Aerosole sind. Weiterhin stehen ebenfalls aktivierte iodhaltige Spezies, wie ICl und IBr, in Verdacht den Ozonzabbau und die Partikelneubildung zu beeinflussen (Vogt et al., 1999). Aufgrund ihrer geringeren Konzentration im ppt-Bereich, ist jedoch die Identifizierung und Quantifizierung von reaktiven Iodspezies in der Atmosphäre immer noch eine große Herausforderung. Im Rahmen dieser Arbeit wird das Flugzeit-Aerosolmassenspektrometer (ToF-AMS) verwendet (Drewnick et al., 2005), um gasförmige Iodspezies in Echtzeit zu identifizieren und quantifizieren. 2. Experimenteller Aufbau Da sich die Iodspezies (I2, IBr und ICl) in der Gasphase befinden, können diese nicht direkt mit einem Aerosolmassenspektrometer untersucht werden. Deshalb müssen die gasförmigen Iodspezies zunächst von der Gas- in die Partikelphase überführt werden. Hierzu werden mit Hilfe eines pneumatischen Zerstäubers kontinuierlich Partikel in eine 10 L Reaktionskammer eingebracht, die die zu untersuchenden Iodspezies selektiv aufnehmen können. Die Iodspezies werden mittels einer temperierten Testgasquelle in die 10 L Reaktionskammer eingeleitet. Nach Verlassen der Reaktionskammer wird das resultierende Aerosol mit einem ToF-AMS analysiert. Das ToF-AMS ermöglicht die Messung der Partikelgröße und der chemischen Zusammensetzung von verdampfbaren Aerosolen in Echtzeit. In Abbildung 1 ist der experimentelle Aufbau schematisch dargestellt.
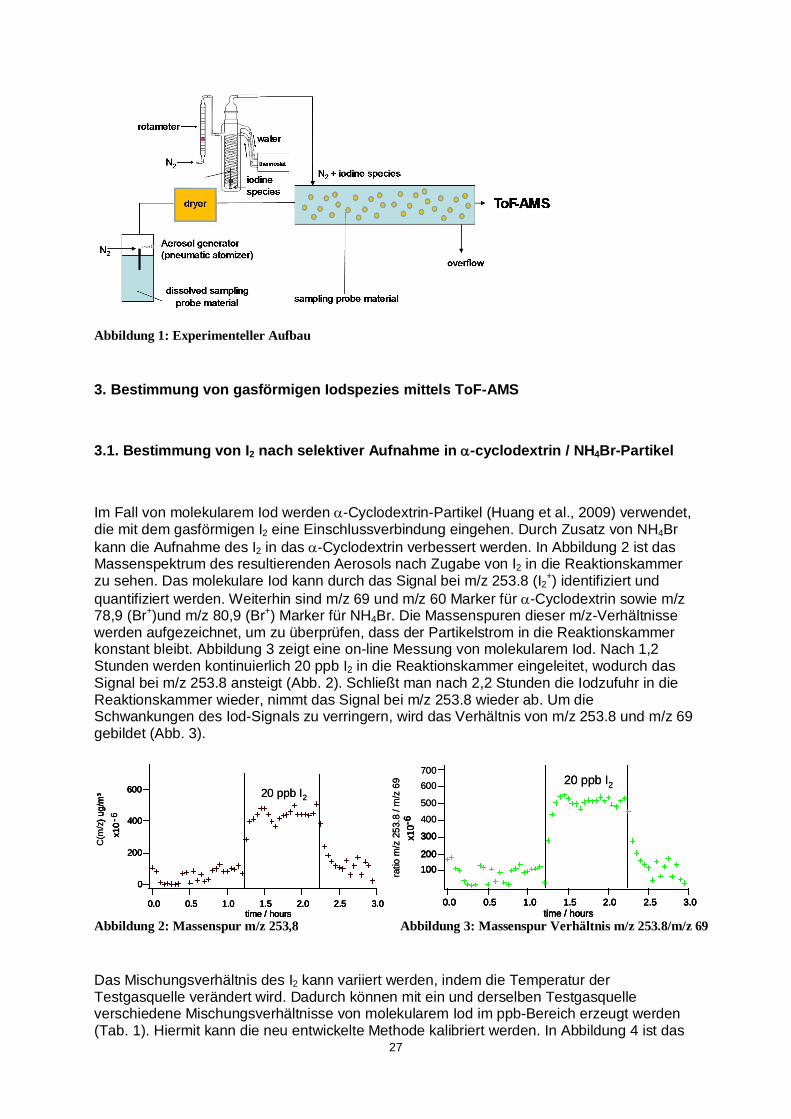
27
Abbildung 1: Experimenteller Aufbau
3. Bestimmung von gasförmigen Iodspezies mittels ToF-AMS
3.1. Bestimmung von I2 nach selektiver Aufnahme in -cyclodextrin / NH4Br-Partikel
Im Fall von molekularem Iod werden -Cyclodextrin-Partikel (Huang et al., 2009) verwendet, die mit dem gasförmigen I2 eine Einschlussverbindung eingehen. Durch Zusatz von NH4Br kann die Aufnahme des I2 in das -Cyclodextrin verbessert werden. In Abbildung 2 ist das Massenspektrum des resultierenden Aerosols nach Zugabe von I2 in die Reaktionskammer zu sehen. Das molekulare Iod kann durch das Signal bei m/z 253.8 (I2+) identifiziert und quantifiziert werden. Weiterhin sind m/z 69 und m/z 60 Marker für -Cyclodextrin sowie m/z 78,9 (Br+)und m/z 80,9 (Br+) Marker für NH4Br. Die Massenspuren dieser m/z-Verhältnisse werden aufgezeichnet, um zu überprüfen, dass der Partikelstrom in die Reaktionskammer konstant bleibt. Abbildung 3 zeigt eine on-line Messung von molekularem Iod. Nach 1,2 Stunden werden kontinuierlich 20 ppb I2 in die Reaktionskammer eingeleitet, wodurch das Signal bei m/z 253.8 ansteigt (Abb. 2). Schließt man nach 2,2 Stunden die Iodzufuhr in die Reaktionskammer wieder, nimmt das Signal bei m/z 253.8 wieder ab. Um die Schwankungen des Iod-Signals zu verringern, wird das Verhältnis von m/z 253.8 und m/z 69 gebildet (Abb. 3).
Abbildung 2: Massenspur m/z 253,8 Abbildung 3: Massenspur Verhältnis m/z 253.8/m/z 69
Das Mischungsverhältnis des I2 kann variiert werden, indem die Temperatur der Testgasquelle verändert wird. Dadurch können mit ein und derselben Testgasquelle verschiedene Mischungsverhältnisse von molekularem Iod im ppb-Bereich erzeugt werden (Tab. 1). Hiermit kann die neu entwickelte Methode kalibriert werden. In Abbildung 4 ist das
600
400
200
0
C(m
/z) u
g/m
³
3.02.52.01.51.00.50.0time / hours
x10
-6
20 ppb I2600
400
200
0
) ug/
m³
3.02.52.01.51.00.50.0time / hours
x10
-
600
400
200
0
C(m
/z) u
g/m
³
3.02.52.01.51.00.50.0time / hours
x10
-6
20 ppb I2600
400
200
0
) ug/
m³
3.02.52.01.51.00.50.0time / hours
x10
-
time / hours
700600
500
400
300
200100
x10-
6
3.02.52.01.51.00.50.0
20 ppb I2
ratio
m/z
253.
8 / m
/z69
time / hours
300
200100
x10-
6
3.02.52.01.51.00.50.0time / hours
700600
500
400
300
200100
x10-
6
3.02.52.01.51.00.50.0
20 ppb I2
ratio
m/z
253.
8 / m
/z69
time / hours
300
200100
x10-
6
3.02.52.01.51.00.50.0
28
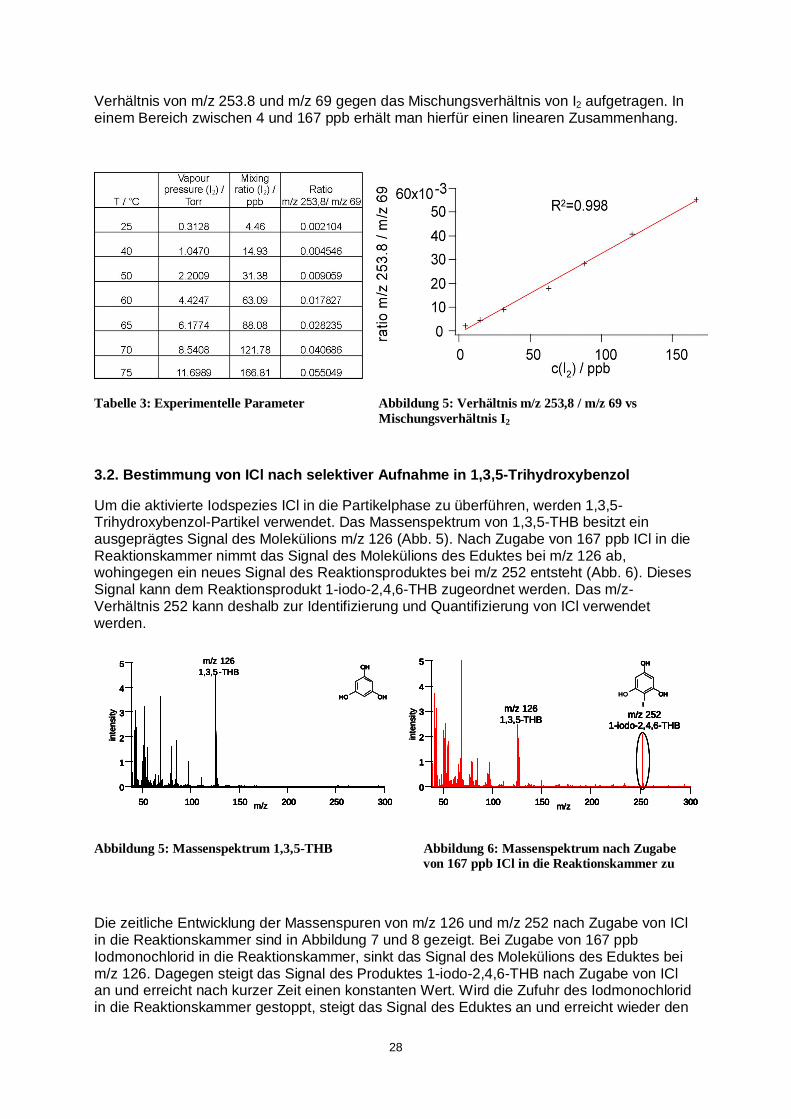
Verhältnis von m/z 253.8 und m/z 69 gegen das Mischungsverhältnis von I2 aufgetragen. In einem Bereich zwischen 4 und 167 ppb erhält man hierfür einen linearen Zusammenhang.
Tabelle 3: Experimentelle Parameter Abbildung 5: Verhältnis m/z 253,8 / m/z 69 vs Mischungsverhältnis I2
3.2. Bestimmung von ICl nach selektiver Aufnahme in 1,3,5-Trihydroxybenzol
Um die aktivierte Iodspezies ICl in die Partikelphase zu überführen, werden 1,3,5-Trihydroxybenzol-Partikel verwendet. Das Massenspektrum von 1,3,5-THB besitzt ein ausgeprägtes Signal des Molekülions m/z 126 (Abb. 5). Nach Zugabe von 167 ppb ICl in die Reaktionskammer nimmt das Signal des Molekülions des Eduktes bei m/z 126 ab, wohingegen ein neues Signal des Reaktionsproduktes bei m/z 252 entsteht (Abb. 6). Dieses Signal kann dem Reaktionsprodukt 1-iodo-2,4,6-THB zugeordnet werden. Das m/z-Verhältnis 252 kann deshalb zur Identifizierung und Quantifizierung von ICl verwendet werden.
Abbildung 5: Massenspektrum 1,3,5-THB Abbildung 6: Massenspektrum nach Zugabe von 167 ppb ICl in die Reaktionskammer zu
Die zeitliche Entwicklung der Massenspuren von m/z 126 und m/z 252 nach Zugabe von ICl in die Reaktionskammer sind in Abbildung 7 und 8 gezeigt. Bei Zugabe von 167 ppb Iodmonochlorid in die Reaktionskammer, sinkt das Signal des Molekülions des Eduktes bei m/z 126. Dagegen steigt das Signal des Produktes 1-iodo-2,4,6-THB nach Zugabe von ICl an und erreicht nach kurzer Zeit einen konstanten Wert. Wird die Zufuhr des Iodmonochlorid in die Reaktionskammer gestoppt, steigt das Signal des Eduktes an und erreicht wieder den
5
4
3
2
1
030025020015010050
OH
OHOH
m/z 1261,3,5-THB
m/z
inte
nsity
5
4
3
2
1
030025020015010050
OH
OHOH
m/z 1261,3,5-THB
m/z
inte
nsity
5
4
3
2
1
030025020015010050
5
4
3
2
1
030025020015010050
OH
OHOH
OH
OHOH
m/z 1261,3,5-THB
m/z
inte
nsity
5
4
3
2
1
030025020015010050
5
4
3
2
1
030025020015010050
OH
OHOH
OH
OHOH
m/z 1261,3,5-THB
m/z
inte
nsity
5
4
3
2
1
030025020015010050
m/z 1261,3,5-THB m/z 252
1-iodo-2,4,6-THB
OH
OHOHI
m/z
inte
nsity
5
4
3
2
1
030025020015010050
m/z 1261,3,5-THB m/z 252
1-iodo-2,4,6-THB
OH
OHI
5
4
3
2
1
030025020015010050
m/z 1261,3,5-THB m/z 252
1-iodo-2,4,6-THB
OH
OHI
m/z
inte
nsity
5
4
3
2
1
030025020015010050
5
4
3
2
1
030025020015010050
m/z 1261,3,5-THB m/z 252
1-iodo-2,4,6-THB
OH
OHOHI
OH
OHOHI
m/z
inte
nsity
5
4
3
2
1
030025020015010050
m/z 1261,3,5-THB m/z 252
1-iodo-2,4,6-THB
OH
OHI
5
4
3
2
1
030025020015010050
m/z 1261,3,5-THB m/z 252
1-iodo-2,4,6-THB
OH
OHI
m/z
inte
nsity

29
Ausgangswert. Im Gegensatz dazu nimmt nach Beendigung der Zufuhr von ICl das Signal des Produktes ab bis der Ausgangswert wieder erreicht wird.
Abbildung 7: Zeitaufgelöste Massenspur m/z 126 Abbildung 8: Zeitaufgelöste Massenspur m/z 252
3.3. Bestimmung von IBr nach selektiver Aufnahme in 1,3,5-Trihydroxybenzol
Für die Überführung der aktivierten Iodspezies IBr in die Partikelphase wird ebenfalls 1,3,5-THB verwendet (Abb. 9). Im Gegensatz zu ICl entsteht bei der Reaktion von IBr und 1,3,5-THB allerdings als Reaktionsprodukt hauptsächlich 1-bromo-2,4,6-THB, das anhand der m/z-Verhältnisse m/z 204 und m/z 206 nachgewiesen werden kann (Abb. 10). Weiterhin ist ein wenig ausgeprägtes Signal bei m/z 252 zu sehen, das dem 1-iodo-2,4,6-THB zugeordnet werden kann. Somit können die Signal m/z 204 und m/z 206 zur Quantifizierung von IBr verwendet werden.
Abbildung 9: Massenspektrum 1,3,5-THB Abbildung 10: Massenspektrum nach Zugabe von 43 ppb IBr in die Reaktionskammer
Der zeitliche Verlauf der Massenspuren des Eduktes (m/z 126) und des Reaktionsproduktes (m/z 204) nach Zugabe von 43 ppb IBr sind in Abbildung 11 und 12 dargestellt.
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
C(m
/z) u
g/m
³
0 0.5 1.0 1.5 2.0time / hours
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
C(m
/z) u
g/m
³
0 0.5 1.0 1.5 2.0time / hours
addition of 167 ppb ICl
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
C(m
/z) u
g/m
³
0 0.5 1.0 1.5 2.0time / hours
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
C(m
/z) u
g/m
³
0 0.5 1.0 1.5 2.0time / hours
addition of 167 ppb ICl
addition of 167 ppb ICl
0.5
0.4
0.3
0.2
0.1
0.0
C(m
/z) u
g/m
³
0 0.5 1.0 1.5 2.0time / hours
0.5
0.4
0.3
0.2
0.1
0.0
C(m
/z) u
g/m
³
0 0.5 1.0 1.5 2.0time / hours
addition of 167 ppb ICl
0.5
0.4
0.3
0.2
0.1
0.0
C(m
/z) u
g/m
³
0 0.5 1.0 1.5 2.0time / hours
0.5
0.4
0.3
0.2
0.1
0.0
C(m
/z) u
g/m
³
0 0.5 1.0 1.5 2.0time / hours
OH
OHOH
Br
5
4
3
2
1
0
30025020015010050
m/z 204, m/z 2061-bromo-2,4,6-THB
m/z 2521-iodo-2,4,6-THB
m/z 1261,3,5-THB OH
OHOH
I
m/z
inte
nsity
OH
OHOH
Br
5
4
3
2
1
0
30025020015010050
m/z 204, m/z 2061-bromo-2,4,6-THB
m/z 2521-iodo-2,4,6-THB
m/z 1261,3,5-THB OH
OHOH
I
OH
OHOH
Br
5
4
3
2
1
0
30025020015010050
m/z 204, m/z 2061-bromo-2,4,6-THB
m/z 2521-iodo-2,4,6-THB
m/z 1261,3,5-THB OH
OHOH
I
m/z
inte
nsity
5
4
3
2
1
0
30025020015010050
OH
OHOH
m/z 1261,3,5 -THB
m/z
inte
nsity
5
4
3
2
1
0
30025020015010050
OH
OHOH
m/z 1261,3,5 -THB
m/z
inte
nsity
5
4
3
2
1
0
30025020015010050
5
4
3
2
1
0
30025020015010050
OH
OHOH
OH
OHOH
m/z 1261,3,5 -THB
m/z
inte
nsity
5
4
3
2
1
0
30025020015010050
5
4
3
2
1
0
30025020015010050
OH
OHOH
OH
OHOH
m/z 1261,3,5 -THB
m/z
inte
nsity
2.0
1.5
1.0
0.5
0.0
C(m
/z) u
g/m
³
2.01.51.00.50time / hours
addition of 43 ppb IBr
2.0
1.5
1.0
0.5
0.0
C(m
/z) u
g/m
³
2.01.51.00.50
2.0
1.5
1.0
0.5
0.0
C(m
/z) u
g/m
³
2.01.51.00.50time / hours
addition of 43 ppb IBr
2.0
1.5
1.0
0.5
0.0
C(m
/z) u
g/m
³
2.01.51.00.50
80x10-3
60
40
20
0
C(m
/z) u
g/m
³
2.01.51.00.50time / hours
addition of43 ppb IBr
80x10-3
60
40
20
0
C(m
/z) u
g/m
³
2.01.51.00.50
80x10-3
60
40
20
0
C(m
/z) u
g/m
³
2.01.51.00.50time / hours
addition of43 ppb IBr
80x10-3
60
40
20
0
C(m
/z) u
g/m
³
2.01.51.00.50

30
Abbildung 11: Zeitaufgelöste Massenspur m/z 126 Abbildung 12: Zeitaufgelöste Massenspur m/z 204
Literatur [1] C.D. O´Dowd, J.L. Jimenez, et al. (2002). Nature, 417, 632–636 [2] G. McFiggans, H. Coe, et al. (2004). Atmos. Chem. Phys., 4, 701-713 [3] F. Küpper, L.J. Carpenter, et al. (2008). Proc. Natl. Acad. Sci., 105 (19), 6954-6958 [4] R. Vogt, R. Sander, et al. (1999). J. Atmos. Chem., 32, 375-395 [5] F. Drewnick, S.S. Hings, et al. (2005). Aerosol Sci. Technol., 39, 637-658 [6] R.-J. Huang and T. Hoffmann (2009). Anal. Chem., 81 (5), 1777-1783 Dauer der Promotion Beginn 01.11.2008 Angaben zu entstandenen Publikationen, Kongressbeiträgen etc. (seit Beginn der Promotion) Posterbeiträge:
• M. Kundel und T. Hoffmann Charakterisierung von Organosulfaten im sekundären organischen Aerosol ANAKON, 17.03-20.03.2009, Berlin
• M.Kundel, M.Ries, M. Schott und T.Hoffmann Application of on-line time-of-flight aerosol mass spectrometry for the determination of molecular iodine European Aerosol Conference (EAC), 06.09.-11.09.2009, Karlsruhe
• M.Kundel, M.Ries, M. Schott und T.Hoffmann Development of an on-line method for the determination of I2 using time-of-flight aerosol mass spectrometry 12th Workshop on Progress in Analytical Methodology for Trace Metal Speciation, 15.09.-18.09.2009, Mainz ( Posterpreis )
Vorträge:
• M.Kundel, M.Ries, M. Schott und T.Hoffmann Method development: Real-time measurement of gaseous I2 by time-of-flight aerosol mass spectrometry Jahrestagung des GRK, 18.10-20.10.2009, Bad Münster am Stein
Zusammenarbeit mit bzw. Bezug zu anderen Projekten (innerhalb und außerhalb des GRK) Bestimmung des Gesamtiodgehalts in Meerwasser und Algen mittels ICP-OES. Zusammenarbeit mit Ute Thorenz und dem Arbeitskreis von Prof. Dr. N. Bings. Diese Untersuchung läuft im Rahmen einer Kooperation mit Udo Nitschke und Dr. Dagmar Stengel vom Martin Ryan Institut der Universität Galway

31
Teilnahme am Studienprogramm (seit Beginn der Promotion)
• 02.11. - 04.11.2008: Jahrestagung des GRK, Bad Münster am Stein • 18.10. - 20.10.2009: Jahrestagung des GRK, Bad Münster am Stein • WS 2009/2010: Ringvorlesung 1 • 08.02-12.02.2010: Blockpraktikum „Mikrobiologische Methoden“, Prof Dr. H. König • 02.03.-04.03.2010: Kurs „ Chemometrik“, Prof Dr. W. Einax
Betreuung im Graduiertenkolleg Betreuer: Prof. Dr. Thorsten Hoffmann Ggf. Auslandsaufenthalte (seit Beginn der Promotion)
• 30.10.2009-02.11.2009: AMS-User-Meeting, Toronto, Kanada Alter bei Eintritt in das Graduiertenkolleg 26

32
Topic and supervising tutor of the PhD-project Scholarship holder: Mr. Ka Hei Lui Speciation of Copper enriched in Agricultural Limespeciation and mobility of arsenic in agricultural lime Supervising tutor: Prof. Dr. Michael Kersten Reporting period: March 2009 – Feburary 2010 Presentation of the PhD-project and results obtained during the reporting period Soils use for cultivation purpose is important for mankind as it is considered to be a non-renewable natural resource. Various human activities such as fertilizers application nowadays have been identified as one of the consequence in terms of gradual increases of many potentially harmful trace element concentrations in agricultural soils. Concerns of long term human health impacts from the society towards exposure of potentially toxic trace elements lead to urgent needs from the government and regulation bodies to set up effective legal limits. It is therefore important for soil scientists to have better understanding about mobility and bioavailability of all these trace elements. There is a natural tendency to reduce in lime status for most of soils. This natural increase in acidity would reduce the soil fertility and damage soil structure. Application of lime to cropland has long been developed to solve this problem and it would invariably increase the crop yield by raising the soil acidity, cation exchange capacity and microbial functionality and hence the soil fertility can be improved. Lime is usually seen as low-value high-bulk products. Around 1.5 million dry tons are used annually for liming of forestry and agriculture with application content in approximately between 2 to 10 tons per unit hectare of land in Germany. With such high level of consumption it is essential to keep the transportation cost as low as possible even if the local lime is in low grade. Low grade limestone usually contains with significant amount of Fe and Mn oxides. Under normal circumstances such heavy metal concentrations present in liming materials do not exceed the threshold limit. However, studies in the past showed that toxic metals and metalloids have the abilities to scavenge with the oxides and possibly leach out from the soils with oxides under severe environmental condition (e.g. waterlogged). It is however vital to investigate the potential risks of soil and ground water contamination when such liming materials present with high level of these oxides. The aim of this project was to determine the main host phases and solid speciation of Cu accumulated in agricultural lime samples. Copper is an essential element for life but can be toxic to ecosystem when it is excessively accumulated. Agricultural liming is one of many ways of accumulating copper in soils and as copper is often accumulated by strongly complexing with soil organic matter. This makes remediation technique such as Ca carbonate amendment not possible. Excessive accumulation of copper would cause Cu phytotoxicity and rhizotoxicity of plants and as a result Cu speciation would be essential on providing necessary information on handling with the Cu contaminated soils in long term future.

33
In modern geochemistry it is well understood that by only analysing the total heavy metal content is not sufficient. Adsorption of heavy metals into the soil system usually greatly depends on the form in which the metal enters the soil (solid particulate in wet or dry deposition or solution), range of metal species, charge on the metal entering the soil, the pH of the receiving environment, the percentage of organic matter in the soil, and the redox condition of the receiving environment. Speciation technique such as sequential extraction method has been applied in order to partition metals in different metal-bearing phases. The technique gives a useful prediction on potential metal mobility upon amendment in soil under acidic or waterlogged (i.e.,reducing) conditions. The technique is often regarded as fractionation by a series of chemical with increasing chemical solution strength removes or dissociates a specific phase with the associated metal bonded to the phases. The geochemical fractions most commonly analysed for classified as exchangeable, bound to carbonates, reducible, oxidisable and residual.
The targeted problem clayey soil (Dystric Cambisol) samples were collected from Permo-Triassic Sedimentary Rock Complex of the Spessart region in Germany which is shown in figure 1.
Fig. 1 location of the collected clayey soil samples:
A modified BCR sequential extraction scheme of Cu fractionation (Davidson et al., 2004) was developed and outlined in the following table 1. Table 1 Table shows sequential extraction procedure for Copper-bearing solid phases Step Extractant Target Phase 1. Acetic acid 1M acetic acid, pH 2.5, 24h, 298K Carbonate matrix 2. HAHC 0.5M hydroxylammonium
hydrochloride, pH 1.5, 24h, 298K Mn hydroxides
3. DCB 0.2M Na-dithionite-citrate-bicarbonate, pH 3, 24h, 298K
Fe hydroxides
4. Aqua regia Concentrated HCl/HNO3, (3:1 Residual fraction

34
ratio) with microwave-assisted Samples of copper concentration in different extraction steps shown in table 1 were measured by flame atomic absorption spectrometry (F-AAS) along with certified reference material (GBW07108/GSR-6, limestone sample from China, Cu content of 23 ± 3 mg/kg) in which served as quality assurance. The recovery rate of Cu for all the sub-samples and the certified reference material were at 100 ± 15% on average (± SD) by comparison with the sum of all extraction steps ( 4 steps), the single hot acid digestions and the X-ray fluorescentce spectroscopy (XRF) analysis yields.The pH of first acetic acid step was stayed below 4 to ensure complete dissolution of carbonate matrix and prevent readsorption of Cu between first two steps. The applied reagent concentrations and soil-to-extractant ratios were 1:40 (1.0g to 40mL) for the first three steps and 1:20 for the last step. The pH of second extraction step was monitored throughout to ensure no transfer of buffer solution from first to second extraction step causing rise of pH of second step.
Fig. 1 Results of BCR sequential extraction analyses of Cu fractionation
26±17 3±2
20±20 2±1
17±21 1±1
2±2
23±7
34±24 3±2
4±2
233±7
1: 1M Acetic acid
2: 0.5M Hydroxylammonium chloride

35
The single concentrated HCl acid digestions showed lime samples (average of six sub-samples) with Fe, Mn and Cu concentrations in 8.5 ± 2.2 g/kg, 5.9 ± 1.8 g/kg and 98.7 ± 69.8 mg/kg respectively. Excessive standard deviation of Cu concentration was due to inhomogeneitites in the lime composition. All sub-samples demonstrated above usual 2-8 mg/kg Cu content in limestone for soil amendment and five out of six sub-samples revealed higher than typica phosphate fertilizers Cu level (1-13 mg/kg). Half of the six-subsamples exceeded soil health tolerance level of 100 mg/kg for Cu. The Cu concentrations of six sub-samples were found to be 12, 41, 69, 137, 140 and 174 mg/kg. All the foundings suggested excessive Cu in lime could be loaded on the uncontaminated soil and cause Cu toxicity to plants. The result of BCR sequential extraction of Cu, Fe, Mn and Ca fractionation was shown in figure 1. The total extracted Cu in the BCR scheme ( 1-4 steps) demonstrated good accordance with the results of single step microwave- assisted aqua regia concentrated HCl acid digestion and XRF analyses.The distribution of Cu in different phases were showed in figure 1 and about 35% of the total Cu which mobilised in the exchangeable and carbonate-bound 1st step was associtated with carbonate minerals as 95% carbonate matrix element of Ca was dissolved in 1st step. In addition, the observed over 30% of dissolved total Fe and 60% of dissloved total Mn in first step could be from the combination of Fe/Mn-bearing carbonates and poor nanocrystalline oxide. The 2nd HAHC step which targetted Mn oxyhydroxides and amorphous Fe oxyhydroxides extracted 18 ± 12% total Cu and released more Fe and Mn. The Cu/Mn ratios were identical for the first and second extraction step but not for the Cu/Fe ratios. The 3rd and 4th step extracted remaining 21 ± 10% and 27 ± 8% of Cu respectively. The DCB step was used to target non-silicate strongly reducible Fe oxides with negligible amount of Mn being found in this step. The copper resided in the silicates and other resistant minerals in the 4th step usually contributed no important role in environmental assessments. The above results in figure 1 suggested that Cu was able to be mobilized acidic and reducing conditons. Cu identified in the first step could be associated with the carbonate matrix or the oxide dendrites alone, or the combination of both. It is important to distinguish Cu loaded between carbonate matrix phase and oxide dendrites phase as Cu from these two phases could be mobilized under different environmental conditon (e.g. reducing). Direct X-ray spectroscopic speciation analysis was considered to be next stage in depth analysis for the Cu speciation. Duration of the PhD-project Since April 2008 List of publications, posters, oral presentations etc. (since the beginning of the PhD-project)
G. T. Schmidt, K. H. Lui, M. Kersten. Speciation and mobility of arsenic in agricultural lime. Journal of Environmental Quality, 38: 2058-2069.
G. T. Schmidt, K. H. Lui, M. Kersten. Speciation and mobility of arsenic in agricultural lime. Journal of Environmental Quality. Oral presentation in Ebernburg GRK 826/3 meeting, November 2008, Mainz, Germany.
D. Zuzaan, K. H. Lui, M. Kersten. Speciation of copper enriched in agricultural lime. Soil Science Society of America Journal. (Submitted, pending for review)
D. Zuzaan, K. H. Lui, M. Kersten. Speciation of copper enriched in agricultural lime. Journal of Environmental Quality. Oral presentation in Ebernburg GRK 826/3 meeting, November 2009, Mainz, Germany.

36
Cooperation within and outside the GRK The major research work has been taking place in Prof. Kersten’s research group. Part of the research work has been undertaken in collaboration with Prof. Wilcke and Prof. Bings’s research group within the Graduiertenkolleg. Moritz Bigalke, Kathrin Schilling, Burkhard Knopf and Jan Niclas Schaper (all GRK-PhD students) have been involved with some of the instrumental analysis (e.g. GF-AAS, ICP-MS and etc.). Participation in the course programme of the GRK (since the beginning of the PhD-project)
02.11 - 04.11.2008 Seminar: “Jahresseminar Des Graduiertenkollegs 826/3 Auf Burg Ebernburg In Bad Münster An Der Nahe"
01.09 - 05.09.2008 Course: ”Analytische Qualitätskontrolle” of Prof. Dr. Wolfhard Wegscheider, Austria
02.11 - 04.11.2009 Seminar: “Jahresseminar Des Graduiertenkollegs 826/3 Auf Burg Ebernburg In Bad Münster An Der Nahe"
02.03 - 04.03.2010 Course: “Einführung in die Elementarstatistik" of Prof. Dr. Einax, Friedrich-Schiller-Universität Jena, Germany
Tutors Supervising tutor: Prof. Dr. Michael Kersten Stay abroad (if applicable) (since the beginning of the PhD-project)
13.07 - 15.07.2009 Course: “Environmental Chemistry of Heavy Metals", Institute of Earth Sciences, University of Heidelberg.

37
Thema und Betreuer des Dissertationsprojekts Stipendiatin: Ute Thorenz, geb. Müller
Entwicklung einer TD-GC-MS Methode zur Messung von Halogenkohlenwasserstoffen aus biotischen und abiotischen Quellen Betreuer: Prof. Dr. T. Hoffmann Berichtszeitraum: August 2009 – April 2010 Darstellung des Dissertationsprojekts und der Forschungsergebnisse im Berichtszeitraum Motivation: Leichtflüchtige Halogenkohlenwasserstoffe in der Atmosphäre übernehmen eine Schlüsselrolle in der Chemie der Troposphäre und der Stratosphäre[1]. Sie sind direkt verbunden mit dem Ozonabbau der Stratosphäre und führen über die Oxidation der Halogene, vor allem des Iods, zur Neubildung von Partikeln. Damit beeinflussen die Halogenkohlenwasserstoffreaktionen den Strahlungshaushalt der Atmosphäre und übernehmen eine wichtige Rolle in klimarelevanten Reaktionen. Einige dieser Reaktionen sind für Iodverbindungen in Abbildung 6 gezeigt.
Abbildung 6: Schema der atmosphärisch relevanten Reaktionen verschiedener Iodspezies
Als Quelle dieser bromierten, chlorierten und/oder iodierten Verbindungen sind seit längerem verschieden Mikro- und Makroalgen bekannt [2,3]. Obwohl diese biotischen Quellen schon seit längerem bekannt sind, ist die direkte Freisetzung aus Algen nur teilweise verstanden und es gibt immer noch Lücken, vor allem die Erforschung verschiedener Regionen und Spezies ist noch lückenhaft. Zusätzlich zu den noch offenen Fragen der biotischen Quellen wurden vor kurzem abiotische, anorganische Bildungsreaktionen der Halogenkohlenwasserstoffe gefunden [4,5]. Diese Bildungswege sind verbunden mit der Reaktion von Ozon an verschiedenen Oberflächen, wie Eis oder Meerwasser. Außerdem wird die Bildung der Halogenkohlenwasserstoffe an Partikeloberflächen vermutet.

38
Aus analytischem Blickwinkel ist es wichtig die Konzentration der einzelnen Spezies zu untersuchen, um dann Rückschlüsse auf deren biotische oder abiotische Quellen zu ziehen. Hierfür ist es sinnvoll sogenannte Tracer Spezies oder Tracer Spezien-Verhältnisse zu finden, anhand derer die Quellen unterscheidbar sind. Nur wenn die Quellen und die Budgets dieser klimarelevanten Verbindungen bekannt sind können zuverlässige Klimamodelle erstellt werden.
Vorgehensweise:
Das Ziel meiner Arbeit ist die Entwicklung einer sensitive Methode zur Untersuchung der halogenierten Kohlenwasserstoffe, um dann mittels Tracer-Verbindungen oder -Verhältnissen zwischen biotischen und abiotischen Quellen zu unterscheiden.
Die Methode der Wahl hierfür ist eine Kombination einer anreichernden Probennahmemethode, da diese Verbindungen in der Atmosphäre in sehr niedrigen Konzentrationen vorliegen, und einer selektiven und sensitiven Nachweißmethode, um die Halogenspezies sicher zu unterscheiden und deren Konzentrationen genau zu bestimmen. Daher ist der in meiner Arbeit verfolgte Weg die Kopplung zwischen der Probenahmen mit Adsorbtionsmitteln und anschließender Thermodesorption (TD) und dem Nachweis mit Gaschromtographie-Massenspektrometrie (GC-MS) in negativer chemischer Ionisation (NCI). Diese Ionisierung wird bevorzugt da aufgrund der hohen Elektronenaffinität halogenierter Verbindungen diese besonders sensitiv ist. Der schematische Aufbau des TD-GC-MS ist in Abbildung 7 gezeigt.
Abbildung 7: Schematischer Aufbau der verwendeten TD-GC-MS
Als Adsorbentien werden sogenannte Dreibett-Desorptionsröhrchen verwendet, die eine für die Zielkomponenten optimierte Mischung von drei verschiedenen Adsorbentien darstellt. Diese Mischung soll sicherstellen dass die unterschiedlich flüchtigen Komponenten zuverlässig adsorbiert werden (kleines Durchbruchsvolumen) und zusätzlich nicht zu stark gebunden werden, damit sie während der Thermodesorption vollständig gelöst werden können ohne die Komponenten zu zerstören. Die anschließende Thermodesorption eignet sich optimal für die Halogenalkane, da auch bei hohen Temperaturen stabil sind. Die Methodenentwicklung umfasst neben dem Temperaturprogramm der TD noch die Optimierung der GC-MS Methode. Dies umfasst die Bestimmung verwendbarer Interner Standards und die Identifizierung der häufigsten Ionen für den SIM-Mode.

39
Die Anwendung auf Realproben, gesammelt an der Atmosphären Forschungsstation in Mace Head, Irland und vom Martin Ryan Institut in Carna, Irland, ist im direkten Anschluss an die fertige Optimierung der Methode geplant. Diese Proben wurden im Rahmen der EASI-AQCIS Kampagne während der Herbst Springflut genommen.
Ergebnisse: Die Ersten Messungen von Standards der Iodalkane zeigen, dass die TD-NCI-GC-MS insgesamt zuverlässige Messungen liefert, wobei die Methodenentwicklung noch ganz am Anfang steht. So macht die deutlich höhere Flüchtigkeit der Halogenalkane im Vergleich zu anderen VOCs sich in den kleinen Durchbruchsvolumina der TD Röhrchen bemerkbar, die in ersten Versuchen auf unter 100 ml bestimmt wurden.
Die Entwicklung des Thermodesortionsprogramms und der GC werden systematisch variiert um die optimalen Bedingungen zu finden. Die Momentanen Parameter liefern das folgende Chromatogramm eines Mischstandards der Iodalkane (Abbildung 8). Dieses zeigt eine bei einigen Peaks unzureichende Auftrennung und eine Konzentration aller Peaks in das vordere Drittel, dies soll durch die Anpassung des Trägergasflusses behoben werden.
Abbildung 8: Chromatogramm eine Mischung verschiedener Iodstandards
Die zugehörigen Massenspektrum zeigen eine stabile Ionisierung der jeweiligen Halogenidionen (CL: 35, 37; Br: 79, 81; I: 127). Die negative Ionisierung zeigt sich somit als äußerst sensitiv aber als wenig selektiv. Die geringe Selektivität soll durch eine Optimierung der Ionisierungsparameter (CI-Gas Druck, Primäre Elektronenenergie etc.) gesteigert werden. Zusätzlich können dann deuterierte oder 14C-markierte interne Standards verwendet werden, um eine hohe Reproduzierbarkeit zu sichern.

40
[1] Katie A. Read, Anoop S. Mahajan, Lucy J. Carpenter, Mathew J. Evans, Bruno V. E. Faria, Dwayne E. Heard, James R. Hopkins, James D. Lee, Sarah J. Moller, Alastair C. Lewis, LuisMendes, James B. McQuaid,Hilke Oetjen, Alfonso Saiz-Lopez, Michael J. Pilling,John M. C.Plane. Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean. Nature letters 2008 (453) 1232-1235
[2] A. Colomb, N.Y. Yasaa, J. Wiliams, I. Peeken, K. Lochte. Screening volatile organic compounds (VOCs) emissions from five marine phytoplankton species by head space gas chromatography/mass spectrometry (HS-GC/MS). J. Environ. Monit. 2008 (10) 325-330.
[3] F. Laturnus, T. Svensson, C. Wiencke, G Öberg. Ultraviolet radiation affects emission of Ozone-depleting substances by marine macroalgae: Results from a laboratory incubation study. Environ. Sci. Technol. 2004 (38) 6605-6609
[4] L.J. Carpenter, J.R. Hoptkins, C. E. Jones, A. C. Lewis, R. Parthipan, D.J. Wevill, L. Poissant, M. Pilote, P. Constant. Abiotic Source of Reactive Organic Halogens in the Sub-Arctic Atmosphere? Environ. Sci. Technol. 2005(39) 8812-8816
[5] Steven L. Manley, John L. de la Cuesta. Methyl iodide production from marine phytoplankton cultures. Limnol. Oceanogr. 42( I ) 1997, 142-147

41
Dauer der Promotion Beginn August 2009, voraussichtlicher Abschluss Herbst 2012. Angaben zu entstandenen Publikationen, Kongressbeiträgen etc. (seit Beginn der Promotion)
U.Müller, Strategies for measuring halocarbons of marine origin using thermodesorption GC-MS. Vortrag, GRK-Jahresseminar 2009, Bad Münster am Stein, 26.06.2006
Zusammenarbeit mit bzw. Bezug zu anderen Projekten (innerhalb und außerhalb des GRK)
Bestimmung des Gesamtiodgehalts in Meerwasser und Algen mittels ICP-OES in Zusammenarbeit mit Michael Kundel und dem Arbeitskreis von Prof. Dr. N. Bings. Diese Untersuchung läuft im Rahmen einer Kooperation mit Udo Nitschke und Dr. Dagmar Stengel vom Martin Ryan Institut der Universität Galway.
Teilnahme am Studienprogramm (seit Beginn der Promotion) Jahresseminar des Graduiertenkollegs auf Burg Ebernburg in Bad Münster am Stein,
18. - 20. 10. 2009 Ringvorlesung I Wintersemester 2009/2010
Blockpraktikum „Mikrobiologische Methoden – Laborpraktikum “ Prof. Dr. H. König 8.-12.02.2010
Kurs „Chemometrik“ Prof. Dr. W. Einax 02.-4.03.2010 Betreuung im Graduiertenkolleg Betreuer: Prof. Dr. T. Hoffmann ggf. Auslandsaufenthalte (seit Beginn der Promotion) Internationale Messkampagne (EASI-AQCIS) an der Atmosphärenmessstation
„Mace Head“ und am Martin Ryan Institute Carna (MRI CARNA) in Carna, Irland vom 15.8.bis zum 15.9.2009.

42
Thema und Betreuer des Dissertationsprojekts Stipendiat: Niklas Schaper, Dipl.-Chem. Thema: Entwicklung eines Picolitertropfengenerators für die emissions- und massenspektrometrische Elementspuren- und Speziesanalytik Betreuer: Prof. Dr. Nicolas H. Bings Berichtszeitraum: Mai 2009 – April 2010 Darstellung des Dissertationsprojekts und der Forschungsergebnisse im Berichtszeitraum Motivation Ein gängiger und zugleich weit verbreiteter Weg des Eintrags flüssiger Proben in analytische Plasmen besteht in der kontinuierlichen Erzeugung eines Aerosols mittels pneumatischer Zerstäubung. Die meisten der heute verwendeten Systeme basieren auf Techniken der Aerosolerzeugung, deren Grundlagen bereits 1879 beschrieben wurden[1]. Ein solches Aerosol weist eine recht breite Tröpfchengrößenverteilung auf, so dass ein Großteil (ca. 95%) des Aerosols über eine Sprühkammer abgeschieden werden muss, da nur kleinste Tröpfchen mit einem aerodynamischen Durchmesser kleiner als dae 10 µm zum Eintrag in ein Plasma geeignet sind. Es existiert eine Vielzahl von Kombinationen unterschiedlicher Zerstäuber und Sprühkammern, die für z.B. maximale Empfindlichkeit, höchstmögliche Präzision oder minimalen Probenverbrauch ausgelegt sind. Gerade Letzteres ist im Falle der Elementspeziesanalytik bei der Kopplung von Trennsystemen wie der Hochleistungs-Flüssigkeitschromatographie (HPLC) oder der Kapillarelektrophorese (CE) mit der induktiv gekoppelten Plasmaemissions-spektrometrie (ICP-OES) oder Plasmamassenspektrometrie (ICP-MS) besonders wichtig. Allerdings sind selbst im Falle der sogenannten Niedrigflusszerstäuber die für einen verlässlichen Betrieb erforderlichen Probenzuführungsraten um ein Vielfaches höher, als sie z.B. von kapillarelektrophoretischen Trennsystemen bereitgestellt werden können. Das Anpassen der Flussrate durch kontinuierliches Zumischen eines weiteren Flüssigkeitsstromes zum Eluenten ist oft unerlässlich, was weitere Probleme hinsichtlich des Nachweisvermögens infolge der Verdünnung und in Bezug auf das erzielbare chromatographische Auflösungsvermögen, bedingt durch eventuelle Totvolumina, bereiten kann. In den vergangenen Jahren wurden im Rahmen der Entwicklung weiterer konventioneller Zerstäubersysteme große Anstrengungen unternommen, um den Probenverbrauch zu begrenzen. Dennoch erfordern moderne Systeme noch immer eine Flussrate von einigen µL / min Analytlösung, um ein stabiles Aerosol zu erzeugen[2]. In der Literatur wurden Systeme beschrieben, um eine Serie von Tropfen mit kleinsten Volumina zu erzeugen. Bisher basierten diese Systeme jedoch auf dem Einsatz piezoakkustischer Aktuatoren oder der gezielten elektrostatischen Ablenkung einzelner Tropfen[3-12]. Diese Einzeltropfengeneratoren wurden überwiegend konstruiert, um Prozesse in der Anregungs- bzw. Ionisationsquelle untersuchen zu können. Ihre Eignung als universelles Probenzuführungssystem wurde jedoch nicht untersucht. In der analytischen Chemie wurden thermische Tintenstrahldrucker bereits eingesetzt, um kleinste Flüssigkeitsmengen auf feste Oberflächen zu dosieren[7]. Basierend auf diesem Ansatz können die Tropfen in der Größenordnung weniger Picoliter auch zur Aerosolerzeugung genutzt werden, was sich aufgrund der schmalen Größenverteilung dieser Tropfen positiv auf die Transporteigenschaften des Aerosols und die resultierenden analytischen Güteziffern bei der ICP-MS auswirken sollte. Außerdem ist zu erwarten, dass sich über die Betriebsparameter der Dosierpatrone die Flussrate unabhängig vom verwendeten Trägergasstrom variieren lässt, was zum einen im Vergleich mit

43
konventionellen pneumatischen Zerstäubern ein völlig neues Konzept zur Erzeugung analytischer Aerosole darstellt und zum anderen für die Methodenoptimierung einen weiteren wertvollen Freiheitsgrad liefert. Vorgehensweise Die in der Literatur[13] beschriebene Modifikation handelsüblicher thermischer Tintenstrahldrucker zur Erzeugung kleinster Tropfen kann nicht auf eine kontinuierliche Aerosolerzeugung übertragen werden, da der computergesteuerte Druckprozess zeilenweise abläuft und zudem durch Papiervorschub- und Düsenreinigungsschritte unterbrochen wird. Um einen kontinuierlichen Betrieb der Dosiereinheit (Tintenpatrone) eines Tintenstrahldruckers zu ermöglichen, wurde ein eigenständiger Mikrocontroller entwickelt. Dieser erlaubt die Kontrolle aller zur thermischen Tropfenerzeugung wichtigen Parameter wie Heizzeit und Wiederholfrequenz. Somit ist es möglich thermische Dosiereinheiten unabhängig von einem Steuerungscomputer zur Tropfenerzeugung zu nutzen. Die Tropfen werden in der Aerosoltransportkammer in einen Argongasstrom dosiert und das entstandene Aerosol wird mit Hilfe dieses Trägergasstromes direkt zur Fackel eines ICP-MS transportiert. Dieser neuartige „Drop-on-Demand“ (DOD) Aerosolgenerator wurde hinsichtlich seiner Leistungsfähigkeit mit dem konventionellen, auf niedrige Probenflussraten optimierten Zerstäuber „MicroMist“™ in Kombination mit einer Zyklon-Sprühkammer verglichen. Neben der Signalstabilität und der erzielbaren Empfindlichkeit bei äußerst geringen Probenflussraten stand dabei die Untersuchung der Möglichkeit zur Kopplungen mit Trennsystemen wie der Kapillarelektrophorese im Vordergrund. Die Fähigkeit, transiente Signale abzubilden wurde an einem, mittels eines Fließinjektionssystems (FIA) erzeugten, transienten Signal demonstriert. Das selbst gebaute Fließinjektionssystem verfügt über eine Probenschleife mit einem Volumen von 3 µL und die Flußrate betrug 0,85 mL/min. Um die Vergleichbarkeit der zwei untersuchten Techniken zu gewährleisten, wurde der pneumatische Zerstäuber über ein T-Stück mit dem Flüssigkeitsstrom des FIA-Systems verbunden und im selbstansaugenden Modus betrieben (Abb. 1).
Abb. 1: Schematischer Aufbau zum Vergleich FIA-DOD-ICP-MS (oben) vs. FIA-ICP-MS (MicroMist™, unten).
Um den DOD-Generator mit der Fließinjektion zu verbinden, wurde wie in Abb. 2 zu sehen die ursprüngliche Geometrie der Dosiereinheit modifiziert. Das Totvolumen des Flüssigkeitsreservoirs wurde durch Anbringen einer Deckelplatte mit Schlauchdurchführungen von etwa 200 µL auf ca. 130 µL verkleinert. Durch die Schläuche kann die Probe mittels konventioneller peristaltischer Pumpen bis zu den Düsen transportiert werden. Nicht benötigte Probe gelangt durch einen zweiten Schlauch in das Abfallgefäß.

44
Abb. 2: Modifizierte DOD-Dosiereinheit zur Kopplung mit FIA und anderen Probenzuführungstechniken.
Soweit nicht anders erwähnt wurden mit 5 % HNO3 angesäuerten Einzelelementlösungen der Elemente Rh, Y oder In der Konzentration c = 100 µg / L für die Versuche verwendet. Die Betriebsparameter des verwendeten Versuchsaufbaus sind in Tab. 1 wiedergegeben.
Tab. 1: Optimierte Betriebsparameter
ICP-MS (Agilent 4500) DOD Generator
Leistung 1250 W Pulsbreite 2,5 - 7,3 µs
Plasmagas 17 L/min Frequenz 0,54 - 2,68 kHZ
Hilfsgas 1 L/min Probenflussrate 2,8 – 8,7 µL/min
Trägergasstrom 1 – 1,3 L/min
i.d. Injektorrohr 2,5 mm
Integrationszeit 100 ms
m/z Isotop 89Y, 103Rh, 115In
"MicroMist"™
FIA System
Probenflussrate 33 µL/min Probenschleife 3 µL
Probenflussrate 0,85 mL/min
Ergebnisse Durch die Verwendung des Mikrocontrollers ist es möglich, die Tropfenbildung gezielt zu beeinflussen. Neben der Aufheizdauer des Heizelementes ist die Wiederholfrequenz der Tropfenbildung entscheidend für das ausgestoßene Volumen, während die Tropfengröße

45
primär vom Durchmesser der Düse der verwendeten Dosiereinheit abhängt. Abb. 3 zeigt, wie zu erwarten, dass mit zunehmender Frequenz der Tropfenbildung auch mehr Flüssigkeit ausgestoßen wird. Die Probenflussrate des neu entwickelten DOD Aerosolgenerators ist somit über einen sehr großen Bereich von einzelnen Tropfen bis hin zu einigen mL/min bei simultaner Verwendung mehrerer Düsen einstellbar, wobei besonders vorteilhaft ist, dass der Trägergasstrom bei einer für das ICP optimalen Flussrate gehalten werden kann, da bei diesem neuartigen Aerosolgenerator-Prinzip die Tropfenbildung vom Gasfluss vollständig unabhängig ist.
Abb. 3: Abhängigkeit der pro Zeitintervall dosierten Flüssigkeitsmenge von der Tropfenbildungsfrequenz.
Da einige Bauteile des Dosiersystems in direktem Kontakt mit der zu untersuchenden Analytlösung stehen, wurde untersucht, inwiefern sich dies auf die Auswahl möglicher Analyten auswirkt, da gegebenenfalls mit erhöhten Blindwerten zu rechnen ist. In einer Studie wurden deshalb Mehrelementstandardlösungen der Konzentration 1 g/L als Analytlösung dosiert, wobei, wie in Abb. 4. erkennbar, bei Al, Si, Ni, Cu und Ta Störungen durch stark erhöhte Blindwerte gefunden wurden. Die vorgestellt Technik ist somit, zumindest unter Einsatz des beschriebenen Dosierpatronentyps, für diese Elemente nur beschränkt tauglich.
Abb. 4: Studie zur Untersuchung der Einsatzmöglichkeit des vorgestellten DOD-Systems zur Dosierung von Elementlösungen einer Konzentration von 1 g/L bei der ICP-MS: geeignete (grün) / bedinge geeignete (rot, erhöhter Blindwert) Analyten.
Auch die Signalstabilität über einen Zeitraum von mehreren Minuten (Abb. 5) ist gegeben, wobei maximale Standardabweichungen von 6-7% auftreten.
H He
Li Be B C N O F Ne
Na Mg Al Si P S Cl Ar
K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr
Rb Sr Y Zr Nb Mo Tc* Ru Rh Pd Ag Cd In Sn Sb Te I Xe
Cs Ba ---- Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po* At* Rn*
Fr* Ra* ----
La Ce Pr Nd Pm* Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
Ac* Th* Pa* U* Np* Pu*

46
Abb. 5: Signalstabilität des vorgestellten DOD-ICP-MS-Systems.
Transiente Signale lassen sich sehr gut mit dem neu entwickelten Aerosolgenerator abbilden, wie in Abb. 6 vergleichend mit Daten eines konventionellen MicroMist™ basierten FIA-ICP-MS Systems (Aufbau s. Abb. 1) zu erkennen ist. Die Signalbreite des konventionellen Systems ist mit 60 s deutlich größer als das 46 s breite Signal des DOD Generators. Es kann somit gefolgert werden, dass das relativ zu einer theoretischen Signalbreite von 0,3 s deutlich ausgedehnte Signal sicher noch auf Totvolumina der Dosiereinheit zurückzuführen ist, dieses aber offenbar kleiner ausfällt als jenes des untersuchten Referenzsystems. Ein Einsatz als Interface zur Kopplung von Fließinjektionssystemen und gegebenenfalls auch von Trennsystemen mit der ICP-MS ist somit - nach weiterer Optimierung - möglich.
Abb. 6: Vergleich eines transienten 115In Signals eines FIA-ICP-MS-Systems (rechts) mit dem eines FIA-DOD-ICP-MS Systems (links). 100 µg/L In, 3 µL Probenschleife. Ansaugrate „MicroMist™“ Zerstäuber: 33 µL/min, DOD-Dosierflussrate 2,73 µL/min.
Ausblick Das in der Dosiereinheit noch vorhandene Totvolumen muss in Zukunft noch weiter verkleinert werden, um eine Kopplung mit einer Kapillarelektrophorese zu ermöglichen. Als sehr vorteilhaft ist in diesem Zusammenhang zu erwähnen, dass das DOD-System auch bei den in der CE üblichen Flußraten eine gleichmäßige Zerstäubung erlaubt und somit auf zusätzliche „make-up“-Flüssigkeitsströme vollständig verzichtet werden kann. Dies wiederum sollte sich vorteilhaft auf das erzielbare Nachweisvermögen und, je nach Güte der CE-DOD-Kopplung, auf das chromatographische Auflösungsvermögen auswirken. Weitere Untersuchungen mit realen Proben sollen die generelle Einsetzbarkeit des neuartigen Aerosolgeneratorsystems aufzeigen. Denn unabhängig vom tatsächlichen Totvolumen der Dosiereinheit kann das bereits bestehende System problemlos in den analytischen Arbeitsfluss integriert werden, da alle standardmäßig vorhandenen

47
Komponenten wie Autosampler und peristaltische Pumpen zum Probentransport eingesetzt werden können. Auch die mit dem neuen DOD-System erzielbaren analytischen Güteziffern wie Nachweisgrenzen und Rauscheinflüsse sollen in weiteren Arbeiten detailliert untersucht werden. Das so charakterisierte, neu entwickelte Probenzuführungssystem soll dann unter anderem zur Speziesanalytik von Aerosol- und Umweltproben verwendet werden, wobei ein besonderes Augenmerk auf metall- und iodhaltigen Partikeln liegt. Diese spielen insbesondere bei Partikelbildungsprozessen in der maritimen Grenzschicht (marine boundary layer, MBL) eine tragende Rolle und haben, wie alle atmosphärischen Aerosole, einen Einfluss auf das Klima der Erde. Das generelle Verständnis dieses Einflusses ist noch sehr begrenzt und bedarf weiter Forschung. Neuere Untersuchungen belegen, dass eine Vielzahl unterschiedlicher iodhaltiger Verbindungen in der Atmosphäre nachweisbar sind. Einige dieser Spezies konnten bereits identifiziert werden, die Identität weiterer Verbindungen ist jedoch noch nicht aufgeklärt[14]. Die Identifizierung und Quantifizierung solcher Verbindungen ist wichtig für das Verständnis ihrer Bildungsprozesse in der Atmosphäre. Darüber hinaus wird im Rahmen der Arbeit zu untersuchen sein, ob ein solcher Tropfengenerator auch in anderen Bereichen der Analytik von Aerosolen eingesetzt werden kann. Denkbar wäre die Erzeugung eines Kalibrieraerosols bekannter Tröpfchengrößenverteilung für Partikelgrößenbestimmungen mit Hilfe moderner Aerosolmassenspektrometer. Literatur: [1] Guoy, CL; Photometric Research on Colored Flames, Ann. Chim. Phys., 18,5-101, 1879 [2] Todoli, JL, Mermet, JM; Spectrochimica Acta Part B, 61, 239-283, 2006 [3] Hieftje, GM, Malmstadt, HV, Analytical Chemistry, 40, 12, 1860-1867, 1968 [4] Childers, AG; Hieftje, GM; Applied Spectroscopy, 40, 5, 688-691, 1986 [5] Wiegand, PM; Crouch, SR; Applied Spectroscopy, 42, 4, 568-571, 1988 [6] French, JB; Etkin, B; Jong, R; Analytical Chemistry, 66, 5, 685-691, 1994 [7] Mahoney, PP; Hieftje, GM; Applied Spectroscopy, 48, 8, 956-958, 1994 [8] Olesik, JW, Hobbs, SE; Analytical Chemistry, 66, 20 ,3371-3378, 1994 [9] Lazar, AC, Farnsworth, PB; Applied Spectroscopy, 51, 5, 617-624 1997 [10] Olesik, JW; Applied Spectroscopy, 51, 5, 158A-175A, 1997 [11] Olesik, JW, Kinzer, JA; Spectrochimica Acta Part B, 61, 696-704, 2006 [12] Steward, II, Olesik, JW; Journal American Society of Mass Spectrometry, 10, 159-174, 1999 [13] Fittschen, U.E.A., Bings, N.H., Hauschild, S, Förster, S, Kiera, A.F., Karavani, E, Frömsdorf, A, Thiele, J, Falkenberg, G; Analytical Chemistry, 80, 1967-77, 2008 [14] Gilfedder, B.S.; Lai, S.C.; Petri, M, Biester, H, Hoffmann, T; Atmospheric Chemistry and Physics, 8, 6069-6084, 2008 Dauer der Promotion Beginn der Promotion: Oktober 2008 Angaben zu entstandenen Publikationen, Kongressbeiträgen etc. (seit Beginn der Promotion) Kongressbeiträge Poster

48
J. Niklas Schaper, Jan Maßmann, Jan H. Petersen, Nicolas H. Bings; „Drop-on-demand aerosol generator for ICP-MS analysis“. Colloquium Spectroscopicum Internationale, Budapest (Ungarn), 2009. (Auszeichnung für bestes Poster der Tagung) J. Niklas Schaper, J. Maßmann, J.H. Petersen, Nicolas H. Bings; “A novel low-flow drop-on-demand aerosol generator: Analytical charactrization and its potential for transient sample introduction into ICP-MS.” Winter Conference on Plasma Spectrochemistry, Fort Myers (USA), 2010. Jan H. Petersen, J. Maßmann, J.N. Schaper, T. Fiedler, N.H. Bings; “Towards a new calibration method for LA-ICP-MS based on dried residues of individual picoliter droplets.” Winter Conference on Plasma Spectrochemistry, Fort Myers (USA), 2010. Jan Maßmann, J.H. Petersen, J.N. Schaper, K. Filsinger, N.H. Bings; “Development and characterization of a drop-on-demand generator for sample introduction of very small volumes for plasma spectrometry.” Winter Conference on Plasma Spectrochemistry, Fort Myers (USA), 2010. (Auszeichnung als bestes Poster der Tagung) Jan H. Petersen, J. Maßmann, J.N. Schaper, N.H. Bings; “Fundamental characterization of a new drop-on-demand aerosol generator: introduction of single droplets into plasma excitation and ionization sources.” TraceSpec 2009, Mainz (Germany), 2009. Jan Maßmann, Jan H. Petersen, J. Niklas Schaper, J.A.C. Broekaert, N.H. Bings; “Development of a drop-on-demand aerosol generator for the introduction of small sample volumes in plasma spectrometry.” Colloquium Spectroscopicum Internationale, Budapest (Ungarn), 2009. Vorträge J. Niklas Schaper, Nicolas H. Bings; “Analytical characterization of a novel low-flow drop-on-demand aerosol generator and its potential for transient sample introduction for speciation analysis.” Annual Meeting of the Interdisciplinary Research Training Group Program (DFG GRK 826), Castle Ebernburg, Bad Münster an der Nahe (Germany), 2009 J. Niklas Schaper, Jan H. Petersen, Jan Maßmann, Nicolas H. Bings; „Entwicklung und Charakterisierung eines neuartigen drop-on-demand Aerosolgenerators für die Plasmaspektrometrie - Einsatzmöglichkeiten in der Speziesanalytik.“ Anwendertreffen: Atomspektrometrie mit Plasmen (ICP-OES, ICP-MS, Mirkowellenplasmen), Universität Hamburg (Deutschland), 2010. Nicolas H. Bings, Jan Maßmann, Jan H. Petersen, J. Niklas Schaper; “New Methods of Sample Introduction for Atomic Spectrometry”, Analytica Conference München, (Germany), 2010. Zusammenarbeit mit bzw. Bezug zu anderen Projekten (innerhalb und außerhalb des GRK) Vorversuche zur Partikelgrößenmessung wurden in Zusammenarbeit mit Dr. Frank Drewnick, Max-Planck-Institut für Chemie, Mainz durchgeführt. Die Bestimmung von Iod und Iodspezies soll in Zusammenarbeit mit der Arbeitsgruppe von Prof. Dr. Thorsten Hoffmann, Institut für Anorganische und Analytische Chemie Johannes Gutenberg-Universität Mainz, erfolgen. Teilnahme am Studienprogramm (seit Beginn der Promotion)
Ringvorlesung I Wintersemester 2009-2010 Blockpraktikum der Mikrobiologie, Prof. Dr. H. König, E. Kaschak,
Februar 2010

49
Sonderkurs „Statistik, Chemometrie und Qualitätssicherung“ Prof. Dr. Jürgen W. Einax, Friedrich-Schiller-Universität Jena
Betreuung im Graduiertenkolleg Betreuer: Prof. Dr. Nicolas H. Bings Weitere Betreuer: Prof. Dr. Thorsten Hoffmann ggf. Auslandsaufenthalte (seit Beginn der Promotion)
XXVI Colloquium Spectroscopicum Internationale, Budapest (Ungarn), 30.08-03.09. 2009
Winter Conference on Plasma Spectrochemistry Fort Myers/Florida, (USA) 04 – 09.01.2010

50
Thema und Betreuer des Dissertationsprojektes Stipendiat: Yvonne Scheller Thema: Detektion von Metallspezies in einzelnen lebenden Zellen mit hoher lateraler Auflösung Betreuer: Prof. Dr. Heinz Duschner Berichtszeitraum: März 2009 – März 2010 Darstellung des Dissertationsprojektes und der Forschungsergebnisse im Berichtszeitraum Der Boden bildet für Pflanzen und viele andere Lebewesen die Lebensgrundlage. Er versorgt sie mit Nährstoffen, Spurenelementen wie z.B. Metalle und Wasser. Metalle dienen oft als Cofaktoren in Enzymen und sind somit essentiell für den Stoffwechsel. Übersteigt die Metall-Konzentration die benötigte Menge oder liegt eine bestimmte Metall-Spezies vor, kann dies toxische Auswirkungen haben. Einige Pflanzen komplexieren die Metalle zur Detoxifizierung. Das gleiche gilt für tierische und humane Zellen. Einige Metalle sind auch für sie essentiell. Sie werden über die Nahrung aufgenommen. Enthält diese toxische Konzentrationen von Metallen, gelangen diese in den Stoffwechsel und verursachen dort möglicherweise die gleichen toxischen Effekte und Schäden wie in Pflanzen. Um diese Prozesse verstehen zu können, benötigt man Hinweise, wo genau in einer einzelnen Zelle die Metalle bzw. bestimmte Spezies gespeichert werden. Bisherige Untersuchungen in der Spurenanalytik beschränken sich auf Methoden wie ICP-MS, die zwar Informationen liefern, welche Spezies in einer Probe vorhanden ist, aber eine Lokalisierung der Spezies in einer einzelnen Zelle ist damit nicht möglich. Hohe laterale Auflösungen innerhalb einer einzelnen Zelle erhält man mit Hilfe der Confocalen Laser Scanning Microscopy (CLSM), die eine 3D-Darstellung der Zelle ermöglicht. Um noch höhere Auflösungen zu erreichen, kann SNOM (Scanning Near-field Optical Microscopy) eingesetzt werden, wodurch Auflösungen bis zu 50 nm erreicht werden können. Der mikroskopische Nachweis von Metall-Spezies könnte Hinweise liefern, wo genau in der Zelle die Spezies gebunden werden und welche spezifischen Eigenschaften dieser Ort aufweist, um die Metall-Spezies zu binden. Zwischen März 2009 und März 2010 beschränkten sich die Versuche auf humane Gingiva-Fibroblasten als Testorganismus. Diese wurden in Zellkulturflaschen mit Basal-Medium Eagles (BME) kultiviert. Für die Versuche wurden die Zellen in 6-Well-Plates oder Petrischalen (Ø 60mm / 15mm) passagiert, sodass man die Zellen direkt im Gefäß mit einem 40x Wasserobjektiv des CLSM Leica TCS SP2 beobachten konnte. Die Zellen wurden mit drei unterschiedlichen Konzentrationen (100µM, 300µM und 500µM) der Metallverbindungen NiCl2, NiSO4, Ni3S2 und CoCl2 für jeweils 16h, 24h und 48h inkubiert. Nach dieser Inkubationszeit wurden die Zellen nach mehrmaligem Waschen mit dem Fluoreszenzfarbstoff Newport Green DCF (7µg/ml) für 60 min inkubiert und die Fluoreszenz anschließend mit dem CLSM untersucht. Um Hinweise zu erhalten, ob geringere Konzentrationen (40µM und 70µM) detektiert werden können, wurden dem Kulturmedium diese Konzentrationen von NiCl2 für 24h und 48h dargeboten. Die maximale extrazelluläre Metallkonzentration war auf 500µM begrenzt. Bei hohen Metallkonzentrationen und langen Inkubationszeiten bis zu 48h zeigten die Zellen teilweise deutliche Veränderungen in ihrer Morphologie. Die langen Zellfortsätze, die charakteristisch für Fibroblasten als Bindegewebszellen sind, schrumpften und es kam zur Abkugelung der Zellen als Folge von Zelltot. Deshalb wurden in den Versuchen die Konzentrationen und Inkubationszeiten auf 500µM bzw. 48h beschränkt.
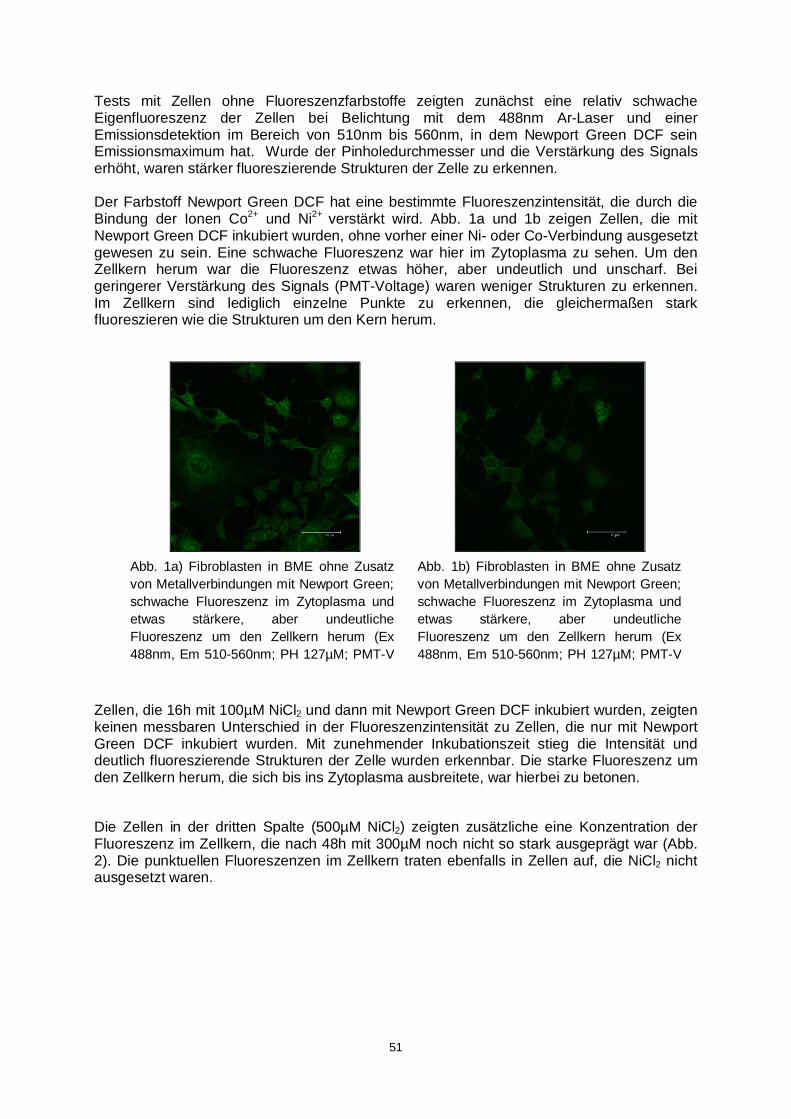
51
Tests mit Zellen ohne Fluoreszenzfarbstoffe zeigten zunächst eine relativ schwache Eigenfluoreszenz der Zellen bei Belichtung mit dem 488nm Ar-Laser und einer Emissionsdetektion im Bereich von 510nm bis 560nm, in dem Newport Green DCF sein Emissionsmaximum hat. Wurde der Pinholedurchmesser und die Verstärkung des Signals erhöht, waren stärker fluoreszierende Strukturen der Zelle zu erkennen. Der Farbstoff Newport Green DCF hat eine bestimmte Fluoreszenzintensität, die durch die Bindung der Ionen Co2+ und Ni2+ verstärkt wird. Abb. 1a und 1b zeigen Zellen, die mit Newport Green DCF inkubiert wurden, ohne vorher einer Ni- oder Co-Verbindung ausgesetzt gewesen zu sein. Eine schwache Fluoreszenz war hier im Zytoplasma zu sehen. Um den Zellkern herum war die Fluoreszenz etwas höher, aber undeutlich und unscharf. Bei geringerer Verstärkung des Signals (PMT-Voltage) waren weniger Strukturen zu erkennen. Im Zellkern sind lediglich einzelne Punkte zu erkennen, die gleichermaßen stark fluoreszieren wie die Strukturen um den Kern herum.
Zellen, die 16h mit 100µM NiCl2 und dann mit Newport Green DCF inkubiert wurden, zeigten keinen messbaren Unterschied in der Fluoreszenzintensität zu Zellen, die nur mit Newport Green DCF inkubiert wurden. Mit zunehmender Inkubationszeit stieg die Intensität und deutlich fluoreszierende Strukturen der Zelle wurden erkennbar. Die starke Fluoreszenz um den Zellkern herum, die sich bis ins Zytoplasma ausbreitete, war hierbei zu betonen.
Die Zellen in der dritten Spalte (500µM NiCl2) zeigten zusätzliche eine Konzentration der Fluoreszenz im Zellkern, die nach 48h mit 300µM noch nicht so stark ausgeprägt war (Abb. 2). Die punktuellen Fluoreszenzen im Zellkern traten ebenfalls in Zellen auf, die NiCl2 nicht ausgesetzt waren.
Abb. 1a) Fibroblasten in BME ohne Zusatz von Metallverbindungen mit Newport Green; schwache Fluoreszenz im Zytoplasma und etwas stärkere, aber undeutliche Fluoreszenz um den Zellkern herum (Ex 488nm, Em 510-560nm; PH 127µM; PMT-V
Abb. 1b) Fibroblasten in BME ohne Zusatz von Metallverbindungen mit Newport Green; schwache Fluoreszenz im Zytoplasma und etwas stärkere, aber undeutliche Fluoreszenz um den Zellkern herum (Ex 488nm, Em 510-560nm; PH 127µM; PMT-V

52
Ähnliches ist in Zellen zu beobachten, die mit NiSO4 und Ni3S2 inkubiert wurden (Abb. 3 und 4). Die Fluoreszenz nach 16h mit 100µM Ni3S2 war schwach und hauptsächlich um den Zellkern zu beobachten. Nach 48 h bzw. durch höhere Konzentrationen wurde die Fluoreszenzintensität im Zytoplasma und in den Strukturen um den Zellkern herum verstärkt und auch hier war eine Konzentration der Fluoreszenz im Zellkern zu beobachten (Abb. 3 und 4; 2. und 3. Spalte).
100µM 300µM 500µM Ni3S2
Abb. 4) Fibroblasten nach Inkubation mit unterschiedlichen Ni3S2-Konzentrationen und Inkubationszeiten; Ni(II)-Detektion mit Newport Green DCF.
Abb. 2) Fibroblasten nach Inkubation mit unterschiedlichen NiCl2-Konzentrationen und Inkubationszeiten; Ni(II)-Detektion mit Newport Green DCF.
100µM 300µM 500µM NiCl2
Abb. 3) Fibroblasten nach Inkubation mit unterschiedlichen NiSO4-Konzentrationen und Inkubationszeiten; Ni(II)-Detektion mit Newport Green DCF.
100µM 300µM 500µM NiSO4

53
Abb. 5 zeigt Fibroblasten, die mit CoCl2 inkubiert wurden. Die Fluoreszenzintensität ist etwas schwächer als in Abb. 2 bis 4. So ist im Zytoplasma die Intensität gering und im Zellkern selbst ist zu keiner getesteten Inkubationszeit und Konzentration Fluoreszenz zu beobachten. Lediglich in den Strukturen um den Kern herum ist Fluoreszenz zu detektieren, die sich länglich zu orientieren scheinen.
Die Detektion und Lokalisation von Ni(II) und Co(II) in einzelnen humanen Zellen durch den Fluoreszenzfarbstoff Newport Green DCF war teilweise bei geringen intrazellulären Konzentrationen schwer. Die schwache, aber detektierbare Eigenfluoreszenz einiger Zellstrukturen erschwerte die Lokalisierung. Bei höheren Konzentrationen war die Intensität des Co(II) oder Ni(II) gebundenen Farbstoffes hoch genug, sodass die Eigenfluoreszenz der Zelle und die Fluoreszenzintensität des ungebundenen Farbstoffes vernachlässigbar wurden. Ein Unterschied zwischen den Zellen mit Ni-Verbindungen und Co-Verbindungen wurde v.a. im Zellkern sichtbar, der bei den Nickel-Verbindungen deutlich fluoreszierte und bei Cobalt nicht. Dabei schienen die Spezies (NiCl2, NiSO4 oder Ni3S2) keinen nennenswerten Einfluss zu nehmen. Um weitere Informationen über die Lokalisation des Farbstoffes in den einzelnen Zellen zu erhalten, wurden Zellkompartimente, die in den vorangegangenen Bildern Hinweise auf mögliche Beteiligung lieferten, mit Fluoreszenzfarbstoffen unterschiedlicher Emissionsmaxima und Fluoreszenzspektren markiert und parallel zu dem metallspezifischen Farbstoff mit dem CLSM detektiert (Abb. 6). So sieht man, dass sich der Fluoreszenzfarbstoff im Zellkern konzentriert, im Zytoplasma befindet und auch mit dem ER-Tracker in der Lokalisation übereinstimmt.
100 µM 300 µM 500 µM CoCl2
Abb. 5) Fibroblasten nach Inkubation mit unterschiedlichen CoCl2-Konzentrationen und Inkubationszeiten; Co(II)-Detektion mit Newport Green DCF.

54
Dauer der Promotion Die Promotion wurde im Juni 2006 begonnen und wird voraussichtlich bis Herbst 2010 andauern. Angaben zu entstandenen Publikationen, Kongressbeiträgen, etc. (seit Beginn der Promotion) Y. Scheller, Posterpräsentation, GRK-Evaluation in Mainz, Oktober 2006 Y. Scheller, H. Duschner, Posterpräsentation auf der Gordon Research Conference „Cell
Biology of Metals“ in Newport, Rhode Island, Juli 2007 Y. Scheller, Posterpräsentation bei dem Workshop „Analytical Methodologies for Trace
Metal Speciation – Trace Spec 2007“, Münster, September 2007; Poster Award (Roland Frei Award)
Y. Scheller, Heinz Duschner, Vortrag auf der Konferenz „Trace Element Speciation in Biomedical, Nutritional and Environmental Sciences“, München, Mai 2008
Y. Scheller, Posterpräsentation bei dem Workshop „Analytical Methodologies for Trace Metal Speciation – Trace Spec 2009“, Mainz, September 2009
Abb. 6: Fibroblasten nach Inkubation mit 300 µM Nicl2; a) Ni(II) Markierung mit Newport Green DCF; b) Membran markierung mit CellMask Deep Red; c) Markierung des Endoplasmatischen Reticulum mit ER Tracker; d) Overlay von a, b und c

55
Zusammenarbeit mit bzw. Bezug zu anderen Projekten Die Zusammenarbeit findet bisher hauptsächlich mit Mitarbeitern von Herrn Stöckigt aus der Pharmazie statt, die Pflanzen in Zellkultur kultivieren. Teilnahme am Studienprogramm (seit Beginn der Promotion) Ringvorlesung II SS 2006 28.08. – 01.09.2006: „Metallomics and Metalloproteomics“ bei Prof. Dr. Ryszard Lobinski,
CRNS Pau, Frankreich 17.07. – 21.7.2006: Praktikum in der Angewandten Struktur- und Mikroanalytik, Prof.
Duschner („Direkte Mineralphasenanalytik und Elementspeziation in Festkörpern mit Hilfe von Laser-Scanning- und Elektronen-Mikroskopiemethoden sowie der Photoelektronen“) (XPS, ESEM, CLSM)
21.05. – 25.05.2007: Praktikum in der Mikrobiologie bei Prof. König, Universität Mainz (Isolierung von Bakterien, Identifizierung von Bakterien anhand der Morphologie und biochemischen Tests)
Ringvorlesung WS 07/08 01.09.2008 – 05.09.02008: Kurs „Analytische Qualitätskontrolle“ bei Prof Dr. Wolfhard
Wegscheider der Montanuniversität Leoben Betreuung im Graduiertenkolleg Betreuer: Prof. Dr. Heinz Duschner Ggf. Auslandsaufenthalte (seit Beginn der Promotion) 6 Tage Aufenthalt in Newport, Rhode Island (USA). Teilnahme an der Gordon Research Conference „Cell Biology of Metals“ Alter bei Eintritt in das Graduiertenkolleg 25

56
Thema und Betreuer des Dissertationsprojekts
Stipendiatin: Kathrin Schilling
Thema: Speziesabhängige Fraktionierung der stabilen Isotope von Selen während der biogenen Alkylierung in Böden
Betreuer: Prof. Dr. Wolfgang Wilcke
Berichtszeitraum: März 2009 – April 2010
Darstellung des Dissertationsprojekts und der Forschungsergebnisse im Berichtszeitraum
Motivation
Selen ist ein essentielles Spurenelement für viele Organismen, jedoch liegen die Schwellengehalte für Mangel und Toxizität eng beieinander. Der natürliche Se-Gehalt in den meisten Böden weltweit beträgt 0,1 bis 2 mg kg-1. Allerdings nimmt durch anthropogene Aktivität infolge von Bergbau und Metallgewinnung die Umweltbelastung durch Se zu. Das führt zu einer Anreicherung von Se in den Böden. Mithilfe von biologischer Sanierung besteht die Möglichkeit, stark Se-kontaminierte Böden zu reinigen.
gelöste Selenverbindungen- SeO42-; SeO3
2-; organisches Selen
Aufnahme
Pilze, Bakterien, Pflanzen
Methylierte Selenverbindungen
Beeinflussende Faktoren:-Methyldonoren-Kohlenstoffquellen; Nährstoffe-Temperatur; Feuchtigkeit; pH
Boden
Atmosphäre
Methylierung
Dimethylselenid (CH3SeCH3)Dimethyldiselenid (CH3SeSeCH3)
Abb. 1: Ausgasung von Methylseleniden aus dem Boden.
Es ist nachgewiesen, dass einige Bakterien, Pilze und Algen die Fähigkeit besitzen, anorganisches Se in volatile organische Se-Verbindungen umzuwandeln. Unter den metabolisierten Se-Verbindungen treten vor allem Dimethylselenid (DMSe) und geringe Mengen an Dimethyldiselenid (DMDSe) auf. Dieser Methylierungsprozess bewirkt sowohl eine Detoxifizierung, als auch eine Abnahme des Se-Gehaltes aufgrund von Verflüchtigung (Abb.1). Da Se in mehreren stabilen Isotopen auftritt, führen unterschiedliche Biomethylierungsprozesse zu unterschiedlichen Isotopensignaturen der flüchtigen Se-Verbindungen. Daraus folgt umgekehrt, dass Stabilisotopenverhältnisse von Se in Methylseleniden als Indikatoren unterschiedlicher Methylierungsprozesse interpretiert werden können. Des Weiteren können die Se-Isotopenverhältnisse über den Fortschritt von Methylierungsreaktionen sowie die physiko-chemischen Bedingungen, unter denen diese Reaktionen ablaufen, informieren.
Das Ziel unserer Arbeit ist es, (1.) den Einfluss unterschiedlicher Quellen (Selenit/Selenat) und physiko-chemischer Bedingungen bei der Methylierung von Se durch den Pilz Alternaria alternata auf die Se-Isotopensignaturen von
flüchtigen Methyl-Seleniden aufzuklären und (2) zu untersuchen, welche Se-Fraktionen in urbanen Böden für die mikrobielle Methylierung zur Verfügung stehen und welche Ausgangs-Isotopenverhältnisse des Selens in diesen Fraktionen vorliegen. Dazu haben wir uns eines vorhandenen, archivierten Probensatzes aus der

57
Stadt Bayreuth in Nordbayern bedient (Abb. 2), für den bereits umfangreiche Informationen zu seinen chemischen Eigenschaften vorliegen.
Abb. 2: Lage der Stadt Bayreuth und und bekannte regionale Se-Gehalte (mg kg-1) in Oberböden.
Methodik
In Mikrokosmosexperimenten wurde die häufig im Boden vorkommende Dematiaceae-Art Alternaria alternata unter verschiedenen pH-Werten (4,0 und 7,0) im Kulturmedium bei Se-Konzentrationen von 5 mg L-1 [als Se(VI) bzw. Se(IV)] 11-15 Tage inkubiert. Anschließend wurden die flüchtigen Methylselenide des Gasraumes beprobt, sowie das Medium und der Organismus aufgeschlossen.
Als geeignete Methode zur qualitativen Gewinnung der produzierten Methylselenide erwiesen sich basische Peroxidfallen (Abb. 3).
Die urbanen Oberbodenproben aus Bayreuth wurden mit verschiedenen Extraktionsmitteln in der Mikrowelle aufgeschlossen. Im Anschluss wurden die Extrakte mittels Hydridsystem aufgereinigt und die entstandenen Se-Hydride in eine basischen Peroxidlösung (ähnlich der Methode zur Rückhaltung von Methylseleniden) überführt und zu Se(VI) oxidiert. Durch diese Methode werden alle Elemente eliminiert, die aus wässrigen Lösungen mit geeigneten Reduktionsmitteln keine gasförmigen, kovalente Hydride bilden. Allerdings bilden neben Se auch Ge und As Hydride. Da die 74Ge und 76Ge-Isotope 74Se und 76Se und das 75AsH-Molekülion 76Se im Massenspektrometer überlagern, war ein weiterer Aufreinigungsschritt mittels Ionenaustauschchromatographie (Harz AG1-X8) notwendig Die Messung der Isotope 78Se, 80Se und 82Se kann außerdem durch Kr-Isotope gleicher Masse gestört werden, die in der Umgebungsluft enthalten sind. Um bei der Isotopenmessung mögliche Interferenzen durch Kr auszuschließen, erfolgte vor der Messung eine Äquilibrierung der Proben mit Luft, sodass in allen Extrakten, den Standards und Referenzmaterialien eine gleichmäßige Kr-Konzentration enthalten war. Drastische Störungen betreffen vor allem das Hauptisotop 80Se durch Argonisotope (40Ar2), welches als Gas zur Plasmaerzeugung im Multicollector (MC)-ICP-MS verwendet wird.
Um die Se-Isotopenverhältnisse in den Methylseleniden, dem verbleibenden Kulturmedium und den aufgeschlossenen Organismen zu bestimmen, wurden zunächst die Se-Konzentrationen ermittelt, um (1) über

58
eine Se-Bilanzierung die quantitative Erfassung des eingesetzten Selens sicher zu stellen und Verluste während der Aufarbeitung, die möglicherweise mit Isotopenfraktionierung verbunden sind, auszuschließen und (2) um für die Isotopenmessungen am MC-ICP-MS das Probenvolumen so zu dimensionieren, dass eine Gesamt-Se-Masse von 100 ng in das Messgerät eingeführt wird. Die Korrektur der gerätebedingten Massenfraktionierung, sowie Fraktionierungen während der Probenaufbereitung, erfolgte über einen Isotopen-Doppelspike von 74Se und 77Se, der vor der Analyse im Verhältnis Probe/Spike von 1:2 hinzugefügt wurde. Das Se-Isotopenverhältnis wurde in der konventionellen „ “-Notation als 76/82Se relativ zu dem internationalen Standard NIST SRM 3149 angegeben (Gl. 1):
Gl. 1: 82/76Se = 1000*1Stan
76/82Pr
76/82
dard
obe
SeSe
A) B)
Fehler! Es ist nicht möglich, durch die Bearbeitung von Feldfunktionen Objekte zu erstellen.
Abb. 3: Messung des Se-Isotopenverhältnisses in Methylseleniden. A) Versuchsanordnung zu quantitativen Rückhaltung der Methylselenide aus Biomethylierung von unterschiedlichen anorganischen Se-Salzen durch A. alternata. B) Schema der Probenaufbereitung.
Ergebnisse
Durch die Inkubation von A. alternata mit Se(IV) kam es zu einer ausgeprägteren Methylierung von Se als durch die Inkubation mit Se(VI) (Abb. 4). Über den Zeitraum von 11-15 Tagen wurden lediglich 2-10% des Se(VI) methyliert, während Se(IV) zu 20-43% als Methylselenid verflüchtigt wurde. Dies spiegelte sich auch in der Se-Assimilation durch den Pilz A. alternata wider, der am Ende der Inkubation mit Se(VI) nur 12-25% des insgesamt zugesetzten Selens enthält, am Ende der Inkubation mit Se(IV) hingegen bis zu 61%. Dementsprechend verbleiben nach der Inkubation mit Se (VI) mehr als 85% des Selens im Medium, während Se(IV) fast vollständig in A. alternata akkumuliert bzw. methyliert wurde.
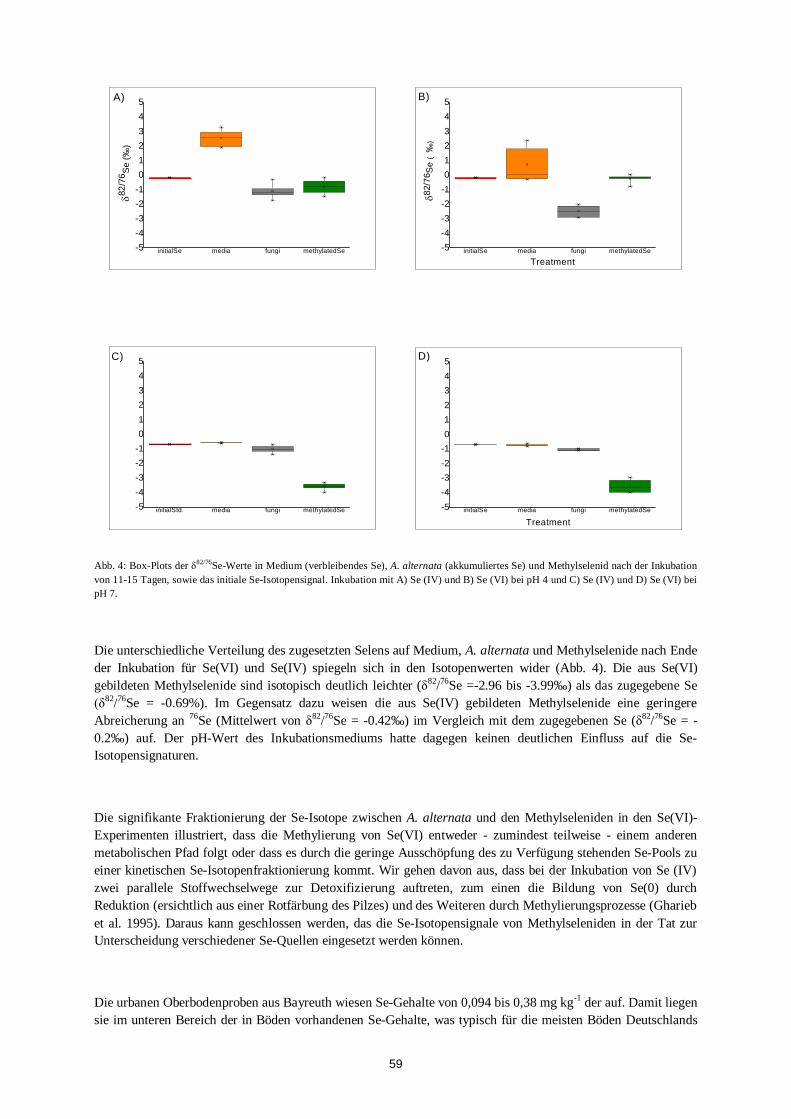
59
initialSe media fungi methylatedSe-5-4-3-2-1012345
82
/76 S
e (‰
)
A)
initialSe media fungi methylatedSe-5-4-3-2-1012345
82
/76 S
e (
‰)
Treatment
B)
initialStd. media fungi methylatedSe-5-4-3-2-1012345C)
initialSe media fungi methylatedSe-5-4-3-2-1012345
Treatment
D)
Abb. 4: Box-Plots der 82/76Se-Werte in Medium (verbleibendes Se), A. alternata (akkumuliertes Se) und Methylselenid nach der Inkubation von 11-15 Tagen, sowie das initiale Se-Isotopensignal. Inkubation mit A) Se (IV) und B) Se (VI) bei pH 4 und C) Se (IV) und D) Se (VI) bei pH 7.
Die unterschiedliche Verteilung des zugesetzten Selens auf Medium, A. alternata und Methylselenide nach Ende der Inkubation für Se(VI) und Se(IV) spiegeln sich in den Isotopenwerten wider (Abb. 4). Die aus Se(VI) gebildeten Methylselenide sind isotopisch deutlich leichter ( 82/76Se =-2.96 bis -3.99‰) als das zugegebene Se
82/76Se = -0.69%). Im Gegensatz dazu weisen die aus Se(IV) gebildeten Methylselenide eine geringere Abreicherung an 76Se (Mittelwert von 82/76Se = -0.42‰) im Vergleich mit dem zugegebenen Se ( 82/76Se = -0.2‰) auf. Der pH-Wert des Inkubationsmediums hatte dagegen keinen deutlichen Einfluss auf die Se-Isotopensignaturen.
Die signifikante Fraktionierung der Se-Isotope zwischen A. alternata und den Methylseleniden in den Se(VI)-Experimenten illustriert, dass die Methylierung von Se(VI) entweder - zumindest teilweise - einem anderen metabolischen Pfad folgt oder dass es durch die geringe Ausschöpfung des zu Verfügung stehenden Se-Pools zu einer kinetischen Se-Isotopenfraktionierung kommt. Wir gehen davon aus, dass bei der Inkubation von Se (IV) zwei parallele Stoffwechselwege zur Detoxifizierung auftreten, zum einen die Bildung von Se(0) durch Reduktion (ersichtlich aus einer Rotfärbung des Pilzes) und des Weiteren durch Methylierungsprozesse (Gharieb et al. 1995). Daraus kann geschlossen werden, das die Se-Isotopensignale von Methylseleniden in der Tat zur Unterscheidung verschiedener Se-Quellen eingesetzt werden können.
Die urbanen Oberbodenproben aus Bayreuth wiesen Se-Gehalte von 0,094 bis 0,38 mg kg-1 der auf. Damit liegen sie im unteren Bereich der in Böden vorhandenen Se-Gehalte, was typisch für die meisten Böden Deutschlands
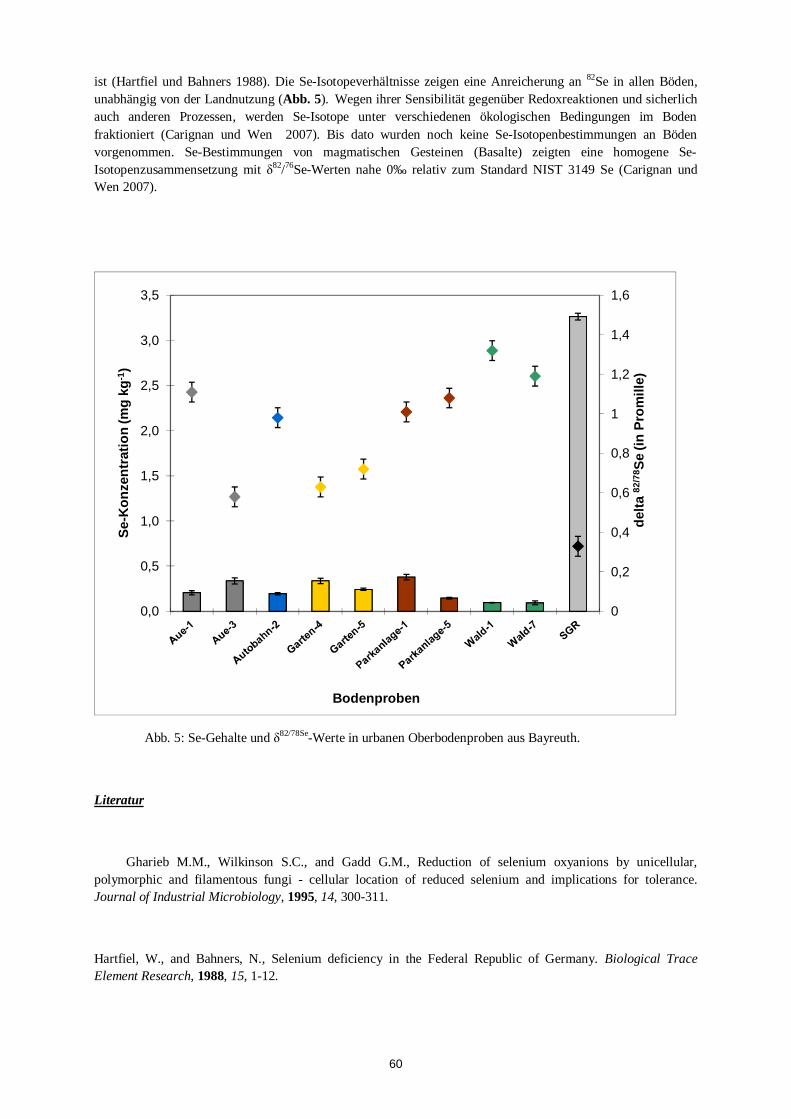
60
ist (Hartfiel und Bahners 1988). Die Se-Isotopeverhältnisse zeigen eine Anreicherung an 82Se in allen Böden, unabhängig von der Landnutzung (Abb. 5). Wegen ihrer Sensibilität gegenüber Redoxreaktionen und sicherlich auch anderen Prozessen, werden Se-Isotope unter verschiedenen ökologischen Bedingungen im Boden fraktioniert (Carignan und Wen 2007). Bis dato wurden noch keine Se-Isotopenbestimmungen an Böden vorgenommen. Se-Bestimmungen von magmatischen Gesteinen (Basalte) zeigten eine homogene Se-Isotopenzusammensetzung mit 82/76Se-Werten nahe 0‰ relativ zum Standard NIST 3149 Se (Carignan und Wen 2007).
Abb. 5: Se-Gehalte und 82/78Se-Werte in urbanen Oberbodenproben aus Bayreuth.
Literatur
Gharieb M.M., Wilkinson S.C., and Gadd G.M., Reduction of selenium oxyanions by unicellular, polymorphic and filamentous fungi - cellular location of reduced selenium and implications for tolerance. Journal of Industrial Microbiology, 1995, 14, 300-311.
Hartfiel, W., and Bahners, N., Selenium deficiency in the Federal Republic of Germany. Biological Trace Element Research, 1988, 15, 1-12.
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
delta
82/7
8 Se
(in P
rom
ille)
Se-K
onze
ntra
tion
(mg
kg-1
)
Bodenproben

61
Carignan, J., and Wen, H. Scaling NIST SRM 3149 for Se isotope analysis and isotopic variations of natural samples. Chemical Geology, 2007, 242, 347-350.
Dauer der Promotion
Beginn der Promotion: August 2007
Angaben zu entstandenen Publikationen, Kongressbeiträgen etc. (seit Beginn der Promotion)
K. Schilling. Fractionation of selenium isotopes during alkylation of inorganic selenium? Vortrag, GRK-Jahresseminar 2007, Bad Münster am Stein, 26.10.2007.
K. Schilling, W. Wilcke. A method to determine the isotopic composition of selenium in volatile organoselenium compounds. Poster, 4th International Conference on Trace Element Speciation in Biomedical, Nutritional and Environmental Sciences, München, 25.-29.05.2008
K. Schilling. Method to determine effects of bio-methylation on selenium isotopes by MC-ICP-MS. Vortrag, GRK-Jahresseminar 2008, Bad Münster am Stein, 04.11.2008
K. Schilling, W. Wilcke. A method to determine the isotopic composition of selenium in volatile organoselenium compounds. Poster, Jahrestagung der Deutschen Bodenkundlichen Gesellschaft, Bonn, 5.-13. 09 2009
K. Schilling, W. Wilcke. A method to determine the isotopic composition of selenium in volatile organoselenium compounds. Poster, 12th Workshop on Progress in Analytical Methodologies for Trace Metal Speciation, Mainz, 5.-13. 09 2009
K. Schilling. Se- isotopic fractionation of biomethylation by fungus Alternaria alternata. Vortrag, GRK-Jahresseminar 2009, Bad Münster am Stein, 19.10.2009
Zusammenarbeit mit bzw. Bezug zu anderen Projekten (innerhalb und außerhalb des GRK)
Eine enge Kooperation besteht mit der Arbeitsgruppe von Prof. Dr. Thomas M. Johnson (University of Illinois –Urbana/Champaign), im Bereich der Probenpräparation (Aufreinigung) und Isotopenmessung von Selen mittels MC-ICP-MS.
Des Weiteren besteht eine Zusammenarbeit mit der Arbeitsgruppe von Prof. Dr. Helmut König (Mikrobiologie und Weinforschung/ Universität Mainz), in dessen Laboren die Mikrokosmos-Versuche durchgeführt wurden.
Darüber hinaus hat mich die Gruppe von Dr. Hans-Peter Jochum (Max-Planck-Institut für Chemie, Mainz) bei der Analyse von Se-Konzentrationen in den Bodenextrakten unterstützt.
Teilnahme am Studienprogramm (seit Beginn der Promotion)
Ringvorlesung Wintersemester 2007/2008
Ringvorlesung Wintersemester 2009/2010

62
Sonderkurs: „Angewandte Statistik mit R für die Biogeochemie“, Dr. Thorsten Pohlert; 20.06.2009
Blockpraktikum „Mikrobiologisches Praktikum“, Prof. Dr. H. König; 08.-12.02.2010
Sonderkurs „Chemometrik“, Prof. Dr. J. W. Einax, S. Prikler 02.-04.03.2010.
Betreuung im Graduiertenkolleg
Betreuer: Prof. Dr. Wolfgang Wilcke
Weitere Betreuer: Prof. Dr. Michael Kersten, Prof. Dr. Helmut König
ggf. Auslandsaufenthalte (seit Beginn der Promotion)
August bis September 2008: University of Illinois- Urbana-Champaign – Se-Isotopenmessungen
Mai bis Juni 2009: University of Illinois- Urbana-Champaign – Se-Isotopenmessungen in den Proben aus den Mikrokosmos-Experimenten
Oktober bis Dezember 2009: University of Illinois- Urbana-Champaign – Se-Isotopenmessungen in Bodenextrakten
März bis Mai 2010: University of Illinois- Urbana-Champaign – abschließende Se-Isotopenmessungen

63
Thema und Betreuer des Dissertationsprojekts Stipendiat: Nils Stöbener Elementspeziation von Neptunium im Ultraspurenbereich Betreuer: Prof. Dr. T. Reich Berichtszeitraum: März 2009 - Februar 2010
Darstellung des Dissertationsprojekts und der Forschungsergebnisse im Berichtszeitraum Motivation
Für die Bewertung der Langzeitsicherheit eines Endlagers für hochradioaktive Abfälle ist ein detailliertes Verständnis des Sorptions- und Migrationsverhaltens von Plutonium und der minoren Actiniden Neptunium, Americium und Curium im vorherrschenden Wirtsgestein essentiell, da langlebige Isotope dieser Elemente die Radiotoxizität von abgebranntem Kernbrennstoff nach 1000 Jahren Abklingzeit maßgeblich bestimmen. Insbesondere für Neptunium, dessen langlebigstes Isotop 237Np (T1/2 = 2,14 106 a) in Kernreaktoren jährlich in kg-Mengen gebildet wird, ist eine genaue Kenntnis der unter verschiedenen Bedingungen vorliegenden Spezies unabdingbar, da die Mobilität des Elements in der Geosphäre stark von seinem Oxidationszustand abhängt. So dominiert unter oxidierenden Bedingungen Np(V), welches eine hohe Löslichkeit im Wässrigen aufweist und damit eine hohe Mobilität besitzt. Das unter reduzierenden Bedingungen vorherrschende Np(IV) ist dagegen weniger löslich und wird stärker an Mineralen und Gesteinen sorbiert. Ausgiebige Studien zur Speziation des Neptuniums unter den im Umfeld eines Endlagers erwarteten Bedingungen sind deshalb für Sicherheitsanalysen unabdingbar. Eine besondere Herausforderung stellen hierbei die niedrigen Konzentrationen von unter 10-10 mol L-1 Neptunium dar, die im Falle der Leckage eines Endlagers im Aquifer erwartet werden. Die Speziesanalytik benötigt hier äußerst empfindliche und selektive Methoden zum Nachweis von Neptunium. Im Institut für Kernchemie wird seit vielen Jahren Resonanzionisations-Massenspektrometrie (RIMS) zum Ultraspurennachweis von langlebigen Isotopen unterschiedlicher Elemente eingesetzt [1]. Für Plutonium können dabei absolute Nachweisgrenzen im Bereich von 106 Atomen (ca. 0,5 fg) in verschiedensten Matrices erreicht werden. RIMS kann darüber hinaus mit chemischen Trennmethoden wie der Kapillarelektrophorese (CE) gekoppelt werden und eignet sich damit auch zur Elementspeziation bei niedrigsten Konzentrationen [2]. Für die Untersuchung des geochemischen Verhaltens von Neptunium unter endlagerrelevanten Bedingungen bietet sich daher die Entwicklung einer auf CE-RIMS basierenden Speziationsmethode an. Methode Zur Vermeidung der bei gängigen massenspektrometrischen Verfahren wie ICP-MS auftretenden isobaren Interferenzen erfolgt die Ionisation der Probenatome bei RIMS durch eine hochselektive, mehrstufige optische Anregung. Durch Laserlicht unterschiedlicher Wellenlänge werden dabei stufenweiße immer höher liegende Energieniveaus der Atome populiert, bis die Gesamtenergie schließlich das Ionisationspotential (IP) des Elements überschreitet. Bedingt durch die Einzigartigkeit optischer Übergänge garantieren solche mehrstufigen Anregungsleitern eine elementselektive Ionisation. Das Laserlicht zur Anregung wird in unserem Aufbau durch drei Titan-Saphir-Laser (Ti:Sa) erzeugt, die gemeinsam von einem Nd:YAG-DPSS-Laser gepumpt werden. Zur Bereitstellung ausreichender Energie für die Ionisation von Neptunium (IP: 50535 cm-1 / 6,27 eV) mit einem

64
dreistufigen Anregungsschema wird das Licht für den ersten Anregungsschritt in einem BBO ( -Bariumborat)-Kristall frequenzverdoppelt. Vorgehensweise Da im Neptunium bisher lediglich für den ersten Schritt geeignete Energieniveaus bekannt sind [3], musste zur Entwicklung von dreistufig-resonanten Anregungsleitern zunächst mit Resonanzionisationsspektroskopie (RIS) nach hochliegenden Zuständen sowie nach autoionisierenden Resonanzen (AI) gesucht werden, die im zweiten bzw. dritten Schritt angeregt werden können. Für die Suche nach geeigneten zweiten Schritten wurden hierbei Neptuniumatome mit dem frequenzverdoppelten Licht eines Lasers zunächst in bereits bekannte Niveaus mit einer Energie um 25000 cm-1 angeregt. Die weitere Anregung der Atome in die zweiten Schritte erfolgte mit einem speziell angepassten Ti:Sa-Laser, der modensprungfrei über einen Wellenlängenbereich von über 200 nm (3000 cm-1) verstimmt werden kann und somit zum Einsatz in der Spektroskopie prädestiniert ist. Da diese zweiten Schritte noch deutlich unterhalb des Ionisationspotentials liegen, wurde während der Messungen Licht eines dritten Ti:Sa-Lasers eingestrahlt, das eine nichtresonante Ionisation der hochangeregten Neptuniumatome ermöglicht. Die auf diesem Weg identifizierten hochliegenden Energieniveaus dienten dann als Ausgangspunkte für die Suche nach autoionisierenden Zuständen, die im dritten Schritt angeregt werden können und in einer Anregungsleiter eine effiziente und selektive Ionisation der Neptuniumatome garantieren. Die Anregung der ersten beiden Schritte erfolgte hierbei mit auf fester Wellenlänge betriebenen Ti:Sa-Lasern, während der Laser zur Anregung des dritten Schritts durchgestimmt wurde. Als Atomstrahlquelle diente bei allen Messungen ein Graphitrohrofen (Mainz Atomic Beam Unit MABU der Arbeitsgruppe Prof. K. Wendt, Institut für Physik), in den Probemengen von ~1015 Atomen 237Np auf Zirkonfolie als Reduktionsmittel eingebracht wurden. Der beim Hochheizen des Ofens erzeugte Atomstrahl wurde kollinear mit den Laserstrahlen überlagert; der Nachweis der gebildeten Np+-Laserionen erfolgte in einem Quadrupol-Massenfilter mit Channeltron-Detektor. Mit den auf diesem Weg identifizierten hochliegenden und autoionisierenden Energieniveaus wurden anschließend mehrere dreistufig-resonante Anregungsleitern entwickelt, die anschließend auf ihre Eignung zum Einsatz in der Ultraspurenanalyse getestet wurden. Hierzu wurde unter anderem untersucht, ob die optischen Übergänge mit der zur Verfügung stehenden Laserleistung gesättigt werden können und wie effizient die Ionisation insgesamt verläuft. Ergebnisse Als Ausgangspunkte für die Suche nach hochliegenden Zuständen, die für den zweiten Schritt einer dreistufigen Anregungsleiter geeignet sind, wurden sechs Niveaus ungerader Parität zwischen 24000 cm-1 und 27000 cm-1 über dem Grundzustand ausgewählt. Von jedem dieser Zustände können zahlreiche Energieniveaus im Abstimmbereich des zur Spektroskopie eingesetzten Lasers populiert werden (s. Abb.1).

65
Abb.1: Spektrum mit hochliegenden Zuständen in Neptunium, die ausgehend von einem Niveau bei 25277,8 cm-1 (J = 9/2) bevölkert werden können.
Ausgehend von einigen der identifizierten zweiten Schritte wurde wie beschrieben nach autoionisierenden Resonanzen gesucht, die eine im Vergleich zur nichtresonanten Ionisation der hochangeregten Atome effizientere und selektivere Ionisation ermöglichen. Auch hier konnte in den jeweils betrachteten Energiebereichen eine Vielzahl geeigneter Zustände gefunden werden (s. Abb. 2).
Abb. 2: Ausschnitt aus dem Spektrum der von einem hochliegenden Energieniveau bei 37558,1 cm-1 zugänglichen autoionisierenden Resonanzen.
Aus den durch Resonanzionisationsspektroskopie erhaltenen Daten zu hochliegenden Zuständen und autoionisierenden Resonanzen können nahezu unbegrenzt viele Schemata zur dreistufig-resonanten Anregung und Ionisation von Neptunium entwickelt werden. Im Rahmen unserer Messungen wurden zunächst fünf verschiedene Anregungsleitern näher

66
betrachtet. In allen Fällen zeigte sich, dass zumindest in den ersten beiden Anregungsschritten eine Sättigung der Übergänge mit moderaten Laserleistungen möglich ist (s. Abb. 3).
Abb. 3: Verschiedene Anregungsleitern für 237Np mit Sättigungsleistungen für einzelne Übergänge. In den Fällen, in denen keine Sättigungsleistung angegeben ist, konnte mit der zur Verfügung stehenden Laserleistung (ca. 1 W) keine Sättigung erreicht werden.
Bei der gewählten kollinearen Überlagerung von Atomstrahl und Laserstrahlen konnten für mehrere Anregungsleitern Effizienzen im einstelligen Promillebereich gemessen werden, bei senkrechter Überlagerung jedoch nur im Bereich von 10-7. Dieser deutliche Unterschied kann primär durch die längere Verweilzeit der Neptuniumatome in der Wechselwirkungsregion bei kollinearer Überlagerung und die daraus resultierende höhere Ionisationswahrscheinlichkeit erklärt werden. Da bei kollinearer Geometrie jedoch keine Unterdrückung durch thermisch aus der Quelle emittierte Ionen gegeben ist, wird im Institut für Kernchemie derzeit ein RIMS-Aufbau mit senkrechter Einkopplung der Laserstrahlen betrieben. Die Auswahl einer geeigneten Anregungsleiter muss hierbei die fehlende Dopplerverbreiterung des Laserlichts und die hieraus eventuell folgende Nichtbevölkerung von Hyperfeinzuständen des 237Np berücksichtigen. Untersuchungen hierzu sind derzeit in Arbeit. Literatur [1] K. Wendt, N. Trautmann. Recent developments in isotope ratio measurements by
resonance ionization mass spectrometry. Int. J. Mass Spectrom., 2005, 242, 161-168. [2] S. Bürger, N. L. Banik, R. A. Buda, J. V. Kratz, B. Kuczewski, N. Trautmann.
Speciation of the oxidation states of plutonium in aqueous solutions by UV/Vis spectroscopy, CE-ICP-MS and CE-RIMS. Radiochim. Acta, 2007, 95, 433-438.
[3] J. Blaise, J.-F. Wyart. selected constants, energy levels and atomic spectra. Tables
Internationales de Constantes, Université P. et M. Curie, Paris 1992. Dauer der Promotion Beginn April 2009, voraussichtlicher Abschluss Frühjahr 2012. Angaben zu entstandenen Publikationen, Kongressbeiträgen etc. (seit Beginn der Promotion)

67
N. Stöbener, T. Gottwald, S. Raeder, G. Passler, T. Reich, N. Trautmann, K. Wendt. Ultraspurenanalyse von Neptunium mit Resonanzionisations-Massenspektrometrie. Poster, GDCh Wissenschaftsforum 2009, Frankfurt, 30.08.-02.09.2009 N. Stöbener, T. Gottwald, S. Raeder, G. Passler, T. Reich, N. Trautmann, K. Wendt. Trace Analysis of Neptunium with Resonance Ionization Mass Spectrometry. Poster, TraceSpec 2009, Mainz, 15.-18.09.2009 Zusammenarbeit mit bzw. Bezug zu anderen Projekten (innerhalb und außerhalb des GRK) Eine enge Kooperation besteht mit der Arbeitsgruppe Prof. K. Wendt, Institut für Physik, vor allem im Bereich der Resonanzionisationsspektroskopie an Neptunium, aber auch zur apparativen Weiterentwicklung der RIMS allgemein. Teilnahme am Studienprogramm (seit Beginn der Promotion) Ringvorlesung Wintersemester 2009/2010 Sonderkurs „Chemometrik“, Prof. J.W. Einax, 02.-04.03.2010