sindrome linfoproliferativos-tratamiento
TRANSCRIPT

SINDROME SINDROME LINFOPROLIFERATIVOSLINFOPROLIFERATIVOS
CRONICOSCRONICOSDRA. NANCY LOAYZA U
HEMATOLOGIA C.

SINDROME SINDROME LINFOPROLIFERATIVO LINFOPROLIFERATIVO
CRONICOCRONICOEl término síndrome linfoproliferativos
crónicos (SLC) incluye una variedad de entidades clínico-biológicas que resultan de la proliferación clonal de un linfocito B o T maduro e inmunocompetente

SINDROME LINFOPROLIFERATIVO SINDROME LINFOPROLIFERATIVO CRONICOCRONICO
El diagnóstico preciso de los diferentes procesos linfoides es importante no sólo desde un punto de vista académico, ya que permite elucidar los posibles mecanismos etiopatogénicos responsables de la neoplasia o proliferación clonal linfoide, sino también clínico, ya que el pronóstico y conducta terapéutica varía ampliamente dependiendo del proceso.

SINDROME LINFOPROLIFERATIVO SINDROME LINFOPROLIFERATIVO CRONICOCRONICO
El diagnóstico preciso de los SLC debe basarse en una constelación de datos clínicos y de laboratorio. Deben incluir: Citología, Histología, Marcadores inmunológicos Estudios citogenéticos y moleculares

SINDROMES SINDROMES LINFOPROLIFERATIVOS BLINFOPROLIFERATIVOS B
Leucemia Linfática crónica (LLC)Leucemia prolinfocítica B (LP-B)Tricoleucemia y variante de tricoleucemiaLeucemia de células plasmáticas

SINDROMES SINDROMES LINFOPROLIFERATIVOS TLINFOPROLIFERATIVOS T
Leucemia prolinfocítica T (LP-T)Leucemia de linfocitos granularesLeucemia de linfocitos “Sesary-like”
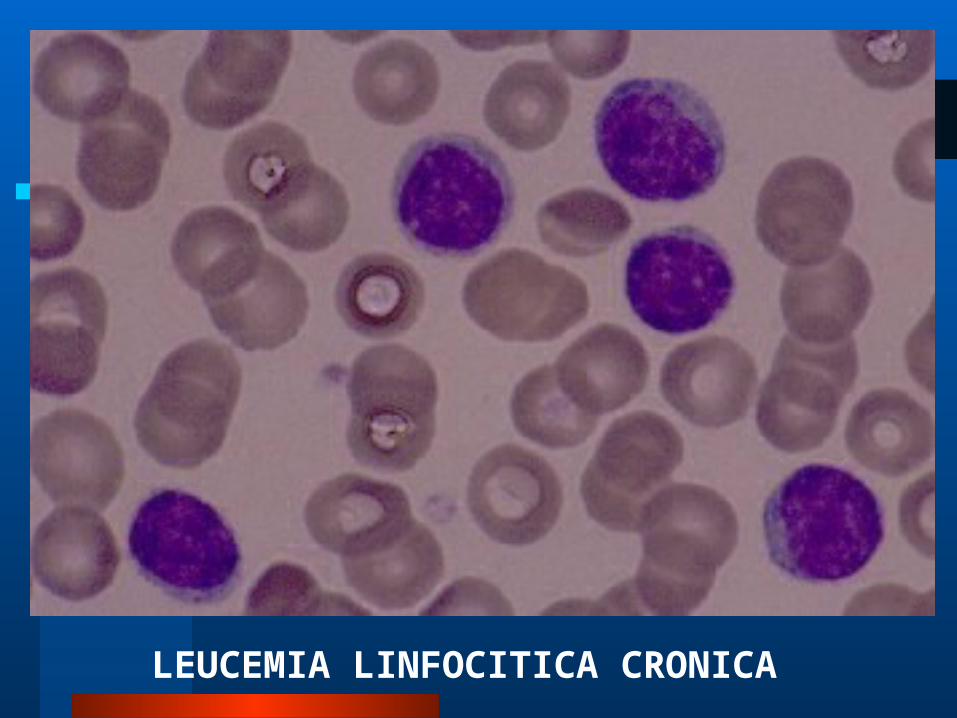
LEUCEMIA LINFOCITICA CRONICA

LEUCEMIA LINFOCITICA LEUCEMIA LINFOCITICA CRONICACRONICA

GENERALIDADESGENERALIDADES Proliferación maligna de linfocitos
maduros.Casi el 90% de casos corresponden a
linfocitos B Es el tipo más común de leucemia (?) y
constituye el 30% de todos los casos.El 90% de casos sucede en mayores de
50 años.

ETIOLOGIAETIOLOGIALos familiares en primer grado tiene un
riesgo casi tres veces mayor que el normal de desarrollar LLC u otras neoplasias malignas.
Ni la radiación ni los retrovirus se conocen como causa de LLC.

CUADRO CLINICOCUADRO CLINICO
El 25% de pacientes son asintomáticos y se diagnostican por pruebas de rutina.
Los síntomas son inespecíficos: malestar, anorexia, hemorragias, etc.
En el 80% de casos aparecen linfadenopatías.
En el 50% a 70% hay esplenomegalia y en < 50% hay hepatomegalia.

DIAGNOSTICODIAGNOSTICO
Es común la presencia de anemia y trombocitopenia con linfocitosis absoluta.
Son características la presencia de sombras de Grumpech en lámina periférica.
El diagnóstico definitivo se hace por verificación de la clonalidad de los linfocitos B mediante CMF: CD19, CD20, CD21 y CD5.
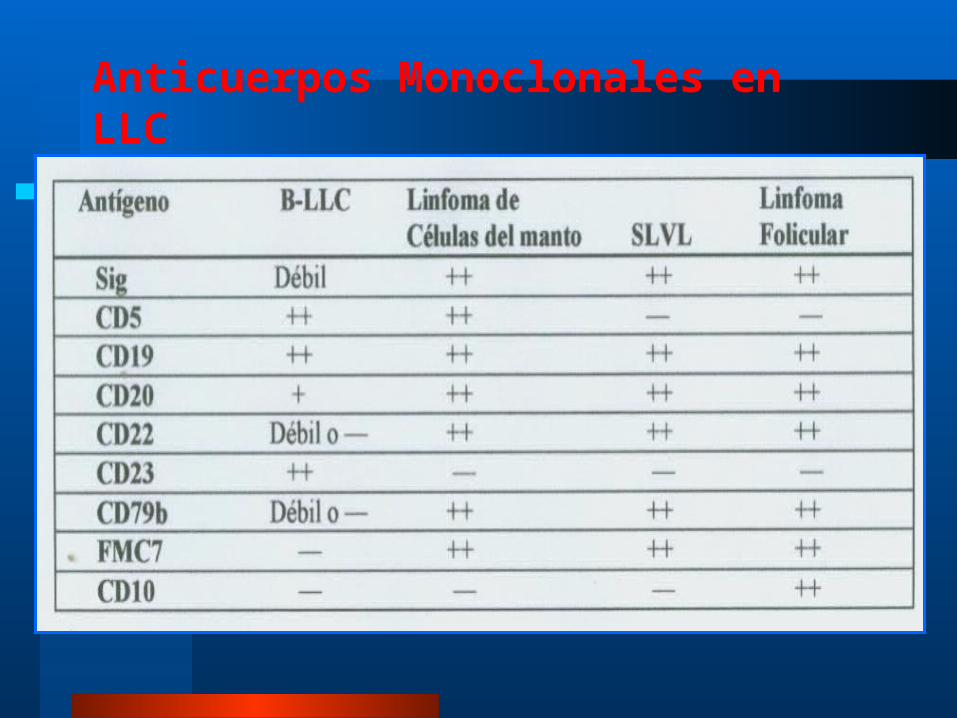
Anticuerpos Monoclonales en LLC
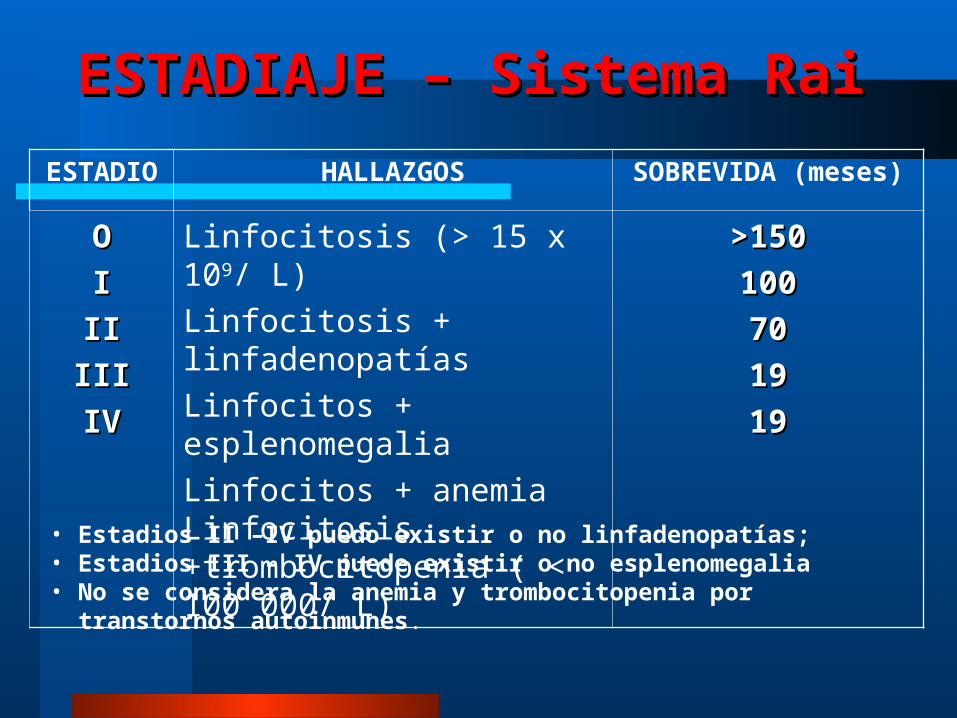
ESTADIAJE – Sistema RaiESTADIAJE – Sistema RaiESTADI
OHALLAZGOS SOBREVIDA
(meses)OOIIIIIIIIIIIIIVIV
Linfocitosis (> 15 x 109/ L)Linfocitosis + linfadenopatíasLinfocitos + esplenomegaliaLinfocitos + anemia Linfocitosis +trombocitopenia ( < 100 000/ L)
>150>150100100707019191919
• Estadios II –IV puedo existir o no linfadenopatías; • Estadios III – IV puede existir o no esplenomegalia• No se considera la anemia y trombocitopenia por transtornos autoinmunes.

ESTADIAJE – BINET e ESTADIAJE – BINET e International Workshop en CLLInternational Workshop en CLL
ESTADIO
HALLAZGOS SOBREVIDA (meses)
AA
BB
CC
Hb ≥ 10 gr/dl Plts ≥ 100 x109/L <3 áreas afectadas*
Hb ≥ 10 gr/dl Plts ≥ 100 x109/L ≥ 3 áreas afectadas
Hb < 10 gr/dl Plts < 100 x109/L
>120>120
6161
3232*Las áreas ganglionares linfoides incluye la cervical, la axilar, la inguinal (uni o bilateral), el bazo y el hígado.

TRATAMIENTOTRATAMIENTO

BAJO RIESGO ( ESTADIO 0 )BAJO RIESGO ( ESTADIO 0 )
No deben tratarseEn principio el conteo de leucocitos
per se no modifica la conducta hasta que no exceda 200 x 109/L

BAJO RIESGO ( ESTADIO 0 )BAJO RIESGO ( ESTADIO 0 )
Actualmente se discute sobre una posible smoldering LLC que puede definirse según ensayo como pacientes con los siguientes elementos: Conteo al debut < de 30 x 109/L de leucocitos Tiempo de doblaje del conteo linfoide mayor
de 12 meses Hb > de 13 g/L Patrón de infiltración medular no difuso
(nodular o intersticial)

RIESGO INTERMEDIO ( l,ll, A,B) RIESGO INTERMEDIO ( l,ll, A,B) Observación cada mes para evaluar progresión
de la enfermedad Duplicación linfocitaria en período menor de un año. Incremento en más 50 % de adenopatías, esplenomegalia y/o
hepatomegalia Disminución de cifras de Hb y plaquetas sin cambio de estadio Presencia de síntomas constitucionales atribuibles a la
enfermedad: fiebre, sudoración, astenia Episodios de sepsis bacteriana a repetición sin otra explicación.
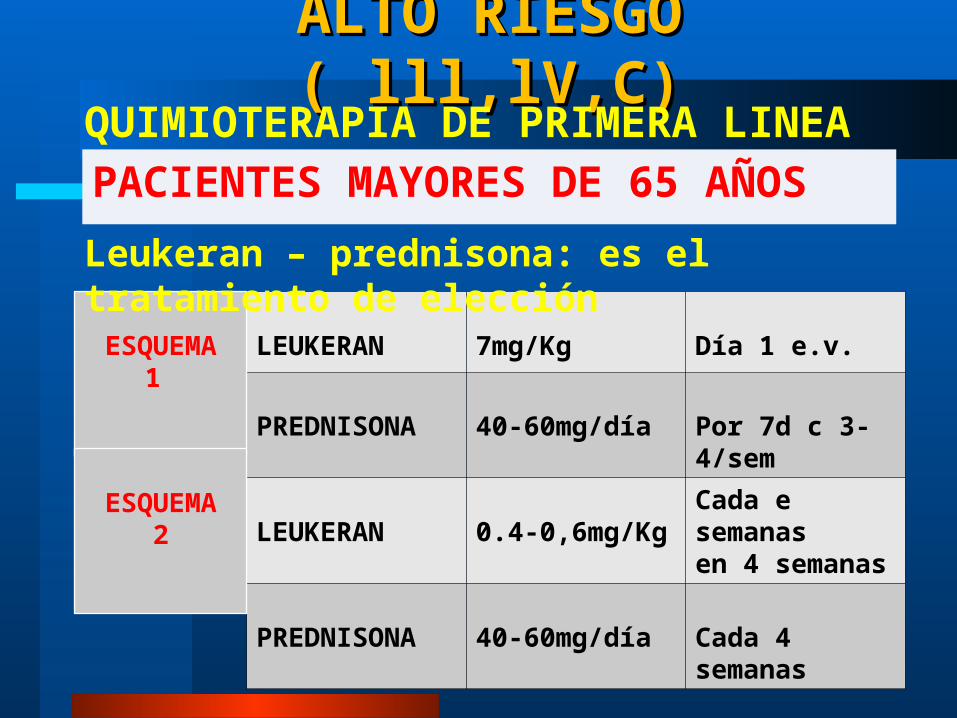
ALTO RIESGO ( lll,lV,C)ALTO RIESGO ( lll,lV,C)
PACIENTES MAYORES DE 65 AÑOSQUIMIOTERAPIA DE PRIMERA LINEA
LEUKERAN 7mg/Kg Día 1 e.v.
PREDNISONA 40-60mg/día Por 7d c 3-4/sem
LEUKERAN 0.4-0,6mg/KgCada e semanasen 4 semanas
PREDNISONA 40-60mg/día Cada 4 semanas
ESQUEMA1
ESQUEMA2
Leukeran – prednisona: es el tratamiento de elección

ALTO RIESGO ( lll,lV,C)ALTO RIESGO ( lll,lV,C)
PACIENTES MAYORES DE 65 AÑOSQUIMIOTERAPIA DE PRIMERA LINEA
ESQUEMA1
ESQUEMA2
Ciclofosfamida-prednisona: es tan efectivo como el anterior, pero puede dejarse como segunda línea en los casos refractarios al leukeran.
CICLOFOSFAMIDA 500-700mg/m2 Dividida en 4 días
PREDNISONA 40-60mg/día Por 7d c 3-4/sem
CICLOFOSFAMIDA 100mg/día Vía oral

ALTO RIESGO ( lll,lV,C)ALTO RIESGO ( lll,lV,C)
PACIENTES MENORES DE 65 AÑOS
Con los siguientes requisitos ECOG menor de 3 No enfermedades asociadas no controladas Sin inmunodeficiencia asociada Sin evento autoinmune activo (AHAI , trombocitopenia)
QUIMIOTERAPIA DE PRIMERA LINEA
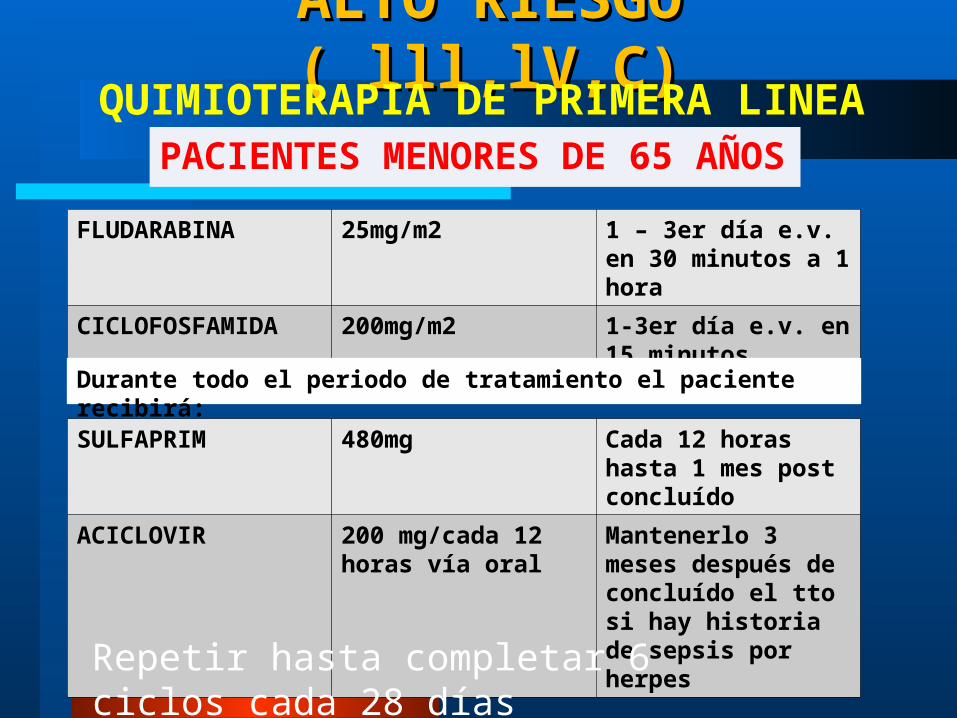
ALTO RIESGO ( lll,lV,C)ALTO RIESGO ( lll,lV,C)
PACIENTES MENORES DE 65 AÑOSQUIMIOTERAPIA DE PRIMERA LINEA
FLUDARABINA 25mg/m2 1 – 3er día e.v. en 30 minutos a 1 hora
CICLOFOSFAMIDA 200mg/m2 1-3er día e.v. en 15 minutos
SULFAPRIM 480mg Cada 12 horas hasta 1 mes post concluído
ACICLOVIR 200 mg/cada 12 horas vía oral
Mantenerlo 3 meses después de concluído el tto si hay historia de sepsis por herpes
Durante todo el periodo de tratamiento el paciente recibirá:
Repetir hasta completar 6 ciclos cada 28 días
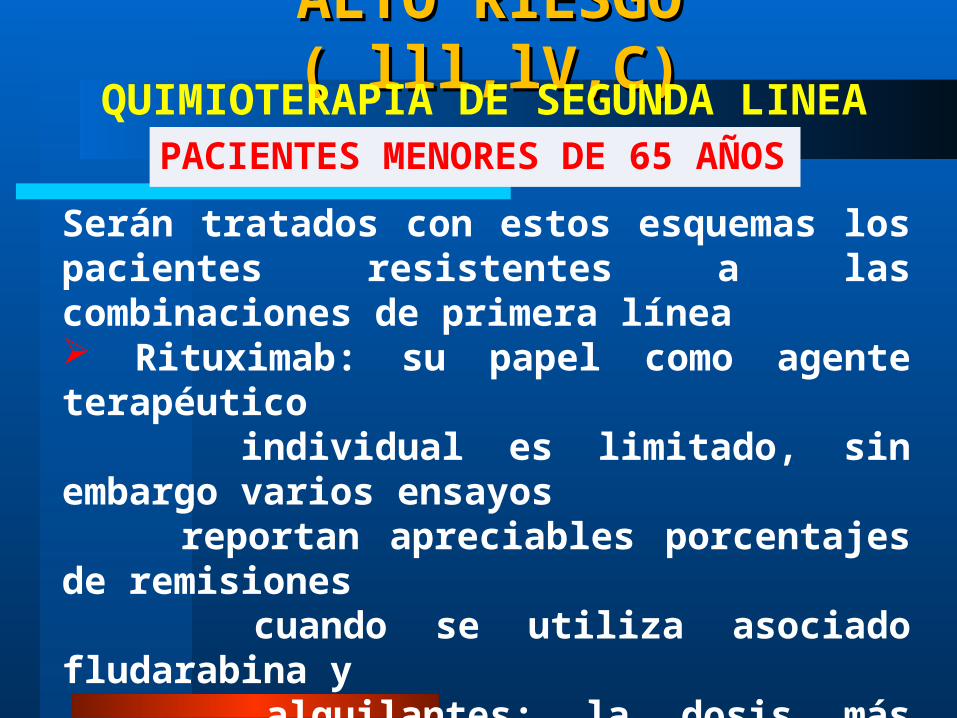
ALTO RIESGO ( lll,lV,C)ALTO RIESGO ( lll,lV,C)
PACIENTES MENORES DE 65 AÑOSQUIMIOTERAPIA DE SEGUNDA LINEA
Serán tratados con estos esquemas los pacientes resistentes a las combinaciones de primera línea Rituximab: su papel como agente terapéutico individual es limitado, sin embargo varios ensayos reportan apreciables porcentajes de remisiones cuando se utiliza asociado fludarabina y alquilantes; la dosis más empleada es de 375 mg / m2 e.v. semanalmente en 4 dosis Altas dosis de Leukeran Mitoxantrone + Leukerán + Dexametasona
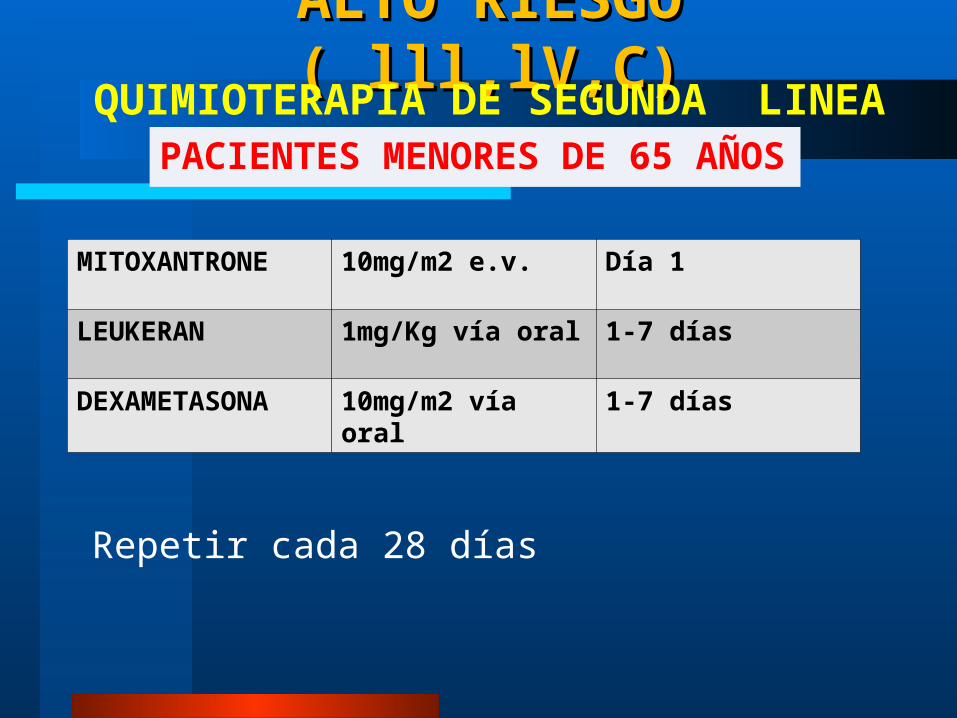
ALTO RIESGO ( lll,lV,C)ALTO RIESGO ( lll,lV,C)
PACIENTES MENORES DE 65 AÑOSQUIMIOTERAPIA DE SEGUNDA LINEA
Repetir cada 28 días
MITOXANTRONE 10mg/m2 e.v. Día 1
LEUKERAN 1mg/Kg vía oral 1-7 días
DEXAMETASONA 10mg/m2 vía oral 1-7 días
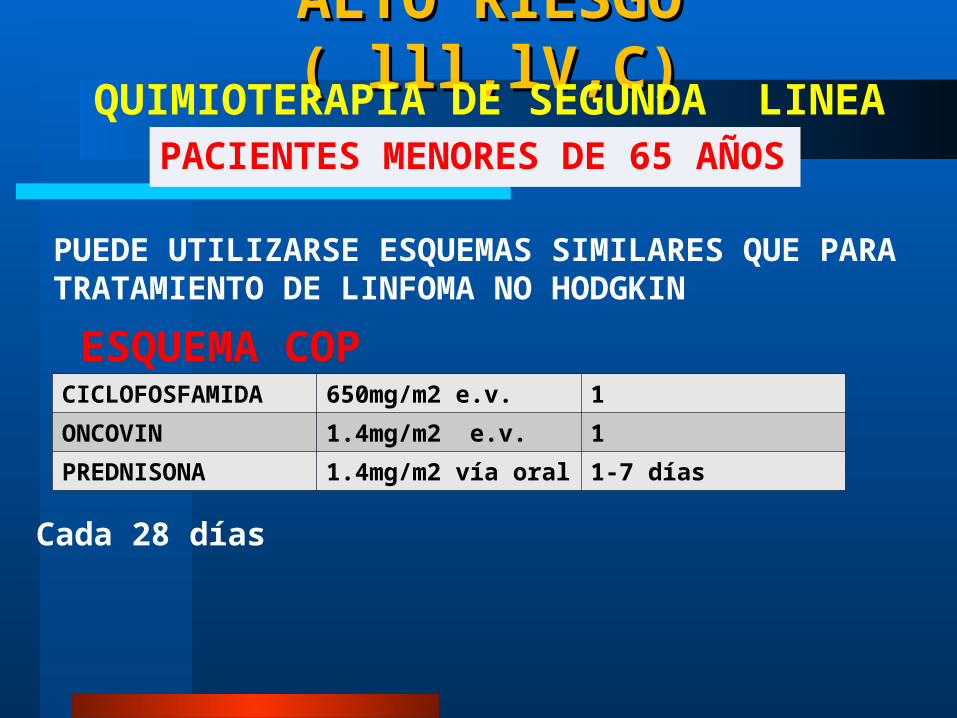
ALTO RIESGO ( lll,lV,C)ALTO RIESGO ( lll,lV,C)
PACIENTES MENORES DE 65 AÑOSQUIMIOTERAPIA DE SEGUNDA LINEA
PUEDE UTILIZARSE ESQUEMAS SIMILARES QUE PARA TRATAMIENTO DE LINFOMA NO HODGKIN
CICLOFOSFAMIDA 650mg/m2 e.v. 1ONCOVIN 1.4mg/m2 e.v. 1PREDNISONA 1.4mg/m2 vía oral 1-7 días
ESQUEMA COP
Cada 28 días

ALTO RIESGO ( lll,lV,C)ALTO RIESGO ( lll,lV,C)
PACIENTES MENORES DE 65 AÑOSQUIMIOTERAPIA DE SEGUNDA LINEA
PUEDE UTILIZARSE ESQUEMAS SIMILARES QUE PARA TRATAMIENTO DE LINFOMA NO HODGKIN
CICLOFOSFAMIDA 750mg/m2 e.v. 1ADRIAMICINA 50 mg/m2 e.v. 1ONCOVIN 1.4mg/m2 e.v. 1PREDNISONA 100mg vía oral 1-5 díasBLEOMICINA 10 U /m2 i.m. 1 y 5 día
ESQUEMA CHOP-BLEOMICINA
Cada 28 días