sÉquenÇage de type rad-seq ... - univ-lille.fr · sÉquenÇage de type rad-seq, prÉsentation et...
TRANSCRIPT

SÉQUENÇAGE DE TYPE RAD-SEQ, PRÉSENTATION ET TRAITEMENT ANALYTIQUE
Yvan Le Bras1, Anthony Bretaudeau1,2, Cyril Monjeaud1
1Plateforme GenOuest & CNRS UMR 6074 IRISA-INRIA, Rennes 2INRA, UMR IGEPP & Plateforme BIPAA , Rennes

Agenda
• Context • Biogenouest network and western France
• Life sciences and environment
• From e-biogenouest project to the CeSGO e-Science center • “Bridging data, metadata and computation”
• RAD-seq • Generalities
• STACKS

Biogenouest
Biogenouest is a network bringing together technological core facilities dedicated to Life and Environmental Sciences in the West of France

Biogenouest
Created in 2002, Biogenouest coordinates 31 technological core facilities based in the regions of Brittany and Pays de la Loire, with the aim to organize and pool interregional resources.
Biogenouest also federates 70 research units involved in thematic research covering 4 areas of activity : Marine resources, Agri-food, Health and Bioinformatics.

GenOuest : Bioinformatics core facility
• Member of the Biogenouest network
• Member of the IFB : French Bioinformatics Institute
• National recognition : IBiSA platform
• Regional strategic facility for INRA (National Institute of Agronomical Research)
• ISO9001:2008 certified
• Established since 2002
• 10 to 12 people
• Computing infrastructure, storage, software development, expertise, R&D projects


Context
Kahn. On the future of genomic data. Science (2011) vol. 331 (6018) pp. 728-9
Now : Genomics : Next Generation Sequencing
Now : Proteomics
Next : Bio-imaging
Digital data
Huge amount
Heterogenous
Critical situation for some laboratories

LE PROJET E-BIOGENOUEST Un environnement virtuel de recherche en sciences de la vie

E-BIOGENOUEST Programme fédérateur Biogenouest co-financé par les Régions Bretagne et Pays de la Loire • 24 mois • Lancé depuis Mai 2012 • Porteur : Olivier Collin (IRISA) – Animateur : Yvan Le Bras (IRISA)
• Tester une approche e-Science en Sciences de la vie
• Proposer une structuration e-Science dans le Grand Ouest

Briser les silos
Défis : Chaque domaine est particulier mais digital!
Solution : Se centrer sur la donnée!
Big Data relatif, collaboration, interdisciplinarité, mutualisation, solutions open source

Optimiser / Standardiser
Code, algorithmes, calcul, stockage, réseau, bonnes pratiques
Prévoir interopérabilité, langage commun, vocabulaire contrôlé, ontologies
Kahn. On the future of genomic data. Science (2011) vol. 331 (6018) pp. 728-9

People paradox
Using Clouds for Metagenomics: A Case Study Wilkening et al. IEEE cluster 2009
Séquençage
Bioinformatique
Défis : Manque RH, Evolution des usages
Solutions : mutualisation, science citoyenne, Environnement virtuel de recherche
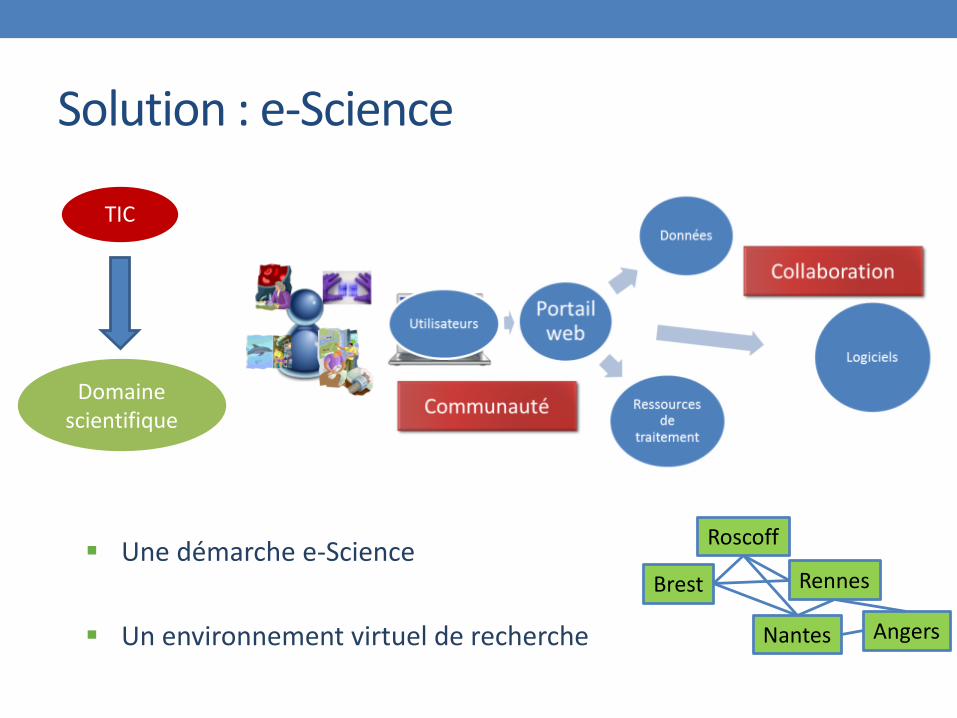
Solution : e-Science
TIC
Domaine scientifique
Une démarche e-Science
Un environnement virtuel de recherche
Brest
Roscoff
Rennes
Nantes Angers

LE VRE Un environnement virtuel de recherche en sciences de la vie

HUBzero : Scientifique collaborative platform
eBGO HUB HUBzero to share knowledge and
manage groups and projects
Informations 264 users 129 projects 56 groups 803 resources … Purdue University M. McLennan, R. Kennell. Comput Sci Eng, 12:48-53, 2010.

ISAtools : Experimental data management
EMME ISAtools suite to store data &
metadata
Fonctionalities -based on biomed ontologies -bridge between existing biomed standards -format publication submission -Pydio to upload data -biological investigation repository (data + metadata) Oxford eResearch Centre P. Rocca-Serra et al. Bioinformatics, 26;254(6), 2010

Galaxy : Data analysis web platform
GALAXY by GenOuest To analyse & share data as
processes and tools
Informations 47140 jobs 125 users More than 800 outils Share - data - histories - workflows - tools Penn state university J. Goecks, A. Nekrutenko, J. Taylor, et al. Genome Biol, 25;11(8):R86, 2010

• Pour les scientifiques en sciences de la vie et environnement • Optimiser son temps (programmation vs compréhension)
• Préserver (données et processus analytiques)
• Accéder, partager et visualiser de n’importe où
• Une aide à la gestion de projet …
• Pour les développeurs • Améliorer l’utilisation des algo et outils : Bioinfo Recherche Service
• Accélérer la mise en production
• Pour la gestion des infrastructures • Optimiser l’utilisation (stockage, calcul et réseaux…)
• Infrastructure pour la donnée infastructure de donnée
I have a dream…

• NGS technology • Bioinformatics research: Optimize NGS raw data treatment
• The Colib’read project
• Life sciences research: RAD-sequencing
• Virtualization • Academic cloud
• IFB / GenOuest Openstack / OpenNebula
• Application & dependencies packaging: Docker
• Innovative methods in science • Citizen science approaches
e-Science focus

LE RADSEQ Voir la présentation de Karim Gharbi du 30/01/2014
edinburgh genomics / University of Edinburgh

Buts

• Allozymes, RAPD, AFLP, Microsatellites, Puces SNPs… NGS
• Baisse du coût de séquençage … pas de l’analyse ;) • Le séquençage de beaucoup d’individus + bcp de marqueurs reste
onéreux
• Besoins de sous-échantillonner le génome
L’arrivée du NGS
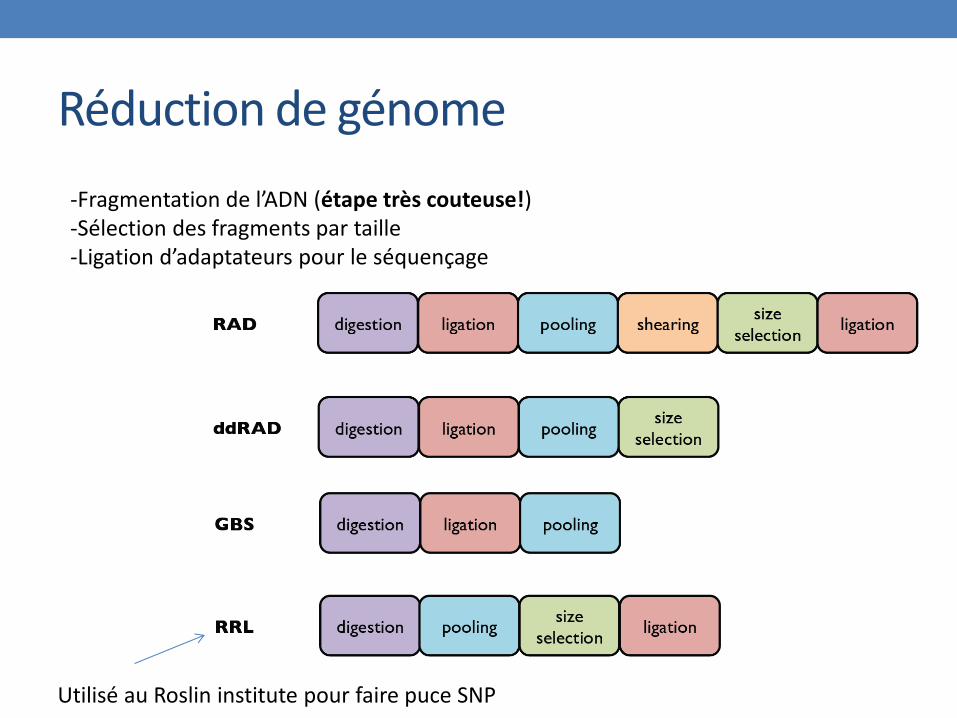
Réduction de génome
-Fragmentation de l’ADN (étape très couteuse!) -Sélection des fragments par taille -Ligation d’adaptateurs pour le séquençage
Utilisé au Roslin institute pour faire puce SNP

Réduction de génome
-Publi initiale : Eric Johnsson, 2008 -Article d’Hohenlohe dans les 5 premiers

Application

Single-end RAD

Single-end RAD

Single-end RAD
Coupure 5’ -> 3’
Brin complémentaire
Hohenlohe et al., PLoS Genetics 2010

Paired-end RAD

Paired-end RAD

Paired-end RAD

Single vs Paired-end RAD

ddRAD

ddRAD

Paired-end ddRAD

RAD vs ddRAD

• RAD simple : séquençage entre site de restriction et cassure par sonication
• ddRAD : séquençage entre 2 sites de restriction donc plus de flexibilité sur les fragments générés, répartitions des lectures
• En ddRAD, le tout adaptateur + barcode + site de restriction + ADN + adaptateur ~500pb
RAD vs ddRAD

Ne rester pas en RAD, ni à sec….

• Pb analyse image quand même nucléotide dans toute les séquences à la même position. Surtout sur HiSeq, moins sur MiSeq
• Mieux vaut utiliser une combinaison de barcodes différents …. Reste pb du site de restriction! Il vaut mieux alors également mélanger les expériences!
Biais

• 250 ng ADN nécessaire, 1 µg demandé à Edinburgh genomics
• Il faut une qualité au top! Pas d’ADN dégradé. Cela peut induire de ne pas utiliser 30 à 40% des données
• PCR 12 à 14 cycles
• Profondeur différente en fonction des tailles de fragments!
• Peut poser pb surtout si mutation au niveau d’un site de restriction chez certains individus
• Licence / Brevet QiaGen?
• Voir Davey et al, Molecular Ecology (2012)
Informations diverses

Demystifying the RAD fad
Johnatan B. Puritz Marine Genomics Laboratory, Texas A&M University-Corpus Christi
Mikhail V. Matz Jesse N. Weber Daniel I. Bolnick Department of Integrative Biology, University of Texas at Austin
Robert J. Toonen Hawai’i Institute of Marine Biology, University of Hawai’i
Christopher E. Bird Department of Life Sciences, Texas A&M University-Corpus Christi

Recent novel approaches for pop. genomics data analysis (Andrews & Luikart, 2014) • RAD sequencing = powerful & useful approach in mol. Ecology
• Several different published methods
• None = best option in all situation

Four different RAD protocols
• Original RAD (mbRAD Miller et al. 2007 and Baird et al. 2008) • Genomic DNA digestion by 1 restriction enzyme (low frequency cutter)
• Ligation of barcode containing adapters onto digested 5’ ends
• Ligated genomic DNA sonication
• Ligation of a 3’ adapter to the sonicated end
• Pool of the samples
• Size-selection of the library
• RAD fragments PCR enrichment

Four different RAD protocols
• Double digest RAD protocol (Peterson et al. 2012) • Genomic DNA digestion by 2 restriction enzymes (low + high frequency
cutter)
• Ligation of barcode P1 adapters (matching the first restriction site) and P2 adapters (matching second restriction site)
• Pool of the samples
• Size-selection of the library
• RAD fragments PCR enrichment + second barcode introduction to increase multiplexing potential
• Extremely similar to GBS (Poland et al. 2013)
• Pros & cons associated with ddRAD also relevant to RESTseq (Stolle & Moritz 2013)

Four different RAD protocols
• ezRAD protocol (Toonen et al. 2013) • Genomic DNA digestion by 2 restriction enzymes (high frequency cutter
on the same cut site)
• Commercially available Illumina TruSeq library preparation kit
• DNA end reparation
• Ligation of single or dual indexing adapters onto genomic fragments
• Pool of samples
• Size selection of the library
• RAD fragments PCR enrichment, or not, depending on the Illumina kit

Four different RAD protocols
• 2bRAD protocol (Wang et al. 2012) • Genomic DNA digestion by 1 restriction enzyme (36-bp fragments
excision recognition site + adjacent 5’ & 3’ base pairs)
• Ligation of dual barcode adapters
• Agarose gel target band excision after PCR enrichment
• No intermediate purification stages
• No size-selection

Potential biases when conducting RADseq
• PCR artefacts
• Restriction fragment size bias
• Heterozygous restriction sites • root cause of allele dropout (ADO) – 1 allele not detected from 1
heterozygous locus
• Fix: Filter any loci that are not represented in all genotyped individuals
• Strand bias • Different genotypes from forward & reverse reads
• Fix: Filter any loci in this case… only possible in 2bRAD

mbRAD advantages
• Random shearing of the 3’ end helps to identify putative PCR duplicates • If identical starting position of the paired-end read: duplicate
• Random shearing improves the distribution of coverage
• Random shearing + larger insert size ranges: de novo assembled RAD loci are of greater length • Critical for identifying function & Gene ontology
• Coverage and quality are fundamental!!! • Distinguishing true SNP from sequencing error: if coverage is low, your
statistical test will no yield significant results!

mbRAD disadvantages
• The most technically challenging and complex protocol!
• Requires non standard lab equipment: sonicator
• Restriction fragment length bias (due to the shearing) • Sequencing at different depth

ddRAD advantages
• Greatest degree of customization • Depending on the enzymes chosen & range of fragment sizes selected
• Allow to have hudreds of SNPs per individual at very low cost or thousands for QTL mapping experiments at moderate cost
• Examine histograms of digested samples early • Identify / exclude excessively frequent fragments (i.e. transposons)

ddRAD disadvantages
• Using fragment size selection to tune the quantity of loci can lead to variable representation of some loci • This can be minimized using precise selection tool (i.e. Pippin Prep)
• ddRAD particularly susceptible to ADO (Arnold et al. 2013) • To be considered when performing sensitive population genetic analyses
• ddRAD requires the highest quality genomic DNA of all RAD methods • Proper fragment ligation relies on completely intact 5’ & 3’ overhangs!

ezRAD advantages
• Illumina TruSeq kit • Extensive manual, customer support & guarantee
• Probably the simplest path to obtain RAD data for small lab without experience / equipment / resources to develop in-house RAD capability
• Combined with an Illumina PCR-Free TruSeq kit, ezRAD is the only RAD protocol that can bypass all potential PCR bias

ezRAD disadvantages
• Illumina TruSeq kit • Simplicity & uniformity but expensive
• However can be used with ½ & 1/3 reaction volumes
• All ezRAD reads start with the same four GATC bases • The first 4-5 nucleotides of Read 1are used to discriminate among
adjacent clusters
• If always the same 4 first bases, difficulty to discriminate to different samples

2bRAD advantages
• Extreme protocol simplicity & cost-efficiency • No intermediate purification stages
• No need for special instrumentation (only PCR + standard agarose gel)
• Lack of biases due to fragment size selection • All endocnuclease recognition sites can be sampled

2bRAD disadvantages
• Difficulties to map 36 bp tags in a unambiguously way • But works well in no or moderately duplicated genomes (i.e. Wang et al
2012 on Arabidopsis)
• 2bRAD fragments cannot be used to build genome contigs
• 2bRAD fragments are less likely to be cross-mappable across large genetic distances, such as across different species

Conclusions
• Most important considerations when selecting a particular RAD protocol are • The facilities & the molecular experience of the researcher applying the
approach
• The biology of the organisms
• The hypotheses being tested
• All RAD protocols are powerful tools for SNP discovery & genotyping of nonmodel species
• It is important to learn about pitfalls inherent to each method & how to adress them

Main Bioinformatics pipelines • STACKS
• Website: http://catchenlab.life.illinois.edu/stacks/ • mbRAD, ddRAD, ezRAD & 2bRAD? • STACKS does not handle INDELS, so any loci near an INDEL is lost • STACKS does not call SNPs from paired end reads natively, and does especially poorly with paired end
fragments that are not of a random length (e.g., ddRAD and ezRAD)
• dDocent
• Website: https://ddocent.wordpress.com/ddocent-pipeline-user-guide/ • ddRAD & ezRAD
• PyRAD
• Website: http://dereneaton.com/software/pyrad/
• mbRAD, ddRAD, PE-ddRAD, GBS, PE-GBS, EzRAD, PE-EzRAD, 2B-RAD • use of an alignment-clustering method (vsearch)
• 2bRAD (Wang et al 2012)
• de novo: https://github.com/z0on/2bRAD_denovo • With reference genome: https://github.com/z0on/2bRAD_GATK • 2bRAD

STACKS

STACKS

LE RADSEQ SOUS GALAXY Utilisation du pipeline Stacks

• Détection de SNP • Etude des parents d’une famille
• Cartographie Génétique • Etude d’une famille avec 93 descendants
• Construction de mini-contig • Données pairées
• Génomique des population • Sans génome de référence
• Avec génome de référence
Programme

• Se familiariser avec l’utilisation de données NGS à partir de librairies réduites (RRL)
• S’initier à l’utilisation • de Galaxy
• du pipeline STACKS
• Apprendre • Préparation de données brutes Illumina RAD
• Alignement de lectures sur un génome de référence
• Assembler des loci RAD
• Détecter des SNP, déterminer génotypes et haplotypes
• Calculer des statistiques en génétique des populations
Buts

• Ceux proposés par Julian dans ces formations
• Jeux de données épinoche de l'article d'Hohenlohe et al. 2010
• Nettoyage et l'analyse des données via Galaxy, le pipeline STACKS et BWA.
• Les jeux de données seront tous produits via des séquenceurs de type Illumina GAII ou HiSeq2000.
• Les logiciels sont tous open source
Jeux de données et outils

Merci de votre attention
eBGO HUB (collaboration) http://www.e-biogenouest.org/
EMME portal (data management ) http://emme.genouest.org/
Galaxy instance (data analysis) http://galaxy.genouest.org/
GO4Bioinformatics (education) http://go4bioinformatics.genouest.org/
Cyril Monjeaud
Olivier Collin
La plate-forme Bio-informatique GenOuest Le groupe Symbiose IRISA/INRIA
GenOuest-Dyliss-Genscale