seqdb: a relational system for high-throughput biology
TRANSCRIPT

SeqDB: a relational system for high-throughput biology
Adrian Dalca and Matt Edwards
{adalca,matted}@mit.edu
Abstract
We propose the use of a Relational DatabaseManagement System (RDBMS) to supportdata storage and scientific analysis for mappedshort DNA reads. For every laboratory utiliz-ing short genomic reads, these data are storedin several of many flat file or binary formats,and are handled with a plethora of makeshiftscripts and toolsets. By utilizing a RDBMS,we can unify the system in which these dataare stored and computed upon. This solutionallows for clean and powerful code integration,compression, and efficient application via re-use of computation. It allows for powerful andflexible analysis in a manner that allows theanalyst to decouple themselves from the phys-ical storage mechanisms of the data (classicalphysical data independence). For validation,we set-up a postgres database for millions ofgenomic reads, and show that with such a sys-tem, working with these data results in greatersimplicity for data management while beingable to perform, in common analysis tasks, onpar or better than currently leading solutions.
1 Introduction
The recent advancements in Next-Generation Se-quencing (NGS) Technologies have transformed thestudy of biology ([9]). NGS technologies can gener-ate hundreds of millions of bases of DNA from oneinstrument in a matter of days. This large amount ofdata is used for many purposes in modern molecularbiology and genetics, including probing the details ofnew species or diseased individuals ([8]). Specifically,each experiment will generate hundreds of millions ofshort DNA snippets called reads (which vary in lengthfrom 100 to 400 bases) that come from a particularsample.
The ever-increasing throughput of these sequenc-ing technologies, currently outpacing Moore’s law ([3]),means that a single laboratory will have to handle aterabyte of data after only a handful of experiments.When the sample has a known genome (e.g. human),specialized software tools, called mappers, are used tofind the approximate original location of each short
read in the whole genome. This can generate a “den-sity map” of short reads, showing where particularDNA fragments are often generated from. In certaintypes of experiments, these density patterns are veryinformative. These include experiments where the se-quenced sample corresponds to some actual local bi-ological process of interest (for example, gene expres-sion). In others, finding small single-base differencesin the sequenced reads (from the single reference) isthe goal. Other common experiments involve sequenc-ing of large groups of patients with a particular diseasealong with corresponding control groups (e.g. [8]).
The typical manner of interacting with these datainvolve large experiments arriving weekly or monthly,being added to some data storage system (usually sim-ple files), and analyzed. Large analyses (e.g. for asingle publication) may rely on many experiments col-lected over several months, and aggregating varioustypes of data from these experiments. From a datamanagement perspective, this amounts to an OLAPworkload - intensive read-only queries along with in-frequent batch inserts of large blocks of data. By us-ing flat files and various individual scripts, this work-load quickly becomes hard to manage, occupies a largeamount of disk space and is very cumbersome to de-velop code for. After a certain amount of time, forexample after a report is published, further analysismay not be performed on a particular dataset. How-ever, users will want to maintain the data in some re-coverable form in order to perform re-analysis, shouldthat become necessary.
2 Current Approaches
As introduced above, current methods for managingand analyzing genetic data for a group of experimentsof a laboratory are largely based on flat file-based ac-cess and custom analysis scripts devised for each par-ticular situation. Due to the flexibility of this approachand lack of standardization, various flat file formatshave emerged in the last few years, such as FASTQ([2]) and Staden chromatogram files ([10]). Each ofthese files maintains different information in differentformats, as various researchers decide for their imme-diate task.
In 2009, Li et al. ([6]) published a paper describ-

Aligned reads file (one per experiment)
Experiment Sequencer + alignment
• Flat file (format varies) • SAM file (standard flat file) • BAM file (standard binary file)
External Applications - SNP calling - Expression profiles - Statistical queries - etc
Re-cooking - Filter on quality - Extract exons - etc
New aligned reads file (one per experiment)
Other organized file
Other Applications
Flat File
Figure 1: Traditional pipeline displaying its intricacies: there are several potential flat or binary files one mightuse, and one may run different applications which are tailored to specific a experimental setup of files, whichagain result in more data files (maybe even read files again). The files, in turn, may go into useful applications.The files and applications may vary in format and specifics, implementation language, and so on. Li et al. hasattempted to standardize the field with the SAM and BAM formats and associated tools, but still suffer fromseveral shortcomings.
ing work aimed at unifying the various formats. Theyproposed the SAM format (Sequence Alignment/Mapformat) joined by another very compact, compressedand indexed binary format called BAM (Binary SAM).The SAM and BAM files are intimately tied together,and for specific operations tend to be incredibly effi-cient. However, the files are very restrictive: for ex-ample, the columns are pre-specified and extra infor-mation is unlikely to be added, the mappings have tobe sorted on mapping position, and unmapped readscannot be stored. Li et al. also provided a set of toolsaimed at working very efficiently on these files for cer-tain tasks that many labs might use, called SamTools.In the past two years, many laboratories have learnedto incorporate SAM and SamTools in their workflow,although most are still utilizing other flat files for var-ious parts of their pipelines.
Aside from immediate storage of read and mappingdata, results from various analyses are usually storedin makeshift flat files of whatever format is needed. Forexample, a coverage file storing the number of readscovering each position will likely store two integers percolumn in a tab-delimited ascii file, with perhaps a fewmore columns that are useful to a very specific task. Aplethora of applications has been developed in variouslanguages working on all of these various files, creatinga messy web of dependencies and requirements. A fig-ure summarizing and clarifying this workflow is found
in Figure 1.A small number of large, OLAP-like databases have
been setup by large research conglomerates for storageand batch retrieval purposes from combined researchgroups around the world (Ensembl [1], Sequence ReadArchive [5]).These databases are optimized for verysimple queries on pre-loaded data in interaction withvery small inputs from users (such as finding a singlesequence or batch retrieval for a single experiment), aswell as visualization. They are not built to be usableon a single laboratory’s data and the more sophisti-cated types of applications these laboratories woulduse, which are normally instead built on flat files andSamTools.
In contrast to these approaches, we propose using aRelational Database Management System (RDBMS)for the storage and handling of genomic data. In sum-mary, this methodology allows for very flexible storage(formats can change and be compressed as needed),application development in a consistent system (viastored procedures and UDFs), storage and manage-ment of processed data (such as the coverage exampleabove), compression, and efficient re-use of computa-tion. In section 3 we introduce our approach, the re-lational schema and indices used, discuss compression,and we detail the application framework and its ad-vantages. In section 4 we discuss the results, startingwith code usability before moving on to discuss per-

formance of query families.
3 Approach
We propose to utilize a RDBMS system to store, man-age and build applications for genomic reads and map-pings. We demonstrate such a system via a Post-greSQL 8.4.5 installation, and all of the implemen-tations we discuss in this paper are run under thissystem.
3.1 Relational schema and indices
Our schema is designed to hold all relevant informa-tion about a sequencing experiment, which is termeda “run.” For a visual representation of this schema,consult Figure 2. Runs are grouped into experimentsas larger coherent units. The main two tables are“reads” and “mappings” table. Each sequencing ex-periment produces a set of reads. Each alignment ofthe sequencing reads (done by a specialized externalprogram, sometimes right after a read is sequenced)produces a set of mappings, for each read. There canbe multiple mappings for each read. As an example,this might occur when multiple alignment programsare each used to find the originated position of each se-quencing read. Experiment-specific metadata is storedin the run and experiment tables in order to allowsimple queries in downstream analysis. This contrastswith a single file per experiment approach where anal-yses that worked across multiple experiments wouldfirst involve a manual step of identifying all relevantexperiment files.
The most intensive queries in our workload involvepositional access to the read positions. This mightentail looking over a set of ranges or over the entiregenome in windowed chunks. We support accesses ofthis type with a multi-column clustered B+tree indexover experiments, genomes, and position. Clusteringis accomplished by sorting the reads prior to insertionin the database. This adds only a small amount ofoverhead, which is even less painful since these inser-tion tasks happen infrequently. Since reads from thesame experiment are always inserted together, the sortorder invariant is maintained.
3.2 Adaptive tables
One of the advantages of working within a declara-tive system like a relational database is the ability toexploit logical independence. In many database ap-plications, extra space can be traded for quicker com-putation time. Our query workload is a good candi-date for this technique, since many types of analysesuse very similar intermediate constructs. In compet-ing solutions based on static binary or full text files,these intermediate quantities would have to be recom-puted for every dependent analysis task. If these in-termediate results are stored, it is typically directly
to disk, sacrificing some amount of space and compu-tation time (in, for example, a naive full text formatdesigned and programmed on-the-fly). Custom sav-ing and parsing code will have to be written for thesetasks. If the intermediate results are not saved in sucha fashion, the core task will impose a (possibly verylarge) overhead on all dependent tasks.
In our relational system, we can make the databasesystem generate and use this precomputed data with-out changing the user’s queries. To implement this,we used postgres UDFs as table sources for commonresults that are frequently used as subqueries. Twoexamples are a precomputed join of read nucleotidesand positions for a specific alignment and experimentand a table reporting the number of reads overlap-ping each genome position. When a user accesses thesequantities through the UDF, the function checks in anauxiliary table to see if precomputed data is availableto answer the specific query. If it is not, it performsthe requisite computation directly. We note that thisis not optimal because the current postgres optimizerdoes not optimize within functions (for example, pusha selective predicate into the function) when they areused as table sources (or written in PL/Python). How-ever, the size of the tables our UDFs return are typ-ically small, and the queries using their results rarelyapply selective predicates that are not already builtin the UDF and that might improve performance dra-matically if pushed within the function.
3.3 Compression
It is important to note that the most space in thedatabase is occupied by the sequence and quality val-ues of a read itself. The specific byte usage varieswith the length of the read, but a 76-long read willhave at least 152 bytes allocated just for the se-quence and quality values, while all other data is muchsmaller. Unfortunately, compressing the quality valuesper-record is likely to be inefficient since the valueshave variations that are close to random but not smallenough for delta encoding. As a proof-of-concept, wetested a per-record zlib compression of these qualityvalues and found little improvement. The sequence ofa particular read also appears random, and thereforehard to compress. A zlib implementation shows thatonly some of the reads are compressible, specificallyin large repeat regions. However, we note that nu-cleotides are represented as chars in almost all formats(with the notable exception being the binary BAM),therefore each using 8 bits. However, the DNA al-phabet only includes 4 or 5 letters: the 4 possible nu-cleotides, and, if the sequencing allows, an N letterto indicate an uncertain base, but this is very rare.Therefore, we most often need only 2 bits per nu-cleotide, giving us approximately 4x compression overthe read sequences. In our setup, we implement a cou-ple of UDFs that can be automatically called when

Reads
Alignment M:N
Genome Name
Ptr to loc on disk
Alignment Quality
Edit string
Sequence
Error probabilities
Position
Name
Run
Metadata
Experiment
Metadata
Mapping method
Metadata
Unclustered Index on Read Name
Clustered B+ tree index on (position, genome, run)
Disk Ref 1. ACGGTGCA Ref 2. ACCGTGTCCG etc…
Figure 2: The schema used in our setup and implementations.
inserting reads or using their sequence (equivalent tocreating a custom postgres type). In general, this im-plementation provides a trade-off between space andperformance: with the compression UDFs in use, in-serting may take longer to process while the storageof those reads will be less. Using the compressed se-quences might be faster, since the system has to readless from disk, or slower, if the system needs to mas-sively decompress all sequences.
This letter compression is an example of a sim-ple scheme that can be used in general, i.e. it isnot tied to the database system, but is very cleanlyimplementable in the RDBMS system as a com-press/decompress UDF to be used with read/writesif necessary. In an external application, working witheither flat or binary files, this compression is still pos-sible, but is very cumbersome to work with, especiallyif different attributes are compressed and others arenot, or are compressed differently.
The tradeoffs navigated by the binary format BAM,along with its resulting storage performance, is pre-sented in section 4.3.
4 Results and Discussion
In this section we discuss at length the usability of thecode, performance of implemented applications, stor-age, and some future directions.
4.1 Usability
By using a RDBMS (Postgres 8.4.5) for our data man-agement and application development, we are buildingcode on top of a already well-designed and in some
cases optimized engine. By using simple languagessuch as SQL and Python, we can build UDFs that ex-tract information with many filters and patterns fromvarious tables, perform joins and more complicated op-erations, and create tables of results with fixed or ar-bitrary attributes all in very few and readable lines ofcode.
These properties bring us two advantages which areof specific importance to the read management prob-lem.
Firstly, note that when working with files, toolsetssuch as SamTools tend to separate data from differ-ent experimental runs in different files, and force con-straints on them such as the need to be sorted. Also,they will most often have a very fixed format whichcannot be extended easily. Through the power of SQLcode and the foundation of a RDBMS, we can maintainour data in large tables without the need to separatetables for each experimental run. This allows us toperform cross-experiment and cross-reference compar-isons with very specific filters and joins that efficientlyutilize indices. Therefore, we have very simple codefor combining datasets and tasks, which although pos-sible with SamTools, is very cumbersome. Similarly,through a RDBMS we can choose to store more meta-data for a read (such as parameters used), an experi-ment (such as the date it was done or the researcher incharge), or a temporary table that we compute (whenwas it computed, using what values). This is muchharder to do with files such as BAM/SAM which haveentire toolsets and extensions built that depend on atheir pre-defined format, while it is trivial to do inpostgre (simply add a column which has some default

value for any existing data) without affecting any ofthe already-written code. Such metadata allows us toperform queries such as Extract all reads by experi-menter X done between these dates, using temporarytables built after some data if available. Such queriesare (almost) impossible on SAM/BAM or other for-mats.
Secondly, the compact nature of SQL allows us todo the selections, joins, aggregations and so on in veryfew lines of code. Such operations, although mundane,may take many lines in large packages with variousinput/output files. As a quick comparison, for theConsensus Calling application (see next section fordetails), we require 5 SQL statments, which can beelgantly written in less than 40 very short lines (this in-cludes a couple of function declarations). The code forthe same application has hundreds of lines in Toolsetssuch as SamTools or Savant [11].
We note an important condition of this simple andpowerful code generation: the user need to know atleast one of SQL, Python or PlPgSQL in order to cre-ate these UDFs. Therefore, there might be a smalltraining time before this type of code can be devel-oped.
4.2 Performance
In this section we compare the running time of a fewstandard (though not necessarily easy) applications,which we have implemented as SQL/Python UDFs inour postgres setup as well as python wrappers runningon top of the SamTools toolset for SAM and BAM forcomparison.
We reiterate from section 3.1 that for most of theapplications we can make use of similar low level com-putations, and therefore storing the results of this com-putation in a temporary table may help improve per-formance. In fact, in several of the implementationswe present results for naive implementations (whichre-compute results) and smarter implementations thatuse temporary stored tables. A concrete example ofsuch a table is an explosion of all the sequences ofall reads from experiment run E that mapped to ref-erence R: exploded E R(position, letter), so that wehave very quick access to specific letters in specific po-sitions or ranges - this computation involves processinga join and performing string operations, which are to-gether handled by a Python UDF. A index on positionis also very helpful here. Such tables may take sometime to compute (a few minutes for 10 million reads),but this computation is only needed one time, and ingeneral the tables will not be very big (a few hundredMb). Note that while it is also possible to pre-computesuch files when using the traditional pipeline (tools likeSamtools), doing so is both inconvenient and may notbe helpful. In particular, much code is developed andrefined with its own algorithm inside the toolset, andforcing it to use a particular temporary file might not
be very efficient due to the need of changing to a less-efficiently-implemented method.
For reference, all benchmarks were performed on a16-core machine (Intel Xeon CPUs at 2.3 Ghz) with48 Gb of main memory. Persistent storage was a localRAID array capable of sustaining reads at 200 Mb/sec.Postgres was allowed to take advantage of this high-memory environment (and few-user OLAP workload)by allowing 4 Gb of temporary buffers and workingmemory (but no artificial increase in speed was done -i.e. tables were not pre-loaded in memory or anythingof the sort). To optimize batch insert, forced filesystemcommits (fsync) were disabled. No optimizer tuningwas attempted.
The following queries were compared:
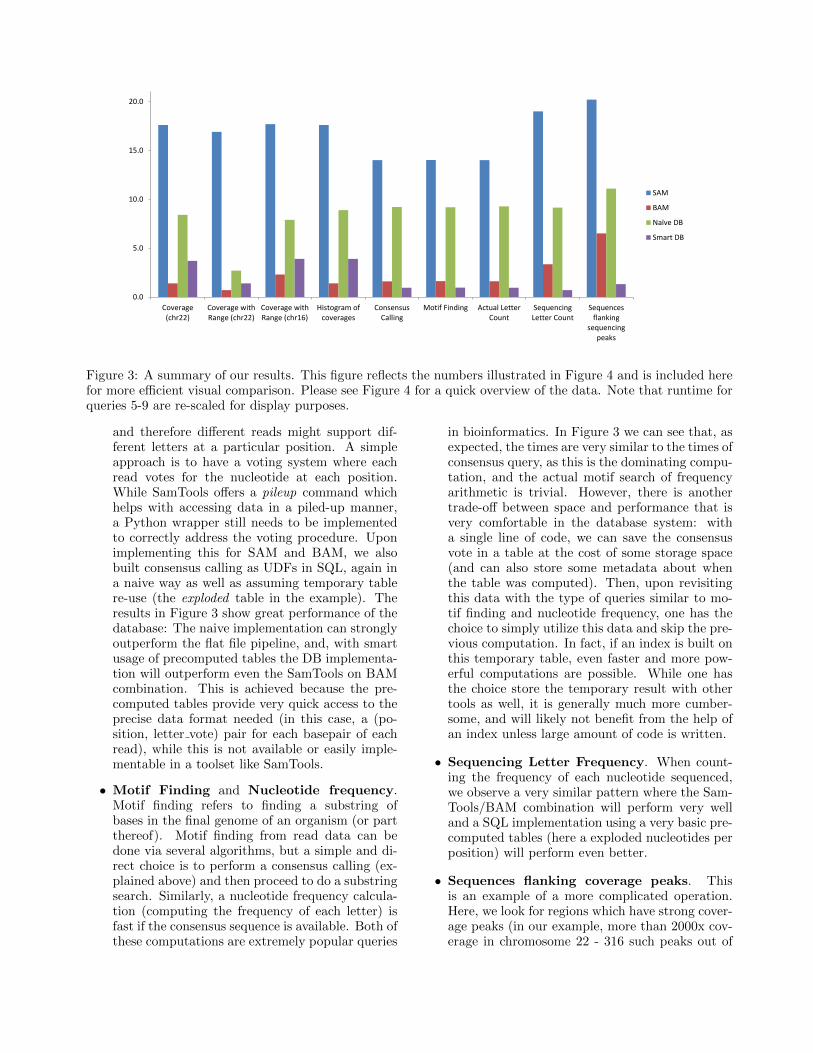
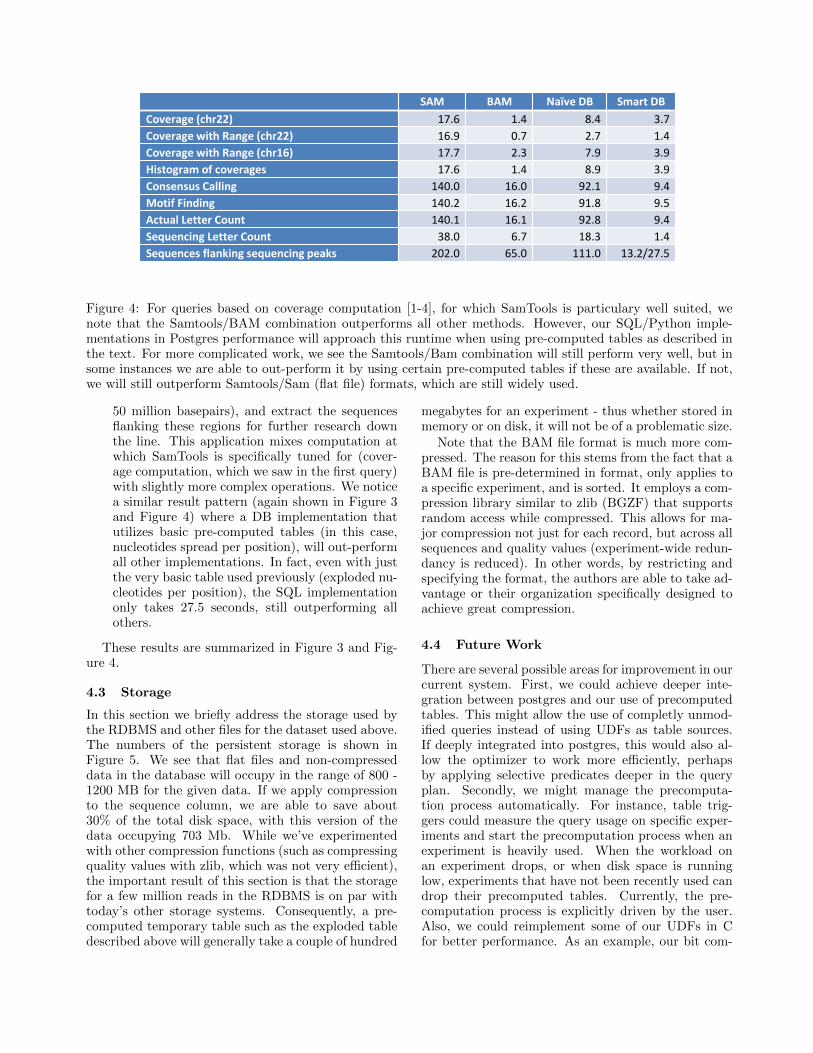
• Simple coverage computation. Samtools isoptimized for this type of query, having a large C-based library and Python wrappers for computingthis computation, for both SAM and BAM files.We implement Python wrappers for using thesetools with our data in both SAM and BAM for-mat. Inside the RDBMS, we implement both anaive implementation that only utilizes the per-sistent data tables, as well as a smarter implemen-tation that uses an exploded sequence temporarytable. From Figure 3 we can see the fastest appli-cation is SamTools running on the binary BAMfiles, as is expected considering the optimizationsperformed for this specific query. However, ournaive implementation will outperform the SAMflat file results alone - and this is achieved bythe fact that postgres utilizes our clusters overread experiments, alignment references, and align-ment position (and is in general perhaps helpedby having to read less data due to compression).If we make use of precomputed tables (specifically,one similar to exploded described above) we canachieve a runtime relatively close to the state-of-the art BAM results. If we wish to only usehalf of the reference (i.e. only work with readsthat align under the 25 million reads position),we achieve a strong improvement in the databaseimplementation runtimes due to the available in-dices, whereas the improvements for SAM/BAMare less dramatic. We note similar results for an-other range query on a different reference as well.
• Coverage Histogram computation. Anotherpopular query is to compute the distribution ofcoverage across a genome. Since this is most sim-ply achieved by computing the coverage first, theresults are very similar to the above query’s re-sults.
• Consensus calling. An important applicationis to decide the winning nucleotide for each po-sition. Sequencers and aligners make mistakes,
0.0
5.0
10.0
15.0
20.0
Coverage(chr22)
Coverage withRange (chr22)
Coverage withRange (chr16)
Histogram ofcoverages
ConsensusCalling
Motif Finding Actual LetterCount
SequencingLetter Count
Sequencesflanking
sequencingpeaks
SAM
BAM
Naïve DB
Smart DB
Figure 3: A summary of our results. This figure reflects the numbers illustrated in Figure 4 and is included herefor more efficient visual comparison. Please see Figure 4 for a quick overview of the data. Note that runtime forqueries 5-9 are re-scaled for display purposes.
and therefore different reads might support dif-ferent letters at a particular position. A simpleapproach is to have a voting system where eachread votes for the nucleotide at each position.While SamTools offers a pileup command whichhelps with accessing data in a piled-up manner,a Python wrapper still needs to be implementedto correctly address the voting procedure. Uponimplementing this for SAM and BAM, we alsobuilt consensus calling as UDFs in SQL, again ina naive way as well as assuming temporary tablere-use (the exploded table in the example). Theresults in Figure 3 show great performance of thedatabase: The naive implementation can stronglyoutperform the flat file pipeline, and, with smartusage of precomputed tables the DB implementa-tion will outperform even the SamTools on BAMcombination. This is achieved because the pre-computed tables provide very quick access to theprecise data format needed (in this case, a (po-sition, letter vote) pair for each basepair of eachread), while this is not available or easily imple-mentable in a toolset like SamTools.
• Motif Finding and Nucleotide frequency.Motif finding refers to finding a substring ofbases in the final genome of an organism (or partthereof). Motif finding from read data can bedone via several algorithms, but a simple and di-rect choice is to perform a consensus calling (ex-plained above) and then proceed to do a substringsearch. Similarly, a nucleotide frequency calcula-tion (computing the frequency of each letter) isfast if the consensus sequence is available. Both ofthese computations are extremely popular queries
in bioinformatics. In Figure 3 we can see that, asexpected, the times are very similar to the times ofconsensus query, as this is the dominating compu-tation, and the actual motif search of frequencyarithmetic is trivial. However, there is anothertrade-off between space and performance that isvery comfortable in the database system: witha single line of code, we can save the consensusvote in a table at the cost of some storage space(and can also store some metadata about whenthe table was computed). Then, upon revisitingthis data with the type of queries similar to mo-tif finding and nucleotide frequency, one has thechoice to simply utilize this data and skip the pre-vious computation. In fact, if an index is built onthis temporary table, even faster and more pow-erful computations are possible. While one hasthe choice store the temporary result with othertools as well, it is generally much more cumber-some, and will likely not benefit from the help ofan index unless large amount of code is written.
• Sequencing Letter Frequency. When count-ing the frequency of each nucleotide sequenced,we observe a very similar pattern where the Sam-Tools/BAM combination will perform very welland a SQL implementation using a very basic pre-computed tables (here a exploded nucleotides perposition) will perform even better.
• Sequences flanking coverage peaks. Thisis an example of a more complicated operation.Here, we look for regions which have strong cover-age peaks (in our example, more than 2000x cov-erage in chromosome 22 - 316 such peaks out of
SAM BAM Naïve DB Smart DB
Coverage (chr22) 17.6 1.4 8.4 3.7
Coverage with Range (chr22) 16.9 0.7 2.7 1.4
Coverage with Range (chr16) 17.7 2.3 7.9 3.9
Histogram of coverages 17.6 1.4 8.9 3.9
Consensus Calling 140.0 16.0 92.1 9.4
Motif Finding 140.2 16.2 91.8 9.5
Actual Letter Count 140.1 16.1 92.8 9.4
Sequencing Letter Count 38.0 6.7 18.3 1.4
Sequences flanking sequencing peaks 202.0 65.0 111.0 13.2/27.5
Figure 4: For queries based on coverage computation [1-4], for which SamTools is particulary well suited, wenote that the Samtools/BAM combination outperforms all other methods. However, our SQL/Python imple-mentations in Postgres performance will approach this runtime when using pre-computed tables as described inthe text. For more complicated work, we see the Samtools/Bam combination will still perform very well, but insome instances we are able to out-perform it by using certain pre-computed tables if these are available. If not,we will still outperform Samtools/Sam (flat file) formats, which are still widely used.
50 million basepairs), and extract the sequencesflanking these regions for further research downthe line. This application mixes computation atwhich SamTools is specifically tuned for (cover-age computation, which we saw in the first query)with slightly more complex operations. We noticea similar result pattern (again shown in Figure 3and Figure 4) where a DB implementation thatutilizes basic pre-computed tables (in this case,nucleotides spread per position), will out-performall other implementations. In fact, even with justthe very basic table used previously (exploded nu-cleotides per position), the SQL implementationonly takes 27.5 seconds, still outperforming allothers.
These results are summarized in Figure 3 and Fig-ure 4.
4.3 Storage
In this section we briefly address the storage used bythe RDBMS and other files for the dataset used above.The numbers of the persistent storage is shown inFigure 5. We see that flat files and non-compresseddata in the database will occupy in the range of 800 -1200 MB for the given data. If we apply compressionto the sequence column, we are able to save about30% of the total disk space, with this version of thedata occupying 703 Mb. While we’ve experimentedwith other compression functions (such as compressingquality values with zlib, which was not very efficient),the important result of this section is that the storagefor a few million reads in the RDBMS is on par withtoday’s other storage systems. Consequently, a pre-computed temporary table such as the exploded tabledescribed above will generally take a couple of hundred
megabytes for an experiment - thus whether stored inmemory or on disk, it will not be of a problematic size.
Note that the BAM file format is much more com-pressed. The reason for this stems from the fact that aBAM file is pre-determined in format, only applies toa specific experiment, and is sorted. It employs a com-pression library similar to zlib (BGZF) that supportsrandom access while compressed. This allows for ma-jor compression not just for each record, but across allsequences and quality values (experiment-wide redun-dancy is reduced). In other words, by restricting andspecifying the format, the authors are able to take ad-vantage or their organization specifically designed toachieve great compression.
4.4 Future Work
There are several possible areas for improvement in ourcurrent system. First, we could achieve deeper inte-gration between postgres and our use of precomputedtables. This might allow the use of completly unmod-ified queries instead of using UDFs as table sources.If deeply integrated into postgres, this would also al-low the optimizer to work more efficiently, perhapsby applying selective predicates deeper in the queryplan. Secondly, we might manage the precomputa-tion process automatically. For instance, table trig-gers could measure the query usage on specific exper-iments and start the precomputation process when anexperiment is heavily used. When the workload onan experiment drops, or when disk space is runninglow, experiments that have not been recently used candrop their precomputed tables. Currently, the pre-computation process is explicitly driven by the user.Also, we could reimplement some of our UDFs in Cfor better performance. As an example, our bit com-

SAM BAM DB DB +
Compression
Fastq
1200 295 1076 703 791
Figure 5: The storage used by various files for the reads of our experiment.
pression function for DNA sequence reads is writtenin PL/Python. A compiled C function would providemuch greater performance. Finally, we might explorea hybrid BAM/postgres solution that uses BAM forstorage at the table level. We explored a initial versionof this using a Python library interfacing with BAMfiles, but the overhead for converting records into post-gres led to very poor performance.
5 Conclusion
In this project we proposed the use of RelationalDatabase Management Systems to maintain and buildapplications for genomic data resulting from Next-Generation Sequencing technologies. These data areplagued in the research field by a plethora of file for-mats and toolsets that together result in very messyand inefficient pipelines. As SamTools is working to-wards unifying formatting standards, many shortcom-ings remain. By utilizing a RDBMS for this data, wecan unify to a system in which all computation anddata is maintained while being flexible and efficientwith the data formats. This solution also allows forseamless compression and use of metadata for queriesthat are otherwise impossible with conventional meth-ods. In terms of application development, the abilityto utilize previously computed data without compli-cated code and overhead allows for very efficient querydevelopment.
To support our proposal and theoretical develop-ment we set up a Postgres 8.4.5 database with 16million actual sequencing reads, and built compres-sion and application UDFs. We showed that while ourstorage requirements are on the same order as those ofother file systems, we can easily outperform even thebest flat-file toolset in most queries, and with carefulre-use of computation we can perform on par (or evenoutperform) even the best binary indexed toolsets. Wetherefore propose that the use of RDBMS for readmanagement is a promising avenue of further researchand of heavy interest to sequencing researchers.
References
[1] Birney et al. “An overview of Ensembl.” GenomeResearch (2004).
[2] Cock et al. “The Sanger FASTQ file for-mat for sequences with quality scores, and the
Solexa/Illumina FASTQ variants.” Nucleic AcidsResearch (2010).
[3] Editorial board. “Metagenomics versus Moore’slaw.” Nature Methods (2009).
[4] Fiume et al. “Savant: genome browser forhigh-throughput sequencing data.” Bioinformatics(2010).
[5] Leinonen et al. “The sequence read archive.” Nu-cleic Acids Research (2010).
[6] Li et al. “The Sequence Alignment/Map formatand SAMtools.” Bioinformatics (2009).
[7] Mardis. “The impact of next-generation sequenc-ing technology on genetics.” Trends in Genetics(2008).
[8] Ng et al. “Targeted capture and massively-parallelsequencing of 12 human exomes.” Nature (2009).
[9] Schuster. “Next-generation sequencing transformstoday’s biology.” Nature Methods (2008).
[10] http://staden.sourceforge.net/manual/formats_unix_2.html