sclerosi laterale amiotrofica (sla) · molte delle malattie neurodegenerative presentano entrambe...
TRANSCRIPT

Sclerosi laterale amiotrofica (SLA)
Descritta per la prima volta nel 1860 dal neurologo francese Jean-Martin Charcot sclerosi = "indurimento” laterale = riferito alla zona del midollo spinale che manifesta le prime alterazioni amiotrofica = a (negazione) + mio ("muscolo") + trofico ("nutrimento") : i muscoli del malato si atrofizzano per un “nutrimento insufficiente”
Jean-Martin Charcot

CLINICA
• Esordio : spinale - debolezza ad uno o più arti (di solito laterale), fascicolazioni, o bulbare – difficoltà di parola e di deglutizione
• Paralisi progressiva dovuta alla degenerazione dei motoneuroni (è anche detta malattia dei motoneuroni) spinali e corticali, ad eccezione di quelli oculomotori e del nucleo di Onuf.
• Perdita progressiva e irreversibile della capacità di deglutizione (disfagia), dell'articolazione della parola (disartria) e del controllo dei muscoli scheletrici, fino alla compromissione dei muscoli respiratori e alla morte.
• La SLA non altera funzioni sensoriali, sessuali e sfinteriali del malato. Le funzioni cognitive sono compromesse solo in una piccola percentuale dei casi (demenza fronto-temporale).

Molte delle malattie neurodegenerative presentano entrambe le classi di sintomi in qualche momento nel corso della progressione della malattia Due tipi principali di sintomi clinici DEMENZE: disordini cognitivi, associativi, caratteriali e di memoria DISORDINI DEL MOVIMENTO: Ipercinesia, acinesia, paralisi Le manifestazioni cliniche dipendono dai sistemi neuronali coinvolti nel corso della malattia

TERAPIA
Non esistono terapie in grado di arrestare il decorso della malattia.
Terapie sintomatiche per fascicolazioni, crampi, effetto pseudobulbare (attacchi di riso o pianto incontrollati), ansia, depressione.
Terapia di supporto per • insufficiente deglutizione/alimentazione (fino a PEG) • insufficiente ventilazione (respiratore, fino a
tracheotomia)
Unico farmaco approvato : riluzolo (estende la vita in media di 3 mesi in una parte dei pazienti).

EPIDEMIOLOGIA
La SLA è la malattia del motoneurone più comune. • Età media di esordio : ~ 50 anni (+ forme giovanili)
• Durata media : 3-4 anni
• Incidenza : 2-8 / 100.000 ( come Sclerosi Multipla)

FATTORI DI RISCHIO EPIDEMIOLOGICI
• Età
• Sesso maschile (lieve prevalenza)
• Pratica agonistica (calcio)
• Traumi cranici (?)
• Esposizione a tossici (metalli) (?)
• Fumo (?)
• Familiarità – 10-20% dei casi la malattia è trasmessa in modalità
Aut/Dom (raramente Aut/Rec) a penetranza completa (tutte le generazioni sono colpite)

THE LANCET Neurology Vol 2 November 2003
Putting the boot in: soccer linked to ALS?
Simone Beretta, Maria Teresa Carrì,
Ettore Beghi, Adriano Chiò, and Carlo
Ferrarese
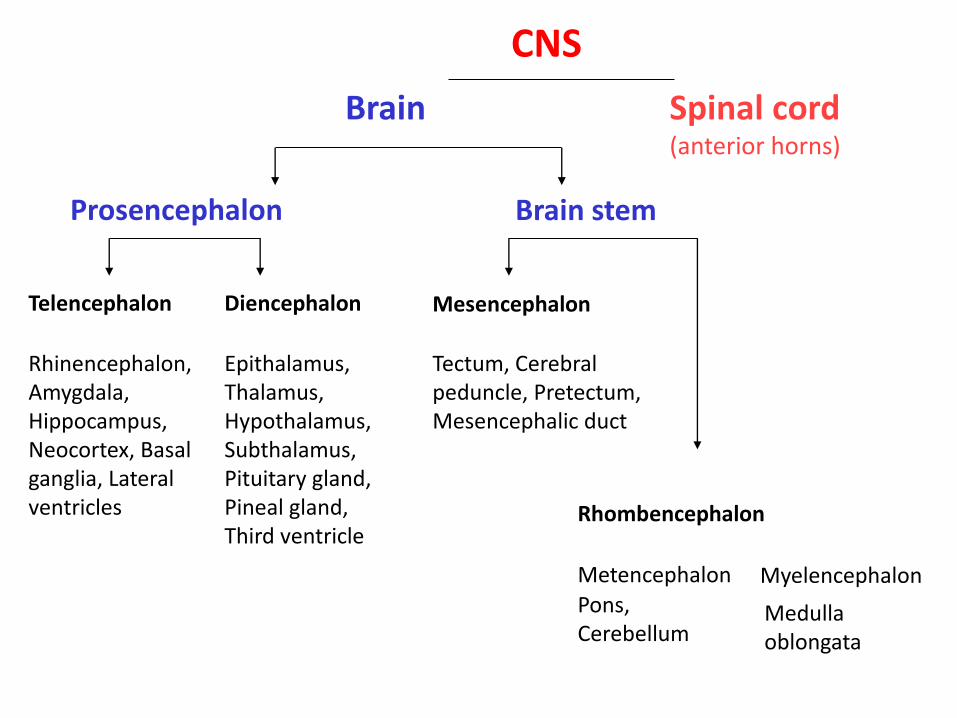
Brain Spinal cord (anterior horns)
Prosencephalon Brain stem
Telencephalon Diencephalon Mesencephalon
Rhombencephalon
Rhinencephalon, Amygdala, Hippocampus, Neocortex, Basal ganglia, Lateral ventricles
Epithalamus, Thalamus, Hypothalamus, Subthalamus, Pituitary gland, Pineal gland, Third ventricle
Tectum, Cerebral peduncle, Pretectum, Mesencephalic duct
Metencephalon Myelencephalon Pons, Cerebellum
Medulla oblongata
CNS

ISTOPATOLOGIA
1. Neurodegenerazione di regioni cerebrali
specifiche – perdita 1° e 2° motoneurone
• Midollo spinale (corna anteriori)
• Corteccia cerebrale (lobo frontale, temporale e parietale)
In stadi avanzati : proliferazione astrocitaria e attivazione microgliale (neuroinfiammazione)
Degenerazione muscolare :
Dying back o dying forward ?
9
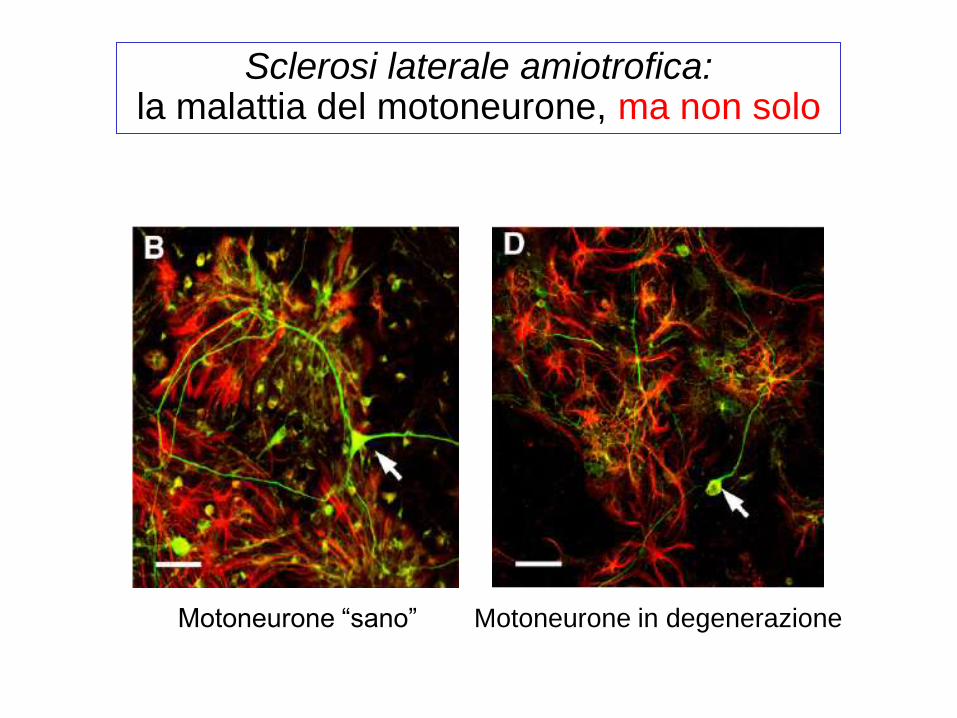
Motoneurone in degenerazione Motoneurone “sano”
Sclerosi laterale amiotrofica: la malattia del motoneurone, ma non solo
La malattia del motoneurone (ma non solo)
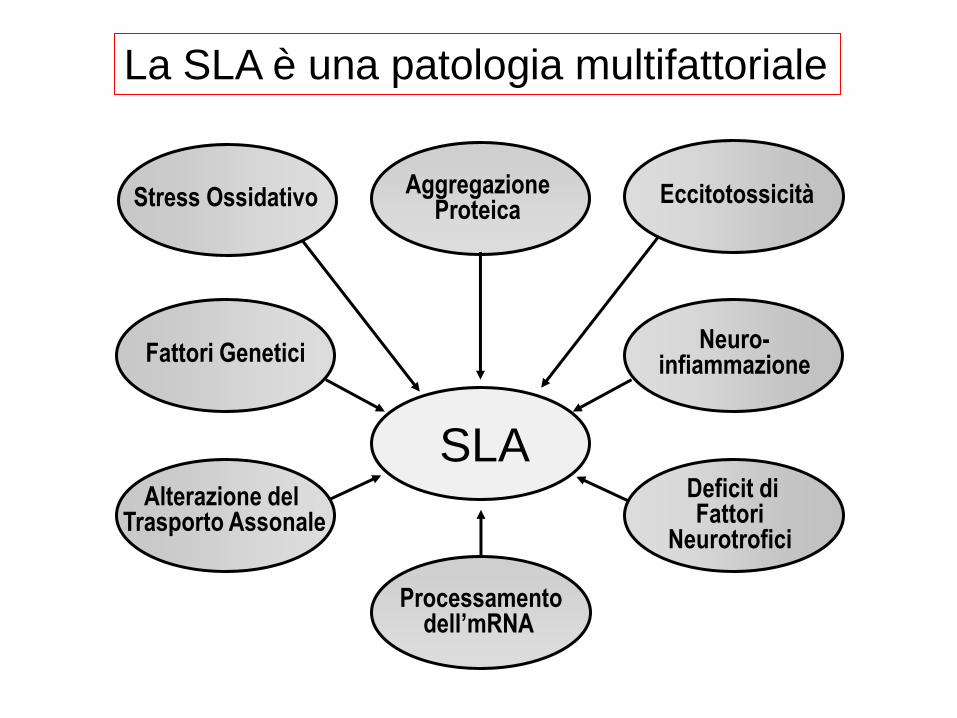
La SLA è una patologia multifattoriale
Deficit di Fattori
Neurotrofici
Eccitotossicità
Neuro- infiammazione
Alterazione del Trasporto Assonale
Fattori Genetici
Stress Ossidativo Aggregazione
Proteica
SLA
Processamento dell’mRNA
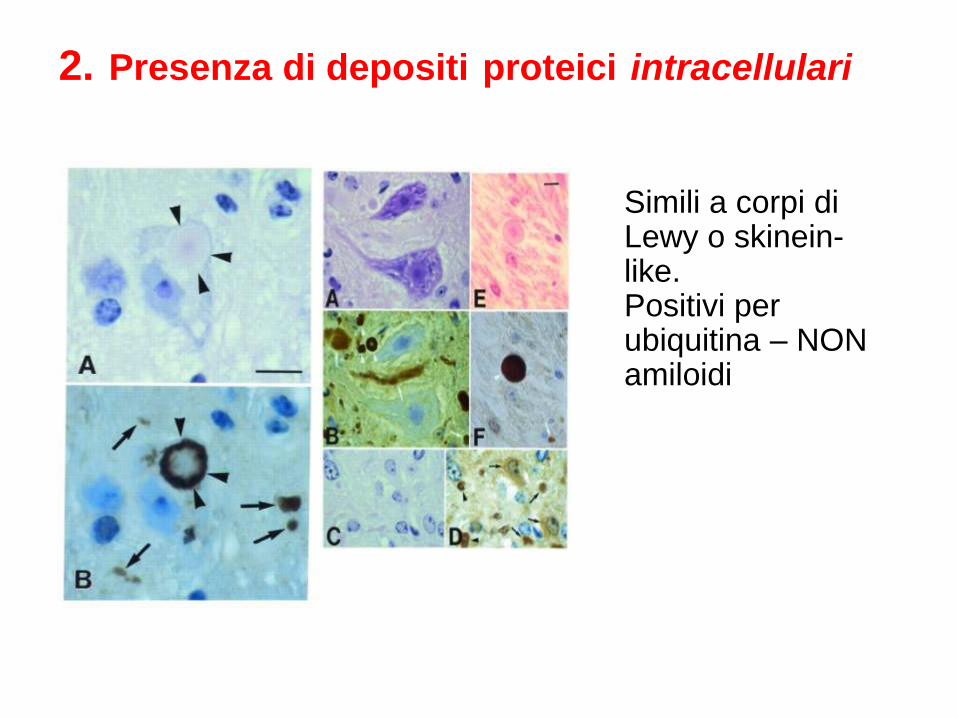
2. Presenza di depositi proteici intracellulari
Simili a corpi di Lewy o skinein-like. Positivi per ubiquitina – NON amiloidi
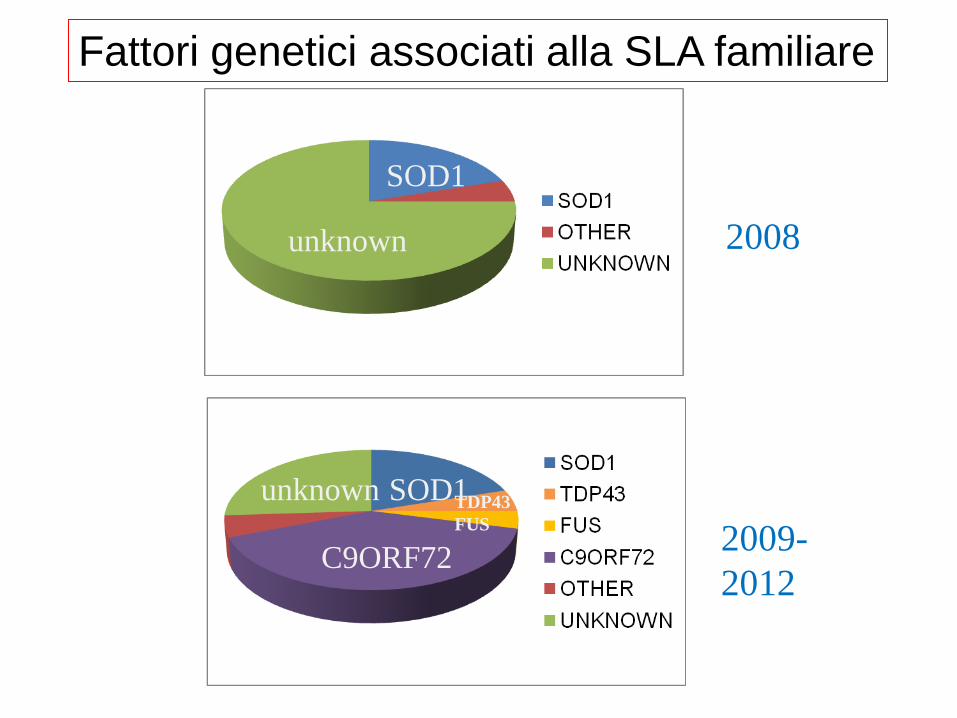
SOD1
unknown 2008
unknown SOD1
C9ORF72
TDP43
FUS 2009-
2012
Fattori genetici associati alla SLA familiare

“La SLA sporadica e la SLA familiare, benchè
presentino una certa
eterogeneità di manifestazione,
sono clinicamente indistinguibili”
Modelli di SLA associata alla
espressione di SOD1 mutante

Experimental models for the
study of SOD1-linked FALS
• Recombinant proteins
• Saccharomyces cerevisiae
• Transfected mouse cell lines
• Transfected human cell lines
• Transgenic Drosophila melanogaster flies
• Zebrafish
• Transgenic mice/rats
C
O
M
P
L
E
X
I
T
Y
Every model has pros and cons….

• About 20% of FALS patients carry point mutations in the gene sod1.
• More than 140 different mutations in sod1 have been described in FALS families.
• Mutations of critical residues alter metal binding ability and/or protein stability, conferring a new function.
SOD1 and FALS

wtSOD1 fALS-SOD1 G37R

> Free protein carbonyls
> Lipid peroxidation adducts
> Mitochondrial DNA oxidation adducts
> Nuclear DNA oxidation adducts
> Cytoplasmatic RNA oxidation adducts
> Protein nitration
Oxidative damage markers found
in ALS spinal cord
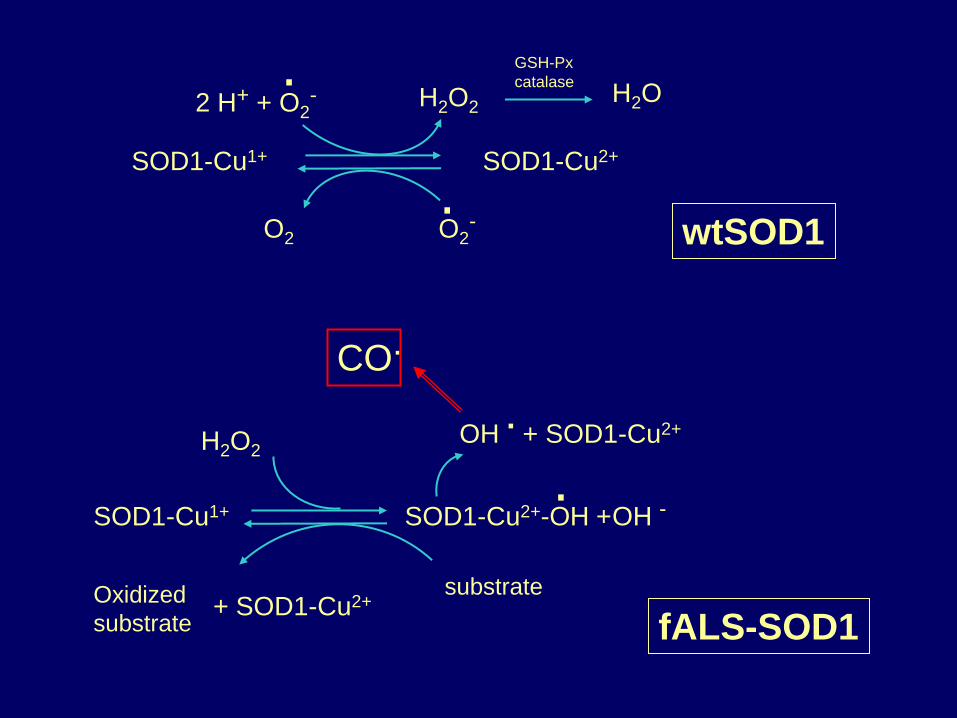
wtSOD1
2 H+ + O2- H2O2 H2O
SOD1-Cu1+ SOD1-Cu2+
O2 O2
-
GSH-Px
catalase
.
.
H2O2
SOD1-Cu1+ SOD1-Cu2+-OH + OH -
Oxidized
substrate
substrate + SOD1-Cu2+
OH . + SOD1-Cu2+
.
fALS-SOD1
CO.
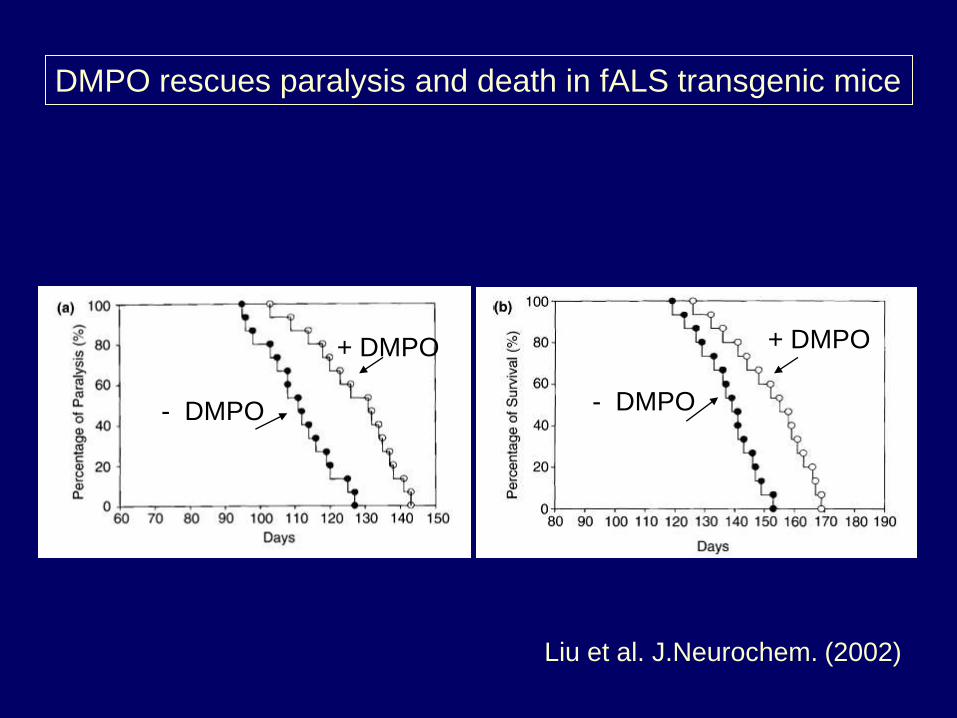
DMPO rescues paralysis and death in fALS transgenic mice
+ DMPO
- DMPO - DMPO
+ DMPO
Liu et al. J.Neurochem. (2002)
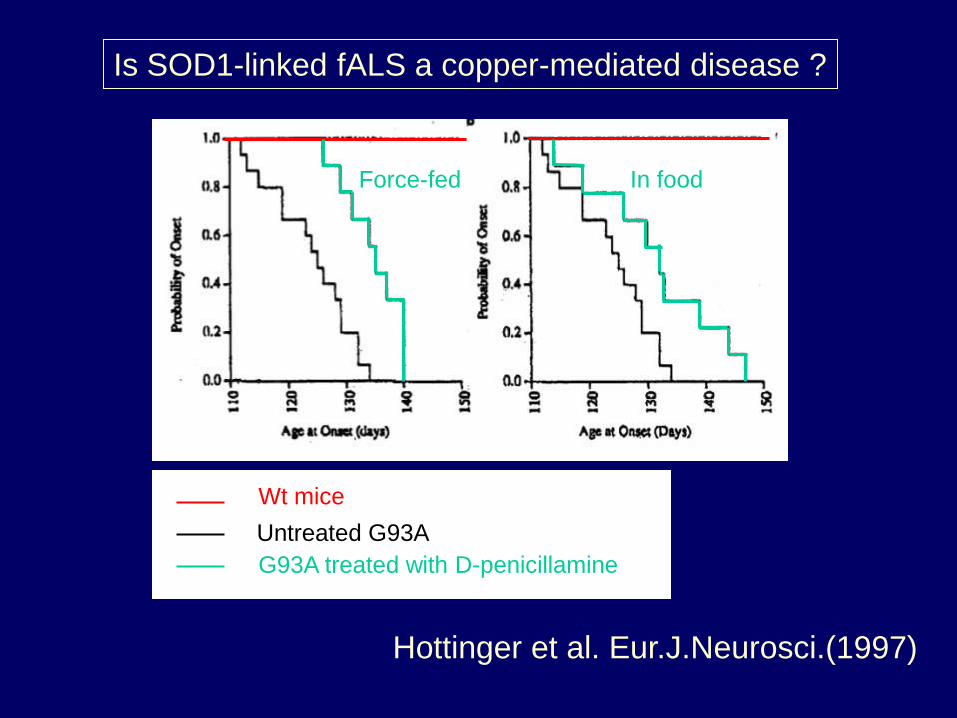
Is SOD1-linked fALS a copper-mediated disease ?
Wt mice
Untreated G93A
G93A treated with D-penicillamine
Force-fed In food
Hottinger et al. Eur.J.Neurosci.(1997)
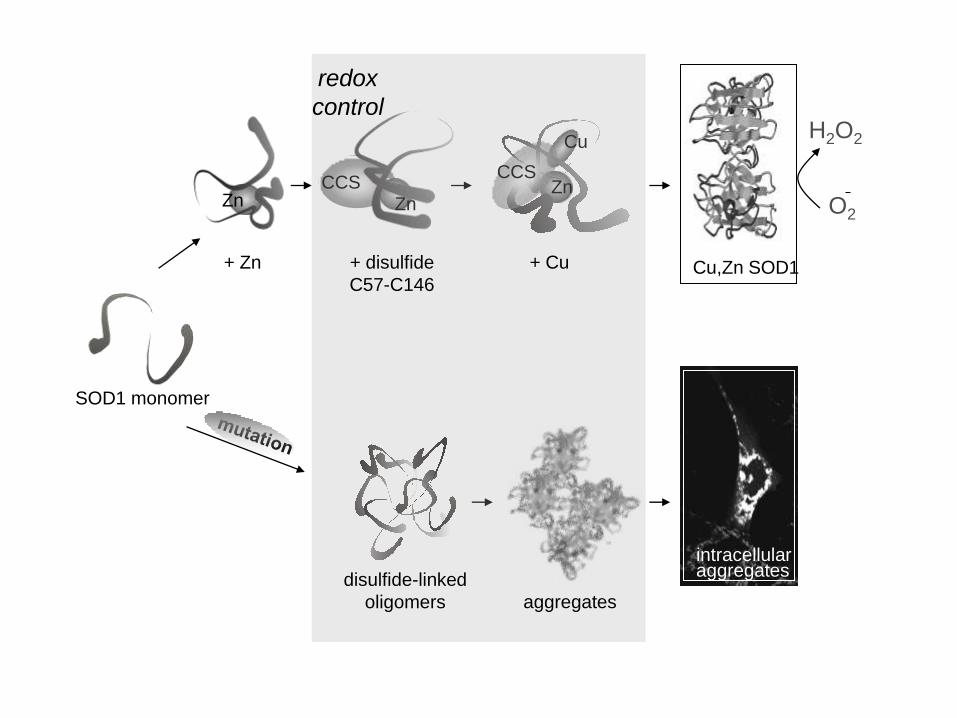
SOD1 monomer
Zn
+ Zn Cu,Zn SOD1
CCS Zn
Cu
Zn CCS
disulfide-linked
oligomers aggregates
H2O2
O2
+ disulfide
C57-C146
+ Cu
redox
control
intracellular aggregates
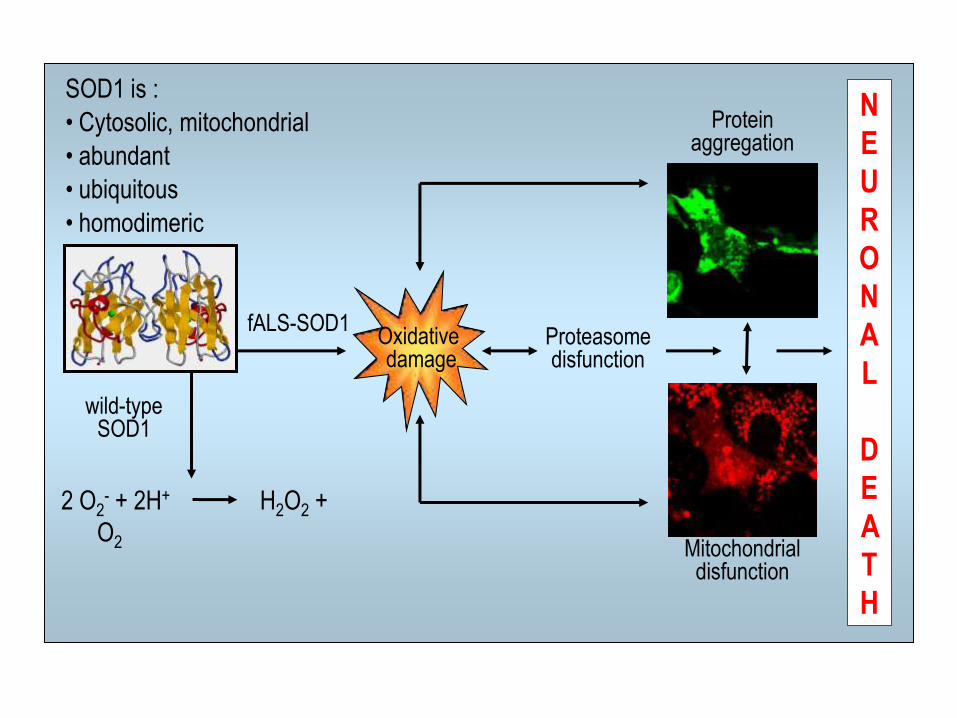
2 O2- + 2H+ H2O2 +
O2
wild-type SOD1
fALS-SOD1
Protein aggregation
Mitochondrial disfunction
N
E
U
R
O
N
A
L
D
E
A
T
H
Oxidative damage
Proteasome disfunction
SOD1 is :
• Cytosolic, mitochondrial
• abundant
• ubiquitous
• homodimeric
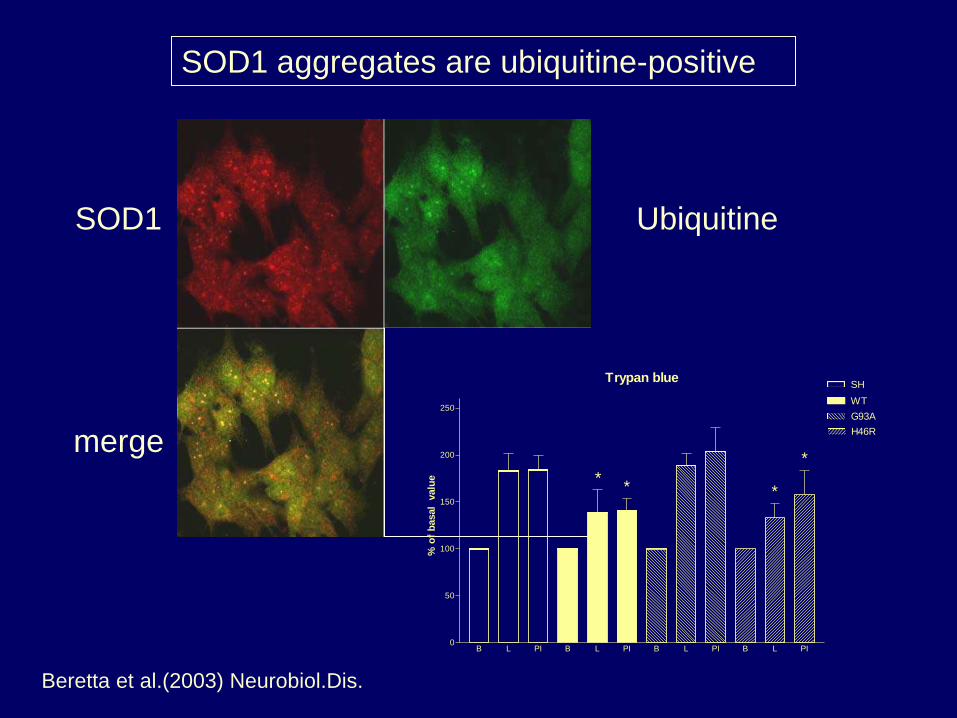
SOD1 aggregates are ubiquitine-positive
SOD1 Ubiquitine
merge
Trypan blue
B L PI B L PI B L PI B L PI0
50
100
150
200
250
SH
WT
G93A
H46R%
of
basal
valu
e *
*
**
Beretta et al.(2003) Neurobiol.Dis.

More than 150 different sod1 mutations have been found in FALS patients.
Do ALL mutant SOD1s share the SAME toxic function ?
Bendotti and Carrì (2004) Trends Mol.Med.
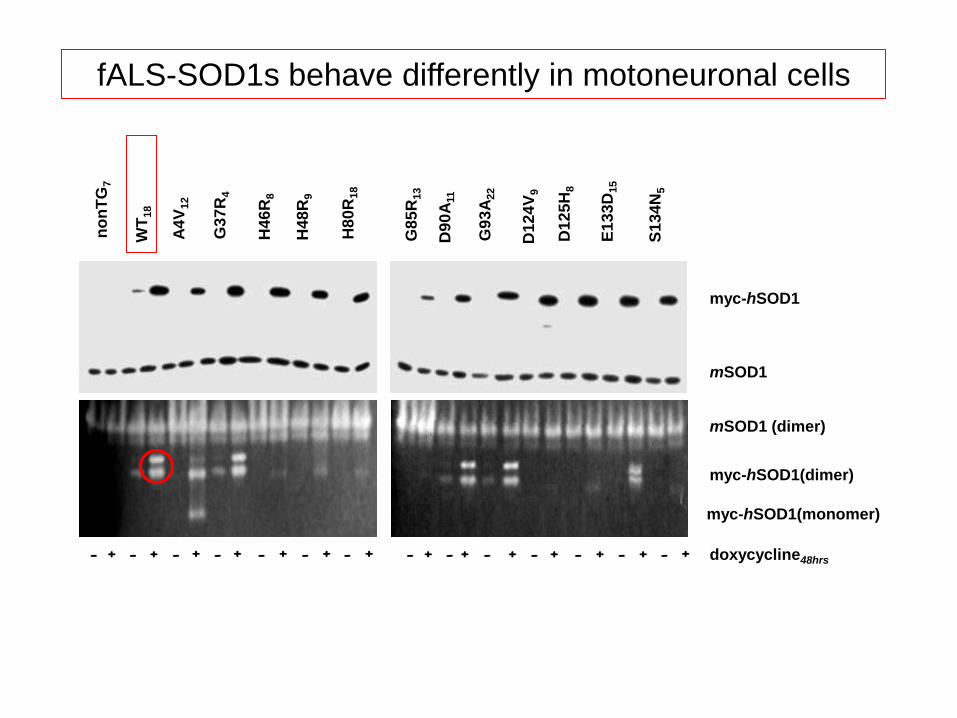
myc-hSOD1
doxycycline48hrs
mSOD1 (dimer)
myc-hSOD1(dimer)
myc-hSOD1(monomer)
E1
33
D15
D90
A11
G9
3A
22
D12
5H
8
S1
34
N5
G8
5R
13
A4
V12
G3
7R
4
H48
R9
H80
R18
H46
R8
no
nT
G7
WT
18
- + - + - + - + - + - + - + - + - + - + - + - + - + - +
mSOD1
D12
4V
9
fALS-SOD1s behave differently in motoneuronal cells
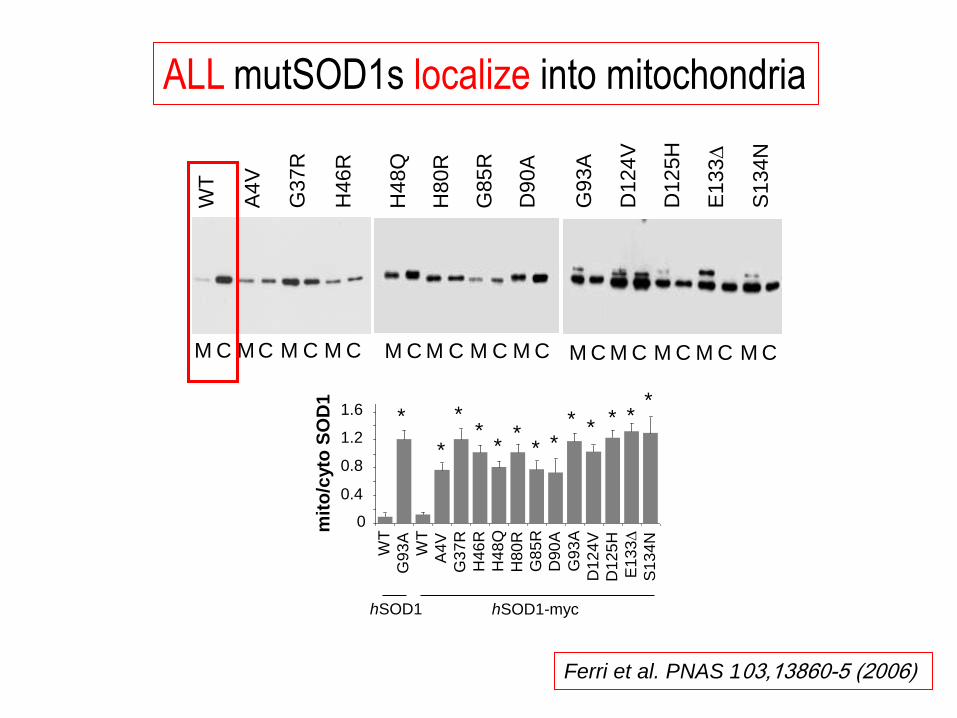
hSOD1-myc hSOD1
mit
o/c
yto
SO
D1
0
0.4
0.8
1.2
1.6 W
T
A4
V
G3
7R
H46
R
H48
Q
H80
R
G8
5R
D90
A
G9
3A
D12
4V
D12
5H
E1
33
D
S1
34
N
WT
G9
3A
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
1,8
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
*
*
* *
* *
* * * * * *
*
ALL mutSOD1s localize into mitochondria
M C M C M C M C M C M C M C M C M C M C M C M C M C
WT
A4V
G37R
H46R
H48Q
H80R
G85R
D90A
G9
3A
D124V
D125H
E133
D
S134
N
Ferri et al. PNAS 103,13860-5 (2006)
Do mutant SOD1s exert their toxic function
INSIDE (or IN ASSOCIATION with) MITOCHONDRIA ?
Mitochondria are damaged early in patients (and models)
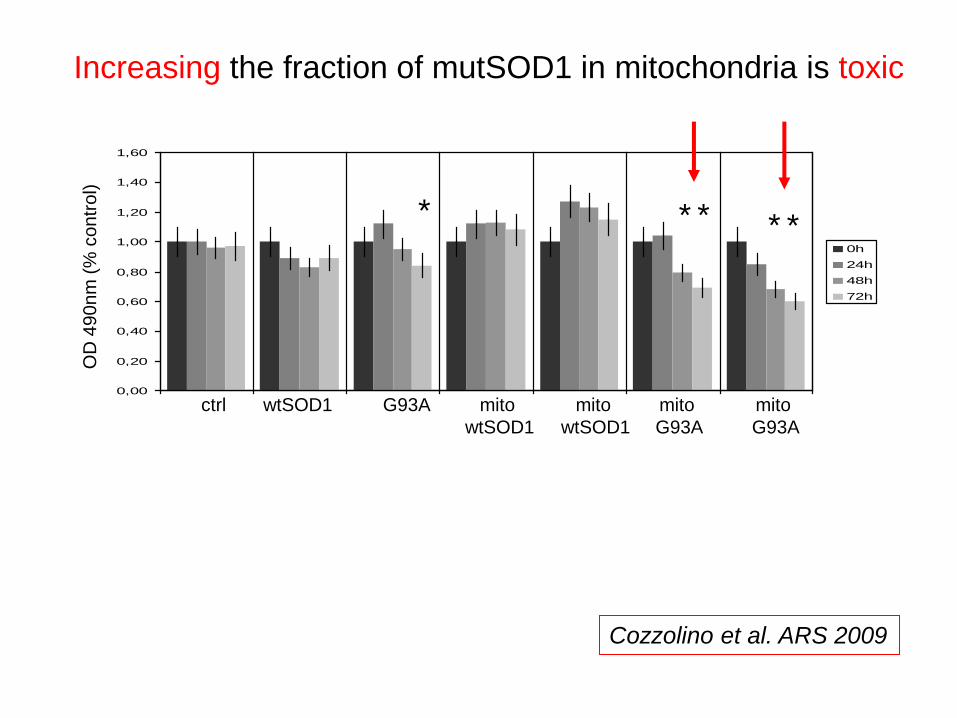
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
1 2 3 4 5 6 7
0h
24h
48h
72h
* * * * *
wtSOD1 G93A mito
wtSOD1
mito
wtSOD1
mito
G93A
mito
G93A
ctrl
OD
49
0n
m (
% c
on
tro
l)
Increasing the fraction of mutSOD1 in mitochondria is toxic
Cozzolino et al. ARS 2009
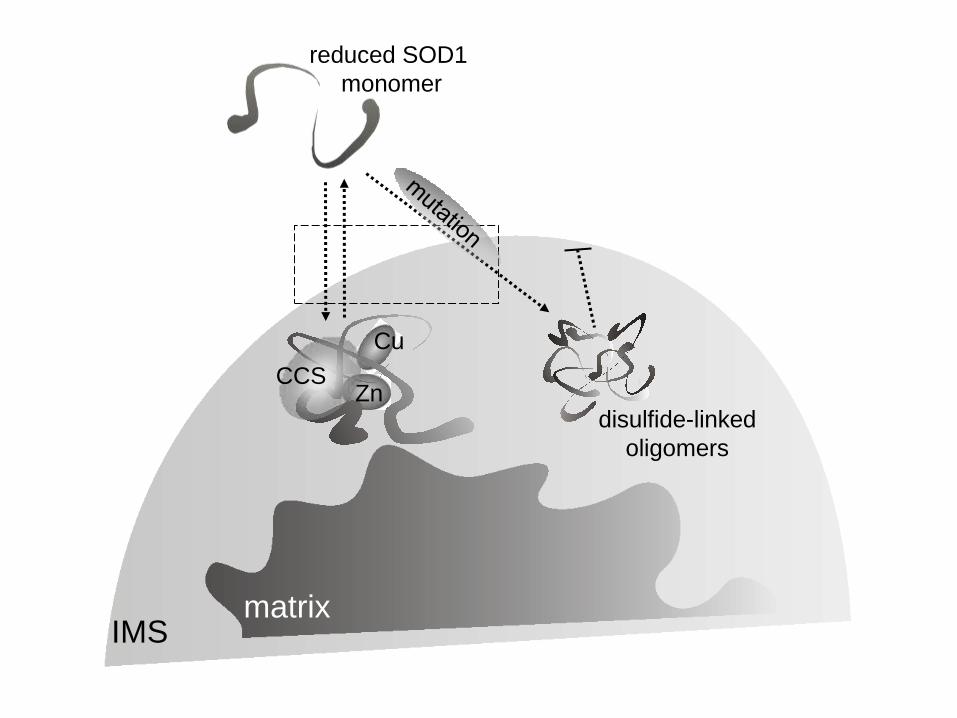
Cu
Zn CCS
reduced SOD1
monomer
IMS matrix
disulfide-linked
oligomers
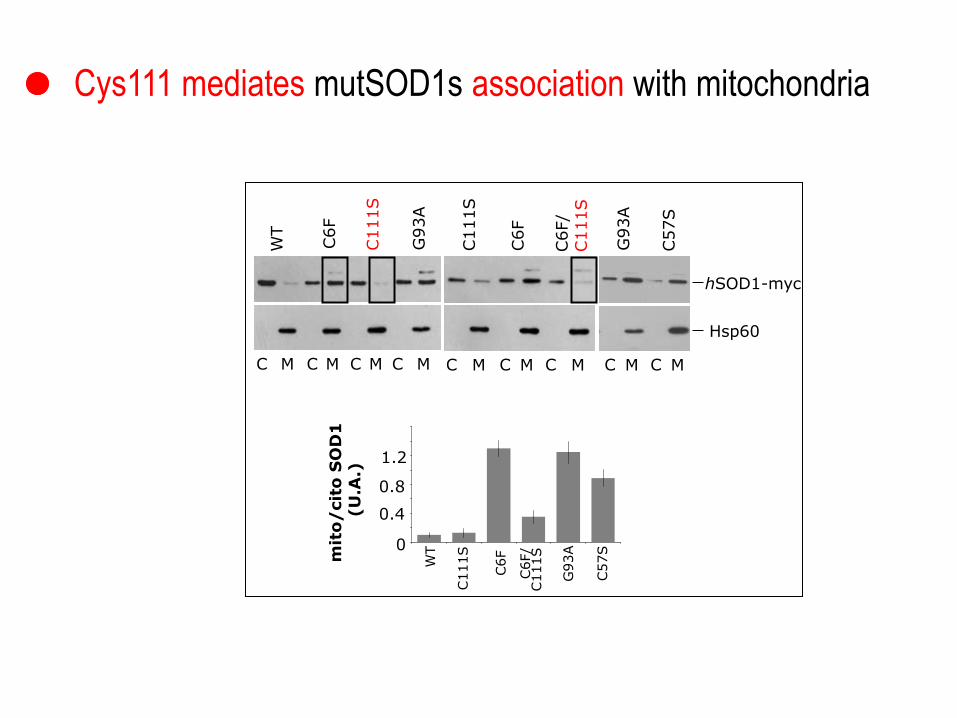
WT
C6F
C111S
G
93A
C6F
C111S
C6F/
C111S
C M C M C M C M C M C M C M
hSOD1-myc
Hsp60
G93A
C57S
C M C M
WT
C111S
C6F
C6F/
C111S
G93A
mit
o/
cit
o S
OD
1
(U
.A.)
0
0.4
0.8
1.2
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
1 2 3 4 5 6
C57S
Cys111 mediates mutSOD1s association with mitochondria
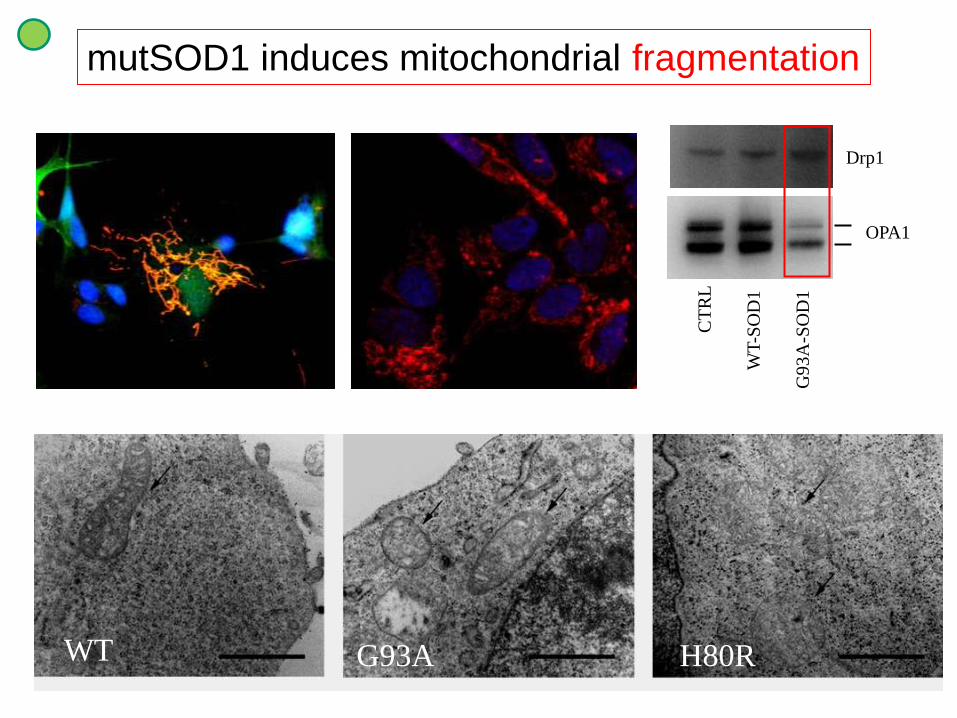
WT G93A H80R
mutSOD1 induces mitochondrial fragmentation
CT
RL
WT
-SO
D1
G9
3A
-SO
D1
OPA1
Drp1
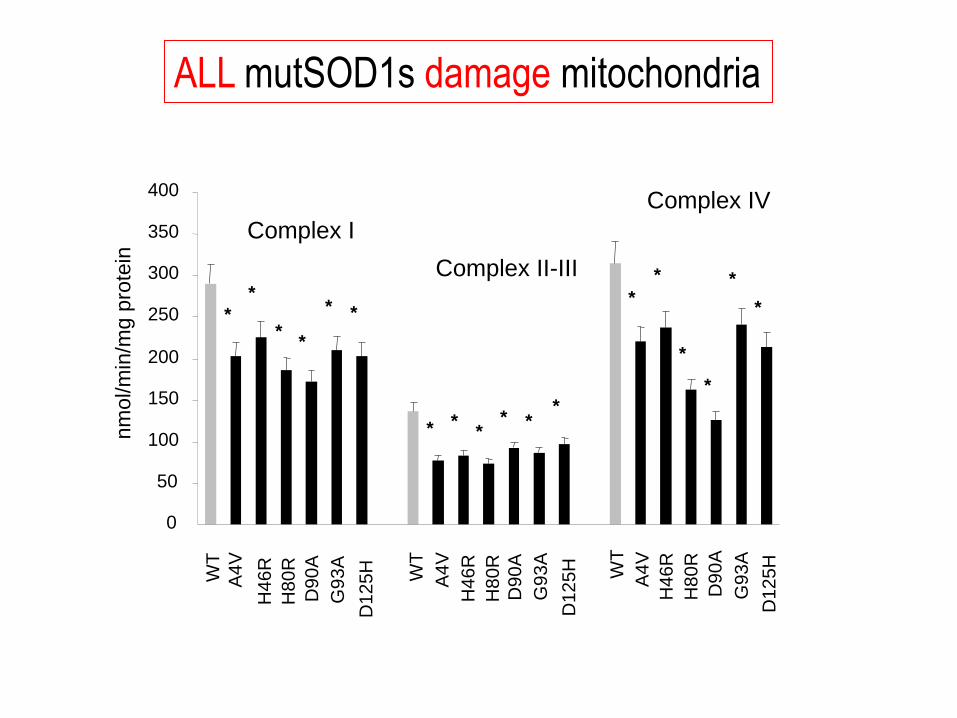
nm
ol/m
in/m
g p
rote
in Complex I
Complex II-III
Complex IV W
T
WT
WT
A4
V
H4
6R
H8
0R
D9
0A
D1
25
H
G93A
A4V
H4
6R
H8
0R
D9
0A
D1
25
H
G93A
A4
V
H4
6R
H8
0R
D9
0A
D1
25
H
G93A
0
50
100
150
200
250
300
350
400
* *
* *
* *
* * *
* * *
*
*
*
*
*
*
ALL mutSOD1s damage mitochondria
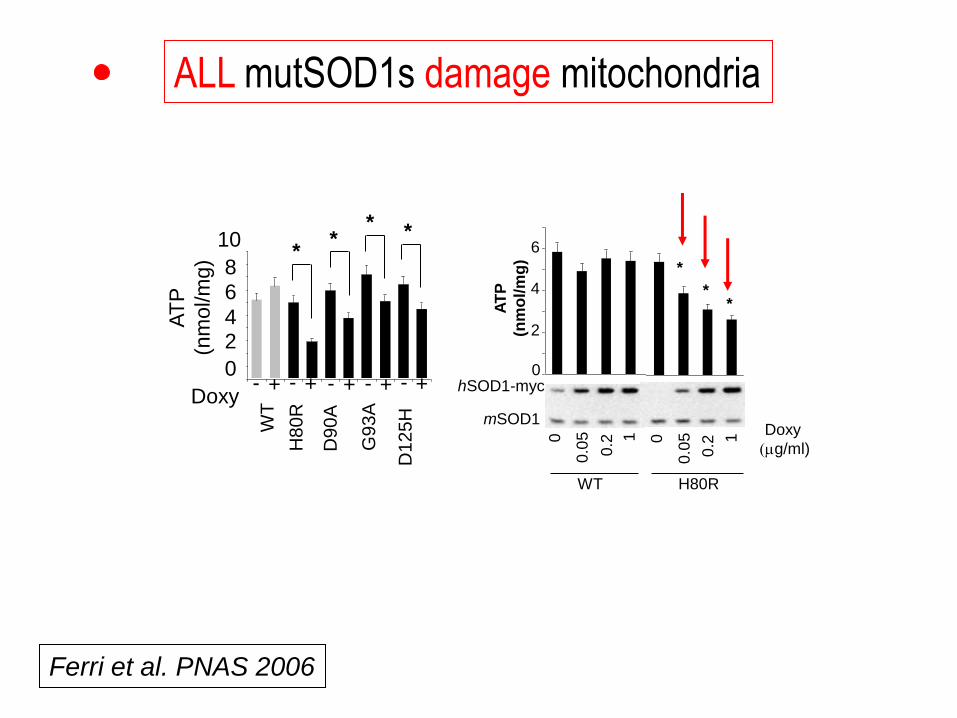
0
1
2
3
4
5
6
7
*
* *
0
2
4
6
AT
P
(nm
ol/
mg
)
0
1
2
3
4
5
6
7
mSOD1 Doxy
(mg/ml)
hSOD1-myc
WT H80R
0
0.0
5
0.2
1
0
0.0
5
0.2
1
Doxy
AT
P
(nm
ol/m
g)
0
1
2
3
4
5
6
7
8
9
10
WT
H8
0R
D9
0A
G93A
D1
25
H
+ - + - + - + - + - 0
2 4
6 8
10 *
* * *
Ferri et al. PNAS 2006
ALL mutSOD1s damage mitochondria
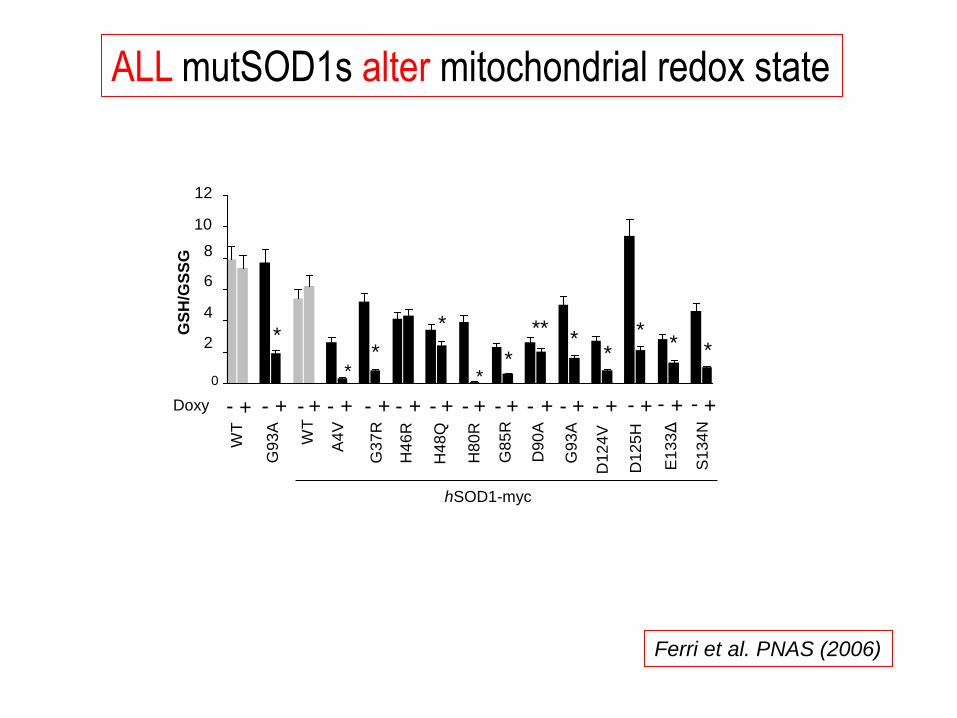
ALL mutSOD1s alter mitochondrial redox state
WT
H80
R
D90
A
G9
3A
D12
5H
A4
V
G3
7R
H46
R
H48
Q
E1
33
Δ
S1
34
N
D12
4V
G8
5R
Doxy
GS
H/G
SS
G
+ + + + + + - - - - - + - + + + + + - - - - - - - + + -
WT
+ -
G9
3A
0
2
4
6
8
10
12
* *
*
* *
* *
* **
*
* *
hSOD1-myc
Ferri et al. PNAS (2006)
ctr
l
wt
G93A
wt
G93A
150
hSOD1 input
mito soluble
SOD1
SOD1
hSOD1
VDAC
30
ctr
l
wt
G93A
wt
G93A
mito insoluble
150 30
-b-ME
+b-ME
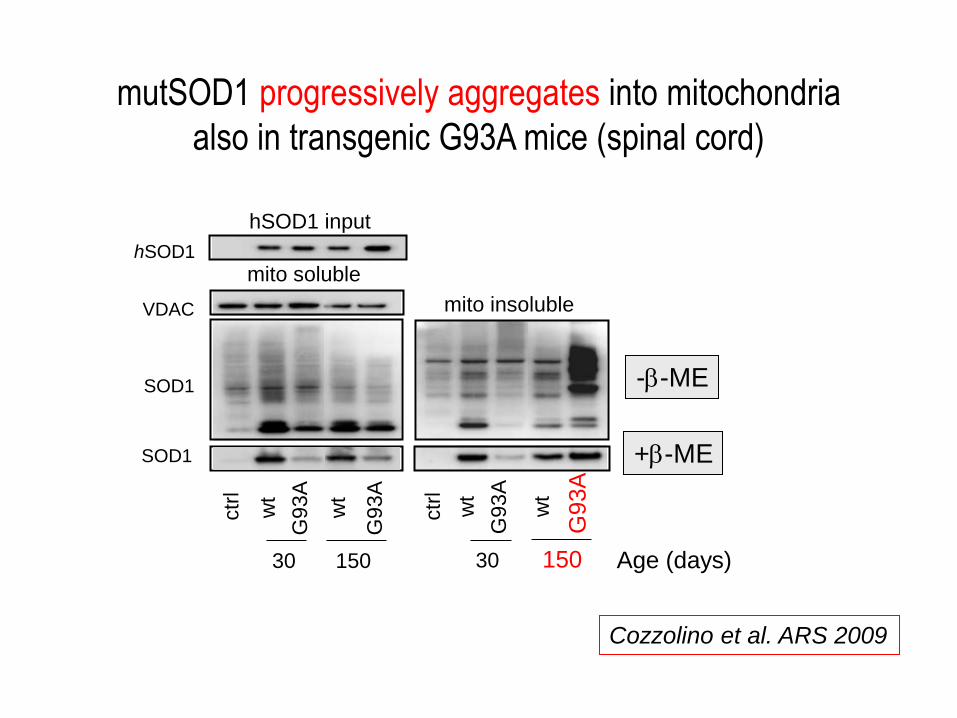
mutSOD1 progressively aggregates into mitochondria
also in transgenic G93A mice (spinal cord)
Age (days)
Cozzolino et al. ARS 2009
effector caspases
Apaf1
cyt c
casp9
apoptosome
SOD1
SOD1
ROS
SOD1
Ca2+ Bcl-2
family caspase-
independent cell death
apoptosis
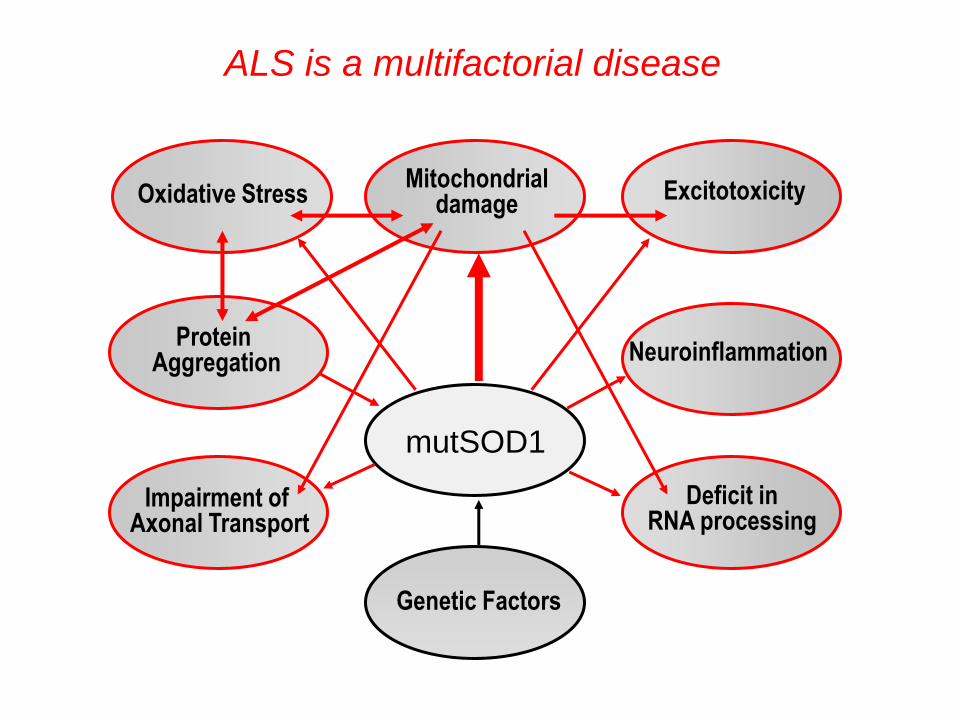
Deficit in RNA processing
Excitotoxicity
Neuroinflammation
Impairment of Axonal Transport
Oxidative Stress
Protein Aggregation
ALS is a multifactorial disease
Mitochondrial damage
Genetic Factors
mutSOD1

inflammatory
mediators
motor neuron
astrocyte
ims
EAAT2
Ca2+
glutamate
motor neuron
astrocyte
SOD1
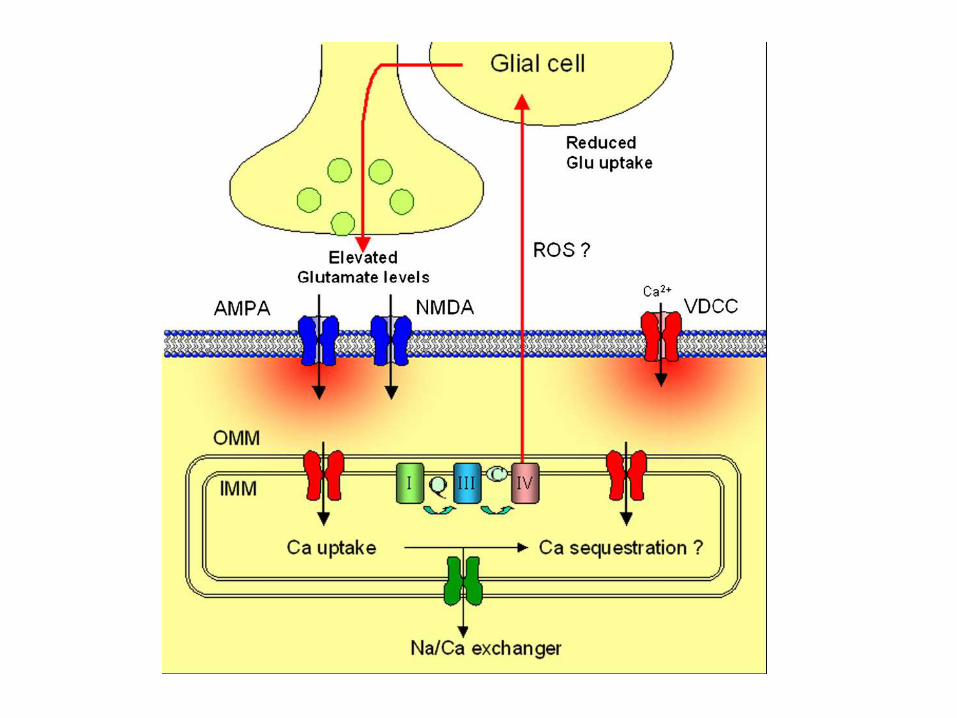

Motor neurons express a large number of Ca2+-permeable receptors lacking the GluR2 subunit. Thus, glutamatergic stimulation increases the intracellular Ca2+ concentration and because of the low Ca2+-buffering capacity of motor neurons part of this Ca2+ will be taken up by mitochondria.
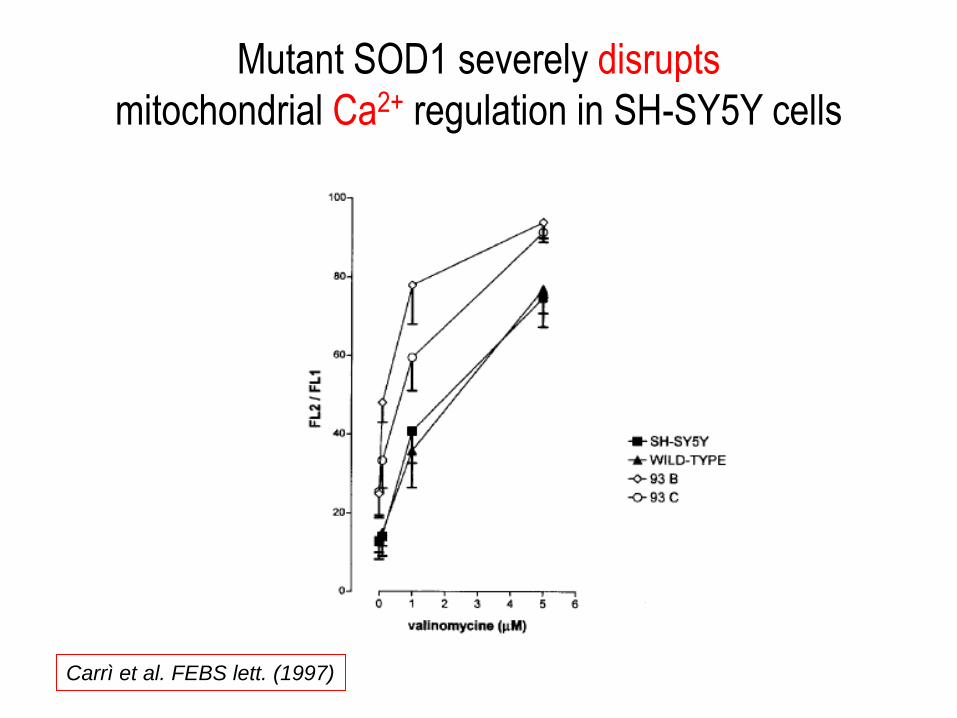
Mutant SOD1 severely disrupts
mitochondrial Ca2+ regulation in SH-SY5Y cells
Carrì et al. FEBS lett. (1997)
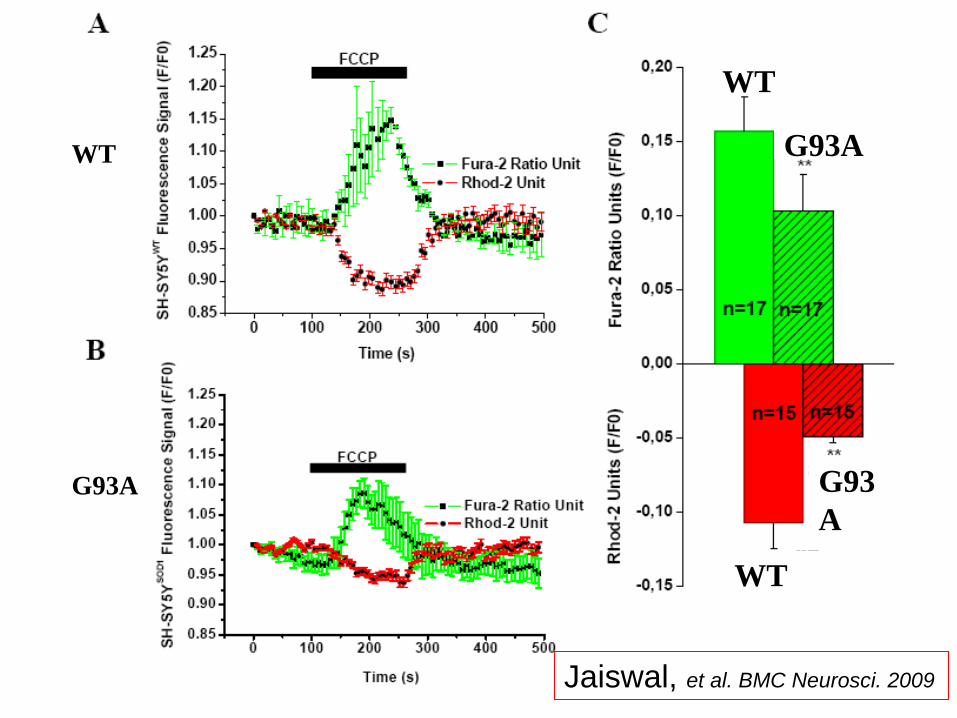
WT
G93A
G93A
G93
A
WT
WT
Jaiswal, et al. BMC Neurosci. 2009

ER stress occurs when the ER–mitochondria calcium cycle is
disturbed and misfolded proteins accumulate in the ER.
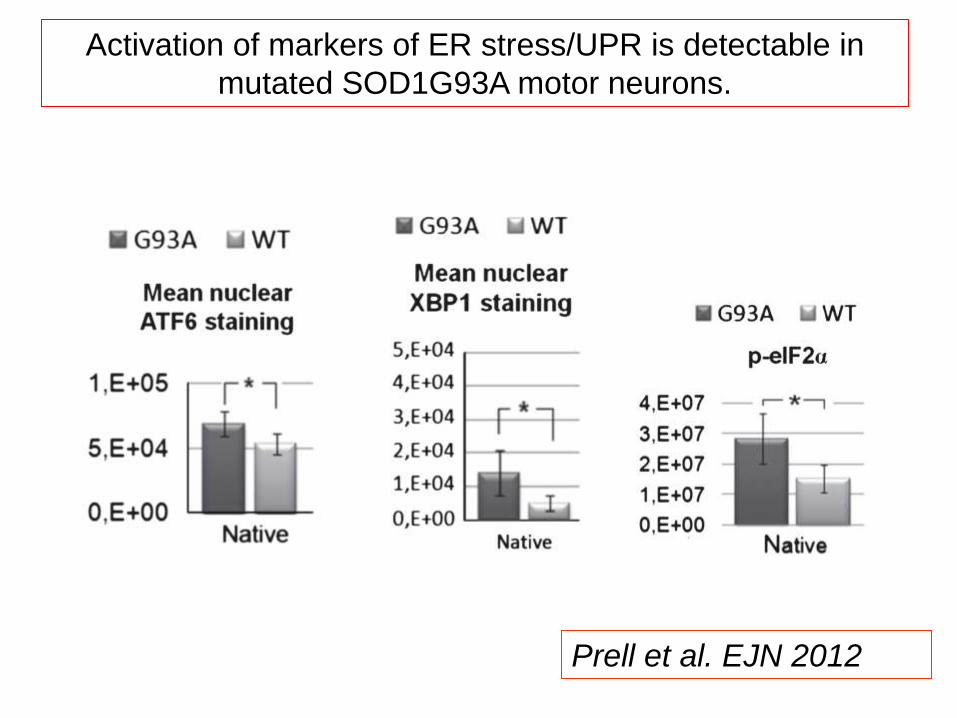
Prell et al. EJN 2012
Activation of markers of ER stress/UPR is detectable in
mutated SOD1G93A motor neurons.
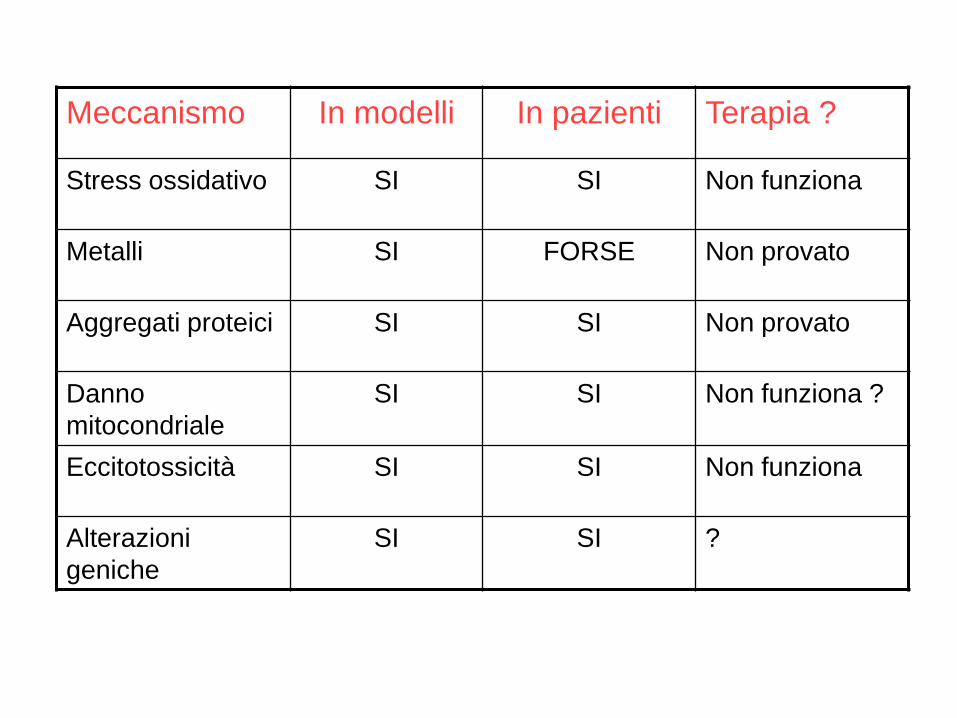
Meccanismo In modelli In pazienti Terapia ?
Stress ossidativo SI SI Non funziona
Metalli SI FORSE Non provato
Aggregati proteici SI SI Non provato
Danno
mitocondriale
SI SI Non funziona ?
Eccitotossicità SI SI Non funziona
Alterazioni
geniche
SI SI ?

Are experimental models “good”
representations of the situation in patients ?

La malattia del motoneurone (ma non solo)

IL FUTURO DELLA TERAPIA PER LA SLA
Terapia “combinata”
Target :
• Aggregazione proteica
• Danno mitocondriale
• Proteine mutanti
• Fattori trofici
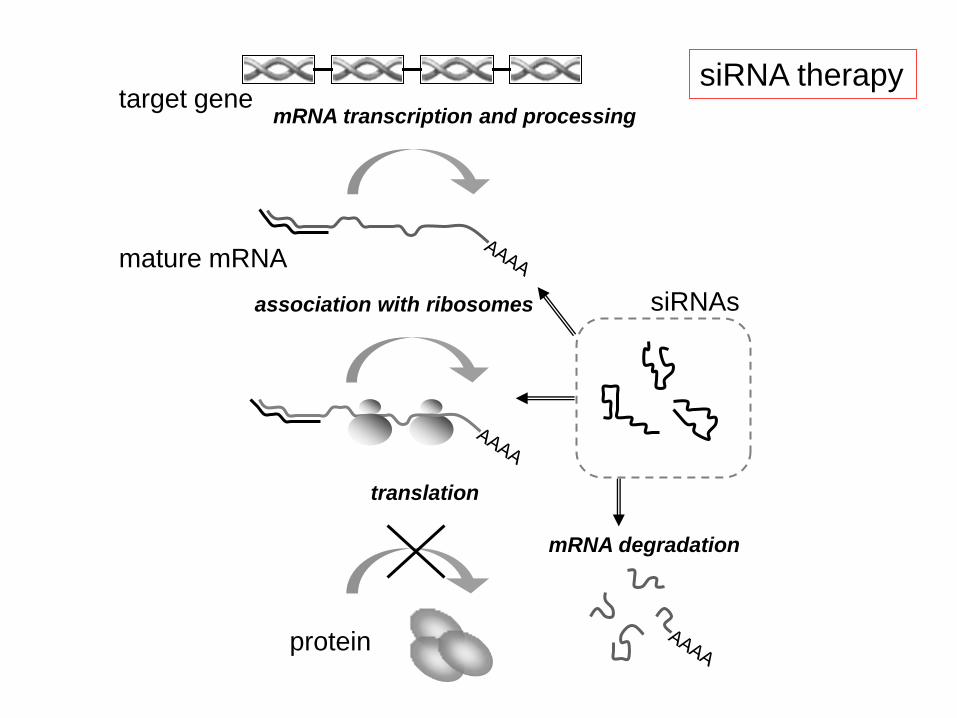
mRNA transcription and processing
mature mRNA
siRNAs
target gene
translation
protein
association with ribosomes
mRNA degradation
siRNA therapy
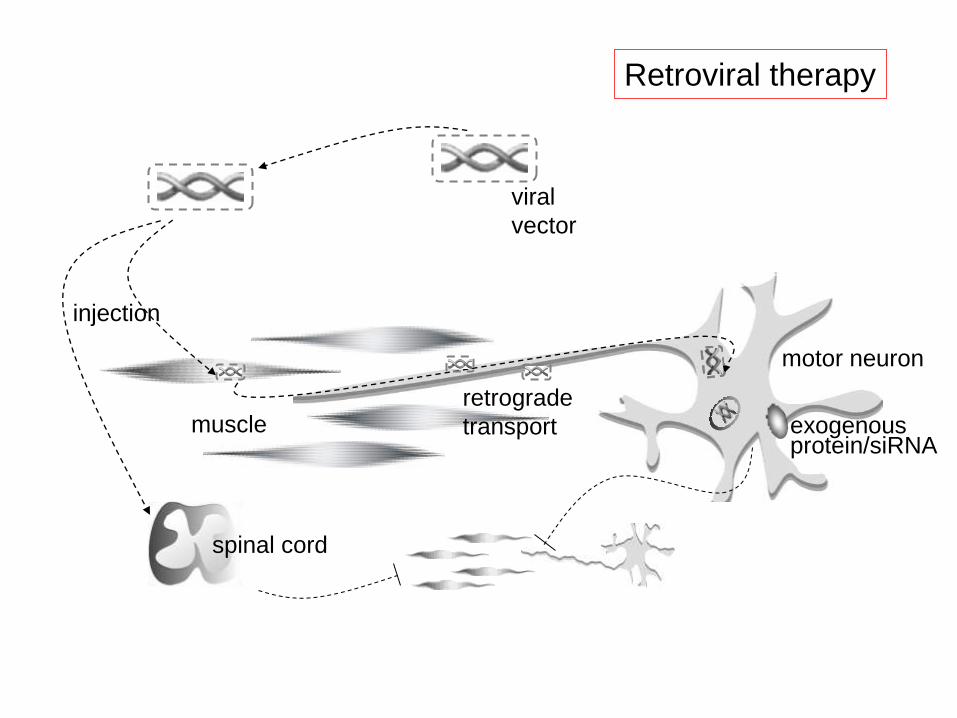
motor neuron
muscle
viral
vector
injection
retrograde
transport exogenous protein/siRNA
spinal cord
Retroviral therapy
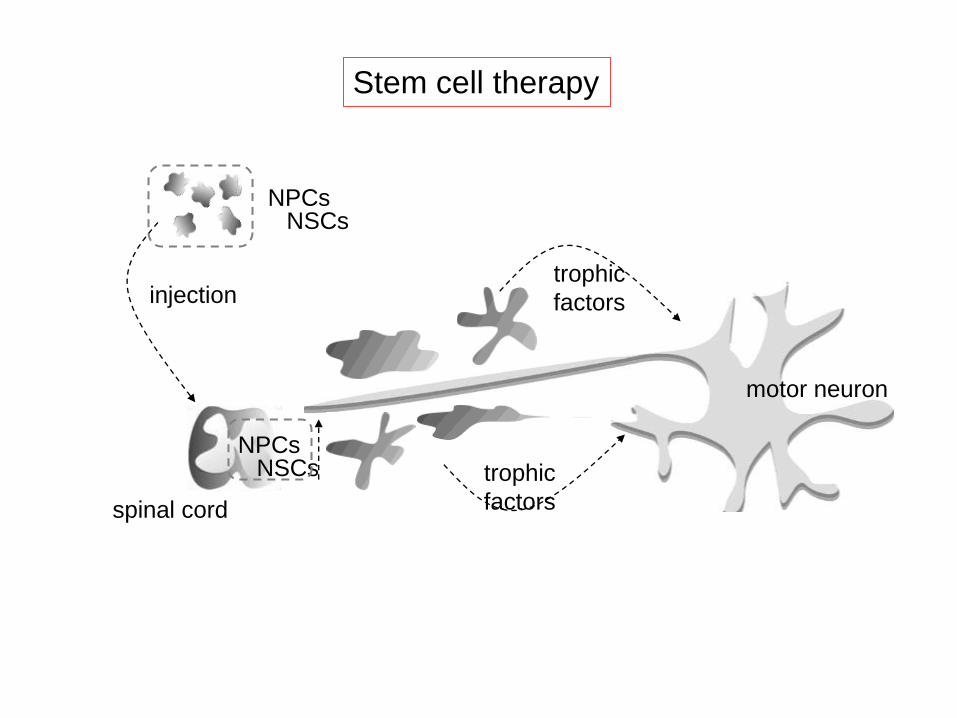
motor neuron
muscle
injection
spinal cord
NPCs NSCs
NPCs NSCs
trophic
factors
trophic
factors
Stem cell therapy

In vivo veritas
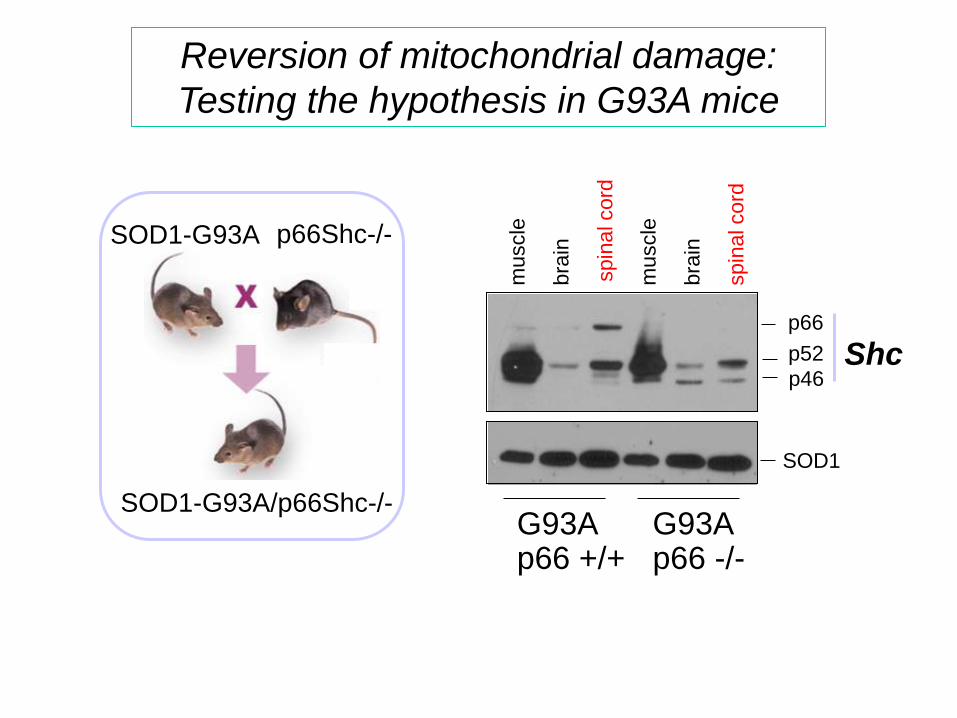
Reversion of mitochondrial damage:
Testing the hypothesis in G93A mice
SOD1-G93A p66Shc-/-
SOD1-G93A/p66Shc-/-
muscle
bra
in
spin
al cord
SOD1
Shc p66
p52
p46
muscle
bra
in
spin
al cord
G93A p66 +/+
G93A p66 -/-
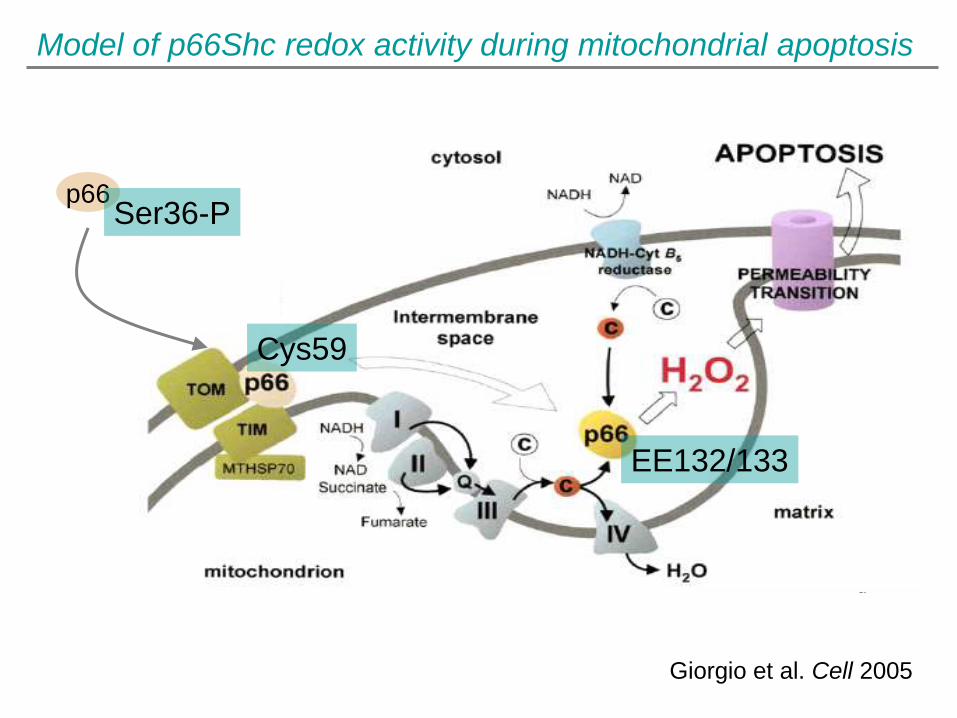
Model of p66Shc redox activity during mitochondrial apoptosis
Giorgio et al. Cell 2005
p66 Ser36-P
Cys59
EE132/133
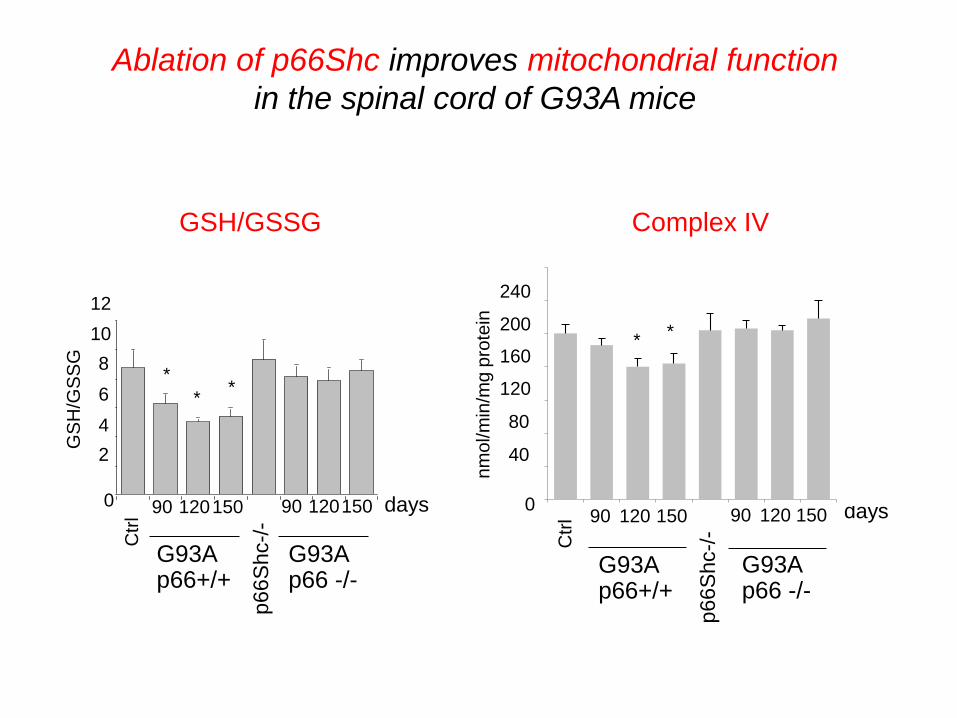
Ablation of p66Shc improves mitochondrial function
in the spinal cord of G93A mice
Complex IV GSH/GSSG
nm
ol/m
in/m
g p
rote
in
0
40
120
160
200
240
80
Ctr
l
p66S
hc-/
-
90 120 150
G93A p66+/+
90 120 150 days 0
20
40
60
80
100
120
140
1
* *
0
2
4
6
8
10
12
G93ACTRL
G93A 90gg
G93A 120gg
G93A 150 gg
shc-/-Ctrl
shc-/- G93A 90gg
shc-/- G93A 120gg
shc-/- G93A 150gg
GS
H/G
SS
G
Ctr
l
p6
6S
hc-/
-
90 120 150 90 120 150 days
* *
*
0
2
6
8
10
12
4
G93A p66 -/-
G93A p66+/+
G93A p66 -/-
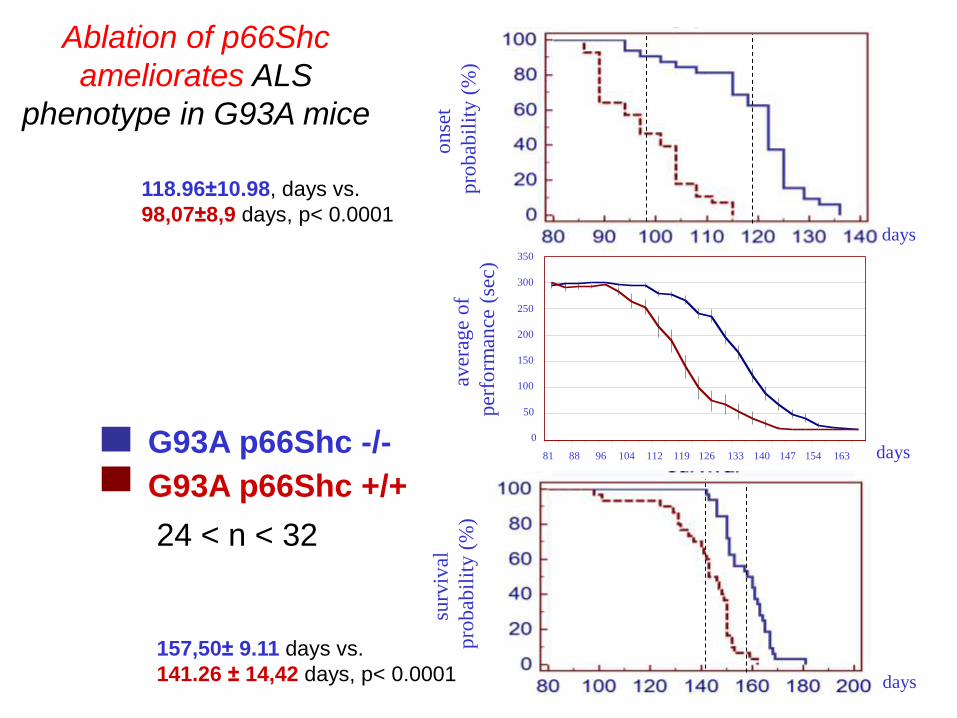
G93A p66Shc -/-
G93A p66Shc +/+
Ablation of p66Shc
ameliorates ALS
phenotype in G93A mice
24 < n < 32
118.96±10.98, days vs.
98,07±8,9 days, p< 0.0001
157,50± 9.11 days vs.
141.26 ± 14,42 days, p< 0.0001
0
50
100
150
200
250
300
350
81 88 96 104 112 119 126 133 140 147 154 163
0
50
100
150
250
200
300
350
81 126 88 104 96 112 119 154 133 140 147 163
days
days
days
aver
age
of
per
form
ance
(se
c)
surv
ival
pro
bab
ilit
y (
%)
on
set
pro
bab
ilit
y (
%)

A putative
mitochondrial
modulator,
dexpramipexol
e
increases the
efficiency of
oxidative
phosphorylation