saarc regional training - saarc agriculture centre diagnosis...saarc regional training on molecular...
TRANSCRIPT


i
SAARC Regional Training
On
Molecular Diagnosis and Laboratory Surveillance of PPR
21-26 July 2019
Editors
Mohammed A Samad
Md. Abu Yousuf
Md. Nure Alam Siddiky
Md. Giasuddin
Nathu Ram Sarker
Ashis Kumar Samanta
S.M. Bokhtiar
Bangladesh Livestock Research Institute
Savar, Dhaka-1341
SAARC Agriculture Centre Dhaka, Bangladesh

Molecular Diagnosis and Laboratory Surveillance of PPR
ii
The SAARC Regional Training on “Molecular Diagnosis and Laboratory Surveillance of
PPR” is held at Bangladesh Livestock Research Institute, Savar, Dhaka-1341 during 21-
26 July 2019. The training is sponsored by SAARC Agriculture Centre, Dhaka,
Bangladesh
Editors
Dr. Mohammed Abdus Samad, SSO, AHRD, BLRI
Dr. Md. Abu Yousuf, SO, AHRD, BLRI
Dr. Md. Nure Alam Siddiky, Consultant, CAMR-ZD in BD, BLRI
Dr. Md. Giasuddin, Head, AHRD, BLRI
Dr. Nathu Ram Sarker, Director General, BLRI
Dr. Ashis Kumar Samanta, SPS, SAC
Dr. S. M. Bokhtiar, Director, SAC
Recommended citation
Samad, M.A., Yousuf, M.A., Siddiky, M.N.A., Giasuddin., M., Sarker, N.R., Samanata,
A.K., & Bokhtiar, S.M. eds. (2019). Training Manual on Molecular Diagnosis and
Laboratory Surveillance of PPR. Bangladesh Livestock Research Institute, Savar, Dhaka-
1341, P. 84
Published by
Bangladesh Livestock Research Institute
Savar, Dhaka 1341, Bangladesh
Tel: +88-02-7791670-2, 7791676, Fax: +88-02-7791675
Email: [email protected]
www.blri.gov.bd
ISBN: 978-984-34-7035-5
All rights reserved
This work is subjected to copy rights. All rights reserved by the publishers, whether
whole or part of the material thereof. The authors are solely responsible for the content of
the abstract papers compiled in this publication. The publisher/editors shall not be
responsible for the views, opinion and materials expressed by the authors.
Printed by
Nathudhara Printing Press
277/3 Elephant Road (1st Floor), Kataban, Dhaka

Molecular Diagnosis and Laboratory Surveillance of PPR
iii
Secretary Ministry of Fisheries and Livestock
Govt. of Bangladesh
Message
It gives me an immense pleasure that Bangladesh Livestock Research Institute is
organizing a regional training on “Molecular Diagnosis and Laboratory Surveillance of
Peste des petits ruminants (PPR)” from 21st to 26
th July 2019 under the aegis of SAARC
Agriculture Centre at BLRI, Savar, Dhaka-1341.
The role of livestock in livelihood, nutritional and food security of millions of
people live in SAARC region is very significant. Formulation of effective disease control
strategies is a daunting task for sustainable development of livestock sector in the region.
PPR is one of the important economic and devastating disease for small ruminants
prevalent in most of the SAARC countries. PPR can easily be transmitted from one
SAARC countries to another due to trans-boundary nature of the disease as well as
porous borders. Regional concerted and coordinated effort is prerequisite to eradicate
PPR in compliance with the global initiatives. Bangladesh has also been developed
national strategic action plan for the eradication of PPR following the guideline of PPR
global eradication campaign. Govt. of Bangladesh has taken different initiatives to
address the PPR eradication in align with global strategy. I believe BLRI has a very good
laboratory capacity as well as skilled human resources to organize the training very
successfully.
I am sure that the participants from SAARC countries would be exposed to many
conventional and new approaches employed for the precise molecular diagnosis of PPR.
Further, this training would also provide a common platform and networking to the
researchers from fellow SAARC countries to discuss strategic plan for the progressive control
of PPR to eradicate by 2030.
I wish this training program a great success.
(Md. Raisul Alam Mondal)

Molecular Diagnosis and Laboratory Surveillance of PPR
iv
Director General
Bangladesh Livestock Research Institute
Message
Bangladesh Livestock Research Institute (BLRI) a pioneer institute has been
entrusted to conduct Research & Development in the field of animal health, production
and different cross cutting issues related to livestock development in the country. BLRI is
hosting Regional Leading Diagnostic Laboratory (RLDL-PPR) for PPR under the aegis
of South Asian Association for Regional Cooperation (SAARC).
Livestock plays an important role in rural livelihood of subsistence farmers
dependent on the sector. The livestock sector in this region has been facing challenges
for transboundary, emerging and re-emerging threats of diseases. PPR is one of the
common notifiable disease for small ruminant prevalent in the region. BLRI has
developed progressive control pathway of PPR eradication model in accordance with
the guideline of OIE. Capacity building in the field of molecular diagnosis,
bioinformatics and surveillance of PPR will be of immensely useful for the participants
to containment disease in the region. There is need for continuous exchange of
knowledge & ideas and disease outbreak information among SAARC countries for early
detection and emergency preparedness of emerging, re-emerging and transboundary
diseases of the region.
It is a matter of pride and responsibility of the Institute to host the SAARC
regional training on ‘Molecular Diagnosis and Laboratory Surveillance of Peste des
petits ruminants” is held from 21st to 26
th July 2019. I hope the deliberations in the
training will be mutually beneficial among the participants as well as to the host
organizations. This training will create an avenue for BLRI to deliver our experience and
expertise in the field of molecular diagnosis of PPR among the participants which will
pave a better way to control the disease within the SAARC region.
I wish the participants a pleasant stay and I wish the training a great success.
(Dr. Nathu Ram Sarker)

Molecular Diagnosis and Laboratory Surveillance of PPR
v
Director
SAARC Agriculture Centre
Message
I am pleased to know that a training manual is published for the SAARC regional
training programme on "Molecular Diagnosis and Laboratory Surveillance of PPR"
jointly organized by SAARC Agriculture Centre (SAC) and Bangladesh Livestock
Research Institute.
SAARC Agriculture Centre (SAC), under the aegis of South Asian Association for
Regional Cooperation (SAARC) has been working for the promotion of agricultural
research & development as well as technology transfer through regional networks among
agricultural research/extension institutions and policy makers in the SAARC region.
BLRI is one of the specialized premier institute undertaking research and developmental
activities on animal health and production for the promotion of livestock sector. Livestock
is one of the important sector for food (milk, meat, egg) production, livelihood
improvement, employment generation and women empowerment. The livestock sector in
the region has been facing numerous challenges such as disease burden, scarcity of feeds
and fodder, poor quality genetic resources and so on. Peste des petits ruminants (PPR) is
endemic in all over the SAARC region with huge economic implications. SAARC has
developed a regional road map for the eradication of PPR by 2025. The regional training
on "Molecular Diagnosis and Laboratory Surveillance of PPR" would provide theoretical
as well as hands-on knowledge to the participants and exposure on different techniques and
technologies for molecular diagnosis and characterization of PPR. I believe the contents of
the manual is certainly the store of information related to advanced research and
development of PPR particularly molecular diagnosis. This manual is unique and store of
knowledge for anyone who is interested in pursuing research on PPR prevention and
control.
I wish all the grand success for this regional training programme and its endeavors.
(Dr. S. M. Bokhtiar)

Molecular Diagnosis and Laboratory Surveillance of PPR
vi
FOREWORD
Bangladesh Livestock Research Institute is organizing a training program on “Molecular
Diagnosis and Laboratory Surveillance of PPR” from 21st to 26
th July 2019, in
collaboration with SAARC Agriculture center, Dhaka, Bangladesh. The participants for the
training program are from Bangladesh, India, Nepal and Sri Lanka. The objective of the
training program is to impart the training to member nations in the domain of molecular
diagnosis and surveillance for PPR which is an important area need to be addressed to foster
the PPR global control strategy. The theme of the training program is appropriately chosen
by the SAARC Agriculture Center considering the urgent need to build capacity in order
to formulate PPR control and prevention strategies.
PPR is also known as goat plague which is increasing importance in Africa and Asia
wherever small ruminants form an important component of agricultural food production.
It threatens the food security and sustainable livelihood of farmers across the region. The
world organization for animal health (OIE) has identified PPR as a noticeable and
economically important trans-boundary viral disease of sheep and goats. The capacity
building in terms of laboratory and skilled human resources are still in meager and scanty
for PPR diagnosis and surveillance. The training encompasses theory followed by hands
on exposure on different novel techniques and technologies on diagnosis and sero-
surveillance. The resource speakers have been chosen very appropriately with their long
experience and devotion in the field of PPR. I hope the course contents of the training
would be immensely useful to the participants from SAARC countries with constructive
exchange of knowledge and experience.
I am sure that the present SAARC Agriculture Centre sponsored training program on
Molecular Diagnosis and Laboratory Surveillance of PPR would be quite useful for the
participants from the SAARC countries and this document will serve them as a reference
for carrying out various analytical procedures in their laboratory for molecular diagnosis
and surveillance. I would like to extent my sincere thanks to the SAARC Agriculture
Centre, Bangladesh for giving us the opportunity to organize hands on training on such
an important aspect. I would also like to thank Director General, Bangladesh Livestock
Research Institute for his support and guidance to conduct this training program
effectively.
(Mohammed A Samad, PhD)
Director
SAARC RLDL for PPR

Molecular Diagnosis and Laboratory Surveillance of PPR
vii
Contents
Message iii
Message iv
Message v
Foreword vi
PPR Eradication Strategy of Bangladesh 1
Making History: Eradicating Peste des petits ruminants –Saving Lives 7
and Livelihoods
Regional Roadmap on Progressive Control of Peste des petits ruminants 16
(PC PPR) for South Asian countries
BLRI Developed PPR Control Model 23
An Overview of General Guidelines for PPR Sample Collection, Preservation 26
and Genomic Analysis
Principles of PCR and Guidelines for Good Laboratory Practices 37
ELISA: An Essential Tool for Surveillance of PPR 42
National Animal Disease Referral Expert System (NADRES) 52
Laboratory Management with Biosafety and Biosecurity Practices 58
RT-PCR: The Technique for the Detection of PPR 60
Detection of PPRV Antibody in Sera by Competitive Elisa (cELISA) 67
Cell Culture for Virus Isolation and Identification 72
List of the Participants 81
List of the Resource Persons 83

Molecular Diagnosis and Laboratory Surveillance of PPR
1
Figure 1. District-wise goat population
distribution for the year 2017-18
PPR ERADICATION STRATEGY OF BANGLADESH
Mohammed A Samad* and Abu Sufian1
Director, SAARC Regional Leading Diagnostic Laboratory for PPR, Bangladesh Livestock
Research Institute, Savar, Dhaka-1341 1Department of Livestock Services, Khamarbar, Farmgate, Dhaka-1215
Email: [email protected]
Livestock is an integral component of the complex farming system in Bangladesh as it
not only a source of meat protein but also a major source of farm power services as well
as employment. The livestock sub-sector provides full time employment for 20% of the
total population and part-time employment for another 50%. The GDP contribution of
this sub-sector has been a modest 1.54% annually in the 2017-18 fiscal year (DLS. 2018)
with the growth rate of livestock 3.40%. However, the sector’s actual contribution has
been consistently underestimated as the value added in draught power used in farm
operation, threshing, sugarcane and oilseed crushing, local transport, dung for cooking
fuel and manure for fertilization of crop fields were not taken into account. An estimate
of the uncounted sectoral contribution of livestock indicates a foregone value of three
times the amount of official GDP attributed to this sector (FAO 1990). Moreover,
livestock products, namely, leather and leather products, hides and skins are important
exportable items. Consequently, given versatile nature of the potential contribution
offered by the livestock sector including curbing of malnutrition prevalent in Bangladesh
also.
Bangladesh is blessed with small ruminant
(SR) population of 29.57 million (goats:
26.10 million and sheep: 3.47 millions). SR
usually kept by landless farmers in
marginal areas and are ranked second to
poultry in the livestock species ladder while
prioritizing the species kept by poor. Out of
the SR, goat represents 47.34% of the total
livestock populations in Bangladesh (DLS,
2017) and are normally reared by the rural
women community in the country. Thus
goat rearing plays an important role in
women empowerment. Nowadays
commercial goat farming
has become popular using intensive
farming system. Commonly available
breeds are Black Bengal goat, exotic breeds
such as the Jamunapari, Sirohi, Beetal and
crossbreds. Most recently, a few exotic

Molecular Diagnosis and Laboratory Surveillance of PPR
2
Figure 2. Spatial distribution of PPR
cases for the year 2017-18
breeds like Boer goat is being adapted as meat type goat in the country. However, Black
Bengal goat is the dominant (about 90%) breed in the country.
Sheep in Bangladesh, stands as third in position after cattle and goat population, and are
used primarily for meat production. Bangladesh possesses 3.40 million sheep at present
(DLS, 2017). Small and landless farmers rear about 38%, medium farmers 40% and large
farmers 22% of total sheep in Bangladesh. They are sparsely distributed throughout the
country but relatively higher concentration (about 32% of total sheep population) are
found in three different ecological zones like, Barind, Jamuna river basin and Coastal
areas, where farmers maintain larger commercial (meat) flocks. Sheep available in
Bangladesh are mostly indigenous non-descript type. Nonetheless, Garoles is one of the
native Bangladeshi sheep which are found in the extreme south-west of Bangladesh
adjacent to the Sundarbans forest in the coastal area and western districts of Bangladesh.
The coat is usually light brown in colour, with some animals having black spots on the
legs and the head region. Garole has an earlier puberty age, produces twin kids and has a
high resistance to internal parasites. They can survive better in saline water than other
sheep populations and the Black Bengal goat.
Peste des petits ruminants (PPR) is
considered as one of the major threats to
SR population in Bangladesh. The first
outbreak of PPR in Bangladesh occurred in
1993 in a bordering district, Meherpur,
south western part of the country. Since
then disease is continuing to occur and has
become endemic. The disease has been
described as the most important single
cause of morbidity and mortality in small
ruminant’s population in Bangladesh. As
per Upazila veterinary hospital based
secondary surveillance program 89,093
cases were identified, of which in Rajshahi
division ranked top position (69,899)
followed by Chattagram and Rangpur
(37,815) and Chattagram division (34,059)
shown in Figure 2. There is no reliable and
published data on the economic losses
incurred due to PPR in Bangladesh. A study
depicted, in 2010, there were 84,000
clinical cases reported, causing an
estimated loss of 1,842 million Taka (US$ 24.6 million). Since secondary surveillance
will not cover entire area of a sub-district (Upazila), for this reason, the actual number of
infected cases may not represent in the data as the cases of remote rural areas are not
included.

Molecular Diagnosis and Laboratory Surveillance of PPR
3
PPR is an acute, highly contagious, world organization for animal health (OIE) notifiable
and economically important transboundary viral disease of sheep and goats associated
with high morbidity and mortality and caused by PPR virus. The disease is capable of
destroying whole of the susceptible host population by infuriating epidemics and
panzootics, thus damaging economy, undermining food security and livelihood of poor
people of the society. PPR virus (PPRV) belongs to the family Paramyxoviridae, genus
Morbillivirus. The PPRV was first identified in Côte d’Ivoire in 1942 and for many
yea’rs it was considered as an African disease localized mainly in Western and Central
Africa. However, over the passage of time it became endemic across the Africa, Arabian
Peninsula, Middle East, Turkey, Pakistan, India, Bangladesh, Nepal, Tajikistan and
Kazakhstan in Central Asia, Mongolia and China. The PPR virus has been classified into
four distinct genotypes/ lineages (I, II, III, and IV). All four lineages are prevalent in
Africa while across Asia only lineage IV has been reported.
Seventeen Sustainable Development Goals (SDGs) of United Nations (UN) integrate the
three dimensions of sustainable development economic, social and environmental and are
targeted 2030 towards fulfilling the such goals, of which nine (9) goals are directly or
indirectly relate to livestock sectors. Goat rearing in Bangladesh has envisaged the SDGs
targets are- no poverty, zero hunger, gender equality, reduced overall inequality. But at
present small goat farmers in Bangladesh are facing massive economic losses due to PPR
as the disease still endemic in Bangladesh. It has been estimated that losses due to PPR
worth 24.6 million US$ annually that has a direct impact to fulfillment of Sustainable
Development Goals (SDGs) of United Nations. Thus, considering the fact, it is very
pragmatic to take immediate intervention to control and finally eradicate the disease by
2030. Since the disease is transboundary in nature, so regional approach to be optimized
to control the disease from country and the region as well.
Based on the country self-assessment using the PPR Monitoring and Assessment Tool
(PMAT), Bangladesh holds the position in Stage 2 of PPR, Global Control and
Eradication Strategy (GCES) and will continue up until 2020 towards reaching the Stage
3 in 2021, finally getting free status by 2025.
A national strategic plan (NSP) for the control and prevention of PPR and its subsequent
eradication by 2025 in Bangladesh has been framed in the light of Global Strategy for the
Control and Eradication of PPR. The methodology proposed in the NSP targets at
institutionalizing the efforts for the progressive control leading to the eradication of PPR
in Bangladesh. These activities will be carried out mainly through a donor funded
program, however, government is implementing the activities for PPR control and
prevention through own resources.
The control strategy consists of following main components:
PPR strategy and technical plans
Legal framework
Stakeholder awareness and engagement
Strengthening veterinary services

Molecular Diagnosis and Laboratory Surveillance of PPR
4
Support to the diagnostic and surveillance
Systems strengthening of laboratory and surveillance capacities
Epidemiology and laboratory networks
Measures toward PPR eradication
Demonstration of PPR freedom
Control of other small ruminant diseases (SRD) in support of PPR Eradication
Coordination, Management and Partnerships
Overall objective of NSP
To control and eradicate PPR from Bangladesh for poverty reduction, livelihoods, food
and nutrition security, resilience, market access and economic development.
Specific objectives of SP
a) To ensure an enabling environment for PPR eradication by improving policies,
awareness, legal framework and veterinary services.
b) To develop a robust diagnostic and surveillance system by improving capacity for
epidemiological assessment, surveillance, diagnosis and networking.
c) To ensure accessibility and availability of quality PPR vaccine and implement
vaccination program for achieving desired level of herd immunity.
d) To ensure in country and transboundary movement control to prevent the spread of
PPR virus and other SRDs.
e) To develop an effective coordination mechanism at national, regional and global
level by developing partnership and networking.
Expected outputs
Output -1: Enabling environment created for PPR Eradication by developing and
implementing National PPR strategy, guidelines, SOPs, awareness raising and improved
veterinary service delivery.
Output -2: A robust diagnostic and surveillance system developed by improving
capacity for epidemiological assessment, surveillance, diagnosis and networking.
Output -3: Accessibility and availability of quality PPR vaccine achieved and
vaccination programme undertaken for achieving desired level of herd immunity.
Output -4: In country and transboundary movement control of animal and animal
products to prevent the spread of PPR virus and other SRDs ensured.
Output - 5: An effective coordination mechanism at national, regional and global level
developed by ensuring partnership and networking.

Molecular Diagnosis and Laboratory Surveillance of PPR
5
Time bound logical framework
NSP description Verifiable indicators
of achievement
Time
Frame
Goal Improvement the socioeconomic conditions of small
ruminant’s producers by ensuring livelihood, poverty
reduction and achieving food security through progressive
control towards eradication of PPR in goat and sheep in
Bangladesh as compliance with SDGs target and obtain PPR free status from OIE
Significant reduction
of PPR incidence with
a positive
socioeconomic impact in the country.
2019-2015
Purpose Bangladesh will move towards eradication stage (Stage #3)
of GCES.
Report and document
have prepared and
submitted.
Output -1 Enabling environment created for PPR Eradication by
developing and implementing National PPR strategy,
guidelines, SOPs, awareness raising and improved veterinary service delivery.
SOPs and Guidelines
developed and
improved veterinary services developed.
2019-2020
Activities Develop and implement national strategic plan for the
control of PPR.
Develop a PPR vaccination and surveillance plan for
PPR and SRDs.
Prepare communication guidelines and materials for
raising awareness on PPR
Develop and Update standard operating
procedures/protocol for the control and eradication of
PPR.
Training of field vets, paraprofessionals and laboratory
staffs on surveillance, epidemiology and diagnosis of
PPR including TADs.
NSP developed
Communication
guidelines developed
Vaccination and
surveillance
strategy/plan developed
Relevant
manpower trained
Output -2 A robust diagnostic and surveillance system developed by
improving capacity for epidemiological assessment,
surveillance, diagnosis and networking.
A real-time
surveillance and robust
diagnostic system developed.
2020-2024
Activities Assess the epidemiological situation relating to PPR
disease distribution, risk factors for endemicity along the value chain and risk analysis.
Support to dedicated Community Animal Health
Workers for PPR Vaccination and primary health care.
Active surveillance, outbreak investigation and report
accordingly to be installed.
Sero-monitoring in immunized herd will be established.
Listing of wild life species along with surveillance of
susceptible species
Establish an Early Warning System (EWS) for TADs
on real time basis.
Strengthen PPR diagnosis capacities of regional
laboratories (FDILs), Central Disease Investigation Lab
(CDIL) and Regional Leading Diagnostic Laboratory.
PPR disease
epidemiology understood
Real-time
surveillance installed
Supported to all
laboratories for
PPR disease
diagnosis
Strengthened the
capacity of district
and upazila level laboratories

Molecular Diagnosis and Laboratory Surveillance of PPR
6
NSP description Verifiable indicators
of achievement
Time
Frame
for PPR (RLDL)
Capacity building of Upazila/ District Veterinary
Hospitals for collection, storing and Shipment of PPR samples.
Output 3 Accessibility and availability of quality PPR vaccine is
achieved and vaccination programme undertaken for achieving desired level of herd immunity.
Vaccine induced
desired level of herd immunity.
2020-2021
Activities Support to current vaccine production in terms of better
vaccine production technology and including quality
control in public and private sector.
Support to thermostable PPR vaccine production for
field use.
Strengthening of LRI in setting up regular testing of all batches of vaccines produced through standard SOPs.
Quality vaccine
production enhanced
and Quality control of
produced vaccine established.
Output 4 In country and transboundary movement control of
susceptible animal and animal products to prevent the spread of PPR virus and other SRDs is ensured.
TADs have prevented
though controlling of
animal movement by
border inspection.
2023-2025
Activity Support to animal quarantine station monitoring animal movement/transportation.
Border guard training on PPR transmission and control.
Animal Quarantine
station established and
Border guard
sensitized on PPR disease.
Output 5 An effective coordination mechanism at national, regional
and global level is developed by ensuring partnership
and networking.
A functional
Coordination
mechanism established
2019-2025
Establish a PPR Eradication Management Unit
(PEPMU) at DLS with accommodating necessary staffs.
A Technical Committee headed by DG DLS for the
periodical oversight of the programme and monitor the progress and keep the programme on right track.
A high level multi-sectoral Steering committee headed
by Secretary, Ministry of Fisheries and Livestock will
be formed drawing members from relevant public
sector agencies, academicians and stakeholders from the private sector and farming community
A functional Central
Coordination mechanism established
Monitoring and evaluation
The government, industry representatives, civil society and non-government
organizations will jointly evaluate the implementation of this strategy work plan every
year. They will jointly conduct annual evaluation of implementation of the strategic
action plan and identify an achieved level of each criteria. The evaluation results will be
presented publicly after approval from the government. For this activity, the PPR
Monitoring and Assessment Tool (PMAT) will also be used.

Molecular Diagnosis and Laboratory Surveillance of PPR
7
MAKING HISTORY: ERADICATING PESTE DES
PETITS RUMINANTS – SAVING LIVES AND
LIVELIHOODS
Bouna Diop Secretary, FAO/OIE PPR Global Secretariat, Viale delle Terme di Caracalla, 00153
Rome, Italy
Email: [email protected]
Introduction
A global strategy to control and eradicate PPR was agreed at an international conference
hosted by FAO and OIE in April 2015 in Abidjan, Côte d’Ivoire. Drawing from their
experience in eradicating rinderpest, FAO and OIE have formed a joint global secretariat
to guide efforts to eradicate PPR worldwide by 2030; this timeframe coincides with the
2030 Agenda for Sustainable Development. This note provides an overview of the state
of play in implementing the global strategy and the challenges encountered.
Importance of small ruminants
Small ruminants – totaling 2.2 billion heads worldwide according to FAOSTAT - are the
primary livestock resource of 300 million poor rural families around the globe, including
subsistence farmers and landless villagers as well as pastoralists. For these households,
sheep and goats are a source of food and regular income, a means to capitalize savings,
and a safety net during times of hardship. Selling animals or their products provides the
necessary resources to access food, as well as educational and social services
Food products derived from sheep and goats are an essential part of the diet for many
people around the world and contribute to overcoming malnutrition. Sheep and goat milk
and meat are of high nutritional value and provide high-quality protein, vitamins and
minerals critical for cognitive development and physical strength, particularly for
children. Small ruminants are well adapted to arid and semi-arid environments, and are
kept in a variety of production systems throughout the world. These include pastoral
areas, where goats and sheep make a mixed flock. Households may totally depend on the
animals for survival, as crop production is almost absent in such arid or desert areas.
Small ruminants are mobile assets; pastoralists move with them in search for water and
new pasture, or in times of climatic stress and volatile security situations. In such pastoral
systems, meat and milk are key for food security and nutrition. Income from sales of live
animals and their products account for between 60 and 80 percent of total household
income. This money is essential for purchasing cereals and other household items,
covering social and financial obligations, paying for school, or dealing with doctors’ fees.
In most pastoralists’ cultures, women are in control of small ruminant operations and the
associated income flow. This favors gender balance and contributes to an equitable
allocation of earnings and animal-source foods within the household. Pastoralism is

Molecular Diagnosis and Laboratory Surveillance of PPR
8
dominant in some large regions in Africa (Sahel region, Afar in Ethiopia, Turkana in
Kenya, Somali region), the Middle East and Central and East Asia. In particular, in the
dry zone in the Sahel region, it is the only way of life.
Peste des petits ruminants
Peste des petits ruminants (PPR) is a highly contagious viral disease of domestic and wild
small ruminants first reported in 1942 in Côte d’Ivoire. The disease is caused by a
morbillivirus, Peste des petits ruminants virus (PPRV) belonging to the genus
Morbillivirus in the family Paramyxoviridae. PPR was first reported in Cote d’Ivoire,
West Africa. For some time, the disease was reported only from West Africa before
expanding through other regions in Africa (East, Central), the Middle and Near East, and
Asian countries extending from West Asia to China. PPR primarily affects sheep and
goats, although cattle, camels, buffaloes and some wild ruminant species can also be
infected, indicating spillover from domestic sheep and goats. Morbidity and mortality
rates in small ruminants vary, but can be as high as 100% and 90%, respectively in
previously unexposed flocks. PPRV also acts as a predisposing factor for secondary
bacterial infections which can contribute to high morbidity and mortality.
A PPR outbreak is an emergency due to its rapid spread and high animal mortality rate.
Fatal diseases of small ruminants, such as PPR, affect the already vulnerable livelihoods
and can decimate the savings of poor populations, in particular in pastoral areas. People
become desperate when they lose their assets. PPR outbreaks, and the desperation due to
the loss, can therefore trigger turmoil, migration, and volatile security situations.
Eradicating PPR will increase sustainability, alleviate poverty, improve the resilience of
poor pastoralists and their communities, enable them to better cope with other shocks and
threats, prevent forced migration and mitigate extremist trends.
Following the world-wide eradication of rinderpest in 2011, a global consensus was
reached on the need to eradicate PPR. The disease can be readily and cost-effectively
diagnosed and a reliable, inexpensive and high quality vaccine is available that confers
lifelong immunity to vaccinated animals after a single dose. The virus also has a
relatively short infectious phase and does not survive for long outside a host, making it an
ideal candidate for a concerted eradication effort. Controlling and eventually eradicating
PPR means fighting rural poverty, ensuring food security and nutrition, and strengthening
resilience and national economies. It will contribute significantly to achieving the
Sustainable Development Goals (SDGs), particularly SDG 1 (no poverty), SDG 2 (zero
hunger), but also SDGs 5 (gender equality) and 8 (decent work and economic growth).
PPR Global Control and Eradication Strategy
The PPR Global Control and Eradication Strategy (GCES) developed by FAO and OIE
was endorsed during an international conference on PPR held in Abidjan, Côte d’Ivoire,
in April 2015, with the vision of a PPR-free world by 2030. The PPR GCES promotes a
Molecular Diagnosis and Laboratory Surveillance of PPR
9
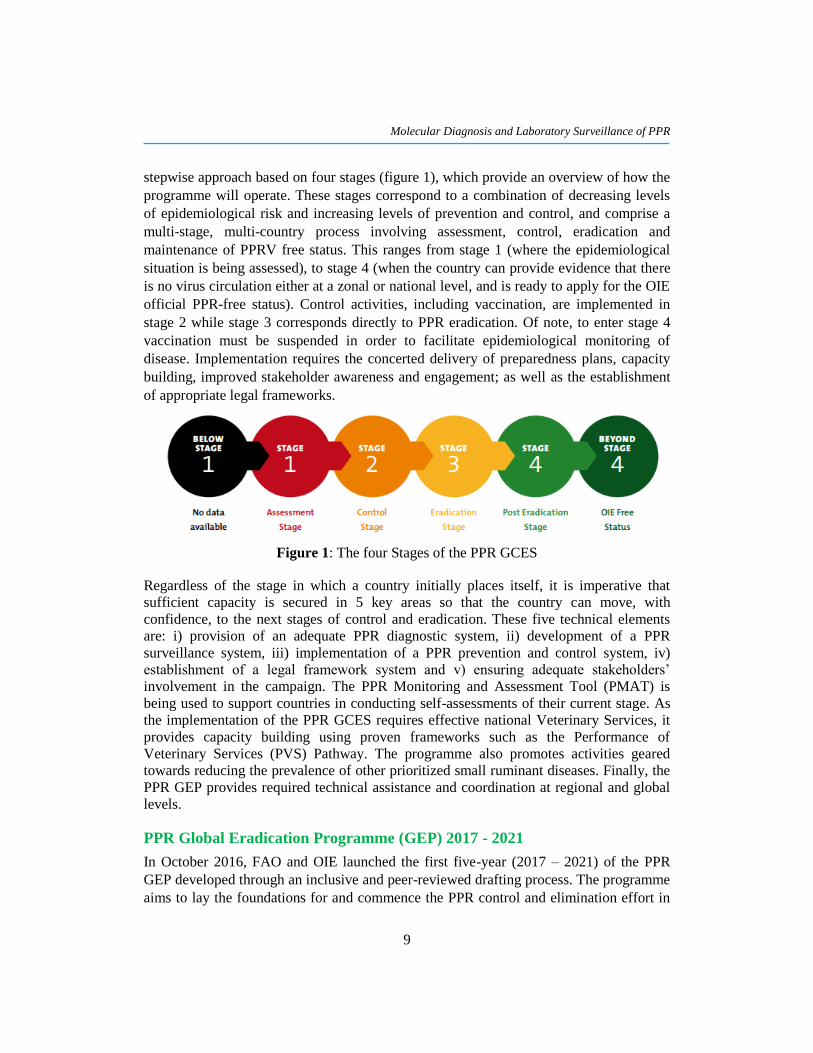
stepwise approach based on four stages (figure 1), which provide an overview of how the
programme will operate. These stages correspond to a combination of decreasing levels
of epidemiological risk and increasing levels of prevention and control, and comprise a
multi-stage, multi-country process involving assessment, control, eradication and
maintenance of PPRV free status. This ranges from stage 1 (where the epidemiological
situation is being assessed), to stage 4 (when the country can provide evidence that there
is no virus circulation either at a zonal or national level, and is ready to apply for the OIE
official PPR-free status). Control activities, including vaccination, are implemented in
stage 2 while stage 3 corresponds directly to PPR eradication. Of note, to enter stage 4
vaccination must be suspended in order to facilitate epidemiological monitoring of
disease. Implementation requires the concerted delivery of preparedness plans, capacity
building, improved stakeholder awareness and engagement; as well as the establishment
of appropriate legal frameworks.
Figure 1: The four Stages of the PPR GCES
Regardless of the stage in which a country initially places itself, it is imperative that
sufficient capacity is secured in 5 key areas so that the country can move, with
confidence, to the next stages of control and eradication. These five technical elements
are: i) provision of an adequate PPR diagnostic system, ii) development of a PPR
surveillance system, iii) implementation of a PPR prevention and control system, iv)
establishment of a legal framework system and v) ensuring adequate stakeholders’
involvement in the campaign. The PPR Monitoring and Assessment Tool (PMAT) is
being used to support countries in conducting self-assessments of their current stage. As
the implementation of the PPR GCES requires effective national Veterinary Services, it
provides capacity building using proven frameworks such as the Performance of
Veterinary Services (PVS) Pathway. The programme also promotes activities geared
towards reducing the prevalence of other prioritized small ruminant diseases. Finally, the
PPR GEP provides required technical assistance and coordination at regional and global
levels.
PPR Global Eradication Programme (GEP) 2017 - 2021
In October 2016, FAO and OIE launched the first five-year (2017 – 2021) of the PPR
GEP developed through an inclusive and peer-reviewed drafting process. The programme
aims to lay the foundations for and commence the PPR control and elimination effort in

Molecular Diagnosis and Laboratory Surveillance of PPR
10
infected countries by developing capacity; understanding the epidemiological situation
and defining appropriate implementation strategies to reduce the prevalence of PPR and
eventually eradicate the disease. For non-infected countries, the programme will assist in
developing capacity to demonstrate the absence of PPR virus and move towards OIE
official PPR free status recognition. The programme will also support countries to reduce
the prevalence of other prioritized small ruminant diseases and strengthen veterinary
systems. But the programme goes beyond disease eradication alone– it also aims to
improve national production models and help herders build the strongest, most resilient
livelihoods with their animal resources.
Overall coordination of the PPR GEP
To drive the PPR eradication effort on a global scale and effectively support countries in
fighting the disease, and building on the efforts of the FAO-OIE Global Framework of
the Progressive Control of Transboundary Animal Diseases (GF-TADs), FAO and OIE
established a Joint PPR Secretariat in March 2016 in FAO Headquarters. The Secretariat
is responsible for the overall coordination of the PPR GEP. The Secretariat is supported
by an Advisory Committee (AC) which provides strategic guidance and oversight on the
execution of the programme while also playing an important advocacy role with policy
makers, donors, national veterinary services and livestock owners. The Global Research
and Expertise Network (PPR GREN) was launched in April 2018 during a meeting
hosted by the Joint FAO/IAEA Division of Nuclear Techniques in Food and Agriculture
in Vienna. PPR GREN is established as a forum for scientific and technical consultations
to foster a science-based and innovative debate on PPR.
What the PPR GEP is doing?
PPR infected-countries are found in nine regions throughout Africa, Asia, the Middle
East and Europe (Map 1). PPR Regional Roadmap Meetings have been organized in all
these regions. The first round of meeting provided the opportunity to present the PPR
GCES and its tools; carry out a first self-assessment of each country’s situation regarding
PPR and the capacity of its Veterinary Services to control the disease; and develop the
regional Roadmap for the region and obtain countries engagement for its implementation.
The meetings also served to identify other small ruminant diseases that could be
controlled together with PPR and set up the Regional Advisory Group (RAG) to oversee
the implementation of PPR control activities in the region. The regional roadmap
meetings are important to ensure continuous assessment and monitoring of the disease
situation, to discuss challenges faced on PPR GEP implementation and progress made
and to promote regional approaches because of the transboundary nature of the disease.
FAO and OIE have also developed partnerships with regional organizations, the African
Union – Inter African Bureau for Animal Resources (AU-IBAR), the African Union Pan
African Veterinary Vaccine Centre (AU-PANVAC) and Regional Economic

Molecular Diagnosis and Laboratory Surveillance of PPR
11
Communities such as AMU1, AOAD
2, ASEAN
3, ECCAS
4, ECO
5, ECOWAS
6, GCC
7,
IGAD8, SAARC
9, SADC
10 as well as with relevant civil society organizations.
FAO, OIE and partners are providing support to countries and regions to formulate their
PPR National and Regional Strategic Plans, which detail the steps for assessing,
controlling, and eradicating PPRV, and maintaining PPRV freedom, as well as the
financial resources required and committed by national and regional authorities to
implement the Plans. Eight out of the nine regions have formulated their respective
regional strategies, which now need to be endorsed by their constituencies. In addition,
68 infected countries have formulated their National Strategic Plans (NSP) in alignment
with the regional and global strategy. The formal endorsement of NSP by national
authorities and the integration of PPR into existing agriculture sector programmes and
activities are essential to make more national budgets available for the PPR eradication
programme.
Meetings of PPR vaccine manufacturers are held every two years since 2014. The 3rd
meeting was organized by FAO and OIE in Amman, Jordan in April 2019 in
collaboration with the Veterinary Services of Jordan and JOVAC. On December 2017,
the PPR Secretariat organized a workshop on thermotolerant PPR vaccines with the
funding support of GALVmed. The workshop reviewed the current research on
thermotolerant PPR vaccines, discussed the parameters for defining thermotolerance
Standard Operating Procedures (SOPs) developed by AU-PANVAC and explored
modalities for the development/production of thermotolerant PPR vaccines.
1 Arab Maghreb Union 2 Arab Organization for Agriculture Development 3 Association of Southeast Asian Nations 4 Economic Community of Central African States 5 Economic Cooperation Organization 6 Economic Community of West African States 7 Gulf Cooperation Council 8 Intergovernmental Authority for Development 9 South Asian Association for Regional Cooperation 10 Southern African Development Community

Molecular Diagnosis and Laboratory Surveillance of PPR
12
FAO and OIE organized in March 2019 in Rome a workshop on Controlling PPR at the
livestock/wildlife interface in collaboration with the Wildlife Conservation Society and
Royal veterinary College.
A Joint PPR Resource Mobilization and Marketing Strategy was developed which
includes a marketing narrative, an analysis of potential funding sources and a detailed
action plan. The marketing narrative is a human centered approach outlining that ending
PPR will greatly contribute to ending rural poverty, ensuring food security and to
strengthening resilience (SDG1 and SDG2). The subsequent market analysis identifies
potential resource partners at global, regional and national levels as well as strategic
alliances. Domestic resources from affected countries will represent a crucial funding
source. In September 2018, FAO, OIE and the European Union organized the Global
Conference ``Partnering and Investing for a PPR-free World’’ in Brussels which resulted
in a ministerial Declaration highlighting the need to fill a funding gap of USD$ 340
million.
PPR global situation
Fifty seven countries are recognized as free from PPR by OIE and one country on zonal
basis (Namibia) (May 2019). Seventhly nine countries engaged in the Regional
Roadmaps.
Figure 2: Global distribution pattern of PPR
PPR Stage 1 2 3 4
Number of countries 30 38 5 6

Molecular Diagnosis and Laboratory Surveillance of PPR
13
Challenges to the PPR GEP implementation
The implementation of the PPR GEP poses a series of challenges that need to be
addressed.
PPR vaccination campaigns conducted by most of the countries are not in line with
the PPR GCES, as not really based on epidemiology assessment, with an insufficient
number of vaccinated animals and an inappropriate PVE (Post-vaccination
evaluation). The coordination of control measures between neighboring countries is
not satisfactory and the regional or epizone approach is not taken into consideration.
A technical meeting to discuss about PPR vaccinations and epidemiological
assessment as well as PVE is scheduled to be held by end of 2019.
The PMAT, a companion tool to the PPR GCES, aiming to categorize countries
according to the four different stages of the PPR GCES and to provide PPR infected
countries guidance, milestones based on epidemiological, and activity-based
evidence is not used correctly. Training and capacity building at country level are
urgently needed on the use of PMAT, in a revised version easier to use, and PVE
Improving epidemiological understanding of PPRV: epidemiological research is
required to better understand PPR transmission dynamics, in particular its spread and
infectivity and the differing roles of wildlife and livestock species, production
systems, ecosystems and viral lineages in this process. The overall goal is to identify
critical control points, and optimal methods for intervention at these points, to
support effective management of eradication. One example is a need to support the
evaluation of R0 values, in relation to the various lineages of PPR and the various
ecosystems it affects. Along these lines evidence of a fully functional disease
reporting system (in order to properly quantify the effectiveness of vaccination) or a
prolonged period of field work in one or more infected countries may help to
generate some of the basic longitudinal data required for computer simulations. The
results of these epidemiological studies will likely be the main guide to decisions
about what levels of progressive control might practically be achieved, as well as the
feasibility or otherwise of timely eradication.
Infection of wildlife and other species: there are now convincing reports
demonstrating the ability of PPRV to cross the species barrier. Indeed, PPRV can
infect animal species other than small ruminants, with dromedaries, pigs and cattle
reportedly being identified with PPRV (Roger et al., 2000; Gopilo et al., 2005;
Munir, 2014). It is currently unclear whether these infections are relevant from an
epidemiological and eradication perspective; however, it is essential to fully
understand the role of wildlife in the spread and potential maintenance of PPRV in
the environment in order to be able to initiate successful control strategies. The
importance of this research was highlighted by a recent outbreak in Mongolia. In
December 2016 the disease was diagnosed in several wildlife populations in the East
of the country, including saiga antelope (Saiga tatarica mongolica), ibex (Capra
sibirica) and goitered gazelle (Gazella subguttorosa), with 50% mortality of the
10,000 highly endangered saiga population (Aguilar et al, 2018). Moving forward, an

Molecular Diagnosis and Laboratory Surveillance of PPR
14
important first step is to ensure that the currently available tests for sero-diagnosis of
PPRV are validated in serum samples from these animal species, e.g. camels, saiga
and ibex. With specific reference to the PPRV eradication campaign, and as
mentioned above, the significance of these infections as a whole should be carefully
evaluated, as they may not significantly affect the ultimate success of the programme.
Applying movement controls: the control of animal movement, including the
imposition of quarantines and other sanitary measures, are integral to most infectious
disease control and eradication programmes. However, strict movement control can
be counter-productive because it can actually stimulate unnecessary movement of
animals in order to bypass quarantines and restriction orders. To this end, movement
controls must be tempered by the experience of local animal health teams who are
better equipped to judge the behaviour of local owners when faced with such
restrictions.
Applying serological monitoring in the field: there is an ongoing debate about the recruitment rate of newly susceptible sheep and goats into small ruminant populations following vaccination, and how this may require more frequent vaccinations than the annual ones that proved so successful during rinderpest eradication. This could be investigated through computer modelling but empirical data would be persuasive. In this context it would be useful to use serological
monitoring to investigate the levels of recruitment and to develop a proposed methodology for risk-based surveillance, which could be translated into useful actions such as targeted re-vaccination. There are wider calls for serological monitoring to be used to assess the success rates of vaccination campaigns or to invigilate the effectiveness of individual vaccination teams. However, in reality this may only be useful if immediate revaccination can be carried out, which is not often
possible. Whilst, it is important to identify and remedy technical or administrative errors or indeed administrator negligence the implementation of penalties or fines is very difficult. Ultimately, the case for detailed serological monitoring may have to be made on a case-by-case basis, depending on a cost-benefit analysis and whether the resultant data can realistically contribute to improve long-term planning.
Accurately assessing socio-economic impact: although there are many parameters available to facilitate the evaluation of the socio-economic impact of a disease, there are several drawbacks to applying these, e.g. they can be subject specific or handle only one major factor at a time, therefore lacking the ability to estimate the cumulative impact of a disease on the economy. Nevertheless, these economy-wide
considerations are crucial in implementing and funding control and eradication strategies for emerging diseases. For PPR a well-planned cost-benefit analysis of PPR comparing policies and responses that include both the direct and indirect impacts associated with PPR additional cost-benefit analysis are needed to better understand PPR impact in all settings. (Muhammad Munir et al., 2013). The annual global impacts of PPR have been estimated at between US$ 1.4 billion to US$ 2.1
billion; cost-benefit studies have also been carried in different countries; losses which clearly justify both national and global PPR eradication programmes being pursued.

Molecular Diagnosis and Laboratory Surveillance of PPR
15
Funding and political will: Many of the countries where PPR is now endemic
simply cannot finance an efficient, effective and sustained control/eradication
programme. Indeed, they will need significant support for operational costs, training
and meetings in order to properly implement PPR GEP. Even if livestock owners
themselves contribute more towards the costs of vaccination, there will still be a
requirement at the regional and global level for international funding to provide
technical and coordination costs, as well as member state support. In this context, it
will be necessary to explore public-private partnerships, such as those that have
proved so effective for polio, measles and malaria control.
Conclusion
PPR eradication stands within our reach and will have a positive impact on the lives of
pastoralist communities in all developing countries, directly supporting global efforts to
end poverty and hunger by 2030. The right political and financial backing coupled with a
dedicated plan of action are key to success.

Molecular Diagnosis and Laboratory Surveillance of PPR
16
REGIONAL ROADMAP ON PROGRESSIVE CONTROL
OF PESTE DES PETITS RUMINANTS (PC PPR) FOR
SOUTH ASIAN COUNTRIES
Nure Alam Siddiky Consultant, CAMR-ZD in BD, Bangladesh Livestock Research Institute, Savar, Dhaka-1341
Email: [email protected]
PPR is a widespread, virulent and devastating transboundary animal disease of domestic
and wild small ruminants. The disease can have significant economic, food security and
livelihood impacts. SAARC region has a small ruminant’s population of about 300
million. The disease is endemic in most of the South Asian countries or reported at least
once in the recent times except Sri Lanka which is free from the disease. The immediate
response to control and contain the disease is based on clear epidemiologically defined
targeted surveillance for early detection and early warning, sound vaccination strategy,
and enhanced capacities in response. It can be further complemented by a medium to
long-term strategy to enhance the capacities of communities and small ruminant owners
so that their assets are protected through improved integrated activities targeting small
ruminant health and productivity.
Table 1: Economic impact of PPR in SAARC countries
Country
Total
Incidence
(M $)
Total
mortality
(M $)
Production
loss (M $):
Due to disease
Treatment
loss (M $):
Due to disease
Overall
loss
(M $)
Bangladesh 4.86 114.4 149.16 24.30 292.72
Bhutan 0.01 0.07 0.28 0.04 0.40
India 43 968.00 1,386.00 215.00 2,612.00
Nepal 1.95 46.14 59.62 9.76 107.47
Pakistan All together 342.00
Total 49.82 1,128.61 1,595.06 249.1 3,012.59
Regional Support Unit (RSU) for SAARC at FAO Sub-regional ECTAD Unit organized
a regional workshop to develop a regional roadmap for progressive control of PPR for
South Asian countries in 2011. The representatives from SAARC countries attending the
meeting and reviewed the status of PPR at global, regional and country level and finally
developed a roadmap for the progressive control of PPR.

Molecular Diagnosis and Laboratory Surveillance of PPR
17
In order to review the progress made so far and challenges to implement the agreed
roadmap by 2011-2025 in the SAARC countries, the RSU organized the second regional
workshop of PC-PPR from 19-20 December 2013 in Kathmandu, Nepal with the support
from the Government of Nepal, SAARC Secretariat and the European Union.
PPR remains endemic in most of the countries in the region except Sri Lanka. Maldives
and Bhutan had sporadic outbreaks. There is high risk of incursion of the virus through
animal movements and imports of small ruminants even in the countries, region and areas
which are free and/or have sporadic assurances. The countries in South Asian region have
varied capacities, capabilities and facilities in the fields of epidemiology, diagnosis and
vaccine production. India has for instance has well advanced capacity in diagnostic facilities
including those developed indigenously over the years and some of which are now well
recognized commercial private vaccine production entities. India also claims to be self-
sufficient in production of live attenuated homologous vaccine using safe and potent
Sungri/96 strain virus. The Regional Leading Diagnostic Laboratory (RLDL) in Dhaka,
Bangladesh developed capacities in performing cELISA, AGID and cEISA for antibody
detection, icELISA and EISA for viral antigen detection, RT-PCR, qPCR and sequencing
for viral genomic material detection, and also virus isolation on vero cells. It has now
started testing samples referred by SAARC countries.
India is implementing PPR control programme in a phased manner. The five states
were covered during first phase of 2007-2011. The entire country is likely to be
covered during the current phase of 2012-2017. Bangladesh, Nepal and Pakistan
have their localized control programmes for PPR. Bangladesh, India, Nepal, Pakistan
has developed national action plan for the eradication of PPR in accordance with global
eradication campaign. SAARC developed a regional roadmap in close consultation with
Member countries for the eradication of PPR by 2025.The salient features of SAARC
regional roadmap for the eradication of PPR has given below:
The consolidated (revised) regional roadmap for the SAARC countries for the duration of
2014-2025 spread over in three phases is as under:
Component Phase-1
(2014-2015)
Phase-2
(2016-2020)
Phase-3
(2021-2025)
Policy Small ruminant sector review
including husbandry system,
population, demographic
factors, livelihoods issue;
(BD; NP; MD*; PK*; SL*;
BH; IN)
Study report
Developing strategic
plan; (NP; MD; PK;
NP; SL***; BH;
IN*)
Developing strategic plan;
(BD; BH)
Plan document
Getting strategic plan
endorsed by
Getting strategic plan
endorsed by competent
Copy government
endorsement

Molecular Diagnosis and Laboratory Surveillance of PPR
18
competent forum;
(PK; SL***; IN*)
forum; (BD; NP; MD; PK;
BH)
Identification of budget sources for
Implementation of strategic plan. (BD; NP; MD; PK; SL***; IN*)
Identification of budget sources for implementation of strategic plan; (BD; NP)
List / minutes of meetings with budgetary sources
Implementing strategic plan; (NP; PK; SL***)
Implementing strategic plan;(BD; NP; MD; PK)
Yearly Work Plan
Considering zoning based on magnitude and severity of risk; (NP; MD; PK; SL***; BH; IN*)
Considering zoning based on magnitude and severity of risk; (BD ; PK)
Stop vaccination but continue surveillance for PPR virus / antibodies; (BD; MD; PK; SL***; IN)
Minute of the meeting / copy of policy decisions
Developing exit strategy; (MD)
Developing exit strategy; (PK)
Developing exit strategy; (BD; PK; SL***; IN)
Strategy document
Institutional setup and capacity building
Assessment of strengths and weakness of veterinary services at national / regional/ local level; (BD; NP; SL; MD*; PK; BH*; IN)
Assessment document
Strengthening of veterinary services;
(PK; BH; IN)
Strengthening of veterinary services; (BD; NP; MD; PK; SL; BH)
Strengthening of veterinary services; (SL; BH)
Work Plan
Conducting training need assessment in terms of area and number; (MD; PK; BH; IN)
Conducting training need assessment in terms of area and number; (BD; NP)
Needs assessment document
Identification and list of the equipment other than cold chain to be procured; (NP; MD; PK**; SL***; BH; IN)
Identification and list of the equipment other than cold chain to be procured; (BD; NP; BH)
Approved list of equipment’s
Outbreak Response
and contingency plan
Conducting training need
assessment in terms of area
and number; (MD; PK; BH;
IN)
Conducting
training need
assessment in
terms of area and
number; (BD; NP)
Needs
assessment
document

Molecular Diagnosis and Laboratory Surveillance of PPR
19
Developing
contingency plan.
(BD; NP; MD; PK;
SL; BH; IN)
Developing Contingency
plan. (BH)
Developing
contingency
plan. (BH)
Approved
contingency
plan document
Allocation of budget
for contingency plan;
(NP; MD; SL; BH;
IN)
Allocation of budget for
contingency
plan; (BD; PK)
Allocation of
budget for
contingency plan;
(PK)
Financial
statement /
Pink Book/
Agreement
withINGO/
NGOs etc.
Implementing
contingency plan;
(IN)
Identification and list of the
equipment
other than cold chain to be
procured; (BD;
NP; BH)
Work plan
Legislation Updating disease control Act
(NP)
Updated bill/Act
etc.
Enforcement of
import regulation
regarding
PPR; (BH)
Copy of import
regulation
Epidemiology/n
surveillance /
outbreak
investigation
Conducting training need
assessment in terms of
area and number; (MD; PK;
BH;IN)
Conducting
training need
assessment in
terms of area and
number; (BD; NP)
Needs
assessment
document
Developing
surveillance plan and
epidemiology, sero
surveillance
/monitoring of PPR;
(NP; MD*; PK; SL;
BH; IN)
Developing surveillance plan
and
epidemiology, sero
surveillance/ monitoring of
PPR; (BD; IN)
Copy of PPR
surveillance Plan
Identification of risk
factors for PPR(BD;
NP; MD*; PK**;
SL*; BH; IN)
Study report
Mapping of key
cross-border routes
and markets and
services and facilities
available; (BD; NP;
MD*; PK*; SL*;
BH; IN)
Study report

Molecular Diagnosis and Laboratory Surveillance of PPR
20
Identification of hot
spots; (BD; NP;
MD*; PK**; SL***;
BH; IN*)
Identification of hot
spots;(NP)
Study Report?
Spatial
maps
Identification and list
of the equipment
other
than cold chain to be
procured; (NP; MD;
PK**; SL***; BH;
IN)
Identification and List of the
equipment
other than cold chain to be
procured; (BD;
NP ; BH)
Approved list of
equipment
Disease investigation
team composition
and
SOPs for disease
investigation (NP;
BH; MD; PK; SL;
BH; IN)
Team composition and SOPs
for disease
investigation; (BD; BH)
Team
composition for
disease
investigation and
SOPs for
investigation;
(BH)
Notification by
competent
authorities.
Identification of risk
factors for area
classification /
zoning (infected,
buffer and free
zones); (MD*; NP;
SL***; BH; IN*)
Identification of risk factors
for area classification / zoning
(infected, buffer and free
zones); (BD)
Study Report
Developing sero
surveillance plan;
(NP; MD*; SL; BH;
IN)
Developing sero surveillance
Plan; (BD; NP; PK; BH)
Developing sero
surveillance plan;
(BH)
Copy of
approved sero
surveillance
plan
Implementing sero
surveillance plan;
(NP; MD;
SL; BH)
Implementing sero
surveillance plan;
(BD; NP; PK; SL; BH; IN)
Implementing
sero
surveillance plan;
(BD); PK; BH)
Work plan
Developing line of
communication;
(BD; NP; MD*; PK;
SL; BH;IN)
Developing line of
communication; (NP)
Approved copy of
organogram
Vaccine and
vaccination
Good quality vaccine, assured
cold chain and SOPs to
ensure cold chain at all level
(storage to
inoculation of vaccine); (NP;
PK**; IN)
Procurement of
Cold chain and
SOPs to ensure
cold chain at all
level (storage to
inoculation of
vaccine); (BD; NP;
MD; PK; BH)
Approved list of
equipment

Molecular Diagnosis and Laboratory Surveillance of PPR
21
Listing of all the
steps for the vaccine
procurement and
vaccination (SOPs);
(NP; PK; SL***;
BH*; IN)
Listing of all the steps for the
vaccine procurement and
vaccination (SOPs);
(BD; NP; MD; PK; BH)
Listing of all the
steps for the
vaccine
procurement and
vaccination
(SOPs); (BH)
Approved copy
of SOPs
Scheduling of the
field activity; (NP;
MD; PK; SL; BH;
IN)
Scheduling of the field
activity; (BD)
Work plan + Time
lines
Conducting training
need assessment in
terms of area and
number; (MD; PK;
BH; IN)
Conducting training need
assessment in terms of area
and number; (BD; NP)
Needs assessment
document
Post vaccination
monitoring with
reference to FAO/
OIE guidelines;
(MD; PK; NP;
SL***)
Post vaccination monitoring
with reference to FAO/ OIE
guidelines; (BD; NP; BH; IN;
PK)
Post vaccination
monitoring with
reference to
FAO/OIE
guidelines; (BD;
PK; BH)
Approved plan
for post
vaccination
monitoring
Diagnosis Identification of labs for
diagnosis and diagnostic tests;
(BD; NP; MD; PK; SL;
IN*)
Identification of
labs for diagnosis
and diagnostic
tests; (NP)
Notification for
designated labs
and diagnostic
tests
Identification and list
of the equipment
other than cold chain
to be procured; (NP;
MD; PK**; SL***;
BH; IN)
Identification and list of the
equipment
other than cold chain to be
procured; (BD;
NP ; BH)
Approved list of
equipment
Conducting training
need assessment in
terms of area and
number; (MD; PK;
BH; IN)
Conducting training need
assessment in
terms of area and number;
(BD; NP)
Needs assessment
document
Impact assessment/
food security/
poverty alleviation
Listing of all of stakeholders
and their respective role; (BD;
NP; MD; PK; SL; BH)
Approved list of
stakeholders
Consultation with
stakeholders; (BD;
NP; MD; PK**; SL;
BH; IN*);
Impact Assessment; (IN) Minutes of
meeting/proceedin
gs of consultation
process/ study
report

Molecular Diagnosis and Laboratory Surveillance of PPR
22
Abbreviations: BD-Bangladesh, BH-Bhutan, NP-Nepal, IN-India, SL-Sri Lanka, MD- Maldives, PK-
Pakistan
Advocacy and
Communication
Seeking political
commitment; (BD; NP; MD;
PK; SL***; BH; IN)
Seeking political
commitment;
(NP; PK)
Advocacy plan
Developing Public
Awareness
Campaigns: (NP;
MD; PK**; SL; BH;
IN)
Developing Public Awareness
Campaigns; (BD; NP; PK;
BH)
Public awareness
plan
Implementing Public
Awareness
Campaigns; (NP;
MD; SL; BH; IN)
Implementing Public
Awareness Campaigns; (BD;
NP; BH)
Work plan /Tools
of awareness
campaign
Monitoring and
evaluation
Developing monitoring and
evaluation system
for respective activity /
intervention for PPR control;
(NP; MD; PK; SL***)
Developing
monitoring and
evaluation system
for respective
activity
intervention for
PPR control; (BD;
NP; BH; IN)
Developing
monitoring and
evaluation
system for
respective
activity /
intervention for
PPR control;
(BH)
Evaluation of PPR
control plan
including
surveillance and
vaccination
outcomes; (MD)
Evaluation of PPR control
plan including surveillance
and vaccination outcomes;
(BD; NP; PK; IN)
Evaluation of PPR
control plan
including
surveillance
and vaccination
outcomes; (BD;
PK)
Evaluation Plan

Molecular Diagnosis and Laboratory Surveillance of PPR
23
BLRI DEVELOPED PPR CONTROL MODEL
Md. Giasuddin Animal Health Research Division, Bangladesh Livestock Research Institute, Savar, Dhaka-1341
Email: [email protected]
Introduction
PPR (Peste des petits Ruminants) is a highly contagious diseases which severely attacks
small ruminants (Goat and Sheep) in almost 70 countries in Africa, the Middle East and
part of Asia (OIE). The disease was first identified in this region in the year 1993. Now
the disease is endemic in this region and considered as one of the most important
transboundary animal disease. It constitutes a threat to livestock production in many
developing countries including Bangladesh. OIE categorized PPR with a group of
economically important animal diseases, which must be notified to the OIE (Diallo et al.,
2007; Sen et al., 2010).
In all regions where PPR is endemic, it constitutes a serious threat to small ruminant
production and thereby influences on the livelihood of poor farmers, the main owners of
sheep and goats (Diallo et al., 2007). Bangladesh has been blessed with an exclusive
breed of goat, the Black Bengal goat which is world famous for the quality of its meat,
skin and proliferation nature. Furthermore, the goat is considered as the poor man’s cow,
and is an important means of livelihood of rural underprivileged people of Bangladesh
(Miazi et al., 2008). In Bangladesh, PPR was first identified during a severe outbreak in
1993. Since then, the disease has become endemic in Bangladesh (Sil et al., 1995)
causing serious economic losses. In 2010, there were 84000 hospital cases, causing an
estimated economic loss of Taka 1842 million (US$ 24.6) (Islam, 2011). The actual
number of cases may be much more as cases from very rural area were not reported to the
hospital.
At present small goat farmers in Bangladesh are facing massive economic losses due to
PPR as the disease is still endemic in Bangladesh. It has been estimated that losses due to
PPR that has a direct impact to fulfillment of Sustainable Development Goals (SDGs) of
United Nations. The volume of the economic losses demands immediate interventions to
be taken for the progressive control leading to eradication of PPR by 2030.
Following issues need to be addressed for successful implementation of the model
a) Identify a laboratory for PPR diagnosis monitoring
b) Quality vaccines
c) Trained manpower
d) Farmers training
e) Resource mobilization

Molecular Diagnosis and Laboratory Surveillance of PPR
24
According to OIE strategic PPR control approach, BLRI researchers implemented some
specific activities to achieve the goal.
Below stage 1: We have selected an area where
There were insufficient and unstructured raw data to understand the true risks for
PPR.
No appropriate epidemiological investigations were undertaken.
No official prevention and control program was present.
Stage-1: To cross the stage-1, we have done following activities
Activities:
Selected an area or region with biological barrier which separate the region from
other areas
A base line survey was conducted for determining the number of susceptible animals.
A work plan has been developed
Farmer’s attitude, farmer’s training, demand of vaccines and trained worker for
vaccination and other health management has been assessed
A comprehensive control strategy developed in this stage
Stage-2: (Control stage): In this stage, following tools has been considered
Good quality PPR vaccine
Improvement of farm biosecurity
Animal identification or introduction of health card to identify the farm or animal
Implementation of animal movement control
Quarantine the sick or new introduced animal
Engagement of administrative and political leader
Involvement of stockholders
Activities –Following activities has been implemented to overcome the stage 2
Farmers and technicians training

Molecular Diagnosis and Laboratory Surveillance of PPR
25
Pre-vaccination antibody assessment
Deworming
Mass vaccination
Post-vaccination antibody assessment to know the level of antibodies
Stage-3 (Eradication stage): Following proper implementation of control stage 2 to 3
years, the area or region or country will enter into eradication stage. We have done
following activities during stage 3-
Activities
Investigation of any outbreak of PPR or PPR like diseases
Mass vaccination of all goat and sheep over the age of 2 months
Regular virological and serological surveillance
Maintain zero circulation of PPR virus in the area
Stage-4 (Post eradication stage): After maintaining 2-3 years of eradication stage, the
area will enter into post eradication stage. In this stage there will be no PPR outbreak in
the area and will be maintain for at least 24 months and apply for free status. In our BLRI
model we executed all activities in accordance with OIE prescribed guidelines.
Beyond stage-4: We have maintained zero circulation of PPR virus in post eradication
stage, now we can claim for PPR free status as with all necessary documents.
Authority may declare the area or region free status.
Requirement for successful implementation of the PPR control model
Good quality PPR vaccine
PPR diagnostic laboratory
Trained manpower
National strategic plan for PPR control
Conclusion
BLRI developed a PPR control model on the basis of the guideline of OIE Global
Strategy for the Control and Eradication of PPR. It is found very effective if the model is
properly implemented.

Molecular Diagnosis and Laboratory Surveillance of PPR
26
AN OVERVIEW OF GENERAL GUIDELINES FOR PPR
SAMPLE COLLECTION, PRESERVATION AND
GENOMIC ANALYSIS
Emdadul Haque Chowdhury Department of Pathology, Faculty of Veterinary Science
Bangladesh Agricultural University, Mymensingh
Email: [email protected]
Peste des Petits Ruminants (PPR) also known as Goat Plague, is a disease of goats, sheep,
and other taxonomically related species. It is caused by a morbillivirus in the family
Paramixoviridae. It is clinically and pathologically similar to Rinderpest (RP), and is the
most economically important viral disease of small ruminants in the areas where it
occurs. In the field PPR virus causes disease in goats and sheep but not in cattle or pigs,
although these latter two species can be infected sub clinically by experimental
inoculation. One outbreak of PPR was also reported in India, but most of the cases
buffalo sero-converts without showing any clinical disease. Goats are usually considered
to be more susceptible than sheep, but this is not always the case. The disease is
characterized by: high fever, discharges from nose, eyes, and mouth; profuse diarrhea;
pneumonia; and, oral erosion with high morbidity and mortality rates. Strategic
vaccination along with biosecurity measures could help to control the disease.
History of outbreaks
First PPR was described in Ivory Coast in West Africa in 1942. The disease has since
been recognized as endemic in West and Central Africa and gradually spread to other part
of the Africa. The Morocco outbreak is the first case of PPR in North Africa which
followed by Tunisia in 2010. In 1987 PPR appeared in the Middle East and has since then
been confirmed in Arabia (1991), Southern India (1989), Bangladesh (1993), Pakistan
(1993), Iraq (2000), Afghanistan (2006), Turkey ( 2004), Nepal (2010), China and
Bhutan (2010) and recently outbreaks has also been reported in some part of Europe; PPR
reported in Kazakstan, 2014; On June 19, 2018, the Bulgarian National Diagnostic
Research Veterinary Medical Institute confirmed PPR in sheep; the Bulgarian PPR
received growing attention in Europe because of its continuing spreads and economic
impacts.
Distribution of virus and phylogenetic relationship
In the recent years gene sequence analysis has facilitated the classification PPRV strains
into four lineages which are prevailing on the different geographical location of the
world. Lineage I is represented mainly by Western African isolates from the 1970s and
recent isolates from Central Africa; lineage II by West African isolates from the Ivory
Coast, Guinea and Burkina Faso; lineage III by isolates from Eastern Africa, the Sudan,

Molecular Diagnosis and Laboratory Surveillance of PPR
27
Yemen and Oman; and lineage IV includes all viruses isolated from recent outbreaks
across the Arabian Peninsula, the Middle East, Southern Asia and recently across several
African territories. Genetic characterization of the Moroccan and Egyptian PPR virus
classified it as a lineage IV virus; this marks the first time this lineage has been detected
in Africa, where all four lineages are now present. Serological detection of antibodies to
PPRV has also been reported in samples from Vietnam. Bhutan is the latest country being
affected with PPR in South Asia and samples submitted to WRLs from Bhutan have
recently been typed as lineage IV virus.
In Bangladesh, A total 5 (five) circulating field isolates of PPR virus during a period of
2015 to 2017 were characterized in our laboratory using molecular techniques. Full
length of N, M and F genes of the five fields PPRV isolates were sequenced and was
constructed a phylogenic tree considering the full length sequence of respective PPRV
genes taken from Gene Bank (NCBI blast) by MEGA 7.0. Phylogenetically, the
Bangladeshi PPRV strains belong to the PPRV lineage IV and formed a separate
subgroup and these are closely related with China-Tibet/07 and Indian/TN/VEL/2015
PPRV isolate. A number of amino acid substitutions fluctuating from one (in 2016 PPRV
field isolate) to four (in 2017 PPRV field isolates) amino acids (AA) were found among
the N genes of five field PPRV isolates. These may indicate that PPR virus in Bangladesh
slowly evolving. The following figure 1 can give an idea about the phylogenetic
relationship of Bangladeshi PPR virus isolates along with others elsewhere.
Figure 1. Phylogenetic analysis of N gene by maximum likelihood method
(92 sequences:1574 bp nucleotides; unpublished data from our laboratory)
PPRV China isolate (2013-2015)
PPRV/Bangladesh/BD2/2008(MG581412.1)
PPRV/Tibet/Bharal/2008(JX217850.1)
PPRV/China/Tib/07(JF939201.1)
PPRV/China/33/2007(KX421388.1)
PPRV/China/Tibet/Geg/07(FJ905304.1)
PPRV/x11(GQ184299.1)
PPRV/Bangladesh/2015-1
PPRV/Bangladesh/2015-2
PPRV/Bangladesh/2017-2
PPRV/Bangladesh/2016
PPRV/Bangladesh/2017-1
PPRV/IND/TN/VEL/2015/03(KT860064.1)
PPRV/IND/Delhi/2016/05(KX033350.1)
PPRV/IND/TN/ED/2015/04(KT860065.1)
PPRV/IND/TN/GIN/2014/01(KT270355.1)
PPRV/IND/TN/VM/2014/02(KT860063.1)
PPRV/India/TN/Gingee/2014(KR261605.1)
PPR/PRADESH/95(JN647694.1)
PPRV/Izatnagar/94(KR140086.1)
PPRV/Sungri/1996/MSD(KJ867542.1)
PPRV/Sungri/96(GQ452013.1)
PPRV/Sungri/96(AY560591.3)
PPRV/Revati/2006(GU014574.1)
PPRV/Jhansi/03(EU344738.1)
PPRV/Jhansi/2003(GU014571.1)
PPRV/Revati/2005(FJ750559.1)
PPRV/Guj/2007(JN632532.1)
PPRV/Morocco/2008(KC609745.1)
PPRV/Morocco/2008(KC594074.1)
PPRV/S15(KY885100.1)
PPRV/Smailia1/2014(KT006588.1)
PPRV/Ethiopia/2010(KJ867541.1)
PPRV/Georgia/Tbilisi/2016(MF737202.1)
PPRV/Sungri/96(KF727981.2)Vaccine
PPRV/Turkey/00(AJ563705.1)
PPRV/2005(AJ849636.2)
Lineage IV
Lineage II
Lineage I
Lineage III99
99
99
93
99
72
82
64
99
92
95
50
99
99
95
30
46
97
4
75
97
82
65
38
46
99
65
96
78
99

Molecular Diagnosis and Laboratory Surveillance of PPR
28
Host Range
PPR is primarily a disease of goats and sheep and goats are usually more severely
affected than sheep. But in India and the Middle East both goats and sheep are affected
with equally devastating consequences. Breed may affect the outcome of PPR virus
infection. Cattle and pigs are known to be a dead end host. Sero neutralization test for the
presence of PPR antibodies detected 4.2% in 142 camels. PPR affects wildlife animals
both under field condition and experimentally. The disease was induced experimentally
in American white deer (Odocoileus virginianus) which was found to be susceptible and
a field outbreak was reported from a zoological collection in Alain. It caused a high
mortality and severe disease in Dorcas Gazelles (Gazella dorcas), Nubian Ibex (Capra
ibex nubiana), Laristan sheep (Ovis orientalis laristani) and gemsbok (Oryx azellaa).
Subclinical involvement of Nigale (Tragelaphinae) was suspected. In another report from
Saudi Arabia, PPR was suspected on clinical and serological base in Gazaelle and deer.
Antelope and other small wild ruminant species can also be severely affected. Sero-
conversion of large ruminants occurs but no clinical disease so far, except only one
outbreak reported in buffalo. Mild clinical disease has also been reported from camel.
Etiology
PPRV is a member of the genus Morbillivirus under the family Paramyxoviridae and
order Mononegavirales. Paramyxoviruses are enveloped animal viruses which are found
almost exclusively in nucleocapsid structures. This virus shares structural, biological,
antigenic and molecular features in common with the other members of the group. It is
closely related to the rinderpest virus (RPV), the measles virus of humans, the distemper
virus of dogs and some wild carnivores, and the morbilliviruses of aquatic mammals. The
genome of PPRV is non-segmented, negative-strand unlike other member of
Morbillivirus. PPRV consist of 15948 nucleotides that encodes eight proteins: the
nucleocapsid protein (N), the phosphoprotein (P), the matrix protein (M), the fusion
protein (F), the haemagglutinin protein (H), the polymerase protein (L) and the two non-
structural proteins, C and V ( Figure 2). PPRV genome is organized into six contiguous,
non-overlapping transcription units corresponding to the gene of the six structural viral
proteins in the order of 3'-N-P-M-F-H-L-5' in the genome sense. They are separated by
short sequences of three nucleotides called intergenic regions (IG) which is CTT in most
cases. In some virus strains the H-L junction sequence is CGT. The main feature of the
genome organization that is unique to morbilliviruses is the existence of a long
untranslated region (UTR) at 3 end of M gene and at the 5 end of F gene.
Epidemiology
The discharges from eyes, nose and mouth, as well as the loose feces, contain large
amounts of the virus. Fine infective droplets are released into the air from these
secretions and excretions, particularly when affected animals cough and sneeze. For PPR
to spread, close contact between infected and susceptible animals is needed. Since the
virus is enveloped, it is extremely sensitive to inactivation by environmental factors such

Molecular Diagnosis and Laboratory Surveillance of PPR
29
as heat, sunlight and chemicals. The virus is very fragile and cannot survive for a long
time outside host. Its half-life has been estimated to be 22 min at 56oC and 3.3 hours at
37oC. No carrier state is known to exist. The appearance of clinical PPR may be
associated with introduction of recently purchased sick animals from markets or contact
in a closed/village flock with sheep and/or goats that had been sent to market but returned
unsold. Trade in small ruminants at markets where animals from different sources are
brought into close contact with one another increases opportunities for PPR transmission,
as does the development of intensive fattening units. Changes in weather also have been
suggested to contribute to outbreaks but the reports from different geographical regions
are not in total agreement. Young animals are more susceptible. Morbidity is an about
70%, mortality also varied from 50-100%.
Clinical signs
The disease usually appears in the acute form, with an incubation period of 4 to 5 days
followed by a sudden rise in body temperature to 104-106° F (40-41° C). Affected
animals appear ill and restless and have a dull coat, dry muzzle, and depressed appetite.
Affected animals breathe fast, sometimes so fast that they exhibit rocking movements
with both the chest and abdominal walls moving as the animal breathes. Severely
affected cases show difficult and noisy breathing marked by extension of the head and
neck, dilation of the nostrils, protrusion of the tongue and soft painful coughs. They have
obvious signs of pneumonia. A clear watery discharge starts to appear from the eyes,
nose and mouth, which progressively becomes mucopurulent. These mucopurulent
discharges tend to dry, causing matting together of the eyelids, obstruction of the nose
and difficulty in breathing. Animals that survived more days developed erosive and
necrotising stomatitis, enteritis and anorexia. At the height of development of oral
lesions, most animals manifest severe diarrhea, often profuse but not hemorrhagic. Body
temperature usually remains high for about 5-8 days, and then slowly returns to normal
prior to recovery or drops below normal before death.
Pathology
The mucosal surfaces of the esophagus, abomasums, large intestine, rectum and cecum
show congestion, severe edema, hemorrhage, necrotic plagues with erosions. Rumen,
reticulum, and omasum rarely may have lesions; erosions on pillars of rumen, congestion
of the abomasums may be seen. Small intestine lesions are usually moderate and include
extensive necrosis of Peyer's patches, resulting in severe ulceration. The large intestine is
usually more severely affected with congestion around the ileocecal valve, at the ceco-
colic junction, and in the rectum. In the posterior part of the colon and the rectum,
discontinuous streaks of congestion ("zebra stripes") form on the crests of the mucosal
folds. Liver may be moderately enlarged and pale. Indented gall bladder may be found. In
some goats rumen, reticulum, omasum and abomasum may filled with foetid watery fluid
and this watery fluid also may be found in the small intestine. Small erosions and
petechiae are visible on nasal mucosa, turbinates, larynx and trachea, while pleuritis may
be seen in lungs, resulting in hydrothorax. Respiratory tract usually contains frothy

Molecular Diagnosis and Laboratory Surveillance of PPR
30
exudates and the lungs are found to be severely congested with areas of consolidation,
consistently in the apical lobes and in antero-ventral lobes. Most lymph nodes throughout
the body particularly mesenteric and mediastinal lymphnodes are found enlarged,
congested, and edematous but spleen may be either enlarged and congested or slightly
atrophied. Ecchymotic and brush paint hemorrhage are seen on the epicardium. Liver and
kidney lesions were very limited earlier, recent outbreaks shows severe liver and kidney
damage (Fig.2 - 3).
Figure 2. Liver shows hemorrhages and
congestion
Figure 3. Kidney shows hemorrhages, congestion
and inflammation.
Hematological profile of PPRV infected goats
Packed cell volume, total erythrocyte count and hemoglobin concentration decrease at
later stage of the disease in the PPR infected goats. On the other hand, total leukocyte
count is increased in PPR infected goats due to significant increase in the absolute
number of lymphocytes and neutrophil. However, the other blood parameters such as
erythrocyte sedimentation rate and absolute monocytes, eosinophils and basophils counts
remain comparable between PPR infected and healthy goats.
Biochemical profile of PPRV infected goats
PPR infected goats show significantly lower level of total protein and albumin in their
sera when compared to healthy goats. No significant difference is found in the amount of
glucose, bilirubin and blood urea nitrogen between PPR infected and healthy goats.
Analysis of the serum enzymes shows significant increase in the level of creatine kinase,
aspartate transaminase and alanine transaminase in the PPR infected goats compared to
the healthy goats.
Pathogenesis
The PPRV has a particular affinity for lymphoid tissues and epithelial tissue of the
gastro-intestinal (GI) and respiratory tracts, where it produces characteristic lesions. The
At 18 dpi (Dead)

Molecular Diagnosis and Laboratory Surveillance of PPR
31
respiratory route is the likely portal of entry to the host. After the entry of the virus
through the respiratory system, it localizes in pharyngeal and mandibular lymph nodes as
well as tonsil and replicates. After initial replication, the virus spread to the local
lymphatic tissues and also enters into circulation (primary viremia). Primary viremia
results in the spread of virus to other lymphoid tissues and other organs including skin,
kidney and GI tract. In these various organs, the virus replicates in the endothelial cells,
epithelial cells and monocytes /macrophages and plenty of viruses enter into blood
(secondary viremia). The secondary viraemia is associated with further rise of body
temperature. Virus appeared to be released through the microvilli of the epithelial cells.
The incubation period of the disease is 4-5 days in a sero-negative herd but may range
between 3-10 days. It is suggested that death is a consequence of the combined effects of
the pathology and immuno-suppression.
Diagnosis
The routine diagnosis of PPR is based on clinical examination, gross pathology,
histopathologic findings and laboratory confirmation which include tests for the detection
of PPR antigen and PPR antibody, viral isolation, viral nucleic acid hybridization and
recently polymerase chain reaction. Either primary lamb kidney cell or vero cells are
usually used to isolate PPR virus. Isolation of PPR virus using vero cell usually takes 3-5
blind passages which may take 10-15 days. However, use of SLAM-Vero cell increases
the isolation efficiency of PPR virus. Very recently our laboratory has developed primary
goat kidney cell and is being used to isolate PPR virus from the field outbreaks.
Control
There is no specific treatment against PPR. Antibiotics may prevent secondary
pulmonary infections but this treatment is too costly in case of an outbreak. Therefore,
vaccination is the preferred method of control. However, vaccination cannot give 100%
guarantees to control the disease. Implementation of biosecurity measures along with
vaccination and early diagnosis of the disease are required for better control of the
disease. However, vaccination before any big festival in which major movements of goats
occurred across the country may limit the spread of the PPR virus in the country. PPR
infected goat that survived from infection becomes resistant against PPR virus but
becomes immuno-suppressed. Therefore may die due to other opportunistic infection and
needs special attention.
Sample collection, preservation and transportation guideline
General considerations in sample collection
The starting point for the laboratory investigation is the collection of samples. Before
taking samples, careful consideration should be given to the purpose for which they are
required. This will determine the type and number of samples needed to provide valid
results. When samples are taken from live animals, care should be taken to avoid injury
or distress to the animal or danger to the operator and attendants. Whenever handling

Molecular Diagnosis and Laboratory Surveillance of PPR
32
biological material, from either live or dead animals, the risk of zoonotic disease should
be kept in mind and precautions taken to avoid human infection.
Points to be considered before collection of sample
What test would be requested for? Or test to be performed.
What are the appropriate samples to collect?
What transport medium and preservative to be used?
What cool chain to be maintained?
What are the packaging and shipment requirements?
Is the laboratory ready to receive the sample?
Laboratory tests used in the investigation of PPR
The following tests usually used to detect or investigate PPR disease: isolation of virus in
laboratory host system; Detection viral antigen in tissues (Agar gel immunodiffusion test,
Antigen-capture ELISA and Immunohistochemistry); Detection of antibodies in serum
(Agar gel immunodiffusion test, Haemagglutination inhibition test, ELISA) and
Detection of viral DNA / RNA (PCR, RT-PCR etc.).
Types of samples collected for PPR investigation
The following samples need to be collected to investigate PPR: swabs (nasal, tracheal,
oropharyngeal, cloacal), blood, tissues, tissue smear, etc.
Requirement for packaging & transport
Primary container depending on sample type
Secondary container (Air tight Box/Bottle/Tube/ polythene pouch and envelope)
Cool box/ vaccine carrier
Wet ice or Ice packs (put ice packs in deep freezer before transport)
Pen or permanent marker for labeling samples
From live animal
Tears/ nasal swab
Swab from conjunctiva or posterior nasal cavity avoiding external secretion. Keeping swabs moist after collection is most important. Swab should be placed in virological transport medium or any sterile isotonic fluid, like phosphate buffered saline (PBS) or common tissue culture medium like Eagle’s MEM or 50% buffered glycerol with antibiotics (penicillin 200 IU/ml and streptomycin 200 μg/ml) can be used. Commercially available kits containing swabs and viral transport media are also acceptable. This sample can be used for isolation, PCR or any other assays related with antigen detection.
Swab/samples can be placed in 100% ethanol if other media is not available. Samples with ethanol can be only used in PCR.
Sampling also possible with filter paper. Filter (Whatman filter paper, Grade1, 5 to 6 cm in length and 0.25 cm in width) paper can be soaked with nasal

Molecular Diagnosis and Laboratory Surveillance of PPR
33
secretions. Air dried the filter paper avoiding direct sun light and put them in eppendorf tube or polyethane pouch. Pieces of filter paper smeared with secretions can be used in RT-PCR as template.
If none of the system available, collect swab in a tube, put the samples in
container in ice and send it to the laboratory quickly. If necessary, replace ice.
Take sample as many as possible (5-6 animals from an outbreak).
Gum debris
This material can be collected by a spatula or finger rubbed across the gum and
inside the upper and lower lips. The material collected is then scraped into a
container containing 50% buffered glycerol or described as above.
Or place the materials in 100% ethanol for PCR
Collect and shift using ice.
Blood sample
Use whole blood for isolation or RT-PCR.
Sampling also possible with filter paper. Filter (Whatman filter paper, Grade1, 5
to 6 cm in length and 0.25 cm in width) paper can be soaked with blood (pour the
blood drop wise from bottom towards the tip). Air dried the filter paper avoiding
direct sun light and put them in eppendorf tube or polyethane pouch. Pieces of
filter paper smeared with secretions can be used in RT-PCR as template.
Blood samples is good when animal is in febrile state, while nasal/other secretion
is good when animal is in febrile and non-febrile state.
Serum samples
For sero- prevalence study, blood sample should be collected considering
different age group. For example- if a herd size is 25 goat, 20 sample should be
collected in the order of, 8 from goats age < 1years old, 8 from 1-2 years old and
4 from >2 years old.
For diagnostic purpose from a PPR suspected herd, paired sample is preferred,
one is at beginning of outbreak (acute phase of the disease) and other is after 10-
15 days later (Convalescent stage).
Tissues from dead animals
The following tissues should be collected during post mortem examination:
Perform post- mortem as early as possible and record all post mortem changes. Then
collect-
Bronchial Lymph nodes, pieces of lungs (mediastinal) and alimentary tract
(mesenteric) for virus isolation, RT-PCR and other immunological assays that
detect antigen.
Collect part of all tissues in 10% neutral buffered formalin for histopathology and
immunohistochemistry (when required).

Molecular Diagnosis and Laboratory Surveillance of PPR
34
Sample preservation
To prevent spoilage due to autolysis (refrigeration, freezing is required)
To prevent bacterial growth (antibiotics, refrigeration, freezing is required)
To prevent desiccation (fluid vehicle is required)
To prevent hypotonic shock (isotonic buffered solution need to be added)
To prevent freezing shock (glycerol, gelatin, albumin, calf serum –
cryoprotectent need to be added)
Precautions for specimen preservation and transport
Specimens for virus isolation should be refrigerated immediately after collection,
if facilities does not permit keep them chilled ( at 2 to 40C) in refrigerator or with
wet ice and shipped to the laboratory as soon as possible (within 48 hours)
Keep swab in chilled condition for short period (Maximum 4 days at 40C) and
don’t store swab sample at -200C. For longer storage keep them at -70
0C.
Filter paper sample can be kept at normal temperature and can be sent to the PPR
lab by normal post.
Serum sample can be kept at 20C or 4
0C for maximum period of two days and for
longer period store the sample at -200C
Specimens for PCR test should be kept in chilled condition in refrigerator or with
wet ice for short time (48 hours), if specimens cannot be processed immediately;
they should be kept -20°C for short period or below at -70°C for longer period.
When refrigeration facilities are unavailable preserve the specimen in Tryzol
reagent or 100% ethanol in screw cap container store at 4°C and shipped to the
laboratory as soon as possible
Use cool box with icepacks for transporting the specimen
Send the specimen preferably through messenger
Always avoid freeze thaw cycle
Labelling and packaging of specimen for transportation and storage
Specimen type/name e.g. blood, swab, PM tissue samples
Put an identification number
Place of collection
Date and time of collection
* Always submit clinical and sample collection history along with the samples. This is
mandatory. Laboratory may have prescribed format for sample collection and
submission. Before sending samples, recipient laboratory should be ready and agree
to receive the samples.

Molecular Diagnosis and Laboratory Surveillance of PPR
35
Table 1: Storage condition
Samples/
Time
Swab Blood/sera Tissue for
isolation or
ELISA
Sample in
filter paper
Formalin
Fixed tissue
>6
months
<-70°C or
liquid
nitrogen
<-70°C or
liquid
nitrogen
<-70°C or
liquid nitrogen
<-70°C (up
to 1 year)
Several years
in room
temperature
Up to 6
months
-20°C -20°C -20°C
1-7 days 4- 8 °C 4- 8 °C 4- 8 °C Room
temp. Shipment Maximum 12
hours in ice
Max 12 hours
in room
temp./ice
Maximum 12
hours in ice
References
Begum, S., Nooruzzaman M., Murshida, M.M., Mohanto, N., Parvin, R., Islam, M.R., and
Chowdhury, E.H. 2018. PPR virus infection showed altered hematological and serum
biochemical profile in Black Bengal goats. Onderstepoort Journal of Veterinary Research
85(1), 1595.
Rahman, M.M., Parvin, Rokshana., Bhuiyan, A.R., Giasuddin, M., Chowdhury, S.M.Z.H., Islam,
M.R., Chowdhury, E.H.2016. Genetic characterization of Peste des Petits ruminants virus
circulating in Bangladesh. British Journal of Virology, 3(4),115-122 ·
Chowdhury, E.H., Bhuiyan, A.R., Rahman, M.M., Alam., Siddique, M.S.A., and Islam, M.R..
2014. Natural Peste des Petits ruminants virus infection in Black Bengal goats: Virological,
pathological and immunohistochemically investigation. BMC Veterinary Research
11/2014; 10(1):263. doi:10.1186/s12917-014-0263-y
Siddiqui, M.S.I., Ahasan, S.A., Islam, N., Kundu., P., and Chowdhury, E.H. 2014. Peste des petits
virus antibodies in goats and cattle of the Saints Martin’s island of Bangladesh. The
Bangladesh Veterinarian, 31, 55-59.
Bhuiyan, A.R., Chowdhury, E.H., Kwaiatek, O., Parvin, R., Rahman, M.M., Islam, M.R., Albina,
E., and Libeau., G. 2014. Dried fluid spots for Peste des Petits ruminants virus load
evaluation allowing for non-invasive diagnosis and genotyping. BMC Veterinary Research,
10/2014; 10(1), 247.,doi:10.1186/s12917-014-0247-y.
Bhuiyan, A.R., Rahman, M.M., Begum, J.A., Islam, M.R., and Chowdhury, E.H. 2012.
Comparison of genes as target for molecular diagnosis of Peste des petits ruminants in
goats. The Bangladesh Veterinarian, 29(2), 56 -62. doi: http://dx.doi.org/10.3329/bvet.v
29i2.14343
Rahman, M.A., Shadmin, I., Noor, N., Parvin, R., Chowdhury, E.H., and Islam, M.R. 2011. Peste
des petits ruminants virus infection of goats in Bangladesh: Pathological investigation,
molecular detection and isolation of the virus. Bangladesh Veterinarian, 28, 1-7. doi:
http://dx.doi.org/10.3329/bvet.v28i1.8808
Rahman, M.M., Bhuiyan, A.R., Parvin,
R., Giasuddin, M., Haque, M.E., Sayem, S.M., Islam,
M.R., and Chowdhury, E.H. 2011. Immune response of goats to thermostable preparation

Molecular Diagnosis and Laboratory Surveillance of PPR
36
of conventional Peste des petits ruminants (PPR) vaccine in Bangladesh. SAARC Journal of
Agriculture, 9, 73-81. www.saarcagri.net/index.php?option=com_content
Islam, M.R., Shamsuddin, M., Rahman, M.A., Das, P.M., and Dewan, M.L. 2001. An outbreak of
Peste des petits ruminants in Black Bengal goats in Mymensingh, Bangladesh. The
Bangladesh Veterinarian, 18, 14-19.
Dialo, A., Barrett, T., Barbron, M., Meyer, G., and Lefevre, P.C. 1994. Cloning of the
nucleocapsid protein gene of PPR virus: relationship to other morbilliviruses. Journal of
Gen. Virology, 75, 233-237.
Pronab, D., Sreenivasa, B.P., Barrett, T., Corteyn, M., Singh, R.P., Bandyopadhyay, S.K., and
Dhar, P. 2002. Recent epidemiology of Peste des petits ruminants virus (PPRV). Veterinary
Microbiology, 88(2), 153-159.
Raj, G.D., Kumar, A.S., Shaila, M.S., Nachimuthu, K., and Palaniswami, K.S. 2003. Molecular
epidemiology of Peste des petits ruminants viruses from southern India. Veterinary Record,
152, 264-6.
Rowland, A.C., Scott, G.R., and Hill, D.H. 1969. The pathology of an erosive stomatitis and
enteritis in West African dwarf goats. Journal of Pathology, 98, 83-87.
Taylor, W.P. 1984. The distribution and epidemiology of Peste des petits ruminants. Preventive
Veterinary Medicine 2, 157-166.

Molecular Diagnosis and Laboratory Surveillance of PPR
37
PRINCIPLES OF PCR AND GUIDELINES FOR GOOD
LABORATORY PRACTICES
Mohammad Rafiqul Islam Senior National Technical Advisor, One Health Education
ECTAD, FAO, Bangladesh
Email: [email protected]
Polymerase chain reaction (PCR)
Polymerase chain reaction is one of the most common and often indispensable tools for
molecular biology research and applications. Kary Mullis, an American biochemist,
received Nobel Prize in 1993 for PCR that he invented almost 10 years earlier. Mullis
summarized the procedure of PCR as follows: "Beginning with a single molecule of the
genetic material DNA, the PCR can generate 100 billion similar molecules in an
afternoon. The reaction is easy to execute. It requires no more than a test tube, a few
simple reagents, and a source of heat." The PCR has now become very sophisticated in
terms of instrumentation and reagents. However, the basic principle still remains the
same, which involves repeated cycles of (i) heat denaturation of double stranded DNA,
(ii) annealing of primers (short pieces of complementary DNA) to the denatured strands
of DNA, and (iii) extension or amplification of new complementary strand of DNA under
the influence of DNA polymerase enzyme (Fig. 1). Amplification of the right fragment of
DNA is confirmed by agarose gel electrophoresis of PCR products.
Figure 1. Basic principle of PCR (left) and an example of a PCR protocol (right)
The invention of PCR
Mullis developed the PCR in 1983, when he was working in Emeryville, California for
Cetus Corporation, one of the first biotechnology companies. There, he was responsible
for synthesizing short chains of DNA. He conceived of PCR while cruising along the
Pacific Coast Highway one night in his car. He was playing in his mind with a new way
of analyzing changes (mutations) in DNA when he realized that he had instead invented a

Molecular Diagnosis and Laboratory Surveillance of PPR
38
method of amplifying any targeted DNA region through repeated cycles of duplication
driven by DNA polymerase. The PCR method relies on thermal cycling, i.e., repeated
heating and cooling of a buffered reaction mixture of template DNA, two oligonucleotide
primers, all the four types of deoxynucleotide triphosphates (dNTPs) and DNA
polymerase enzyme. During each of the thermal cycles the two strands of DNA first
denature or separate, the primers anneal to the complimentary sequences in the denatured
template and then as primed by the annealed primers and driven by polymerase enzyme
new strands of complimentary DNA are synthesized through incorporation of dNTPs. On
each thermal cycle the number of targeted DNA increases exponentially leading to the
synthesis of thousands, millions or billions of copies of the same DNA fragment which
can be visualized on agarose gel electrophoresis and staining.
The legacy of PCR
Although Mullis is credited for the invention of PCR, he in fact had put together several
known principles of biochemistry in a noble approach of repeated cycling to obtain a
noble product, a visible quantity of targeted DNA fragment. The invention of PCR has a
long legacy. James Watson and Francis Crick received Nobel Prize in 1953 for their
discovery of DNA structure founding the field of molecular genetics. Another Nobel
laureate Arthur Kornberg identified DNA polymerase enzyme in 1957. Hor Gobind
Khorana, who received Nobel Prize for elucidation of genetic codes, also used
oligonucleotide primers and polymerase. In 1977 Frederick Sanger reported a method for
determining the sequence of DNA. The technique employed an oligonucleotide primer,
DNA polymerase, and dNTPs (native and modified). For this innovation he was awarded
the Nobel Prize in 1980. In 1971 Kjell Kleppe, a researcher in Khorana's lab, envisioned
a process very similar to PCR and he described how a two-primer system might lead to
replication of a specific segment of DNA. However, it is Kary Mullis who demonstrated
the use of two primers, template, dNTPs and polymerase in repeated cycles of heating
and cooling to synthesize copies of targeted DNA.
Refinement of PCR
The DNA polymerases initially employed for experimental PCR were unable to
withstand high temperatures needed for DNA denaturation. So the early procedures for
DNA replication were very inefficient and time consuming, and required large amounts
of DNA polymerase to be replenished at each cycle. The discovery in 1976 of Taq
polymerase - a DNA polymerase purified from the thermophilic bacterium, Thermus
aquaticus, which naturally lives in hot (50 to 80°C) environments such as hot springs -
paved the way for dramatic improvements of the PCR method. The DNA polymerase
isolated from T. aquaticus is stable at high temperatures remaining active even after DNA
denaturation, thus obviating the need to add new DNA polymerase after each cycle. This
allowed an automated thermocycler-based process for DNA amplification. Most
commonly used thermal cycling profile for PCR includes denaturation of DNA at 94°C,
primer annealing at 45-65°C and synthesis (elongation) of new strand at 68-72°C.

Molecular Diagnosis and Laboratory Surveillance of PPR
39
Relatively more recent advancement in PCR is the introduction of real-time PCR, which
does not require post-PCR analysis of the products by agarose gel electrophoresis.
Instead, the amplification of DNA can be observed real time on computer screen as
amplification curve generated by the signals of fluorescent dyes incorporated into the
newly amplified DNA.
Reverse transcription polymerase chain reaction (RT-PCR)
The classical PCR technique can be applied only to DNA strands. RT-PCR is a variant of
PCR, where with the help of reverse transcriptase enzyme, RNA can be transcribed into DNA
(complementary DNA, or cDNA), thus making PCR analysis of RNA molecules possible.
Reverse transcriptase was discovered in 1970 by Howard Temin at the University of
Wisconsin–Madison and David Baltimore at MIT from RNA tumour viruses
(retroviruses). For their achievements, both shared the 1975 Nobel Prize in Physiology or
Medicine (with Renato Dulbecco).
Good laboratory practices (GLPs) for PCR laboratory
PCR is an extremely sensitive procedure and hence is very prone to contamination.
Accidental contamination with DNA or RNA or the source of DNA/RNA, similar to that
expected in the test sample, could lead to a false positive reaction. The source of
contamination could be another sample, the environment, the positive control or the DNA
amplified earlier in the laboratory. Another type of contamination which is particularly
important for RT-PCR is RNase enzyme. RNase is a highly stable enzyme with
ubiquitous distribution, virtually present on every surface including human hands,
laboratory benches, glassware and plastic ware and air exposed to dusts (containing
microbes). RNase destroys RNA giving false negative results in RT-PCR, which is a
nightmare for any laboratory diagnostician. Hence to avoid unwanted false positive or
false negative reactions and to ensure laboratory reproducibility there is no alternative but
to adhere to strict GLPs. The building blocks of GLP in a PCR laboratory are (i) proper
work flow, (ii) clean working area, (iii) laboratory discipline, (iv)quality control and (v)
good housekeeping.
Work flow
A PCR involves several steps including extraction of nucleic acid, assembly of test
reaction (master-mix preparation and template addition), amplification of DNA and
finally analysis of PCR products. Activities prior to amplification can be identified as
pre-PCR steps and analysis of PCR products as the post-PCR step. It is very important to
physically separate the pre-PCR, amplification and post-PCR activities (Figure 2). This
demands designation of isolated laboratory spaces for nucleic acid extraction, reaction
assembly, amplification and PCR product analysis. However, mere separation of
activities might not bring expected benefits if a unidirectional work flow is not
established. Ideally there must not be any back flow from the post-PCR areas to the pre-
PCR areas.

Molecular Diagnosis and Laboratory Surveillance of PPR
40
Figure 2. Work flow in a PCR laboratory
Clean work area
The nucleic acid extraction room should meet the standard of a virology lab in terms of
biosafety and aseptic measures. A Class II biosafety cabinet should be available for
extraction of nucleic acids. The PCR reaction assembly room should meet a “clean room”
standard and should have at least a PCR hood. PCR laboratories need to be kept clean
and tidy. There should be routine cleaning and decontamination schedule. Sodium
hypochlorite solution or commercially available DNA decontaminating agents can be
used. Ethanol could clean the surface and wipe out microorganisms but would not destroy
DNA residues if remained on the surface. UV irradiation would destroy DNA but only to
a certain distance. All pipettes, tips, gloves and tubes should be protected from dust while
not in use.
Laboratory discipline
A laboratory designed for maintaining clean environment and good work flow would be
of little value unless a stringent laboratory discipline could be established. Laboratory
workers should be well informed about the laboratory rules and should follow them
rather religiously. Extraction of DNA/RNA from infectious materials must be done at a
designated place (ideally in a Class II biosafety cabinet). Dedicated and separate sets of
equipment and appliances should be used in different sections of PCR lab and they
should be clearly marked and preferably colour coded. Consumables, reagents and water
for pre- and post-PCR activities should be stored separately. Appliances, consumables,
reagents, water, marker, pen, note books, etc. must not be moved between pre- and post-
PCR sections. Separate lab coats should be used for pre- and post-PCR sections.
Disposable gloves must be used for all activities and they should be changed frequently,
particularly after touching any contaminated surface. All surfaces should be considered
potentially contaminated.
Work flow should never be reversed. If possible, it is useful to establish a daily schedule
for pre- and post PCR activities for routine diagnostic activities. One should plan and
organize work before beginning a test, such as making all calculations, filling in

Molecular Diagnosis and Laboratory Surveillance of PPR
41
worksheets, ensuring availability of all reagents and supplies at respective work areas.
Reagents and water should be aliquoted in convenient volumes with due precautions. It is
advisable to pulse centrifuge tubes to collect reagents at the bottom before opening the
tubes. Any reagent must not be left on the bench any longer than necessary. Pipetting
steps should be minimized by preparing master-mix, whenever applicable and any
leftover master mix must be discarded. Use of filter tips is suggested in all steps
particularly at least prior to amplification.
Quality control
PCR and RT-PCR involves the use of a number of reagents which are highly perishable
and prone to contamination. False positive and false negative results could be a matter of
great concern. It is therefore essential to check the reproducibility of PCR and RT-PCR.
This requires the use of appropriate controls such as positive control, negative control,
reagent control (no template but water), internal positive control, etc.
Apart from these, PCR laboratories should participate in quality assurance programme at
regular intervals.
Good housekeeping
Good housekeeping is an integral part of good laboratory practices. All consumables
should be stored systematically. All reagents should be properly labeled with name, date
and batch number and stored in appropriate place keeping in mind proper work flow. Log
book should be maintained where appropriate.
Proper disposal of laboratory waste is also very important. Used tips and tubes should be
discarded carefully in designated and labeled receptacles avoiding possible contamination
of work areas and then these should be disposed by incineration. Ethidium bromide used
for staining DNA is a potential carcinogen. Special precaution should be taken and
appropriate procedures should be followed in disposing agarose gel and buffer containing
ethidium bromide.

Molecular Diagnosis and Laboratory Surveillance of PPR
42
ELISA: AN ESSENTIAL TOOL FOR
SURVEILLANCE OF PPR
Jahangir Alam National Institute of Biotechnology, Ganakbari, Ashulia, Dhaka-1349, Bangladesh
Email: [email protected]
Historical background
Enzyme-linked immunosorbent assay (ELISA) and Enzyme immunoassay (EIA) became
very familiar names for medical and veterinary laboratories, diagnostic product
manufacturers, regulatory bodies, quality assessment & proficiency-testing organizations,
etc. Three scientific research groups independently and simultaneously developed
immunolabelled technique and demonstrate its feasibility. The ELISA technique
developed by Peter Perlmann and Eva Engvall at Stockholm University, Sweden
published in September 1971 while Avrameas & Guilbert from the Pasteur Institute
published in December 1971. On the other hand the EIA technique developed by Bauke
van Weemen and Anton Schuurs at the Research Laboratories of NV Organon, Oss, The
Netherlands published article on June 1971. Radio immune assay (RIA) was first
described in July, 1960 for measurement of endogenous plasma insulin by Solomon
Berson and Rosalyn Yalow of the Veterans Administration Hospital in New York. Yalow
would later be awarded the 1977 Nobel Prize for Medicine for “the development of the
RIA for peptide hormones”, but because of his untimely death in 1972, Berson could not
share the award.
Basic principles of ELISA
ELISA is a plate-based assay
technique designed for detecting and
quantifying substances such as
peptides, proteins, antibodies,
hormones, etc. by changing
antibodies and color to identify a
substance. In an ELISA, an antigen
must be immobilized on a solid
surface and then combined with an
antibody that is linked to an enzyme.
Detection is accomplished by assessing the conjugated enzyme activity via incubation
with a substrate to produce a measureable product (Figure 1). The most crucial element
of the detection strategy is a highly specific antibody-antigen interaction. It is a common
test that detects and measure antibodies in blood.
Figure 1. Basic Procedure of ELISA (Source: https://www.cusabio.com/c-20659.html)

Molecular Diagnosis and Laboratory Surveillance of PPR
43
Type of ELISA
As its name implies, ELISA involves the use of enzymes and the specific binding of
antibody and antigen. According to how it works, ELISA can be divided into four major
types: direct, indirect, sandwich and competitive. Each type has advantages as well as
disadvantages (Table 1). On the basis of whether ELISA can quantify the level of the
target molecule, ELISA can be divided into two types, qualitative and quantitative.
Qualitative ELISA provides a simple positive or negative result for a sample, while
quantitative ELISA reflects the concentration of the target molecule in a sample via a
standard curve.
Table 1: Comparison of direct, indirect, sandwich, and competitive ELISA
Type Advantages Disadvantages
Direct ELISA Simple protocol, time-
saving, and reagents-
saving.
No cross-reactivity from
secondary antibody.
High background.
No signal amplification, since only a primary
antibody is used and a secondary antibody is
not needed.
Low flexibility, since the primary antibody
must be labeled.
Indirect ELISA Signal amplification, since
one or more secondary
antibodies can be used to
bind to the primary
antibody.
High flexibility, since the
same secondary antibody
can be used for various
primary antibodies.
Complex protocol compared with direct
ELISA.
Cross-reactivity from secondary antibody.
Sandwich
ELISA
High flexibility.
High sensitivity.
High specificity, since
different antibodies bind
to the same antigen for
detection.
The antigen of interest must be large enough so
that two different antibodies can bind to it at
different epitopes.
It's sometimes difficult to find two different
antibodies that recognize different epitopes on
the antigen of interest and cooperate well in a
sandwich format.
Competitive
ELISA
High flexibility.
High sensitivity.
Best for the detection of
small antigens, even when
they are present in low
concentrations.
Relatively complex protocol.
Needs the use of inhibitor antigen.
Source: https://www.cusabio.com/c-20659.html; https://www.thermofisher.com/bd/en/home/life-
science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-
methods/overview-elisa.html (Accessed in 11 July 2019)

Molecular Diagnosis and Laboratory Surveillance of PPR
44
ELISA and its application
Since its conception in the
early 1970’s the ELISA has
been a primary method of
analyte detection. In its four
decades it has become a
fundamental tool in a wide
range of scientific fields, its
diverse nature demonstrated by
its range of uses. It was
extensively utilized with great
success in laboratory diagnosis
and research in the medical
field, veterinary medicine,
agricultural sciences,
biotechnology and in many
fields of research where it
could be applied. In addition to
molecular technologies, there is a need to use serological confirmatory methods in a dual
approach to directly identify and characterize disease agents and to assess disease
prevalence through the measurement of specific antibodies. The use of ELISA methods
in testing the environment and animal or plant products are reported to as safe for human
and animal consumption is also a rapidly evolving area for ELISA. Although
immunoassays are both highly sensitive and specific, false positive and negative results
may occur. False-negative results may be caused by improper sample storage, reagent
deterioration, improper washing technique or prozone effect. False-positive results have
been reported for samples containing small fibrin strands that adhere to the solid phase
matrix or due to substances in the blood or urine that cross-react or bind to the antibody
used in the test. Crowther, (2009) scanned the literature involving ELISA mentioned in
all science areas from 1976 to 2004 (Figure 2) elucidates continuous rise in the number of
works using ELISA methods. He also categorized the ELISA related publication in field
wise (Table 2) and demonstrate the major areas of use in medicine and dentistry;
immunology and microbiology, molecular biology, and genetics and biotechnology. He
mentioned that the earliest exploitation of ELISA was done in the field of immunology
and microbiology and molecular biology and biotechnology.
PPR and its diagnosis
Peste des Petits Ruminants (PPR), is an acute, highly contagious, notifiable and
economically important transboundary viral disease of sheep and goats associated with
high morbidity and mortality. Clinically, the disease resembles rinderpest (RP) in cattle
and is characterized by high fever (pyrexia), conjunctivitis, oculo-nasal discharges,
necrotizing and erosive stomatitis, diarrhea, and bronchopneumonia followed by either
death of the animal or recovery from the disease. The causative agent, PPR virus (PPRV)
Figure 2. Number of literature in Science Direct
database for ELISA (1976-2004). Source:
Crowther, 2009.

Molecular Diagnosis and Laboratory Surveillance of PPR
45
is an enveloped RNA virus belongs to the genus Morbillivirus of the family
Paramyxoviridae (sub family Paramyxovirinae) under the order Mononegavirales. The
PPRV is genetically grouped into four lineages (I, II, III, and IV) based on the F and N
gene sequences analyses. Lineages I–III circulate in Africa, while lineage IV is generally
found in Asia. However, lineage IV is recently reported from Morocco. PPR is tentatively
diagnosed by clinical observations, characteristic symptoms, epidemiology, postmortem
lesions, and laboratory confirmation by using various serological and molecular
techniques. Serological tests include agar gel immuno-diffusion test (AGID)/AGPT,
counter- immuno-electrophoresis (CIE) and ELISA, etc.
Table 2: Subject wise distribution of published literature in science groups
Subject Year
1980-84 1985-89 1990-94 1995-99 2000-04
Agriculture and biological sciences 87 274 615 804 827
Molecular biology, genetics and
biotechnology
374 1329 1762 1845 2096
Chemistry 8 29 77 208 279
Environmental science 4 13 52 125 162
Immunology and microbiology 514 1584 2128 2450 2772
Medicine and dentistry 280 971 1639 2875 3372
Neurosciences 21 124 198 380 484
Pharmacology and toxicology 24 108 247 397 497
Veterinary sciences 71 219 522 769 853
(Source: Crowther, 2009)
Application of ELISA in PPR
For early and specific diagnosis of PPR, researchers are working towards the
development of molecular diagnostics tools. However, as a rapid, simple and sensitive
assay, ELISA has been widely used in serological profiling of PPRV in mass screening of
samples for sero-monitoring, sero-surveillance or clinical prevalence. Various workers
have used MAb produced against PPRV and RPV for detection of antibodies and
antigens in ELISA. Several researchers used neutralizing MAbs against H protein of
PPRV for specific detection of PPRV antibodies in competitive ELISA (c-ELISA) and
blocking ELISA (B-ELISA) for antibody detection. Use of the virus neutralizing MAb
was reported to more advantageous as it produced better correlation between VNT and
ELISA. The sensitivity and specificity of a B-ELISA was reported to be 90.4 and 98.8%,
respectively when compared to VNT. MAb-based c-ELISA for measurement of
antibodies to PPR and RP viruses in sheep, goat and cattle are in common use.
Researcher also used anti-‘N’ MAb to PPRV in a c-ELISA, which was reported to 94.5%

Molecular Diagnosis and Laboratory Surveillance of PPR
46
sensitive and 99.4% specific in comparison to VNT. Researchers also developed a MAb-
based c-ELISA for the detection of PPRV antibodies using a virus neutralizing MAb
directed against an epitope in ‘H’ protein specific MAb (4B11) to PPRV and cell culture
propagated vaccine virus antigen. This c-ELISA had high diagnostic specificity (99.8%)
and sensitivity (90.5%) for detection of PPRV antibody in convalescent sera, when
compared with VNT and also the commercially available kit, where, it had high
diagnostic sensitivity (92.2%) and specificity (98.4%). This assay is currently being
employed extensively throughout India for monitoring/sero-surveillance of PPR. Further,
a polyclonal antibody based indirect ELISA was reported for detection of antibodies to
PPRV in the serum samples of goats and sheep using Vero cell culture propagated
purified PPRV antigen with a high degree of specificity and sensitivity when compared
with c-ELISA and VNT, respectively. This may be a good alternative tool to c-ELISA for
sero-epidemiological surveys. Further, MAb-based immunocapture ELISA and sandwich
ELISA (s-ELISA) have been used extensively for detection of PPRV antigen in clinical
specimens. The immunocapture ELISA was developed in the world reference laboratory
(CIRAD–EMVT, France) and is an internationally accepted assay for PPRV antigen
detection. This assay uses a biotinylated anti-‘N’ MAb against a cross-reactive epitope of
RP/PPRV to capture or detection of PPRV antigen in clinical samples. Similarly, the s-
ELISA kit developed at Division of Virology, IVRI, Mukteswar, India used a MAb (4G6)
directed against an epitope of N protein of PPRV, which is the routinely being used for
clinical prevalence or detection of PPRV antigen in clinical specimens in India. This
assay was efficacious, with diagnostic sensitivities (89%) and specificities (93%),
comparable to the immunocapture-ELISA. Further, assay using multiple antigenic
peptides (MAPs), as isotope, for detection of PPRV antibodies in serum samples has also
been developed for serosurveillance and seromonitoring (Balamurugan et al., 2014).
Problems in ELISA
The ELISA results are generally based on the color depth of the chromogenic substrate.
The color reaction is required to be carried out at 37 °C for about 10 minutes, then the
color reaction is terminated with a stop solution, and the absorbance at a specific
wavelength is monitored with a microplate reader. Since the detection path of each
reaction well is perpendicular to the microplate reader, the bottom of each reaction well
should be kept clean when testing, and the amount of chromogenic substrate and stop
solution should be accurate. The amount of liquid will affect the final degree. There are
various problem in ELISA experiment including abnormal color reaction, standard curve,
data analysis, the repeatability of experiment, etc. Some causes and solutions related to
color development are depicted in table 3.

Molecular Diagnosis and Laboratory Surveillance of PPR
47
Table 3: The problems of the color reaction
Cause Solution
No signal
Assay set up incorrectly, used
incorrect reagents or incorrect
wavelength
Review protocol. Repeat assay using a positive control
and check plate reader for wavelength, filters, gain etc.
Not enough antibody used Try different concentration of the primary and/or
secondary antibody.
Incubation time too short Incubate samples overnight at 4°C or follow the
manufacturer guidelines.
Antibody stored at 4°C for several
weeks or subjected to repeated
freeze/thaw cycles
Use a fresh aliquot of antibody that has been stored at -
20°C or below.
Recognition of epitope impeded
by adsorption to plate
To enhance detection of a peptide by direct or indirect
ELISA, conjugate peptide to a large carrier protein before
coating onto the microtiter plate.
Slow color development of
enzymatic reaction
Prepare substrate solution immediately before use. Ensure
the stock solution has not expired and is not
contaminated. Allow longer incubation.
Low sensitivity or weak signal
Improper storage of ELISA kit Store all reagents as recommended. Please note that all
reagents may not have identical storage requirements.
Plate reader settings incorrect Check plate reader for wavelength, filters, gain, etc.
Inactive detection reagent or
detection reagent too dilute
Ensure reporter enzyme/flour has the expected activity, or
use a higher concentration of detection reagent.
Insufficient amount of antigen was
coated to microtiter plate
Use more antigen for coating or very coating buffer.
Not enough antibody used Increase concentration of the primary and/or secondary
antibody. Optimize antibody concentrations for your assay.
Mixing or substituting reagents
from different kits
Avoid mixing components from different kits.
Incubation temperature too low Optimize the incubation temperature for your assay.
Reagents should be at room temperature before beginning
the assay.
Assay plates were compromised or
previously used.
Be sure to refrigerate plates in sealed bags with a desiccant
to maintain stability. Prevent condensation from forming
on plates by allowing them to equilibrate to room
temperature while in the packaging. If partial plates are
used, you must be sure to label used wells to prevent reuse;
cover them with sealing tape and use the remaining wells

Molecular Diagnosis and Laboratory Surveillance of PPR
48
Cause Solution
as soon as possible. Do not store partially used plates with
other plates. Include a desiccant in the storage bag.
High background signal
Too much antibody used Optimize antibody concentrations for your assay with
different dilutions.
Too much detection reagent Ensure the reagent has been diluted properly or repeat
assay with a higher dilution of detection reagent.
Incubation temperature too high Try different temperature for optimizing your assay.
Reaction not stopped Use stop solution to prevent overdevelopment.
Waiting too long to read plate after
adding stop solution
Read plate immediately after adding stop solution.
Incubation with substrate carried
out in the light
Perform substrate incubation in the dark.
Non-specific binding of antibody Use a suitable blocking buffer or use an affinity-purified
antibody.
Dirty plate Clean the plate bottom carefully and reread.
Slow color development
Incubation temperature is wrong Ensure plates and reagents are kept at room temperature.
Contaminated solutions Make fresh solutions.
Detection reagent too old,
contaminated or used at the wrong
pH
Use fresh detection reagents at the correct pH.
Wrong conjugate was used,
conjugate was prepared incorrectly
or has deteriorated.
Be sure that the conjugate used is the one that came with
the kit. All conjugates are kit- and lot-specific. If
preparation of a working conjugate is needed, be sure that
the concentrate and diluent are mixed in correct volumes.
Do not prepare the working solution too far in advance
and do not save any unused portion for future use. If no
conjugate preparation is necessary, be sure to pour out
only the amount required for immediate use and do not
return any unused portion to the stock bottle.
Wash buffer contains sodium azide. Avoid sodium azide in the wash buffer.
(Source: (i) https://www.cusabio.com/c-15091.html. (ii) https://www.thermofisher.com/bd/en/home/life-
science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-
methods/overview-elisa/elisa-troubleshooting-guide.html (Accessed in 11 July 2019)

Molecular Diagnosis and Laboratory Surveillance of PPR
49
Factors to be considered for selection of ELISA kit
Given its convenience and effectiveness, ELISA has wide applications in disease
diagnosis, biomedical research, and various industries. Virtually any type of molecule
(protein, lipid, carbohydrate, nucleic acid, etc.) can be detected by the method of ELISA.
Choosing a commercially available right ELISA kit is very important. Several fractors
should take into consideration while choosing an ELISA kit. Some factors for choosing
the right ELISA kit are mentioned below (https://www.cusabio.com/c-20299.html)
(Accessed on 11 July 2019)
The species to be studied
If the sample is from a classical model such as human, mouse and rat, it is relatively easy
to find a validated ELISA kit. But if the sample is from a non-classical model such as
monkey, there are limited numbers of commercial ELISA kits available. In this case, you
may have to choose a kit validated on species that shows homology with the species of
your sample.
The analyte to be detected
It needs clear understanding about what kind of analyte (usually protein) to detect. A
sandwich ELISA is generally suitable for detecting large proteins with multiple epitopes
such as a cytokine. A competitive ELISA is appropriate for detecting small molecules
like hapten. Most commercial ELISA kits are validated on serum/plasma and culture
supernatants. It is important to read the product instructions in detail to ensure that the kit
is compatible with your sample. For example, the way that plasma samples are collected
(heparin or EDTA) can affect which ELISA kit should be chosen. Besides, other factors
such as hemolysis and the presence of lipids in the sample can interfere with assay
performance.
Purpose of the analysis
ELISA is a tool that can be used for both qualitative and quantitative analyses.
Qualitative ELISA provides a simple positive or negative result for a sample, while
quantitative ELISA reflects the concentration of the analyte in a sample via a standard
curve. Based on the purpose of the analysis either qualitative ELISA or quantitative kit
should be chosen.
Type of antibodies
Need to clearly know what types of antibodies are used in the kit: monoclonal or
polyclonal antibody. In sandwich ELISA, it is sometimes helpful to use a polyclonal
antibody for capture and a monoclonal antibody for detection.
Requirement of sensitivity
If there is no hint about the concentration of the analyte in the sample, ELISA kits with a
broad detection range is a better choice. If the concentration of the analyte in the sample

Molecular Diagnosis and Laboratory Surveillance of PPR
50
is very low, ELISA kits with high sensitivity will be better. If the concentration of the
analyte is too high, dilution of samples may be required to adapt to the detection range of
the ELISA kit.
Sample size
ELISA kits usually require from 100ul down to 10ul sample. If the amount of sample is
very small or sample is very precious, better choose ELISA kits that require less amount
of sample.
Recovery and linearity data
Recovery and linearity experiments are used to assess the performance of ELISA kits.
Recovery helps determine whether analyte detection is affected by differences in sample
matrices. High recovery is better. The linearity of dilution determines the extent to which
the dose-response of the analyte is linear in a particular diluent. Ideally, the concentration
of the samples should be similar for all dilutions. Most suppliers provide recovery and
linearity data on product specifications. Besides, other important parameters such as
sensitivity and dynamic range are also provided. ELISA kits of different manufacturers
may have different parameter data. Need careful comparison of these parameter data,
particularly recovery and linearity data, to choose a right ELISA kit.
Detection system
There are several different detection systems in ELISA, including colorimetric,
fluorescent, and luminescent methods. All ELISA involves the immobilization of the
analyte to a surface as well as the use of an enzyme label and a matching substrate.
Choosing an appropriate enzyme and a matching substrate is important. Moreover,
enzyme-substrate reaction conditions, the microplate, and the detection device should be
properly chosen.
The experimental protocol
ELISA kits with simple protocols, convenient operation and short experiment time will
make it easier to do an ELISA test.
Reference
ELISA kits that have been used by other researchers and reported in the literature are
typically more trustworthy. Additionally, manufacturers and products that have received
certification are generally more reliable.
Cost
The price is always a factor for selection, particularly when the budget is limited.
Shipping cost should also be taken into consideration.

Molecular Diagnosis and Laboratory Surveillance of PPR
51
Conclusion
ELISA is a rapid, simple and powerful method, offering a host of advantages over other
common proteomics techniques. However, it requires careful selection of kit for
obtaining best results from an experiment. Hopefully, this document will give some
insight about ELISA and helped to decide whether ELISA is the right method for
intended experiment.
References
Avrameas., S., and Guilbert, B. 1971. Enzymo-immunological determination of proteins with the
aid of immunoadsorbants and enzyme-labelled antigens. C R Acad Sci Hebd Seances Acad
Sci D. 1971 Dec 20; 273(25):2705-7.
Balamurugan, V., Hemadri, D., Gajendragad, M.R., Singh, R.K., and Rahman, H. 2014. Diagnosis
and control of peste des petits ruminants: a comprehensive review. Virus Diseases, 25(1),
39–56; doi. 10.1007/s13337-013-0188-2
Engvall, J.R., and Perlamann, P.1971.Enzyme-linked immunosorbent assay (ELISA). Quantitative
assay of immunoglobulin G. Immunochemistry. Sep; 8(9), 871-874.
Rosalyn, S. Yalow., and Solomon, A.B.1960. Immunoassay of endogenous plasma insulin in
man. Journal of Clinical Investestment, 1960 Jul; 39(7), 1157–1175. doi:
10.1172/JCI104130
Wild, D. 2000. Immunoassay Handbook. 2nd ed. London: Nature Publishing Group. Zweig MH,
Csako G.(1990). High-dose hook effect in a two site IRMA for measuring thyrotropin. Ann
Clin Biochem 27, 494-495

Molecular Diagnosis and Laboratory Surveillance of PPR
52
NATIONAL ANIMAL DISEASE REFERRAL
EXPERT SYSTEM (NADRES)
Parimal Roy ICAR-National Institute of Veterinary Epidemiology & Disease Informatics, Bengaluru, India
Email: [email protected]
During 1987, The Indian Council of Agricultural Research (ICAR) established an All
India Coordinated Research Project on Animal Disease Monitoring and Surveillance,
(AICRP on ADMAS). On 1st April 2000, the AICRP on ADMAS was upgraded to Project
Directorate on Animal Disease Monitoring and Surveillance (PD_ ADMAS) (during IX
Plan). The Directorate got further impetus with the addition of five more collaborating
units in X plan and two mission mode NATP projects viz., Animal Health Information
System and Data monitoring System (AHIS_DMS) and Weather based Animal Disease
Forecasting (WB_ADF) having 17 and 20 collaborating units respectively. Combining the
input from AHIS_DMS and WB_ADF, an interactive, dynamic online animal disease
forewarning system called NADRES (National Animal Disease Referral Expert System)
was developed with overall aim to improve the early warning and response capacity to
animal disease threats in the country for the benefit of farmers. Presently NIVEDI is
having 31 AICRP centres.
Early warning of disease incidence or outbreaks and the capacity of prediction of risk of
spread to new areas is an essential pre-requisite for the effective containment and control
of epidemic animal diseases, including zoonosis. Early warning is based on the concept that
dealing with a disease epidemic in its early stages is easier and more economical than
having to deal with it once it is wide spread. From the public health prospective, early
warning of disease outbreaks with a known zoonotic potential will enable control
measures that can prevent human morbidity and mortality. National Institute of Veterinary
Epidemiology & Disease Informatics developed the software application, NADRES that
systematically collect, verify, analyse and respond to the information from designated
AICRP-ADMAS, unofficial media reports and informal networks. NADRES builds on
the added value combining the alert and response mechanisms of different organizations
like state animal husbandry departments, Departments from universities, department of
Animal husbandry, Dairying and Fisheries, AICRP on ADMAS and other agencies
including NGOS, enhancing the capacity for the benefit of the farmers in the country and
other stakeholders to assist in prediction, prevention and control of animal disease threats,
including zoonosis, through sharing information, epidemiological analysis and joint
missions to assess and control the outbreak, whenever needed. For Zoonotic disease events,
alerts of animal outbreaks or incidence can provide the direct early warning so that human
surveillance could be enhanced and preventive action can be taken. Similarly there may
be cases where human surveillance is more sensitive and alerts of human cases precede
known animal occurrence of disease. Sharing assessments of an outbreak will enable a
joint and comprehensive analysis of the disease event and its possible consequences. Joint

Molecular Diagnosis and Laboratory Surveillance of PPR
53
dissemination will furthermore allow harmonised communication by the Central and state
Animal departments, ICAR-NIVEDI, regarding disease control strategies.
Regarding the joint response to disease emergencies, the three organizations will be able to
respond to a larger number and cover a wider range of outbreaks or exceptional
epidemiological events with the provision of a wider range of expertise. This will improve
the national preparedness for epidemics and provide rapid, efficient and coordinated
assistance in developing disease control strategies.
Specific objectives of NADRES
Allow state and central animal husbandry departments to better prepare themselves
to prevent incursion of animal diseases/infection and enable their rapid containment.
Increase timeliness and sensitivity of alerts
Improve the detection of exceptional epidemiological events at country level
Improve the transparency among different stakeholders
Improve the national surveillance and monitoring systems and strengthen the networks of
veterinary laboratories working in the country.
Improve national preparedness for animal and zoonotic epidemics and provide rapid,
efficient and coordinated assistance to states experiencing them.
Provide the technical support to states on issues at the animal/human interface of
outbreak control.
Disease outbreak data base
Database on disease outbreaks were collected though the networks of AICRP on ADMAS
with 31 centres across the country, provide the regular outbreak information along with
date and location of outbreaks, susceptible population, deaths, attacks etc., Disease data
obtained on a format is entered in to NADRES database in a double data-entry validation
mode to achieve to zero error entry. Database contains the disease events since 1990 was
further improved by inclusion of additional 16 AICRP centres.
Risk factors database
Risk factors such as weather parameters from different sources includes the monthly
precipitation(mm), sea level pressure (millibar),minimum temperature (0C) maximum
temperature(0C) wind speed (m/s), vapour pressure (millibar), soil moisture(%) ,
perceptible water(mm), potential evaporation transpiration (mm), cloud cover(%) etc.,
extracted from National Centre for environmental prediction (NCEP), Indian Meteorological
Department(IMD),National Innovations Climate Resilient Agriculture (NICRA) and
other sources. The remote sensing variables like Normalised Difference vegetative index
(NDVI) and Land Surface temperature were extracted from MODIS/LANDSAT/LISS III
or IV satellite images. The livestock population and densities were extracted from Livestock
census 2012.

Molecular Diagnosis and Laboratory Surveillance of PPR
54
Statistical model
A multivariate logistic regression model was used to predict the probability disease risk in
relation to weather parameters, remote sensing variables and livestock population or
densities. The goal of logistic regression is to find the best fitting (yet biological
reasonable) model to describe the relationship between dichotomous characteristic of
interest (disease outbreak) and a set of predictors (weather parameters, RS variables and
demographics) Logistic regression generates the co-efficients (and its standard error and
significance level) of a formula to predict a logit transformation of the probability of
presence of the characteristics of interest.
where p is the probability of presence of the characteristic of interest. The logit
transformation is defined as the logged odds
ather than choosing parameters that minimize the sum of squared errors (like in
ordinary regression), estimation in logistic regression chooses parameters that maximize
the likelihood of observing the sample values.
Overall fit of model
The null model −2 Log Likelihood is given by −2 * ln (L0) where L0 is the likelihood of
obtaining the observations if the independent variables had no effect on the outcome. The
full model −2 Log Likelihood is given by −2 * ln (L) where L is the likelihood of
obtaining the observations with all independent variables incorporated in the model.
The difference of these two yields a Chi-Squared statistic which is a measure of how well
the independent variables affect the outcome or dependent variable. If the P-value for the
overall model fit statistic is less than the conventional 0.05 then there is evidence that at
least one of the independent variables contributes to the prediction of the outcome. Cox &
Snell R2 and Nagelkerke R
2 are other goodness of fit measures known as pseudo R-
squares. Note that Cox & Snell’s pseudo R-squared has a maximum value that is not 1.
Nagelkerke R2 adjusts Cox & Snell’s so that the range of possible values extends to 1.
Hosmer-Lemeshow test
The Hosmer-Lemeshow test is a statistical test for goodness of fit for the logistic
regression model. The data are divided into approximately ten groups defined by
increasing order of estimated risk. The observed and expected number of cases in each
group is calculated and a Chi-squared statistic is calculated as follows:

Molecular Diagnosis and Laboratory Surveillance of PPR
55
with O, Eg and ng the observed events, expected events and number of observations for the
gth risk decile group, and G the number of groups. The test statistic follows a Chi-squared
distribution with G−2 degrees of freedom.
A large value of Chi-squared (with small p-value < 0.05) indicates poor fit and small Chi-
squared values (with larger p-value closer to 1) indicate a good logistic regression model
fit.
Early warning system (EWS)
Early identification of an infectious disease outbreak is an important first step towards
implementing effective disease interventions and reducing resulting mortality and
morbidity. The geographic and seasonal distribution of many infectious diseases are
associated with climate and therefore the possibility of using seasonal climate forecasts as
predictive indicators in disease early warning system (EWS) is an interest of focus.
Geographic Information system (GIS), remote sensing (RS) and Global Positioning system
(GPS) are the three commonly used veterinary geo-informatics technologies employed in
this digital era for rapid communication of data for better management of animal diseases.
Early warning systems are combinations of tools and process embedded within
institutional structures coordinated by national or international agencies. These systems
are composed of four elements depending upon they focus on specific hazard or many,
namely, knowledge of risk, a technical monitoring and warning services, dissemination of
meaningful warnings to at-risk areas, and farmers awareness and preparedness to act.
Warning services lie at the core of these systems, and how well they operate depends on
having a sound scientific basis for predicting and forecasting. As early warning systems grow
in geographical coverage and sophistication, false alarms to in rise. High false alarms can
undermine the public confidence, breed mistrust, dilute the impact of alerts and reduce the
credibility of future warnings.
Classification table
The classification table is another method to evaluate the predictive accuracy of the
logistic regression model.
In this table the observed values for the dependent outcome and the predicted values (at
a user defined cutoff value, for example p=0.50) are cross-classified and provides the
accuracy index for assessment model performance in prediction.

Molecular Diagnosis and Laboratory Surveillance of PPR
56
Table 1: Accuracy of Prediction
Sl. No. Diseases Accuracy (%)
1 Anthrax 95.52
2 Babesiosis 96.14
3 Black Quarter 92.43
4 Bluetongue 97.99
5 Enterotoxemia 95.98
6 Fasciolosis 97.06
7 Foot and mouth disease 86.72
8 Haemorrhagic septicaemia 90.58
9 Peste des petits ruminants 90.12
10 Sheep & Goat pox 95.52
11 Swine fever 95.37
12 Theileriosis 96.60
13 Trypanosomosis 96.45
Internal Accuracy was performed using 10 years of data. Accuracy obtained was > 90%
except Foot and mouth disease (86.72%).
The probability of disease outbreak was categorized in 6 risk levels- No risk (NR), Very
low risk (VLR), Low risk (LR), Moderate risk (MR), High risk (HR) and Very high risk
(VHR) for enabling the stake holders to take appropriate control measures by suitably
allocating available resources.
Table 2: Probability distribution of risk
interpretations
Figure 1.Risk prediction of Anthrax for the month of June 2018
Sl.
No.
Probabilit
y of risk
Interpretation
1 0 No risk/No or inadequate
data 2 0-0.20 Very low risk
3 0.21-0.40 Low risk
4 0.41-0.60 Moderate risk
5 0.61-0.80 High risk
6 0.8-1.0 Very high risk

Molecular Diagnosis and Laboratory Surveillance of PPR
57
NADRES outlook
The ICAR-NIVEDI website consists of details regarding NADRES database, Monthly
prediction bulletin, Risk maps, disease maps and Disease trend analysis.
LDF-mobile application
To extend the reach of the NADRES forewarning bulletin among the various stakeholders,
a Mobile Application (app) “LDF-Mobile App” was developed. The forewarning
methodology adapted in the “mobile app” remains the same as monthly bulletin. In
addition to forewarning, the LDF-Mobile App also provides the details of clinical samples
to be collected in case of outbreaks of the listed diseases for laboratory confirmation.
Immediate preventive measures to be taken up in case of positive prediction/disease
confirmation. The LDF mobile app is available at ICAR-NIVEDI website. It will also be
made available on Google play store.

Molecular Diagnosis and Laboratory Surveillance of PPR
58
LABORATORY MANAGEMENT WITH BIOSAFETY
AND BIOSECURITY PRACTICES
Asadulghani Head, Biosafety and BSL-3 Laboratory, icddr,b, Mohakhali, Dhaka-1212
Email: [email protected]
Safety-first” is to keep the workers and
the environment safe. In biomedical
research and diagnostic laboratories,
either in human or in animal sector,
biosafety and biosecurity (BSBS) issues
have to be prioritized as part of
laboratory management system.
Prioritizing BSBS issues, the
laboratorians’ responsibility is to ensure
Biosafety & Biosecurity
Accuracy Timeline
accuracy/ assure quality (QA) of laboratory diagnosis and research. The intended
diagnosis or research has to be completed by a defined timeline (TL). Thus, the three
priorities of biomedical and microbiological research and diagnostic laboratories, in
general, are BSBS, QA and TL. Any violation in those three may have a serious human,
animal and environmental health impact.
Biosafety is to protect not just humans from harm but Earth’s entire biosphere as well. On
this regard we follow two principles – Risk Assessment and Containment. Risk
assessment identifies biohazardous and non-biohazardous materials, infectious and non-
infectious biohazards, and among the infectious biohazards the risk group of the
infectious agents may be present in the specimens under study. While we contain the
biohazard following the risk assessment, we ensure optimized personal practices, use
safety equipment to manipulate biohazardous materials within, and we work in a
dedicated and properly designed laboratory facility.
Laboratory
Priorities

Molecular Diagnosis and Laboratory Surveillance of PPR
59
As a consequence, implementing biosafety policies and procedure, we mitigate
unintentional release of biohazards from the laboratory. Whereas, biosecurity is to limit
access to facilities, biological materials, and related information to protect intentional
release of biohazard. All the diagnostic and research work have to be conducted
following approved standard operating procedures. Relevant staff members have to be
trained not only to develop SOPs but also to work following SOPs strictly. To ensure
safety in quality and vice versa, SOPs have to de developed for all laboratories
procedures and operations, validated, and strictly followed.
Early diagnosis and detection of infectious diseases is the key parameter for robust
control of spread of infectious diseases. Thus, scientists are moving from culture based
techniques to culture free techniques, which are faster, more sensitive and specific. This
ultimately helps the scientists to detect and control the diseases outbreak within shortest
possible time. Research and development for disease diagnosis, control and eradication
must also have specific goals, to achieve within clearly defined and determined timeline,
to contribute in the control of spread and ultimately eradicate the infectious disease if
applicable.
This is an established fact that human errors, poor technique, and contravening the
standards contribute to unnecessary exposure and compromise the best safeguards set
into place for protection. Thus, it is necessary to implement and accordingly follow the
policies and procedures of biosafety in microbiological and biomedical research and
diagnostics to mitigate human errors, poor techniques, as part of standard practices,
optimizing safeguard at the workplace.

Molecular Diagnosis and Laboratory Surveillance of PPR
60
RT-PCR: THE TECHNIQUE FOR
DETECTION OF PPR
Md. Abu Yousuf*, Mohammed A Samad, Mahmudul Hasan and Md. Giasuddin Animal Health Research Division, Bangladesh Livestock Research institute, Savar, Dhaka-1341
Email: [email protected]
Introduction
Peste des petits ruminants is a highly contagious and infectious viral disease of domestic wild small ruminants. PPRV is a member of the genus Morbillivirus under the family Paramyxoviridae and order Mononegavirales. It is closely related to the rinderpest virus (RPV), the measles virus (MV) of humans, the canine distemper virus (CDV) of dogs and some wild carnivores, and the morbilliviruses of aquatic mammals. PPR is primarily a disease of goats and sheep and goats are usually more severely affected than sheep. The genome of PPRV is non-segmented, negative-strand enveloped virus. PPRV consist of 15948 nucleotides that encodes eight proteins: the nucleocapsid protein (N), the phosphoprotein (P), the matrix protein (M), the fusion protein (F), the haemagglutinin protein (H), the polymerase protein (L) and the two nonstructural proteins, C and V. PPRV genome is organized into six contiguous, non-overlapping transcription units corresponding to the gene of the six structural viral proteins in the order of 3'-N-P-M-F-H-L-5' in the genome sense. It is an economically significant disease of small ruminants such as sheep and goats. In many areas of Asia, small ruminant production and therefore the livelihoods of poor farmers is threatened by transboundary animal diseases (TADs) like Peste des petits ruminants (PPR).The disease causes severe losses to small ruminant production and is presently considered as one of the major threats to about 22 million small ruminant population of Bangladesh where mortality may reach up to 100% in an outbreak. Because of its high mortality rate, PPR affects food security directly by reducing the availability of meat and milk for family consumption and of funds for purchasing other commodities and foods as these animals are the main assets for the poor. Thus its control is a major goal for the programs aimed at poverty alleviation. Rapid diagnosis is essential for effective control measures. In South Asia Global Framework for the Progressive Control of Transboundary Animal Diseases (GFTADs) has recognized three priority diseases: FMD, PPR and HPAI. Considering the importance of PPR disease, SARRC regional cooperation and other international organization like FAO came forward to establish specialized laboratory on PPR in BLRI premises to diagnosis the disease, increase coordination among the laboratory and to conduct other research related to this disease.
Bio-safety
Containment Level BSL–II should be used for overall laboratory practice. Handling of
PPR suspected samples should be carried out under a Biosafety Cabinet Class II. Though
PPR is not zoonotic disease but sample must be handled with care using Personal
Protection Equipment (PPE) like lab coat, disposable gloves.

Molecular Diagnosis and Laboratory Surveillance of PPR
61
Conventional PCR
One-step single tube reverse transcription PCR assay incorporating primer specific for N
of PPR virus is used to detect the virus. This assay utilizes the Nexus, eppendorf, double
house PCR machine and operations according to standard operating procedures. In house
developed internal quality control (IQC materials are used in both extraction processes
and during test run. Results inference is drawn after electrophoresis using 100kb DNA
ladder. The sequential steps of PCR has given below:
annealing 60°C
denaturation 95°C
extension 72°C
Gel electroforesis
1
2
4
8
2n
n cycles
Specificity determined by 2 primers
Traditional PCR (Semi-quantitative)
RNA isolation (Extraction)
Viral RNA will be extracted from the tissue suspension or swab samples using RNeasy
Kit (PureLink RNA mini Kit , Ambion, USA) as recommended by the manufacturer.
Add 400 μl RLT buffer (add 10 μl of β- mercaptoehthanol to 1 ml RLT buffer) and
300 μl tissue suspensions in an Eppendorf tube, vortex and incubate for 3 minutes at
room temperature.
Then add 700 μl of 70% ethanol to the supernatant, mix and transfer to an RNeasy
spin column place in a 2 ml collection tube.

Molecular Diagnosis and Laboratory Surveillance of PPR
62
Transfer 700 μl in spin column from Eppendorf tube, centrifuge at 12000-13000 rpm
for 30 sec, discard the flow-through at the collection tube and set again in the
collection tube
Take the rest 700 μl and repeat the procedure as above
Add 700 μl of wash buffer-1 to the column, centrifuge as above and discard the flow-
through.
Add 500 μl of wash buffer-2 (add 4 volumes ethanol to 1 volume of RPE) to the spin
column and centrifuge for 30 sec at 12000-13000 rpm.
After removal of the flow-through, add again 500 μl of wash buffer-2 in the column
and centrifuge for 2 min at 12000-13000 rpm.
Place the spin column in a 1.5 ml Eppendorf tube and add 50 μl of RNAse free water
in to the centre of the column and centrifuge for 1 min as above.
Discard the RNeasy spin column and label Eppendorf tube containing RNA and store
at -20ºC or at -70ºC for short term and long term storage, respectively.
Master mix preparation
Total Sample (including positive and negative control)
Ambion , the RNA company Quantity/ sample Sample no.
Nuclease free water (μl) 5
2x RT- PCR buffer ( μl) 13
Primer F (100 pmol/ μl) (μl) 0.5
Primer R (100 pmol/ μl) 0.5
25x RT-PCR Enzyme Mix (μl) 1.0
Total Master mix 20.0
Template RNA (μl) 5
Final Reaction Volume (μl) 25
N gene specific primer
Gene Primer Sequence Position Size References
N
NP3 5´-TCTCGGAAATCGCCTCACAGACTG-3´ 1232-
1255 351
bp
Couacy-
Hymann, et
al,2002 NP4 5´-CCTCCTCCTGGTCCTCCAGAATCT -3´ 1583-
1560

Molecular Diagnosis and Laboratory Surveillance of PPR
63
Picture 1. Preparation of master mix and performing conventional RT-PCR
Thermal profile
RT step Initial
denaturation
Denaturation Annealing Elongation No. of
cycles
Final
elongation
Temp 500 C 95
0 C 94
0 C 55
0 C 72
0 C
35
720 C
Time 30 min 15 min 30 sec 30 sec 30 sec 10 min
PC NC S10 S9 S8 S7 S6 S5 S4 S3 S2 S1 M
Figure 1. Agarose gel electrophoresis of PCR products (351 bp) amplified with NP3 and NP4,
PPR specific primers. Lane M:100bp DNA molecular weight marker; Lane PC: Positive
control; Lane NC: Negative control; Lane S1-S10: Field samples
351bp

Molecular Diagnosis and Laboratory Surveillance of PPR
64
Analyses of RT-PCR products by agarose gel electrophoresis
Prepare agarose gels 1 to1.5% (w/v) in 1x TAE or TBE buffer
Add ethidium bromide to the agarose solution at 0.5 μg/ml
Pour the agarose solution containing ethidium bromide into the gel casting tray
When the gel set completely, remove the comb and transfer the gel into the
electrophoresis tank and cover with 1x TAE buffer
Mix DNA samples with DNA loading buffer (5 vol. DNA solutions with 1 vol. of
Bromophenol blue/ xylene cyanol as loading buffer) and load them into individual slots
DNA marker mixing with loading buffer
Perform electrophoresis at 100 V for 30 min
Place on the UV transilluminator in the dark chamber of the image viewing and
documentation system
The image will be shown in the monitor and to be captured to analyses the result
Trouble shooting of PCR
Observation Possible cause Solution
No product
Incorrect annealing temperature Recalculate primer Tm values test
annealing temperature gradient starting at
50 C below the lower Tm of the primer pair.
Poor primer design Verify that primers are non-
complementary both internally and to each
other.
Poor primer specificity Verify that oligos are complementary to
proper target sequence
Missing reaction component Repeat reaction setup
Suboptimal reaction conditions Optimize annealing temperature by testing
an annealing temperature gradient, starting at
50 C below the lower Tm of the primer pair.
Poor temperature quality Check 260/280 ratio of DNA template
Presence of inhibitor in reaction Further purify starting temperature by
alcohol precipitation, drop analysis or
commercial clean up kit decrease sample
volume.
Contamination of reaction tubes
or solutions
Reaction tubes prior to use to eliminate
biological inhibitors. Prepare fresh
solutions or use new reagents and new
tubes.

Molecular Diagnosis and Laboratory Surveillance of PPR
65
Special practices for PCR lab
Keep pre-PCR (extraction), PCR (master mix & amplification) and post-PCR
(analysis) work as isolate as possible.
Strictly adhere to the work flow for pre-PCR, PCR and post-PCR activities. Take
utmost precaution to avoid carry-over or cross contamination of PCR reagents with
(a) previously amplified PCR products, or (b) extracted DNA/RNA.
Use separate sets of equipment, appliances and supplies for different steps of PCR.
Do not move pipette, tips, tubes, marker, pen, note books, etc. between pre-PCR,
PCR and post-PCR sections.
Store consumables, reagents and water separately for pre-PCR, PCR and post-PCR
work. Aliquot reagents and water in convenient volumes.
Sharing of reagents and supplies among a group of researchers must be based on
mutual trust and understanding under strict control of a single leader. Always use
Incorrect temperature
concentration
For low complexity temperature (plasmid,
lambda, BAC DAN), use 1 pg-10 ng of
DNA per 50 μl reaction.
For Higher temperature complexity
templates (genomic DNA), use 1 ng- μg of
DNA per 50 μl reaction.
Multiple or
non-specific
products
Primer annealing temperature
too low
Increase annealing temperature.
Poor primer design Verify that primers are non-complementary
both internally and to each other.
Excess primer Primer concentration can range from 005
– 1 μM in the reaction.
Incorrect temperature
concentration
For Higher temperature complexity
templates (genomic DNA), use 1 ng- μg of
DNA per 50 μl reaction.
Concentration with exogenous
DNA
Setup dedicated work area and pipettor for
reaction setup. Wear gloves during
reaction setup.
Incorrect
product size
Incorrect annealing temperature Recalculate primer Tm values
Mispriming Verify that primers have no additional
complementary regions within the
template DNA.
Improper Mg ++
concentration Adjust Mg++
concentration in 0.2-1 mM
increments.
Nuclease concentration Repeat reactions using fresh solutions.

Molecular Diagnosis and Laboratory Surveillance of PPR
66
disposable gloves and change gloves frequently based on logical judgment. Consider
all surfaces potentially contaminated and avoid touching them wearing gloves
Plan and organize work before beginning a test. Do all calculations; fill in
worksheets; ensure availability of all reagents and supplies at respective work areas
before start.
Never reverse work-flow. Ideally, nothing should come back from the post-PCR
section to the pre-PCR or PCR sections without proper decontamination. Never bring
cDNA to the extraction and master-mix room.
Clean and wipe all work surfaces with 70% alcohol before and after each session
briefly pulse-centrifuge tubes to collect reagents at the bottom before opening.
Do not leave reagents on the bench (even on ice) any longer than necessary.
Use only freshly prepared ice flakes. Do not freeze ice for re-use.
Archive all information about new clones and newly synthesized primers.
Work wrap-up
Check all your samples, buffer, media and reagents in the refrigerator, freezer and
shelves.
Discard the materials that are no longer required (consult with your supervisor).
Handover the materials to the supervisor, along with a list, if these worth keeping
(isolates, clones, selected samples for future study, developed/prepared reagents, etc.)
Don’t forget to label properly
Clean all glassware and return to respective place/person, as appropriate
Return books, dissertations, reprints, etc. that you might have borrowed

Molecular Diagnosis and Laboratory Surveillance of PPR
67
DETECTION OF PPRV ANTIBODY IN SERA BY
COMPETITIVE ELISA (cELISA)
Md. Abu Yousuf* Mohammed A Samad, M Rafiqul Islam1 and Md. Giasuddin
Animal Health Research Division, Bangladesh Livestock Research Institute, Savar, Dhaka-1341 1Livestock Division, Bangladesh Agricultural Research Council, Farmgate, Dhaka-1215
Email: [email protected]
Introduction
Competitive ELISA (cELISA) based on monoclonal antibodies specific for N –protein
and H-protein developed for detection of antibodies in animal sera. Goats and sheep
experienced PPRV infection at younger age remained sero-positive for 1-2 years
following exposure. cELISA has been widely used to detect PPR antibodies in many
countries. In the N-protein cELISA, the serum antibodies and the MAB complete on
specific epitope on nucleoprotein generated through recombinant technology using
baculovirus expression vector system. The sera samples were tested by c-ELISA. (ID vet.
Innovative Diagnostics, France) according to the instruction.
Principle of cELISA
The wells are coated with purified recombinant PPR nucleoprotein (NP).The samples to
be tested and the controls are added to the micro wells. Anti-NP antibodies, if present
form an antigen-antibody complex which masked the NP epitopes. An anti-NP
peroxidase (PO) conjugate was added to the micro wells. It fixed to the remaining free
NP epitopes, forming an antigen conjugate peroxidase complex. After washing in order to
eliminate the excess conjugate, the substrate solution (TMB) is added. The resulting
coloration depend on the quantity of specific antibodies present in the sample to be
tested:
- In the absence of antibodies, a blue solution appeared which becomes yellow
after the addition of the stop solution
- In the presence of antibodies, no coloration appeared.
- The absorbance is read at 450 nm.

Molecular Diagnosis and Laboratory Surveillance of PPR
68
Requirements for sample collection
70% Alcohol/ iodine swab
5 ml sterilized disposable syringe
Eppendorf tube
Centrifuge machine (Rotor for eppendorf tube )
Pipette ( 100µl -1000 µl capacity with tips)
Wet ice or ice pack
Vaccine carrier or suitable carrier
Refrigerator
Blood sample collection and serum separation
The goat blood is collected in early morning. After controlling the goat vein was
detected. (2-3) ml of fresh blood sample was collected from each of the animal
aseptically by puncturing jugular vein after swabbing with 70% alcohol or iodine swab in
a gentle manner. The loaded syringe was remaining in inverted condition at (30-40) min
for blood clotting. In that way blood sample was taken from different aged goats. After
clotting of blood, serum was separated into eppendorf tube, numbering and packaging
was done remain in freeze condition (2-8) for further use.
Figure1. Collection of blood and performed cELISA
Sample labeling
Specimen type/name. e.g. serum, blood, swab etc.
Unique identification number
Place of collection (location)
Date of collection

Molecular Diagnosis and Laboratory Surveillance of PPR
69
Equipment
Samples (Serum)
cELISA kit component
96 well Micro plate
Positive control
Negative control
Tips (200 1000 , 2000 ) Single channel pipette
Wash solution
Stop solution
ELISA washer
ELISA reader
Thermomixer compact (Eppendorf, Germany)
Minimum glass-distilled or deionized water
Incubator
Micropipettes (Multichannel, Single channel )
Graduated cylinders (10-2000ml)
Graduated pipettes (1-20 ml) with suitable bulbs
Storage bottles with closures (1-100m1)
Dilution tubes (2-4 ml.)
Reagents
The competitive ELISA kit is developed by ID. Vet Innovative Diagnostics Montpellier,
France.
Kit components
Screen® PPR Competition Kit, ID vet Innovative Diagnostics contain the following
reagents and chemicals
Micro plates coated with PPR recombinant nucleoprotein
Anti-NP-HRP concentrated conjugate (10X)
Positive control
Negative control
Dilution buffer 13
Dilution buffer 4
Wash concentrate 20X)
Substrate solution
Stop solution (H2SO4 0.5 M)
Sample preparation
In order to avoid differences in incubation times between samples, prepared a 96 well
plate containing the test and control samples, before transferring them into ELISA micro

Molecular Diagnosis and Laboratory Surveillance of PPR
70
plate using a multi-channel pipette.
Wash solution preparation
If necessary, brought the wash concentrate (20X) to room temperature (21°C ± 5°C) and mix
thoroughly to ensure the wash concentrate is completely solubilized. Prepare the wash
solution (1X) by diluting the wash concentrate (20X) in distilled water.
Test procedure of cELISA
Allow all reagents to come at room temperature (21 0c ±5
0c) before use. Homogenize all
reagents by inversion or vortex.
1. Add:
a. 25 μl of dilution buffer 13 to each well
b. 25 μl of the positive control to well A1 and B1
c. 25 μl of the negative control to wells C1 and D1.
d. 25 μl of each sample to be tested to the remaining wells.
2. Incubate 45 min + 4 min at 370C (± 3
0C)
3. Wash each well 3 times with approximately 300 μl of the wash solution. Avoid
drying of the well between washings.
4. Prepare the conjugate 1x by diluting the conjugate 10x to 1/10 in dilution buffer.
5. Add 100 µl of the conjugate 1x to each well.
6. Incubate 30 min ± 3 min at 210 C ( ±5
0 C)
7. Wash each well 3 times with approximately 300 µl of the wash solution. Avoid
drying of the wells between washings.
8. Add 100µl of the substrate solution to each well.
9. Incubate 15 min ± 2 min 210C (±5
0 C) in the dark.
10. Add 100 µl of the stop solution to each well in order to stop the reaction.
11. Read and record the O.D. at 450 nm with the help of ELISA plate reader.
12. Finally the reading data is place into data sheet of Microsoft Excel program and
save it in the computer hard.
Validation
The test is validated if:
The mean value of the negative control O.D (ODNC) is greater than 0.7.
ODNC >0.700
The mean value of the positive control (ODPC) is less than 30% of the OD.
ODPC /ODNC <0.3

Molecular Diagnosis and Laboratory Surveillance of PPR
71
Interpretation
For each sample, the competition percentage is calculate using the following formula
S/N % =
x 100
Sample presenting a S/N%:
- Less than or equal to 50% are considered positive
- Greater than 50% and less than or equal to 60% are considered doubtful
- Greater than 60% are considered negative
Result Status
S/N % ≤ 50 % Positive
50 % < S/N % ≤ 60% Doubtful
S/N % > 60% Negative
Precautions
Do not pipette by mouth.
The substrate solution can be irritating to the skin.
The stop solution (H2SO4 0.5M) can cause serious burns (R35). In the event of
contact with skin or eyes, wash immediately and abundantly with water and consult a
doctor (S26).
The conjugate, the controls and the substrate solution must be stored at 5 ( 3 )
The other reagents can be stored between +2 and +26
Components bearing the same name (wash solution, dilution buffers) can be used for
the entire ID vet product range.
Do not expose the substrate solution to bright light nor to oxidizing agents.
Decontaminate all reagents before elimination.

Molecular Diagnosis and Laboratory Surveillance of PPR
72
CELL CULTURE FOR VIRUS ISOLATION
AND IDENTIFICATION
Md. Giasuddin*, Md. Abu Yousuf, Amal Kumar Saha Animal Health Research Division, Bangladesh Livestock Research Division, Savar, Dhaka-1341
Email: [email protected]
Introduction
The scientists started try to alive animal cell in vitro before near about 140 years. Now a
days cell culture is a very vital tools of many biological research. Once it was very
difficult to test any microbes in live animal but cell culture has made it easy to testing this
type of experiment. Cell culture also has been used for vaccine development. Somewhere
it is vigorously using to heal human diseases and burn injured patient. Specially stem cell
commonly using for human patient. Cell culture is doing for investigate the normal
metabolic pathways can be investigated by applying radioactively substrates and looking
at products. It can be used for effects of compounds on specific cell types such hormones,
growth factors may be evaluated by cell culture. Virus isolation in cell culture is very
important for preservation of the isolates and its subsequent use.
Regional Leading Diagnostic Laboratory for PPR, Bangladesh is using cell culture for
PPR virus isolation, identification and vaccine development.
Vero Cell culture procedure
Equipment
1. Bio safety cabinet- class-II
2. Incubator with CO2 facilities
3. Centrifuge machine with minimum capacity 5000 RPM and 15 ml falcon tube holder.
4. Kitchen refrigerator
5. -800 deep fridge
6. Liquid Nitrogen container
7. Inverted Microscope
8. Hot air oven
9. Water bath
10. Autoclave machine
11. Distilled water plant
Prime chemical reagents of (vero/ BHK21) cell culture
1. Minimum Essential Midium (MEM) or Dulbecco Minimum Essential Midium
2. Fetal calf serum/ Bovine calf serum
3. L-Glutamin
4. Sodium Bicarbonate

Molecular Diagnosis and Laboratory Surveillance of PPR
73
5. Antibiotic (Penicillin Streptomycin)
6. HEPES
7. Trypsin
8. Cell storage media (DMSO)
Essential auxiliary chemicals
1) Distilled deionized water
2) PBS
3) Alcohol (As cleaning reagent)
4) Disinfectant
Plastic ware
1. Tissue culture flasks (25cm2,75cm
2)
2 . Pipettes and Pipette holder
3. Tips
4. Falcon tubes
5. Syringe
6. Syringe filter (.22 micron & .45 micron)
7. 96 well flat bottom plate
8. Tub
Glassware
1. Graduated bottle (500 ml, 250 ml, 1000 ml)
2. Measuring cylinder
3. Biker (250 ml, 500 ml, 1000 ml)
5. Cell counter slide
6. Petri dish
Other consumables
1. Hand gloves
2. Apron
3. Masks and hair cover
4. Permanent marker
5. Aluminum foil
6. Cotton
7. Tissue paper
8. Autoclable polythene bag
9. Lab. shoe
Chemical reagents storage temperature
1. MEM at + 40C
2. Fetal calf serum/ Bovine calf serum at -200
to -800C

Molecular Diagnosis and Laboratory Surveillance of PPR
74
3. L-Glutamin at -200C
4. Sodium Bicarbonate at +40 C
5. Antibiotic (Penicillin Streptomycin) at -200C
6. HEPES at +40 C
7. Trypsin at -200C
8. Cell storage media (DMSO) at -200C
** Growth and Maintenance media have to be kept at +40 C
N.B. a) All the stored chemical reagents are have to be kept in water bath at 370C for
melting to bring in working condition before any cell culture related tasks.
b) For cleaning all the bottle of ingredients have to be wiped by tissue paper and then
wash by 70% alcohol and keep in bio safety cabinet.
Necessary reagents for Cell growth and maintenance media preparation
1. Minimum Essential Midium (MEM) or Dulbecco Minimum Essential Midium
2. Fetal calf serum/ Bovine calf serum
3. L-Glutamin
4. Sodium Bicarbonate
5. Antibiotic (Penicillin Streptomycin)
6. HEPES
7. Double distilled sterile water
Cell culture procedure
Picture 1. Cell culture in the laboratory
Start biosafety cabinet and switch on UV light. Stop the UV light after recommended
time (3-5 minutes) and then start the airflow and normal light. At first a preserved cryo
tube of cell has to pull out from liquid nitrogen and inoculate in growth media. And keep
in 370 C incubator for 24 hours and for betterment of cell growth cell inoculated flask can
be washed by PBS and add GM again in flask at next day. It will take 3 days for 80-90%
confluent condition.

Molecular Diagnosis and Laboratory Surveillance of PPR
75
After 80-90% confluent of flask bottom it may ready for further tasks especially for virus
isolation, identification and vaccine development and cell multiplication (Sub-culture).
Sub culture
Confluent cell culture flask pull out from incubator and wash and clean at the same
way.
Pour out growth media from flask and wash 2-3 times by PBS.
Trypsinized and allocate in several flask and keep it for growth.
Picture 2. Cell culture in the laboratory
Add 200-300 micro liter trypsin for 252cm flask for trypsinization (trypsin quantity
will depend on flask size).
After trypsinization add some growth media and shake or pipetting for cell cluster
uniformly distribution in media. Add more GM in flask according to expected
concentration and flasks.
And keep in incubator at 370Cfor growth.
Cell counting by hemocytometer
Step 1
Take a hemocytometer which has 9 square chambers. We shall count of cell total 5
chambers among 9 chambers according to table given be low.
1 2
5
3 4
Keep 100 micro litre cell in one chamber
Add 100 micro litre typan blue in chamber

Molecular Diagnosis and Laboratory Surveillance of PPR
76
Count every square separately
Count viable and non-viable cell differently
Step 2
We have to count some parameters for cell number determination. Those are-
Total viable cells
Total nonviable cell
Average number of cell per square
Dilution factor
Concentration (Viable cells /ml)
Suppose we have got 54 viable cells and 3 nonviable cells. In that case calculation will
be,
1. % of viable cells = # viable cells/ Total # cells x 100 = 54/57= 0.947x100= 94.7%
2. Average # of cells/ square= Viable cells/No. Square= 54/5=10.8
3. Dilution factor= Final volume /volume of cell= 200 micro litre/100 micro litre=2
*(Cell 100 micro litre and Typan blue 100 micro litre)
4. Concentration (viable cell) = average number of cell/Dilution factor x 104
= 10.8 x 2
x 104 = 216000 cells/ ml. In scientific notation 2.16 x10
5 cell/ml
Sample collection
Many types of viral samples may come from field level. After a certain processing we
can use these collected samples for our purposes like virus isolation and identification.
Samples types and collection procedures have described below-
Swab samples
Use a cotton/dacron swabs to collect the specimen. Collect swab from posterior nasal
cavity or Conjunctiva, avoiding external secretion. Keeping swabs moist after collection
is most important. Place swab in 3-4 ml Viral Transport Media (VTM). Any sterile
isotonic fluid, like phosphate buffered saline (PBS) with antibiotics (penicillin 200
International Units/ml and streptomycin 200 μg/ml or Gentamycin 500 ug/ml), or
common tissue culture medium like Eagle’s MEM can be used. Swabs may be broken off
to fit within the capped tube containing the VTM and shipped. Alternatively,
whirl/agitate the swab in the media for several minbefore removing it and shipping the
suspension.
Commercially available kits containing swabs and viral transport media are acceptable.
Blood sample
Collect blood aseptically from Jugular vein in heparini zed tubes when the animal is in an
acute febrile stage of the disease and store at chilled condition.

Molecular Diagnosis and Laboratory Surveillance of PPR
77
PM tissue samples
Collect lung, mesenteric and bronchial lymph nodes aseptically from PPR suspected dead
goats during post-mortem.
Transportation of the samples
Samples should be transported in chilled condition with ice packs or with wet ice. They
should be received by the testing laboratory within 48 hours of collection. If shipment is
delayed and facilities are available, the specimens should be frozen at -70o C and shipped
on dry ice. Otherwise, store specimens in refrigerator (freezing at -20o C reduces viability
of virus).
Preparation of inoculums for virus isolation
Swab samples
Draw 0.5 ml of sample suspension into 1 cc syringe. Attach 0.2um syringe filter
(such as an Acrodisc 13) and push sample through filter directly onto cells or in an
eppendorf tube. The drawback is that some samples will not pass through the filter.
To overcome clogging the filter make sure suspension is well homogenized before
drawing into the syringe.
Blood samples
Centrifuges heparinized blood at 1000 rpm for 5 minute and collect the buffy coat.
Wash the white blood cell in Minimal Essential Medium (MEM) containing
antibiotics (penicillin 200 International Units/ml and streptomycin 200 μg/ml).
Resuspend the pellet cells in 1.0 ml of MEM plus antibiotics. Inoculate 0.5 ml of the
cell suspension into confluent Vero cells grown in 25 cm2 tissue culture flask.
Tissue samples
Use individual organ or pool the collected tissues (lung, lymph nodes) collected from
the same animal, weigh and macerate them with sterile mortar and pestle while the
tissues are still frozen.
Add PBS to make 20% (w/v) suspension and collect the suspension in a sterile tube.
Centrifuge tissue suspension at 3000 rpm for 10 minutes. Collect the supernatant in
fresh sterile Falcon tubes and add antibiotic (Gentamycin) at 500 μg/ml and store at -
700C.
Prior to inoculation, filter the suspension, using Acrodisc® Syringe filter (13 mm
diameter, 0.2 μm pore size) [Sigma-Aldrich or similar] and use the filtrate as
inoculums.
Inoculation in vero cells
Maintain the Vero cell in media M199 with bovine fetal serum (10% for growth and
5% for maintenance).
Inoculate the flask when Vero cell monolayer becomes confluent.

Molecular Diagnosis and Laboratory Surveillance of PPR
78
Discard the spent medium, wash the flask/s with 10 ml sterile pre-warmed PBS for 2
times.
Inoculate the cells with the prepared sample suspension at 200 μl/25 sq. cm flasks.
Run positive (with PPR Vaccine virus, 200 ul/ per flask) and negative control (200 μl
PBS)
Spreads the inoculums all over the cell sheet and incubate the flasks at 37°C for 1
hour for adsorption of the virus. Spread the inoculums by tilting the flasks every 15
minutes. After one hour, add 5ml of growth medium containing 10% fetal calf serum
to the flasks without removing the excess inoculums.
Incubate the flasks at 370 and examine twice daily for the appearance of cytopathic
effects (CPE).
Harvesting of virus
Harvest the culture medium at 5thday of each passage or when the maximum CPE
manifested.
Collect the infected tissue culture by freezing and thawing for 4 times, and centrifuge
at 2500 rpm for 5 minutes, Collected the suspension in sterile 50ml Falcon tubes.
Use the collected suspension for further passage in fresh Vero cells.
Special practice for tissue culture lab
Use laminar flow cabinet for preparation/handling of fresh and uninfected cell culture
and the biosafety cabinet for handling infectious materials.
Wipe all work surfaces liberally with a disinfectant (70% alcohol) before and after
each session of work.
Wipe hands with 70% alcohol before commencement of the sterile work.
Use only sterilized glassware/plastic ware. Decontaminate the surfaces of all
glassware with 70% alcohol before placing into the laminar flow cabinet or biosafety
cabinet.
Flame all bottles at around the neck before and after opening and before re-closing.
(Note – Use of spirit lamp or Bunsen burner is not allowed in the biosafety cabinet.
Rely on decontamination with 70% alcohol)
Store stock and working media/reagents in a separate freezer designated for tissue
culture reagents only. Divide all reagents into suitable aliquots whenthey are opened
for the first time.
Store viruses and infected cultures in clearly labeled vials and place them in separate
designated freezer.
Collect all used glassware in disinfectant (Sodium hypochlorite/Virkon) and follow
the procedure of cleaning and disinfection.
Clean and disinfect all surfaces daily at the end of the day.

Molecular Diagnosis and Laboratory Surveillance of PPR
79
Collect all used glassware and plastic ware (except tips and Eppendorf tubes) in a
bucket containing disinfectant (sodium hypochlorite, virkonS), leave overnight and
proceed for cleaning & sterilization or disposal. Cleaning & sterilization of used
glassware must be completed in the shortest possible time.
Waste disposal
Collect all sharp items (needle, blade, broken ampoule) in sharp disposal cans. When
full, dispose the can by incineration.
Solid waste such as flasks, micropipette tips, eppendorp tubes centrifuge tubes,
contaminated gloves, tissues, etc., should be placed inside heavy-duty Poly bag for
contaminated waste and incinerated.
Collect the biodegradable solid waste in large volume of freshly prepared disinfectant
(sodium hypochlorite, virkon S), leave overnight and dispose as garbage.
Decontaminate infectious liquid waste in large volume of freshly prepared
disinfectant (sodium hypochlorite, virkon S), leave overnight and dispose in sanitary
sewer.
Cell preservation procedure
Select confluent and healthy cell flask from incubator. Discard media and has to be
washed for 2-3 times by PBS. And then cell to be trypsinized by trypsin (300 micro litre
for 252 cm flask). After 2-3 minwe will get floating cell. Add some growth media. For
more uniform cell concentration in media has to be performed 1-4 times pipetting. Cells
allocate in 15 ml falcon tube and then it has to be centrifuged 10 minin 1500 rpm. After
centrifuge supernant has to be discarded and add supplied cell storage media and then
allocate in cryo tube for preservation. In second phase, allocated cell direct to be kept in -
800C for overnight and then it can be transported into liquid N2 for long time storage.
TCID50 Determination
Step-1
Take cell containing flask
Discard media, wash 3 times with PBS
Trypsinization of monolayer flask for cell detachment (.5ml for 252 cm flask)
Add growth media
Transfer tryposinized cell to a sterile petridish
Distribution of 100 micro litre of cell with growth media in per column for TCID50
using 96 wells flate bottom plate (NUNC plate)
Incubate at 370 C for 24-48 hrs for confluent growth of cell

Molecular Diagnosis and Laboratory Surveillance of PPR
80
Step-2
Thawing of virus sample
Make 10 fold dilution of virus
Take cultured cell plate from
incubator and check cell
under microscope
Discard media and wash 3
times with PBS
Add 30 micro liter virus
dilution in 96 well flat bottom
plate except last 2 columns
(Control)
Incubate for 40-45 minat 370 C
Then 200 micro litre maintenance media is added in each well. Incubate at 370C in a
incubator for 24-72 hrs.
Calculation
After completion of incubation time cell plate has to be observed for CPE under
microscope in every well to calculate TCID50 according to Reed and Munch method.
Virus dilution 10-5
10-6
10-7
10-8
Positive 10 8 4 2
Well inoculated 10 10 10 10
Percentage of infection 100 80 40 20
Calculating proportionate distance (PD)
PD = % positive at or above 50%- 50%
% positive at or above 50% - % Positive below 50%
PD= [(80-50/80-40)]
= 30/40
= .75
Calculating TCID50 /ml
50% in point titre (TCID50 per 0.1ml) =10 log total dilution above 50% - [PD*log (dilution factor)]
= 10-6-(0.75* log 10)
= 10-6-0.75*1
= 10 -6.75
TCID50= 107.75
/ml viral titre

Molecular Diagnosis and Laboratory Surveillance of PPR
81
List of the Participants
Dr. Md. Golam Azam Chowdhury Upzila Livestock Officer
Central Disease Investigation Laboratory
Dhaka, Bangladesh
Cell: +880-1716940769
Email: [email protected]
Dr. Shukes Chandra Badhy Upzila Livestock Officer
Central Disease Investigation Laboratory
Dhaka, Bangladesh
Cell: +880-1718129848
Email: [email protected]
Dr. Sonia Akter Scientific Officer
Goat and Sheep Production Research Division
Bangladesh Livestock Research Institute
Savar, Dhaka-1341
Cell: +88 01717871911
Email: [email protected]
Dr. Md. Habibur Rahman Scientific Officer
Goat and Sheep Production Research Division
Bangladesh Livestock Research Institute
Savar, Dhaka-1341
Cell: +88 01716286487
Email: [email protected]

Molecular Diagnosis and Laboratory Surveillance of PPR
82
Dr. V. Balamurugan
Principal Scientist (Veterinary Microbiologist)
ICAR-NIVEDI, Post Box No. 6450,
Yelahanka, Bengaluru-560064,
Karnataka, India
Mobile: +91- 9481807438; 9108427438
Email: [email protected]
Dr. D. Muthuchelvan
Principal Scientist
In-Charge, PPR Laboratory
Indian Veterinary Research Institute
Mukteswar, Nainital Dist
Uttarakhand
Phone: +91 05942-286346
Email: [email protected]
Dr. Krishna Raj Pandey Senior Veterinary Officer
Veterinary Laboratory
Surkhet, Nepal
Phone: 9849337320
Email: [email protected]
Dr. (Mrs.) G. A. Gunawardana Veterinary Research Officer
Veterinary Research Institute
Sri Lanka
Mobile: 00-94-717895190
Telephone no.: 00-94-81-2388125
Email: [email protected]

Molecular Diagnosis and Laboratory Surveillance of PPR
83
List of the Resource Persons
Dr. Nathu Ram Sarker Director General
Bangladesh Livestock Research Institute
Savar, Dhaka-1341, Bangladesh
Tel: +88027791683
Email: [email protected]
Dr. Asadulghani Head, Biosafety and BSL-3 Laboratory
Biosafety Office, Icddr,b
Mohakhali, Dhaka-1212
Email: [email protected]
Dr. Mohammad Rafiqul Islam
Professor
Department of Pathology, Faculty of Veterinary
Science
Bangladesh Agricultural University,
Mymensingh-2202
Email: [email protected]
Bouna Diop Secretary
FAO/OIE PPR Global Secretariat
Viale delle Terme di Caracalla, 00153
Rome, Italy
Tel: +39 06570 55667
Email: [email protected]
Dr. Emdadul Haque Chowdhury Professor
Department of Pathology
Faculty of Veterinary Science
Bangladesh Agricultural University
Mymensingh -2202, Bangladesh
Cell: +88 01712017381
Email: [email protected]
Dr. Mohammed Abdus Samad Senior Scientific Officer &
Director, RLDL for PPR
Bangladesh Livestock Research
Institute
Savar, Dhaka-1341, Bangladesh
Call: +88 01717047877
Email: [email protected]
Dr. Md. Giasuddin Head, Animal Health Research Division &
Director, National Reference Laboratory for
Avian Influenza
Bangladesh Livestock Research Institute
Savar, Dhaka-1341, Bangladesh
Call: +88 01711055597
Email: [email protected]
Dr. Md. Nure Alam Siddiky National Consultant (Laboratory)
Combating the Threats of Antimicrobial
Resistance and Zoonotic Diseases to
Achieve the GHSA in Bangladesh
BLRI, Savar, Dhaka-1341
Cell: +88 01716475486
E-mail: [email protected]
Dr. Jahangir Alam Chief Scientific Officer
Animal Biotechnology Division
National Institute of Biotechnology
Ganakbari, Ashulia
Savar, Dhaka-1349, Bangladesh
Email: [email protected]
Cell:+88 01712819098
Dr. Md. Abu Yousuf Scientific Officer (Bacteriology)
Animal Health Research Division
Bangladesh Livestock Research
Institute
Savar, Dhaka-1341.
Cell: +88 01717449845
Email: [email protected]

Molecular Diagnosis and Laboratory Surveillance of PPR
84
Dr. Mohammad Rafiqul Islam Principal Scientific Officer
Livestock Division
Bangladesh Agricultural Research Council
Farmgate, Dhaka-1215, Bangladesh
Cell: +88 01716350628
Email: [email protected]
Dr. Mahmudul Hasan Scientific Officer
BLZAC Project
Bangladesh Livestock Research
Institute
Savar, Dhaka
Cell: +88 01737152465
Email: [email protected]
Dr. Parimal Roy Director
ICAR - National Institute of Veterinary
Epidemiology and Disease Informatics
(NIVEDI)
Indian Council of Agricultural Research
Post Box # 6450, Yelahanka
Bengaluru - 560064, India
Email: [email protected]
