role of lymphotoxin in experimental models of infectious … · role of lymphotoxin in experimental...
TRANSCRIPT

INFECTION AND IMMUNITY, Nov. 2005, p. 7077–7088 Vol. 73, No. 110019-9567/05/$08.00�0 doi:10.1128/IAI.73.11.7077–7088.2005Copyright © 2005, American Society for Microbiology. All Rights Reserved.
MINIREVIEWS
Role of Lymphotoxin in Experimental Models of Infectious Diseases:Potential Benefits and Risks of a Therapeutic Inhibition of the
Lymphotoxin-� Receptor PathwayThomas W. Spahn,1* Hans-Pietro Eugster,2 Adriano Fontana,2 Wolfram Domschke,1
and Torsten Kucharzik1
Department of Medicine B, Munster University Hospital, Munster, Germany,1 and Section of ClinicalImmunology, University Hospital Zurich, Zurich, Switzerland2
The immune system provides different mechanisms to pro-tect organisms against pathogens, most of which are infectiousagents. Simultaneously, immune activation secondary to ge-netic factors and/or environmental signals can induce detri-mental autoimmunity. The effector pathways in host defenseand autoimmunity use similar cytokines and chemokines. Ac-cordingly, tumor necrosis factor alpha (TNF-�), for instance, issimilarly important in the control of various infections and inthe induction of autoimmunity.
Hallmarks of the adaptive immune system are antigen-spe-cific cellular and humoral immune responses. Secondary lym-phoid organs serve as sites of contact between antigen-present-ing cells (APCs) and immune effector T and B lymphocytes.Chemokines and cytokines serve as messengers determiningthe type of immune response to a given antigen. The TNFfamily cytokine lymphotoxin (LT) plays a pivotal role in thedevelopment of secondary lymphoid organs.
The chronic and relapsing course of many autoimmune dis-eases calls for new biological agents capable of suppressing theunderlying inflammatory disorders. Recent studies indicatethat inhibition of LT� receptor (LT�R)-mediated signaling inadult animals suppresses autoimmunity by modulating the cel-lular structure of secondary lymphoid organs (reviewed in ref-erence 22). Because of the wide range of autoimmune diseasespositively influenced by this treatment, blockade of the LT�Rmight serve as a new treatment principle for human autoim-mune diseases. However, immune responses to infectiouspathogens are also altered in mice with disrupted LT�R sig-naling. While the course of virus- and lipopolysaccharide(LPS)-induced shock, experimental Trypanosoma brucei infec-tion, cerebral malaria, and experimental prion disease are lesssevere, inhibition of the LT�R is also associated with exacer-bation of mycobacterial infection and infectious colitis. Thisreview summarizes the findings of studies using mice withdisrupted LT�R signaling in models of infectious diseases anddiscusses the relevance of these observations in consideringLT�R blockade as a potential treatment for human autoim-mune diseases.
THE LYMPHOTOXIN AND LIGHT LIGAND/RECEPTORSYSTEM AND ITS ROLE IN LYMPHOID ORGANARCHITECTURE AND AUTOIMMUNE DISEASES
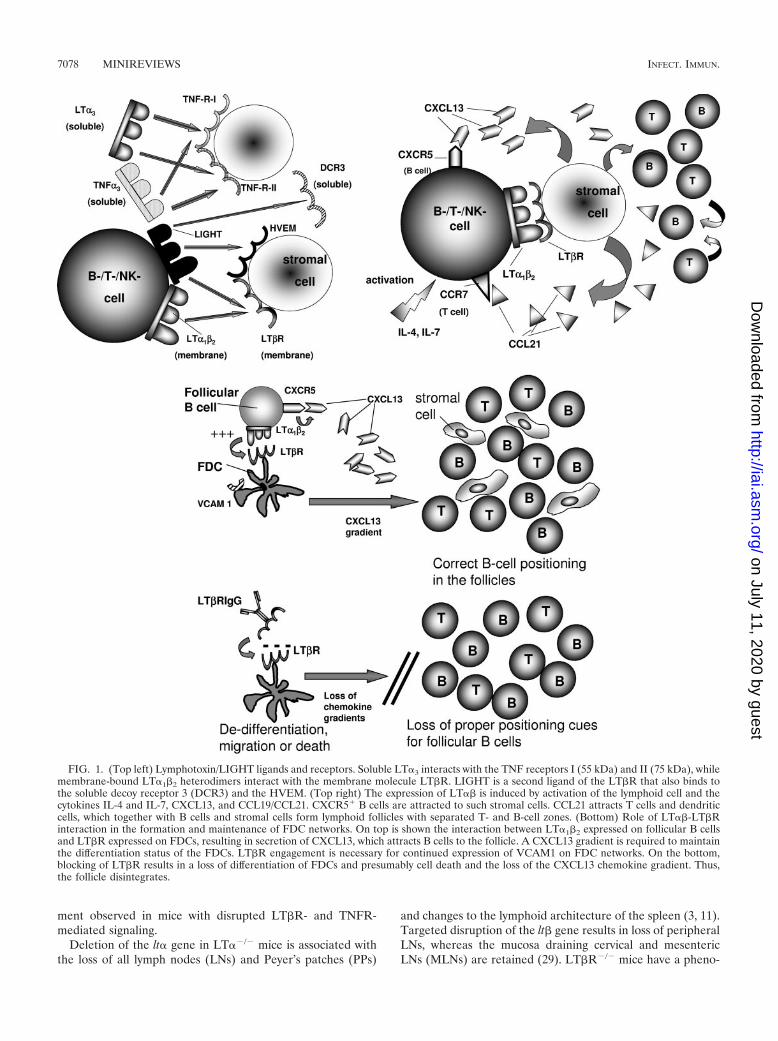
Expression and regulation of ligands and receptors. Lym-photoxin is a TNF family cytokine. The seminal discovery ofimpaired secondary lymphoid organ formation in LT� gene-deficient (�/�) mice (11) has shed new light on the biologicalfunctions of LT, which was long considered to be a redundantcytokine for TNF-�. Figure 1, top left, describes the LT/LIGHT ligands and receptors. Soluble LT�3 is a secreted pro-tein that interacts with the TNF receptors I (55 kDa) and II (75kDa) (TNFR-I and -II) (reviewed in reference 68). LT� iscoexpressed with the membrane protein LT� as LT�� het-erodimers, which are tethered to the cell membrane. LT�1�2
binds to a TNF family receptor known as LT�R. LIGHT is asecond ligand interacting with the LT�R. LIGHT also binds tothe TNF family receptors herpesvirus entry mediator (HVEM)and decoy receptor 3. Activated lymphocytes and a subset ofresting B cells express LT. The LT�R is expressed mainly onnonhematopoietic and myeloid lineage cells (reviewed in ref-erence 22). The expression of LT�� and LIGHT is induced byactivation of lymphoid cells and certain cytokines and chemo-kines, including interleukin 4 (IL-4), IL-7, CXC chemokineligand 13 (CXCL13), and CCL19/CCL21 (22). While regula-tion of LT�R expression remains to be defined, HVEM ex-pression is induced during T-cell activation (22). Figure 1, topright, depicts the factors, chemokines, and cytokines involvedin LT�� regulation and regulated by LT�R activation. Expres-sion of LT on lymphocytes provides signals necessary for stro-mal cells to secrete CXCL13. CXC chemokine receptor 5�
(CXCR5�) B cells are attracted to such stromal cells. CCL21attracts T cells and dendritic cells, which together with B cellsand stromal cells form lymphoid follicles with separated T- andB-cell zones, high endothelial venules, and follicular dendriticcell (FDC) networks.
LT ligand/LT receptor LIGHT gene-deficient and trans-genic mice. The roles of the LT/LIGHT ligands and receptorshave been characterized in gene-deficient mice. Gestationaland postgestational inhibition of the LT�R allows us to distin-guish between the specific functions of the LT��/LIGHT-LT�R pathway at different developmental time points. Table 1summarizes the defects in intestinal lymphoid organ develop-
* Corresponding author. Present address: Department of GeneralInternal Medicine and Gastroenterology, Marienhospital Osnabruck,Johannisfreiheit 2-4, 49074 Osnabruck, Germany. Phone: 49-541-326-4102. Fax: 49-541-326-4656. E-mail: [email protected].
7077
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from
ment observed in mice with disrupted LT�R- and TNFR-mediated signaling.
Deletion of the lt� gene in LT��/� mice is associated withthe loss of all lymph nodes (LNs) and Peyer’s patches (PPs)
and changes to the lymphoid architecture of the spleen (3, 11).Targeted disruption of the lt� gene results in loss of peripheralLNs, whereas the mucosa draining cervical and mesentericLNs (MLNs) are retained (29). LT�R�/� mice have a pheno-
FIG. 1. (Top left) Lymphotoxin/LIGHT ligands and receptors. Soluble LT�3 interacts with the TNF receptors I (55 kDa) and II (75 kDa), whilemembrane-bound LT�1�2 heterodimers interact with the membrane molecule LT�R. LIGHT is a second ligand of the LT�R that also binds tothe soluble decoy receptor 3 (DCR3) and the HVEM. (Top right) The expression of LT�� is induced by activation of the lymphoid cell and thecytokines IL-4 and IL-7, CXCL13, and CCL19/CCL21. CXCR5� B cells are attracted to such stromal cells. CCL21 attracts T cells and dendriticcells, which together with B cells and stromal cells form lymphoid follicles with separated T- and B-cell zones. (Bottom) Role of LT��-LT�Rinteraction in the formation and maintenance of FDC networks. On top is shown the interaction between LT�1�2 expressed on follicular B cellsand LT�R expressed on FDCs, resulting in secretion of CXCL13, which attracts B cells to the follicle. A CXCL13 gradient is required to maintainthe differentiation status of the FDCs. LT�R engagement is necessary for continued expression of VCAM1 on FDC networks. On the bottom,blocking of LT�R results in a loss of differentiation of FDCs and presumably cell death and the loss of the CXCL13 chemokine gradient. Thus,the follicle disintegrates.
7078 MINIREVIEWS INFECT. IMMUN.
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

type very similar to that of LT��/� mice (19). LIGHT, thesecond ligand of the LT�R, is predominantly expressed on Tcells and serves as a costimulatory molecule (reviewed in ref-erence 66). In contrast to LT��/� mice, LIGHT�/� mice de-velop intact lymphoid organs, indicating a predominant rolefor the LT��-LT�R interaction in the formation of secondarylymphoid organs (61). Conversely, transgenic expression ofLIGHT in LT��/� mice restores splenic T- and B-cell segre-gation, FDCs, and germinal-center (GC) formation but notmarginal-zone (MZ) formation, suggesting that LIGHT cancompensate for the loss of certain LT�� functions in the pres-ence of the LT�R (66). CD8� cells from LIGHT�/� miceshow decreased proliferative responses, while the IL-2 secre-tion is decreased in CD4� cells from these mice (61), indicat-ing a role for LIGHT as a costimulatory molecule. Conversely,transgenic expression of LIGHT on T cells is associated with ahyperactivated enlarged T-cell compartment and spontaneousautoimmunity, including inflammatory bowel disease (67).
Gestational and postgestational inhibition of LT�R-medi-ated signaling. Treatment with soluble LT�R-immunoglobulinG (IgG) fusion protein (LT�RIgG) similarly blocks the inter-action of LT�� and LIGHT with the LT�R. Therefore, theeffects observed in LT�RIgG-treated animals are a result ofthe inhibition of both ligands. Gestational treatment withLT�RIgG prevents the formation of PPs and LNs (56). Post-gestational LT�R signaling during the first 6 weeks after birthis critical for the development of intestinal lamina propria Bcells, IgA secretion, and isolated lymphoid follicles of the in-testine (38, 48).
Mode of action of LT�R inhibition in adult mice. The lym-phoid microenvironment is defined as the local interplay be-tween mobile lymphocytes and the fixed reticular/stromal cellsand includes cell adhesion, trafficking, chemokine function,and cellular positioning (22). Secondary lymphoid organs arestructures with a high degree of plasticity. Inhibition of LT�Rsignaling in adult mice alters the lymphoid microenvironment.As shown in Fig. 1, bottom, FDC networks and GCs disinte-grate and B-cell follicles disappear in the absence of LT�Rsignaling. Figure 2 depicts the lymphoid microarchitecture of amurine wild-type (wt) spleen and describes the changes ob-served in mice with blocked LT�R signaling. Table 2 and Fig.3 summarize the changes to the lymphoid microenvironmentobserved in mice with disrupted LT�1�2-LT�R signaling. FDCnetworks consist of a scaffold of specialized reticular fibro-
blasts that retain and present intact antigen to B cells. MemoryB cells develop during the GC reaction. Permanent LT�1�2-LT�R interaction is required to maintain a CXCL13 chemo-kine gradient, which attracts CXCR5� B cells to the follicleand is also required to maintain the differentiation status of therecruited B cells and the FDCs in the network. Similarly,LT�R engagement is required for continued expression of thevascular-cell adhesion molecule 1 (VCAM1) by the FDC net-work (23, 25).
The expression of VCAM1 is similarly reduced in FDCnetworks and splenic MZs following LT�RIgG treatment.Splenic MZs are located between the red and white pulp of thespleen and consist of a reticular matrix harboring B cells,macrophages, and dendritic cells (DCs). Blood-borne antigenis captured in the MZ and presented to B cells. Marginal zonesare critical for immunity against T-cell-independent bacterialantigens (24). MZ markers disappear following LT�RIgGtreatment.
Inhibition of the LT�R also blocks the migration or matu-ration of the cysteine-rich domain of the mannose receptor(CR-Fc)-positive DCs (42, 71) Figure 3 describes three poten-tial mechanisms by which inhibition of the LT�R alters auto-immunity and host defense.
PPs, isolated lymphoid follicles, cryptopatches, and colonicpatches are organized lymphoid aggregates of the intestine.The number and cellular contents of these aggregates are re-duced in adult mice undergoing anti-LT�R treatment (13).
The potential mode of action in anti-LT�R treatment is toimpair immune function by preventing proper placement of Tcells, B cells, and APCs in secondary lymphoid organs, thuspreventing the induction of appropriate antigen-specific im-mune responses. In contrast, selective inhibition of LIGHTsignaling results in a loss of LIGHT-mediated costimulatorystimuli. Soluble HVEM-Fc fusion protein selectively blocksLIGHT-HVEM interactions, inhibits CD3-induced T-cell pro-liferation, and reduces the frequency of spontaneous diabetesin nonobese diabetic mice (67).
Ectopic “tertiary” lymphoid organs and inflammatory dis-eases. A number of human inflammatory and autoimmunedisorders are associated with the formation of ectopic lym-phoid structures at the site of the inflamed organ which resem-ble secondary lymphoid organs (69; reviewed in reference 60).It is likely that immune responses to self antigens expand inthese de novo lymphoid organs, as they allow colocalization of
TABLE 1. Mice with defects in organized GALT development induced by gene defects or by gestational or postgestational treatmenta
Gene-deficient/treated mice PPb MLNb Reference
LT��/�, LT�R�/� � � 3, 11, 19LT��/� � � 2, 29, 33Light�/� � LT��/� � Less MLN than LT��/� 61TNF�/� Reduced number � 30, 52TNFR-I (55 kDa)�/� Reduced number � 47, 52LT��/� � LT��/� � � 28Gestational LT�RIgG treatment � � 56Gestational LT�RIgG and TNFRIgG treatment � � or few 57Postgestational LT�RIgG treatment Fewer cells lost
after long-termtreatment
� 13
a Modified from Reference 43a. GALT, gut-associated lymphoid tissue.b �, organ(s) present; �, organ(s) not detectable.
VOL. 73, 2005 MINIREVIEWS 7079
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

FIG. 2. Splenic lymphoid architecture and effects of inhibition of LT�R-mediated signaling. The spleen consists of a red and a white pulp, whichare segregated by marginal zones that contain macrophages. Marginal zones are not detectable in LT��/� and LT�R�/� mice. Within the whitepulp (inset with magnification), T and B cell areas are clearly separated in mice with physiological LT�R-mediated signaling. The borders betweenT- and B-cell zones are ill defined in LT��/�, LT�R�/�, and LT��/� mice. B-cell follicles are absent in these gene-deficient mice. FDCs can bedetected within B-cell follicles of wild-type mice. Within days after inhibition of LT�R-mediated signaling, FDCs disappear in adult mice. Similarly,a proportion of CD11c� dendritic cells disappear in the spleens of mice with blocked LT�R-mediated signal transduction.
TABLE 2. Changes to lymphoid microenvironment secondary to inhibition of TNFR/LT�R during pregnancy or after gestation
Organ
Developmenta with:
TNF/LT��/�, LT��/� LT�R�/� TNF��/� LT��/�LT�RIgG
Gestationalb Postgestationalc
SpleenPrimary B-cell follicles � � � � � �Marginal zone � � � � � AlteredGerminal centers � � � � � �Follicular dendritic cells � � � � � �Dendritic cells Reduced Reduced NDd Reduced ND Reduced
Mesenteric LN � � � � � �Primary B-cell follicles � � ND �/� � �Germinal centers � � ND �/� � �Follicular dendritic cells � � ND � � �
Lamina propria B cells � � ND ND � �
a �, structure present; �, structure not detectable.b Treatment with 200 �g LT�RIgG on gestational days 16 and 18.c As described in references 42 and 49.d ND, not determined.
7080 MINIREVIEWS INFECT. IMMUN.
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

antigen-specific T and B cells with APCs. In mice, ectopiclymphoid structures can be induced by transgenic overexpres-sion of LT� (31), and they are called tertiary lymphoid tissues.Data from animal models of autoimmune diseases associatedwith the formation of ectopic lymphoid tissue indicate thatautoimmunity is less severe, cured, or prevented if the LT�R isblocked in these conditions (reviewed in reference 22). A po-tential role for microbial pathogens in the pathogenesis ofinflammatory disorders, such as rheumatoid arthritis or inflam-matory bowel disease, has been discussed (18). Granulomasare common histological hallmark of Crohn’s disease and my-cobacterial infections and serve as sites where T cells, APCs,and antigens collocate (44). Granulomas resemble tertiarylymphoid tissues. The recruitment of T cells to granulomas wasimpaired in LT��/� (donor) 3 (transfer) wt (recipient) bonemarrow chimeras infected with Mycobacterium tuberculosis(58). It is therefore possible that anti-LT�R targeted therapymight also shut down common pathways of host defense andinflammatory responses that might lead to autoimmunity ingenetically predisposed persons.
Lymphotoxin and LIGHT contribute to central immune tol-erance in mice. While a stimulatory role for LT�R signaling inthe induction of peripheral autoimmune disease has been dem-onstrated by effective treatment of such conditions by anti-LT�R therapy, the thymic expression of LT�R and LT�Rligands contributes to central tolerance. Lymphotoxin signaling
is required for the expression of Aire, which is a key mediatorof central tolerance for peripherally restricted antigens. Simi-larly, LT�R ligand expression on thymic epithelial cells is re-quired for proper differentiation of thymic medullary epithelialcells. LT��/� and LT�R�/� mice show infiltration of liver,lung, pancreatic islands, and kidney with activated lympho-cytes, and autoantibodies can be detected in these mice (7, 10).Thus, LT�R signaling plays different roles in the peripheraland central control of immune tolerance.
THE ROLE OF LT�R-MEDIATED SIGNALING ININFECTIOUS DISEASES
Bacterial infections. Both types of animal models predomi-nantly requiring cellular immunity and T-cell-mediated hu-moral immunity for clearance of bacteria have been investi-gated. Host defense against intracellular bacteria eliminated bycellular immune mechanisms in mice with disrupted LT�R-mediated signaling was severely impaired in most studies. Theimmune response against bacterial pathogens inducing com-bined T- and B-cell immunity varied in mice with blockedLT�R activation with the different models tested, suggestingthat certain bacterial antigens (Citrobacter rodentium) requiredLT�R-mediated signaling while others (Salmonella enterica)were cleared in an LT�R-independent fashion.
FIG. 3. Biological effects of inhibition of LT�1�2/LIGHT-LT�R interaction in autoimmune and infectious diseases. Soluble LT�RIgG fusionprotein blocks LT�R-mediated signaling. Three biological mechanisms (circles) have been described: loss of CXCL13 gradient in folliculardendritic cell networks, loss of adhesion molecule expression, and loss of proper and timely positioning of T cells in lymphoid organs. Thesemechanisms contribute to changes in secondary lymphoid organ formation and microarchitecture, as indicated by the four boxes. The alteredlymphoid organs are associated with changes in the courses of autoimmune and infectious inflammatory diseases. Animal models in whichLT�RIgG has been used in adult mice, a situation comparable to potential human treatment, are indicated by italic font and underlining. Diseasemodels which are not altered in mice with blocked LT�R have not been depicted. ind., induced.
VOL. 73, 2005 MINIREVIEWS 7081
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

(i) Intracellular mycobacterial infections: BCG and M. tu-berculosis. While a central role for TNF-� in immunity againstmycobacterial infections has been well characterized (1, 20, 27,64), the contribution of soluble LT�3 as the second TNFR-Iligand in antimycobacterial host defense was unknown. There-fore LT��/� mice and TNF-� and LT� double-gene-deficient(TNF/LT��/�) mice were infected with Mycobacterium bovisbacillus Calmette-Guerin (BCG) (8, 26, 51) or Mycobacteriumtuberculosis (14). Studies investigating the role of LT in exper-imental mycobacterial disease are of clinical relevance consid-ering the reactivation of tuberculosis observed in patientstreated with TNF-� antagonists (21). Table 3 summarizes stud-ies investigating bacterial infections in mice with blockedLT�R signaling. The course of mycobacterial infection waslethal in TNF/LT��/� and LT��/� mice. Survival was longerin BCG-infected LT��/� mice (182 days) and in TNF-��/�
mice (56 days) than in TNF/LT��/� mice (35 days), indicatingthat the absence of TNF-� in this infection leads to broaderimmunodeficiency than the absence of LT�. The impairedantimycobacterial immune response in mice without TNF-�was aggravated by the simultaneous lack of LT� (8). Similarly,
the bacterial loads of the lung on day 28 of the infection were1,000-, 40-, and 1.4-fold higher in TNF/LT��/�, TNF-��/�,and LT��/� mice than in the respective wt mice. Introductionof an LT� transgene in TNF/LT��/� mice delayed diseaseonset but failed to restore resistance to BCG infection, sug-gesting a transient protective effect exerted by LT� in thisdisease model. Roach et al. generated LT��/� 3 wt bonemarrow chimeras in order to investigate the role of solubleLT�3 in antimycobacterial immunity (58). There was lethaldisease in wt mice with lymph nodes and LT��/� bone mar-row, indicating a critical role for soluble LT� in antimycobac-terial immunity.
The proinflammatory cytokine TNF-� is secreted as a solu-ble TNF-�3 molecule and is also tethered to the cell mem-brane. Olleros et al. investigated the role of membrane-boundnoncleavable TNF-� by creating membrane TNF-� transgenicmice on the TNF/LT��/� background, thus specifically study-ing the role of membrane TNF-� in the absence of solubleTNF-� and LT�. Following inoculation with BCG, the infec-tion was controlled in membrane TNF�/� transgenic TNF/LT��/� mice, though the bacterial load was higher in these
TABLE 3. Course of experimental bacterial infections in mice with disrupted LT�R signaling
Pathogen LT/TNF ligand/receptor�/�
mouse/inhibitor Outcome Reference
Mycobacterium bovisBCG
BALB/c wt treated withLT�RIgG/humanIgG
Reduced number of splenic granulomas, splenic eosinophil infiltrate, higherbacterial burden in LT�RIgG-treated mice
39
TNF/LT��/� Increased bacterial burden, lethal course of infection, impaired and delayedgranuloma formation
26
TNF/LT��/�, TNF/LT��/� LT��/�
transgenic, LT��/�
Lethal course of disease with pulmonary necrosis and increased bacterialburden in TNF/LT��/� mice; prolonged survival in LT��/� transgenicTNF/LT��/� mice; resistance to LPS-induced shock in TNF/LT��/�
mice
8
TransgenictransmembraneTNF�/� expression inTNF/LT��/� mice,TNF/LT��/�, TNF-��/�, LT��/�
TNF/LT��/� mice with transgenic membrane TNF expression controlmycobacterial infection with higher bacterial burden than in wt mice;TNF/LT��/�, TNF��/�, LT��/� mice succumb
51
Mycobacteriumtuberculosis
LT��/� 3 RAG1�/�
bone marrowchimera
Lethal course of infection, formation of enlarged granulomas 58
LT��/� 3 RAG1�/�
bone marrowchimera
Clearing of infection at the same rate as wt mice
LT��/�, LT��/�,LT�R�/�, LIGHT�/�
LT��/�, LT��/�, higher bacterial loads in lungs and livers; LT�R�/�,lethal course of infection, formation of enlarged granuloma; LIGHT�/�,clearing of infection at the same rate as wt mice
14
Listeriamonocytogenes
TNF/LT��/� Lethal course of infection, higher bacterial burden in liver and spleen,hepatic necrosis
17
LT�R�/� Lethal course of infection 14
Salmonella entericaserovarTyphimurium
TNF/LT��/� Resistance to lethal LPS challenge, lethal course of infection, higher organbacterial load
12
Salmonella enterica LT�R�/� Course of colitis induced following streptomycin or water treatment issimilar in wt and LT�R�/� mice
4
Citrobacterrodentium
LT��/�, LT��/�,LT�R�/�, LT�RIgG-treated
Lethal course of infection in all LT ligand/receptor gene-deficient micewith spread of bacteria to liver and spleen; more severe course inLT�RIgG-treated wt mice
65
7082 MINIREVIEWS INFECT. IMMUN.
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

mice than in wt animals. Thus, membrane TNF alone is capa-ble of controlling BCG infections (51). As TNF-��/� mice alsosuccumb to BCG infection, the expression of LT� in the ab-sence of TNF is not sufficient to control this mycobacterialinfection (8).
Causes for the impaired antimycobacterial immunity in LT�and TNF/LT� gene-deficient mice varied in the different mod-els studied. The granulomatous responses to BCG infectionwere similarly delayed and impaired in TNF/LT��/� and TNF-��/� mice (8, 26). There were fewer macrophages with re-duced inducible nitrite oxide synthase (iNOS) and acid phos-phatase expression. Fewer T cells could be detected in theselesions. These observations indicate a central role for TNF inthe recruitment of T cells and macrophages to granulomatouslesions, which cannot be compensated for by the presence ofLT�. Conversely, transgenic expression of noncleavable mem-brane TNF-� in TNF/LT��/� mice resulted in a two- to four-fold increase in the number of hepatic granulomas, which wereof smaller size and predominantly consisted of macrophages(51).
In LT��/� 3 wt bone marrow chimeras infected with M.tuberculosis, there was normal recruitment of T cells to thelungs (58). However, pulmonary T cells remained in theperivascular and peribronchial areas and failed to collocatewith the macrophages in granulomas.
There are controversial findings generated in different sys-tems regarding the role of LT�1�2-LT�R interaction in thecontrol of mycobacterial infections (14, 39, 58). Wild-type miceinfected with BCG and undergoing LT�RIgG treatment andLT�R�/� mice infected with M. tuberculosis suffered a moresevere course of disease (14, 39). Similarly, the bacterial loadsin livers and lungs of LT��/� mice infected with M. tubercu-losis were elevated (14). LT�R�/� 3 wt bone marrow chime-ras failed to control M. tuberculosis infection (14). The im-paired immune response against mycobacteria in mice withdisrupted LT�R was associated with decreased iNOS activityin the lung and spleen (14, 39). In contrast, Roach reportednormal clearance of mycobacterial infections in LT��/�3 wtbone marrow chimeras. The reasons for these discrepant ob-servations are unknown and might be related to a differentlymphoid microenvironment in LT��/�3 wt and LT�R�/�3wt bone marrow chimeras. LIGHT, a second ligand of theLT�R, is not involved in the control of disease, as LIGHT�/�
mice cleared M. tuberculosis infections at the same rate as wtmice did (14).
The course of experimental murine listeriosis was more se-vere in TNF/LT��/� and LT�R�/� mice, suggesting that inaddition to TNF-�, LT� and engagement of the LT�R arecritical for control of this intracellular pathogen (27, 53, 59).
Most studies indicate that interaction of LT�3-TNFR and ofLT� with the LT�R is required for control of infections withthe intracellular pathogens Mycobacterium and Listeria. Theelimination of these pathogens depends strongly on cellularimmunity. Only one study utilized LT�RIgG in adult mice(39), a situation comparable to human treatment of autoim-mune diseases. This study showed a significant but moderateincrease in the number of acid-fast bacilli (three- to fourfold)in LT�RIgG-treated mice compared to 10- to 1,000-fold in-creases observed in the studies utilizing gene-deficient mice.However, the biological relevance of this observation in terms
of disease-related mortality was not investigated in this study,as all mice were sacrificed for in vitro analysis 4 weeks afterinfection, while most gene-deficient mice used in other studiesdied after day 30 following mycobacterial infection.
Experimental infectious colitis. (i) Salmonella enterica sero-var Typhimurium and Salmonella enterica. Infection of LTfamily gene-deficient mice with Salmonella enterica serovarTyphimurium has been utilized to investigate the roles of LT�and TNF-� in the regulation of anti-Salmonella immunity.
Oral infection of TNF/LT��/� mice with S. enterica serovarTyphimurium results in a lethal course of infection comparedto mild disease in wt mice. This difference was most likely dueto reduced recruitment of neutrophils to the site of infection,as well as reduced intracellular killing of S. enterica serovarTyphimurium by granulocytes (12).
Mice undergoing oral pretreatment with streptomycin de-velop infectious colitis, which closely resembles human S. en-terica-induced colitis, following oral infection with S. entericaserovar Typhimurium. The development of S. enterica-inducedcolitis was not affected by the presence of PP, MLN, or theLT�R, as the courses of the infection in wt and LT�R�/� micewithout PP and MLN were similar. Infection of mice with S.enterica without antibiotic treatment induced a typhoid type ofdisease with bacterial expansion in PP and MLN. Interestingly,the typhoid type of S. enterica infection was also similar in wtand LT�R�/� mice, indicating that while S. enterica mighthome to intestinal lymphoid organs, PP, MLN, and LT�R arenot required for antibacterial immunity against this invasivepathogen (4).
(ii) Citrobacter rodentium. We have recently investigated therole of LT �1�2-LT�R interactions in the course of infectiouscolitis induced by Citrobacter rodentium (65). Infection of micewith the gram-negative bacterium C. rodentium serves as ananimal model of human infection with enteropathogenic andenterohemorrhagic Escherichia coli (36). In adult and immune-competent mice, there is only mild transient colitis with hyper-plasia of infected colonic epithelial cells. The course of C.rodentium-induced colitis was more severe in LT�RIgG-treated mice, with increased disease-related mortality (65).Similarly, there was nearly 100% disease-related mortality inC. rodentium-infected LT��/�, LT��/�, and LT�R�/� mice,suggesting a critical role for LT�1�2-LT�R interactions in anti-Citrobacter immunity. In mice with disrupted LT�R signaling,there were fewer splenic CD11c� dendritic cells following oralinfection. FDCs were absent in the spleens of LT�RIgG-treated mice. Similarly, there were fewer colonic lymphoidfollicles in LT�RIgG-treated mice and in the gene-deficientmice used. In LT�R�/� mice, anti-Citrobacter IgG2a antibodytiters were reduced while IgG1 titers were increased. Similarly,there was increased Citrobacter-induced secretion of IL-4 inLT�R�/� mice. These observations indicate that the loss oflocal intestinal lymphoid organs and changes to antigen-pre-senting functions of the spleen are associated with impairedimmunity against this noninvasive pathogen.
(iii) LPS-induced systemic shock. A number of studiesshowed resistance of TNF/LT��/� mice against lethal endo-toxemia induced by intravenous LPS injection (12, 17), de-pending on the bacterial origin of the LPS. Eugster describedresistance to shock induced by coadministration of D-galac-tosamine and E. coli-derived LPS (17). Netea et al. demon-
VOL. 73, 2005 MINIREVIEWS 7083
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

strated increased resistance of TNF/LT��/� mice to lethalendotoxemia induced by E. coli and K. pneumoniae LPS com-pared to S. enterica serovar Typhimurium LPS (46). Thesedifferences were associated with increased IL-1 and gammainterferon secretion following injection of the lethal S. entericaserovar Typhimurium LPS. BCG-sensitized TNF/LT��/� andTNF-��/� mice were completely resistant to E. coli LPS-in-duced shock, whereas LT��/� mice showed prolonged survivalcompared to wt mice (8). Thus, LT� contributes to septicshock, although TNF-� appears to be more potent in theinduction of LPS shock than LT�.
Viral infections. A number of studies have investigated therole of LT in viral infections, most of them studying influenzavirus, herpesvirus, and lymphocytic choriomeningitis virus in-fections in gene-deficient mice with anatomical defects. Table4 summarizes studies of experimental viral infections in mice.Except for two studies (37, 55), all of them utilized mice withgenetic defects of the LT ligands. Similar to LPS-inducedshock models, virus-induced systemic shock was less severe inmice with impaired LT�R, most likely due to a depletion ofvirus-specific CD8� T cells following LT�RIgG treatment.Overall antiviral cytotoxic-T-cell immune responses were moreor less impaired, and the clearance of the virus was slowed
down or inhibited, leading to a lethal course in influenza Avirus (40), murine cytomegalovirus (MCMV) (5), and Theiler’svirus (37) infections. In the extensively studied lymphocyticchoriomeningitis virus infection model, the defective antiviralimmune response was secondary to the loss of the marginalzone in the spleen (6, 45) but not due to the absence of LT�itself. Similarly, treatment of adult wt mice with LT�RIgG didnot affect immunity against Theiler’s virus infection, whileLT��/� and LT�R�/� mice failed to mount appropriate anti-viral cytotoxic-T-cell responses (37), suggesting that changes tosplenic and lymph node architecture, but not the presence ofLT�, were critical for clearing of the infection.
Parasite infections. Studies investigating the role of LT inparasite infections are summarized in Table 5.
(i) Toxoplasma gondii. Schluter et al. compared the course ofexperimental toxoplasmosis in wt, TNF-��/�, LT��/�, andTNF/LT��/� mice in order to dissect the roles of both ligandsof the TNF receptors in this infection (62). TNFR-I plays apredominant role in experimental toxoplasmosis. TNF-� in-duces toxoplasmastatic gamma interferon secretion in macro-phages and microglial cells in the central nervous system (9,34).
All gene-deficient mice tested in this study failed to control
TABLE 4. Course of experimental viral infections in mice with disrupted LT�R signaling
Pathogen LT/TNF ligand/receptor�/� mouse/inhibitor Outcome Reference
Influenza virus A LT��/� Delayed clearance of low-dose infection, lethal course of high-doseinfection
40
LT��/�, TNF/LT��/�, TNF-��/�,TNFR-I�/�, TNFR-II�/�, TNFR-I/II�/�
TNF-� and LT� are required for loss of bone marrow derived Bcells during infection
63
MCMV LT��/�, LT�R-human IgG transgenic(�/�)
Increased susceptibility and lethality to MCMV in LT��/� andLT�R-human IgG�/� mice
5
Theiler’s virus(Daniel’s strain)
LT��/�, LT�R�/�, LT�RIgGtreatment, TNF-��/�, TNFR-I�/�,TNFR-II�/�
Virus persistence and demyelination in LT��/� and LT�R�/� butnot in TNF-��/�, TNFRI�/�, or TNFRII�/� mice; failure tomount virus-specific CTL response in LT��/� and LT�R�/�
mice; LT�RIgG treatment does not impair antiviral immunity
37
Herpes simplex virus(HSV)
LT��/� Impaired cytotoxic and cytokine-mediated CD8� T-cell effectorfunctions enhanced susceptibility to HSV-inducedencephalopathy
32
Murinegammaherpesvirus 68 (MHV-68)
LT��/� Delayed clearance of infection 35
Vaccinia virus TNF/LT��/� Slightly reduced primary antiviral CTL response 17
Lymphocyticchoriomeningitisvirus (LCMV)
TNF/LT��/� Strongly reduced primary antiviral CTL response, reduced day 8antiviral CTL response; clearing of infection from spleen andliver
17
LCMV LT��/�, LT��/� 3 wt bone marrowchimera
Diminished antiviral CTL responses due to disorganized splenicstructure
6
LT��/�, TNF-��/�, TNF/LT��/� B-cell 3 RAG1�/� bone marrowchimera
Intact splenic marginal zone is required for LCMV replication inthe spleen and for CTL induction
45
LCMV-13 (virus-induced systemicshock)
LT�RIgG Reduced and delayed disease-related mortality associated withreduction of virus-specific CD8� T cells
65
7084 MINIREVIEWS INFECT. IMMUN.
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

intracerebral T. gondii and succumbed to acute necrotizingToxoplasma encephalitis. The lethal course of disease was as-sociated with reduced intracerebral expression of iNOS andlower splenic NO levels. Experiments with bone marrow re-constitution chimeras demonstrated an exclusive role ofTNF-�- and LT�-producing hematopoietic cells for survivingtoxoplasmosis.
(ii) Leishmania. Infection of LT��/� mice with Leishmaniamajor was associated with a fatal course of disease with visceralspread of parasites despite the resistant genetic background ofthe C57BL/6 mice used in this study (70). The impaired anddelayed cellular and humoral anti-L. major immune responsein LT��/� mice was secondary to changes in the lymphoidarchitecture. Reconstitution of LT��/� mice with wt bonemarrow failed to restore effective antiparasite immunity,whereas wt mice receiving LT��/� bone marrow were notimmunocompromised.
Murine Leishmania donovani infection induces visceralleishmaniasis and is more severe in both TNF-��/� andLT��/� mice (15). Experiments with bone marrow radiationchimeras indicated a critical role for liver-generated LT� in themigration of leukocytes from periportal to sinusoidal areas,while T-cell-generated TNF-� and LT� were required for thecontrol of parasite growth.
(iii) Trypanosoma brucei. Infection of LT��/� and TNF/LT��/� double-gene-deficient mice with the extracellular par-asite Trypanosoma brucei was associated with control of diseaseand slightly prolonged survival of LT��/� mice following in-fection (43). Trypanosoma-specific IgM and IgG2a serum an-tibody titers were increased in LT��/� mice, indicating that
germinal centers and FDC networks were not required for thisantiparasite humoral immune response.
(iv) Cerebral malaria. Infection of mice with Plasmodiumberghei serves as an animal model for human cerebral malaria.LT��/� mice were protected against cerebral malaria, as theydid not develop perivascular cerebral hemorrhage. Bone mar-row chimera experiments indicated that a radioresistant cere-bral cell population is the source of the LT� required forextravasation of malaria-infected erythrocytes (16).
Prion disease/scrapie. Transmissible spongiform encepha-lopathies (TSEs), or “prion diseases,” are chronic neurodegen-erative diseases that affect humans and animals. Most TSEs,including human variant Creutzfeldt-Jakob disease and exper-imental prion disease in mice, are transmitted by peripheralexposure. TSE infection results in conversion of normal prionprotein (PrPc) to the disease-associated form, PrPsc. Intrace-rebral or peripheral administration of prions to mice induces arise of infectivity in the spleen and in other lymphoid organslong before the development of neuropathological changes.PrPsc migrates from the lymphoid compartments to the centralnervous system by neuronal transport. FDCs in the germinalcenters of lymphoid organs have been implicated as initial sitesof accumulation of PrPsc. FDCs trap antigen-antibody com-plexes. Studies using intraperitoneal (i.p.) (41, 54) and oral(50) routes of scrapie infection provided different results re-garding the role of FDCs and LT ligands/receptors in thisinfection. LT��/�, LT��/�, TNF/LT��/�, and LT�R�/� micewith disrupted LT�1�2-LT�R signaling undergoing i.p. inocu-lation resisted infection and contained no infectivity in spleensand lymph nodes (54). Similarly, pretreatment of wt mice with
TABLE 5. Course of experimental parasite and prion infections in mice with disrupted LT�R signaling
Pathogen/disease LT/TNF ligand/receptor�/� mouse/inhibitor Outcome Reference
Toxoplasma gondii LT��/�, TNF-��/�, TNF/LT��/� Lethal course of acute necrotizing Toxoplasma encephalitis, reducedearly gamma interferon secretion, diminished IgM and IgG titers
62
Leishmania major LT��/�, LT��/� 3 wt bone marrowchimera, wt 3 LT��/� bone marrowchimera
Lethal course of leishmaniasis in resistant mouse strain in theabsence of local lymph nodes; local lymph nodes required foranti-leishmania immune response
70
Leishmania donovani LT��/�, TNF��/�, LT��/� 3 wt bonemarrow chimera; TNF-��/� 3 wtbone marrow chimera
Increased susceptibility of LT��/� and TNF-��/� mice, LT� andTNF-� from hematopoietic cells regulate migration of leukocytesin liver; LT� controls migration from periportal areas; TNF-�regulates leukocyte recruitment to liver
15
Cerebral malaria LT��/�, TNF-��/�, wt 3 LT��/�
bone marrow chimeraLoss of susceptibility to cerebral malaria in LT��/� mice, secretion
of LT� by radioresistant cell population in the brain16
Trypanosoma brucei LT��/�, TNF/LT��/� Early trypanosomiasis unaltered in LT��/� mice, increased survivalof LT��/� mice, and increased IgM/IgG2a serum antibody titers;control of infection in TNF/LT��/� mice with reduced infection-induced pathology
43
Scrapie LT��/�, LT��/�, LT�R�/�,TNF/LT��/�
Resistance to i.p. infection 54
TNFR-I�/� TNFR-II�/�, TNF-��/� Susceptibility to i.p. infectionLT��/�, LT��/� Susceptibility to oral infection in LT��/� mice, resistance to oral
infection in LT��/� mice50
Treatment with LT�RIgG Reduced prion accumulation, reduction of disease susceptibilityfollowing i.p. infection, prevention of prion accumulation inPeyer’s patches and mesenteric lymph nodes and ofneuroinvasion following oral infection
41
VOL. 73, 2005 MINIREVIEWS 7085
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

LT�RIgG prior to i.p. scrapie infection blocked early PrPsc
accumulation in the spleen and reduced disease susceptibility.In contrast, LT��/� mice orally infected with scrapie weresusceptible to disease while LT��/� mice were resistant (50).However, pretreatment of wt mice with LT�RIgG prior to oralinfection with scrapie blocked PrPsc in PP and MLN and pre-vented neuroinvasion (41).
As FDCs were similarly absent in TNF-��/�, TNFR-I�/�,and LT�, LT��/� mice but only LT gene-deficient mice wereprotected against experimental scrapie, FDCs are not requiredfor the replication of scrapie in lymphoid tissue following i.p.infection. More likely, some other yet-undefined effect of im-paired LT�R-mediated signaling is critical for control of theexpansion of scrapie protein in lymphoid tissues. The suscep-tibility of LT��/� mice to oral scrapie and the resistance ofLT��/� mice and LT�RIgG-pretreated mice to oral infectionare two controversial observations which require further inves-tigation.
SUMMARY AND CONCLUSIONS
The studies of experimental infectious diseases summarizedin this review reveal the complex biological functions of theLT��/LIGHT-LT�R pathway in immunity to infectiousagents. As the courses of the respective infections were atten-uated, unchanged, or even more severe, the role of LT�Rsignaling in host defense depends on the respective pathogens.Animal models predominantly requiring cellular immunity orT helper cell-mediated humoral immunity for clearance of therespective infectious agent have been investigated. Overall, thecontribution of the LT�R pathway to host defense against therespective pathogen depended on the antigenic properties ofthe pathogen, but not on the type of immune response inducedby it.
Host defense against bacterial intracellular pathogens suchas mycobacteria and Listeria mediated by cellular immunemechanisms in mice with disrupted LT�R-mediated signalingwas severely impaired in most studies. Similarly, in most mod-els of viral infections, cytotoxic-T-cell responses were dimin-ished, although the defect in host defense observed was sec-ondary to changes in lymphoid microarchitecture and notcaused by the absence of LT. The elimination of the obligateintracellular parasite Toxoplasma gondii depends on T-cell re-sponses and on the presence of LT�. Conversely, the clearanceof the intracellular parasite Leishmania major depends on Thelper 1-mediated cellular immunity and is independent ofLT�R-mediated signaling, while the extracellular parasiteTrypanosoma brucei is similarly cleared in LT��/� mice and inwild-type mice (43), indicating that there is no common patternfor LT�R signaling in the host defense against intracellular orextracellular parasites. The immune responses against bacte-rial pathogens inducing combined T- and B-cell immunity var-ied in mice with blocked LT�R activation with the differentmodels tested, suggesting that certain bacterial antigens(Citrobacter rodentium) required LT�R mediation while others(Salmonella enterica) were cleared in an LT�R-independentfashion.
The beneficial effects of anti-LT�R therapy observed in ex-perimental virus- and LPS-induced shock, cerebral malaria,and prion disease call for further studies of the role of LT�R
signaling in the pathogeneses of similar human disease condi-tions and suggest that anti-LT�R therapy might also be afuture treatment for these diseases.
Few studies using bone marrow chimeras and soluble antag-onist LT�RIgG fusion protein in wt mice have demonstrateddifferential roles of secondary lymphoid organs and the cyto-kines LT�3 and its membrane-bound heterodimers. Thus, sol-uble LT�3 and the LT�R play pivotal roles in immunity againstmycobacterial infections and C. rodentium-induced colitis. Thepresence of the LT�R on bone marrow-derived cells is re-quired to clear these infections in mice. In contrast, LT�1�2/LIGHT-LT�R interactions are not required to clear experi-mental L. major infection, while a normal splenic, PP, and LNmicroenvironment is required to overcome experimental leish-maniasis (70). Similarly, an intact splenic microenvironment isrequired for the induction of appropriate antiviral immuneresponses in the lymphocytic choriomeningitis virus model (6,45).
Inhibition of the LT�R is a future therapeutic concept intreatment of autoimmune diseases (22). The effects of suchtreatment are secondary to changes to the lymphoid microen-vironment and have also been demonstrated in the spleens ofnonhuman primates (23). Compared to the effects observed inLT gene-deficient mice, changes following short-termLT�RIgG treatment are moderate (Table 1). However, long-term treatment with LT�RIgG in mice also deletes PPs andcolonic patches and reduces the number of intestinal DCs (13).
The treatment of adult mice with anti-LT�R agents is, con-sidering the substantial differences between human and murineimmune systems, a situation comparable to the treatment ofhumans with anti-LT�R therapy. Impaired host defense fol-lowing LT�RIgG treatment has been observed in the BCG andC. rodentium models, while Theiler’s virus infection was notaffected by LT�RIgG treatment (37, 39, 65). Thus, bearing inmind the different modes of action in experimental murineinfections and spontaneous infections of humans, immunityagainst mycobacterial infections and infectious colitis inducedby enteropathogenic and enterohemorrhagic E. coli might beimpaired in humans undergoing anti-LT�R treatment. Theimmunosuppressive and thus host defense-suppressive effect ofanti-LT�R therapy will probably depend on the dose and du-ration of such treatments.
Gestational treatment of mice with LT�RIgG results in per-manent changes to the development of lymphoid organs (56).Similar to other potent immune-modulating therapies, thetreatment of pregnant women should be strictly prohibited,and preventive measures, such as the use as of oral contracep-tives, should be mandatory in sexually active women undergo-ing such treatment.
Considering the need for new and effective treatment mo-dalities of human inflammatory and autoimmune diseases,LT�R blockade might be a potent biological tool which has tobe carefully tested in clinical trials, considering the delicatebalance between sufficient host defense and the suppression ofautoimmunity.
REFERENCES
1. Adams, L. B., C. M. Mason, J. K. Kolls, D. Scollard, J. L. Krahenbuhl, andS. Nelson. 1995. Exacerbation of acute and chronic murine tuberculosis byadministration of a tumor necrosis factor receptor-expressing adenovirus.J. Infect. Dis. 171:400–405.
7086 MINIREVIEWS INFECT. IMMUN.
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

2. Alimzhanov, M. B., D. V. Kuprash, M. H. Kosco-Vilbois, A. Luz, R. L.Turetskaya, A. Tarakhovsky, K. Rajewsky, S. A. Nedospasov, and K. Pfeffer.1997. Abnormal development of secondary lymphoid tissues in lymphotoxinbeta-deficient mice. Proc. Natl. Acad. Sci. USA 94:9302–9307.
3. Banks, T. A., B. T. Rouse, M. K. Kerley, P. J. Blair, V. L. Godfrey, N. A.Kuklin, D. M. Bouley, J. Thomas, S. Kanangat, and M. L. Mucenski. 1995.Lymphotoxin-alpha-deficient mice. Effects on secondary lymphoid organdevelopment and humoral immune responsiveness. J. Immunol. 155:1685–1693.
4. Barthel, M., S. Hapfelmeier, L. Quintanilla-Martinez, M. Kremer, M. Rohde,M. Hogardt, K. Pfeffer, H. Russmann, and W.-D. Hardt. 2003. Pretreatmentof mice with streptomycin provides a Salmonella enterica serovar Typhi-murium colitis model that allows analysis of both pathogen and host. Infect.Immun. 71:2839–2858.
5. Benedict, C. A., T. A. Banks, L. Senderowicz, M. Ko, W. J. Britt, A. Angulo,P. Ghazal, and C. F. Ware. 2001. Lymphotoxins and cytomegalovirus coop-eratively induce interferon-beta, establishing host-virus detente. Immunity15:617–626.
6. Berger, D. P., D. Naniche, M. T. Crowley, P. A. Koni, R. A. Flavell, and M. B.Oldstone. 1999. Lymphotoxin-beta-deficient mice show defective antiviralimmunity. Virology 260:136–147.
7. Boehm, T., S. Scheu, K. Pfeffer, and C. C. Bleul. 2003. Thymic medullaryepithelial cell differentiation, thymocyte emigration, and the control of au-toimmunity require lympho-epithelial cross talk via LT�R. J. Exp. Med.198:757–769.
8. Bopst, M., I. Garcia, R. Guler, M. L. Olleros, T. Rulicke, M. Muller, S. Wyss,K. Frei, M. Le Hir, and H. P. Eugster. 2001. Differential effects of TNF andLT� in the host defense against M. bovis BCG. Eur. J. Immunol. 31:1935–1943.
9. Chao, C. C., S. Hu, G. Gekker, W. J. Novick, Jr., J. S. Remington, and P. K.Peterson. 1993. Effects of cytokines on multiplication of Toxoplasma gondiiin microglial cells. J. Immunol. 150:3404–3410.
10. Chin, R. K., J. C. Lo, O. Kim, S. E. Blink, P. A. Christiansen, P. Peterson,Y. Wang, C. Ware, and Y. X. Fu. 2003. Lymphotoxin pathway directs thymicAire expression. Nat. Immunol. 4:1121–1127.
11. De Togni, P., J. Goellner, N. H. Ruddle, P. R. Streeter, A. Fick, S. Mari-athasan, S. C. Smith, R. Carlson, L. P. Shornick, J. Strauss-Schoenberger,et al. 1994. Abnormal development of peripheral lymphoid organs in micedeficient in lymphotoxin. Science 264:703–707.
12. Dharmana, E., M. Keuter, M. G. Netea, I. C. Verschueren, and B. J. Kull-berg. 2002. Divergent effects of tumor necrosis factor-alpha and lympho-toxin-alpha on lethal endotoxemia and infection with live Salmonella typhi-murium in mice. Eur. Cytokine Netw. 13:104–109.
13. Dohi, T., P. D. Rennert, K. Fujihashi, H. Kiyono, Y. Shirai, Y. I. Kawamura,J. L. Browning, and J. R. McGhee. 2001. Elimination of colonic patches withlymphotoxin beta receptor-Ig prevents Th2 cell-type colitis. J. Immunol.167:2781–2790.
14. Ehlers, S., C. Holscher, S. Scheu, C. Tertilt, T. Hehlgans, J. Suwinski, R.Endres, and K. Pfeffer. 2003. The lymphotoxin beta receptor is criticallyinvolved in controlling infections with the intracellular pathogens Mycobac-terium tuberculosis and Listeria monocytogenes. J. Immunol. 170:5210–5218.
15. Engwerda, C. R., M. Ato, S. Stager, C. E. Alexander, A. C. Stanley, and P. M.Kaye. 2004. Distinct roles for lymphotoxin-alpha and tumor necrosis factorin the control of Leishmania donovani infection. Am. J. Pathol. 165:2123–2133.
16. Engwerda, C. R., T. L. Mynott, S. Sawhney, J. B. De Souza, Q. D. Bickle, andP. M. Kaye. 2002. Locally up-regulated lymphotoxin alpha, not systemictumor necrosis factor alpha, is the principal mediator of murine cerebralmalaria. J. Exp. Med. 195:1371–1377.
17. Eugster, H. P., M. Muller, U. Karrer, B. D. Car, B. Schnyder, V. M. Eng, G.Woerly, M. Le Hir, F. di Padova, M. Aguet, R. Zinkernagel, H. Bluethmann,and B. Ryffel. 1996. Multiple immune abnormalities in tumor necrosis factorand lymphotoxin-alpha double-deficient mice. Int. Immunol. 8:23–36.
18. Fourneau, J. M., J. M. Bach, P. M. van Endert, and J. F. Bach. 2004. Theelusive case for a role of mimicry in autoimmune diseases. Mol. Immunol.40:1095–1102.
19. Futterer, A., K. Mink, A. Luz, M. H. Kosco-Vilbois, and K. Pfeffer. 1998. Thelymphotoxin beta receptor controls organogenesis and affinity maturation inperipheral lymphoid tissues. Immunity 9:59–70.
20. Garcia, I., Y. Miyazaki, G. Marchal, W. Lesslauer, and P. Vassalli. 1997.High sensitivity of transgenic mice expressing soluble TNFR1 fusion proteinto mycobacterial infections: synergistic action of TNF and IFN-gamma in thedifferentiation of protective granulomas. Eur. J. Immunol. 27:3182–3190.
21. Gardam, M. A., E. C. Keystone, R. Menzies, S. Manners, E. Skamene, R.Long, and D. C. Vinh. 2003. Anti-tumour necrosis factor agents and tuber-culosis risk: mechanisms of action and clinical management. Lancet Infect.Dis. 3:148–155.
22. Gommerman, J., and J. L. Browning. 2003. Lymphotoxin/LIGHT, lymphoidmicroenvironments and autoimmune disease. Nat. Rev. Immunol. 3:642–655.
23. Gommerman, J. L., F. Mackay, E. Donskoy, W. Meier, P. Martin, and J. L.Browning. 2002. Manipulation of lymphoid microenvironments in nonhu-
man primates by an inhibitor of the lymphotoxin pathway. J. Clin. Investig.110:1359–1369.
24. Guinamard, R., M. Okigaki, J. Schlessinger, and J. V. Ravetch. 2000. Ab-sence of marginal zone B cells in Pyk-2-deficient mice defines their role inthe humoral response. Nat. Immunol. 1:31–36.
25. Husson, H., S. M. Lugli, P. Ghia, A. Cardoso, A. Roth, K. Brohmi, E. G.Carideo, Y. S. Choi, J. Browning, and A. S. Freedman. 2000. Functionaleffects of TNF and lymphotoxin �1�2 on FDC-like cells. Cell Immunol.203:134–143.
26. Jacobs, M., N. Brown, N. Allie, and B. Ryffel. 2000. Fatal Mycobacteriumbovis BCG infection in TNF-LT-alpha-deficient mice. Clin. Immunol. 94:192–199.
27. Kindler, V., A. P. Sappino, G. E. Grau, P. F. Piguet, and P. Vassalli. 1989.The inducing role of tumor necrosis factor in the development of bactericidalgranulomas during BCG infection. Cell 56:731–740.
28. Koni, P. A., and R. A. Flavell. 1999. Lymph node germinal centers form inthe absence of follicular dendritic cell networks. J. Exp. Med. 189:855–864.
29. Koni, P. A., R. Sacca, P. Lawton, J. L. Browning, N. H. Ruddle, and R. A.Flavell. 1997. Distinct roles in lymphoid organogenesis for lymphotoxinsalpha and beta revealed in lymphotoxin beta-deficient mice. Immunity6:491–500.
30. Korner, H., M. Cook, D. S. Riminton, F. A. Lemckert, R. M. Hoek, B.Ledermann, F. Kontgen, B. Fazekas de St Groth, and J. D. Sedgwick. 1997.Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organo-genesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 27:2600–2609.
31. Kratz, A., A. Campos-Neto, M. S. Hanson, and N. H. Ruddle. 1996. Chronicinflammation caused by lymphotoxin is lymphoid neogenesis. J. Exp. Med.183:1461–1472.
32. Kumaraguru, U., I. A. Davis, S. Deshpande, S. S. Tevethia, and B. T. Rouse.2001. Lymphotoxin alpha�/� mice develop functionally impaired CD8� Tcell responses and fail to contain virus infection of the central nervoussystem. J. Immunol. 166:1066–1074.
33. Kuprash, D. V., M. B. Alimzhanov, A. V. Tumanov, A. O. Anderson, K.Pfeffer, and S. A. Nedospasov. 1999. TNF and lymphotoxin beta cooperate inthe maintenance of secondary lymphoid tissue microarchitecture but not inthe development of lymph nodes. J. Immunol. 163:6575–6580.
34. Langermans, J. A., M. E. Van der Hulst, P. H. Nibbering, P. S. Hiemstra, L.Fransen, and R. Van Furth. 1992. IFN-gamma-induced L-arginine-depen-dent toxoplasmastatic activity in murine peritoneal macrophages is mediatedby endogenous tumor necrosis factor-alpha. J. Immunol. 148:568–574.
35. Lee, B. J., S. Santee, S. Von Gesjen, C. F. Ware, and S. R. Sarawar. 2000.Lymphotoxin-alpha-deficient mice can clear a productive infection with mu-rine gammaherpesvirus 68 but fail to develop splenomegaly or lymphocyto-sis. J. Virol. 74:2786–2792.
36. Levine, M. M. 1987. Escherichia coli that cause diarrhea: enterotoxigenic,enteropathogenic, enteroinvasive, enterohemorrhagic, and enteroadherent.J. Infect. Dis. 155:377–389.
37. Lin, X., X. Ma, M. Rodriguez, X. Feng, L. Zoecklein, Y. X. Fu, and R. P.Roos. 2003. Membrane lymphotoxin is required for resistance to Theiler’svirus infection. Int. Immunol. 15:955–962.
38. Lorenz, R. G., D. D. Chaplin, K. G. McDonald, J. S. McDonough, and R. D.Newberry. 2003. Isolated lymphoid follicle formation is inducible and de-pendent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin beta re-ceptor, and TNF receptor I function. J. Immunol. 170:5475–5482.
39. Lucas, R., F. Tacchini-Cottier, R. Guler, D. Vesin, S. Jemelin, M. L. Olleros,G. Marchal, J. L. Browning, P. Vassalli, and I. Garcia. 1999. A role forlymphotoxin beta receptor in host defense against Mycobacterium bovis BCGinfection. Eur. J. Immunol. 29:4002–4010.
40. Lund, F. E., S. Partida-Sanchez, B. O. Lee, K. L. Kusser, L. Hartson, R. J.Hogan, D. L. Woodland, and T. D. Randall. 2002. Lymphotoxin-alpha-defi-cient mice make delayed, but effective, T and B cell responses to influenza.J. Immunol. 169:5236–5243.
41. Mabbott, N. A., J. Young, I. McConnell, and M. E. Bruce. 2003. Folliculardendritic cell dedifferentiation by treatment with an inhibitor of the lympho-toxin pathway dramatically reduces scrapie susceptibility. J. Virol. 77:6845–6854.
42. Mackay, F., G. R. Majeau, P. Lawton, P. S. Hochman, and J. L. Browning.1997. Lymphotoxin but not tumor necrosis factor functions to maintainsplenic architecture and humoral responsiveness in adult mice. Eur. J. Im-munol. 27:2033–2042.
43. Magez, S., B. Stijlemans, G. Caljon, H. P. Eugster, and P. De Baetselier.2002. Control of experimental Trypanosoma brucei infections occurs inde-pendently of lymphotoxin-alpha induction. Infect. Immun. 70:1342–1351.
43a.Mebius, R. E. 2003. Organogenesis of lymphoid tissues. Nat. Rev. Immunol.3:292–303.
44. Mielke, M. E., C. Peters, and H. Hahn. 1997. Cytokines in the induction andexpression of T-cell-mediated granuloma formation and protection in themurine model of listeriosis. Immunol. Rev. 158:79–93.
45. Muller, S., L. Hunziker, S. Enzler, M. Buhler-Jungo, J. P. Di Santo, R. M.Zinkernagel, and C. Mueller. 2002. Role of an intact splenic microarchitec-
VOL. 73, 2005 MINIREVIEWS 7087
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

ture in early lymphocytic choriomeningitis virus production. J. Virol. 76:2375–2383.
46. Netea, M. G., L. J. van Tits, J. H. Curfs, F. Amiot, J. F. Meis, J. W. van derMeer, and B. J. Kullberg. 1999. Increased susceptibility of TNF-alpha lym-photoxin-alpha double knockout mice to systemic candidiasis through im-paired recruitment of neutrophils and phagocytosis of Candida albicans.J. Immunol. 163:1498–1505.
47. Neumann, B., A. Luz, K. Pfeffer, and B. Holzmann. 1996. Defective Peyer’spatch organogenesis in mice lacking the 55-kD receptor for tumor necrosisfactor. J. Exp. Med. 184:259–264.
48. Newberry, R. D., J. S. McDonough, K. G. McDonald, and R. G. Lorenz. 2002.Postgestational lymphotoxin/lymphotoxin beta receptor interactions are es-sential for the presence of intestinal B lymphocytes. J. Immunol. 168:4988–4997.
49. Ngo, V. N., H. Korner, M. D. Gunn, K. N. Schmidt, D. S. Riminton, M. D.Cooper, J. L. Browning, J. D. Sedgwick, and J. G. Cyster. 1999. Lymphotoxinalpha/beta and tumor necrosis factor are required for stromal cell expressionof homing chemokines in B and T cell areas of the spleen. J. Exp. Med.189:403–412.
50. Oldstone, M. B., R. Race, D. Thomas, H. Lewicki, D. Homann, S. Smelt, A.Holz, P. Koni, D. Lo, B. Chesebro, and R. Flavell. 2002. Lymphotoxin-alpha-and lymphotoxin-beta-deficient mice differ in susceptibility to scrapie: evi-dence against dendritic cell involvement in neuroinvasion. J. Virol. 76:4357–4363.
51. Olleros, M. L., R. Guler, N. Corazza, D. Vesin, H. P. Eugster, G. Marchal,P. Chavarot, C. Mueller, and I. Garcia. 2002. Transmembrane TNF inducesan efficient cell-mediated immunity and resistance to Mycobacterium bovisbacillus Calmette-Guerin infection in the absence of secreted TNF andlymphotoxin-alpha. J. Immunol. 168:3394–3401.
52. Pasparakis, M., L. Alexopoulou, M. Grell, K. Pfizenmaier, H. Bluethmann,and G. Kollias. 1997. Peyer’s patch organogenesis is intact yet formation ofB lymphocyte follicles is defective in peripheral lymphoid organs of micedeficient for tumor necrosis factor and its 55-kDa receptor. Proc. Natl. Acad.Sci. USA 94:6319–6323.
53. Pfeffer, K., T. Matsuyama, T. M. Kundig, A. Wakeham, K. Kishihara, A.Shahinian, K. Wiegmann, P. S. Ohashi, M. Kronke, and T. W. Mak. 1993.Mice deficient for the 55 kd tumor necrosis factor receptor are resistant toendotoxic shock, yet succumb to L. monocytogenes infection. Cell 73:457–467.
54. Prinz, M., F. Montrasio, M. A. Klein, P. Schwarz, J. Priller, B. Odermatt, K.Pfeffer, and A. Aguzzi. 2002. Lymph nodal prion replication and neuroinva-sion in mice devoid of follicular dendritic cells. Proc. Natl. Acad. Sci. USA99:919–924.
55. Puglielli, M. T., J. L. Browning, A. W. Brewer, R. D. Schreiber, W. J. Shieh,J. D. Altman, M. B. Oldstone, S. R. Zaki, and R. Ahmed. 1999. Reversal ofvirus-induced systemic shock and respiratory failure by blockade of thelymphotoxin pathway. Nat. Med. 5:1370–1374.
56. Rennert, P. D., J. L. Browning, R. Mebius, F. Mackay, and P. S. Hochman.1996. Surface lymphotoxin alpha/beta complex is required for the develop-ment of peripheral lymphoid organs. J. Exp. Med. 184:1999–2006.
57. Rennert, P. D., D. James, F. Mackay, J. L. Browning, and P. S. Hochman.
1998. Lymph node genesis is induced by signaling through the lymphotoxinbeta receptor. Immunity 9:71–79.
58. Roach, D. R., H. Briscoe, B. Saunders, M. P. France, S. Riminton, and W. J.Britton. 2001. Secreted lymphotoxin-alpha is essential for the control of anintracellular bacterial infection. J. Exp. Med. 193:239–246.
59. Rothe, J., W. Lesslauer, H. Lotscher, Y. Lang, P. Koebel, F. Kontgen, A.Althage, R. Zinkernagel, M. Steinmetz, and H. Bluethmann. 1993. Micelacking the tumour necrosis factor receptor 1 are resistant to TNF-mediatedtoxicity but highly susceptible to infection by Listeria monocytogenes. Nature364:798–802.
60. Ruddle, N. H. 1999. Lymphoid neo-organogenesis: lymphotoxin’s role ininflammation and development. Immunol. Res. 19:119–125.
61. Scheu, S., J. Alferink, T. Potzel, W. Barchet, U. Kalinke, and K. Pfeffer. 2002.Targeted disruption of LIGHT causes defects in costimulatory T cell acti-vation and reveals cooperation with lymphotoxin beta in mesenteric lymphnode genesis. J. Exp. Med. 195:1613–1624.
62. Schluter, D., L. Y. Kwok, S. Lutjen, S. Soltek, S. Hoffmann, H. Korner, andM. Deckert. 2003. Both lymphotoxin-alpha and TNF are crucial for controlof Toxoplasma gondii in the central nervous system. J. Immunol. 170:6172–6182.
63. Sedger, L. M., S. Hou, S. R. Osvath, M. B. Glaccum, J. J. Peschon, N. vanRooijen, and L. Hyland. 2002. Bone marrow B cell apoptosis during in vivoinfluenza virus infection requires TNF-alpha and lymphotoxin-alpha. J. Im-munol. 169:6193–6201.
64. Senaldi, G., S. Yin, C. L. Shaklee, P. F. Piguet, T. W. Mak, and T. R. Ulich.1996. Corynebacterium parvum- and Mycobacterium bovis bacillus Calmette-Guerin-induced granuloma formation is inhibited in TNF receptor I (TNF-RI) knockout mice and by treatment with soluble TNF-RI. J. Immunol.157:5022–5026.
65. Spahn, T. W., C. Maaser, L. Eckmann, J. Heidemann, A. Lugering, R.Newberry, W. Domschke, H. Herbst, and T. Kucharzik. 2004. The lympho-toxin-beta receptor is critical for control of murine Citrobacter rodentium-induced colitis. Gastroenterology 127:1463–1473.
66. Wang, J., A. Foster, R. Chin, P. Yu, Y. Sun, Y. Wang, K. Pfeffer, and Y. X.Fu. 2002. The complementation of lymphotoxin deficiency with LIGHT, anewly discovered TNF family member, for the restoration of secondarylymphoid structure and function. Eur. J. Immunol. 32:1969–1979.
67. Wang, J., J. C. Lo, A. Foster, P. Yu, H. M. Chen, Y. Wang, K. Tamada, L.Chen, and Y. X. Fu. 2001. The regulation of T cell homeostasis and auto-immunity by T cell-derived LIGHT. J. Clin. Investig. 108:1771–1780.
68. Ware, C. F. 2005. Network communications: lymphotoxins, LIGHT, andTNF. Annu. Rev. Immunol. 23:787–819.
69. Weyand, C. M., P. J. Kurtin, and J. J. Goronzy. 2001. Ectopic lymphoidorganogenesis: a fast track for autoimmunity. Am. J. Pathol. 159:787–793.
70. Wilhelm, P., D. S. Riminton, U. Ritter, F. A. Lemckert, C. Scheidig, R. Hoek,J. D. Sedgwick, and H. Korner. 2002. Membrane lymphotoxin contributes toanti-leishmanial immunity by controlling structural integrity of lymphoidorgans. Eur. J. Immunol. 32:1993–2003.
71. Yu, P., Y. Wang, R. K. Chin, L. Martinez-Pomares, S. Gordon, M. H.Kosco-Vilbois, J. Cyster, and Y. X. Fu. 2002. B cells control the migration ofa subset of dendritic cells into B cell follicles via CXC chemokine ligand 13in a lymphotoxin-dependent fashion. J. Immunol. 168:5117–5123.
Editor: J. B. Kaper
7088 MINIREVIEWS INFECT. IMMUN.
on July 11, 2020 by guesthttp://iai.asm
.org/D
ownloaded from