revista chilena de epilepsiarevistachilenadeepilepsia.cl/wp-content/uploads/2015/11/rev-epil... ·...
TRANSCRIPT

1
REVISTA CHILENA DE EPILEPSIAPublicación Oficial de la Sociedad de Epileptología de Chile
Capítulo Chileno de la ILAEhttp: //www.epilepsiadechile.com / E-mail: [email protected]
Año 15, Nº 2, Agosto 2015ISSN 0717-5337
Editorial / EditorialInmunología y Epilepsia / Immunology and Epilepsy 3
Trabajos Originales / Original Works• Comorbilidad psiquiátrica en adolescentes con epilepsia Psychiatric comorbidity in adolescents with epilepsy Karina Rosso, Yairet Soto, Enzo Rivera, Fernando Pinochet 5
Actualizaciones / Updates• Electroencefalografía neonatal normal. Normal neonatal electroencephalography Loreto Ríos, Catalina Torres 12• Electroencefalografía neonatal. Registro anormal. Record abnormal Neonatal electroencephalography Loreto Ríos, Jovanka Pavlov 23• Espiga Ondas continuas del sueño lento Continous Spike-wave during sleep syndrome (CSWS) Antonia Mena, Perla David, Kathleen Batalla 36• Síndrome de Landau-Kleffner Landau-Kleffner Syndrome Kathleen Batalla, Antonia Mena, Perla David 40
Caso Clínico / Clinical Report• Status super- refractario del lóbulo temporal: ¿Existe una secuencia abreviada y bidireccional de cambios EEG? Análisis de más de 900 crisis durante casi un año. Super status refractory temporal lobe: Exists an abbreviated sequence EEG changes? Analysis of 900 crisis for almost a year Cayetano Napolitano 52
Crónica / Chronicle• Memoria Año 2014 / Memory 2014 57• Cursos, Congresos y Actividades 2015 / Courses, Congress and Activities 2015 60• Programa XIV Jornadas Invernales de Epilepsia 2015 / Winter days of Epilepsy 62• Declaración de Intereses / Interest declaration 64• Sugerencias para las contribuciones a los autores / Suggestions to authors of contributions 67

2
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Directorio ILAE 2013 - 2017
PresidentEmilio Perucca
Vice PresidentTatsuya Tanaka
Secretary- GeneralHelen Cross
TreasurerSam Wiebe
Past PresidentSalomon Moshé
IBE PresidentAthanasios Covanis
IBE Secretary-GeneralSari Tervonen
IBE TreasurerRobert Cole
Dirección:Av. Providencia 2315, Of. 215Fonos: 2231 0172, 2235 1470, Fax: 2234 0671Providencia, Santiago, Chile.E-Mail: [email protected] ó [email protected]
DiseñoGráfico:Juan Silva / [email protected] / 9799 5964
SOCIEDAD DE EPILEPTOLOGÍA DE CHILE
Capítulo Chileno de la Liga Internacional contra la Epi-lepsiaFundada el 13 de Marzo de 1999
Directorio de la Sociedad de Epileptología de Chile
PresidenteDr. Cayetano Napolitano
VicepresidenteDra. Daniela Triviño
Secretaria GeneralDra. Carla Manterola
TesoreroDr. Juan Moya
DirectorDr. Marcelo Devilat
Past PresidentDr. Darío Ramírez
Editores de PublicacionesDrs. Perla David, Ledia Troncoso, Marcelo Devilat
Delegados ANLICHEDr. Tomás MesaDr. Jorge Förster
Comité EditorialDr. Cayetano NapolitanoDr. Juan SalinasDra. Julia SantinDra. Alejandra HernándezDra. Verónica BurónDr. Juan MoyaDra. Francisca LópezDra. Scarlet WittingDra. Loreto Ríos
Comité InternacionalProf. Dr. Roberto Caraballo, ArgentinaProf. Dr. Pedro Serrano, EspañaProf. Dr. Eduardo Barragán, MéxicoProf. Dr. Jaderson Da Costa, BrasilProf. Dra. Magda Lahorges, BrasilProf. Dra. Elza Yacubian, Brasil
Comité Revisión de ParesDra. Ledda AguileraDr. Jaime GodoyDr. Rodrigo Salinas

3
Inmunología y EpilepsiaReinaldo Uribe San MartínEpileptólogo Red Salud UC-Christus y Hospital Sotero del Río.Director GDT Epilepsia Sonepsyn.
Editorial
I. INTRODUCCIÓN
Tan antigua como la aparición del sistema nervioso central, estructura casi puramente eléctrica, la epi-lepsia ha sido descrita desde los primeros registros médicos dada su importante prevalencia y capricho-sa aparición, la cual merma la calidad de vida de quien la padece.
A pesar del enorme esfuerzo de múltiples investi-gadores, las crisis epilépticas continúan sin un tra-tamiento curativo específico, siendo hasta el mo-mento, su mejor abordaje, la intención de resecar el foco epiléptico que no en pocos casos es un pro-cedimiento frustro o meramente paliativo. Lo com-plejo del mecanismo epileptogénico subyacente ha llevado a diversos autores a indagar en cada vez más complejas vías de aparición de la fenomenolo-gía ictal, como alteraciones metabólicas, genéticas o al humilde juicio del autor, la inmunología.
Esta nueva veta en la epileptología moderna, trans-versal a varias enfermedades, afecta a toda célula implicada en la generación de crisis y que tratare-mos de detallar brevemente en esta editorial.
II. CONCEPTO DE EPILEPTOGÉNESIS
Tres son los hitos descritos en la formación de cir-cuitos epileptogénicos: 1) la aparición de una lesión o noxa precipitante, 2) un periodo latente, libre de crisis, pero estructurador o epileptogénico y 3) la real fase epiléptica, con aparición de crisis espon-táneamente recurrentes y que al igual que el apren-dizaje, se fortalece como red y permite la autoper-petuación.
Basados en el concepto de que previo a la aparición de una primera crisis existen cambios molecula-res, celulares y organizacionales, se ha investigado cómo el sistema inmune modula en especial este periodo de latencia, encontrándose implicados al-
gunos particulares tipos de citoquinas, anticuerpos, células inflamatorias, modificándose a su vez las sinapsis y las uniones en hendidura (gap junctions).
III. MECANISMOS INFLAMATORIOS EN EPI-LEPSIA
Citoquinas
Si bien existen varios tipos de citoquinas implicadas, algunas consideradas pro y otras anti inflamatorias, no necesariamente reflejando una respuesta pro o anti epiléptica, haremos énfasis en la interleukina 1 beta (IL-1b) dado su reciente reporte como biomar-cador en epilepsia post traumática. Diamond M et al., describieron una cohorte de 256 pacientes que sufrieron un traumatismo encefalocraneano (TEC) moderado o severo. El análisis multivariado demos-tró que índices elevados de IL-1b (proporción entre líquido cefalorraquídeo y suero) se asociaron con un mayor riesgo de presentar epilepsia post TEC y que además existiría un genotipo particular que ex-presa índices aún más elevados. Interesantemente, la activación de los receptores de IL-1 / Toll-like receptors aumenta la conductancia de calcio en los canales NMDA, conocido por su rol pro epileptogé-nico en modelos animales.
Anticuerpos
Las encefalopatías autoinmunes mediadas por anti-cuerpos, se dividen entre los de superficie (anti ca-nales o receptores como VGKC, NMDAR, AMPA, GABA-B, m-GluR5) y los intracelulares (GAD65, ANNA-1, Ma1/2), cada uno con un síndrome cada vez mejor definido que incluye no sólo manifes-taciones epilépticas, sino también trastornos del movimiento y neuropsiquiátricos. En pacientes con estado epiléptico, donde más importante que la terapia antiepiléptica clásica es el tratamiento de la etiología subyacente, la detección de estos auto-anticuerpos demuestran una causa inflamatoria es-

4
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
pecífica y precisa, pudiendo presentarse de manera primaria como en el lupus eritematoso sistémico (LES) o secundaria, como en los casos paraneoplá-sicos. La presencia de estos anticuerpos es espe-cialmente frecuente en los difíciles casos de estado epiléptico refractario o donde éste es una condición de novo (llamados NORSE del acrónimo en inglés New Onset Refractory Status Epilepticus). Así, se instauró un nuevo arsenal terapéutico con el inicio precoz de esteroides, inmunoglobulinas, plasmafé-resis o inmunosupresores, ya utilizado por la mayo-ría de los epileptólogos. En los casos en que existe relación con una neoplasia, el tratamiento de ésta está indicado no sólo per se, sino también para el control del estado epiléptico.
Uniones en hendidura
Maravillosamente, nuestro sistema nervioso cen-tral se edifica estructurado, conformando en cada sitio una red exquisita y única que no podría de-sarrollarse sin la ajustada y directa comunicación intercelular otorgada por las uniones en hendidura. La conocida gliosis descrita en piezas de pacientes sometidos a cirugía resectiva de epilepsia, refleja la activación inflamatoria tanto de neuronas como de la glía, con proliferación de astrocitos y micro-glías. Por ejemplo, en esclerosis hipocampal se ob-serva una marcada sobreexpresión de uniones en hendidura, factores inflamatorios y receptores glu-tamatérgicos por sobre gabaérgicos. Diversos blo-queadores de uniones en hendidura o hemicanales (conexinas) se han utilizado en modelos animales, los que están dando resultados prometedores para ser utilizados en humanos.
IV. DESÓRDENES AUTOINMUNES
Dos son los grupos de epilepsias relacionadas a la autoinmunidad:- Epilepsias inmuno-mediadas: como la encefali-
tis de Rasmussen, el sindrome de West, el Landau Kleffner, el de espiga onda continua del sueño, las encefalitis límbicas, la hemiconvulsión-hemi-plejia y la enfermedad de Batten.
- Enfermedades sistémicas y neurológicas auto-inmunes asociadas a crisis o epilepsia: donde encontramos el LES, la enfermedad celíaca, la Encefalitis de Hashimoto o la Esclerosis Múlti-ple.
Diversas series han demostrado que la principal enfermedad autoinmune relacionada a epilepsia corresponde al síndrome antifosfolípido, especial-mente asociado a LES. Aquí se mezclan diversos mecanismos fisiopatológicos, con sobreexpresión de citoquinas proinflamatorias, anticuerpos especí-ficos, infiltración y gliosis inflamatoria secundaria. Evidentemente y aparte de ofrecer una terapia con antiepilépticos, la correcta inmunosupresión ayuda-rá a un mejor control de la epilepsia en estos casos.
V. REFLEXIÓN FINAL
No siempre es evidente que un paciente con epi-lepsia padezca de una patología autoinmune subya-cente y además que ésta influya activamente en el control de las crisis. En este sentido, Toledano M et al., evaluaron la respuesta a terapias inmunes en una cohorte de 29 pacientes con epilepsia refracta-ria a los cuales se les investigó exhaustivamente la presencia de anormalidades autoinmunes (historia, citoquinas, anticuerpos, etc.). De esta cohorte, 23 pacientes recibieron metilprednisolona y 6 recibie-ron inmunoglobulinas como primera terapia, de los cuales el 52% respondió. Al invertir el tratamiento ofrecido en un principio a los pacientes que no ha-bían logrado control de crisis, se alcanzó además un 43% de respuesta. Es de esperar que en el fu-turo, nuevos ensayos sean realizados en pacientes con epilepsias refractarias y en especial, tratando de identificar los mecanismos inmunes implicados en este proceso, con la intención tal vez no de su-primir, sino de modular la respuesta inmune, di-señando nuevas estrategias que ayuden a evitar la aparición de epilepsia, con el objetivo de atacar la piedra filosofal del epileptólogo, o sea deteniendo la epileptogénesis.

5
Trabajo Original
Comorbilidad Psiquiátrica en Adolescentes con Epilepsia.Karina Rosso1, Yairet Soto2, Enzo Rivera3, Fernando Pinochet4.
ABSTRACT
Objective: To present and compare psychiatric co-morbidity among adolescents with and without epi-lepsy.Methodology: Analytical cross sectional study. We applied the M.I.N.I. Kid, a structured diagnostic interview to 30 adolescents aged 13-18 years with active epilepsy and 30 adolescents without epilepsy. Results: The comparison between adolescents with and without epilepsy yielded the following results: with at least one psychiatric disorder 60% versus 43% (p=NS); two or more psychiatric disorders, 43% versus 13% (p=0.02); history of year repe-tition, 40% versus 13% (p=0.04); depressive syn-drome, 16% versus 3%, (p=NS); previous suicide risk, 50% versus 20% (p=0.02); risk of suicide at the time of evaluation, 20% versus 0% (p=0.02); ADHD, 43% versus 10% (p <0.01), predominantly inattentive type (54%).Conclusion: There is a clear tendency toward more psychiatric disorders in adolescents with epilepsy than in those without epilepsy, most of them without a diagnosis or treatment. The high frequency of sui-cidal risk is highlightedKeywords: Epilepsy, adolescence, psychiatric co-morbidity, psychopathology, MINI Kid.
RESUMEN
Objetivo: Dar a conocer y comparar la comorbilidad psiquiátrica entre adolescentes con y sin epilepsia.Metodología: Estudio analítico transversal. Se in-cluyeron los pacientes entre 13-18 años con epilep-sia activa, con al menos una crisis epiléptica durante los 12 meses previos. Se evaluaron 30 adolescentes con epilepsia y 30 adolescentes sin epilepsia. Se les aplicó el M.I.N.I. Kid, entrevista estructurada para los principales diagnósticos psiquiátricos.Resultados: Al comparar los adolescentes con epi-lepsia versus aquellos sin epilepsia se encontró: con al menos un trastorno psiquiátrico en 60% versus 43% (p=NS); dos o más trastornos psiquiátricos, 43% versus 13% (p=0,02); antecedente de repiten-cia escolar en 40% versus 13% (p=0,04); síndrome depresivo en 16% versus 3% (p=NS); riesgo de sui-cidio antiguo, 50% versus 20% (p=0,02); riesgo de suicidio al momento de la evaluación, 20% versus 0% (p=0,02); trastorno de ansiedad de separación, 16% versus 0 (p=NS); síndrome de déficit atencio-nal, 43% versus 10%, (p<0,01), predominantemen-te del tipo inatento (54%). Había sólo tres pacientes con diagnóstico y tratamiento de síndrome depre-sivo al momento de la evaluación. No hubo diag-nóstico o tratamiento previo de otras patologías. Conclusión: Existe una clara tendencia a presentar más trastornos psiquiátricos en adolescentes con epilepsia que en aquellos sin epilepsia, la gran ma-yoría de ellos sin diagnóstico, ni tratamiento previo a la evaluación. Destaca la alta frecuencia de riesgo de suicidio. Palabras claves: Epilepsia, adolescencia, comorbili-dad psiquiátrica, psicopatología, MINI Kid.
INTRODUCCIÓN
Durante las últimas décadas han cambiado los ob-jetivos del tratamiento de la epilepsia. Si bien ante-riormente el tratamiento se focalizaba en la reduc-ción del número de crisis, hoy en día se considera
1. Neuropediatra, Unidad de Neurología Infantíl, Hospital Car-los Van Buren, Universidad de Valparaíso.
2. Psiquiatra Infantil. Unidad de Neuropsiquiatría Infantil, Hosp. Carlos Van Buren. Unidad Infanto-Juvenil, Hosp. del Salva-dor. Cátedra de Pediatría-Neurología y Psiquiatría Infanto-Juvenil, Univ. de Valparaíso. Valparaíso, Chile.
3. Neurólogo, Epileptólogo. Serv. de Neurología, Hosp. Carlos Van Buren. Cátedra de Neurología Univ. de Valparaíso.
4. Médico Bioestadístico. Universidad de Valparaíso. Corres-pondencia Dra. Karina Rosso, Vista Hermosa 94, Cerro Cas-tillo, Viña del Mar, mail: [email protected]. Estudio realizado en Hospital Carlos Van Buren, Unidad de Neurop-siquiatría Infantil HCVB, Unidad Infanto-Juvenil, Hospital del Salvador, Cátedra de Pediatría-Neurología y Psiquiatría Infanto-Juvenil, Universidad de Valparaíso. Valparaíso, Chile.
Declara no tener conflicto de intereses. Recibido 1/09/2014. Aceptado 20/09/2014.

6
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
que el manejo debe ir dirigido a la calidad de vida de estos pacientes, determinado en gran parte por la frecuencia de las crisis, pero sin dejar de lado otros aspectos relevantes como son los efectos colaterales de los anticonvulsivantes, las comorbilidades aso-ciadas o la inserción social y laboral.
La epilepsia se ha descrito desde tiempo remotos, Hipócrates (400 A.C.), en su obra “De Morbo Sa-cro” (“sobre la enfermedad sagrada”) describe: “los melancólicos normalmente se hacen epilépticos y los epilépticos melancólicos: lo que determina la preferencia es la dirección que la enfermedad toma, si va al cuerpo se produce la epilepsia, si a la inteli-gencia la melancolía” (1).
La adolescencia es un período de alta vulnerabilidad emocional, marcado por la formación de la identi-dad y consolidación del desarrollo de la personali-dad, período en el cual los trastornos psiquiátricos son de mayor prevalencia. En niños con epilepsia, la prevalencia de trastornos de la salud mental varía entre el 16% y 77%. (2, 3, 4) Esta gran variabili-dad entre los distintos estudios se debe a diferen-cias metodológicas tales como nosología, métodos de evaluación de casos, marco temporal, tipos de epilepsia, etc.
Existen múltiples teorías respecto de la asociación entre epilepsia y trastornos mentales; se han descri-to diversos factores de riesgo, pero con resultados inconsistentes entre los estudios. (2) La mayoría de los estudios no han encontrado asociación entre edad de inicio, frecuencia o tipo de crisis, laterali-zación del foco epiléptico y psicopatología en niños con epilepsia. (4,5)
Los trastornos psiquiátricos más frecuentemente descritos en la literatura en niños y adolescentes con epilepsia son: trastornos del ánimo, trastornos de ansiedad y trastorno por déficit de atención con hiperactividad (TDAH)(2, 6, 7).
Un estudio en niños con epilepsia entre 5 y 16 años encontró que a pesar de que el 60% de los casos cumplía algún diagnóstico a partir de los criterios DSM IV (Diagnostic and Statistical Manual of Mental Disorders, versión IV), sólo el 33% había recibido un tratamiento psiquiátrico. (5,8).
M.I.N.I. Kid (Mini International Neuropsychiatric Interview for Children and Adolescents), es una en-
trevista psiquiátrica estructurada, para el diagnós-tico de los trastornos mentales más frecuentes del eje I del DSM IV y CIE 10. Tiene tres versiones, el M.I.N.I. Kid es la forma extensa para aplicación desde 6 hasta 17 años 11 meses. Se aplica al niño o adolescente sólo o acompañado por su padre. La validación del MINI Kid mostró que es una bue-na herramienta diagnóstica para los trastornos del ánimo, trastorno de ansiedad, TDAH y trastorno de conducta (área bajo la curva = 0.81–0.96, κ = 0.56–0.87). Para los trastornos psicóticos tenía me-nor sensibilidad, pero con especificidad aceptable. (8) La entrevista está separada por módulos, donde cada ítem incuye 2- 4 preguntas de tamizaje, que de ser positivas, se aplican todas las preguntas del tema. Todas las respuestas están en formato si /no. La aplicación de la entrevista, tiene una duración de 30-45 minutos.
Los diagnósticos que incluye el MINI Kid son: evaluación del riesgo de suicidabilidad, episodio depresivo mayor, trastorno distímico, episodio ma-níaco, trastorno de angustia, agorafobia, trastorno de ansiedad de separación, fobia social, fobia es-pecífica, trastorno obsesivo compulsivo, estado por estrés postraumático, abuso y/o dependencia de al-cohol, abuso y/o dependencia de drogas, trastorno de tics, trastorno por déficit de atención e hiperac-tividad (TDAH), trastorno de la conducta, trastorno oposicionista desafiante (TOD), trastornos psicóti-cos, trastorno de ansiedad generalizada y trastorno adaptativo.
El objetivo de este trabajo es describir y comparar la comorbilidad psiquiátrica en adolescentes porta-dores de epilepsia activa versus adolescentes sin epilepsia.
PACIENTES Y MÉTODO
Se trata de un estudio descriptivo con un grupo control. A partir de la base estadística del Hospital Carlos van Buren (HCVB) de Valparaíso, se revisa-ron las fichas médicas de todos los pacientes adoles-centes con diagnóstico de epilepsia en control entre enero y diciembre del año 2010. Se conversó con los padres o cuidadores de cada adolescente para completar y actualizar los datos, caracterizando así la fecha de última crisis.
Para evaluar la comorbilidad psiquiátrica tanto en el grupo casos como control se utilizó como instru-

7
Comorbilidad psiquiátrica en adolescentes con epilepsia Karina Rosso et al.
mento el M.I.N.I. Kid, “mini international neurop-sychiatric interview for children and adolescents”.
Criterios inclusión: Sujetos de entre 13 y 18 años de edad, portadores de epilepsia activa, definida como la ocurrencia de al menos una crisis en los 12 meses previos.
Criterios de exclusión: Ausencia de crisis epilép-tica durante los últimos 12 meses, déficit intelec-tual con coeficiente intelectual menor o igual a 70, trastorno generalizado del desarrollo (debido a la imposibilidad de obtener respuestas confiables en la encuesta M.I.N.I. Kid), rechazo de consentimiento informado. Se definieron estos criterios con el obje-tivo de hacer el estudio comparable a otros estudios en la literatura.
Grupo de estudio (o con epilepsia):
Selección de los individuos: de 95 pacientes entre 13 y 18 años de edad que se hallaban en control con diagnóstico de epilepsia en el HCVB, se des-cartaron 31 por no haber tenido crisis durante los 12 meses previos a la evaluación. Otros 30 pacientes eran portadores de epilepsias asociadas a deficien-cia intelectual o trastorno generalizado del desarro-llo. De esta manera 34 cumplieron los criterios de inclusión, dos de ellos no aceptaron participar en el estudio, refiriendo que tenían dificultad para trans-portarse hasta el HCVB y otros dos habían cam-biado de domicilio. Finalmente el grupo de estudio quedó constituido por 30 casos (Figura 1).
Grupo de comparación: Treinta adolescentes sin diagnóstico de epilepsia, pareados por edad y gé-nero, seleccionados por muestreo aleatorio estrati-ficado a partir de Liceo Eduardo de la Barra de Val-paraíso. Se les aplicó la misma entrevista M.I.N.I. Kid, con la misma evaluadora para ambos grupos.
Variables analizadas: Según la ficha médica y los datos aportados por los apoderados de cada pa-ciente, se registraron las siguientes variables: edad, sexo, comorbilidades psiquiátricas, tipo de epilep-sia, tipo y frecuencia de crisis, fármacos antiepilép-ticos en uso y respuesta al tratamiento farmacológi-co (refractaria o no).
Procedimiento: Los adolescentes que cumplieron con los criterios de inclusión fueron contactados telefónicamente para una evaluación junto a sus pa-
dres con el objetivo de explicarles directamente en qué consistía el estudio y solicitar su aprobación. Luego se les aplicó la encuesta M.I.N.I. Kid direc-tamente a los adolescentes, y a partir de su resultado se obtuvo los siguientes diagnósticos, todos como categóricas dicotómicas, con resultado presente o ausente: Episodio depresivo mayor, Trastorno distímico, Episodio (hipo) maníaco, Trastorno de angustia, Agorafobia, Trastorno de ansiedad de separación, Fobia social, Fobia específica, Tras-torno obsesivo compulsivo, Abuso y dependencia de alcohol, Trastorno asociado al uso de sustan-cias psicoactivas, Trastorno por déficit de atención con hiperactividad, Trastorno de conducta disocial, Trastorno negativista desafiante, Trastornos psicó-ticos, Trastorno de ansiedad generalizada y Tras-torno adaptativo. Además este cuestionario incluye un módulo que evalúa el riesgo de suicidio pasado con preguntas respecto al deseo de estar muerto, los intentos por hacerse daño y los intentos de quitarse la vida antiguos; el riesgo de suicidio actual se re-fiere a si en el último mes ha tenido deseos de estar muerto, hacerse daño, pensar en quitarse la vida, en cómo quitársela o intentos frustros, cada una de es-tas preguntas con distinto puntaje, así se puede cla-sificar en riesgo de suicidio leve, moderado o alto.
Estudio estadístico: Se realizó estadística descripti-va, utilizándose como medidas de resumen la media y la desviación estándar (D.E.) en el caso de aque-llas variables con distribución normal, o mediana y rango intercuartílico (R.I.C) para aquellas sin dis-tribución normal. Se usó prueba exacta de Fisher para ver la asociación entre dos variables dicotó-micas entre sí. Se realizó comparación de medias de las variables cuantitativas, usándose prueba t de Student para el caso de las variables con distri-bución normal y prueba U de Mann-Whitney para aquéllas con distribución no normal. Para comparar las distintas variables cuantitativas según el tipo de epilepsia se utilizó la prueba de Análisis de Varian-za (ANOVA) de una vía, donde se comparan las me-dias de los grupos. Para evaluar qué grupos poseen las diferencias más significativas, se utilizó la prue-ba Post-hoc de Bonferroni. Se utilizó el Coeficiente de Correlación de Pearson para evaluar el grado de asociación entre las variables cuantitativas. Se con-sideró estadísticamente significativo p < 0,05.
Consideraciones éticas: Sin conflicto de intereses, el proyecto fue aprobado por el Comité de Bioéti-ca de la Facultad de Medicina de la Universidad de

8
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Valparaíso. Se aplicaron consentimientos informa-dos tanto a los padres como a los adolescentes, en ambos grupos.
Por ser un estudio descriptivo, observacional, analí-tico, transversal, no hubo ninguna modificación del tratamiento. A partir del resultado de la entrevista M.I.N.I. Kid, en el grupo de adolescentes con epi-lepsia se puso al tanto al médico tratante y en ambos grupos se derivó a psicólogo y/o psiquiatra en caso de ser necesario.
RESULTADOS
Caracterización de los grupos de adolescentes con y sin epilepsia:
En este grupo, el promedio de edad de inicio de la epilepsia fue a los 8 años (rango: 0,7 a 14 años), con un promedio de duración de la epilepsia de 6,5 años.
Ocho pacientes presentaban epilepsias sintomáti-cas, las causas fueron: secuela de encefalitis aguda, trastorno de migración neuronal, esclerosis mesial temporal, esclerosis tuberosa y traumatismo encéfa-lo craneano, 1 de etiología criptogénica y 22 de los 30 pacientes se clasificaron como epilepsias idiopá-ticas. 10 correspondían a epilepsia generalizada y 20 a epilepsia focal.
La frecuencia de las crisis epilépticas, según lo re-portado por el paciente y/o cuidadores fueron: 4 referían crisis diarias, 6 correspondían a crisis se-manal, 4 pacientes con crisis mensual y menos que 1 crisis al mes en 16 de los 30 pacientes. Respecto al tratamiento con fármacos antiepilépti-cos, 20 estaban en monoterapia y 10 con 2 o más fármacos, 9 de los cuales estaban catalogados como epilepsia refractaria a tratamiento médico.
La tabla 1 muestra las características de edad, gé-nero y antecedente de repitencia escolar en los 2
Figura 1: Diagrama de flujo de pacientes, para selección del Grupo de estudio.

9
Comorbilidad psiquiátrica en adolescentes con epilepsia Karina Rosso et al.
grupos. Un 40% del grupo total de adolescentes con epilepsia tenía antecedente de repitencia es-colar, versus el 13% en el grupo de comparación (p=0,04). Considerando la epilepsia como factor de riesgo para repitencia, el OR fue de 4,33 (IC: 1,06 –20,93). Según las evoluciones en la ficha médica, un 23% de los adolescentes con epilepsia tenía el antecedente de TDAH, sin embargo, al momento de la evaluación, ninguno estaba recibiendo algún tipo de tratamiento para el TDAH. Un 10% de los ado-lescentes con epilepsia tenía registrado en su ficha
clínica al menos 1 episodio depresivo previo a la evaluación. Cinco adolescentes tenían pseudocrisis asociadas a la epilepsia. En tres pacientes estaba el registro de bullying previo. Una paciente tenía el antecedente de intento de suicidio e intoxicación con carbamazepina que requirió hospitalización en cuidados intensivos. En el grupo control, se-gún lo referido por los mismos alumnos, ninguno tenía diagnóstico, ni había recibido tratamiento por TDAH ni trastorno de conducta al momento de la evaluación.
Tabla Nº 1. Características de los grupos
Epilepsia Sin epilepsia p1
Mediana edad (RIC) 14 (2) 14 (2) Sexo (fem : masc) 17: 13 17:13 Antecedente de Repitencia escolar (%) 12/30 4/30 0,04
1. Prueba exacta de Fisher. RIC: rango intercuartílico.
La tabla 2 muestra los diagnósticos que arrojó la entrevista M.I.N.I. Kid en el grupo de adolescentes con epilepsia y sin epilepsia, y su comparación. Se aplicó prueba exacta de Fisher para la comparación de proporciones.
Al comparar presencia o ausencia de diagnósticos psiquiátricos, el 60% de los pacientes con epilep-sia presentó algún trastorno psiquiátrico, mientras que entre adolescentes sin epilepsia esto ocurrió en un 43%, diferencia no estadísticamente significati-va. Sin embargo, al comparar aquéllos con más de un trastorno psiquiátrico, la comorbilidad estuvo presente en el 43% de los pacientes con, versus el 13% en el grupo sin epilepsia (p= 0,02). Tomando en cuenta aquellos con al menos un diagnóstico psi-quiátrico, el grupo sin epilepsia tuvo una mediana de un diagnóstico, mientras que en aquellos con epilepsia, la mediana fue de 2,5 diagnósticos, sien-do ésta una diferencia estadísticamente significativa (p < 0,01).
La evaluación de riesgo de suicidio actual consis-tió en preguntas considerando el último mes, tales como: ¿Deseaste estar muerto? ¿Pensaste en quitar-te la vida? ¿Pensaste en cómo quitarte la vida? ¿Tra-taste de quitarte la vida? Los seis casos con riesgo de suicidio al momento de la evaluación fueron derivados a psicólogo y psiquiatra inmediatamente tras la evaluación.
Los diagnósticos, cuya presencia resultó estadísti-camente significativa (p < 0,05) al comparar entre el grupo de adolescentes con epilepsia y sin epilepsia fueron: trastorno de ansiedad de separación, fobia social y TDAH. Cabe destacar que en el resto de los diagnósticos, pese a que no hubo diferencia estadís-ticamente significativa, hubo una clara tendencia a ser mayor en el grupo con epilepsia. En el grupo con epilepsia predomina el TDAH sub-tipo inatento, siendo de 54% en el grupo con epilep-sia versus un 34% en el grupo sin epilepsia, con una diferencia estadísticamente significativa (p < 0,05).
DISCUSIÓN
Tanto a nivel general, como en el desglose de los trastornos psiquiátricos, se vio una clara tendencia a tener más trastornos mentales entre los adolescentes con epilepsia. En TDAH, trastorno de ansiedad de separación y fobia social, esta diferencia fue esta-dísticamente significativa. Cabe destacar el efecto de diagnósticos conglomerados en esta población, en que el 27% de los adolescentes con epilepsia te-nía más de tres diagnósticos psiquiátricos.
La evaluación también incluyó la estimación del riesgo de suicidio, tanto antiguo como al momento de la evaluación, siendo éste uno de los aspectos de mayor relevancia dentro de los resultados, eviden-ciando que entre los adolescentes con epilepsia, la

10
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Tabla Nº 2. Diagnósticos Comorbilidad psiquiátrica en grupo epilepsia y sin epilepsia.
Epilepsia n (%) Controles n (%) p1
Episodio depresivo mayor 5 (16,6% ) 1 (3,3%) 0.19Riesgo de suicidio antiguo 15 (50%) 6 (20%) 0.02Riesgo de suicidio actual 6 (20%) 0 0,02Trastorno distímico 3 (10%) 1 (3,3%) 0,33Episodio (hipo) maníaco 4 (13,3%) 2 (6,6%) 0,67Trastorno de angustia actual 4 (13,3%) 1 (3,3%) 0,35Trastorno de angustia de por vida 6 (20%) 3 (10%) 0,47Trastorno de ansiedad de separación 5 (16,6%) 0 0,05Fobia social 5 (16,6%) 0 0,05Abuso de alcohol 0 2 (6,6%) 0,49Trastorno por déficit de atención / hiperactividad 13 (43%) 3 (10%) <0,01Trastorno de la conducta 5 (16,6%) 1 (3,3%) 0,19Trastorno negativista desafiante 5 (16,6%) 1 (3,3%) 0,19Trastornos psicóticos 2 (6,6%) 0 0,49
1. Test exacto de Fisher.
mitad había tenido riesgo de suicidio en el pasado y 20% presentaba riesgo de suicidio al momento de la evaluación. Por otro lado, hubo una asociación significativa entre la presencia de riesgo de suici-dio pasado y actual, el riesgo de suicidio actual con depresión y trastorno de conducta. Esto demuestra las consecuencias de estas comorbilidades en el de-sarrollo de la autoestima y la calidad de vida del paciente en este sentido, llama la atención que la mitad de los pacientes haya considerado la posibili-dad de suicidarse previo a la evaluación.
Entre adolescentes con epilepsia, existe mayor fra-caso escolar, lo cual se evidenció en este estudio por el antecedente de repitencia en el 40% de los casos (p=0,04). La comorbilidad psiquiátrica más frecuente fue el TDAH, presente en el 43% de los adolescentes con epilepsia. Se podría decir que pro-bablemente al tratar el déficit atencional en estos pacientes, mejoraría el pronóstico escolar. (12,13)
En el grupo de adolescentes sin epilepsia, hubo dos casos de abuso de alcohol (6,6%) y un caso de abuso de drogas (3,3%), versus en el grupo con epilepsia en que no hubo ningún caso de abuso de sustancias. Considerando el sesgo que ocurre al evaluar abu-so de sustancias, esta diferencia se podría atribuir a que los pacientes en general están en conocimiento de que el uso y abuso del alcohol y otras drogas puede descompensar la epilepsia. Durante las en-
trevistas, a modo de descripción cualitativa, la gran mayoría de los adolescentes evaluados contestaron con una rotunda negativa frente a la pregunta si consume alcohol.
En una revisión de 10 años de los estudios de tras-tornos psiquiátricos en pacientes con epilepsia, se mostró que existe un rango de 33 a 77% de comor-bilidad psiquiátrica, siendo en la presente investiga-ción de 60% (2, 11).
En nuestra práctica diaria, en pacientes con epilep-sia, debemos buscar estos trastornos, principalmen-te TDAH, trastorno del ánimo y ansiedad, trastor-nos de conducta y sobretodo signos indicadores de suicidio. Pese a grandes avances en aspectos técnicos en el área de la epilepsia; vemos que los adolescentes portadores de epilepsia presentan comorbilidades psiquiátricas frecuentes, que en nuestra realidad, al igual que en otros países, están siendo sub-diag-nosticadas.
REFERENCIA BIBLIOGRÁFICAS
1. Kanner AM. Psychiatric issues in epilepsy: The complex relation of mood, anxiety dis-orders and epilepsy. Epilepsy Behav 2009 May;15(1):83-7.
2. Plioplys S., Dunn D., Caplan R. 10 year re-

11
Comorbilidad psiquiátrica en adolescentes con epilepsia Karina Rosso et al.
search update review: Psychiatric problems in children with epilepsy. J Am Acad Child Ado-lesc Psychiatry. 2007 Nov;46(11):1389-402.
3. Baker GA., Spector S, McGrath Y, Soteri-ou H. Impact of epilepsy in adolescence: a UK controlled study. Epilepsy Behav. 2005 Jun;6(4):556-62.
4. Caplan R, Siddarth P, Gurbani S, Ott D, San-kar R, Shields WD, et al. Psychopathology and pediatric complex partial seizures: seizure-re-lated, cognitive, and linguistic variables. Epi-lepsia. 2004 Oct;45(10):1273-81.
5. Ott D, Siddarth P, Gurbani S, Koh S, Tournay A, Shields WD, Behavioral disorders in pediat-ric epilepsy: unmet psychiatric need. Epilepsia. 2003 Apr;44(4):591-7.
6. Sheehan DV, Sheehan KH, Shytle RD, Janavs J, Bannon Y, Rogers JE., et al. A population survey of mental health problems in children with epilepsy. Dev Med Child Neurol. 2003 May;45(5):292-5.
7. Dunn DW, Kronenberger WG. Childhood epi-lepsy, attention problems, and ADHD: review and practical considerations. Semin Pediatr Neurol. 2005 Dec;12(4):222-8.
8. Sheehan DV, Sheehan KH, Shytle RD, Janavs J, Bannon Y, Rogers JE. Reliability and valid-ity of the Mini International Neuropsychiatric Interview for children and Adolescents (MINI
KID). J Clin Psychiatry. 2010 Mar;71(3):313-26.
9. Sheehan DV, Lecrubier Y, Sheehan KH, Am-orim P, Janavs J, et al. The Mini Internation-al Neuropsychiatric Interview (M.I.N.I.): The Development and validation of a Structured Diagnostic Psychiatric Interview for DSM IV and ICD- 10. J Clin Psychiatry. 1998; 59 Suppl 20:22-33;quiz 34-57.
10. Richer LP, Shevell MI, Rosenblatt BR. Epi-leptiform abnormalities in children with at-tention-deficit-hyperactivity disorder. Pediatr Neurol. 2002 Feb;26(2):125-9.
11. Jones JE, Watson R, Sheth R, Caplan R, Koehn M, Seidenberg M. Psychiatric comorbidity in children with new onset epilepsy. Dev Med Child Neurol. 2007 Jul;49(7):493-7.
12. Hermann B, Jones J, Dabbs K, Allen CA, Sheth R, Fine J. The frequency, complications and aetiology of ADHD in new onset paediatric epilepsy. Brain. 2007 Dec;130(Pt 12):3135-48. Epub 2007 Oct 18.
13. Fastenau PS, Jianzhao Shen, Dunn DW, Austin JK. Academic underachievement among chil-dren with epilepsy: proportion exceeding psy-chometric criteria for learning disability and associated risk factors. J Learn Disabil. 2008 May-Jun;41(3):195-207.

12
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015Actualización
Electroencefalografía Neonatal NormalLoreto Ríos1, Catalina Torres2
ABSTRACT
Neonatal EEG monitoring is the most accurate tool to determine the maturity of the central nervous system , being even superior to the clinical exa-mination in the detection and prognosis of various brain dysfunctions. For its proper interpretation is it necessary to know in detail the brain maturatio-nal changes that are expressed at each gestational age, the parameters which will be analyzed and the optimal technical conditions. The aim of this article is to give a practical and complete approach on the normal electroencephalography of the neonatal period.Key words: normal neonatal electroencephalogra-phy
RESUMEN
La electroencefalografía neonatal es la herramienta más precisa para determinar la madurez del sistema nervioso central, siendo incluso superior al examen clínico en la detección y pronóstico de diversas dis-funciones cerebrales, pero para su correcta interpre-tación es necesario conocer en detalle los cambios madurativos cerebrales que se expresan a cada edad gestacional, los parámetros a analizar y las condi-ciones técnicas óptimas. El objetivo de este artículo es dar una aproximación lo más práctica y completa sobre la electroencefalografía normal en el período neonatal.Palabras clave: electroencefalografía normal neo-natal.
INTRODUCCIÓN
En el período de recién nacido (RN) el examen neurológico tiene una baja sensibilidad dado que
la mayoría de los reflejos evaluados son de origen espinal y de tronco cerebral, explicándose así que la respuesta frente a diferentes noxas, desde un punto de vista neurológico, sea inespecífica y no siempre se correlacione con la gravedad de ésta. El estudio imagenológico, en especial la resonancia magnéti-ca, considerada el examen de elección para el estu-dio de epilepsia en niños y adultos, particularmente en la evaluación de malformaciones del desarrollo cortical, tiene en el RN una utilidad algo limitada dada por la escasa mielinización del sistema ner-vioso central y por la necesidad de inmovilizar al paciente lo cual en recién nacidos críticamente en-fermos o de pretérmino extremo es de compleja im-plementación. Considerando que la maduración del sistema ner-vioso central (SNC) ocurre independientemente a igual velocidad si el proceso se realiza intra o extrauterinamente, la electroencefalografía (EEG) en el período del RN ha demostrado ser altamente superior al examen clínico en la detección y pro-nóstico de disfunciones cerebrales, siendo además hasta ahora el único examen que permite realizar una documentación continua funcional cerebral al lado de la cama del paciente en forma no invasiva, persistiendo, a pesar de los avances tecnológicos, como la herramienta más precisa en determinar la ontogenia o madurez del SNC , llegando a esta-blecer en forma consistente y con una precisión ± dos semanas la edad concepcional (EC) en el recién nacido de pretérmino (RN PreT) y de ± una sema-na en el de término (RNT). Sin embargo, durante este período son tantos los cambios madurativos cerebrales que se expresan en el EEG, que para su correcta interpretación, el reconocimiento de patro-nes normales y anormales exige tener un acabado entrenamiento y dilatada experiencia. Por todo lo anterior, es que la electroencefalogra-fía neonatal es considerada una rama aparte de la electroencefalografía pediátrica, por lo que este
1. Neuróloga Infantil. Liga Chilena Contra la Epilepsia. Centro Epilepsia Clínica Las Condes.2. Neuróloga adultos, Clínica Santa María.Recibido 1-5-15. Aceptado 30-8-15.
13
Electroencefalografía neonatal normal Loreto Ríos et al.
capítulo tiene como pretensión sólo entregar una guía práctica básica y didáctica para el lector no especialista.
CARACTERÍSTICAS DEL REGISTRO La duración y registro neonatal tendrán característi-cas diferentes, dependiendo si el objetivo es definir ontogénesis o disfunción cerebral aguda.
Si el objetivo es definir madurez cerebral es fun-damental obtener como mínimo un ciclo de sueño completo (sueño activo [SA] + sueño quieto [SQ]) y vigilia, considerándose necesario mínimo 2 horas como tiempo para obtener una óptima informa-ción. En un RNT, un ciclo de sueño dura aproxi-madamente 45-60 minutos, y comprende aproxima-damente 25 minutos de SA, 20 minutos de SQ y 10-15 minutos de sueño indeterminado (también re-ferido como sueño transicional). Hay que recordar que la mayor información se logra con el paciente durmiendo, dado que la vigilia frecuentemente va acompañada de llanto y movimiento que imposibi-litan la lectura. Si el objetivo es el estudio de disfunción cerebral aguda, por ejemplo en casos de encefalopatía hi-póxico isquémica, trastornos metabólicos u otros la duración puede ser menor, pero no menos de 1 hora como mínimo. En casos de sospecha de crisis epi-lépticas que en este grupo etario son principalmente subclínicas es recomendado video EEG de 24 hrs para su detección.
El registro neonatal debe ser con estudio poligráfi-co e idealmente con video asociado, el cual registre además de actividad EEG, movimientos oculares, respiración (flujo nasal y abdominal), actividad muscular (EMG de mentón) y ECG. De no existir la posibilidad de video, e incluso habiendo video, a experiencia personal de las autoras, lo óptimo es que durante el registro, el monitoreo esté presencia-do y registrado por profesional debidamente entre-nado, capaz de diferenciar movimientos fisiológi-cos de los anormales, como por ejemplo diferenciar movimientos oculares fisiológicos del SA y chupe-teo normal de crisis sutiles. Parámetros de Registro Neonatal: Las guías de registro EEG Neonatal han sido es-tablecidas por la Sociedad Americana de Neuro-
fisiología Clínica (Epstein 2006), que consiste en electrodos ubicados según sistema internacional 10-20 con algunas modificaciones para neonatos en su colocación, en los montajes sugeridos y en los parámetros de análisis.
Tabla 1: Montajes neonatales sugeridos
Montaje 1 Montaje 2 Montaje 3
Fp1-T3 Fp1-T3 Fp1-F7T3-01 T3-01 F7-T3Fp1-C3 Fp1-C3 T3-T5C3-01 C3-01 Fp1-F3Fp2-T4 Fp2-T4 F3-C3T4-O2 T4-O2 C3-P3Fp2-C4 FP2-C4 P3-01C4-02 C4-O2 Fp2-F8T3-C3 T3-C3 F8-T4C3-CZ C3-CZ T4-T6CZ-C4 CZ-C4 T6-O2C4-T4 C4-T4 Fp2-F4 F4-C4FZ-CZ FZ-CZ C4-P4CZ-PZ CZ-PZ P4-O2 FZ-CZ CZ-PZ EOG Izq EOG Izq EOG izqEOG Der EOG Der EOG der EMG EMG Nasal Abdominal ECG ECG ECG
En casos seleccionados, con alto riesgo de presen-tar crisis, dado lo focal y restringido que pueden ser éstas en este grupo etario es que a criterio de las autoras se recomienda utilizar montaje de doble banana cabeza completa, a fin de no dejar descu-biertas zonas cerebrales tales como temporales an-teriores, posteriores y parietales , lo cual es posible con electrodos especiales para RN, idealmente de plata u oro con introducción paulatina de compati-bilidad para Resonancia Magnética y Tomografía axial computada. Con respecto a los parámetros de registro para una correcta interpretación es imprescindible utilizar los filtros sugeridos para neonatos, insistiendo en el filtro de baja frecuencia debe ser a 0,5 hz y la velo-cidad a 15mm/segundo.

14
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Tabla 2. Parámetros EEG Digital
Sensibilidad 7uv/mmConstante de tiempo 0,3 segsFiltro de baja 0,5 hzFiltro de alta 70 hzNotchfilter(60-70hz) offVelocidad 15 mm/seg
Parámetros poligráficos equipo análogo
Constante Filtro Amplitud de tiempo
EOG 0,3-0,2 35 10EMG 0,3 35 10NASAL 3 70 5-10ABDOMINAL 0,6 10 1-10
Información necesaria para una correcta inter-pretación:
La interpretación no puede ser realizada si no se registra en forma certera la siguiente información: - Fecha de nacimiento, edad gestacional y días de
vida. - Asegurar que el registro tenga un ciclo completo
(SA y SQ).- Anotar hora de inicio y término.- Medicamentos con efecto en sistema nervioso
central y momento de administración.- Si se encuentra en hipotermia y etapa de la misma
(registrar en el examen cambios de la temperatura corporal).
- Registrar durante todo el examen, apneas, sustos, movimientos gruesos del cuerpo, llanto, gemi-dos, gestos faciales, intervenciones externas, etc.
INTERPRETACIÓN DEL REGISTRO La interpretación se basa en el conocimiento del pa-trón normal esperado para cada edad concepcional (EC). Ya se señaló que la madurez cerebral ocurre independientemente si es intra o extrauterinamente por lo que dos niños con la misma EC deben tener trazados similares. La EC se define como edad gestacional (EG) cal-culada al nacer más los días de vida extrauterina. Por ejemplo, en un RN PreT (32 semanas) con 1 mes de vida, su edad concepcional al momento del
EEG es de 36 semanas. Exigiremos que su madurez eléctrica sea compatible con esta edad de 36 + 2 se-manas y no de 1 mes de vida. La imprecisión de dos semanas se compensa con la posibilidad de realizar EEG seriados (semanales), que permitirá acercarse con máxima precisión a la edad concepcional.
¿QUÉ PARÁMETROS SE DEBEN ANALIZAR EN LAS DIFERENTES EDADES?
Trazado de fondo: diferenciar si se trata de un tra-zado continuo o discontinuo. Analizar su frecuen-cia y voltaje.Sincronía: definida como una diferencia menor a 1,5 seg. entre salvas ínter- hemisféricas, durante trazado discontinuo o alternante. Ésta varía según la EC (Figura 1).OrganizaciónTopográfica: grafo-elementos espe-cíficos de cada estadio según EC.Labilidad: cambios en la actividad de fondo que se generan en forma espontánea (cambios de estados).Reactividad: cambios del trazado electroencefalo-gráfico frente a estimulación del neonato.
Figura 1.
ONTOGENIA DEL ELECTROENCEFALOGRA-MA NEONATAL NORMAL
Edad gestacional < 28 semanas:Aproximadamente a las 28 semanas de gestación, los surcos cerebrales principales recién están co-menzando a formarse.
A esta edad existe muy poca motilidad espontánea, de ocurrir son principalmente movimientos tónicos. Los movimientos mandibulares clónicos aislados o repetitivos son frecuentes durante el período de prematurez y deben diferenciarse de actividad ic-tal. Los ojos se abren muy ocasionalmente frente

15
Electroencefalografía neonatal normal Loreto Ríos et al.
estímulos vigorosos y el patrón respiratorio es con-tinuamente irregular.
Patrón EEG: El trazado es siempre discontinuo, se caracteriza por un trazado de base plano inte-rrumpido por salvas (Burst) de ondas mixtas, predo-minantemente delta de mediano y alto voltaje (25-200uV) a 0,3-1,5 hz. de duración breve, menor a 15 segundos, en regiones parasagitales y occipitales (Figura 2). Una atenuación bitemporal puede obser-varse y refleja la falta de desarrollo del giro frontal
inferior y giro temporal superior.
Los períodos de atenuación de voltaje interdescar-gas, propios del trazado discontinuo son llamados intervalos interdescarga, los cuales a esta edad tie-nen su máxima duración y se acortan paulatinamen-te a mayor EC y durante los momentos de mayor actividad motora. En promedio estos períodos tie-nen una duración entre 8 y 16 segundos pudiendo llegar hasta un máxima promedio de 35 segundos (Tabla 3).
Figura 2: RN de 28 semanas. Se observa un trazado discontinuo durante todo el registro. Durante atenua-ción de voltaje se observa theta rítmico occipital. S: 7uV/mm, HF: 70 hz, LF: 0,5 hz, V: 15mm/s.
Tabla 3: EC e intervalo interdescarga
Edad Concepcional Máximo intervalo(semanas) Interdescarga (segundos)
< 30 35 30-33 20 34-36 10 37-40 6
Lo característico de ver a esta edad es el patrón del-ta brush, que consiste en que sobre las ondas delta
antes descritas, se sobreimponen brotes cortos de actividad rítmica rápida en rango de 8-22 hz de amplitud, que no excede los 60-75uV. La frecuencia con que se observan y su ubicación depende direc-tamente de la edad del paciente y el estado del mis-mo. A esta edad su presentación es difusa predomi-nantemente central.
Trenes de actividad rítmica alfa y/o theta pueden observarse independiente de los delta brush a esta edad, principalmente en regiones occipitales (Figu-ra 2).

16
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
La sincronía interhemisférica es alta, siendo apro-ximadamente entre un 90 y 100%.
Edad gestacional 29-31 semanas:A esta edad comienzan a aparecer diferencias de es-tado (vigilia – sueño). Aumenta la motilidad, se re-conocen movimientos oculares rápidos (MOR) y el estímulo para la apertura ocular es menor. La respi-ración adopta por cortos períodos un patrón regular. Comienza además, a esbozarse el reconocimiento de dos estadios del sueño: activo y quieto.
Patrón EEG: Continúa siendo preferentemente dis-continuo con intervalos interdescargas mas breves y segmentos de trazado continuo que marcan el inicio del SA. La sincronía interhemisférica presenta un marcado descenso, cayendo a aproximadamente un 50-70% debido a que el hipersincronismo previo,
proveniente de impulsos del tronco, comienzan a ser dominados por actividad cortical en pleno pro-ceso de sinaptogénesis y mielinización.
Los delta brush persisten siendo muy abundantes en zonas centrales, occipitales y temporales, teniendo una mejor configuración en regiones centrales y en SA.
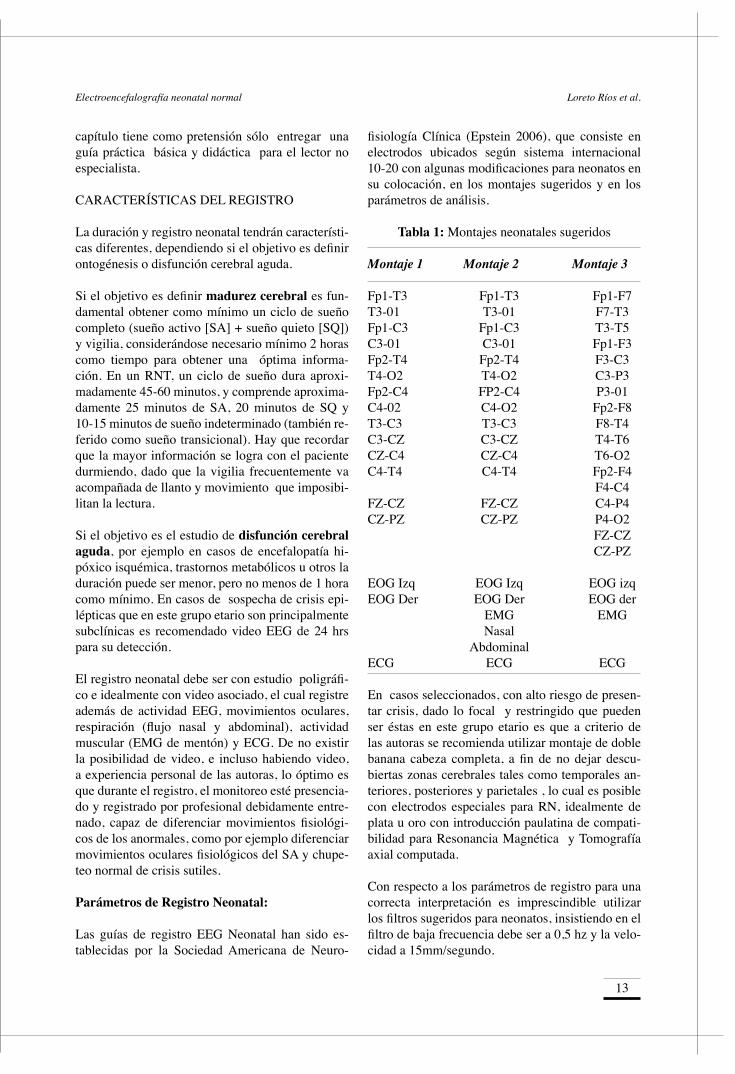
A esta edad, aparecen las transientes agudas tempo-rales o también denominadas theta rítmico temporal caracterizado por actividad theta rítmica aguda (4-7 hz) con amplitud aproximada de 25 a 120 uV que ocurre frecuentemente en regiones temporales. Son de aparición simétrica, bilateral sincrónica y asin-crónica. Rara vez se ven antes de las 29 semanas y después de las 32 semanas. Por lo tanto son de gran utilidad para estimar EC (Figura 3 y 4).
Tips < 28 semanas: Máxima discontinuidad, delta brush, alta sincronía.
Figura 3: RN de 30 semanas. Se observa un trazado discontinuo con delta brush (*) e inicio de theta rítmico temporal. S: 7uV/mm, HF: 70 hz, LF: 0,5 hz, V: 15mm/s. EEG Neonatal ALADE 2013 Dra. M. Yacubián.

17
Electroencefalografía neonatal normal Loreto Ríos et al.
Edad gestacional 32-34 semanas: Los movimientos corporales y oculares son más fá-sicos que tónicos. Se pueden reconocer ambos esta-dios de sueño, aunque lo que predomina es el sueño indeterminado, en el que es muy difícil establecer en cual de los dos estadios se encuentra. Sus perío-dos de respiración regular son más frecuentes.
Patrón EEG: En vigilia quieta y SA ya se puede observar un patrón EEG continuo. Durante SQ aún persiste el patrón discontinuo, pero con períodos de inactividad más breves, que nunca exceden los 15 segundos.
Los delta brushes y el theta rítmico, se presentan en ambos estadios de sueño, predominantemente sobre regiones temporales y occipitales.La sincronía interhemisférica en SQ es aproximada-mente entre un 60-80%.
A esta edad ocurren dos hallazgos importantes a considerar:1. Aparición de transientes agudas (TA) multifoca-
les en todos los estadios, que deben diferenciar-se de actividad epileptiforme. Estas se observan principalmente en regiones temporales y centra-les, sincrónicas y asincrónicas, aisladas o en tre-
Figura 4: RN de 31 semanas. Se observa aparición de theta rítmico temporal (*) y persistencia de theta occipital (**).S: 7uV/mm, HF: 70 hz, LF: 0,5 hz, V: 15mm/s. EEG Neonatal ALADE 2013 Dra. M. Yacubián.
Tips 29-31 semanas: Inicio SA como continuo, theta rítmico temporal, baja sincronía interhemisférica.

18
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
nes. Es importante diferenciar estas TA centrales de
artefactos secundarios a la pulsación de fontanela o excursión ventilatoria.
2. Aparición de reactividad en EEG, principalmente frente a estímulo auditivo.
Tips 32-34 semanas: SQ discontinuo intervalo breve, SA y vigilia continuo. Transientes agu-
das multifocales y reactividad.
Edad gestacional 35-37 semanas:El patrón de comportamiento de vigilia y sueño se reconoce claramente, existiendo una franca reac-tividad frente a estímulos externos, si bien aún no existe un patrón cíclico bien establecido. Los mo-vimientos tónicos son remplazados en su totalidad por movimientos fásicos del cuerpo que se mani-fiestan principalmente durante sueño activo. No se observa motilidad durante sueño quieto a excepción
de movimientos mioclónicos generalizados y movi-mientos faciales.
Patrón EEG: Clara diferenciación entre ambos es-tadios. El SA se caracteriza por aparición de MOR, con movimientos verticales de globos oculares y disminución de actividad tónica EMG a nivel de mentón y el SQ se reconoce fácilmente por apari-ción de largos períodos de respiración regular.
El sueño deja de ser discontinuo en SQ, donde se observa trazado alternante caracterizado por brotes de actividad theta y delta de gran amplitud (200uV) entremezclados con actividad rápida de alta fre-cuencia alfa y beta de 3 a 10 segundos de dura-ción, con periodos alternantes de similar duración durante los cuales se observa actividad theta y alfa de amplitud promedio de 50 uV. Este patrón mas frecuentemente observado durante SQ a esta edad y muestra casi un 100% de sincronía (Figura 5).
Figura 5: RN de 37 semanas. Se registra SQ con trazado alternante. S: 7uV/mm, HF: 70 hz, LF: 0,5 hz, V: 15mm/s.
19
Electroencefalografía neonatal normal Loreto Ríos et al.
Se mantiene entonces el trazado continuo en todos los estadios y en vigilia es posible incluso reconocer cierta gradiente antero posterior de amplitud, predo-minando mayor amplitud en regiones posteriores. Los delta brushes en esta edad comienzan a dismi-nuir hasta desaparecer a las 42 semanas, siendo más frecuentes durante sueño quieto y sobre regiones occipitales. La sincronía interhemisférica en SQ es aproximada-mente entre un 70-85 %.
Es así como se consideran hallazgos característicos a esta edad:1. Reemplazo progresivo del trazado discontinuo
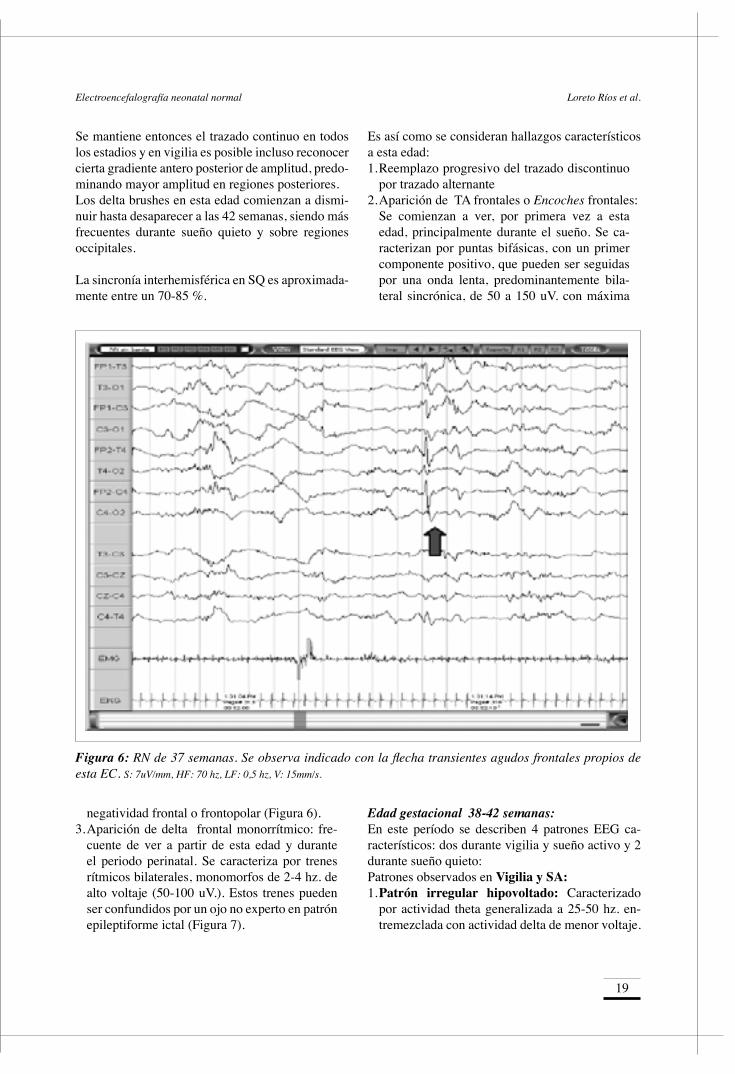
por trazado alternante2. Aparición de TA frontales o Encoches frontales: Se comienzan a ver, por primera vez a esta
edad, principalmente durante el sueño. Se ca-racterizan por puntas bifásicas, con un primer componente positivo, que pueden ser seguidas por una onda lenta, predominantemente bila-teral sincrónica, de 50 a 150 uV. con máxima
Figura 6: RN de 37 semanas. Se observa indicado con la flecha transientes agudos frontales propios de esta EC. S: 7uV/mm, HF: 70 hz, LF: 0,5 hz, V: 15mm/s.
negatividad frontal o frontopolar (Figura 6).3. Aparición de delta frontal monorrítmico: fre-
cuente de ver a partir de esta edad y durante el periodo perinatal. Se caracteriza por trenes rítmicos bilaterales, monomorfos de 2-4 hz. de alto voltaje (50-100 uV.). Estos trenes pueden ser confundidos por un ojo no experto en patrón epileptiforme ictal (Figura 7).
Edad gestacional 38-42 semanas:En este período se describen 4 patrones EEG ca-racterísticos: dos durante vigilia y sueño activo y 2 durante sueño quieto:Patrones observados en Vigilia y SA:1. Patrón irregular hipovoltado: Caracterizado
por actividad theta generalizada a 25-50 hz. en-tremezclada con actividad delta de menor voltaje.

20
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
2. Patrón mixto: Semejante al anterior, pero con la presencia además de ondas delta a 2-4 hz. de ma-yor voltaje entremezcladas.
Patrones observados en SQ:-Patrón lento hipervoltado o patrón de onda len-ta continua: Caracterizado por actividad delta con-tinua a 50-150 hz.-Trazado alternante ya anteriormente descrito. La desaparición gradual de este trazado ocurre entre las 3 y 6 semanas de vida de un RN de término.
Los delta Brushes son muy escasos, pudiéndose ver exclusivamente en SQ.
Las TA frontales y el delta frontal monorrítmico aun persisten, principalmente durante SQ. Las TA multifocales pueden observarse sólo ocasio-nalmente sobre regiones centrales y temporales. La
presencia de TA se consideran normales hasta las 46 semanas de EC.
Resumen de Hitos de Evolutividad: (Figura 8):- Menor a 30 semanas siempre es discontinuo con
intervalos interdescargas progresivamente más cortos.
- Continuidad se observa inicialmente en SA a las 30 semanas, luego en vigilia a las 34 semanas y finalmente en SQ como trazado alternante a las 36-38 semanas.
- Desde las 29 semanas, la sincronía va aumentan-do pero siempre es más asincrónico en SQ.
- La reactividad está presente desde las 32 sema-nas.
Figura 7: RN de 38 semanas. Se observa aparición de delta frontal monorítmico S: 7uV/mm, HF: 70 hz, LF: 0,5 hz, V: 15mm/s.
Tips 35-37 semanas: Trazado alternante en SQ, transientes agudas frontales, delta frontal monorít-mico.

21
Electroencefalografía neonatal normal Loreto Ríos et al.
Actividad delta: - Delta Brushes: • Aparecen a las 26 semanas, predominantemente
en áreas centrales. • 28-32 semanas: son muy frecuentes, predominio
difuso, en SA. • 32-34 semanas: son más posteriores (temporo-
occipitales), predominio SA y SQ. • 34-37 semanas: predominan en sueño SQ y a
partir de las 36 semanas se localizan típicamente en región occipital.
- Delta rítmicos en cuadrantes posteriores • 29-31 semanas: generalmente síncronos que de-
ben diferenciarse de los movimientos de cabeza o respiratorios.
- Actividad delta rítmica frontal. (ADRF) • > 37 semanas: Deben desaparecer a las 40 se-
manas.
TAF y ADRF: orientan a RNT, se ven frecuente-mente después de las 35 semanas y desaparecen el primer mes de vida en vigilia y puediendo persistir durante el sueño hasta los 2 meses de vida. TA Temporales: orientan a prematuridad. Apare-cen a las 26-28 semanas. Máxima expresión es a las 30-32 semanas. Se pueden encontrar hasta las 37-38 semanas. No deben existir después de las 44 semanas.
DISCONTINUO DISCONTINUO-CONTINUO CONTINUO < 29 Sem 29-31 Sem 32-34 Sem 35-37 Sem 38-40 Sem RNT Discontinuo máxi- Discontinuo Discontinuo Continuo Se registran 4 80% inician mo intervalo Inicio SA como solo en SQ Alternante patrones: en SA inter-descarga continuo en SQ
Sincronía 100% Sincronía 50-70% Sincronía 70% Sincronía 80% Vigilia y SA: Sincronía 100% -DT bajo voltaje -DT mixto en voltaje Delta brushes Delta brushes Delta brushes < Delta brushes SQ: > 44 semanas se abundantes y en SQ <TA -Trazado ve Sueño onda más frecuentes alternante delta en SQ en SA -Sueño ondas delta Delta occipital TA Temporales TA multifocales TAF TA disminuyen Delta frontal en vigilia monorítmico Theta rítmico Reactividad Existe traslape Theta rítmico temporal frecuente entre TA central y delta brushes
BIBLIOGRAFÍA
1. Volpe JJ. Neurology of the Newborn. 3rd. ed. Philadelphia, PA: WB Saunders; 2001.
2. Sarnat HB. Anatomic and physiologic correlates of neurological development in prematurity. In: Sarnat HB, editor. Topics in neonatal neurology,
Grune & Stratton, 1984, pp. 1-25.3. Lombroso CT 1985a. Neonatal polygraphy in
full-term and preterm infants. A review of nor-mal and abnormal findings. J Clin Neurophysiol 2: 105-155.
4. Hrachovy RA, Mizrahi EM, and Kellaway P. 1990. Electroencephalography of the New born.

22
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
In Daly DD, Pedley TA (eds) Current Practice of Clinical Electroencephalography, 2nd ed., New York. pp. 201-242.
5. Epstein CM, Guidelines Two: minimum techni-cal standards for pediatric electroencephalogra-phy. J Clin Neurophysiol 2006;23:92-96.
6. Renée A. Shellhaas, Taeun Chang, Tammy Tsuchida et al. The American Clinical Neuro-physiology Society´s Guideline on Continuous Electroencephalography Monitoring in Ne-onates. Journal of Clinical Neurophysiology 2011;28 (6),611-617.
7. Elion Shany and Itai Berger. Neonatal Elecoen-cephalography: Review of a practical approach. Journal of Child Neurology 2011; 26 (3) 341-355.
8. Tammy N. Tsuchida, Courtney J. Wusthoff, Renée A. Shellhaas et al. American Clinical Neurophysiology Society Standardized EEG Terminology and Categorization for the descrip-tion of continuous EEG Monitoring in Neonates:
Report of the American Clinical Neurophysiol-ogy Society Critical Care Monitoring Commit-tee. J Clin Neurophysiol 2013;30:161-173.
9. Mizrahi EM, Hrachovy RA, Kellaway P. Atlas of Neonatal Electroencephalography. Third Edi-tion. Lippicott Williams & Wilkins. 2004.
10. Lombroso CT. 1979. Quantified electrographic scales on 10 pre-term healthy newborns fol-lowed up to 40-43 weeks of conceptional age by serial polygraphic recordings. Electroencepha-logr Clin Neurophysiol 46:460-474.
11. Sher MS, and Barmada M. 1987. Estimation of gestational age by electrographic, clinical and anatomical criteria. Pediatr Neruol 3: 256-262.
12. Lombroso CT. Neonatal EEG Polygraphy in Normal and Abnormal Newborns. Electroen-cephalography. Basic Principles, Clinical Ap-plications and Related Fields, E. Niedermeyer and F. H. Lopes da Silva, Eds., chapter 48, pp. 803-875, Williams and Wilkins, Baltimore, Md, USA, 1993.

23
ABSTRACT
The neurological examination has low sensitivity in this period implying that the response to various insults from the neurological point of view is non-specific and not always correlate with the severity of this. That is why the neonatal electroencepha-lography has proved highly superior to clinical ex-amination in the detection and prognosis of brain dysfunction. Besides being so far the only test that allows a continuous brain functional documenta-tion bedside patient noninvasively. The resolution or persistence of abnormal patterns has important prognostic implications.The evaluation of an abnormal trace is important to analyze separately four variables:I. Tracing abnormalities base.II. No ictal paroxysmal abnormal patterns.III. Ictal paroxysmal abnormal patterns.IV. Abnormalities in the organization of states and maturational.Keywords: EEG, high sensitivity, diagnosis, out-come.
RESUMEN
El examen neurológico tiene una baja sensibilidad en este período implicando que la respuesta frente a diferentes noxas desde el punto de vista neurológi-co sea inespecífica y no siempre se correlaciona con la gravedad de ésta. Es por esto que la electroen-cefalografía neonatal ha demostrado ser altamente superior al examen clínico en la detección y pro-nóstico de disfunciones cerebrales. Siendo además hasta ahora, el único examen que permite realizar una documentación funcional continua cerebral al lado de la cama del paciente en forma no invasiva. La resolución o persistencia de patrones anormales
tiene implicancias pronósticas muy importantes.
En la evaluación de un trazado anormal es impor-tante analizar cuatro variables por separado:I. Anomalías del trazado de base.II. Patrones paroxísticos anormales no ictales.III. Patrones paroxísticos anormales ictales.IV. Anormalidades en la organización de los estados y maduracional.Palabras clave: alta sensibilidad, diagnóstica, evo-lutiva y pronóstica.
INTRODUCCION
Como se mencionó en el capítulo de electroence-falografía (EEG) neonatal normal, el examen neu-rológico tiene una baja sensibilidad en este perío-do implicando que la respuesta frente a diferentes noxas desde el punto de vista neurológico sea ines-pecífica y no siempre se correlaciona con la grave-dad de ésta. Es por esto que la electroencefalografía neonatal ha demostrado ser altamente superior al examen clínico en la detección y pronóstico de dis-funciones cerebrales. Siendo además hasta ahora, el único examen que permite realizar una documenta-ción funcional continua cerebral al lado de la cama del paciente en forma no invasiva. La resolución o persistencia de patrones anormales tiene implican-cias pronósticas muy importantes.
En la evaluación de un trazado anormal es impor-tante analizar cuatro variables por separado:I. Anomalías del trazado de base.II. Patrones paroxísticos anormales no ictales.III. Patrones paroxísticos anormales ictales.IV. Anormalidades en la organización de los estados y maduracional.
I. ANORMALIDADES DEL TRAZADO DE BASESi bien los patrones electroencefalográficos anor-males rara vez son patognomónicos de patologías
Electroencefalografía Neonatal.Registro Anormal.Loreto Ríos1, Jovanka Pavlov2
1. Neuróloga Infantil. Centro Avanzado de Epilepsia, Clínica Las Condes, Liga Chilena Contra la Epilepsia.
2. Neuróloga Infantil. Laboratorio Electroencefalografía Hospi-tal Clínico de La Florida, Hospital Carabineros de Chile.
Recibido 1-8-15. Aceptado 21-8-15.
Actualización

24
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Figura 1: RNT (38 semanas), con asfixia neonatal severa, APGAR:0-0-5. EEG: Patrón hipovoltado indi-ferenciado, tomado a las 24 horas de vida. S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s
específicas, si son altamente sensibles. Es importan-te tener en cuenta que los cambios electroencefalo-gráficos en el neonato pueden ser muy rápidos, la normalización del trazado puede ocurrir incluso en pacientes en que persisten severas secuelas neuro-lógicas, por esto el seguimiento periódico es fun-damental, ya sea a través de video monitoreo conti-nuo, seguimiento diario o semanal, dependiendo del estado clínico. El trazado de base provee el índice pronóstico. Nunca hay que olvidar que es muy im-portante considerar durante el registro, la labilidad y reactividad del trazado, dado que la ausencia de estos dos parámetros da un factor de gravedad y mal pronóstico.
A continuación se detallan brevemente, según la clasificación de Lombroso las diferentes anomalías del trazado de base y patrones paroxísticos epilépti-cos y no epilépticos:
1. Patrón Electroencefalográfico Inactivo (Iso-eléctrico).
No existe un consenso universal aceptado para de-finir este patrón, según diversos autores se exige voltaje < 5 uV o < 2 uV de actividad continua, con una sensibilidad de registro 2 uV/mm y distancia interelectrodo 10 cm, sin reactividad a estímulos. Tiene valor pronóstico ominoso, pero no implica muerte cerebral. Si existe este patrón, debe repe-tirse un electrocefalograma dentro de 24-48 horas. Si persiste debe realizarse potenciales evocados de tronco, que de estar muy alterados, la sobrevida es poco probable.
2. Patrón Hipovoltado Indiferenciado
Se define como actividad entre 5-15 uV durante vi-gilia y sueño activo y 10-15 uV en sueño quieto. En
25
Electroencefalografía neonatal. Registro anormal. Loreto Ríos et al.
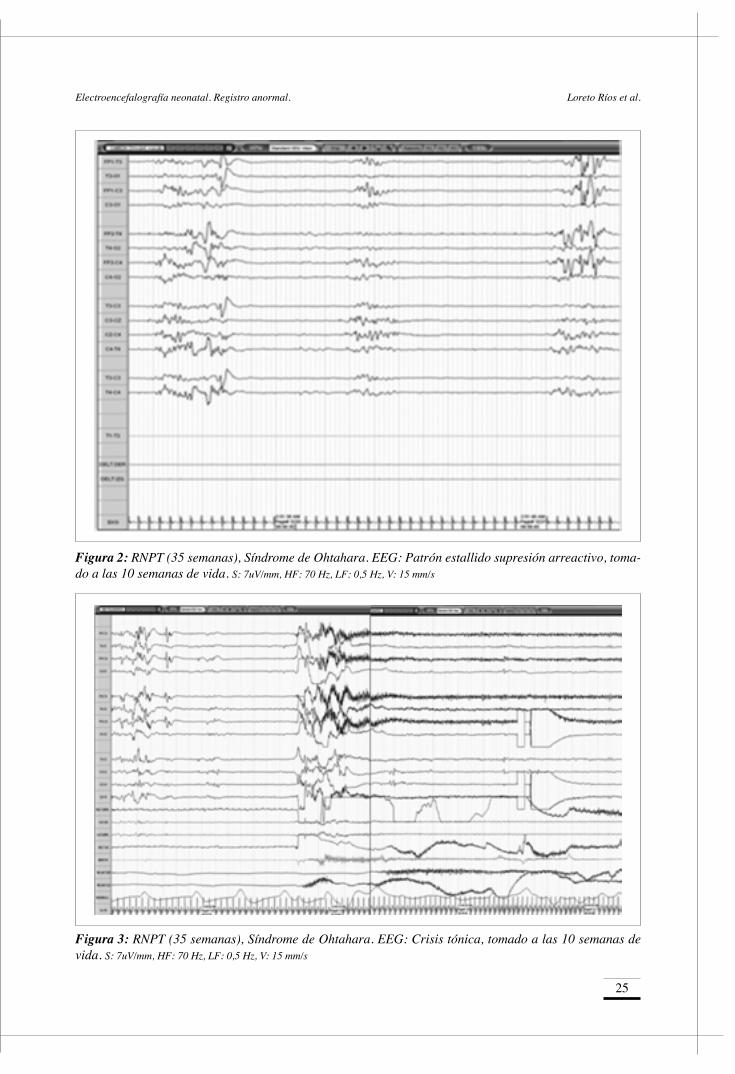
Figura 2: RNPT (35 semanas), Síndrome de Ohtahara. EEG: Patrón estallido supresión arreactivo, toma-do a las 10 semanas de vida. S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s
Figura 3: RNPT (35 semanas), Síndrome de Ohtahara. EEG: Crisis tónica, tomado a las 10 semanas de vida. S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s

26
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
términos generales se describe como un patrón < 20 uV en forma continua, sin diferenciación entre los diferentes estadíos. La causa más frecuente de este patrón es la hipoxia.
Su persistencia por más de 2 semanas en un recién nacido de término (RNT), se considera criterio de alto riesgo de anormalidad neurológica.3. Patrón Estallido Supresión (E.S.)
Se caracteriza por períodos de inactividad (< 5 uV) de 2-20 segundos de duración, más constantes que en trazado discontinuo, interrumpidos por paroxis-mos de 1 a 10 segundos, sincrónicos o asincrónicos compuestos de actividad mixta de ondas delta-theta con espigas y puntas multifocales intercaladas de alto voltaje. Frecuentemente es arreactivo, descri-biéndose en escasas ocasiones la persistencia de
reactividad frente a la estimulación vigorosa del recién nacido. La no reactividad es un signo de mal pronóstico. Puede aparecer sólo en sueño, especial-mente sueño quieto, con un trazado de vigilia igual-mente anormal. En casos más extremos se presenta en todos los estadíos.
Este patrón es altamente orientador de dos encefa-lopatías epilépticas precoces tales como, el Síndro-me de Ohtahara (S.O.) y la Encefalopatía mioclóni-ca infantil precoz (E.M.I.). Si bien el S.O. se asocia más frecuentemente a malformaciones cerebrales y la E.M.I. a una etiología de origen metabólico (por ejemplo hiperglicinemia no cetócica), muchas de sus características se superponen y de no tener cla-ro el origen, debe considerarse el estudio de errores innatos del metabolismo.
Tabla 1: Características EEG estallido supresión en encefalopatía epilépticas precoces:
Síndrome de Ohtahara Encefalopatía mioclónica infantil precoz
Presente en vigilia y sueño Aumentado en sueño Intervalo inter-descarga regular Intervalo inter-descarga irregular (estallidos más breves y períodos más prolongados de supresión)
Otras causas de este patrón son anoxia, coma bar-bitúrico, intoxicación por litio y otras drogas que pudiesen provocar trazado E.S. iatrogénico.Este patrón cambia frecuentemente a un patrón hip-sarrítmico después de las 42-48 semanas.
4. Patrón Excesivamente Discontinuo
Definido cuando el intervalo interdescarga del tra-zado discontinuo o alternante es mayor a lo espe-rado para la edad concepcional (EC). Se considera una duración máxima de intervalo interdescarga según edad de:
Tabla 2: EC e intervalo interdescarga
Edad Concepcional Máximo intervalo (semanas) Interdescarga (segundos) < 30 35 30-33 20 34-36 10 37-40 6

27
Electroencefalografía neonatal. Registro anormal. Loreto Ríos et al.
Figura 4: RNT (39 semanas). Accidente vascular arteria cerebral media izquierda. EEG: Asimetría inter-hemisférica. Flecha: hipovoltaje hemisférico izquierdo. S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s
5. Patrón Discontinuo Permanente
Se considera discontinuidad permanente, cuando la duración del intervalo interdescarga mide >35 se-gundos, con una amplitud < 10 uV.
6.DeltaDifuso
Es un trazado muy raro de ver. Consiste en actividad principalmente entre 0,5-1 Hz. tanto en sueño como en vigilia, con muy poca actividad en banda theta. Su persistencia después de 2 semanas de vida en RNT es de muy mal pronóstico. Si existe actividad crítica en base lenta indica pronóstico muy grave.
7. Asimetría de Amplitud Interhemisférica
Se define como asimetrías inter-hemisféricas per-sistentes sobre un 25- 50% en amplitud. Este patrón
frecuentemente se asocia a anomalías estructurales. Existe además el patrón de asimetría interhemisfé-rica transitoria, la cual como su nombre señala, es transitoria y sin mayor implicancia clínica. 8. Atenuación Regional Focal
Es un patrón difícil de evaluar por tener muchos falsos positivos y negativos. Sin embargo si la ate-nuación es persistente debe sospecharse una lesión estructural, debido a que se describe que tendría una especificidad de hasta 85%.
9. Asincronía de las Descargas Interhemisférica
Se define como una desviación de la sincronía espe-rada para una E.C. determinada. Se habla de asin-cronía cuando existe una separación interdescarga >1,5 segundos.

28
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
II. PATRONES PAROXÍSTICOS ANORMALES NO ICTALES.
Estas alteraciones, al ojo no experto son una de las más difíciles de diferenciar de transientes agudas normales propias de cada edad evolutiva del siste-ma nervioso central. A criterio de los autores es uno de los grafoelementos que deben interpretarse con más cuidado.A continuación se detallan algunas de estas:
1. Transientes Agudos (T.a):
Los criterios de anormalidad son varios a consi-
derar, si bien la morfología puede ser similar a las transientes fisiológicas, deben considerarse como anormales cuando:1) Son repetitivas, más que al azar.2) Excesivamente frecuentes para la EC (>3 min).3) Consistentemente focales.4) Ocurren en brotes breves.
Su aparición es durante los períodos de discontinui-dad o atenuación de voltaje en período alternante y/o la persistencia en vigilia y sueño quieto en re-cién nacido > 37 semanas. Cuando ocurren en bro-tes breves su morfología puede ser polifásica.
Figura 5: RNPT 31 semanas. Apneas en estudio. EEG tomado a los 60 días de vida (EC: 39 semanas). Asincronía interhemisférica > 1,5 seg. S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s
SE CONSIDERAN CON PRONÓSTICO GRAVE Y DESFAVORABLE EL PATRÓN INAC-TIVO, EXCESIVAMENTE DISCONTINUO Y EL PATRÓN DELTA DIFUSO, CUANDO PERSISTE DESPUÉS DE LAS SEGUNDA SEMANA DE VIDA.
Tips: Frecuencias de transientes agudas >3/min es ANORMAL

29
Electroencefalografía neonatal. Registro anormal. Loreto Ríos et al.
Transientes agudas NORMAL ANORMAL
Polaridad Siempre negativa Prominente fase positiva inicial.
Amplitud Entre mayor amplitud y duración es más sospechosa.
Excede los 150 uV y 150 mseg anormal.
Localización Normales ocurren al azar. Frecuentes en misma zona, en sucesión y trenes es anormal (≥3 min).
Morfología Mono o difásica Polifásicas y seguido de onda lenta de muy alto voltaje. Aparición Prematuro en cualquier etapa pero en Si aparecen en vigilia es más probable RNT más comunes en sueño transición. anormal. Persistencia después de las 44 semanas.
Nemotecnia: PALMA. Gentileza Dra.Catalina Torres
Si ocurren en frecuencia mayor a 10 segundos con-figuran un patrón ictal. Su etiología es diversa y poco específica.
El hallazgo de estas transientes agudas, mejora su especificidad si existe persistencia de ellos en EEG seriados de no menos de 1 semana de intervalo.
A continuación se describen aquellas transientes agudas anormales interictales más características:
T.A. ROLÁNDICAS POSITIVAS: Son transientes agudas de polaridad positiva, que aparecen en re-giones rolándicas con amplitud entre 20 a 200 mV en montajes bipolares, uni o bilaterales, sincrónicas o no y a veces aisladas en Cz. Son fuertes predic-tores de hemorragia intraventricular y/o lesión en sustancia blanca con pobre pronóstico. Son cuanti-ficadas por su frecuencia de aparición. La aparición de 1 a 2 por minuto se relacionan con lesiones de sustancia blanca (leucomalasia periventricular) y discapacidad motora.
Figura 6: RNT(38 semanas). Hemorragia intraventricular. Transientes agudas centrales (der: montaje bipolar izq: montaje derivado a Cz). . S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s

30
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
T.A. TEMPORALES POSITIVAS: Son transientes agudas de polaridad positiva en la región temporal, con amplitud sobre 50 uV y una duración de 70 a 400 mseg. Más frecuentes en RNPT que han sufrido insultos hipóxicos con alteraciones cerebrales tales como hemorragia intraventricular grado III y leu-comalacia periventricular. Tiene un valor predictivo desfavorable cuando se acompaña de un patrón de base anormal. Si el patrón de base es normal su va-lor tiene menor especificidad.
2. Patrón de Descargas Periódicas de Baja Fre-cuencia.
Son descargas estereotipadas, rítmicas o casi- rít-micas, de puntas de base ancha de 0,5 a 2 Hz. Pue-den ser focales, hemigeneralizadas o generalizadas. Dentro de ellas se describen las descargas periódi-cas de baja frecuencia de morfología dicrótica y las descargas epileptiformes lateralizadas periódicas.
En general se describen juntas, dado que muchas veces son muy difíciles de diferenciar, compartien-do similitudes morfológicas y espaciales (ubica-ción más frecuente en regiones parietotemporales o temporales). Ambos eventos ocurren en pacientes encefalopáticos, con lesiones estructurales agudas. Su presencia corresponde a un signo de muy mal pronóstico. 3. Patrón de Descargas Multifocales Periódicas o Cuasiperíodicas.
Se caracteriza por descargas con morfología y ran-gos de repetición (c/4 a 14 seg.) constantes para cada foco. La actividad para el resto del EEG no se ve afectada por otras descargas focales de otras áreas. Se ha descrito como patognomónico de En-cefalitis Herpética Neonatal con un líquido cefalo-rraquídeo alterado.
Figura 7: RNT 39 semanas. Asfixia neonatal severa. Patrón de descargas periódicas de baja frecuencia, con ondas dicróticas. S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s

31
Electroencefalografía neonatal. Registro anormal. Loreto Ríos et al.
4. Patrón Alterante “Theta Pointu”
También llamado de las convulsiones del quinto día. Se caracteriza por actividad theta interictal, dis-continua y frecuentemente asincrónica, arreactiva y mezclada con transientes agudas. Sin las caracterís-ticas fisiológicas del recién nacido de término.
III. PATRONES PAROXÍSTICOS ANORMALES ICTALES.
Para hablar de un patrón ictal, este debe durar más de 10 segundos y en él debe diferenciarse entre pa-trones ictales focales, multifocales y generalizados.
- Patrones Focales: Dada la escasa mielinización a esta edad las crisis frecuentemente son focales y restringidas con escasa o nula propagación a áreas vecinas. Es por esto que se recomienda a criterio de las autoras utilizar montaje de doble banana cabeza completa, a fin de no dejar descubiertas zonas cere-brales tales como temporales anteriores, posteriores y parietales. Su localización más habitual es en re-giones rolándicas, seguidas de áreas temporales y menos frecuente en regiones posteriores.
Este patrón de descarga se asocia a crisis clínicas, más comúnmente a patrones clónicos contralatera-les. Los patrones ictales focales se subdividen en no rítmicos, rítmicos y pseudo-rítmicos.
- Patrones no rítmicos: Aparecen en forma de ondas agudas o puntas, las que deben considerar-se anormales si son repetitivas y excesivas para la EC, consistentemente focales, en salvas, polifásicas o cuando se registran en períodos de atenuación del trazado alternante.
- Patrón rítmico o pseudo-rítmico: Se definen como descargas rítmicas de morfología similar, que cambian en evolución con respecto a frecuencia, amplitud y campo eléctrico. Este patrón puede di-fundir hacia áreas homólogas pero muy excepcio-nalmente se generalizan.
- Patrones multifocales: Descargas similares al patrón focal en morfología y frecuencia, que emer-gen en 2 o más localizaciones, independientemente. Con gran variabilidad en frecuencia o en morfolo-gía entre diferentes focos o dentro de la misma lo-calización.
Su presentación clínica puede variar de tipo clóni-co a tónico, a menudo con características fragmen-tarias, saltatorias y anárquicas, con sacudidas que migran rápidamente de un lado a otro y se pueden confundir con crisis generalizadas.
- Patrón Generalizado: El característico y patog-nomónico es el patrón de HIPSARRITMIA NEO-NATAL. Este patrón aparece sólo en sueño quieto, pudiendo estar ausente aún en vigilia y sueño acti-
Figura 8: RNPT 31 semanas. Patrón rítmico de origen temporal derecho (T4). S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s

32
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
vo. Se observa en neonatos severamente compro-metidos, usualmente post-asfixia o en el curso de una meningoencefalitis. Se caracteriza por desorga-nización severa hipervoltada (>200 uV) de la activi-dad basal, entremezclada con actividad epileptifor-me tipo espiga y poliespiga onda lenta, multifocal
independiente, asociada a períodos de depresión o supresión de voltaje.
Si existen crisis estas son multifocales, anárquicas o sutiles. Puede comprometer sólo un hemisferio (hemi-hipsarritmia) y es casi siempre visto en el
Figura 9: RNT. 38 semanas. Crisis eléctrica central (Cz). ). S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s
Figura 10: RNT 38 semanas. Asfixia neonatal. Patrón multifocal de inicio en C3 y propagación a Cz.S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s

33
Electroencefalografía neonatal. Registro anormal. Loreto Ríos et al.
contexto de daño cerebral severo. En estos casos debe sospecharse malformaciones cerebrales, en especial, síndrome de Aicardi.
- Patrón de descargas ictales breves: Son descar-gas ictales de pocos segundos de duración. Denomi-nadas brief rhytmic discharges (BRD) en la litera-tura anglosajona.
Son de significado incierto y son vistas usualmen-te en el contexto de un EEG con actividad de base anormal y/o con crisis electrográficas.
-EEGendéficitdepiridoxina: Es un trastorno in-frecuente, pero que por su fácil tratamiento y exce-lente pronóstico, cuando su diagnóstico es precoz, debe estar incluido en el diagnóstico diferencial en las crisis neonatales. El patrón ictal se caracteriza por brotes repetitivos de ondas de alto voltaje a 1-4 Hz entremezclados con espigas, de presentación generalizada o hemisférica alternante. El patrón in-terictal es de bajo voltaje o de ondas lentas difusas,
con o sin descargas interictales.
La administración de piridoxina endovenosa en do-sis de 100-200 mg. induce una mejoría electroence-falográfica rápida y significativa.
IV. ANORMALIDADES EN LA ORGANIZA-CIÓN DE LOS ESTADOS Y MADURACIONAL.
Esta sección sin lugar a dudas, es imposible reali-zar sin el conocimiento claro y acabado de todo lo anteriormente descrito. El análisis de las anormali-dades en la organización de los estados y madura-cional escapan a los objetivos de este capítulo, sin embargo es importante conocerlos. A continuación se enumeran:
1. Desviaciones del estado de sueño: Consiste en alteraciones en la labilidad y duración de los esta-díos del sueño. Esta condición debe ser establecida con monitoreos prolongados y realizados con inter-valo de 1 semana. Sin embargo, el no registrar cam-
Figura 11: RNT 38 semanas. Encefalopatía hipóxico isquémica. Descarga ictal breve de ritmos theta a nivel T3. S: 7uV/mm, HF: 70 Hz, LF: 0,5 Hz, V: 15 mm/s

34
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
bios en el estadío de sueño en el registro de 1 hora exige repetir el estudio sospechando esta condición. Su detección incide en el pronóstico futuro.
2. Estadíos no reconocibles: En este caso no es po-sible diferenciar los diferentes estadíos en registros prolongados y en recién nacidos >34 semanas. Mu-chas veces no es posible diferenciar vigilia y sueño. En estos casos debe descartarse factores inductores de esta condición como: hipotermia, factores tóxi-cos reversibles, niveles de fármacos antiepilépticos elevados entre otros. La detección de este patrón debe interpretarse con valor pronóstico expectante.
3. Excesivo estadío de transición: Consiste en un porcentaje de sueño transicional mayor a lo espe-rado. Su valor pronóstico es inespecífico y no es ominoso.
4. Labilidad de los estadios: Consiste en cambio rápidos de los estadíos mayor de lo esperado para edad concepcional. Se ha descrito en recién nacidos hiperexcitables de madres consumidoras de drogas.5. Dismadurez: EEG retrazado en más de 2 semanas en relación a EC. Este a su vez reúne dos conceptos, el de heterocronismo (diferentes edades electroen-cefalográficas en un mismo trazado) y anacronis-mos (EEG no compatible con EC). Existe una for-ma transitoria y otra persistente, lo que es definido según registros electroencefalográficos semanales.
Clasificación video EEG de las crisis neonatales:
Crisis clínica con sello electrocortical consisten-te:Fisiopatología-epiléptica Focal clónica Unifocal Multifocal Hemiconvulsiva Axial Focal tónica Postura troncal asimétrica Postura de extremidades Desviación ocular sostenida Mioclónica Generalizada Focal
Crisis clínica sin un sello electrocortical consis-tente:Fisiopatología-presumiblemente no epiléptica Mioclónica Generalizada Focal Fragmentaria Generalizada tónica Automatismos Motores Movimientos Oro-buco-linguales Signos oculares Movimientos de progresión
Crisis eléctrica sin actividad crítica clínica: Fisiopatología-epiléptica
Mizrahi EM, Kellaway P. Diagnosis and management of neona-tal seizures. Philadelphia: Lippincott-Raven, 1998.
En conclusión, luego de analizar los distintos patrones electroencefalográficos posibles de reconocer en el período neonatal y en conocimiento de la existencia en este período de crisis tanto electrográfi-cas como electroclínicas la sugerencia de las autoras es realizar un VideoEEG de 2 horas de duración en busca de alteraciones, y de existir crisis complementar con Video monitoreo prolongado de 24 horas, con montaje cabeza completa para precisar localización y definir pronóstico.

35
Electroencefalografía neonatal. Registro anormal. Loreto Ríos et al.
BIBLIOGRAFÍA
1. Lombroso CT. Neonatal EEG Polygraphy in Normal and Abnormal Newborns. Electroen-cephalography. Basic Principles, Clinical Appli-cations and Related Fields, E. Niedermeyer and F.H Lopes da Silva, Eds.,chapter 48, pp. 803-875, Williams and Wilkins, Baltimore, Md, USA, 1993.
2. Shany E., Berger I., neonatal Electroencephalo-graphy: Review of a Practical Approach. J Child Neurol 2011 26:341.
3. Lombroso CT. Neonatal electroencephalogra-phy. In: Niedemeyer E., Lopez da Silva F, Eds. Electroencephalography: Basic principles, cli-nical applications and related fields. Baltimore: Urban & Schwarzenberg, 1989:599-637.
4. Bláthnaid Mc., Hahn C. Continuous EEG Mo-nitoring in the Neonatal Intensive Care Unit. J Clin Neurophysiol 2013; 30: 106–114).
5. André M., et al, Electroencephalography in pre-mature and full-term infants. Developmental features and glossary. Neurophysiologie Clini-que/Clinical Neurophysiology (2010) 40, 59—124.
6. Tsuchida T. et al, American Clinical Neurophy-siology Society Standardized EEG Terminology and Categorization for the Description of Conti-nuous EEG Monitoring in Neonates: Report of the American Clinical Neurophysiology Socie-ty Critical Care Monitoring Committee. J Clin Neurophysiol 2013; 30: 161–173.
7. Nash KB. et al., Video-EEG monitoring in new-borns with hypoxic-ischemic encephalopathy treated with hypothermia. Neurology 2011; 76: 556-562.
8. Yamatogia Y. Ohtaharab S. Early-infantile epi-leptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain & Development 2002;24: 13–23.
9. Kidokoro H. et al, Chronologic Changes in Neo-natal EEG Findings in Periventricular Leuko-malacia. Pediatrics 2009; 124; e468.
10. Shellhaas R., Continuous Electroencephalogra-phy Monitoring in Neonates, Curr Neurol Neu-rosci Rep (2012) 12:429–435.
11. McCoy B. and Hahn C.Continuous EEG Mo-nitoring in the Neonatal Intensive Care Unit. J Clin Neurophysiol 2013; 30:106-114.

36
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
ABSTRACT
Continous Spike-wave during sleep syndrome (CSWS) is a severe encephalopathy that presents with clinical seizures, neurocognitive regression and an electroencephalogram (EEG) patter of electrical epileptic during 25-85% of the no-REM phase of sleep. Occurs mainly in children between 4 to 6 years with a 0, 5% of prevalence. Objective: Review of recent studies to check etiolo-gy, new concepts and treatment.Method: We reviewed studies which were found under Pubmed system and chapters of books pub-lished between year 2005 to 2014.Results: The studies reviewed have shown that eti-ology is still unclear, though there is genetic predis-position and brain structures injury involved. The diagnosis is made by an accurate clinical history and findings in the electroencephalogram. There is little evidence about treatment, but experts recom-mend the use of benzodiazepines associated to cor-ticosteroids. The prognosis in relation to seizures and EEG is generally good, but neurocognitive im-paired are left.Conclusions: We reviewed different studies were some concepts, etiology and treatment could be cleared out. Anyhow, the pathogenesis and treat-ment is still unclear showing the need of further studies to improve our children therapy and out-come.Key words: continuous spike-wave during sleep, epileptic encephalopaties, Clobazam, CSWS, ESES.
RESUMEN
El síndrome de espigas de ondas continuas del sue-ño lento se caracteriza por una encefalopatía severa asociada a un electroencefalograma (EEG) con un patrón de status epilépticos durante 25 a 85% de la
actividad no-REM del sueño, convulsiones y regre-sión en al menos un área del desarrollo neurocogni-tivo y posterior cese de estas convulsiones. Ocurren en niños entre 4 y 6 años principalmente con una prevalencia de 0,5%.Objetivo: Revisión de publicaciones recientes para aclarar etiología, nuevos conceptos y tratamientos del cuadro.Método: Se revisaron publicaciones a través de sistema Pubmed y libros publicados entre los años 2005 a 2014.Resultados: En todos los estudios revisados la etiopatogenia aún no se encuentra clara, pero se ha visto una relación genética y asociaciones a daños cerebrales estructurales. El diagnóstico se realiza en base a clínica y hallazgos al electroencefalograma. Existe poca evidencia respecto al tratamiento, pero se recomiendo el uso de benzodiacepinas junto a corticoides. El pronóstico generalmente es bueno en cuanto a las crisis convulsivas y EEG, pero suele dejar secuelas en el área neurocognitiva.Conclusiones: Se revisaron distintos estudios donde se pudo aclarar nuevos conceptos y ciertos hallazgos sobre la etiología y tratamiento de este cuadro, lo que permite darnos cuenta la necesidad de nuevos estudios, para así poder tratar de mejor manera a nuestros pacientes.Palabras clave: Espigas lentas, encefalopatías epi-lépticas, CSWS, ESES, Clobazam.
El síndrome de espigas de ondas continuas del sue-ño lento fue primeramente descrito en 1971 (1). Se caracteriza por una encefalopatía severa asociada a un electroencefalograma (EEG) con un patrón de status epilépticos durante 25 a 85% de la actividad no-REM del sueño, convulsiones y regresión en al menos un área del desarrollo neurocognitivo y pos-terior ausencia de convulsiones (2,3).
EPIDEMIOLOGÍA
Los datos sobre la epidemiología de este cuadro son escasos. CSWS es una patología poco frecuen-
Espigas de ondas continuas del sueño lento(CSWS)Antonia Mena1,2, Perla David1,2, Kathleen Batalla1,3
Correo: [email protected]
Actualizaciones
1. Clínica Dávila.2. Universidad de Los Andes3. Universidad del DesarrolloRecibido 15-8-15. Aceptado 15-9-15.

37
Espigas de ondas continuas del sueño lento (CSWS) Antonia Mena et al.
te, que se presenta en niños y adolescentes, princi-palmente en la primera década de la vida (2,4). La prevalencia es de 0,5%-0,6% de todas las epilepsias infantiles, aunque se cree que está subestimado de-bido a la existencia de variables definiciones y cri-terios de inclusión inconsistentes (2,4,5).
En cuanto a la distribución de género se ha visto que es más frecuente en hombres (alrededor de un 60%) (4,5).
Este síndrome se caracteriza por ser edad depen-diente. Se presenta mayormente en niños prees-colares y escolares, con inicio de crisis entre los 2 meses y 12 años con un peak entre los 4 y 6 años y desaparición de las convulsiones alrededor de los 6-9 años (2,4,5,6).
ETIOLOGÍA
La etiología de este síndrome aún no se conoce. Sin embargo, se han visto factores asociados, tal como daños durante el desarrollo de la corteza, atrofia de la corteza unilateral o difusa, lesiones vasculares perinatales, hemiplejias congénitas, crisis neona-tales e hidrocefalia. No hay estudios que muestren asociación genética significativa, pero en varios es-tudios se ha visto la presencia de hasta un 10% de convulsiones como antecedente familiar.(2,3,4,5,6).
Por otro lado, la fisiopatología de este cuadro aún no se entiende por completo. Se cree que las descar-gas epileptiformes son causadas por un foco único primario, ya sean lesiones corticales u otros, que disminuyen la actividad de neurotransmisores que conectan el sistema retículo-talámico con sincroni-zación bilateral a través del cuerpo calloso, produ-ciendo así alteraciones en las funciones del sueño (2,4).
CLÍNICA
Este síndrome se caracteriza por presentar 4 etapas. Primero, la fase de inicio clínico donde se presentan convulsiones con un electroencefalográfico (EEG) con un patrón de status epilépticos durante más del 85% de la actividad no-REM del sueño. Luego la fase de estado se presenta 2 a 4 años posterior al inicio epiléptico donde se ve regresión en al me-nos una de las áreas del desarrollo neurocognitivo, aumentando la frecuencia de convulsiones y final-mente la fase de ausencia de convulsiones con las
consecuencias del deterioro neurocognitivo, la que se presenta 3 años después de la fase de estado.
En cuanto a las convulsiones, suelen aparecer entre los 2 y 5 años de edad, (5,6). Se presentan gene-ralmente como convulsiones nocturnas tónico-cló-nicas unilaterales con un bajo progreso a un status epiléptico unilateral (6%) (2,5). A la vez, se pueden presentar convulsiones de tipo ausencias miocló-nicas, atónicas o convulsiones parciales simples o complejas.
En un 20% de los casos se presentan convulsiones de dos o más tipos en un mismo paciente (5). No se presentan convulsiones tónicas en este cuadro, pu-diendo de esta manera diferenciarlo de otros síndro-mes. Estas convulsiones generalmente desaparecen al llegar a la pubertad (4).
En relación a la regresión neurocognitiva se puede afectar un amplio espectro de áreas en distintos gra-dos, pero mayormente de manera severa. Tiene un inicio insidioso y un progreso subagudo y el área afectada dependerá de la localización de las descar-gas epileptiformes.
Las descargas epileptiformes a nivel frontal se aso-cian mayoritariamente a compromiso de funciones cognitivas y ejecutivas con hiperactividad, desin-hibición, comportamiento impulsivo, agresividad y agitación. Si las descargas son a nivel temporal producen alteración en lenguaje pudiendo producir afasias, alteración en la orientación temporo-espa-cial como problemas de memoria a corto plazo. A la vez se han visto niños con inestabilidad emocio-nal, distonías, ataxias, dispraxias como también al-teraciones en las capacidades motoras finas como gruesas (4,5).
Se ha visto que al cesar las convulsiones y las al-teraciones en el electroencefalograma (EEG), suele mejorar el área neurocognitiva, pero dejando ciertas secuelas de menor o mayor grado.
EXÁMENES
Electroencefalograma (EEG)
En la fase de inicio clínica, en el EEG en desvelo se ven frecuentemente espigas multifocales en la zona rolándica y/o frontocentral, la cuales presentan ma-yor frecuencia y activación durante el sueño.

38
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
En la Fase de Estado, el EEG de sueño se caracte-riza por un trazado con actividad paroxística con-tinua, bilateral o lateralizada, difusa con descargas generalizadas de punta-onda lenta a 1,5-2,5Hz que aparecen desde el inicio del sueño y se presentan entre un 25 a 85% en la actividad no-REM. Durante la fase de estado, aumentan las descargas epilepti-formes (4,5,6).
Imágenes
Resonancia Magnética Cerebral:En la mayoría de los pacientes se encuentra en con-diciones normales, pero un tercio de los pacientes presenta anormalidades estructurales como displa-sia cortical, hidrocefalia o daño cortical focal (4).
PET con fluorodesoxiglucosa: Muestra aumento de su metabolismo en el área con descargas epilepti-formes (4).
Cintigrama con 99m Tc-hexametil propilen amino oxima (HMPAO): muestra hipoperfusión en el área de descarga epileptiforme (4).
DIAGNÓSTICO DIFERENCIAL
El diagnóstico diferencial debe realizarse con otros síndromes epilépticos que ocurren en preescolares y escolares, como el síndrome de Lennox Gastaut, el síndrome de Doose, entre otros. Como también de todo aquel cuadro en que se presente una regresión en el desarrollo neurocognitivo y alteración en el EEG durante el sueño, incluyendo el Síndrome de Landau-Kleffner, las formas atípicas de epilepsia parcial benigna rolándica, la epilepsia parcial se-cundaria debida a lesiones estructurales rolándicas, estatus epiléptico orofaciales, entre otras.
TRATAMIENTO
El tratamiento de este cuadro se enfoca en normali-zar la actividad del EEG de sueño para así mejorar las capacidades neurocognitivas. El objetivo será eliminar las descargas epilépticas en el sueño en los dos años posterior al diagnóstico, ya que es el pe-riodo crítico para evitar secuelas cognitivas graves.
A pesar de esto, existe poca evidencia para guiar eltratamiento de este cuadro, por lo que se siguen los consejos de expertos o lo visto en series de casos.
A la vez se recomienda, dentro de lo posible, no usar politerapia, a pesar que en la mayoría de las ocasiones esto es muy difícil, debido a la severidad de este síndrome.
Dependiendo en el tipo de convulsión se recomien-da como primera línea el uso de corticoides con benzodiacepinas como el Clobazam, como también es posible utilizar Etosuximida asociado a altas do-sis de Ácido Valproico o Sultiamo (4,6,7,8,9).
Para el uso de corticoides se aconseja Prednisona en dosis de 2-5mg/kg/día o Metilprednisolona en dosis de 20mg/kg/día por 3 días, siendo los que han mos-trado mayor mejoría, pero aun así faltan estudios para definir combinaciones y dosis.
La fenitoína, carbamazepina, oxcarbazepina, topi-ramato y barbitúricos están contraindicados ya que empeoran las anormalidades en el EEG (4,7,9).
Por otro lado, se ha evaluado el uso de de ACTH (80UI al día por 3 meses), Inmonuglobulinas, dieta cetogénica y cirugías, las cuales aún no han mostra-do resultados estadísticamente significativos.
PRONÓSTICO
El pronóstico de la epilepsia en este cuadro, de forma global es positivo, ya que las convulsiones y anormalidades en el EEG suelen desaparecer de forma gradual en la pubertad, recuperando la es-tructura del sueño. Sin embargo, en el área neuro-cognitiva suelen quedar secuelas graves, especial-mente déficit intelectual moderado a severos, sin capacidad de ser independientes.
CONCLUSIONES
El síndrome de espigas de ondas continuas del sue-ño lento(CSWS) es una encefalopatía severa aso-ciada a un EEG con un patrón de status epilépticos durante 25 a 85% de la actividad no-REM del sue-ño, convulsiones de distintos tipos y regresión en al menos un área del desarrollo neurocognitivo y posterior ausencia de convulsiones.
A pesar de que la etiología de este síndrome aún no queda clara se ha visto cierta relación genética y asociaciones a daños cerebrales estructurales.
Existe poca evidencia respecto a cuál tratamien-

39
Espigas de ondas continuas del sueño lento (CSWS) Antonia Mena et al.
to es el indicado para este cuadro, por lo cual se requieren mayores estudios y entendimiento en la etiología de esta patología para así poder manejarlo mejor y evitar secuelas neurocognitivas que sufren los pacientes con este cuadro.
REFERENCIAS
1. Raha S, Urvashi S, Vrajesh U. Neurocognitive and neurobehavioral disabilities in Epilepsy with Electrical Status Epilepticus in slow sleep (ESES) and related syndromes. Epilepsy and Behaviour 2012; 25: 381-5.
2. Sánchez Fernández I, Chapman K, Peters J, Harini C, Rotenberg A, Loddenkemper T. Con-tinous spikes and waves during sleep: elec-troclinical presentation and suggestions for management. Epilepsy research and treatment. 2013; 1-12.
3. Sánchez Fernández I, Peters J, Hadjiloizou S, Prabhu S, Zarowski M, Stannard K, Takeoka M, Rotenberg A, Kothare S, Loddenkemper T. Clinical staging and electroencephalograph-ic evolution of continuous spikes and waves during sleep. Epilepsia. 2012; 53(7):1185-1195.
4. Striano P, Capovilla G. Epileptic encephalop-athy with continous spikes and waves during sleep. Curr Neurol Neurosci Rep. 2013; 13 (360): 1-6.
5. Loddenkemper T, Sánchez Fernández I, Peters J. Continous Spike and waves during sleep and electrical status epilepticus in sleep. Journal of Clinical Neurophysiology. 2011; 28 (2): 1-11.
6. García-Peñas J. Disfunción neurocognitive en el síndrome de estado de mal eléctrico durante el sueño lento: ¿podemos modificar la evolución natural del síndrome con un tratamiento farma-cológico precoz?. Rev Neurol 2010;50 (3):37-47.
7. Caraballo R, Veggiotti P, Kaltenmeiera M, Pi-azza E, Gambonic B, Lopez MF, Noli D, Adic
J, Cersosimo R. Encephalopathy with status epilepticus during sleep or continous spikes and waves during slow sleep syndrome: A mul-ticenter, long-term follow-up study of 117 pa-tients. Epilepsy Research 2013;105:164-173.
8. Veggiotti P, Pera M, Teutonico F, Brazzo D, Ba-lottin U, Tassinari C. Therapy of encephalopa-thy with status epilepticus during sleep (ESE/CSWS syndrome): an update. EpilepticDisord 2012; 14 (1): 1-11.
9. Mctague A, Cross JH. Treatment of epileptic en-cephalopathies. CNS Drugs. 2013;27:175-184.
10. Singhal N, Sullivan J. Continous spike-wave during slow wave sleep and related conditions. ISRN Neurology. 2014: 1-6.
11. De Tiege X, Trotta N, Op de beeck M, Bour-guignon M, Marty B, Wens V, Nonclerq A, Goldman S, Van Bogaert P. Neurophysiological activity underlying altered brain metabolism in epileptic encephalopathies with CSWS. Epilep-sy Research. 2013; 1-10.
12. Heinzle Bolsterli B, Fattinger S, Kurth S, Lebourgeois M, Ringli M, Bast T, Critelli H, Schmitt B, Huber R. Spike wave location and density disturb sleep slow waves in patients with CSMWS (continuous spike waves during sleep). Epilepsia. 2014; 55 (4): 584-91.
13. Bolsterli B, Schmitt B, Bast T, Critelli H, Heinzle, Jenni O, Huber R. Impaired slow wave sleep downscaling in encephalopathy with sta-tus epilepticus during sleep (ESES). Clinical Neurophysiology. 2011; 122: 1779-87.
14. Seri S, Ndoc Thai J, Brazzo D, Pisani F, Cerqui-glini A. Neurophysiology of CSWS-associated cognitive dysfunction. Epilepsia. 2009; 50 (7): 33-36.
15. Nikanorova M, Miranda M, Atkins M, Sahl-hoildt L. Ketogenic diet in the treatment of refractory continous spikes and waves during slow sleep. Epilepsia. 2009; 50 (5): 1127-1131.

40
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Síndrome de Landau-KleffnerKathleen Batalla1,3, Perla David1,2, Antonia Mena1,2
Actualizaciones
ABSTRACT
Landau Kleffner Syndrome was first described by Landau Kleffner and 1957, where six described previously healthy patients develop loss of recep-tion / expression language associated with seizures. The International League Against Epilepsy (ILAE) classifies as an age-dependent epileptic encepha-lopathy. It is characterized by global or mixed ac-quired verbal agnosia, aphasia variable evolution and paroxysmal abnormalities EEGcon spike-wave during non-REM sleep in auditory cortical areas, sometimes with seizures and behavioral disorders. Studies show that aphasia is closely associated with EEG abnormalities, electrical status epilepti-cus during sleep (ESES) SLK favors a maladaptive plasticity; the non-REM sleep disruption not only prevents new learning but can alter corticalesya developed structures. It has raised a possible role of the thalamus and the reticular activating sys-tem (RAS) in developing the SLK and ESES, whose study is crucial.
Recently it has been considered the genetic factor in the continuous spectrum from BECTS, autoimmune CSWS.También LKS and theories, based on the dis-covery of autoantibodies described.
LandauKleffner Syndrome (SLK) is a rare condition but poses several challenges. Key is early diagno-sis as an aggressive early treatment of the disease translates better prognosis. However only the first manifestations pesquisan with EEG, which are non-specific. The treatment is controversial given the limited knowledge of the pathophysiology, at pres-ent there is not an effective treatment.º
Key Words: Diagnosis, Pathogenesis and Treat-ment.
RESUMEN
El Síndrome de Landau Kleffner fue inicialmente descrito por Landau y Kleffner en 1957, donde se describen seis pacientes previamente sanos, desa-rrollan pérdida de recepción/expresión del lenguaje asociado a crisis epilépticas. La liga internacional contra la epilepsia (ILAE) la clasifica como una en-cefalopatía epiléptica edad dependiente. Se carac-teriza por agnosia verbal adquirida global o mixta, con afasia de evolución variable y anomalías paro-xísticas en el EEGcon espiga-onda durante el sueño no-REM en áreas corticales auditivas, a veces con crisis epilépticas y trastornos de conducta.
Estudios muestran que la afasia está íntimamente asociada a las alteraciones del EEG, el estado epi-léptico eléctrico durante el sueño (ESES)del SLK favorece una plasticidad mal adaptativa; la inte-rrupción del sueño no REM no sólo a evita nuevos aprendizajes sino que puede alterar estructuras cor-ticalesya desarrolladas. Se ha planteado un posible rol del tálamo y el Sistema reticular activante (SRA) en el desarrollo del SLK y ESES, cuyo estudio sería fundamental.
Recientemente se ha considerado el factor genético en el espectro del continuo desde BECTS, LKS y CSWS.También se describen teorías autoinmunes, basadas en el descubrimiento de auto-anticuerpos.El Síndrome de LandauKleffner (SLK) es una con-dición infrecuente pero plantea diversos desafíos. Es clave realizar un diagnóstico precoz dado que un tratamiento agresivo en etapa temprana de la enfermedad traduce mejor pronóstico. Sin embar-go sus primeras manifestaciones solo se pesquisan con EEG, las que son inespecíficas. El tratamiento es controvertido dado el escaso conocimiento de la fisiopatología, en la actualidad no se cuenta con un tratamiento eficaz.
El desafío entonces no sólo es en hallar una terapia adecuada en base a la fisiopatología de este cuadro
1. Clínica Dávila.2. Universidad de Los Andes3. Universidad del Desarrollo

41
Síndrome de Landau-Kleffner XXxxxxx et al.
aún no bien comprendida, sino también en hacer un diagnóstico precoz.
Palabras clave: Diagnóstico, Etiopatogenia y Tra-tamiento.
INTRODUCCIÓN
El síndrome de Landau Kleffner (SLK), también conocida como agnosia auditiva epiléptica es la más frecuente de las afasias adquiridas en niños3. La Liga internacional contra la Epilepsia (ILAE) la clasifica como una encefalopatía epiléptica4 re-lacionada con la edad, presenta regresión severa del lenguaje, anormalidades electroencefalográficas y que infrecuentemente se asocian a crisis epilépti-cas9. Inicialmente descrito por Landau y Kleffner en 1957 en un reporte de 6 niños sanos que de forma aguda o progresiva presentan pérdida de recepción y/o expresión del lenguaje asociado a anomalías paroxísticas en el electroencefalograma1-2; la publi-cación de nuevos casos ha ido matizando las carac-terísticas clínicas, electroencefalográficas y evoluti-vas de este síndrome.
La regresión del lenguaje abarca tanto las habilida-des receptivas y expresivas en diversos grados5, y las manifestaciones electroencefalográficas inicia-les serían las ondas puntas centrotemporales y crisis rolándicas, posteriormente en periodo interictal con espigas parieto-occipitales, temporales posteriores o centrotemporales bilaterales que son más intensas en la fase del sueño No REM, llevando a un modelo de estado epiléptico eléctrico en el sueño. La etiopatogenia todavía no es plenamente com-prendida. Y dado las manifestaciones iniciales pu-diera corresponder al grupo de Epilepsia parcial benigna (o Epilepsia Rolándica), epilepsia parcial benigna atípica, síndrome de punta-onda continua del sueño lento o del Síndrome de Landau-Kleff-ner13-14; la búsqueda de la terapia racional para estos niños continúa ampliando el desafío aún más.
EPIDEMIOLOGÍA El síndrome de Landau Kleffner es una encefalo-patía epiléptica edad dependiente (infancia y ado-lescencia), el inicio de la regresión del lenguaje se presenta en niños entre 18 meses a 13 años (mayo-ría entre 3 y 7 años), la edad específica de inicio es difícil de evaluar dado la heterogenicidad de crite-
rios de inclusión y por el inicio sub-agudo10-12; hasta el 75% de los casos se inicia antes de los 7 años, con una relación hombres a mujeres 2:17,10-12. Algunos autores describen la incidencia en población entre 5-14 años es de 1/1.000.0006. La prevalencia de esta encefalopatía probablemente se subestima, repre-sentando alrededor del 1% de la epilepsia infantil8, en centros especializados se reportan 1-2 casos por año. No hay asociación geográfica ni étnica, y se presen-ta el antecedente familiar de epilepsia en el 12%. La tardanza diagnóstica se presenta por confusio-nes con sordera y retrasos del lenguaje y porque los estudios electroencefalográficos rutinarios no inclu-yen el registro de sueño.
DIAGNOSTICO Se considera que debe haber indemnidad neuroló-gica previa a la presentación del síndrome, agnosia verbal adquirida global o mixta, anomalías paroxís-ticas electroencefalográficas (EEG), convulsiones en el 70% de los casos, con trastornos de conduc-ta, regresión autista y evolución variable de la afa-sia15-16.
La ILAE la clasifica entre los síndromes electroclí-nicos como encefalopatía epiléptica. Lo que implica la noción de que la actividad epiléptica en sí misma puede contribuir a producir alteraciones cognitivas y de comportamientos graves, más allá de lo que podría esperarse de la patología subyacente17.
Respecto a los trastornos lingüísticos La manifestación más importante del síndrome de Landau-Kleffner es la pérdida del lenguaje recepti-vo y más tarde del expresivo. La afasia es la primera señal clínica en el 40% de los casos y será el rasgo prominente15; caracterizado por una regresión pro-gresiva en el lenguaje en la forma de una agnosia verbal auditiva que incluye fallo del reconocimien-to semántico de señales acústicas, inicialmente no comprende palabras, con posterior reducción de la expresión espontánea, con pérdida severa en el 90% de los niños, llegando al mutismo en ocasiones20; la agnosia verbal puede ser tan severa y progresar a agnosia de los sonidos no lingüísticos; el trastorno lingüístico es de curso fluctuante con remisiones y exacerbaciones16.

42
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Seguimientos de largo plazo neurofisiológicos y neuropsicológicos, muestran que la afasia está ín-timamente asociada a las alteraciones del EEG con descargas continuas del sueño lento18-19. Después de un tiempo variable de unos pocos meses a diferen-tes años, la afasia estabiliza y por lo general mejora antes de la edad adulta.
Aunque el resultado es usualmente peor en el len-guaje que para la epilepsia, menos del 25% de los pacientes con LKS permanecen severamente afási-cos luego de la normalización del EEG9.
El pronóstico de este síndrome está fuertemente relacionado con la edad de inicio y el lenguaje ad-quirido previamente, es más, la correlación entre la edad de inicio de regresión lingüística y el resultado final del lenguaje es más alta que la correlación con la edad de inicio de las crisis epilépticas. El resulta-do suele ser deficiente en los niños que desarrolla-ron regresión idioma antes de los 5 años, tal como ESES también un importante factor de mal pronós-tico, en varios estudios la recuperación completa de la función del lenguaje normal se limita a aquellos en quienes este estado duró menos de 3 años9,21-22.
Pacientes que recuperan satisfactoriamente el nivel del lenguaje, en pruebas de escucha dicótica revelan una extinción contralateral a la corteza temporal, donde se generaron las descargas epilépticas du-rante el período activo de LKS18, lo que explica el deterioro en test verbales de memoria a corto plazo, incluso cuando hay buena recuperación lingüísti-ca23-25.
Se ha sugerido que la actividad epiléptica contínua en la corteza temporal podría alterar su normal di-ferenciación para el procesamiento del lenguaje, resultando en una reorganización funcional hemis-férica. Este deterioro persiste hasta 10 años después de la recuperación de la afasia epiléptica, presen-tando asimetrías en las áreas asociativas sugirien-do disfunción permanente en el sistema auditivo verbal, aunque los componentes del área auditiva primaria recuperan su normal latencia y amplitud.La actividad electroencefalograma en LKS16.
Alteracioneselectroencefalográficasycrisisepi-lépticas
El EEG puede evolucionar a partir desde epilep-sias idiopáticas benignas a ESES, observado en
el 50% de los pacientes con LKS, que se asocian con las ausencias atípicas, mioclono negativo, y los trastornos cognitivos. El EEG típicamente muestra actividad repetitiva, puntas aguda-onda, y espiga-onda de regiones temporal o parietal-oc-cipital15,25. La actividad epiléptica puede preceder a la afasia o presentarse conjuntamente. Un 20-30% de los niños nunca presentarán crisis, pero el EEG siempre será anormal. Este patrón elec-troencefalográfico se caracteriza por una signifi-cativa activación de las descargas epileptiformes durante el sueño (ESES), que tiende a ser unila-teral o claramente lateralizado9. Es probable que en los primeros compases de la enfermedad los pa-roxismos sólo se recojan en registro de sueño3.
Clínicamente, las crisis ocurren en 75% de los pa-cientes, son poco frecuentes y de buen pronóstico. El inicio es entre 4 y 6 años. Un 20% continúan con crisis epilépticas después de los 10 años, es ex-cepcional la aparición de crisis después de los 15 años16. Las crisis son principalmente nocturnas y heterogéneas. Destacan las convulsiones motoras focales y CTCG, pero además se describen las au-sencias atípicas, automatismos menores, convulsio-nes atónicas con Generalización secundaria. Con-vulsiones tónicas son probablemente incompatibles con LKS.
La frecuencia y severidad de las crisis no están de-terminadas por la gravedad de las anomalías del EEG o del trastorno lingüístico y conductual.
Trastorno cognitivo-conductual
Alteraciones conductuales son comunes al inicio del cuadro (50-70%), el deterioro generalmente es paralelo al deterioro lingüístico, inicialmente pue-den estar relacionados con ansiedad del niño con capacidad de comprensión de lo que pasa.
Alteraciones cognitivas y conductuales se presentan en más del 75% de los pacientes con LKS, desde gran hiperactividad y déficit de atención, que son comunes, con progresión a la desinhibición severa y psicosis en raras ocasiones; hasta síntomas claros de regresión autista16, 20. La gravedad relativa de los problemas lingüísticos, conductuales y cognitivos puede variar con el tiempo en el mismo niño y entre los niños.
Estudios de seguimiento a largo plazo han demos-

43
trado que LKS no siempre se asocia con el deterioro intelectual, y que de hecho presente un CI, en test no verbales, normal e incluso alto3.Trastornos de la memoria son comunes, porque las descargas continuas en el sueño no permiten la con-solidación de la memoria 9, 26-28.
ETIOPATOGENIA: NEUROBIOLOGIA DE SLK
El síndrome Landau-Kleffner, se caracteriza por espiga-onda durante el sueño no-REM en las áreas corticales auditivas9, 27. El trastorno comienza y fi-naliza a casi el mismo tiempo que los períodos crí-ticos del desarrollo se inician y terminan. (Hubel, Wiesel)11-14. El estado epiléptico eléctrico durante el sueño favorece una plasticidad mal adaptativa. La interrupción del sueño no REM no sólo evita nuevos aprendizajes, sino que puede alterar estruc-turas corticales desarrolladas. La pérdida de la es-timulación fisiológica cortical en período temprano resulta en cambio anatómico, ampliando la respues-ta del territorio sano y disminuyendo el deprivado, con un reordenamiento del circuito tálamo–cortical. La estructura del cerebro al final del período crítico queda sellada por el resto de la vida del niño. Los períodos crítico corticales se desarrollan con dife-rentes cursos de tiempo; en el contexto de ESES, importa la dominancia hemisférica y la edad, ya que define la ventana terapéutica durante el cual el trata-miento puede minimizar la afasia. Aunque el rango de edad exacta que comprende el período crítico del lenguaje se debate (23, 24 25), la mayoría de los casos se diagnostican en el pico las edades del desarrollo del lenguaje (menores de 7 años). El cese de las cri-sis electrográficas es menos precisa, pero la resolu-ción típica es en la adolescencia. Destacando que a mayor edad de inicio de la enfermedad mejor el resultado27 consistente con la plasticidad más débil. No está claro si los cambios neuronales o molecu-lares que terminan el período crítico también fina-lizan la actividad epiléptica electroencefalográfica, o si las convulsiones se detienen por alguna otra ra-zón no relacionada. En LKS, la función del lenguaje se pierde a pesar de que la capacidad del cerebro, en teoría, podría cambiar dominancia hemisférica o re-distribuir las funciones a través de diferentes áreas corticales.
Esto sostiene que la actividad eléctrica anormal en las áreas del lenguaje por si sola no podría causar pérdida del lenguaje, sino que el efecto ESES en los
circuitos activados (quizás a través de un efecto so-bre poda dendríticas) la que evita la redistribución de la función a la otra área cortical.
Espigas Interictales (EI): Existe una amplia litera-tura que muestra que convulsiones recurrentes en el cerebro en desarrollo resultan a largo plazo en con-secuencias adversas. Las EI interfieren la función de la memoria en el hipocampo durante los períodos críticos, con disminución del recuento de células hipocampales. Una crisis puede resultar en un de-terioro transitorio, sin embargo las EI alteran espe-cíficamente los circuitos neuronales, especialmente cuando se presenta el estímulo, correspondiente a la fase de codificación de la tarea. Habiendo deterio-ros cognitivos que preceden a la epilepsia como en SLK. Incluso si el habla se recupera paralela al EEG esto no prueba que la actividad epileptiforme pro-voca la afasia, mas bien es posible que la disminu-ción actividad epileptiforme y la recuperación del habla simplemente refleja la resolución de lesiones a las áreas del lenguaje.
El papel del sueño en período crítico de plasti-cidad
Puede ser sorprendente que la anormal actividad eléctrica nocturna tenga efectos tan devastadores en los procesos que se aprenden y se utilizan durante la vigilia. En adultos la privación sueño impide la consolidación de recuerdos31. Se encontró que el re-cuerdo de los niños con ESES empeoraba después de dormir en lugar de mejorar. En un paciente tra-tado con esteroides, con detención de la actividad eléctrica anormal durante la noche, la consolidación de la memoria dependiente del sueño regresó. El déficit de lenguaje observado representa problemas de discriminación auditivo fonológico33, debido a la reorganización fundamental de la estructura cortical durante los períodos críticos auditivos o del lengua-je, comprometiendo la consolidación de la memoria declarativa, necesaria para adquirir nuevo vocabu-lario en el día a día (Urbain et al). La cantidad de plasticidad durante el sueño varía linealmente con la cantidad de sueño no-REM.
La organización cortical permiten la respuesta de los circuitos neuronales de manera efectiva al me-dio ambiente con selectividad focal de la corteza, Tassinari y Rubboli propusieron que la interrupción focal del sueño onda lenta podría ser la base del deterioro de la función cognitiva en pacientes con
Síndrome de Landau-Kleffner XXxxxxx et al.

44
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
ESES. Por ej. el aumento de actividad delta-gama se correlaciona con el aprendizaje espacial (corteza parietal) y temporal, sólo evidente en la noche des-pués de aprender una tarea35. La interrupción en las áreas auditivas de la corteza parece impedir el nor-mal desarrollo de los circuitos de reconocimiento de fonemas, lo que lleva a la agnosia auditiva (ref).
Deterioro después del desarrollo inicial
La definición de esta epilepsia incluye la pérdida de la función después de haberla adquirido. Esto pare-ce contradictorio dado el vasto potencial de adap-tación del cerebro joven. Se explicaría por el com-promiso de los mecanismos de plasticidad normal, apareciendo nuevas correlaciones no funcionales en la actividad neuronal, y la arquitectura cortical que se desarrollaría no sería adaptada al entorno senso-rial real presente durante vigilia.
Un posible papel de tálamo en SLK y ESES
Identificar el origen de las descargas punta-onda es fundamental para el desarrollo de tratamientos. Monteiro et al.43 y Guzzetta et al.44 propusieron que los daños en el tálamo podrían ser la razón de acti-vidad punta-onda continua que se produce durante el sueño, basado en la similitud de la actividad eléc-trica desarrollada en pacientes con daño talámico y encontrada en los animales de experimentación que carecen de un tálamo, incluso en ausencia de daño cortical.
Lafisiologíadel tálamo en la retransmisióndeinformación
El tálamo juega un papel central en la regulación cortical actividad mediante la determinación de cuándo y cuánta información se transmite a la corte-za cerebral. Las neuronas del Tálamo, se cree, fun-cionan en un modo tónico en el estado de vigilia (o atención) y un modo de disparo en el sueño (o falta de atención) .50,51 En el modo de tónico, las neu-ronas de relevo modulan su tasa de disparo fielmen-te basado en el estímulo que reciben. En el modo de disparo, la neurona relevo está generalmente en silencio (hiperpolarizadas), a menos que se presen-te un fuerte estímulo, entonces dispara una ráfaga corta rápida de potenciales de acción en respuesta a un estímulo repentino e intenso para “despertar” su objetivo cortical.
El Sistema Reticular activador (SRA) como un posible locus de patología.
¿Por qué la ausencia de actividad normal del tálamo convertiría a la corteza en epileptógenica?. Durante el sueño normal de ondas lentas, las neuronas del tálamo se mantienen hiperpolarizadas por la activi-dad de la SRA. La actividad epileptiforme en sueño impide que las descargas tálamo-cortical activen la corteza Guzzetta et al.44.
Se ha observado que daños locales de un núcleo re-levo o de SRA, podrían resultar en la disfunción lo-calizada durante el sueño en el circuito mediada por el núcleo específico, llano. Asi el daño a la SRA ad-yacente al núcleo geniculado medial, podría causar interrupción del sueño en relación al sistema auditi-vo, causando potencialmente los síntomas de LKS.
Circuitos del desarrollo y la expresión del recep-tor Entre los factores probables que podrían determinar epileptogenicidad esta la expresión regulada por el desarrollo de receptores de excitación e inhibición de los canales iónicos en el neocórtex. Los canales de expresión inhibitoria aumenta durante el período crítico, con supresión de la actividad espontánea, permitiendo que la actividad cortical estímulo in-ducida de forma a las conexiones corticales60. Estos canales parecen mediar la plasticidad sueño-depen-diente; 61 por lo que su regulación es fundamental para las funciones perturbadas por ESES.
ETIOLOGÍA La etiología de este síndrome se desconoce, aunque los casos idiopáticos pueden sugerir una predispo-sición genética. Las lesiones estructurales son extremadamente infrecuentes, y no ha sido demostrada la relación con las manifestaciones clínicas29 aunque hay ca-sos descritos asociados a las infecciones del SNC, tumores de localización temporal, traumatismo cra-neal, y enfermedad desmielinizante inflamatoria3.
Se han postulado participación genética y autoin-mune, pero aún está lejos de demostrarse30.

45
Factores genéticos Recientemente se ha considerado el factor ge-nético en el espectro del continuo desde BECTS, LKS y CSWS, en que diversos autores30 (Tassinari et al.,2000; Nickels & Wirrell, 2008); entre otros encuentran historia familiar de epilepsia tres veces mayor en los con evolución espigas continuas del sueño.
La alta proporción de trastornos del lenguaje repor-tados en parientes de pacientes con afasias adqui-ridas es de interés, como lo describió Lesca et al 201230, el mayor estudio genético respecto a LKS o ECSWS, donde encontró que el 45% de los pacien-te con LKS o ECSWS tenían historia familiar de crisis febriles, epilepsia o alteraciones cognitivas, del lenguaje y lectura. Esta observación clave pone de relieve la contribución genética probable de es-tos fenotipos complejos y superpuestos.
En este mismo estudio se demuestra la heterogenei-dad genética de SLK o ECSWS, se describen algu-nas lesiones nuevas, que pudieran relacionar LSK, ECSWS, trastornos lingüísticos, y trastornos cog-nitivos. Alteraciones genéticas heredada de interés potencial se enumeran en las tablas (Tablas 1 y 2)30. Pero la implicación fisiopatológica de tales eventos genómicos heredados aún no ha sido demostrada30. Se estudió la base genética del síndrome de Landau-Kleffner (LKS) en una cohorte de gemelos monoci-góticos discordantes dos (MZ) pares de gemelos y 11 casos aislados. Se identificó una sola mutación en el gen GRIN2A, en el sitio de unión del gluta-mato.
Factores autoinmunidad Al realizar tratamiento con inmunoglobulina intra-venosa se evidencia una respuesta clínica y elec-troencefalográfica transitoria. Antes del tratamiento con inmunoglobulina puede haber un índice de IgG elevado en el líquido cefalorraquídeo (LCR), pero el examen del LCR no muestra signos de infección del sistema nervioso central20. Teorías autoinmunes se han descrito, basadas en el descubrimiento de auto-anticuerpos IgG anti-cerebrales en plasma en un 45% de los niños con SLK frente a 2% de la población sana31, también se describe incremento autoanticuerpos IgM anti-
células endoteliales del cerebro en niños con LKS comparación con los controles, hallazgo que com-parten niños con autismo, el trastorno desintegrati-vo infantil y epilepsia20, 31. La autoinmunidad puede también estar implicada en los trastornos del desarrollo neurológico, que in-cluyen déficits del lenguaje.
Se detectó reacción autoinmune a la mielina en los niños con síndrome de Landau-Kleffner, los au-toanticuerpos que reaccionan con el tejido cerebral se han encontrado niños con una variante atípica del síndrome de Landau-Kleffner y autismo en compa-ración con los controles. Un gen en particular CNT-NAP2 (contactina asociado proteinlike, 2), está implicado en la reacción autoinmune a la mielina detectado en los niños con SLK.
NEUROIMÁGENES Los estudios de neuroimagen son generalmente normales32. Estudios de volumen del cerebro mues-tran una reducción del área de perisilviana bilate-ral, correspondiente al área receptiva del lenguaje33. Todavía no se sabe si esto es una causa o una con-secuencia de la actividad epiléptica9. Se ha encon-trado reducción asimétrica de volumen en el lóbulo temporal izquierdo incluyendo circunvolución de Heschl, plano temporal y giro temporal superior. Esta atrofia en el área de Wernicke es consistente con la pérdida neuronal, gliosis y un mal pronóstico para la recuperación del lenguaje33.
Estudios funcionales por Tomografía Emisión de Fotón único Computarizada (SPECT) y la Tomo-grafía por Emisión de Positrones con 18F-fluro-deoxyglucose (FDG-PET) han revelado consisten-temente aumento o disminución de la perfusión temporo-parietal dependiendo del momento clínico del estudio33. El FDG - PET en LKS presentan un metabolismo más alto del manto cortical al contra-rio de las estructuras subcorticales, tálamo y ano-malías restringidas a regiones focales corticales; esto sugeriría un circuito cortico-talámico inhibido. Anomalías en la SPECT y PETSCAN muestra una fuerte evidencia a favor de la disfunción cerebral focal, que implica la corteza temporal durante la fase activa de la enfermedad. El hipermetabolismo se encuentra en las zonas topográficas donde las descargas punta-onda son predominantes9. Las al-
Síndrome de Landau-Kleffner XXxxxxx et al.
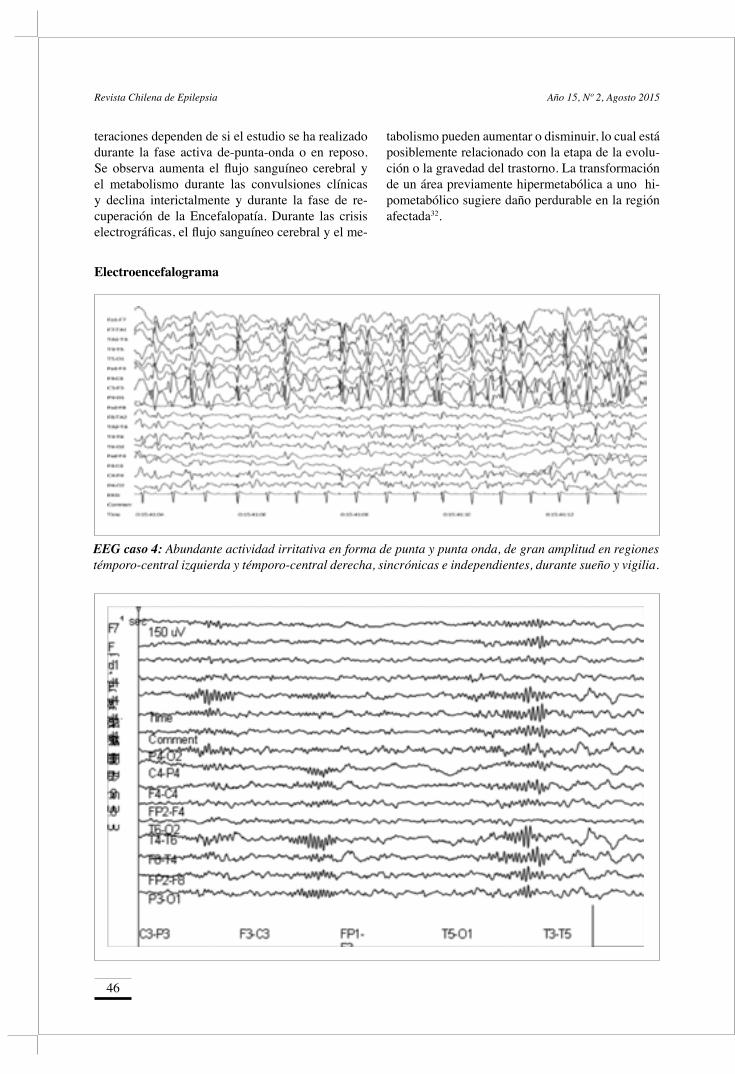
46
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
teraciones dependen de si el estudio se ha realizado durante la fase activa de-punta-onda o en reposo. Se observa aumenta el flujo sanguíneo cerebral y el metabolismo durante las convulsiones clínicas y declina interictalmente y durante la fase de re-cuperación de la Encefalopatía. Durante las crisis electrográficas, el flujo sanguíneo cerebral y el me-
tabolismo pueden aumentar o disminuir, lo cual está posiblemente relacionado con la etapa de la evolu-ción o la gravedad del trastorno. La transformación de un área previamente hipermetabólica a uno hi-pometabólico sugiere daño perdurable en la región afectada32.
EEG caso 4: Abundante actividad irritativa en forma de punta y punta onda, de gran amplitud en regiones témporo-central izquierda y témporo-central derecha, sincrónicas e independientes, durante sueño y vigilia.
Electroencefalograma

47
Control Monitoreo Video EEG caso 1. Sueño no REM, Normal.
Estudios de seguimiento longitudinal de 8,2 a 10,8 años, con de resonancia Magnética funcional aso-ciada a pruebas neuropsicológicas y EEG 24 horas, han demostrado reorganización cerebral de la re-presentación de lenguaje en niños con epilepsia be-nigna con puntas centro-temporales (BECTS) que evoluciona a síndrome de Landau-Kleffner (LKS) y que luego revierte a BECTS.
PRONÓSTICO
Durante el periodo de la enfermedad las crisis epi-lépticas son de fácil control, no así las alteraciones al EEG. Las crisis epilépticas y alteraciones EEG remiten alrededor de los 15 años, espontáneamente; junto con esto mejoran gradualmente el lenguaje y las alteraciones neuropsicológicas. Se ha visto que los pacientes afectos del síndro-me de Landau-Kleffner de larga evolución tienen un pronóstico muy desfavorable en relación con la recuperación del lenguaje, solo el 50% de los pa-cientes logran vivir una vida relativamente normal, con 10-20% que logran recuperación completa, el otro 50% queda con secuelas que hasta en un 25% son graves. Algunos autores opinan que el intervalo evolutivo con la posibilidad de que el problema sea reversible es inferior a 3 años.
Esta situación ha llevado a plantear actitudes agre-sivas en aquellos pacientes que llevan una evolu-ción prolongada, y los han sometido a transecciones subpiales múltiples del córtex cerebral involucrado, los resultados han sido alentadores, tanto en la evo-lución del lenguaje como en el control del trazado EEG de sueño, con desaparición del POCS. Según series relativamente largas, habida cuenta de lo ex-cepcional del problema, el resultado es favorable, y se insiste en la necesidad de una intervención tem-prana para un mejor pronóstico.
ESTRATEGIAS TERAPÉUTICAS El tratamiento es controvertido, más aún dado su fisiopatología no se comprende en su totalidad y no se cuenta con un tratamiento eficaz.
Las bases del tratamiento actual se sustentan en controlar la descargas epileptiformes, suponiendo éstas pudieran ser responsables no sólo de las crisis epilépticas, sino también del resto de las manifesta-ciones clínicas, como el tr. Neurolingüístico; y en el tratamiento de los trastornos neuropsicológicos y lingüísticos resultantes. Los tratamientos utilizados de forma empírica abar-can: Fármacos antiepilépticos, corticoides, ACTH,
Síndrome de Landau-Kleffner XXxxxxx et al.

48
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015Ta
ble
I. Li
st o
f CN
Vs d
etec
ted
in C
SWSS
and
LKS
pat
ient
s an
d re
late
d w
ith e
pile
ptic
, aut
ism
spe
ctru
m, c
ogni
tive
and/
or s
peec
h an
d la
ngua
ge d
isor
ders
Patie
nt (g
ende
r) Di
agno
sis
Chro
moso
mal lo
catio
n Siz
e (kb
) CN
V typ
e (inh
erita
nce)
Gene
cont
ent.a
Relat
ionsh
ip of
CNV/
gene
with
coor
dinate
s (nt
, buil
d37)
(R
eferen
ce)
De no
vo C
NVs
1=18
13 (M
) i-C
SWSS
14
q21.3
,Chr1
4:475
9257
8-476
6239
4 70
Lo
ss (d
e nov
o) MD
GA2 (
part)
ASD
(Buca
n et a
l., 20
09)
EA85
(F)
LKS-
woES
ES
16p1
3.2,C
hr16:1
0246
239-1
0354
862
109
Loss
(de n
ovo)
GRIN
ZA (p
art)
RE, E
E, ES
ES-lik
e (En
dele
et al,
2010
; Reu
tlinge
r et a
l, 201
0)DZ
52(F)
i-C
SWSS
16
q23.3
,Chr1
6:835
9949
8-838
5738
2 25
8 Lo
ss (d
e nov
o) CD
HI3 (
part),
HSBP
I SL
I(SLI
Cons
ortium
, 200
4)Po
ssibl
y de n
ovo C
NVs
DY34
(M)b
s-CSW
SS
22q1
3.32-3
3,Chr2
2:493
4669
7-512
1900
9 1,8
72
Loss
(abs
ent in
moth
er)
SHAN
K3, H
DACI
0, PL
XNB2
AS
D, SP
I (Dura
nd et
al., 2
007)
(+3
7 add
itiona
l gen
es)b
EC08
(F)
i-CSW
SS
10q2
1.3,C
hr10:6
8438
375-6
8506
557
68
Loss
(ND)
CT
NNA3
(Intro
nic)
AZD
(Mya
shita
et al
., 200
7)DX
94 (F
) s-C
SWSS
13
q21.2
,Chr
13:60
4196
03-60
6475
21
228
Loss
(ND)
DI
APH3
(part
) AS
D (Vo
rstma
n et a
l, 201
1)Inh
erite
d CNV
s.un
repor
ted pr
eviou
slyDZ
77 (M
) i-C
SWSS
Sp
14.1,
Chr5:
2863
4980
-2883
7425
20
2 Lo
ss (P
at)
Betw
een C
DH9 a
nd C
DH6
ASD
(Ma e
t al.,
2009
; Wan
g et a
l, 200
9)EB
99 (F
) i-C
SWSS
10
q21.3
,Chr1
0:682
5153
5-684
9686
6 24
5 Lo
ss (M
at)
CTNN
A3 (p
art)
AZD
(Mya
shita
et al
., 200
7)EA
25 (M
) i-C
SWSS
10
q21.3
,Chr
10:68
5504
81-68
6680
09
118
Loss
(Mat)
CT
NNA3
(intro
nic)
AZD
(Mya
shita
et al
., 200
7)10
A603
(M)
i-CSW
SS
20p1
2.1,C
hr20:1
4491
297-1
4591
133
100
Loss
(Pat)
MA
CROD
2 (int
ronic)
AS
D (An
ney e
t al.,
2010
)3-1
9 (F)
LKS-
wiES
ES
1q44
,Chr1
:2453
2097
8-245
4100
54
89
Gain
(Mat)
KIF
268 (
intron
ic)
SPI, E
SZ (C
alebe
et al
., 201
0)7-1
514 (
M)
i-CSW
SS
3q28
-q29
,Chr3
:1920
6752
0-192
1222
31
54
Gain
(Mat)
FG
FI2 (p
art)
ASD,
SLI (K
wasn
icka-C
rawfor
d
et
al., 2
005)
7-151
4 (M)
i-C
SWSS
3q
29,C
hr3:19
2212
953-1
9235
2465
13
9 Ga
in (Pa
t) PG
FI 2 (
part)
7-151
4 (M)
i-C
SWSS
3q
29,C
hr3:19
2870
621-1
9338
5022
51
4 Ga
in (M
at)
HRAS
LS, A
TPI3A
5, AT
P
13A4
, OPA
I (part
)12
-522 (
M)
LKS-
woES
ES
20p1
2.1,C
hr20:1
4395
797-1
4464
507
69
Gain
(Pat)
MACR
OD2 (
intron
ic)
ASD
(Anne
y et a
l., 20
10)
EA85
(F)
LKS-
woES
ES
20q1
3.3,C
hr20:6
0015
337-6
0078
775
63
Loss
(Pat)
CD
H4 (in
tronic
) CD
H4: c
adhe
rinInh
erited
CNV
s,pre
vious
ly kn
own
DZ29
(M)
LKS-
wiES
ES
10q2
1.3,C
hr10:6
8087
319-6
8110
043
23
Loss
(Mat)
CT
NNA3
(intro
nic)
AZD
(Mya
shita
et al
., 200
7)12
-522 (
M)
LKS-
woES
ES
7q35
,Chr7
:1462
2625
8-146
2548
37
29
Gain
(Mat)
CN
TNAP
2 (int
ronic)
AS
D, ES
Z, SL
I, SCZ
; FOX
P2 ta
rget
(Verne
s ert a
l., 20
08; P
eñag
arika
no
et
al., 2
011)
ESES
, elec
trical
status
epile
pticu
s duri
ng lo
w-wa
ve sl
eep;
CSWS
S, co
ntinu
ous s
pike a
nd w
aves
durin
g slow
-wav
e slee
p syn
drome
(i: id
iopati
c, s:
symp
tomati
c); LK
S, La
ndau
-Klef
fner s
yndro
me; L
KS-w
iESES
, LKS
with
elec
trical
status
epile
pticu
s duri
ng sl
ow-w
ave s
leep;
LKS-
woES
ES, L
KS w
ithou
t elec
trical
status
epile
pticu
s duri
ng sl
ow-w
ave s
leep;
EE, e
pilep
tic el
ceph
alopa
thy; R
E, rol
andic
epile
psy;
ESZ,
epile
ptic s
eizure
s; AS
D, au
tism
spec
trum
disord
er;
AZD,
Alzh
eimer’
s dise
ase;
SLI, s
pecifi
c lan
guag
e imp
airme
nt; SP
I, spe
ech i
mpair
ment;
ADHD
, atte
ntion
-defi
cithy
perac
tivity
diso
rder; S
CZ, s
chizo
phren
ia; C
NV, c
opy n
umbe
r vari
ation
; nt, n
ucleo
tide;
kb, k
iloba
se; C
hr, ch
romos
ome;
part,
partia
l dele
tion o
r dup
licati
on; N
D, no
t dete
rmina
ted; P
at, pa
terna
lly inh
erited
; Mat,
mate
rnally
inheri
ted. A
ll eve
nts in
cludin
g trun
catin
g dele
tions
conc
erned
codin
g seq
uenc
es w
ithin
the co
rresp
ondin
g gen
es. F
, fema
le; M
, male
.Ch
romos
omal
locati
ons a
re giv
en ac
cordi
ng to
the F
ebrua
ry 20
09 hu
man r
eferen
ce se
quen
ce (b
uild3
7) (ht
tp://g
enom
e.ucs
c.edu
l).a G
enes
are l
isted
acco
rding
to th
eir ch
romos
omal
locati
on fro
m pte
l (telo
mere
of sh
ort ar
m) to
qtel
(telom
are of
long
arm)
.b U
naffe
cted m
other
not c
arrier
, una
ffecte
d fath
er no
t teste
d; the
very
large
size
of th
e dele
tion a
nd th
e larg
e num
ber o
f gen
es co
ntaine
d with
in str
ongly
indic
ates d
e nov
o orig
in.

49
Tabl
e 2.
Lis
t of o
ther
rare
CN
Vs o
f int
eres
t det
ecte
d in
CSW
SS a
nd L
KS p
atie
nts
Patie
nt (g
ende
r) Di
agno
sis
Chro
moso
mal lo
catio
n coo
rdina
tes
Size (
kb)
CNV t
ype
Gene
cont
ent.a
Comm
ent (R
eferen
ce)
(nt, b
uild3
7)
(inhe
ritan
ce)
De no
vo C
NVs
4-914
(M)
i-CSW
SS
8p23
.1,Ch
r8:95
9822
6-107
8779
2 1,1
89
Loss
(de n
ovo)
TNKS
(part
), MR1
24-1,
MSRA
,PRSS
55,
MIR1
24-1:
micr
o RNA
invo
lved i
n neu
ronal
RPILI
, SOX
7, PIN
OCI, X
KR6 (
part)
differ
entia
tion (
Yoo e
t al.,
2011
)02
5a (M
) i-C
SWSS
8q
21,C
hr8:89
1020
84-89
3982
98
296
Loss
(de n
ovo)
MMP1
6 (pa
rt) MM
P16:
matrix
meta
llopro
teina
seCN
Vs, u
ndete
rmine
dwh
ether
de no
voor
inheri
ted02
9a (M
) i-C
SWSS
1q
25.3,
Chr1:
1835
9453
2-183
8207
90
226
Gain
(abse
nt in
ARPC
S, AP
OBEC
4, RG
LI (pa
rt) -
the m
other)
029a
(M)
i-CSW
SS
3q25
,Chr3
:1581
8331
3-158
2966
41
113
Loss
(abs
ent in
RS
RCI (p
art), M
LFI (p
art)
-
the
moth
er)DX
94 (F
) s-C
SWSS
3q
26.32
-33,C
hr3:17
8969
064-1
7915
0965
18
2 Ga
in (ab
sent
in KO
NMB3
(part
) KC
NMB3
: pota
ssium
chan
nel s
ubun
it
the
moth
er)
ZNF6
39, M
FNI, G
NB4 (
part)
DV95
(F)
i-CSW
SS
Xp22
.12,C
hrX:21
5236
73-21
5583
29
35
Loss
(Pare
nts
CNKS
R2 (p
art)
-
no
t ava
ilable
)
Inheri
ted C
NVs,
unrep
orted
previo
usly
09A1
613 (
M)
i-CSW
SS
3q25
,Chr3
:1543
9545
4-154
7883
05
393
Loss
(Pat)
MM
E (pa
rt) MM
E: en
keph
alina
se1-1
813 (
M)
i-CSW
SS
5q11
.2,Ch
r5:58
5712
92-58
7451
39
174
Loss
(Pat)
PD
E4D
(intro
nic)
-DY
14 (M
) LK
S-wi
ESES
6q
27,C
hr6:16
7355
901-1
6737
3534
18
Lo
ss (M
at)
RNAS
ET2 (
part)
RNAS
ET2:
autos
omal
reces
sive
leuco
ence
phalo
graph
y (He
nnck
e et a
l., 20
09)
13-13
(M)
i-CSW
SS
7q22
,Chr7
:1072
1419
3-107
2625
39
48
Loss
(Mat)
DU
S4L (
part),
BCAP
29 (p
art)
-13
-19 (M
) i-C
SWSS
8p
22,C
hr8:14
5535
53-14
5723
70
19
Loss
(Mat)
SG
CZ (in
tronic
) SG
CZ: s
arcog
lycan
, zeta
ED31
(M)
s-CSW
SS
8q22
.3,Ch
r8:10
2849
359-1
0286
8211
19
Lo
ss (P
at)
NCAL
D (in
tronic
) NC
ALD:
reuro
nal c
alcium
sens
or ne
uroca
lcin
delta
: EF-
hand
calci
um bi
nding
doma
in4-5
2 (M)
i-C
SWSS
1p
21.2-
21.1,
Chr1:
1021
2309
9-103
0996
62
977
Gain
(Mat)
OL
MF3
OLMF
3: do
wnstr
eam
targe
t of P
AX6
003a
(M)
i-CSW
SS
3p11
.2,Ch
r3:87
9178
10-88
7788
3 29
Ga
in (M
at)
HTRIF
, CGG
BPI,Z
NF65
4,C3o
rf38
HTRIF
: sero
tonin
recep
tor01
6a (M
) s-C
SWSS
3q
29,C
hr3:19
4088
557-1
9413
0145
21
7 Ga
in (M
at)
LRRC
I5 (pa
rt),GP
S,ATP
I3A3,
Neigh
borin
g ATP
I3A4:
ASD,
SLI
LO
C100
1315
51
(Kwas
nicka
-Craw
ford e
t al.,
2005
)3-8
1 (F)
LKS-
wiES
ES
8q11
.23,C
hr8:53
3971
26-53
8089
53
412
Gain
(Pat)
FAM1
50A,R
BICCI
-
EA85
(F)
LKS-
woES
ES
9p13
.2,Ch
r9:37
2990
58-37
4516
97
153
Gain
(Mat)
ZC
CHC7
(part
), GRH
PR,
-
ZBTB
5 (pa
rt)DY
64 (M
) i-C
SWSS
10
q21.1
,Chr1
0:560
3442
6-560
8944
2 55
Ga
in (Pa
t) PC
DH15
(part
) Pr
otoca
dheri
nDY
61 (P
) s-C
SWSS
14
q21.3
,Chr1
4:465
2400
8-471
6126
3 63
7 Ga
in (M
at)
RPL1
0L
RPL1
0 muta
tions
repo
rted i
n ASD
patie
nts (K
lauck
et al
., 200
6)
Co
ntinu
ed
Síndrome de Landau-Kleffner XXxxxxx et al.

50
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
inmunoglobulinas intravenosas, y procedimientos quirúrgicos.
Los anticonvulsivantes como el Ácido Valproico, solo o asociado con Clobazam, Etosuximida, y Lamotrigina muestran eficacia transitoria; se reco-mienda asociación con corticoides, Metilpredniso-lona en pulsos ev. (20mg/kg/dia), por 3 días, segui-do de Prednisona oral (2mg/kg/dia, dosis inicial por 1 mes), luego retiro gradual por 6 meses. La aplicación de la dieta cetogénica y el tratamien-to quirúrgico con múltiples transecciones subpia-les30,35, tienen resultados variables. Actualmente, los corticosteroides, hormona corticotropina (ACTH) son la terapia más utilizada, habiéndose ensayado con inmunoglobulinas con resultados también va-riables. Se recomienda evitar el uso de Carbamazepina, Ox-carbazepina, Fenitoina y fenobarbital por su asocia-ción a exacerbaciones de la actividad epileptiforme durante el sueño, facilitando la aparición de ESES y el déficit neuropsicológico. Respecto al tratamiento no farmacológicos no exis-ten trabajos específicos acerca de tratamientos mul-tidisciplinarios en pacientes con SLK y sus resulta-dos en el trastorno neuropsicológicos, conductuales y lingüísticos. Pero la experiencia en otras patolo-gías que compromenten estos trastornos y estudios en pacientes con afasia, de múltiples causas, avalan lo que un abordaje multidisciplinario hace para lo-grar el resultado mas óptimo.
BIBLIOGRAFÍA
1. Landau WM, Kleffner FR. Syndrome of ac-quired aphasia with convulsive disorder in chil-dren. Neurology 1957;7:523-530.
2. Epileptic encephalopathy of late childhood: Landau-Kleffner syndrome and the syndrome of continuous spikes and waves during slow-wave sleep. Review article. Smith MC, et al. J Clin Neurophysiol. 2003.
3. M. Nieto-Barrera. Afasias adquiridas: síndrome de Landau-Kleffner y otros.
4. The Organization of the Epilepsies: Report of the ILAE Commission on Classification and Terminology Ingrid E Scheffer, Samuel F Berkovic, Giuseppe Capovilla, Mary B Connol-ly, Laura Guilhoto, Edouard Hirsch, Solomon
L Moshe, Douglas Nordli, Yue-Hua Zhang, Sa-meer M Zuberi.
5. Anne Marie Tharpe*, Barbara J. Olson F,. Lan-dau-Kleffner Syndrome: Acquired Epileptic Aphasia in Children.
6. Epidemiological study of Landau-Kleffner syndrome (LKS) in Japan. Kaga M1, Inagaki M2, Ohta R2.
7. Beaumanoir A. Le syndrome de Landau-Kleff-ner. En: Roger J, Bureau M, Dravet Ch, Dreifuss FE, Perret A, Wolf P, eds. Les syndromes épilep-tiques de l’enfant et de l’adolescent. 2 ème edn.London: John Libbey; 1992. p. 231-44.
8. A review of the relationships between Lan-dau-Kleffner syndrome, electrical status epilep-ticus during sleep, and continuous spike-waves during sleep. Review article. Hughes JR, et al. Epilepsy Behav. 2011.
9. Electrical Status Epilepticus in Sleep : Clinical Presentation and Pathophysiology Iván Sánchez Fernández MD a,b,1, Tobias Loddenkemper MD a,1, Jurriaan M. Peters MD a, Sanjeev V. Kothare MD.
10. Bureau M. Outstanding cases of CSWS and LKS: Analysis of the data sheets provided by the participants. In: Beaumanoir A, Bureau M, Deonna L, Mira L, Tassinari CA, editors. Con-tinuous spikes and waves during slow sleep. London: John Libbey; 1995. p. 213 e6.
11. Yan Liu X, Wong V. Spectrum of epileptic syn-dromes with electrical status epilepticus during sleep in children. Pediatr Neurol. 2000;22:371-9.
12. Massa R, de Saint-Martin A, Hirsch E, et al. Landau-Kleffner syndrome: Sleep EEG char-acteristics at onset. Clin Neurophysiol. 2000; 111(Suppl. 2):S87 e93.
13. Fejerman N, Caraballo R, Dalla Bernardina B. Evoluciones atípicas de las epilepsias focales benignas de la niñez. En Fejerman N, Caraballo R, eds.
14. Epilepsias focales benignas en lactantes, niños y adolescentes. Buenos Aires: Panamericana; 2008. p. 175-208.
15. Lúcia H. Coutinho dos Santos1, Sérgio A An-toniuk1, Marcelo Rodrigues2, Sílvio Bruno2, Isac Bruck1. Landau-Kleffner Syndrome: Study of four cases. Arq Neuropsiquiatr 2002; 60(2-A):239-241
16. Landau-Kleffner syndrome. Epileptic encepha-lopathies in infancy and early childhood. C. P. Panayiotopoulos.

51
17. Terminología y conceptos revisados para la Or-ganización de Crisis y Epilepsias: Informe de la Comisión de la ILAE sobre clasificación y ter-minología, 2005-2009 Anne T. Berg, Samuel F. Berkovic, Martin J. Brodie, Jeffrey Buchhalter, J. Helen Cross, Walter van Emde Boas, Jerome Engel, Jacqueline French, Tracy A. Glauser, Gary W. Mathern, Solomon L. Moshe, Douglas Nordli, Perrine Plouin, and Ingrid E. Scheffer.
18. Marie-Noëlle Metz-Lutz and Melissa Filippini. Neuropsychological Findings in Rolandic Epi-lepsy and Landau-Kleffner Syndrome Epilep-sia, 47(Suppl. 2):71–75, 2006.
19. Maquet P, Hirsch E, Metz-Lutz MN, et al. Re-gional cerebral glucose metabolism in children with deterioration of one or more cognitive functions and continuous spike-and-wave dis-charges during sleep. Brain 1995;118:1492-520.
20. The pathophysiological mechanisms of cog-nitive and behavioral disturbances in children with Landau-Kleffner syndrome or epilepsy with continuous spike-and-waves during slow-wave sleep Lotte Nieuwenhuis a,*,1 , Joost Nicolai b,1.
21. Robinson R, Baird G, Robinson G, Simonoff E. Landau-Kleffner Syndrome: course and cor-relates with outcome. Dev Med. Child Neurol 2001; 43:243-7.
22. Morrell F, Whisler WW, Smith MC, Hoepnner TJ, de ToledoMorrell L, Pierre-Louis SJ, et al. Landau-Kleffner syndrome: treatment with subpial intracortical transection. Brain 1995; 118:1529-46.
23. Metz-Lutz MN, de Saint Martin A, Hirsch E, et al. Auditory verbal processing following Lan-dau and Kleffner syndrome. Brain Lang 1996; 55:147-50.
24. Metz-Lutz MN, De Saint Martin A, Hirsch E, et al. Impairments in auditory verbal processing and dichotic listening after recovery of epilepsy in Landau and Kleffner syndrome. Brain Cogn 1999;40:193-7.
25. Aicardi J. El síndrome de Landau-Kleffner. Rev Neurol 1999;29:380-385.
26. Las evoluciones atípicas de la epilepsia rolándi-ca son complicaciones predecibles Gabriela
Pesántez-Ríos, Antonio Martínez-Bermejo, Joa-quín Arcas, Milagros Merino-Andreu, Arturo Ugalde-Canitrot.
27. Holmes GL, Lenck-Santini PP. Role of interictal epileptiform abnormalities in cognitive impair-ment. Epilepsy Behav 2006;8:504 e15.
28. Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci 2010;11:114 e26.
29. Loddenkemper T, Sánchez Fernández I, Peters JM. Continuous spike and waves during sleep and electrical status epilepticus in sleep. J Clin Neurophysiol 2011;28:154 e64.
30. Epileptic encephalopathies of the Landau-Klef-fner and continuous spike and waves during slow-wave sleep types: Genomic dissection makes the link with autism Gaetan Lesca, Gabri-elle Rudolf, Audrey Labalme, Edouard Hirsch, Alexis Arzimanoglou, Pierre Genton, Jacques Motte, Anne de Saint Martin, Maria-Paola Val-enti, Clotilde Boulay, Julitta De Bellescize, Pas-cale Ke´o-Kosal, Nadia Boutry-Kryza, Patrick Edery, Damien Sanlaville, and Pierre Szepe-towski.
31. Connolly AM, Chez MG, Pestronk A, Arnold ST, Mehta S, Deuel RK. Serum autoantibod-ies to brain in Landau-Kleffner variant, autism, and other neurologic disorders. J Pedriatr 1999; 134:607-13.
32. Loddenkemper T, Sánchez Fernández I, Peters JM. Continuous spike and waves during sleep and electrical status epilepticus in sleep. J Clin Neurophysiol 2011;28:154 e64.
33. Takeoka M, Riviello JJ Jr, Duffy FH, et al. Bilat-eral volume reduction of the superior temporal areas in Landau-Kleffner syndrome. Neurology 2004;63:1289 e92.
34. Intervención multidisciplinar en afasias. Raúl Villodre Campos y Amparo Morant Gimeno. Instituto de Neuro-rehabilitación y afasia, INIA NEURAL, Valencia.
35. Epileptic Disord 2012; 14 (1): 1-11 Therapy of encephalopathy with status epilepticus during sleep (ESES/CSWS syndrome): an update. Pierangelo Veggiotti, Maria Carmela Pera, Fed-erica Teutonico, Daniela Brazzo, Umberto Ba-lottin, Carlo Alberto Tassinari.
Síndrome de Landau-Kleffner XXxxxxx et al.

52
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Status súper-refractario del lóbulo temporal: ¿Existe una secuencia abreviada y bidireccional de cambios EEG? Análisis de más de 900 crisis durante casi un año.Cayetano [email protected]
Caso Clínico
ABSTRACT
Video EEG study in ICU for 10 months, a patient of 23 years with a super refractory bi temporal sta-tus, our aim was to: Identify the steps described by Treiman et al, see if there is a sequence and obser-ve the behavior of the stages identified through the months. We recorded more than 900 electro-clinical seizures and yet we could not identify all the steps described by Treiman. . If coinciding with Treiman et al, a sequence was identified, but abbreviated, the sequence begins with stage 1 to stage 2 then go directly to step 5. This sequence is also observed in reverse; from stage 5-1 and 2. We postulate that the abbreviated EEG sequence is determined by the ag-gressive and early treatment of status. Bidirectional evolution of EEG and the patient’s response when we finally stop thiopental make us raise the stage 5 is not the final stage of an epileptic status. The circuit described and tends to be self-perpetuating mechanism can be a super refractoriness.
Key Words: Super refractory status epilepticus, Vi-deo EEG monitoring in ICU, EEG stages.
RESUMEN Estudiamos con EEG video en UCI, durante 10 me-ses, a un paciente de 23 años con un status super refractario bi temporal, nuestro objetivo fue: iden-tificar las etapas descritas por Treiman et al., ver si existe una secuencia y observar el comportamiento de las etapas identificadas a través de los meses. Registramos más de 900 crisis electro-clínicas y a pesar de ello no pudimos identificar todas las etapas descritas por Treiman. Sí coincidiendo con Treiman et al., se identificó una secuencia, pero abreviada, la secuencia se inicia con la etapa 1 luego la etapa 2 para pasar directamente a la etapa 5. Esta secuen-cia se observó también en sentido inverso; desde
etapa 5 a 1 y 2. Postulamos que la secuencia EEG abreviada está determinada por la agresividad y precocidad del tratamiento del status. La evolución bidireccional del EEG y la respuesta del paciente cuando finalmente pudimos suspender la anestesia con tiopental nos hacen plantear que la etapa 5 no es la etapa final de un status epiléptico. El circuito que se describe y que tiende a auto-perpetuarse pue-de ser un mecanismo de super refractariedad. Palabras clave: Status epiléptico super refractario, Monitoreo EEG video en UCI, Etapas de Trieman.
La incorporación del EEG en el estudio de pacientes en UCI, especialmente los registros continuos, ha sido un aporte significativo, no sólo en el diagnós-tico de los status no convulsivos, sino que también en optimizar el manejo terapéutico; especialmente en los casos de status refractarios los cuales pueden prolongarse en el tiempo, con el uso de múltiples fármacos y la potencial falla de diferentes sistemas (Drislane et al., 2008). Treiman et al., en 1990, describen una secuencia de cambios EEG durante un status convulsivo gene-ralizado. También sugieren que la última etapa de su secuencia EEG (etapa 5) con descargas genera-lizadas epileptiformes periódicas (DGEP) tiene un peor pronóstico en relación a las etapas iniciales y que las DGEP representan una etapa tardía de un status epiléptico.
Si bien los estudios de status experimental en ani-males han corroborado los hallazgos de Treiman (Wang et al., 2009)(Gao et al., 2007), existe bastan-te controversia con respecto a estudios con pacien-tes en status convulsivo, en los cuales la existen-cia de las etapas EEG han sido negadas en algunos trabajos (Nei et al., 1999)(Lowenstein et al., 1992), poniendo también en duda que la etapa de DGEP sea realmente una etapa final de un status epiléptico (Garzón et al., 2001). Hospital Militar de Santiago
Recibido 30-7-15. Aceptado 12-8-15.

53
A propósito de un caso clínico con status super-refractario bitemporal, el cual seguimos con EEG video en UCI durante 10 meses, con registro de más de 900 crisis electro-clínicas revisamos sus registros EEG videos (EEGV), para responder los siguientes puntos:1. Es posible identificar las etapas descritas por Treiman et al., y si existe alguna secuencia evolu-tiva. y2. Qué ocurre con las etapas EEG con el paso de los meses.
CASO CLINICO
El paciente es un hombre de 23 años sin antece-dentes médicos previos, su afección se inicia 4 días antes del ingreso a nuestro hospital, con cefa-lea persistente asociada a cuadro febril seguido de crisis convulsivas generalizadas las que se repiten en varias oportunidades en las horas siguientes. Al ingreso se utiliza Fenitoina e.v, seguido de Valcote e.v, ante la persistencia de crisis convulsivas gene-ralizadas se transfiere a UCI iniciándose coma indu-cido con Midazolam e.v. luego asociado a Propofol e.v; el registro precoz con VEEG en UCI (al cuarto día de iniciado el cuadro) muestra persistencia de crisis las que se originan en forma independiente en ambos lóbulos temporales por lo cual se decide iniciar Tiopental, manejando las dosis hasta lograr paroxismo supresión, con lo cual se logra controlar las crisis, pero estas se reinician al intentar retirar-lo. El paciente recibió durante su hospitalización múltiples esquemas de anticonvulsivantes orales, sin control adecuado de sus crisis. En base a los estudios realizados (RNM cerebro seriadas, LCR, anticuerpos antineuronales, marcadores tumorales, estudio de cáncer etc) se plantea el diagnóstico de una Encefalitis límbica seronegativa motivo por lo cual se utilizó durante la evolución del cuadro tra-tamiento con solumedrol e.v., inmunoglobulinas y plasmaféresis, sin modificar significativamente el curso de su afección (detalles del estudio clínico y de laboratorio del paciente en Napolitano & Orriols, 2013).
Finalmente a los 270 días en UCI se logra retirar el tiopental, (previamente se había decidido no reini-ciarlo); el paciente sorprendentemente despierta y se relaciona con la familia y el terapeuta, se mantie-ne en esa condición 4-5 días para luego comprome-terse progresivamente de conciencia, con períodos de agitación psicomotora, alucinaciones visuales y
luego sopor profundo, con evidencia de sacudidas nistágmicas ocasionales y movimientos oculares episódicos y alejadas crisis convulsivas generaliza-das, sin que se registre en el EEG actividad ictal comicial evidente, sólo lentitud theta bilateral con ocasionales series lentas monomorfas bilaterales. Se sospecha persistencia de status límbico, logran-do finalmente, después de semanas, estabilizar al paciente, pero cuando logra vigilia se evidencia escaso contacto con el medio, emite sólo sonidos y con incapacidad de comprender el lenguaje ver-bal. El paciente fallece dos años después postrado, a consecuencia de una sepsis de origen óseo.
Realizamos registros diarios de VEEG en UCI using a 64-channel NicVue 2.6 Nicolet system (Ni-colet, Madison,WI). The recordings were not conti-nuous, lasting from 12 to 17 h per day (starting at approximately 3:00 p.m. and ending at approxima-tely 8:00 a.m. the following day). The international 10–20 electrode system was used without additio-nal electrodes. The data were digitalized at 200 Hz using 1-Hz low-frequency and 70-Hz high-frequen-cy filters. Other types of filters were occasionally used for better analysis of some seizures.
We used primarily longitudinal and transversal bi-polar montages to analyze the seizures. All of the recordings were analyzed visually (C.N.), and the electroclinical seizures were identified using the cri-teria described by Young et al. (Young et al., 1996).
En el material de VEEG se buscaron los siguientes patrones EEG:1) discrete seizures with interictal slowing2) merging seizures with waxing and waning am-
plitude3) continuous ictal activity4) continuous ictal activity punctuated by low vol-
taje flat periods 5) periodic epileptiform discharges on a flat bac-
kground (Treiman et al., 1990 ) y se analizó la existencia de alguna secuencia entre los patro-nes EEG descritos; comparando los hallazgos EEG observados al inicio del status en relación a lo observado después de meses de evolución.
DISCUSIÓN
Nuestro paciente tiene características particulares, presentó un status epiléptico, con una duración ex-cepcionalmente prolongada al cual le hicimos re-
Status súper-refractario del lóbulo temporal Cayetano Napolitano

54
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
gistros de VEEG en UCI, durante casi un año. Ha pesar que registramos en total más de 900 crisis electro-clínicas no pudimos observar todas las eta-pas ni la secuencia completa descrita por Treiman et al. (1990).
Si coincidiendo con Treiman et al. durante la evo-lución de nuestro caso se identifica una secuencia EEG, pero abreviada; se identifica la etapa 1 y 2, para luego evolucionar directamente a la etapa 5, de descargas generalizadas epileptiformes periódicas; una secuencia abreviada también ha sido descrita por otros autores (Nei et al., 1999). Esta secuencia a veces pudimos observarla en sentido inverso, es-tando con un patrón de DGEP y sin crisis evidentes intercalar crisis aisladas o que se agrupan, o sea pa-sar de un patrón 5 a un patrón 1 o 2. En el trabajo seminal de Treiman et al, la secuencia que el describe es unidireccional y el tratamiento con fármacos anticonvulsivantes intenta que no progrese a la etapa siguiente y desde el punto de vista EEG obtener un trazado post-ictal con lentitud y depresión de voltaje. En la secuencia EEG que estamos describiendo se combina una evolución unidireccional en un mo-mento de la secuencia (paso de etapa 1 a 2 y luego 5) para luego seguir con una etapa en que existe una evolución inversa (bidireccional) (paso de etapa 5 a 1 y luego 2). Esta secuencia pensamos (al igual que Akman & Riviello, 2011) lo más probable está determinada en la primera parte por efecto del trata-miento endovenoso y la segunda parte por disminu-ción de su efecto con ingreso y reingreso al círculo descrito. Por lo tanto, a diferencia de lo que describió Trei-man et al., la secuencia EEG que observamos no sólo es abreviada sino que también puede ser bidi-reccional. En este sentido Akman & Riviello ya ha-bían destacado lo dinámico de la secuencia EEG en su serie de niños con status refractario, y destacan que esos cambios son bidireccionales. A diferencia de lo observado por Akman & Riviello, en nuestro paciente observamos en ocasiones que al disminuir la anestesia endovenosa pueden reaparecer crisis aisladas o que se agrupan (etapa 1 y 2) sin pasar necesariamente por un patrón de descargas genera-lizadas periódicas (etapa 5).
El hecho que la secuencia de cambios EEG sea bi-
direccional plantea dudas sobre si verdaderamente la etapa de descargas generalizadas epileptiformes periódicas (etapa 5 de Treiman) es la etapa final de la evolución de un status convulsivo. Todo apunta a que este patrón EEG, en el contexto de un status refractario, indica una probabilidad cierta a man-tener crisis; y si no se logra cortar el círculo que se establece y se auto-perpetúa entre el patrón de descargas generalizadas epileptiformes periódicas y la reemergencia de crisis aisladas o que se van agrupando para volver nuevamente al patrón de descargas periódicas, el pronóstico ciertamente es ominoso, y este circuito que se auto perpetúa puede ser un mecanismo de super refractariedad (Fig.1). La presencia de crisis cíclicas electrográficas en pacientes con enfermedades críticas puede tener un mecanismo patofisiológico similar al descrito en nuestro paciente con la aparición de circuitos de re-entrada de crisis, lo cual está evidenciando fallas en los mecanismos de finalización de crisis (Friedman et al., 2008). La etapa 5 la observamos en repetidas oportunida-des durante la evolución del status de nuestro pa-ciente, a veces por periodos prolongados, con incre-mentos sin relación a crisis evidentes y en ocasiones asociada a crisis electro-clínicas espontáneas o bien inducidas por estímulos. La secuencia abreviada y bidireccional descrita la observamos al inicio del status y también después de meses de evolución. Tenemos la impresión que en nuestro paciente la presencia de un patrón de descargas generalizadas epileptiformes periódicas en el EEG no fue el fac-tor determinante para explicar el pronóstico omi-noso final. Cuando finalmente suspendimos el tio-pental, después de 9 meses en UCI; y se decide no reiniciarlo, el paciente permanece algunos días sin crisis, logra despertar, se relaciona con su familia y el terapeuta, para agravarse progresivamente al no lograrse un control de las crisis, determinando pos-teriormente un daño cerebral severo y permanente. El desenlace negativo nos parece está más bien re-lacionado con la etiología del status que impidió un control efectivo y precoz de las crisis. Existe cierta evidencia que sugiere que las DGEP en el EEG (etapa 5) corresponde a un patrón que puede ser inducido por diferentes causas: sepsis, status epiléptico, trastornos metabólicos o tóxicos, hipoxia cerebral etc., (Foreman et al., 2012) y por

55
lo tanto está indicando noxa cerebral, y en conse-cuencia si el agente o la causa se controla, el patrón EEG puede mostrar una evolución hacia un registro de menor disfunción cortical (regresión del daño) o a la inversa si la causa se agrava puede evolucio-nar hacia un patrón de mayor gravedad como puede ser depresión de voltaje global y finalmente trazado isoeléctrico (Pender & Losey, 2012). En el trabajo clásico de Treiman et al. (1990), ellos plantean que la secuencia de EEG descrita repre-senta la historia natural de la evolución EEG en status convulsivo generalizado no tratado, nosotros planteamos que la imposibilidad en diversas series y también en nuestro caso, de registrar la secuencia completa descrita por Treiman et al., tiene directa relación con la mayor precocidad y la agresividad del tratamiento actual del status convulsivo refrac-tario (Lowenstein et al., 1999) con el uso de fár-macos e.v. sucesivos y a veces asociados los cuales son capaces de frenar las crisis epilépticas aisladas o que se van repitiendo, y de esa manera eliminar las etapas siguientes (etapas 3 y 4 de Treiman et al.); pero si el proceso que generó el status refracta-rio es muy epileptogénico (como en nuestro caso) y ese potencial solo está “frenado”, la expresión EEG
siguiente es la aparición de un patrón generalizado epileptiforme periódico, el cual, como plantea Ak-man & Riviello, 2011, indica una situación de un continuum o reemergencia de un estado ictal activo desde una actividad de base frenada (estado inter-ictal), y en el contexto de un status epiléptico sólo presagia nuevas crisis, las cuales pueden reaparecer al disminuir los fármacos anticonvulsivantes o aún frente a estímulos diversos, lo cual observamos fre-cuentemente en nuestro paciente. Si nuestro planteamiento es acertado, es difícil que se puedan observar en series recientes la evolución completa EEG descrita por Treiman et al., y más bien apreciar una secuencia abreviada de cambios EEG. Pender & Losey, 2012, describen un caso excep-cional de status epiléptico con una secuencia EEG completa de Treiman et al., en una paciente que fa-llece en menos de 24 horas, lo cual en vez de contra-decir nuestro planteamiento lo apoya, tomando en cuenta que esa paciente en la práctica tuvo una evo-lución espontánea de su status, casi sin tratamiento. Es imposible saber si la secuencia EEG que des-
Figura 1.
Status súper-refractario del lóbulo temporal Cayetano Napolitano

56
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
cribimos es aplicable sólo a nuestro caso clínico, o pudiera ser observado en status refractarios que se prolongan en el tiempo o en status super-refracta-rios de otro origen.
REFERENCES
1. Akman CI and Riviello Jr. JJ. (2011) Gene-ralized Periodic Epileptiform Discharges in Critically ill Children: A Continuum of Status Epilepticus or an Epiphenomenon. J Clin Neu-rophysiol28:366-372
2. Drislane FW, Lopez MR, Blum AS and Scho-mer DL. (2008) Detection and Treatment of Re-fractory Status Epilepticus in the Intensive Care Unit. J Clin Neurophysiol 25:181-186.
3. Foreman B, Claassen J, Khaled KA, Jirsch J, Alschuler DM, Wittman J, Emerson RG, Hirsch LJ. (2012) Generalized periodic discharges in the critically ill: a case-control study of 200 pa-tients. Neurology 79(19):1951-1960.
4. Friedman DE, Shevon C, Emerson RG and Hirsch LJ. (2008) Cyclic electrografic seizures in critically ill patients. Epilepsia 49:281-287.
5. Gao XG, Liu Y, Liu XZ.( 2007) Treatment of late lithium-pilocarpine-induced status epilepti-cus with diazepam. Epilepsy Research 74:126-130.
6. Garzon E, Franca Fernandez RM and Sakamo-to AC. (2001) Serial EEG during human status epilepticus. Evidence for PLED as an ictal pat-
tern. Neurology 57:1175-1183.7. Young GB, Jordan KG, Gordon SD.(1996) An
assessment of nonconvulsive seizures in the in-tensive care unit using continuous EEG moni-toring: an investigation of variables associated with mortality. Neurology 47:83-89.
8. Lowenstein DH, Aminoff MJ. (1992) Clinical and EEG features of status epilepticus in coma-tose patients. Neurology 35:100-110.
9. Lowenstein DH, Bleck T, Macdonald RL. (1999) It”s time to revise the definition of status epilepticus. Epilepsia 40:120-122.
10. Napolitano CE and Orriols M. (2013) Changing patterns of propagation in a super-refractory status of the temporal lobe. Over 900 seizures recorded over nearly one year. Epilepsia & Be-havior Case Report 1:126-131.
11. Nei M, Lee JM, Shanker VL, Sperling MR. (1999) The EEG and prognosis in status epilep-ticus. Epilepsia 40:157-163.
12. Pender RA and Losey TE. (2012) A rapid cour-se through the five electrografic stages of status epilepticus. Epilepsia 53(11)e193-e195.
13. Treiman DM, Walton NY, Kendrick C. (1990) A progressive sequence of electroencephalogra-phic changes during generalized convulsive sta-tus epilepticus. Epilepsy Res 5:49-60.
14. Wang NC, Good LB, Marsh ST and Treiman DM. (2009) EEG stages predict treatment res-ponse in experimental status epilepticus. Epilep-sia 50:949-952.

57
Crónica
Memoria Anual del año 2014
REUNIONES DE TRABAJO
Realizadas los segundos sábados de cada mes du-rante el año 2014.
Reunión de Trabajo Nº 143Sábado 12 de Abril de 2014 a las 9.30 hrs.Síndrome Dress. A propósito de un Caso Clínico.Dra. Carolina GallegosNeuróloga Hospital de Carabineros
Reunión de Trabajo Nº 144Sábado 10 de Mayo de 2014 a las 9.30 hrs.Síndrome de Dravet.Dra. Joanna BoraxNeuróloga InfantilHospital de Carabineros
Reunión de Trabajo Nº 145Sábado 12 de Julio de 2014 a las 9.30 hrs.Presentación Caso Clínico: Status Convulsivo y Porfiria.Dr. Luis Espinoza MartínezNeurólogoClínica Bicentenario
Reunión de Trabajo Nº 146Sábado 09 de Agosto de 2014 a las 9.30 hrs.Cirugía de Epilepsia Lóbulo Temporal sin Le-sión, Presentación de un caso con Monitoreo In-vasivo.Dra. Karina Rosso A.Neurología InfantilPontificia Universidad Católica
Reunión de Trabajo Nº 147Sábado 13 de Septiembre de 2014 a las 9.30 hrs.Guías Prácticas de Epilepsia.Dra. Lilian Cuadra O.Ministerio de Salud
Reunión de Trabajo N°148Sábado 11 de Octubre a las 9.30 hrs.Cirugía de Epilepsia en Edad Pediátrica.Dr. Christian Cantillano M. Neurocirujano del Hospital Sótero del Río
Reunión de Trabajo N°149Sábado 08 de Noviembre a las 9.30 hrs.Epilepsia y Música.Dr. Claudia Riffo A.Neuróloga Infantil Pontificia Universidad Católica de Chile
Reunión de Trabajo N°150Sábado 13 de Diciembre a las 9.30 hrs.Epilepsia y Enfermedades Mitocondriales.Dr. Juan MoyaNeurólogo Infantil Hospital Luis Calvo Mackenna
Reunión de Trabajo N°151Sábado 10 de Enero de 2015.Actualización en el Tratamiento de Apnea del Sueño y Epilepsia.Dr. Mauricio BravoHospital Militar
Epilepsia Super Resistente. Tratamiento Com-pasivo y de Acompañamiento con Cannabis. Presentación de un Caso.Dr. Marcelo Devilat; Dra. Carla Manterola; Dr. Juan Moya.Servicio de Neurología y Psiquiatría Hospital Luis Calvo Mackenna.
Reunión de Trabajo N°152Asamblea General Ordinaria, Sábado 14 de Marzo 2015.Elección de Directorio.Conmemoración XVI años Sociedad de Epilep-tología de Chile.

58
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
ACTIVIDADES AÑO 2014
•XIVJornadasInvernalesdeEpilepsia20146 y 7 de Junio 2014Hotel Neruda, Santiago.
•VIIICongresoLatinoamericanodeEpilepsia.17 al 20 de Septiembre de 2014, Buenos Aires, Ar-gentina.
•Sonepsyn:LXIXCongresoChilenodeNeuro-logía, Psiquiatría y Neurocirugía9 al 11 de Octubre, Hotel Patagónico, Puerto Varas.
•SOPNIA:XXXIICongresoAnualdelaSocie-dad de Psiquiatría y Neurología de la Infancia y Adolescencia. “Al Sur del Mundo: Dilemas y De-safíos en Psiquiatría y Neurología”15,16 y 17 de Octubre de 2014, Hotel Dreams, Pun-ta Arenas.
•SociedaddeNuerofisiologíaClínica:XIIJorna-dasdeNeurofisiologíaSábado 13 de Diciembre 2014, Santiago, Chile.
REVISTA CHILENA DE EPILEPSIA
La Revista Chilena de Epilepsia, publicación ofi-cial de la Sociedad, cuenta con la Dra. Perla Da-vid como editora y la Dra. Ledia Troncoso y el Dr. Marcelo Devilat como coeditores. Este año se está editando el 15º volumen de esta revista. En él, se presentarán trabajos originales, casos clínicos, tra-bajos de revisión y actualizaciones. Se encuentra actualmente aceptada en SciELO con tres edicio-nes por año. La revista se encuentra online hace dos años, además en un sitio web propio: www.revista-chilenadeepilepsia.cl
PÁGINA WEB
Nuestra página Web, www.epilepsiadechile.com, está a disposición de los socios y del público en general. En ella encontramos información sobre la Sociedad y temas relacionados a la Epilepsia, en las siguientes secciones:1) Inicio2) Presentación de la Sociedad3) Actividades (Jornadas Invernales de Epilepsia,
Reuniones de Trabajo)4) Apuntes (Actualizaciones en epilepsia, Historia
de la epilepsia)
5) Publicaciones (Revista Chilena de Epilepsia y Normas Técnicas)
6) Noticias7) Directorio8) Socios9) Vínculos web de relevancia (incluyendo ILAE;
IBE; WHO y MINSAL)10) Contacto
El sitio Web ha cumplido una importante labor, principalmente para estudiantes universitarios y profesionales; éstos son los usuarios más frecuen-tes, solicitando referencias e información acerca de las epilepsias. Con el espíritu de expandir los co-nocimientos en relación a la epilepsia, se decidió incluir, previa autorización de los autores, presen-taciones realizadas tanto en las Jornadas Invernales como en las Reuniones de Trabajo. Además, existe una importante consulta de familiares de pacientes con epilepsia, con el consiguiente impacto en la di-fusión de información hacia este grupo.
CONTACTOS NACIONALES
La Sociedad mantiene contactos con la Sociedad de Neurología, Psiquiatría y Neurocirugía, con la Sociedad de Psiquiatría y Neurología de la Infancia y Adolescencia, la Sociedad Chilena de Pediatría y la Asociación de Ligas contra la Epilepsia de Chile (ANLICHE).
Miembros de la Sociedad han participado en el Gru-po Normativo de Epilepsia, del Ministerio de Salud de Chile y en la elaboración de la guía práctica clí-nica en epilepsia. Así mismo, miembros de nuestra entidad participaron en el XXXII Congreso Anual de la SOPNIA.
CONTACTOS INTERNACIONALES
La Sociedad es miembro de la Comisión de Asuntos Latinoamericanos de la Liga Internacional contra la Epilepsia.
CELEBRACIÓN DEL DÍA LATINOAMERICA-NO DE LA EPILEPSIA
El día 9 de Septiembre se celebró el Día Latinoame-ricano de la Epilepsia. Uno de los eventos que cele-bran esta fecha, es el organizado por la Sociedad de Epileptología de Chile, que contó con la presencia del

59
Memoria Anual del año 2014 Crónica
Dr. Cayetano Napolitano, presidente de la Sociedad de Epileptología de Chile, en la sede de la sociedad.
REVISTA EPILEPSIA La Revista Epilepsia, publicación oficial de la ILAE, se recibe desde 1999 hasta la fecha, y se en-cuentra a disposición de los socios en la sede de la Sociedad de Epileptología de Chile, además de en-contrarse online.
SECRETARÍA
Actualmente es la Sra. Luisa Esparza con un nue-vo horario, los días lunes, miércoles y viernes, de 18.30 a 20.30 hrs. fono: 02-22310172 Fax: 02-22340671, e-mail [email protected]
NUEVOS SOCIOS AÑO 2014 El año 2014 se incorporaron como nuevos socios:
Dra. Carolina Gallegos (Abril 2014)Dra. Joanna Borax (Mayo 2014)Dra. Carla Manterola (Junio 2014)Dr. Luis Espinoza (Julio 2014)Dra. Karina Rosso (Agosto 2014)Dr. Christian Cantillano (Octubre 2014)Dra. Claudia Riffo (Noviembre 2014)Dr. Reinaldo Poblete (Julio 2015)Dr. Claudio Lühr (Agosto 2015)
BIBLIOTECA
La biblioteca, ubicada en la sede de la Sociedad, re-cibe la Revista Epilepsia y la edición de la Revista Chilena de Epilepsia, además de otras publicacio-nes que se encuentran a disposición de los socios.
ACREDITACIÓN 2015
Les recordamos que el directorio está recibiendo los antecedentes de los socios para la acreditación de la Sociedad de Epileptología de Chile, de acuerdo a su participación en actividades de la Sociedad, en libros y revistas científicas, en actividades interna-cionales y electrónicas. Se adjunta formulario y re-glamento en curso.
ACTIVIDADES PARA EL 2015
•XVJornadasInvernalesdeEpilepsiaLos días 05 y 06 de Junio de 2015, se realizarán las XV Jornadas Invernales de Epilepsia, tituladas: “Epilepsia desde una Mirada Moderna”. El Comi-té Organizador, invita a participar a los interesados, mediante trabajos originales, que pueden ser envia-dos al sitio Web de la Sociedad de Epileptología de Chile.
AGRADECIMIENTOS
El directorio agradece a la Industria Farmacéutica y Tecnológica la colaboración que han realizado a la Sociedad durante el año 2014: Abbott Laboratories de Chile, Laboratorio Drugtech de la Corporación Farmacéutica Recalcine, Laboratorios Tecnofarma, Laboratorios Saval, GlaxoSmithKline y ATI Bios-can/Chile.
Dra. Carla ManterolaSecretaria General
Dr. Cayetano NapolitanoPresidente

60
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Jornadas Invernales de Epilepsia. Santiago, Chile, 05 y 06 de Junio de 2015.“Epilepsia desde una mirada moderna”
Crónica
VIERNES 05 JUNIO 2015
08:15-08:30 Introducción. Dra. Ledia Troncoso.
Módulo I: Epileptogénesis: desde la neurocien-cia a la clínica.
08:30-09:00 Epigenética-optogenética. Dr. Andrés Barrios.
09:00-09:30 Nuevos síndromes epilépticos, nue-vos genes, canalopatías.
Dr. Alvaro Retamales.
09:30-10:00 En la búsqueda etiológica: afecciones heredometabólicas y genéticas con epilepsia como síntoma marcador.
Dr. Felipe Castro.
10:00-10:30 Café
Módulo II: Visión actualizada en el estudio de las epilepsias.
10:30-11:00 Electrofisiología. Dr. Roberto Caraballo.
11:00-11:30 Neuroimagenología. Dr. Salvador Camelio.
11:30-12:00 Mesa redonda - módulos 1-11. Preside: Dr. Roberto Caraballo. Participan: Expositores de ambos
módulos.
13:00-14:00 Simposio almuerzo Abbott. Neuroes-timulación: estimulador del Nervio Vago, estimulador de núcleo anterior talámico, hipotermia cerebral, perfu-sión de drogas localizadas, estimula-ción óptica Cortex cerebral.
Dra. Loreto Ríos, Dr. Hernán Aceve-do.
14:00-15:30 Presentación de pósters. Dirigen: Dra. Maritza Carvajal, Dr.
Enzo Rivera, Dra. Perla David, Dra. Viviana Venegas.
Módulo III: Manejo moderno de las epilepsias. Simposio Abbott.
15:30-16:00 Monoterapia en fármacos antiepilép-ticos de segunda generación.
Dra. Daniela Triviño.
16:00-16:30 Politerapia: fármacos antiepilépticos y uso racional.
Dra. Daniela Aguilera.
16.30-17:00 Fármacos antiepilépticos de tercera generación: Lacosamida, Rufinami-da, Ezogabina, Retigabina, Perampa-nel.
Dr. Roberto Caraballo.
17:00-17:30 Café
17:30-18:00 Terapia con inmunomoduladores. Dr. Reinaldo Uribe.
18:00-18:30 Real utilidad de antiepilépticos vía parenteral (endov.) vs vía oral / Dieta cetogénica.
Dr. Juan Moya.
18.30-19.45 Una mirada moderna del manejo te-rapéutico del paciente con epilepsia y trastornos psiquiátricos.
Dr. Jaime Godoy / Dr. Fernando Iva-novic-Zuvic.

61
SÁBADO 06 DE JUNIO 2015
Módulo III (continuación): Cirugía de la Epi-lepsia.
09:00-09:30 Estimulación cerebral profunda. Dr. David Aguirre.
09:30-10:00 Gammaknife en epilepsia. Dr. Claudio Lühr.
10:00-10:30 Cirugía no lesional. Dr. Christian Cantillano. 10:30-11:00 Mesa redonda. Preside: Dr. Arturo Zuleta. Participan: Dr. Manuel Campos, Dr.
Claudio Lühr, Dr. Christian Cantilla-no, Dr. David Aguirre.
Módulo IV: Una mirada moderna desde la clí-nica.
11:00-11:30 Convulsiones neonatales: diagnósti-co y manejo actualizado.
Dra. Karla Henríquez.
11:30-12:00 Esclerosis tuberosa: un modelo de epilepsia refractaria.
Dra. María Francisca López.
12:30-13:00 Estado epiléptico super-refractario. Dr. Cayetano Napolitano.
13:00-13:15 Cierre. Comité organizador XV Jornadas In-
vernales de Epilepsia.
Jornadas Invernales de Epilepsia 2015 Crónica

62
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Cursos, Congresos y Actividades 2015Crónica
SOCIEDAD DE EPILEPTOLOGÍA DE CHILE.5 y 6 de Junio 2015XV Jornadas Invernales de Epilepsia, “Tratamiento Modernos de las Epilepsias”.Hotel Neruda, Santiago.
SONEPSYN: LXIX CONGRESO CHILENO DE NEUROLOGÍA, PSIQUIATRIA Y NEUROCIRU-GIA.9 al 11 de OctubreHotel Patagónico, Puerto Varas.
SOPNIA: CONGRESO ANUAL DE LA SOCIE-DAD DE PSIQUIATRIA Y NEUROLOGÍA DE LA INFANCIA Y ADOLESCENCIA.11, 12 y 13 de Noviembre de 2015,La Serena.
SOCIEDAD DE NUEROFISIOLOGÍA CLÍNICA XIII Jornadas de Neurofisiología,Sábado 12 de Diciembre 2015En Auditorio Clínica Dávila, Santiago.
FECHAS MÁS IMPORTANTES DE CONGRE-SOS INTERNACIONALES.
• 31º Congreso Internacional de Epilepsia de Es-tambul, Turquía.5-9 Septiembre 2015.13-16 mayo, 2016.
• 11º Congreso de Epilepsia de Asia y Oceanía.Hong KongSitio web: www.epilepsyhongkong2016.org
• 12º Congreso Europeo de Epilepsia.11-15 septiembre, 2016.Centro de Congresos de Praga, República ChecaSitio web: www.epilepsyprague2016.orgConvocatoria de la CEPE ForosFecha límite de presentación 25 de agosto.
• Sexto Congreso de la Asociación China contra la Epilepsia (CAAE).29 octubre-1 noviembre 2015Foro Internacional de la EpilepsiaShanghai, China.Interactúa con más de 1.000 neurólogos y epilep-tólogos de China y asistir a programas científicos sobre la epilepsia.Información: [email protected]
• 69a Reunión Anual de la Sociedad Americana de Epilepsia4 al 8 diciembre, 2015Pennslyvania Convention Center, Filadelfia, PA
• 53o Congreso Anual de la Liga Alemana contra la Epilepsia.Marzo 3 al 5, 2016Jena, AlemaniaMás: www.epilepsie2016.de
• Congreso Epilepsia brasileña9 - 11 de junio de 2016Recife, Brasil.
CALENDARIO DE REUNIONES DE TRABAJO SOCIEDAD DE EPILEPTOLOGÍA DE CHILE AÑO 2015: SEDE SOCIEDAD DE EPILEPTO-LOGIA
• REUNIÓN DE TRABAJO N°151.Sábado 10 de Enero.- Actualización en tratamiento de Apnea de Sueño
y Epilepsia. Dr. Mauricio Bravo.
- Epilepsia super resistente. Tratamiento compasi-vo y de acompañamiento con cannabis. Presenta-ción de un caso.
Dr. Marcelo Devilat, Dra. Carla Manterola, Dr. Juan Moya.

63
REUNIÓN DE TRABAJO N°152.Sábado 14 de Marzo.Asamblea General Ordinaria y Elección Nuevo Di-rectorio.
REUNION DE TRABAJO Nº 153.Sábado 11 de Abril.“Epilepsia en Pacientes Mayores de 15 años con Síndrome de Down”.Dr. Darío Ramírez, Hospital Salvador.
REUNION DE TRABAJO Nº 154.Sábado 9 de Mayo.Revisión en Panayotopolous.Dra. Patricia AlfaroNeuróloga InfantilHospital San Juan de Dios.
REUNION DE TRABAJO Nº 155.Sábado 11 de Julio.“Status Súper Refractario, Cuánto Esperar? Alter-nativa Quirúrgica”.Dr. Hernán Acevedo
REUNION DE TRABAJO Nº 156.Sábado 08 de Agosto.Asociación entre Síndrome de Encefalopatía Poste-rior Reversible (PRES), Crisis Convulsivas y Epi-lepsia. Descripción de un Caso Clínico.Dra. Kathleen Batalla FalconServicio de Urgencia, Hospital Padre Hurtado.
REUNION DE TRABAJO N°156.Sábado 12 de SeptiembreInmunidad y EpilepsiaDr. Reinaldo Uribe
REUNION DE TRABAJO N°157.Sábado 17 de Octubre.Gamma Niffe y EpilepsiaDr. Claudio Lühr
REUNION DE TRABAJO N°158.Sábado 14 de Noviembre
REUNION DE TRABAJO N°159.Sábado 12 de Diciembre
Cursos, Congresos y Actividades 2015 Crónica

64
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
Declaración de Interés
Crónica
SUBSECRETARÍA DE SALUD PÚBLICADIVISIÓN DE PREVENCIÓN Y CONTROL DE
ENFERMEDADES
Las consideraciones de salud pública tienen una im-portancia primordial en todo el trabajo técnico del Ministerio de Salud. Es preciso que se adopten me-didas para garantizar que se efectúe la mejor eva-luación posible de los datos científicos, en una at-mósfera independiente exenta de presiones directas o indirectas. Por lo tanto, para preservar la integri-dad técnica y la imparcialidad del trabajo del Minis-terio de Salud, es necesario prevenir situaciones en las cuales el resultado de ese trabajo pudiera verse afectado por intereses financieros o de otra índole.
Por consiguiente, se pide a cada experto(a) que de-clare si es parte interesada en algo que, en lo refe-rente a su participación en el trabajo que realiza en el Ministerio de Salud, podría dar lugar a un con-flicto real, potencial o aparente de intereses entre (1) entidades comerciales y el participante perso-nalmente o (2) entidades comerciales y la unidad administrativa para la cual trabaja el participante. Por “entidad comercial” se entiende cualquier em-presa, asociación, organización u otra entidad, sea cual fuere su naturaleza, que tenga intereses comer-ciales.
¿Quéesunconflictodeintereses?
Hay conflicto de intereses si:
1. El experto(a) o su pareja (“por pareja” se entien-de un cónyuge u otra persona con la cual el experto mantiene una estrecha relación personal de natura-leza semejante), o la unidad administrativa para la cual trabaja el experto, tienen un interés financiero o de otra índole que podría afectar indebidamente a la posición del experto, en lo concerniente al asunto que se está considerando.2. Hay conflicto aparente de intereses cuando un in-
terés, que no necesariamente influiría en el experto, podría dar lugar a que otros cuestionasen la objeti-vidad de éste.
3. Existe un conflicto potencial de intereses toda vez que una persona razonable se pregunta si debe o no informar acerca de un interés.
Se puede prever diferentes tipos de intereses finan-cieros o de otra índole, bien sea personal o rela-cionado con la unidad administrativa para la cual trabaja el experto, y la siguiente lista, que no es ex-haustiva, puede servir de orientación. Por ejemplo, deben declararse los siguientes tipos de situaciones:
a. toda participación patrimonial vigente en una sustancia, una tecnología o un proceso (por ejemplo la propiedad de una patente), que se examinarán en la reunión o en el trabajo o que están relacionados de otra manera con el tema correspondiente;
b. todo interés financiero vigente, por ejemplo la posesión de valores bursátiles tales como acciones u otros títulos de una entidad comercial que sea par-te interesada en el asunto por examinar en la reu-nión o el trabajo (Ej.: Industria Farmacéutica);
c. todo empleo, consultoría, cargo de dirección u otra posición, remunerados o no, en el curso de los 4 años precedentes en cualquier entidad comercial que sea parte interesada en el tema de la reunión/trabajo, o una negociación en curso sobre un posi-ble empleo u otra asociación con una entidad co-mercial semejante;
d. todo trabajo o investigación remunerados realiza-dos en el curso de los 4 últimos años por encargo de una entidad comercial que sea parte interesada en el tema de tas reuniones o del trabajo;
e. todo pago u otra forma de apoyo recibidos en el curso de los 4 últimos años, o cualquier expectativa

65
de apoyo futuro de una entidad comercial que sea parte interesada en el tema de las reuniones o del trabajo, aunque no beneficie al experto personal-mente sino a su puesto o a la Unidad Administrativa para la cual trabaja el experto, por ejemplo una sub-vención, una beca u otro tipo de pago, por ejemplo para financiar un puesto o una consultoría,
En relación con lo anterior, se debe declarar igual-mente si uno es parte interesada en una sustancia, una tecnología o un proceso competidores, o en al-gún trabajo realizado para, en asociación con o con apoyo de una entidad comercial que tenga un inte-rés competidor directo.
Cómo se rellena esta declaración:
Debe declarar cualquier interés financiero o de otra índole que pudiera dar lugar a situaciones de con-flicto real, potencial o aparente de intereses:
1) En relación con usted mismo o su pareja, así como2) en relación con la unidad administrativa para la cual trabaja usted.
Debe revelar solamente el nombre de la entidad co-mercial y la naturaleza del interés; no es necesario especificar ninguna cantidad (aunque usted lo pue-de hacer si considera que esa información es perti-nente para evaluar el interés). En lo concerniente a los puntos 1 y 2 de la lista precedente, el interés sólo se debe declarar si es vigente. Con respecto a los puntos 3, 4 y 5, se debe declarar cualquier interés existente en el curso de los 4 últimos años. Si el interés ya no es vigente, sírvase declarar el año en que dejó de serlo.
Declaración:
¿Tiene usted o tiene su pareja un interés financiero o de otra índole en el tema de la reunión o en el traba-jo en el cual usted participará, y puede considerarse que ello dará lugar a un conflicto real, potencial o aparente de intereses?
Sí: No: En caso afirmativo, sírvase especificar.____________________________________________________________________________________________________________________________________________________________________
___________________________________________________________________________________________________________________________
Por favor responda las siguientes preguntas, en rela-ción a los últimos 24 meses:
¿Ha recibido usted honorarios por dictar conferen-cias?
Sí: No:En caso afirmativo, sírvase especificar._______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
¿Ha recibido apoyos económicos e invitaciones para asistir a congresos y otras actividades cien-tíficas?
Sí: No:En caso afirmativo, sírvase especificar._______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
¿Ha recibido fondos para realizar investigaciones?
Sí: No:En caso afirmativo, sírvase especificar._______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
¿Ha recibido honorarios por consultorías?
Sí: No: En caso afirmativo, sírvase especificar.__________________________________________________________________________________
Declaración de Interés Crónica

66
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
_____________________________________________________________________________________________________________________________________________________________________________________________________________
¿Hay algo más que podría afectar a su objetividad o independencia o en el trabajo que Ud. Realiza en el Ministerio de Salud, o la impresión que otros podrían tener de la objetividad e independencia de usted?_______________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
Por la presente, declaro que la información revelada es correcta y que no tengo conocimiento de ninguna otra situación de conflicto real, potencial o aparente de intereses. Me comprometo a notificar cualquier cambio al respecto, o incluso si se plantea una cues-
tión pertinente durante el curso mismo del trabajo que realizó en el Ministerio de Salud.
Nombre:__________________________________________________________________________________
Especialidad:__________________________________________________________________________________
Institución:__________________________________________________________________________________
Firma:
_________________________________________
Fecha:_________________________________________

67
Sugerencias para las contribuciones a los autores
Las contribuciones podrán tener la forma de traba-jos originales de investigación clínica o experimen-tal, de medicina social y salud pública relacionadas con las epilepsias, revisiones de temas, casos clíni-cos, crónica y cartas al editor.
Las colaboraciones deberán ser enviadas a la se-cretaría de la Sociedad de Epileptología de Chile y revisadas por el Comité Editorial.
Los artículos se entregarán mecanografiados en pa-pel tamaño carta con doble espacio, con un máximo de 26 líneas por página, con un margen de 2.5 cm en todos sus bordes, escritos con letra Arial nivel 12. La extensión máxima para los artículos originales y de revisión será de 16 páginas, de 8 para los casos clínicos y de 3 para los artículos de crónica y cartas al editor. Se incluirá un original con dos fotocopias y un archivo en CD utilizando programa Word para PC.
Se aceptarán figuras (dibujos y gráficos) enviados en forma de copia fotográfica en papel satinado blanco y negro de 10 x 15 cm. La lectura de las fi-guras se hará en hoja separada. En el dorso de cada figura se marcará el número que la identifica y una flecha con su orientación con lápiz de carbón. En el texto se indicará dónde debe ser intercalada.
Las tablas (cuadros o tablas) se enviarán mecano-grafiados y numerados según orden de aparición en el texto, en el cual se señalará su ubicación.
Se aceptará un máximo de 5 elementos (figuras o tablas) por artículo.
El título deberá ser claro y conciso. Se incluirá el nombre de los autores con el primer apellido, el títu-lo profesional de cada uno de ellos y el lugar donde se realizó el trabajo. Las referencias bibliográficas deben limitarse a un máximo de 15. Se sugiere re-ferir y citar bibliografía latinoamericana y chilena y
al terminar mencionar el e-mail del autor principal.
Clasificacióndelascontribuciones:1. Trabajo original. Realizado según el siguiente
esquema: a) Introducción, donde se plantea la situación
general del problema; b) Objetivos, donde se plantean los antecedentes y los problemas que se quiere resolver; c) Material o Pacientes y Mé-todos, en el que se hacen explícitas las caracte-rísticas del universo y cómo se instrumentalizó; d) Resultados, donde se expone la situación ob-tenida; e)Discusión, en la que se comentan los resultados con relación a los problemas plantea-dos o a la información proporcionada por otros autores; f) Resumen de 200 palabras en español e inglés.
2. Trabajos de revisión. Se trata de una revisión bibliográfica acerca de un tema específico, pre-sentado según las instrucciones de longitud y referencias bibliográficas ya señaladas.
3. Casos clínicos. Presentación de casos de interés práctico, según el esquema de trabajo original.
4. Actualidades. Revisión de capítulos de interés especial, realizadas por profesionales que ten-gan experiencia en el tema y contribuyan a cla-rificar conceptos.
5. Crónica. Espacio destinado a noticias de interés en el campo de la clínica, neurofisiología, imá-genes, Salud Pública o administración. Presen-tación según instrucciones detalladas más arri-ba.
6. Cartas al editor, cuyo objetivo es ser una tribuna abierta de la Revista a sus lectores.
7. Enviar resumen en español e inglés.
Crónica

68
Revista Chilena de Epilepsia Año 15, Nº 2, Agosto 2015
8. Debe consignar fecha de envío del trabajo ya que será recibida y enviada a dos revisores ex-pertos anónimos, para revisión aprobación y/o rechazo o modificación.
9. Se debe declarar conflictos de intereses de los autores.
PresentacióndelasreferenciasbibliográficasDeben enumerarse en el texto en forma consecuti-va, en el mismo orden en que aparecen citadas por primera vez, y acompañarse la lista total de ellas.
En caso de haber más de 5 autores, se colocará la palabra “et al” para incluir los restantes. Cada refe-rencia de revista debe anotarse en el orden siguien-te: Apellido paterno del autor con la primera inicial del nombre, título del trabajo, revista en que apa-rece el artículo según “Index Medicus”, año, volu-men, página inicial y final del texto. Las referencias de libros se anotarán así: título del libro, ciudad en que fue publicado, editorial, año. Se usarán comas para separar a los autores entre si. Ejemplos: Pérez J, Santos G. Serotonina humana. Rev Med Chile 1967;45:12-14.